
p53: Twenty five years understanding the mechanism of genome protection
21
Correspondence to J. Jordán (Tel.: +34 967 599 000; Fax: +34 967 599 327; e-mail: joaquin.jordan@uclm. es). p53: Twenty five years understanding the mechanism of genome protection M. Gomez-Lazaro, F. J. Fernandez-Gomez and J. Jordán Centro Regional de Investigaciones Biomédicas, Facultad de Medicina, Universidad de Castilla-La Mancha, Avda. Almansa, 02006 Albacete, Spain (Received on November 10, 2004) M. GOMEZ-LAZARO, F. J. FERNANDEZ-GOMEZ and J. JORDÁN. p53: Twenty five years understanding the mechanism of genome protection (minireview). J. Physiol. Biochem., 60 (4), 287-308, 2004. This year the p53 protein, also known as “guardian of the genome”, turns twenty five years old. During this period the p53 knowledge have changed from an initial pro-oncogene activity to the tumorsupressor p53 function. p53 is activated upon stress signals, such as gamma irradiation, UV, hypoxia, virus infection, and DNA damage, leading to protection of cells by inducing target genes. The molecules acti- vated by p53 induce cell cycle arrest, DNA repair to conserve the genome and apop- tosis. The regulation of p53 functions is tightly controlled through several mecha- nisms including p53 transcription and translation, protein stability, post-translation- al modifications, and subcellular localization. In fact, mutations in p53 are the most frequent molecular alterations detected in human tumours. Furthermore, in some degenerative processes, fragmentation and oxidative damage in DNA take place, and in these situations p53 is involved. So, p53 is considered a pharmacological target, p53 overexpression induces apoptosis in cancer and its expression blockage protects cells against lethal insults. Key words: Apoptosis, Necrosis, mdm2, Cancer, Tumor suppressor, Mutation. J. Physiol. Biochem., 60 (4), 287-308, 2004 Twenty-five years ago a 53KDa protein with previously unknown properties was discovered. This event was the culmina- tion of two kind of studies: virologic and serologic. In the virologic studies, SV40- transformed cells were used. It was found that a 55kDa protein co-precipitated with the large T antigen of simian virus 40 (SV40) (21, 82, 87, 95, 108). This protein was also found overexpressed in several murine SV40 transformed cells, and in embryonic carcinoma cells (82, 95). On the other hand, the serologic approach showed how antisera prepared from BALB/c Meth A sarcoma in syngeneic or compatible F1 mice, recognized a protein
Transcript of p53: Twenty five years understanding the mechanism of genome protection

Correspondence to J. Jordán (Tel.: +34 967 599 000;Fax: +34 967 599 327; e-mail: [email protected]).
p53: Twenty five years understandingthe mechanism of genome protection
M. Gomez-Lazaro, F. J. Fernandez-Gomez and J. Jordán
Centro Regional de Investigaciones Biomédicas, Facultad de Medicina, Universidad deCastilla-La Mancha, Avda. Almansa, 02006 Albacete, Spain
(Received on November 10, 2004)
M. GOMEZ-LAZARO, F. J. FERNANDEZ-GOMEZ and J. JORDÁN. p53:Twenty five years understanding the mechanism of genome protection (minireview).J. Physiol. Biochem., 60 (4), 287-308, 2004.
This year the p53 protein, also known as “guardian of the genome”, turns twentyfive years old. During this period the p53 knowledge have changed from an initialpro-oncogene activity to the tumorsupressor p53 function. p53 is activated uponstress signals, such as gamma irradiation, UV, hypoxia, virus infection, and DNAdamage, leading to protection of cells by inducing target genes. The molecules acti-vated by p53 induce cell cycle arrest, DNA repair to conserve the genome and apop-tosis. The regulation of p53 functions is tightly controlled through several mecha-nisms including p53 transcription and translation, protein stability, post-translation-al modifications, and subcellular localization. In fact, mutations in p53 are the mostfrequent molecular alterations detected in human tumours. Furthermore, in somedegenerative processes, fragmentation and oxidative damage in DNA take place, andin these situations p53 is involved. So, p53 is considered a pharmacological target, p53overexpression induces apoptosis in cancer and its expression blockage protects cellsagainst lethal insults.
Key words: Apoptosis, Necrosis, mdm2, Cancer, Tumor suppressor, Mutation.
J. Physiol. Biochem., 60 (4), 287-308, 2004
Twenty-five years ago a 53KDa proteinwith previously unknown properties wasdiscovered. This event was the culmina-tion of two kind of studies: virologic andserologic. In the virologic studies, SV40-transformed cells were used. It was found
that a 55kDa protein co-precipitated withthe large T antigen of simian virus 40(SV40) (21, 82, 87, 95, 108). This proteinwas also found overexpressed in severalmurine SV40 transformed cells, and inembryonic carcinoma cells (82, 95). Onthe other hand, the serologic approachshowed how antisera prepared fromBALB/c Meth A sarcoma in syngeneic orcompatible F1 mice, recognized a protein

with an apparent molecular weight of 53kDa in extracts of BALB/c cells (32). Ani-mals suffering several types of tumorsexhibited an immune response specific forthis protein (82, 108, 132). Later studiesrevealed the presence of antibodies againstthis protein in 9% of sera from breast can-cer patients (29) as well as in sera fromchildren with a wide variety of cancer (20,63).
During the first decade after p53 wasdiscovered, this protein was suggested toact as an oncogene. Indeed, p53 was impli-cated in positive regulation of cell growth(129) and in cell transformation (43, 125).In 1989, Levine and colleagues establishedits real physiological function: the tumorsuppressor action, acting negatively toblock transformation (45). It then becameclear that mutant forms of p53 exhibitedoncogenic activities (43, 46, 59). In addi-tion, p53 mutations were found very fre-quent in colon cancer (6). This was seenlater in most of the common types ofhuman cancers (61). Since these observa-tions were done, a large amount of studieshave demonstrated that: i) inactivatingmutations and/or deletions of the p53gene are a common event in differenttypes of human cancer (130, 139), ii) thewild-type p53 protein acts as a transcrip-tion factor, while some p53 proteinmutant forms fail to exhibit this biochem-ical function (42, 44, 77) and iii) the targetgene products induced by wild-type p53mediate its tumor-suppressor functionplaying a direct role in modulating growtharrest, apoptosis, membrane signalling,protein degradation or oxidative stress(127, 168, 170) (Table I). All these evi-dence established turning point in thisprotein short life, as pointed out by thegrowing number of publications referringto it.
We now know that p53 function isessential in the cellular response againstcell damage or genome mutation. Becauseof this, in the beginning of the 90’s it wascalled “The Guardian of the Genome”(86).
p53 structure
In humans the p53 gene locus is locatedin the shorter arm of chromosome 17(17p1.3). Its mRNA is translated into a393 amino acid (aa) protein that compris-es several domains, including an acidic N-terminal region containing the transacti-vation domain, a core holding thesequence-specific DNA-binding domainand a complex C-terminal domain withmultiple functions.
– The N-terminal domain (aa 1–93)holds a strong transcription activation sig-nal (156), and is involved in transcription-al target gene activation, and is subdividedinto the transcription activation subdo-main (aa 1–42, aa 43–63) and the proline-rich subdomain (aa 62–93). N-terminalalso contains the highly conserved BOX-Itransactivation domain that directs thebinding of p53 to the transcriptionaladapter protein p300 (5) .
– The central core domain (aa 102-292)contains the specific DNA bindingregion, and is the most preserved region ofthe protein amongst vertebrates. Thestress-regulated transactivation functionof p53 is driven by its sequence-specificDNA-binding domain and is co-ordinat-ed by specific protein-protein interactionsthat can in turn be modulated by covalentand non-covalent modifications. The coredomain is organized into a large b-sand-wich that forms a scaffold for a loop-b-strand-helix motif and two loops that co-ordinate with zinc ion (25). The directcontact with the DNA is through residues
M. GOMEZ-LAZARO, F. J. FERNANDEZ-GOMEZ AND J. JORDÁN288
J. Physiol. Biochem., 60 (4), 2004

TWENTY FIVE YEARS AROUND p53 289
J. Physiol. Biochem., 60 (4), 2004
Table I. Functions of p53 and targets genes.
Gene/protein Synonims Role
Cell cycle inhibition
p21WAF1 CDK-interacting protein 1, MDA-6 Cyclin-dependent kinase inhibitor14-3-3s KCIP-1, Protein 1054 Tethers cyclin B1-CDK1 complexes in the cytoplasm.
G2 cycle arrestGADD45b MyD118 Growth and apoptosis.
Activation of stress-responsive MTK1/MEKK4 MAPKKKBTG2/TIS21 NGF-inducible anti-proliferative General transduction
protein PC3 Post-translational modifications.Anti-proliferative protein.Transcription regulation mediated by ESR1
MIC-1 Colon cancer-associated Growth factor inhibitorprotein Mic1
IGFBP3 None Growth factor inhibitorMDM2 Transformed 3T3 double Inhibits p53 and p73- mediated cell cycle arrest
minute 2 and apoptosis. Ubiquitin ligase E3p53-binding protein Mdm2 Nuclear export of p53 and targets for proteolysis
TGF-a EGF-like TGF, ETGF, TGF type 1 Growth factor inhibitorCyclin B1 None Cell cycle control (G2/M transition)Cyclin D1 PRAD1 oncogene, Cell cycle control (G1/S transition)
BCL-1 oncogeneCyclin E None Cell cycle control (G1/S, start, transition)Cyclin B2 None Cell cycle control (G2/M transition)Cyclin D2 None Cell cycle control (G1/S transition)Cyclin A None Cell cycle control (G2/M transition)CDK4 PSK-J3 Cell cycle control. CDK2 p33 protein kinase Cell cycle control. Interacts with cyclins A, B3, D, or E.
Activity of CDK2 is maximal during S phase and G2DCK1 Doublecortin-like and CAM Calcium-signaling neuronal migration
kinase-like 1TOPO-IIa None Topological states of DNA by transient breakage and
subsequent rejoining of DNA strands.Double-strand breaks
PLK PLK-1, STPK13 Cell division and G1 or S phasePISSLRE Serine/threonine, e-protein kinase Cdc2 related protein kinaseBRCA-1 None DNA repair. E2-dependent ubiquitation. Cdc6 CDC6-related protein, p62(cdc6), DNA replication
HsCDC6, HsCDC18 Checkpoint controls that ensure DNA replicationGOS2 None Probably involved in translation
ApoptosisBax None Programmed cell deathApaf-1 None Activation of pro-caspase-9PUMA None Mitochondrial cytochrome c releaseP53AIP1 None Lost of DYm and apoptosisPIDD None Mitochondrial cytochrome c release

