
Textural, rheological and sensory properties and oxidative stability of nut spreads-a review
Upload
independentCategory
view
3download
0
Oxidative stress-induced apoptosis of cochlear sensory cells:otoprotective strategies
Tina Huanga, Alan G. Chenga, Howard Stupaka, Wei Liua, Ana Kima,Hinrich Staeckerc, Philippe P. Lefebvred, Brigitte Malgranged, Richard Kopkee,
Gustave Moonend, Thomas R. Van De Watera,b,d,*aDepartment of Otolaryngology, Albert Einstein College of Medicine, Bronx, New York, NY, USAbDepartment of Neuroscience, Albert Einstein College of Medicine, Bronx, New York, NY, USA
cDepartment of Otolaryngology, University of Maryland, Baltimore, MD, USAdDepartment of Human Physiology and Pathophysiology, University of Liege, Liege, Belgium
eDepartment of The Army, Naval Medical Center, San Diego, CA, USA
Received 3 March 1999; received in revised form 3 November 1999; accepted 4 November 1999
Abstract
Apoptosis is an important process, both for normal development of the inner ear and for removal of oxidative-stress damaged
sensory cells from the cochlea. Oxidative-stressors of auditory sensory cells include: loss of trophic factor support, ischemia-reperfusion, and ototoxins. Loss of trophic factor support and cisplatin ototoxicity, both initiate the intracellular production ofreactive oxygen species and free radicals. The interaction of reactive oxygen species and free radicals with membrane
phospholipids of auditory sensory cells creates aldehydic lipid peroxidation products. One of these aldehydes, 4-hydroxynonenal,functions as a mediator of apoptosis for both auditory neurons and hair cells. We present several approaches for the preventionof auditory sensory loss from reactive oxygen species-induced apoptosis: 1) preventing the formation of reactive oxygen species;(2) neutralizing the toxic products of membrane lipid peroxidation; and 3) blocking the damaged sensory cells' apoptotic
pathway. 7 2000 ISDN. Published by Elsevier Science Ltd. All rights reserved.
Keywords: Apoptosis; Auditory system; Neurons; Hair cells; Reactive oxygen species; Caspases; Calpains; Cisplatin; Trophic factor withdrawal;
Ischemia/hypoxia
1. Introduction
Apoptosis, or programmed cell death (PCD), is anactive form of cell death that often requires the syn-thesis of new proteins as is evidenced by the ability of
protein synthesis inhibitors such as cycloheximide to
block many apoptotic pathways. It occurs during nor-
mal development as well as when cells are exposed to
various insults such as toxins, ischemia/hypoxia, or
radiation. An apoptotic cell is de®ned by both its mor-
phologic and biochemical features. Such features
include: nuclear fragmentation, chromatin conden-
sation, cell shrinkage, surface blebbing with the main-
tenance of membrane integrity, formation of apoptotic
bodies, and DNA proteolysis into a distinct ``ladder''
pattern when run on an agarose gel. The remains of
the cell are eventually phagocytosed by neighboring
cells or macrophages. This is in contrast to necrosis
which is a passive form of cell death involving cellular
Int. J. Devl Neuroscience 18 (2000) 259±270
0736-5748/00/$20.00 7 2000 ISDN. Published by Elsevier Science Ltd. All rights reserved.
PII: S0736-5748(99 )00094 -5
www.elsevier.com/locate/ijdevneu
* Corresponding author. Tel.: +1-718-430-4082; fax: +1-718-430-
4258.
E-mail address: [email protected] (T.R. Van De Water).
Abbreviations: Lac, b galactosidase; BDNF, brain derived growth
factor; GSH, glutathione; GSHe, glutathione diethyl esther; HSV,
Herpes amplicon vector; HNE, 4-hydroxynonenal; Met, methionine;
NGF, nerve growth factor; NT-3, neurotrophin 3; PCD, pro-
grammed cell death; ROS, reactive oxygen species; VCN, ventral
cochlear nucleus.

swelling, organelle damage, plasma membrane break-down, and the spillage of cell contents into the inter-cellular environment. The DNA gel pattern of a celldying by necrosis consists of a smear of DNA frag-ments which is in marked contrast to the orderly lad-dering of DNA fragments seen in apoptotic DNAproteolysis Table 1 [6].
Programmed cell death can be a useful processduring morphogenesis and has been shown to be anessential process for the normal development of thevertebrate inner ear [11,13,38]. If this process of pro-grammed cell death is perturbed by transfecting thedeveloping inner ear with an anti-apoptotic molecule(e.g. a retroviral vector expressing human bcl-2) thendysmorphogenesis of the membranous labyrinth willoccur (e.g. malformed or absent semicircular ducts)[11]. There also appears to be a temporal sequence ofprogrammed cell death within the mammalian mem-branous labyrinth with programmed cell death a�ect-ing ®rst the vestibule and then the auditory sensoryepithelium with a correlation between periods of activecell proliferation and periods of apoptosis [38]. Thepattern of programmed cell death with areas of devel-oping neuroepithelial structures also appears to be cor-related with expression patterns of the low a�nityneurotrophin receptor, p75NGFR, which is a memberof the tumor necrosis factor family of cell death recep-tors. This expression pattern strongly implicates thesphingomyelin pathway as a modulator of cell deathwithin the neuroepithelium in the early stage of oticdevelopment and morphogenesis [13]. A signi®cantprogrammed cell death of ventral cochlear nucleus(VCN) neurons has also been demonstrated which co-incides with the onset of hearing immediately followinga period of rapid increase in the number of VCN neur-ons, and may be a mechanism for balancing auditoryinput of the VIIIth nerve projection and its receptor®eld within the VCN [52]. So it is evident from these
examples, i.e. programmed cell death in otic develop-ment [11,13,38], and maturation [52] that this processis indeed a naturally occurring phenomena that is arequirement for normal formation of the vertebrateinner ear and its central targets. However, once themembranous labyrinth and its central nervous systemnuclei (e.g. VCN) have completed their process ofmaturation, then the loss of auditory sensory cells dueto oxidative stress-induced apoptosis (e.g. amikacinototoxicity) can result in a permanent de®cit since theresponse of the mammalian cochlea to the apoptoticloss of sensory hair cells does not result in a successfulrenewal of the lost sensory hair cells [53], in contrastto the successful renewal of these cells that occurs fol-lowing similar ototoxic damage to the avian cochlea(basilar papilla). It is, therefore, of particular import-ance in the mammalian inner ear to preserve the exist-ing cochlear sensory cells (i.e. both hair cells andneurons) from loss due to oxidative stress-inducedapoptosis.
We review several initiators of the apoptotic path-way as it pertains to the auditory hair cells of theorgan of Corti and the auditory neurons of the spiralganglion. Three examples of oxidative stress-inducedapoptosis of auditory sensory cells are discussed: (1)neurotrophin withdrawal-induced apoptosis of thespiral ganglion neurons, (2) cisplatin exposure-inducedapoptosis of hair cells and spiral ganglion neurons,and (3) hypoxia/ischemia-induced apoptosis of haircells and spiral ganglion neurons. Otoprotective strat-egies against these di�erent types of oxidative stress-induced apoptosis are presented as well as possiblefuture investigations.
