
Separation of phenyl acetic acid and 6-aminopenicillanic acid ...
Upload
independentCategory
view
1download
0
This article was published in an Elsevier journal. The attached copyis furnished to the author for non-commercial research and
education use, including for instruction at the author’s institution,sharing with colleagues and providing to institution administration.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Kinetics and mechanism of oxidation of S(IV)by the ethylenebisbiguanide Ag(III) ion q
Rupa Shah, Vimal Soni, Aditya Prakash, Raj N. Mehrotra *
Department of Chemistry, JNV University, Jodhpur 342 005, India
Received 17 February 2007; accepted 14 June 2007Available online 4 July 2007
Abstract
The reaction, first-order both in Ag(H2L)3+ and S(IV), is not affected by dissolved oxygen and the rate is retarded by the hydrogenion. The rate law in Eq. (i) is deduced from the mechanism given below where kobs is the pseudo first-order rate constant([S(IV)]� [Ag(H2L)]3+), and k1 and k2 are composite rate constants.
kobs ¼ ðk1 þ k2½Hþ��1Þ½SðIVÞ� ðiÞ
SO2ðaqÞ�Ka1
HSO�3 þHþ ðiiÞ
AgðH2LÞ3þ þ SO2ðaqÞ�K1
AgðH2LÞSO3þ2 ðiiiÞ
AgðH2LÞ3þ þHSO�3 �K2
AgðH2LÞHSO2þ3 ðivÞ
AgðH2LÞSO3þ2 !
k0HSO�3 þAgðH2LÞ2þ þHþ ðvÞ
AgðH2LÞHSO2þ3 !
k00HSO�3 þAgðH2LÞ2þ ðviÞ
AgðH2LÞ2þ þHSO�3 þH2O!fastAgðH2LÞþ þHSO�4 þ 2Hþ ðviiÞ
where k1 ¼ k0K1 and k2 ¼ k00K1K2 in Eq. (i). The inner-sphere mechanism is supported by similar activation parameters reported in otheroxidations for similar reaction pathways.� 2007 Elsevier Ltd. All rights reserved.
Keywords: Kinetics; Mechanism; Oxidation; Sulfite; Redox reaction; Ethylenebis(biguanide)Ag(III)-complex
1. Introduction
Silver(III) is stabilised in the [Ag(H2IO6)2]3�, [Ag-(H2TeO6)2]5� and [Ag(OH)4]� ions in alkaline medium[1]. The square planar [Ag(OH)4]� ion has been studiedextensively [2]. Ag(III)-tetraglycinate [3] and Ag(III)-dim-
ethylglyoximate [4], prepared from [Ag(OH)4]�, are lessstudied because of their limited stability.
The ethylexnebis(biguanide)silver(III) nitrate, [Ag(C6N10-H16)](NO3)3, hereafter abbreviated to [Ag(H2L)]3+, is sta-ble in both strongly acidic and alkaline media. It is freelysoluble at pH < 4.5 and sparingly soluble at 4.5 < pH > 8[5]. It is a milder oxidant than Ag(I) [6], and has been usedin the oxidation of both organic and inorganic molecules[6–9].
The redox chemistry of S(IV) is interesting. Transitionmetal ions are known to catalyse the oxidation and the
0277-5387/$ - see front matter � 2007 Elsevier Ltd. All rights reserved.
doi:10.1016/j.poly.2007.06.020
q This work was supported by the UGC vide grant F.12-59/1997 andF.12-147/2001.
* Corresponding author. Tel.: +91 2912721478.E-mail address: [email protected] (R.N. Mehrotra).
www.elsevier.com/locate/poly
Polyhedron 26 (2007) 4809–4817

Author's personal copy
subject has been reviewed [10]. The SO42� anion is the
exclusive product in the oxidation with substitution inertcomplexes whereas both SO4
2� and S2O62� ions are the
products in the oxidation by labile complexes [11]. The oxi-dation of S(IV) has been extensively studied mainly by one-electron oxidants1 The oxidation by Os(bpy)3
3+ [12],Cu(III) [13], dodecatungstocobaltate(III) [14] and Fe3+(aq)[15] are sensitive to dissolved oxygen in the distilled waterwhereas no such effect is observed in the oxidations byalkaline MnO4
� [16], [Mn(acac)3] (Hacac = acetylacetone)[17], [Mn(III)(cydta)] [18] and in the presence of excessFe3+(aq) ion [19].
Oxidation with the Tl3+(aq) ion [20] involved the forma-tion of weak sulfite-complexes whereas the oxidation withAuCl4
� and AuCN2X2� (X = Cl, Br) ions [21] proceeded
directly. These ions did not participate in any catalyticcycle [22,23], and a free radical chain was not initiated asin the photo induced auto-oxidation of S(IV) [24]. It wasconsidered imperative to examine the oxidation by the[Ag(H2L)]3+ ion with the view of whether it binds itselfwith S(IV), since its electron affinity is high [25] and struc-turally it can have axial coordination [26] as reported in theoxidation of H2O2 [27] and aliphatic and aromatic alcohols[28].
2. Experimental
2.1. Materials
The orange red ethylenebis(biguanide)silver(III) nitrate,Ag(H2L)(NO3)3, was prepared using the method of Rayand Chakravorty [29]. The purity of the prepared sampleand its concentration in solution was checked with aHP8452A spectrophotometer2 (kmax 372 nm, e = 1205;kmax 370 nm, e = 1210 in solution 0.01 < [H+] < 0.1mol dm�3 [30]).
The suitably diluted HClO4 (E. Merck, GR) was stan-dardised against a standard alkali solution. The LiClO4
solution was prepared by neutralising LiOH (Sigma) bystandard HClO4 and standardised as described [31]. Thesolutions of Na2SO3 (Baker, AR) were freshly preparedand standardised as described earlier [18]. The estimatedconcentration includes the concentrations of the SO2(aq),HSO3
� and SO32� species and is represented by [S(IV)].
All the chemicals used were either analytical or purifiedgrade and all the solutions were prepared in twice-distilledwater.
2.2. Kinetic measurements
Initially solutions of Ag(H2L)3+ and Na2SO3 werefreshly prepared in distilled water degassed with nitrogen
for a sufficient time and diluted suitably with stock solu-tions of degassed HClO4 and LiClO4 to have the desiredconcentrations. However, the solutions were not degassedlater since the rate constants from the degassed and non-degassed solutions agreed within the experimental limits(3–4%). The S(IV) solution was used within 2 h of prepara-tion because such a solution in perchloric acid is stable overthis period [20]. The ionic strength was maintained at0.5 mol dm�3 with LiClO4 unless stated otherwise.
