
PI 3-kinase p110β: a new target for antithrombotic therapy
8
NATURE MEDICINE VOLUME 11 | NUMBER 5 | MAY 2005 507 PI 3-kinase p110β: a new target for antithrombotic therapy Shaun P Jackson 1 , Simone M Schoenwaelder 1,5 , Isaac Goncalves 1,5 , Warwick S Nesbitt 1 , Cindy L Yap 1 , Christine E Wright 2 , Vijaya Kenche 1,4 , Karen E Anderson 1 , Sacha M Dopheide 1 , Yuping Yuan 1 , Sharelle A Sturgeon 2,4 , Hishani Prabaharan 1,4 , Philip E Thompson 1,4 , Gregg D Smith 1,4 , Peter R Shepherd 3 , Nathalie Daniele 3 , Suhasini Kulkarni 1 , Belinda Abbott 1 , Dilek Saylik 1,4 , Catherine Jones 2,4 , Lucy Lu 2,4 , Simon Giuliano 1 , Sascha C Hughan 1 , James A Angus 2 , Alan D Robertson 4,5 & Hatem H Salem 1,5 Platelet activation at sites of vascular injury is essential for the arrest of bleeding; however, excessive platelet accumulation at regions of atherosclerotic plaque rupture can result in the development of arterial thrombi, precipitating diseases such as acute myocardial infarction and ischemic stroke. Rheological disturbances (high shear stress) have an important role in promoting arterial thrombosis by enhancing the adhesive and signaling function of platelet integrin α IIb β 3 (GPIIb-IIIa). In this study we have defined a key role for the Type Ia phosphoinositide 3-kinase (PI3K) p110β isoform in regulating the formation and stability of integrin α IIb β 3 adhesion bonds, necessary for shear activation of platelets. Isoform-selective PI3K p110β inhibitors have been developed which prevent formation of stable integrin α IIb β 3 adhesion contacts, leading to defective platelet thrombus formation. In vivo, these inhibitors eliminate occlusive thrombus formation but do not prolong bleeding time. These studies define PI3K p110β as an important new target for antithrombotic therapy. Acute myocardial infarction and stroke represent the leading causes of death and disability in the industrialized world. A common precipi- tating factor for these diseases is the sudden rupturing or fissuring of advanced atherosclerotic lesions, leading to excessive platelet and fibrin accumulation. A number of factors contribute to the heightened throm- bogenic potential of ruptured plaques, including the high reactivity of adhesive substrates in the plaque 1,2 , the presence of tissue factor 3–5 and the indirect platelet-activating effects of high shear caused by narrowing of the vessel lumen by the atherothrombotic process 6–8 . Reductions in the luminal diameter of blood vessels, owing to the combined effects of atherosclerosis, vasoconstriction and thrombus development, result in a marked increase in blood flow velocity and a concomitant increase in shear stress. High shear stress and shear gradients are characteristic features of arterial thrombosis and analysis of thrombus formation in vivo has shown a major role for rapid increases in shear in promoting platelet activation and thrombus growth 9 . Platelet thrombus formation is crucially dependent on the adhesive and signaling functions of integrin α IIb β 3 (platelet GPIIb-IIIa), a major platelet integrin that mediates platelet–vessel wall and platelet-plate- let adhesive interactions through engagement of multiple adhesive ligands, including von Willebrand factor (vWf), fibrinogen, fibronec- tin and soluble CD40L 10 . All existing antiplatelet agents target one or more key steps in the thrombotic mechanism, which ultimately downregulate the adhesive function of integrin α IIb β 3 ; however, a con- siderable limitation of current therapies is their deleterious effects on hemostasis, leading to bleeding complications 11 . An ideal antithrom- botic therapy would selectively target an event crucial for pathological thrombus formation without affecting hemostasis. One such strategy would be to attenuate the ability of platelets to form stable integrin α IIb β 3 adhesion contacts under high shear stress by targeting shear- sensitive signaling processes in platelets. To date, the identification of signaling elements crucial for shear activation of platelets, but not for hemostasis, have remained elusive. An important signaling pathway promoting shear activation of plate- lets involves the PI3Ks 12,13 . These lipid kinases are divided into three distinct classes (Class I, II and III) based on their primary structure, mode of regulation and substrate specificity 14,15 . The Class I PI3Ks are responsible for agonist-induced production of the second messengers phosphoinositide (PI) (3,4,5)P 3 and PI(3,4)P 2 , and participate in the regulation of a broad range of functional platelet responses, including activation of integrin α IIb β 3 16 . The Class I enzymes are further divided into α, β, δ and γ isoforms based on their distinct p110 catalytic sub- units and modes of regulation. The Class Ia isoforms (p110α, p110β, p110δ) are classically regulated by tyrosine kinases, whereas the Class Ib isoform (p110γ) is typically activated by G-protein-coupled recep- tors 17 . Platelets contain all Class I PI3K isoforms, although the levels of 1 Australian Centre for Blood Diseases, Monash University, 6 th Floor Burnet Building Alfred Medical Research and Education Precinct (AMREP), 89 Commercial Road, Prahran, Victoria, Australia 3181. 2 Department of Pharmacology, Level 8, Medical Building, Corner of Grattan Street and Royal Parade, University of Melbourne, Victoria, Australia 3010. 3 Department of Biochemistry and Molecular Biology, University College of London, Gower Street, London, WC1E6BT, UK. 4 Kinacia Pty Ltd, Cerylid Biosciences Ltd, 576 Swan Street, Richmond, Victoria, Australia 3121. 5 These authors contributed equally to this work. ADR and HHS are equal senior authors. Correspondence should be addressed to S.P.J. ([email protected]). Published online 17 April 2005; doi:10.1038/nm1232 ARTICLES © 2005 Nature Publishing Group http://www.nature.com/naturemedicine
Transcript of PI 3-kinase p110β: a new target for antithrombotic therapy

NATURE MEDICINE VOLUME 11 | NUMBER 5 | MAY 2005 507
PI 3-kinase p110β: a new target for antithrombotic therapyShaun P Jackson1, Simone M Schoenwaelder1,5, Isaac Goncalves1,5, Warwick S Nesbitt1, Cindy L Yap1, Christine E Wright2, Vijaya Kenche1,4, Karen E Anderson1, Sacha M Dopheide1, Yuping Yuan1, Sharelle A Sturgeon2,4, Hishani Prabaharan1,4, Philip E Thompson1,4, Gregg D Smith1,4, Peter R Shepherd3, Nathalie Daniele3, Suhasini Kulkarni1, Belinda Abbott1, Dilek Saylik1,4, Catherine Jones2,4, Lucy Lu2,4, Simon Giuliano1, Sascha C Hughan1, James A Angus2, Alan D Robertson4,5 & Hatem H Salem1,5
Platelet activation at sites of vascular injury is essential for the arrest of bleeding; however, excessive platelet accumulation at regions of atherosclerotic plaque rupture can result in the development of arterial thrombi, precipitating diseases such as acute myocardial infarction and ischemic stroke. Rheological disturbances (high shear stress) have an important role in promoting arterial thrombosis by enhancing the adhesive and signaling function of platelet integrin αIIbβ3 (GPIIb-IIIa). In this study we have defined a key role for the Type Ia phosphoinositide 3-kinase (PI3K) p110β isoform in regulating the formation and stability of integrin αIIbβ3 adhesion bonds, necessary for shear activation of platelets. Isoform-selective PI3K p110β inhibitors have been developed which prevent formation of stable integrin αIIbβ3 adhesion contacts, leading to defective platelet thrombus formation. In vivo, these inhibitors eliminate occlusive thrombus formation but do not prolong bleeding time. These studies define PI3K p110β as an important new target for antithrombotic therapy.
