
Phenotypic variations of TRAIL sensitivity in cloned populations of prostate cancer cells
13
Journal of Cellular Biochemistry 104:1452–1464 (2008) Phenotypic Variations of TRAIL Sensitivity in Cloned Populations of Prostate Cancer Cells N.A. Cross,* E.A. Waterman, N. Jokonya, A. Fowles, C.H. Buckle, J. Phillips, I. Holen, F.C. Hamdy, and C.L. Eaton Academic Urology Unit, University of Sheffield Medical School, Sheffield, UK Abstract Factors that regulate the induction of apoptosis of tumour cells are potential candidates for therapeutic intervention for the majority of cancers. Studying modifiers of apoptotic responses, such as members of the tumour necrosis factor receptor superfamily, may give clues as to how induction of apoptosis in tumours could be maximized to enhance the benefit of treatment regimes. Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) is a promising anti-tumour molecule since its activity is specific for tumour cell populations. TRAIL binds to death receptors, inducing apoptosis in susceptible cells. The mechanisms which determine whether tumour cells are susceptible to TRAIL are unclear, and several mechanisms have been proposed, including expression of osteoprotegerin (OPG), decoy receptors, and factors that affect intracellular signalling of pro-apoptotic molecules, such as c-FLIP. Here we show that experiments to modulate the activity of one of these factors, OPG, by over-expression and also by stable knockdown of OPG expression, alters the TRAIL sensitivity of PC3 prostate cancer cells. However we show that some observed effects, which appear to support the hypothesis that OPG prevents TRAIL-induced apoptosis of tumour cells, may be due to variation of the TRAIL response of sub-clones of tumour cells, even within a cloned population. These results highlight potential limitations of experiments designed to test contribution of factors affecting intrinsic apoptosis susceptibility using cloned tumour cell populations. J. Cell. Biochem. 104: 1452–1464, 2008. ß 2008 Wiley-Liss, Inc. Key words: osteoprotegerin; RANKL; TRAIL; apoptosis; clonal variation; prostate cancer; osteolytic; bone metastasis Tumour necrosis factor-related apoptosis- inducing ligand (TRAIL) is a member of the tumour necrosis factor (TNF)-superfamily [Wiley et al., 1995; Pitti et al., 1996] and is a potent inducer of apoptosis. TRAIL has poten- tial as an anti-tumour agent since its activity appears to specifically target tumour cells while not affecting most normal cell types [Lawrence et al., 2001]. Induction of apoptosis in suscep- tible cells in response to TRAIL is mediated by its binding to specific Death Receptors, DR4 and DR5 [Sheridan et al., 1997; Pan et al., 1997a,b] initiating apoptosis via an established signal- ling cascade. Activation of the latter by TRAIL- Death Receptor complexes is abrogated by decoy receptors on the cell surface, which bind TRAIL but do not contain functional intracellular signalling domains [Sheridan et al., 1997; Pan et al., 1997b]. TRAIL sensitivity is tightly regu- lated and can be affected both by intracellular and extracellular factors. For example intra- cellular signalling of TRAIL responses may be inhibited by over-expression of c-FLIP, an inhi- bitor of initiator caspases [Zhang et al., 2004]. Secretion of osteoprotegerin (OPG), which acts as a soluble decoy receptor for TRAIL, prevents binding of TRAIL to the Death Receptors [Emery et al., 1998]. OPG also acts as a soluble decoy receptor for Receptor Activator of NFkB (RANK) Ligand (RANKL), preventing osteo- blast-derived RANKL binding and activating RANK on the surface of osteoclasts, thus affect- ing bone turnover [Simonet et al., 1997]. Our recent studies have shown that prostate cancer cells and breast cancer cells are able to secrete biologically relevant levels of OPG ß 2008 Wiley-Liss, Inc. Abbreviations used: OPG, osteoprotegerin; RANK, receptor activator of NFkB; TRAIL, tumour-necrosis factor-related apoptosis-inducing ligand. Grant sponsor: Yorkshire Cancer Research; Grant sponsor: European Framework 6 award (FP6-PRIMA); Grant spon- sor: Medical Research Council (MRC-PROMPT). *Correspondence to: N.A. Cross, Academic Urology Unit, Du10, University of Sheffield Medical School, Beech Hill Road, Sheffield S10 2RX, UK. E-mail: [email protected] Received 14 December 2006; Accepted 9 January 2008 DOI 10.1002/jcb.21721
Transcript of Phenotypic variations of TRAIL sensitivity in cloned populations of prostate cancer cells

Journal of Cellular Biochemistry 104:1452–1464 (2008)
Phenotypic Variations of TRAIL Sensitivity inCloned Populations of Prostate Cancer Cells
N.A. Cross,* E.A. Waterman, N. Jokonya, A. Fowles, C.H. Buckle, J. Phillips, I. Holen,F.C. Hamdy, and C.L. Eaton
Academic Urology Unit, University of Sheffield Medical School, Sheffield, UK
Abstract Factors that regulate the induction of apoptosis of tumour cells are potential candidates for therapeuticintervention for the majority of cancers. Studying modifiers of apoptotic responses, such as members of the tumournecrosis factor receptor superfamily, may give clues as to how induction of apoptosis in tumours could be maximized toenhance the benefit of treatment regimes. Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) is a promisinganti-tumour molecule since its activity is specific for tumour cell populations. TRAIL binds to death receptors, inducingapoptosis in susceptible cells. The mechanisms which determine whether tumour cells are susceptible to TRAIL areunclear, and several mechanisms have been proposed, including expression of osteoprotegerin (OPG), decoy receptors,and factors that affect intracellular signalling of pro-apoptotic molecules, such as c-FLIP. Here we show that experimentsto modulate the activity of one of these factors, OPG, by over-expression and also by stable knockdown of OPGexpression, alters the TRAIL sensitivity of PC3 prostate cancer cells. However we show that some observed effects, whichappear to support the hypothesis that OPG prevents TRAIL-induced apoptosis of tumour cells, may be due to variation ofthe TRAIL response of sub-clones of tumour cells, even within a cloned population. These results highlight potentiallimitations of experiments designed to test contribution of factors affecting intrinsic apoptosis susceptibility using clonedtumour cell populations. J. Cell. Biochem. 104: 1452–1464, 2008. � 2008 Wiley-Liss, Inc.
Key words: osteoprotegerin; RANKL; TRAIL; apoptosis; clonal variation; prostate cancer; osteolytic; bone metastasis
Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) is a member of thetumour necrosis factor (TNF)-superfamily[Wiley et al., 1995; Pitti et al., 1996] and is apotent inducer of apoptosis. TRAIL has poten-tial as an anti-tumour agent since its activityappears to specifically target tumour cells whilenot affecting most normal cell types [Lawrenceet al., 2001]. Induction of apoptosis in suscep-tible cells in response to TRAIL is mediated byits binding to specific Death Receptors, DR4 and
DR5 [Sheridan et al., 1997; Pan et al., 1997a,b]initiating apoptosis via an established signal-ling cascade. Activation of the latter by TRAIL-Death Receptor complexes is abrogated by decoyreceptors on the cell surface, which bind TRAILbut do not contain functional intracellularsignalling domains [Sheridan et al., 1997; Panet al., 1997b]. TRAIL sensitivity is tightly regu-lated and can be affected both by intracellularand extracellular factors. For example intra-cellular signalling of TRAIL responses may beinhibited by over-expression of c-FLIP, an inhi-bitor of initiator caspases [Zhang et al., 2004].Secretion of osteoprotegerin (OPG), which actsas a soluble decoy receptor for TRAIL, preventsbinding of TRAIL to the Death Receptors[Emery et al., 1998]. OPG also acts as a solubledecoy receptor for Receptor Activator of NFkB(RANK) Ligand (RANKL), preventing osteo-blast-derived RANKL binding and activatingRANK on the surface of osteoclasts, thus affect-ing bone turnover [Simonet et al., 1997].