A photoelectric SFA 11 colorimeter, working on theprinciple of a stopped-flow instrument and connected toa programmable printer to print the absorbance at fixedintervals of time, was used to measure the rate. The temper-ature of the stock syringes, filled with the reactant solu-tions, and the cell compartment was maintained at thedesired temperature (±0.1 �C) by circulating water from aHaake D8G refrigerated circulatory water bath. The reac-tant solutions were manually pushed into the reaction cell.The start switch was activated by the piston of the syringecollecting the waste solution from the reaction cell.
The kinetics, under pseudo first-order reaction condi-tions ([S(IV)]� [Ag(H2L)]3+), were studied by followingthe disappearance of Ag(H2L)3+ at 420 nm [17], at whichthe Ag(III) complex absorbs and the S(IV) solution istransparent. The absorbance of the reaction mixtures atinfinite time was measured the next day.
Each run was studied at least thrice. The plots ofln(At � A1) against time, obtained by using the EXCEL PRO-
GRAM, were linear for more than three half-lives of the reac-tion. The kobs (s�1) value was computed from the slope ofthe plots. The deviation between the mean kobs value,reported in the Tables, and those obtained from multipleruns was within ±3–4%.
2.3. Test for free radical
Acrylonitrile (1–2 ml) was added to each of the reactantsolutions purged with nitrogen. The solutions, remainingclear over few minutes, were mixed to initiate the reaction.The appearance of a white precipitate in the reaction mix-ture indicated the formation of free radicals during thecourse of the reaction.
2.4. Stoichiometry
In one set of experiments the stoichiometry was studiedunder the kinetic conditions (excess of [S(IV)]) and in theother set [Ag(H2L)3+] was taken in slight excess over[S(IV)]. Each set consisted of several reaction mixtures withdifferent [H+], and these were left for sufficient time in glassstoppered flasks to ensure completion of the reaction. Theunreacted S(IV) and Ag(H2L)3+ were estimated volumetri-cally [18], and spectrophotometrically at 370 nmrespectively.
Reaction mixtures with similar concentrations were pre-pared in another set of experiments and after the comple-tion of the reaction, nitrogen was bubbled through the
1 The exhaustive reference to the studies by various one-electronoxidants is given in Ref. [20].
2 The instrument could not be used for the kinetic study because its cellcompartment could not be thermostated for a constant temperature.
4810 R. Shah et al. / Polyhedron 26 (2007) 4809–4817

Author's personal copy
spent reaction mixtures for sufficient time to expel excessSO2 Æ H2O. Thereafter a slight excess of BaClO4 solutionwas added to precipitate the SO4
2� ion for its gravimetricestimation. The estimated amount of SO4
2� ion from thesesets agreed within (99.8 ± 0.6)%.
The results based on the estimation of unreacted S(IV)and Ag(III) and SO4
2� ion indicated a stoichiometric ratio,D [Ag(enbbg)3+]/D[S(IV)], of 1.02 ± 0.02 and therefore thestoichiometry is represented by Eq. (1).
AgðH2LÞ3þ þH2SO3 þH2O! AgðH2LÞþ þ SO2�4 þ 4Hþ
ð1Þ
2.5. Absence of Ag+ and free ethylenebis(biguanide) in the
spent reaction mixtures
The addition of NaCl solution to the spent reaction mix-tures did not result in the precipitation of AgCl indicatingthat the free Ag+ ion was absent as a reaction product. Asimilar absence of a precipitate in the reaction mixturestreated with CuSO4 solution was an indication that freeethylenebis(biguanide) was also absent in the spent reac-tion mixture. Hence, it is concluded that the reductionproduct of Ag(H2L)3+ is the Ag(H2L)+ ion.
2.6. Absence of S2O62� in the reaction product
The spent reaction mixtures with a deficient amount ofS(IV) were treated with a known amount of Cr(VI) solu-tion in 1 mol dm�3 H2SO4. The Cr(VI) in these reactionmixtures was estimated at 348 nm [32] after some time.The results of estimation indicated that no Cr(VI) was usedby the reaction mixtures. It was, therefore, concluded thatno detectable amount of S2O6
2� ion was produced duringthe reaction. This method is suitable for the detection ofS2O6
2� concentrations as low as 5 · 10�6 mol dm�3.
2.7. Spectral studies
The rapid scanning of the reaction mixture (105[Ag-(H2L)]3+ = 7.0, 103[S(IV)] = 1.5 and [HClO4] = 0.2 moldm�3) on a HP UV–Vis 8452A spectrophotometer was sim-ilar to that of Ag(H2L)3+ solution of the same concentrationat 370 and 420 nm. This indicated that either there is no for-mation of complex(es) between [Ag(H2L)]3+ and [S(IV)] orthe complexes were weak if such complexes were formed.
2.8. Distribution of S(IV) species
The three forms of S(IV) which are in equilibrium inaqueous solution are considered in the equilibria (2)–(4).The relative concentrations of these species are pH depen-dent and the hydrated species, (SO2 Æ H2O), is the dominantspecies at pH values <2 [33].
