
Nature review 2014 - Dendritic cell and macrophage in kidney
19
NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 1 Vascular Medicine Institute (N.M.R., J.S.I.), Thomas E. Starzl Transplantation Institute (N.M.R., J.S.I., A.W.T.), University of Pittsburgh School of Medicine, W1544 Biomedical Science Tower, 200 Lothrop Street, Pittsburgh, PA 15261, USA. MRC Centre for Inflammation Research, Queen’s Medical Research Institute, University of Edinburgh, 47 Little France Crescent, Edinburgh EH16 4TJ, UK (D.A.F., J.H.). Correspondence to: A.W.T. [email protected] Dendritic cells and macrophages in the kidney: a spectrum of good and evil Natasha M. Rogers, David A. Ferenbach, Jeffrey S. Isenberg, Angus W. Thomson and Jeremy Hughes Abstract | Renal dendritic cells (DCs) and macrophages represent a constitutive, extensive and contiguous network of innate immune cells that provide sentinel and immune-intelligence activity; they induce and regulate inflammatory responses to freely filtered antigenic material and protect the kidney from infection. Tissue-resident or infiltrating DCs and macrophages are key factors in the initiation and propagation of renal disease, as well as essential contributors to subsequent tissue regeneration, regardless of the aetiological and pathogenetic mechanisms. The identification, and functional and phenotypic distinction of these cell types is complex and incompletely understood, and the same is true of their interplay and relationships with effector and regulatory cells of the adaptive immune system. In this Review, we discuss the common and distinct characteristics of DCs and macrophages, as well as key advances that have identified the renal-specific functions of these important phagocytic, antigen-presenting cells, and their roles in potentiating or mitigating intrinsic kidney disease. We also identify remaining issues that are of priority for further investigation, and highlight the prospects for translational and therapeutic application of the knowledge acquired. Rogers, N. M. et al. Nat. Rev. Nephrol. advance online publication 30 September 2014; doi:10.1038/nrneph.2014.170 Introduction The manifestations of kidney disease are protean and include acute kidney injury (AKI) due to ischaemia– reperfusion or direct tubular cytotoxicity; autoimmune glomerulonephritis; and rejection of kidney transplants through mechanisms that affect the glomerular, inter- stitial and vascular compartments. Increasing molec- ular evidence now demonstrates the pivotal role of nonparenchymal cells in determining both renal tissue injury and subsequent reparative responses following diverse insults. In particular, advances in technology and the understanding of innate and adaptive immuno- pathological cellular responses have facilitated identifica- tion of dendritic cells (DCs) and macrophages, as well as assessment of their function, in nonlymphoid solid organs such as the kidney. Both of these cell types reside within the renal interstitium, and possess the capacity to activate and regulate both protective and deleterious renal pathology. DCs have long been established as indis- pensable antigen-presenting cells (APCs), which act as systemic sentinels capable of responding to endogenous and exogenous ‘danger’ signals to initiate and propagate immune responses to inciting antigens or induce immune tolerance. 1–3 Macrophages have been defined as a distinct, but related population of APCs (albeit less potent than DCs), whose primary functions are maintenance of tissue homeostasis and phagocytic clearance of various native and foreign bodies. In this Review, we examine the archetypal components of the nonparenchymal compartment in the kidney. We consider the advances that have been made in understand- ing the discrete but overlapping roles of both DCs and macrophages—a difficult task owing to the substantial functional and phenotypic similarity between these mono- nuclear phagocyte populations (Figure 1). We also discuss how innovations, such as the identification and assessment of distinct lineage markers and gene expression profiles, transgenic mouse models and cell ablation techniques, have facilitated the discrimination of DC and macro- phage subsets that function in tissue homeostasis and immune tolerance, acute and chronic inflammationand alloimmunity. We note, however, that considerable limi- tations remain for many of these approaches, particu- larly related to the ability to accurately target DCs and/ or macrophages to modulate disease outcome, in addi- tion to questions regarding their relevance to human kidney disease. Defining DCs and macrophages Considerable flexibility, heterogeneity and complexity is recognized within the myeloid–monocyte developmental lineage. 4 For example, both DCs and macrophages originate from common progenitor cells in the bone marrow under the influence of key growth factors (Figure 2): colony- stimulating factor 1 (CSF-1; also known as macrophage colony-stimulating factor [M-CSF]), fms-like related tyros- ine kinase 3 ligand (FLT3LG), and granulocyte-macrophage Competing interests J.S.I. is Chair of the Scientific Advisory Boards of Radiation Control Technologies and Vasculox. A.W.T. is co-inventor of a US patent (6,224,859 B1) for the generation of tolerogenic dendritic cells to promote transplant tolerance. The other authors declare no competing interests. REVIEWS © 2014 Macmillan Publishers Limited. All rights reserved
Transcript of Nature review 2014 - Dendritic cell and macrophage in kidney

NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 1
Vascular Medicine Institute (N.M.R., J.S.I.), Thomas E. Starzl Transplantation Institute (N.M.R., J.S.I., A.W.T.), University of Pittsburgh School of Medicine, W1544 Biomedical Science Tower, 200 Lothrop Street, Pittsburgh, PA 15261, USA. MRC Centre for Inflammation Research, Queen’s Medical Research Institute, University of Edinburgh, 47 Little France Crescent, Edinburgh EH16 4TJ, UK (D.A.F., J.H.).
Correspondence to: A.W.T. [email protected]
Dendritic cells and macrophages in the kidney: a spectrum of good and evilNatasha M. Rogers, David A. Ferenbach, Jeffrey S. Isenberg, Angus W. Thomson and Jeremy Hughes
Abstract | Renal dendritic cells (DCs) and macrophages represent a constitutive, extensive and contiguous network of innate immune cells that provide sentinel and immune-intelligence activity; they induce and regulate inflammatory responses to freely filtered antigenic material and protect the kidney from infection. Tissue-resident or infiltrating DCs and macrophages are key factors in the initiation and propagation of renal disease, as well as essential contributors to subsequent tissue regeneration, regardless of the aetiological and pathogenetic mechanisms. The identification, and functional and phenotypic distinction of these cell types is complex and incompletely understood, and the same is true of their interplay and relationships with effector and regulatory cells of the adaptive immune system. In this Review, we discuss the common and distinct characteristics of DCs and macrophages, as well as key advances that have identified the renal-specific functions of these important phagocytic, antigen-presenting cells, and their roles in potentiating or mitigating intrinsic kidney disease. We also identify remaining issues that are of priority for further investigation, and highlight the prospects for translational and therapeutic application of the knowledge acquired.
Rogers, N. M. et al. Nat. Rev. Nephrol. advance online publication 30 September 2014; doi:10.1038/nrneph.2014.170
IntroductionThe manifestations of kidney disease are protean and include acute kidney injury (AKI) due to ischaemia–reperfusion or direct tubular cytotoxicity; autoimmune glomerulonephritis; and rejection of kidney transplants through mechanisms that affect the glomerular, inter-stitial and vascular compartments. Increasing molec-ular evidence now demonstrates the pivotal role of nonparenchymal cells in determining both renal tissue injury and subsequent reparative responses following diverse insults. In particular, advances in technology and the understanding of innate and adaptive immuno-pathological cellular responses have facilitated identifica-tion of dendritic cells (DCs) and macrophages, as well as assessment of their function, in nonlymphoid solid organs such as the kidney. Both of these cell types reside within the renal interstitium, and possess the capacity to activate and regulate both protective and deleterious renal pathology. DCs have long been established as indis-pensable antigen-presenting cells (APCs), which act as systemic sentinels capable of responding to endogenous and exo genous ‘danger’ signals to initiate and propagate immune responses to inciting antigens or induce immune tolerance.1–3 Macrophages have been defined as a distinct, but related population of APCs (albeit less potent than DCs), whose primary functions are maintenance of tissue
homeostasis and phagocytic clearance of various native and foreign bodies.
In this Review, we examine the archetypal components of the nonparenchymal compartment in the kidney. We consider the advances that have been made in understand-ing the discrete but overlapping roles of both DCs and macrophages—a difficult task owing to the substantial functional and phenotypic similarity between these mono-nuclear phagocyte populations (Figure 1). We also discuss how innovations, such as the identification and assessment of distinct lineage markers and gene expression profiles, transgenic mouse models and cell ablation techniques, have facilitated the discrimination of DC and macro-phage subsets that function in tissue homeo stasis and immune tolerance, acute and chronic inflammationand allo immunity. We note, however, that considerable limi-tations remain for many of these approaches, particu-larly related to the ability to accurately target DCs and/or macro phages to modulate disease outcome, in addi-tion to questions regarding their relevance to human kidney disease.
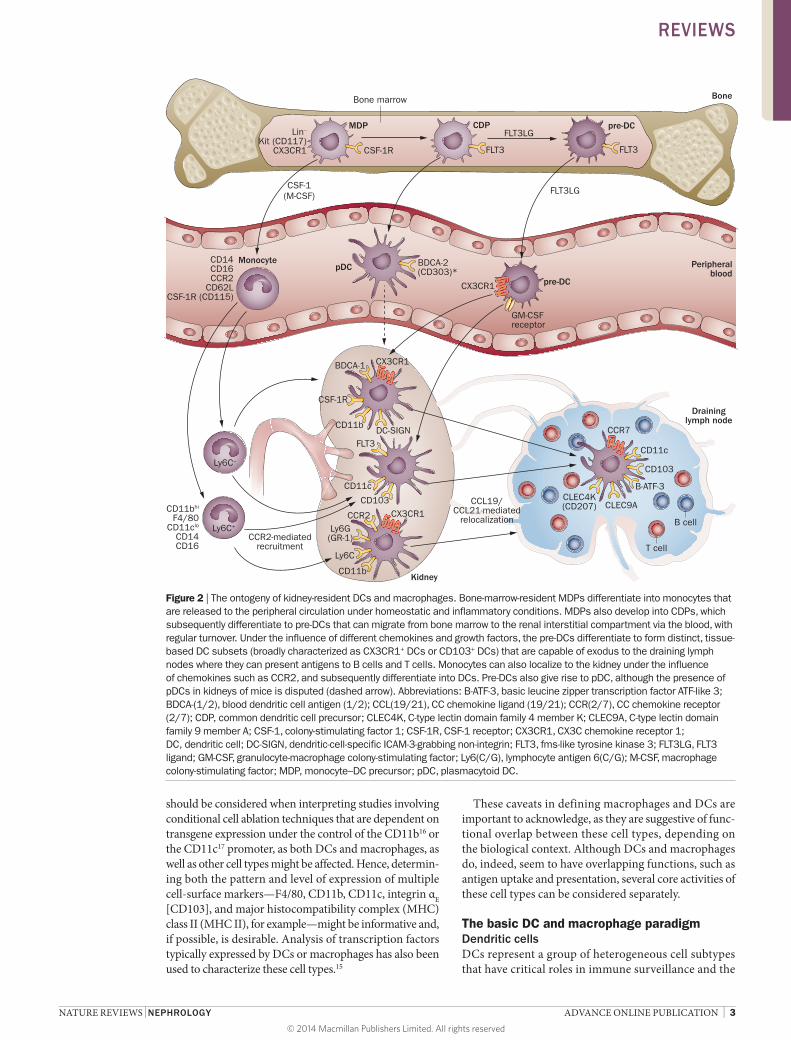
Defining DCs and macrophages Considerable flexibility, heterogeneity and complexity is recognized within the myeloid–monocyte developmental lineage.4 For example, both DCs and macrophages originate from common progenitor cells in the bone marrow under the influence of key growth factors (Figure 2): colony-stimulatin g factor 1 (CSF-1; also known as macro phage c olony-stimulatin g factor [M-CSF]), fms-like related tyros-ine kinase 3 ligand (FLT3LG), and granulocyte- macrophage
Competing interestsJ.S.I. is Chair of the Scientific Advisory Boards of Radiation Control Technologies and Vasculox. A.W.T. is co-inventor of a US patent (6,224,859 B1) for the generation of tolerogenic dendritic cells to promote transplant tolerance. The other authors declare no competing interests.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

2 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
colony-stimulating factor (GM-CSF).5–7 Furthermore, monocytes recruited from the circulation can differenti-ate into tissue macrophages or inflammatory DCs during disease processes.8,9 Thus, how best to define DC versus macrophage cell types and activities (Figure 1) has been
Key points
■ Dendritic cells (DCs) and macrophages are distinct cell types, but demonstrate similarities in terms of ontogeny, phenotype, and function
■ Both of these cell types are present within the renal interstitium and are critical to homeostatic regulation of the kidney environment
■ The numbers of DCs and macrophages in the kidney increase following renal injury ■ A variety of DC and macrophage subtypes exist, each with distinct phenotypes
and activities, and many can be identified based on panels of cellular markers ■ The manifestation of glomerular or tubular kidney disease, as well as disease
outcome in preclinical models, is determined by the subtype of DC and/or macrophage involved
■ A number of pharmacological or genetic approaches are available to deplete or eliminate DCs or macrophages in preclinical models, but the effects of such interventions are not renal-specific
an important area of contention, and this issue remains controversial (Box 1).10–13 Currently, no definitive cel-lular demarcation has been established, and studies have tended to investigate either DCs or macro phages, with segregation based on traditional interpretations of identi-fication. Specifically, such research efforts—predominantly in mouse models—have relied on the supposition that macro phages or DCs can be accurately identified accord-ing to expression levels of the ‘macrophage’ markers F4/80 (also known as EGF-like module-containing mucin-like hormone receptor-like 1 [EMR1]) and CD11b (integrin αM) or the ‘DC marker’ CD11c (integrin αx). How ever, these markers are not exclusive to their respective cell types (Figure 1); bone-marrow-derived macrophages can express CD11c in vitro, whereas DCs can express F4/80 and CD11b, suggesting that these markers do not identify unique cell populations.14,15 The incomplete restriction of such cell-surface markers to specific cell types compli-cates the interpretation of experimental data based on this method of cell identification. In addition, this limitation
Functions Tissue surveillanceSource of chemokines and cytokinesPhagocytosis of debris and pathogensCytotoxicityFibrosis and remodelling of ECM
Markers of in�ammatoryM1 macrophages
Markers ofanti-in�ammatoryM2 macrophages
CSF-1R (CD115)
CD14
CD62L
Ly6G(Gr-1)
Ly6C
CD86(B7.2)
MHC II
CD11b(integrin αX)
F4/80(EMR1)CD11b
(integrin αM)
CD80(B7.1)
IL-4R/IL-10R
CD206
CD163
CD68
FcγRII (CD32)
*ICAM-1 (CD54)
FcγRIII (CD16)
SIRPα
DC-SIGN(CD209)*
BDCA-1 (CD1c)*
FLT3 (CD135)
CX3CR1
CD103 (integrin αE)
CCR7 (maturation)
Macrophage
Shared antigens
Dendritic cell
ID-2, IRF8,ZBTB46,B-ATF-3
IRF5
IRF4STAT3
FunctionsTissue surveillance
Source of chemokines and cytokinesPhagocytosis
Antigen presentation and T-cell stimulationInduction of immune toleranceIL-6, IL-10, IL-12,TNF, TGF-β
Figure 1 | The heterogeneous but overlapping phenotype and functions of renal DCs and macrophages. DCs are traditionally described as mediators of immune surveillance and antigen presentation, and as the primary determinants of responses to antigens—through initiation of either immune effector-cell functions or the development of tolerance. Macrophages also function as innate immune cells, predominantly through phagocytosis and production of toxic metabolites. However, the classical paradigm of DC versus macrophage phenotypes and functions is increasingly indistinct within the kidney, as these cells exhibit overlapping surface markers, functional capabilities, and ontogenic pathways. This molecular and phenotypic overlap between cell types and subsets complicates their identification and evaluation. *Marker described only in humans. Abbreviations: B-ATF-3, basic leucine zipper transcription factor ATF-like 3; BDCA-1, blood dendritic cell antigen 1; CCR, CC chemokine receptor; CSF-1R, colony-stimulating factor 1 receptor; CX3CR1, CX3C chemokine receptor 1; DC, dendritic cell; DC-SIGN, dendritic-cell-specific ICAM-3-grabbing non-integrin; ECM, extracellular matrix; EMR1, EGF-like module-containing mucin-like hormone receptor-like 1; FcγR(II/III), low affinity IgG Fc region receptor (II/III); FLT3, fms-like tyrosine kinase 3; Gr-1, granulocyte-differentiation antigen-1; ICAM-1, intercellular adhesion molecule 1; ID-2, inhibitor of DNA binding 2; IL-4R, IL-4 receptor; IL-10R, IL-10 receptor; IRF, interferon regulatory factor; Ly6(C/G), lymphocyte antigen 6(C/G); SIRPα, signal-regulatory protein α (also known as tyrosine-protein phosphatase nonreceptor type substrate 1); STAT3, signal transducer and activator of transcription 3; ZBTB46, zinc finger and BTB domain containing protein 46.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved
NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 3
should be considered when interpreting studies involving conditional cell ablation techniques that are dependent on transgene expression under the control of the CD11b16 or the CD11c17 promoter, as both DCs and macrophages, as well as other cell types might be affected. Hence, determin-ing both the pattern and level of expression of multiple cell-surface markers—F4/80, CD11b, CD11c, integrin αE [CD103], and major histocompatibility complex (MHC) class II (MHC II), for example—might be informative and, if possible, is desirable. Analysis of transcription factors typically expressed by DCs or macrophages has also been used to characterize these cell types.15
These caveats in defining macrophages and DCs are important to acknowledge, as they are suggestive of func-tional overlap between these cell types, depending on the biological context. Although DCs and macrophages do, indeed, seem to have overlapping functions, such as antigen uptake and presentation, several core activities of these cell types can be considered separately.