Our recent studies have shown that prostatecancer cells and breast cancer cells are ableto secrete biologically relevant levels of OPG
� 2008 Wiley-Liss, Inc.
Abbreviations used: OPG, osteoprotegerin; RANK, receptoractivator of NFkB; TRAIL, tumour-necrosis factor-relatedapoptosis-inducing ligand.
Grant sponsor: Yorkshire Cancer Research; Grant sponsor:European Framework 6 award (FP6-PRIMA); Grant spon-sor: Medical Research Council (MRC-PROMPT).
*Correspondence to: N.A. Cross, Academic Urology Unit,Du10, University of Sheffield Medical School, Beech HillRoad, Sheffield S10 2RX, UK. E-mail: [email protected]
Received 14 December 2006; Accepted 9 January 2008
DOI 10.1002/jcb.21721

in vitro, reducing the pro-apoptotic effects ofTRAIL [Holen et al., 2002, 2005], an effect thatis reversed by soluble RANKL. Similar findingshave been observed in other tumour types,suggesting that OPG secretion from tumourcells may represent a mechanism allowingtumour cells to bypass the pro-apoptotic effectsof TRAIL [Naumann et al., 2004]. FurthermoreOPG derived from cells of the bone marrow,which represents the most common singlemetastatic site for breast cancer, prostatecancer and multiple myeloma, secrete highlevels of OPG in vitro which can inhibitTRAIL-induced apoptosis of tumour cells [Ship-man and Croucher, 2003; Neville-Webbe et al.,2004; Nyambo et al., 2004]. OPG is expressed inthe bone marrow by stromal cells, osteoblastsand potentially by endothelial cells. Work in ourlaboratories and elsewhere has also shown thatserum levels of OPG are raised in patients withadvanced prostate cancer compared to patientswith organ confined disease [Brown et al., 2001;Jung et al., 2001; Eaton et al., 2004] and thatelevated OPG levels may be an early indicationof relapse in those patients receiving androgenablation treatments [Eaton et al., 2004]. Takentogether, these studies highlight OPG as amarker of progression in prostate cancer thatmay reflect disturbances in bone turnoverassociated with skeletal metastasis. Equally,raised serum OPG in prostate cancer patients,could also promote escape from anti-tumourmechanisms such as TRAIL-induced apoptosisresulting in disease progression.
Recent studies using the LNCaP prostatecancer cell line suggest that over-expression ofOPG in these cells can alter the bone micro-environment when implanted in bone [Coreyet al., 2005]. In contrast to PC3 cells, whichinduce osteolytic tumours in vivo, LNCaP cellsinduce osteosclerotic lesions. PC3 cells inducelytic bone disease most probably as a resultof high DKK1 expression [Hall et al., 2005],which would be predicted to inhibit osteoblastmaturation. In PC3 xenografts it is thereforelikely that osteoclast-mediated bone resorptionremains unopposed resulting in predominantlylytic lesions. Indeed, this concept has beenrecently tested and it has been shown that inthe absence of DKK1, PC3 cells induce osteo-sclerotic lesions [Hall et al., 2005]. Over-expres-sion of OPG in LNCaP cells increases thebone mineral density of tumour-affected bone,and decreases active osteoclast numbers,
suggesting that OPG derived from prostatetumours can affect the bone marrow [Coreyet al., 2005]. However LNCaP cells are largelyresistant to TRAIL, and therefore correlatesbetween OPG secretion and protection fromTRAIL-induced apoptosis were not conclusivefrom this study.
To further elucidate the relevance of secretedOPG to the prevention of TRAIL-inducedapoptosis of tumour cells, we designed experi-ments that would allow us to test the hypothesisthat tumour-derived OPG protects prostatetumour cells from the effects of TRAIL bothin vitro and in vivo. We engineered PC3 prostatecancer cells, which are sensitive to TRAILand secrete sufficient levels of OPG to inhibitTRAIL-induced apoptosis in vitro, to expresshigh levels of OPG and tested whether thesecells now displayed enhanced resistance toTRAIL. Conversely we used shRNA knockdownof endogenously produced OPG in PC3 cells andtested whether TRAIL sensitivity was increasedin these cells. We show that the generation ofstable cell lines with either over-expression,or knockdown of OPG expression by shRNA,results in some clones of cells with phenotypesconsistent with our hypothesis that PC3tumour-derived OPG can protect cells fromTRAIL-induced apoptosis. However we alsoshow that the phenotypes observed may occurvia mechanisms independent of secreted OPG.
METHODS
Cell Lines and Cultures
The human metastatic, androgen independ-ent cell line PC3 was obtained from ATCC. Cellswere cultured in DMEM medium containingGlutamax (Invitrogen, Paisley, UK) supple-mented with 10% Fetal calf serum (Invitrogen)and routinely grown in 5% CO2 at 378C. Prior totransfection experiments, single clones of PC3cells were isolated by serial dilution. These cellswere shown to express OPG and were TRAILsensitive. All cultures were confirmed negativeof mycoplasma spp. infection using EZ-PCRmycoplasma system (Geneflow, Staffordshire,UK). In addition to studies with PC3 cell linesand sub-clones, two breast cancer cell lines,MDA-MB-231 and MDA-MB-436, were used insome experiments. Both of these cell lines havebeen shown to produce OPG and be sensitive toTRAIL [Holen et al., 2005].
Phenotypic Variations of TRAIL Sensitivity 1453

Over-Expression of OPG
A cloned population of PC3 cells was stablytransfected with the pTracer vector (Invitrogen)containing full length OPG (nucleotides 168–1,644, NM_002546.2), or the empty vectorcontaining no insert. The sequence of the insertwas confirmed by DNA sequencing to match thereference sequence for OPG. Cells were stablytransfected using the profection transfectionsystem (Promega, Southamptom, UK). Stabletransformed cell populations were selectedusing 50 mg/ml Blasticidin (Invitrogen). Indi-vidual colonies were picked using cloning disks(Sigma, Poole, UK).
Knockdown of Endogenous OPG
Knockdown of endogenous OPG was per-formed using the pSilencer vector system(Ambion, Huntingdon, UK). shRNA insertswere designed to OPG using the shRNA designtools (www.ambion.com) and were synthesisedby MWG-Biotech (Ebersberg, Germany) asfollows: Forward 50-GAT CCC ACA GCT CACAAG AAC AGA TTC AAG AGA TCT GTT CTTGTG AGC TGT GTT A-30 and reverse 50-AGCTTA ACA CAG CTC ACA AGA ACA GAT CTCTTG AAT CTG TTC TTG TGA GCT GTG G-30.Inserts were cloned into pSilencerCMV4.0 NeoR
vector and transfected into cloned PC3 cellsusing the Profection transfection system (Prom-ega). Stable transformed cells were selectedusing 0.5 mg/ml Geneticin (Invitrogen) andstable transfected clones were isolated asdescribed earlier.