M. GOMEZ-LAZARO, F. J. FERNANDEZ-GOMEZ AND J. JORDÁN290
J. Physiol. Biochem., 60 (4), 2004
NOXA None Mitochondrial cytochrome c releaseKILLER/DR5 TRAIL-R2 Receptor for the cytotoxic ligand TNFSF10/TRAILBid None Pro-apoptoticTRAF4 Cysteine-rich domain Activation of NF-kappa-B and JNK
associated with RING andTraf domains protein 1,Malignant 62
Fas/Apo1 TNFR6, FASL receptor, Receptor for TNFSF6/FASLApoptosis-mediating surfaceantigen FAS, Apo-1 antigen,CD95 antigen
BAK Apoptosis regulator BAK, Binding and antagonizing the repressor Bcl-2 or E1BBCL2-like 7 protein 19k protein
Bcl-6 Zinc finger protein 51 Transcriptional regulator with an important roleLAZ-3 protein, BCL-5, in lymphomagenesisZinc finger and BTB domaincontaining protein 27
Wig1 Zinc finger protein WIG1 TP53-dependent growth regulatory pathwayCaspase 2 CASP-2, ICH-1 protease, Activation cascade of caspases for apoptosis
ICH-1L/1SCaspase 3 Apopain, Apoptosis execution
Cysteine protease CPP32,Yama protein, SREBP, SCA-1
Caspase 9 ICE-like apoptotic protease 6 Binding to Apaf-1 leads to activation of the proteaseICE-LAP6, cascadeApoptotic protease Mch-6,Apoptotic protease activating factor 3
MIHB BIRC-2 Apoptosis inhibitorMIHC Brush border myosin I, Movement of organelles along actin filaments
BBM-I, BBMI,Myosin I heavy chain, Myosin Ia
PDCD2 Zinc finger protein Rp-8 DNA-binding protein with a regulatory functionZinc finger MYND domaincontaining protein 7
DNA repairP53R2 None DNA damaged repairPCNA None Auxiliary protein of DNA polymerase delta.
Control of eukaryotic DNA replication. DDB2 Damage-specific DNA binding Repair of UV-damaged DNA. Binds to pyrimidine dimers
protein 2, DDB p48 subunit,DDBb, UV-damagedDNA-binding protein 2
LIG1 DNA ligase I Seals nicks in double-stranded DNAPolydeoxyribonucleotidesynthase [ATP]
ERCC5 DNA excision repair-related DNA excision repairgene, DNA-repair proteincomplementing XP-G cells,
Gene/protein Synonims Role
Table I. (Continuation).

TWENTY FIVE YEARS AROUND p53 291
J. Physiol. Biochem., 60 (4), 2004
Xeroderma pigmentosum groupG complementing protein,DNA excision repair proteinERCC-5
RPA1 RF-A, Absolutely required for simian virus 40 DNAReplication factor-A protein 1, replication in vitroSingle-stranded DNA-bindingprotein
XRCC9 FACG protein, Post-replication repairDNA-repair protein XRCC9
RFC4 Replication factor Elongation of the multiprimed DNA templateC 37 kDa subunit,A1 37 kDa subunit,RF-C 37 kDa subunit, RFC37
Angiogenesis and metastasis inhibitorsMaspin Protease inhibitor 5 Serine protease inhibitor
KAI1 Metastasis suppressor homolog Metastasis suppressor protein
Senescence Ras Transduction of mitogenic signals
Raf Raf-1, C-RAF, cRaf Transduction of mitogenic signals
P14/ARF None Regulator of MDM2
MAPK Extracellular signal- Phosphorylates microtubule-associated protein-2 regulated kinase 2 (MAP2). Myelin basic protein (MBP) and Elk-1Mitogen-activated protein kinase 2,p42-MAPK, ERT1
E2F1 RBBP-3, Control of cell-cycle progression from G1 to S phasePRB-binding protein E2F-1,RBAP-1
P16 P16INK4A tumor suppressor Cyclin-dependent kinase inhibitorprotein
PML None Cell proliferation and apoptosis
TranscriptionATF3 None Represses transcription
Signal transductionDUSP5 Dual specificity protein Displays phosphatase activity
phosphatase hVH3
DGKA Diglyceride kinase, Converts diacylglycerol into phosphatidate andDGK-alpha, DAG kinase alpha, attenuates protein kinase C activity80 kDa diacylglycerol kinase
CDC25C M-phase inducer phosphatase 3 Inducer in mitotic control
Oxidative stressPIG3 Quinone oxidoreductase Oxidative stress
homolog
Gene/protein Synonims Role
Table I. (Continuation).

R148 and R273, while other residues arenecessary for the integrity of the struc-ture.
– The C-terminus (aa 293–393) is a flex-ible region, which has a basic end and atetramerization domain that ensuresassembly of p53 into conformationallyactive tetramers. It is subdivided in thetetramerization domain (326–353) and theregulatory domain (CTRD; 363–393).Flanking the p53 tetramerization domain,there is a negative regulatory domainwhose post-translational modificationmay play an important role in modulatingthe specific activity of p53 in vivo. Onefunction for this regulatory C-terminaldomain is to maintain p53 protein in alatent state for specific DNA binding, butalso: nuclear import, nuclear export, non-specific DNA binding and homooligo-merization. In the p53 latent form, thisregion is tucked back inside the proteincentral region preventing the binding toits DNA target sequence.
p53 family
p53 protein belongs to a family of tran-scription factors composed by at least twoother members: p73 and p63 (also knownas KET, p40, p51 or p73L). All of them
share substantial sequence homology, andp63 and p73 can, at least when overpro-duced, activate p53-responsive promotersand induce apoptosis. They contain verylarge introns (98) and while split in theirprimary sequence, present divergences intheir C-terminal regions and also in exonnumber, functions, regulation and tissuedistribution. On the basis of phylogeneticanalysis, it has been postulated that thep53 gene may have evolved from an ances-tral gene similar to p73/p63 (75, 166). Thefact that no other p53 family membershave been identified within the Drosophi-la or the Caenorhabditis elegans genomesuggests that the p63/p73 subfamilyevolved after the division of the arthropodand vertebrate lineages (121).
The N-domain is poorly conserved,while the DNA binding domain presentsthe highest homologies. Therefore, theessential residues for p53 protein foldingare conserved among its family, for thisreason p73 and p63 can bind to p53 DNAbinding sequences. Strikingly, p63 showsthe highest preservation of primary aminoacid sequence in the N-terminal domainbetween human and mouse with 99%identity (166). This high conservation ofthe transactivation domain is possiblyrelated to the fact that p63 plays a uniquerole among the p53 family members with
M. GOMEZ-LAZARO, F. J. FERNANDEZ-GOMEZ AND J. JORDÁN292
J. Physiol. Biochem., 60 (4), 2004
Gene/protein Synonims Role
Table I. (Continuation).
Protein metabolism
LOXL1 Lysyl oxidase-like protein 1 Lysyl oxidase familyLOL
Cell-cell signalingEDN2 ET-2, Endothelins are endothelium-derived vasoconstrictor
Preproendothelin-2, peptidesPPET2
Protocadherin-1 None Cell to cell adhesion and communication,Extracellular matrix

