
Pharmacological Inhibition of Protein Kinase C Attenuates Cardiac Fibrosis and Dysfunction in...
14
Pharmacological Inhibition of -Protein Kinase C Attenuates Cardiac Fibrosis and Dysfunction in Hypertension-Induced Heart Failure Koichi Inagaki, Tomoyoshi Koyanagi, Natalia C. Berry, Lihan Sun, Daria Mochly-Rosen Abstract—Studies on genetically manipulated mice suggest a role for -protein kinase C (PKC) in cardiac hypertrophy and in heart failure. The potential clinical relevance of these findings was tested here using a pharmacological inhibitor of PKC activity during the progression to heart failure in hypertensive Dahl rats. Dahl rats, fed an 8% high-salt diet from the age of 6 weeks, exhibited compensatory cardiac hypertrophy by 11 weeks, followed by heart failure at 17 weeks and death by the age of 20 weeks (1233 days). Sustained treatment between weeks 11 and 17 with the selective PKC inhibitor V1-2 or with an angiotensin II receptor blocker olmesartan prolonged animal survival by 5 weeks (V1-2: 1547 days; olmesartan: 1495 days). These treatments resulted in improved fractional shortening (V1-2: 582%; olmesartan: 532%; saline: 416%) and decreased cardiac parenchymal fibrosis when measured at 17 weeks without lowering blood pressure at any time during the treatment. Combined treatment with V1-2, together with olmesartan, prolonged animal survival by 5 weeks (37 days) relative to olmesartan alone (from 1605 to 19714 days, respectively) and by 11 weeks (74 days) on average relative to saline-treated animals, suggesting that the pathway inhibited by PKC inhibition is not identical to the olmesartan-induced effect. These data suggest that an PKC-selective inhibitor such as V1-2 may have a potential in augmenting current therapeutic strategies for the treatment of heart failure in humans. (Hypertension. 2008;51:1565-1569.) Key Words: heart failure protein kinase C ventricular remodeling hypertrophy fibrosis A lthough two-thirds of cases of heart failure in the United States are because of myocardial infarction, hyperten- sion is a major contributor to this morbidity. Therefore, we studied hypertension-induced heart failure using hypertensive salt-sensitive Dahl rats. 1,2 Because many of the signaling events associated with heart failure involve activation of protein kinase C (PKC), 1,3 we determined whether PKC regulation affects disease progres- sion. We focused on PKC, because there are conflicting reports on its role in cardiac hypertrophy and heart failure based on genetic manipulation of mice. 1,4–6 Because the enzyme may have different roles during heart development, we used a pharmacological approach to selectively inhibit it at a defined time during disease. We previously designed isozyme-selective peptide regula- tors of PKC, which function by inhibiting or activating PKC translocation. These peptide regulators are linked to memb- rane-permeable peptides, TAT 47–57 , to enable their effective intracellular delivery and are, therefore, useful pharmacolog- ical tools. Using the Dahl salt-sensitive hypertension-induced heart failure rat model, 1,2 we determined here whether sus- tained pharmacological inhibition of PKC with the above- mentioned peptide or with an angiotensin receptor blocker, olmesartan, delays progression to heart failure. Methods Hypertension-Induced Heart Failure Model Animal protocols were approved by the Stanford University Institu- tional Animal Care and Use Committee. Male Dahl salt-sensitive rats on an 8% NaCl-containing diet or on a low-salt diet, as described previously, 1,4–6 were treated between the ages of 11 and 17 weeks with V1-2 or V1-1 (3 mg/kg per day), with equimolar concentra- tions of TAT (1.6 mg/kg per day; synthesized as described 4,7 ) or with saline, using osmotic pumps implanted subcutaneously (3 mg/kg per day provides a maximal effect on PKC translocation without causing any adverse effects 7 ). A fourth group was treated with olmesartan (3 mg/kg per day in 0.5% carboxymethylcellulose) by daily gavage (Figure S1A, available online at http://hyper.ahajournals.org). The osmotic pumps were replaced every 2 weeks but were discontinued after the age of 17 weeks, because half of the hypertensive control rats died by that age. Additional groups were treated with both V1-2 and olmesartan or with olmesartan alone from 11 to 19 weeks of age (Figure S1B). Cardiac functions and blood pressure did not differ between these groups before drug treatment (Figure S1C and S1D). Systolic blood pressure was measured by the tail-cuff method (BP-2000, Visitech Systems), and fractional shortening (FS) was measured by transthoracic echocardiography (Vivid 7, GE). Left Received January 7, 2008; first decision January 23, 2008; revision accepted March 21, 2008. From the Department of Chemical and Systems Biology, Stanford University School of Medicine, Stanford, Calif. Current address (K.I.): Otsu Red-Cross Hospital, Shiga, Japan. The first 2 authors contributed equally to this study. Correspondence to Daria Mochly-Rosen, Department of Chemical and Systems Biology, Stanford University School of Medicine, CCSR, Room 3145A, 269 Campus Dr, Stanford, CA 94305-5174. E-mail [email protected] © 2008 American Heart Association, Inc. Hypertension is available at http://hyper.ahajournals.org DOI: 10.1161/HYPERTENSIONAHA.107.109637 1565 by guest on September 29, 2015 http://hyper.ahajournals.org/ Downloaded from by guest on September 29, 2015 http://hyper.ahajournals.org/ Downloaded from by guest on September 29, 2015 http://hyper.ahajournals.org/ Downloaded from by guest on September 29, 2015 http://hyper.ahajournals.org/ Downloaded from by guest on September 29, 2015 http://hyper.ahajournals.org/ Downloaded from by guest on September 29, 2015 http://hyper.ahajournals.org/ Downloaded from by guest on September 29, 2015 http://hyper.ahajournals.org/ Downloaded from by guest on September 29, 2015 http://hyper.ahajournals.org/ Downloaded from by guest on September 29, 2015 http://hyper.ahajournals.org/ Downloaded from by guest on September 29, 2015 http://hyper.ahajournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Pharmacological Inhibition of Protein Kinase C Attenuates Cardiac Fibrosis and Dysfunction in...