SO2H2O�Ka1
HSO�3 þHþ ð2Þ
HSO�3 �Ka2
SO2�3 þHþ ð3Þ
2HSO�3 �Kd
S2O2�5 þH2O ð4Þ
The fractional concentrations of these species, based onthe equilibria (2) and (3), were calculated for a total[S(IV)] = 0.02 mol dm�3 over 0.01 6 [H+] 6 0.5 mol dm�3
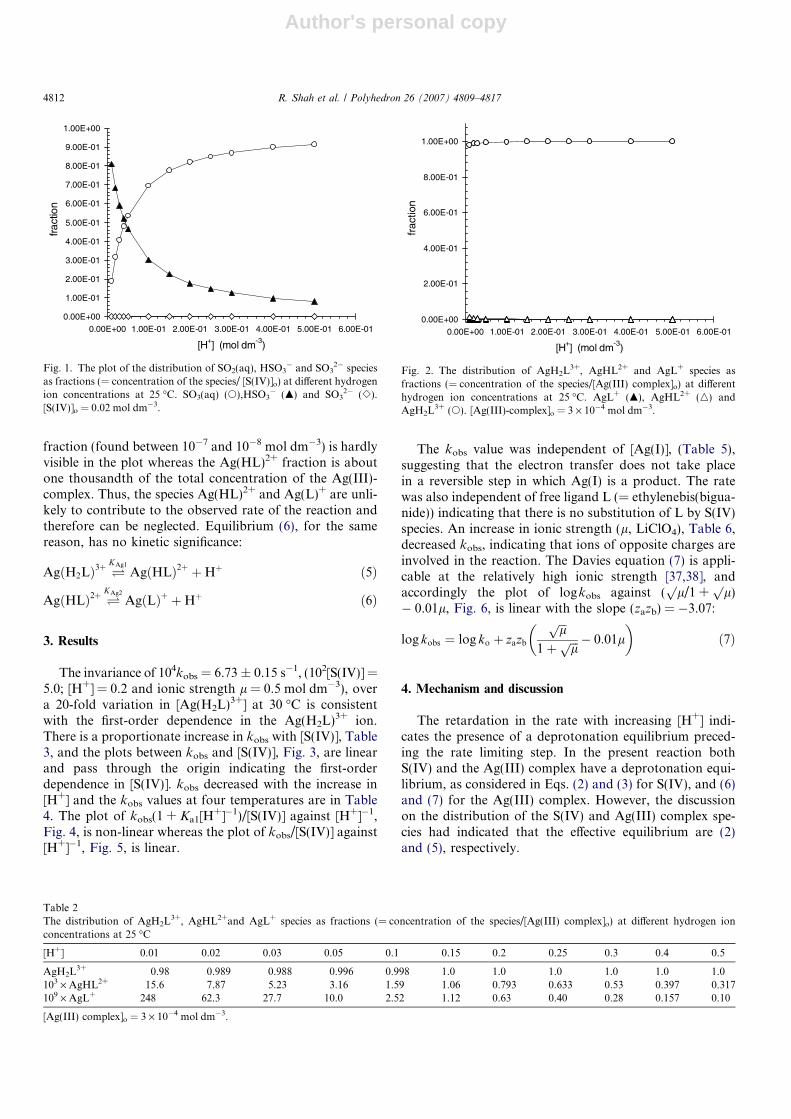
using the equilibrium constants Ka1 = 4.30 · 10�2 andKa2 = 4.57 · 10�7 mol dm�3, respectively, at 25 �C [34].The fraction is equal to the quotient of the concentrationof the particular species divided by the total [S(IV)]. Thecalculated values of the fraction for each of the speciesare given in Table 1 and the distribution is graphically illus-trated in Fig. 1. From the distribution plot it is inferredthat not only the fraction concentration of SO3
2� (foundbetween 10�6 and 10�8 mol dm�3) but its significance asa possible reaction partner is negligible. Hence, the reactiveS(IV) species are SO2 Æ H2O and the HSO3
� ion.The reason for neglecting equilibrium (4), Kd = 8.8 ·
10�2 dm3 mol�1 at 25 �C [35], while calculating the concen-trations of the above species was circumscribed by the factthat no second-order dependence in S(IV) was observedand S2O6
2� was not the reaction product. Hence, equilib-rium (4) has no kinetic significance in the presentinvestigation.
2.9. Distribution of Ag(III)-complex species
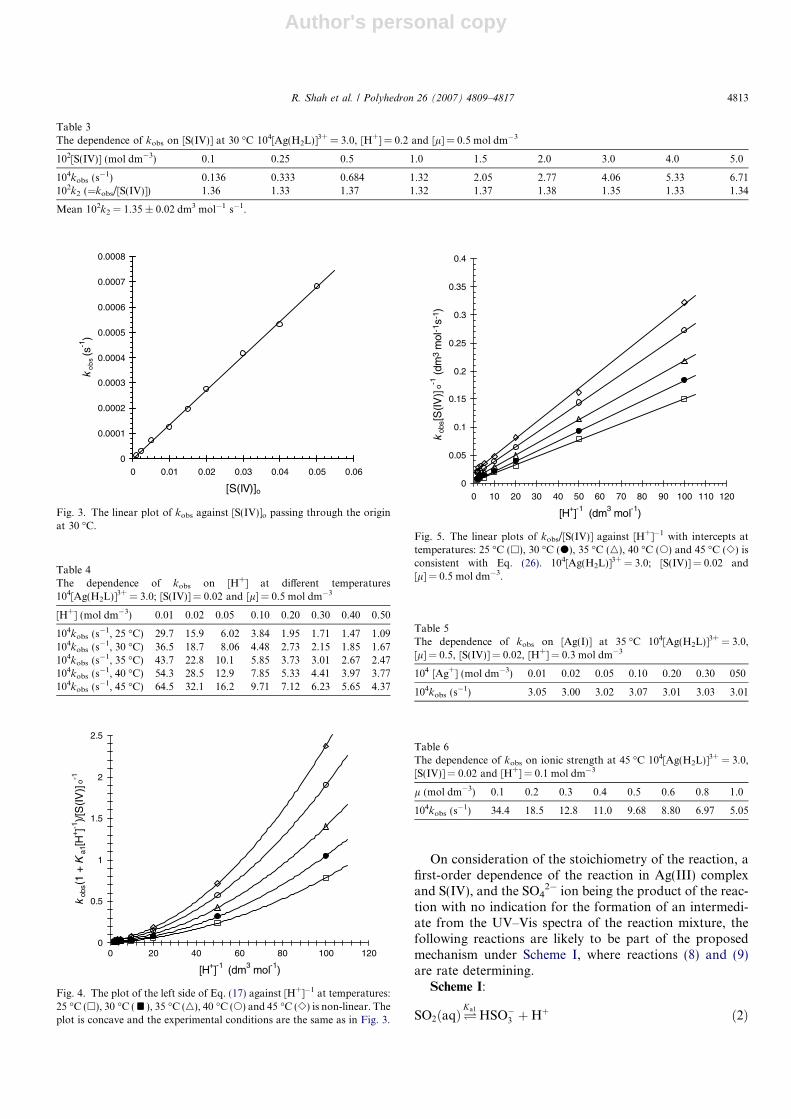
The distribution of the fractions of Ag(III)-complex spe-cies, governed by equilibria (5) and (6), was similarly calcu-lated for [Ag(III)-complex] = 3 · 10�4 mol dm�3 over thesame range of [H+] as in the case of S(IV) usingKAg1a = 1.58 · 10�4 and KAg2a = 1.58 · 10�7 mol dm�3 at25 �C [36]. The values, in Table 2, are plotted in Fig. 2 fromwhich it is contingent that the fraction of the Ag(H2L)3+
species is �1, even at the lowest hydrogen ion concentra-tion. The fraction values of Ag(HL)2+ and Ag(L)+ decreasewith increase in hydrogen ion concentration. The Ag(L)+
Table 1The distribution of H2SO3, HSO3
� and SO32� species as fractions (= concentration of the species/[S(IV)]o) at different hydrogen ion concentrations at
25 �C
[H+] 0.01 0.02 0.03 0.04 0.05 0.1 0.15 0.2 0.25 0.3 0.4 0.5
H2SO3 0.187 0.315 0.408 0.478 0.535 0.695 0.775 0.82 0.85 0.87 0.9 0.915HSO3
� 0.81 0.685 0.59 0.52 0.465 0.302 0.225 0.178 0.148 0.126 0.098 0.08106 · SO3
2� 37 15.5 8.95 5.9 4.23 1.37 0.68 0.406 0.27 0.191 0.111 0.0725
[S(IV)]o = 0.02 mol dm�3.