2. The role of oxidative stress damage in apoptosis
In the inner ear, damage caused by oxidative stress
Table 1
Working criteria for distinguishing apoptosis from necrosis [6]
Criterion Apoptosis Necrosis
Descriptive
Morphology Cell shrinkage with membrane preservation Formation of
apoptotic bodies
Cell swelling with early membrane
failure
DNA fragmentation Early internucleosomal cleavage Late non-speci®c breakdown
[Ca2+] Typically remains low until late in the cell death process Early increases
Interventional
Raise [Ca2+] Protective No e�ect or death promoting
Lower [Ca2+] Death promoting Protective
Block protein synthesis (e.g. with
cycloheximide)
Often protective No e�ect
Block ICE family members (e.g. with ZVAD-
FMK)
Often protective No e�ect
Alter bcl-2, bax, bad gene expression bcl-2 protective; bax and bad death promoting No e�ect
Administer neurotrophins Often protective (if receptors present) Pretreatment is death promoting
T. Huang et al. / Int. J. Devl Neuroscience 18 (2000) 259±270260

can induce apoptosis in the cochlear hair cells as wellas in the neurons of the spiral ganglion. Insults suchas neurotrophin withdrawal, ototoxin exposure, andhypoxia/ischemia lead to oxidative stress-induceddamage which then leads to apoptosis. Reactive oxy-gen species (ROS) and free radicals are formed asbyproducts of several metabolic pathways and thesemolecules can themselves cause cell damage by react-ing with cellular proteins. The reaction of ROS withthe plasma membrane leads to the formation of phos-pholipid membrane peroxidation products such as 4-hydroxynonenal (HNE), an aldehydic lipid peroxi-dation product, which is highly reactive and can leadto sensory cell damage and death [26]. Cells protectthemselves against oxidative stress via an antioxidantdefense system which utilizes free radical scavengersand other enzymes that maintain the appropriateredox state of cellular proteins. These free radical sca-vengers and antioxidants enzymes include superoxidedismutase, catalase, glutathione peroxidase, and thethiol tripeptide glutathione (GSH). Glutathione acts toreduce proteins which have been oxidized by ROS andis normally responsible for the detoxi®cation of toxicproducts of this process such as HNE. Oxidative stressto PC12 cells and mature primary hippocampal cellcultures caused a buildup of HNE intracellularly andHNE applied directly to these cells caused mitochon-drial damage and rapid apoptotic cell death of theexposed cells [26]. Other aldehydic products of mem-brane peroxidation (e.g. maliondialdehyde) did notcause the cells to exhibit signs of apoptosis [26]. HNEapplied to auditory hair cells and spiral ganglion neur-ons also caused cell death via apoptosis (unpublisheddata). Antioxidants (Ebselen) protected the cells fromoxidative stress-induced death [25], but not fromHNE-induced death indicating that HNE is down-stream from the formation of ROS. A cell permeantform of glutathione (GSHe) was able to rescue theseand other cells exposed to oxidative stress. ROS-induced damage has also been implicated in the apop-totic death of cochlear sensory cells exposed to neuro-trophin withdrawal, cisplatin administration, hypoxia/ischemia, and sound trauma. Cell permeant GSHe wasalso protective in several of these models [25].
3. The role of caspase in apoptosis
Caspases are cysteine proteases which cleave onlyafter aspartic acid residues. They are normally foundin an inactive pro-form and require cleavage by eitheranother member of the caspase family or granzyme Bfor activation. Caspases were initially linked to apop-tosis after the discovery in C. elegans of two pro-apop-totic genes critical to its developmental programmedcell death, i.e. ced-3 and ced-4, and a suppressor of
ced-3/ced-4 dependent PCD, i.e. ced-9 [14]. Bcl-2 is themammalian homologue of ced-9 [50]. Support for therole of caspases in apoptosis include the ®ndings thatcaspase activity increases in in vitro models of apopto-sis [20], overexpression of caspases induces apoptosisin mammalian cells, and caspase inhibitors can protectcells from apoptotic death [14,43].
Thirteen members of the caspase family have beenidenti®ed thus far and perhaps the reason there are somany is because of the cleavage site speci®city of thecaspases. Caspase-8 can be activated by cell deathreceptors (e.g. Fas) while caspase-9 is activated bycytotoxins and requires interaction with mitochondrialcytochrome c. Both of these caspases are thought toactivate caspase-3 which has been shown to be activein several di�erent models of apoptosis. Caspase-3 actsas a cell death e�ector molecule that fully commits thecell to the apoptotic pathway [1,51]. The fact that cas-pases are so prevalent and crucial in apoptosis [18] hasmade it an appealing target for inhibition. Not onlycan caspase inhibitors prevent apoptosis due to severaldi�erent insults, but caspase inhibition has been shownto protect neurons from direct oxidative stress-inducedapoptosis [10,20,54]. However, a recent study has ques-tioned the e�ectiveness of caspase inhibition as a neu-roprotective strategy since caspases may bedownstream of the release of mitochondrial cyto-chrome c into the cytosol [48].
4. The role of calpains in apoptosis
Another protease group that has been implicated inapoptosis are the calpains. Calpain are calcium acti-vated proteases which under normal conditions arepresent in a pro-form, and are believed to be activatedvia autolysis although some evidence shows that auto-lysis may not be necessary [45]. Calpains exist in twoforms, i.e. calpain I, activated by low calcium concen-trations (mM), and calpain II, activated by higher con-centrations (mM) of calcium in vitro [34]. While thecalcium concentrations needed in vitro for activationare higher than those found physiologically, it hasbeen found that lower calcium concentrations may beneeded to activate calpain when DNA or membranephospholipids are present [4,45]. Substrates of calpainsinclude cytoskeletal proteins (e.g. fodrin and microtu-bule associated proteins), membrane bound proteins(e.g. Ca2+-ATPase), and other cytosolic and nuclearproteins (e.g. protein kinase C, p53).