Pharmacological Inhibition of �-Protein Kinase CAttenuates Cardiac Fibrosis and Dysfunction in
Hypertension-Induced Heart FailureKoichi Inagaki, Tomoyoshi Koyanagi, Natalia C. Berry, Lihan Sun, Daria Mochly-Rosen
Abstract—Studies on genetically manipulated mice suggest a role for �-protein kinase C (�PKC) in cardiac hypertrophyand in heart failure. The potential clinical relevance of these findings was tested here using a pharmacological inhibitorof �PKC activity during the progression to heart failure in hypertensive Dahl rats. Dahl rats, fed an 8% high-salt dietfrom the age of 6 weeks, exhibited compensatory cardiac hypertrophy by 11 weeks, followed by heart failure at �17weeks and death by the age of �20 weeks (123�3 days). Sustained treatment between weeks 11 and 17 with theselective �PKC inhibitor �V1-2 or with an angiotensin II receptor blocker olmesartan prolonged animal survival by �5weeks (�V1-2: 154�7 days; olmesartan: 149�5 days). These treatments resulted in improved fractional shortening(�V1-2: 58�2%; olmesartan: 53�2%; saline: 41�6%) and decreased cardiac parenchymal fibrosis when measured at17 weeks without lowering blood pressure at any time during the treatment. Combined treatment with �V1-2, togetherwith olmesartan, prolonged animal survival by 5 weeks (37 days) relative to olmesartan alone (from 160�5 to 197�14days, respectively) and by �11 weeks (74 days) on average relative to saline-treated animals, suggesting that thepathway inhibited by �PKC inhibition is not identical to the olmesartan-induced effect. These data suggest that an�PKC-selective inhibitor such as �V1-2 may have a potential in augmenting current therapeutic strategies for thetreatment of heart failure in humans. (Hypertension. 2008;51:1565-1569.)
Key Words: heart failure � protein kinase C � ventricular remodeling � hypertrophy � fibrosis
Although two-thirds of cases of heart failure in the UnitedStates are because of myocardial infarction, hyperten-
sion is a major contributor to this morbidity. Therefore, westudied hypertension-induced heart failure using hypertensivesalt-sensitive Dahl rats.1,2
Because many of the signaling events associated with heartfailure involve activation of protein kinase C (PKC),1,3 wedetermined whether PKC regulation affects disease progres-sion. We focused on �PKC, because there are conflictingreports on its role in cardiac hypertrophy and heart failurebased on genetic manipulation of mice.1,4–6 Because theenzyme may have different roles during heart development,we used a pharmacological approach to selectively inhibit itat a defined time during disease.
We previously designed isozyme-selective peptide regula-tors of �PKC, which function by inhibiting or activating PKCtranslocation. These peptide regulators are linked to memb-rane-permeable peptides, TAT47–57, to enable their effectiveintracellular delivery and are, therefore, useful pharmacolog-ical tools. Using the Dahl salt-sensitive hypertension-inducedheart failure rat model,1,2 we determined here whether sus-tained pharmacological inhibition of �PKC with the above-
mentioned peptide or with an angiotensin receptor blocker,olmesartan, delays progression to heart failure.
MethodsHypertension-Induced Heart Failure ModelAnimal protocols were approved by the Stanford University Institu-tional Animal Care and Use Committee. Male Dahl salt-sensitive ratson an 8% NaCl-containing diet or on a low-salt diet, as describedpreviously,1,4–6 were treated between the ages of 11 and 17 weekswith �V1-2 or �V1-1 (3 mg/kg per day), with equimolar concentra-tions of TAT (1.6 mg/kg per day; synthesized as described4,7) or withsaline, using osmotic pumps implanted subcutaneously (3 mg/kg perday provides a maximal effect on PKC translocation without causingany adverse effects7). A fourth group was treated with olmesartan (3mg/kg per day in 0.5% carboxymethylcellulose) by daily gavage(Figure S1A, available online at http://hyper.ahajournals.org). Theosmotic pumps were replaced every 2 weeks but were discontinuedafter the age of 17 weeks, because half of the hypertensive controlrats died by that age. Additional groups were treated with both �V1-2and olmesartan or with olmesartan alone from 11 to 19 weeks of age(Figure S1B). Cardiac functions and blood pressure did not differbetween these groups before drug treatment (Figure S1C and S1D).Systolic blood pressure was measured by the tail-cuff method(BP-2000, Visitech Systems), and fractional shortening (FS) wasmeasured by transthoracic echocardiography (Vivid 7, GE). Left
Received January 7, 2008; first decision January 23, 2008; revision accepted March 21, 2008.From the Department of Chemical and Systems Biology, Stanford University School of Medicine, Stanford, Calif. Current address (K.I.): Otsu
Red-Cross Hospital, Shiga, Japan.The first 2 authors contributed equally to this study.Correspondence to Daria Mochly-Rosen, Department of Chemical and Systems Biology, Stanford University School of Medicine, CCSR, Room
3145A, 269 Campus Dr, Stanford, CA 94305-5174. E-mail [email protected]© 2008 American Heart Association, Inc.
