
Photodissociation of HI and DI: Testing models for electronic structure via polarization of atomic...
10
Photodissociation of HI and DI: Testing models for electronic structure via polarization of atomic photofragments David N. Jodoin and Alex Brown a Department of Chemistry, University of Alberta, Edmonton AB T6G 2G2, Canada Received 23 May 2005; accepted 7 June 2005; published online 5 August 2005 The photodissociation dynamics of HI and DI are examined using time-dependent wave-packet techniques. The orientation and alignment parameters a Q K p are determined as a function of photolysis energy for the resulting ground-state I 2 P 3/2 and excited-state I 2 P 1/2 atoms. The a Q K p parameters describe the coherent and incoherent contributions to the angular momentum distributions from the A 1 1 , a 3 1 , and t 3 1 electronic states accessed by perpendicular excitation and the a 3 0 + state accessed by a parallel transition. The outcomes of the dynamics based on both shifted ab initio results and three empirical models for the potential-energy curves and transition dipole moments are compared and contrasted. It is demonstrated that experimental measurement of the a Q K p parameters for the excitation from the vibrational ground state v =0 would be able to distinguish between the available models for the HI potential-energy curves and transition dipole moments. The differences between the a Q K p parameters for the excitation from v = 0 stand in sharp contrast to the scalar properties, i.e., total cross section and I * branching fraction, which require experimental measurement of photodissociation from excited vibrational states v 0 to distinguish between the models. © 2005 American Institute of Physics. DOI: 10.1063/1.1989327 I. INTRODUCTION The hydrogen halides, HX X =F, Cl, Br, and I, repre- sent some of the simplest systems that dissociate to yield open-shell fragments. They provide model systems for study- ing molecular photodissociation dynamics on multiple potential-energy curves PECs including the effects of nona- diabatic coupling. In particular, the photodissociation process HX + h → H 2 S + X 2 P 3/2 → H 2 S + X 2 P 1/2 to yield the ground and spin-orbit excited-state halogen at- oms has been of both experimental and theoretical interest. 1–35 Following standard nomenclature, the two spin- orbit states of the halogen atom, X 2 P 3/2 and X 2 P 1/2 , are referred to as X and X * , respectively. For HF, HCl, HBr, and HI, the spin-orbit splittings between the two product chan- nels are 404, 882, 3685, and 7603 cm -1 , 36 respectively, and thus, the effects of increasing spin-orbit coupling on the dis- sociation process can be examined systematically. The goals of the experimental and theoretical studies have been to understand the roles of various electronic states involved in the excitation and the possible nonadiabatic tran- sitions that could take place between the excited PECs as the molecule fragments. In order to achieve these goals, the more recent experimental and theoretical studies have fo- cused on determining the branching fraction , which pro- vides the yield of spin-orbit excited atoms X * relative to the total yield, and on the lowest-order K =0 anisotropy param- eter , which provides information on the parallel and/or perpendicular nature of the electronic transitions contributing to the dissociation. While the measurement of the scalar properties, i.e., total cross section and , and the lowest- order vector property provide a wealth of information on the potential-energy curves, transition dipole moments, and, if applicable, nonadiabatic couplings underlying the dynam- ics, recent experiments 37–39 have determined the alignment of chlorine and bromine atoms resulting from the dissocia- tion of HCl and HBr, respectively. Also, calculations of the alignment and orientation of the resulting halogen atoms have been carried out for the photodissociation of HF, 40 HCl, 38,41 and HI. 42 The resulting alignment and orientation are extremely sensitive to the details of the potential-energy curves and transition dipole moments and, as discussed in Sec. III for HI, provide more detailed information than mea- surement of the total cross section, branching fraction, or the anisotropy parameter . The halogen atoms can have a preferred orientation and/or alignment in space since they possess angular mo- menta. The spatial distribution of the photofragment angular momenta can be described by the contributions from disso- ciation on a single PEC and from the interference from dis- sociation via multiple PECs. 43 The a Q K p parameters 44 can fully describe the polarization of the resulting atomic photo- fragments in the molecular frame. K and Q refer to the spa- tial distributions in the molecular frame. The symmetry of the transition dipole moments from the ground electronic state to the dissociating states is given by p and can be , , or , corresponding to the pure parallel, pure perpendicu- lar, or mixed parallel/perpendicular excitation. An equivalent set of anisotropy parameters describing the dissociation in the laboratory frame has also been introduced. 45,46 a Electronic mail: [email protected] THE JOURNAL OF CHEMICAL PHYSICS 123, 054301 2005 0021-9606/2005/1235/054301/10/$22.50 © 2005 American Institute of Physics 123, 054301-1 Downloaded 05 Aug 2005 to 129.128.168.123. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Photodissociation of HI and DI: Testing models for electronic structure via polarization of atomic...

Photodissociation of HI and DI: Testing models for electronic structurevia polarization of atomic photofragments
David N. Jodoin and Alex Browna�
Department of Chemistry, University of Alberta, Edmonton AB T6G 2G2, Canada
�Received 23 May 2005; accepted 7 June 2005; published online 5 August 2005�
The photodissociation dynamics of HI and DI are examined using time-dependent wave-packettechniques. The orientation and alignment parameters aQ
�K��p� are determined as a function ofphotolysis energy for the resulting ground-state I�2P3/2� and excited-state I�2P1/2� atoms. TheaQ
�K��p� parameters describe the coherent and incoherent contributions to the angular momentumdistributions from the A 1�1, a 3�1, and t 3�1 electronic states accessed by perpendicular excitationand the a 3�0+ state accessed by a parallel transition. The outcomes of the dynamics based on bothshifted ab initio results and three empirical models for the potential-energy curves and transitiondipole moments are compared and contrasted. It is demonstrated that experimental measurement ofthe aQ
�K��p� parameters for the excitation from the vibrational ground state �v=0� would be able todistinguish between the available models for the HI potential-energy curves and transition dipolemoments. The differences between the aQ
�K��p� parameters for the excitation from v=0 stand in sharpcontrast to the scalar properties, i.e., total cross section and I* branching fraction, which requireexperimental measurement of photodissociation from excited vibrational states �v�0� todistinguish between the models. © 2005 American Institute of Physics. �DOI: 10.1063/1.1989327�
I. INTRODUCTION
The hydrogen halides, HX �X=F, Cl, Br, and I�, repre-sent some of the simplest systems that dissociate to yieldopen-shell fragments. They provide model systems for study-ing molecular photodissociation dynamics on multiplepotential-energy curves �PECs� including the effects of nona-diabatic coupling. In particular, the photodissociation process
HX + h� → H�2S� + X�2P3/2�
→ H�2S� + X�2P1/2�
to yield the ground and spin-orbit excited-state halogen at-oms has been of both experimental and theoreticalinterest.1–35 Following standard nomenclature, the two spin-orbit states of the halogen atom, X�2P3/2� and X�2P1/2�, arereferred to as X and X*, respectively. For HF, HCl, HBr, andHI, the spin-orbit splittings between the two product chan-nels are 404, 882, 3685, and 7603 cm−1,36 respectively, andthus, the effects of increasing spin-orbit coupling on the dis-sociation process can be examined systematically.
The goals of the experimental and theoretical studieshave been to understand the roles of various electronic statesinvolved in the excitation and the possible nonadiabatic tran-sitions that could take place between the excited PECs as themolecule fragments. In order to achieve these goals, themore recent experimental and theoretical studies have fo-cused on determining the branching fraction �, which pro-vides the yield of spin-orbit excited atoms �X*� relative to thetotal yield, and on the lowest-order �K=0� anisotropy param-eter �, which provides information on the parallel and/orperpendicular nature of the electronic transitions contributing
to the dissociation. While the measurement of the scalarproperties, i.e., total cross section and �, and the lowest-order vector property � provide a wealth of information onthe potential-energy curves, transition dipole moments, and,if applicable, nonadiabatic couplings underlying the dynam-ics, recent experiments37–39 have determined the alignmentof chlorine and bromine atoms resulting from the dissocia-tion of HCl and HBr, respectively. Also, calculations of thealignment and orientation of the resulting halogen atomshave been carried out for the photodissociation of HF,40
HCl,38,41 and HI.42 The resulting alignment and orientationare extremely sensitive to the details of the potential-energycurves and transition dipole moments and, as discussed inSec. III for HI, provide more detailed information than mea-surement of the total cross section, branching fraction, or theanisotropy parameter �.