TWENTY FIVE YEARS AROUND p53 293
J. Physiol. Biochem., 60 (4), 2004
respect to normal murine and humandevelopment.
In addition, gene processing of p53 isdifferent as well. While p63 and p73 pre-sent different proteic isoforms, two p53mRNA isoforms (normal splice, NS, andalternative splice, AS) can be detected inthe mouse. ASp53, but not NSp53, fails tobind non-specifically to single-strandednucleic acids and is constitutively activefor specific DNA binding (8). These pro-teins are generated at different times dur-ing the cell cycle and also at different lev-els between different tissues. With respectto p63, there are at least three differentprotein isoforms, differing in the C-termi-nal end (a, b and g forms) that may varyalso at the transactivation domain becauseof the cryptic promoter located at intron 3that generates additional transcripts(DNp63a, DNp63b and DNp63g) (166).In the case of p73 there are six differentvariants of the protein (a, b, g, d and eforms) (89). On the other hand, theexpression pattern is also different, there-fore, while p53 is ubiquitously expressed,p73 is located in the epidermis, sinuses,inner ear and brain and p63 in epidermisproliferating basal cells , cervix, urotheli-um and prostate.
There are also differences in theupstream signalling pathways involved inthe activation of these genes. Both cellularand viral oncoproteins can discriminatebetween p53 and the rest of its family(105). p53 activators like dactinomicineand UV radiation are not able to activatep73, although both proteins are accumu-lated in response to DNA damage.
However, the greatest differencebetween these proteins is their mutationalprevalence in human cancer. While p53mutations are found in a high percentageof cancers mutations on p63 and p73 areunusual. Besides, and in contrast to what
happens in p53-/- mice (64) the absence ofp63 or p73 is not involve in tumor devel-opment in animal models. Instead, theabsence of one of these two proteins isresponsible for developmental abnormali-ties.
In summary, p53, p63 and p73 appearto have overlapping but also distinct func-tions: p53 regulates the stress response forsuppressing tumors; p63 is essential forectoderm development; and p73 mightregulate both the stress response anddevelopment.
p53 protein level regulation
p53 is a short-lived protein with a half-life of ~5-20 min in most cell types stud-ied. The levels of this protein are undersevere control (123, 131). Stressful situa-tions including genotoxic (irradiation,chemical carcinogens, cytotoxic drugs,cyclobutan pirimidin dimmers) (47, 56,76, 94, 119), non-genotoxic (telomere ero-sion, hypoxia, hyperthermia, depletion ofribonucelotides, nitric oxide), and onco-genic activation of growth signalling cas-cades are all responsible for the stabiliza-tion and activation of the p53 protein, byincreasing the half-life of protein by sev-eral fold (102, 128), yielding to an increasein protein levels. These increments aremainly a consequence of post-translation-al mechanisms, although small changes atthe transcriptional level are also involved.The post-translational modifications thatp53 can suffer are phosphorylations,dephosphorylation, acetylation, sumoyla-tion and changes in redox state (Table II).
The phosphorylation of residues in theC-terminal and N-terminal regions mod-ulates p53 function. The enzymes withkinase activity that can phosphorylate p53are: casein kinase I (Ser6 y Ser 9) and II(Ser392), DNA-PK (Ser15 y Ser37), ATM
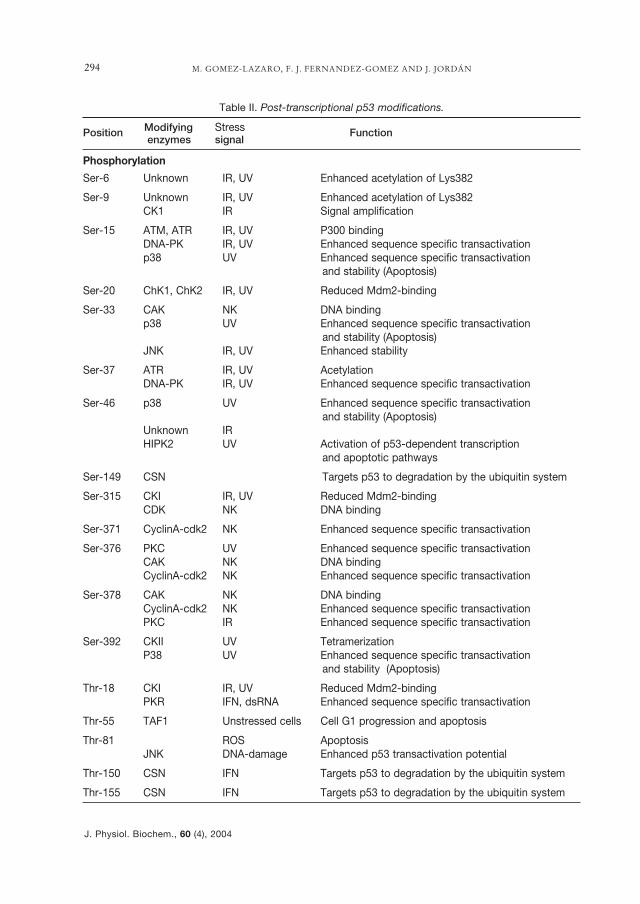
M. GOMEZ-LAZARO, F. J. FERNANDEZ-GOMEZ AND J. JORDÁN294
J. Physiol. Biochem., 60 (4), 2004
Table II. Post-transcriptional p53 modifications.
Position Modifying Stress Functionenzymes signal
Phosphorylation
Ser-6 Unknown IR, UV Enhanced acetylation of Lys382
Ser-9 Unknown IR, UV Enhanced acetylation of Lys382CK1 IR Signal amplification
Ser-15 ATM, ATR IR, UV P300 bindingDNA-PK IR, UV Enhanced sequence specific transactivationp38 UV Enhanced sequence specific transactivation
and stability (Apoptosis)
Ser-20 ChK1, ChK2 IR, UV Reduced Mdm2-binding
Ser-33 CAK NK DNA bindingp38 UV Enhanced sequence specific transactivation
and stability (Apoptosis)JNK IR, UV Enhanced stability
Ser-37 ATR IR, UV AcetylationDNA-PK IR, UV Enhanced sequence specific transactivation
Ser-46 p38 UV Enhanced sequence specific transactivationand stability (Apoptosis)
Unknown IRHIPK2 UV Activation of p53-dependent transcription
and apoptotic pathways
Ser-149 CSN Targets p53 to degradation by the ubiquitin system
Ser-315 CKI IR, UV Reduced Mdm2-bindingCDK NK DNA binding
Ser-371 CyclinA-cdk2 NK Enhanced sequence specific transactivation
Ser-376 PKC UV Enhanced sequence specific transactivationCAK NK DNA bindingCyclinA-cdk2 NK Enhanced sequence specific transactivation
Ser-378 CAK NK DNA bindingCyclinA-cdk2 NK Enhanced sequence specific transactivationPKC IR Enhanced sequence specific transactivation
Ser-392 CKII UV TetramerizationP38 UV Enhanced sequence specific transactivation
and stability (Apoptosis)
Thr-18 CKI IR, UV Reduced Mdm2-bindingPKR IFN, dsRNA Enhanced sequence specific transactivation
Thr-55 TAF1 Unstressed cells Cell G1 progression and apoptosis
Thr-81 ROS ApoptosisJNK DNA-damage Enhanced p53 transactivation potential
Thr-150 CSN IFN Targets p53 to degradation by the ubiquitin system
Thr-155 CSN IFN Targets p53 to degradation by the ubiquitin system

TWENTY FIVE YEARS AROUND p53 295
J. Physiol. Biochem., 60 (4), 2004
Dephosphorylation
Ser-315 Unknown IR Increase binding to 14-3-3Cdc14 phosphatase NK Nuclear localization
Acetylation
Lys-320 PCAF IR, UV DNA binding
Lys-373 P300/CBP IR, UV DNA bindingp33(ING1b) NK Apoptosis
Lys-382 p33(ING1b) NK ApoptosisP300/CBP IR, UV DNA binding
Sumoylation
Lys-386 E1 and hUbc9 UV Stabilization
Ribosylation
PARP IR Enhanced sequence specific transactivation and stability
dsRNA, double stranded RNA; IR, ionizing radiation; IFN, interferon; NK, not known; UV, ultraviolet.
Position Modifying Stress Functionenzymes signal
and ATR (Ser15), CDK-activating kinase(CAK; Ser 33), cdc2/cdk2 (Ser315) andPKC (Ser378) (97, 111). In addition to allthese proteins that regulate the stabilityand function of p53 through phosphory-lation, a functionally distinct group ofproteins, is now emerging. This newgroup of proteins appears to operate ascofactors stimulating one or more of thewild-type properties of p53. One suchfamily, which is possibly involved inbreast cancer, is the apoptosis stimulatingprotein of p53 (ASPP) (134).
p53 redox regulation involves two clus-ters of cysteine residues in the centraldomain of the protein, one of them (aa121, 132, 138 and 272) being located nearthe loop-sheet-helix region of p53 thatcontacts with the consensus DNAsequences. Oxidized p53 loses itssequence-specific DNA-binding capabili-ties (56), and divalent metal cations,whose binding to p53 is dependent on the
reduced state of certain cysteine residues,are critical for p53 function (33, 56, 155).Furthermore, nitric oxide and thioredoxinreductase affect p53 conformation and/ortranscriptional activity (17, 126). Thedemonstration that the redox/repair pro-tein, Ref-1, activates the DNA-bindingand transactivation activities of p53 (68)further stresses the potential functionalimportance of the redox state of p53.
Like other proteins, p53 is degraded.The major degradation pathway of p53 isthe ubiquitin-26S proteosome (100), but itcan also be hydrolysed by cysteine pro-teases such as calpains (83). Because p53degradation takes place entirely in thecytoplasm, a return of p53 from the nucle-us to the cytoplasm is necessary. Thisreturn occurs via the CRN1 pathway.Three systems involve in the signalling forp53 degradation have been described: thesignalosome COP9 (CSN) that phospho-rylates p53 at Thr155 and the neighbour-
Table II. (Continuation).

M. GOMEZ-LAZARO, F. J. FERNANDEZ-GOMEZ AND J. JORDÁN296
J. Physiol. Biochem., 60 (4), 2004
ing residues (9); the JNK kinase that phos-phorylates p53 at Thr81; and the MDM2protein (58), a phosphoprotein with E3ubiquitin protein ligase activity, which isable to interact with the p53 amino-termi-nal end (67). The most important of theseis the one mediated by the oncogenicMDM2 protein (15, 99), which can guidep53 to the cytoplasm where it is degraded(62, 146). The ability of p53 to bind andupregulate the transcription of MDM2gene (7), further increases the significanceof this pathway. Therefore, the rise of p53protein levels results in the increase inMDM2, which in turns guides p53 to itsproteolitic degradation. In this context,MDM2 could be responsible for the lowlevels of p53 seen in cells that are notunder stress conditions (122). In some sit-uations, MDM2 transcription is inducedlater than other p53 target genes, whichfavours the presence of a time period thatallows p53 to exert its activity before itsdegradation takes place (165). This idea issupported by the observation that in situ-ations where MDM2 protein function iscancelled, such as in MDM2-/- mice, anupregulation of p53 takes place. This islethal in embryonic stages, but can bereversed if p53 is inactivated (72). Thep14 ARF protein (murine p19ARF) modu-lates p53 half-life by downregulation ofthe MDM2 action on p53. p14ARF actioninvolves the kidnapping of MDM2 in thenucleolus, so as to prevent the MDM2-p53 interaction in the nucleoplasm (160)or even the inhibition of MDM2 activity(67). Recent studies of the ubiquitin path-way have resulted in the identification oftwo new ubiquitin ligases E3 for p53,the constitutively photomorphogenic 1(Cop1) and Pirh2 proteins. Thus, negativeregulation of p53 seems to occur throughat least four E3 or E3-like proteins(COP1, Pirh2, Mdm2, Mdm4) (37) and
three of these, COP1, Pirh2 and Mdm2,are p53-induced genes. This creates a verycomplex and responsive control circuitable to fine-tune the p53 response. Anoth-er new gene is the deubiquitinatingenzyme herpesvirus-associated ubiquitin-specific protease (HAUSP, also calledUSP7) that in human interacts with p53protein, and removes the ubiquitin fromubiquitinated p53. Thus, human HAUSPstabilizes the status of p53 and inducesp53-dependent cell growth repression andapoptosis (91).
The role of phosphorylation in modu-lating p53 function becomes apparent dur-ing senescence, quiescence and after expo-sure to DNA damaging agents, wheresteady-state phosphorylation of p53increases at Ser15 (161). Phosphorylationat Ser15 increases binding to CBP (85) andp300 (39), and simultaneously decreasesbinding to MDM2 (85). Two phosphoa-ceptor sites recently identified are: atThr18 in the BOX-I domain, which ismodified in human breast cancer (28),induced during senescence (161) or tran-siently following ionizing radiation (133).The second site at Ser20 is modified con-stitutively in normal human fibroblastsand oxidant stresses can result in de-phos-phorylation at this site (27). In addition,an intact Ser20 residue is required foreffective p53 activity (151) and the ioniz-ing irradiation-induced form of p53 pro-tein is phosphorylated at Ser20 by aChk2-dependent pathway (137, 138).
Acetylation processes are also involvedin the p53 and MDM2 regulation. p53acetylation not only controls its transcrip-tional activity but also inhibits its MDM2-mediated ubiquitination, while MDM2acetylation prevents its activity. In the lat-ter mechanism two proteins are involved:the CREB binding protein (CBP) and, ina less extent, p300 (159). Phosphorylation

