
Phthalocyanine- and Calixarene-Templating Effect on the Catalytic Performance of Solid Supported...
13
1 23 Catalysis Letters ISSN 1011-372X Catal Lett DOI 10.1007/ s10562-011-0662-7 Phthalocyanine- and Calixarene- Templating Effect on the Catalytic Performance of Solid Supported Vanadates Grigoriy Sereda, Taejin Kim, Aubrey Jones, Hari Khatri, Christopher L. Marshall, H. Subramanian & Ranjit T. Koodali
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Phthalocyanine- and Calixarene-Templating Effect on the Catalytic Performance of Solid Supported...

1 23
Catalysis Letters ISSN 1011-372X Catal LettDOI 10.1007/s10562-011-0662-7
Phthalocyanine- and Calixarene-Templating Effect on the CatalyticPerformance of Solid SupportedVanadates
Grigoriy Sereda, Taejin Kim, AubreyJones, Hari Khatri, ChristopherL. Marshall, H. Subramanian & RanjitT. Koodali

1 23
Your article is protected by copyright and
all rights are held exclusively by Springer
Science+Business Media, LLC. This e-offprint
is for personal use only and shall not be self-
archived in electronic repositories. If you
wish to self-archive your work, please use the
accepted author’s version for posting to your
own website or your institution’s repository.
You may further deposit the accepted author’s
version on a funder’s repository at a funder’s
request, provided it is not made publicly
available until 12 months after publication.

Phthalocyanine- and Calixarene-Templating Effecton the Catalytic Performance of Solid Supported Vanadates
Grigoriy Sereda • Taejin Kim • Aubrey Jones •
Hari Khatri • Christopher L. Marshall •
H. Subramanian • Ranjit T. Koodali
Received: 22 April 2011 / Accepted: 28 June 2011
� Springer Science+Business Media, LLC 2011
Abstract Catalytic performance of solid supported mono-
vanadate catalysts toward oxidation of propane is signifi-
cantly affected by the templating effect of: (1) pretreatment
of the solid support with calixarene derivatives before
deposition of ammonium vanadate, (2) direct deposition of
vanadyl phthalocyanine as the vanadate precursor. This
effect is believed to be linked to the accessibility of the
active sites, and was studied in comparison with a reference
mesoporous support.
Keywords Supported catalysts � Oxidation �Heterogeneous catalysis � Catalysis
1 Introduction
Vanadium-based catalysts have been the center of attention
due to their activity toward industrially important synthetic
processes such as manufacturing sulfuric acid and alkenes.
The wide range of available coordination numbers and
oxidation states of vanadium (ranging from -3 to ?5)
significantly expands the potential of vanadium-based
catalysts toward various oxidative and reductive organic
transformations [1]. Catalysts containing vanadium oxide
supported on metal oxides, Al2O3, SiO2, TiO2, and ZrO2,
are employed in several industrial heterogeneous catalytic
processes, such as partial oxidation of methanol to form-
aldehyde, oxidation of o-xylene to phthalic anhydride,
ammoxidation of alkyl aromatics, oxidation of sulfur
dioxide to sulfur trioxide, selective catalytic reduction
(SCR) of nitrogen oxides (NOx) with ammonia to nitrogen
and water, and oxidative dehydrogenation of alkanes to
alkenes [2–7]. Mechanical strength, thermal stability, better
control of the catalyst’s structure, and therefore good
selectivity are the attributes of supported vanadium oxide
catalysts over bulk V2O5. The metal oxide-support inter-
actions in supported vanadium oxide catalysts affect both
the redox properties and the dispersion of the active sites
[8, 9]. Catalytic studies of vanadium oxide supported by
ZrO2, Al2O3, and SiO2 toward oxidative dehydrogenation
of methanol and alkanes have shown that the nature of the
solid support has a significant influence on the catalytic
performance of the material [10]. At low (\0.52 V/nm2)
catalyst loadings, vanadium oxide is more likely to be
highly dispersed, forming after calcination in air isolated
tetrahedral vanadate (V?5) species, while with the
increased loading, vanadium oxide species may form
polymeric (dinuclear, trinuclear, etc.) two-dimensional
networks of distorted tetrahedral and square-pyramidal
G. Sereda (&) � A. Jones � H. Khatri � H. Subramanian �R. T. Koodali
Department of Chemistry, The University of South Dakota,
414 E. Clark St., Vermillion, SD 57069, USA
e-mail: [email protected]
A. Jones
e-mail: [email protected]
H. Khatri
e-mail: [email protected]
H. Subramanian
e-mail: [email protected]
R. T. Koodali
e-mail: [email protected]
T. Kim � C. L. Marshall
Chemical Sciences and Engineering Division, Argonne National
Laboratory, 9700 S. Cass Avenue, Argonne, IL 60439, USA
e-mail: [email protected]
C. L. Marshall
e-mail: [email protected]
123
Catal Lett
DOI 10.1007/s10562-011-0662-7
Author's personal copy

oxovanadium clusters. At high loadings, three dimensional
V2O5 crystallites in octahedral coordination become abun-
dant [11]. It has been documented that monolayer- or sub-
monolayer of oxidized vanadium contains active sites
towards many reactions, whereas multilayer coverages are
inactive [12]. It was shown [13], that the V–O-solid support
bond is more significantly involved in the catalytic cycle of
oxidation, compared to V–O–V and V=O bonds. The dis-
persion of vanadium oxides in sub-monolayer to monolayer
quantities depends on the nature of solid support [13], nature
of sample preparation method (incipient wetness impreg-
nation [14, 15], grafting [16, 17], chemical vapor deposition
[18]), strength of support-vanadium oxide interactions,
loading density of vanadium oxide precursors [19], and the
type of ligand attached to the vanadium atom [20].
The distribution of the deposited vanadium oxide clus-
ters over the surface of the solid support can be charac-
terized by UV–vis Diffuse Reflectance Spectroscopy
(DRS), based on the known quantitative dependence of the
effective band gap energy on the number of vanadium
atoms in the clusters [21], and 51V solid state NMR [14],
which provide detailed information about the local coor-
dination number of the two dimensional surface vanadate
species on an oxide support. Deconvolution of the UV–vis
DRS Kubelka–Munk function with Gaussian functions was
introduced as an additional valuable tool for studying the
nature of vanadium oxide and vanadate species on the
surface of silica and mesoporous silica supports [22, 23].
Distribution of vanadium oxide deposited on zeolites was
studied by Temperature Programmed Reduction (TPR)
with H2 [24], Electron Spin Resonance (ESR), X-ray
Absorption Near Edge Structure (XANES), and photolu-
minescence spectroscopy [25]. There have been several
reported attempts to control deposition of vanadium oxide
by varying the nature of the precursor employed. In 2001, a
series of vanadium–calixarene complexes was synthesized
as molecular models for supported vanadate [26]. Recently,
the group of Alexander Katz has grafted mono- and dinu-
clear calixarene-bound vanadates to silica [27]. Calcination
of these materials led to silica-supported vanadates, whose
‘‘Edge energy’’ determined by UV–vis DRS depended on
the number of vanadium atoms in the calixarene-ligated
precursor. Unfortunately, catalytic performance of these
materials was not tested. Though the ‘‘Edge energy’’ shift
was most likely driven by at least partial formation of
calixarene-templated formation of divanadates during cal-
cination, different vanadium loadings used in the study led
to some ambiguity to the interpretation of the true nature of
the supported vanadia species. Further, a study of possible
mobility of once-deposited vanadium oxide (especially, in
the presence of moisture [28]) suggests that the structure of
the final material does not depend on the vanadium oxide
precursor [29]. It inspired us to thoroughly explore vanadia
supported catalysts with the same vanadium loadings, but
prepared differently. A recent 51V-NMR study [14]
revealed that the vanadium precursor affects the strength of
interaction between c-Al2O3 and deposited vanadate spe-
cies at the density of 4 V-atoms/nm2. Interestingly, neither
the structure of the surface vanadate species nor their
reducibility with H2 was significantly affected. Unfortu-
nately, catalytic performance of the prepared series of
materials was not tested toward any other reactions. While
a large body of research is done on c-alumina, practical
application of its more thermally stable h-form as solid
support for catalysts is worthy of a detailed study.