The halogen atoms can have a preferred orientationand/or alignment in space since they possess angular mo-menta. The spatial distribution of the photofragment angularmomenta can be described by the contributions from disso-ciation on a single PEC and from the interference from dis-sociation via multiple PECs.43 The aQ
�K��p� parameters44 canfully describe the polarization of the resulting atomic photo-fragments in the molecular frame. K and Q refer to the spa-tial distributions in the molecular frame. The symmetry ofthe transition dipole moments from the ground electronicstate to the dissociating states is given by p and can be �, �,or ��, �� corresponding to the pure parallel, pure perpendicu-lar, or mixed parallel/perpendicular excitation. An equivalentset of anisotropy parameters describing the dissociation inthe laboratory frame has also been introduced.45,46a�Electronic mail: [email protected]
THE JOURNAL OF CHEMICAL PHYSICS 123, 054301 �2005�
0021-9606/2005/123�5�/054301/10/$22.50 © 2005 American Institute of Physics123, 054301-1
Downloaded 05 Aug 2005 to 129.128.168.123. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp
In the present work, the aQ�K��p� parameters �K�3� de-
scribing the alignment/orientation of iodine fragments, bothI and I*, produced from the photodissociation of HI and DIare reported. They are determined from a quantum-mechanical time-dependent wave-packet calculation basedon recently published21,22 empirical potential-energycurves and transition dipole moments as well as the bestavailable ab initio data.24 For the empirical models, the mo-lecular parameters were determined by fitting the experimen-tally measured scalar properties, i.e., total cross section andbranching fractions, for the HI and DI photofragmentationprocesses. Calculations based on them were then shown toreproduce the lowest-order anisotropy parameter �. Theab initio PECs and transition dipole moments have beenshown24,47,48 to reproduce the experimentally measuredscalar properties. Here we demonstrate that measurement ofthe aQ
�K��p� parameters for the excitation from v=0 can dis-tinguish between the various models for the electronic struc-ture of HI.
The paper is organized in the following manner. SectionII A outlines the underlying electronic structure as it relatesto the two lowest-energy asymptotes H�2S1/2�+I�2P3/2� andH�2S1/2�+I�2P1/2�, which are the only ones of interest here. Abrief description of the aQ
�K��p� parameters relevant to HI �DI�dissociation is presented in Sec. II B. The methods used todetermine the aQ
�K��p� parameters from a time-dependentwave-packet treatment of the dissociation dynamics are out-lined in Sec. II B. The aQ
�K��p� parameters describing theI�2P3/2� and I�2P1/2� fragments arising from the photodisso-ciation of HI from its vibrational ground state �v=0� arediscussed in Sec. III A. In Sec. III B, the corresponding re-sults for DI are presented and contrasted to those for HI.Finally, some conclusions and proposals for the experimentalverification of these results are given in Sec. IV. TheaQ
�K��p� parameters for vibrationally mediated photodissocia-tion are also discussed, although detailed results are notpresented here.
II. THEORY
The first step required in a theoretical investigation ofphotodissociation is to determine the underlying potential-energy curves or surfaces for polyatomic molecules, and thecorresponding electronic transition dipole moments connect-ing the ground state to the excited electronic states. The cou-plings between the electronic states may also be required butthe current models for HI do not invoke coupling betweenstates. Once the electronic structure is known, a methodol-ogy for treating the dynamics is required and, in the presentwork, a time-dependent wave-packet treatment is utilized.From the dynamics, the photofragmentation T-matrixelements can be determined and used to obtain theaQ
�K��p� parameters via a well-established theoreticalframework.43,44,46,49 Since much of the theory has been pre-sented elsewhere, including a detailed discussion for the pho-todissociation of HI,42 each of these elements is discussedonly briefly.
A. Electronic structure
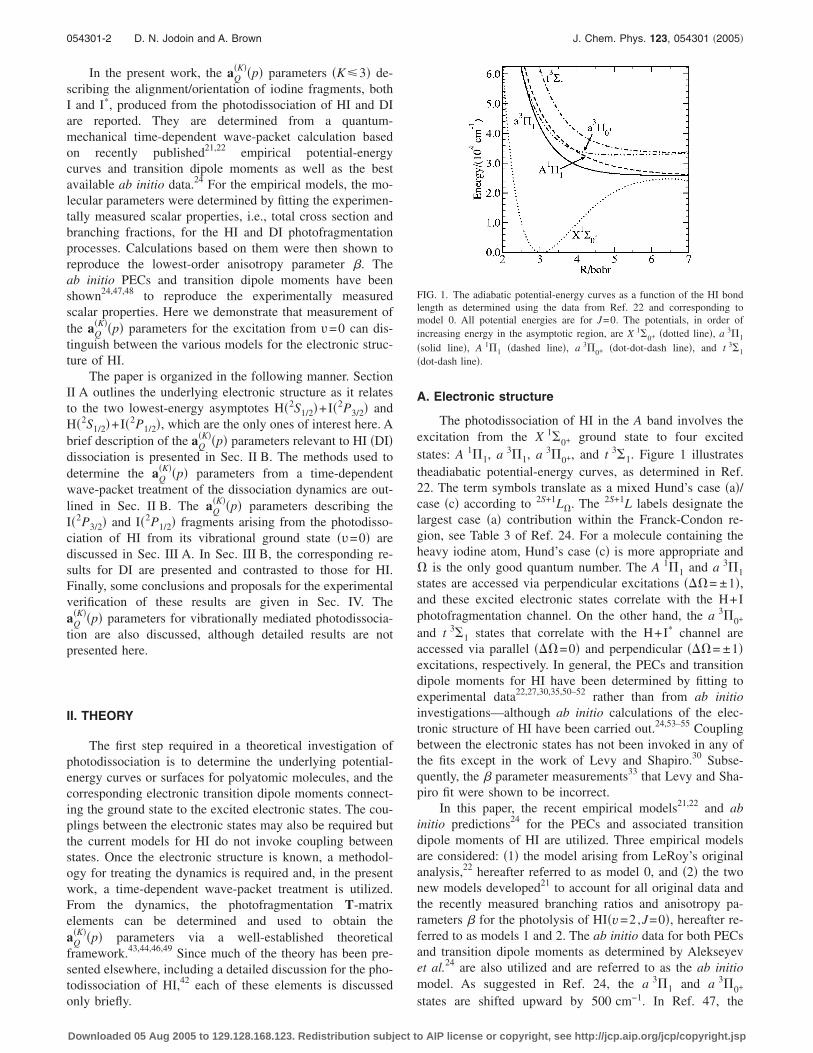
The photodissociation of HI in the A band involves theexcitation from the X 1�0+ ground state to four excitedstates: A 1�1, a 3�1, a 3�0+, and t 3�1. Figure 1 illustratestheadiabatic potential-energy curves, as determined in Ref.22. The term symbols translate as a mixed Hund’s case �a�/case �c� according to 2S+1L�. The 2S+1L labels designate thelargest case �a� contribution within the Franck-Condon re-gion, see Table 3 of Ref. 24. For a molecule containing theheavy iodine atom, Hund’s case �c� is more appropriate and� is the only good quantum number. The A 1�1 and a 3�1states are accessed via perpendicular excitations ��= ±1�,and these excited electronic states correlate with the H+Iphotofragmentation channel. On the other hand, the a 3�0+
and t 3�1 states that correlate with the H+I* channel areaccessed via parallel ��=0� and perpendicular ��= ±1�excitations, respectively. In general, the PECs and transitiondipole moments for HI have been determined by fitting toexperimental data22,27,30,35,50–52 rather than from ab initioinvestigations—although ab initio calculations of the elec-tronic structure of HI have been carried out.24,53–55 Couplingbetween the electronic states has not been invoked in any ofthe fits except in the work of Levy and Shapiro.30 Subse-quently, the � parameter measurements33 that Levy and Sha-piro fit were shown to be incorrect.
In this paper, the recent empirical models21,22 and abinitio predictions24 for the PECs and associated transitiondipole moments of HI are utilized. Three empirical modelsare considered: �1� the model arising from LeRoy’s originalanalysis,22 hereafter referred to as model 0, and �2� the twonew models developed21 to account for all original data andthe recently measured branching ratios and anisotropy pa-rameters � for the photolysis of HI�v=2,J=0�, hereafter re-ferred to as models 1 and 2. The ab initio data for both PECsand transition dipole moments as determined by Alekseyevet al.24 are also utilized and are referred to as the ab initiomodel. As suggested in Ref. 24, the a 3�1 and a 3�0+
states are shifted upward by 500 cm−1. In Ref. 47, the
FIG. 1. The adiabatic potential-energy curves as a function of the HI bondlength as determined using the data from Ref. 22 and corresponding tomodel 0. All potential energies are for J=0. The potentials, in order ofincreasing energy in the asymptotic region, are X 1�0+ �dotted line�, a 3�1
�solid line�, A 1�1 �dashed line�, a 3�0+ �dot-dot-dash line�, and t 3�1
�dot-dash line�.
