
The new mutation, E46K, of ?-synuclein causes parkinson and Lewy body dementia
Upload
astrazenecaCategory
view
7download
0
Biochemical and Biophysical Research Communications 309 (2003) 679–684
www.elsevier.com/locate/ybbrc
BBRC
Parkin suppresses wild-type a-synuclein-induced toxicityin SHSY-5Y cells
Yemisi Oluwatosin-Chigbu,a Alan Robbins,a Clay W. Scott,b Jeffrey L. Arriza,a
John D. Reid,a and John R. Zyska,*
a Department of Molecular Science, AstraZeneca Pharmaceuticals LP, 1800 Concord Pike, Wilmington, DE 19850-5437, USAb Department of Lead Discovery, AstraZeneca Pharmaceuticals LP, 1800 Concord Pike, Wilmington, DE 19850-5437, USA
Received 14 August 2003
Abstract
Current hypotheses concerning the mechanism of neuronal cell death in Parkinson’s disease (PD) and related synucleopathies
propose a functional interaction between parkin and a-synuclein (aS). Recently parkin was shown to suppress mutant aS-inducedtoxicity in primary neurons, providing a basis for an association between these proteins and neuronal loss [Neuron 36 (2000) 1007–
1019]. We have asked if a similar association could be made between wild-type (wt) aS and parkin. We examined inducible over-
expression of aS in SHSY-5Y cells through adenoviral infection under conditions which produce cellular toxicity through a
reduction in ATP levels. We demonstrate that parkin suppresses toxicity induced by mutant (A53T) and wt aS. Parkin over-ex-
pression was also associated with the appearance of higher molecular weight aS-immunoreactive bands by Western blot analysis.
These data, consistent with a protective role for parkin, extend previous findings to include a functional association between parkin
and the wt form of aS.� 2003 Elsevier Inc. All rights reserved.
Keywords: Parkin; a-Synuclein; Parkinson’s disease; Synucleopathies; Adenoviral gene delivery; Dopaminergic cell line
Parkinson’s disease is a neurodegenerative conditionresulting from a loss of dopaminergic neurons of the
substantia nigra [1]. Efforts to understand this mecha-
nism at the molecular level have involved the study of
genetic factors in rare familial forms of PD. One such
condition, autosomal recessive juvenile Parkinsonism
(ARJP), involves loss-of-function mutations in the E3
ubiquitin ligase parkin [2–5]. Another familial form of
PD results from autosomal dominant mutations in aS, aprotein of unknown function and a major constituent of
Lewy bodies (LB), a hallmark of the predominant,
sporadic disease [6,7, see 8 for review].
Studies involving parkin and aS suggest that an
association may exist between these proteins which
could be linked to a mechanism of cellular toxicity.
Over-expression of wt aS or its mutants is frequently
* Corresponding author. Fax: 1-302-886-4983.
E-mail address: [email protected] (J.R. Zysk).
0006-291X/$ - see front matter � 2003 Elsevier Inc. All rights reserved.
doi:10.1016/j.bbrc.2003.08.059
associated with cytotoxicity or an increased sensitivityto oxidative stress [9–11]. By contrast, parkin appears
to subserve a protective role at the cellular level,
ubiquitinating proteins destined for clearance by the
26S proteosome and responding to unfolded protein-
induced stress [4]. Parkin has also been shown to
moderate oxidative stress at the cellular level, whereas
ARJP mutations in parkin result in increased oxidative
cellular damage and sensitization of cells to death in-duced by other insults [12]. Co-localization studies
suggest an association between aS and parkin in PD,
and other synucleopathies [13,14]. Recently, Petrucelli
and co-workers demonstrated that parkin rescues pri-
mary neurons from mutant aS-induced toxicity and
insult by proteosome inhibitors [15].
In the present study, we have asked if parkin over-
expression affects wt aS-induced cellular toxicity in anacute cell-based model, since this is the form of syn-
uclein associated with the predominant sporadic form
of PD. In order to address this question, we have
680 Y. Oluwatosin-Chigbu et al. / Biochemical and Biophysical Research Communications 309 (2003) 679–684
employed adenoviral constructs to produce levels of aSexpression in dopaminergic SHSY-5Y cells sufficient to
induce cellular toxicity as measured by a reduction in
cellular ATP levels. We report that overexpression of
parkin suppresses mutant and wt aS-induced cellular
toxicity and that this cytoprotection is associated with
the appearance of higher molecular mass aS-immuno-
reactive proteins. These results are consistent with a
cytoprotective role for parkin, confirm earlier findingsbetween parkin and autosomal dominant synuclein,
and indicate that a functional association exists be-
tween parkin and the native form of the protein as
well.