Acute myocardial infarction and stroke represent the leading causes of death and disability in the industrialized world. A common precipi-tating factor for these diseases is the sudden rupturing or fissuring of advanced atherosclerotic lesions, leading to excessive platelet and fibrin accumulation. A number of factors contribute to the heightened throm-bogenic potential of ruptured plaques, including the high reactivity of adhesive substrates in the plaque1,2, the presence of tissue factor3–5 and the indirect platelet-activating effects of high shear caused by narrowing of the vessel lumen by the atherothrombotic process6–8. Reductions in the luminal diameter of blood vessels, owing to the combined effects of atherosclerosis, vasoconstriction and thrombus development, result in a marked increase in blood flow velocity and a concomitant increase in shear stress. High shear stress and shear gradients are characteristic features of arterial thrombosis and analysis of thrombus formation in vivo has shown a major role for rapid increases in shear in promoting platelet activation and thrombus growth9.
Platelet thrombus formation is crucially dependent on the adhesive and signaling functions of integrin αIIbβ3 (platelet GPIIb-IIIa), a major platelet integrin that mediates platelet–vessel wall and platelet-plate-let adhesive interactions through engagement of multiple adhesive ligands, including von Willebrand factor (vWf), fibrinogen, fibronec-tin and soluble CD40L10. All existing antiplatelet agents target one or more key steps in the thrombotic mechanism, which ultimately
downregulate the adhesive function of integrin αIIbβ3; however, a con-siderable limitation of current therapies is their deleterious effects on hemostasis, leading to bleeding complications11. An ideal antithrom-botic therapy would selectively target an event crucial for pathological thrombus formation without affecting hemostasis. One such strategy would be to attenuate the ability of platelets to form stable integrin αIIbβ3 adhesion contacts under high shear stress by targeting shear-sensitive signaling processes in platelets. To date, the identification of signaling elements crucial for shear activation of platelets, but not for hemostasis, have remained elusive.
An important signaling pathway promoting shear activation of plate-lets involves the PI3Ks12,13. These lipid kinases are divided into three distinct classes (Class I, II and III) based on their primary structure, mode of regulation and substrate specificity14,15. The Class I PI3Ks are responsible for agonist-induced production of the second messengers phosphoinositide (PI) (3,4,5)P3 and PI(3,4)P2, and participate in the regulation of a broad range of functional platelet responses, including activation of integrin αIIbβ3
16. The Class I enzymes are further divided into α, β, δ and γ isoforms based on their distinct p110 catalytic sub-units and modes of regulation. The Class Ia isoforms (p110α, p110β, p110δ) are classically regulated by tyrosine kinases, whereas the Class Ib isoform (p110γ) is typically activated by G-protein-coupled recep-tors17. Platelets contain all Class I PI3K isoforms, although the levels of
1Australian Centre for Blood Diseases, Monash University, 6th Floor Burnet Building Alfred Medical Research and Education Precinct (AMREP), 89 Commercial Road, Prahran, Victoria, Australia 3181. 2Department of Pharmacology, Level 8, Medical Building, Corner of Grattan Street and Royal Parade, University of Melbourne, Victoria, Australia 3010. 3Department of Biochemistry and Molecular Biology, University College of London, Gower Street, London, WC1E6BT, UK. 4Kinacia Pty Ltd, Cerylid Biosciences Ltd, 576 Swan Street, Richmond, Victoria, Australia 3121. 5These authors contributed equally to this work. ADR and HHS are equal senior authors. Correspondence should be addressed to S.P.J. ([email protected]).
Published online 17 April 2005; doi:10.1038/nm1232
A R T I C L E S©
2005
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
emed
icin
e

508 VOLUME 11 | NUMBER 5 | MAY 2005 NATURE MEDICINE
p110δ are much lower in platelets than in other hemopoietic cells18–20. To date, the role of the individual PI3K isoforms in regulating platelet functional responses has been poorly defined and there is currently no information on the role of individual PI3K isoforms in regulating shear activation of platelets.
RESULTSPI3K regulates integrin αIIbβ3 adhesive bond stabilityPrevious studies have defined a key role for PI3K in promoting the initial formation of integrin αIIbβ3 adhesive bonds under high shear12,21 and there is evidence that PI3Ks operate downstream of each of the major platelet receptors involved in shear-induced platelet activation, notably
glycoprotein (GP) Ib22,23, integrin αIIbβ321 and
the ADP purinergic receptor P2Y1213,24. The
specific PI3K isoforms involved in this process and the importance of these enzymes in main-taining the stability of formed integrin αIIbβ3 adhesion bonds have not been established. To investigate this, platelets were perfused through fibrinogen-coated microcapillary tubes at low shear to enable formation of integrin αIIbβ3
adhesion bonds, then exposed to sudden accelerations in blood flow, thereby increasing tensile stress on formed bonds. Of platelets adher-ent to fibrinogen, 63.8 ± 6.0% were able to resist the detaching effects of rapid increases in shear, whereas 27.4 ± 6.3% of platelets pretreated with the pharmacological PI3K inhibitor LY294002 were able to maintain firm adhesion (Fig. 1a). Previous studies have established an impor-tant role for integrin αIIbβ3–dependent calcium flux, in combination with released ADP, in promoting shear-resistant platelet adhesion25. Consistent with an important role for PI3Ks in this process, LY294002 completely inhibited calcium flux in fibrinogen-adherent platelets (Fig. 1b), either in the presence or absence of ADP receptor antago-nists (data not shown). To investigate the identity of the PI3K isoforms
a b c
d e f
a b
c d
Figure 1 PI3K promotes cytosolic calcium flux and stable platelet adhesion on immobilized fibrinogen following exposure to rapid increases in shear. Calcium dye–loaded human platelets (a,b) or those isolated from PI3K p110γ–containing (γ+), p110γ–deficient (γ–) (c,d) or PI3K p110δ–deficient (δ–) (d,e) mice were pretreated with vehicle alone (Me2SO), the non-isoform-selective PI3K inhibitor LY294002 (LY, 25 µM), and/or ADP inhibitors (ØADP: apyrase (1.5 U/ml), MRS-2179 (100 µM), AR-C69931MX (10 µM)), as indicated. The results represent the percentage of platelets (a,c,e) becoming displaced from the point of initial contact, and (b,d) undergoing a sustained oscillatory calcium response following an increase in shear from 150 s–1 to 1,800 s–1 for human and 600 s-1 to 10,000 s–1 for mouse platelets (arrow) (mean ± s.e.m., n = 3).
Figure 2 Development of a new PI3K p110β isoform–selective inhibitor. (a) Chemical structure of LY294002. Key features for PI3K activity and selectivity include the chromone nucleus (i), the 2-morpholinyl substitution (ii) and the pendant aryl ring (iii)42. These features were systematically modified in structure-function studies. (b) Chemical structures and PI3K p110β inhibitory potency of 2-morpholinyl-pyrido-[1,2-a] pyrimidinones. Successively more potent inhibitors of the PI3K p110β isoform were identified, such as TGX-126 and TGX-221 (Supplementary Fig. 1 online). (c) Relative dose-response inhibition of PI3K p110α, p110β and p110γ (expressed as percent activity relative to Me2SO control) by TGX-221 (p110α and γ, n = 5; p110β, n = 10, mean ± s.e.m.). (d) Effect of TGX-221 (221, 0.1 µM) on PI(4)P, PI(3)P, PI(4,5)P2 and PI(3,4)P2 levels in shear-activated platelets. Histograms represent the level of each lipid expressed as a percent of total lipid (all groups n = 3, mean ± s.e.m., ***P < 0.0001; ns, P > 0.1, Student’s t-test).