054301-2 D. N. Jodoin and A. Brown J. Chem. Phys. 123, 054301 �2005�
Downloaded 05 Aug 2005 to 129.128.168.123. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp
a 3�1←X 1�0+ and A 1�1←X 1�0+ transition dipole mo-ments are scaled by 1.05 to reduce the rms discrepancy be-tween the calculations and experimental measurements forthe I* branching fraction for the excitation from v=2. Sincethis effect is notably weaker than shifting the PECs, we havechosen to use the unscaled transition dipole moments for theab initio model. The existing experimental data cannot dis-tinguish between models 1 and 2 while model 0 does notproperly account for the measurements from v=2. Theshifted ab initio data properly accounts for both the v=0measurements24,48 and the v=2 experiments.47 Model 0 isincluded in order to compare the new results for the aQ
�K��p�parameters determined using model 1, model 2, and the abinitio model with those of the previous investigation.42
All three empirical models utilize the ground electronicpotential-energy functions for HI and DI from Coxon andHajigeorgiou.56 The potential-energy functions for the ex-cited A 1�1, a 3�1, and t 3�1 states are represented by thesimple exponential function
Vexp�r� = Ds + As exp�− ��y8��r − rx�� . �1�
The a 3�0+ state is represented by the extended Morse oscil-lator �EMO� function
VEMO = �Ds − De� + De�exp�− ��y8��r − rx�� − 1�2. �2�
In Eqs. �1� and �2�, Ds is the asymptotic energy of electronicstate s. For the A 1�1 and a 3�1 states correlating with theground-state iodine fragments, Ds is set at 25 778 cm−1. Forthe a 3�0+ and t 3�1 states correlating with the excited-stateiodine, Ds is 33 781 cm−1. The difference between theseasymptotic energies, 7603 cm−1, reflects the spin-orbit split-ting in atomic iodine. In Eq. �2�, rx is a reference distance,and the exponent coefficient ��y8� is defined as an expansionin the dimensionless radial variable y8= �r8−rx
8� / �r8+rx8�, i.e.,
��y8� = �0 + �1y8 + �2y82 + ¯ . �3�
In the EMO function used for the a 3�0+ state, the well depthof the shallow minimum, De, is fixed at 600 cm−1.
The three empirical models use two types of transitiondipole moment functions �r�: �1� linearly scaled versions ofthe ab initio functions24 and �2� empirical expansions inpowers of the radial variable y8:
�r� = �i=0
ciy8i . �4�
The parameters defining the excited-state potential-energycurves and the transition dipole moment functions are givenin Tables I, II, and III for models 0, 1, and 2, respectively.
In a recently published comment,47 Alekseyev et al.,who generated the ab initio data we have used in this paper,speak strongly against model 2 being viable based on plau-sible physical reasoning. Firstly, in model 2, the a 3�0+ anda 3�1 states lie notably lower than the corresponding ab ini-tio potentials. This contradicts the argument based on theintrinsic properties of the computational method that thesePECs should be shifted upwards in energy. Secondly, model2 uses a a 3�0+←X 1�0+ transition dipole moment that in-creases with decreasing bond length. All computationalmethods indicate otherwise, i.e., the transition dipole mo-ment decreases with decreasing bond length, see Fig. 3 ofRef. 24. For these reasons model 2 should most likely bediscounted. So, while the primary comparison for the pur-poses of encouraging experiments to measure these param-eters should be between model 1 and the ab initio model,calculations based on models 0 and 2 are included for com-pleteness, and to demonstrate the sensitivity of the aniso-tropy parameters to differences in the PECs and transitiondipole moments.
A detailed description of the correspondence betweenthe photofragment atomic states and the adiabatic molecular
TABLE I. Parameters defining the potential energy �in cm−1� and transitiondipole moment �in Debye� functions for model 0 �Ref. 22�.
a3�1 A1�1 a3�0+ t3�1+
Form Exponential Exponential EMO Exponentialrx /Å 1.609 1.609 2.7 1.609
As / cm−1 15714 21161 a 26890�0 /Å−1 2.949 2.128 1.54659 2.60�1 /Å−1
¯ −0.30 0.067 ¯
�2 /Å−1¯ ¯ −0.074 ¯
c0b 1.468 0.9368 1.014 0.93
aEMO potential form, Eq. �2�, with a fixed well depth of De=600 cm−1.bTransition dipole moment defined as a cubic spline through the ab initiovalues from Ref. 24, multiplied by the scaling factor c0.
TABLE II. Parameters defining the potential energy �in cm−1� and transitiondipole moment �in Debye� functions for model 1 �Ref. 21�.
a3�1 A1�1 a3�0+ t3�1+
Form Exponential Exponential EMO Exponentialrx /Å 1.609 1.609 2.63892 1.609
As / cm−1 15188 20873 a 27690�0 /Å−1 2.965 2.304 1.6323 2.60�1 /Å−1
¯ −0.437 0.056 ¯
c0 1.329b 0.978b 0.4072c 1.0b
c1 0.069
aEMO potential form, Eq. �2�, with a fixed well depth of De=600 cm−1.bTransition dipole moment defined as a cubic spline through the ab initiovalues from Ref. 24, multiplied by the scaling factor c0.cTransition dipole moment defined by Eq. �4� with the coefficients c0 and c1.
TABLE III. Parameters defining the potential energy �in cm−1� and transi-tion dipole moment �in Debye� functions for model 2 �Ref. 21�.
a3�1 A1�1 a3�0+ t3�1+
Form Exponential Exponential EMO Exponentialrx 1.609 1.609 2.7 1.609
As / cm−1 12823 20384 a 26830�0 /Å−1 2.495 2.945 1.51592 2.60�1 /Å−1
¯ −0.260 ¯ ¯
c0 0.8234b 0.46234c 0.4131c 1.00b
c1 −0.613 −0.130
aEMO potential form, Eq. �2�, with a fixed well depth of De=600 cm−1.bTransition dipole moment defined as a cubic spline through the ab initiovalues from Ref. 24, multiplied by the scaling factor c0.cTransition dipole moment defined by Eq. �4� with the coefficients c0 and c1.
054301-3 Photodissociation of HI and DI J. Chem. Phys. 123, 054301 �2005�
Downloaded 05 Aug 2005 to 129.128.168.123. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp

states is given in Ref. 40. The adiabatic treatment impliesthat four electronic states, i.e., the doubly degenerate a 3�1and A 1�1 states, contribute to the angular distribution�orientation/alignment� of the ground-state I�2P3/2� fragment.These states correlate adiabatically as
HI�a3�1;�� = ± 1� → H�mH = ± 1/2� + I�mI = ± 1/2�
�5�
and
HI�A1�1;�� = ± 1� → H�mH = � 1/2� + I�mI = ± 3/2� .
�6�
Within the present adiabatic models for the photodissocia-tion, the ground-state X 1�0+ does not contribute to the pro-duction of I�2P3/2�. However, in principle, this state couldgain population via nonadiabatic recoupling from the a 3�0+
state—although experimental measurements26,27,29,31,34,35 of� suggest that there is no parallel contribution to the produc-tion of I�2P3/2�.
Three excited states, i.e., the nondegenerate a 3�0+ stateand the doubly degenerate t 3�1 state, contribute to the an-gular distribution of the excited state I�2P1/2� fragment. Thea 3�0+ state correlates equally with both mH= ±1/2 andmI* = �1/2 states, i.e.,
HI�a3�0+;�� = 0� → H�mH = ± 1/2� + I*�mI = � 1/2� .
�7�
The t 3�1 state correlates as
HI�t3�1;�� = ± 1� → H�mH = ± 1/2� + I*�mI = ± 1/2� .
�8�
The molecular wave functions corresponding to these as-ymptotes are given in Table II of Ref. 40. The long-rangecorrelations given in Eqs. �5�–�8� are required for the deter-mination of the orientation and alignment parameters,aQ
�K��p�.
B. Determination aQ„K…
„p… parameters via time-dependent wave-packet dynamics
The primary goal of this work is to determine theaQ
�K��p� parameters describing the orientation and alignmentof the iodine atomic fragments in the molecular frame. Thetheoretical and/or experimental determination of aQ
�K��p� pa-rameters provides the most detailed understanding of thephotodissociation dynamics. For these parameters, K and Qrefer to the spatial distribution in the molecular frame and prefers to the symmetry of the transition connecting theground electronic state to the dissociative excited states. Thesymmetry p can be pure perpendicular ���, pure parallel ���,or mixed parallel/perpendicular ��,��. The scalar and lowest-order vector properties, including the total cross section, theI* branching fraction, and the anisotropy parameter �, havebeen determined previously for these PECs and transitiondipole moments.21,22,24,47,48 Therefore, we do not discussthese parameters in the present work.
In the following description of the anisotropy param-eters, the nuclear spins of the photofragments have been ne-glected. Since the duration of the dissociation process is typi-cally much smaller than the Heisenberg uncertainty timet=� / �2E� associated with the hyperfine splitting in theatoms, this assumption is justified. While the nuclear spinsdo not affect the photodissociation dynamics, the hyperfineinteraction in the final fragments results in the partial depo-larization of the fragment’s electron angular momenta.57 Forthe dissociation of the HI molecule considered here, any or-bital alignment of the I-atom photofragment will degrade to23% of its nascent value through coupling with theI=5/2 nuclear spin.
The dimensionless anisotropy parameters aQ�K��p� are
normalized combinations of the dynamical functionsfK�q ,q��. For the dissociation of HI resulting in the produc-tion of iodine and hydrogen atoms having angular momentajI and jH, respectively, the dynamical functions for the iodineatom are defined as
fK�q,q�� = �n,�,�I,n�,��,�I�
�− 1�K+jI+�I� jI jI K
− �I �I� q − q��
�TjI�IjH�H
n� �*TjI�I�jH�H
n���
��n,�− �R,E� dq ��i
�*��n�,��− �R,E� dq� ��i
� .