In this paper, we report how deposition of vanadate on
h-Al2O3, TiO2, and SiO2 is affected by three modes of
organic templating: (I) Use of vanadyl phthalocyanine 1 as
the vanadate precursor. (II) Complexation of the NH4VO3-
borne vanadium with octa-tert-butylcalix[8]arene 2, pre-
physisorbed on a solid support, and (III) Complexation of
the NH4VO3-borne vanadium with sodium calyx[8]arene-
octa-sulfonate 3, covalently pre-attached to a solid support.
2 Experimental Section
2.1 Preparation of Catalysts
Vanadyl phthalocyanine 1, solid supports, other chemicals
and solvents were obtained from commercial sources.
Octa-tert-butylcalix[8]arene 2 and sodium calix[8]arene-
octasulfonate 3 were synthesized by known procedures [30,
31]. For the non-templated reference materials (A01C,
A02C, T01C, and S01C, Table 1), vanadate was deposited
on the corresponding solid support, using NH4VO3–
H2C2O4 as vanadate precursor, according to the known
technique [32].
Vanadium loadings of 0.25 or 0.5 V/nm2 were targeted.
The prepared materials were dried in air at 200 �C for 2 h
and calcined in static air at 500 �C for 4 h. The actual
vanadium loads were calculated on the basis of surface
analysis of the final catalyst and presented in Table 2.
2.1.1 Deposition of Vanadyl Phthalocyanine
For the catalysts, templated by vanadyl phthalocyanine 1, a
calculated (for 0.25 or 0.5 V/nm2) amount of 1 in a frit
funnel was placed inside a Soxhlet extraction chamber. The
appropriate solid support (1 g) in 100 mL of chloroform
was placed in the evaporation part of the Soxhlet extractor.
Due to the low solubility of 1 in chloroform, the full
extraction typically required about 200 cycles (100 h). The
prepared materials were dried in air at 140 �C until the
constant weight (typically 1 h) and finally calcined in air at
500 �C for 4 h.
G. Sereda et al.
123
Author's personal copy
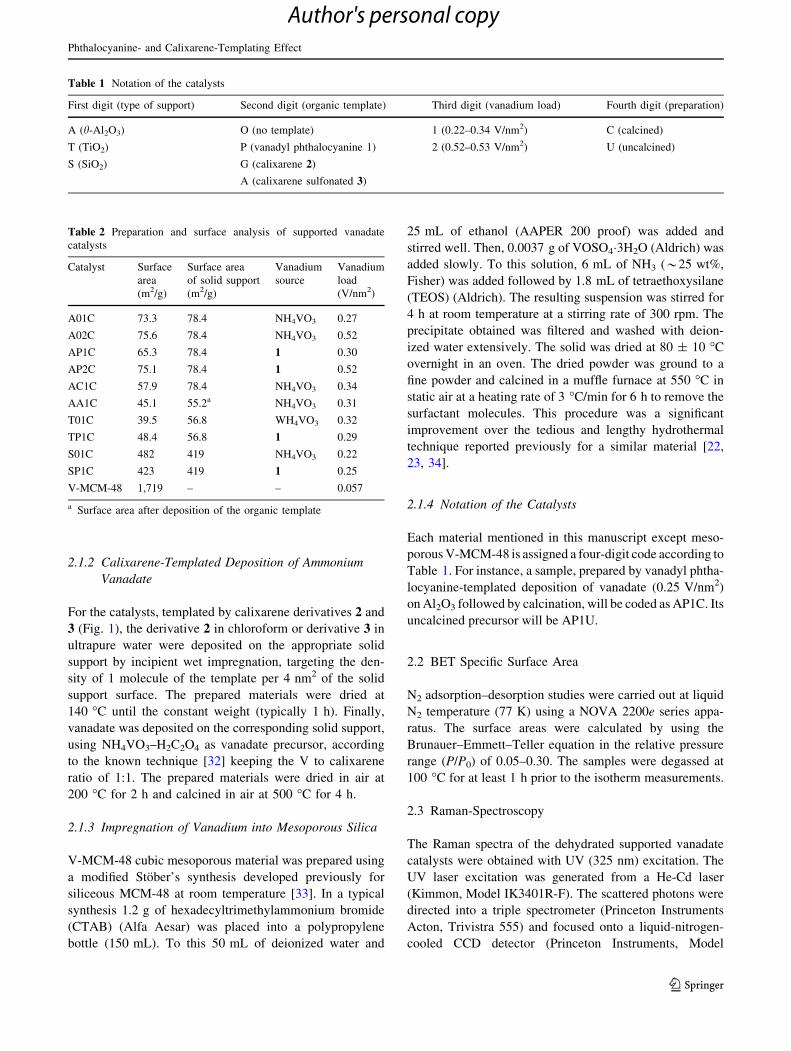
2.1.2 Calixarene-Templated Deposition of Ammonium
Vanadate
For the catalysts, templated by calixarene derivatives 2 and
3 (Fig. 1), the derivative 2 in chloroform or derivative 3 in
ultrapure water were deposited on the appropriate solid
support by incipient wet impregnation, targeting the den-
sity of 1 molecule of the template per 4 nm2 of the solid
support surface. The prepared materials were dried at
140 �C until the constant weight (typically 1 h). Finally,
vanadate was deposited on the corresponding solid support,
using NH4VO3–H2C2O4 as vanadate precursor, according
to the known technique [32] keeping the V to calixarene
ratio of 1:1. The prepared materials were dried in air at
200 �C for 2 h and calcined in air at 500 �C for 4 h.
2.1.3 Impregnation of Vanadium into Mesoporous Silica
V-MCM-48 cubic mesoporous material was prepared using
a modified Stober’s synthesis developed previously for
siliceous MCM-48 at room temperature [33]. In a typical
synthesis 1.2 g of hexadecyltrimethylammonium bromide
(CTAB) (Alfa Aesar) was placed into a polypropylene
bottle (150 mL). To this 50 mL of deionized water and
25 mL of ethanol (AAPER 200 proof) was added and
stirred well. Then, 0.0037 g of VOSO4�3H2O (Aldrich) was
added slowly. To this solution, 6 mL of NH3 (*25 wt%,
Fisher) was added followed by 1.8 mL of tetraethoxysilane
(TEOS) (Aldrich). The resulting suspension was stirred for
4 h at room temperature at a stirring rate of 300 rpm. The
precipitate obtained was filtered and washed with deion-
ized water extensively. The solid was dried at 80 ± 10 �C
overnight in an oven. The dried powder was ground to a
fine powder and calcined in a muffle furnace at 550 �C in
static air at a heating rate of 3 �C/min for 6 h to remove the
surfactant molecules. This procedure was a significant
improvement over the tedious and lengthy hydrothermal
technique reported previously for a similar material [22,
23, 34].
2.1.4 Notation of the Catalysts
Each material mentioned in this manuscript except meso-
porous V-MCM-48 is assigned a four-digit code according to
Table 1. For instance, a sample, prepared by vanadyl phtha-
locyanine-templated deposition of vanadate (0.25 V/nm2)
on Al2O3 followed by calcination, will be coded as AP1C. Its
uncalcined precursor will be AP1U.
2.2 BET Specific Surface Area
N2 adsorption–desorption studies were carried out at liquid
N2 temperature (77 K) using a NOVA 2200e series appa-
ratus. The surface areas were calculated by using the
Brunauer–Emmett–Teller equation in the relative pressure
range (P/P0) of 0.05–0.30. The samples were degassed at
100 �C for at least 1 h prior to the isotherm measurements.
2.3 Raman-Spectroscopy
The Raman spectra of the dehydrated supported vanadate
catalysts were obtained with UV (325 nm) excitation. The
UV laser excitation was generated from a He-Cd laser
(Kimmon, Model IK3401R-F). The scattered photons were
directed into a triple spectrometer (Princeton Instruments
Acton, Trivistra 555) and focused onto a liquid-nitrogen-
cooled CCD detector (Princeton Instruments, Model
Table 1 Notation of the catalysts
First digit (type of support) Second digit (organic template) Third digit (vanadium load) Fourth digit (preparation)
A (h-Al2O3) O (no template) 1 (0.22–0.34 V/nm2) C (calcined)
T (TiO2) P (vanadyl phthalocyanine 1) 2 (0.52–0.53 V/nm2) U (uncalcined)
S (SiO2) G (calixarene 2)
A (calixarene sulfonated 3)
Table 2 Preparation and surface analysis of supported vanadate
catalysts
Catalyst Surface
area
(m2/g)
Surface area
of solid support
(m2/g)
Vanadium
source
Vanadium
load
(V/nm2)
A01C 73.3 78.4 NH4VO3 0.27
A02C 75.6 78.4 NH4VO3 0.52
AP1C 65.3 78.4 1 0.30
AP2C 75.1 78.4 1 0.52
AC1C 57.9 78.4 NH4VO3 0.34
AA1C 45.1 55.2a NH4VO3 0.31
T01C 39.5 56.8 WH4VO3 0.32
TP1C 48.4 56.8 1 0.29
S01C 482 419 NH4VO3 0.22
SP1C 423 419 1 0.25
V-MCM-48 1,719 – – 0.057
a Surface area after deposition of the organic template
Phthalocyanine- and Calixarene-Templating Effect
123
Author's personal copy
7439-0002). The laser power delivered to the sample was
2 mW. The catalysts were maintained in loose powder
form in a fluidized bed reactor. The catalysts where ini-
tially dehydrated at 500 �C for 1 h in flowing 5% O2/N2
(60 mL/min, Linde gas) and the Raman spectra of
the dehydrated samples were collected after cooling the
catalysts back to room temperature in the flowing He
(60 mL/min, Airgas, ultrapure carrier grade). Due to the
low vanadium loads in studied materials, we increased the
signal/noise ratio by performing 6 scans at the acquisition
time of 10 min.