Assessment of OPG Secretion Into Medium
Cells were plated at 10,000 cells/cm2 in24-well plates, and after 2 days, the mediumwas changed. Cell numbers were determinedby Coulter counter and conditioned mediumcollected at 24-h intervals for up to 4 days.Conditioned medium was stored at �208C untilanalysis as follows. Briefly, ELISA plates werecoated with 2 mg/ml mouse monoclonal antihu-man OPG (MAB 8051, R&D Systems, Abingdon,Oxfordshire). The OPG standard curve wasgenerated using recombinant human OPG(R&D Systems) at concentrations from 2,000to 31.25 pg/ml. The secondary antibody wasbiotinylated antihuman OPG (BAF 805, R&DSystems) at 200 ng/ml, and detection was doneusing streptavidin-horseradish peroxidase(R&D Systems) in combination with a 3,30,5,50-
tetramethylbenzidine substrate (Sigma). Thereaction was stopped after 5–20 min incubationin the dark by addition of 50 ml 2 M H2SO4. Theplate was read at 450 nm on a Dynex platereader using Revelation1 software (Dynex,Worthing, Sussex, UK).
Evaluation of OPG Expression by Western Blotting
OPG was immuno-precipitated from condi-tioned medium from PC3 cells and PC3-OPG/EV clones as follows. Two micrograms ofMAB8051 mouse anti-OPG (R&D systems)was added to 1 ml of conditioned medium for2 h at 48C. To this, 40 ml of hydrated ProteinA sepharose (Pharmacia Biotech) was addedand incubated at 48C for 18 h. After washingthree times in PBS, samples were addedto SDS–PAGE loading buffer and run onNuPAGE 4–12% Bis-TRIS polyacrylamide gels(Invitrogen). Proteins were then blotted ontoPVDF membrane at 50 V for 1 h. To ensureeven transfer, membranes were stained withPonceau S (Sigma). Antigens were detected onthe casein-blocked membrane using mousemonoclonal primary antibodies against humanOPG (MAB 8051). Detection was carried outusing a peroxidase labelled anti-mouse secon-dary antibody (Amersham Biosciences, Buck-inghamshire, UK) with ECL detection kit(Amersham Biosciences).
Assessment of Gene Expression byReal-Time RT-PCR
Total cellular RNA was isolated using Tri-Reagent (Sigma) which was reverse transcribedusing Superscript II reverse transcriptase(Invitrogen) and random primers (Invitrogen).Real-timeRT-PCR wasperformedonanABI7900(Applied Biosystems, Warrington, UK) usingUniversal PCR mastermix (Applied Biosystems).Briefly, each reaction consisted of 5 ml of 2�Universal Taqman mastermix, 3.5 ml H2O, 0.5 mlAssay-on-Demand primer/probe mix (AppliedBiosystems). Assays used were GAPDH(Hs99999905 m1), OPG (Hs00171068 m1) DR5(Hs00187196 m1) and (OAS1 Hs00242943 m1).All reactions were performed in triplicate in384-well plates.
RT-PCR data were analysed using SDS 2.0software (Applied Biosystems). Basal geneexpression was first expressed as DCT, namelythe cycle threshold (CT) at which fluorescencewas detectable above background for the inter-nal standard (GAPDH) minus CT of the test
1454 Cross et al.

gene. Data for OPG levels and OAS1 levelswere expressed as relative expression calcu-lated by the comparative CT or DDCT method(Applied Biosystems recommendations). Rela-tive expression was determined by applying theformula 2�DDCT , where DCT¼CT of test gene—CT of reference gene (GAPDH) and DDCT¼DCT
of test gene (knockdown cells)—DCT of test gene(control cells). To enable the use of the com-parative CT calculation, all primer/probe setswere shown to have an efficiency of 100% asdetermined by template dilution experiments(Applied Biosystems recommendations) inwhich a 10-fold dilution of cDNA resulted in a3.3-fold (�0.1) increase in CT over at least threeorders of magnitude.
Assessment of Apoptosis
Apoptosis levels in response to the presence orabsence of TRAIL was determined by assess-ment of nuclear morphology following DAPIstaining. Briefly, cells were seeded into 24-wellplates as described previously. Cells wereallowed to grow for 4 days, then medium waschanged and the cells allowed to grow for afurther 4 days. Cells were then challenged with25 ng/ml TRAIL (R&D Systems) for OPG over-expression studies, and 10 ng/ml TRAIL forOPG knockdown studies in fresh media or in thepresence of 4-day conditioned medium for 24 h.Cells were harvested and fixed in 4% form-aldehyde. In some experiments, cells werechallenged with TRAIL in conditioned mediain the absence and presence of at least a 10-foldmolar excess of sRANK ligand (PeproTech,London, UK). The fixed cells were cytospunonto glass slides and stained with DAPI (1 mg/ml; Sigma). The slides were mounted usingCitiflour (Agar Scientific, Essex, UK) and cellswere counted using UV light microscopy. Inother experiments, cells were plated at lowdensity in 100 mm dishes and discrete coloniesallowed to grow until 2–3 mm diameter, allow-ing assessment of the range of TRAIL sensitiv-ities in a given cell culture. Colonies weremarked prior to challenge with TRAIL (25 ng/ml) in fresh medium, and apoptosis in individ-ual colonies was assessed by adding 1 mg/mlHoechst 33342 (Sigma) prior to assessment ofapoptosis by fluorescence microscopy.
Intratibial Implantation of PC3-OPG Cells
PC3-OPG and PC3-EV cells were harvestedwhen 80% confluent and detached using tryp-
sin/EDTA All procedures were performed onmale MF1 athymic mice (aged 6–8 weeks;Harlan laboratories, Bicester, UK). Intratibialinjections were performed using a modificationof the method of Corey et al. [2002]. Prior to andduring surgery, mice were anaesthetised using1–2% Isofluorane. An incision was made infront of the right tibia and two holes drilledinto the bone marrow cavity using a 27-gaugeneedle. The bone marrow cavity was thenflushed with sterile saline prior to injection of10 ml of culture medium containing 1� 105 cells.Finally, holes in the bone were filled with bonewax. Prior to closure, the area washed withsterile water to kill any tumour cells that spilledout of the holes in the bone, thus reducing theincidence of sub-cutaneous tumour growth inthe leg.
Tartrate Resistant Acid PhoshphataseStaining of Activated Osteoclasts
After sacrifice of animals, legs were fixed in4% paraformaldehyde followed by decalcifica-tion for 96 h in 0.15 M EDTA at 458C. Tissueswere then assessed by standard histology.TRAP-staining was performed on sections usingstandard histological procedures.
