
On the Nature of Actinide- and Lanthanide-Metal Bonds in Heterobimetallic Compounds
29
1 November 20, 2012 1 On the Nature of Actinide- and Lanthanide-Metal Bonds in 2 Heterobimetallic Compounds 3 Bess Vlaisavljevich, a Pere Miró, a Christopher J. Cramer, a Laura Gagliardi a * 4 a Department of Chemistry, University of Minnesota and Minnesota Supercomputing Institute, 207 Pleasant St. 5 SE, Minneapolis, MN 55455, USA 6 7 Ivan Infante b 8 b Kimika Fakultatea, Euskal Herriko Unibertsitatea and Donostia International Physics Center (DIPC), P.K. 9 1072, 20080 Donostia, Euskadi (Spain) 10 11 Stephen T. Liddle c 12 c School of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, U.K. 13 14 AUTHOR EMAIL ADDRESS ([email protected]) 15 RECEIVED DATE (to be automatically inserted after your manuscript is accepted if required 16 according to the journal that you are submitting your paper to) 17 CORRESPONDING AUTHOR FOOTNOTE [email protected] 18
-
Upload
spanalumni -
Category
Documents
-
view
0 -
download
0
Transcript of On the Nature of Actinide- and Lanthanide-Metal Bonds in Heterobimetallic Compounds

1
November 20, 2012 1
On the Nature of Actinide- and Lanthanide-Metal Bonds in 2
Heterobimetallic Compounds 3
Bess Vlaisavljevich,a Pere Miró,a Christopher J. Cramer,a Laura Gagliardia* 4
aDepartment of Chemistry, University of Minnesota and Minnesota Supercomputing Institute, 207 Pleasant St. 5 SE, Minneapolis, MN 55455, USA 6
7
Ivan Infanteb 8
bKimika Fakultatea, Euskal Herriko Unibertsitatea and Donostia International Physics Center (DIPC), P.K. 9 1072, 20080 Donostia, Euskadi (Spain) 10
11
Stephen T. Liddlec 12
cSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, U.K. 13
14
AUTHOR EMAIL ADDRESS ([email protected]) 15
RECEIVED DATE (to be automatically inserted after your manuscript is accepted if required 16
according to the journal that you are submitting your paper to) 17
CORRESPONDING AUTHOR FOOTNOTE [email protected] 18

2
1
ABSTRACT 2
3
Eleven experimentally characterized complexes containing heterobimetallic bonds between f-block and 4
other elements were examined by quantum chemical methods: [(η5-C5H5)2(THF)LuRu(η5-C5H5)(CO)2], [(η5-5
C5Me5)2(I)ThRu(η5-C5H5)(CO)2], [(η5-C5H5)2YRe(η5-C5H5)2], [{N(CH2CH2NSiMe3)3}URe(η5-C5H5)2], 6
[Y{Ga(NArCh)2}{C(PPh2NSiH3)2}(CH3OCH3)2], [{N(CH2CH2NSiMe3)3}U{Ga(NArCH)2}(CH3OCH3)], [(η5-7
C5H5)3UGa(η5-C5Me5)], [Yb(η5-C5H5){Si(SiMe3)3(THF)2}], [(η5-C5H5)3U(SnPh3)], [(η5-C5H5)3U(SiPh3)], and 8
(Ph[Me]N)3USi(SiMe3)3. Geometries in good agreement with experiment were obtained at the density 9
functional level of theory. The multiconfigurational complete active space self-consistent field method 10
(CASSCF) and subsequent corrections with second order perturbation theory (CASPT2) were applied to 11
further understand the electronic structure of the Lanthanide/Actinide-metal (or metal-metalloid) bonds. 12
Fragment calculations and energy decomposition analyses were also performed and indicate that charge 13
transfer occurs from one supported metal fragment to the other, while the bonding itself is always dominated 14
by ionic character. 15
16
KEYWORDS: heterobimetallic, energy decomposition analysis, CASSCF, DFT, lanthanide, actinide, f-17
elements, metal-metal bond 18
19
1. Introduction 20
Chemists have pursued the challenge of synthesizing a wide variety of metal-metal bonds for the past 21
fifty years with motivations ranging from fundamental interest1-4 to more practical applications e.g. molecular 22
wires5 and small molecule activation.6 Researchers have asked why stable metal-metal bonding interactions 23
form, what is their reactivity, and what potential uses in catalysis, metal surface chemistry, or bioinorganic 24
reactivity may be possible.7 While challenging, many attempts to isolate complexes containing main group 25
homodinuclear metal-metal bonds have proven successful and as a result this type of bonding is now generally 26

3
well understood.1 In contrast, transition metal heterobimetallic metal-metal bonds are less prevalent since they 1
present more synthetic challenges; however, the highly ionic bonds in such compounds provide opportunities 2
for very different chemistry than are found for their homobimetallic counterparts. Heterobimetallic bonds have 3
inequivalent metal centers. As a result, one metal center can be tailored to be electron rich or electron deficient 4
and thus exploited for catalysis.6, 8 5
Moving beyond transition metals, metal-metal bonding in the f-block, as a whole, is uncommon and 6
offers even greater synthetic challenges, particularly for actinides.7 In fact, in f-element chemistry even 7
homodinuclear metal-metal bonding has been elusive, but there have been several actinide-actinide dimers 8
reported in the gas phase. For example, Th2 was detected in both the gas phase and a rare gas matrix while U2 9
has been detected in the gas phase but not yet isolated.9-13 However, many challenges remain regarding the 10
preparation of molecules containing actinide-actinide bonds, particularly in moving beyond the diatom towards 11
the synthesis of complexes that can be characterized by single-crystal diffraction. In fact, complexes 12
containing homodinuclear unsupported actinide-actinide or lanthanide-lanthanide bonds have not been 13
observed to date, but heterobimetallic metal-metal complexes containing one f-element have been. A 14
comprehensive list of f-element-metal bonds was compiled by Liddle and Mills in a recent review.7 They 15
categorized the systems into Ln-M and An-M (Ln=lanthanide and An=actinide) bonds where M is a transition 16
metal, a group 13, a group 14, or a group 15 metals or metalloids respectively (selected elements in groups 13-17
15 are sometimes referred to as metalloids). However, no actinide-group 15 bonds have been observed. 18
Theoretical investigations can provide insight into the electronic structure of metal-metal bonding and 19
help with the interpretation of experiment. While it is clear that these systems have a large ionic contribution to 20
their bonding, the degree to which covalent interactions may also play a role in the metal-metal bond bears 21
consideration. DFT calculations have been performed on several An/Ln-M systems.14-17 Two previous DFT 22
studies of particular interest addressed complexes containing a U-Ga bond18 and a U-Re bond,14 respectively. 23
In both cases, DFT calculations suggested that the bonds exhibit minor but significant orbital contributions 24
(~30%); not only was a σ-donation between metal centers observed, but visualization of the Kohn-Sham 25
frontier orbitals suggested the presence of weak π-donation from the metal ligands to uranium. These results 26

4
spurred our interest in examining systems with unsupported heterobimetallic metal-metal bonds using a high 1
level of theory (CASSCF/CASPT2) to provide a detailed description of the electronic structure of these 2
interactions. 3
Since our interest focuses on the covalency of these interactions, we limited our study to systems containing 4
unsupported metal-metal bonds. We considered experimentally characterized systems with an actinide or a 5
lanthanide bond to a single transition metal, group 13, or group 14 element: Th-Ru, Lu-Ru, U-Re, Y-Re, U-Ga, 6
Y-Ga, U-Si, and Yb-Si bonds. All the systems have ligands supporting the individual metals. While it is not a 7
lanthanide, yttrium is classified as a rare earth element along with scandium and the lanthanides; therefore, Y-8
M bonds are included in this study. Specifically, calculations were performed for [(η5-C5H5)2(THF)LuRu(η5-9
C5H5)(CO)2],19 [(η5-C5Me5)2(I)ThRu(η5-C5H5)(CO)2],20 [(η5-C5H5)2YRe(η5-C5H5)2],15 10
[{N(CH2CH2NSiMe3)3}URe(η5-C5H5)2],14 [Y{Ga(NArCh)2}{C(PPh2NSiH3)2}(CH3OCH3)2],17 11
[{N(CH2CH2NSiMe3)3}U{Ga(NArCH)2}(CH3OCH3)],18 [(η5-C5H5)3UGa(η5-C5Me5)],16 [Yb(η5-12
C5H5){Si(SiMe3)3(THF)2}],21 [(η5-C5H5)3U(SnPh3)],22 [(η5-C5H5)3U(SiPh3)],23 and (Ph[Me]N)3USi(SiMe3).24 13
The ligand choice for each system is described in detail in the respective experimental papers. 14
For the sake of expedience, abbreviations will be used in the following discussion. For example, [(η5-15
C5H5)2(THF)LuRu(η5-C5H5)(CO)2] will be referred to simply as Lu-Ru. Other systems will be referred to in an 16
analogous manner. More than one system was studied containing a U-Si or a U-Ga bond. These will be 17
referred to U-Si-1, U-Si-2, U-Ga-1, and U-Ga-2. The molecules, abbreviations, and formal oxidation states are 18
given in Figures 1 and 2. Based on the formal oxidation states, one can predict closed shell spin states for all 19
of the cases (with the exception of uranium containing species). For UIII a quartet spin state is expected, while 20
UIV is a triplet. 21
22
2. Theoretical Calculations 23
Calculations were performed using density functional theory (DFT) and multiconfigurational methods 24
(CASSCF/CASPT2).25-27 Density functional theory (DFT) geometry optimizations of the experimentally 25
synthesized clusters were performed using the TURBOMOLE 5.10 package28 with the Perdew-Burke-26