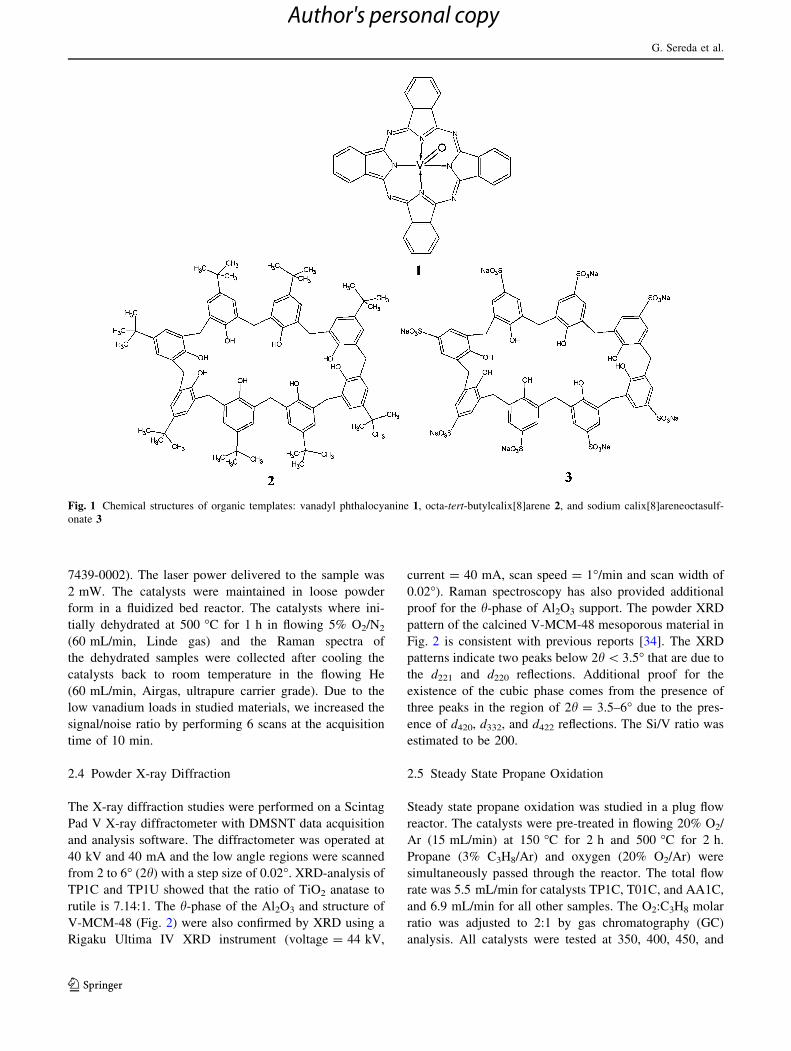
2.4 Powder X-ray Diffraction
The X-ray diffraction studies were performed on a Scintag
Pad V X-ray diffractometer with DMSNT data acquisition
and analysis software. The diffractometer was operated at
40 kV and 40 mA and the low angle regions were scanned
from 2 to 6� (2h) with a step size of 0.02�. XRD-analysis of
TP1C and TP1U showed that the ratio of TiO2 anatase to
rutile is 7.14:1. The h-phase of the Al2O3 and structure of
V-MCM-48 (Fig. 2) were also confirmed by XRD using a
Rigaku Ultima IV XRD instrument (voltage = 44 kV,
current = 40 mA, scan speed = 1�/min and scan width of
0.02�). Raman spectroscopy has also provided additional
proof for the h-phase of Al2O3 support. The powder XRD
pattern of the calcined V-MCM-48 mesoporous material in
Fig. 2 is consistent with previous reports [34]. The XRD
patterns indicate two peaks below 2h\ 3.5� that are due to
the d221 and d220 reflections. Additional proof for the
existence of the cubic phase comes from the presence of
three peaks in the region of 2h = 3.5–6� due to the pres-
ence of d420, d332, and d422 reflections. The Si/V ratio was
estimated to be 200.
2.5 Steady State Propane Oxidation
Steady state propane oxidation was studied in a plug flow
reactor. The catalysts were pre-treated in flowing 20% O2/
Ar (15 mL/min) at 150 �C for 2 h and 500 �C for 2 h.
Propane (3% C3H8/Ar) and oxygen (20% O2/Ar) were
simultaneously passed through the reactor. The total flow
rate was 5.5 mL/min for catalysts TP1C, T01C, and AA1C,
and 6.9 mL/min for all other samples. The O2:C3H8 molar
ratio was adjusted to 2:1 by gas chromatography (GC)
analysis. All catalysts were tested at 350, 400, 450, and
Fig. 1 Chemical structures of organic templates: vanadyl phthalocyanine 1, octa-tert-butylcalix[8]arene 2, and sodium calix[8]areneoctasulf-
onate 3
G. Sereda et al.
123
Author's personal copy
500 �C. Reactions were carried out in a quartz tube and the
reactor was packed with quartz wool above and below the
catalyst bed. The catalyst bed temperature was maintained
with external temperature control and the catalysts bed
temperature was measured with a thermocouple in contact
with the sample. The reaction products were analyzed
using a GC equipped with a TCD or FID. For all samples
the space velocity was maintained in the range of
0.011–0.013 mol C3H8 mol V-1 s-1. For samples TP1C,
T01C, and AA1C, the propane flow rate was 0.16 mL/min.
For all other samples, propane flow rate was 0.21 mL/min.
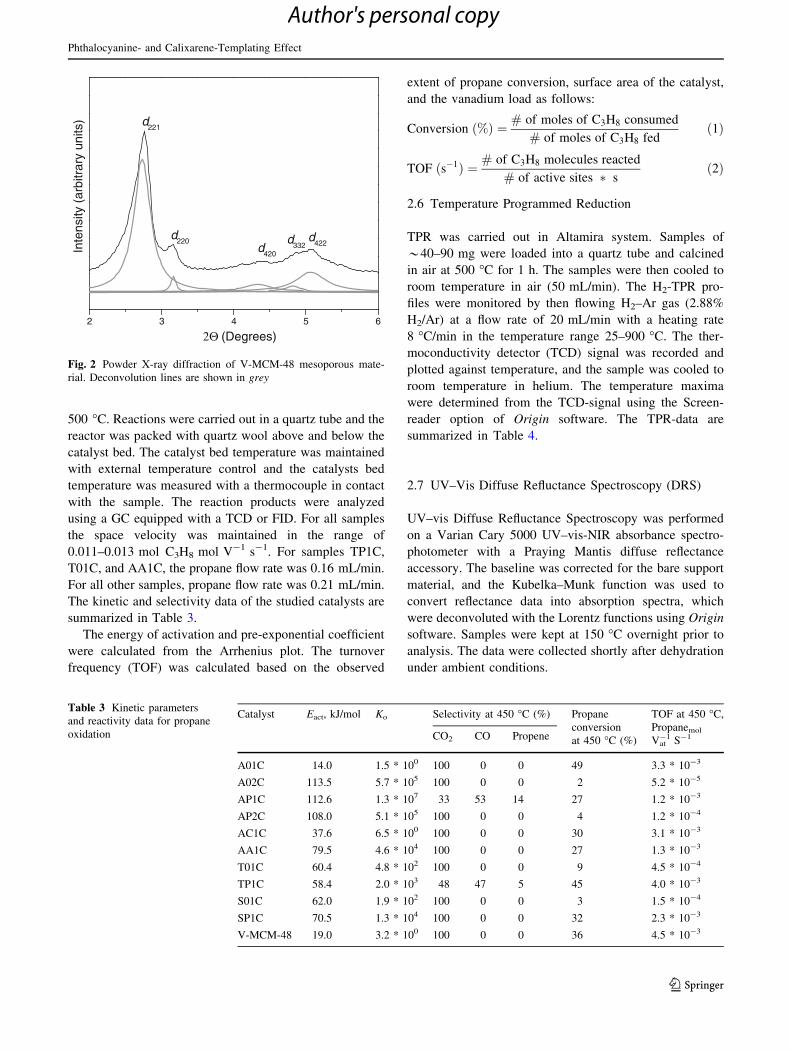
The kinetic and selectivity data of the studied catalysts are
summarized in Table 3.