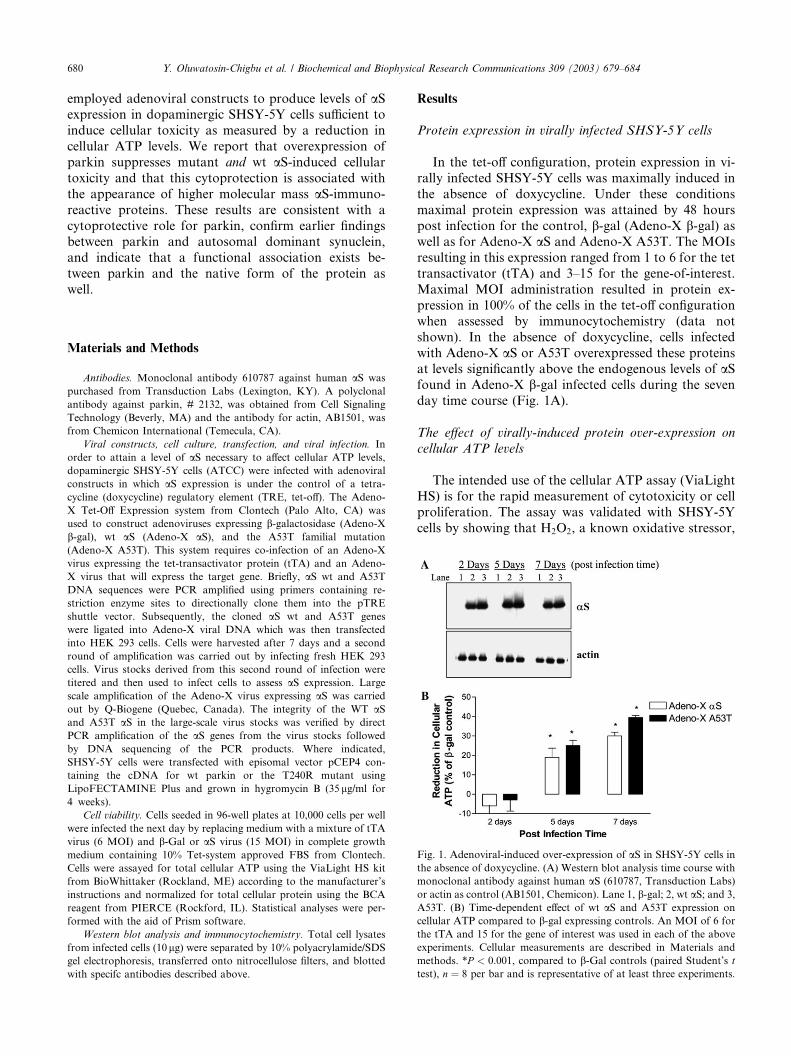
Fig. 1. Adenoviral-induced over-expression of aS in SHSY-5Y cells in
the absence of doxycycline. (A) Western blot analysis time course with
monoclonal antibody against human aS (610787, Transduction Labs)
or actin as control (AB1501, Chemicon). Lane 1, b-gal; 2, wt aS; and 3,
A53T. (B) Time-dependent effect of wt aS and A53T expression on
cellular ATP compared to b-gal expressing controls. An MOI of 6 for
the tTA and 15 for the gene of interest was used in each of the above
experiments. Cellular measurements are described in Materials and
methods. *P < 0:001, compared to b-Gal controls (paired Student’s t
test), n ¼ 8 per bar and is representative of at least three experiments.
Materials and Methods
Antibodies. Monoclonal antibody 610787 against human aS was
purchased from Transduction Labs (Lexington, KY). A polyclonal
antibody against parkin, # 2132, was obtained from Cell Signaling
Technology (Beverly, MA) and the antibody for actin, AB1501, was
from Chemicon International (Temecula, CA).