�9�
The expression ��n,�− �R ,E� dq ��i
� is the energy-dependentphotofragmentation T-matrix element associated with chan-nel n and is the critical quantity that must be determinedfrom the dynamics calculation �see below�. The indices qand q� are the vector spherical harmonic components57,58 ofthe molecular electric dipole moment with respect to the re-coil axis. These indices can take the value of 0 correspondingto a parallel electronic transition or the values ±1 corre-sponding to perpendicular electronic transitions. The initialand final z components of the total electronic angular mo-mentum about the molecular axis are related by �=�i+q.The diagonal elements of the dynamical functions fK�q ,q��with q=q� correspond to incoherent excitation of perpen-dicular or parallel transitions. The off-diagonal elements withq�q� correspond to coherent excitation of different molecu-lar continua. The expression for the dynamical functions forthe hydrogen atom can be obtained from Eq. �9� by exchang-ing the subscripts I and H.
For the ground-state iodine photofragments I�2P3/2�, therelevant dynamical functions range from K=0 to 3, while forthe excited-state iodine I�2P1/2�, they correspond to K=0 or1. Since the dynamical functions are directly related to theangular momentum state multipoles,40 K is referred to as themultipole rank. For the ground-state iodine fragment, thecomplete set of state multipoles �dynamical functions� con-tains K=0 �population�, K=1 �orientation, dipole moment�,K=2 �alignment, quadrupole moment� and K=3 �orientation,octopole moment�. The description for the excited-state io-dine fragment requires only K=0 and K=1 dynamical func-tions.
054301-4 D. N. Jodoin and A. Brown J. Chem. Phys. 123, 054301 �2005�
Downloaded 05 Aug 2005 to 129.128.168.123. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp

In order to clarify the meaning of the anisotropy param-eters that are utilized for describing the ground-state iodineI�2P3/2� and the excited-state iodine I�2P1/2� fragments, theirrelationships to the dynamical functions are described below.The zeroth-rank �K=0� anisotropy parameter is the well-known � parameter and is given by43
� =2�f0�0,0� − f0�1,1��2f0�1,1� + f0�0,0�
. �10�
The � parameters for the iodine atoms are not discussed hereas they have been examined previously for models 0, 1, and2 �Refs. 21 and 22� and for the ab initio model.24 Theground-state I-atom products result exclusively from the per-pendicular transitions to the a3�1 and A1�1 states and�=−1 independent of the photolysis energy. On the otherhand, the I* products result primarily from the parallel tran-sition to the a3�0+ state below 50 000 cm−1 and from theperpendicular transition to t3�1 above 50 000 cm−1. Thus,the � parameter describing the production of I* changes from+2 at low energies to −1 at high energies.
As in our previous study of HI and DI based on model0,42 the a0
�1����, a0�2����, a0
�3����, a2�2����, and a2
�3���� aniso-tropy parameters are computed for the ground-state frag-ments, and the a0
�1���� and a1�1��� , � � parameters are deter-
mined for the excited-state fragments. The a0�K����
parameters describe the incoherent perpendicular excitation,the a2
�K���� parameters describe the coherent perpendicularexcitation, and the a1
�1��� , � � parameter describes the coher-ent parallel and perpendicular excitation. In principle, thereare additional parameters describing the coherent paralleland perpendicular excitations for the production of ground-state iodine, i.e., aQ
�K��� , � �, 2�K�3. However, within thepresent adiabatic treatment of the dynamics, there is no par-allel contribution to I�2P3/2� fragments and therefore allaQ
�K��� , � � parameters are identically zero. Only a single stateaccessed by parallel excitation correlates with the excited-state asymptote, and thus no parallel only parametersaQ
�K���� are computed. Since the role of the t3�1 state is neg-ligible in A band, the previous ab initio studies24 have omit-ted it from discussions and we choose to do the same for theab initio model in the present work. Hence, only results forthe parameters describing ground-state iodine fragments arepresented based on the ab initio PECs and transition dipolemoments.
The parameters describing the incoherent perpendicularexcitation are related to the dynamical functions Eq. �9� by
a0�1���� =
f1�1,1�f0�1,1�
, �11�
a0�2���� = V2�jI�−1 f2�1,1�
f0�1,1�, �12�
and
a0�3���� = V3�jI�−1 f3�1,1�
f0�1,1�. �13�
For the ground-state iodine photofragment, the parametersV2�jI� and V3�jI� are given by
V2�jI = 3/2� = � jI�jI + 1��2jI + 3��2jI − 1��1/2
=�15
4�3�14�
and
V3�jI = 3/2� =jI�jI + 1�
��jI − 1��jI + 2��2jI − 1��2jI + 3��1/2
=15
4�21. �15�
Since K=0 or 1 for the excited-state iodine fragmentI�2P1/2�, only the a0
�1���� parameter is required for the de-scription of incoherent perpendicular excitation.
The description of the angular distribution of the ground-state I�2P3/2� fragment also requires two parameters for co-herent perpendicular excitation, i.e.,
a2�2���� = −
1
2V2�jI�−1 f2�1,− 1�
f0�1,1��16�
and
a2�3���� =
i
2V3�jI�−1 f3�1,− 1�
f0�1,1�. �17�
For the excited-state iodine fragment, the parametera1
�1��� , � � describing coherent parallel and perpendicular ex-citations is
a1�1��� , � � =
− 3�2f1�1,0�2f0�1,1� + f0�0,0�
. �18�
The parameter describing coherent parallel and perpendicularexcitations usually has its real and imaginary parts presentedseparately.44
In order to determine the dynamical functions requiredto calculate the anisotropy parameters, the photofragmenta-tion T-matrix elements must be obtained from the dynamics.A time-dependent wave-packet formulation for thedynamics59–64 is used in the present work. The time-dependent approach is based upon the solution of the nuclearSchrödinger equation
i����R,t�
�t= H�R���R,t� = �−
�2
2
d2
dR2 + V�R����R,t� .
�19�
In Eq. �19�, is the reduced mass of the molecule, ��R , t� isa column vector describing the time-dependent wave func-tion on each of the excited electronic states of the molecule,and V�R� is a diagonal matrix of the adiabatic potential en-ergies. No off-diagonal couplings are considered in thepresent adiabatic treatment of the dynamics. The rotationalpart of the nuclear kinetic-energy operator is neglected inEq. �19�. As in our previous studies of hydrogen halidedissociation,2,4,40,65 the axial recoil approximation is assumedto be valid, which is equivalent to the neglect of the overallrotation of the molecule.66
In the time-dependent approach, initial wave-packetpackets �n�R , t=0� are prepared by multiplying the initialnuclear vibrational wave function of the molecule, ��i
�R�,
054301-5 Photodissociation of HI and DI J. Chem. Phys. 123, 054301 �2005�
Downloaded 05 Aug 2005 to 129.128.168.123. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp

by the corresponding adiabatic transition dipole moment be-tween the ground state and the nth excited state dq�
n �R�. Theindex q� is the vector spherical harmonic component of thetransition dipole moment function and it is determined by thesymmetries of the ground and excited electronic states. Theinitial ground-state nuclear wave functions have been deter-mined using the Fourier grid Hamiltonian method.59,67,68
Once the initial wave packets are defined, they are propa-gated in time under the influence of the excited-statepotential-energy surfaces. The time propagation is performedusing the Chebychev expansion technique.69,70
In order to determine the photofragmentation T-matrixelements, the time-evolving wave packets are analyzed ateach time step as they pass through an analysis line definedat a large fixed value of the bond length �R=R��. By takingthe Fourier transform over time of these cuts through thetime-dependent wave packets �n�R� , t�, energy-dependentcoefficients2,4,60 An�R� ,E� are then obtained, where
An�R�,E� =1
2��
0
�
�n�R�,t�exp�i�Ei + h��t/��dt . �20�
As has been discussed previously,40,60 the photofragmenta-tion T-matrix elements are related to these energy-dependentcoefficients by
��n,�− �R,E� dq ��i
� = i h2kv
2��1/2
exp�− ikvR��An�R�,E� .
�21�
Once the T-matrix elements are known, the dynamical func-tions fK�q ,q�� can be determined via Eq. �9�, and then theanisotropy parameters can be obtained using the relation-ships given in Eqs. �11�–�18�.
III. RESULTS AND DISCUSSION
In the following sections, the focus is on the determina-tion of the anisotropy parameters aQ
�K��p� within the currentadiabatic models21,22,24 for dissociation. The calculationsdemonstrate that the experimental measurement of these pa-rameters for the excitation from v=0 can distinguish be-tween the current empirical models and the ab initio results.In order to distinguish between the present models via scalarproperties, e.g., the total cross section or the I* branchingfraction, measurements must be made for v�0 using vibra-tionally mediated photodissociation.21 The measurement ofthe aQ
�K���� parameters allows the determination of the rela-tive roles of the a3�1 and A1�1 states. The relative contri-butions of these states cannot be determined via measure-ment of either the I* branching fraction, since both correlatewith ground-state fragments or the � parameter, since bothare accessed via perpendicular transitions. The sensitivity ofthe a1
�1��� , � � parameter to the relative amounts of paralleland perpendicular excitation is also demonstrated.
