
BIM Guidelines Inform Facilities Management Databases - MDPI
Upload
independentCategory
view
0download
0
Notch3 Cooperates With the EGFR Pathway to ModulateApoptosis Through The Induction Of Bim
Jun Konishi1,*, Fuming Yi1,*, Xinping Chen1, Huan Vo1, David P. Carbone1, and Thao P.Dang1
1Division of Hematology and Medical Oncology, Vanderbilt University Medical Center, Nashville,TN, USA
AbstractNotch signaling is a highly conserved pathway important for normal embryonic development andas well as cancer. We previously demonstrated a role for Notch3 in lung cancer pathogenesis.Notch3 inhibition resulted in tumor apoptosis and growth suppression. In vitro, these effects wereenhanced when the EGFR pathway was also inhibited, suggesting significant crosstalk betweenthese two pathways. How Notch3 and EGFR/MAPK pathways cooperate in modulating apoptosisis not yet known. In this study, we provide evidence that Notch3 regulates Bim, a BH-3-onlyprotein via MAPK signaling. Furthermore, loss of Bim expression prevents tumor apoptosisinduced by Notch3 inhibition. Using γ-secretase inhibitor and erlotinib in a xenograft model, Biminduction and tumor inhibition were enhanced compared to either agent alone, consistent with ourprevious observation of significant synergism between Notch and EGFR/ras/MAPK signaling.Thus, our data support the hypothesis that Notch3 not only plays a crucial role in lung cancerthrough regulating apoptosis but also cooperates with the EGFR/MAPK pathway in modulatingBim.
KeywordsNotch3; Bim; Apoptosis; Lung Cancer
INTRODUCTIONNotch3 belongs to a family of proteins essential for cellular differentiation in a variety ofdeveloping tissues. In mammals, there are four Notch receptors (Notch1 to Notch4) and twofamilies of ligands, Jagged (Jagged1, 2) and Delta-like (Dll1, −3, −4). Ligand binding resultsin two successive proteolytic cleavages by an ADAM-type metalloprotease and by a γ-secretase, respectively (for a recent review see (Roy et al., 2007)). The released Notchintracellular domain (NICD) translocates to the nucleus, binds to transcription factor CSL(CBF1, Sel, Lag-1), and induces expression of target genes, such as the Hairy-enhancer ofSplit (HES) and hairy and Enhancer-of-split related with YRPW motif (Hey) gene families.
Aberrant activation of Notch proteins is associated with cancer phenotypes (Das et al., 2004;Curry et al., 2005; Duechler et al., 2005; Reedijk et al., 2005). Notch3 is overexpressed inabout 40% of non-small cell lung cancers (NSCLC), and suppression of Notch3 results in
Users may view, print, copy, download and text and data- mine the content in such documents, for the purposes of academic research,subject always to the full Conditions of use: http://www.nature.com/authors/editorial_policies/license.html#terms
Correspondence to: T.P. Dang, Vanderbilt-Ingram Cancer Center, 658 PRB, Nashville, TN 37232, USA. [email protected].*Both authors contributed equally to the paper
NIH Public AccessAuthor ManuscriptOncogene. Author manuscript; available in PMC 2010 July 28.
Published in final edited form as:Oncogene. 2010 January 28; 29(4): 589–596. doi:10.1038/onc.2009.366.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

the loss of the malignant phenotype both in vitro and in vivo models (Haruki et al., 2005;Konishi et al., 2007). Tumor suppression is enhanced when Notch inhibition is combinedwith epidermal growth factor (EGF) pathway inhibitors in vitro. These observations supportthe findings in both the development and the cancer literature that Notch and EGFR/ras/MAPK pathways are interdependent. Whether ras enhances or antagonizes Notch signalingin development appears to be context-dependent. In cancer, however, many studies havesuggested that the ability of ras or Notch to transform cells depends on the cooperativerelationship between them (Dievart et al., 1999; Fitzgerald et al., 2000; Weijzen et al., 2002;Haruki et al., 2005).
The role of the Notch pathway in tumorigenesis also involves the regulation of the apoptoticpathway. The Bcl-2-related proteins are key regulators of apoptosis. There are threesubfamilies, the pro-survival Bcl-2 like proteins, such as Bcl-2, Bcl-xL and Mcl-1, the pro-apoptotic proteins Bax and Bak, and the BH3-only proteins, which share homology with theBcl-2 family only in the BH3 region (Heiser and Hebrok, 2004). The BH3-only proteinsfunction mainly as initiators of apoptosis. They are activated by cellular stress, such as DNAdamage in the case of Noxa and Puma, or by growth factor deprivation in the case of Hrkand Bim. Other BH3-only proteins such as Bid are activated by death receptors viacaspase-8 (Borner, 2003). Recently, it has been reported that Bim is required to induceapoptosis in several cancer types, including lung cancer (Gomez-Bougie et al., 2005; Gonget al., 2007). Little is known about the relationship between the Notch3 pathway and Bimregulation. Since Bim is induced by growth factor deprivation, we hypothesize that thecrosstalk between EGFR/ras and Notch3 modulates apoptosis through the regulation of Bim.
In this study, we demonstrate that Notch3 modulates apoptosis through the Bcl-2 proteinfamily. We also provide evidence that the Notch3 and MAPK pathways modulate Bim toregulate apoptosis. Although Notch3 is known to regulate apoptosis through other pathwayssuch as the NF- κB pathway, this is the first study linking Bim to Notch3-dependentapoptosis in lung cancer.