Viral constructs, cell culture, transfection, and viral infection. In
order to attain a level of aS necessary to affect cellular ATP levels,
dopaminergic SHSY-5Y cells (ATCC) were infected with adenoviral
constructs in which aS expression is under the control of a tetra-
cycline (doxycycline) regulatory element (TRE, tet-off). The Adeno-
X Tet-Off Expression system from Clontech (Palo Alto, CA) was
used to construct adenoviruses expressing b-galactosidase (Adeno-X
b-gal), wt aS (Adeno-X aS), and the A53T familial mutation
(Adeno-X A53T). This system requires co-infection of an Adeno-X
virus expressing the tet-transactivator protein (tTA) and an Adeno-
X virus that will express the target gene. Briefly, aS wt and A53T
DNA sequences were PCR amplified using primers containing re-
striction enzyme sites to directionally clone them into the pTRE
shuttle vector. Subsequently, the cloned aS wt and A53T genes
were ligated into Adeno-X viral DNA which was then transfected
into HEK 293 cells. Cells were harvested after 7 days and a second
round of amplification was carried out by infecting fresh HEK 293
cells. Virus stocks derived from this second round of infection were
titered and then used to infect cells to assess aS expression. Large
scale amplification of the Adeno-X virus expressing aS was carried
out by Q-Biogene (Quebec, Canada). The integrity of the WT aSand A53T aS in the large-scale virus stocks was verified by direct
PCR amplification of the aS genes from the virus stocks followed
by DNA sequencing of the PCR products. Where indicated,
SHSY-5Y cells were transfected with episomal vector pCEP4 con-
taining the cDNA for wt parkin or the T240R mutant using
LipoFECTAMINE Plus and grown in hygromycin B (35 lg/ml for
4 weeks).
Cell viability. Cells seeded in 96-well plates at 10,000 cells per well
were infected the next day by replacing medium with a mixture of tTA
virus (6 MOI) and b-Gal or aS virus (15 MOI) in complete growth
medium containing 10% Tet-system approved FBS from Clontech.
Cells were assayed for total cellular ATP using the ViaLight HS kit
from BioWhittaker (Rockland, ME) according to the manufacturer’s
instructions and normalized for total cellular protein using the BCA
reagent from PIERCE (Rockford, IL). Statistical analyses were per-
formed with the aid of Prism software.
Western blot analysis and immunocytochemistry. Total cell lysates
from infected cells (10lg) were separated by 10% polyacrylamide/SDS
gel electrophoresis, transferred onto nitrocellulose filters, and blotted
with specifc antibodies described above.
Results
Protein expression in virally infected SHSY-5Y cells
In the tet-off configuration, protein expression in vi-
rally infected SHSY-5Y cells was maximally induced in
the absence of doxycycline. Under these conditions
maximal protein expression was attained by 48 hours
post infection for the control, b-gal (Adeno-X b-gal) aswell as for Adeno-X aS and Adeno-X A53T. The MOIs
resulting in this expression ranged from 1 to 6 for the tet
transactivator (tTA) and 3–15 for the gene-of-interest.
Maximal MOI administration resulted in protein ex-
pression in 100% of the cells in the tet-off configuration
when assessed by immunocytochemistry (data not
shown). In the absence of doxycycline, cells infected
with Adeno-X aS or A53T overexpressed these proteinsat levels significantly above the endogenous levels of aSfound in Adeno-X b-gal infected cells during the seven
day time course (Fig. 1A).