Hypertension is available at http://hyper.ahajournals.org DOI: 10.1161/HYPERTENSIONAHA.107.109637
1565 by guest on September 29, 2015http://hyper.ahajournals.org/Downloaded from by guest on September 29, 2015http://hyper.ahajournals.org/Downloaded from by guest on September 29, 2015http://hyper.ahajournals.org/Downloaded from by guest on September 29, 2015http://hyper.ahajournals.org/Downloaded from by guest on September 29, 2015http://hyper.ahajournals.org/Downloaded from by guest on September 29, 2015http://hyper.ahajournals.org/Downloaded from by guest on September 29, 2015http://hyper.ahajournals.org/Downloaded from by guest on September 29, 2015http://hyper.ahajournals.org/Downloaded from by guest on September 29, 2015http://hyper.ahajournals.org/Downloaded from by guest on September 29, 2015http://hyper.ahajournals.org/Downloaded from

ventricle specimens from 17-week-old rats were fixed with 10%buffered formalin, embedded in paraffin, and two 5-�m sectionsfrom 3 to 4 animals were stained with Masson’s trichrome. Anti-transforming growth factor (TGF)-�1 and anticollagen I goat poly-clonal antibodies, anti-GAPDH, and �-actin mouse monoclonalantibodies (Santa Cruz Biotechnology), as well as anti-tissue inhib-itor of matrix metalloproteinase (TIMP) 2 rabbit polyclonal antibody(Chemicon), followed by horseradish peroxidase–conjugated goatantirabbit, mouse, or goat IgG antibody were used for immunoblot-ting. Data were normalized to GAPDH or �-actin. Matrix metallo-proteinase (MMP)-2 activity was measured by in-gel zymography, asdescribed previously,2 and quantified using ImageJ 1.35s software(http://rsb.info.nih.gov/ij/).
Collagen Secretion From Primary CardiacCultured FibroblastsConfluent neonatal cultured fibroblasts8 were serum starved for 48hours before the addition of TGF-�1 (R&D Systems, 10 ng/mL).�V1-2 (1 �mol/L) was administered every 4 hours and at 15 minutesbefore the TGF-�1 addition. Culture media were collected after 24hours, and collagen content was determined using a Sircol solublecollagen assay kit (Biocolor).
StatisticsData are means�SEMs. All of the statistical analyses were assessedby 1-way factorial ANOVA with Fisher’s test, except for thestandard Kaplan-Meier analysis with log-rank test for survival(Figures 1A and 4A) and 2-way repeated ANOVA for systolic bloodpressure (Figure S1C and S1D).
ResultsIn hypertensive Dahl rats, �PKC levels increase by �2-foldduring the adaptive hypertrophy (between 11 and 17 weeks)and decline to the levels of normotensive animals thereafter.1
To determine whether �PKC plays a positive or negative rolein heart failure and death, we treated hypertensive rats with�V1-2 (3 mg/kg per day) or with saline (hypertensive control)from the age of 11 weeks to 17 weeks (Figure S1A). A thirdgroup was treated with the �PKC-selective inhibitor �V1-1 asa negative control, because we found that �PKC levels andactivity do not change in this model.1 A fourth group ofhypertensive rats was treated with the angiotensin II receptor
blocker olmesartan, commonly used for the prevention andtreatment of heart failure in humans. However, we chose adose of olmesartan (3 mg/kg per day) that ameliorates heartfailure without affecting systolic blood pressure, the etiologyof the disease in this model (Figure S1C).
Sustained delivery of �V1-2 or olmesartan (delivered bydaily gavage) between weeks 11 to 17 improved animalsurvival and improved or normalized FS and other parametersof cardiac function (Figures 1 and 2 and Table S1). Therelative increase in cardiac weight was attenuated by olme-sartan but not by �V1-2 (Figure 1D). (The number of animalsin the control-treated group decreased over time, becausefewer animals survived without treatment with �V1-2.) Asexpected, because �PKC levels do not change with thedisease in this animal model,1 �PKC inhibition had no effecton the disease progression (Figure 1), indicating the selectiveeffect of �PKC inhibition (we confirmed that the peptideinhibitors selectively inhibited the translocation of theircorresponding isozymes and not other PKC isozymes in thesehearts; Figure S2). Therefore, while on treatment with the�PKC inhibitor or with olmesartan, cardiac dysfunctionbecause of pressure-overload in hypertensive rats diminished.
Heart failure is associated with increased parenchymalfibrosis,9 and sustained treatment with �V1-2 (and, to a lesserextent, treatment with olmesartan) reduced left ventriclefibrosis relative to hypertensive-control hearts (Figure 3A).The perivascular fibrosis increase in hypertensive rats wasgreatly attenuated by treatment with �V1-2 but not witholmesartan (Figure 3A, right). Furthermore, collagen I levelsin hearts of 17-week–old rats were significantly decreased inthe �V1-2-treated group when compared with the hyperten-sive control group (Figure 3B).