A. Anisotropy parameters for iodine fragments fromthe dissociation of HI
The angular distribution for the ground-state iodine frag-ment I�2P3/2� is described by both orientation �K=1 and
K=3� and alignment �K=2� parameters. On the other hand,the excited-state I�2P1/2� fragment’s angular distribution isfully described by orientation �K=1� parameters only. Theanisotropy parameters determined from the four models forthe electronic structure are discussed first for the ground-state iodine fragment and then for the excited-state atom. Theprimary comparison is between the results based on model 1and the ab initio model, as model 0 has been discounted dueto its inability to reproduce the results of the vibrationallymediated photodissociation experiments,21 while model 2 isunlikely due to physical arguments based on the best avail-able ab initio results.47 However, the results based on theselatter two models are included for comparison and to dem-onstrate the sensitivity of the aQ
�K���� parameters to changesin the PECs and the transition dipole moments. It should benoted that when dissociating with linearly polarized light, thespatial distribution of the angular momentum of the en-semble of I�2P1/2� photofragments is dependent only on thea0
�2���� and a2�2���� parameters as the excitation is of perpen-
dicular character only. On the other hand, when dissociatingwith circularly polarized light, the angular distribution of theiodine fragments reflects contributions from the odd Kparameters.44 Thus, while all of the aQ
�K���� parameters maybe determined from our calculations, their measurement re-quires experiments with both linearly and circularly polar-ized light.
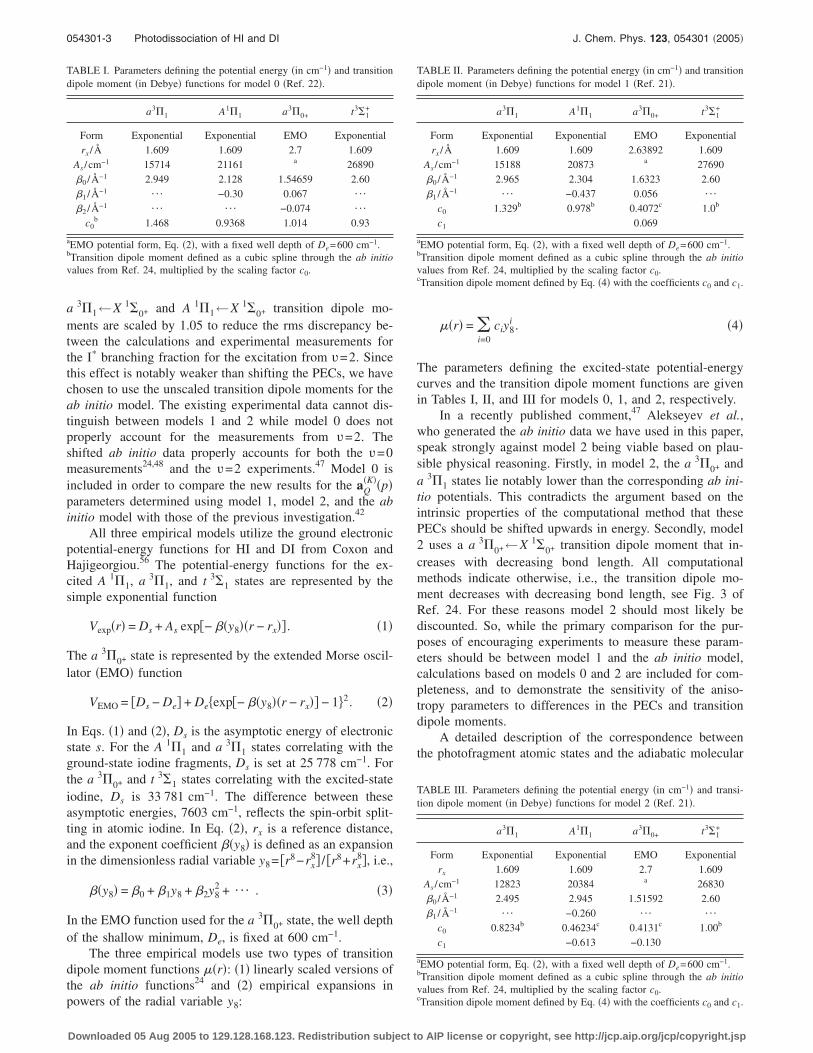
Figure 2 illustrates the anisotropy parameters describingthe incoherent perpendicular excitation, a0
�K����, for theground-state I�2P3/2� fragment produced from the photodis-sociation of HI as a function of photolysis wavelength. Theresults are for the excitation from the rotationless ground�v=0� vibrational state. Results are presented for the threeempirical models and the ab initio model. As discussed inSec. II A, the primary comparison is between model 1 andthe ab initio model and Fig. 2 illustrates that there are cleardifferences between the a0
�K���� parameters for these twomodels. For example, the difference between a0
�2���� formodel 1 and the ab initio model is greater than 0.1 for pho-tolysis energies between 37 000 and 47 000 cm−1 and themaximum difference of 0.26 occurs at approximtely
FIG. 2. Incoherent anisotropy parameters �a� a0�1����, �b� a0
�2����, and �c�a0
�3����, for the production of ground state I�2P3/2� as a function of photonenergy for the photodissociation of HI initially in the rotationless ground�v=0� vibrational state. The results were determined from model 0 �solidline�, model 1 �dotted line�, model 2 �dashed line�, and the ab initio model�dot-dash line�, respectively.
054301-6 D. N. Jodoin and A. Brown J. Chem. Phys. 123, 054301 �2005�
Downloaded 05 Aug 2005 to 129.128.168.123. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp
41 000 cm−1. For HCl and HBr, the a0�K���� parameters for
K=1 and 2 have been measured37,38 with an uncertainty of±0.2 and, thus, which of the models, if either, provide acorrect prediction should be amenable to experimental mea-surement. Also, the measurements21,23,25–29,31,32,35 of the I*
branching fractions and � parameters have been made be-tween 34 772 and 48 780 cm−1 indicating that the energyrange exhibiting the largest difference between the models isaccessible experimentally.
Within the adiabatic model of dissociation, the a0�K����
parameters are directly related to the excitation probabilityto, and relative yields from, the A1�1 and a3�1 states.40
Therefore, the differences in the a0�K���� parameters reflect
the differences in the energies of these two excited states andthe magnitudes of the transition dipole moments to the twostates. In fact, the low-energy and high-energy limiting val-ues of the a0
�K���� parameters reflect the near 100% yieldwith respect to the total I production from a3�1 �0% yieldfrom A1�1� at low energies and the 0% �100%� yield at highenergies. The fact that the transition between the limitingvalues of the a0
�K���� parameters occurs at higher energy formodel 1 versus the ab initio model can primarily be attrib-uted to the scaling of the transition dipole moments; thePECs are very similar, see Fig. 10 of Ref. 22. For model 1,the ab initio transition dipole moment to the a3�1 state isscaled by 1.329, increasing its contribution at all photolysisenergies. On the other hand, the transition dipole moment toA1�1 is scaled by 0.978, and therefore, the contribution ofthis state is very similar for both model 1 and the ab initiomodel. The similarities between the results for models 0 and1 are not surprising given that the PECs and transition dipolemoments for the a3�1 and A1�1 states are very similar, seeTables I and II. The significant difference between model 2and the other models reflects two phenomena. First, there isa much larger difference between the vertical excitation en-ergies to the a3�1 and A1�1 states as compared to othermodels, i.e., 7600 cm−1 for model 2 versus 5500 cm−1
for other models. Second, model 2 decreases the contributionof the a3�1 state by scaling the ab initio transition dipolemoment by 0.8234 and has a completely different functionalform of the transition dipole moment to the A1�1 state,which increases its contribution at lower photolysis energies.