TWENTY FIVE YEARS AROUND p53 297
J. Physiol. Biochem., 60 (4), 2004
of p53 in the transactivation domain atSer15 activates p53 by an ATM-dependentpathway. Adjacent phosphorylation atThr18 or Ser20 by CHK2 activates p53 bystabilizing the binding of p300 to p53 (36).
CBP/p300-mediated acetyl-transferaseactivity is critical to play its role incatalysing p53 acetylation and activatingp53-mediated function during stressresponse. In fact p300/CBP-mediatedacetylation of p53 is negatively regulatedby MDM2 (66). Interestingly, two addi-tional regulators have also been identifiedin the p53 acetylation pathway:PID/MTA2 and Sir2alpha. PID/MTA2induces p53 deacetylation by recruitingthe histone deacetylase complex 1(HDAC1), and Sir2alpha, a NAD-depen-dent histone deacetylase, attenuates p53transcriptional activity through deacetyla-tion. Both enzymes are critical for p53-dependent stress response (54).
p53 is also subjected to modification byconjugation of SUMO-1 (22). Sumoyla-tion of p53 by the ubiquitin-like protein,SUMO-1/sentrin/PIC1, has been shownto stimulate its transcriptional activationactivity.
Besides the interactions referred abovein which p53 acts as a substrate, p53 pro-tein interacts with other cellular and viralproteins. These include the adenovirusE1B-55kDa (which inhibits the transcrip-tional activity of p53) (14, 120), the twosubunits of the TFIID complex (impor-tant for transmitting activation signalsbetween p53 and the initiation complex ofthe RNA polymerase) (88, 96), TBP(involved in both activation and repres-sion of transcription) (24), the singlestranded DNA binding protein RP-A andthe p62 subunit of the transcription/repairfactor TFIIH (40), and BRCA1 a tran-scriptional coactivator for p53.
p53 functions
From a mechanistical point of view, thep53 protein acts as a cellular stress sensor(49) and the rise in p53 levels causes thecell to undergo either one of two possiblefates: a) arrest in the G1 phase of the cellcycle or, b) apoptosis or geneneticallyprogrammed cell death (88). G1 arrest ispart of a checkpoint response wherebycells with sustained DNA damage pausein G1 so as to allow for DNA repairbefore cell cycle progresses. By this mech-anism, the propagation of potentiallyoncogenic mutations is limited. The p53-dependent apoptotic pathway is alsoinduced by DNA damage in certain celltypes, as well as in cells undergoing inap-propriate proliferation.
Originally, it was postulated that p53was not essential for a normal cell func-tion. This idea was based in the observa-tion that in some cell types its latent formin physiological conditions is almostundetectable by Western Blot or inmuno-cytochemiscal techniques. Furthermore,first studies in mice with a mutant form ofp53 showed normal embryogenesis (35).However, later studies revealed that asmall number of p53-deficient embryosdisplayed developmental irregularitiessuch as abnormal neural tube closure (3,116).
p53 induces cell cycle arrest in the G1/Stransition step through a mechanisminvolving genes such as p21WAF1/CIP1,retinoblastoma, E2F, PCNA or GADD45(Fig. 1A). The p21WAF1/CIP1 gene is adirect transcriptional target for the p53protein. It is widely accepted that p21 is acyclin-dependent kinase (cdks) inhibitorthat can influence cell cycle progressionby controlling the activity of cdks. Thiskinase can act on retinoblastoma tumoursuppressor protein which, in a hypophos-

phorylated state, associates with E2Ftranscription factors to prevent the activa-tion of genes required for progression intoS phase (41).
Once the cell cycle arrest has beeninduced, p53 contributes to DNA damagerestoration processes (51) in cooperation
with p21WAF1/CIP1 and GADD45 path-way (that forms complexes with PCNAprotein) (52). Moreover, GADD45 regu-lates the transcription of the nucleotidereductase R2 subunit necessary for DNAsynthesis and repair (145), and also regu-lates the transcription of genes directly
M. GOMEZ-LAZARO, F. J. FERNANDEZ-GOMEZ AND J. JORDÁN298
J. Physiol. Biochem., 60 (4), 2004
Fig. 1. Functions of p53: The protein p53 is activated by several types of stress signals, such as DNA damage,hypoxia or nucleotide depletion. As a consequence of p53 activation, either cell cycle arrest or apoptosis pro-grammes are activated. A. Cell cycle arrest: p53 activation turns on the transcription of p21CIP1, that inhibitscyclin-dependent kinases, causing hypophosphorylation of retinoblastoma, preventing the release of E2F andblocking the G1-S transition. B. Apoptosis: If the cell is unable to restore DNA damage, p53 might upregulatep53 target genes such as Bax, Noxa or PUMA which induce the release of mitochondrial apoptogenic factors.

TWENTY FIVE YEARS AROUND p53 299
J. Physiol. Biochem., 60 (4), 2004
involved in the nucleotide excision repairof DNA.
However, when the cell is unable torestore the DNA damage, p53 induces theactivation of the apoptotic signalling path-ways, with or without de novo transcrip-tion (Fig. 1B). Among the apoptotic genescontrolled by p53 are members of the Bcl-2 family (including PUMA, NOXA, bax),the p53-induced Gene family (PIG) anddeath receptor genes expression (CD95,DR5 or PIDD) (4, 93).
The Bcl-2 family includes proteins withpro- and anti– apoptotical function.PUMA (p53 upregulated modulator ofapoptosis) is an essential mediator of p53-dependent and –independent apoptosis.In vivo (69) PUMA is quickly induced byp53, due to the existence of a p53 bindingsequence in PUMA first intron. PUMAmediates p53-induced cell death throughthe cytochrome c/Apaf-1-dependentpathway, and the inhibition of the expres-sion of its antisense reduces the apoptoticresponse to p53 (114). NOXA encodes aBcl-2 homology 3 (BH3)-only member ofthe Bcl-2 protein family. When ectopical-ly expressed, NOXA undergoes BH3motif-dependent localization to mito-chondria and interacts with anti-apoptot-ic Bcl-2 family members (such as Bcl-2and Bcl-xL), but not with pro-apoptoticproteins like bax. Bax protein is alsoinduced by p53. Under normal physiolog-ical conditions Bax is located in thecytosol, but when an apoptotical signal isreceived bax translocates to the outermitochondrial membrane (34, 57, 70),where it interacts with the transitionalpermeability pore. Bax ablation generatesa partial or complete resistance againstp53 –mediated apoptotic stimuli (169).
The p53-inducible gene 3 (PIG3) wasoriginally discovered in a serial analysis ofgene expression designed to identify genes
induced by p53 before the onset of apop-tosis (127). There are some evidences thatsuggest that the PIG3 protein is involvedin the generation of ROS (127), which areimportant downstream mediators of thep53-dependent apoptotic response (71,92). First, PIG3 expression precedes theappearance of ROS in p53-induced apop-tosis (127). Second, PIG3 has sequencesimilarity with numerous NAD(P)Hquinone oxidoreductases (127). Third,certain p53 mutants capable of inducingcell cycle arrest but not apoptosis retainthe ability to activate target genes such asp21WIF1, but not PIG3 (19, 154). Howev-er, PIG3 expression alone is insufficient tocause apoptosis (127).
p53 regulates the membrane expressionof the death receptors such as CD95/Fas/APO-1 and TRAIL receptor 2/KILLER/DR5 in a transcriptional ornon- transcriptional manner (124, 163).
Nevertheless, p53 not only acts bymeans of the transcriptional activation ofits target genes (136, 167), but alsothrough its ability to interact with otherproteins in order to modify their functionand to induce apoptosis in the presence oftranscriptional and protein synthesisinhibitory drugs (16, 115). For example, inthe absence of other proteins, p53 directlyactivates Bax to permeabilize the mito-chondrion and engage the apoptotic pro-gram in tumor cells. In these cells a frac-tion of p53 protein localizes to the mito-chondria (104, 109, 110). Indeed, p53directly interacts with the antiapoptoticproteins bcl-xl and bcl-2, allowing baxand bac liberation, which causes changesin the mitochondrial membrane perme-ability resulting in cell death (109).
Further, ASPP1 and ASPP2 (apoptosisstimulating of p53 proteins 1 and 2) stim-ulate the apoptotic function of p53 andalso induce apoptosis independently of