The basic DC and macrophage paradigmDendritic cellsDCs represent a group of heterogeneous cell subtypes that have critical roles in immune surveillance and the
T cell
B cell
pDCMonocyte
Ly6C–
CCR2-mediatedrecruitment
Ly6C+
pre-DC
Peripheralblood
Draininglymph node
Kidney
CCR7
CD11c
CD103
CLEC4K(CD207) CLEC9ACCL19/
CCL21-mediatedrelocalization
CX3CR1
GM-CSFreceptor
BDCA-2(CD303)*
pre-DC
FLT3FLT3
FLT3LGCDP
CSF-1R
MDP
CSF-1(M-CSF) FLT3LG
BoneBone marrow
CD11c
FLT3
CD103
CD11b
CSF-1R
CX3CR1
DC-SIGN
BDCA-1
Lin–
Kit (CD117)CX3CR1
CD11bhi
F4/80CD11clo
CD14CD16
CD14CD16CCR2
CD62LCSF-1R (CD115)
B-ATF-3
CD11b
Ly6C
CX3CR1
Ly6G(GR-1)
CCR2
Figure 2 | The ontogeny of kidney-resident DCs and macrophages. Bone-marrow-resident MDPs differentiate into monocytes that are released to the peripheral circulation under homeostatic and inflammatory conditions. MDPs also develop into CDPs, which subsequently differentiate to pre-DCs that can migrate from bone marrow to the renal interstitial compartment via the blood, with regular turnover. Under the influence of different chemokines and growth factors, the pre-DCs differentiate to form distinct, tissue-based DC subsets (broadly characterized as CX3CR1+ DCs or CD103+ DCs) that are capable of exodus to the draining lymph nodes where they can present antigens to B cells and T cells. Monocytes can also localize to the kidney under the influence of chemokines such as CCR2, and subsequently differentiate into DCs. Pre-DCs also give rise to pDC, although the presence of pDCs in kidneys of mice is disputed (dashed arrow). Abbreviations: B-ATF-3, basic leucine zipper transcription factor ATF-like 3; BDCA-(1/2), blood dendritic cell antigen (1/2); CCL(19/21), CC chemokine ligand (19/21); CCR(2/7), CC chemokine receptor (2/7); CDP, common dendritic cell precursor; CLEC4K, C-type lectin domain family 4 member K; CLEC9A, C-type lectin domain family 9 member A; CSF-1, colony-stimulating factor 1; CSF-1R, CSF-1 receptor; CX3CR1, CX3C chemokine receptor 1; DC, dendritic cell; DC-SIGN, dendritic-cell-specific ICAM-3-grabbing non-integrin; FLT3, fms-like tyrosine kinase 3; FLT3LG, FLT3 ligand; GM-CSF, granulocyte-macrophage colony-stimulating factor; Ly6(C/G), lymphocyte antigen 6(C/G); M-CSF, macrophage colony-stimulating factor; MDP, monocyte–DC precursor; pDC, plasmacytoid DC.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

4 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
instigation of immunity or tolerance.1,18 As mentioned, DCs originate from myeloid haematopoietic progeni-tor cells in the bone marrow, via developmental path-ways that exhibit considerable plasticity;19 however, DC ontogenesis is governed predominantly by the haemato-poietic growth factor FLT3LG (Figure 2).20–22 They are rare cells that are present in the peripheral circulation (<0.1% of total circulating leucocytes), but also reside in virtually all tissues (especially near portals of entry), and display broad maturational and functional diver-sity.1,18 In keeping with their role as potent APCs, DCs are highly proficient at both internalizing and process-ing antigens;23 through a highly-orchestrated molecu-lar process, the cells sub sequently present peptide fragments of processed foreign or self-antigen in the context of MHC class I or class II molecules to T cells that express complementary T-cell receptors (TCRs).18 Once activated in the periphery, DCs exhibit enhanced migratory capacity and emigrate to secondary lymphoid organs to convey the processed antigens for T-cell stim-ulation, which requires both MHC–TCR inter actions and reinforcing signals induced by co-stimulatory molecules provided by the DC.24 DC activation, matur-ation and migration are triggered by a wide array of cell-surface receptors for Toll-like receptor (TLR) ligands, C-type lectins, cytokines and chemokines involved in
inflammatory responses (extensively reviewed else-where3,20,25). Through these mechanisms, DCs control the initiation of naive and memory T-cell responses,26 and mediate a critical link between the innate and a daptive immunological systems.1,18
MacrophagesSimilarly to DCs, macrophages comprise a diverse group of innate immune cell subsets that are particu-larly rich in lysosomes, and are adept at phagocytosis of tissue debris and infectious material. In addition, macrophages can regulate other cell types by serving as APCs, and also play a part in wound healing via the pro-duction of an array of cytokines, chemokines and growth factors.27 The lineage profile of macrophage precursor populations originating in the bone marrow is similar to that of DCs, although macrophage development, dif-ferentiation and proliferation are primarily governed by CSF-1.7,28 Interestingly, postnatal administration of CSF-1 to mice increased kidney weight and tissue macro-phage number.29 Macrophages arise at an early stage of organogenesis during fetal development;30 in the kidney specifically, their presence precedes the appearance of nephrons, and macrophage cell types seem to have a predominantly trophic role at prenatal stages of kidney development.30 Although the commonly accepted tenet is that macrophage renewal within the interstitial com-partment of solid tissues is driven by differentiation of haematopoietic stem cells, emerging evidence suggests a contribution from embryonic progenitor cells,31,32 as well as in situ proliferation of tissue macrophages.9,33
The recognition that macrophages can adopt several phenotypes led to the ‘M1’ and ‘M2’ paradigm of macro-phage development. According to this paradigm, M1 (also termed classically activated) macrophages are regarded as proinflammatory.34 By contrast, M2 (alterna-tively activated) macrophages are considered to promote both wound healing and tissue fibrosis.34 These pheno-types have been defined by in vitro experiments, and although undoubtedly simplistic, the M1–M2 macro-phage concept emphasizes the fact that macrophages, similar to DCs, have inherent maturational and func-tional plasti city, suggesting that the phenotype of indi-vidual cells and cell populations can change and evolve over time in vivo.35
DCs/macrophages in homeostatic kidneys Identification of renal DCs and macrophages A range of mononuclear cells have been identified within the renal interstitium. In addition to DCs and macrophages, CD3+, CD4+ and CD8+ T cells have been isolated from unmanipulated rodent kidneys,36 as have CD19+ B cells, CD3-NK1.1+ cells, CD4+CD25+ T regula-tory (TREG) cells37 and γδ T cells.36,38 Although murine DCs were first described 40 years ago in the seminal papers by Steinman and Cohn,39–41 information on the localization, phenotype and functional characteristics of this cell type was confined initially to DCs within secondary lymphoid tissue. Although DCs, as well as multiple DC subtypes, have now been described within
Box 1 | Sources of confusion in the characterization of DCs versus macrophages
Defining independent populations of macrophages and DCs (or subsets of these cell types) for study in vivo or in vitro is often difficult, and this limitation is relevant when considering the available data on these cell types. The factors confounding characterization of independent macrophage and DC populations are as follows:
Nonexclusive cellular markers ■ For example, CD11c, F4/80 (EMR1), CD11b, and MHC class II molecules are
all co-expressed in these cell types15
■ Furthermore, CD11c expression is induced by inflammation in macrophages,60 and neutrophils,196 as well as DCs
■ CSF-1R (a traditional macrophage marker) is expressed by classic DCs57
Shared cell lineage and developmental pathway ■ Similar growth factors promote development of macrophage and DC subsets;
differentiation of these mononuclear cell types from common progenitors requires CSF-1, GM-CSF and FLT3LG5,7
■ Cell phenotype in vitro demonstrates plasticity depending on the growth factors used
■ CSF-1 and CSF-1R mutations cause marked depletion of DC populations in experimental models57
■ Injection of CSF-1 into mice causes expansion of CD11c+ cells, including macrophages, conventional DCs and plasmacytoid DCs197
■ GM-CSF promotes DC development in vitro; GM-CSF produces alternative macrophages198,199 that are associated with a ‘mature DC-type’ cytokine profile
Functional similarity ■ DCs and macrophages are both involved in tissue surveillance and are highly
phagocytic ■ Many immunoregulatory factors, including cytokines and chemokines, can be
produced by both cell types15
■ Although antigen-presenting capacity is typically attributed to DCs, macrophages can suppress T-cell activation in an antigen-specific manner15,200
Abbreviations: CSF-1, colony-stimulating factor 1 (also known as, macrophage colony-stimulating factor); CSF-1R, CSF-1 receptor; DC, dendritic cell; EMR1; EGF-like module-containing mucin-like hormone receptor-like 1; FLT3LG, fms-like tyrosine kinase 3 ligand; GM-CSF, granulocyte-macrophage colony-stimulating factor.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 5
virtually all lymphoid and nonlymphoid tissues, formal identification of renal DCs, particularly CD11c+ DCs, had been disputed historically. The renal DC population was thought initi ally to be localized within the glomer-ular compartment and, therefore, to mediate the patho-genesis of glomerulonephritides. This assumption was supported by the isolation of rodent glomerular cells that were phenotypically distinct from mesangial cells, which expressed MHC II (I-A subclass) and exhibited T-cell allostimulatory capa city in primary mixed leucocyte cultures.42–45 Concurrent rodent and human studies con-firmed the presence of cells positive for MHC II mol-ecules within the cortical interstitium of the kidney.46,47 Furthermore, electron microscopic examination of rodent peritubular interstitium enabled differentiation of fibroblasts from immune cells with features sugges-tive of DCs (high expression of MHC II) or macro-phages (abundant primary and secondary lysosomes).48 Immunohistochemical identification of CD11c+ DCs requires minimization of protein denaturation by avoid-ing high temperatures;49 this technique has facilitated the detection and localization of these cells proximate to peritubular capillaries.50
Phenotypic characterization In healthy mice, the renal CD11c+ DC population is hetero geneous, expressing MHC II and low or inter-mediate levels of CD11b and F4/80.50 Although these markers can also be expressed by macrophages, the renal DCs identified in mice more closely resembled splenic dendritic cells than peritoneal macrophages with regard to morpho logical, molecular (co-stimulatory molecule expression) and functional properties.50 Furthermore, these cells exhibited allogeneic T-cell stimulatory capac-ity, although to a lesser degree than observed with splenic CD11c+ DCs.50 Extensive phenotypic characterization revealed that conventional CD11c+ DCs (cDCs) had an immature phenotype (low CD80 and CD86, and neg-ligible CD40 expression) and substantial phagocytic capacity—in addition to the absence of nonconventional plasmacytoid DC (pDC) markers (CD8α and B220).50 CD11c+MHC II+ renal DCs can be segregated into two distinct subsets: integrin αEβ7 expressing and, therefore, CD103+ (CD103+CD11bloCX3CR1−F4/80−SIRP-α−) cells; and CD11b+ (CD103−CD11b+CX3CR1+F4/80+SIRP-α+) cells (Figure 2).51 Both of these renal DC subsets appear to undergo cell division and, therefore, proliferation in homeostatic renal tissue.51 CD103+ renal DCs arise pri-marily from bone-marrow-derived precursors, pre-cDC, and express higher levels of DNA-binding protein inhibi-tor ID-2 and interferon regulatory factor 8 (IRF8) than the CD11b+ subset, as well as FLT3.51 Indeed, both the FLT3 receptor and its ligand, FLT3LG, are an absolute requirement for the development of this DC subset.51 CD11b+ DCs express CSF-1 receptor (CSF-1R; also known as CD115), in keeping with a developmental bias towards this growth factor, but are also dependent on FLT3LG for complete reconstitution.51
Although improvements in renal DC characterization have been made, the absolute numbers of these cells that
can be isolated from the kidney remain relatively low compared with secondary lymphoid organ DC popula-tions;50 however, systemic administration of FLT3LG in mice52 and nonhuman primates53 enables expansion of both renal cDC and pDC populations in vivo. Ex vivo, these mobilized DCs, when freshly isolated, retained an immature phenotype and promoted the develop-ment of IL-10-producing TREG cells in mixed leuco-cyte reactions.52 Low CC chemokine receptor (CCR1, CCR2, CCR5, and CCR7) transcript levels in these DCs reflect a failure to migrate in vitro in response to chemokines (CC chemokine ligand 3 [CCL3], CCL5, and CCL20);52,54 however, their capacity to migrate to the lymphoid-tissue-homing chemokines CCL19 and CCL20 could be augmented ex vivo by exposure to ba cterial l ipopolysaccharide (LPS).54
Although no candidate precursor has been for-mally identified, renal-resident DCs are thought to be derived from common DC precursors that arise from bone marrow progenitors and subsequent blood-borne pre-DC precursors (Figure 2)55—as demonstrated for CD103+ DCs.51 Nevertheless, lymphocyte antigen 6C (Ly6C)– ‘patrolling’ circulating monocytes might also contribute to renal-resident DC populations; Ly6C+ potential DC precursors infiltrate the kidney under inflammatory conditions,7,9 influenced by the CX3C chemo kine receptor 1 (CX3CR1; also known as fractalkine receptor) and CCR2.56