5
Ernzerhof (PBE) exchange-correlation functional29 and triple-zeta valence plus polarization (def2-TZVPP)30 1
basis sets on all atoms, except for actinides and lanthanides where the (def-TZVPP)31 basis was used. Small 2
core quasi-relativistic pseudopotentials were used for the Th, Lu, U, Y, Yb, Ru, and Re atoms.32, 33 The 3
resolution of the identity (RI) approximation was used to speed the calculation of the Coulomb integrals.33-35 4
Multiconfigurational complete active space (CASSCF)25 calculations followed by second-order perturbation 5
theory (CASPT2)26, 27 were performed on the DFT optimized geometries with the unsupported metal-metal 6
bond distance reoptimized at the CASPT2 level. Thus, a numerical procedure was followed according to which 7
single-point energy calculations were computed for structures for which the metal-metal distance was varied in 8
0.05 Å increments while each supported metal fragment remained fixed at its DFT optimized geometry. Using 9
a second-order fit, the minimum metal-metal bond distance was obtained. For systems where 10
multiconfigurational effects were negligible, energies were calculated at the second order Møller-Plesset 11
perturbation (MP2)36, 37 level in place of CASSCF/CASPT2 (see below). 12
Scalar relativistic effects were included at the CASSCF/CASPT2 level using the 2nd order Douglas–Kroll–13
Hess Hamiltonian38 and relativistic all electron ANO-RCC basis sets. The metals were treated with the triple-ζ 14
quality basis set (ANO-RCC-VTZP)39-41 with the following contractions: In Th and U, the primitive sets were 15
contracted to 9s8p6d4f2g1h. In Y, the contraction was 7s6p4d2f1g while in Lu and Yb 8s7p4d3f2g1h and 16
8s7p4d3f2g were used, respectively. For the transition metals, the Ru contraction was 7s6p4d2f1g while the Re 17
contraction was 8s7p5d3f2g. Lastly, the Ga contraction was 6s5p3d2f1g, Sn was 7s6p4d2f1g, and Si was 18
5s4p2d1f. The main group atoms bonded to the metals were treated with double-ζ basis sets, while the 19
peripheral atoms were treated with minimal basis sets. The ground states of these compounds were always well 20
separated from their lowest excited states and thus spin-orbit effects were not included. 21
The CASSCF/CASPT2 and MP2 calculations were performed with the MOLCAS 7.5 package.42 The 22
computational costs arising from the two-electron integrals were drastically reduced by employing the 23
Cholesky decomposition technique43-45 for the CASSCF/CASPT2 level combined with the Local Exchange 24
screening.46 Furthermore, the CASSCF/CASPT2 approach has proven to be successful for many actinide-25
containing systems.47-50 26

6
The character of the metal-metal bond was further investigated by performing fragment calculations with 1
energy decomposition as implemented in the Amsterdam Density Functional program (ADF2009)51 using the 2
local VWN exchange-correlation potential52 and nonlocal Perdew-Burke-Ernzerhof (PBE)29 exchange-3
correlation corrections. A triple-ζ basis set plus two polarization functions was used and relativistic corrections 4
were added using the scalar relativistic zero-order regular approximation (ZORA).53 No core electrons were 5
frozen. 6
7
2.1 Active Space Choice 8
The active space choice is essential in CASSCF calculations and should include nearly degenerate orbitals in 9
a balanced way. The molecular orbitals in the frontier regions of these compounds consist primarily of linear 10
combinations of metal valence atomic orbitals. As such, we chose to include all molecular orbitals with 11
significant contributions from the valence orbitals of the two metals. Specifically, the valence orbitals close in 12
energy to the HOMO and LUMO were localized on the An/Ln and M centers and were put into the active 13
space. (Active orbitals are provided in the supporting information). 14
The active spaces for the Lu-Ru and Th-Ru systems contain eight electrons in eight orbitals, denoted as 15
(8,8). Both complexes have closed-shell singlet ground states. The eight orbitals are linear combinations of 4d 16
and 5s orbitals on Ru and 5d and 6s orbitals on Lu (6d and 7s for Th). The 4f orbitals on Lu are highly 17
localized on Lu and quite low in energy; therefore, there they were not included in the active space (a typical 18
situation for 4f electrons). Similarly, the active space for Y-Re contains orbitals consisting of contributions 19
from the Y 4d and 5s orbitals and the Re 6d and 6s orbitals, resulting in a (6,6) active space. Since the ground 20
state of U-Re is a triplet, the six analogous orbitals to Y-Re were included in the active space along with two 21
additional orbitals localized on uranium and contain the two unpaired electrons, resulting in an (8,8) active 22
space. The singly occupied molecular orbitals (SOMOs) were linear combinations of U 5f orbitals. The active 23
spaces for U-Ga-1 and U-Ga-2 contain molecular orbitals that are linear combinations of U 7s, 6d, and 5f and 24
Ga 3s and 3p orbitals. The two systems differ in the formal oxidation state of uranium, U(IV) and U(III) for U-25
Ga-1 and U-Ga-2, respectively. For U-Ga-1, the doubly occupied orbitals were well separated from the 26

7
unpaired electrons; therefore, a (2,4) active space was used. For U-Ga-2, a (5,7) active space was used. In this 1
case, three orbitals are essentially singly occupied and one orbital is nearly doubly occupied while the 2
remaining orbitals are nearly empty. However, for the Y-Ga system, the HOMO-LUMO gap at the HF level is 3
very large (185 kcal/mol). The occupied orbitals with Y contributions are much lower in energy than the 4
HOMO. As a result, this complex was studied exclusively with MP2. (HOMO and LUMO are shown in the 5
supporting information). Like Y-Ga, Yb-Si was also essentially single reference and was studied with both 6
CASSCF/CASPT2 and MP2. Only a small active space was required and a (2,5) space composed of orbitals 7
that are linear combinations of Yb 6s and Si 3s and 3p orbitals was used. In fact, a (2,2) space may have been 8
large enough, but three additional virtual orbitals in the valence region were included since they contained 9
contributions from Yb and Si as well. These three orbitals had occupation numbers very close to zero. The last 10
two complexes studied contained U-Si bonds, denoted U-Si-1 and U-Si-2. The active space was chosen to 11
include orbitals that have contributions from U 7s, 6d, and 5f orbitals and Si 3s and 3p orbitals. The best active 12
space for the triplet ground state of U-Si-1 was a (6,6) space. For U-Si-2, the best active space for the triplet 13
ground state was (4,4). 14
CASSCF calculations were not performed for U-Sn since the energy decomposition results (described later 15
in this work) gave the same bonding picture for U-Sn as with its isostructural complex U-Si-1 and we have no 16
reason to suspect CASSCF would show anything different. In the subsequent CASPT2 calculations all orbitals 17
up to the sub-core were frozen. For example, in U-Re, there were 214 doubly occupied orbitals not included in 18
the active space and 104 of these orbitals were frozen in the CASPT2 calculations. In the CASSCF 19
calculations, no orbitals were frozen. 20
21
3. Results and discussion 22
Several complexes containing either metal-lanthanide or metal-actinide bonds were studied. In choosing 23
systems to compare, we recognize that ligand effects are extremely important in the stability of these systems; 24
therefore, we studied experimentally characterized systems to ensure the relevance of these compounds. 25
26