RESULTSNotch3 modulates apoptosis through the regulation of pERK and Bcl-2-related proteinsbut has no effect on the Akt/PI3K pathway
Consistent with our earlier work, loss of Notch3 activity resulted in the downregulation ofpERK, a member of the MAPK family. Interestingly, while Notch3 modulated the MAPKpathway, no effect on phospho-Akt, PI3K, PKC-α and Foxo3a was observed (Figure 1A).Notch3 affects the PI3K-Akt pathway in some experimental systems but not others,suggesting that, unlike the MAPK pathway, cellular context is more important for theNotch3 modulation of the PI3K-pAkt pathway (Campos et al., 2002; Wang et al., 2007).Using Notch3 siRNA, we also observed that Notch regulates both pro-apoptotic and pro-survival proteins (Figure 1B). Although no change was detected in Mcl-1, phospho-Bcl-2(pBCl2) and Bcl-xL were downregulated in HCC2429 when the cells were transfected withNotch3 siRNA, compared to control siRNA. Conversely, the pro-apoptotic protein Bax wasupregulated. Taken together, these observations are consistent with the hypothesis thatNotch3 is oncogenic in lung cancer.
BH3-only proteins is a subset of the Bcl-2 family. They function as damage sensors andrespond to distinct forms of cellular stress or DNA damage (Borner, 2003). Since the Notchpathway is known to crosstalk with the ras/MAPK pathway, and pro-apoptotic Bim isinduced by growth factor deprivation, we hypothesized that Bim is the mediator in Notch3regulation of apoptosis. This hypothesis is consistent with our earlier observation thatNotch3-mediated apoptosis is enhanced in the presence of growth factor deprivation (Haruki
Konishi et al. Page 2
Oncogene. Author manuscript; available in PMC 2010 July 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

et al., 2005). In the present study, Bim expression was increased after transfection withNotch3 siRNA (Figure 1C). Phospho-BAD, a pro-apoptotic protein that is modulated bycytokines/growth factors withdrawal, was similarly affected. Puma functions as sensor forDNA damage and has been shown to be a direct target of p53-mediated apoptosis. On theother hand, Bid is a target of the death receptor ligands. No change in the levels of Puma andBid was observed when tumor cells were transfected with Notch3 siRNA, suggesting that insome lung cancers Notch3-dependent apoptosis is not mediated by p53 or the deathreceptors, but by the modulation of growth factors and cytokines. We detected significantlyhigher mRNA expression of Bim in tumor cells transfected with Notch3 siRNA, comparedto siRNA control (Figure 1D). It is known that the Bim promoter possesses binding sites forFoxo3a, Mybs, and c-Jun, and mutation of any one of these sites abolishes Bim transcriptionin response to growth factor deprivation (Biswas et al., 2007). Since we have shown thatNotch3 has no effect on Foxo3a, transcriptional regulation of Bim by Notch3 is indirect andmore likely through c-jun and the ras/MAPK pathway.
Inhibition of Notch3 with siRNA induces apoptosisInhibition of the Notch pathway by either a dominant-negative receptor (DN) or by the γ-secretase-inhibitor MRK-003 results in tumor apoptosis (Haruki et al., 2005). Since both theDN receptor and γ-secretase-inhibitor potentially target all of the Notch family receptors,we used the small interference RNA (siRNA) system to determine whether theseobservations are specific to Notch3. Apoptosis markers such as cleaved PARP andcytochrome-c are induced by Notch3 siRNA (Figure 2A). Furthermore, caspase-3 activitywas enhanced in lung cancer cells treated with Notch3 siRNA (Fig. 2B). While alterations inapoptotic markers PARP and cytochrome-c were observed, no change in apoptosis wasdetected with annexin V staining when cells were maintained in 10% FCS (Figure 2C).However, in the presence of growth deprivation, the percentage of apoptotic cells wassignificantly increased in Notch3 siRNA-treated cells compared with vector control. Thisobservation is consistent with our previous studies with the γ-secretase inhibitor MRK-003and Notch3 DN, in which apoptosis was seen in serum-free conditions (Haruki et al., 2005;Konishi et al., 2007). These results demonstrate that while Notch3 can modulate manycomponents of apoptosis such as Bcl-xL and Bax in full-serum conditions, a noticeableeffect on apoptosis requires growth factor deprivation.
Bim is necessary for induction of apoptosis in Notch3 knockout cellsTo determine whether Bim is necessary for Notch3-dependent apoptosis, we used BimsiRNA to suppress Bim in HCC2429 cells transfected with Notch3 siRNA. The loss of Bimabrogated the induction of PARP by Notch3 siRNA (Figure 3A). In further support of ourhypothesis, we observed that Bim knockdown abolished Notch3 siRNA induced apoptosisin the absence of serum compared with vector control suggesting that the effect of Notch3on apoptosis is dependent on Bim (Figure 3B). A small increase in apoptosis was detected inBim negative cells in both serum-free and 10% FCS conditions. Interestingly, Bimdownregulation also resulted in downregulation of Notch3 suggesting a feedback loopexisted between the two pathways.