A R T I C L E S©
2005
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
emed
icin
e

NATURE MEDICINE VOLUME 11 | NUMBER 5 | MAY 2005 509
involved in this process, flow-based adhesion assays were performed on mouse platelets deficient in the Type IA PI3K p110δ or Type 1B PI3K p110γ isoform. Similar to human platelets, maintenance of mouse inte-grin αIIbβ3 adhesive bonds on a fibrinogen matrix is dependent on the cooperative signaling function of integrin αIIbβ3 and released ADP; how-ever, in the mouse system, the contribution of integrin αIIbβ3–dependent signaling processes to sustained adhesion is most clearly manifested in the presence of ADP receptor antagonists25. This species difference pro-bably reflects differences in the binding kinetics of mouse versus human integrin αIIbβ3 to human fibrinogen. In the presence of ADP receptor antagonists, 71.4 ± 3.2% of wild-type mouse platelets maintained stable adhesion contacts with fibrinogen after exposure to rapid increases in shear (Fig. 1c). In contrast, 39.8 ± 5% of wild-type platelets treated with LY294002 and ADP receptor antagonists were able to maintain stable adhesive interactions under the same experimental conditions. As shown in Fig. 1, analysis of PI3K p110δ–deficient and PI3K p110γ–deficient platelets showed no significant difference in shear-resistant platelet adhesion relative to wild-type matched controls (Fig. 1c,e). Similarly, deficiency of PI3K p110δ and PI3K p110γ had no signifi-cant effect on integrin αIIbβ3–dependent cytosolic calcium flux, in the presence or absence of ADP receptor antagonists (Fig. 1d,f), excluding an important role for these isoforms in maintaining integrin αIIbβ3 adhesive contacts under high shear.
Development of isoform-selective PI3K p110β inhibitorsEstablishing the roles of the PI3K p110α and p110β isoforms in sig-naling pathways is complicated by the lack of available mouse models and specific isoform-selective inhibitors. To overcome this problem we developed β isoform–selective PI3K inhibitors based on detailed structure and function analysis of LY294002 (Fig. 2a). Replacing the chromone nucleus with the pyrido[1,2-a]pyrimidin-4-one nucleus and elaboration of the pendant aryl ring yielded compounds with increased potency and selectivity against the Type I PI3K p110β isoform (Fig. 2b and Supplementary Fig. 1 online). One of the most potent members of this group (TGX-221) has an IC50 of 5 nM against the purified kinase (phosphatidylinositol as substrate in the presence of 50 µM ATP; IC50 ∼10 nM with PI(4,5)P2 as substrate) and shows ∼1,000-fold selectivity over PI3K p110α (Fig. 2c). TGX-221 had minimal inhibitory effect on Type II PI3K C2α and PI4K and showed >1,000-fold selectivity for PI3K p110β over a broad range of protein kinases (Supplementary Table 1 online), and similar to LY294002, had minimal effect on global protein phosphorylation in thrombin-stimulated platelets (data not shown). Similar to LY294002, the inhibitory effects of TGX-221 were influenced by the concentration of ATP. At physiologically relevant cytosolic ATP levels (typically ≥1,000 µM) the IC50 against PI3K p110β increased from 5 nM to ∼50 nM, indicating that TGX-221 was probably acting as a competitive ATP antagonist. Analysis of the effects of TGX-221 on PI3K lipid product generation in vivo showed that TGX-221 inhib-ited shear-induced PI(3,4)P2 production in a dose-dependent manner
(Fig. 2d and Supplementary Fig. 1 online), without altering the cellular levels of PI(3)P and the conventional phosphoinositides, PI(4)P and PI(4,5)P2 (Fig. 2d). Increases in PI(3,4,5)P3 levels were not detected under these experimental conditions, presumably as a result of the rapid dephosphorylation of PI(3,4,5)P3 to PI(3,4)P2 by SHIP-1 (ref. 26). The IC50 for inhibiting PI(3,4)P2 production in platelets (∼50 nM) was ∼50–100-fold lower than that observed with LY294002 (2.5–5 µM), which correlated well with the relative IC50s for these com-pounds against PI3K p110β at physiological ATP concentrations. These studies show that TGX-221 is a potent cell-permeable inhibitor of PI3K p110β and furthermore, show an important role for PI3K p110β in promoting shear-dependent PI(3,4)P2 generation.
PI3K p110β regulates integrin αIIbβ3 bond stabilityAnalysis of the effects of TGX-221 on shear-resistant adhesion of human platelets on a fibrinogen matrix showed a defect in integrin
a b
c d
e f
g hFigure 3 PI3K p110β promotes cytosolic calcium flux and stable platelet adhesion in response to rapid increases in shear. Calcium dye–loaded human platelets (a,b), or those isolated from PI3K p110γ–deficient (γ–) (c,d) or PI3K p110δ–deficient (δ–) mice (e,f), were pretreated with vehicle alone (Me2SO) or 0.5 µM TGX-221 (221). The results represent the percentage of platelets displaced (a,c,e) and the percentage of platelets exhibiting a sustained oscillatory calcium response (b,d,f) after the shear increase (arrow) (mean ± s.e.m., n = 3). (g) Differential interference contrast images of platelet thrombi formed on a vWf matrix in the presence (221) or absence (Me2SO) of TGX-221. (h) Quantification of the effect of TGX-221 on thrombus volume on a vWf matrix (mean ± s.e.m., n = 7).
A R T I C L E S©
2005
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
emed
icin
e

510 VOLUME 11 | NUMBER 5 | MAY 2005 NATURE MEDICINE
αIIbβ3-dependent calcium flux and firm platelet adhesion similar in magnitude to that observed with LY294002-treated platelets (Fig. 3a,b). Similarly, PI3K p110δ–deficient and PI3K p110γ–deficient mouse plate-lets were equally sensitive to the inhibitory effects of TGX-221 and LY294002, defining a major role for PI3K p110β in promoting integrin αIIbβ3–dependent calcium flux (Fig. 3c–f). To investigate the importance of PI3K p110β in initiating the formation of integrin αIIbβ3 adhesive bonds under high shear, flow-based adhesion studies were performed on an immobilized human vWf matrix. TGX-221 abolished stable plate-let adhesion and thrombus formation on a vWf substrate at elevated shear rates (1,800 s–1; Fig. 3g,h). This reduction in firm platelet adhesion was associated with inhibition of cytosolic calcium flux and integrin αIIbβ3 activation (data not shown). Similarly, shear-induced aggrega-tion of platelets in suspension (5,000 s–1) was inhibited by TGX-221 by >90% (data not shown). Recent studies have defined an important role for rapid increases in shear (temporal shear gradients) in promoting integrin αIIbβ3–dependent calcium flux, necessary for efficient platelet activation on a vWf matrix27. As shown in Figure 4, TGX-221 com-pletely eliminated cytosolic calcium flux (Fig. 4a,c) and integrin αIIbβ3 activation (Fig. 4f) induced by temporal shear gradients (0–10,000 s–1), confirming a major role for PI3K p110β in integrin αIIbβ3 mechano-transduction. In control studies, a PI3K p110δ inhibitor (D-010 (ref. 28); Supplementary Fig. 2 online) had no inhibitory effects on platelet activation induced by high shear stress (data not shown) or by tem-poral shear gradients (Supplementary Fig. 2 online). Similar find-ings were found with PI3K p110γ–deficient platelets (Supplementary Fig. 2 online). Furthermore, the effects of TGX-221 on calcium flux were selective, as it had no inhibitory effect on GPIb-dependent calcium
signals (Fig. 4b) or on calcium signals induced by a range of physiologi-cal agonists (Fig. 4d,e and data not shown). These studies define a major role for PI3K p110β in promoting the formation and maintenance of integrin αIIbβ3 adhesive bonds under high shear through regulation of integrin αIIbβ3–dependent calcium flux.