8
3.1 Geometry Optimization 1
DFT Results 2
Geometries were optimized at the DFT level for all structures shown in Figures 1 and 2. In general, PBE 3
bond distances and angles are in good agreement with experimental values. Some differences between 4
experimental and calculated geometries are expected to occur because the calculations were performed in the 5
gas phase; however, bond distances vary at most by 0.1 Å and bond angles by 1-6°. (See the details in the 6
supporting information). 7
8
CASPT2 Results 9
CASPT2 calculations were performed on the PBE optimized geometries to further study the interactions 10
contributing to the Ln/An-M bonds. All of the systems studied have electronic ground states well separated 11
from their first excited states and in every case one configuration contributed at least 90% towards the total 12
CASSCF wavefunction. Specifically, the CASSCF wavefunction for Lu-Ru, Th-Ru, U-Ga-1, U-Ga-2, and U-13
Si-1 contain 92% one configuration, while U-Si-2 is 99% one configuration. Y-Re and U-Re are 96% one 14
configuration, and Yb-Si has one configuration contributing 97%. Y-Ga is a closed shell singlet with a well 15
separated HOMO and LUMO. As a result, Y-Ga is, for all purposes, single-reference and was treated with 16
second order Moller-Plesset perturbation theory (MP2) instead of CASPT2. For comparison, MP2 calculations 17
were preformed for Yb-Si as well. 18
While a full CASPT2 geometry optimization is not feasible, the unsupported Ln-M, or An-M, bond distances 19
were optimized at the CASPT2 level by the numerical procedure described in the methods section. By 20
optimizing the CASPT2 distance, we can check if the qualitative description of the bonding is sensitive to 21
small changes in the geometry. For these systems, no significant changes were observed. The results are given 22
in Table 1 along with the experimental and PBE distances. 23
In Table 1, the metal-metal bond distances determined via PBE, CASPT2, and MP2 are compared. Also 24
included in the table are the distances determined via X-ray diffraction14-24 as well as with the sum of the 25
covalent radii. The covalent radii in Table 1 are taken from the reference by Mantina et al.54 The authors report 26

9
two sets of covalent radii, those by Cordero et al.55 and those by Pyykkö and Atsumi56 and they recommend 1
using an average of these values. Comparison with the sum of the covalent radii has been used as a criterion 2
for the existence of a direct metal-metal bond. Looking at Table 1, five of the Ac/Ln-M distances from X-ray 3
diffraction are shorter than the sum of the covalent radii while five are longer (no crystal was isolated for U-Si-4
1). While this could suggest that these types of bonds are borderline with respect to being single bonds, recall 5
the uncertainty in the atomic radii is on the order of 0.1 Å, and all the experimental bonds fall within this 6
margin of error. Moreover, the systems in question contain highly ionic bonds and through the use of theory, 7
the nature of the bonding can be further understood. 8
The DFT optimized metal-metal bond distances agree quite well with experiment. The mean unsigned 9
difference (MUD) between the X-ray and DFT distances is 0.003 Å while the MUD between CASPT2 and 10
experiment is 0.087 Å (it should be recalled that at the CASPT2 level only one degree of freedom is 11
optimized). Nevertheless, a particularly interesting result occurred for the U-Ga-2 complex. The crystal 12
structure contained two geometries with U-Ga distances of 3.065 Å and 3.080 Å. Our results with PBE gave a 13
distance of 3.065 Å, agreeing perfectly with the first structure, while the CASPT2 bond distance of 3.083 Å 14
was very close to the second structure. In all of the other structures, the PBE An/Ln-M distance was longer 15
than the CASPT2 distance. For the U-Re bond, the procedure used in the CASPT2 optimization was repeated 16
with DFT. The fully optimized DFT geometry had a U-Re distance of 3.043 Å. When only the U-Re distance 17
was varied, the optimized distance was 3.074 Å. Given that only one distance is varied, the distances are in 18
good agreement. Additionally, the energy difference between the structure at 3.074 Å and the optimized 19
structure is 1.1 kcal/mol. 20
21
3.2 Discussion of the CASSCF results 22
For homobimetallic bonds, the metal-metal bond is necessarily covalent and often is characterized by a high 23
bond order. For heterobimetallic compounds, one expects important ionic contributions to the bonding due to 24
differences in atomic electronegativities; however, covalent contributions to the bonding may be involved as 25
well. As a rule, the active space in multireference calculations should contain sets of bonding and antibonding 26

10
orbitals. In previous work,14, 18 not only were U-Ga-1 and U-Re characterized experimentally, they were also 1
studied using fragment analysis as implemented in the Amsterdam Density Functional (ADF) program. The 2
calculations indicated a predominantly ionic bond but noted that the orbital interaction provides a significant 3
contribution to the bonding. In the case of U-Ga-1, the orbital interaction contributes 28% of the total bond 4
energy, while in U-Re the orbital contribution is 32%. But most notably, the DFT calculations suggested σ- 5
and modest π-donation from rhenium (or gallium) to uranium originating from polarization of ligand orbitals 6
towards uranium. 7
The potential existence of a π-interaction was a very exciting idea, and our objective was to further 8
understand its significance. The CASSCF results for both U-Ga-1 and U-Re do not, however, exhibit any 9
bonding and antibonding pairs of orbitals corresponding to a uranium-metal bond, but orbital interactions 10
between the two metal centers are observed. Unfortunately, defining these orbitals as bonds simply from the 11
qualitative shape of the molecular orbital is not possible. In fact, the CASSCF results suggest that the manner 12
in which we classify the orbital-types in highly ionic bonds must be approached carefully. In all cases, no 13
pairs of bonding and antibonding orbitals are present and it is thus difficult to talk about σ or π bonds. This 14
holds true for all of the complexes reported in this study. Nevertheless, the lack of a σ and π bond does not 15
conflict with the previous calculations.11,15 Although there are no bonding and antibonding pairs, the previous 16
DFT calculations suggest that orbital interactions contribute about thirty percent to the total bonding energy. 17
The covalent interactions were described as “σ and π donations”. This means that an occupied orbital on one 18
fragment interacts with an empty orbital on the other fragment transferring electron density from one metal to 19
the other. For all of the systems studied, an orbital corresponding to a σ-donation is observed. U-Ga-1 and U-20
Re will be discussed in more detail, but for all of the remaining systems no significant additional orbital 21
interactions are observed. 22
Turning to U-Re and its lanthanide counterpart Y-Re, the most interesting natural orbitals from the U-Re and 23
Y-Re CASSCF calculations are shown in Figure 3. The U-Re orbital on the bottom left (occupation number 24
1.97) is dominated by the Re 5dz2 orbital with a small uranium contribution. An analogous orbital is also 25
present in Y-Re (occupation number 1.97) and both orbitals are examples of a σ-donation. Figure 3 also 26

11
includes a natural orbital from both U-Re and Y-Re that is primarily composed of the Re 5dxy orbital. This 5d 1
orbital is oriented in the correct direction to participate in a π-interaction; however, and in agreement with the 2
previous calculation, the orbital, as in the case with the σ-interaction, is mainly localized on the Re atom. The 3
analogous Y-Re orbital (occupation number 1.97) has no contribution for yttrium, while the U-Re orbital 4
(occupation number 1.96) has a small uranium contribution. If we plot this orbital at a lower isodensity (0.02 5
as opposed to 0.04) we see an increased “π-like” appearance; however, interpreting the significance of this 6
orbital is not straightforward. 7
Likewise in U-Ga-1, CASSCF results showed an example of σ-donation (Figure 4). Although in this system 8
the active space contained only the orbitals corresponding to the two unpaired electrons, larger active spaces 9
were tested but proved to be unnecessary. Looking at the DFT results from the previous work15, we observe the 10
same orbital ordering in our inactive space as in DFT and can identify the two molecular orbitals in our 11
calculations that were previously described: the σ-donating orbital and possible π-donating orbital (Figure 4). 12
In this case, the π-orbital is localized on Ga and the neighboring N atoms even at the lower isodensity value 13
leading us to conclude that a π-interaction is not observed at the CASSCF level. 14
All in all, the CASSCF results indicate that, while a π interaction is clearly not present in U-Ga-1, it may be 15
possible in U-Re. Since we have reoptimized the geometry, we performed additional DFT calculations, in 16
analogy with those performed previously,14, 18 to compute the energy decomposition to make further 17
comparisons with CASSCF and the previous studies. 18
19
3.3 Energy Decomposition Analysis Results 20
These systems are well suited to the application of the energy decomposition method since they are formed 21
from individually supported fragments. Indeed, the synthesis proceeds by combining the An/Ln fragment and 22
the metal fragment with the final complex formed by salt or alkane elimination. 14, 15, 17-21, 24 The only exception 23
is represented by U-Ga-216 where no salt is eliminated since the two fragments are directly combined. During 24
the fragment analysis, each species is divided into two fragments by breaking the total complex along the 25
An/Ln-M bond. For U-Ga-2, the fragments, as in the experimental fragments, have zero charges. In the 26