Effect of Notch3 on Bim depends on intact MAPK signalingTo better define the interaction between Notch3 and EGFR/ras/MAPK pathways in Bimmodulation, we examined the effect of the MEK inhibitor U0126 and the EGFR inhibitorAG1478 on lung cancer cells treated with Notch3 siRNA. Interestingly, Bim expression isinduced with the MEK inhibitor U0126, while the added loss of Notch3 signaling did notaugment Bim expression (Figure 4A). One explanation for this observation is that the effectNotch3 has on Bim is dependent on intact MAPK signaling and that inhibiting Notch3 bysiRNA is not of added benefit. On the other hand, when HCC2429 cells were treated with
Konishi et al. Page 3
Oncogene. Author manuscript; available in PMC 2010 July 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

both Notch3 siRNA and AG1478 at 5 µM, Bim expression was induced significantlycompared to Notch3 siRNA, U0126 or AG1478 alone (Figure 4B). The Akt-PI3K pathwayregulates Bim through phosphorylation of the transcription factor Foxo3a (Urbich et al.,2005). Thus, inhibiting both Notch3 and EGFR synergistically affects Bim through bothFoxo3a and MAPK signaling.
To confirm that Notch3 regulates Bim is through MAPK, we transfected activated Notch3(N3DA) into COS cells treated with either U0126 or AG1478. As expected, U0126prevented Bim suppression by activated Notch3, further supporting our contention that theanti-apoptotic effect of Notch3 is MAPK-dependent (Figure 4C). On the other hand,inhibition of EGFR with AG1478 could only partially reverse the effect of activated Notch3on Bim expression despite near suppression of pERK, suggesting that Bim is also modulatedby a MAPK-independent pathway (Figure 4D).
Gamma-secretase and EGFR tyrosine kinase inhibition enhance anti-tumor activity in vivoand Bim expression
We previously observed that in vitro the combination of γ-secretase inhibitor MRK-003 andthe EGFR tyrosine kinase inhibitor (AG1478) was more effective in inhibiting tumor growththan either agent alone (Konishi et al., 2007). To determine whether the combination of γ-secretase inhibitor MRK-003 and the EGFR tyrosine kinase inhibitor erlotinib also enhancestumor activity in vivo, we utilized a xenograft model. The combination of MRK-003 anderlotinib significantly reduced tumor size compared with either single agent alone (Fig. 5A).We also noted that the combination of MRK-003 and erlotinib resulted in greater expressionof Bim in the mouse tumors, compared to either agent alone (Figure 5B). Given the role ofNotch pathway in angiogenesis, we tumor measured microvessels density (MVD) (data notshown). No change in tumor MVD was observed, supporting the hypothesis that mechanismof tumor inhibition in vivo involves Bim modulation. Finally, whether this effect is due toNotch inhibition is uncertain due to the non-specificity of GSI. However, we previouslyhave demonstrated that loss of Notch3 rendered the GSI ineffective, suggesting that at leastour lung cancer model, the antitumor effect may be Notch3 dependent (Konishi et al., 2007).
DISCUSSIONLike Notch, the Bcl-2 protein family members play central roles in both development andcancer, facilitating strict organ morphogenesis during embryonic development andmaintenance of tissue homeostasis. These proteins are regulators of programmed cell deaththrough the integration of diverse extra- and intracellular death signals. In this study, wedemonstrated that loss of Notch3 resulted in downregulation of the pro-survival proteins,Bcl-2 and Bcl-xL and upregulation of the pro-apoptotic protein Bax, as well as the BH3-only proteins Bim and Bad. In tumorigenesis, activated Notch3 has been shown to induce T-cell leukemia through the constitutive activation of NF-κB (Bellavia et al., 2000). ActivatedNotch1 has been shown to inhibit p53-mediated apoptosis (Bocchetta et al., 2003; Beverly etal., 2005). In this study, we examined the effect of Notch3 on both the NF-κB pathway andp53 using Notch3 siRNA. While our findings indicate that Bim is a target of Notch3signaling, we were unable to discern appreciable changes in the levels of p53 or NF-κB-related proteins (data not shown), suggesting that the role of Notch3 in apoptosis is distinct,and context dependent.
Bim is a BH3-only member of the Bcl-2-like family of proteins. Loss or withdrawal ofcytokines and growth factors specifically induce its expression. Once Bim is activated, itbinds and inactivates Bcl-2-like pro-survival proteins, leading to cytochrome c release frommitochondria and caspase activation. Furthermore, Bim is required to mediate EGFRinhibitor-induced apoptosis in lung cancer cells, also supporting a significant interaction
Konishi et al. Page 4
Oncogene. Author manuscript; available in PMC 2010 July 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

between Bim and the EGFR/ras/MAPK pathway (Costa et al., 2007; Gong et al., 2007;Wang et al., 2007). In this paper, we demonstrate not only that the loss of Notch3 results inupregulation of Bim, but moreover that Bim expression is further enhanced when both theEGFR and Notch3 pathways were inhibited. This finding provides additional evidence forthe crosstalk between the EGFR and Notch3 in modulating apoptosis.
While Notch signaling regulates apoptosis through the NF-κB and p53 pathways in somecells, Bim appears to be necessary for the induction of Notch3-dependent apoptosis in lungcancer. This finding is supported by the recent observation that Notch inhibition with a γ-secretase inhibitor upregulates Bim in malignant melanomas (Qin et al., 2004). While it ispossible that this effect is unrelated to Notch inhibition due to the potential lack ofspecificity of the γ-secretase inhibitors, our siRNA data suggest that Bim upregulationresults from specific Notch3 knockdown, and that Notch-induced apoptosis is indeeddependent on this upregulation of Bim.