Calpains have been found to be active in severalapoptotic models including stroke, myocardial ische-mia, cytotoxin-induced apoptosis, and irradiation. Cal-pain inhibitors were also found to be protective inthese models [2,5]. The role of caspases in apoptosis iswell-established and ubiquitous in almost all cell types,
T. Huang et al. / Int. J. Devl Neuroscience 18 (2000) 259±270 261
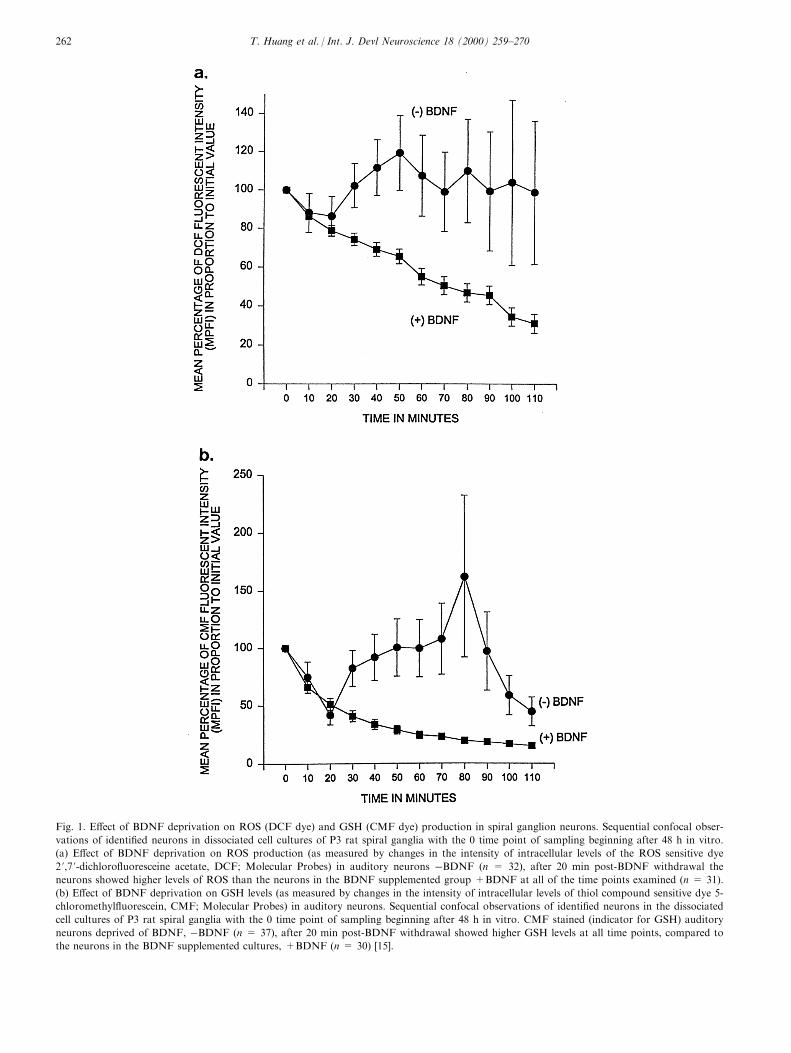
Fig. 1. E�ect of BDNF deprivation on ROS (DCF dye) and GSH (CMF dye) production in spiral ganglion neurons. Sequential confocal obser-
vations of identi®ed neurons in dissociated cell cultures of P3 rat spiral ganglia with the 0 time point of sampling beginning after 48 h in vitro.
(a) E�ect of BDNF deprivation on ROS production (as measured by changes in the intensity of intracellular levels of the ROS sensitive dye
2 ',7 '-dichloro¯uoresceine acetate, DCF; Molecular Probes) in auditory neurons ÿBDNF (n = 32), after 20 min post-BDNF withdrawal the
neurons showed higher levels of ROS than the neurons in the BDNF supplemented group +BDNF at all of the time points examined (n = 31).
(b) E�ect of BDNF deprivation on GSH levels (as measured by changes in the intensity of intracellular levels of thiol compound sensitive dye 5-
chloromethyl¯uorescein, CMF; Molecular Probes) in auditory neurons. Sequential confocal observations of identi®ed neurons in the dissociated
cell cultures of P3 rat spiral ganglia with the 0 time point of sampling beginning after 48 h in vitro. CMF stained (indicator for GSH) auditory
neurons deprived of BDNF, ÿBDNF (n = 37), after 20 min post-BDNF withdrawal showed higher GSH levels at all time points, compared to
the neurons in the BDNF supplemented cultures, +BDNF (n = 30) [15].
T. Huang et al. / Int. J. Devl Neuroscience 18 (2000) 259±270262

while the role of calpains in apoptosis may be morecell-speci®c [36,37]. One of the di�culties in establish-ing the role of calpains in apoptosis is the lack of cal-pain speci®c cell-permeant inhibitors. Calpastatin, theendogenous inhibitor, is extremely speci®c, but lackscell permeability. Newer synthetic inhibitors are morepermeant, but lack speci®city, especially when used athigh concentrations.
5. Neurotrophin withdrawal-induced apoptosis
Neurotrophins are necessary for neuronal survivalboth in the peripheral and the central nervous systems.Neurotrophins are thought to regulate neuronal survi-val and target innervation during development viadi�erential expression of neurotrophins by targetorgans and cells. NGF withdrawal from dissociatedrat superior cervical ganglia neurons results in the nearcomplete apoptotic death of these neurons. In the cen-tral nervous system, Kew et al. showed that septalcholinergic neurons undergo apoptosis when subjectedto NGF withdrawal [24]. Dugan et al. found thatNGF withdrawal produced an increased production ofROS in both superior cervical ganglia neuronal cul-tures as well as in a central nervous system-derivedneuronal cell line (GT1-1 trk) which was quickly inhib-ited by the reapplication of NGF [9]. ROS productionwas also found to increase after NGF deprivation inPC12 cells [20,43]. Of interest was the observation thattreatment of the NGF deprived PC12 cells with anantioxidant and a free radical scavenger was able toblock the NGF withdrawal-induced apoptosis. Wefound that neurotrophin withdrawal (i.e.ÿBDNF) indissociated spiral ganglion cell cultures resulted inhigher levels of intercellular ROS in the neurons thanin the neurons of the control (+BDNF) cultures andthat there was a later rise in GSH production thatoccurred within the neurons in response to an el-evation in the levels of ROS (Fig. 1) [15]. GSH acts toneutralize the deleterious e�ects of ROS within an oxi-datively stressed cell. Caspase inhibitors are also ableto block neurotrophin withdrawal-induced apoptosisin neurons. Several studies have shown protection withcaspase inhibitors in a variety of cell lines [19,43,54].In addition, not only are caspase inhibitors able toblock apoptosis, they are also able to block the for-mation of ROS [43]. This suggests that the activationof caspase is upstream to the production of ROS withthe caveat that they may be downstream of irreversiblechanges in mitochondrial viability [48]. Both neurotro-phin type 3 (NT-3) and BDNF are produced by haircells of the developing cochlea, and exert their actionsin paracrine fashion [17]. Acute withdrawal of neuro-trophins in this system leads to apoptosis [47].
Several strategies have been used in the mammalian
cochlea to prevent neurotrophin withdrawal-inducedapoptosis. Our laboratory has demonstrated that directinfusion of suprapharmalogical levels of either NT-3or BDNF into the scala tympani could rescue thespiral ganglion neurons after loss of trophic support(i.e. auditory hair cells), but the osmotic pump has alimited lifespan. The use of a Herpes simplex viral vec-tor (HSV) with a bdnf gene and a gene for the markerprotein b galactosidase (i.e. HSVbdn¯ac ) was thenused to deliver BDNF to the neurons of the spiralganglion resulting in prolonged protection of the audi-tory neurons after destruction of the cochlear hair cells(Fig. 2) [47]. Transduction of these ototoxin-damagedmouse cochleae with a marker protein gene therapyvector (i.e. HSVlac ) but lacking a gene that encodesfor a therapeutic protein (e.g. bdnf ) did not supportthe survival of the spiral ganglion neurons. An ad-ditional proof of the gene therapy vector delivery ofthe therapeutic protein (i.e. BDNF) to the neurons ofthe spiral ganglion is immunostaining of these neuronswith an anti-BDNF antibody demonstrating the pre-sence of BDNF within the somas of the HSVbdn¯actransduced neurons. Transduced neurons were ident-i®ed by the presence of the marker protein b galactosi-dase. Caspase inhibitors as well as calpain inhibitorsprotect dissociated spiral ganglion neuronal cell cul-tures when applied concurrently with the neurotrophinwithdrawal (Cheng et al., in press). Current work isaimed at determining the activity levels, types, andsequence of activation of caspases and calpains in-itiated by a loss of neurotrophic support (i.e. auditoryhair cells).