The energy of activation and pre-exponential coefficient
were calculated from the Arrhenius plot. The turnover
frequency (TOF) was calculated based on the observed
extent of propane conversion, surface area of the catalyst,
and the vanadium load as follows:
Conversion ð%Þ ¼ # of moles of C3H8 consumed
# of moles of C3H8 fedð1Þ
TOF ðs�1Þ ¼ # of C3H8 molecules reacted
# of active sites � sð2Þ
2.6 Temperature Programmed Reduction
TPR was carried out in Altamira system. Samples of
*40–90 mg were loaded into a quartz tube and calcined
in air at 500 �C for 1 h. The samples were then cooled to
room temperature in air (50 mL/min). The H2-TPR pro-
files were monitored by then flowing H2–Ar gas (2.88%
H2/Ar) at a flow rate of 20 mL/min with a heating rate
8 �C/min in the temperature range 25–900 �C. The ther-
moconductivity detector (TCD) signal was recorded and
plotted against temperature, and the sample was cooled to
room temperature in helium. The temperature maxima
were determined from the TCD-signal using the Screen-
reader option of Origin software. The TPR-data are
summarized in Table 4.
2.7 UV–Vis Diffuse Refluctance Spectroscopy (DRS)
UV–vis Diffuse Refluctance Spectroscopy was performed
on a Varian Cary 5000 UV–vis-NIR absorbance spectro-
photometer with a Praying Mantis diffuse reflectance
accessory. The baseline was corrected for the bare support
material, and the Kubelka–Munk function was used to
convert reflectance data into absorption spectra, which
were deconvoluted with the Lorentz functions using Origin
software. Samples were kept at 150 �C overnight prior to
analysis. The data were collected shortly after dehydration
under ambient conditions.
2 3 4 5 6
d332d
420
d422
d220
d221
2Θ (Degrees)
Inte
nsity
(ar
bitr
ary
units
)
Fig. 2 Powder X-ray diffraction of V-MCM-48 mesoporous mate-
rial. Deconvolution lines are shown in grey
Table 3 Kinetic parameters
and reactivity data for propane
oxidation
Catalyst Eact, kJ/mol Ko Selectivity at 450 �C (%) Propane
conversion
at 450 �C (%)
TOF at 450 �C,
Propanemol
Vat-1 S-1CO2 CO Propene
A01C 14.0 1.5 * 100 100 0 0 49 3.3 * 10-3
A02C 113.5 5.7 * 105 100 0 0 2 5.2 * 10-5
AP1C 112.6 1.3 * 107 33 53 14 27 1.2 * 10-3
AP2C 108.0 5.1 * 105 100 0 0 4 1.2 * 10-4
AC1C 37.6 6.5 * 100 100 0 0 30 3.1 * 10-3
AA1C 79.5 4.6 * 104 100 0 0 27 1.3 * 10-3
T01C 60.4 4.8 * 102 100 0 0 9 4.5 * 10-4
TP1C 58.4 2.0 * 103 48 47 5 45 4.0 * 10-3
S01C 62.0 1.9 * 102 100 0 0 3 1.5 * 10-4
SP1C 70.5 1.3 * 104 100 0 0 32 2.3 * 10-3
V-MCM-48 19.0 3.2 * 100 100 0 0 36 4.5 * 10-3
Phthalocyanine- and Calixarene-Templating Effect
123
Author's personal copy
2.8 Elemental Analysis
Elemental analyses were performed on the Exeter Analyt-
ical, Inc CE-440 elemental analyzer configured for the C,
N, S mode (oxygen combustion oven at 1,080 �C and
copper reduction oven at 820 �C).
3 Results and Discussion
3.1 h-Al2O3-Supported Vanadate
The specific catalytic activity, expressed as TOF, product
selectivity, and kinetics parameters for steady state pro-
pane oxidation over the V2O5/h-Al2O3 catalysts are
summarized in Table 3. First, we found that catalytic
activity and selectivity of supported vanadate catalysts at
very low vanadium loading (0.27–0.30 V/nm2) for the
oxidation of propane significantly depends on the vana-
date precursor used for the deposition. A reference sample
(A01C) prepared by incipient wet impregnation of aque-
ous NH4VO3–H2C2O4 effectively catalyzed full combus-
tion of propane to CO2 with high conversion (49%). Such
behavior corresponds to low values of apparent activation
energy (*14 kJ/mol) and low pre-exponential coefficient
(*1.5) characteristic for the presence of highly active
metal sites with low accessibility. Presence of different
types of Al–OH binding sites on the surface of h-Al2O3 is
consistent with a reported theoretical and experimental
study [35], which suggested that at the surface density of
0.16 V/nm2, vanadium tends to bind with the terminal
Al–OH configuration, which is the most basic among all
possible AlmOH configurations (m = 1, 2, 3). The possi-
bility of different modes of vanadate binding to c-Al2O3
at higher surface densities (4 V/nm2) was also proved by
the 51V-NMR [14].
3.1.1 Vanadyl Phthalocyanine Precursor
We selected to deposit a monomolecular or submonomo-
lecular density layer of this precursor to prevent formation
of vanadium clusters during calcination and focus on the
interaction of the isolated vanadate species with the solid
support. Elemental analysis did not reveal any significant
amounts of residual carbon and nitrogen in the catalysts.
Specifically, for sample AP1C (%C: 0.09, 0.03; %N: 0.01,
0.00), and AP2C (%C: 0.11, 0.12; %N: 0.01, -0.04).
Organic template effect of the phthalocyanine ligand led to
dramatic increases in both the activation energy and the
pre-exponential coefficient with corresponding decreases in
the propane conversion and TOF of the calcined sample
(sample AP1C, Table 3). Most importantly, the templating
effect of the phthalocyanine ligand significantly changed
the selectivity for propane oxidation. While A01C cata-
lyzed only combustion to CO2, at 450 �C, AP1C showed
53% and 14% selectivity for CO and propene, respectively.
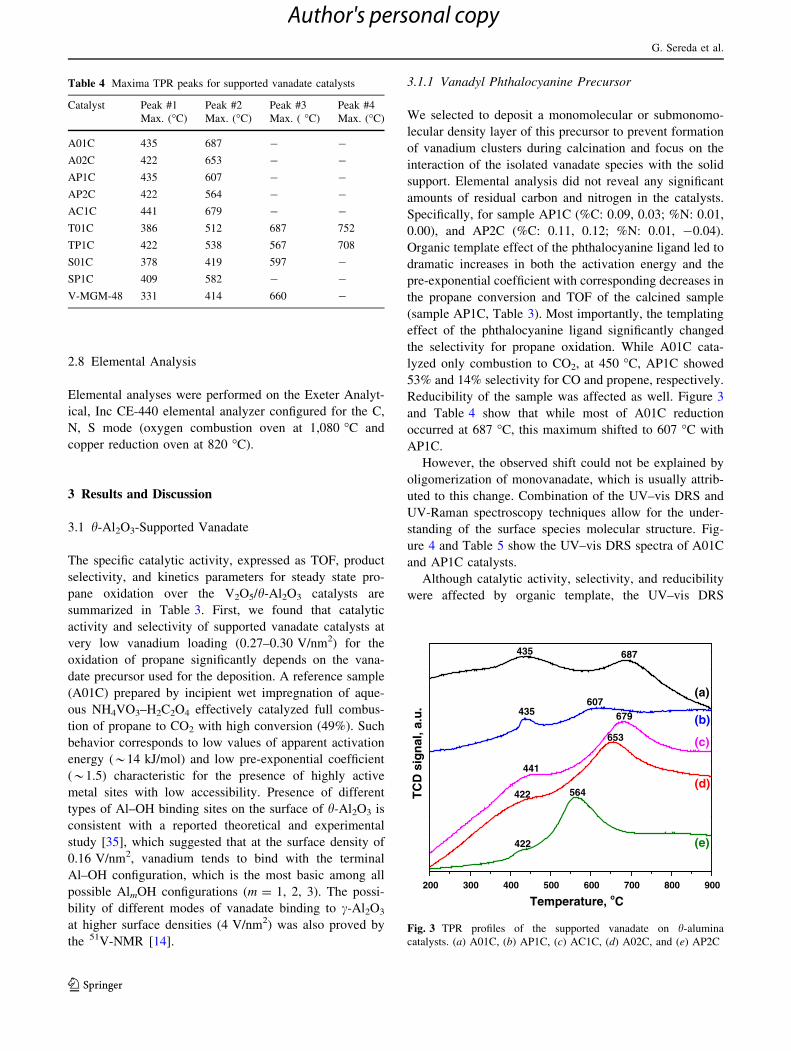
Reducibility of the sample was affected as well. Figure 3
and Table 4 show that while most of A01C reduction
occurred at 687 �C, this maximum shifted to 607 �C with
AP1C.