Withdrawal of growth factors results in induction of Bim through either of the two majorEGFR-dependent pathways: the Raf/MAPK and Akt/PI3K (Shinjyo et al., 2001). The Akt/PI3K pathway is known to negatively affect Bim through downregulation of Foxo3a. In ourstudy, no change in pAkt or pFoxoa3 was noted in the lung cancer cell lines transfected withNotch3 siRNA, indicating that Notch3 regulates Bim expression through the MAPKpathway and not the Akt pathway. Activated Notch3 was unable to rescue U0126 inducedapoptosis, providing further evidence that Notch3 modulation may be entirely dependent onMAPK pathway. This contention is consistent with our previous findings that Notch3regulates the MAPK activation through modulating MAPK phosphatase.
In summary, we have characterized one mechanism by which Notch3 plays an importantrole in the biology of cancer through the modulation of apoptosis. While the crucial role ofNotch signaling in apoptosis has been well-established in the context of cancer as well asdevelopment, the mechanism by which Notch3 affects apoptosis in lung cancer remainedobscure to date. In this study, we provided evidence that Bim is necessary for Notch3-dependent apoptosis and that the effect of Notch3 on Bim is through MAPK regulation.
Finally, we previously observed a cooperative relationship between the Notch3 pathway andthe EGFR pathway in maintaining the tumor phenotype. Our data have shown that thiscooperative relationship also involves the modulation of Bim. Interestingly, Bim expressionis upregulated in lung cancer cell lines sensitive to inhibitors of EGFR signaling, but not inresistant cell lines (Gong et al., 2007). Taken together, these observations suggest that theconcomitant inhibition of Notch and EGFR pathways represents a rational strategy forpromoting apoptosis in lung cancer and potentially overcoming treatment resistance.
MATERIALS AND METHODSCell lines and inhibitors
The Notch3 expressing lung cancer cell line, HCC2429, was established as previouslydescribed (Dang et al., 2000). The NSCLC cell line H460 and COS cell line were obtainedfrom American Type Culture Collection (ATCC) and maintained in RPMI with 10% FCS.Activated Notch3 was created previously and cloned into pBabe retroviral vector. AG1478and U0126 were obtained from Calbiochem (La Jolla, CA), and erlotinib was obtained fromGenetech, Inc (San Francisco, CA). The formulation and the in vivo dosing of γ-sectretaseinhibitor, MRK-003, was provided by Merck & Co., Inc and was described previously(Lewis et al., 2007).
Konishi et al. Page 5
Oncogene. Author manuscript; available in PMC 2010 July 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Notch3 and Bim Small interfering RNAsNotch3-overexpressing cell lines HC2429 and H460 were seeded at 1.5 × 105 cells per 6-well plate the day before transfection. The Notch3 siRNA sequence is 5’-CACCUAUAACUGCCAGUGC-3’ and was synthesized by Qiagen. Cells were transfectedwith 100 nmol/L siRNA in Opti-MEM medium (Invitrogen, Calsbad, CA) usingLipofectAMINE 2000 (Invitrogen, Calsbad, CA) according to the manufacturer’srecommendation. The efficiency of siRNA transfection was measured with Western blotanalysis. Bim siRNA (catalog number M-004383-01-0010) was purchased from DharmaconResearch (Lafayette, CO). An unspecific (non-silencing) siRNA against the target sequence5′ AAT TCT CCG AAC GTG TCA CGT TT 3′ (cat. no. 80-11310, Xeragon) was used ascontrols.
Caspase-3 Cellular ActivityHCC2429 cells was transfected with Notch3 siRNA or control as described above. After 24or 48 hours, the cells were harvested in lysis buffer, and the cell extracts were used todetermine caspase-3 activity, using the Caspase-3 Cellular Activity Assay Kit (Calbiochem,La Jolla, CA). Caspase-3 activity was measured in cell lysates in the presence and absenceof Caspase-3 inhibitor at 0.1 µM final concentration.
Apoptosis AnalysisCells transfected with Notch3 siRNA for 48 hours were maintained in 10% FCS-RPMI orserum-free medium. These cells were stained with FITC-conjugated annexin V andpropidium iodide using Annexin V-FITC Apoptosis Detection kit (Calbiochem, La Jolla,CA). The percentage of apoptotic cells was determined with a Beckman Coulter FACSCalibur Flow Cytometer. In dual Notch3 and Bim siRNA transfection experiments, the cellswere transfected with Bim siRNA twenty-four hours after Notch3 siRNA transfection.Twenty-four hours after Bim siRNA transfection, the cells were harvested and analyzed forapoptosis, as described above. Caspase-3 activity was measured using the Caspase-3Cellular Activity Assay Kit (Calbiochem, La Jolla, CA) according to manufacture’srecommendation.
RNA Extraction and Real-Time PCRTotal RNA was extracted with Trizol (Invitrogen, Carlsbad, CA) from HCC2429 cells 24hrs after transfection of either Notch3 siRNA or control siRNA. RNA was reversetranscribed with the iScript cDNA synthesis kit (Bio-RAD, Hercules, CA). Real-time PCRwas performed with the iQ5 multicolor Real-Time PCR detection system (Bio-RAD). Fiftyµl mixture was used for reaction, which contains 5 µl cDNA sample (0.5–1 µg/µl), 300 nMforward primers for Bim (GCAGATATGCGCCCAGAGAT) or β-actin(ATGGCTCCGGTATGTGCAA), 300 nM reverse primers for Bim(AAGCGTTAAACTCGTCTCCGATA) or β-actin (TGTCTTTCTGGCCCATACCAA),and 25 µl SYBR Green Supermix (Bio-RAD). After incubation at 50°C for 2 min followedby 95°C for 10 min, the reaction was carried out for 40 cycles of the following: 95°C for 15sec and 60°C for 1 min. The threshold cycle value (Ct) was obtained using the iCyclerOptical system interface software. Mean Ct of Bim was calculated from triplicatemeasurements and normalized with the mean Ct of the control gene for β-actin.