R. Shah et al. / Polyhedron 26 (2007) 4809–4817 4811
Author's personal copy
fraction (found between 10�7 and 10�8 mol dm�3) is hardlyvisible in the plot whereas the Ag(HL)2+ fraction is aboutone thousandth of the total concentration of the Ag(III)-complex. Thus, the species Ag(HL)2+ and Ag(L)+ are unli-kely to contribute to the observed rate of the reaction andtherefore can be neglected. Equilibrium (6), for the samereason, has no kinetic significance:
AgðH2LÞ3þ �KAg1
AgðHLÞ2þ þHþ ð5Þ
AgðHLÞ2þ �KAg2
AgðLÞþ þHþ ð6Þ
3. Results
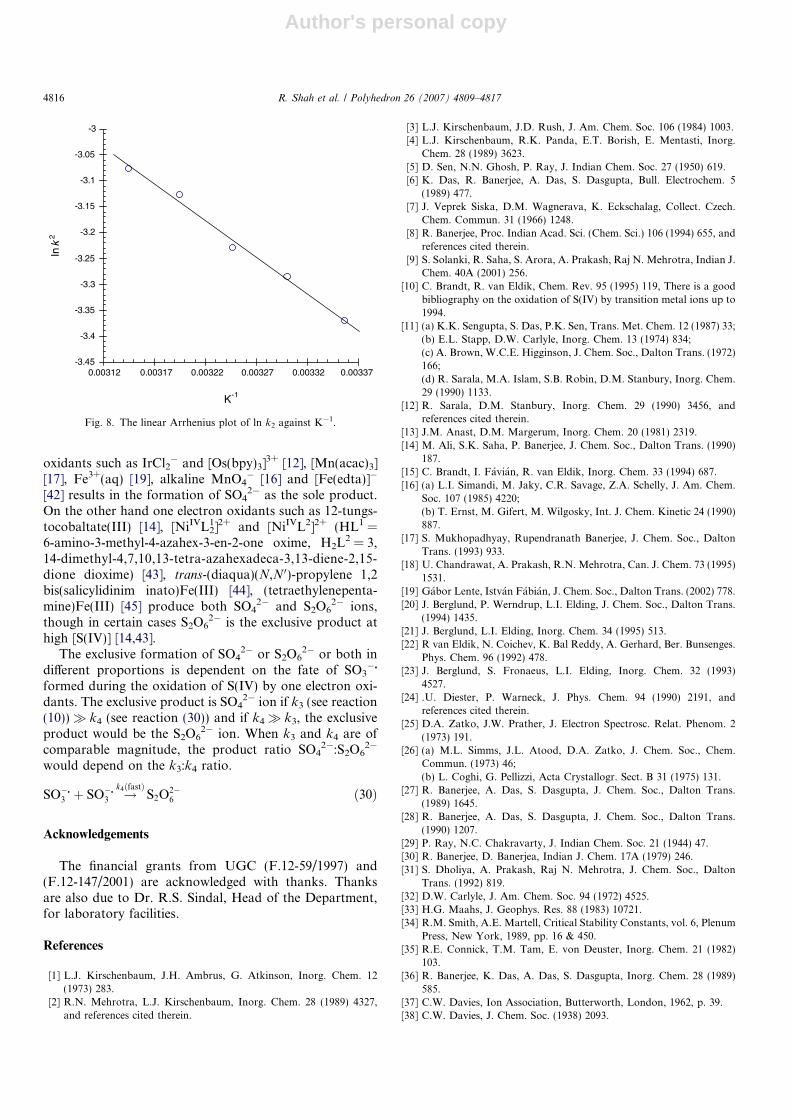
The invariance of 104kobs = 6.73 ± 0.15 s�1, (102[S(IV)] =5.0; [H+] = 0.2 and ionic strength l = 0.5 mol dm�3), overa 20-fold variation in [Ag(H2L)3+] at 30 �C is consistentwith the first-order dependence in the Ag(H2L)3+ ion.There is a proportionate increase in kobs with [S(IV)], Table3, and the plots between kobs and [S(IV)], Fig. 3, are linearand pass through the origin indicating the first-orderdependence in [S(IV)]. kobs decreased with the increase in[H+] and the kobs values at four temperatures are in Table4. The plot of kobs(1 + Ka1[H+]�1)/[S(IV)] against [H+]�1,Fig. 4, is non-linear whereas the plot of kobs/[S(IV)] against[H+]�1, Fig. 5, is linear.
The kobs value was independent of [Ag(I)], (Table 5),suggesting that the electron transfer does not take placein a reversible step in which Ag(I) is a product. The ratewas also independent of free ligand L (= ethylenebis(bigua-nide)) indicating that there is no substitution of L by S(IV)species. An increase in ionic strength (l, LiClO4), Table 6,decreased kobs, indicating that ions of opposite charges areinvolved in the reaction. The Davies equation (7) is appli-cable at the relatively high ionic strength [37,38], andaccordingly the plot of logkobs against (
pl/1 +
pl)
� 0.01l, Fig. 6, is linear with the slope (zazb) = �3.07:
log kobs ¼ log ko þ zazb
ffiffiffilp
1þ ffiffiffilp � 0:01l
� �ð7Þ
4. Mechanism and discussion
The retardation in the rate with increasing [H+] indi-cates the presence of a deprotonation equilibrium preced-ing the rate limiting step. In the present reaction bothS(IV) and the Ag(III) complex have a deprotonation equi-librium, as considered in Eqs. (2) and (3) for S(IV), and (6)and (7) for the Ag(III) complex. However, the discussionon the distribution of the S(IV) and Ag(III) complex spe-cies had indicated that the effective equilibrium are (2)and (5), respectively.