However, the observed shift could not be explained by
oligomerization of monovanadate, which is usually attrib-
uted to this change. Combination of the UV–vis DRS and
UV-Raman spectroscopy techniques allow for the under-
standing of the surface species molecular structure. Fig-
ure 4 and Table 5 show the UV–vis DRS spectra of A01C
and AP1C catalysts.
Although catalytic activity, selectivity, and reducibility
were affected by organic template, the UV–vis DRS
Table 4 Maxima TPR peaks for supported vanadate catalysts
Catalyst Peak #1
Max. (�C)
Peak #2
Max. (�C)
Peak #3
Max. ( �C)
Peak #4
Max. (�C)
A01C 435 687 - -
A02C 422 653 - -
AP1C 435 607 - -
AP2C 422 564 - -
AC1C 441 679 - -
T01C 386 512 687 752
TP1C 422 538 567 708
S01C 378 419 597 -
SP1C 409 582 - -
V-MGM-48 331 414 660 -
200 300 400 500 600 700 800 900
679
441
(c)653
422(d)
(e)
564
422
607435
435 687
(a)
(b)
TC
D s
ign
al, a
.u.
Temperature, oC
Fig. 3 TPR profiles of the supported vanadate on h-alumina
catalysts. (a) A01C, (b) AP1C, (c) AC1C, (d) A02C, and (e) AP2C
G. Sereda et al.
123
Author's personal copy
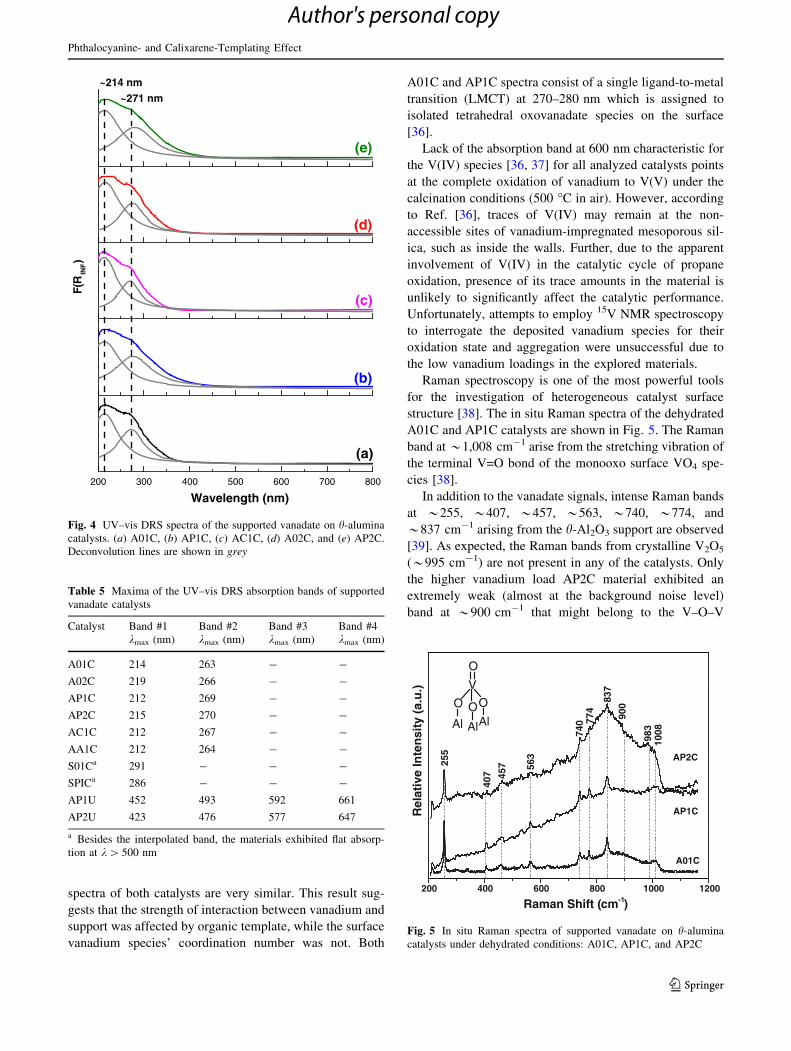
spectra of both catalysts are very similar. This result sug-
gests that the strength of interaction between vanadium and
support was affected by organic template, while the surface
vanadium species’ coordination number was not. Both
A01C and AP1C spectra consist of a single ligand-to-metal
transition (LMCT) at 270–280 nm which is assigned to
isolated tetrahedral oxovanadate species on the surface
[36].
Lack of the absorption band at 600 nm characteristic for
the V(IV) species [36, 37] for all analyzed catalysts points
at the complete oxidation of vanadium to V(V) under the
calcination conditions (500 �C in air). However, according
to Ref. [36], traces of V(IV) may remain at the non-
accessible sites of vanadium-impregnated mesoporous sil-
ica, such as inside the walls. Further, due to the apparent
involvement of V(IV) in the catalytic cycle of propane
oxidation, presence of its trace amounts in the material is
unlikely to significantly affect the catalytic performance.
Unfortunately, attempts to employ 15V NMR spectroscopy
to interrogate the deposited vanadium species for their
oxidation state and aggregation were unsuccessful due to
the low vanadium loadings in the explored materials.
Raman spectroscopy is one of the most powerful tools
for the investigation of heterogeneous catalyst surface
structure [38]. The in situ Raman spectra of the dehydrated
A01C and AP1C catalysts are shown in Fig. 5. The Raman
band at *1,008 cm-1 arise from the stretching vibration of
the terminal V=O bond of the monooxo surface VO4 spe-
cies [38].
In addition to the vanadate signals, intense Raman bands
at *255, *407, *457, *563, *740, *774, and
*837 cm-1 arising from the h-Al2O3 support are observed
[39]. As expected, the Raman bands from crystalline V2O5
(*995 cm-1) are not present in any of the catalysts. Only
the higher vanadium load AP2C material exhibited an
extremely weak (almost at the background noise level)
band at *900 cm-1 that might belong to the V–O–V
200 300 400 500 600 700 800
~214 nm~271 nm
F(R
INF)
Wavelength (nm)
(e)
(d)
(b)
(a)
(c)
Fig. 4 UV–vis DRS spectra of the supported vanadate on h-alumina
catalysts. (a) A01C, (b) AP1C, (c) AC1C, (d) A02C, and (e) AP2C.
Deconvolution lines are shown in grey
Table 5 Maxima of the UV–vis DRS absorption bands of supported
vanadate catalysts
Catalyst Band #1
kmax (nm)
Band #2
kmax (nm)
Band #3
kmax (nm)
Band #4
kmax (nm)
A01C 214 263 - -
A02C 219 266 - -
AP1C 212 269 - -
AP2C 215 270 - -
AC1C 212 267 - -
AA1C 212 264 - -
S01Ca 291 - - -
SPICa 286 - - -
AP1U 452 493 592 661
AP2U 423 476 577 647
a Besides the interpolated band, the materials exhibited flat absorp-
tion at k[ 500 nm
200 400 600 800 1000 1200
1008
900
AP2C
AP1C
457
407
563
740 77
4
983
837
255
A01C
Rel
ativ
e In
ten
sity
(a.
u.)