Antibodies and Western Blot AnalysisNotch3 was detected with a rabbit polyclonal antibody at 1:500 dilution (Orbigen, Inc., SanDiego, CA). The rabbit antibodies to PARP, Bcl-xL, phosho-Bcl-2, Puma, BAX, phospho-extracellular signal-regulated kinase (pERK), ERK, phospho-Akt, Akt, phosphoinositide 3-kinase (PI3K), and PKA C-α, were purchased from Cell signaling Technology (Danvers,
Konishi et al. Page 6
Oncogene. Author manuscript; available in PMC 2010 July 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

MA). The rabbit antibodies to Mcl-1 and Bim were purchased from Santa CruzBiotechnology (Santa Cruz, CA) and Calbiochem (La Jolla, CA), respectively. The rabbitantibodies to phospho-Foxo3a and Foxo3a were from Millipore (Billerica, MA).Monoclonal cytochrome C antibody was obtained from BD Biosciences (San Jose, CA).Proteins were stained with Ponceau S to confirm equal loading of in each analysis. ForWestern blot analysis of mitogen-activated protein kinase (MAPK), Akt, PI3K and PKA-Cαactivation, the cells were transfected with Notch3 siRNA before serum deprivation andmaintained in serum-free medium for 24 hours before the addition of serum. The cells wereharvested after designated time intervals.
In vivo tumorigenicityAthymic 4- to 6-week-old female nude mice (nu+/nu+) were used for the tumor xenograftmodels. H460 (1 × 106 cells in the volume of 200 µl of PBS) was inoculated subcutaneously(s.c.) into the right posterior legs of the mice. Treatment was initiated when tumors werepalpable. Erlotinib (100 mg/kg) was administered orally every other day to the mice alone orin combination with MRK-003 (150 mg/kg) given daily for 3 days followed by 4 days off.Erlotinib was diluted in 1% methylcellulose/0.1%Tween 80. MRK-003 was diluted in 0.5%methylcellulose. Tumors were measured every 2 days with a caliper. Tumor Volume (TV)was calculated with the formula: TV = (Length) × (Width)2 / 2. Percentage tumor volume(% TV) on day X was calculated as: %TV = (tumor volume on day X / tumor volume on day1) × 100.
Statistical analysesThe size of implanted tumors at different time points after treatment was compared with thatof control groups. Unless specifically stated, statistical inference in comparative experimentsboth in vivo and in vitro was obtained using unpaired, two-sided Student’s t test. For alldeterminations, the differences were considered significant when P value is < 0.05.
AcknowledgmentsGrant Support: This study was supported by NCI 1R01 CA115707
REFERENCES1. Roy M, Pear WS, Aster JC. Curr Opin Genet Dev. 2007; 17:52–59. [PubMed: 17178457]
2. Curry CL, Reed LL, Golde TE, Miele L, Nickoloff BJ, Foreman KE. Oncogene. 2005; 24:6333–6344. [PubMed: 15940249]
3. Das I, Craig C, Funahashi Y, Jung KM, Kim TW, Byers R, Weng AP, Kutok JL, Aster JC,Kitajewski J. J Biol Chem. 2004; 279:30771–30780. [PubMed: 15123653]
4. Duechler M, Shehata M, Schwarzmeier JD, Hoelbl A, Hilgarth M, Hubmann R. Leukemia. 2005;19:260–267. [PubMed: 15565166]
5. Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, Lockwood G, Egan SE. CancerRes. 2005; 65:8530–8537. [PubMed: 16166334]
6. Haruki N, Kawaguchi KS, Eichenberger S, Massion PP, Olson S, Gonzalez A, Carbone DP, DangTP. Cancer Res. 2005; 65:3555–3561. [PubMed: 15867348]
7. Konishi J, Kawaguchi K, Vo H, Haruki N, Gonzalez A, Carbone DP, Dang TP. Cancer Res. 2007
8. Dievart A, Beaulieu N, Jolicoeur P. Oncogene. 1999; 18:5973–5981. [PubMed: 10557086]
9. Fitzgerald K, Harrington A, Leder P. Oncogene. 2000; 19:4191–4198. [PubMed: 10980592]
10. Weijzen S, Rizzo P, Braid M, Vaishnav R, Jonkheer SM, Zlobin A, Osborne BA, Gottipati S, AsterJC, Hahn WC, Rudolf M, Siziopikou K, Kast WM, Miele L. Nat Med. 2002; 8:979–986.[PubMed: 12185362]