The effect of virally-induced protein over-expression on
cellular ATP levels
The intended use of the cellular ATP assay (ViaLight
HS) is for the rapid measurement of cytotoxicity or cell
proliferation. The assay was validated with SHSY-5Y
cells by showing that H2O2, a known oxidative stressor,
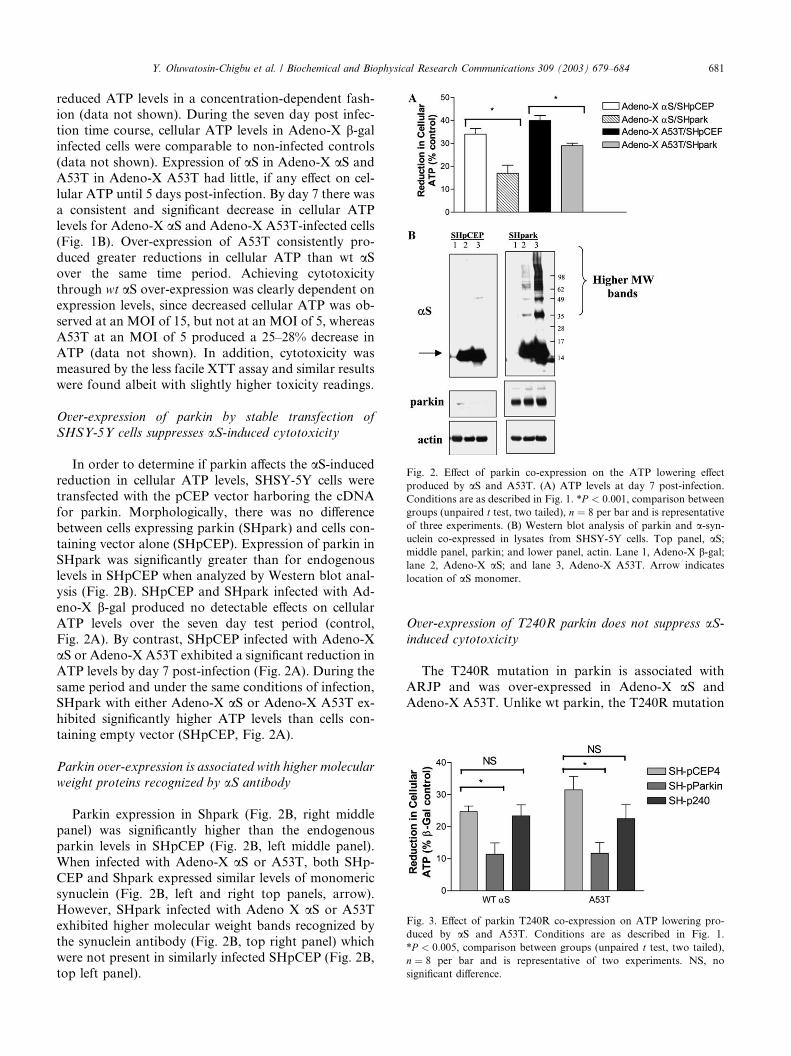
ig. 2. Effect of parkin co-expression on the ATP lowering effect
roduced by aS and A53T. (A) ATP levels at day 7 post-infection.
onditions are as described in Fig. 1. *P < 0:001, comparison between
roups (unpaired t test, two tailed), n ¼ 8 per bar and is representative
f three experiments. (B) Western blot analysis of parkin and a-syn-clein co-expressed in lysates from SHSY-5Y cells. Top panel, aS;iddle panel, parkin; and lower panel, actin. Lane 1, Adeno-X b-gal;ne 2, Adeno-X aS; and lane 3, Adeno-X A53T. Arrow indicates
cation of aS monomer.
Fig. 3. Effect of parkin T240R co-expression on ATP lowering pro-
duced by aS and A53T. Conditions are as described in Fig. 1.
*P < 0:005, comparison between groups (unpaired t test, two tailed),
n ¼ 8 per bar and is representative of two experiments. NS, no
significant difference.
Y. Oluwatosin-Chigbu et al. / Biochemical and Biophysical Research Communications 309 (2003) 679–684 681
reduced ATP levels in a concentration-dependent fash-ion (data not shown). During the seven day post infec-
tion time course, cellular ATP levels in Adeno-X b-galinfected cells were comparable to non-infected controls
(data not shown). Expression of aS in Adeno-X aS and
A53T in Adeno-X A53T had little, if any effect on cel-
lular ATP until 5 days post-infection. By day 7 there was
a consistent and significant decrease in cellular ATP
levels for Adeno-X aS and Adeno-X A53T-infected cells(Fig. 1B). Over-expression of A53T consistently pro-
duced greater reductions in cellular ATP than wt aSover the same time period. Achieving cytotoxicity
through wt aS over-expression was clearly dependent on
expression levels, since decreased cellular ATP was ob-
served at an MOI of 15, but not at an MOI of 5, whereas
A53T at an MOI of 5 produced a 25–28% decrease in
ATP (data not shown). In addition, cytotoxicity wasmeasured by the less facile XTT assay and similar results
were found albeit with slightly higher toxicity readings.