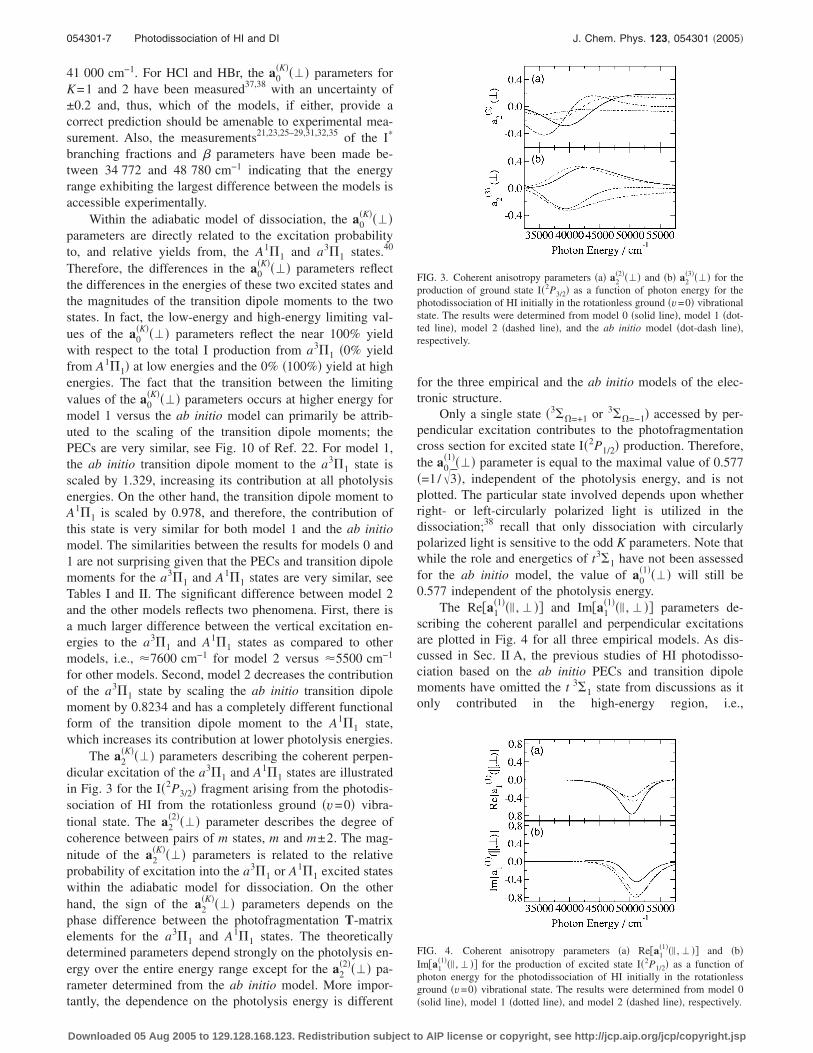
The a2�K���� parameters describing the coherent perpen-
dicular excitation of the a3�1 and A1�1 states are illustratedin Fig. 3 for the I�2P3/2� fragment arising from the photodis-sociation of HI from the rotationless ground �v=0� vibra-tional state. The a2
�2���� parameter describes the degree ofcoherence between pairs of m states, m and m±2. The mag-nitude of the a2
�K���� parameters is related to the relativeprobability of excitation into the a3�1 or A1�1 excited stateswithin the adiabatic model for dissociation. On the otherhand, the sign of the a2
�K���� parameters depends on thephase difference between the photofragmentation T-matrixelements for the a3�1 and A1�1 states. The theoreticallydetermined parameters depend strongly on the photolysis en-ergy over the entire energy range except for the a2
�2���� pa-rameter determined from the ab initio model. More impor-tantly, the dependence on the photolysis energy is different
for the three empirical and the ab initio models of the elec-tronic structure.
Only a single state �3��=+1 or 3��=−1� accessed by per-pendicular excitation contributes to the photofragmentationcross section for excited state I�2P1/2� production. Therefore,the a0
�1���� parameter is equal to the maximal value of 0.577�=1/�3�, independent of the photolysis energy, and is notplotted. The particular state involved depends upon whetherright- or left-circularly polarized light is utilized in thedissociation;38 recall that only dissociation with circularlypolarized light is sensitive to the odd K parameters. Note thatwhile the role and energetics of t3�1 have not been assessedfor the ab initio model, the value of a0
�1���� will still be0.577 independent of the photolysis energy.
The Re�a1�1��� , � �� and Im�a1
�1��� , � �� parameters de-scribing the coherent parallel and perpendicular excitationsare plotted in Fig. 4 for all three empirical models. As dis-cussed in Sec. II A, the previous studies of HI photodisso-ciation based on the ab initio PECs and transition dipolemoments have omitted the t 3�1 state from discussions as itonly contributed in the high-energy region, i.e.,
FIG. 3. Coherent anisotropy parameters �a� a2�2���� and �b� a2
�3���� for theproduction of ground state I�2P3/2� as a function of photon energy for thephotodissociation of HI initially in the rotationless ground �v=0� vibrationalstate. The results were determined from model 0 �solid line�, model 1 �dot-ted line�, model 2 �dashed line�, and the ab initio model �dot-dash line�,respectively.
FIG. 4. Coherent anisotropy parameters �a� Re�a1�1��� , � �� and �b�
Im�a1�1��� , � �� for the production of excited state I�2P1/2� as a function of
photon energy for the photodissociation of HI initially in the rotationlessground �v=0� vibrational state. The results were determined from model 0�solid line�, model 1 �dotted line�, and model 2 �dashed line�, respectively.
054301-7 Photodissociation of HI and DI J. Chem. Phys. 123, 054301 �2005�
Downloaded 05 Aug 2005 to 129.128.168.123. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp
�50 000 cm−1. According to the ab initio calculations,24 theonset of the t 3�1←X 1�0+ absorption is underestimated. Inaddition, the data suggest that a diabatic representation maybe more appropriate for describing the t 3�1 transition. Forthese reasons, this state was omitted in the discussion of thephotodissociation in the original paper24 and we choose toomit it here for the ab initio model. Hence, no a1
�1��� , � �parameters are determined for the ab initio model. For theempirical models, the a1
�1��� , � � parameters show a strongdependence on the photolysis energy in the region whereexcitations to both a 3�0+ and t 3�1 are feasible, i.e., ap-proximately 41 000–56 000 cm−1.
As discussed in Ref. 42, the a1�1��� , � � parameters are
more sensitive to the amount of parallel and perpendicularexcitations than the corresponding � parameters. Experimen-tal measurements of the a1
�1��� , � � parameters below ap-proximately 45 000 cm−1 should be able to provide evidencefor or against the possibility of nonadiabatic coupling be-tween the t 3�1 state and the a 3�1 and A 1�1 states. It isclear that direct excitation to t 3�1 is not feasible in thatenergy range,24 see Fig. 1, and therefore, any perpendicularcontribution to the I* dissociation channel must be fromnonadiabatic coupling. Measurement of the a1
�1��� , � � pa-rameters would also allow a more definitive assignment ofthe energy of the t 3�1 state and will help in the refinementof the electronic structure models.
From the results of Figs. 2–4 it is clear that the experi-mental measurement of the aQ
�K��p� parameters for the iodinefragment resulting from HI photodissociation would providean alternative to the measurement of scalar properties fromvibrationally mediated photodissociation as a mean to distin-guish between the empirical models, and, of course, to deter-mine the accuracy of the ab initio results. It may also beuseful to measure the aQ
�K��p� parameters for the deuteratedspecies DI as this would provide an additional test for theempirical models and the ab initio data. The results for DIare discussed below.
B. Anisotropy parameters for iodine fragments fromthe dissociation of DI
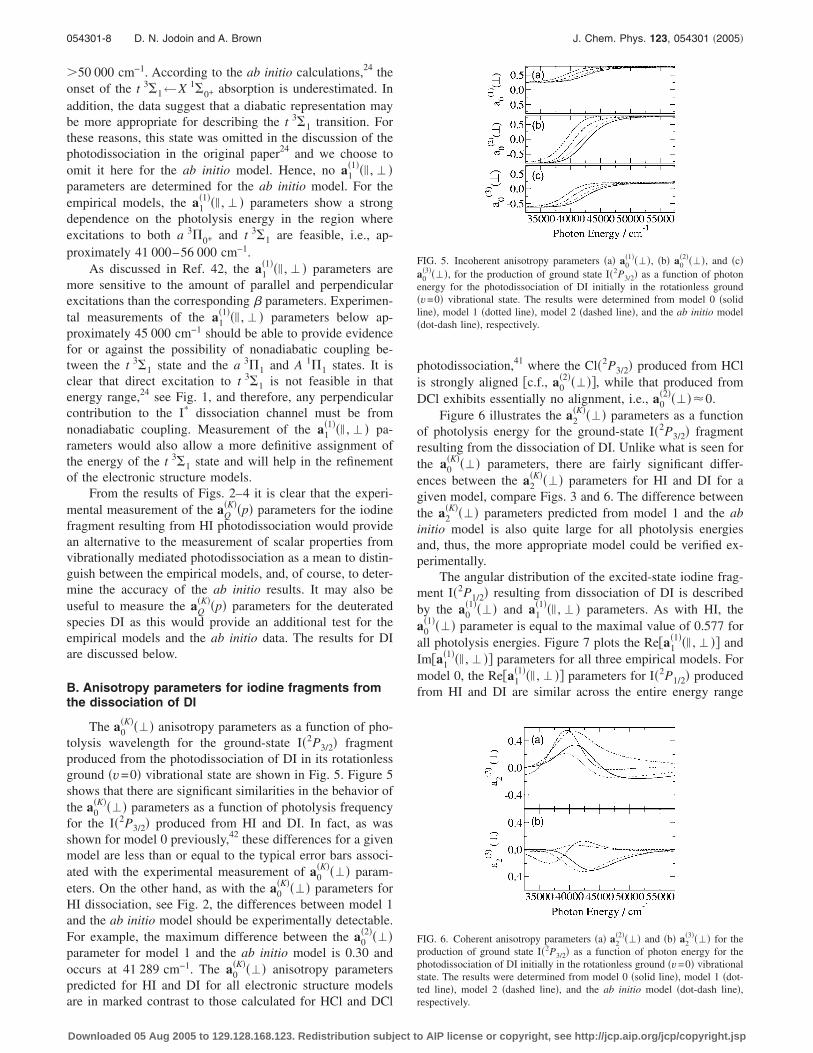
The a0�K���� anisotropy parameters as a function of pho-
tolysis wavelength for the ground-state I�2P3/2� fragmentproduced from the photodissociation of DI in its rotationlessground �v=0� vibrational state are shown in Fig. 5. Figure 5shows that there are significant similarities in the behavior ofthe a0
�K���� parameters as a function of photolysis frequencyfor the I�2P3/2� produced from HI and DI. In fact, as wasshown for model 0 previously,42 these differences for a givenmodel are less than or equal to the typical error bars associ-ated with the experimental measurement of a0
�K���� param-eters. On the other hand, as with the a0
�K���� parameters forHI dissociation, see Fig. 2, the differences between model 1and the ab initio model should be experimentally detectable.For example, the maximum difference between the a0
�2����parameter for model 1 and the ab initio model is 0.30 andoccurs at 41 289 cm−1. The a0
�K���� anisotropy parameterspredicted for HI and DI for all electronic structure modelsare in marked contrast to those calculated for HCl and DCl
photodissociation,41 where the Cl�2P3/2� produced from HClis strongly aligned �c.f., a0
�2�����, while that produced fromDCl exhibits essentially no alignment, i.e., a0
�2����0.Figure 6 illustrates the a2
�K���� parameters as a functionof photolysis energy for the ground-state I�2P3/2� fragmentresulting from the dissociation of DI. Unlike what is seen forthe a0
�K���� parameters, there are fairly significant differ-ences between the a2
�K���� parameters for HI and DI for agiven model, compare Figs. 3 and 6. The difference betweenthe a2
�K���� parameters predicted from model 1 and the abinitio model is also quite large for all photolysis energiesand, thus, the more appropriate model could be verified ex-perimentally.