6. Cisplatin-induced apoptosis
Cisplatin is a widely used chemotherapeutic agent,but its use is limited by dose-related side e�ects suchas ototoxicity which can lead to irreversible sensori-neural hearing loss.
Cisplatin-induced hearing loss is most frequent inthe higher frequencies which is consistent with the baseto apex pattern of cochlear hair cell damage. The mostsevere damage is present in the third row of outer haircell stereocilia bundles at the basal turn of the organof Corti. The stereocilia tip links become damaged,and the stereocilia then become disarrayed and/orfused, and decrease in number [7]. At even more higherdoses, the remaining rows of outer hair cells and theinner hair cells become damaged, shrink and disap-pear. At even higher doses of cisplatin, the stria vascu-laris becomes damaged and the entire organ of Cortiand Reissner's membrane collapses [29]. Additionally,changes in auditory brainstem-evoked responses fol-lowing cisplatin exposure show signi®cant increases inhearing thresholds [29].
T. Huang et al. / Int. J. Devl Neuroscience 18 (2000) 259±270 263

Fig. 2. Toluidine blue-stained cochlea sections from control, neomycin-injected, and neomycin- injected and HSVbdn¯ac transduced (Herpes vec-
tor expressing bdnf and b galactosidase genes) mouse inner ears. (A) Control, untreated cochlea. Note that Rosenthal's canal (arrows) is ®lled
with toludine blue stained auditory neurons. The organ of Corti (arrowhead) appears normal. (B) Cochlea exposed to intracochlear injection of
neomycin showed destruction of Corti's organ (arrowhead) with loss of hair cells, and damage to the stria vascularis (S). In this cochlear speci-
men there was a signi®cant decrease in the density of auditory neurons in Rosenthal's canal (arrows) with the surviving neurons appearing
unhealthy, i.e. poorly stained with toludine blue. (C) Cochlea of a mouse that was injected with neomycin followed by injection of HSFbdn¯ac
gene therapy vector. Four weeks after injection, the spiral ganglion neurons in Rosenthal's canal (arrows) appear similar in both density and
toludine blue staining characteristics to those in Rosenthal's canal of the control mice (A). Note that the organ of Corti (arrowhead) and stria
vascularis (S) are both extensively damaged with loss of hair cells from Corti's organ [47].
T. Huang et al. / Int. J. Devl Neuroscience 18 (2000) 259±270264

Oxidative stress has also been shown to be involvedin cisplatin-induced apoptosis [15,25,41,42]. Cisplatinexposure in vivo caused an increase in lipid peroxi-dation byproducts and decreased levels of antioxidantenzyme activities in the cochlea which were normalizedby antioxidants and free radical scavengers [41,42].Ford et al. found that there may be a dissociation
between acute cisplatin ototoxicity as evidenced byendocochlear potentials and compound action poten-tials and lipid peroxidation which occurs after theacute functional damage [12]. Our laboratory foundthat cisplatin exposure of cochlear sensory cells invitro led to increased amounts of ROS and decreasedlevels of antioxidant enzyme activities [15,16,25]. Ad-
Fig. 3. P3 rat organ of Corti explants after 72 h in vitro. FITC-phalloidin-stained stereocilia bundles and cuticular plates of wholemount organo-
typic cultures viewed with a Zeiss Axiophot epi¯uorescent microscope. (a) Control explant demonstrating orderly row of IHCs with organized
stereocilia bundles (white arrow) and three rows of OHCs with V-shaped stereocilia bundles (white arrowhead). (b) An explant exposed to cispla-
tin (10 mg/ml), demonstrating a loss of a majority of its hair cells, a disrupted pattern of cytoarchitecture, and a loss of all stereocilia bundles. A
large white arrow points to the cuticular plate of a remaining hair cell. (c) An explant exposed to cisplatin plus D-Met (10ÿ2 mol/l). Three orderly
rows of OHCs are evident, as are a row of IHCs. The stereocilia bundles of both the IHCs (white arrow) and OHCs (white arrowhead) appear
to have a normal morphology. (d) An explant exposed to cisplatin plus D-Met (10ÿ3 mol/l). Overall cytoarchitecture has remained organized, but
IHCs are missing from some segments of the cochlear duct as indicated by an area bracketed by the black arrows. The white arrowhead points
to an OHC with fused and missing stereocilia. Otoprotection by D-Met at this concentration is reduced compared with the higher concentration
(i.e. 10ÿ2 mol/l) seen in the photomicrograph of (c). (e) In an explant exposed to cisplatin plus L-Nac (10ÿ2 mol/l), three rows of normal appear-
ing OHCs are evident. A row of IHCs shows some areas with missing hair cells as indicated by an area bracketed by the black arrows. The
white arrowhead points to an OHC that has lost its stereocilia bundle. (f) An explant exposed to cisplatin plus L-Nac, 10ÿ2 mol/l plus RPIA
(100 mM/l). Note the intact orderly rows of IHCs and OHCs with normal stereocilia bundles (white arrow and white arrowhead, respectively).
(Bar in (e) = 20 mm for the photomicrographs in (a)±(f) [25].
T. Huang et al. / Int. J. Devl Neuroscience 18 (2000) 259±270 265

dition of glutathione diethyl ester (GSHe) lowered theROS to control levels and resulted in signi®cant pro-tection [25]. The addition of BDNF to dissociatedspiral ganglion cell cultures, exposed to cisplatin,resulted in the protection of the auditory neurons fromapoptosis, as well as a reduction in their levels of ROS[15]. Finally, HNE was found to be a mediator of cis-platin-induced apoptosis for both auditory hair cellsand spiral ganglion neurons (unpublished data).
Cisplatin exposure of the kidney resulted in caspaseactivation, while overexpression of bcl-2 had a protec-tive e�ect on the kidney tubule cells, but caspase in-hibitors were not attempted in this model of cisplatinnephrotoxicity [49]. Our laboratory has shown thatcaspase inhibitors o�ers signi®cant protection from cis-platin-induced apoptosis for both the hair cells inorgan of Corti explants and the neurons in the disso-ciated spiral ganglion cell cultures [31]. However, treat-ment with caspase inhibitors did not protect theauditory sensory epithelium from HNE-induced apop-tosis (unpublished data). Another caveat to be con-sidered when using caspase inhibitors as anotoprotective strategy is that these inhibitors may onlyprovide short term protection from apoptotic celldeath and that the caspase inhibitor-protected cellsmay eventually die via another cell death pathway ifthey have sustained irreversible damage to their mito-chondria [48].