Macrophages, conventionally defined as CD11b+, express greater levels of CSF-1R compared with the CD11c+ DC subset.57 The CSF-1R–enhanced green fluor escent protein (eGFP) transgenic-reporter mouse (the so-called ‘MacGreen’ mouse; Table 1) has been used to track postnatal macrophage development. In this model, cells with active gene expression driven by the CSF-1R promoter were demonstrated to be present before nephro genesis, in close apposition to developing renal tubules, and increased in number after administra-tion of CSF-1.30 In adult kidneys, resident macrophages are believed to originate from bone-marrow-resident monocyte precursors,58 which are characterized as lineage negative (Lin−) cells that express CX3CR1 and the mast/stem cell growth factor receptor Kit (also known as CD117); additional recruitment of Ly6C+ cells can occur under the influence of CCR2 (Figure 2).58,59 Tissue macrophages derived from infiltrating monocytes can undergo differentiation into the broad M1 and M2 cate-gories depending on context. Classically activated M1 macrophages are typically induced through encounter with danger-associated molecular patterns (DAMPs) or proinflammatory cytokines, and produce IL-12 and IL-23 (as do DCs) to promote CD4+ T-helper (TH) cell polarization.60,61 Alternatively activated M2 macrophages can arise through deactivation and differentiation of M1 macrophages, or de novo, directly from infiltrating mono-cytes;60 this subset is immunoregulatory, and produces anti-inflammatory IL-10 as well as Wnt7B.62
Studies in CX3CR1eGFP/+ transgenic-reporter mice (Table 1),63 as well as the MacGreen model,64 have pro-vided evidence of DC–macrophage network within
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

6 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
the kidney. The CX3CR1eGFP/+ mouse model enables mapping of DCs and/or macrophages derived from CX3CR1+ monocytes emigrated from the bone marrow. This study confirmed the homeostatic presence of renal DCs throughout the entire interstitial scaffold, encasing glomeruli, as well as low numbers of these cells within the mesangial matrix.63 In the MacGreen mouse,64 macro-phages were visualized surrounding glomeruli and encas-ing renal tubules within the medulla. The complexity of this renal-resident DC–macrophage network has been elegantly reinforced in a study demonstrating multi-ple discrete cell subsets, distinguished by cell-surface markers, cytokine production, and transcription factor and chemokine receptor expression, in the kidney;15 the cell populations were initially defined according to expression levels of CD11b and CD11c.15 Two defined subsets were found to exhibit a DC phenotype, with the ability to robustly stimulate CD4+ T-cell prolifera-tion, including the induction of forkhead box protein P3 (FOXP3)+ TREG cells.15 However, these cell types also phagocytosed latex beads, and expressed the macrophage marker CD68 in conjunction with developmental and reparative growth factors, such as insulin-like growth factor 1, platelet derived growth factor and Wnt7B.15 By contrast, two other cell subsets displayed a macrophage phenotype, characterized by phagocytic capability but an absent or limited capacity to induce T-cell prolifera-tion.15 These macrophage cell types also expressed growth factors and produced IL-10 after LPS stimulation.15 Thus, these findings reiterate the concept that overlapping DC and macrophage characteristics should be considered in experimental studies or transgenic mice that use a single cell marker such as CD11b, CD11c, CX3CR1 or CSF-1R, as these are also all variably expressed by DC and macro-phages in the normal kidney. A further limitation of current studies examining DCs and macrophages is that characterization of populations of these cells is based pre-dominantly on whole kidney, rather than compartmen-talized tissue digests. Different renal microenvironments could potentially be associated with distinct DC and macrophage subtypes, leading to erroneous conclusions regarding cell localization when whole kidney samples are analysed; for example, glomerular macrophages
do not express F4/80, but can be identified based on CD68 positivity.65,66
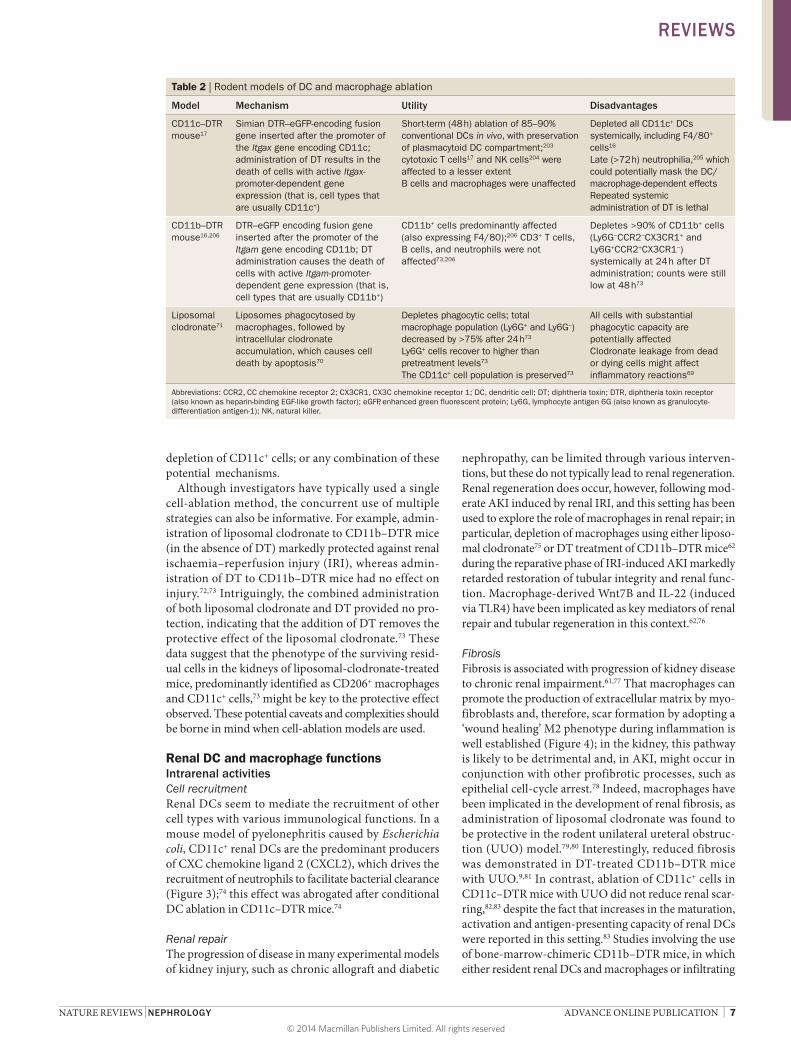
Models of macrophage and DC ablationVarious methods have been used to conditionally ablate macrophages and/or DCs in vivo, with the aim of dissect-ing their function in disease (Table 2). In early studies, liposomal clodronate formed the basis for cell ablation; this agent is toxic to many phagocytic cell types, but particularly macrophages, and systemic administra-tion profoundly ablated macrophage populations in the kidney, as well as liver and spleen, such that the effects of cell ablation might have been secondary to intra-renal or extrarenal effects.67–71 Despite these caveats, this approach has been used to deplete DCs and macrophages in multiple mouse and rat models of renal disease.
Selective cell ablation has been attempted using trans-genic mice that express the human diphtheria toxin receptor (DTR; also known as heparin-binding EGF-like growth factor [HB-EGF]) under the control of the CD11b16 or CD11c17 promoter, to deplete macrophages or DCs, respectively, following diphtheria toxin (DT) administration. Although overlapping expression of these markers in DC or macrophage subsets as well as other cell types suggests that multiple cell populations are inevitably depleted (Table 2), CD11b–DTR and CD11c–DTR mice have, nonetheless, proved highly informative. Despite the apparent simplicity of such cell-depletion studies, multiple caveats—in addition to the technical caveats discussed—limit their interpretation as many possible explanations are available for the observed results. For example, reduced tissue injury after depletion of CD11c+ cells could potentially be manifest through a number of mechanisms: production of injurious factors by the resident or infiltrating renal CD11c+ cells that were depleted; production of protective factors by the resid-ual surviving renal CD11c+ cells, other haemato poietic cells or parenchymal cells subsequent to interaction with the apoptotic corpses of ablated cells, for example; promotion of systemic protective effects of renal or extra-renal CD11c+ cell depletion (that is, skewing of immune responses); a protective effect attributable to regeneration of cell populations following transient
Table 1 | Summary of DC and macrophage reporter mice
Mouse model Mechanism Utility Cells types identified Disadvantages Ref(s)
CX3CR1eGFP/+ An exon of one allele of Cx3cr1 is replaced by the open reading frame encoding eGFP
Fate mapping of CX3CR1+ DCs and macrophages that differentiate from bone-marrow-derived monocytes of the same lineage
CD11b+MHCII+CX3CR1+CD11c+/−
F4/80+/−CD103− (approximately 90% of the total renal DC/macrophage population)
Does not identify the remaining CD11b−
MHCII+CX3CR1−
CD11c+F4/80−
CD103+cell population (estimated 5% of renal-resident DCs)
201
MacGreen An eGFP encoding gene driven by a CSF-1R gene (csf1r) promoter sequence is engineered within the first intron
Identification of macrophages
Bone marrow: 50% of eGFP+ cells are also F4/80+ and CSF-1R+
Peripheral blood: all eGFP+ cells express F4/80 and CD11bKidney: peri-epithelial macrophage-like cells correlate with F4/80 expression in a predominantly medullary location
Does not distinguish cell subsets
64, 65, 202
Abbreviations: CSF-1R, colony-stimulating factor-1 receptor; CX3CR1, CX3C chemokine receptor 1; DC, dendritic cell; eGFP, enhanced green fluorescent protein.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved
NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 7
depletion of CD11c+ cells; or any combination of these potential mechanisms.
Although investigators have typically used a single cell-ablation method, the concurrent use of multiple strat egies can also be informative. For example, admin-istration of liposomal clodronate to CD11b–DTR mice (in the absence of DT) markedly protected against renal ischaemia– reperfusion injury (IRI), whereas admin-istration of DT to CD11b–DTR mice had no effect on injury.72,73 Intriguingly, the combined administration of both liposomal clodronate and DT provided no pro-tection, indicating that the addition of DT removes the protective effect of the liposomal clodronate.73 These data suggest that the phenotype of the surviving resid-ual cells in the kidneys of liposomal-clodronate-treated mice, predominantly identified as CD206+ macrophages and CD11c+ cells,73 might be key to the protective effect observed. These potential caveats and complexities should be borne in mind when cell-ablation models are used.
Renal DC and macrophage functionsIntrarenal activitiesCell recruitmentRenal DCs seem to mediate the recruitment of other cell types with various immunological functions. In a mouse model of pyelonephritis caused by Escherichia coli, CD11c+ renal DCs are the predominant producers of CXC chemokine ligand 2 (CXCL2), which drives the recruitment of neutrophils to facilitate bacterial clearance (Figure 3);74 this effect was abrogated after conditional DC ablation in CD11c–DTR mice.74
Renal repairThe progression of disease in many experimental models of kidney injury, such as chronic allograft and diabetic
nephropathy, can be limited through various interven-tions, but these do not typically lead to renal regeneration. Renal regeneration does occur, however, following mod-erate AKI induced by renal IRI, and this setting has been used to explore the role of macrophages in renal repair; in particular, depletion of macrophages using either liposo-mal clodronate75 or DT treatment of CD11b–DTR mice62 during the reparative phase of IRI-induced AKI markedly retarded restoration of tubular integrity and renal func-tion. Macrophage-derived Wnt7B and IL-22 (induced via TLR4) have been implicated as key mediators of renal repair and tubular regeneration in this context.62,76
FibrosisFibrosis is associated with progression of kidney disease to chronic renal impairment.61,77 That macrophages can promote the production of extracellular matrix by myo-fibroblasts and, therefore, scar formation by adopting a ‘wound healing’ M2 phenotype during inflammation is well established (Figure 4); in the kidney, this pathway is likely to be detrimental and, in AKI, might occur in conjunction with other profibrotic processes, such as epithelial cell-cycle arrest.78 Indeed, macrophages have been implicated in the development of renal fibrosis, as administration of liposomal clodronate was found to be protective in the rodent unilateral ureteral obstruc-tion (UUO) model.79,80 Interestingly, reduced fibrosis was demonstrated in DT-treated CD11b–DTR mice with UUO.9,81 In contrast, ablation of CD11c+ cells in CD11c–DTR mice with UUO did not reduce renal scar-ring,82,83 despite the fact that increases in the maturation, activation and antigen-presenting capacity of renal DCs were reported in this setting.83 Studies involving the use of bone-marrow-chimeric CD11b–DTR mice, in which either resident renal DCs and macrophages or infiltrating
Table 2 | Rodent models of DC and macrophage ablation
Model Mechanism Utility Disadvantages
CD11c–DTR mouse17
Simian DTR–eGFP-encoding fusion gene inserted after the promoter of the Itgax gene encoding CD11c; administration of DT results in the death of cells with active Itgax-promoter-dependent gene expression (that is, cell types that are usually CD11c+)
Short-term (48 h) ablation of 85–90% conventional DCs in vivo, with preservation of plasmacytoid DC compartment;203 cytotoxic T cells17 and NK cells204 were affected to a lesser extentB cells and macrophages were unaffected
Depleted all CD11c+ DCs systemically, including F4/80+ cells16
Late (>72 h) neutrophilia,205 which could potentially mask the DC/macrophage-dependent effectsRepeated systemic administration of DT is lethal
CD11b–DTR mouse16,206
DTR–eGFP encoding fusion gene inserted after the promoter of the Itgam gene encoding CD11b; DT administration causes the death of cells with active Itgam-promoter-dependent gene expression (that is, cell types that are usually CD11b+)
CD11b+ cells predominantly affected (also expressing F4/80);206 CD3+ T cells, B cells, and neutrophils were not affected73,206
Depletes >90% of CD11b+ cells (Ly6G−CCR2−CX3CR1+ and Ly6G+CCR2+CX3CR1−) systemically at 24 h after DT administration; counts were still low at 48 h73
Liposomal clodronate71
Liposomes phagocytosed by macrophages, followed by intracellular clodronate accumulation, which causes cell death by apoptosis70
Depletes phagocytic cells; total macrophage population (Ly6G+ and Ly6G−) decreased by >75% after 24 h73
Ly6G+ cells recover to higher than pretreatment levels73
The CD11c+ cell population is preserved73
All cells with substantial phagocytic capacity are potentially affectedClodronate leakage from dead or dying cells might affect inflammatory reactions69
Abbreviations: CCR2, CC chemokine receptor 2; CX3CR1, CX3C chemokine receptor 1; DC, dendritic cell; DT; diphtheria toxin; DTR, diphtheria toxin receptor (also known as heparin-binding EGF-like growth factor); eGFP, enhanced green fluorescent protein; Ly6G, lymphocyte antigen 6G (also known as granulocyte-differentiation antigen-1); NK, natural killer.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

8 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
bone-marrow-derived cells were depleted, demonstrated that resident renal non parenchymal cells proliferate in UUO, but do not actively contribute to fibrosis;9 the cells with a profibrotic transcriptional profile expressed low levels of Ly6C and had matured from kidney-infiltrating ‘Ly6Chigh’ monocytes.9 In addition, persistent pro-inflammatory macrophage activation has been shown to promote tubular atrophy and fibrosis after AKI.84 Collectively, these data suggest that macrophages, rather than DCs, drive renal scarring.