p53 by binding to p63 and p73. Thesesproteins are the first common activatorsof all p53 family members yet identified(12), and selectively induce the expressionof pro-apototic genes such as Bax, PIG2and PUMA but not mdm2 or p21.
p53 can function as a regulator of thesenescence growth arrest as well (18). p53contributes to the reversible growth fac-tor-dependent arrest of quiescence (G0).Proliferating, senescent and quiescentcells exhibit different p53 phosphoryla-tion patterns (161), suggesting that p53acts differently in these growth states. p53binding and transactivating activities areenhanced in quiescent cells and the abro-gation of p53 function delays the entryinto G0 following growth factor with-drawal (65).
There is an important question that stillremains to be answered is: How does thecell choose between cell cycle arrest andapoptosis? Different answers to this ques-tion have been proposed: i) a preferentialgene induction model, in which p53induces pro-apoptotic genes after deathstimulation, while pro-apoptotic genes arenot activated in p53 cell cycle arrestinduction; ii) a model in which p53 alwaysinduces the same set of genes and the p53-mediated apoptosis requires an additionaland independent signal and finally, amodel in which; iii) the transcriptionalactivation of the different target genes isnot the same and this transcriptiondepends on the p53 activation rate. There-fore in response to DNA damage thephosphorylation is achieved for Chk2,ATM or ADN-PK kinases in the follow-ing residues: Ser6, Ser9, Ser15, Ser29 andSer37. This phosphorylation stimulatesthe expression of the genes related withrestoration or cell cycle arrest, whilephosphorylation mediated by HIPK2 or
p38 kinases (a kinase that phosphorylatesp53 in Ser46) induces apoptotic genes.
On the other hand, p53 function couldbe related to its localization. The latentform of p53 protein presents a diffuse celllocalization and, in some cases is placed inthe cytoplasm bound to proteins such asParc (for p53-associated parkin-like cyto-plasmic protein) (117). Once activated orstabilized, p53 translocates to the nucleus(81), where acts as a multi-functional tran-scription factor (157). This nuclear local-ization is essential for its tumor suppres-sor function. Recently, it has been shownthat p53 is located in the mitochondrionduring cell death processes.
p53 in pathological situations
Since p53 integrates numerous signalsthat control cell life and death its disrup-tion has severe consequences. Because ofthe central role of p53 in the regulation ofcell cycle, mutations in the p53 gene arethe most frequently observed geneticlesions in human cancers. Therefore, incancer cells p53 function is impairedeither because of mutation of the gene orfollowing binding of viral or cellularoncogene-derived proteins. This causesp53 modification and genetic alterationsaccumulate rapidly leading to tumor for-mation.
Depending on the location of the mod-ification alterations in p53 gene have dif-ferent effects on the activity of the gene.Thus, mutations may occur in regulatoryregions that control often, and when, thegene is transcribed. Likewise, a mutationin the promoter region can result in adecrease or absence of p53 in the cell.Mutations in one allele assert a dominant-negative effect over the remaining wild-type allele, and a pernicious effect on thep53 function (13). Some mutations retain
M. GOMEZ-LAZARO, F. J. FERNANDEZ-GOMEZ AND J. JORDÁN300
J. Physiol. Biochem., 60 (4), 2004

TWENTY FIVE YEARS AROUND p53 301
J. Physiol. Biochem., 60 (4), 2004
wild-type activities toward a subset ofpromoters, whereas others are pro-onco-genic (140). Inactivating mutations in p53at over 200 different aa positions withinthis core DNA-binding domain have beendetected in human cancers. Biochemicaland biophysical characterization of thesep53 mutant forms have led to the sugges-tion of the existence of at least three dis-tinct classes of mutants with unique bio-chemical defects in tetramerization, con-formational regulation and intrinsic fold-ing and stability. Tumor-specific pointmutations occur in many forms of humancancer and as many as 50% of cancerscontaining a p53 mutation, affecting p53-DNA interactions in the 90% resulting ina partial or complete loss of transactiva-tion functions. 20% of mutations are con-centrated at five ‘hotspot’ codons whichare located in the DNA-binding “core”domain. These loci presnt five clusters ofhighly conserved residues found insequence analysis comparisons of p53sequences from different species (158).
Inherited p53 mutations are rare. How-ever, germ line mutations have been foundin some families with Li-Fraumeni syn-drome (101, 142). This results in an inher-ited predisposition to a wide spectrum ofcancers including breast cancer, osteosar-comas, soft tissue sarcoma, melanoma,adenocortical carcinomas, and leukaemias,all of which appear at an early stage.
Today we have more evidence that bio-logical variations between mutants mayhave clinical importance and this is thereason why individual patients respond ina different way to cytotoxic therapy. Forexample, breast cancer patients withmutations within the DNA bindingdomain appear to have increased resis-tance to doxorubicin but not to taxol (1,48).
In some degenerative processes frag-mentation and oxidative damage of DNAtakes place. In these situations, whichinclude atherosclerotic lesions (107) andneurodegenerative disorders (73, 144,149), p53 is involved. Recent studies usingmice lacking both p53 and apolipoproteinE (ApoE) (p53-/-apoE-/- mice) indicatethat p53 is important for atherogenesis;p53-/-apoE-/- mice developed larger ather-osclerotic lesions as compared to apoE-/-
mice (55). In vitro studies show that p53facilitates apoptosis of smooth musclecells isolated from human atheroscleroticlesions (10, 11) and that p53 is involved inoxidized LDL-induced apoptosis ofhuman macrophages (78).
p53 levels are high in the frontal andtemporal lobes in Alzheimer´s diseasepatients (79) and immunohistochemicalstudies show p53 colocalization withinapoptotic cells in cortical neurons andwhite glial cells (31). Further, b-amyloidpeptide [25-35] induces cell death in neu-ronal cultures by mechanisms that involvep53 (153).
p53 protein plays a role also in Parkin-son’s disease. In experimental models 6-hydroxydopamine (164) and MPTP (103,147) act as an activator of p53, and theinhibition of p53 reduces the degenerationprocess in dopaminergic neurons (38,148). Indeed, this disorder is accompaniedby widespread occurrence of intracyto-plasmic Lewy bodies, formed from fibril-lary a-synuclein and hyperphosphorylat-ed neurofilament proteins (50). Theinvolvement of sinuclein in the neurode-generative processes is a source of contro-versy. Thus, the a-sinuclein is able toblock p53 expression and its transcrip-tional activity, but it is not helpful againsttoxicity induced by 6-OHDA (2). How-ever, the b-synuclein reduces cell deathinduced by 6-OHDA and staurosporine.

In these conditions a dramatic drop in p53levels has been observed. This probably,by means of a post-transcriptional processin which MDM2 and p38 might beinvolved (30).
Other systems where apoptosis isinduced in a p53-dependent manner are:the methamphetamine-induced apoptosisin the dopaminergic neurons (60), oligo-dendrocytes from patients with multiplesclerosis (162); patients with amyotrophiclateral sclerosis (106) and excitotoxicmodels (112).
p53 as pharmacological target
Pharmacologists have made someattempts to target the p53 pathway. Majoradvances have been made in the oncologyfield where gene therapy represents a newmeans for cancer therapy. This therapeu-tical approach is mainly focused on genereplacement of defective p53 individuals.
Furthermore, it should be feasible torestore some functional activity of p53mutants by enhancing the stability of theprotein or by supplying additional DNAcontacts (13, 118, 140). Modified aden-oviruses provide a potent vector for deliv-ering p53 into cancer cells. The insertionof p53 can arrest cancer cell growth andinduce apoptosis and tumor regression inadvanced cancers such as squamous cellcarcinoma of the head and neck, and non-small cell lung cancer. Reduction of 50%in head and neck cancers has beenachieved with adenoviral-p53 treatments(26). In nasopharyngeal carcinoma p53gene therapy combined with ionizingradiation appears to interact in a synergis-tic manner (90) and promising results havebeen reported on prostate cancer accord-ing to preliminary clinical data. Therefore,the p53 therapy may provide an alterna-tive treatment to surgical radiotherapy
treatments for these patient population.Although still in a developmental phase,other pharmacological research lines pro-pose to use mimetic peptides of the activep53 regions, in order to obtain antitu-moral effects.
A promising approach to activate p53 isthe inhibition of the p53-MDM2 interac-tion with compounds including nutlin,chalcones and chlorofusin. Nutlinbehaves as a p53-MDM2 interactionblocker, interfering in protein-proteininteractions that yield in MDM2 failure toguide p53 degradation and causing anincrease in p53 protein levels (152). Chal-cones (1,3-diphenyl-2-propen-1-ones) arealso MDM2 inhibitors that bind to a sub-site of the p53 binding cleft of MDM2 anddisrupt the MDM2/p53 protein complex,releasing p53 from both the p53/MDM2and DNA-bound p53/MDM2 complexes(143). Cells treated with these compoundssuffered cell cycle arrest and apoptosis.The fungal metabolite, chlorofusin alsoantagonizes the p53/MDM2 interaction(143).
On the other hand, cancer chemopre-ventive selenium compounds in the formof selenomethionine (SeMet) activate p53by a redox mechanism that requires theredox factor. SeMet induces sequence-specific DNA binding and transactivationby p53 and shows promising results in theprevention of prostate and other humancancers (135, 141).
Finally, since p53 up-regulation hasbeen described as a common feature ofseveral degenerative disorders, p53inhibitors can be used in these patholo-gies. The blockade of p53 expression bymeans of the use of antisense oligonu-cleotide prevents neuronal cultures fromexcitotoxic processes almost completely(74, 84, 150). Recent studies identify thetetrahydrobenzothiazole analogue,
M. GOMEZ-LAZARO, F. J. FERNANDEZ-GOMEZ AND J. JORDÁN302
J. Physiol. Biochem., 60 (4), 2004