Another molecule shown to be protective against cis-platin-induced apoptosis is the thiol-containing aminoacid, methionine (Met), i.e. both the D- and the L-iso-forms (Reser et al. Neurotoxicology, 1999, 20, 731±748;Fig. 3) [3,16,25]. D-Met and L-Met, free radical scaven-gers, may act by several di�erent mechanisms includ-ing direct binding to cisplatin itself or by reversing thebinding of cisplatin to GSH [25]. A combination of D-Met and BDNF provided better protection than eitherof these agents by themselves [16]. However, a caveatwhen considering the use of either L-Met or D-Met asan otoprotective strategy for cisplatin chemotherapy isits apparent ability to directly bind to cisplatin. Thismay limit the e�ectiveness of systemic administrationof this otoprotective aminoacid, however, local appli-cation (e.g. L-Met round window membrane) may bean e�ective otoprotective strategy that does not inter-fere with cisplatin's chemotherapeutic value while pro-viding protection against cisplatin ototoxic damage(unpublished results).
7. Ischemia/hypoxia and noise-induced apoptosis
It has long been thought that neuronal cell death-induced by ischemia was due to necrosis. The neuronsin the core of the ischemic infact undergo hyperpolar-ization and a calcium in¯ux which is thought to be
mediated by the release of excitotoxic molecules. Theischemic neurons exhibited swelling of their somas anddendrites with breakdown of the plasma and internalcellular organelle membranes. However, recent evi-dence also points to a role for apoptosis in ischemic/hypoxic cell death, particularly in the area of thepenumbra, i.e. the area surrounding the core of theinfarct. Apoptosis, most likely, is also responsible fordelayed neuronal death following transient globalischemia as well as neuronal death in transient focalischemia [35,40]. Gottron et al. found that cortical cellcultures exposed to conditions of transient ischemiadisplayed characteristics of both excitotoxicity-inducednecrosis as well as ischemia-induced apoptosis [19].Ischemia in vitro is usually mimicked by combininghypoxia with glucose deprivation of the exposed cells.Mild hypoxic insults result in the apoptosis of exposedneurons, and more closely mimic the conditions withinthe ischemic penumbra [5].
Oxidative stress has been shown to be involved inthe damage to nervous system cellular componentssuch as neurons, microglia, and astrocytes in ischemia/hypoxia-induced apoptosis during reperfusion[32,33,46]. An increase in ROS was found when tissueswere reperfused in vivo or returned to normoxic con-ditions in vitro. Studies on transgenic mice expressingincreased levels of antioxidants (i.e. SOD+) showed anattenuation of DNA degradation, membrane lipid per-oxidation, and neuronal death [8,23]. Kruman et al.found evidence that HNE production occurred duringmembrane lipid peroxidation phase of cell damage in-itiated by oxidative stress (e.g. ROS), and was a me-diator of resultant apoptosis [26]. As a corollary tothis ®nding, Kuntsmann et al. demonstrated that capil-lary endothelial cells in the brain have an increasedability to degrade HNE which they propose may be aneuroprotective strategy in the CNS against ischemiaand hypoxia [5,27].
Caspases and calpains have both been implicated inthe ischemic/hypoxic apoptotic pathway. Caspase in-hibitors have been shown to be e�ective in protectingprimary cultures of rat cortical neurons from hypoxiaas well as in in vivo models of transient global andfocal ischemia [36,49]. Calpain inhibitors have beenshown to be protective in both in vitro as well as invivo models of ischemia/hypoxia [25,21].
In the ear, hypoxia is used to model some of thedeleterious e�ect on the ear produced by noise-inducedtrauma. Lamm and Arnold found that the partialpressure of oxygen as well as cochlear blood ¯owdecreased during broad-band noise exposure and Pujolet al. showed that the acute damage caused in theorgan of Corti by either hypoxia or loud noise ex-posure was similar [28,39]. Noise exposure has beenlinked to oxidative stress [28], in particular the acti-vation of the GSH defense system [30]. GSH was
T. Huang et al. / Int. J. Devl Neuroscience 18 (2000) 259±270266
found to increase in the lateral wall of the cochleaafter loud noise exposure and an inhibitor of GSHsynthesis increased hearing loss while a GSH replen-isher (N6 phenylisopropyl adenosine, RPIA) decreasedhearing loss [55,56]. Our laboratory has shown thatGSH antioxidant enzyme systems are activated duringsound conditioning. Recent work has demonstrated
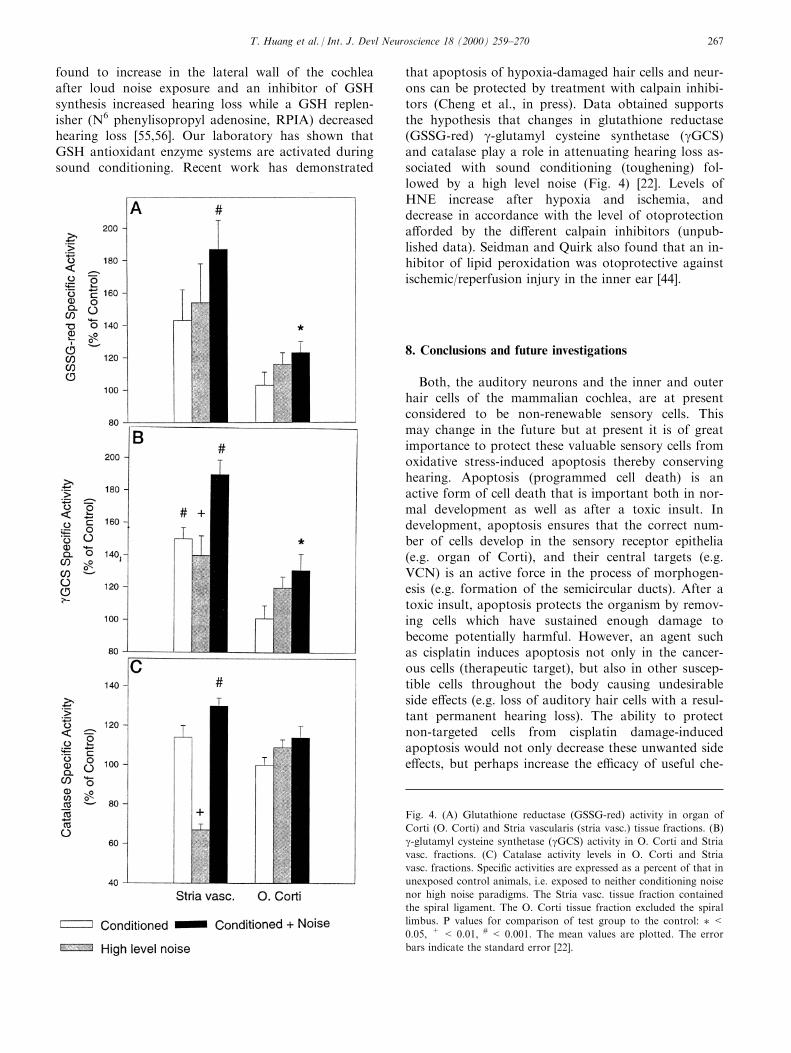
that apoptosis of hypoxia-damaged hair cells and neur-ons can be protected by treatment with calpain inhibi-tors (Cheng et al., in press). Data obtained supportsthe hypothesis that changes in glutathione reductase(GSSG-red) g-glutamyl cysteine synthetase (gGCS)and catalase play a role in attenuating hearing loss as-sociated with sound conditioning (toughening) fol-lowed by a high level noise (Fig. 4) [22]. Levels ofHNE increase after hypoxia and ischemia, anddecrease in accordance with the level of otoprotectiona�orded by the di�erent calpain inhibitors (unpub-lished data). Seidman and Quirk also found that an in-hibitor of lipid peroxidation was otoprotective againstischemic/reperfusion injury in the inner ear [44].