12
remaining systems, the fragments used in the calculation are charged since in the experiment the fragment 1
charges are balanced by the presence of the counterions that lead to salt formation. In particular, during the 2
fragment analysis the An/Ln fragment is charged +1, while the metal fragment has a charge of −1. 3
The total bonding energy and the charge transfer (based on multipole expansion charges57) were calculated 4
and the results are reported in Table 2. All of these systems, with the exception of U-Ga-2, show qualitatively 5
similar bond energies. The bonding energy in U-Ga-2 is an order of magnitude smaller than the other species; 6
this result is expected since, through simple Coulomb interactions, bringing a positively charged fragment and 7
a negatively charged fragment together should result in a substantially more favorable bonding energy than the 8
combination of two neutral fragments. Furthermore, a charge transfer is observed showing that electron density 9
is transferred from the negatively charged metal fragment to the positively charged An/Ln fragment (with the 10
exception of U-Ga-2). The negatively charged metal fragments present lone pair orbitals that are localized on 11
the metal centers. Inspection of these orbitals in the fragment calculations (see Figure 5), indicates lone pair 12
orbitals that contribute to the interactions that Liddle et al14, 18 described as σ-donation. Looking more closely at 13
U-Re, an example representative of the σ-donation present in all of the systems can be seen (Figure 5). The 14
doubly occupied orbital from the Re fragment calculation (right) is the lone pair on Re. This lone pair interacts 15
with the unoccupied orbital on the U fragment (left) to form the “σ-like” molecular orbital (center). This 16
qualitative picture, in combination with the charge transfer, supports the idea that a σ-donation is occurring in 17
this complex. Analogous orbitals are seen in all the systems with charged fragments. As the final electronic 18
structure exhibits nearly complete transfer of this charge density to the An/Ln fragment, as opposed to 19
retention of a dative bond, the nature of the metal-metal interaction is more ionic than covalent. 20
U-Ga-2 is an exception with respect not only to the bonding energy but also to charge transfer. The lack of 21
charge transfer between the fragments in this compound is a consequence of using neutral fragments during the 22
analysis. For the other systems, a large charge transfer is observed, as expected, since two oppositely charged 23
fragments are joined together. However, the nature of the bond in U-Ga-2 is the same as in the other systems, 24
i.e. a σ-donation occurs. Furthermore, the percentage of orbital interaction in U-Ga-2 is similar to the one 25
present in the other systems. If we used neutral fragments for the other species, we would see almost no charge 26

13
transfer as well. Additionally, according to the CASSCF results, the orbitals in the active space are 1
qualitatively similar to those in the HOMO-LUMO region of fragment calculations. For these reasons, the U-2
Ga bond in U-Ga-2 can be considered similar to the Ac/Ln-M bonds present in the other systems. 3
Returning to the presence of π-donation in U-Re and U-Ga-1,14, 18 the previously suggested possibility of an, 4
albeit weak, π-donation was based on the visualization of the Kohn-Sham frontier orbitals and the breakdown 5
of their compositions as computed by DFT. The fragment analysis in the current study and those referenced14, 18 6
were performed at slightly different levels of theory on different geometries. Specifically, the fragment 7
analysis by Liddle et al.14, 18 was performed using the BP86 functional with a triple-ζ basis set (TZP) whereas 8
we used PBE with a triple-ζ plus two polarization-functions (TZ2P) basis. 9
Our energy decomposition studies further supported the conclusions derived from the CASSCF results: U-10
Re had some very small orbital interaction reminiscent of a π-bond (even less than in our CASSCF results), but 11
U-Ga-1 did not. Initially we were concerned that our results conflicted with the previous studies, but in fact 12
they do not. For U-Ga-1, different models were used in the previous work and this study. The previous work 13
truncated the SiMe3 groups to SiH3 accounting for the difference in our results. With the larger model (U-Ga-1) 14
we see the same picture at the CASSCF and DFT levels. For U-Re, the full experimental structure was studied 15
both previously and here, but with different levels of theory. As a comparison, we performed the fragment 16
analysis for U-Re at the geometry reported by Liddle et al.14 with PBE/TZ2P and reproduced the previous 17
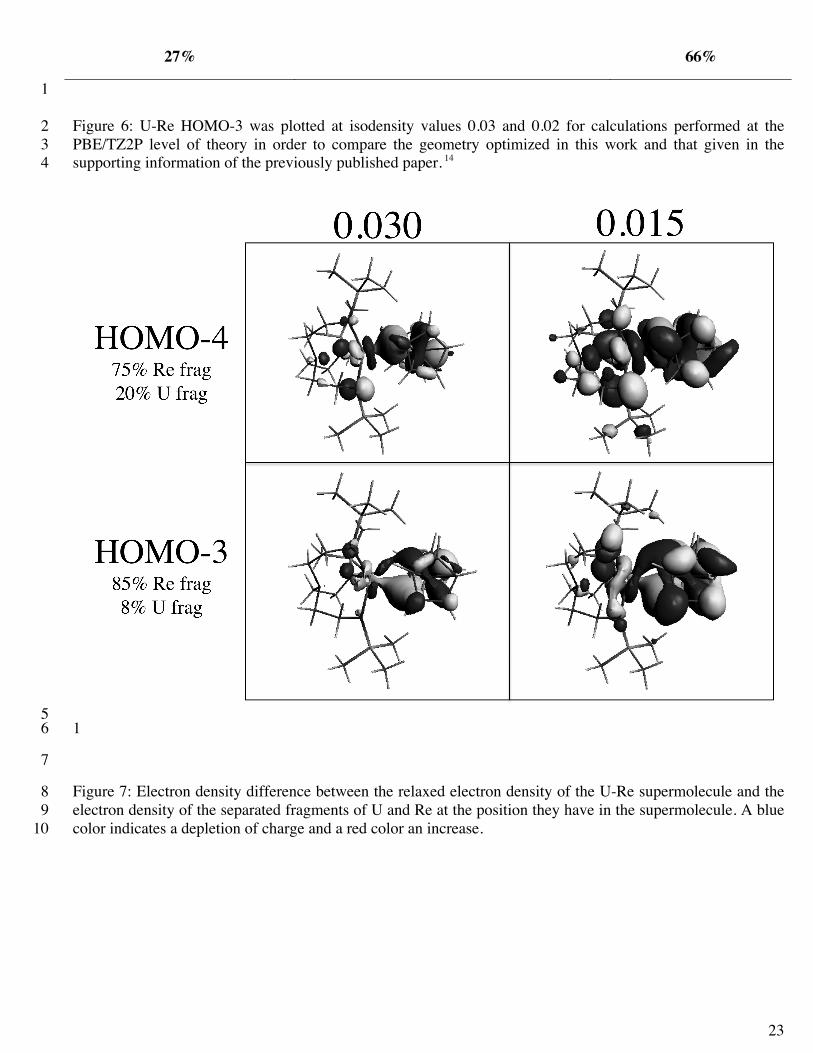
results. For the geometry optimized in this study, at an isodensity value of 0.03, we did not observe the same π 18
shaped orbital as seen for the geometry from the previous work; however, if we lower our isodensity to 0.02 19
our orbitals resemble those shown previously (see Figure 6). However, at the lower isodensity, we also saw 20
orbitals in the other systems, like Y-Re, for example, that began to look “π-like”. For this reason, we are 21
hesitant to interpret the Kohn-Sham orbitals visually and retain our interpretations based on the higher-level 22
CASSCF results. 23
As for the differences between our geometry and those previously reported, they vary only slightly. For 24
example, the U-Re distances in our calculation and Liddle et al.’s are 3.043 and 3.062 Å respectively in 25