We next measured the levels of one of the major profibroticcytokines, TGF-�1.10 The ratio of active:latent TGF-�1 was2-fold higher in the hypertensive group relative to thenormotensive age-matched rats. Six weeks of treatment with�V1-2, but not with olmesartan, partially inhibited TGF-�1
A
Olm
εV1-2
—
—
HT
NT
B
D
LVW
/TL
(g/m
m)
0
.01
.02
.03 * * * *
— —TA
TδV
1-1
εV1-
2O
lm
NT HT
Age (weeks)
0
0.2
0.4
0.6
0.8
1.0
15 17 19 21 23 25 27 29†‡†‡
OlmεV1-2δV1-1TATSaline
11
C
Lung
W/T
L (g
/mm
) *
†‡†‡
— —TA
TδV
1-1
εV1-
2O
lm
**
NT HT
0.02.04.06.08.10.12
Frac
tiona
l Sho
rteni
ng (%
)
— —TA
TδV
1-1
εV1-
2O
lm
NT HT
010203040506070
*†‡
†‡**
E
Figure 1. �PKC inhibitor or olmesartanslows down the progression of heartfailure. A, Survival rate of rats withhypertension-induced heart failure. Ratswere treated for 6 weeks with eithersaline control (hypertensive control[HT-C], n�12) or equimolar concentra-tions of TAT carrier peptide control (TAT;n�13), the �PKC inhibitor, �V1-2 (n�10),�V1-1 (n�13), or an angiotensin II recep-tor blocker, olmesartan (Olm; n�12). Thegray area represents treatment duration.B, Shown are examples of morphologi-cal changes in the tissue (scale bars,10 mm; left) of echocardiograms (right).Lung weight to tibial length (LungW/TL;C) and left ventricle weight to tibiallength (LVW/TL; D) were measured in17-week-old rats (n�6 per group), andaveraged FS (E) data from each ratgroup at the age of 17 weeks (n�6).*P�0.05 vs normotensive-control (NT-C);†P�0.05 vs HT-C; ‡P�0.05 vs hyperten-sive TAT (HT-TAT).
1566 Hypertension June 2008
by guest on September 29, 2015http://hyper.ahajournals.org/Downloaded from
activation (Figure 3C). We also found that the activity ofMMP2 levels, one of the major MMPs regulating fibrosis andheart failure,2 increased in the 17-week–old hypertensive rats(234�19% compared with normotensive control rats). Thisincrease was greatly inhibited after �PKC inhibition but notby olmesartan (Figure 3D). We next determined whetherTIMP2, the major endogenous tissue inhibitor of MMP-2,11
was affected by �PKC inhibition (Figure 3D). The level ofTIMP2 increased in the �V1-2-treated group relative to theother groups.
We next determined whether �PKC regulation affectscollagen secretion using cultured cardiac fibroblasts (Figure3E). Collagen secretion increased with TGF-�1 treatment,and �V1-2 inhibited it. Because �V1-2 treatment did notaffect collagen secretion under basal conditions (data notshown), the data suggest that �PKC may further contribute toheart failure progression, at least in part, by enhancingTGF-�1–induced collagen release.
Olmesartan treatment did not affect the activity or levels of�PKC (Figure S2A and S2C), and �V1-2 treatment normal-ized perivascular fibrosis but olmesartan did not (Figure 3).Yet, either treatment increased the life span of the hyperten-sive rats to a similar extent and decreased the rate of cardiacdysfunction development (Figure 1). These results suggestthat the mechanism leading to protection by these 2 agentsmay be different and that treatment of hypertensive rats with
both agents should be additive. Indeed, treatment of thehypertensive rats with both �V1-2 and olmesartan up to week17 increased the life span of the animals by up to anadditional 13 weeks as compared with the treatment ofolmesartan alone (average survival was 160�5 days aftertreatment with olmesartan versus 197�14 days after treat-ment with olmesartan together with �V1-2; Figure 4A).Again, the high blood pressure remained unchanged (FigureS1D), yet �V1-2 plus olmesartan-treated animals maintainedFS similar to the normotensive control rats (Figure 1E andTable S1), even at the age of 24 weeks, 5 weeks after thetreatment with �V1-2 and olmesartan terminated (Figure 4Band Table S2). Although the second study did not include agroup of animals treated with �V1-2 alone, it is likely that thecombined treatment of �V1-2, together with olmesartan (acommonly used drug for patients with heart failure), issuperior to treating with each of these agents alone. There-fore, without affecting hypertension, the cause of the disease,treatment with both the �PKC inhibitor and an angiotensin IIreceptor blocker between weeks 11 and 19 prolonged thelives of the rats by �17 weeks with the 2 inhibitors togetherand enhanced the therapeutic effect obtained by each treat-ment alone. The molecular basis for this prolonged longevityremains to be determined.
DiscussionUsing a rat model of hypertension-induced heart failure, wedemonstrated that pharmacological inhibition of �PKC dur-ing the transition from compensatory cardiac hypertrophy toheart failure slowed the progression of heart failure. Therewas a similar benefit to using the angiotensin II receptorblocker olmesartan. Furthermore, combination treatment withthe �PKC inhibitor and olmesartan during transition to heartfailure appears to provide a sustained effect; animals survived�38 weeks, although the treatment was terminated by week19. Importantly, these therapeutic effects were not associatedwith a reduction in blood pressure.