RESULTS
Over-Expression of OPG in PC3 Cells
A cloned population of PC3 cells was trans-fected with pTracer vector containing fulllength human OPG. Stable cell clones wereisolated and OPG ELISA performed as shown inFigure 1a. OPG levels in conditioned mediumfrom empty vector clones and untransfectedPC3 cells were less than 1 ng/ml after 4 days inculture. In conditioned medium from clones ofcell over-expressing OPG, over 35 ng/ml wasdetected. Over-expression of OPG did notcorrelate with altered proliferation of trans-fected cell clones, although some variation inproliferation rates of individual OPG clones wasevident, however this did not correlate withthe amount of OPG secreted (Fig. 1b). Real-timeRT-PCR analysis of PC3-OPG clones showedapproximately 1000-fold increase in OPG tran-script levels compared to untransfected PC3cells and PC3 cell clones transfected with emptyvector (Fig. 1c). Conditioned medium from PC3cell cultures was analysed for the presence ofOPG by western blotting (Fig. 1d). Conditioned
Phenotypic Variations of TRAIL Sensitivity 1455
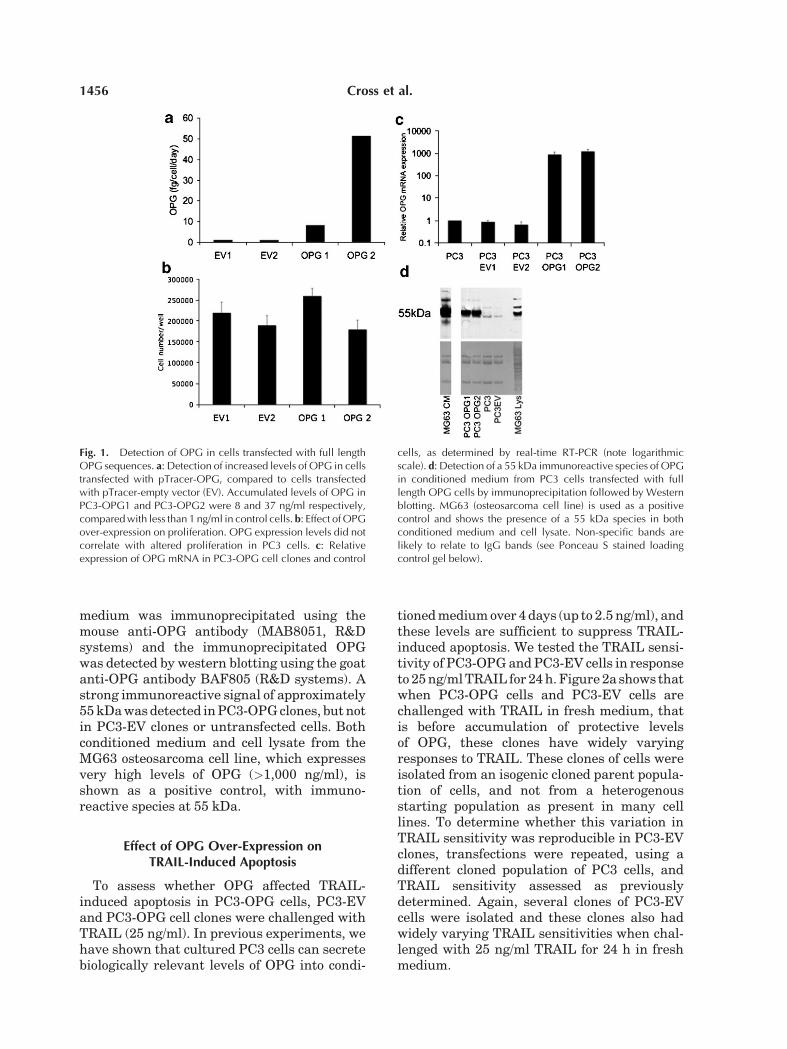
medium was immunoprecipitated using themouse anti-OPG antibody (MAB8051, R&Dsystems) and the immunoprecipitated OPGwas detected by western blotting using the goatanti-OPG antibody BAF805 (R&D systems). Astrong immunoreactive signal of approximately55 kDa was detected in PC3-OPG clones, but notin PC3-EV clones or untransfected cells. Bothconditioned medium and cell lysate from theMG63 osteosarcoma cell line, which expressesvery high levels of OPG (>1,000 ng/ml), isshown as a positive control, with immuno-reactive species at 55 kDa.
Effect of OPG Over-Expression onTRAIL-Induced Apoptosis
To assess whether OPG affected TRAIL-induced apoptosis in PC3-OPG cells, PC3-EVand PC3-OPG cell clones were challenged withTRAIL (25 ng/ml). In previous experiments, wehave shown that cultured PC3 cells can secretebiologically relevant levels of OPG into condi-
tioned medium over 4 days (up to 2.5 ng/ml), andthese levels are sufficient to suppress TRAIL-induced apoptosis. We tested the TRAIL sensi-tivity of PC3-OPG and PC3-EV cells in responseto 25 ng/ml TRAIL for 24 h. Figure 2a shows thatwhen PC3-OPG cells and PC3-EV cells arechallenged with TRAIL in fresh medium, thatis before accumulation of protective levelsof OPG, these clones have widely varyingresponses to TRAIL. These clones of cells wereisolated from an isogenic cloned parent popula-tion of cells, and not from a heterogenousstarting population as present in many celllines. To determine whether this variation inTRAIL sensitivity was reproducible in PC3-EVclones, transfections were repeated, using adifferent cloned population of PC3 cells, andTRAIL sensitivity assessed as previouslydetermined. Again, several clones of PC3-EVcells were isolated and these clones also hadwidely varying TRAIL sensitivities when chal-lenged with 25 ng/ml TRAIL for 24 h in freshmedium.
Fig. 1. Detection of OPG in cells transfected with full lengthOPG sequences. a: Detection of increased levels of OPG in cellstransfected with pTracer-OPG, compared to cells transfectedwith pTracer-empty vector (EV). Accumulated levels of OPG inPC3-OPG1 and PC3-OPG2 were 8 and 37 ng/ml respectively,compared with less than 1 ng/ml in control cells. b: Effect of OPGover-expression on proliferation. OPG expression levels did notcorrelate with altered proliferation in PC3 cells. c: Relativeexpression of OPG mRNA in PC3-OPG cell clones and control
cells, as determined by real-time RT-PCR (note logarithmicscale). d: Detection of a 55 kDa immunoreactive species of OPGin conditioned medium from PC3 cells transfected with fulllength OPG cells by immunoprecipitation followed by Westernblotting. MG63 (osteosarcoma cell line) is used as a positivecontrol and shows the presence of a 55 kDa species in bothconditioned medium and cell lysate. Non-specific bands arelikely to relate to IgG bands (see Ponceau S stained loadingcontrol gel below).
1456 Cross et al.

In these experiments, TRAIL sensitivity didnot correlate with OPG levels, potentiallyindicating the expression of an inappropriateform of OPG from these cells when expression isforced at high levels. To determine whetherOPG was able to prevent TRAIL-induced apop-tosis, as shown previously for the PC3 cell line[Holen et al., 2002], PC3-OPG clones werechallenged with TRAIL in the presence of freshmedium or growth medium that had beenconditioned over these cells for up to 4 days(Fig. 2b). In this system, OPG over-expressiondid not further prevent TRAIL-induced apopto-sis. Data are show for the PC3-OPG2 clone, butsimilar effects were observed in numerous PC3-OPG clones, suggesting that this secreted OPGis unable to prevent TRAIL-induced apoptosisin these clones.
To establish that cloned populations of cellswere responding to TRAIL in a similar mannerto previous experiments, PC3 cells, PC3-OPGcells and PC3-EV cells were challenged withTRAIL in the presence or absence of 20 ng/mlrecombinant human OPG (Fig. 2c). All threestrains of cells were significantly protected from
TRAIL by the addition of rhOPG, consistentwith our previous studies [Holen et al., 2002].