8. Conclusions and future investigations
Both, the auditory neurons and the inner and outerhair cells of the mammalian cochlea, are at presentconsidered to be non-renewable sensory cells. Thismay change in the future but at present it is of greatimportance to protect these valuable sensory cells fromoxidative stress-induced apoptosis thereby conservinghearing. Apoptosis (programmed cell death) is anactive form of cell death that is important both in nor-mal development as well as after a toxic insult. Indevelopment, apoptosis ensures that the correct num-ber of cells develop in the sensory receptor epithelia(e.g. organ of Corti), and their central targets (e.g.VCN) is an active force in the process of morphogen-esis (e.g. formation of the semicircular ducts). After atoxic insult, apoptosis protects the organism by remov-ing cells which have sustained enough damage tobecome potentially harmful. However, an agent suchas cisplatin induces apoptosis not only in the cancer-ous cells (therapeutic target), but also in other suscep-tible cells throughout the body causing undesirableside e�ects (e.g. loss of auditory hair cells with a resul-tant permanent hearing loss). The ability to protectnon-targeted cells from cisplatin damage-inducedapoptosis would not only decrease these unwanted sidee�ects, but perhaps increase the e�cacy of useful che-
Fig. 4. (A) Glutathione reductase (GSSG-red) activity in organ of
Corti (O. Corti) and Stria vascularis (stria vasc.) tissue fractions. (B)
g-glutamyl cysteine synthetase (gGCS) activity in O. Corti and Stria
vasc. fractions. (C) Catalase activity levels in O. Corti and Stria
vasc. fractions. Speci®c activities are expressed as a percent of that in
unexposed control animals, i.e. exposed to neither conditioning noise
nor high noise paradigms. The Stria vasc. tissue fraction contained
the spiral ligament. The O. Corti tissue fraction excluded the spiral
limbus. P values for comparison of test group to the control: � <0.05, + < 0.01, # < 0.001. The mean values are plotted. The error
bars indicate the standard error [22].
T. Huang et al. / Int. J. Devl Neuroscience 18 (2000) 259±270 267

motherapeutic agents such as cisplatin, thereby widen-ing its therapeutic window.
Oxidative stress with the production of ROS andfree radicals and the activation of apoptotic proteases,e.g. caspases and calpains, have been shown to beinvolved in apoptosis. These mechanisms have severaldi�erent points at which inhibition could be targetedto protect cells from apoptosis. To prevent oxidativestress-induced apoptosis, strategies for protectioninclude: (1) preventing the formation of ROS either bybinding the toxin (e.g. L-Met interaction with cisplatin)or replacing the missing neurotrophic factors (e.g.HSVbdn¯ac, a gene therapy vector) in the case of neu-rotrophin withdrawal, (2) preventing the toxic e�ectsof lipid peroxidation byproducts (e.g. GSHe neutraliz-ation of HNE), (3) adding exogenous free radical sca-vengers and antioxidant enzymes and molecules (e.g.overexpressing SOD) to prevent ROS interactions withcellular proteins, and (4) increasing the activity of theendogenous antioxidant system (e.g. treatment with L-Nac). Caspase and calpain activation may be inhibitedby adding exogenous inhibitors of these cell death-e�ector proteases or by using gene therapy to upregu-late anti-apoptotic gene products such as bcl-2 (e.g.HSVbcl2 gene therapy vector). In the inner ear, insultssuch as neurotrophin withdrawal, sound trauma, hy-poxia/ischemia, ototoxic antibiotics, and ototoxic che-motherapeutic agents have all been shown to induceapoptosis both in the auditory hair cells as well as inthe neurons of the spiral ganglion. Previous exper-iments have shown that all the strategies outlinedabove can be used to protect the auditory system fromapoptosis.
Future studies include de®ning the apoptotic path-ways activated by di�ering insults to the auditory sys-tem (e.g. Fas pathway vs. JNK/c-Jun pathway) in anattempt to ®nd other targets for inhibition. We havefound that caspases and calpains may be involved indi�erent apoptotic pathways or that they may be acti-vated at di�erent points along the apoptotic pathway(Cheng et al., Brain Research, 1999, 850, 234±243).Since the cysteine proteases are thought to act as acascade, we hope to ®nd the most upstream caspase inthe cascade (e.g. caspase 8) and target that caspase forinhibition by a speci®c inhibitor. Also, while inhibitorsare often added prior to or concurrent with the insult,most clinical interventions occur after an insult. Thenext step after in vitro experiments is to move to invivo experiments in pursuit of the ®nal goal of design-ing therapeutic interventions for human use. We arehopeful that some of the molecular strategies outlinedabove will eventually be useful for protection againstoxidative stress-induced human hearing loss. Rescue ofdamaged cochlear sensory cells that still possess thepotential for long term survival may also provide theopportunity to use growth factor therapy in combi-
nation with otoprotection to initiate self-repair andsubsequently return the damaged sensory cell to fullfunction.
Acknowledgements
This work was supported by the Hearing ResearchFund of Monte®ore Medical Center to TRV, andAlbert Einstein College of Medicine NeuropathologyTraining grant NS07098 support to Tina Huang, AlanG. Cheng, Howard Stupak, and Ana Kim.
References
[1] Barinaga, M., Death by dozens of cuts. Science, 1998, 280, 32±
34.
[2] Bartus, R. T., Hayward, N. J., Elliott, P. J., Sawyer, S. D.,
Baker, K. L., Dean, R. L., Akiyama, A., Straub, J. A.,
Harbeson, S. L., Li, Z. and Powers, J., Calpain inhibitor
AK295 protects neurons from focal brain ischemia. Stroke,
1994, 25, 2265±2270.
[3] Campbell, K. C., Rybak, L. P., Meech, R. P. and Hughes, L.,
D-methionine provides excellent protection from cisplatin oto-
toxicity in the rat. Hear. Res, 1996, 102, 90±98.
[4] Carafoli, E. and Molinari, M., Calpain: a protease in search of
a function? Biochem. Biophys. Res. Comm, 1998, 247, 193±203.
[5] Chen, Z., Schottler, F. and Lee, K. S., Neuronal recovery after
moderate hypoxia is improved by the calpain inhibitor
MDL28170. Brn. Res, 1997, 769, 188±192.
[6] Choi, D., Ischemia-induced neuronal apoptosis. Curr. Opin.