TWENTY FIVE YEARS AROUND p53 303
J. Physiol. Biochem., 60 (4), 2004
pifithrin-alpha, as a p53 inhibitor, whichis effective in protecting neuronal cellsagainst a variety of lethal insults andreducing the side effects of anticancerdrugs (80, 171). Furthermore, the neuro-protective actions of lithium and antioxi-dant drugs like OPC-14117 in excitotoxicprocesses can be due to a modulator effectin both mRNA expression levels and p53protein (23, 113).
Acknowledgements
This work has been supported by SAF2002-04721 from CICYT and SEF-Almirall Prodesfarmato J.J. M. G-L and F.J. F-G are fellows from JCCM.We thank Prof. E. Nava for helpful discussion andcritical reading of the manuscript.
M. GOMEZ-LAZARO, F. J. FERNAN-DEZ-GOMEZ y J. JORDÁN. p53: Veinticin-co años acerca del mecanismo de protección delgenoma (minirrevisión). J. Physiol. Biochem.,60 (4), 287-308, 2004.
En este año, la proteína p53, también cono-cida como “el guardián del genoma”, cumpleveinticinco años. Durante este periodo, elconocimiento sobre las funciones desempeña-das por p53 ha ido cambiando desde una acti-vidad pro-oncogénica hasta su función onco-supresora. Esta proteína se activa en respuestaa estímulos de estrés como radiaciones gammay ultravioleta, hipoxia, infección vírica y dañoen el ADN, protegiendo a la célula medianteacción sobre sus genes diana. Las moléculasactivadas por p53 inducen parada en ciclo celu-lar y reparación del ADN con el fin de conser-var el genoma o de inducir apoptosis. La regu-lación de las funciones de p53 está controladaestrechamente a través de varios mecanismosque incluyen la transcripción, traducción, esta-bilidad de la proteína por modificaciones post-transcripcionales y su localización subcelular.Una de las alteraciones moleculares detectadascon más frecuencia en los tumores humanosson las mutaciones en p53. Mas aún, en algunosprocesos degenerativos, donde tiene lugar la
fragmentación y el daño oxidativo en el ADN,la p53 está implicada. Así, se considera a la p53como una posible diana farmacológica, ya quesu sobreexpresión induce apoptosis en célulascancerosas y el bloqueo de su expresión prote-ge a las células contra daños letales.
Key words: Apoptosis, Necrosis, mdm2, Cáncer,Supresor tumoral, Mutación.
References
1. Aas, T., Borresen, A. L., Geisler, S., Smith-Sorensen, B., Johnsen, H., Varhaug, J. E.,Akslen, L. A. and Lonning, P. E. (1996): Nat.Med., 2, 811-814.
2. Alves Da Costa, C., Paitel, E., Vincent, B. andChecler, F. (2002): J. Biol. Chem., 277, 50980-50984.
3. Armstrong, J. F., Kaufman, M. H. Harrison, D.J. and Clarke, A. R. (1995): Curr. Biol., 8, 937-943.
4. Ashkenazi, A. and Dixitm, V. M. (1998): Science,281, 1305-1308.
5. Avantaggiati, M. L., Ogryzko, V., Gardner, K.,Giordano, A., Levine, A. S. and Kelly, K. (1997):Cell, 89, 1175-1184.
6. Baker, S. J., Fearon, E. R., Nigro, J. M., Hamil-ton, S. R., Preisinger, A. C, Jessup, J. M., vanTu-inen, P., Ledbetter, D. H., Barker, D. F., Naka-mura, Y., et al. (1989): Science, 244, 217-221.
7. Barak, Y., Juven, T., Haffner, R. and Oren, M.(1993): EMBO J., 12, 461-468.
8. Bayle, J. H., Elenbaas, B. and Levine, A. J.(1995): Proc. Natl. Acad. Sci. USA, 92, 5729-5733.
9. Bech-Otschir, D., Kraft, R., Huang, X., Hen-klein, P., Kapelari, B., Pollmann, C. and Dubiel,W. (2001): EMBO J., 20, 1630-1639.
10. Bennett, M. R., Evan, G. I. and Schwartz, S. M.(1995): Circ. Res., 77, 266–273.
11. Bennett, M. R., Littlewood, T. D., Schwartz, S.M. and Weissberg, P. L. (1997): Circ. Res., 81,591–599.
12. Bergamaschi, D., Samuels, Y., Jin, B., Duraising-ham, S., Crook, T. and Lu, X. (2004): Mol. CellBiol., 24, 1341-1350.
13. Brachmann, R. K., Yu, K., Eby, Y., Pavletich, N.P. and Boeke, J. D. (1998): EMBO J., 17, 1847-1859.

14. Braithwaite, A. W., Blair, G. E., Nelson, C. C.,McGovern, J. and Bellett, A. J. (1991): Onco-gene, 6, 781-787.
15. Burch, L. R., Midgley, C. A., Currie, R. A.,Lane, D. P. and Hupp, T. R. (2000): FEBS Lett.,472, 93-98.
16. Caelles, C., Helmberg, A. and Karin, M. (1994):Nature, 370, 220-223.
17. Calmels, S., Hainaut, P. and Ohshima, H.(1997): Cancer Res., 57, 3365-3369.
18. Campisi, J. (2000): In Vivo, 14, 183-188. 19. Campomenosi, P., Monti, P., Aprile, A., Abbon-
dandolo, A., Frebourg, T., Gold, B., Crook, T.,Inga, A., Resnick, M. A., Iggo, R. and Fronza, G.(2001) Oncogene, 20, 3573–3579
20. Caron de Fromentel, C., May-Levin, F.,Mouriesse, H., Lemerle, J., Chandrasekaran, K.and May, P. (1987): Int. J. Cancer, 39, 185-189.
21. Chang, C., Simmons, D. T., Martin, M. A. andMora, P. T. (1979): J Virol., 31, 463-471.
22. Chen, L. and Chen, J. (2003): Oncogene, 22,5348-5357.
23. Chen, R. W. and Chuang, D. M. (1999): J. Biol.Chem., 274, 6039-6042.
24. Chen, X., Farmer, G., Zhu, H., Prywes, R. andPrives, C. (1993): Genes Dev. 7, 1837-1849.Erratum in: (1993): Genes Dev., 7(12B), 2652.
25. Cho, Y., Gorina, S., Jeffrey, P. D. and Pavletich,N. P. (1994): Science, 265, 345-355.
26. Clayman, G. L., el-Naggar, A. K., Lippman, S.M., Henderson, Y. C., Frederick, M., Merritt, J.A., Zumstein, L. A., Timmons, T. M., Liu, T. J.,Ginsberg, L., Roth, J. A., Hong, W. K., Bruso, P.and Goepfert, H. (1998): J. Clin. Oncol., 16,2221-2232.
27. Craig, A. L., Blaydes, J. P., Burch, L. R.,Thompson, A. M. and Hupp, T. R. (1999):Oncogene, 18, 6305-6312.
28. Craig, A. L., Burch, L., Vojtesek, B., Mikutows-ka, J., Thompson, A. and Hupp, T. R. (1999):Biochem. J., 342, 133-141.
29. Crawford, L. V., Pim, D. C. and Bulbrook, R.D. (1982): Int. J. Cancer, 30, 403-408.
30. Da Costa C. A., Masliah, E. and Checler, F.(2003): J. Biol. Chem., 278, 37330-37335.
31. De la Monte, S. M., Sohn, Y. K. and Wands, J. R.(1997): J. Neurol. Sci., 152, 73-83.
32. De Leo, A. B., Jay, G., Appella, E., Dubois, G.C, Law, L. W. and Old, L. J. (1979): Proc. Natl.Acad. Sci. USA, 76, 2420-2424.
33. Delphin, C., Cahen, P., Lawrence, J. J. andBaudier, J. (1994): Eur. J. Biochem., 223, 683-692.
34. Dewson, G., Cohen, G. M. and Wardlaw, A. J.(2001): Blood, 98, 2239-2247.
35. Donehower, L. A., Harvey, M., Slagle, B. L.,McArthur, M. J., Montgomery, C. A. Jr., Butel,J. S. and Bradley, A. (1992): Nature, 356, 215-221.
36. Dornan, D. and Hupp, T. R. (2001): EMBORep., 2, 139-144.
37. Dornan, D., Bheddah, S., Newton, K., Ince, W.,Frantz, G. D., Dowd, P., Koeppen, H., Dixit, V.M. and French, D. M. (2004): Cancer Res., 64,7226-7230.
38. Duan, W., Zhu, X., Ladenheim, B., Yu, Q. S.,Guo, Z., Oyler, J., Cutler, R. G., Cadet, J. L.,Greig, N. H. and Mattson, M. P. (2002): Ann.Neurol., 52, 597–606.
39. Dumaz, N. and Meek, D. W. (1999): EMBO J.,18, 7002-7010.
40. Dutta, A., Ruppert, J. M., Aster, J. C. and Win-chester, E. (1993): Nature, 365, 79-82.
41. El-Deiry, W. S., Tokino, T., Velculescu, V. E.,Levy, D. B., Parsons, R., Trent, J. M., Lin, D.,Maercer, W. E., Kinzler, K.W. and Vogelstein,B. (1993): Cell, 75, 817-825.
42. El-Deiry, W.S., Kern, S. E., Pietenpol, J. A.,Kinzler, K. W. and Vogelstein, B. (1992): Nat.Genet., 1, 45-49.
43. Eliyahu, D., Raz, A., Gruss, P., Givol, D. andOren, M. (1984): Nature, 312, 646-649.
44. Fields, S. and Jang, S. K. (1990): Science, 249,1046-1049.
45. Finlay, C. A., Hinds, P. W. and Levine, A. J.(1989): Cell, 57, 1083-1093.
46. Finlay, C. A., Hinds, P. W., Tan, T. H., Eliyahu,D., Oren, M, and Levine, A. J. (1988): Mol. CellBiol., 2, 531-539.
47. Forrester, K., Ambs, S., Lupold, S. E., Kapust,R. B., Spillare, E. A., Weinberg, W. C., Felley-Bosco, E., Wang, X. W., Geller, D. A., Tzeng,E., Billiar, T. R. and Harris, C. C. (1996): Proc.Natl. Acad. Sci. U S A, 93, 2442-2447.
48. Geisler, S., Lonning, P. E., Aas, T., Johnsen, H.,Fluge, O., Haugen, D. F., Lillehaug, J. R.,Akslen, L. A. and Borresen-Dale, A. L. (2001):Cancer Res., 61, 2505-2512.
49. Giaccia, A. J. and Kastan, M. B. (1999): GenesDev., 12, 2973-2983.
50. Giasson, B. I., Ischiropoulos, H., Lee, V. M. andTrojanowski, J. Q. (2002): Free Radic. Biol.Med., 32, 1264–1275.
51. Gottifredi, V., McKinney, K., Poyurovsky, M.V. and Prives, C. (2004): J. Biol. Chem., 279,5802-5810.
52. Gottlieb, T. M. and Oren, M. (1996): Biochem.Biophys. Acta, 1287, 77-102.
53. Graeber, T. G., Osmanian, C., Jacks, T., Hous-man, D. E., Koch, C. J., Lowe, S. W. and Giac-cia, A. J. (1996): Nature, 379, 88-91.
M. GOMEZ-LAZARO, F. J. FERNANDEZ-GOMEZ AND J. JORDÁN304
J. Physiol. Biochem., 60 (4), 2004