Over-expression of parkin by stable transfection of
SHSY-5Y cells suppresses aS-induced cytotoxicity
In order to determine if parkin affects the aS-inducedreduction in cellular ATP levels, SHSY-5Y cells were
transfected with the pCEP vector harboring the cDNA
for parkin. Morphologically, there was no difference
between cells expressing parkin (SHpark) and cells con-
taining vector alone (SHpCEP). Expression of parkin in
SHpark was significantly greater than for endogenouslevels in SHpCEP when analyzed by Western blot anal-
ysis (Fig. 2B). SHpCEP and SHpark infected with Ad-
eno-X b-gal produced no detectable effects on cellular
ATP levels over the seven day test period (control,
Fig. 2A). By contrast, SHpCEP infected with Adeno-X
aS or Adeno-X A53T exhibited a significant reduction in
ATP levels by day 7 post-infection (Fig. 2A). During the
same period and under the same conditions of infection,SHpark with either Adeno-X aS or Adeno-X A53T ex-
hibited significantly higher ATP levels than cells con-
taining empty vector (SHpCEP, Fig. 2A).
Parkin over-expression is associated with higher molecular
weight proteins recognized by aS antibody
Parkin expression in Shpark (Fig. 2B, right middle
panel) was significantly higher than the endogenous
parkin levels in SHpCEP (Fig. 2B, left middle panel).
When infected with Adeno-X aS or A53T, both SHp-
CEP and Shpark expressed similar levels of monomeric
synuclein (Fig. 2B, left and right top panels, arrow).However, SHpark infected with Adeno X aS or A53T
exhibited higher molecular weight bands recognized by
the synuclein antibody (Fig. 2B, top right panel) which
were not present in similarly infected SHpCEP (Fig. 2B,
top left panel).
F
p
C
g
o
u
m
la
lo
Over-expression of T240R parkin does not suppress aS-induced cytotoxicity
The T240R mutation in parkin is associated with
ARJP and was over-expressed in Adeno-X aS and
Adeno-X A53T. Unlike wt parkin, the T240R mutation

682 Y. Oluwatosin-Chigbu et al. / Biochemical and Biophysical Research Communications 309 (2003) 679–684
was not associated with suppression of synuclein-in-duced cytotoxicity (Fig. 3). Expression levels for T240R
were approximately three-fold higher than endogenous
parkin levels despite repeated attempts to further in-
crease expression. By contrast, expression levels for wt
parkin were at least four times higher than for T240R
(data not shown).
Discussion
The objectives of this communication were to de-
velop a dopaminergic, cell-based model of aS over-
expression with the capability of inducing wt and
mutant-dependent cytotoxicity and to determine if this
is affected by parkin co-expression. To this end, we
have devised a system using adenoviral infection, ca-pable of producing cytotoxicity with either mutant or
wt aS in a time and dose-dependent manner. Interest-
ingly, while maximal overexpression of aS was
achieved by day two post infection and remained stable
up to day seven, we did not detect significant cyto-
toxicity until day five, suggesting a cummulative effect
of synuclein overexpression. Cell viability was similar
in non-infected and control infected (b-gal-expressing)cells, indicating that toxicity in aS over-expressors is
specific and not virally-induced. We also demonstrated
that parkin co-expression provides cytoprotection
against both mutant and wt synuclein extending earlier
findings [15]. As in previous studies, we have observed
an enhancement in the toxicity of over-expressed aS in
the presence of stressors, notably MPPþ, rotenone,
and, to a lesser extent, lactacystin (data not shown).However, one of the goals of this study was to deter-
mine if over-expressed wt aS is capable of inducing
toxicity in the absence of external stressors and under
normal cell culture conditions.
It is generally accepted that the mechanism of toxicity
of aS in PD involves dopamine, a toxic compound in its
own right, which is linked to oxidative damage [16–18].
Indeed, in non-dopaminergic cortical neurons, aS ex-hibits neuroprotective characteristics [16]. So although
the absence of added stressors was an important objec-
tive of the toxicity model, a dopaminergic cell line
(SHSY-5Y) was deemed relevant to this study. Mito-
chondrial dysfunction is also associated with PD
through a decrease in complex I activity and an atten-
dant diminution in cellular ATP [11,19]. In fact, inhib-
itors of complex I, such as MPTP and rotenone areknown inducers of Parkinsonism [20,21]. Thus, total
cellular ATP was chosen as a readout for cellular tox-
icity because of its relevance to the disease process as
well as the central role it plays in cellular metabolism
and maintenance of cell viability [22]. The biolumines-
cent assay was also implemented because of its facility,
wide dynamic range, high sensitivity, and validation
against other assays for cell viability and cytotoxicity[23,24].