Neurobiol, 1996, 6, 667±672.
[7] Comis, S. D., Rhys-Evans, P. H., Osborne, M. P., Pickles, J.
O., Je�ries, D. J. R. and Pearse, H. A. C., Early morphological
and chemical changes induced by cisplatin in the guinea pig
organ of Corti. J. Laryngol. Otol, 1986, 100, 1375±1383.
[8] Copin, J. C., Reola, L. F., Chan, T. Y., Li, Y., Epstein, C. J.
and Chan, P. H., Oxygen deprivation but not a combination of
oxygen, glucose, and serum deprivation induces DNA degra-
dation in mouse cortical neurons in vitro: attenuation by trans-
genic overexpression of Cu Zn-superoxide dismutase. J.
Neurotrauma, 1996, 13, 233±244.
[9] Dugan, L. L., Creedon, D. J., Johnson, E. M. Jr. and
Holtzman, D. M., Rapid suppression of free radical formation
by nerve growth factor involves the mitogen-activated protein
kinase pathway. Proc. Natl. Acad. Sci. USA, 1997, 94, 4086±
4091.
[10] Endres, M., Namura, S., Shimizu-Sasamata, M., Waeber, C.,
Zhang, L., Gomez-Isla, T., Hyman, B. T. and Moskowitz, M.
A., Attenuation of delayed neuronal death after mild focal
ischemia in mice by inhibition of the caspase family. J. Cerebral
Blood Flow and Met, 1998, 18, 238±247.
[11] Fekete, D. M., Homburger, S. A., Waring, M. T., Riedl, A. E.
and Garcia, L. F., Involvement of programmed cell death in
morphogenesis of the vertebrate inner ear. Development, 1997,
124, 2451±2461.
[12] Ford, M. S., Nie, Z., Whitworth, C., Rybak, L. P. and
Ramkumar, V., Up-regulation of adenosine receptors in the
cochlea by cisplatin. Hear. Res, 1997, 111, 143±152.
[13] Frago, L. M., Leon, Y., de la Rosa, E., Gomez-Nunoz, A. and
Varela-Nieto, I., Nerve growth factor and ceramides modulate
cell death in early developing inner ear. J. Cell Sci, 1998, 111,
549±556.
T. Huang et al. / Int. J. Devl Neuroscience 18 (2000) 259±270268

[14] Fraser, A., McCarthy, N. and Evans, G. I., Biochemistry of cell
death. Curr. Opin. Neurobiol, 1996, 6, 71±80.
[15] Gabaizadeh, R., Staecker, H., Liu, W. and Van De Water, T.
R., BDNF protection of auditory neurons from cisplatin
involves changes in intracellular levels of both reactive oxygen
species and glutathione. Mol. Brn. Res, 1997, 50, 71±78.
[16] Gabaizadeh, R., Staecker, H., Liu, W., Kopke, R. D.,
Malgrange, B., Lefebvre, P. P. and Van De Water, T. R.,
Protection of both auditory hair cells and auditory neurons
from cisplatin-induced damage. Acta Otolaryngol. (Stockholm),
1997, 117, 232±238.
[17] Garrido, J. J., Alonso, M. T., Lim, F., Carnicero, E., Giraldez,
F. and Schimmang, T., De®ning responsiveness of avian
cochlear neurons to brain-derived neurotrophic factor and nerve
growth factor by HSV-1-mediated gene transer. J. Neurochem,
1998, 70, 2336±2346.
[18] Gorman, A. M., Orrenius, S. and Ceccatelli, S., Apoptosis in
neuronal cells: role of caspase. NeuroReport, 1998, 9, R49±R55.
[19] Gottron, F. J., Ying, H. S. and Choi, D. W., Caspase inhibition
selectively reduces the apoptotic component of oxygen-glucose
deprivation-induced cortical neuronal cell death. Mol. Cell.
Neurosci, 1997, 9, 159±169.
[20] Haviv, R., Lindenboim, L., Li, H., Yuan, J. and Stein, R.,
Need for caspases in apoptosis of trophic factor-deprived PC12
cells. J. Neurosci. Res, 1997, 50, 69±80.
[21] Hong, S., Goto, Y., Lanzino, G., Soleau, S., Kassell, N. F. and
Lee, K. S., Neuroprotection with a calpain inhibitor in a model
of focal cerebral ischemia. Stroke, 1994, 25, 663±669.
[22] Jacono, A. A., Bohua, H., Kopke, R. D., Henderson, D., Van
De Water, T. R. and Steinman, H. M., Changes in cochlear
antioxidant enzyme activity after sound conditioning and noise
exposure in the chinchilla. Hear. Res, 1998, 117, 31±38.
[23] Keller, J. N., Kindy, M. S., Holtsberg, F. W., StClair, D. K.,
Yen, H. C., Germeyer, A., Steiner, S. M., Bruce-Keller, A. J.,
Hutchins, J. B. and Mattson, M. P., Mitochondrial manganese
superoxide dismutase prevents neural apoptosis and reduces
ischemic brain injury: suppression of peroxynitrite production,
lipid peroxidation, and mitochondrial dysfunction. J. Neurosci,
1998, 18, 687±697.
[24] Kew, J. N. C., Smith, D. W. and Sofroniew, M. V., Nerve
growth factor withdrawal induces the apoptotic death of devel-
oping septal cholinergic neurons in vitro: protection by cyclic
AMP analogue and high potassium. Neurosci, 1996, 70, 329±
339.
[25] Kopke, R. D., Liu, W., Gabaizadeh, R., Jacono, A., Feghali, J.,
Spray, D., Garcia, P., Steinman, H., Malgrange, B., Ruben, R.
J., Rybak, L. and Van De Water, T. R., Use of organotypic cul-
tures of Corti's organ to study the protective e�ects of antioxi-
dant molecules on cisplatin-induced damage of auditory hair
cells. Am. J. Otol, 1997, 18, 559±571.
[26] Kruman, I., Bruce-Keller, A. J., Bredesen, D., Waeg, G. and
Mattson, M. P., Evidence that 4-hydroxynonenal mediates oxi-
dative stress-induced apoptosis. J. Neurosci, 1997, 17, 5089±
5100.
[27] Kuntsmann, S., Mertsch, K., Blasig, L. E. and Grune, T., High
metabolic rates of 4-hydroxynonenal in brain capillary endo-
thelial cells during hypoxia/reoxygenation. Brn. Res, 1996, 740,
353±355.
[28] Lamm, K. and Arnold, W., Noise-induced cochlear hypoxia is
intensity dependent, correlates with hearing loss and precedes
reduction of cochlear blood ¯ow. Audiol. Neurootol, 1996, 1,
148±160.
[29] Laurell, G., Teixeira, M., Sterkers, O. and Ferrary, E., E�ect of
cisplatin administration on the electrochemical composition of
endolymph in the rat cochlea. Hear. Res, 1995, 87, 16±20.
[30] Lautermann, J., Crann, S. A., McLaren, J. and Schacht, J.,
Glutathione-dependent antioxidant systems in the mammalian
inner ear: e�ects of aging, ototoxic drugs and noise. Hear. Res,
1997, 114, 75±82.