11. Heiser PW, Hebrok M. Cell Cycle. 2004; 3:270–272. [PubMed: 14726662]
Konishi et al. Page 7
Oncogene. Author manuscript; available in PMC 2010 July 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

12. Borner C. Mol Immunol. 2003; 39:615–647. [PubMed: 12493639]
13. Gomez-Bougie P, Oliver L, Le Gouill S, Bataille R, Amiot M. Oncogene. 2005; 24:8076–8079.[PubMed: 16091744]
14. Gong Y, Somwar R, Politi K, Balak M, Chmielecki J, Jiang X, Pao W. PLoS Med. 2007; 4:e294.[PubMed: 17927446]
15. Campos AH, Wang W, Pollman MJ, Gibbons GH. Circ Res. 2002; 91:999–1006. [PubMed:12456485]
16. Wang T, Holt CM, Xu C, Ridley C, P OJR, Baron M, Trump D. Cell Signal. 2007; 19:2458–2467.[PubMed: 17822871]
17. Biswas SC, Shi Y, Sproul A, Greene LA. J Biol Chem. 2007; 282:29368–29374. [PubMed:17702754]
18. Urbich C, Knau A, Fichtlscherer S, Walter DH, Bruhl T, Potente M, Hofmann WK, de Vos S,Zeiher AM, Dimmeler S. FASEB J. 2005; 19:974–976. [PubMed: 15824087]
19. Bellavia D, Campese AF, Alesse E, Vacca A, Felli MP, Balestri A, Stoppacciaro A, Tiveron C,Tatangelo L, Giovarelli M, Gaetano C, Ruco L, Hoffman ES, Hayday AC, Lendahl U, Frati L,Gulino A, Screpanti I. Embo J. 2000; 19:3337–3348. [PubMed: 10880446]
20. Bocchetta M, Miele L, Pass HI, Carbone M. Oncogene. 2003; 22:81–89. [PubMed: 12527910]
21. Beverly LJ, Felsher DW, Capobianco AJ. Cancer Res. 2005; 65:7159–7168. [PubMed: 16103066]
22. Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, Tenen DG, KobayashiS. PLoS Med. 2007; 4:1669–1679. discussion 1680. [PubMed: 17973572]
23. Wang YF, Jiang CC, Kiejda KA, Gillespie S, Zhang XD, Hersey P. Clin Cancer Res. 2007;13:4934–4942. [PubMed: 17652623]
24. Qin JZ, Stennett L, Bacon P, Bodner B, Hendrix MJ, Seftor RE, Seftor EA, Margaryan NV,Pollock PM, Curtis A, Trent JM, Bennett F, Miele L, Nickoloff BJ. Mol Cancer Ther. 2004;3:895–902. [PubMed: 15299072]
25. Shinjyo T, Kuribara R, Inukai T, Hosoi H, Kinoshita T, Miyajima A, Houghton PJ, Look AT,Ozawa K, Inaba T. Mol Cell Biol. 2001; 21:854–864. [PubMed: 11154272]
26. Dang TP, Gazdar AF, Virmani AK, Sepetavec T, Hande KR, Minna JD, Roberts JR, Carbone DP.J Natl Cancer Inst. 2000; 92:1355–1357. [PubMed: 10944559]
27. Lewis HD, Leveridge M, Strack PR, Haldon CD, O'Neil J, Kim H, Madin A, Hannam JC, LookAT, Kohl N, Draetta G, Harrison T, Kerby JA, Shearman MS, Beher D. Chem Biol. 2007; 14:209–219. [PubMed: 17317574]
Konishi et al. Page 8
Oncogene. Author manuscript; available in PMC 2010 July 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
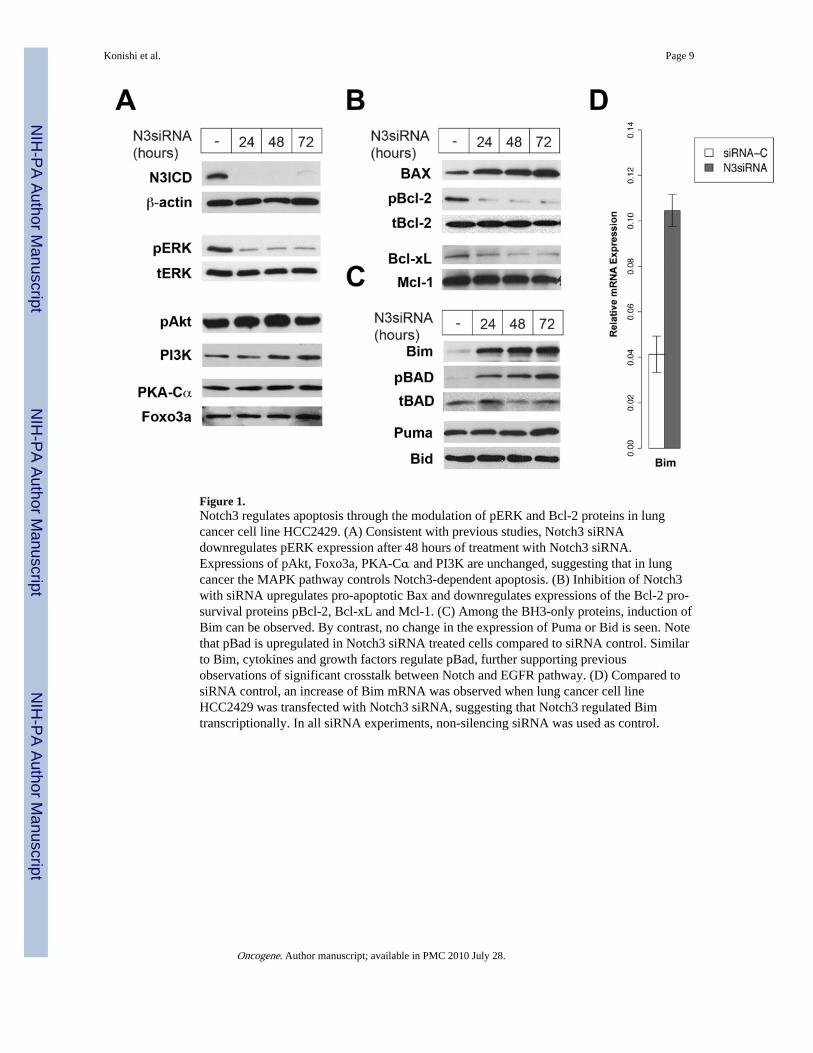
Figure 1.Notch3 regulates apoptosis through the modulation of pERK and Bcl-2 proteins in lungcancer cell line HCC2429. (A) Consistent with previous studies, Notch3 siRNAdownregulates pERK expression after 48 hours of treatment with Notch3 siRNA.Expressions of pAkt, Foxo3a, PKA-Cα and PI3K are unchanged, suggesting that in lungcancer the MAPK pathway controls Notch3-dependent apoptosis. (B) Inhibition of Notch3with siRNA upregulates pro-apoptotic Bax and downregulates expressions of the Bcl-2 pro-survival proteins pBcl-2, Bcl-xL and Mcl-1. (C) Among the BH3-only proteins, induction ofBim can be observed. By contrast, no change in the expression of Puma or Bid is seen. Notethat pBad is upregulated in Notch3 siRNA treated cells compared to siRNA control. Similarto Bim, cytokines and growth factors regulate pBad, further supporting previousobservations of significant crosstalk between Notch and EGFR pathway. (D) Compared tosiRNA control, an increase of Bim mRNA was observed when lung cancer cell lineHCC2429 was transfected with Notch3 siRNA, suggesting that Notch3 regulated Bimtranscriptionally. In all siRNA experiments, non-silencing siRNA was used as control.