ARJP-associated loss-of-function mutations in par-
kin and the attendant loss of dopaminergic neurons in
the disease state suggest that parkin plays an important
role in neuronal viability. By itself, the stable, over-ex-
pression of wt parkin in SHSY-5Y cells had no overt
effect on cell viability reflecting similar findings in SK-N-
MC and NT-2 cells [12]. The positive effect associatedwith parkin on cell viability was only observed during
aS over-expression. A similar parkin-associated cyto-
protective effect was found in SHSY-5Y cells over-ex-
pressing Pael-receptor, a parkin substrate and
constituent of LBs [25] and more recently in a Dro-
sophila model [26]. The failure of the A240T form of
parkin to provide cytoprotection from aS-induced tox-
icity reflects the findings of Petrucelli and coworkerswho showed that R42P parkin, another loss-of-function
mutant, was ineffective against insult by lactacystin [15].
Another possible reason for a lack of protection is the
lower fold-expression of the mutant relative to wt over-
expression.
The tendency of aS to aggregate is associated with the
formation of LBs in sporadic PD and familial forms of
the disease involving autosomal dominant mutations[6,7]. Parkinsonism resulting from ARJP is character-
ized by a lack of inclusions, suggesting that functional
parkin is necessary for LB formation [2,3,27]. Thus, the
argument has been made that LBs are protective in
nature or at least not inherently cytotoxic [28]. There is
also evidence to suggest that aS exists as a protofibril
multimer which exists in equilibrium with LBs [9,21].
We found no evidence of aS-immunosensitive LB-likeinclusions in SHpark or SHpCEP cells over-expressing
either wt or mutant synuclein by immunocytochemistry
(although diffuse cytoplasmic staining of aS was evident,
data not shown). However, Lewy-like inclusions in a
chronic disease state would probably take longer to
form than in the acute cell model used in our study. We
therefore examined whole cell extracts in order to de-
termine if aS aggregation occurs and if parkin co-ex-pression affects this phenomenon. In fact, detectable,
higher molecular weight a-synuclein immunoreactive
bands were only found in SHpark-infected cells, sug-
gesting that parkin plays a role in regulating synuclein
aggregation and/or processing. If these bands are ag-
gregates of aS, they could represent precursors to in-
clusion formation [9,11]. Although speculative, a lack of
higher molecular weight aS immunoreactive bands incells not over-expressing parkin is consistent with a role
for parkin in processing synuclein as a less toxic species.
It should be mentioned that the SHSY-5Y cells in this
study express low levels of endogenous parkin (Fig. 2B)
which would be overwhelmed by virally induced aSexpression. The higher molecular weight species of aSdescribed in the present study were not recognized with

Y. Oluwatosin-Chigbu et al. / Biochemical and Biophysical Research Communications 309 (2003) 679–684 683
ubiquitin or parkin antibodies, suggesting that thesebands are neither parkin substrates nor parkin–synuc-
lein complexes (data not shown). The further charac-
terization of these higher molecular weight species will
probably be necessary in defining the nature of the
parkin/aS association and attendant mechanisms of
toxicity and protection.
In this study, we have used virally transfected SHSY-
5Y cells as a starting point to understand synucleopa-thies at the molecular level. Adenoviral delivery offers
the advantages of inducible, high level expression and
high efficiency of transfection without inherent toxicity
under the conditions described. Adenoviral vectors also
enable the delivery of up to 8 kb of DNA lending flexi-
bility to the size of the gene under consideration. The
facile nature of this system may be useful in defining
pathways and identifying targets involved in aS-relatedtoxicity. Co-expression of selected candidate genes and
screening of enhancers or supressors of cytotoxicity
provides a platform for examining protein-protein in-
teractions and cell-based mechanisms. Parkin co-ex-
pression and suppression of wt aS-induced cytotoxicity
as a proof-of-principle indicates that this is a viable
approach to understanding the nature of synucleopa-
thies at the molecular level. The validation of potentialtargets derived from this system may be accomplished
using suitable transgenic and genetic models currently
being developed [29].