The precise mechanism by which macrophages promote fibrosis in the kidney remains unclear. Trans-forming growth factor β1 (TGF-β1) is implicated in this process, as a key profibrotic cytokine (Figure 4); however, mice lacking expression of this cytokine specifically in macrophages exhibited comparable levels of fibrosis to wild-type mice with IRI-induced AKI or UUO, suggest-ing that macrophage-derived TGF-β1 is not critical to fibrosis in these disease models.85 Macrophage-derived mediators, such as the lectin galectin 3,81 might be
important to renal fibrosis, but further work is needed to clarify their identity and role in this process.
Extrarenal DC functionFollowing activation, renal DCs are known to traffic to draining lymph nodes (Figure 3), where they present antigens. Several studies have provided additional insights into the behaviour of these DCs when exposed to antigenic material. CD11c+ renal DCs preferentially and rapidly endocytose small (<40 kDa) molecules, compared with splenic DCs that predominantly endo-cytose larger antigenic material (>500 kDa).86 DCs in the kidney-draining lymph nodes also endocytose low- molecular-weight antigens that are transported to them by cell-independent mechanisms. Renal lymph-n ode-based DCs, which express CD8, chemokine XC receptor 1 (XCR1; also known as the lymphotactin receptor), and basic leucine zipper transcriptional factor ATF-like 3 (B-ATF-3), are capable of antigen cross-presentation to subsequently delete cytotoxic T cells in a programmed death ligand 1 (PD-L1)-dependent manner.87 These find-ings imply a necessary tolerogenic function for renal DCs that are continually exposed to innocuous antigens, such as serum and food proteins. This hypothesis is further supported by the finding that proteasomal process-ing of albumin by rat bone-marrow-derived renal DCs in vitro enables these cells to prime and subsequently activate CD8+ T cells.88 Similar observations have been made in rats subjected to five-sixths nephrectomy to induce focal segmental glomerulosclerosis, which resulted in interstitial inflammation, including infiltra-tion of CD11c+CD103+ DCs.88 The cell infiltrates in the kidney were reduced in animals treated with the pro-teasome inhibitor bortezomib.88 In this study, DCs iso-lated from kidney-draining lymph nodes in five-sixths nephrectomized mice activated syngeneic CD8+ T cells in a manner that was time-dependent and reduced by bortezomib treatment.
Systemic administration of LPS in mice initiates DC efflux from the kidney to the draining lymph nodes over 24–72 h.89,90 Renal subcapsular placement of ovalbumin and subsequent LPS administration results in DO11.10 (ovalbumin-specific) CD4+ T-cell proliferation in the ipsilateral renal lymph node, with antigen presentation mediated by CD11c+ renal DCs.89,90 A similar DC migra-tory response has been seen following IRI.89,90 These find-ings are relevant mechanistically to the ex vivo analysis of FLT3LG-mobilized renal DCs that upregulate CCR7 in response to LPS (Figure 1),54 a necessary requirement for DC migration to draining lymph nodes, which is m ediated by CCL19 and CCL21 (Figure 2).
DCs/macrophages—roles in renal diseaseMultiple rodent models of diverse clinical renal diseases are available (Table 3), and can provide mechanistic insights into glomerular and tubular pathophysiology (reviewed elsewhere91). We discuss a number of stand-ard immunological and cytotoxin-based models of renal disease in which the roles of DCs and/or macrophages have been investigated extensively.
Anti-in�ammatoryDC activity
Mitigation of AKI (IRIand cisplatin)
Amelioration of NTN
Neutrophilrecruitment
Gramnegativebacilli
Migratingmonocyte
Migration to lymph node andpresentation of antigens to T cells
DC exodusin response
to IRILPS
and otherstimuli
Interstitium
Lymphatics
Peritubular capillary
Endothelium
Tubular lumen
Albumin oralbumin
fragments
Tightjunction
Proin�ammatory DC activityCytokine secretion in IRI
Protein uptake andantigen presentation
IL-12 secretion, intrarenalattraction and expansion ofT cells and B cells in SLEIntrarenal T-cell activation
Induction of TH17 cells in UUO
Interactions with tissue-residentcells and the in�ux of proteins
and immune cells dictates DC maturation
CXCL2
CD45CD11c
Neutrophil
Renal tubualepithelial cell
Figure 3 | Renal DC function in health and disease. DCs perform homeostatic functions, including induction of tolerance to peripheral antigens typically cleared by the glomerulus, such as albumin, and anti-infection immunosurveillance. Interaction of DCs with bacteria causes them to generate chemokines to attract effector cells, such as neutrophils. The kidney-resident DCs also operate to exacerbate (proinflammatory DCs) or mitigate (anti-inflammatory DCs) a wide range of parenchymal disease, and the role of these cells in disease might be determined by either tissue-resident cells or influxing cells and antigens. For example, the responsiveness and maturation state of DCs might be regulated by ongoing interactions with tubular epithelial cells. Abbreviations: AKI, acute kidney injury; DC, dendritic cell; IRI, ischaemia–reperfusion injury; LPS, lipopolysaccharide; NTN, nephrotoxic nephritis; SLE, systemic lupus erythematosus; TH17, type 17 T-helper; UUO, unilateral ureteral obstruction.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 9
GlomerulonephritidesNephrotoxic nephritisMice injected with antiserum produced in sheep, rabbits or goats after vaccination with mouse renal cortex develop rapid and progressive glomerular damage, termed nephrotoxic nephritis. This condition mimics the crescentic glomerulonephritis seen in human auto-immune disease.92 The glomerular-based antigen– antibody complexes that are generated in this model are captured by both DCs and macrophages, and are sub-sequently presented to T cells (Table 3). The relevant APCs within the kidney during the initial ‘injury’ phase of nephrotoxic nephritis are typically CD11c+CD11b+ cells with a maturing phenotype. Of note, depletion of these cells in CD11c–DTR mice at this early stage (day 4) exacer bated disease, suggesting an anti-inflammatory role for renal DCs;93 however, depletion of CD11c+ cells at a later stage (day 7) reduced the numbers of effector DCs and T cells, and attenuated disease,93 supporting a pro-inflammatory role of DCs as they mature and implying
a biphasic role for DCs in this disease process.93 Renal DCs isolated from these mice induced CD4+ T‑cell pro‑liferation ex vivo, and promoted concurrent secretion of interferon (IFN)‑γ and IL‑10 in co‑culture experi‑ments; IL‑10 levels were elevated at day 4 and increased over time whereas proinflammatory cytokine expression only seemed to be increased at day 10, again support‑ing the proinflammatory and anti‑inflammatory roles of DCs.93 Ly6G– renal DCs, rather than cells derived from infiltrating Ly6G+ monocytes, seem to produce most of the proinflammatory tumour necrosis factor (TNF) and IL12/23p40 in this model.93 Tubulointerstitial DCs have also been shown to release the proinflammatory cytokine IL‑1β, via NACHT, LRR and PYD domains‑containing protein 3 (NLRP3)‑inflammasome–caspase‑1 pathway activation.94 DC depletion using CD11c–DTR mice provided additional evidence supporting a protective role for renal DCs in nephrotoxic nephritis, by demon‑strating that deletion of these cells at days 4 or 10 after induction of disease aggravates the extent of injury.95 The mechanism of DC‑mediated renal protection in nephrotoxic nephritis seems to centre on IL‑10 produc‑tion and regulation of type 1 TH (TH1)‑cell responses.96,97 Initial evidence suggested that this effect might be medi‑ated via expression of inducible co‑stimulatory molecule ICOS‑L specifically on renal DCs, which could promote IL‑10 secretion by T cells that express its co‑receptor.98 Interestingly, expression of this protein by renal DCs has been shown to decrease concomitant with increased production of proinflammatory cytokines.93
Macrophage ablation in CD11b–DTR mice has been shown to attenuate glomerular disease and tubular injury, and decrease effector CD4+ T‑cell populations in crescen‑tic glomerulonephritis models.99 Other methods of abro‑gating macrophage function through blockade of factors involved in either recruitment or activation of these cells, such as CCL2, are also protective in glomerulone phritis, suggesting a proinflammatory role for renal macro‑phages.100,101 Further evidence of a proinflammatory role for renal macrophages in glomerulonephritis comes from a rat mesangioproliferative glomerulone phritis model. In this model, injection of mouse monoclonal antibody targeting thymocyte antigen 1a (Thy1.1) results in mesangial‑ based macrophage infiltration.102 Other models of glomerulonephritis induced by antibodies targeting the glomerular basement membrane antibody also demonstrate ingress (or localization of adoptively transferred cells) and proliferation of monocyte‑derived macro phages within glomeruli, correlating with histo‑pathological severity;103,104 these phenomena can be reversed by inhibiting CSF‑1,105 or treatment with an anti‑CD80/CD86 monoclonal antibody.106
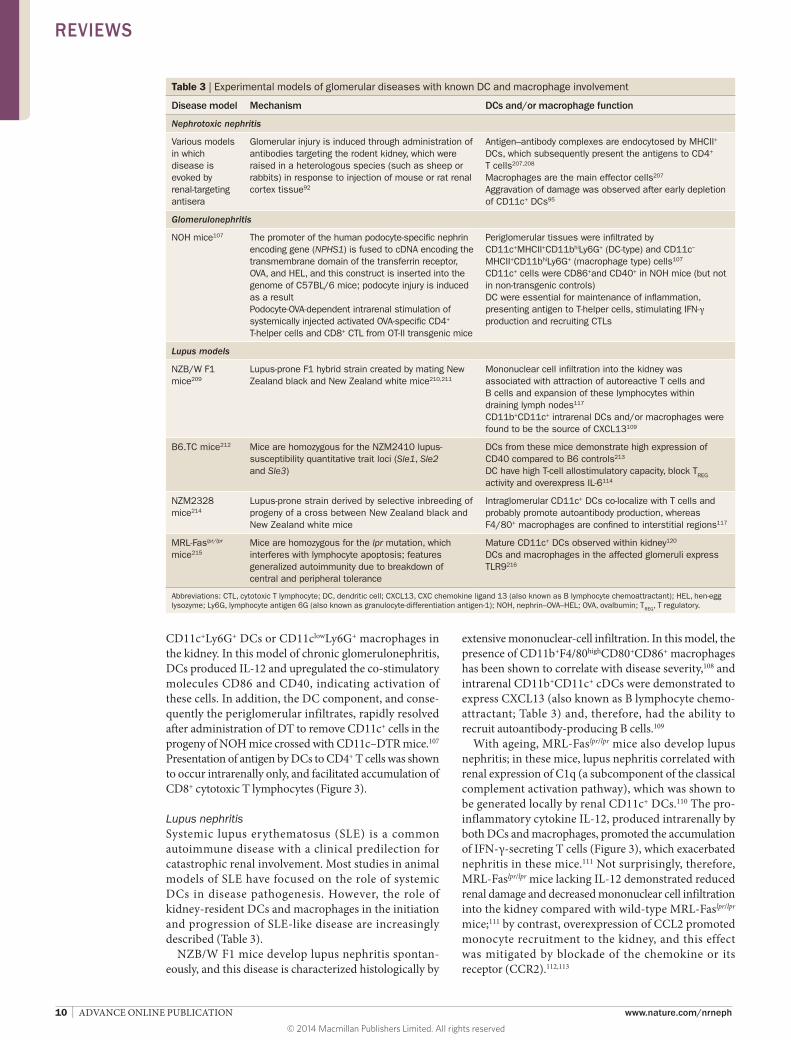
Podocyte immunopathology in NOH miceIn a mouse model of podocyte immunopathology, in which antigens are expressed in glomerular podocytes (so‑called NOH mice; Table 3),107 glomerular infiltrates observed in the experimental disease setting revealed increased numbers of CD11c+CD11bintermediate/high DCs, and CD11c−CD11b+ macrophages, as well as proinflammatory
Stimulation of pericyte accumulation and activation,myo�broblast differentiation, and production of ECM
Myo�broblastsMatrix
depositionPericyte
Tubular lumen
Tightjunction
M1 macrophage M2 macrophage
Repairof AKI
Tissuerepair
Directtubularinjury
Presentationof antigensto T cells
Monocyte recruitedto kidney interstitium
CSF-1 (M-CSF), IL-10
Macrophagereprogramming
Wnt7bIL-22
HO-1IL-10
Increased adhesionand proin�ammatoryactivators (ICAM-1,
osteopontin)
IL-12IL-23
Pro�brotic factors(TGF-β, PDGF,galectin 3)
Figure 4 | Macrophages in renal disease. Tissue-resident macrophages or infiltrating proinflammatory monocytes can become classically activated by exposure to danger-associated molecular patterns or proinflammatory cytokines to take on an M1 phenotype, associated with production of IL-12 and IL-23, engagement of T cells for antigen presentation, activation or exacerbation of profibrotic parenchymal changes, and direct and indirect tissue injury. M1 macrophages can be reprogrammed to become alternatively activated M2 macrophages by stimulation with anti-inflammatory cytokines, such as IL-10 or CSF-1, or ingestion of apoptotic cells. M2 macrophages might facilitate and coordinate restoration of tubular cell and, therefore, kidney tubule integrity following injury. M2 macrophages can also express anti-inflammatory mediators, such as HO-1 and IL-10, which act to limit tissue injury and promote resolution of inflammation, but might also drive pericyte and myofibroblast activation through production of TGF-β, galectin 3 and PDGF. Abbreviations: AKI, acute kidney injury; CSF-1, colony-stimulating factor 1; ECM, extracellular matrix; HO-1, haem oxygenase-1; ICAM-1, intercellular adhesion molecule 1; M-CSF, macrophage colony-stimulating factor; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor β; Wnt7b, wingless-related MMTV integration site 7B.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved
10 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
CD11c+Ly6G+ DCs or CD11clowLy6G+ macrophages in the kidney. In this model of chronic glomerulonephritis, DCs produced IL-12 and upregulated the co-stimulatory mol ecules CD86 and CD40, indicating activation of these cells. In addition, the DC component, and conse-quently the periglomerular infiltrates, rapidly resolved after administration of DT to remove CD11c+ cells in the progeny of NOH mice crossed with CD11c–DTR mice.107 Presentation of antigen by DCs to CD4+ T cells was shown to occur intrarenally only, and facilitated accumulation of CD8+ cytotoxic T lymphocytes (Figure 3).