Assessment of OPG-Over Expression onTumour Growth In Vivo
To elucidate why OPG derived from PC3-OPGcells did not protect PC3 cells from TRAIL-induced apoptosis, we tested whether this OPGcould interfere with RANK-RANKL interac-tions. Previous studies have shown that over-expression of OPG in LNCaP prostate cancercells can reduce osteoclast activation in bone bydisrupting RANK–RANKL interactions andhence, osteoclast activation [Corey et al.,2005]. PC3 cells induce osteolytic tumours inbone, which are associated with activatedosteoclasts. PC3-OPG2 cells were injecteddirectly into the tibia of nude mice andbone sections assessed by staining for Tartrateresistant acid phosphatase (TRAP) to deter-mine whether the tumour cells supportedosteoclast differentiation. Figure 3 shows thatPC3-OPG2 cells supported osteoclast activationin bone, suggesting that that OPG derived fromthese cells was not functional.
Fig. 2. a: PC3-EV and PC3-OPG clones were challengedwith 25 ng/ml TRAIL for 24 h in fresh medium. Markedvariation between clones of cells was evident and no correlationbetween OPG expression and TRAIL sensitivity was observed.b: Challenge of PC3-OPG2 cells with 25 ng/ml TRAIL in freshmedium and in medium conditioned (over PC3-OPG2 cellsfor 48 h). Conditioned medium containing high levels
(approximately 25 ng/ml) of accumulated OPG did not protectthe cells from TRAIL. c: Challenge of PC3 cells, PC3-OPG2 andPC3-EV2 cells with TRAIL in the presence or absence of rhOPG(20 ng/ml). rhOPG protected all cells from TRAIL (P< 0.05 t-test).All data shown are representative experiments of three inde-pendent analyses.
Phenotypic Variations of TRAIL Sensitivity 1457

Knockdown of EndogenousOPG in PC3 Cells
We tested whether knockdown of endogenousOPG affected TRAIL sensitivity using stableexpression of shRNA directed to OPG. Again,starting from a cloned population of PC3 cellswhich expressed OPG and was TRAIL sensitive,
stable clones of cells were isolated with approx-imately 50% reduction of OPG secretion asdetermined by OPG ELISA (Fig. 4b). Real-timeRT-PCR confirmed knockdown of OPG mRNAin these clones of cells (Fig. 4a).
Effect of OPG Knockdown onTRAIL-Induced Apoptosis
To test whether knockdown of OPG affectedTRAIL sensitivity, cells were challenged with10 ng/ml TRAIL for 24 h in fresh medium (i.e.,before accumulation of OPG) and apoptosislevels determined by evaluation of nuclearmorphology following DAPI staining (Fig. 5).The level of OPG knockdown in the clones wasvery similar, however TRAIL sensitivity wasincreased in knockdown clone 1 (OPG KD1),whereas no increase in TRAIL sensitivity wasobserved in knockdown clone 2 (OPG KD2).Increased TRAIL-sensitivity was also observedin several other OPG knockdown clones whenchallenged in fresh medium. To assess whetherdifferential expression of the TRAIL receptorDR5 could account for differences in TRAIL
Fig. 3. TRAP staining of activated osteoclasts surrounding PC3-OPG2 cells implanted into the tibia of nude mice, showing thepresence of numerous activated osteoclasts (red) in areas oferoded bone.
Fig. 4. a: Knockdown of OPG mRNA expression by shRNA asdetermined by real-time RT-PCR. Analyses were performed usingtriplicate wells and data obtained from duplicate independentexperiments. b: Knockdown of OPG protein secretion in PC3cells. Cells were cultured for up to 4 days and OPG levels inconditioned medium determined by ELISA. Reduced levelsof secreted OPG was not due to alterations to cell proliferation.
After 72 h, conditioned medium from control cells contained835 pg/ml of accumulated OPG, whereas knockdown cellsexpressed 400 pg/ml. c: Analysis of Death Receptor 5 mRNAlevels in OPG-knockdown cells. OPG-knockdown cells demon-strated raised DR5 mRNA levels as determined by real-timeRT-PCR.
1458 Cross et al.

sensitivity, real-time RT-PCR for DR5 mRNAlevels was performed. OPG KD clones 1 and 2exhibited raised DR5 levels (Fig. 4c), howeversince only OPG KD clone 1 demonstratedincreased TRAIL-sensitivity, DR5 levels alonewere unable to fully explain the differences inTRAIL sensitivity between two knockdownclones with very similar OPG knockdown.
Our previous studies show that the accumu-lation of OPG in conditioned medium protectsPC3 cells against TRAIL-induced apoptosis,and this activity is inhibited using sRANKL[Holen et al., 2002]. To confirm that OPG levelswere insufficient to protect PC3 cells whenchallenged in fresh medium, that is increasedTRAIL sensitivity of OPG-KD clones was notdue to reduced OPG secretion, parental PC3cells were challenged with TRAIL in freshmedium in the presence and absence ofsRANKL, which binds OPG preventing anti-TRAIL activity of OPG (Fig. 6). sRANKL had noeffect on TRAIL-induced apoptosis of parentalPC3 cells in fresh medium and had no effect onapoptosis in the absence of TRAIL, confirmingthat altered OPG levels cannot explain alteredTRAIL sensitivity when challenged in freshmedium, and that increased TRAIL-sensitivityin OPG knockdown cells must be independentof secreted OPG levels.
Interferon Response Is Not Induced by OPGshRNA Expression
Some studies using RNA interference haveencountered problems due to an interferonresponse [Bridge et al., 2003; Fish and Kruithof,2004]. To test whether interferon response wasthe likely cause of increased TRAIL-inducedapoptosis in OPG KD clone 1, cells were testedfor the expression of Oligo Adenylate Synthase-1 (OAS1), a marker of interferon response[Pebernard and Iggo, 2004]. Using quantitativeRT-PCR, all clones of cells were assessed forOAS1 levels. Figure 7 shows that all clones of
Fig. 5. TRAIL-induced apoptosis of cells with reduced OPGexpression. Cells were challenged with 10 ng/ml TRAIL for 24 hin fresh medium. Lower levels of TRAIL were used in theseexperiments, compared to OPG over-expression studies, toallow observation of small changes in TRAIL sensitivity, whichmay be due to alterations of low levels of endogenous OPG. Allclones demonstrated a significant induction of apoptosis inresponse to TRAIL (P<0.05, t-test). OPG Knockdown clone 1demonstrated significantly increased TRAIL sensitivity comparedcontrol cells. Knockdown clone 2, with comparable OPGknockdown to clone 1, demonstrated similar TRAIL sensitivityto control cells. All data shown are representative experiments ofthree independent analyses.
Fig. 6. Increased TRAIL-sensitivity of cells with reduced OPGexpression in fresh medium is not due to secreted OPG. PC3 cellswere cultured challenged with 0, 1, or 10 ng/ml TRAIL for 24 h inthe presence or absence of sRANKL (100 ng/ml). sRANKL did notaffect apoptosis in the absence of TRAIL, and did not affect TRAILsensitivity in fresh medium, suggesting that OPG levels in freshmedium after 24 h are insufficient to affect TRAIL-inducedapoptosis in normal PC3 cells. These findings confirm that theincreased TRAIL sensitivity of PC3 OPG KD clone 1 in freshmedium cannot be attributed to decreased OPG expression.