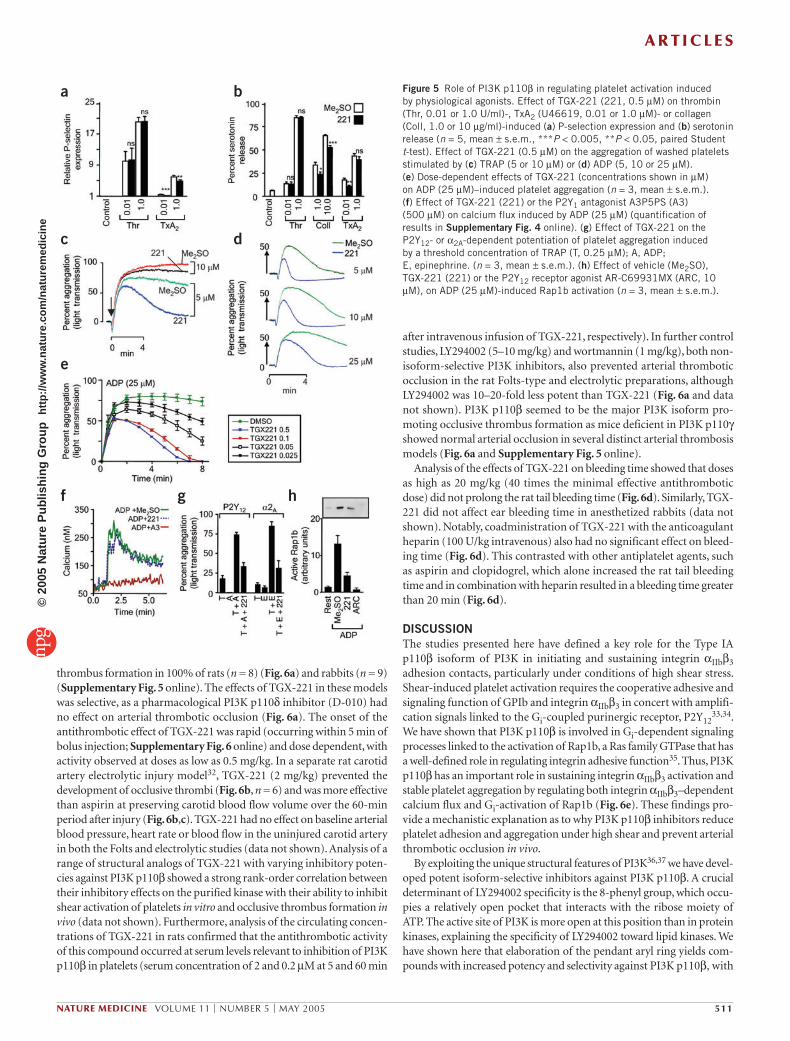
PI3K p110β involvement in platelet aggregation and secretionTo investigate the importance of PI3K p110β in promoting platelet activation by physiological agonists, we performed platelet aggre-gation and granule release studies. TGX-221 had no inhibitory effects on platelet aggregation and alpha- and dense-granule release induced by thrombin, high-dose collagen or the TxA2 analog U46619 (Fig. 5a–c, Supplementary Fig. 3 online and data not shown). Furthermore, TGX-221 had no inhibitory effect on the ability of these agonists to stimulate integrin αIIbβ3 activation (as assessed by PAC-1 binding; data not shown). But inhibition of PI3K 110β did inhibit sustained platelet aggregation induced by threshold concentrations of thrombin receptor agonist peptide (TRAP; Fig. 5c and Supplementary Fig. 3 online), collagen (Supplementary Fig. 3 online) and U46619 (data not shown), although alpha- and dense-granule release was only marginally affected by this inhibitor (Fig. 5a,b). These studies show that PI3K p110β has an important role in sustaining platelet aggregation in response to weak agonist stimulation.
Platelet aggregation induced by threshold concentrations of agonists is partially dependent on the granule release of ADP29. TGX-221 inhibited ADP-induced platelet aggregation over a dose range consistent with the inhibition of PI3K p110β. The inhibitory effects of TGX-221 were apparent over a broad range of ADP concentrations and were primarily manifested as an inability of ADP to sustain platelet aggregation (Fig. 5d,e). Inhibition of PI3K p110β had no effect on the ability of ADP to stimulate P2Y1-dependent cytosolic calcium flux (Fig. 5f and Supplementary Fig. 4 online) or platelet shape change (Supplementary Fig. 4 online), suggesting a potentially important role for PI3K p110β in P2Y12 signaling. Consistent with this, TGX-221 inhibited P2Y12/Gi-dependent potentiation of platelet aggregation induced by threshold concentrations of other agonists (Fig. 5g). Similar findings were apparent when the Gi-linked α2Α adrenergic receptor agonist epinephrine was used to potentiate platelet aggregation (Fig. 5g), suggesting an important role for PI3K p110β in platelet Gi signaling processes. Consistent with this, Gi-dependent Rap1b activation, a Ras family GTPase that potentiates integrin αIIbβ3 activation30 was inhibited by >70% by TGX-221 (Fig. 5h). These studies define an important role for PI3K p110β in P2Y12/Gi signaling processes linked to sustained platelet aggregation.
Antithrombotic effect of PI3K p110β inhibitorsTo investigate the antithrombotic potential of TGX-221 in vivo, a modi-fied ‘Folts-type’ thrombosis model was used in anesthetized rats and rabbits31. TGX-221 (2 mg/kg intravenous bolus) abolished occlusive
a b
d
f
e
c
Figure 4 Role of PI3K p110β in promoting platelet activation in response to fluid shear stress or soluble agonists. Representative single platelet recordings showing the effects of TGX-221 on (a) integrin αIIbβ3–dependent calcium responses induced by temporal shear gradients or (b) GPIb-dependent [Ca2+]c responses occurring under steady-state shear conditions. (c) The proportion of platelets showing an integrin αIIbβ3– or GPIb-dependent calcium response was quantified and expressed as mean ± s.e.m. (n = 6, mean ± s.e.m.). (d,e) Population-based analysis of ∆[Ca2+]c in platelets activated with (d) TxA2 (U46619, 50 nM) or (e) thrombin receptor agonist peptide (TRAP, 0.4 µM). (f) Effect of TGX-221 (221) on integrin αIIbβ3 activation (assessed by PAC-1 binding, PAC-1) induced by temporal shear gradients.