0.00E+00
1.00E-01
2.00E-01
3.00E-01
4.00E-01
5.00E-01
6.00E-01
7.00E-01
8.00E-01
9.00E-01
1.00E+00
0.00E+00 1.00E-01 2.00E-01 3.00E-01 4.00E-01 5.00E-01 6.00E-01
[H+] (mol dm-3)
fract
ion
Fig. 1. The plot of the distribution of SO2(aq), HSO3� and SO3
2� speciesas fractions (= concentration of the species/ [S(IV)]o) at different hydrogenion concentrations at 25 �C. SO3(aq) (s),HSO3
� (m) and SO32� (e).
[S(IV)]o = 0.02 mol dm�3.
Table 2The distribution of AgH2L3+, AgHL2+and AgL+ species as fractions (= concentration of the species/[Ag(III) complex]o) at different hydrogen ionconcentrations at 25 �C
[H+] 0.01 0.02 0.03 0.05 0.1 0.15 0.2 0.25 0.3 0.4 0.5
AgH2L3+ 0.98 0.989 0.988 0.996 0.998 1.0 1.0 1.0 1.0 1.0 1.0103 · AgHL2+ 15.6 7.87 5.23 3.16 1.59 1.06 0.793 0.633 0.53 0.397 0.317109 · AgL+ 248 62.3 27.7 10.0 2.52 1.12 0.63 0.40 0.28 0.157 0.10
[Ag(III) complex]o = 3 · 10�4 mol dm�3.
0.00E+00
2.00E-01
4.00E-01
6.00E-01
8.00E-01
1.00E+00
0.00E+00 1.00E-01 2.00E-01 3.00E-01 4.00E-01 5.00E-01 6.00E-01
[H+] (mol dm-3)
frac
tion
Fig. 2. The distribution of AgH2L3+, AgHL2+ and AgL+ species asfractions (= concentration of the species/[Ag(III) complex]o) at differenthydrogen ion concentrations at 25 �C. AgL+ (m), AgHL2+ (n) andAgH2L3+ (s). [Ag(III)-complex]o = 3 · 10�4 mol dm�3.
4812 R. Shah et al. / Polyhedron 26 (2007) 4809–4817
Author's personal copy
On consideration of the stoichiometry of the reaction, afirst-order dependence of the reaction in Ag(III) complexand S(IV), and the SO4
2� ion being the product of the reac-tion with no indication for the formation of an intermedi-ate from the UV–Vis spectra of the reaction mixture, thefollowing reactions are likely to be part of the proposedmechanism under Scheme I, where reactions (8) and (9)are rate determining.
Scheme I:
SO2ðaqÞ�Ka1
HSO�3 þHþ ð2Þ
Table 3The dependence of kobs on [S(IV)] at 30 �C 104[Ag(H2L)]3+ = 3.0, [H+] = 0.2 and [l] = 0.5 mol dm�3
102[S(IV)] (mol dm�3) 0.1 0.25 0.5 1.0 1.5 2.0 3.0 4.0 5.0
104kobs (s�1) 0.136 0.333 0.684 1.32 2.05 2.77 4.06 5.33 6.71102k2 (=kobs/[S(IV)]) 1.36 1.33 1.37 1.32 1.37 1.38 1.35 1.33 1.34
Mean 102k2 = 1.35 ± 0.02 dm3 mol�1 s�1.
0
0.0001
0.0002
0.0003
0.0004
0.0005
0.0006
0.0007
0.0008
0 0.01 0.02 0.03 0.04 0.05 0.06
[S(IV)]o
ko
bs (
s-1)
Fig. 3. The linear plot of kobs against [S(IV)]o passing through the originat 30 �C.
Table 4The dependence of kobs on [H+] at different temperatures104[Ag(H2L)]3+ = 3.0; [S(IV)] = 0.02 and [l] = 0.5 mol dm�3
[H+] (mol dm�3) 0.01 0.02 0.05 0.10 0.20 0.30 0.40 0.50
104kobs (s�1, 25 �C) 29.7 15.9 6.02 3.84 1.95 1.71 1.47 1.09104kobs (s�1, 30 �C) 36.5 18.7 8.06 4.48 2.73 2.15 1.85 1.67104kobs (s�1, 35 �C) 43.7 22.8 10.1 5.85 3.73 3.01 2.67 2.47104kobs (s�1, 40 �C) 54.3 28.5 12.9 7.85 5.33 4.41 3.97 3.77104kobs (s�1, 45 �C) 64.5 32.1 16.2 9.71 7.12 6.23 5.65 4.37
0
0.5
1
1.5
2
2.5
0 20 40 60 80 100 120
[H+]-1 (dm3 mol-1)
kob
s(1
+ K
a1[H
+ ]-1)/[
S(IV
)] o-1
Fig. 4. The plot of the left side of Eq. (17) against [H+]�1 at temperatures:25 �C (h), 30 �C (�), 35 �C (n), 40 �C (s) and 45 �C (e) is non-linear. Theplot is concave and the experimental conditions are the same as in Fig. 3.
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
0 10 20 30 40 50 60 70 80 90 100 110 120
[H+]-1 (dm3 mol-1)
kob
s[S
(IV)]
o-1 (dm
3 m
ol-1
s-1 )
Fig. 5. The linear plots of kobs/[S(IV)] against [H+]�1 with intercepts attemperatures: 25 �C (h), 30 �C (d), 35 �C (n), 40 �C (s) and 45 �C (e) isconsistent with Eq. (26). 104[Ag(H2L)]3+ = 3.0; [S(IV)] = 0.02 and[l] = 0.5 mol dm�3.