[31] Liu, W., Staecker, H., Stupak, H., Malgrange, B., Lefebvre, P.
and Van De Water, T. R., Caspase inhibitors prevent cisplatin-
induced apoptosis of auditory sensory cells. NeuroReport, 1998,
9, 2609±2614.
[32] Maeda, Y., Matsumoto, M., Hori, O., Kuwabara, K., Ogawa,
S., Yan, S. D., Ohtsuki, T., Kinoshita, T., Kamada, T. and
Stern, D. M., Hypoxia/reoxygenation-mediated induction of
astrocyte interleukin 6: a paracrine mechanism potentially
enhancing neuron survival. J. Exp. Med, 1994, 180, 2280±2297.
[33] McCarthy, M. J., Rubin, L. L. and Philpott, K. L.,
Involvement of caspases in sympathetic neuron apoptosis. J.
Cell Sci, 1997, 110, 2165±2173.
[34] Mehdi, S., Cell-penetrating inhibitors of calpain. TIBS, 1991,
16, 150±153.
[35] Namura, S., Zhu, J., Fink, K., Endres, M., Srinivasan, A.,
Tomaselli, K. J., Yuan, J. and Moskowitz, M. A., Activation
and cleavage of caspase-3 in apoptosis induced by experimental
cerebral ischemia. J. Neurosci, 1998, 18, 3659±3668.
[36] Nath, R., Probert, A. Jr., McGinnis, K. M. and Wang, K. K.,
Evidence for activation of caspase-3-like protease in excitotoxin-
and hypoxia/hypoglycemia-injured neurons. J. Neurochem,
1998, 71, 186±195.
[37] Nath, R., Raser, K. J., McGinnis, K., Nadimpalli, R., Sta�ord,
D. and Wang, K. K., E�ects of ICE-like protease and calpain
inhibitors on neuronal apoptosis. NeuroReport, 1996, 8, 249±
255.
[38] Nishizake, K., Anniko, M., Orita, Y., Karita, K., Masuda, Y.
and Yoshino, T., Programmed cell death in the developing epi-
thelium of the mouse inner ear. Acta Otolaryngol. (Stockholm),
1998, 118, 96±100.
[39] Pujol, R., Rebillard, G., Puel, J. L., Lenoir, M., Eybalin, M.
and Recasens, M., Glutamate neurotoxicity in the cochlea: a
possible consequence of ischaemic or anoxic conditions occur-
ring in ageing. Acta Otolaryngol. (Stockholm), 1990, 476, 32±
36.
[40] Rosenbaum, D. M., Michaelson, M., Batter, D. K., Doshi, P.
and Kessler, J. A., Evidence for hypoxia-induced, programmed
cell death of cultured neurons. Ann. Neurol, 1994, 36, 864±870.
[41] Rybak, L. P., Husain, K., Evenson, L., Morris, C., Whitworth,
C. and Somani, S. M., Protection by 4-methylthiobenzioc acid
against cisplatin-induced ototoxicity: antioxidant system.
Pharmacol. and Toxicol, 1997, 81, 173±179.
[42] Rybak, L. P., Ravi, R. and Somani, S. M., Mechanism of pro-
tection by diethyldithiocarbamate against cisplatin ototoxicity:
antioxidant system. Fund. and App. Toxicol, 1995, 26, 293±300.
[43] Schulz, J. B., Bremen, D., Reed, J. C., Lommatzsch, J.,
Takayama, S., Wullner, U., Loschmann, P., Klockgether, T.
and Weller, M., Cooperative interception of neuronal apoptosis
by Bcl-2 and Bag-1 expression: prevention of caspase activation
and reduced production of reactive oxygen species. J.
Neurochem, 1997, 69, 2075±2086.
[44] Seidman, M. D. and Quirk, W. S., The protective e�ects of tiri-
lated mesylate (U74006F) on ischemic and reperfusion-induced
cochlear damage. Otolaryngol. H and N Surg, 1991, 105, 511±
516.
[45] Sorimachi, H., Ishiura, S. and Suzuki, K., Structure and physio-
logical function of calpains. Biochem. J, 1997, 328, 721±732.
[46] Spranger, M., Kiprianova, I., Krempien, S. and Schwab, S.,
Reoxygenation increases the release of reactive oxygen inter-
mediates in murine microglia. J. Cerebral Blood Flow and Met,
1998, 18, 670±674.
[47] Staecker, H., Gabaizadeh, R., Federo�, H. and Van De Water,
T. R., Brain-derived neurotrophic factor gene therapy prevents
spiral ganglion degeneration after hair cell loss. Otolaryngol. H
and N Surg, 1998, 119, 7±13.
T. Huang et al. / Int. J. Devl Neuroscience 18 (2000) 259±270 269

[48] Stefanis, L., Park, D. S., Friedman, W. and Greene, L. A.,
Caspase-dependent and -independent death of camptothecin-
treated embryonic cortical neurons. J. Neurosci, 1999, 19, 6235±
6247.
[49] Takeda, M., Kobayashi, M., Shirato, I., Osaki, T. and Endou,
H., Cisplatin-induced apoptosis of immortalized mouse proxi-
mal tubule cells is mediated by interleukin-1 beta converting
enzyme (ICE) family of proteases but inhibited by overexpres-
sion of Bcl-2. Arch. Toxicol, 1997, 71, 612±621.
[50] Tamatani, M., Ogawa, S. and Tohyama, M., Roles of Bcl-2 and
caspases in hypoxia-induced neuronal cell death: a possible neu-
roprotective mechanism of peptide growth factors. Mol. Brn.
Res, 1998, 58, 27±39.
[51] Thornberry, N. A. and Lazebnik, Y., Caspases: enemies within.
Science, 1998, 281, 1312±1316.
[52] Tierney, T. S. and Moore, D. R., Naturally occurring neuron
death during postnatal development of the gerbil ventral
cochlear nucleus begins at the onset of hearing. J. Comp.
Neurol, 1997, 387, 421±429.
[53] Vago, P., Humbert, G. and an Lenoir, M., Amikacin intoxi-
cation induces apoptosis and cell proliferation in rat organ of
Corti. NeuroReport, 1998, 9, 431±436.
[54] Troy, C. M., Stefanis, L., Prochiantz, A., Greene, L. A.,
Shelanski, Villa P., Kaufmann, S. H. and Earnshaw, W. C.,
Caspases and caspase inhibitors. TIBS, 1997, 22, 328±393.
[55] Yamasoba, T., Harris, C., Shoji, F., Lee, R. J., Nuttall, A. L.
and Miller, J. M., In¯uence of intense sound exposure on
glutathione synthesis in the cochlea. Brn. Res, 1998, 804,
72±78.
[56] Yamasoba, T., Nuttall, A. L., Harris, C., Raphael, Y. and
Miller, J. M., Role of glutathione in protection against noise-
induced hearing loss. Brn. Res, 1998, 784, 82±90.
T. Huang et al. / Int. J. Devl Neuroscience 18 (2000) 259±270270
Copyright © 2022 FDOKUMEN




