Lupus nephritisSystemic lupus erythematosus (SLE) is a common autoimmune disease with a clinical predilection for catastrophic renal involvement. Most studies in animal models of SLE have focused on the role of systemic DCs in disease pathogenesis. However, the role of kidney- resident DCs and macrophages in the initiation and progression of SLE-like disease are increasingly described (Table 3).
NZB/W F1 mice develop lupus nephritis spontan-eously, and this disease is characterized histologically by
extensive mononuclear-cell infiltration. In this model, the presence of CD11b+F4/80highCD80+CD86+ macrophages has been shown to correlate with disease severity,108 and intrarenal CD11b+CD11c+ cDCs were demonstrated to express CXCL13 (also known as B lympho cyte chemo-attractant; Table 3) and, therefore, had the ability to recruit autoantibody-producing B cells.109
With ageing, MRL-Faslpr/lpr mice also develop lupus nephritis; in these mice, lupus nephritis correlated with renal expression of C1q (a subcomponent of the classical complement activation pathway), which was shown to be generated locally by renal CD11c+ DCs.110 The pro-inflammatory cytokine IL-12, produced intrarenally by both DCs and macrophages, promoted the accumu lation of IFN-γ-secreting T cells (Figure 3), which exacer bated nephritis in these mice.111 Not surprisingly, therefore, MRL-Faslpr/lpr mice lacking IL-12 demonstrated reduced renal damage and decreased mononuclear cell infiltration into the kidney compared with wild-type MRL-Faslpr/lpr mice;111 by contrast, overexpression of CCL2 promoted monocyte recruitment to the kidney, and this effect was mitigated by blockade of the chemokine or its receptor (CCR2).112,113
Table 3 | Experimental models of glomerular diseases with known DC and macrophage involvement
Disease model Mechanism DCs and/or macrophage function
Nephrotoxic nephritis
Various models in which disease is evoked by renal-targeting antisera
Glomerular injury is induced through administration of antibodies targeting the rodent kidney, which were raised in a heterologous species (such as sheep or rabbits) in response to injection of mouse or rat renal cortex tissue92
Antigen–antibody complexes are endocytosed by MHCII+ DCs, which subsequently present the antigens to CD4+ T cells207,208
Macrophages are the main effector cells207
Aggravation of damage was observed after early depletion of CD11c+ DCs95
Glomerulonephritis
NOH mice107 The promoter of the human podocyte-specific nephrin encoding gene (NPHS1) is fused to cDNA encoding the transmembrane domain of the transferrin receptor, OVA, and HEL, and this construct is inserted into the genome of C57BL/6 mice; podocyte injury is induced as a resultPodocyte-OVA-dependent intrarenal stimulation of systemically injected activated OVA-specific CD4+ T-helper cells and CD8+ CTL from OT-II transgenic mice
Periglomerular tissues were infiltrated by CD11c+MHCII+CD11bhiLy6G+ (DC-type) and CD11c–
MHCII+CD11bhiLy6G+ (macrophage type) cells107
CD11c+ cells were CD86+and CD40+ in NOH mice (but not in non-transgenic controls)DC were essential for maintenance of inflammation, presenting antigen to T-helper cells, stimulating IFN-γ production and recruiting CTLs
Lupus models
NZB/W F1 mice209
Lupus-prone F1 hybrid strain created by mating New Zealand black and New Zealand white mice210,211
Mononuclear cell infiltration into the kidney was associated with attraction of autoreactive T cells and B cells and expansion of these lymphocytes within draining lymph nodes117 CD11b+CD11c+ intrarenal DCs and/or macrophages were found to be the source of CXCL13109
B6.TC mice212 Mice are homozygous for the NZM2410 lupus-susceptibility quantitative trait loci (Sle1, Sle2 and Sle3)
DCs from these mice demonstrate high expression of CD40 compared to B6 controls213
DC have high T-cell allostimulatory capacity, block TREG activity and overexpress IL-6114
NZM2328 mice214
Lupus-prone strain derived by selective inbreeding of progeny of a cross between New Zealand black and New Zealand white mice
Intraglomerular CD11c+ DCs co-localize with T cells and probably promote autoantibody production, whereas F4/80+ macrophages are confined to interstitial regions117
MRL-Faslpr/lpr mice215
Mice are homozygous for the lpr mutation, which interferes with lymphocyte apoptosis; features generalized autoimmunity due to breakdown of central and peripheral tolerance
Mature CD11c+ DCs observed within kidney120
DCs and macrophages in the affected glomeruli express TLR9216
Abbreviations: CTL, cytotoxic T lymphocyte; DC, dendritic cell; CXCL13, CXC chemokine ligand 13 (also known as B lymphocyte chemoattractant); HEL, hen-egg lysozyme; Ly6G, lymphocyte antigen 6G (also known as granulocyte-differentiation antigen-1); NOH, nephrin–OVA–HEL; OVA, ovalbumin; TREG, T regulatory.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 11
B6.TC mice also develop spontaneous glomerulo-nephritis concomitant with the production of auto-antibodies. Compared to DCs isolated from B6 control mice, DCs isolated from B6.TC mice have higher T-cell allostimulatory capacity and block the suppressive func-tion of CD4+CD25+ TREG cells via overproduction of IL-6,114 in addition to facilitating B-cell proliferation and autoantibody production (Table 3).115 NZM2328 mice also demonstrate a proliferative, gender-biased (female predilection, as in SLE) glomerulonephritis.116 In this model, renal histology is characterized by intraglomer-ular CD11c+ DCs that co-localize with T cells,117 whereas F4/80+ macrophages are confined to peri-glomerular regions, suggesting that intrarenal DCs might mediate the presentation of (auto)antigen, thereby promoting disease progression (Table 3).
Evidence from preclinical therapeutic studiesAdministration of the immunosuppressant cyclophospha-mide and/or blockade of co-stimulatory signalling (using CTLA4-Ig fusion proteins or anti-CD40-ligand mono-clonal antibodies) in NZB/W F1 mice was associated with a decrease in the number of CD11c+ renal DCs and remis-sion of lupus.118 Amelioration of disease in MRL-Faslpr/lpr mice, which was associated with reduced macrophage and T-cell infiltration into the kidney, has been demonstrated using a p38 mitogen-activated protein kinase inhibitor;119 p38-inhibitor treatment also decreased the abundance of renal CD11c+ DCs, including those with a mature pheno-type that expressed high-mobility group protein B1.120 pDCs have also been implicated in lupus pathogenesis by amelioration of lupus pathology in NZB mice genetically engineered to lack pDCs through del etion of the gene encoding IRF8, and in mice carrying a mutation in the Slc15a4 gene, which retain pDCs incapable of producing type I IFN.121 In perinuclear antineutrophil cytoplasmic antibody (ANCA)-positive SCG/Kj mice that develop rapidly progressive glomerulo nephritis,122 a novel tri-azolopyrimidine derivative (NK026680) that inhibits DC function was demonstrated to suppress multiple disease manifestations and the development of autoanti bodies, providing additional evidence of the role of DCs in glomerulonephritis.123
Manipulation of the humoral immune response through DC-mediated induction of immune tolerance might also represent an approach to mitigating disease in glomerulonephritis. Ligation of Fc receptors (particu-larly low affinity IgG Fc region receptor IIb [FcγRIIb]) by immunoglobulins, including endogenous antibodies and immune complexes derived from lupus-prone mice, have been shown to provide an inhibitory signal that prevents TLR-mediated DC maturation and enhances the tolero-genicity of this cell type.124 Furthermore, adoptive trans-fer of immature DCs overexpressing FcγRIIb before or after the onset of clinical lupus attenuated lupus nephritis in MRL-Faslpr/lpr mice.124 Conversely, MRL-Faslpr/lpr mice lacking FcγRIIb displayed greater B-cell activity and higher levels of DC-derived IL-12.125
The finding that administration of C-reactive protein reverses renal manifestations of nephrotoxic nephritis
in a macrophage-dependent manner suggests that this cell type might also be a valid target in glomerulo-nephritis.126 Interestingly, immune complexes have also been shown to suppress macrophage inflammatory responses via FcγRIIb,127 and this effect was protective in a cryoglobulin- associated membranoproliferative glomerulonephritis mouse model.128 However, binding of tissue-bound antibody or immune complexes might also activate macrophages to produce proinflammatory mediators via activation of various signal transduction pathways, including spleen tyrosine kinase (SYK). In keeping with this possibility, the SYK inhibitor fosta-matinib markedly ameliorated nephrotoxic nephritis in rats and limited macrophage activation following treatment with aggregated IgG.129
Acute kidney injuryIRI and cisplatin-induced kidney injuryBoth IRI and administration of cisplatin can induce tubular epithelial cell coagulative necrosis, and both approaches have been used to study the role of renal DCs and macrophages in AKI. CD11blowF4/80high DC and CD11bhighF4/80low macrophages (both differenti-ated from Ly6Chigh infiltrating cells) could be identified early (3 h) following IRI, with the numbers of these cells peaking at 24 h after IRI, and developing a predomi-nantly macrophage phenotype.56 Although ablation of DCs exacerbates cisplatin-induced AKI,130 a similar DC-mediated protective effect has been disputed in models of IRI (Table 4).72,131 On the other hand, macro-phage ablation provided protection from renal injury in an IRI model,132–134 and this protective effect was reversed by adoptive transfer of the mouse macrophage cell line, RAW 264.7.75,133 However, macrophages also seem to have distinct phenotypes depending on time post-injury in an IRI model: M1 characteristics predominated ini-tially (1–3 days following reperfusion); whereas M2 skewing was observed during the proliferative repair phase (dependent on Wnt7b62 and CSF-1135 produc-tion).60 Interestingly, adoptive-transfer and cell-tracking experiments revealed phenotype switching of IFN-γ-stimulated proinflammatory (M1) macro phages injected after IRI to M2 macrophages at the onset of tissue repair in the kidney.60 The relevance of each of these macro-phage subsets to AKI is reflected in loss-of-function experiments, which demonstrated that clodronate- induced depletion of (M1) cells before or early after IRI reduces kidney injury, whereas cell depletion at later time points (when M2 macrophages predominate) delays tissue restoration.60,136
In a variation of the DTR models described, mice that selectively express the human DTR under the control of the promoter region of γ glutamyl transferase 1, a gene that is specifically expressed in proximal tubule cells, develop AKI in response to injection of DT.135 In this model, AKI is associated with renal infiltration of a CD11b+F4/80+CD11c+CX3CR1+CD86+ DC/macrophage cell type,135 and administration of liposomal clodronate before DT exposure exacerbated renal injury, increased mortality, and prolonged renal repair, as did concomitant
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

12 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
DTR-mediated depletion of both CD11c+ DCs, macro-phages and proximal renal tubular epithelial cells.135 These data further demonstrate the role of DC and/or macrophages for tubular recovery after AKI.
Ablation of CD11c+F4/80+ DC/macrophages using liposomal clodronate administered 1 day after renal IRI aggravated injury and retarded tissue regeneration, seem-ingly by preventing intrinsic IL-10 release by these cells, which was associated with increased proinflammatory
cytokine levels and persistence of an inflammatory milieu.136 In the same model, injury was partly allevi-ated by adoptive transfer of CD11c+ DCs.136 The protec-tive effects of IL-10 generated by renal DCs have been seen in other models of AKI, including cisplatin-induced nephrotoxicity: renal DCs were demonstrated to express higher levels of IL-10 following cisplatin treatment (with concomitant increases in IL-10 receptor 1 expression).137 Conversely, enhanced cisplatin-mediated injury was
Table 4 | Rodent models of renal injury and the effect of DC or macrophage depletion
Disease model Renal phenotype DC and/or macrophage involvement
Effect of DC and/or macrophage depletion on disease (by depletion strategy)
CD11c–DTR CD11b–DTR Liposomal clodronate
Acute kidney injury
Unilateral ureteral obstruction
Fibrosis Interstitial infiltration of CD11b+Ly6G+ or F4/80+ DCs
No change in fibrosis82,83
Reduced fibrosis9 Reduced fibrosis80
Decreased IFN-γ and IL-17 T cells152
IRI Coagulative necrosis of renal tubular epithelial cells
Interstitial infiltration of neutrophils, macrophages and CD11c+ DCsDepletion of macrophages before IRI is protective; depletion at 3–5 days post-IRI causes defective repair60
Exacerbatory72 Protective131
Exacerbatory72
No change73
Exacerbatory136 Protective72,73,75,132
Decreased TNF production147
Cisplatin Coagulative necrosis of renal tubular epithelial cells; renal excretion of cisplatin results in concentration of drug in cortex resulting in damage predominantly to S3 segment of the proximal tubule
Not described Cell depletion at time of cisplatin treatment exacerbated injury130
Not studied Not studied
Adriamycin Glomerular capillary permeability; ROS-mediated tubular damage
Adoptive transfer of LPS-treated pDC ameliorates disease217
Not studied Not studied Not studied
Glomerulonephritis
Nephrotoxic nephritis
Crescentic glomerulonephritis
DCs present renal antigens to T cells
Cell depletion at day 4 and day 10 after induction of nephritis exacerbated renal injury;95 depletion at day 7 reduced injury93
Decreased glomerular crescents and proteinuria; improved renal function99
Decreased proteinuria;218 CRP treatment negated this protective effect126
NOH mice107 Podocyte-related glomerular pathology
Glomerular infiltration of CD11c+CD11b+Ly6G+ DCs/macrophages
Decreased CD11c+CD11b+Ly6G+ cell infiltration into the glomerulus107
Not studied Not studied
Lupus nephritis
Lupus-prone NZB/W mice
Severe glomerulonephritis after polyI:C administration219
Glomerular infiltration of CD11b+Ly6G−F4/80+ DCs/macrophages220
Not studied Not studied Decreased glomerular accumulation of macrophages and glomerular injury219
MRL-Faslpr/lpr mice Glomerular crescent formation, granular IgG and C3 deposition within capillaries, proteinuria
DC produce C1q;110 treatment with p38 MAPK inhibitor decreases CD11c+ DC infiltrates;120 CSF-1 deficiency protects against lupus development221
Decreased autoantibody production and disease severity222
Not studied Not studied
Infectious disease
Pyelonephritis Escherichia coli-induced renal injury74
DC-mediated production of CXCL2 causes recruitment of neutrophils74
Decreased neutrophil recruitment
Not studied Not studied
Abbreviations: CRP, C-reactive protein; CXCL2, CXC chemokine ligand 2; DC, dendritic cell; IRI, Ischaemia–reperfusion injury; LPS, lipopolysaccharide; Ly6G, lymphocyte antigen 6G (also known as granulocyte-differentiation antigen-1); pDC, plasmacytoid DC; ROS, reactive oxygen species; TLR9, Toll-like receptor 9.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 13
observed in mice lacking IL-10 expression, as well as in chimeric mice lacking IL-10 production only in DCs.137
CD11c+ DCs might also provide protection against AKI by modulating the effects of additional cell popu-lations. Findings in cisplatin-mediated AKI are con-sistent with this hypothesis, suggesting that IL-10 modulates increased expression of ICOS-L on renal DCs.130 Although not exclusively demonstrated within the kidney, these data imply that the presence of IL-10 may regulate CD4+ T-cell responses.95 Indeed, the pro-tective role of TREG cells (both CD4+CD25+FOXP3+ and CD4+CD25+IL-10+)138 induced by CD11c+ DCs follow-ing renal IRI has been well- recognized.139–142 Moreover, infusion of mesenchymal stem cells abrogated renal IRI (reviewed in143), via an effect that was partly mediated by CD11c+ DCs.144 In particular, an immature phenotype of tissue-resident DCs and intrarenal FOXP3+ expression were associated with this renoprotective effect of mesen-chymal stem cells, and these features were lost after DT-treatment of CD11c–DTR mice; partial restoration of these characteristics was achieved by the adoptive transfer of CD11c+ DCs, although not if they lacked the capacity to produce IL-10.