Table 5The dependence of kobs on [Ag(I)] at 35 �C 104[Ag(H2L)]3+ = 3.0,[l] = 0.5, [S(IV)] = 0.02, [H+] = 0.3 mol dm�3
104 [Ag+] (mol dm�3) 0.01 0.02 0.05 0.10 0.20 0.30 050
104kobs (s�1) 3.05 3.00 3.02 3.07 3.01 3.03 3.01
Table 6The dependence of kobs on ionic strength at 45 �C 104[Ag(H2L)]3+ = 3.0,[S(IV)] = 0.02 and [H+] = 0.1 mol dm�3
l (mol dm�3) 0.1 0.2 0.3 0.4 0.5 0.6 0.8 1.0
104kobs (s�1) 34.4 18.5 12.8 11.0 9.68 8.80 6.97 5.05
R. Shah et al. / Polyhedron 26 (2007) 4809–4817 4813

Author's personal copy
AgðH2LÞ3þ �KAg1
AgðHLÞ2þ þHþ ð5Þ
AgðH2LÞ3þ þ SO2ðaqÞ!k SO��3 þAgðH2LÞ2þ þ 2Hþ ð8Þ
AgðH2LÞ3þ þHSO�3 !ko
SO��3 þAgðH2LÞ2þ þHþ ð9Þ
AgðH2LÞ2þ þ SO��3 þH2O !k3ðfastÞAgðH2LÞþ þ SO2�
4 þ 2Hþ
ð10Þ
The free radical SO3�� in aqueous solution is supported
by esr spectroscopy [39]. The rate law in view of the reac-tions (2), (5) and (8)–(10) is expressed by the followingequation:
rate ¼ �d½AgðIIIÞ�odt
¼ ðk þ koK1½ Hþ��1Þ½SO2ðaqÞ�½AgðH2 LÞ3þ� ð11Þ
The correlation of the concentrations of SO2(aq) withthe analytical concentration [S(IV)]o in view of equilibria(2) and (3) is expressed by Eq. (12), which changes toEq. (13) on the assumption that (1 + Ka1[H+]�1)�Ka2Ka1[H+]�2 which is consistent with the fraction of therespective species shown in Fig. 1 over a given range ofhydrogen ion concentrations:
½SO2� ¼½SðIVÞ�o
1þ Ka1½Hþ��1 þ Ka1Ka2½Hþ��2ð12Þ
or
½SO2ðaqÞ� ¼ ½SðIVÞ�oð1þ Ka1½Hþ��1Þ�1 ð13ÞSimilarly, the correlation of the concentration of
Ag(H2L)3+ with its analytical concentration, [Ag(III) com-plex]o, in view of equilibria (5) and (6) is given by Eq. (14),which with the assumption that 1� KAga1 [H+]�1 + KAga1-KAga2 [H+]�2 is reduced to Eq. (15). Eq. (15) is consistentwith the plot in Fig. 2 where the fraction value of Ag-(H2L)3+ � 1 even at the lowest hydrogen ion concentration:
½AgðH2LÞ3þ� ¼ ½AgðIIIÞ-complex�o1þ KAg1a½Hþ��1 þ KAg1aKAg2a½Hþ��2
ð14Þ
½AgðH2LÞ3þ� ¼ ½AgðIIIÞ-complex�o ð15Þ
Eq. (16) is obtained on substituting the respective valuesof [SO2(aq)] and [Ag(H2L)3+] from Eqs. (13) and (15) intoEq. (11):
�d½AgðIIIÞ�dt
¼ k þ koKa1½Hþ��1
1þKa1½Hþ��1
!½SðIVÞ�o½AgðIIIÞ-complex�o
ð16Þor
kobsð1þ Ka1½Hþ��1Þ=½SðIVÞ�o ¼ ðk þ koKa1½Hþ��1Þ ð17ÞThe desired plot of the left side of Eq. (17) against
[H+]�1 is illustrated in Fig. 4, which is concave indicatingthat kobs(1 + Ka1[H+]�1)/ [S(IV)] increased more rapidlythan the [H+]�1 value. Since the term (1 + Ka1[H+]�1) can-not be neglected, Scheme I is unlikely to represent themechanism of the reaction.
The alternate Scheme II considers the involvement ofthe weak intermediates most likely. The intermediatesevaded both kinetic and spectrophotometric detectionsbecause of their weak nature.
Scheme II
SO2ðaqÞ�Ka1
HSO�3 þHþ ð2Þ
AgðH2LÞ3þ �KAg1
AgðHLÞ2þ þHþ ð5Þ
AgðH2LÞ3þ þ SO2ðaqÞ�K1
AgðH2LÞSO3þ2 ð18Þ
AgðH2LÞ3þ þHSO�3 �K2
AgðH2LÞHSO2þ3 ð19Þ
AgðH2LÞSO3þ2 !
k0AgðH2LÞ2þ þ SO��3 ð20Þ
AgðH2LÞHSO2þ3 !
k00AgðH2LÞ2þ þ SO��3 þHþ ð21Þ
AgðH2LÞ2þ þ SO��3 þH2O !k3ðfastÞAgðH2LÞþ þ SO2�
4 þ 2Hþ
ð10Þ
The proposed inner-sphere mechanism has similaritywith those of alcohols [28], H3PO2 [40] and ascorbic acid[41], and Tl3+(aq) oxidation of SO2(aq) under similar con-ditions of H+ ions [20]. The rate in terms of reactions (17)–(20) and (14) is given by the following equation:
rate ¼ �d½AgðIIIÞ�odt
¼ ðk0K1 þ k00K2K1½Hþ��1Þ½AgðH2LÞ3þ�½SO2ðaqÞ� ð22Þ
The correlation between [Ag(H2L)]3+ and [Ag(III) com-plex]o in terms of Eqs. (18) and (19) is given by Eq. (23),whereas Eq. (13) holds good for the correlation between[SO2(aq)] and [S(IV)]:
½AgðH2LÞ3þ� ¼ ½AgðIIIÞ�o1þ K1½SO2ðaqÞ� þ K1K2½SO2ðaqÞ�½Hþ��1
ð23Þ
0.6
0.8
1
1.2
1.4
1.6
0.22 0.26 0.3 0.34 0.38 0.42 0.46 0.5
(√μ/1+√μ)−0.01μ
4 +
log
k obs
Fig. 6. The linear plot of log kobs against (p
l/(1 +p
l) � 0.01l) has aslope = �3.07 ± 0.16 104[Ag(H2L)]3+ = 3.0, [S(IV)] = 0.02 and [H+] =0.1 mol dm�3 at 45 �C.