Konishi et al. Page 9
Oncogene. Author manuscript; available in PMC 2010 July 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
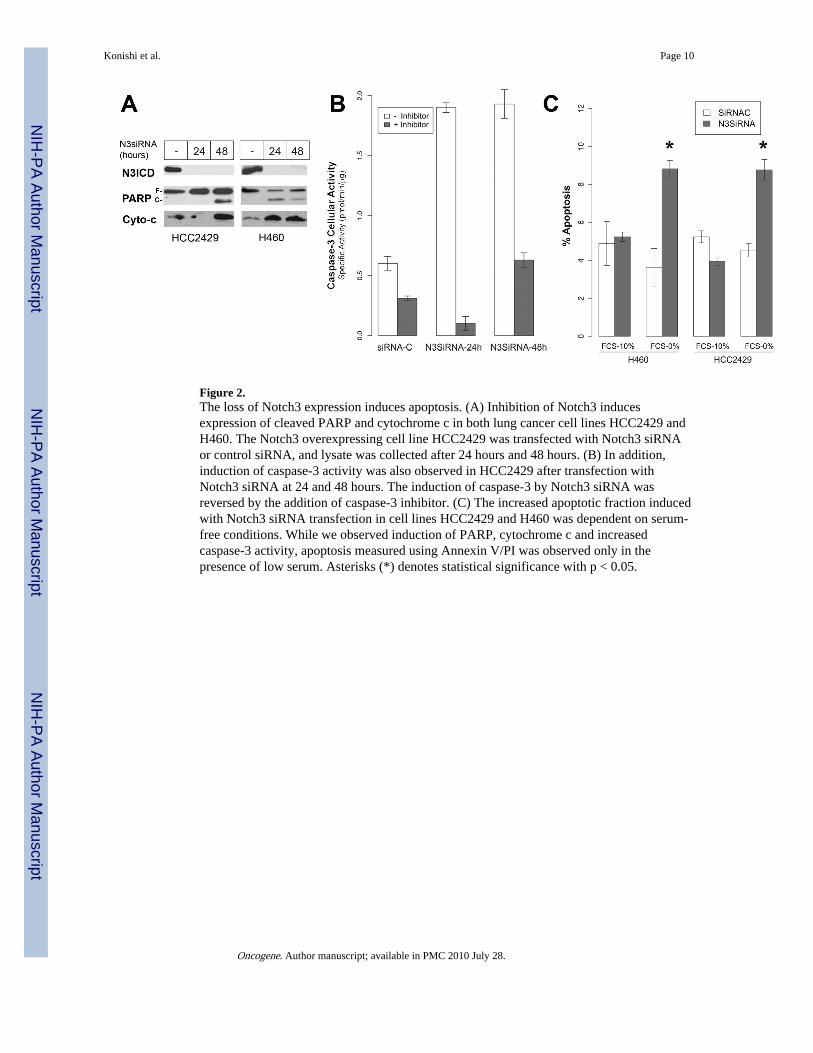
Figure 2.The loss of Notch3 expression induces apoptosis. (A) Inhibition of Notch3 inducesexpression of cleaved PARP and cytochrome c in both lung cancer cell lines HCC2429 andH460. The Notch3 overexpressing cell line HCC2429 was transfected with Notch3 siRNAor control siRNA, and lysate was collected after 24 hours and 48 hours. (B) In addition,induction of caspase-3 activity was also observed in HCC2429 after transfection withNotch3 siRNA at 24 and 48 hours. The induction of caspase-3 by Notch3 siRNA wasreversed by the addition of caspase-3 inhibitor. (C) The increased apoptotic fraction inducedwith Notch3 siRNA transfection in cell lines HCC2429 and H460 was dependent on serum-free conditions. While we observed induction of PARP, cytochrome c and increasedcaspase-3 activity, apoptosis measured using Annexin V/PI was observed only in thepresence of low serum. Asterisks (*) denotes statistical significance with p < 0.05.