A R T I C L E S©
2005
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
emed
icin
e
NATURE MEDICINE VOLUME 11 | NUMBER 5 | MAY 2005 511
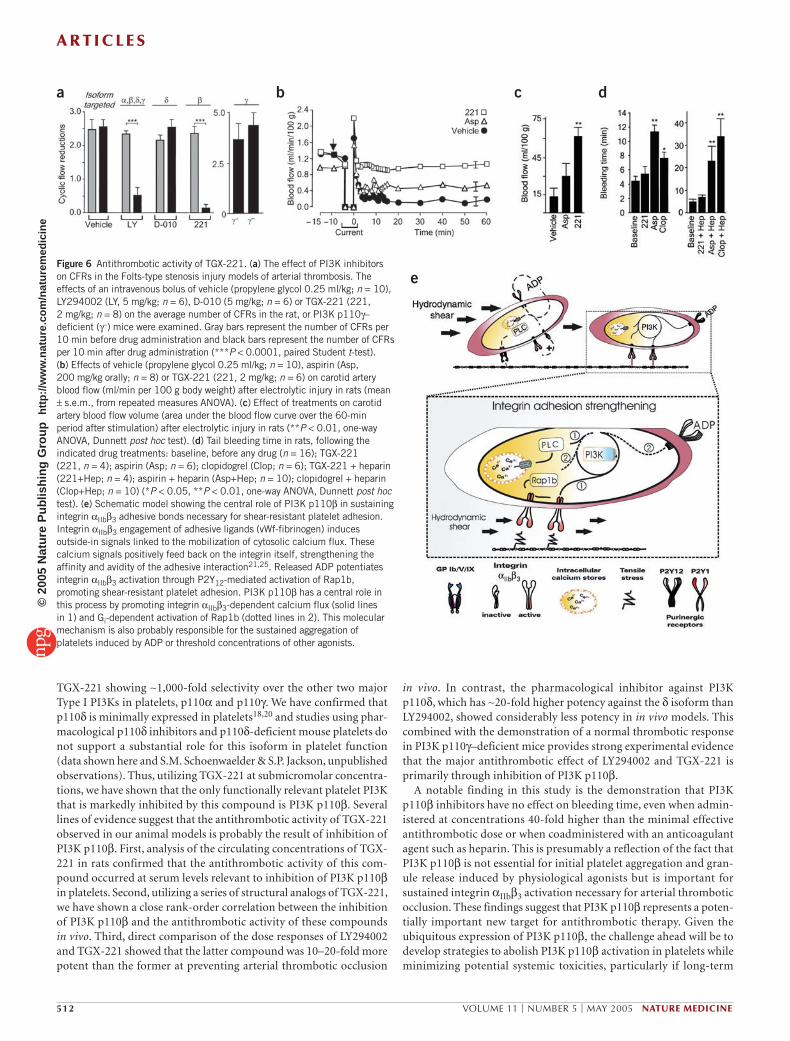
thrombus formation in 100% of rats (n = 8) (Fig. 6a) and rabbits (n = 9) (Supplementary Fig. 5 online). The effects of TGX-221 in these models was selective, as a pharmacological PI3K p110δ inhibitor (D-010) had no effect on arterial thrombotic occlusion (Fig. 6a). The onset of the antithrombotic effect of TGX-221 was rapid (occurring within 5 min of bolus injection; Supplementary Fig. 6 online) and dose dependent, with activity observed at doses as low as 0.5 mg/kg. In a separate rat carotid artery electrolytic injury model32, TGX-221 (2 mg/kg) prevented the development of occlusive thrombi (Fig. 6b, n = 6) and was more effective than aspirin at preserving carotid blood flow volume over the 60-min period after injury (Fig. 6b,c). TGX-221 had no effect on baseline arterial blood pressure, heart rate or blood flow in the uninjured carotid artery in both the Folts and electrolytic studies (data not shown). Analysis of a range of structural analogs of TGX-221 with varying inhibitory poten-cies against PI3K p110β showed a strong rank-order correlation between their inhibitory effects on the purified kinase with their ability to inhibit shear activation of platelets in vitro and occlusive thrombus formation in vivo (data not shown). Furthermore, analysis of the circulating concen-trations of TGX-221 in rats confirmed that the antithrombotic activity of this compound occurred at serum levels relevant to inhibition of PI3K p110β in platelets (serum concentration of 2 and 0.2 µM at 5 and 60 min
after intravenous infusion of TGX-221, respectively). In further control studies, LY294002 (5–10 mg/kg) and wortmannin (1 mg/kg), both non-isoform-selective PI3K inhibitors, also prevented arterial thrombotic occlusion in the rat Folts-type and electrolytic preparations, although LY294002 was 10–20-fold less potent than TGX-221 (Fig. 6a and data not shown). PI3K p110β seemed to be the major PI3K isoform pro-moting occlusive thrombus formation as mice deficient in PI3K p110γ showed normal arterial occlusion in several distinct arterial thrombosis models (Fig. 6a and Supplementary Fig. 5 online).
Analysis of the effects of TGX-221 on bleeding time showed that doses as high as 20 mg/kg (40 times the minimal effective antithrombotic dose) did not prolong the rat tail bleeding time (Fig. 6d). Similarly, TGX-221 did not affect ear bleeding time in anesthetized rabbits (data not shown). Notably, coadministration of TGX-221 with the anticoagulant heparin (100 U/kg intravenous) also had no significant effect on bleed-ing time (Fig. 6d). This contrasted with other antiplatelet agents, such as aspirin and clopidogrel, which alone increased the rat tail bleeding time and in combination with heparin resulted in a bleeding time greater than 20 min (Fig. 6d).
DISCUSSIONThe studies presented here have defined a key role for the Type IA p110β isoform of PI3K in initiating and sustaining integrin αIIbβ3 adhesion contacts, particularly under conditions of high shear stress. Shear-induced platelet activation requires the cooperative adhesive and signaling function of GPIb and integrin αIIbβ3 in concert with amplifi-cation signals linked to the Gi-coupled purinergic receptor, P2Y12
33,34. We have shown that PI3K p110β is involved in Gi-dependent signaling processes linked to the activation of Rap1b, a Ras family GTPase that has a well-defined role in regulating integrin adhesive function35. Thus, PI3K p110β has an important role in sustaining integrin αIIbβ3 activation and stable platelet aggregation by regulating both integrin αIIbβ3–dependent calcium flux and Gi-activation of Rap1b (Fig. 6e). These findings pro-vide a mechanistic explanation as to why PI3K p110β inhibitors reduce platelet adhesion and aggregation under high shear and prevent arterial thrombotic occlusion in vivo.