Fig. 7. Real-time RT-PCR determination of OAS1 levels in cellswith reduced OPG expression. OAS1, a marker of interferonresponse, is not significantly altered between clones of cells withreduced OPG expression but with altered TRAIL sensitivities,suggesting that increased TRAIL sensitivity of Clone 1 is not dueto induction of interferon by expression of shRNA.
Phenotypic Variations of TRAIL Sensitivity 1459

cells expressed very similar levels of OAS1,suggesting that increased TRAIL-sensitivity ofOPG knockdown clones was not likely to be dueto an interferon response.
Assessment of TRAIL Sensitivity ofIndividual Cell Colonies
To assess the range of TRAIL-sensitivities ofindividual cell populations within cell lines andclonal cell lines, cells were plated at low densityand discrete colonies allowed to develop. Colo-nies were photographed and position refer-enced. These colonies were challenged withTRAIL and apoptosis determined by Hoechst33342 staining and assessment of nuclearmorphology. Using untransfected PC3 cells,colonies with widely varying TRAIL sensitiv-ities were observed, ranging from as high as35% apoptosis to less than 5% apoptosis over
24 h (Fig. 8a). In parallel experiments, cellswere treated with TRAIL for longer periods andcolonies that were unaffected by TRAIL readilyproliferated, whereas neighbouring colonieswere completely killed. Similar variations inTRAIL sensitivity were also observed withsingle cell clones of PC3 cells, demonstratingthat even within cloned cell lines, there isconsiderable variation in TRAIL-sensitivitybetween individual cells (Fig. 8b). Similarvariation was observed in the PC3-OPG clonalcell lines (Fig. 8c). To establish whether thisphenomenon was restricted to PC3 cells, wetested two breast cancer cell lines, MDA-MB-231 and MDA-MB-436, both shown to be TRAILsensitive in our previous studies [Holen et al.,2005]. Similarly, cloned cell strains from bothof these cell lines demonstrated a wide rangeof TRAIL-sensitivities in individual colonies(Fig. 8d,e).
Fig. 8. Determination of the range of TRAIL-sensitivities within clonal and non-clonal cell lines. a: PC3(parental cell line, uncloned). b: PC3 (cloned). c: PC3-OPG (cloned). d: MDA231 breast cancer cells(cloned).e: MDA436 breast cancer cells (cloned).Considerable variationof TRAIL sensitivity exists with bothcloned and uncloned cell lines.
1460 Cross et al.

DISCUSSION
Bone and regional lymph nodes are the mostcommon sites for prostate cancer metastasis,and it is likely that interactions betweentumour cells and bone marrow cells make bonea suitable environment for the growth andsurvival of prostate cancer and other tumourtypes. OPG is a bone-derived factor that couldpotentially contribute to prostate cancer growthand survival in bone by two mechanisms: (1) bymodulation of the bone remodelling, and (2) bypromoting prostate cancer cell survival. Theaim of the present study was to develop asuitable system that would allow the testing ofthe hypothesis that endogenously producedOPG from prostate cancer cells could preventTRAIL-induced apoptosis in vivo, as a continu-ation of in vitro findings with this and otherOPG-producing cell lines [Holen et al., 2002,2005]. By modulating OPG expression in PC3cells by over-expression and knockdown, fol-lowed by challenge with TRAIL in vivo, theeffects of tumour-derived OPG, including thatproduced endogenously, would be assessed onpreventing the cytotoxic effects of TRAIL.To minimise any effects of clonal variation, alltransfections were carried out using clonedpopulations of PC3 cells. This would reducethe random isolation of TRAIL sensitive andinsensitive cells strains that develop in hetero-geneous populations of PC3 and other cell lines.We successfully isolated strains of cells fromthe cloned parent population with either OPGover-expression, or OPG knockdown by stableexpression of shRNA directed to OPG. Thesecells were tested for TRAIL sensitivity andwe found in both cases, evidence for clonalvariation of TRAIL responses. We found widevariations of TRAIL sensitivities betweenstrains of cells transfected with empty vectors,suggesting that variation could not be as aresult of OPG secretion. We also found varia-tions in TRAIL sensitivity between strains ofcells with apparently similar levels of OPGknockdown, again suggesting that increasedTRAIL-sensitivity was not necessarily dueto decreased OPG expression. Furthermore,experiments using sRANKL to inactivate OPGprovided evidence that OPG levels did notdetermine altered TRAIL-sensitivity in OPGknockdown cells.
Observations in the current study are con-sistent with clonal variation of TRAIL sensitiv-
ity in cloned populations of PC3 cells. The useof cloned populations of cells was specificallydesigned to reduce the occurrence of sucheffects, however clonal cell populations mayevolve during the time between initial clonalisolation, transfection and subsequent analysis.Clearly, the differing TRAIL-sensitivities ofempty vector cell strains provides the potentialfor considerable variation between PC3-OPG cells and PC3-EV cells, variation thatcould be misinterpreted as effects due to geneexpression.
The predicted effect of OPG over-expressionin TRAIL-sensitive cells would be a reduction inTRAIL sensitivity, which would be furtherenhanced in the presence of accumulatedOPG. Our studies suggest that over-expressedOPG derived from PC3 cells did not function inthe same way as recombinant OPG in modulat-ing responses to TRAIL. In the present study,recombinant OPG was able to suppress TRAIL-induced apoptosis as reported previously [Holenet al., 2002], however OPG derived from PC3-OPG cells did not appear to have any TRAIL-suppressive activity, even when present atlevels that would be expected to be stronglyinhibitory based on studies with recombinantOPG. Furthermore, injection of PC3-OPG2 cellsinto the tibia of nude mice resulted in wide-spread osteolytic disease, with tumour cellssurrounded by active osteoclasts. This situationobserved in our study differs from the reportedeffects of rhOPG on prostate cancer cell growthin bone [Zhang et al., 2001], and also the effectsin bone of over-expression of OPG from LNCaPcells [Corey et al., 2005], where OPG inhibitedosteoclast activation in bone. We conclude fromour studies that OPG over-expression from PC3cells does not produce biologically active oravailable OPG. The findings of the currentstudy mirror observations using breast cancercells [Fisher et al., 2006], whereby over-expres-sion of OPG in MCF-7 cells resulted in highlyosteolytic tumours surrounded by increasednumbers of activated osteoclasts, suggestingthat OPG derived from tumour cells differs inits activity compared to recombinant OPGdelivered therapeutically. Furthermore, over-expression of OPG in MCF-7 cells did not affectTRAIL-induced apoptosis in vitro [Fisher et al.,2006], suggesting clear differences betweenrecombinant human OPG and OPG expressedat high levels from some tumour cell lines.Why secreted OPG possessed no apparent
Phenotypic Variations of TRAIL Sensitivity 1461

biological activity when over-expressed in PC3cells in the present study is unclear, howeveraberrant post-translational modification in PC3cells is possible. Our own studies confirm thatthe same cloned OPG mRNA sequence usedin the present study was able to producebiologically active OPG, as shown by decreasedosteoclast activity, in an alternate biologicalsystem [Rabin et al., 2007].