Increasing evidence suggests that the cytokine milieu associated with sterile inflammation established after IRI is modulated by resident renal cells, including DCs and/or macrophages and RTECs, to drive pathological and reparative processes. In particular, RTECs have been shown to express CSF-1, which promotes macrophage proliferation in situ.135,145 However, TLR-activated RTECs limited classical macrophage activation in vitro, and the results of antibody-based inhibition experiments sug-gesting a role for IL-10 in this regulatory process.146 Furthermore, a series of in vitro and in vivo experiments demonstrated that RTECs could induce nonprogrammed, quiescent macrophages or programmed proinflammatory M1 macrophages to adopt a M2 phenotype that limited acute renal inflammation and promoted repair.60 Resident CD11c+F4/80+ renal DC/macrophage numbers remained static after IRI to the kidney, whereas the numbers of infil-trating F4/80– DCs and/or macrophages with a mature phenotype increased.147 TNF, as well as IL-6 and CCL2 were produced in greater quantities by the resident renal DCs isolated from ischaemic kidneys than from control kidneys, and in vivo depletion of DCs diminished total TNF secretion within the renal CD45+ cell compart-ment.147 Interestingly, renal IRI-mediated induction of interferon regulatory factor 4 (IRF4, an inducible inhibitor of TLR2 and TLR4 signalling) is localized to CD45+CD11c+ DCs/macrophages, and mice lacking IRF4 demonstrated increased renal damage after IRI, which was associated with increased TNF expression and abrogated by liposomal clodronate, suggesting that IRF4 coordinates immunosuppressive effects of these cells by restricting TNF production.148 These findings suggest that resident renal cells dictate the immune responses that occur in AKI through creation and m odification of the cytokines present within the injured tissues.
Cell-surface receptors that have a role in mediat-ing AKI are often expressed concomitantly on renal
parenchymal and interstitial cells. For example, single Ig IL-1-related receptor (SIGIRR; also known as Toll–IL-1 receptor 8), modulates TLR signalling responses, particularly APC function, in response to LPS challenge and ischaemic renal damage.149,150 In particular, SIGIRR-deficient mice demonstrate increased susceptibility to tissue damage, including renal IRI, with increased production of IL-6, CXCL2 and CCL2, compared with wild-type mice.149 This response is abrogated in wild-type mice transplanted with Sigirr–/– bone marrow cells after renal IRI, as well as in Sigirr–/– mice treated with lipo-somal clodronate,149 suggesting that SIGIRR represses the response of renal myeloid cells to IRI, rather than RTECs. Transplantation of Sigirr–/– renal allografts in a fully MHC-mismatched mouse model has also been associ-ated with expansion and maturation of CD11b+CD11c+ resident DCs/macrophages that prime T cells and impede the development of CD4+CD25+FOXP3+ TREG cells, supporting a negative modulatory role for this protein in renal DCs/macrophages.151
Unilateral ureteral obstructionUUO is a well-characterized model in which early inflam-mation is followed by renal fibrosis. CD11c+ DCs exhibit phenotypic maturation, and enhanced antigen presen-tation to and activation of T cells following UUO, with accompanying interstitial infiltration by CD11b+Ly6G+ or F4/80+ DCs (depending on the cell-surface marker used).82,83,152 DC depletion in CD11c–DTR mice at various times after induction of UUO does not affect the subsequent development of renal fibrosis,82,83 although this feature is ameliorated by macrophage depletion in CD11b–DTR mice,9 or by administration of liposomal clodronate (Table 4).80 However, additional studies suggest a proinflammatory role for DCs in UUO, as decreased IFN-γ and IL-17 production by T cells was observed after DC depletion.152 Three distinct populations of CD11b+ macrophages that express either high, intermediate or low levels of Ly6C have been identified to differentiate infiltrating bone-marrow-derived monocytes from tissue-resident macrophages.9 Recruited Ly6Chi cells differentiate within the kidney becoming Ly6Cint and then Ly6Clo cells, with the latter demonstrating a profibrotic transcriptional profile. This finding is in keeping with data demonstrat-ing that blockade of CCL2 or its receptor, CCR2, and thus recruitment of inflammatory-type Ly6Chi monocytes impedes macrophage accumulation.153
TransplantationThe involvement of DCs and macrophages in renal transplantation has been studied,154 but limited infor-mation is available concerning the effects of standard immunosuppressive agents on renal DC or macrophage numbers and function. Both donor macrophages and DCs are transferred within the allograft at the time of transplantation, and subsequently host DCs and macro-phages are recruited into the transplanted kidney.155,156 Macrophages proliferate intensely within kidney allo-grafts in the absence of immunosuppression, a process that is promoted by CSF-1.157 Early animal studies
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

14 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
revealed a lack of MHC II antigen induction in kidney allografts when recipients received ciclosporin, compared with untreated transplant recipients.158 Paradoxically, ciclosporin increased DC number and maturation status in rats.159–162 Warm ischaemia alone leads to loss of the CD11c+CD11b+CD103+ DC subset from syngeneic grafts within 10 weeks, whereas cold ischaemia results in addi-tional loss of the CD103− DC subset and their replace-ment by host CD11b+CD11c+CD103+TNF+ DCs, which is associated with progressive T-cell accumulation and IFN-γ production.161
The ability of DCs to subvert the alloimmune response following transplantation has been documented exten-sively in the literature (reviewed in20). Given their typi-cally immature phenotype,52 renal DCs represent an ideal potential source of regulatory DCs; however, use of these cells is limited by inherent difficulties in isolat-ing adequate populations. Nevertheless, mobilization of mouse renal DCs using FLT3LG increased the absolute numbers that could be isolated for subsequent infusion before cardiac transplantation, which prolonged allograft survival in the absence of immunosuppression.52
Macrophages are also recognized as important contrib-utors to both acute and chronic allograft injury in animal models. In loss-of-function studies, administration of liposomal clodronate mitigated functional impairment and tissue injury in a rat renal transplant model without affecting the number or activation status of lympho-cytes.162 Similarly, depletion of CD11b+ cells in CD11b–DTR mice protected renal allografts from acute injury and rejection.163 Furthermore, the pharmacological antag-onism of the CSF-1–CSF-1R pathway reduced macro-phage infiltration, T-cell activation within the allograft, acute cellular rejection and tubulointerstitial injury.164,165 Interestingly, such treatment had no effect on systemic activation of T cells and B cells or humoral rejection that occurred later in the renal transplant model.165 Persistent macrophage infiltration also characterizes the develop-ment of chronic allograft nephropathy in rodents,166 and correspondingly, this manifestation is ameliorated by blockade of macrophage development or activity.167,168
Renal DCs and macrophages in humansThe studies in rodent models described in the previous sections have provided important insights in the vari-able and plastic characteristics of renal DCs and macro-phages, and provided a basis for increased phenotypic and functional understanding of such cells in humans. However, consensus has not been reached on the identi-fication of distinct cell subtypes in rodents or humans and, therefore, comparison of cell types and extrapo-lation of data from animal models to human disease is difficult. These issues provide avenues for future research (see Box 2). Nevertheless, our knowledge of renal DCs and macrophages in the healthy and diseased human kidney has expanded.
In the healthy kidneyThe DC and macrophage populations present in normal human kidneys have been studied less extensively than those in rodents. To date, most studies in human kidneys have focused on demonstrating the presence of DCs within renal parenchyma using immunohistochemistry (mainly according to expression of blood DC antigen [BDCA] molecules, CD68 and DC-specific ICAM-3-grabbing non-integrin [DC-SIGN; also known as CD209]). The putative function of these cells has been extrapo-lated based on findings in animal models. Both cDCs (BDCA-1[CD1c]+DC-SIGN+CD68+ and BDCA1+DC-SIGN−CD68−) and pDCs (BDCA-2+DC-SIGN−) have been demonstrated within the normal renal interstitium and are rarely present within glomeruli in humans.169
In renal allografts Examination of human renal allograft biopsy samples during acute cellular rejection has revealed an increased number of DC-SIGN+ DCs, particularly intraglomer-ular cells.169 Cells positive for BDCA-1, BDCA-2 and DC lysosomal associated membrane protein (DC-LAMP, also known as CD208) have also been identified within renal allografts during acute rejection.169 CCR1+DC-SIGN+ DCs have been described in the kidney after transplantation.170 Macrophages (CD68+ cells) have also been identified in renal allografts with histological evidence of antibody-mediated,171,172 acute cellular173 and chronic rejection.174 Furthermore, reduced estimated glomerular filtration rate correlated with macrophage-related gene expression panels in renal transplant tissues obtained in biopsies performed for various clinical indications.175 Histological alterations reflecting interstitial fibrosis and/or tubular atrophy as well as inflammation were also associated with a macrophage signature in microarray analyses.176,177 Whether these data reflect a subset of macrophages associ-ated with acute rejection or a reparative phase to injury remains unclear; nevertheless, glomerular178,179 or inter-stitial180,181 macrophage infiltration is a poor prognostic sign and marker of unfavourable graft outcome.
Diseases of the native kidneyMacrophage infiltration has also been shown to be associ-ated with adverse outcomes in native kidney disease; for example, CD68+ macrophage infiltration is an independent
Box 2 | Renal DCs and macrophages—future aims
■ Consensus must be reached regarding differences and similarities between DCs and macrophages, to enable appropriate identification and classification of the following types: glomerular versus tubulointerstitial; tissue-resident versus infiltrating; immunogenic versus tolerogenic; fibrotic versus reparative
■ Efforts are needed to standardize the terminology between mouse and human cells
■ Exploration of RTEC–DC–macrophage crosstalk is important
■ Expansion of understanding of DCs and macrophages in non-immunological and chronic disease processes, such as diabetic kidney disease, is required
■ Development of models with kidney-specific DC and/or macrophage ablation should be a key goal
■ Emphasis should be placed on translational research and the applicability of findings to clinical disease
Abbreviations: DC, dendritic cell; RTEC, renal tubular epithelial cell.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 15
risk factor for progression to end-stage renal disease in patients with membranous nephropathy.182 Evaluation of biopsy tissues from patients with a diverse range of chronic kidney diseases demonstrated that macrophage infiltration correlated with both the degree of chronic damage and serum creatinine levels,183 and that these cells co-localized to areas of capillary rarefaction, implicating a role for macrophages in microvasculature injury.184
Early studies detected CD1b+ DCs within areas of active interstitial inflammation and glomerular crescent forma-tion.185 Observations regarding DC infiltration have also been made in the kidneys of patients with IgA nephrop-athy, with the findings indicating that the number and anatomical location of these cells alters in the disease setting.169 In patients with lupus nephritis, increased numbers of putative cDC that express BDCA-1, BDCA-3 (also known as CD141 and thymomodulin), and BDCA-4 (CD304; neuropilin-1) have been reported.186 BDCA-1+ myeloid DCs were also associated with C1q in kidney tissues from patients with severe lupus nephritis, with concordant animal model data suggesting that intrinsic renal myeloid DCs contribute to local C1q synthesis.110 In human glomerulonephritis, mononuclear-cell subtypes might be compartmentally separated, with tubulointer-stitial cell accumulation of CD68+DC-SIGN+ DCs cells and intraglomerular localization of CD68+DC-SIGN− macro phages. A correlation between the number of tubulo interstitial DCs and degree of renal injury has also been suggested.187 Mature DC-LAMP+ DCs are not present in healthy kidneys but are evident at low numbers in the kidneys of patients with renal disease, including lupus nephritis.169,188
In addition, a study in patients with chronic kidney disease has demonstrated increased BDCA-3highCLEC9A+ and BDCA-1+CD1a−DC-SIGN− myeloid DCs, as well as pDCs, in human kidney biopsy samples with histologi-cal evidence of fibrosis, compared with biopsy samples without fibrosis or healthy tissue samples.189 The myeloid DC phenotype of some of the renal DCs observed in this study suggests that they were derived from the peripheral circulation, and these cells were implicated as the pre-dominant source of the elevated TGF-β levels in biopsy samples from fibrotic kidneys.189
Although the presence of pDC within the diseased kidney has been demonstrated in mouse models, the role of these cells in human disease has been disputed. The contribution of pDC to renal pathology has been demonstrated most consistently in lupus nephritis, where secretion of IL-18 by resident glomerular cells is a potent chemoattractant for IL-18-receptor-expressing pDC.190,191 BDCA-2+ChemR23 (Chemokine receptor-like 1)+ pDC have been demonstrated to be present in the kidney in the context of severe lupus nephritis.186 pDC are also reported to be evident in increased numbers in kidney transplants with delayed graft function, com-pared with the setting of nephrotoxicity associated with calcineurin inhibitors, in which the myeloid DCs are preponderant.192
Cross-talk between T cells and DCs is well-recognized in mouse models, and evidence from human kidney
samples suggests that a similar inter-relationship is involved in the initiation of immunological responses in the kidney. In particular, biopsy tissues from patients with ANCA-associated vasculitis revealed a close prox-imity between CD3+ T cells and DC-SIGN+ DCs.193 Further more, clusters of CD21+ follicular DCs that express the B-lymphocyte-specific chemokine CXCL13 have also been demonstrated in tertiary lymphoid struc-tures that develop within the renal parenchyma follow-ing chronic antigen stimulation (in the setting of renal allografts),194 and neolymphangiogenesis with admixed CD4+ T cells, CD8+ T cells, CD20+ B cells, S-100+ cDCs and pDCs.195 These observations suggest that intra-renal antigen presentation by DCs to T cells and B cells promotes chronic, immune-cell-mediated inflamma-tory responses in various kidney diseases; thus, renal DCs (and/or macrophages) probably have key roles in mediating kidney disease.
