
Patients with an Inherited Syndrome Characterized by Immunodeficiency, Microcephaly, and Chromosomal...
8
Am. J. Hum. Genet. 42:066-073, 1988 Patients with an Inherited Syndrome Characterized by Immunodeficiency, Microcephaly, and Chromosomal Instability: Genetic Relationship to Ataxia Telangiectasia N. G. J. Jaspers,* R D. F. M. Taalman,t and C. Baan* *l.aboratory of Cell Biology and Genetics, Erasmus University, Rotterdam; and tthe Department of Human Genetics, Catholic University, Nijmegen Summary Fibroblast cultures from six unrelated patients having a familial type of immunodeficiency combined with microcephaly, developmental delay, and chromosomal instability were studied with respect to their response to ionizing radiation. The cells from five of them resembled those from individuals with ataxia telangiec- tasia (AT) in that they were two to three times more radiosensitive on the basis of clonogenic cell survival. In addition, after exposure to either X-rays or bleomycin, they showed an inhibition of DNA replication that was less pronounced than that in normal cells and characteristic of AT fibroblasts. However, the pa- tients are clinically very different from AT patients, not showing any signs of neurocutaneous symptoms. Genetic complementation studies in fused cells, with the radioresistant DNA synthesis used as a marker, showed that the patients' cells could complement representatives of all presently known AT complementa- tion groups. Furthermore, they were shown to constitute a genetically heterogeneous group as well. It is concluded that these patients are similar to AT patients with respect to cytological parameters. The clinical differences between these patients and AT patients are a reflection of genetic heterogeneity. The data indi- cate that the patients suffer from a chromosome-instability syndrome that is distinct from AT. Introduction The inherited syndrome ataxia telangiectasia (AT) is clinically characterized by progressive cerebellar ataxia, oculocutaneous telangiectasia, frequent im- munodeficiency, and chromosomal instability. Pa- tients with the disorder have an enhanced risk of developing neoplasia, particularly in the lympho- reticular system (for reviews, see Sedgwick and Boder 1972; Gatti et al. 1982). Cultured cells from AT patients are hypersensitive to the cytotoxic and clastogenic action of ionizing radiation and of radiomimetic chemical agents such as bleomycin, adriamycin, neocarzinostatin, and hydrogen perox- ide (Taylor 1978; Bridges and Harnden 1982; Shiloh Received March 24, 1987; final revision received June 17, 1987. Address for correspondence and reprints: Dr. N. G. J. Jaspers, The Laboratory of Cell Biology and Genetics, Erasmus University, P.O. Box 1738, 3000 DR Rotterdam, The Netherlands. C 1988 by The American Society of Human Genetics. All rights reserved. 0002-9297/88/4201-0009$02.00 66 et al. 1983; Gatti and Swift 1985). An almost con- sistent biochemical abnormality found in AT cells is a relative resistance of DNA synthesis to the agents mentioned (Cramer and Painter 1981; Painter 1981; Jaspers et al. 1982; Shiloh et al. 1982). Complemen- tation analysis using the abnormal pattern of DNA replication as a marker has revealed that AT is genet- ically heterogeneous (Jaspers and Bootsma 1982; Murnane and Painter 1982). Thus far, five different complementation groups have been identified (Jas- pers et al. 1985; N. G. J. Jaspers and C. Baan, unpub- lished data). In recent years a number of patients have been described as suffering from an inherited syndrome resembling AT in some respects but not presenting one or more of the cardinal clinical symptoms of AT (Daneshbod-Skibba et al. 1980; Weemaes et al. 1981; Sperling 1983; Byrne et al. 1984; Helmerhorst et al. 1984; Seemanova et al. 1985; Conley et al. 1986). The subjects of this report are patients in this category who show immunological impairment and
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Patients with an Inherited Syndrome Characterized by Immunodeficiency, Microcephaly, and Chromosomal...