Since the above studies suggested that ele-vated OPG production in PC3 cells did not affectTRAIL-induced apoptosis, we decided to knock-down endogenous OPG production in clonedPC3 cells. Endogenous OPG produced by theparent PC3 cell line had been shown previouslyto suppress TRAIL-induced apoptosis. Analysisof OPG production by cloned cells before trans-fection and control cells after transfectionindicated that the level of production (max-imum �1 ng/ml) was substantially less thanthat produced by the heterogenous parent PC3cell line (�2.5 ng/ml, Holen et al., 2002). Studiesusing recombinant OPG indicate that concen-trations between 1 and 10 ng/ml are sufficientto inhibit TRAIL-induced apoptosis in PC3cells. This being the case, it may be that eventhough the knockdown experiments success-fully depressed OPG levels in clones, there wasnot sufficient OPG made by these cloned cellsprior to knockdown or in control cells trans-fected with a control vector to inhibit TRAIL.However other non-OPG related differences inapoptosis regulation could again be responsiblefor the lack of consistent correlations betweenOPG secretion and TRAIL responses. We testedwhether Death Receptor expression mightaccount for observed differences betweenTRAIL-sensitive and less-sensitive OPG knock-down clones. Since Death Receptors areresponsible for TRAIL-signalling, alterationsto Death Receptor expression could potentiallypromote TRAIL-induced apoptosis in thesecells. Death Receptor 5 expression was indeedraised in all knockdown clones tested, how-ever not all OPG knockdown clones were moresensitive to TRAIL than control cells (Fig. 4c).Whereas it is possible that raised DeathReceptor levels observed in OPG-knockdowncells could potentially be due to off-targeteffects of constitutive shRNA expression tar-geted to OPG, we cannot conclude that raisedDeath Receptor expression in OPG knockdowncells is correlated with increased TRAIL-sensitivity in these cells.
To establish whether the wide variation inTRAIL-sensitivity observed in transfected,cloned cell populations was due to the effectsof modulation of OPG expression or a naturalphenomenon in cloned cell population, therange of TRAIL-sensitivities was also deter-mined for cloned populations of cells withoutmodulation of OPG expression. In these studies,it was clear that considerable natural variationof TRAIL-sensitivity exists in cloned cell cul-tures. This observation was also observed inMDA-MB-231 and MDA-MB-436 breast cancercells, confirming that it was not a phenomenonrestricted to PC3 cells. Interestingly, the resultsdemonstrate that whereas apoptosis frequen-cies of 10–40% are often reported when cells arechallenged with TRAIL, certain populations arekilled by TRAIL, whereas other colonies are notaffected by TRAIL. These findings have impor-tant implications for studies of TRAIL-sensitivecell lines.
Comparisons of cell lines from differentsources demonstrate widespread heterogeneityin genetically related cell lines, and in addition,heterogeneity exists within individual popula-tions of cells [Nugoli et al., 2003]. Analysis ofosteosarcoma cells over a considerable numberof consecutive passages predictably revealswidespread changes of both genotype andphenotype [Hausser and Brenner, 2005]. Theisolation of highly invasive populations from auveal melanoma cell line over a much shorterperiod revealed populations of geneticallyrelated cells with highly unstable karyotypes,potentially accounting for their invasive pheno-type [Cross et al., 2005]. Previous studies haveshown that analysis of transfected populationsof cells show biological variation in geneexpression, as determined by oligonucleotidearrays of cell lines [Oh et al., 2003]. The use ofcloned or isogenic populations of cells is com-monly used in transfection studies to circum-vent such phenotypic variations betweenselected clonal cell populations. However, evi-dence from the present study and elsewheresuggest that marked heterogeneity in thecloned populations can develop, over relativelyshort periods of time, with several potentialmechanisms. The preferential isolation of clonesof cells, during cloning, with high levels ofgenetic instability may be intrinsic to the pro-cess and may account for variation of observedphenotypes [Cross et al., 2005]. Alternatively, itwas shown recently that adenocarcinoma cells,
1462 Cross et al.

including prostate cancer cells, contain intrinsicstem-like characteristics, facilitating asymmet-rical cell division [Locke et al., 2005]. Pheno-typic drift occurs in cloned populations of cellsin the absence of selective pressure, even afterseveral successive clonal isolations [Barneset al., 2006]. Clonal variation could be reducedbut not abolished after further rounds of clonalisolation, suggesting that isolation of cells witha more limited capacity for phenotypic drift isachievable, but isolation of phenotypically iden-tical cells would not be possible [Barnes et al.,2006]. However, the inadvertent isolation ofclones of cells with altered gene expressionprofiles, in many situations, may not adverselyaffect the outcomes of experiments, as alteredgene expression may not relate to the biologicaloutcome in question.
In summary, we have demonstrated markedbiological variation of TRAIL sensitivity withintransfected populations of cells in the absence ofselection for this phenotype by TRAIL. Theseresults have important implications for studiesof this type. Although the use of cloned, trans-fected populations of cells has proven useful fordetermining the biological effect of gene over-expression and gene silencing in many situa-tions, such studies are dependent on the use ofstringent controls. This study demonstratesthat adequate controls are not always possibleusing standard transfection procedures, andhighlights how easily observations using trans-fected clonal cell populations can ‘fit’ thehypothesis being tested in the absence of testingsufficient numbers of control and test clones.Caution must therefore be observed in inter-preting data where cloned populations of cellsare assumed to have identical phenotypes.
ACKNOWLEDGMENTS
This work was funded by a grant fromYorkshire Cancer Research (NAC), a EuropeanFramework 6 award (FP6-PRIMA; FCH andCLE) and a grant from the Medical ResearchCouncil (MRC-PROMPT; FCH and CLE).
REFERENCES
Barnes LM, Moy N, Dickson AJ. 2006. Phenotypic variationduring cloning procedures: Analysis of the growthbehaviour of clonal cell lines. Biotechnol Bioeng 94:530–537.
Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R.2003. Induction of an interferon response by RNAivectors in mammalian cells. Nat Genet 34:263–264.
Brown JM, Vessella RL, Kostenuik PJ, Dunstan CR, LangePH, Corey E. 2001. Serum osteoprotegerin levels areincreased in patients with advanced prostate cancer. ClinCancer Res 7:2977–2983.
Corey E, Quinn JE, Bladou F, Brown LG, Roudier MP,Brown JM, Buhler KR, Vessella RL. 2002. Establishmentand characterization of osseous prostate cancer models:Intra-tibial injection of human prostate cancer cells.Prostate 52:20–33.
Corey E, Brown LG, Kiefer JA, Quinn JE, Pitts TE, BlairJM, Vessella RL. 2005. Osteoprotegerin in prostatecancer bone metastasis. Cancer Res 65:1710–1718.
Cross NA, Rennie IG, Murray AK, Sisley K. 2005. Theidentification of chromosome abnormalities associatedwith the invasive phenotype of uveal melanoma in vitro.Clin Exp Metastasis 22:107–113.
Eaton CL, Wells JM, Holen I, Croucher PI, Hamdy FC.2004. Serum osteoprotegerin (OPG) levels are associatedwith disease progression and response to androgenablation in patients with prostate cancer. Prostate 59:304–310.