The angular distribution of the excited-state iodine frag-ment I�2P1/2� resulting from dissociation of DI is describedby the a0
�1���� and a1�1��� , � � parameters. As with HI, the
a0�1���� parameter is equal to the maximal value of 0.577 for
all photolysis energies. Figure 7 plots the Re�a1�1��� , � �� and
Im�a1�1��� , � �� parameters for all three empirical models. For
model 0, the Re�a1�1��� , � �� parameters for I�2P1/2� produced
from HI and DI are similar across the entire energy range
FIG. 5. Incoherent anisotropy parameters �a� a0�1����, �b� a0
�2����, and �c�a0
�3����, for the production of ground state I�2P3/2� as a function of photonenergy for the photodissociation of DI initially in the rotationless ground�v=0� vibrational state. The results were determined from model 0 �solidline�, model 1 �dotted line�, model 2 �dashed line�, and the ab initio model�dot-dash line�, respectively.
FIG. 6. Coherent anisotropy parameters �a� a2�2���� and �b� a2
�3���� for theproduction of ground state I�2P3/2� as a function of photon energy for thephotodissociation of DI initially in the rotationless ground �v=0� vibrationalstate. The results were determined from model 0 �solid line�, model 1 �dot-ted line�, model 2 �dashed line�, and the ab initio model �dot-dash line�,respectively.
054301-8 D. N. Jodoin and A. Brown J. Chem. Phys. 123, 054301 �2005�
Downloaded 05 Aug 2005 to 129.128.168.123. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp

illustrated while there are fairly significant differences formodels 1 and 2 for photolysis energies between 47 500 and52 500 cm−1. For models 1 and 2, the maximal value ofRe�a1
�1��� , � ��, occurring at approximately 50 000 cm−1, islarger for DI than for HI. The calculated Im�a1
�1��� , � �� pa-rameters from HI and DI exhibit opposite signs for model 0while they are of the same sign for models 1 and 2. Thedifference between Im�a1
�1��� , � �� parameters for HI and DIcould be measured experimentally. The Im�a1
�1��� , � �� pa-rameter is dependent on the sine of the phase difference ofthe radial wave functions created on the a 3�0+ and t 3�1
states, i.e., sin �, modulated by the transition amplitudesfor these transitions.43,44 On the other hand, theRe�a1
�1��� , � �� parameter has a cos � dependence. There-fore, the differences between HI and DI reflect these differ-ences and measurement of the a1
�1��� , � � parameters will pro-vide a detailed information regarding the a 3�0+ and t 3�1
states.The calculated differences between HI and DI await ex-
perimental verification. If verified, they could provide addi-tional confirmation of one of the current models of adiabaticphotodissociation for HI or would allow refinement of themodels.
IV. CONCLUSIONS
We have reexamined the photodissociation of HI and DIusing time-dependent wave-packet dynamics based on therecent empirical models �models 1 and 2, see Tables II andIII� for the potential-energy curves and transition dipole mo-ment functions of Camden et al.21 and based on the bestavailable ab initio data.24 In particular, the focus has been ondetermining the �near� complete set of anisotropy parametersaQ
�K��p� describing the I�2P3/2� and I�2P1/2� angular momen-tum distributions arising from the photodissociation of HIand DI as a function of photon energy.71 These results havebeen compared to each other and contrasted to ones42 deter-mined using the original model22 of LeRoy et al. �model 0,see Table I�. We demonstrate that measurement of the aniso-tropy parameters for the excitation from v=0 will be able to
distinguish between the two empirical models that were re-cently developed to account for the measurements from v=2 and the ab initio data; their measurement could also dis-tinguish model 0, which did not properly reproduce the datafor vibrationally mediated photodissociation. While there areexperimental difficulties associated with the measurement ofaQ
�K��p�, including the depolarization of the iodine fragments,they will provide an alternative to the vibrationally mediatedphotodissociation experiments proposed.21 In our recentstudy of HI and DI photodissociation,42 predictions weremade regarding the production of both highly spin-polarizedH and D atoms from the dissociation of HI and DI. Withinthe current adiabatic treatment of the dissociation dynamics,all models predict that spin-polarized hydrogen and deute-rium can be produced for energies below approximately37 500 cm−1 and between 47 500 and 52 500 cm−1, see dis-cussion in Ref. 42 for further details.
Calculations of the anisotropy parameters have also beenperformed for photodissociation of vibrationally excited HIand DI in the v=1, 2, 3, and 4 states, although the results thathave been presented are only for the excitation from v=0.Sharp changes in the vector properties discussed in this workare observed over narrow energy ranges due to the nodes inthe initial vibrational state as have been theoretically deter-mined for other properties, e.g., branching fractions and totalcross sections, for the vibrationally mediated photodissocia-tion of the hydrogen halides.4,12,15,19,21,24,72 As with the scalarproperties,21 there are clear differences between the predic-tions of the aQ
�K��p� parameters based on various models forthe electronic structure. To date, no experimental measure-ments of the aQ
�K��p� parameters from vibrationally mediatedphotodissociation have been obtained for the hydrogen ha-lides.
The recent experimental work of Camden et al. on HIhas shown that vibrationally mediated photodissociation pro-vides a sensitive probe of repulsive excited states. However,the present study clearly demonstrates the sensitivity of vec-tor properties to the details of the electronic structure. Thefour models of the excited states and transition dipole mo-ments, each of which gives similar results for the scalarproperties for the excitation from v=0, exhibit significant,and, more importantly, experimentally measurable differ-ences in their vector properties.
ACKNOWLEDGMENTS
The authors thank the Natural Sciences and EngineeringResearch Council of Canada and the University of Albertafor financial support. One of the authors �A.B.� thanks Dr. A.Alekseyev �Bergische Universität Wuppertal� for providingthe ab initio potential-energy data.
1 J. Zhang, C. W. Riehn, M. Dulligan, and C. Wittig, J. Chem. Phys. 104,7027 �1996�.
2 A. Brown and G. G. Balint-Kurti, J. Chem. Phys. 113, 1870 �2001�.3 B. M. Cheng, C. Y. Chung, M. Bahou, Y. P. Lee, and L. C. Lee, J. Chem.Phys. 117, 4293 �2002�.
4 P. M. Regan, D. Ascenzi, A. Brown, G. G. Balint-Kurti, and A. J. Orr-Ewing, J. Chem. Phys. 112, 10259 �2000�.
5 S. Lee and K. H. Jung, J. Chem. Phys. 112, 2810 �2000�.
FIG. 7. Coherent anisotropy parameters �a� Re�a1�1��� , � �� and �b�
Im�a1�1��� , � �� for the production of excited state I�2P1/2� as a function of
photon energy for the photodissociation of DI initially in the rotationlessground �v=0� vibrational state. The results were determined from model 0�solid line�, model 1 �dotted line�, and model 2 �dashed line�, respectively.
054301-9 Photodissociation of HI and DI J. Chem. Phys. 123, 054301 �2005�
Downloaded 05 Aug 2005 to 129.128.168.123. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp

6 Y. Li, O. Bludsky, G. Hirsch, and R. J. Buenker, J. Chem. Phys. 112, 260�2000�.
7 P. M. Regan, S. R. Langford, D. Ascenzi, P. A. Cook, A. J. Orr-Ewing,and M. N. R. Ashfold, Phys. Chem. Chem. Phys. 1, 3247 �1999�.