Am. J. Hum. Genet. 42:066-073, 1988
Patients with an Inherited Syndrome Characterizedby Immunodeficiency, Microcephaly, and ChromosomalInstability: Genetic Relationship to Ataxia TelangiectasiaN. G. J. Jaspers,* R D. F. M. Taalman,t and C. Baan*
*l.aboratory of Cell Biology and Genetics, Erasmus University, Rotterdam; and tthe Department of Human Genetics,Catholic University, Nijmegen
Summary
Fibroblast cultures from six unrelated patients having a familial type of immunodeficiency combined withmicrocephaly, developmental delay, and chromosomal instability were studied with respect to their responseto ionizing radiation. The cells from five of them resembled those from individuals with ataxia telangiec-tasia (AT) in that they were two to three times more radiosensitive on the basis of clonogenic cell survival.In addition, after exposure to either X-rays or bleomycin, they showed an inhibition of DNA replicationthat was less pronounced than that in normal cells and characteristic of AT fibroblasts. However, the pa-tients are clinically very different from AT patients, not showing any signs of neurocutaneous symptoms.Genetic complementation studies in fused cells, with the radioresistant DNA synthesis used as a marker,showed that the patients' cells could complement representatives of all presently known AT complementa-tion groups. Furthermore, they were shown to constitute a genetically heterogeneous group as well. It isconcluded that these patients are similar to AT patients with respect to cytological parameters. The clinicaldifferences between these patients and AT patients are a reflection of genetic heterogeneity. The data indi-cate that the patients suffer from a chromosome-instability syndrome that is distinct from AT.
Introduction
The inherited syndrome ataxia telangiectasia (AT)is clinically characterized by progressive cerebellarataxia, oculocutaneous telangiectasia, frequent im-munodeficiency, and chromosomal instability. Pa-tients with the disorder have an enhanced risk ofdeveloping neoplasia, particularly in the lympho-reticular system (for reviews, see Sedgwick andBoder 1972; Gatti et al. 1982). Cultured cells fromAT patients are hypersensitive to the cytotoxic andclastogenic action of ionizing radiation and ofradiomimetic chemical agents such as bleomycin,adriamycin, neocarzinostatin, and hydrogen perox-ide (Taylor 1978; Bridges and Harnden 1982; Shiloh
Received March 24, 1987; final revision received June 17, 1987.Address for correspondence and reprints: Dr. N. G. J. Jaspers,
The Laboratory of Cell Biology and Genetics, Erasmus University,P.O. Box 1738, 3000 DR Rotterdam, The Netherlands.C 1988 by The American Society of Human Genetics. All rights reserved.0002-9297/88/4201-0009$02.00
66
et al. 1983; Gatti and Swift 1985). An almost con-sistent biochemical abnormality found in AT cells isa relative resistance of DNA synthesis to the agentsmentioned (Cramer and Painter 1981; Painter 1981;Jaspers et al. 1982; Shiloh et al. 1982). Complemen-tation analysis using the abnormal pattern of DNAreplication as a marker has revealed that AT is genet-ically heterogeneous (Jaspers and Bootsma 1982;Murnane and Painter 1982). Thus far, five differentcomplementation groups have been identified (Jas-pers et al. 1985; N. G. J. Jaspers and C. Baan, unpub-lished data).
In recent years a number of patients have beendescribed as suffering from an inherited syndromeresembling AT in some respects but not presentingone or more of the cardinal clinical symptoms of AT(Daneshbod-Skibba et al. 1980; Weemaes et al.1981; Sperling 1983; Byrne et al. 1984; Helmerhorstet al. 1984; Seemanova et al. 1985; Conley et al.1986). The subjects of this report are patients in thiscategory who show immunological impairment and

Variants of Ataxia Telangiectasia
chromosomal instability resembling that in AT butno clinical signs of neurocutaneous involvement. Inaddition, these patients have microcephaly and devel-opmental delay as a common feature. This type ofdisorder was first described by Weemaes et al.(1981), who tentatively called it the "Nijmegenbreakage syndrome" (NBS).
In the present paper we report the results of studieson cultured fibroblasts of another five such patientsfrom various countries, whose clinical characteristics,as published by others (Weemaes et al. 1981; Sperl-ing 1983; Seemanova et al. 1985; Conley et al.1986), resemble those of the original NBS patient inmany respects, although there are variable clinicaldifferences as well. The response to X-rays was inves-tigated, and the cell strains found to be hypersensitivewere subjected to complementation analysis. Thedata suggest that the patients in the present studyresemble AT patients with respect to cellular radio-sensitivity but comprise a separate genetic category;
moreover, they could be assigned to two separatecomplementation groups.