Emery JG, McDonnell P, Burke MB, Deen KC, Lyn S,Silverman C, Dul E, Appelbaum ER, Eichman C,DiPrinzio R, Dodds RA, James IE, Rosenberg M, LeeJC, Young PR. 1998. Osteoprotegerin is a receptor for thecytotoxic ligand TRAIL. J Biol Chem 273:14363–14367.
Fish RJ, Kruithof EK. 2004. Short-term cytotoxic effectsand long-term instability of RNAi delivered usinglentiviral vectors. BMC Mol Biol 5:9.
Fisher JL, Thomas-Mudge RJ, Elliott J, Hards DK, SimsNA, Slavin J, Martin TJ, Gillespie MT. 2006. Osteopro-tegerin overexpression by breast cancer cells enhancesorthotopic and osseous tumor growth and contrastswith that delivered therapeutically. Cancer Res 66:3620–3628.
Hall CL, Bafico A, Dai J, Aaronson SA, Keller ET. 2005.Prostate cancer cells promote osteoblastic bone meta-stases through Wnts. Cancer Res 65:7554–7560.
Hausser HJ, Brenner RE. 2005. Phenotypic instability ofSaos-2 cells in long-term culture. Biochem Biophys ResCommun 333:216–222.
Holen I, Croucher PI, Hamdy FC, Eaton CL. 2002.Osteoprotegerin (OPG) is a survival factor for humanprostate cancer cells. Cancer Res 62:1619–1623.
Holen I, Cross SS, Neville-Webbe HL, Cross NA, Balasu-bramanian SP, Croucher PI, Evans CA, Lippitt JM,Coleman RE, Eaton CL. 2005. Osteoprotegerin (OPG)expression by breast cancer cells in vitro and breasttumours in vivo–A role in tumour cell survival? BreastCancer Res Treat 92:207–215.
Jung K, Lein M, von Hosslin K, Brux B, Schnorr D, LoeningSA, Sinha P. 2001. Osteoprotegerin in serum as a novelmarker of bone metastatic spread in prostate cancer. ClinChem 47:2061–2063.
Lawrence D, Shahrokh Z, Marsters S, Achilles K, Shih D,Mounho B, Hillan K, Totpal K, DeForge L, Schow P,Hooley J, Sherwood S, Pai R, Leung S, Khan L, Gliniak B,Bussiere J, Smith CA, Strom SS, Kelley S, Fox JA,Thomas D, Ashkenazi A. 2001. Differential hepatocytetoxicity of recombinant Apo2L/TRAIL versions. Nat Med7:383–385.
Phenotypic Variations of TRAIL Sensitivity 1463

Locke M, Heywood M, Fawell S, Mackenzie IC. 2005.Retention of intrinsic stem cell hierarchies in carcinoma-derived cell lines. Cancer Res 65:8944–8950.
Naumann U, Wick W, Beschorner R, Meyermann R, WellerM. 2004. Expression and functional activity of osteopro-tegerin in human malignant gliomas. Acta Neuropathol(Berl) 107:17–22.
Neville-Webbe HL, Cross NA, Eaton CL, Nyambo R, EvansCA, Coleman RE, Holen I. 2004. Osteoprotegerin (OPG)produced by bone marrow stromal cells protects breastcancer cells from TRAIL-induced apoptosis. BreastCancer Res Treat 86:269–279.
Nugoli M, Chuchana P, Vendrell J, Orsetti B, Ursule L,Nguyen C, Birnbaum D, Douzery EJ, Cohen P, TheilletC. 2003. Genetic variability in MCF-7 sublines: Evidenceof rapid genomic and RNA expression profile modifica-tions. BMC Cancer 3:13.
Nyambo R, Cross N, Lippitt J, Holen I, Bryden G, HamdyFC, Eaton CL. 2004. Human bone marrow stromal cellsprotect prostate cancer cells from TRAIL-induced apop-tosis. J Bone Miner Res 19:1712–1721.
Oh MK, Scoles DR, Haipek C, Strand AD, Gutmann DH,Olson JM, Pulst SM. 2003. Genetic heterogeneity ofstably transfected cell lines revealed by expressionprofiling with oligonucleotide microarrays. J Cell Bio-chem 90:1068–1078.
Pan G, O’Rourke K, Chinnaiyan AM, Gentz R, Ebner R, NiJ, Dixit VM. 1997a. The receptor for the cytotoxic ligandTRAIL. Science 276:111–113.
Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. 1997b. Anantagonist decoy receptor and a death domain-containingreceptor for TRAIL. Science 277:815–818.
Pebernard S, Iggo RD. 2004. Determinants of interferon-stimulated gene induction by RNAi vectors. Differentia-tion 72:103–111.
Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A,Ashkenazi A. 1996. Induction of apoptosis by Apo-2ligand, a new member of the tumor necrosis factorcytokine family. J Biol Chem 271:12687–12690.
Rabin N, Kyriakou C, Coulton L, Gallagher OM, Buckle C,Benjamin R, Singh N, Gassford J, Otsuki T, Nathwani
AC, Croucher PI, Yong KL. 2007. A new xenograft modelof myeloma bone disease demonstrating the efficacy ofhuman mesenchymal stem cells expressing osteoprote-gerin by lentiviral gene transfer. Leukemia 21:2181–2191.
Sheridan JP, Marsters SA, Pitti RM, Gurney A, SkubatchM, Baldwin D, Ramakrishnan L, Gray CL, Baker K,Wood WI, Goddard AD, Godowski P, Ashkenazi A.1997. Control of TRAIL-induced apoptosis by a familyof signaling and decoy receptors. Science 277:818–821.
Shipman CM, Croucher PI. 2003. Osteoprotegerin is asoluble decoy receptor for tumor necrosis factor-relatedapoptosis-inducing ligand/Apo2 ligand and can functionas a paracrine survival factor for human myeloma cells.Cancer Res 63:912–916.
Simonet WS, Lacey DL, Dunstan CR, Kelley M, ChangMS, Luthy R, Nguyen HQ, Wooden S, Bennett L, BooneT, Shimamoto G, DeRose M, Elliott R, Colombero A,Tan HL, Trail G, Sullivan J, Davy E, Bucay N,Renshaw-Gegg L, Hughes TM, Hill D, Pattison W,Campbell P, Sander S, Van G, Tarpley J, Derby P, LeeR, Boyle WJ. 1997. Osteoprotegerin: A novel secretedprotein involved in the regulation of bone density. Cell89:309–319.
Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP,Nicholl JK, Sutherland GR, Smith TD, Rauch C, SmithCA. 1995. Identification and characterization of a newmember of the TNF family that induces apoptosis.Immunity 3:673–682.
Zhang J, Dai J, Qi Y, Lin DL, Smith P, Strayhorn C,Mizokami A, Fu Z, Westman J, Keller ET. 2001.Osteoprotegerin inhibits prostate cancer-induced osteo-clastogenesis and prevents prostate tumor growth in thebone. J Clin Invest 107:1235–1244.
Zhang X, Jin TG, Yang H, DeWolf WC, Khosravi-Far R,Olumi AF. 2004. Persistent c-FLIP(L) expression isnecessary and sufficient to maintain resistance to tumornecrosis factor-related apoptosis-inducing ligand-medi-ated apoptosis in prostate cancer. Cancer Res 64:7086–7091.
1464 Cross et al.




