By exploiting the unique structural features of PI3K36,37 we have devel-oped potent isoform-selective inhibitors against PI3K p110β. A crucial determinant of LY294002 specificity is the 8-phenyl group, which occu-pies a relatively open pocket that interacts with the ribose moiety of ATP. The active site of PI3K is more open at this position than in protein kinases, explaining the specificity of LY294002 toward lipid kinases. We have shown here that elaboration of the pendant aryl ring yields com-pounds with increased potency and selectivity against PI3K p110β, with
a b
c d
e
f g h
Figure 5 Role of PI3K p110β in regulating platelet activation induced by physiological agonists. Effect of TGX-221 (221, 0.5 µM) on thrombin (Thr, 0.01 or 1.0 U/ml)-, TxA2 (U46619, 0.01 or 1.0 µM)- or collagen (Coll, 1.0 or 10 µg/ml)-induced (a) P-selection expression and (b) serotonin release (n = 5, mean ± s.e.m., ***P < 0.005, **P < 0.05, paired Student t-test). Effect of TGX-221 (0.5 µM) on the aggregation of washed platelets stimulated by (c) TRAP (5 or 10 µM) or (d) ADP (5, 10 or 25 µM). (e) Dose-dependent effects of TGX-221 (concentrations shown in µM) on ADP (25 µM)–induced platelet aggregation (n = 3, mean ± s.e.m.). (f) Effect of TGX-221 (221) or the P2Y1 antagonist A3P5PS (A3) (500 µM) on calcium flux induced by ADP (25 µM) (quantification of results in Supplementary Fig. 4 online). (g) Effect of TGX-221 on the P2Y12- or α2A-dependent potentiation of platelet aggregation induced by a threshold concentration of TRAP (T, 0.25 µM); A, ADP; E, epinephrine. (n = 3, mean ± s.e.m.). (h) Effect of vehicle (Me2SO), TGX-221 (221) or the P2Y12 receptor agonist AR-C69931MX (ARC, 10 µM), on ADP (25 µM)-induced Rap1b activation (n = 3, mean ± s.e.m.).
A R T I C L E S©
2005
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
emed
icin
e
512 VOLUME 11 | NUMBER 5 | MAY 2005 NATURE MEDICINE
a b c
e
d
TGX-221 showing ∼1,000-fold selectivity over the other two major Type I PI3Ks in platelets, p110α and p110γ. We have confirmed that p110δ is minimally expressed in platelets18,20 and studies using phar-macological p110δ inhibitors and p110δ-deficient mouse platelets do not support a substantial role for this isoform in platelet function (data shown here and S.M. Schoenwaelder & S.P. Jackson, unpublished observations). Thus, utilizing TGX-221 at submicromolar concentra-tions, we have shown that the only functionally relevant platelet PI3K that is markedly inhibited by this compound is PI3K p110β. Several lines of evidence suggest that the antithrombotic activity of TGX-221 observed in our animal models is probably the result of inhibition of PI3K p110β. First, analysis of the circulating concentrations of TGX-221 in rats confirmed that the antithrombotic activity of this com-pound occurred at serum levels relevant to inhibition of PI3K p110β in platelets. Second, utilizing a series of structural analogs of TGX-221, we have shown a close rank-order correlation between the inhibition of PI3K p110β and the antithrombotic activity of these compounds in vivo. Third, direct comparison of the dose responses of LY294002 and TGX-221 showed that the latter compound was 10–20-fold more potent than the former at preventing arterial thrombotic occlusion
in vivo. In contrast, the pharmacological inhibitor against PI3K p110δ, which has ∼20-fold higher potency against the δ isoform than LY294002, showed considerably less potency in in vivo models. This combined with the demonstration of a normal thrombotic response in PI3K p110γ–deficient mice provides strong experimental evidence that the major antithrombotic effect of LY294002 and TGX-221 is primarily through inhibition of PI3K p110β.
A notable finding in this study is the demonstration that PI3K p110β inhibitors have no effect on bleeding time, even when admin-istered at concentrations 40-fold higher than the minimal effective antithrombotic dose or when coadministered with an anticoagulant agent such as heparin. This is presumably a reflection of the fact that PI3K p110β is not essential for initial platelet aggregation and gran-ule release induced by physiological agonists but is important for sustained integrin αIIbβ3 activation necessary for arterial thrombotic occlusion. These findings suggest that PI3K p110β represents a poten-tially important new target for antithrombotic therapy. Given the ubiquitous expression of PI3K p110β, the challenge ahead will be to develop strategies to abolish PI3K p110β activation in platelets while minimizing potential systemic toxicities, particularly if long-term
Figure 6 Antithrombotic activity of TGX-221. (a) The effect of PI3K inhibitors on CFRs in the Folts-type stenosis injury models of arterial thrombosis. The effects of an intravenous bolus of vehicle (propylene glycol 0.25 ml/kg; n = 10), LY294002 (LY, 5 mg/kg; n = 6), D-010 (5 mg/kg; n = 6) or TGX-221 (221, 2 mg/kg; n = 8) on the average number of CFRs in the rat, or PI3K p110γ–deficient (γ–) mice were examined. Gray bars represent the number of CFRs per 10 min before drug administration and black bars represent the number of CFRs per 10 min after drug administration (***P < 0.0001, paired Student t-test). (b) Effects of vehicle (propylene glycol 0.25 ml/kg; n = 10), aspirin (Asp, 200 mg/kg orally; n = 8) or TGX-221 (221, 2 mg/kg; n = 6) on carotid artery blood flow (ml/min per 100 g body weight) after electrolytic injury in rats (mean ± s.e.m., from repeated measures ANOVA). (c) Effect of treatments on carotid artery blood flow volume (area under the blood flow curve over the 60-min period after stimulation) after electrolytic injury in rats (**P < 0.01, one-way ANOVA, Dunnett post hoc test). (d) Tail bleeding time in rats, following the indicated drug treatments: baseline, before any drug (n = 16); TGX-221 (221, n = 4); aspirin (Asp; n = 6); clopidogrel (Clop; n = 6); TGX-221 + heparin (221+Hep; n = 4); aspirin + heparin (Asp+Hep; n = 10); clopidogrel + heparin (Clop+Hep; n = 10) (*P < 0.05, **P < 0.01, one-way ANOVA, Dunnett post hoc test). (e) Schematic model showing the central role of PI3K p110β in sustaining integrin αIIbβ3 adhesive bonds necessary for shear-resistant platelet adhesion. Integrin αIIbβ3 engagement of adhesive ligands (vWf-fibrinogen) induces outside-in signals linked to the mobilization of cytosolic calcium flux. These calcium signals positively feed back on the integrin itself, strengthening the affinity and avidity of the adhesive interaction21,25. Released ADP potentiates integrin αIIbβ3 activation through P2Y12-mediated activation of Rap1b, promoting shear-resistant platelet adhesion. PI3K p110β has a central role in this process by promoting integrin αIIbβ3-dependent calcium flux (solid lines in 1) and Gi-dependent activation of Rap1b (dotted lines in 2). This molecular mechanism is also probably responsible for the sustained aggregation of platelets induced by ADP or threshold concentrations of other agonists.
A R T I C L E S©
2005
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
emed
icin
e

NATURE MEDICINE VOLUME 11 | NUMBER 5 | MAY 2005 513
thromboprophylactic therapy is contemplated. Such strategies have previously been used successfully against other ubiquitous targets; the most notable involves irreversible acetylation of cyclo-oxygenases by aspirin.
Overall, our findings provide further evidence for functional specialization of different PI3K isoforms and show for the first time an important role for PI3K p110β in platelet function and thrombosis. Given the important roles of each of the Type I PI3K family members in various pathophysiological processes (PI3K p110α in oncogenesis, PI3K p110β in thrombosis, PI3K p110δ in immune function and PI3K p110γ in inflam-mation38–40) isoform-selective PI3K inhibitors may ultimately have con-siderable therapeutic benefit in a wide range of human diseases.
METHODSMaterials and knockout mice. All materials were obtained from sources described elsewhere21,26,41. The synthesis of TGX-221 and other compounds used in this study have been described42 or will be reported in detail elsewhere. PI3K p110γ–deficient mice were generated by D. Wu (University of Rochester, New York). PI3K p110δ–deficient mice were provided by M. Turner (Babraham Institute, UK). All protocols involving the use of animals were approved by the Alfred Medical Research Education Precinct Animal Ethics Committee, Alfred Medical Research Education Precinct, Victoria, Australia.