Material and Methods
Cell Strains and Culture ConditionsA list of fibroblast strains used is given in table 1.
The complementation-group assignments of the AT
Table I
Ust of Cell Strains Used
Patient cell strains:79RD27, (NBS) Weemaes et al. 19812239, Sperling 19831129W, patient 7 in Seemanova et al. 19851217G, patient 2 in Seemanova et al. 19851129X, patient 9 in Seemanova et al. 1985GM7166, Conley et al. 1986
AT:Group A: AT3BI, ATSLAGroup B: AT2BEGroup C: AT4BI, AT3LAGroup D: ATSBI, AT6BIGroup E: AT8BI
Normal cell strains:CSRO, 77RD224, 77RD218
cell strains were either those reported elsewhere (Jas-pers and Bootsma 1982; Jaspers et al. 1985) or basedon our unpublished observations. The clinical char-acteristics of the patients under study are summarizedin table 2. Some of the patients' cell strains were
supplied to us by Drs. E. Seemanova, E. Passarge, andR. D. Wegner. GM7166 was obtained from the Hu-man Genetic Repository in Camden, NJ.
Cells were routinely cultured in Ham's F10 me-
dium supplemented with 15% fetal bovine serum
and antibiotics. All fibroblast cultures were free of
Table 2
Patient Characteristics
CELL STRAIN
2239 79RD27 1129W 1217G 1129X GM7166
Patient initials, nationalitya (sex) .. M.H., G (F) H.H., NL (M) J.Z., CZ (M) M.D., CZ (F) D.L., CZ (M) D.M., USA (F)Characteristic:
Telangiectasia .............- - - - -Cerebellar ataxia ............- - -Mental retardation ............ - -+ -+ -Microcephaly ................ + + + + + +Growth retardation ....... .... + + + + + +Peculiar face ................. + + + + + +Infections .................... + + + + + +IgA deficiency ................ + + + + + +Impaired blast transformation .. + + + + + +Malignancies ................. b _ bChromosomal instability ....... + + ? ? ? +Elevated alpha-fetoprotein ...... - -
SOURCE.-Literature data and R. D. Wegner (University of Berlin), personal communication.a G = Germany; NL = The Netherlands; and CZ = Czechoslovakia.b Affected sibs have developed fatal lymphoreticular neoplasia.
67

Jaspers et al.
contamination with mycoplasma, as checked withHoechst 33258 fluorescence microscopy.
Inhibition of DNA SynthesisThe method for determining the rate ofDNA repli-
cation was as described elsewhere (Jaspers et al.1982). In short, cells were prelabeled with 14C-thymidine and exposed to graded doses of X-rays orbleomycin. Then they were cultured for 4 h in thepresence of tritiated thymidine and harvested. Theratio of 3H to 14C radioactivities was taken as a mea-sure of the rate of DNA synthesis and was expressedas a percentage of the values found in untreated con-trols.
X-rays were delivered from a Philips machine oper-ating at 300 kV, giving a dose rate of 1.75 Gy/min.Bleomycin stock solutions were stored at -20 C inaliquots to be used only once. Incubation with thedrug was for 1 h.
Survival ExperimentsCellular survival was measured using a thin-feeder-
layer technique described elsewhere (Jaspers et al.1982). Exponentially growing cultures were exposedto X-rays, trypsinized, and seeded onto feederfibroblasts. Colonies of 50 cells visible after 11-18days were stained with Coomassie blue and counted.Cloning efficiencies in unirradiated cells varied from5% to 33%.
Complementation AnalysisComplementation experiments were carried out
according to a protocol described in a previous report(Jaspers and Bootsma 1982). In short, two cell strainswere preloaded with polystyrene microspheres of dif-ferent sizes for 3 days, trypsinized, mixed, and fusedusing inactivated Sendai virus. One day later theywere exposed to X-rays, cultured with 3H-thymidinefor 2 h, and subsequently fixed and processed forautoradiography. Silver grains were counted above-40 nuclei in S-phase both in the heterodikaryonsand in the two types of homodikaryons, identified onthe basis of their contents of plastic beads. The ex-pected average number of grains in the heterokaryonswas approximated using the following formulas: ex-pected mean = (M1 + M2)/2 and expected SEM =SQRT [(S2 + S22)/2 + (M1 - M2)2/(4N - 2)], whereM and S = the values obtained in the two types ofhomodikaryons and N = the number of nucleicounted in the heterodikaryons.