Raman Shift (cm-1)
AlAlAl
OOO
V
O
Fig. 5 In situ Raman spectra of supported vanadate on h-alumina
catalysts under dehydrated conditions: A01C, AP1C, and AP2C
Phthalocyanine- and Calixarene-Templating Effect
123
Author's personal copy

stretch. However, assignment of this band in literature is
controversial. While originally this band was attributed
to the V–O–V stretching vibration [40], recent research
[41, 42] supports its assignment to the V–O-support group.
The 325 nm excitation line is located sufficiently away
from the ligand-to-vanadium charge transfer occurring at
222 nm to cause significant resonance enhancement of the
monovanadate V=O vibration that might skew the con-
clusions. It is reported [43] that even excitation at 244 nm
allows to observe the V–O–V Raman bands, albeit leading
to resonance enhancement of one out of three V=O bands.
The results of the Raman spectra coincide with UV–vis
DRS results and provide no evidence of vanadate oligo-
merization, which is not surprising for such low vanadate
loads. Therefore, presence of the phthalocyanine ligands
does not make polymerized oxovanadium, but rather
affects local structure of the deposited reaction sites. The
templating effect of the phthalocyanine ligands was found
to be much less significant at twice the vanadium load
(0.52 V/nm2), when initial active sites on the alumina
surface become saturated, and selectivity of vanadate
deposition decreases. Thus, neither the activation energy
nor the pre-exponential coefficient or selectivity for oxi-
dation of propane were significantly different for the A02C
(reference) and AP2C (phthalocyanine-templated catalyst,
Table 3). In contrast to the AP1C catalyst, the catalytic
activity and selectivity of AP2C catalyst exhibits lower
activity (conversion and TOF) and 100% selectivity to
CO2. The TPR-profiles of AP2C and AO2C (Fig. 3) show
that most of A02C reduction occurred at 653 �C and this
maximum shifted to 564 �C with AP2C. The maximum at
564 �C for AP2C is also much lower than for AP1C. In
conclusion, a small change of local structure created by
increasing the vanadium surface density dramatically
affected the kinetics values, catalytic activity, and reduc-
ibility of the catalyst. However, the UV–vis DRS and
Raman spectra showed that both A02C and AP2C have a
similar band (*270 nm) with A01C and AP1C and the
Raman spectrum of AP2C possesses similar bands with the
AP1C spectrum shown in Fig. 5. Based on the UV–vis
DRS and Raman spectra, we can conclude that the series of
phthalocyanine-templated catalysts possess only the iso-
lated tetrahedral oxovanadate species and these surface
structures are very similar to the reference samples. From a
separate experiment, we can infer that the difference in the
templating effect on the vanadate deposition for the
0.27–0.30 V/nm2 (catalysts A01C, AP1C) and 0.52 V/nm2
(catalysts A02C, AP2C) was not caused by the aggregation
of the deposited vanadyl phthalocyanine precursors
(materials AP1U and AP2U). UV–vis DRS-spectra of
AP1U and AP2U contain a very weak Q-band (*650 nm)
of the same intensity, which is not characteristic for the
phthalocyanine aggregation in solution (Table 5) [44].
3.1.2 Calixarene–Ammonium Vanadate Precursor
Next, we explored how pretreatment of the alumina surface
with macrocyclic octa-tert-butylcalix[8]arene 2 at the
density of 1 molecule/4 nm2 before the wet impregnation
with NH4VO3 (0.25 V/nm2) affects formation of the van-
adate active sites. As opposed to a pre-prepared vanadium
macrocyclic complex (vanadyl phthalocyanine), complex-
ation of the phenol groups of the physisorbed 2 with
vanadium is less likely to prevent clustering of the vana-
date species during calcination, but was expected to alter
the mode of vanadium interaction with the solid support.
We employed the calixarene–ammonium monovanadate
system as the precursor alternative to vanadyl phthalocy-
anine only for the h-Al2O3 solid support, whose basicity
strengthens interaction with acidic calixarenes. Comparing
to AP1C (vanadyl phthalocyanine), the activation energy
and pre-exponential coefficient of AC1C are much lower
and only CO2 was produced on both catalysts. Neither TPR
nor UV–vis DRS revealed significant changes in the
characteristics of AC1C versus the reference A01C.
In addition to macrocyclic octa-tert-butylcalix[8]arene 2
and vanadyl phthalocyanine 1 organic templates, we
investigated calixarene sulfonate (AA1C) effect on the
catalytic activity and kinetic parameters of oxidation.
Elemental analysis of the AA1C catalyst did not reveal any
significant amounts of residual carbon and sulfur (%C:
0.28, 0.23; %S: 0.06, 0.08). As expected, sodium
calix[8]areneoctasulfonate 3, which is able to bind to alu-
mina chemically due to the salt formation, showed higher
activation energy and pre-exponential coefficient values
than the physisorbed octa-tert-butylcalix[8]arene catalyst
(AC1C). However, these values were much lower than for
the phthalocyanine-templated catalyst (AP1C), and neither
CO nor propene was detected among the reaction products
at 450 �C. The UV–vis DRS spectra for this series of
materials were very similar, which suggests the presence of
exclusively isolated tetrahedral oxovanadate on the surface
(Table 5, spectra not shown for brevity). In summary,
activity of supported vanadate on h-alumina samples for
propane oxidation decreased in the following order:
A01C [ AP1C, AC1C, AA1C [ A02C, AP2C, while for-
mation of CO and propene was observed only for the
catalyst templated by the phthalocyanine ligand (AP1C).
3.2 TiO2-Supported Vanadate
3.2.1 Vanadyl Phthalocyanine Precursor
Titania-supported vanadate catalysts were studied to com-
pare the templating effect with the Al2O3-series. Similarly,
the vanadyl phthalocyanine precursor was selected to
G. Sereda et al.
123
Author's personal copy
minimize oligomerization of monovanadates. Elemental
analysis of TP1C did not reveal any significant amounts of
residual carbon and nitrogen in the catalysts (%C: 0.01,
0.03; %N: -0.10, 0.02). Table 3 shows that the activation
energy for TP1C catalyst, prepared with the phthalocyanine
precursor, is close to the one of T01C (no template), but the
pre-exponential coefficient had nearly quadrupled. The
catalytic activity increased from 9 to 45% with introduction
of the phthalocyanine template. The lack of significant
template effect on the activation energy and increased
propane conversion and TOF for the TiO2-supported cat-
alysts was quite contrasting with its presence in the Al2O3-
series. Importantly, the selectivity trend for CO2, CO, and
propene was similar to the Al2O3 supported catalyst. Thus,
selectivity at 450 �C had shifted from 100% for the refer-
ence combustion catalyst (T01C) to 47% CO and 5%
propene (Table 3), which was similar to the Al2O3-series
trend. Therefore, shift of the reaction selectivity toward
formation of propene and CO can be clearly linked to the
templating effect of vanadyl phthalocyanine.
Figure 6 shows the reduction profiles of titania sup-
ported vanadate catalysts. The TPR profile of T01C sample
is added as a reference. The profile of TP1C shows
reduction peaks at 422, 538, 567, and 708 �C. These values
are higher than for the reference T01C, which suggests
lower reducibility of TP1C versus the reference T01C.