4814 R. Shah et al. / Polyhedron 26 (2007) 4809–4817

Author's personal copy
The substitution of the proper values of [SO2(aq)] fromEq. (13) and that of [Ag(H2L)3+] from Eq. (23) into Eq.(22) gives the following equation:
The terms (1 + Ka1[H+]�1), appearing both in thenumerator and denominator, cancel each other and there-fore Eq. (24) is transformed into Eq. (25):
�d½AgðIIIÞ�dt
¼ ðk0K1 þ k00K2K1½Hþ��1Þ½AgðIIIÞ � complex�o½SðIVÞ�o
ð1þ ðK1 þ K1K2½Hþ��1ÞÞ½SðIVÞ�oð25Þ
Since 1� (K1 + K1K2[H+]�1)[S(IV)]o because of themicroscopic values of K1 and K2, the Eq. (25) is reducedto Eq. (26) where k1 ¼ k0K1 and k2 ¼ k00K2Ka1:
kobs=½SðIVÞ�o ¼ ðk1 þ k2½Hþ��1Þ ð26ÞEq. (26) is consistent with the linear plot of kobs/[S(IV)]o
against [H+]�1 in Fig. 5, with intercepts on the rate ordi-nate. The inner-sphere mechanism is thereby supported.The formation of the intermediate Ag(H2L)HSO3
2+
through reaction (19) is preferred because it is consistentwith the slope value (zazb = �3.07) of the plot in Fig. 6.The alternate route in Eq. (27) is kinetically indistinguish-able from the reaction in Eq. (19) because of the identitythat K2Ka1 ¼ K 02K1, as shown below:
AgðH2LÞSO2ðaqÞ3þ�K 0
2AgðH2LÞHSO2þ
3 þHþ ð27Þor
½AgðH2LÞHSO2þ3 � ¼ K 02K1½AgðH2LÞ3þ�½SO2ðaqÞ�½Hþ��1
ðfrom ð27Þ and ð18ÞÞ ð28Þ¼ K2Ka1½AgðH2LÞ3þ�½SO2ðaqÞ�½Hþ��1
ðfrom ð19Þ and ð2ÞÞ ð29Þ
The values of the estimated rate constants k1 and k2
from the least square values of the intercepts and slopes,at different temperatures, are given in Table 7. It is noted
that at lower temperatures k2 > k1, but the difference nar-rows as the temperature increases. The Arrhenius plots cor-responding to k1 and k2 are given in Figs. 7 and 8,
respectively. The significance of the activation parametersis not discussed because these are based on the productof the rate limiting and equilibrium constants and the val-ues of the equilibrium constants remain indeterminate.However, it seems that the combination of reactions (18)and (20) is enthalpy controlled (DH�
k1 = 70 ± 3 kJ mol�1
and DSIk1 = �42 ± 10 J K�1 mol�1), and the combination
of reactions (2), (19) and (21) is driven by the entropy,(DHI
k2 = 9 ± 1 kJ mol�1 and DSIk2 = �225 ± 2 J K�100
mol�1). The trend that DSIk1 > DSI
k2 is similar to thatobserved in the oxidation of alcohols for which DH�
k1 andDHI
k2 are fairly similar [28]. Both DHIk2 and DSI
k2 are almostof the same magnitude as in the oxidation of H3PO2 for asimilar path [40]. Such a similarity is a pointer and support-ive of the proposed inner-sphere mechanism.
The coordination number in Ag(III) (d8) could be eitherfour (planar) or six (octahedral). The diamagnetic[Ag(H2L)]3+ complex is square planar and the Ag(III) ionis tetra-coordinated by four nitrogen atoms of the ethylene-bisbiguanidinium ion. Thus SO2(aq) has to be axially coor-dinated. A water molecule is also axially coordinated tomake the sulfito-complex octahedral.
It seems that the generalisation that the one or two-elec-tron oxidation of S(IV) by metal ions and their complexesresults in the formation of S2O6
2� or SO42�, respectively
[11] needs reconsideration. The oxidation by one electron
Table 7Values of k1 and k2 (dm3 mol�1 s�1)
Temp (�C) 25 30 35 40 45
103k1 (dm3 mol�1 s�1) 3.23 4.73 8.27 13.4 18.9103k2 (dm3 mol�1 s�1) 34.4 37.4 39.6 43.9 46.1
DH�k1 = 70 ± 3.
DH�k2 = 9 ±1 kJ mol�1.
DS�k1 = �42 ± 10.
D S�k2 = �225 ± 2 J K�1 mol�1.
�d½AgðIIIÞ�dt
¼ ðk0K1 þ k00K2K1½Hþ��1Þ½AgðIIIÞ � complex�o½SðIVÞ�o=ð1þ Ka1½Hþ��1Þ
ð1þ ðK1 þ K1K2½Hþ��1ÞÞ½SðIVÞ�o=ð1þ Ka1½Hþ��1Þð24Þ
-6
-5.5
-5
-4.5
-4
0.00312 0.00317 0.00322 0.00327 0.00332 0.00337
K-1
lnk
1
Fig. 7. The linear Arrhenius plot of lnk1 against K�1.
R. Shah et al. / Polyhedron 26 (2007) 4809–4817 4815
Author's personal copy
oxidants such as IrCl2� and [Os(bpy)3]3+ [12], [Mn(acac)3]
[17], Fe3+(aq) [19], alkaline MnO4� [16] and [Fe(edta)]�
[42] results in the formation of SO42� as the sole product.
On the other hand one electron oxidants such as 12-tungs-tocobaltate(III) [14], [NiIVL1
2]2+ and [NiIVL2]2+ (HL1 =6-amino-3-methyl-4-azahex-3-en-2-one oxime, H2L2 = 3,14-dimethyl-4,7,10,13-tetra-azahexadeca-3,13-diene-2,15-dione dioxime) [43], trans-(diaqua)(N,N 0)-propylene 1,2bis(salicylidinim inato)Fe(III) [44], (tetraethylenepenta-mine)Fe(III) [45] produce both SO4
2� and S2O62� ions,
though in certain cases S2O62� is the exclusive product at
high [S(IV)] [14,43].The exclusive formation of SO4
2� or S2O62� or both in
different proportions is dependent on the fate of SO3��
formed during the oxidation of S(IV) by one electron oxi-dants. The exclusive product is SO4
2� ion if k3 (see reaction(10))� k4 (see reaction (30)) and if k4� k3, the exclusiveproduct would be the S2O6
2� ion. When k3 and k4 are ofcomparable magnitude, the product ratio SO4
2�:S2O62�
would depend on the k3:k4 ratio.
SO��3 þ SO��3 !k4ðfastÞS2O2�
6 ð30Þ
Acknowledgements
The financial grants from UGC (F.12-59/1997) and(F.12-147/2001) are acknowledged with thanks. Thanksare also due to Dr. R.S. Sindal, Head of the Department,for laboratory facilities.
References
[1] L.J. Kirschenbaum, J.H. Ambrus, G. Atkinson, Inorg. Chem. 12(1973) 283.
[2] R.N. Mehrotra, L.J. Kirschenbaum, Inorg. Chem. 28 (1989) 4327,and references cited therein.
[3] L.J. Kirschenbaum, J.D. Rush, J. Am. Chem. Soc. 106 (1984) 1003.[4] L.J. Kirschenbaum, R.K. Panda, E.T. Borish, E. Mentasti, Inorg.