Platelet preparation and aggregation. Preparation of human and mouse washed platelets, platelet-rich plasma (PRP), and performance of platelet aggregations using PRP, were all performed as described previously21,26,41. We obtained informed consent from all human subjects donating blood. All pro-cedures involving collection of blood from human subjects were approved by the Monash University Standing Committee on Ethics in Research Involving Humans, Monash University, Victoria, Australia.
Flow cytometry. We measured surface expression of P-selectin using a P-selectin-specific monoclonal antibody (AK6, 1 µg/ml), and analyzed it on a FACScalibur (Becton Dickinson).
Serotonin release. Dense granule release was measured in 14C-serotonin labeled platelets as reported previously43.
Measurement of intracellular calcium mobilization. Calcium mobilization in resting and ADP-stimulated washed platelets (200 × 109/L) loaded with Fura-2 AM (5 µM) was monitored over time using a fluorimeter (LS-50B; Supplementary Methods online). We performed measurements in the absence or presence of the indicated inhibitors (TGX-221, 0.5 µM; A3P5PS, 300 µM), with dual excitation at 340 and 380 nm and detected emission at 510 nm. Calcium measurements were converted to nM calcium using computer-assisted analysis (FL WinLAB).
Measurement of active Rap1b. The level of GTP-bound Rap1b in human platelet lysates was measured by selective precipitation of GTP-bound Rap1b using GST-RalGDS bound to glutathione-Sepharose, as described previously44,45.
In vitro flow studies. Flow-based assays using whole blood or washed plate-lets were performed as described previously41. Analysis of platelet adhesion and cytosolic calcium flux on a fibrinogen matrix under flow conditions were moni-tored according to published methods21,25,41, and details are in Supplementary Methods online. Studies examining adhesion strength at high shear, along with temporal shear gradient experiments, were performed as described in Supplementary Methods online. Measurement of thrombus formation on a vWf matrix at 1,800 s–1 was performed and analyzed as described previously12.
PI3K lipid kinase assay. Kinase assays were performed according to a previously described method46.
Measurement of 32P lipids in platelets. We applied 32P-loaded washed human platelets to a cone-and-plate viscometer and subjected them to a shear rate of 6,000 s–1 for a period of 2–5 min. The levels of PI(4)P, PI(3)P, PI(4,5)P2 and PI(3,4)P2 in platelets was measured as described previously26.
In vivo thrombosis models. We performed a Folts-type stenosis injury model31 and an electrolytic model32 (Supplementary Methods online) on rats, rabbits and mice.
Tail bleeding studies in rats. Tail bleeding time was measured in anesthetized rats (halothane in O2) 15 min before and 5 and 30 min after drug administration. We administered the following drug concentrations: TGX-221 (2 mg/kg intra-venous), aspirin (200 mg/kg orally), clopidogrel (10 mg/kg orally) and heparin (100 U/kg intravenous). We administered clopidogrel orally 25 h before tail bleeding. Aspirin was administered twice orally: the first dose 25 h before the experiment and the second dose 1 h before the start of the experiment. For experi-ments involving pretreatment with aspirin or clopidogrel or both, tail bleeding time was also measured before the first gavage dose at 25 h before tail bleeding. Incisions 5 mm long and 1 mm deep47 were made in the tail at each time point and bleeding was monitored by blotting with filter paper48 every 30 sec until it had ceased (tail bleeding time).
Statistical analysis. All individual aggregation traces, calcium traces, and images were taken from one experiment representative of at least three independent experiments. All numerical results are presented as mean ± s.e.m. The significance between two data tests was tested using unpaired t-tests in most instances, unless otherwise indicated. Statistical significance was considered when P < 0.05.
Note: Supplementary information is available on the Nature Medicine website.
AUTHOR CONTRIBUTIONSAlan D. Robertson and Hatem H. Salem contributed equally to this work.
ACKNOWLEDGMENTSThis work was supported by the National Health and Medical Research Council and the National Heart Foundation of Australia. Kinacia Pty Ltd also contributed financial support. We would like to thank L. Stephens, P. Hawkins and Z. Ruggeri for discussions, and D. Williamson, P. Mangin, K. Heel, D. Dunstan, I. Harper, and G. Currie, P. Freeman, M. Mulchandani, T. Domagala, M. Wang, N. Mistry, V. Strangis, S. Turnbull and T. Hinds for technical assistance and advice during the preparation of this manuscript.
COMPETING INTERESTS STATEMENTThe authors declare competing financial interests (see the Nature Medicine website for details).
Received 9 September 2004; accepted 9 March 2005Published online at http://www.nature.com/naturemedicine/
1. Baumgartner, H.R., Tschopp, T.B. & Weiss, H.J. Platelet interaction with collagen fibrils in flowing blood. II. Impaired adhesion-aggregation in bleeding disorders. A comparison with subendothelium. Thromb. Haemost. 37, 17–28 (1977).
2. Weiss, H.J. et al. Fibrinogen-independent platelet adhesion and thrombus formation on subendothelium mediated by glycoprotein IIb-IIIa complex at high shear rate. J. Clin. Invest. 83, 288–297 (1989).
3. van Zanten, G.H. et al. Increased platelet deposition on atherosclerotic coronary arteries. J. Clin. Invest. 93, 615–632 (1994).
4. Wilcox, J.N., Smith, K.M., Schwartz, S.M. & Gordon, D. Localization of tissue factor in the normal vessel wall and in the atherosclerotic plaque. Proc. Natl. Acad. Sci. USA 86, 2839–2843 (1989).
5. Toschi, V. et al. Tissue factor modulates the thrombogenicity of human atherosclerotic plaques. Circulation 95, 594–599 (1997).
6. Goto, S., Ikeda, Y., Saldivar, E. & Ruggeri, Z.M. Distinct mechanisms of platelet aggrega-tion as a consequence of different shearing flow conditions. J. Clin. Invest. 101, 479–486 (1998).
7. Ruggeri, Z.M. Mechanisms of shear-induced platelet adhesion and aggregation. Thromb. Haemost. 70, 119–123 (1993).
8. Jander, S. et al. Expression of tissue factor in high-grade carotid artery stenosis: associa-tion with plaque destabilization. Stroke 32, 850–854 (2001).
9. Strony, J., Beaudoin, A., Brands, D. & Adelman, B. Analysis of shear stress and hemo-dynamic factors in a model of coronary artery stenosis and thrombosis. Am. J. Physiol. 265, H1787–H1796 (1993).
10. Ruggeri, Z.M. Platelets in atherothrombosis. Nat. Med. 8, 1227–1234 (2002).11. Jackson, S.P. & Schoenwaelder, S.M. Antiplatelet therapy: in search of the ‘magic bullet’.
Nat. Rev. Drug Discov. 2, 775–789 (2003).12. Yap, C.L. et al. Essential role for phosphoinositide 3-kinase in shear-dependent signaling
between platelet glycoprotein Ib/V/IX and integrin alpha(IIb)beta(3). Blood 99, 151–158 (2002).
13. Resendiz, J.C. et al. Purinergic P2Y12 receptor blockade inhibits shear-induced platelet phosphatidylinositol 3-kinase activation. Mol. Pharmacol. 63, 639–645 (2003).