ConclusionsRenal DCs and macrophages are phenotypically and functionally heterogeneous cells that regulate tissue responses to renal injury and disease. The considerable overlap between DCs and macrophages represents a con-tinuum of phenotype, as well as plasticity of cells of the myeloid–monocytic lineage both in vivo and in vitro. Our knowledge of renal DC and macrophage function lags behind that described for these cell types in other organs; however, progress has been made in addressing this limi-tation through advances in cell isolation, identification, in vivo propagation and the use of innovative genetically-modified mouse models. The central role of renal DCs and macrophages in homeostasis, as well as their capacity to modulate physiological function to drive immune or nonimmune disorders, is increasingly recognized. These cells provide a reservoir for ongoing immunosurveil-lance within the renal tubulointerstitium extending to the draining lymph nodes, in addition to affording an immune privileged site within the glomerulus.
Extensive small-animal work has provided an impor-tant basis for increased phenotypic and functional understanding; however, a consensus regarding precise cell identification and extrapolation to human kidney disease is lacking, providing avenues for future research. Importantly, after several decades of research the ques-tion of whether renal DCs and macrophages constitute sufficiently specific targets for therapeutic intervention to potentially ameliorate kidney disease progression remains, necessitating further study.
Review criteria
A search for original, peer-reviewed articles was performed in MEDLINE and PubMed in November 2013 and again in February 2014. The search terms used, alone and in combination, were “kidney”, “dendritic cells”, “macrophages”, “glomerulonephritis”, “acute kidney injury”, “lupus”, “fibrosis” and “transplantation”. All articles identified were English-language, full-text papers. The reference lists of the identified articles were searched for further relevant papers.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

16 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
1. Banchereau, J. & Steinman, R. M. Dendritic cells and the control of immunity. Nature 392, 245–252 (1998).
2. Geissmann, F. The origin of dendritic cells. Nat. Immunol. 8, 558–560 (2007).
3. Shortman, K. & Liu, Y. J. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2, 151–161 (2002).
4. Nelson, P. J. et al. The renal mononuclear phagocytic system. J. Am. Soc. Nephrol. 23, 194–203 (2012).
5. Lyman, S. D. et al. Molecular cloning of a ligand for the flt3/flk-2 tyrosine kinase receptor: a proliferative factor for primitive hematopoietic cells. Cell 75, 1157–1167 (1993).
6. Wu, L. & Liu, Y. J. Development of dendritic-cell lineages. Immunity 26, 741–750 (2007).
7. Geissmann, F. et al. Development of monocytes, macrophages, and dendritic cells. Science 327, 656–661 (2010).
8. Shortman, K. & Naik, S. H. Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 7, 19–30 (2007).
9. Lin, S. L., Castano, A. P., Nowlin, B. T., Lupher, M. L. Jr & Duffield, J. S. Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J. Immunol. 183, 6733–6743 (2009).
10. Miller, J. C. et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat. Immunol. 13, 888–899 (2012).
11. Hume, D. A. Plenary perspective: the complexity of constitutive and inducible gene expression in mononuclear phagocytes. J. Leukoc. Biol. 92, 433–444 (2012).
12. Hume, D. A. Macrophages as APC and the dendritic cell myth. J. Immunol. 181, 5829–5835 (2008).
13. Hume, D. A., Mabbott, N., Raza, S. & Freeman, T. C. Can DCs be distinguished from macrophages by molecular signatures? Nat. Immunol. 14, 187–189 (2013).
14. Ferenbach, D. & Hughes, J. Macrophages and dendritic cells: what is the difference? Kidney Int. 74, 5–7 (2008).
15. Kawakami, T. et al. Resident renal mononuclear phagocytes comprise five discrete populations with distinct phenotypes and functions. J. Immunol. 191, 3358–3372 (2013).
16. Cailhier, J. F. et al. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. J. Immunol. 174, 2336–2342 (2005).
17. Jung, S. et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 17, 211–220 (2002).
18. Steinman, R. M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 9, 271–296 (1991).
19. Naik, S. H. Demystifying the development of dendritic cell subtypes, a little. Immunol. Cell Biol. 86, 439–452 (2008).
20. Morelli, A. E. & Thomson, A. W. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat. Rev. Immunol. 7, 610–621 (2007).
21. Steinman, R. M. & Banchereau, J. Taking dendritic cells into medicine. Nature 449, 419–426 (2007).
22. Liu, K. & Nussenzweig, M. C. Origin and development of dendritic cells. Immunol. Rev. 234, 45–54 (2010).
23. Mellman, I. & Steinman, R. M. Dendritic cells: specialized and regulated antigen processing machines. Cell 106, 255–258 (2001).
24. Kapsenberg, M. L. Dendritic-cell control of pathogen-driven T-cell polarization. Nat. Rev. Immunol. 3, 984–993 (2003).
25. Bianchi, M. E. DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5 (2007).
26. Diebold, S. S. Determination of T-cell fate by dendritic cells. Immunol. Cell Biol. 86, 389–397 (2008).
27. Mosser, D. M. & Edwards, J. P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 (2008).
28. Hume, D. A. & MacDonald, K. P. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood 119, 1810–1820 (2012).
29. Alikhan, M. A. et al. Colony-stimulating factor-1 promotes kidney growth and repair via alteration of macrophage responses. Am. J. Pathol. 179, 1243–1256 (2011).
30. Rae, F. et al. Characterisation and trophic functions of murine embryonic macrophages based upon the use of a Csf1r-EGFP transgene reporter. Dev. Biol. 308, 232–246 (2007).
31. Schulz, C. et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90 (2012).
32. Sieweke, M. H. & Allen, J. E. Beyond stem cells: self-renewal of differentiated macrophages. Science 342, 1242974 (2014).
33. Hashimoto, D. et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38, 792–804 (2013).
34. Rees, A. J. Monocyte and macrophage biology: an overview. Semin. Nephrol. 30, 216–233 (2010).
35. Sica, A. & Mantovani, A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795 (2012).
36. Ascon, D. B. et al. Phenotypic and functional characterization of kidney-infiltrating lymphocytes in renal ischemia reperfusion injury. J. Immunol. 177, 3380–3387 (2006).
37. Paust, H. J. et al. Regulatory T cells control the Th1 immune response in murine crescentic glomerulonephritis. Kidney Int. 80, 154–164 (2011).
38. Wu, H. et al. Depletion of γδ T cells exacerbates murine adriamycin nephropathy. J. Am. Soc. Nephrol. 18, 1180–1189 (2007).
39. Steinman, R. M. & Cohn, Z. A. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J. Exp. Med. 137, 1142–1162 (1973).
40. Steinman, R. M. & Cohn, Z. A. Identification of a novel cell type in peripheral lymphoid organs of mice. II. Functional properties in vitro. J. Exp. Med. 139, 380–397 (1974).
41. Steinman, R. M., Lustig, D. S. & Cohn, Z. A. Identification of a novel cell type in peripheral lymphoid organs of mice. 3. Functional properties in vivo. J. Exp. Med. 139, 1431–1445 (1974).
42. Schreiner, G. F., Kiely, J. M., Cotran, R. S. & Unanue, E. R. Characterization of resident glomerular cells in the rat expressing Ia determinants and manifesting genetically restricted interactions with lymphocytes. J. Clin. Invest. 68, 920–931 (1981).
43. Schreiner, G. F., Cotran, R. S. & Unanue, E. R. Modulation of Ia and leukocyte common antigen expression in rat glomeruli during the course of glomerulonephritis and aminonucleoside nephrosis. Lab. Invest. 51, 524–533 (1984).
44. Schreiner, G. F. & Cotran, R. S. Localization of an Ia-bearing glomerular cell in the mesangium. J. Cell Biol. 94, 483–488 (1982).
45. Gieseler, R. et al. Enrichment and characterization of dendritic cells from rat renal mesangium. Scand. J. Immunol. 46, 587–596 (1997).
46. Hart, D. N. & Fabre, J. W. Major histocompatibility complex antigens in rat kidney, ureter, and bladder. Localization with monoclonal antibodies and demonstration of Ia-positive dendritic cells. Transplantation 31, 318–325 (1981).
47. Hart, D. N. et al. Localization of HLA-ABC and DR antigens in human kidney. Transplantation 31, 428–433 (1981).
48. Kaissling, B., Hegyi, I., Loffing, J. & Le Hir, M. Morphology of interstitial cells in the healthy kidney. Anat. Embryol. (Berl.) 193, 303–318 (1996).
49. Pajak, B. et al. Immunohistowax processing, a new fixation and embedding method for light microscopy, which preserves antigen immunoreactivity and morphological structures: visualisation of dendritic cells in peripheral organs. J. Clin. Pathol. 53, 518–524 (2000).
50. Kruger, T. et al. Identification and functional characterization of dendritic cells in the healthy murine kidney and in experimental glomerulonephritis. J. Am. Soc. Nephrol. 15, 613–621 (2004).
51. Ginhoux, F. et al. The origin and development of nonlymphoid tissue CD103+ DCs. J. Exp. Med. 206, 3115–3130 (2009).
52. Coates, P. T. et al. In vivo-mobilized kidney dendritic cells are functionally immature, subvert alloreactive T-cell responses, and prolong organ allograft survival. Transplantation 77, 1080–1089 (2004).
53. Morelli, A. E. et al. Growth factor-induced mobilization of dendritic cells in kidney and liver of rhesus macaques: implications for transplantation. Transplantation 83, 656–662 (2007).
54. Coates, P. T. et al. CCR and CC chemokine expression in relation to Flt3 ligand-induced renal dendritic cell mobilization. Kidney Int. 66, 1907–1917 (2004).
55. Schraml, B. U. et al. Genetic tracing via DNGR-1 expression history defines dendritic cells as a hematopoietic lineage. Cell 154, 843–858 (2013).
56. Li, L. et al. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int. 74, 1526–1537 (2008).
57. MacDonald, K. P. et al. The colony-stimulating factor 1 receptor is expressed on dendritic cells during differentiation and regulates their expansion. J. Immunol. 175, 1399–1405 (2005).
58. Geissmann, F., Jung, S. & Littman, D. R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19, 71–82 (2003).
59. Sunderkotter, C. et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J. Immunol. 172, 4410–4417 (2004).
60. Lee, S. et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J. Am. Soc. Nephrol. 22, 317–326 (2011).
61. Anders, H. J. & Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 80, 915–925 (2011).
62. Lin, S. L. et al. Macrophage Wnt7b is critical for kidney repair and regeneration. Proc. Natl Acad. Sci. USA 107, 4194–4199 (2010).
63. Soos, T. J. et al. CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int. 70, 591–596 (2006).
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 17
64. Sasmono, R. T. et al. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood 101, 1155–1163 (2003).
65. Hume, D. A. & Gordon, S. Mononuclear phagocyte system of the mouse defined by immunohistochemical localization of antigen F4/80. Identification of resident macrophages in renal medullary and cortical interstitium and the juxtaglomerular complex. J. Exp. Med. 157, 1704–1709 (1983).
66. Masaki, T., Chow, F., Nikolic-Paterson, D. J., Atkins, R. C. & Tesch, G. H. Heterogeneity of antigen expression explains controversy over glomerular macrophage accumulation in mouse glomerulonephritis. Nephrol. Dial. Transplant. 18, 178–181 (2003).
67. van Rooijen, N. Liposomes as an in vivo tool to study and manipulate macrophage function. Res. Immunol. 143, 177–178 (1992).
68. van Rooijen, N. Liposome-mediated elimination of macrophages. Res. Immunol. 143, 215–219 (1992).
69. van Rooijen, N. & Sanders, A. Elimination, blocking, and activation of macrophages: three of a kind? J. Leukoc. Biol. 62, 702–709 (1997).
70. van Rooijen, N., Sanders, A. & van den Berg, T. K. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. J. Immunol. Methods 193, 93–99 (1996).
71. van Rooijen, N. & van Nieuwmegen, R. Elimination of phagocytic cells in the spleen after intravenous injection of liposome-encapsulated dichloromethylene diphosphonate. An enzyme-histochemical study. Cell Tissue Res. 238, 355–358 (1984).
72. Lu, L. et al. Depletion of macrophages and dendritic cells in ischemic acute kidney injury. Am. J. Nephrol. 35, 181–190 (2012).
73. Ferenbach, D. A. et al. Macrophage/monocyte depletion by clodronate, but not diphtheria toxin, improves renal ischemia/reperfusion injury in mice. Kidney Int. 82, 928–933 (2012).
74. Tittel, A. P. et al. Kidney dendritic cells induce innate immunity against bacterial pyelonephritis. J. Am. Soc. Nephrol. 22, 1435–1441 (2011).
75. Vinuesa, E. et al. Macrophage involvement in the kidney repair phase after ischaemia/reperfusion injury. J. Pathol. 214, 104–113 (2008).
76. Kulkarni, O. P. et al. Toll-like receptor 4-induced IL-22 accelerates kidney regeneration. J. Am. Soc. Nephrol. 25, 978–989 (2014).
77. Kaissling, B., Lehir, M. & Kriz, W. Renal epithelial injury and fibrosis. Biochim. Biophys. Acta 1832, 931–939 (2013).
78. Yang, L., Besschetnova, T. Y., Brooks, C. R., Shah, J. V. & Bonventre, J. V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 16, 535–543 (2010).
79. Sung, S. A., Jo, S. K., Cho, W. Y., Won, N. H. & Kim, H. K. Reduction of renal fibrosis as a result of liposome encapsulated clodronate induced macrophage depletion after unilateral ureteral obstruction in rats. Nephron Exp. Nephrol. 105, e1–e9 (2007).
80. Kitamoto, K. et al. Effects of liposome clodronate on renal leukocyte populations and renal fibrosis in murine obstructive nephropathy. J. Pharmacol. Sci. 111, 285–292 (2009).
81. Henderson, N. C. et al. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am. J. Pathol. 172, 288–298 (2008).
82. Machida, Y. et al. Renal fibrosis in murine obstructive nephropathy is attenuated by depletion of monocyte lineage, not dendritic cells. J. Pharmacol. Sci. 114, 464–473 (2010).
83. Snelgrove, S. L. et al. Renal dendritic cells adopt a pro-inflammatory phenotype in obstructive uropathy to activate T cells but do not directly contribute to fibrosis. Am. J. Pathol. 180, 91–103 (2012).
84. Lech, M. et al. Macrophage phenotype controls long-term AKI outcomes—kidney regeneration versus atrophy. J. Am. Soc. Nephrol. 25, 292–304 (2014).
85. Huen, S. C., Moeckel, G. W. & Cantley, L. G. Macrophage-specific deletion of transforming growth factor-β1 does not prevent renal fibrosis after severe ischemia-reperfusion or obstructive injury. Am. J. Physiol. Renal Physiol. 305, F477–F484 (2013).
86. Lukacs-Kornek, V. et al. The kidney-renal lymph node-system contributes to cross-tolerance against innocuous circulating antigen. J. Immunol. 180, 706–715 (2008).