8 D. Ascenzi, P. M. Regan, and A. J. Orr-Ewing, Chem. Phys. Lett. 310,477 �1999�.
9 H. M. Lambert, P. J. Dagdigian, and M. H. Alexander, J. Chem. Phys.108, 4460 �1998�.
10 J. Zhang, M. Dulligan, and C. Wittig, J. Chem. Phys. 107, 1403 �1997�.11 T. Duhoo and B. Pouilly, J. Chem. Phys. 103, 182 �1995�.12 I. H. Gersonde, S. Henning, and H. Gabriel, J. Chem. Phys. 101, 9558
�1994�.13 M. H. Alexander, B. Pouilly, and T. Duhoo, J. Chem. Phys. 99, 1752
�1993�.14 K. Tonokura, Y. Matsumi, M. Kawasaki, S. Tasaki, and R. Bersohn, J.
Chem. Phys. 97, 8210 �1992�.15 S. C. Givertz and G. G. Balint-Kurti, J. Chem. Soc., Faraday Trans. 2 82,
1231 �1986�.16 E. van Dishoeck, M. van Hemert, and A. Dalgarno, J. Chem. Phys. 77,
3693 �1982�.17 R. Baumfalk, U. Buck, C. Frischkorn, N. H. Nahler, and L. Hüwel, J.
Chem. Phys. 111, 2595 �1999�.18 P. M. Regan, S. R. Langford, A. J. Orr-Ewing, and M. N. R. Ashfold, J.
Chem. Phys. 110, 281 �1999�.19 B. Pouilly and M. Monnerville, Chem. Phys. 238, 437 �1998�.20 G. Peoux, M. Monnerville, T. Duhoo, and B. Pouilly, J. Chem. Phys.
107, 70 �1997�.21 J. P. Camden, H. A. Bechtel, D. J. A. Brown, A. E. Pomerantz, R. N.
Zare, and R. J. LeRoy, J. Phys. Chem. A 108, 7806 �2004�.22 R. J. LeRoy, G. T. Kraemer, and S. Manzhos, J. Chem. Phys. 117, 9353
�2002�.23 S. Manzhos, H. P. Loock, B. L. G. Bakker, and D. H. Parker, J. Chem.
Phys. 117, 9347 �2002�.24 A. B. Alekseyev, H. P. Liebermann, D. B. Kokh, and R. J. Buenker, J.
Chem. Phys. 113, 6174 �2000�.25 P. M. Regan, D. Ascenzi, C. Clementi, M. N. R. Ashfold, and A. J.
Orr-Ewing, Chem. Phys. Lett. 315, 187 �1999�.26 S. R. Langford, P. M. Regan, A. J. Orr-Ewing, and M. N. R. Ashfold,
Chem. Phys. 231, 245 �1998�.27 D. J. Gendron and J. W. Hepburn, J. Chem. Phys. 109, 7205 �1998�.28 A. J. R. Heck and D. W. Chandler, Annu. Rev. Phys. Chem. 46, 335
�1995�.29 T. N. Kitsopoulos, M. A. Buntine, D. P. Baldwin, R. N. Zare, and D. W.
Chandler, Proc. SPIE 1858, 2 �1993�.30 I. Levy and M. Shapiro, J. Chem. Phys. 89, 2900 �1988�.31 Z. Xu, B. Koplitz, and C. Wittig, J. Phys. Chem. 92, 5518 �1988�.32 C. A. Wight and S. R. Leone, J. Chem. Phys. 79, 4823 �1983�.33 G. N. A. van Veen, K. A. Mohamed, T. Baller, and A. E. deVries, Chem.
Phys. 80, 113 �1983�.34 R. Schmiedl, H. Dugan, W. Meier, and K. H. Welge, Z. Phys. A 304, 137
�1982�.35 R. D. Clear, S. J. Riley, and K. R. Wilson, J. Chem. Phys. 63, 1340
�1975�.36 C. E. Moore, Atomic Energy Levels �U.S. Government Printing Office,
Washington, DC, 1971�.37 T. P. Rakitzis, P. C. Samartzis, R. L. Toomes et al., Chem. Phys. Lett.
364, 115 �2002�.38 T. P. Rakitzis, P. C. Samartzis, R. L. Toomes, T. N. Kitsopoulos, A.
Brown, G. G. Balint-Kurti, O. S. Vasyutinskii, and J. A. Beswick,Science 300, 1936 �2003�.
39 T. P. Rakitzis, P. C. Samartzis, R. L. Toomes, and T. N. Kitsopoulos, J.Chem. Phys. 121, 7222 �2004�.
40 G. G. Balint-Kurti, A. J. Orr-Ewing, J. A. Beswick, A. Brown, and O. S.
Vasyutinskii, J. Chem. Phys. 116, 10760 �2002�.41 A. Brown, G. G. Balint-Kurti, and O. S. Vasyutiniskii, J. Phys. Chem. A
108, 7790 �2004�.42 A. Brown, J. Chem. Phys. 122, 084301 �2005�.43 L. D. A. Siebbeles, M. Glass-Maujean, O. S. Vasyutinskii, J. A. Beswick,
and O. Roncero, J. Chem. Phys. 100, 3610 �1994�.44 T. P. Rakitzis and R. N. Zare, J. Chem. Phys. 110, 3341 �1999�.45 B. V. Picheyev, A. G. Smolin, and O. S. Vasyutinskii, J. Phys. Chem. A
101, 7614 �1997�.46 E. R. Wouters, M. Ahmed, D. S. Peterska, A. S. Bracker, A. G. Suits, and
O. S. Vasyutinskii, in Imaging in Chemical Dynamics, edited by A. G.Suits and R. E. Continetti �American Chemical Society, Washington, DC,2000�, p. 238.
47 A. B. Alekseyev, D. B. Kokh, and R. J. Buenker, J. Phys. Chem. A 109,3094 �2005�.
48 N. Balakrishnan, A. B. Alekseyev, and R. J. Buenker, Chem. Phys. Lett.341, 594 �2001�.
49 A. S. Bracker, E. R. Wouters, A. G. Suits, and O. S. Vasyutinskii, J.Chem. Phys. 110, 6749 �1999�.
50 C. F. Goodeve and A. W. C. Taylor, Proc. R. Soc. London A154, 181�1936�.
51 J. Romand, Ann. Phys. �Paris� 4, 527 �1948�.52 J. F. Ogilvie, Trans. Faraday Soc. 67, 2205 �1971�.53 D. A. Chapman, K. Balasubramanian, and S. H. Lin, Chem. Phys. Lett.
118, 192 �1985�.54 D. A. Chapman, K. Balasubramanian, and S. H. Lin, J. Chem. Phys. 87,
5325 �1987�.55 D. A. Chapman, K. Balasubramanian, and S. H. Lin, Phys. Rev. A 38,
6098 �1988�.56 J. A. Coxon and P. G. Hajigeorgiou, J. Mol. Spectrosc. 150, 1 �1991�.57 R. N. Zare, Angular Momentum �World Scientific, New York, 1988�.58 A. R. Edmonds, Angular Momentum in Quantum Mechanics �Princeton
University Press, Princeton, 1960�.59 G. G. Balint-Kurti, R. N. Dixon, and C. C. Marston, Int. Rev. Phys.
Chem. 11, 317 �1992�.60 G. G. Balint-Kurti, R. N. Dixon, and C. C. Marston, J. Chem. Soc.,
Faraday Trans. 86, 1741 �1990�.61 E. J. Heller, J. Chem. Phys. 68, 2066 �1978�.62 E. J. Heller, J. Chem. Phys. 68, 3891 �1978�.63 E. J. Heller, Acc. Chem. Res. 14, 368 �1981�.64 G. G. Balint-Kurti, Adv. Chem. Phys. 128, 249 �2003�.65 A. Brown and G. G. Balint-Kurti, J. Chem. Phys. 113, 1879 �2001�.66 In recent work �V. V. Kuznetsov and O. S. Vasyutinskii, J. Chem. Phys.
123, 034307 �2005��, the role of molecular axis rotation on the photo-fragment angular momentum distributions has been considered. For lowto moderate rotational states, the Coriolis nonadiabatic interaction shouldbe negligible for the hydrogen halides, see, for example, Refs. 13 and 19.Neglecting the Coriolis nonadiabatic interaction, the molecular frame an-isotropy parameters do not depend on the rotation of the molecular axis.
67 C. C. Marston and G. G. Balint-Kurti, J. Chem. Phys. 91, 3571 �1989�.68 G. G. Balint-Kurti, C. L. Ward, and C. C. Marston, Comput. Phys.
Commun. 67, 285 �1991�.69 H. Tal-Ezer and R. Kosloff, J. Chem. Phys. 81, 3967 �1984�.70 R. Kosloff, J. Phys. Chem. 92, 2087 �1988�.71 See EPAPS Document No. E-JCPSA6-123-024527 for the numerical data
contained in Figs. 2–7, i.e., the anisotropy parameters for I�2P3/2�,I�2P1/2�, and H/D produced from the photodissociation of HI and DI as afunction of photolysis energy. This document can be reached via a directlink in the online article’s HTML reference section or via the EPAPShomepage �http://www.aip.org/pubservs/epaps.html�.
72 C. Kalyanaraman and N. Sathyamurthy, Chem. Phys. Lett. 209, 52�1993�.
054301-10 D. N. Jodoin and A. Brown J. Chem. Phys. 123, 054301 �2005�
Downloaded 05 Aug 2005 to 129.128.168.123. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp




