A R T I C L E S©
2005
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
emed
icin
e

514 VOLUME 11 | NUMBER 5 | MAY 2005 NATURE MEDICINE
14. Vanhaesebroeck, B., Leevers, S.J., Panayotou, G. & Waterfield, M.D. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem. Sci. 22, 267–272 (1997).
15. Anderson, K.E. & Jackson, S.P. Class I phosphoinositide 3-kinases. Int. J. Biochem. Cell Biol. 35, 1028–1033 (2003).
16. Jackson, S.P., Yap, C.L. & Anderson, K.E. Phosphoinositide 3-kinases and the regula-tion of platelet function. Biochem. Soc. Trans. 32, 387–392 (2004).
17. Vanhaesebroeck, B. & Waterfield, M.D. Signaling by distinct classes of phosphoinosit-ide 3-kinases. Exp. Cell Res. 253, 239–254 (1999).
18. Vanhaesebroeck, B. et al. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc. Natl. Acad. Sci. USA 94, 4330–4335 (1997).
19. Zhang, J., Vanhaesebroeck, B. & Rittenhouse, S.E. Human platelets contain p110delta phosphoinositide 3-kinase. Biochem. Biophys. Res. Commun. 296, 178–181 (2002).
20. Watanabe, N. et al. Functional phenotype of phosphoinositide 3-kinase p85alpha-null platelets characterized by an impaired response to GP VI stimulation. Blood 102, 541–548 (2003).
21. Nesbitt, W.S. et al. Distinct glycoprotein Ib/V/IX and integrin alpha IIbbeta 3-depen-dent calcium signals cooperatively regulate platelet adhesion under flow. J. Biol. Chem. 277, 2965–2972 (2002).
22. Jackson, S.P. et al. Adhesion receptor activation of phosphatidylinositol 3-kinase. von Willebrand factor stimulates the cytoskeletal association and activation of phos-phatidylinositol 3-kinase and pp60c-src in human platelets. J. Biol. Chem. 269, 27093–27099 (1994).
23. Kasirer-Friede, A. et al. Signaling through GP Ib-IX-V activates alpha IIb beta 3 inde-pendently of other receptors. Blood 103, 3403–3411 (2004).
24. Kauffenstein, G. et al. The P2Y(12) receptor induces platelet aggregation through weak activation of the alpha(IIb)beta(3) integrin--a phosphoinositide 3-kinase-dependent mechanism. FEBS Lett. 505, 281–290 (2001).
25. Goncalves, I. et al. Integrin alpha IIb beta 3-dependent calcium signals regulate platelet-fibrinogen interactions under flow. Involvement of phospholipase C gamma 2. J. Biol. Chem. 278, 34812–34822 (2003).
26. Maxwell, M.J. et al. SHIP1 and Lyn Kinase Negatively Regulate Integrin alpha IIb beta 3 signaling in platelets. J. Biol. Chem. 279, 32196–32204 (2004).
27. Goncalves, I., Nesbitt, W.S., Yuan, Y. & Jackson, S.P. Importance of temporal flow gradients and integrin alpha IIb beta 3 mechanotransduction for rapid shear-activa-tion of platelets. J. Biol. Chem. published online 8 February 2005 (doi: 10.1074/jbc.M410235200).
28. Sadhu, K. et al. Inhibitors of human phosphatidylinositol 3-kinase delta. US Patent No. 6,518,277 (2003).
29. Gachet, C. et al. Activation of ADP receptors and platelet function. Thromb. Haemost. 78, 271–275 (1997).
30. Bertoni, A. et al. Relationships between Rap1b, affinity modulation of integrin alpha IIbbeta 3, and the actin cytoskeleton. J. Biol. Chem. 277, 25715–25721 (2002).
31. Folts, J.D., Stamler, J. & Loscalzo, J. Intravenous nitroglycerin infusion inhibits cyclic blood flow responses caused by periodic platelet thrombus formation in stenosed canine
coronary arteries. Circulation 83, 2122–2127 (1991).32. Bush, L.R. & Shebuski, R.J. In vivo models of arterial thrombosis and thrombolysis.
FASEB J. 4, 3087–3098 (1990).33. Goto, S., Tamura, N., Eto, K., Ikeda, Y. & Handa, S. Functional significance of
adenosine 5 -diphosphate receptor (P2Y(12)) in platelet activation initiated by binding of von Willebrand factor to platelet GP Ibalpha induced by conditions of high shear rate. Circulation 105, 2531–2536 (2002).
34. Nesbitt, W.S. et al. Distinct glycoprotein Ib/V/IX and integrin alpha IIbbeta 3-dependent calcium signals cooperatively regulate platelet adhesion under flow. J. Biol. Chem. 277, 2965–2972 (2002).
35. Bos, J.L. et al. The role of Rap1 in integrin-mediated cell adhesion. Biochemical Society Transactions 31, 83-6 (2003).
36. Walker, E.H. et al. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 6, 909–919 (2000).
37. Djordjevic, S. & Driscoll, P.C. Structural insight into substrate specificity and regulatory mechanisms of phosphoinositide 3-kinases. Trends Biochem. Sci. 27, 426–432 (2002).
38. Okkenhaug, K. et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 297, 1031–1034 (2002).
39. Sasaki, T. et al. Function of PI3Kgamma in thymocyte development, T cell activa-tion, and neutrophil migration. Science 287, 1040–1046 (2000).
40. Stein, R.C. & Waterfield, M.D. PI3-kinase inhibition: a target for drug develop-ment? Mol. Med. Today 6, 347–357 (2000).
41. Yap, C.L. et al. Synergistic adhesive interactions and signaling mechanisms oper-ating between platelet glycoprotein Ib/IX and integrin alpha IIbbeta 3. Studies in human platelets ans transfected Chinese hamster ovary cells. J. Biol. Chem. 275, 41377–41388 (2000).
42. Jackson, S.P. et al. Preparation of morpholinyl- and pyridinyl-substituted hetero-bicyclic ketones as selective inhibitors of phosphoinositide 3-kinase beta for use against thrombosis. (International, WO 2003-IB4177, 2004).
43. Schoenwaelder, S.M. et al. Tyrosine kinases regulate the cytoskeletal attachment of integrin alpha IIb beta 3 (platelet glycoprotein IIb/IIIa) and the cellular retraction of fibrin polymers. J. Biol. Chem. 269, 32479–32487 (1994).
44. Lova, P. et al. A selective role for phosphatidylinositol 3,4,5-trisphosphate in the Gi-dependent activation of platelet Rap1B. J. Biol. Chem. 278, 131–138 (2003).
45. Woulfe, D., Jiang, H., Mortensen, R., Yang, J. & Brass, L.F. Activation of Rap1B by G(i) family members in platelets. J. Biol. Chem. 277, 23382–23390 (2002).
46. Foukas, L.C. et al. Direct effects of caffeine and theophylline on p110 delta and other phosphoinositide 3-kinases. Differential effects on lipid kinase and protein kinase activities. J. Biol. Chem. 277, 37124–37130 (2002).
47. Bang, C.J., Berstad, A. & Talstad, I. Incomplete reversal of enoxaparin-induced bleeding by protamine sulfate. Haemostasis 21, 155–160 (1991).
48. Herbert, J.M., Bernat, A. & Maffrand, J.P. Aprotinin reduces clopidogrel-induced prolongation of the bleeding time in the rat. Thromb. Res. 71, 433–441 (1993).
A R T I C L E S©
2005
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
emed
icin
e



