14
comparison with the experimental distance of 3.0475(4) Å (see Table 3 for a complete comparison of the 1
differences between our structures). Nevertheless, it appears that the shape of the orbital corresponding to a π-2
interaction is very sensitive to even small geometry changes. This supports the previously suggested idea that 3
the π-donation is a minor component of bonds that are dominated by electrostatics, and therefore sensitive to 4
small fluctuations in the geometry; it is thus unlikely to contribute to the reactivity of the U-Re and U-Ga-1 5
systems where these types of metal-metal bonds are described as “elastic” and easily disturbed by crystal 6
packing.18 7
Bonding energies cannot be uniquely described in terms of pure orbital interactions (resonance); in addition, 8
steric and electrostatic contributions must be considered. Fragment analysis expresses the total interaction 9
energy as 10
11
ΔEint = ΔEelectrostatic + ΔEorbital + ΔEPauli (1) 12
13
where the terms on the right-hand-side refer to electrostatic interactions, orbital interactions, and Pauli 14
repulsion (the energy cost associated with orthogonalizing the various fragment orbitals as they are brought 15
into contact with one another). 16
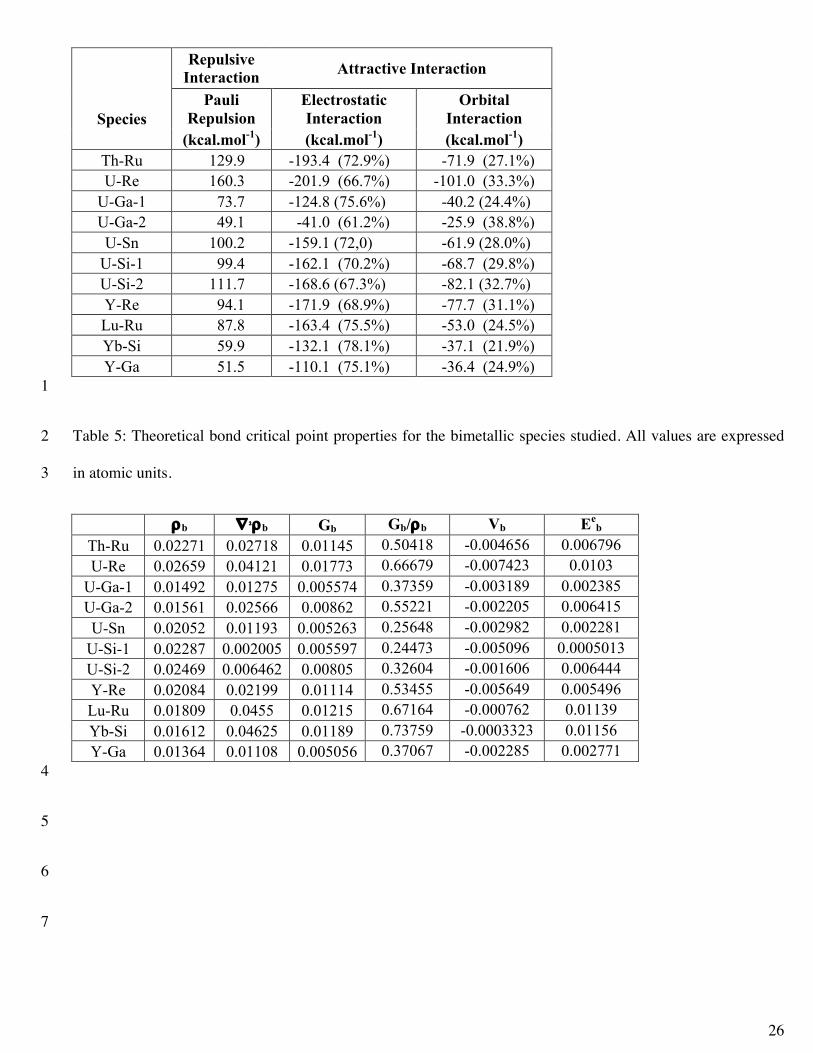
The energy decomposition results obtained using Equation 1 are reported in Table 4. The percentage of the 17
total attractive interaction of electrostatic and orbital interactions is shown in parentheses and clearly indicates 18
that the most important portion of the attractive interaction is electrostatic. This means that these bonds should 19
be thought of as highly ionic since the ionic contributions are stronger than the orbital contributions. However, 20
orbital interactions are present to a non-trivial degree. While the lanthanide containing systems have slightly 21
smaller orbital contributions than do the actinide containing systems, in general all of the An/Ln-M bonds are 22
very similar. Y-Re was described as an donor-acceptor interaction by Butovskii et al.15 In our results, not only 23
the Y-Re bond, but all of the Ln/An-M bonds have the same type of donor-acceptor interaction (i.e. σ-24
donation). The orbital contribution to the U-Re system is nearly the same as the Y-Re system, 33.3% and 25

15
31.1% respectively. Again, we emphasize that these interactions are dominated by a σ-donation from the metal 1
ligand center to the An or Ln center in all cases. 2
To further stress this point, we note that in the energy decomposition analysis implemented in ADF, a 3
favorable orbital interaction is not always associated with a charge transfer between the two fragments. 4
Instead, when a strong charge rearrangement occurs in one fragment as a result of its interaction with the 5
charge distribution of the other fragment (i.e., internal polarization), then the orbital contribution may still be 6
high despite there being no effective charge transfer between the two moieties. One example of this is the U-7
Ga-2 complex, which shows the highest percentage of orbital interaction but lacks significant charge transfer. 8
This means that in this particular case, the polarization effect not only is dominant but also is the only 9
contribution to the stabilization of the orbital interaction term upon relaxation of the U-Ga-2 total electron 10
density. 11
12
3.4 Topological Analysis of the Electron Density 13
To further assess the character of the chemical bonds involved in these heterobimetallic systems, we decided 14
to apply the quantum theory of atoms in molecules developed by Bader58, 59 and analyze the topology of the 15
electron density. In AIM theory, a chemical bond exists if a line of locally maximum electron density links two 16
neighboring atoms (along a so-called bond path) and a bond critical point (BCP) exists, which is defined as the 17
minimum in the density along the locally maximal line. At a BCP, the gradient of the electron density ρ is zero 18
while the Laplacian, ∇2ρ, is the sum of two negative and one positive eigenvalues of the density Hessian 19
matrix, and thus may have either a net positive or net negative value. A positive Laplacian means a local 20
depletion of charge, while a negative value is a sign of a local concentration of charge. The latter is a strong 21
condition for a covalent bond, in the sense that a negative Laplacian indicates always a “shared interaction” of 22
electron density between two linking atoms, while a “closed-shell” interaction is associated with a positive 23
Laplacian, consistent with a type of bond that has moved away from covalency due to depletion of charge at 24
the location of the BCP. Since there exists a continuum between a covalent and a non-covalent bond, Bianchi 25

16
et al.60 have suggested the classification of the bond between two “closed shell” interacting atoms according 1
also to a second condition, the total electronic energy density at the BCP, Eeb. This term is defined as the sum 2
of the kinetic energy density, Gb, which usually dominates in a non-covalent bond, and the potential energy 3
density Vb, which is usually negative and associated with accumulation of charge between the nuclei. In clear 4
covalent bonds both the Laplacian and Eeb are negative. In less clear cases, where the Laplacian is slightly 5
positive, the value of Eeb can be used to make a further classification of the bond, from being slightly covalent 6
to purely ionic/non-bonded. In this classification, with ∇2ρ > 0, if Eeb is negative, the bond is called dative; if 7
Eeb is close to zero, the bond is metallic; if Ee
b is positive, the bond can be either ionic or Van der Waals. 8
In Table 5. we have listed all the parameters needed for a detailed topological analysis of the chemical bond 9
in the heterobimetallic systems considered here. The most salient feature is that for all the complexes the 10
density at the BCP is small and the Laplacian is positive, indicating that in none of the cases is a covalent 11
interaction indicated. Furthermore, Eeb is always slightly positive, suggesting that the bond can be classified as 12
between metallic and ionic. In Figure 7, we have also plotted the electron density difference between the 13
relaxed total electron density of the U-Re cluster and the electron density of the isolated fragments at the 14
position they have in the supermolecule. The blue certifies a local depletion of charge and the red a local 15
accumulation. What is evident from Figure 6, is that approximately at the BCP between U and Re, there is a 16
depletion of charge, i.e. the electrons move away from the center of the bond path that links the two atoms, a 17
clear sign that a closed-shell interaction and not a shared interaction is occurring. Furthermore, it is also clear 18
that there is a strong charge rearrangement on the uranium fragment in the supermolecule as compared to the 19
isolated fragment. This charge rearrangement is associated with the internal polarization described above for 20
the U-Ga-2 case that exhibits an orbital interaction term without significant charge transfer between the two 21
initially neutral fragments. 22
23
4. Conclusions 24