TWENTY FIVE YEARS AROUND p53 305
J. Physiol. Biochem., 60 (4), 2004
54. Gu, W., Luo, J., Brooks, C. L., Nikolaev, A. Y.and Li, M. (2004): Novartis Found Symp., 259,197-205.
55. Guevara, N. V., Kim, H. S., Antonova, E. I. andChan, L. (1999): Nat. Med., 5, 335–339.
56. Hainaut, P. and Milner, J. (1993): Cancer Res.,53, 1739-1742.
57. Harada, H., Becknell, B., Wilm, M., Mann, M.,Huang, L. J., Taylor, S. S., Scott J. D. andKorsmeyer, S. J. (1999): Mol Cell, 3, 413-422.
58. Haupt, Y., Barak, Y. and Oren, M. (1996):EMBO J., 15, 1596-1606.
59. Hinds, P., Finlay, C. and Levine, A.J. (1989): JVirol, 63, 739-746.
60. Hirata, H. and Cadet, J. L. (1997): J. Neu-rochem., 69, 780-790.
61. Hollstein, M., Rice, K., Greenblatt, M. S., Sous-si, T., Fuchs, R., Sorlie, T., Hovig, E., Smith-Sorensen, B., Montesano, R. and Harris, C. C.(1994) Nucleic Acids Res., 22, 3551-3555.
62. Honda, R., Tanaka, H. and Yasuda, H. (1997):FEBS Lett., 420, 25-27.
63. http://p53.curie.fr/p53%20site%20ver-sion%202.0/p53_story_disco.html
64. Irwin, M. S. and Kaelin, W. G. Jr. (2001): Apop-tosis, 6, 17-29.
65. Itahana, K., Dimri, G. P., Hara, E., Itahana, Y.,Zou, Y., Desprez, P. Y. and Campisi, J. (2002): J.Biol. Chem., 277, 18206-18214.
66. Ito, A., Kawaguchi, Y., Lai, C. H., Kovacs, J. J.,Higashimoto, Y., Appella, E. and Yao, T. P.(2002): EMBO J., 21, 6236-6245.
67. Iwakuma, T. and Lozano, G. (2003): Mol. Can-cer Res., 1, 993-1000.
68. Jayaraman, L., Moorthy, N. C., Murthy, K. G.,Manley, J. L., Bustin, M. and Prives, C. (1998):Genes Dev., 12, 462-472.
69. Jeffers, J. R., Parganas, E., Lee, Y., Yang, C.,Wang, J., Brennan, J., MacLean, K. H., Han, J.,Chittenden, T., Ihle, J. N., McKinnon, P. J.,Cleveland, L. and Zambetti, G. P. (2003): Can-cer Cell, 4, 321-328.
70. Jia, L., Patwari, Y., Srinivasula, S. M., Newland,A. C., Fernandes-Alnemri, T., Alnemri, E. S. ansKelsey, S. M. (2001): Oncogene, 20, 4817-4826.
71. Johnson, T. M., Yu, Z. X., Ferrans, V. J., Lowen-stein, R. A. and Finkel, T. (1996) Proc. Natl.Acad. Sci. U. S. A. 93, 11848–11852
72. Jones, S. N., Roe, A. E., Donehower, L. A. andBradley, A. (1995): Nature, 378, 206-208.
73. Joo, C. K., Choi, J. S., Ko, H. W., Park, K. Y.,Sohn, S., Chun, M. H., Oh, Y. J. and Gwag, B. J.(1999): Invest. Ophthalmol. Vis. Sci., 40, 713-720.
74. Jordan, J., Galindo, M. F., Gonzalez-Garcia, C.and Cena, V. (2003): Neuroscience, 122, 707-715.
75. Kaghad, M., Bonnet, H., Yang, A., Creancier, L.,Biscan, J. C., Valent, A., Minty, A., Chalon, P.,Lelias, J. M., Dumont, X., Ferrara, P., McKeon,F. and Caput, D. (1997): Cell, 90, 809-819.
76. Kastan, M. B., Onyekwere, O., Sidransky, D.,Vogelstein, B. and Craig, R. W. (1991): CancerRes., 51, 6304-6311.
77. Kern, S. E., Kinzler, K. W., Bruskin, A., Jarosz,D., Friedman, P., Prives, C. and Vogelstein, B.(1991): Science, 252, 1708-1711.
78. Kinscherf, R., Deigner, H. P., Usinger, C., Pill,J., Wagner, M., Kamencic, H., Hou, D., Chen,M., Schmiedt, W., Schrader, M., Kovacs, G.,Kato, K. and Metz, J. (1997): FASEB J., 11,1317–1328.
79. Kitamura, Y., Shimohama, S., Kamoshima, W.,Matsuoka, Y., Nomura, Y. and Taniguchi, T.(1997): Biochem. Biophys. Res. Commun., 232,418-421.
80. Komarov, P. G., Komarova, E. A., Kondratov,R. V., Christov-Tselkov, K., Coon, J. S., Cher-nov, M. V. and Gudkov, A. V. (1999): Science,285, 1733-1737.
81. Komarova, E. A., Zelnick, C. R., Chin, D.,Zeremski, M., Gleiberman, A. S., Bacus, S. S.And Gudkov, A. V. (1997): Cancer Res., 57,5217-5220.
82. Kress, M., May E., Cassingena, R. and May, P.(1979): J. Virol., 31, 472-483.
83. Kubbutat, M. H. and Vousden, K. H. (1997):Mol. Cell Biol., 17, 460-468.
84. Lakkaraju, A., Dubinsky, J. M., Low, W. C. andRahman, Y. E. (2001): J. Biol. Chem., 276,32000-32007.
85. Lambert, P. F., Kashanchi, F., Radonovich, M.F., Shiekhattar, R. and Brady, J. N. (1998): J.Biol. Chem., 273, 33048-33053.
86. Lane, D. P. (1992): Nature, 358, 15-16.87. Lane, D. P. and Crawford, L. V. (1979): Nature,
278, 261-263.88. Levine, A. J. (1997): Cell, 88, 323-331.89. Levrero, M., De Laurenzi, V., Costanzo, A.,
Gong, J., Wang, J.Y. and Melino, G. (2000): J.Cell Sci., 113, 1661-1670.
90. Li, J. H., Lax, S. A., Kim, J., Klamut, H. and Liu,F. F. (1999): Int. J. Radiat. Oncol. Biol. Phys., 43,607-616.
91. Li, M., Chen, D., Shiloh, A., Luo, J., Nikolaev,A. Y., Qin, J. and Gu, W. (2002): Nature, 416,648-653.
92. Li, P. F., Dietz, R. and von Harsdorf, R. (1999)EMBO J. 18, 6027–6036
93. Lin, Y., Ma, W. and Benchimol, S. (2000):Nature Genetics, 26, 122-127.