Konishi et al. Page 10
Oncogene. Author manuscript; available in PMC 2010 July 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 3.Notch3-mediated apoptosis is dependent on Bim in vitro. (A) Twenty-four hours afterNotch3 siRNA transfection, lung cancer cell line HCC2429 was transfected with BimsiRNA. Cells were harvested after an additional 24 hours. Reduction of Bim expression wasobserved after 24 hours as compared control siRNA. The loss of Bim prevented induction ofPARP by Notch3 siRNA. (B) In serum free media, the loss of Bim abrogates apoptosis,suggesting that Bim is necessary for Notch3-dependent apoptosis. No significant change isobserved in 10% FCS condition. In all siRNA experiments, non-silencing siRNA was usedas control. The difference between SiRNA-C and Bim SiRNA in N3SiRNA treated cellswere statistical significance with p < 0.05.
Konishi et al. Page 11
Oncogene. Author manuscript; available in PMC 2010 July 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
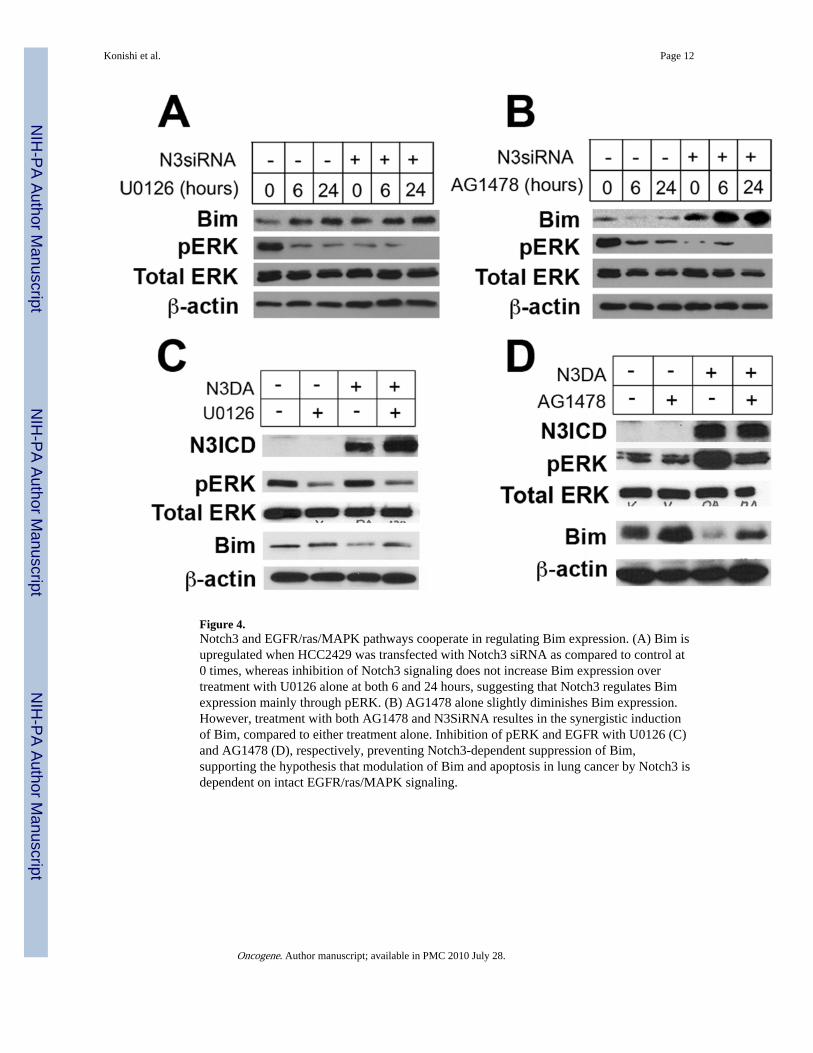
Figure 4.Notch3 and EGFR/ras/MAPK pathways cooperate in regulating Bim expression. (A) Bim isupregulated when HCC2429 was transfected with Notch3 siRNA as compared to control at0 times, whereas inhibition of Notch3 signaling does not increase Bim expression overtreatment with U0126 alone at both 6 and 24 hours, suggesting that Notch3 regulates Bimexpression mainly through pERK. (B) AG1478 alone slightly diminishes Bim expression.However, treatment with both AG1478 and N3SiRNA resultes in the synergistic inductionof Bim, compared to either treatment alone. Inhibition of pERK and EGFR with U0126 (C)and AG1478 (D), respectively, preventing Notch3-dependent suppression of Bim,supporting the hypothesis that modulation of Bim and apoptosis in lung cancer by Notch3 isdependent on intact EGFR/ras/MAPK signaling.
Konishi et al. Page 12
Oncogene. Author manuscript; available in PMC 2010 July 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 5.Combination of Notch inhibition with MRK-003 and EGFR inhibition with erlotinib hasgreater anti-tumor effect in vivo and enhances Bim expression. (A) H460 cells wereinoculated subcutaneously into nude mice and treatment was initiated when tumors werepalpable. Treatment with the combination of erlotinib and MRK-003 resulted in the lowestrate of tumor growth, compared to treatment with either alone. The asterisk (*) denotesstatistical significance, p < 0.05 when comparing treatment with the combination vs.MRK-003 alone. The difference between erlotinib alone and the combination is statisticallysignificant across all time points except for Day 1. (B) Bim expression was markedly higherin tumor treated with MRK-003 and erlotinib than either MRK-003 or erlotinib alone.
Konishi et al. Page 13
Oncogene. Author manuscript; available in PMC 2010 July 28.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
Copyright © 2022 FDOKUMEN




