17
Several systems containing heterobimetallic bonds involving one f-block element were studied 1
computationally. Specifically the following molecules were investigated: [(η5-C5H5)2(THF)LuRu(η5-2
C5H5)(CO)2],19 [(η5-C5Me5)2(I)ThRu(η5-C5H5)(CO)2],20 [(η5-C5H5)2YRe(η5-C5H5)2],15 3
[{N(CH2CH2NSiMe3)3}URe(η5-C5H5)2],14 [Y{Ga(NArCh)2}{C(PPh2NSiH3)2}(CH3OCH3)2],17 4
[{N(CH2CH2NSiMe3)3}U{Ga(NArCH)2}(CH3OCH3)],18 [(η5-C5H5)3UGa(η5-C5Me5)],16 [Yb(η5-5
C5H5){Si(SiMe3)3(THF)2}],21 [(η5-C5H5)3U(SnPh3)],22 [(η5-C5H5)3U(SiPh3)],23 and (Ph[Me]N)3USi(SiMe3).24 6
Density functional calculations predict geometries in good agreement with experiment. CASSCF/CASPT2 7
calculations were also performed in order to better understand the nature of the interactions between the two 8
metal centers. The CASSCF wavefunction indicated that the electronic structure of these systems is not 9
multiconfigurational, and the CASSCF/CASPT2 and DFT results give the same description of the An/Ln-M 10
bond. Even though the actinides are known to have more diffuse f-electrons than the lanthanides, the f-11
electrons are localized on the Ln/An center and do not engage in bonding in any of the systems studied. 12
Consequently, the metal-metal interaction is qualitatively the same in all cases and no clear differences in 13
bonding can be identified between the 4f and 5f elements. From this perspective, the lanthanides and actinides 14
in these systems can be thought of as hard d-elements. 15
The results have demonstrated that: (i) the An/Ln-M bonds are primarily ionic; (ii) no ‘true’ σ and π bonds 16
are present in any of these systems, but rather σ-type donation from the metal ligand fragment to the Ln/An 17
fragment is seen in all cases; (iii) a π-donation is possible in the system containing a U-Re bond, but not 18
observed in the other systems, which reinforces the notion, as suggested previously, that this interaction is 19
extremely weak and not expected to alter the reactivity of the species. 20
21
ACKNOWLEDGMENT 22
This research was supported by Director, Office of Basic Energy Sciences, U.S. Department of Energy under 23
Contract n. USDOE/DE-SC002183, and the Materials Science of Actinides Center, an Energy Frontier 24
Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences 25

18
under Award Number DE-SC0001089. We also acknowledge support from the U.S. National Science 1
Foundation (CHE-0952054). 2
SUPPORTING INFORMATION The coordinates of the optimized structures are reported in Supporting 3
Information and are available free of charge. 4
5

19
1
Figure 1: Actinide containing systems. (Ph = C6H5, DME = dimethyl ether, TMS = SiMe3, and Cp* = C5Me5 2
Ar=2,6-dimethylphenyl). Formal oxidation states are indicated in superscript. 3
ThIV-RuII
UIV-ReI
UIV-GaI-1
UIII-GaI-2
UIV-Sn
UIV-Si-1
UIV-Si-2
4
5

20
1 Figure 2: Lanthanide containing systems. (Ph=C6H5, DME= dimethyl ether, TMS = SiMe3, and 2
THF=tetrahydrofuran). Formal oxidation states are indicated in superscript. 3
LuIII-RuII
YIII-ReI
YIII-GaI
YbII-Si
4
5
Figure 3: Select orbitals from the Y-Re and U-Re CASSCF calculations. The σ-donating orbitals are on the 6
left and the possible π-donating orbitals are on the right. 7

21
1
Figure 4: Select doubly occupied orbitals from the U-Ga-1 CASSCF calculation. The σ-donating orbital is 2
on the left and possible π-donating orbital is on the right. 3

22
1
2
3
Figure 5: σ-donation in U-Ga-1. Center: The orbital from U-Ga-1 fragment calculation with the highest 4 percent contribution from the both the U and Ga fragments. Left: The contributing orbital from the U 5 fragment. Right: The contributing orbital from the Ga fragment. 6
U Fragment U-Ga-1 Ga Fragment
Virtual
Occupied
23
27% 66%
1
Figure 6: U-Re HOMO-3 was plotted at isodensity values 0.03 and 0.02 for calculations performed at the 2 PBE/TZ2P level of theory in order to compare the geometry optimized in this work and that given in the 3 supporting information of the previously published paper. 14 4
5 1 6
7
Figure 7: Electron density difference between the relaxed electron density of the U-Re supermolecule and the 8 electron density of the separated fragments of U and Re at the position they have in the supermolecule. A blue 9 color indicates a depletion of charge and a red color an increase. 10

24
1
2
3
Table 1: Optimized bond distances in angstroms. 4
Species PBE CASPT2 (MP2) X-ray diffraction Sum of the Covalent Radii54
Lu-Ru 3.015 2.912 2.995 3.10
Th-Ru 3.070 2.910 3.028 3.26
Y-Re 2.998 2.983 2.962 3.17
U-Re 3.043 2.917 3.048 3.24
Y-Ga 3.119 (2.901) 3.1757 2.99
U-Ga-1 3.242 N/A 3.221/3.298 3.06
U-Ga-2 3.065 3.083 3.065/3.080 3.06
Yb-Si 3.022 2.912 (2.900) 3.032 2.92
U-Sn 3.238 N/A 3.166 3.23
U-Si-1 3.106 2.965 No Crystal Isolated 2.97
U-Si-2 3.003 N/A 3.091 2.97
5
Table 2: Bonding energy and charge transfer between the Ln/An and M fragments. 6

25
Bonding Energy
Charge Transfer Species
(kcal.mol-1) An/Ln to M Th-Ru -135.3 0.88 Lu-Ru -128.6 0.67 U-Re -128.2 0.74 Y-Re -156.2 0.89
U-Ga-1 -91.3 0.53 U-Ga-2 -17.8 -0.06 Y-Ga -95.0 0.46 U-Sn -120.8 0.49
U-Si-1 -131.4 0.50 U-Si-2 -139.1 0.57 Yb-Si -109.3 0.54
1
Table 3: Comparison between calculated geometries for U-Re from the current study and previous work. 14 2
Bond Length (Å) Current Study Calculated14 Experiment14
U-Re 3.043 3.062 3.048 U-N(2) 2.260 2.322 2.283 U-N(4) 2.272 2.307 2.274 U-N(1) 2.289 2.292 2.242 U-N(3) 2.775 2.766 2.681
Angles (°) N(4)-U-N(2) 103.0 106.0 107.1 N(1)-U-N(2) 110.3 112.4 112.6 N(3)-U-N(2) 69.1 68.1 69.4 Re-U-N(2) 110.7 111.7 110.7
N(1)-U-N(4) 108.6 103.9 105.4 N(3)-U-N(4) 69.2 68.6 69.6 Re-U-N(4) 121.3 120.3 119.1
N(3)-U-N(1) 67.3 68.3 69.5(2) Re-U-N(1) 102.9 102.7 101.83(16) Re-U-N(3) 168.4 169.3 169.74(14)
3
Table 4: Energy decomposition performed using Equation 1. The percentage of the total attractive interaction 4
of electrostatic and orbital interactions is given in parentheses. 5
26
Repulsive Interaction Attractive Interaction
Pauli Repulsion
Electrostatic Interaction
Orbital Interaction Species
(kcal.mol-1) (kcal.mol-1) (kcal.mol-1) Th-Ru 129.9 -193.4 (72.9%) -71.9 (27.1%) U-Re 160.3 -201.9 (66.7%) -101.0 (33.3%)
U-Ga-1 73.7 -124.8 (75.6%) -40.2 (24.4%) U-Ga-2 49.1 -41.0 (61.2%) -25.9 (38.8%) U-Sn 100.2 -159.1 (72,0) -61.9 (28.0%)
U-Si-1 99.4 -162.1 (70.2%) -68.7 (29.8%) U-Si-2 111.7 -168.6 (67.3%) -82.1 (32.7%) Y-Re 94.1 -171.9 (68.9%) -77.7 (31.1%) Lu-Ru 87.8 -163.4 (75.5%) -53.0 (24.5%) Yb-Si 59.9 -132.1 (78.1%) -37.1 (21.9%) Y-Ga 51.5 -110.1 (75.1%) -36.4 (24.9%)
1
Table 5: Theoretical bond critical point properties for the bimetallic species studied. All values are expressed 2
in atomic units. 3
ρb ∇ 2ρb Gb Gb/ρb Vb Eeb
Th-Ru 0.02271 0.02718 0.01145 0.50418 -0.004656 0.006796 U-Re 0.02659 0.04121 0.01773 0.66679 -0.007423 0.0103
U-Ga-1 0.01492 0.01275 0.005574 0.37359 -0.003189 0.002385 U-Ga-2 0.01561 0.02566 0.00862 0.55221 -0.002205 0.006415 U-Sn 0.02052 0.01193 0.005263 0.25648 -0.002982 0.002281
U-Si-1 0.02287 0.002005 0.005597 0.24473 -0.005096 0.0005013 U-Si-2 0.02469 0.006462 0.00805 0.32604 -0.001606 0.006444 Y-Re 0.02084 0.02199 0.01114 0.53455 -0.005649 0.005496 Lu-Ru 0.01809 0.0455 0.01215 0.67164 -0.000762 0.01139 Yb-Si 0.01612 0.04625 0.01189 0.73759 -0.0003323 0.01156 Y-Ga 0.01364 0.01108 0.005056 0.37067 -0.002285 0.002771
4
5
6
7