Chem. 28 (1989) 3623.[5] D. Sen, N.N. Ghosh, P. Ray, J. Indian Chem. Soc. 27 (1950) 619.[6] K. Das, R. Banerjee, A. Das, S. Dasgupta, Bull. Electrochem. 5
(1989) 477.[7] J. Veprek Siska, D.M. Wagnerava, K. Eckschalag, Collect. Czech.
Chem. Commun. 31 (1966) 1248.[8] R. Banerjee, Proc. Indian Acad. Sci. (Chem. Sci.) 106 (1994) 655, and
references cited therein.[9] S. Solanki, R. Saha, S. Arora, A. Prakash, Raj N. Mehrotra, Indian J.
Chem. 40A (2001) 256.[10] C. Brandt, R. van Eldik, Chem. Rev. 95 (1995) 119, There is a good
bibliography on the oxidation of S(IV) by transition metal ions up to1994.
[11] (a) K.K. Sengupta, S. Das, P.K. Sen, Trans. Met. Chem. 12 (1987) 33;(b) E.L. Stapp, D.W. Carlyle, Inorg. Chem. 13 (1974) 834;(c) A. Brown, W.C.E. Higginson, J. Chem. Soc., Dalton Trans. (1972)166;(d) R. Sarala, M.A. Islam, S.B. Robin, D.M. Stanbury, Inorg. Chem.29 (1990) 1133.
[12] R. Sarala, D.M. Stanbury, Inorg. Chem. 29 (1990) 3456, andreferences cited therein.
[13] J.M. Anast, D.M. Margerum, Inorg. Chem. 20 (1981) 2319.[14] M. Ali, S.K. Saha, P. Banerjee, J. Chem. Soc., Dalton Trans. (1990)
187.[15] C. Brandt, I. Favian, R. van Eldik, Inorg. Chem. 33 (1994) 687.[16] (a) L.I. Simandi, M. Jaky, C.R. Savage, Z.A. Schelly, J. Am. Chem.
Soc. 107 (1985) 4220;(b) T. Ernst, M. Gifert, M. Wilgosky, Int. J. Chem. Kinetic 24 (1990)887.
[17] S. Mukhopadhyay, Rupendranath Banerjee, J. Chem. Soc., DaltonTrans. (1993) 933.
[18] U. Chandrawat, A. Prakash, R.N. Mehrotra, Can. J. Chem. 73 (1995)1531.
[19] Gabor Lente, Istvan Fabian, J. Chem. Soc., Dalton Trans. (2002) 778.[20] J. Berglund, P. Werndrup, L.I. Elding, J. Chem. Soc., Dalton Trans.
(1994) 1435.[21] J. Berglund, L.I. Elding, Inorg. Chem. 34 (1995) 513.[22] R van Eldik, N. Coichev, K. Bal Reddy, A. Gerhard, Ber. Bunsenges.
Phys. Chem. 96 (1992) 478.[23] J. Berglund, S. Fronaeus, L.I. Elding, Inorg. Chem. 32 (1993)
4527.[24] .U. Diester, P. Warneck, J. Phys. Chem. 94 (1990) 2191, and
references cited therein.[25] D.A. Zatko, J.W. Prather, J. Electron Spectrosc. Relat. Phenom. 2
(1973) 191.[26] (a) M.L. Simms, J.L. Atood, D.A. Zatko, J. Chem. Soc., Chem.
Commun. (1973) 46;(b) L. Coghi, G. Pellizzi, Acta Crystallogr. Sect. B 31 (1975) 131.
[27] R. Banerjee, A. Das, S. Dasgupta, J. Chem. Soc., Dalton Trans.(1989) 1645.
[28] R. Banerjee, A. Das, S. Dasgupta, J. Chem. Soc., Dalton Trans.(1990) 1207.
[29] P. Ray, N.C. Chakravarty, J. Indian Chem. Soc. 21 (1944) 47.[30] R. Banerjee, D. Banerjea, Indian J. Chem. 17A (1979) 246.[31] S. Dholiya, A. Prakash, Raj N. Mehrotra, J. Chem. Soc., Dalton
Trans. (1992) 819.[32] D.W. Carlyle, J. Am. Chem. Soc. 94 (1972) 4525.[33] H.G. Maahs, J. Geophys. Res. 88 (1983) 10721.[34] R.M. Smith, A.E. Martell, Critical Stability Constants, vol. 6, Plenum
Press, New York, 1989, pp. 16 & 450.[35] R.E. Connick, T.M. Tam, E. von Deuster, Inorg. Chem. 21 (1982)
103.[36] R. Banerjee, K. Das, A. Das, S. Dasgupta, Inorg. Chem. 28 (1989)
585.[37] C.W. Davies, Ion Association, Butterworth, London, 1962, p. 39.[38] C.W. Davies, J. Chem. Soc. (1938) 2093.
-3.45
-3.4
-3.35
-3.3
-3.25
-3.2
-3.15
-3.1
-3.05
-3
0.00312 0.00317 0.00322 0.00327 0.00332 0.00337
K-1
lnk
2
Fig. 8. The linear Arrhenius plot of ln k2 against K�1.
4816 R. Shah et al. / Polyhedron 26 (2007) 4809–4817

Author's personal copy
[39] (a) R.O.C. Norman, P.M. Storey, J. Chem. Soc. B (1971) 1009;(b) B.D. Flockhart, K.J. Irvin, R.C. Pink, B.D. Sharma, J. Chem.Soc., Chem. Commun. (1971) 339.
[40] R. Banerjee, R. Das, S. Mukhopadhyay, J. Chem. Soc., DaltonTrans. (1992) 1317.
[41] S. Dasgupta, E. Herlinger, W. Linert, J. Chem. Soc., Dalton Trans.(1993) 567.
[42] M. Dellert-Ritter, R. van Eldik, J. Chem. Soc., Dalton Trans. (1992)1045.
[43] S. Bhattacharya, M. Ali, S. Gangopadhyay, P. Banerjee, J. Chem.Soc., Dalton Trans. (1994) 3733.
[44] A. Das, A.C. Dash, Indian J. Chem. 40A (2001) 65.[45] A.C. Dash, A.K. Patnaik, N. Das, Indian J. Chem. 36A (1997)
268.
R. Shah et al. / Polyhedron 26 (2007) 4809–4817 4817
Copyright © 2022 FDOKUMEN




