% DNA synthesis
100
80
60
4ATs
0[77RD224normal - 079RD200
LAC5RO40
[VGM71 66A2239
patients- *1129W.1217G
201 *1 129X
5 10 15 20
Dose of X-rays (Gy)
Figure I Inhibition of DNA synthesis by X-rays. Cells pre-labeled with 14C-thymidine were exposed to graded doses of X-rays and cultured in the presence of 3H-thymidine for 4 h. The rateof DNA synthesis was estimated by the ratio of 3H to 14C radioac-tivity and expressed as a percentage of untreated cells. Open sym-bols denote cells from normal individuals; closed symbols denotecells from patients. The shaded area represents the responses offour different AT cell strains (ATSBI, AT6BI, AT3LA, andAT4BI). The patient data are means of at least two independentexperiments.
Results
Inhibition ofDNA SynthesisIn previous experiments (Taalman et al. 1983) it
had been shown that cells from the patient with NBSwere hypersensitive to X-rays in terms of cellular sur-vival. This cell strain (79RD27) also showed a rela-tively radioresistant pattern of DNA replication, typ-ical of AT cell strains. To test whether the fibroblastsfrom the present series of patients (who have clinicalsymptoms similar to those of the NBS patient) arealso sensitive, the inhibition of DNA synthesis by X-rays was tested. Figure 1 shows that the cell strains1217G, 1129W, 2239, and GM7166 all exhibited anextent of DNA synthesis inhibition that was withinthe range observed for four different AT cell strains
68

Variants of Ataxia Telangiectasia
% DNA synthesis
100
80-
60-
40 '
20 <
Relative survival(%)
V~~~~
VAT5BI3ATs: o GS-K IM
AAT6B I
normal E0 79RD2004* 77RD 218vGM7166
patients- * 121 7G129X
1 0 20 30 40 50Concentration of Bleomycin (pg/ml)
Figure 2 Inhibition of DNA synthesis by bleomycin. Experi-mental conditions are as given in fig. 1. Treatment with bleomycinlasted 60 min. Open symbols denote cells from normal individualsor AT patients. Closed symbols denote cells from patients. Cellstrain GS-KIM is identical to AT3LA.
tested in the same experiment. 1129X cells showedan inhibition that was intermediate between those ofAT and normal cells.The inhibition ofDNA synthesis by bleomycin was
also less pronounced in the patients' cells than in cellsfrom normal individuals and was comparable to thatin AT fibroblasts (see fig. 2). As an exception, the cellstrain 1 129X could not be discriminated from thetwo normal cell strains after treatment with thisagent.
Cellular SurvivalThe colony-forming abilities of the patients' cells
were measured and compared with those of AT andnormal cells. The results, shown in figure 3, indicatethat the strains GM7166, 2239, 1129W, and 1217Gwere as sensitive to X-rays as was the AT cell strainAT5BI and two to three times more sensitive thanwere three normal control strains. The Do valueswere within the range reported for AT elsewhere by
Figure 3 Clonogenic cell survival after exposure to X-rays.Open symbols denote cells from AT patients or normal individ-uals. Closed symbols denote cells from patients. All patient datarepresent the mean of at least two independent triplicate experi-ments. Error bars are given wherever space allows and representthe SEM. The Do values were calculated from the linear part of thesemilogarithmic plots by means of least-squares regression.
us (Taalman et al. 1983) and others (Bridges andHarnden 1982). Again, the 1129X cells showed an
exceptional behavior in that their radiosensitivitywas only slightly different from that of the normalcontrols.