Acknowledgments
We thank Steve Culp for aiding in the statistical analyses, and Jude
Prosser, Dee Wilkins, Helen Manley, and Jeanine Olie for technical
assistance.
References
[1] P. Jenner, C.W. Olanow, Understanding cell death in Parkinsons-
disease, Ann. Neurol. 44 (1998) S72–S84.
[2] T. Kitada, S. Asakawa, N. Hattori, H. Matsumine, Y. Yamam-
ura, S. Minoshima, M. Yokochi, Y. Mizuno, N. Shimizu,
Mutations in the parkin gene cause autosomal recessive juvenile
Parkinsonism, Nature 392 (1998) 605–608.
[3] H. Shimura, N. Hattori, S. Kubo, Y. Mizuno, S. Asakawa, S.
Minoshima, N. Shimizu, K. Iwai, T. Chiba, K. Tanaka, T. Suzuki,
Familial Parkinson disease product, parkin, is a ubiquitin–protein
ligase, Nat. Genet. 25 (2000) 302–305.
[4] Y. Imai, M. Soda, R. Takahashi, Parkin supresses unfolded
protein stress-induced cell death through its E3 ubiquitin–protein
ligase activity, J. Biol. Chem. 275 (2000) 35661–35664.
[5] Y. Zhang, J. Gao, K.K. Chung, H. Huang, V.L. Dawson, T.M.
Dawson, Parkin functions as an E2-dependent ubiquitin–protein
ligase and promotes the degradation of the synaptic vesicle-
associated protein, CDCrel-1, Proc. Natl. Acad. Sci. USA 97
(2000) 13354–13359.
[6] M.H. Polymeropoulos, C. Lavedan, E. Leroy, S.E. Ide, A.
Dehejia, A. Dutra, B. Pike, H. Root, J. Rubenstein, R. Boyer,
E.S. Stenroos, S. Chandrasekharappa, A. Athanassiadou, T.
Papapetropoulos, W.G. Johnson, A.M. Lazzarini, R.C. Duvoisin,
G. Di Iorio, L.I. Golbe, R.L. Nussbaum, Mutation in the a-synuclein gene identified in families with Parkinson’s disease,
Science 276 (1997) 2045–2047.
[7] R. Kruger, W. Kuhn, T. Muller, D. Woitalla, M. Graeber, S.
Kosel, H. Przuntek, J.T. Epplen, L. Schols, O. Reiss, Ala30Pro
mutation in the gene encoding alpha-synuclein in Parkinson’s
disease, Nat. Genet. 18 (1998) 106–108.
[8] C.B. L€uucking, A. Brice, Alpha-synuclein and Parkinson’s disease,
Cell. Mol. Life Sci. 57 (2000) 1894–1908.
[9] M.S. Goldberg, P.T. Lansbury, Is there a cause-and-effect
relationship between a-synuclein fibrillization and Parkinson’s
disease?, Nat. Cell Biol. 2 (2000) E115–E119.
[10] B.I. Giasson, J.E. Duda, I.V.J. Murray, Q. Chen, J.M. Souza, H.I.
Hurtig, H. Ischiropoulos, J.Q. Trojanowski, V.M.-Y. Lee,
Oxidative damage linked to neurodegeneration by selective
a-synuclein nitration in synucleinopathy lesions, Science 290
(2000) 985–989.
[11] H.-J. Lee, S.Y. Shin, C. Choi, Y.H. Lee, S.-J. Lee, Formation and
removal of a-synuclein aggregates in cells exposed to mitochon-
drial inhibitors, J. Biol.Chem. 277 (2002) 5411–5417.
[12] D.-H. Hyun, MH. Lee, N. Hattori, S.-I. Kubo, Y. Mizuno,
B. Halliwell, P. Jenner, Effect of wild-type or mutant parkin on
oxidative damage, antioxidant defenses, and the proteosome,
J. Biol. Chem. 277 (2002) 28572–28577.
[13] P. Choi, N. Golts, H. Snyder, M. Chong, L. Petrucelli, J. Hardy,
D. Sparkman, E. Cochran, J.M. Lee, B. Wolozin, Co-association
of parkin and a-synuclein, Mol. Neurosci. 12 (2001) 2839–2843.