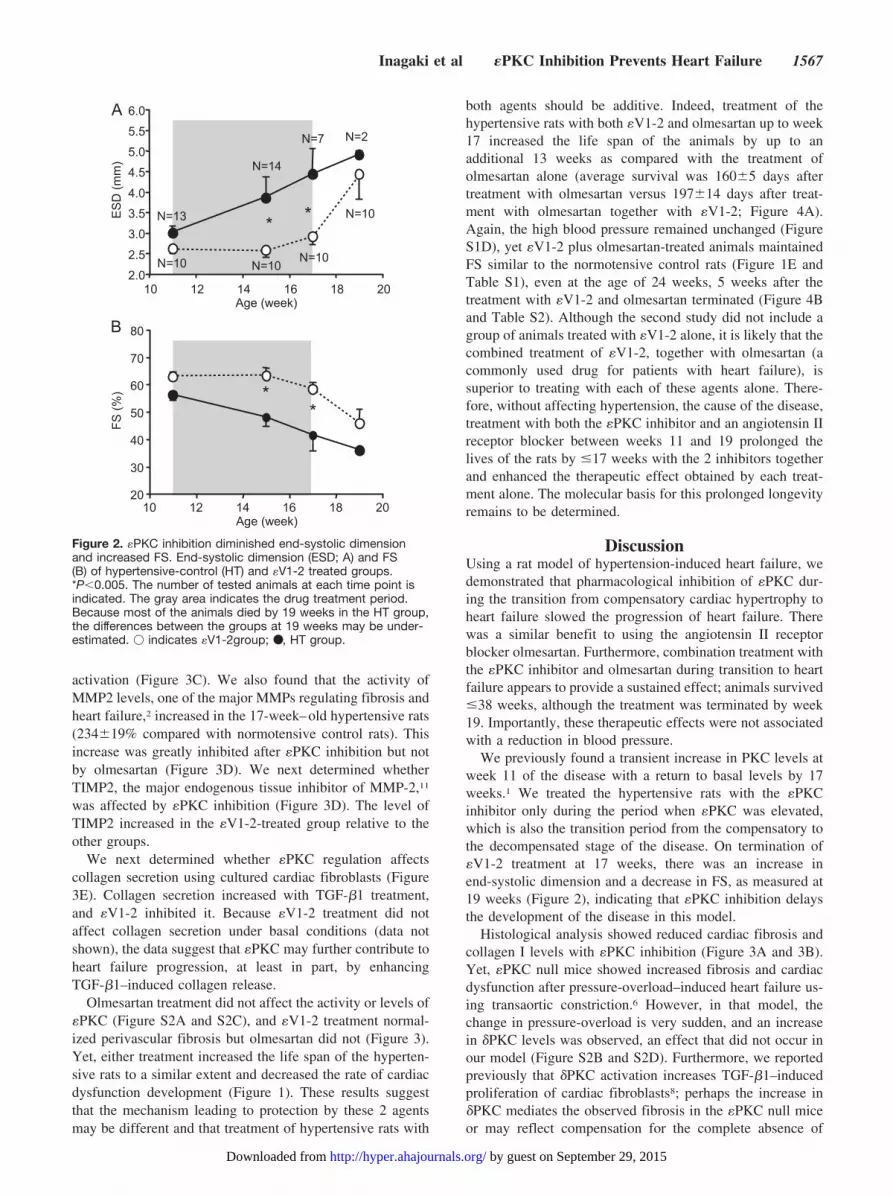
We previously found a transient increase in PKC levels atweek 11 of the disease with a return to basal levels by 17weeks.1 We treated the hypertensive rats with the �PKCinhibitor only during the period when �PKC was elevated,which is also the transition period from the compensatory tothe decompensated stage of the disease. On termination of�V1-2 treatment at 17 weeks, there was an increase inend-systolic dimension and a decrease in FS, as measured at19 weeks (Figure 2), indicating that �PKC inhibition delaysthe development of the disease in this model.
Histological analysis showed reduced cardiac fibrosis andcollagen I levels with �PKC inhibition (Figure 3A and 3B).Yet, �PKC null mice showed increased fibrosis and cardiacdysfunction after pressure-overload–induced heart failure us-ing transaortic constriction.6 However, in that model, thechange in pressure-overload is very sudden, and an increasein �PKC levels was observed, an effect that did not occur inour model (Figure S2B and S2D). Furthermore, we reportedpreviously that �PKC activation increases TGF-�1–inducedproliferation of cardiac fibroblasts8; perhaps the increase in�PKC mediates the observed fibrosis in the �PKC null miceor may reflect compensation for the complete absence of
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
10 12 14 16 18 20Age (week)
ES
D (m
m)
20
30
40
50
60
70
80
10 12 14 16 18 20Age (week)
FS (%
)
**N=13
N=14
N=7 N=2
N=10 N=10N=10
N=10
**
A
B
Figure 2. �PKC inhibition diminished end-systolic dimensionand increased FS. End-systolic dimension (ESD; A) and FS(B) of hypertensive-control (HT) and �V1-2 treated groups.*P�0.005. The number of tested animals at each time point isindicated. The gray area indicates the drug treatment period.Because most of the animals died by 19 weeks in the HT group,the differences between the groups at 19 weeks may be under-estimated. � indicates �V1-2group; ●, HT group.