Complementation AnalysisSince most of the patients' cell strains are now
found to exhibit a type of radioresistant DNA syn-thesis, comparable to that observed in AT, there isthe possibility of using the rate ofDNA replication as
a marker in complementation analysis, as has beendone in the genetic analysis of AT (Jaspers andBootsma 1982; Murnane and Painter 1982; Jasperset al. 1985). To this end the rate of 3H-thymidineincorporation in single binucleated cells was mea-
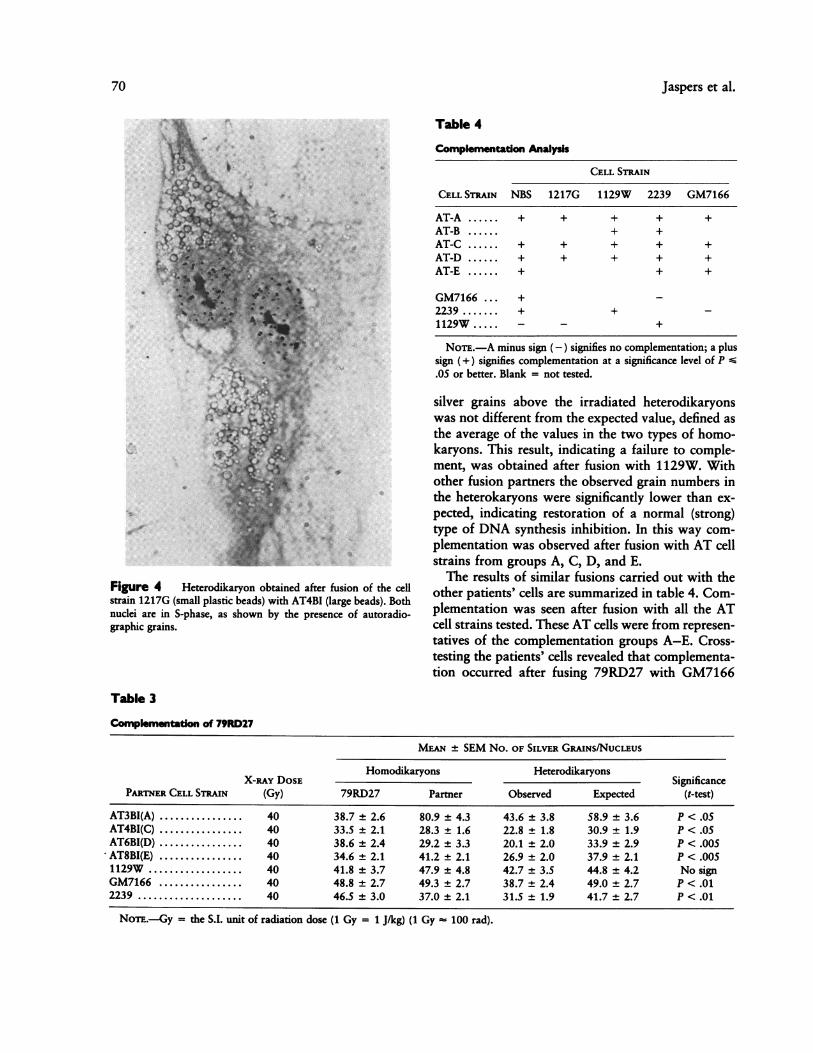
sured using autoradiography and the different typesof fused cells were discriminated on the basis of theircytoplasmic beads. Figure 4 shows an example of a
heterodikaryon.Table 3 presents the results of a series of fusions
with cell strain 79RD27. In some cases the number of
Do( Gy)o 1.28o 1.27A 1.43V 0.56v 0.50A 0.59* 0.49* 0.55* 1.12
3 4X-ray dose (Gy)
69
Jaspers et al.
Figure 4 Heterodikaryon obtained after fusion of the cellstrain 1217G (small plastic beads) with AT4BI (large beads). Bothnuclei are in S-phase, as shown by the presence of autoradio-graphic grains.
Table 4
Complementation Analysis
CELL STRAIN
CELL STRAIN NBS 1217G 1129W 2239 GM7166
AT-A ...... + + + + +AT-B + +AT-C ...... + + + + +AT-D ...... + + + + +AT-E ...... + + +
GM7166 ... +2239 ....... + + -1129W ..... - - +
NoTE.-A minus sign (-) signifies no complementation; a plussign (+) signifies complementation at a significance level of P S.05 or better. Blank = not tested.