[14] M.G. Schlossmacher, M.P. Frosch, W.P. Gai, M. Medina,
N. Sharma, L. Forno, T. Ochiishi, H. Shimura, R. Sharon,
N. Hattori, J.W. Langston, Y. Mizuno, B. Hyman, D. Selkoe,
K.S. Kosik, Parkin localizes to the Lewy bodies of Parkinson’s
disease and dementia with Lewy bodies, Am. J. Pathol. 160 (2002)
1655–1667.
[15] L. Petrucelli, C. O’Farell, P.J. Lockhart, M. Baptista, K. Kehoe,
L. Vink, P. Choi, B. Wolozin, M. Farrer, J. Hardy, M.R.
Cookson, Parkin protects against the toxicity associated with
mutant a-synuclein: proteosome dysfunction selectively affects
catecholaminergic neurons, Neuron 36 (2002) 1007–1019.
[16] J. Xu, S.-Y. Kao, F.J.S. Lee, W. Song, L.-W. Jin, B.A. Yankner,
Dopamine-dependent neurotoxicity of a-synuclein: a mechanism
for selective neurodegeneration in Parkinson disease, Nat. Med. 8
(2002) 600–606.
[17] M.-F. Chesselet, Dopamine and Parkinson’s disease: is the killer
in the house? Mol. Psychol. 8 (2003) 369–370.
[18] W. Zhou, J. Schaack, W.M. Zawada, C.R. Freed, Overexpression
of human a-synuclein causes dopamine neuron death in primary
human mesencephalic culture, Brain Res. 926 (2002) 42–50.
[19] M.F. Beal, Energetics in the pathogenesis of neurodegenerative
diseases, Trends Neurosci. 23 (2000) 298–304.
[20] W.J. Nicklas, I. Vyas, R.E. Heikkila, Inhibition of NADH-linked
oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a
metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahy-
dropyridine, Life Sci. 36 (1985) 2503–2508.
[21] R. Betarbet, T.B. Sherer, G. MacKenzie, M. Garcia-Osuna, A.V.
Panov, J.T. Greenamyre, Chronic systemic pesticide exposure
reproduces features of Parkinson’s disease, Nat. Neurosci. 3
(2000) 1301–1306.
[22] S. Crouch, R. Kozlowski, K. Slater, J. Fletcher, The use of ATP
bioluminescence as a measure of proliferation and cytotoxicity, J.
Immunol. Methods 160 (1993) 81–88.
[23] K. Slater, Cytotoxicity tests for high throughput drug discovery,
Curr. Opin. Biotechnol. 12 (2001) 70–74.
[24] J. Obrien, I. Wilson, T. Orton, F. Pognan, Investigation of the
alamar blue (resazurin) fluorescent dye for the assessment of
mammalian cell cytotoxicity, Eur. J. Biochem. 267 (2000)
5421–5426.

684 Y. Oluwatosin-Chigbu et al. / Biochemical and Biophysical Research Communications 309 (2003) 679–684
[25] Y. Imai, M. Soda, H. Inoue, N. Hattori, Y. Mizuno, R.
Takahashi, An unfolded putative transmembrane polypeptide,
which can lead to endoplasmic reticulum stress, is a parkin
substrate, Cell 105 (2001) 891–902.
[26] Y. Yang, I. Nishimura, Y. Imai, R. Takahashi, B. Lu, Parkin
suppresses dopaminergic neuron-selective neurotoxicity induced
by Pael-R in Drosophila, Neuron 37 (2003) 911–924.
[27] H. Shimura, M.G. Schlossmacher, N. Hattori, M.P. Frosch, A.
Trockenbacher, R. Schneider, Y. Mizuno, K.S. Kosik, D.J.
Selkoe, Ubiquitination of a new form of a-synuclein by parkin
from human brain: implications for Parkinson’s disease, Science
293 (2001) 263–268.
[28] K.K.K. Chung, V.L. Dawson, T.M. Dawson, The role of the
ubiquitin–proteosomal pathway in Parkinson’s disease and other
neurodegenerative disorders, Trends Neurol. Sci. 24 (2001)
S7–S14.
[29] R. Betarbet, T.B. Sherer, J.T. Greenamyre, Animal models of
Parkinson’s disease, BioEssays 24 (2002) 308–318.
Copyright © 2022 FDOKUMEN




