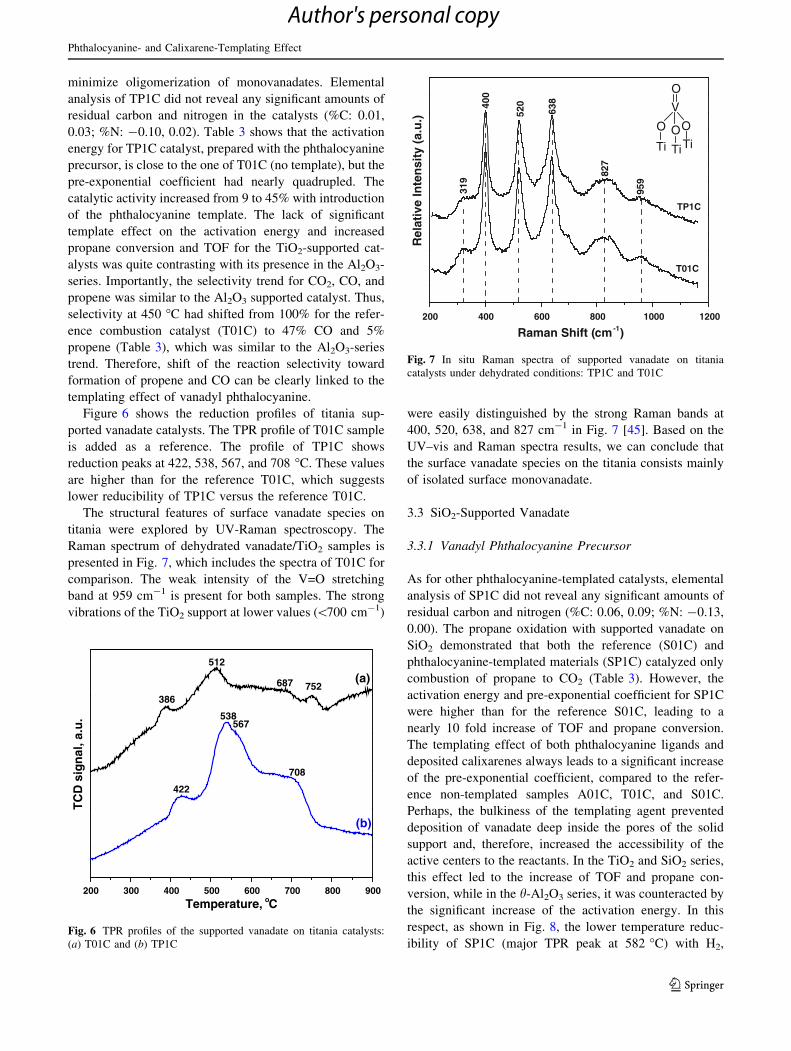
The structural features of surface vanadate species on
titania were explored by UV-Raman spectroscopy. The
Raman spectrum of dehydrated vanadate/TiO2 samples is
presented in Fig. 7, which includes the spectra of T01C for
comparison. The weak intensity of the V=O stretching
band at 959 cm-1 is present for both samples. The strong
vibrations of the TiO2 support at lower values (\700 cm-1)
were easily distinguished by the strong Raman bands at
400, 520, 638, and 827 cm-1 in Fig. 7 [45]. Based on the
UV–vis and Raman spectra results, we can conclude that
the surface vanadate species on the titania consists mainly
of isolated surface monovanadate.
3.3 SiO2-Supported Vanadate
3.3.1 Vanadyl Phthalocyanine Precursor
As for other phthalocyanine-templated catalysts, elemental
analysis of SP1C did not reveal any significant amounts of
residual carbon and nitrogen (%C: 0.06, 0.09; %N: -0.13,
0.00). The propane oxidation with supported vanadate on
SiO2 demonstrated that both the reference (S01C) and
phthalocyanine-templated materials (SP1C) catalyzed only
combustion of propane to CO2 (Table 3). However, the
activation energy and pre-exponential coefficient for SP1C
were higher than for the reference S01C, leading to a
nearly 10 fold increase of TOF and propane conversion.
The templating effect of both phthalocyanine ligands and
deposited calixarenes always leads to a significant increase
of the pre-exponential coefficient, compared to the refer-
ence non-templated samples A01C, T01C, and S01C.
Perhaps, the bulkiness of the templating agent prevented
deposition of vanadate deep inside the pores of the solid
support and, therefore, increased the accessibility of the
active centers to the reactants. In the TiO2 and SiO2 series,
this effect led to the increase of TOF and propane con-
version, while in the h-Al2O3 series, it was counteracted by
the significant increase of the activation energy. In this
respect, as shown in Fig. 8, the lower temperature reduc-
ibility of SP1C (major TPR peak at 582 �C) with H2,
200 300 400 500 600 700 800 900
567
708
422
512
538
386
687 752(a)
(b)
TC
D s
ign
al, a
.u.
Temperature, oC
Fig. 6 TPR profiles of the supported vanadate on titania catalysts:
(a) T01C and (b) TP1C
200 400 600 800 1000 1200
TiTiTi
OOO
V
959
827
319
638
520
TP1C
T01C
Rel
ativ
e In
ten
sity
(a.
u.)
Raman Shift (cm-1)
400 O
Fig. 7 In situ Raman spectra of supported vanadate on titania
catalysts under dehydrated conditions: TP1C and T01C
Phthalocyanine- and Calixarene-Templating Effect
123
Author's personal copy
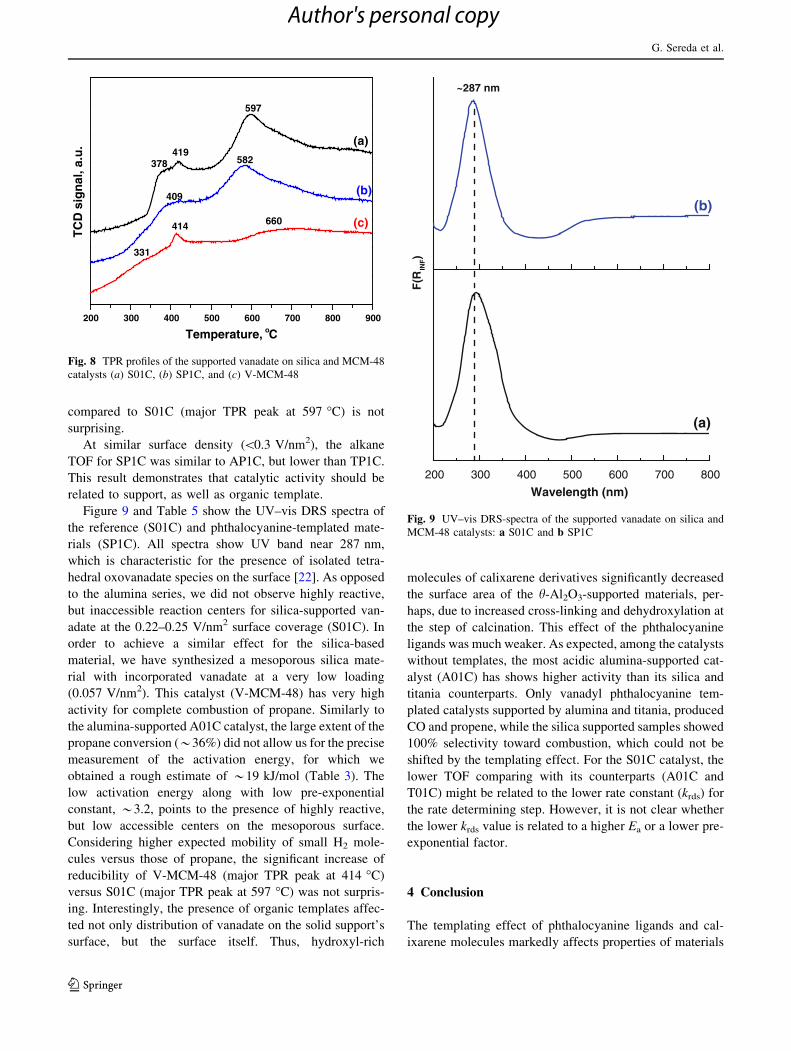
compared to S01C (major TPR peak at 597 �C) is not
surprising.
At similar surface density (\0.3 V/nm2), the alkane
TOF for SP1C was similar to AP1C, but lower than TP1C.
This result demonstrates that catalytic activity should be
related to support, as well as organic template.
Figure 9 and Table 5 show the UV–vis DRS spectra of
the reference (S01C) and phthalocyanine-templated mate-
rials (SP1C). All spectra show UV band near 287 nm,
which is characteristic for the presence of isolated tetra-
hedral oxovanadate species on the surface [22]. As opposed
to the alumina series, we did not observe highly reactive,
but inaccessible reaction centers for silica-supported van-
adate at the 0.22–0.25 V/nm2 surface coverage (S01C). In
order to achieve a similar effect for the silica-based
material, we have synthesized a mesoporous silica mate-
rial with incorporated vanadate at a very low loading
(0.057 V/nm2). This catalyst (V-MCM-48) has very high
activity for complete combustion of propane. Similarly to
the alumina-supported A01C catalyst, the large extent of the
propane conversion (*36%) did not allow us for the precise
measurement of the activation energy, for which we
obtained a rough estimate of *19 kJ/mol (Table 3). The
low activation energy along with low pre-exponential
constant, *3.2, points to the presence of highly reactive,
but low accessible centers on the mesoporous surface.