silver grains above the irradiated heterodikaryonswas not different from the expected value, defined asthe average of the values in the two types of homo-karyons. This result, indicating a failure to comple-ment, was obtained after fusion with 1129W. Withother fusion partners the observed grain numbers inthe heterokaryons were significantly lower than ex-pected, indicating restoration of a normal (strong)type of DNA synthesis inhibition. In this way com-plementation was observed after fusion with AT cellstrains from groups A, C, D, and E.The results of similar fusions carried out with the
other patients' cells are summarized in table 4. Com-plementation was seen after fusion with all the ATcell strains tested. These AT cells were from represen-tatives of the complementation groups A-E. Cross-testing the patients' cells revealed that complementa-tion occurred after fusing 79RD27 with GM7166
Table 3
Complementatlon of 79RD27
MEAN +*- SEM No. OF SILVER GRAINS/NucLEus
X-RAy DOSE Homodikaryons Heterodikaryons SignificancePARTNER CELL STRAIN (Gy) 79RD27 Partner Observed Expected (t-test)
AT3BI(A) .40 38.7 ± 2.6 80.9 ± 4.3 43.6 ± 3.8 58.9 ± 3.6 P < .05AT4BI(C) .40 33.5 ± 2.1 28.3 ± 1.6 22.8 ± 1.8 30.9 ± 1.9 P < .05AT6BI(D) .40 38.6 ± 2.4 29.2 ± 3.3 20.1 ± 2.0 33.9 ± 2.9 P < .005AT8BI(E) .40 34.6 ± 2.1 41.2 ± 2.1 26.9 ± 2.0 37.9 ± 2.1 P < .0051129W.40 41.8 ± 3.7 47.9 ± 4.8 42.7 ± 3.5 44.8 ± 4.2 No signGM7166 .40 48.8 ± 2.7 49.3 ± 2.7 38.7 ± 2.4 49.0 +2.7 P < .012239 .40 46.5 ± 3.0 37.0 ± 2.1 31.5 ± 1.9 41.7 ± 2.7 P < .01
NoTE.-Gy = the S.I. unit of radiation dose (1 Gy = 1 J/kg) (1 Gy 100 rad).
70

Variants of Ataxia Telangiectasia
Table 5
Complementation Groups
Group V1 ......................... NBS, 1129W, 1217GGroup V2 ......................... 2239, GM7166
and with 2239 (also see table 3). 1129W fails to com-plement 79RD27, indicating that these two cellstrains belong to the same complementation group,
different from the one with GM7166 and 2239, thatdo not complement each other. The two Czecho-slovakian cell strains 1129W and 1217G also do notcomplement. Altogether the results indicate that thepatients can be assigned to two different complemen-tation groups, designated as V1 and V2 (table 5), thatare different from the five AT groups.
Discussion
The patients in the present study were indepen-dently identified by different clinicians on the basis ofa familial pattern of immunodeficiency combinedwith developmental delay, microcephaly and some
other variable features. In some of these patients alsochromosomal instability and the presence of lym-
phocytic clones with translocations involving thechromosomes 7 and 14 were demonstrated. In thisrespect a parallel was drawn with the well-knowngenetic disorder AT. To further investigate the rela-tionship to AT we studied the response to ionizingradiation of cultured fibroblasts from these patients.In an earlier report we described radiosensitivity interms of cellular survival, induction of chromosomalbreakage, and DNA synthesis inhibition in the cellstrain 79RD27 from the NBS patient (Taalman et al.1983). The present data show that cellular radiosen-sitivity is also a characteristic of the five patients inthe present study. Both clonogenic cell survival andthe inhibition of DNA synthesis were abnormal andin the range observed in cells from AT patients. Therate of DNA synthesis was relatively resistant tobleomycin, as has been reported for AT as well(Cramer and Painter 1981; Jaspers et al. 1982; Shilohet al. 1983). In one of the cell strains the radiosen-sitivity was much less pronounced and was inter-mediate between that in AT and normal control cells.The cellular survival was at the borderline for ATpatients, and the DNA synthesis inhibition was inter-mediate between AT and normal cells after X-raysand within the normal range after exposure to
bleomycin. Further studies by one of us (R. D. F. M.Taalman) (to be published elsewhere) have shownthat in this patient there were neither consistent im-munological disturbances (except for a lowered IgD)nor signs of chromosomal instability. Furthermore,the inherited nature of the disease had not yet beenestablished (Seemanova et al. 1985). It seems prob-able that some other disease condition is present inthis patient.The present data indicate that on the cytological