94. Linke, S. P., Clarkin, K. C., Di Leonardo, A.,Tsou, A. and Wahl, G. M. (1996): Genes Dev.,10, 934-947.
95. Linzer, D. I. H. and Levine, A. J. (1979): Cell, 1,43-52.
96. Lu, H. and Levine, A. J. (1995): Proc. Natl. Acad.Sci. USA, 92, 5154-5158.
97. Lu, H., Fisher, R. P., Bailey, P. and Levine, A. J.(1997): Mol. Cell Biol., 17, 5923-5934.
98. Mai, M., Huang, H., Reed, C., Qian, C., Smith,J. S., Alderete, B., Jenkins, R., Smith, D. I. andLiu, W. (1998): Genomics, 51, 359-363.
99. Maki, C. G. (1999): J. Biol. Chem., 274, 16531-16535.
100. Maki, C. G., Huibregtse, J. M. and Howley, P.M. (1996): Cancer Res., 56, 2649-2654.
101. Malkin, D., Li, F. P., Strong, L. C., Fraumeni, J.F. Jr, Nelson, C. E., Kim, D. H., Kassel, J.,Gryka, M. A., Bischoff, F. Z., Tainsky, M. A. etal. (1990): Science, 250, 1233-1238. Erratum in:(1993): Science, 259, 878.
102. Maltzman, W. and Czyzyk, L. (1984): Mol. CellBiol., 4, 1689-1694.
103. Mandir, A. S. Simbulan-Rosenthal, C. M.,Poitras, M. F., Lumpkin, J. R., Dawson, V. L.,Smulson, M. E. and Dawson T. M. (2002): J.Neurochem., 83, 186–192.
104. Marchenko, N. D., Zaika, A. and Moll, U. M.(2000): J. Biol. Chem., 275, 16202-16212.
105. Marin, M. C., Jost, C. A., Irwin, M. S.,DeCaprio, J. A., Caput, D.and Kaelin, W. G.Jr.(1998): Mol. Cell Biol., 18, 6316-6324.
106. Martin, L. J. (2000): Neurobiol. Dis., 7, 613-622. 107. Martinet, W., Knaapen, M. W., De Meyer, G.
R., Herman, A. G. and Kockx, M. M. (2002):Circulation, 106, 927-932.
108. Melero, J. A., Stitt, D. T., Mangel, W. F. andCarroll, R. B. (1979): J. Virol., 93, 466-480.
109. Mihara, M., Erster, S., Zaika, A., Petrenko, O.,Chittenden, T., Pancoska, P. and Moll, U. M.(2003): Mol. Cell., 11, 577-590.
110. Mihara, M. and Moll, U. M. (2003): MethodsMol. Biol., 234, 203-209.
111. Milne, D. M., McKendrick, L., Jardine, L. J.,Deacon, E., Lord, J. M. and Meek. D. W.(1996): Oncogene, 13, 205-211.
112. Morrison, R. S., Wenzel, H. J., Kinoshita, Y.,Robbins, C. A., Donehower, L. A. andSchwartzkroin, P. A. (1996): J. Neurosci., 16,1337-1345.
113. Nakai, M., Qin, Z. H., Wang, Y. and Chase, T.N. (1999): Brain Res. Mol. Brain Res., 64, 59-68.
114. Nakano, K. and Vousden, K. H. (2001): Mol.Cell, 7, 683-694.
115. Nechushtan, A., Smith, C. L., Lamensdorf, I.,Yoon, S. H. and Youle, R. J. (2001): J. Cell.Biol., 153, 1265-1276.
116. Nicol, C. J., Harrison, M. L., Laposa, R. R.,Gimelshtein, I. L. and Wells P.G. (1995): Nat.Genet., 10, 181-187.
117. Nikolaev, A. Y., Li, M., Puskas, N., Qin, J. andGu, W. (203): Cell, 112, 29-40.
118. Nikolova, P. V., Wong, K. B., DeDecker, B.,Henckel, J. and Fersht, A. R. (2000): EMBO J.;19, 370-378.
119. Ohnishi, T., Wang, X., Ohnishi, K., Matsumo-to, H. and Takahashi, A. (1996): J. Biol. Chem.,271, 14510-14513.
120. Oliner, J. D., Pietenpol, J. A., Thiagalingam, S.,Gyuris, J., Kinzler, K. W. and Vogelstein, B.(1993): Nature, 362, 857-860.
121. Ollmann, M., Young, L. M., Di Como, C. J.,Karim, F., Belvin, M., Robertson, S., Whittaker,K., Demsky, M., Fisher, W. W., Buchman, A.,Duyk, G., Friedman, L., Prives, C. andKopczynski, C. (2000): Cell, 101, 91-101.
122. Oren, M. (1999): J. Biol. Chem., 274, 36031-36034.
123. Oren, M., Maltzman, W. and Levine, A. J.(1981): Mol. Cell Biol., 1, 101-110.
124. Owen-Schaub, L. B., Zhang, W., Cusack, J. C.,Angelo, L. S., Santee, S. M., Fujiwara, T., Roth,J. A., Deisseroth, A. B., Zhang, W. W., Kruzel,E. et al. (1995): Mol. Cell Biol., 15, 3032-3040.
125. Parada, L. F., Land, H., Weinberg, R. A., Wolf,D. and Rotter, V. (1984): Nature, 312, 649-651.
126. Pearson, G. D. and Merrill, G. F. (1998): J. Biol.Chem., 273, 5431-5434.
127. Polyak, K., Xia, Y., Zweier, J. L., Kinzler, K.W. and Vogelstein, B. (1997): Nature, 389, 300-305.
128. Price, B. D. and Calderwood, S. K. (1993):Oncogene, 8, 3055-3062.
129. Reich, N. C. and Levine, A. J. (1984): Nature,308, 199-201.
130. Rodrigues, N. R., Rowan, A., Smith, M.E.,Kerr, I. B., Bodmer, W. F., Gannon, J. V. andLane, D. P. (1990): Proc. Natl. Acad. Sci. U S A.,87, 7555-7559.
131. Rogel, A., Popliker, M., Webb, C. G. and Oren,M. (1985): Mol. Cell Biol., 5, 2851-2855.
132. Rotter, V., Witte, O. N., Coffman, R. and Bal-timore, D. (1980): J Virol., 36, 547-555.
133. Sakaguchi, K., Saito, S., Higashimoto, Y., Roy,S., Anderson, C. W. and Appella, E. (2000): J.Biol. Chem., 275, 9278-9283.
134. Samuels-Lev, Y., O’Connor, D. J., Bergam-aschi, D., Trigiante, G., Hsieh, J. K., Zhong, S.,
M. GOMEZ-LAZARO, F. J. FERNANDEZ-GOMEZ AND J. JORDÁN306
J. Physiol. Biochem., 60 (4), 2004

TWENTY FIVE YEARS AROUND p53 307
J. Physiol. Biochem., 60 (4), 2004
Campargue, I., Naumovski, L., Crook, T. andLu, X. (2001): Mol. Cell, 8, 781–794.
135. Seo, Y. R., Kelley, M. R. and Smith, M. L.(2002): Proc. Natl. Acad. Sci. USA, 99, 14548-14553.
136. Shaw, P., Bovey, R., Tardy, S., Sahli, R., Sordat,R. and Costa, J. (1992): Proc. Natl. Acad. Sci.USA., 89, 4495-4499.
137. Shieh, S. Y., Ahn, J., Tamai, K., Taya, Y. andPrives, C. (2000): 14, 289-300. Erratum in:(2000): Genes Dev., 14, 750.
138. Shieh, S. Y., Ikeda, M., Taya, Y. and Prives, C.(1997): Cell, 91, 325-334.
139. Sidransky, D., Mikkelsen, T., Schwechheimer,K., Rosenblum, M. L., Cavanee, W. and Vogel-stein, B. (1992): Nature, 355, 846-847.
140. Sigal, A. and Rotter, V. (2000): Cancer Res., 60,6788-6793.
141. Smith, M. L., Lancia, J. K. and Mercer, T.(2004): Anticancer Res., 24, 1401-1408.
142. Srivastava, S., Zou, Z. Q., Pirollo, K., Blattner,W. and Chang, E. H. (1990): Nature, 348, 747-749.
143. Stoll, R., Renner, C., Hansen, S., Palme, S.,Klein, C., Belling, A., Zeslawski, W., Kamion-ka, M., Rehm, T., Muhlhahn, P., Schumacher,R., Hesse, F., Kaluza, B., Voelter, W., Engh, R.A. and Holak, T. A. (2001): Biochemistry, 40,336-344.
144. Su, J. H., Anderson, A. J., Cummings, B. J. andCotman, C. W. (1994): Neuroreport., 5,2529–2533.
145. Tanaka, H., Arakawa, H., Yamaguchi, T., Shi-raishi, K., Fukuda, S., Matsui, K., Takei, Y. andNakamura, Y. (2000): Nature, 404, 42-49.
146. Tao, W. and Levine, A. J. (1999): Proc. Natl.Acad. Sci. USA, 96, 3077-3080.
147. Thornborrow, E. C., Patel, S., Mastropietro, A.E., Schwartzfarb, E. M. and Manfredi, J. J.(2002): Oncogene, 21, 990–999.
148. Trimmer, P. A., Smith, T. S., Jung, A. B. andBennett, J. P. Jr. (1996): Neurodegeneration, 5,233–239.
149. Uberti, D., Ferrari, Toninelli, G. and Memo, M.(2003): Toxicol. Lett., 139, 99-105.
150. Uberti, D., Grilli, M. and Memo, M. (2000): Int.J. Dev. Neurosci., 18, 447-454.
151. Unger, T., Sionov, R. V., Moallem, E., Yee, C.L., Howley, P. M., Oren, M. and Haupt, Y.(1999): Oncogene, 18, 3205-3212.
152. Vassilev, L. T., Vu, B. T., Graves, B., Carvajal,D., Podlaski, F., Filipovic, Z., Kong, N.,Kammlott, U., Lukacs, C., Klein, C., Fotouhi,N. and Liu, E. A. (2004): Science, 303, 844-848.
153. Velez-Pardo, C., Ospina, G. G. and Jimenez delRio, M. (2002): Neurotoxicology, 23, 351-365.
154. Venot, C., Maratrat, M., Dureuil, C., Con-seiller, E., Bracco, L. and Debussche, L. (1998):EMBO J. 17, 4668–4679.
155. Verhaegh, G. W., Richard, J. and Hainaut, P.(1997): Mol. Cell. Biol., 17, 5699-5706.
156. Vogelstein, B. and Kinzler, K. W. (1994):Nature, 408, 307-310.
157. Vogelstein, B., Lane, D. and Levine, A. J.(2000): Nature, 408, 307-310.
158. Walker, D., Bond, J., Tarone, R., Harris, C.,Makalowski, W., Boguski, M. and Greenblatt,M. (1999): Oncogene, 18, 211-218.
159. Wang, X., Taplick, J., Geva, N. and Oren, M.(2004): FEBS Lett., 561, 195-201.
160. Weber, J. D., Taylor, L. J., Rousssel, M. F.,Sherr, C. J. and Bar-Sagi, D. (1999): Nat. CellBiol., 1, 20-26.
161. Webley, K., Bond, J. A., Jones, C. J., Blaydes, J.P., Craig, A., Hupp, T. and Wynford-Thomas,D. (2000): Mol. Cell Biol., 20, 2803-2808.
162. Wosik, K., Antel, J., Kuhlmann, T., Bruck, W.,Massie, B. and Nalbantoglu, J. (2003): J. Neu-rochem., 85, 635-644.
163. Wu, G. S., Burns, T. F., McDonald, E.R. 3rd,Jiang, W., Meng, R., Krantz, I. D., Kao, G.,Gan, D. D., Zhou, J. Y., Muschel, R., Hamilton,S. R., Spinner, N. B., Markowitz, S., Wu, G. andel-Deiry, W. S. (1997): Nat. Genet., 17, 141-143.
164. Wu, H. and Lozano, G. (1994): J. Biol. Chem.,269, 20067-20074.
165. Wu, L. and Levine, A. J. (1997): Mol Med., 3,441-451.
166. Yang, A., Kaghad, M., Wang, Y., Gillet, E.,Fleming, M. D., Dotsch, V., Andrews, N. C.,Caput, D. and McKeon, F. (1998): Mol. Cell., 2,305-316.
167. Yonish-Rouach, E., Borde, J., Gotteland, M.,Mishal, Z., Viron, A. and May, E. (1994): CellGrowth Differ., 1, 39-47.
168. Yu, J., Zhang, L., Hwang, P. M., Rago, C., Kin-zler, K. W. and Vogelstein, B. (1999): Proc.Natl. Acad. Sci. USA, 96, 14517-14522.
169. Zhang L., Yu, J., Park, B. H., Kinzler, K. W.and Vogelstein, B. (2000): Science, 290, 989-992.
170. Zhao, R., Gish, K., Murphy, M., Yin, Y., Not-terman, D., Hoffman, W. H., Tom, E., Mack,D. H. and Levine, A. J. (2000): Genes Dev., 14,981-993.
171. Zhu, X., Yu, Q. S., Cutler, R. G., Culmsee, C.W., Holloway, H. W., Lahiri, D. K., Mattson,M. P. and Greig, N. H. (2002): J. Med. Chem.,45, 5090-5097.




