Considering higher expected mobility of small H2 mole-
cules versus those of propane, the significant increase of
reducibility of V-MCM-48 (major TPR peak at 414 �C)
versus S01C (major TPR peak at 597 �C) was not surpris-
ing. Interestingly, the presence of organic templates affec-
ted not only distribution of vanadate on the solid support’s
surface, but the surface itself. Thus, hydroxyl-rich
molecules of calixarene derivatives significantly decreased
the surface area of the h-Al2O3-supported materials, per-
haps, due to increased cross-linking and dehydroxylation at
the step of calcination. This effect of the phthalocyanine
ligands was much weaker. As expected, among the catalysts
without templates, the most acidic alumina-supported cat-
alyst (A01C) has shows higher activity than its silica and
titania counterparts. Only vanadyl phthalocyanine tem-
plated catalysts supported by alumina and titania, produced
CO and propene, while the silica supported samples showed
100% selectivity toward combustion, which could not be
shifted by the templating effect. For the S01C catalyst, the
lower TOF comparing with its counterparts (A01C and
T01C) might be related to the lower rate constant (krds) for
the rate determining step. However, it is not clear whether
the lower krds value is related to a higher Ea or a lower pre-
exponential factor.
4 Conclusion
The templating effect of phthalocyanine ligands and cal-
ixarene molecules markedly affects properties of materials
200 300 400 500 600 700 800 900
331
(c)660414
582419
378
409
597
(a)
(b)
TC
D s
ign
al, a
.u.
Temperature, oC
Fig. 8 TPR profiles of the supported vanadate on silica and MCM-48
catalysts (a) S01C, (b) SP1C, and (c) V-MCM-48
200 300 400 500 600 700 800
F(R
INF)
(b)
(a)
Wavelength (nm)
~287 nm
Fig. 9 UV–vis DRS-spectra of the supported vanadate on silica and
MCM-48 catalysts: a S01C and b SP1C
G. Sereda et al.
123
Author's personal copy

prepared by deposition of vanadia on h-Al2O3, TiO2, and
SiO2 with the density about 0.25 V/nm2. In all experi-
ments, we observed increasing pre-exponential coefficient
toward oxidation of propane due to the increasing acces-
sibility of the introduced vanadate reaction centers. The
trends of all kinetic parameters and selectivity in the series
h-Al2O3, TiO2, and SiO2 are significantly influenced by
the templating effect. Using vanadyl phthalocyanine as
the vanadate precursor significantly shifts selectivity of
h-Al2O3, TiO2-supported propane combustion catalysts
toward CO and propene. This effect is sensitive to the
vanadium load. A calixarene derivative covalently attached
to the solid support affects the deposition of vanadate more
than its physisorbed counterpart. However, the templated
effect of the phthalocyanine ligands in the vanadyl phtha-
locyanine precursor was the strongest observed. Incorpo-
ration of the monovanadate centers into the mesoporous
silica framework led to an efficient combustion catalyst
with a very low activation energy. This material is a
promising target for the further research on the templated
vanadate catalysts.
Acknowledgments This work has been supported by the Director,
Office of Science, Office of Biological & Environmental Research,
Biological Systems Science Division, of the U.S. Department of
Energy under Contract No. DE-FG02-08ER64624, NSF-CHE-
0722632, NSF (EPSCoR Grants No. 0554609 and 0903804), and by
the State of South Dakota, University of South Dakota (Research
Excellence Development award, U.Discover Program), NSF NPURC
Grant 0532242 (support for Aubrey Jones and purchase of the ele-
mental analyzer). We thank Mr. Bruce Gray for performing elemental
analyses. Taejin Kim and Christopher L. Marshall acknowledge the
U.S. Department of Energy, Office of Basic Energy Science, under
Contract W-31-109-ENG-38 and DE-FG02-03-ER15457.
References
1. Hirao T (1997) Chem Rev 97:2708
2. Forzatti P, Tronconi E, Busca G, Tittarelli P (1987) Catal Today
1:209
3. Nikolov V, Kissurski D, Anastasov A (1991) Catal Rev Sci Eng
33:1
4. Sanati M, Anderson A (1990) J Mol Catal 59:233
5. Prins WI, Numinga ZL (1993) Catal Today 16:187
6. Blasco T, Galli A, Nieto JML, Trifiro F (1997) J Catal 169:203
7. Mamedov EA, Corberan VC (1995) Appl Catal A 1:127
8. Deo G, Wachs IE, Haber J (1994) Crit Rev Surf Chem 4:87
9. Deo G, Wachs IE (1994) J Catal 146:323
10. Ertl G, Knozinger H, Weitkamp J (eds) (1999) Preparation of
solid catalysts. Wiley VCH, Weinheim
11. Arena F, Giordano N, Parmaliana A (1997) J Catal 166:66
12. Arena F, Fusteri F, Parmaliana A (1999) Appl Catal 176:177
13. Wachs IE, Weckhuysen BM (1997) Appl Catal A 157:67
14. Reddy BM, Reddy EP, Srinivas ST, Matikin VM, Nosov AV,
Lapina OB (1992) J Phys Chem 96:7076
15. Xie Sh, Iglesia E, Bell AT (2000) Langmuir 16:7162
16. Cozzolino M, Tesser R, Di Serio M, D’Onofrio P, Santacesaria E
(2007) Catal Today 128:191
17. Blates M, Collart O, Van Der Voort P, Vamsamt E (1999)
Langmuir 15:5841
18. Inamura K, Misono M, Okuhara T (1997) Appl Catal A 149:133
19. Zou H, Li M, Shen J, Auroux A (2003) J Therm Anal Calorim
72:209
20. Lewandowska AE, Banares MA, Khabibulin DF, Lapina OB
(2009) J Phys Chem C 113:20648
21. Kim H, Youn MH, Jung JCh, Song IK (2006) J Mol Catal A
252:252
22. Baltes M, Cassiers K, Van Der Voort P, Weckhuysen BM,
Schoonheydt RA, Vansant EF (2001) J Catal 197:160
23. Capek L, Adam J, Grygar T, Bulanek R, Vradman L, Kosova-
Kucerova G, Cicmanec P, Knotek P (2008) Appl Catal A 342:99
24. Roozeboom F, Miltlermeljer-Hazeleger MC, Moulljn JA,
Medema J, de Beer VHJ, Gellings PJ (1980) J Phys Chem
84:2783
25. Anpo M, Higashimoto S, Matsuoka M, Zhanpeisov N, Shioya Y,
Dzwigaj S, Che M (2003) Catal Today 78:211
26. Limberg Ch, Ziemer B, Magge C (2006) J Mol Catal A 251:34
27. de Silva N, Hwang SJ, Durkin KA, Katz A (2009) Chem Mater
21:1852
28. Jehng JM, Deo G, Weckhuysen BM, Wachs IE (1996) J Mol
Catal A 110:41
29. Machej T, Haber J, Turek AM, Wachs IE (1991) Appl Catal
70:115
30. Gutsche CD, Dhawan B, Hyun K, Muthukrishnan R (1981) J Am
Chem Soc 103:3782
31. Shinkai S, Araki K, Tsubaki T, Arimura T, Manabe O (1987)
J Chem Soc Perkin Trans I 2297
32. Reddy EP, Varma RS (2004) J Catal 221:93
33. Boote B, Subramanian H, Koodali RT (2007) Chem Commun
4543
34. Chang Z, Krishna RM, Xu J, Koodali RT, Kevan L (2001) Phys
Chem Chem Phys 3:1699
35. Kim HS, Zygmunt SA, Stair PC, Zapol P, Curtiss LA (2009)
J Phys Chem C 113:8836
36. Gao X, Wachs IE (2000) J Phys Chem B 104:1261
37. Luan Z, Xu J, He H, Klinowski J, Kevan L (1996) J Phys Chem
100:19595
38. Kim T, Wachs IE (2008) J Catal 255:197
39. Vuurman MA, Hardcastle FD, Wachs IE (1993) J Mol Catal
84:193
40. Went GT, Oyama ST, Bell AT (1990) J Phys Chem 94:4240
41. Wu Z, Kim H-S, Stair PC, Rugmini S, Jackson D (2005) J Phys
Chem B 109:2793
42. Wu ZL, Dai S, Overbury SH (2010) J Phys Chem C 114:412
43. Chua YT, Stair PC, Wachs IE (2001) J Phys Chem B 105:8600
44. Monahan AR, Brado JA, Peluca AF (1979) J Phys Chem 76:1994
45. Zhand J, Li M, Feng Z, Chen J, Li C (2006) J Phys Chem B
110:927
Phthalocyanine- and Calixarene-Templating Effect
123
Author's personal copy




