level these patients cannot be easily distinguishedfrom AT patients. However, clinically they appeardefinitely distinct in that they do not show the neuro-cutaneous hallmarks of AT. In addition, micro-cephaly and developmental delay are symptoms notregarded to be part of the AT syndrome (Sedgwickand Boder 1972). There is the possibility that thepatients are a reflection of an extensive clinicalheterogeneity that may exist in AT. Alternatively,they may suffer from one or more new and differentgenetic syndromes. To distinguish between these twopossibilities we have undertaken a genetic comple-mentation study. This approach is feasible becausethese patients and AT patients share the feature ofradioresistant DNA synthesis, which can be used as amarker in such experiments, as has been reportedelsewhere (Jaspers et al. 1985). Our data show thatthe fibroblasts from all the patients can complementrepresentatives from all presently known com-plementation groups in AT. This result supports theidea that the presently described disease conditionsare genetically distinct from AT. One cell strain,1129X, could not be studied in this respect becauseof its weak radiosensitivity.AT has been shown to be genetically heterogene-
ous. Thus far five different complementation groups,designated A-E, have been identified in experimentsusing various aspects of radiosensitivity as a marker(Jaspers et al. 1985; N. G. J. Jaspers, and C. Baan,unpublished data). Cross-fusions with different cellstrains from the patients in the present study alsoshow genetic heterogeneity. Two separate com-plementation groups could be discerned, tentativelydesignated V1 and V2 (V for variant). Group V1harbors patients originating from The Netherlandsand Czechoslovakia. Sibs of the Czechoslovakian pa-tients have died from lymphoreticular neoplasia at ayoung age (Seemanova et al. 1985). It is to be ex-pected that the NBS patient who belongs to the samegroup has a high risk of developing malignancies aswell. In addition, the Czechoslovakian patients ex-
71

72 Jaspers et al.
hibited a chromosomal instability that closely re-sembled that in the patient with the NBS (R. D. F. M.Taalman, unpublished data). In group V2 there are apatient identified in Germany and an American pa-tient who was reported to be of Eastern-Europeanancestry (Conley et al. 1986). It is not knownwhether patients with these clinical characteristicscan also be found in other geographical locations orethnic groups. In this respect, patients discribed insome other recent reports (Marachio et al. 1986;Teebi et al. 1987) and resembling those reported herewould warrant further study.
In conclusion, the present data support the ideathat patients characterized by a familial type ofimmunodeficiency combined with early-onset mi-crocephaly and developmental delay, as first de-scribed in the NBS patient, suffer from a syndromethat is genetically distinct from AT. By its nature thisconclusion remains preliminary. More extensivecomplementation studies on patients with either ATor AT-like variant syndromes may produce a differ-ent picture; e.g., a classical AT patient could perhapsin the future be assigned to one of the groups V1 orV2. Whatever the final picture may be, our data fur-ther point to a high complexity in the cellular re-sponse to ionizing radiation and to radiomimeticchemical agents. Basic defects in this responsecan be reflected by a wide spectrum of clinical ab-normalities.
AcknowledgmentsThis research was supported by a grant from the Ataxia
Telangiectasia Medical Research Fund and by Euratomcontract BI6-E-141-NL. The authors wish to thank Drs. E.Seemanova, E. Passarge, R. A. Gatti, and R. D. Wegner fortheir essential help in obtaining the fibroblasts.
ReferencesBridges, B. A., and D. G. Harnden, eds. 1982. Ataxia
telangiectasia: a molecular link between cancer, neuro-pathology and immune deficiency. Wiley & Sons,Chichester, United Kingdom.
Byrne, E., J. F. Hallpike, J. I. Manson, G. R. Sutherland,and Y. H. Thong. 1984. Progressive multisystem degen-eration with IgE deficiency and chromosomal instability.J. Neurol. Sci. 66:307-317.
Conley, M. E., M. B. Spinner, B. S. Emanuel, P. C. Nowell,and W. W. Nichols. 1986. A chromosome breakagesyndrome with profound immunodeficiency. Blood67:1251-1256.
Cramer, P., and R. B. Painter. 1981. Bleomycin-resistantDNA synthesis in ataxia telangiectasia cells. Nature291:671-672.
Daneshbod-Skibba, G., E. Therman, and N. T. Shahidi.1980. A boy with congenital malformations and chro-mosome breakage. Am. J. Hum. Genet. 5:315-320.