Inagaki et al �PKC Inhibition Prevents Heart Failure 1567
by guest on September 29, 2015http://hyper.ahajournals.org/Downloaded from

�PKC during development. We also showed that the levels ofactive TGF-�1, a major fibroblast-regulating factor, werereduced by �PKC inhibition in vivo. �PKC inhibition alsoabrogated TGF-�1–induced collagen secretion in vitro. Thesedata are consistent with a profibrotic effect of �PKC in failingheart that we observed in vivo. We also found that �PKC maymodulate cardiac fibrosis by regulating metalloprotease ac-tivity (Figure 3D). However, �PKC inhibition does not affectfibroblast proliferation.8 Therefore, the changes in activeTGF-�1 levels, collagen secretion, and metalloprotease ac-
tivity may lead to the decreased collagen accumulation andfibrosis that we have observed in the hypertensive rats treatedwith the �PKC inhibitor. Consistent with other studies,systolic dysfunction develops in these hypertensive rats at 17weeks and is normalized after treatment with the �PKCinhibitor or olmesartan (Table S1 and Figure S2). Becausediastolic function was not measured, it remains to be deter-mined whether the decrease in fibrosis by �PKC inhibitionimpacts this function. Kidney dysfunction occurring in theseanimals12 may further contribute to heart failure. Because the
—O
lmεV
1-2
A—
Hyp
erte
nsiv
eN
orm
oten
sive
C
(Rat
io to
HT-
cont
rol)
Active TGFβ1Latent TGFβ1
‡ ‡
0
0.5
1.0
— — εV1-2 OlmNT HT
CollagenGAPDH
NT HT
B
— — εV1-2 Olm(Rat
io to
HT-
cont
rol)
0
0.5
1.0 *† *†
Col
lage
n I
Activ
e/La
tent
TG
Fβ1
TGFβ1εV1-2
E
(Rat
io to
no
treat
men
t)
— —00.51.01.52.02.5
|| ||
Secr
eted
Col
lage
nM
MP
2/TI
MP2
D
TIMP2MMP2
NT HTεV1-2— — Olm(R
atio
to H
T-co
ntro
l)
0
0.5
1.0 §§
Figure 3. �PKC regulates fibrosis in hypertensive (HT) Dahl rats. A, Masson’s trichrome staining assessing cardiac fibrosis (blue area).Shown are representative micrographs of parenchymal area (bar�200 �m) or of arteries (bar�100 �m; n�4 per group). Arrowheadsindicate areas of tissue fibrosis. B, Collagen expression in left ventricle tissue in the same samples as in Figure 1 (n�6). *P�0.05 vsHT; †P�0.05 vs normotensive (NT). Bottom, Representative blots. C, The levels of active TGF-�1 presented as a ratio of active dimerform (25-kDa) to latent form (39-kDa; n�6). ‡P�0.05 vs NT. Bottom, Representative blots. D, Net MMP2 activity presented as a ratio ofMMP2 activity (obtained by zymography) divided by the levels of TIMP2 (obtained by Western blot analysis; n�6). A GAPDH level wasused as a loading control for TIMP2 measurement. Bottom, Representatives of each measurement. §P�0.05 vs HT. E, Collagen secre-tion from cultured primary cardiac fibroblasts stimulated with TGF-�1, presented as a ratio to collagen secretion in the absence ofTGF-�1 treatment (n�3). �P�0.05 vs negative control with TGF-�1.
A
0
20
40
60
80
Olm
Olm
+ εV
1-2
Olm
Olm
+εV
1-2
*
Frac
tiona
l sho
rteni
ng (%
)
B19 weeks 24 weeks
Age (weeks)
Olm+εV1-2Olm
*
0
0.2
0.4
0.6
0.8
1.0
17 19 21 23 25 27 29 31 33 35 37 3911 13
Figure 4. Combination treatment with olmesar-tan and the �PKC inhibitor has an additive pro-tective effect in a hypertension (HT)-inducedheart failure model. A, Survival rate of rats withHT-induced heart failure treated with the �PKCinhibitor, �V1-2, together with an angiotensin IIreceptor blocker, olmesartan (Olm; n�12) orwith Olm alone (n�13). The gray area repre-sents the duration of drug treatments. B, FSwas determined in 19. *P�0.05 vs Olm alone.
1568 Hypertension June 2008
by guest on September 29, 2015http://hyper.ahajournals.org/Downloaded from

�PKC inhibitor was delivered systemically, it may benefitother organs, which, in turn, may prevent progression to heartfailure.
Although acute activation of �PKC before ischemia andreperfusion injury in ex vivo rat heart1,13 or before heterotopicheart transplantation14 models are cardioprotective, we al-ready found that a 4-week inhibition of �PKC is protectiveagainst chronic rejection in the heterotopic cardiac transplan-tation model.15 Therefore, �PKC activation is beneficial inacute response to ischemia but is detrimental in chroniccardiac diseases, demonstrating the importance of the use ofselective pharmacological tools that can be administered atdefined time points and for a defined period.
PerspectivesHeart failure is a complex syndrome with multiple etiologies.Whether our findings in these hypertensive rats are translat-able to other models of hypertension and whether the samebenefits can be seen also in myocardial infarction–inducedmodels of heart failure have yet to be determined. Further-more, most of the analyses were carried out at a single timepoint (17 weeks), when some of the treatment groups hadonly 50% survival. The data from the surviving animals mayrepresent events related to a slower disease progression orbetter compensatory mechanisms relative to animals that hadsuccumbed to the disease. We also find it unlikely thatfibrosis inhibition is the sole mechanism leading to thebenefits afforded by �V1-2. The molecular pathways in bothcardiac myocytes and nonmyocytes remain to be elucidated.Finally, we did not determine whether sustained treatmentwill remain beneficial or whether �PKC inhibition and/or anangiotensin receptor blocker provide only a short-term ben-efit. Nevertheless, because the period of treatment coveredthe time from compensated to decompensated hypertrophy inthis model, our study suggests that sustained inhibition of�PKC early during decompensatory hypertrophy may delayor inhibit the development of heart failure. Together, our datasuggest that an �PKC inhibitor, such as �V1-2, may augmentcurrent therapeutic strategies for the treatment of heart failurein humans.
AcknowledgmentWe are grateful to Dr Dan Bernstein for helpful discussionsand advice.
Sources of FundingThis study was supported by National Institutes of Health grantHL076675 to D.M.-R. K.I. was supported in part by a fellowshipgrant from Sankyo Co. N.C.B. was a research fellow supported bythe Stanley J. Sarnoff Cardiovascular Research Foundation, Inc. L.S.was supported in part by a Stanford Graduate Fellowship.
DisclosuresD.M.-R. is the founder of KAI Pharmaceuticals, Inc, a company thatplans to bring protein kinase C regulators to the clinic. However,none of the work described in this study is based on or supported bythe company. The other authors have no disclosures.