27
1. Collman, J. P.; Boulatov, R., Heterodinuclear transition-metal complexes with multiple metal-metal 1 bonds. Angewandte Chemie-International Edition 2002, 41 (21), 3948-3961. 2 2. Cotton, F. A.; Curtis, N. F.; Johnson, B. F. G.; Mague, J. T.; Wood, J. S.; Harris, C. B.; Robinson, W. 3 R.; Lippard, S. J., Mononuclear + Polynuclear Chemistry of Rhenium (3) - Its Pronounced Homophilicity. 4 Science 1964, 145 (363), 1305-&. 5 3. Cotton, F. A., Metal-Metal Bonding in [Re2x8]2- Ions and Other Metal Atom Clusters. Inorganic 6 Chemistry 1965, 4 (3), 334-&. 7 4. Cotton, F. A.; Walton, R. A., Multiple Bonds between Metal Atoms. 2nd ed.; Clarendon Press: Oxford, 8 1993. 9 5. Chisholm, M. H.; Patmore, N. J., Studies of electronic coupling and mixed valency in metal-metal 10 quadruply bonded complexes linked by dicarboxylate and closely related ligands. Accounts of Chemical 11 Research 2007, 40 (1), 19-27. 12 6. Greenwood, B. P.; Rowe, G. T.; Chen, C. H.; Foxman, B. M.; Thomas, C. M., Metal-Metal Multiple 13 Bonds in Early/Late Heterobimetallics Support Unusual Trigonal Monopyramidal Geometries at both Zr and 14 Co. Journal of the American Chemical Society 2010, 132 (1), 44-+. 15 7. Liddle, S. T.; Mills, D. P., Metal-metal bonds in f-element chemistry. Dalton Transactions 2009, (29), 16 5592-5605. 17 8. Nolan, S. P.; Porchia, M.; Marks, T. J., Organo-F-Element Thermochemistry - Actinide Group-14 18 Element and Actinide Transition-Element Bond Disruption Enthalpies and Stoichiometric Catalytic Chemical 19 Implications Thereof in Heterobimetallic Tris(Cyclopentadienyl)Uranium(Iv) Compounds. Organometallics 20 1991, 10 (5), 1450-1457. 21 9. Roos, B. O.; Malmqvist, P. A.; Gagliardi, L., Exploring the actinide-actinide bond: Theoretical studies 22 of the chemical bond in Ac-2, Th-2, Pa-2, and U-2. Journal of the American Chemical Society 2006, 128 (51), 23 17000-17006. 24 10. Raab, J.; Lindh, R. H.; Wang, X. F.; Andrews, L.; Gagliardi, L., A combined experimental and 25 theoretical study of uranium polyhydrides with new evidence for the large complex UH4(H-2)(6). Journal of 26 Physical Chemistry A 2007, 111 (28), 6383-6387. 27 11. Infante, I.; Raab, J.; Lyon, J. T.; Liang, B.; Andrews, L.; Gagliardi, L., Experimental and theoretical 28 evidence for U(C6H6) and Th(C6H6) complexes. Journal of Physical Chemistry A 2007, 111 (47), 11996-29 12000. 30 12. Wang, X. F.; Andrews, L.; Infante, I.; Gagliardi, L., Infrared spectra of the WH4(H-2)(4) complex in 31 solid hydrogen. Journal of the American Chemical Society 2008, 130 (6), 1972-1978. 32 13. Wang, X. F.; Andrews, L.; Gagliardi, L., Infrared spectra of ThH2, ThH4, and the hydride bridging 33 ThH4(H-2)(x) (x=1-4) complexes in solid neon and hydrogen. Journal of Physical Chemistry A 2008, 112 (8), 34 1754-1761. 35 14. Gardner, B. M.; McMaster, J.; Lewis, W.; Liddle, S. T., Synthesis and structure of 36 [{N(CH2CH2NSiMe3)(3)}URe(eta(5)-C5H5)(2)]: a heterobimetallic complex with an unsupported uranium-37 rhenium bond. Chemical Communications 2009, (20), 2851-2853. 38 15. Butovskii, M. V.; Tok, O. L.; Wagner, F. R.; Kempe, R., Bismetallocenes: Lanthanoid-transition-metal 39 bonds through alkane elimination. Angewandte Chemie-International Edition 2008, 47 (34), 6469-6472. 40 16. Minasian, S. G.; Krinsky, J. L.; Rinehart, J. D.; Copping, R.; Tyliszczak, T.; Janousch, M.; Shuh, D. K.; 41 Arnold, J., A Comparison of 4f vs 5f Metal-Metal Bonds in (CpSiMe3)(3)M-ECp* (M = Nd, U; E = Al, Ga; 42 Cp* = C5Me5): Synthesis, Thermodynamics, Magnetism, and Electronic Structure. Journal of the American 43 Chemical Society 2009, 131 (38), 13767-13783. 44 17. Liddle, S. T.; Mills, D. P.; Gardner, B. M.; McMaster, J.; Jones, C.; Woodul, W. D., A 45 Heterobimetallic Gallyl Complex Containing an Unsupported Ga-Y Bond. Inorganic Chemistry 2009, 48 (8), 46 3520-3522. 47 18. Liddle, S. T.; McMaster, J.; Mills, D. P.; Blake, A. J.; Jones, C.; Woodul, W. D., sigma and pi 48 Donation in an Unsupported Uranium-Gallium Bond. Angewandte Chemie-International Edition 2009, 48 (6), 49 1077-1080. 50