Gatti, R. A., M. Bick, C. F. Tam, M. A. Medici, V. A.Oxelius, M. Holland, A. L. Goldstein, and E. Boder.1982. Ataxia telangiectasia: a multiparameter analysisof eight families. Clin. Immunol. Immunopathol. 23:501-516.
Gatti, R. A. and M. Swift, eds. 1985. Ataxia telangiectasia:genetics, immunology and neuropathology of a degen-erative disease of childhood. Kroc Foundation Series,vol. 19. Alan R. Liss, New York.
Helmerhorst, F. M., D. C. Heaton, P. C. Crossen,A. E. G. K. von dem Borne, C. P. Engelfriet, and A. T.Natarajan. 1984. Familial thrombocytopenia associatedwith platelet autoantibodies and chromosomal breakage.Hum. Genet. 65:252-256.
Jaspers, N. G. J., and D. Bootsma. 1982. Genetic hetero-geneity in ataxia telangiectasia studied by cell fusion.Proc. Natl. Acad. Sci. USA 79:2641-2644.
Jaspers, N. G. J., J. de Wit, M. R. Regulski, D. Bootsma.1982. Abnormal regulation of DNA replication and in-creased lethality in ataxia telangiectasia cells exposed tocarcinogenic agents. Cancer Res. 42:335-341.
Jaspers, N. G. J., R. B. Painter, M. C. Paterson, C. Kidson,and T. Inoue. 1985. Complementation analysis of ataxiatelangiectasia. Pp. 147-162 in Gatti and Swift (1985).
Marachio, P., D. Peretti, S. Lambais, F. Lo Curto, D.Caufin, L. Gargantini, L. Minoli, and C. Zuffardi. 1986.A new chromosomal breakage disorder. Clin. Genet.30:353-365.
Murnane, J. P., and R. B. Painter. 1982. Complementationof the defects in DNA synthesis in irradiated and unir-radiated ataxia telangiectasia cells. Proc. Natl. Acad. Sci.USA 79:1960-1963.
Painter, R. B. 1981. Radioresistant DNA synthesis: an in-trinsic feature of ataxia telangiectasia. Mutat. Res.84:183-190.
Sedgwick, R. P., and E. Boder. 1972. Ataxia telangiectasia.Pp. 267-339 in P. V. Vinken and G. W. Bruyn, eds.Handbook of clinical neurology. Vol 14. North-Holland,Amsterdam.
Seemanova, E., E. Passarge, D. Beneskova, J. Houstek, P.Kasal, and M. Sevcikova. 1985. Familial microcephalywith normal intelligence immunodeficiency and risk forlymphoreticular malignancies. Am. J. Med. Genet.20:639-648.
Shiloh, Y., E. Tabor, and Y. Becker. 1982. The response ofataxia telangiectasia homozygous and heterozygous skinfibroblast to neocarzinostatin. Carcinogenesis 3:815-820.
. 1983. Abnormal response of ataxia telangiectasia

Variants of Ataxia Telangiectasia 73
cells to agents that break the deoxyribose moiety ofDNA via a targeted free-radical mechanism. Carcino-genesis 4:1317-1322.
Sperling, K. 1983. Analisi dell' eterogeneita nell'uomo.Prospettive Pediatr. 49:53-66.
Taalman, R. D. F. M., N. G. J. Jaspers, J. M. J. C. Scheres,J. de Wit, and T. W. J. Hustinx. 1983. Hypersensitivityin vitro in a new chromosomal instability syndrome, theNijmegen breakage syndrome. Mutat. Res. 112:23-32.
Taylor, A. M. R. 1978. Unrepaired strand breaks in ir-
radiated ataxia telangiectasia lymphocytes suggestedfrom cytogenetic observations. Mutat. Res. 50:407-418.
Teebi, A. S., S. A. Al Alawi, and A. G. White. 1987.Autosomal recessive non-syndromal microcephaly withnormal intelligence. Am. J. Med. Gen. 26:355-359.
Weemaes, C. M. R., T. W. J. Hustinx, J. M. J. C.Scheres, P. J. J. Van Munster, J. A. J. M. Bakkeren, andR. D. F. M. Taalman. 1981. New chromosome instabil-ity disorder: the Nijmegen breakage syndrome. ActaPaediatr. Scand. 70:557-562.




