References1. Inagaki K, Iwanaga Y, Sarai N, Onozawa Y, Takenaka H, Mochly-Rosen
D, Kihara Y. Tissue angiotensin II during progression or ventricularhypertrophy to heart failure in hypertensive rats; differential effects onPKC epsilon and PKC beta. J Mol Cell Cardiol. 2002;34:1377–1385.
2. Iwanaga Y, Aoyama T, Kihara Y, Onozawa Y, Yoneda T, Sasayama S.Excessive activation of matrix metalloproteinases coincides with leftventricular remodeling during transition from hypertrophy to heart failurein hypertensive rats. J Am Coll Cardiol. 2002;39:1384–1391.
3. Sabri A, Steinberg SF. Protein kinase C isoform-selective signals thatlead to cardiac hypertrophy and the progression of heart failure. Mol CellBiochem. 2003;251:97–101.
4. Dorn GW II, Souroujon MC, Liron T, Chen CH, Gray MO, Zhou HZ,Csukai M, Wu G, Lorenz JN, Mochly-Rosen D. Sustained in vivo cardiacprotection by a rationally designed peptide that causes epsilon proteinkinase C translocation. Proc Natl Acad Sci U S A. 1999;96:12798–12803.
5. Gu X, Bishop SP. Increased protein kinase C and isozyme redistributionin pressure-overload cardiac hypertrophy in the rat. Circ Res. 1994;75:926–931.
6. Klein G, Schaefer A, Hilfiker-Kleiner D, Oppermann D, Shukla P, QuintA, Podewski E, Hilfiker A, Schroder F, Leitges M, Drexler H. Increasedcollagen deposition and diastolic dysfunction but preserved myocardialhypertrophy after pressure overload in mice lacking PKCepsilon. CircRes. 2005;96:748–755.
7. Inagaki K, Begley R, Ikeno F, Mochly-Rosen D. Cardioprotection byepsilon-protein kinase C activation from ischemia: continuous deliveryand antiarrhythmic effect of an epsilon-protein kinase C-activatingpeptide. Circulation. 2005;111:44–50.
8. Braun MU, Mochly-Rosen D. Opposing effects of delta- and zeta-proteinkinase C isozymes on cardiac fibroblast proliferation: use of isozyme-selective inhibitors. J Mol Cell Cardiol. 2003;35:895–903.
9. Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast:therapeutic target in myocardial remodeling and failure. Ann RevPharmacol Toxicol. 2005;45:657–687.
10. Booz GW, Baker KM. Molecular signalling mechanisms controllinggrowth and function of cardiac fibroblasts. Cardiovasc Res. 1995;30:537–543.
11. Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metallopro-teinases: evolution, structure and function. Biochim Biophys Acta. 2000;1477:267–283.
12. Tobian L. Salt and hypertension. Lessons from animal models that relateto human hypertension. Hypertension. 1991;17:I52–I58.
13. Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, Cavallaro G,Banci L, Guo Y, Bolli R, Dorn GW II, Mochly-Rosen D. Opposingcardioprotective actions and parallel hypertrophic effects of delta PKCand epsilon PKC. Proc Natl Acad Sci U S A. 2001;98:11114–11119.
14. Tanaka M, Terry RD, Mokhtari GK, Inagaki K, Koyanagi T, Kofidis T,Mochly-Rosen D, Robbins RC. Suppression of graft coronary arterydisease by a brief treatment with a selective epsilonPKC activator and adeltaPKC inhibitor in murine cardiac allografts. Circulation. 2004;110:II194–I199.
15. Koyanagi T, Noguchi K, Ootani A, Inagaki K, Robbins RC,Mochly-Rosen D. Pharmacological inhibition of epsilon PKC suppresseschronic inflammation in murine cardiac transplantation model. J Mol CellCardiol. 2007;43:517–522.
Inagaki et al �PKC Inhibition Prevents Heart Failure 1569
by guest on September 29, 2015http://hyper.ahajournals.org/Downloaded from

Manuscript Number; HYPERTENSION/2007/109637
Pharmacological inhibition of εPKC attenuates cardiac fibrosis and
dysfunction in hypertension-induced heart failure Short title: εPKC inhibition prevents heart failure
*†Koichi Inagaki, MD, PhD; *Tomoyoshi Koyanagi, PhD; Natalia C. Berry; Lihan Sun;
Daria Mochly-Rosen, PhD
Department of Chemical and Systems Biology, Stanford University School of Medicine,
Stanford, CA 94305-5174
Correspondence to: *Daria Mochly-Rosen, Ph.D. Department of Chemical and Systems Biology Stanford University School of Medicine CCSR, Rm 3145A, 269 Campus Drive Stanford, CA 94305-5174 Tel: 650-725-7720 Fax: 650-723-2253 E-mail: [email protected] *KI and TK contributed equally to this study. †Current address; Otsu Red-Cross Hospital, Shiga, Japan, 520-8511

REFERENCES
1. Inagaki K, Iwanaga Y, Sarai N, Onozawa Y, Takenaka H, Mochly-Rosen D,
Kihara Y. Tissue angiotensin II during progression or ventricular hypertrophy to
heart failure in hypertensive rats; differential effects on PKC epsilon and PKC
beta. J Mol Cell Cardiol. Oct 2002;34(10):1377-1385.
4. Dorn GW, 2nd, Souroujon MC, Liron T, Chen CH, Gray MO, Zhou HZ, Csukai
M, Wu G, Lorenz JN, Mochly-Rosen D. Sustained in vivo cardiac protection by a
rationally designed peptide that causes epsilon protein kinase C translocation.
Proceedings of the National Academy of Sciences of the United States of America.
1999;96(22):12798-12803.