28
19. Beletskaya, I. P.; Voskoboynikov, A. Z.; Chuklanova, E. B.; Kirillova, N. I.; Shestakova, A. K.; 1 Parshina, I. N.; Gusev, A. I.; Magomedov, G. K. I., Bimetallic Lanthanide Complexes with Lanthanide 2 Transition-Metal Bonds - Molecular-Structure of (C4h8o)(C5h5)2lu-Ru(Co)2(C5h5) - the Use of La-139 Nmr-3 Spectroscopy. Journal of the American Chemical Society 1993, 115 (8), 3156-3166. 4 20. Sternal, R. S.; Brock, C. P.; Marks, T. J., Metal Metal Bonds Involving Actinides - Synthesis and 5 Characterization of a Complex Having an Unsupported Actinide to Transition-Metal Bond. Journal of the 6 American Chemical Society 1985, 107 (26), 8270-8272. 7 21. Corradi, M. M.; Frankland, A. D.; Hitchcock, P.; Lappert, M. F.; Lawless, G. A., Synthesis, structure 8 and reactivity of [Yb(eta-C(5)Me(5)){Si(SiMe(3))(3)}(thf)(2)]. Chemical Communications 1996, (20), 2323-9 2324. 10 22. Porchia, M.; Casellato, U.; Ossola, F.; Rossetto, G.; Zanella, P.; Graziani, R., Synthesis and Crystal-11 Structure of Triscyclopentadienyl(Triphenyltin)Uranium - the 1st Example of a Uranium Tin Bond. Journal of 12 the Chemical Society-Chemical Communications 1986, (13), 1034-1035. 13 23. Porchia, M.; Brianese, N.; Casellato, U.; Ossola, F.; Rossetto, G.; Zanella, P.; Graziani, R., Tri(Eta-14 Cyclopentadienyl)Uranium(Iv) Silyl and Siloxide Compounds - Crystal-Structure of [U(Eta-5-15 C5h5)3(Osiph3)] - Insertion of Isocyanide into a Uranium-Silicon Bond. Journal of the Chemical Society-16 Dalton Transactions 1989, (4), 677-681. 17 24. Diaconescu, P. L.; Odom, A. L.; Agapie, T.; Cummins, C. C., Uranium-group 14 element single bonds: 18 Isolation and characterization of a uranium(IV) silyl species. Organometallics 2001, 20 (24), 4993-4995. 19 25. Roos, B. O.; Taylor, P. R., A Complete Active Space Scf Method (Casscf) Using a Density-Matrix 20 Formulated Super-Ci Approach. Chemical Physics 1980, 48 (2), 157-173. 21 26. Andersson, K.; Malmqvist, P. A.; Roos, B. O.; Sadlej, A. J.; Wolinski, K., 2nd-Order Perturbation-22 Theory with a Casscf Reference Function. Journal of Physical Chemistry 1990, 94 (14), 5483-5488. 23 27. Andersson, K.; Malmqvist, P. A.; Roos, B. O., 2nd-Order Perturbation-Theory with a Complete Active 24 Space Self-Consistent Field Reference Function. Journal of Chemical Physics 1992, 96 (2), 1218-1226. 25 28. Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmel, C., Electronic-Structure Calculations on 26 Workstation Computers - the Program System Turbomole. Chemical Physics Letters 1989, 162 (3), 165-169. 27 29. Perdew, J. P.; Burke, K.; Ernzerhof, M., Generalized gradient approximation made simple. Physical 28 Review Letters 1996, 77 (18), 3865-3868. 29 30. Hellweg, A.; Hattig, C.; Hofener, S.; Klopper, W., Optimized accurate auxiliary basis sets for RI-MP2 30 and RI-CC2 calculations for the atoms Rb to Rn. Theoretical Chemistry Accounts 2007, 117 (4), 587-597. 31 31. Schafer, A.; Huber, C.; Ahlrichs, R., Fully Optimized Contracted Gaussian-Basis Sets of Triple Zeta 32 Valence Quality for Atoms Li to Kr. Journal of Chemical Physics 1994, 100 (8), 5829-5835. 33 32. Cao, X. Y.; Dolg, M., Valence basis sets for relativistic energy-consistent small-core lanthanide 34 pseudopotentials. Journal of Chemical Physics 2001, 115 (16), 7348-7355. 35 33. Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R., Auxiliary basis sets for main row atoms and 36 transition metals and their use to approximate Coulomb potentials. Theoretical Chemistry Accounts 1997, 97 37 (1-4), 119-124. 38 34. Eichkorn, K.; Treutler, O.; Ohm, H.; Haser, M.; Ahlrichs, R., Auxiliary Basis-Sets to Approximate 39 Coulomb Potentials. Chemical Physics Letters 1995, 240 (4), 283-289. 40 35. Von Arnim, M.; Ahlrichs, R., Performance of parallel TURBOMOLE for density functional 41 calculations. Journal of Computational Chemistry 1998, 19 (15), 1746-1757. 42 36. Moller, C.; Plesset, M. S., Note on an approximation treatment for many-electron systems. Physical 43 Review 1934, 46 (7), 0618-0622. 44 37. Bartlett, R. J., Many-Body Perturbation Theory and Coupled Cluster Theory for Electron Correlation in 45 Molecules. Ann. Rev. Phys. Chem. 1981, 32, 359-401. 46 38. Hess, B. A., Relativistic Electronic-Structure Calculations Employing a 2-Component No-Pair 47 Formalism with External-Field Projection Operators. Physical Review A 1986, 33 (6), 3742-3748. 48 39. Roos, B. O.; Lindh, R.; Malmqvist, P. A.; Veryazov, V.; Widmark, P. O.; Borin, A. C., New 49 Relativistic Atomic Natural Orbital Basis Sets for Lanthanide Atoms with Applications to the Ce Diatom and 50 LuF3. Journal of Physical Chemistry A 2008, 112 (45), 11431-11435. 51

29
40. Roos, B. O.; Lindh, R.; Malmqvist, P. A.; Veryazov, V.; Widmark, P. O., New relativistic ANO basis 1 sets for transition metal atoms. Journal of Physical Chemistry A 2005, 109 (29), 6575-6579. 2 41. Roos, B. O.; Lindh, R.; Malmqvist, P. A.; Veryazov, V.; Widmark, P. O., New relativistic ANO basis 3 sets for actinide atoms. Chemical Physics Letters 2005, 409 (4-6), 295-299. 4 42. Aquilante, F.; De Vico, L.; Ferre, N.; Ghigo, G.; Malmqvist, P. A.; Neogrady, P.; Pedersen, T. B.; 5 Pitonak, M.; Reiher, M.; Roos, B. O.; Serrano-Andres, L.; Urban, M.; Veryazov, V.; Lindh, R., Software News 6 and Update MOLCAS 7: The Next Generation. Journal of Computational Chemistry 2010, 31 (1), 224-247. 7 43. Aquilante, F.; Gagliardi, L.; Pedersen, T. B.; Lindh, R., Atomic Cholesky decompositions: A route to 8 unbiased auxiliary basis sets for density fitting approximation with tunable accuracy and efficiency. Journal of 9 Chemical Physics 2009, 130 (15), 154107. 10 44. Aquilante, F.; Lindh, R.; Pedersen, T. B., Analytic derivatives for the Cholesky representation of the 11 two-electron integrals. Journal of Chemical Physics 2008, 129 (3), 034106. 12 45. Aquilante, F.; Malmqvist, P. A.; Pedersen, T. B.; Ghosh, A.; Roos, B. O., Cholesky decomposition-13 based multiconfiguration second-order perturbation theory (CD-CASPT2): Application to the spin-state 14 energetics of Co-III(diiminato)(NPh). Journal of Chemical Theory and Computation 2008, 4 (5), 694-702. 15 46. Aquilante, F.; Pedersen, T. B.; Lindh, R., Low-cost evaluation of the exchange Fock matrix from 16 Cholesky and density fitting representations of the electron repulsion integrals. Journal of Chemical Physics 17 2007, 126 (19), 194106. 18 47. Gagliardi, L.; La Manna, G.; Roos, B. O., On the reaction of a uranium atom with a nitrogen molecule: 19 a theoretical attempt. Faraday Discussions 2003, 124, 63-68. 20 48. La Macchia, G.; Brynda, M.; Gagliardi, L., Quantum chemical calculations predict the diphenyl 21 diuranium compound [PhUUPh] to have a stable (1)A(g) ground state. Angewandte Chemie-International 22 Edition 2006, 45 (37), 6210-6213. 23 49. Gagliardi, L.; Heaven, M. C.; Krogh, J. W.; Roos, B. O., The electronic spectrum of the UO2 molecule. 24 Journal of the American Chemical Society 2005, 127 (1), 86-91. 25 50. Gagliardi, L.; Roos, B. O., Multiconfigurational quantum chemical methods for molecular systems 26 containing actinides. Chemical Society Reviews 2007, 36 (6), 893-903. 27 51. Velde, G. T.; Bickelhaupt, F. M.; Baerends, E. J.; Guerra, C. F.; Van Gisbergen, S. J. A.; Snijders, J. 28 G.; Ziegler, T., Chemistry with ADF. Journal of Computational Chemistry 2001, 22 (9), 931-967. 29 52. Vosko, S. H.; Wilk, L.; Nusair, M., Accurate Spin-Dependent Electron Liquid Correlation Energies for 30 Local Spin-Density Calculations - a Critical Analysis. Canadian Journal of Physics 1980, 58 (8), 1200-1211. 31 53. Vanlenthe, E.; Baerends, E. J.; Snijders, J. G., Relativistic Total-Energy Using Regular 32 Approximations. Journal of Chemical Physics 1994, 101 (11), 9783-9792. 33 54. M. Mantina, R. V., C. J. Cramer, D. G. Truhlar, "Atomic Radii of the Elements" in the CRC Handbook 34 of Chemistry and Physics. 91st ed.; CRC Press: Boca Raton, FL, 2010-2011. 35 55. Cordero, B.; Gomez, V.; Platero-Prats, A. E.; Reves, M.; Echeverria, J.; Cremades, E.; Barragan, F.; 36 Alvarez, S., Covalent radii revisited. Dalton Transactions 2008, (21), 2832-2838. 37 56. Pyykko, P.; Atsumi, M., Molecular Single-Bond Covalent Radii for Elements 1-118. Chemistry-a 38 European Journal 2009, 15 (1), 186-197. 39 57. Swart, M.; Van Duijnen, P. T.; Snijders, J. G., A charge analysis derived from an atomic multipole 40 expansion. Journal of Computational Chemistry 2001, 22 (1), 79-88. 41 58. Bader, R. F. W., Atoms in molecules : a quantum theory. Clarendon Press: Oxford ; New York, 1990; p 42 xviii, 438 p. 43 59. Bader, R. F. W., A bond path: A universal indicator of bonded interactions. Journal of Physical 44 Chemistry A 1998, 102 (37), 7314-7323. 45 60. Bianchi, R.; Gervasio, G.; Marabello, D., Experimental electron density analysis of Mn-2(CO)(10): 46 Metal-metal and metal-ligand bond characterization. Inorganic Chemistry 2000, 39 (11), 2360-2366. 47 48 49




