7. Inagaki K, Begley R, Ikeno F, Mochly-Rosen D. Cardioprotection by epsilon-
protein kinase C activation from ischemia: continuous delivery and
antiarrhythmic effect of an epsilon-protein kinase C-activating peptide.
Circulation. Jan 4 2005;111(1):44-50.

Supplemental Figure 1: (A): Scheme of the treatment protocol of the experiments
described in Figures 1-3. (B): Scheme of the treatment protocol of the experiments
described in Figure 4. (C): Systolic blood pressure was measured at the age of 11, 13, 15
and 17 weeks (n=6-17 per group) for animals treated as in the protocol in A (left) or in B
(right). (D): Systolic blood pressure was measured at the age of 11, 15, 19 and 24 weeks
for the animals treated as in the protocol in B. (E): Initial cardiac functions were the same
for all treatment groups at the start of the treatment (e.g., data for the protocol in A).
Averaged fractional shortening from each group at the age of 11 weeks before any drug
treatment has begun. εV1-2 (an εPKC inhibitor, derived from the V1 region of the
enzyme, amino acids 14–21 [EAVSLKPT]) and the δPKC inhibitor, δV1-1 (derived from
the V1 region of δPKC, amino acid 8-17 [SFNSYELGSL]) were synthesized and
conjugated to TAT (carrier peptide, amino acids 47–57 [YGRKKRRQRRR]) via a
disulfide bond at American Peptides (Sunnyvale, CA).]4,7

Supplemental Figure 2: The selective effect of sustained treatment with εV1-2 on
εPKC.
Western blot analyses of the total LV lysates (A and B, respectively) or soluble and
particulate fractions (C and D, respectively; n=6 per group) of ε and δPKC in each
fraction. Quantitative data are presented as a ratio to low-salt control (NT). β-actin or
GAPDH were used as loading controls for the particulate (P) and the soluble (S) fractions,
respectively. n=6 for each. *P<0.05 vs. NT, †P<0.05 vs. HT. The levels and translocation
of ε and δPKC in LV tissues from 17-week-old rats were determined by western blot
analysis, as previously reported.1 All data were normalized by internal controls of
GAPDH or β-actin.

Supplemental Table 1. Body weight and in vivo echocardiographic data.
Group n BW
(g)
PWT
(mm)
EDD
(mm)
ESD
(mm)
FS
(%)
NT-Control 28 424±5* 1.6±0.0 7.0±0.1 2.9±0.2* 60±2*
− 7 285±18† 1.5±0.1 7.4±0.4 4.4±0.6† 41±6†
εV1-2 10 382±7* 1.6±0.1 7.0±0.1 2.9±0.2* 58±2* HT
Olm 20 377±8*† 1.5±0.0 7.5±0.2† 3.5±0.2*† 53±2*†
Body weight (BW) and echocardiographic data (PWT; diastolic posterior LV wall
thickness, EDD; end-diastolic dimension, ESD; end-systolic dimension, FS; fractional
shortening) were measured at 17 weeks of age, when the pharmacological treatments
were terminated. Values are mean ± SEM. †P < 0.05 vs. NT-C; *P < 0.05 vs. HT-Control
(±0.0 denotes SEM smaller than 0.05)

Supplemental Table 2: Body weight and in vivo echocardiographic data of
combination treatment and of olmesartan alone.
Group n BW
(g)
PWT
(mm)
EDD
(mm)
ESD
(mm)
FS
(%)
Olm 6 423±15 1.5±0.0 7.9±0.2 4.9±0.5 38±5
Olm+εV1-2 7 433±13 1.5±0.0 7.2±0.1* 3.4±0.3* 54±4*
The same parameters as in Table 1 were measured at 24 weeks of age (5 weeks after the
pharmacological treatments were terminated). Values are mean ± SEM. *P < 0.05 vs.
Olm. (±0.0 denotes SEM smaller than 0.05)

0
20
40
60
80
— —
NT HT
Supplemental Figure 1: Inagaki et al.
Age (weeks) Low salt High salt diet
6 111
15 17 29
Saline/TAT/δV1-1/εV1-2/Olm
A
C
Systo
lic b
loo
d p
ressu
re
(mm
Hg)
Systo
lic b
loo
d p
ressu
re
(mm
Hg)
Age (weeks) 0
50 100
150
200
250
300
11 13 15 17
Systolic blood pressure
Olm/Olm+εV1-2
Low salt High salt diet
6 11 24 19 39 Age
(weeks)
D
Olm Olm+εV1-2
0 50
100
150
200
250
300
11 15 199
24 Age (weeks)
Systolic blood pressure (mmHg)
E
B
Olm εV1-2 V1-1
TAT Saline
TAT Olm
δV1-1 εV1-2

Supplemental Figure 2 Inagaki et al.

Koichi Inagaki, Tomoyoshi Koyanagi, Natalia C. Berry, Lihan Sun and Daria Mochly-RosenDysfunction in Hypertension-Induced Heart Failure
-Protein Kinase C Attenuates Cardiac Fibrosis andεPharmacological Inhibition of
Print ISSN: 0194-911X. Online ISSN: 1524-4563 Copyright © 2008 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Hypertension doi: 10.1161/HYPERTENSIONAHA.107.1096372008;51:1565-1569; originally published online April 14, 2008;Hypertension.
http://hyper.ahajournals.org/content/51/6/1565World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://hyper.ahajournals.org/content/suppl/2008/04/14/HYPERTENSIONAHA.107.109637.DC1.htmlData Supplement (unedited) at:
http://hyper.ahajournals.org//subscriptions/
is online at: Hypertension Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialHypertensionin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on September 29, 2015http://hyper.ahajournals.org/Downloaded from




