87. Gottschalk, C. et al. Batf3-dependent dendritic cells in the renal lymph node induce tolerance against circulating antigens. J. Am. Soc. Nephrol. 24, 543–549 (2013).
88. Macconi, D. et al. Proteasomal processing of albumin by renal dendritic cells generates antigenic peptides. J. Am. Soc. Nephrol. 20, 123–130 (2009).
89. Dong, X. et al. Antigen presentation by dendritic cells in renal lymph nodes is linked to systemic and local injury to the kidney. Kidney Int. 68, 1096–1108 (2005).
90. Roake, J. A. et al. Dendritic cell loss from nonlymphoid tissues after systemic administration of lipopolysaccharide, tumor necrosis factor, and interleukin 1. J. Exp. Med. 181, 2237–2247 (1995).
91. Kurts, C., Heymann, F., Lukacs-Kornek, V., Boor, P. & Floege, J. Role of T cells and dendritic cells in glomerular immunopathology. Semin. Immunopathol. 29, 317–335 (2007).
92. Assmann, K. J., Tangelder, M. M., Lange, W. P., Schrijver, G. & Koene, R. A. Anti-GBM nephritis in the mouse: severe proteinuria in the heterologous phase. Virchows Arch. A Pathol. Anat. Histopathol. 406, 285–299 (1985).
93. Hochheiser, K. et al. Kidney dendritic cells become pathogenic during crescentic glomerulonephritis with proteinuria. J. Am. Soc. Nephrol. 22, 306–316 (2011).
94. Lichtnekert, J. et al. Anti-GBM glomerulonephritis involves IL-1 but is independent of NLRP3/ASC inflammasome-mediated activation of caspase-1. PLoS ONE 6, e26778 (2011).
95. Scholz, J. et al. Renal dendritic cells stimulate IL-10 production and attenuate nephrotoxic nephritis. J. Am. Soc. Nephrol. 19, 527–537 (2008).
96. Kitching, A. R., Tipping, P. G., Huang, X. R., Mutch, D. A. & Holdsworth, S. R. Interleukin-4 and interleukin-10 attenuate established crescentic glomerulonephritis in mice. Kidney Int. 52, 52–59 (1997).
97. Kitching, A. R., Tipping, P. G., Timoshanko, J. R. & Holdsworth, S. R. Endogenous interleukin-10 regulates Th1 responses that induce crescentic glomerulonephritis. Kidney Int. 57, 518–525 (2000).
98. Witsch, E. J. et al. ICOS and CD28 reversely regulate IL-10 on re-activation of human effector T cells with mature dendritic cells. Eur. J. Immunol. 32, 2680–2686 (2002).
99. Duffield, J. S. et al. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am. J. Pathol. 167, 1207–1219 (2005).
100. Lloyd, C. M., Dorf, M. E., Proudfoot, A., Salant, D. J. & Gutierrez-Ramos, J. C. Role of MCP-1 and RANTES in inflammation and progression to fibrosis during murine crescentic nephritis. J. Leukoc. Biol. 62, 676–680 (1997).
101. Wada, T. et al. Intervention of crescentic glomerulonephritis by antibodies to monocyte chemotactic and activating factor (MCAF/MCP-1). FASEB J. 10, 1418–1425 (1996).
102. Yamamoto, T. & Wilson, C. B. Quantitative and qualitative studies of antibody-induced mesangial cell damage in the rat. Kidney Int. 32, 514–525 (1987).
103. Ikezumi, Y., Hurst, L. A., Masaki, T., Atkins, R. C. & Nikolic-Paterson, D. J. Adoptive transfer studies demonstrate that macrophages can induce proteinuria and mesangial cell proliferation. Kidney Int. 63, 83–95 (2003).
104. Lan, H. Y., Nikolic-Paterson, D. J., Mu, W. & Atkins, R. C. Local macrophage proliferation in the progression of glomerular and tubulointerstitial injury in rat anti-GBM glomerulonephritis. Kidney Int. 48, 753–760 (1995).
105. Han, Y., Ma, F. Y., Tesch, G. H., Manthey, C. L. & Nikolic-Paterson, D. J. c-fms blockade reverses glomerular macrophage infiltration and halts development of crescentic anti-GBM glomerulonephritis in the rat. Lab. Invest. 91, 978–991 (2011).
106. Odobasic, D. et al. Glomerular expression of CD80 and CD86 is required for leukocyte accumulation and injury in crescentic glomerulonephritis. J. Am. Soc. Nephrol. 16, 2012–2022 (2005).
107. Heymann, F. et al. Kidney dendritic cell activation is required for progression of renal disease in a mouse model of glomerular injury. J. Clin. Invest. 119, 1286–1297 (2009).
108. Schiffer, L. et al. Activated renal macrophages are markers of disease onset and disease remission in lupus nephritis. J. Immunol. 180, 1938–1947 (2008).
109. Ishikawa, S. et al. Aberrant high expression of B lymphocyte chemokine (BLC/CXCL13) by C11b+CD11c+ dendritic cells in murine lupus and preferential chemotaxis of B1 cells towards BLC. J. Exp. Med. 193, 1393–1402 (2001).
110. Castellano, G. et al. Infiltrating dendritic cells contribute to local synthesis of C1q in murine and human lupus nephritis. Mol. Immunol. 47, 2129–2137 (2010).
111. Kikawada, E., Lenda, D. M. & Kelley, V. R. IL-12 deficiency in MRL-Fas(lpr) mice delays nephritis and intrarenal IFN-γ expression, and diminishes systemic pathology. J. Immunol. 170, 3915–3925 (2003).
112. Kulkarni, O. et al. Spiegelmer inhibition of CCL2/MCP-1 ameliorates lupus nephritis in MRL-(Fas)lpr mice. J. Am. Soc. Nephrol. 18, 2350–2358 (2007).
113. Perez de Lema, G. et al. Chemokine receptor Ccr2 deficiency reduces renal disease and prolongs survival in MRL/lpr lupus-prone mice. J. Am. Soc. Nephrol. 16, 3592–3601 (2005).
114. Wan, S., Xia, C. & Morel, L. IL-6 produced by dendritic cells from lupus-prone mice inhibits CD4+CD25+ T cell regulatory functions. J. Immunol. 178, 271–279 (2007).
115. Wan, S., Zhou, Z., Duan, B. & Morel, L. Direct B cell stimulation by dendritic cells in a mouse model of lupus. Arthritis Rheum. 58, 1741–1750 (2008).
116. Bagavant, H., Thompson, C., Ohno, K., Setiady, Y. & Tung, K. S. Differential effect of neonatal thymectomy on systemic and organ-specific autoimmune disease. Int. Immunol. 14, 1397–1406 (2002).
117. Bagavant, H., Deshmukh, U. S., Wang, H., Ly, T. & Fu, S. M. Role for nephritogenic T cells in lupus glomerulonephritis: progression to renal failure is accompanied by T cell activation and expansion in regional lymph nodes. J. Immunol. 177, 8258–8265 (2006).
118. Schiffer, L. et al. Short term administration of costimulatory blockade and cyclophosphamide induces remission of systemic lupus
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

18 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
erythematosus nephritis in NZB/W F1 mice by a mechanism downstream of renal immune complex deposition. J. Immunol. 171, 489–497 (2003).
119. Iwata, Y. et al. p38 Mitogen-activated protein kinase contributes to autoimmune renal injury in MRL-Fas lpr mice. J. Am. Soc. Nephrol. 14, 57–67 (2003).
120. Iwata, Y. et al. Dendritic cells contribute to autoimmune kidney injury in MRL-Faslpr mice. J. Rheumatol. 36, 306–314 (2009).
121. Baccala, R. et al. Essential requirement for IRF8 and SLC15A4 implicates plasmacytoid dendritic cells in the pathogenesis of lupus. Proc. Natl Acad. Sci. USA 110, 2940–2945 (2012).
122. Kinjoh, K., Kyogoku, M. & Good, R. A. Genetic selection for crescent formation yields mouse strain with rapidly progressive glomerulonephritis and small vessel vasculitis. Proc. Natl Acad. Sci. USA 90, 3413–3417 (1993).
123. Saiga, K. et al. NK026680, a novel suppressant of dendritic cell function, prevents the development of rapidly progressive glomerulonephritis and perinuclear antineutrophil cytoplasmic antibody in SCG/Kj mice. Arthritis Rheum. 54, 3707–3715 (2006).
124. Zhang, Y. et al. Immune complex enhances tolerogenecity of immature dendritic cells via FcγRIIb and promotes FcγRIIb-overexpressing dendritic cells to attenuate lupus. Eur. J. Immunol. 41, 1154–1164 (2011).
125. McGaha, T. L., Karlsson, M. C. & Ravetch, J. V. FcγRIIB deficiency leads to autoimmunity and a defective response to apoptosis in Mrl-MpJ mice. J. Immunol. 180, 5670–5679 (2008).
126. Rodriguez, W. et al. C-reactive protein-mediated suppression of nephrotoxic nephritis: role of macrophages, complement, and Fcγ receptors. J. Immunol. 178, 530–538 (2007).
127. Zhang, Y. et al. Immune complex/Ig negatively regulate TLR4-triggered inflammatory response in macrophages through Fc γ RIIb-dependent PGE2 production. J. Immunol. 182, 554–562 (2009).
128. Muhlfeld, A. S. et al. Deletion of the Fcγ receptor IIb in thymic stromal lymphopoietin transgenic mice aggravates membranoproliferative glomerulonephritis. Am. J. Pathol. 163, 1127–1136 (2003).
129. Smith, J. et al. A spleen tyrosine kinase inhibitor reduces the severity of established glomerulonephritis. J. Am. Soc. Nephrol. 21, 231–236 (2010).
130. Tadagavadi, R. K. & Reeves, W. B. Renal dendritic cells ameliorate nephrotoxic acute kidney injury. J. Am. Soc. Nephrol. 21, 53–63 (2009).
131. Li, L. & Okusa, M. D. Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin. Nephrol 30, 268–277 (2010).
132. Jo, S. K., Sung, S. A., Cho, W. Y., Go, K. J. & Kim, H. K. Macrophages contribute to the initiation of ischaemic acute renal failure in rats. Nephrol. Dial. Transplant. 21, 1231–1239 (2006).
133. Day, Y. J., Huang, L., Ye, H., Linden, J. & Okusa, M. D. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: role of macrophages. Am. J. Physiol. Renal Physiol. 288, F722–F731 (2005).
134. Ko, G. J., Boo, C. S., Jo, S. K., Cho, W. Y. & Kim, H. K. Macrophages contribute to the development of renal fibrosis following ischaemia/reperfusion-induced acute kidney injury. Nephrol. Dial. Transplant. 23, 842–852 (2008).
135. Zhang, M. Z. et al. CSF-1 signaling mediates recovery from acute kidney injury. J. Clin. Invest. 122, 4519–4532 (2012).
136. Kim, M. G. et al. Depletion of kidney CD11c+ F4/80+ cells impairs the recovery process in ischaemia/reperfusion-induced acute kidney injury. Nephrol. Dial. Transplant. 25, 2908–2921 (2010).
137. Tadagavadi, R. K. & Reeves, W. B. Endogenous IL-10 attenuates cisplatin nephrotoxicity: role of dendritic cells. J. Immunol. 185, 4904–4911 (2010).
138. Kinsey, G. R., Huang, L., Vergis, A. L., Li, L. & Okusa, M. D. Regulatory T cells contribute to the protective effect of ischemic preconditioning in the kidney. Kidney Int. 77, 771–780 (2010).
139. Kinsey, G. R. et al. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J. Am. Soc. Nephrol. 20, 1744–1753 (2009).
140. Gandolfo, M. T. et al. Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int. 76, 717–729 (2009).
141. Kim, M. G. et al. IL-2/anti-IL-2 complex attenuates renal ischemia-reperfusion injury through expansion of regulatory T cells. J. Am. Soc. Nephrol. 24, 1529–1536 (2013).
142. Cho, W. Y. et al. The role of Tregs and CD11c+ macrophages/dendritic cells in ischemic preconditioning of the kidney. Kidney Int. 78, 981–992 (2010).
143. Cantaluppi, V. et al. Rationale of mesenchymal stem cell therapy in kidney injury. Am. J. Kidney Dis. 61, 300–309 (2013).
144. Kim, M. G. et al. CD11c+ cells partially mediate the renoprotective effect induced by bone-marrow-derived mesenchymal stem cells. PLoS ONE 8, e72544 (2013).
145. Menke, J. et al. CSF-1 signals directly to renal tubular epithelial cells to mediate repair in mice. J. Clin. Invest. 119, 2330–2342 (2009).
146. Wang, Y., Tay, Y. C. & Harris, D. C. Proximal tubule cells stimulated by lipopolysaccharide inhibit macrophage activation. Kidney Int. 66, 655–662 (2004).
147. Dong, X. et al. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int. 71, 619–628 (2007).
148. Lassen, S. et al. Ischemia reperfusion induces IFN regulatory factor 4 in renal dendritic cells, which suppresses postischemic inflammation and prevents acute renal failure. J. Immunol. 185, 1976–1983 (2009).
149. Lech, M. et al. Resident dendritic cells prevent postischemic acute renal failure by help of single Ig IL-1 receptor-related protein. J. Immunol. 183, 4109–4118 (2009).
150. Garlanda, C. et al. Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc. Natl Acad. Sci. USA 101, 3522–3526 (2004).
151. Noris, M. et al. The Toll-IL-1R member Tir8/SIGIRR negatively regulates adaptive immunity against kidney grafts. J. Immunol. 183, 4249–4260 (2009).
152. Dong, X., Bachman, L. A., Miller, M. N., Nath, K. A. & Griffin, M. D. Dendritic cells facilitate accumulation of IL-17 T cells in the kidney following acute renal obstruction. Kidney Int. 74, 1294–1309 (2008).
153. Kitagawa, K. et al. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am. J. Pathol. 165, 237–246 (2004).
154. Chadban, S. J., Wu, H. & Hughes, J. Macrophages and kidney transplantation. Semin. Nephrol. 30, 278–289 (2010).
155. Penfield, J. G. et al. Transplant surgery injury recruits recipient MHC class II-positive leukocytes into the kidney. Kidney Int. 56, 1759–1769 (1999).
156. Penfield, J. G. et al. Syngeneic renal transplantation increases the number of renal dendritic cells in the rat. Transpl. Immunol. 7, 197–200 (1999).
157. Le Meur, Y., Jose, M. D., Mu, W., Atkins, R. C. & Chadban, S. J. Macrophage colony-stimulating factor expression and macrophage accumulation in renal allograft rejection. Transplantation 73, 1318–1324 (2002).
158. Milton, A. D., Spencer, S. C. & Fabre, J. W. The effects of cyclosporine on the induction of donor class I and class II MHC antigens in heart and kidney allografts in the rat. Transplantation 42, 337–347 (1986).
159. Ahn, K. O. et al. Influence of angiotensin II on expression of toll-like receptor 2 and maturation of dendritic cells in chronic cyclosporine nephropathy. Transplantation 83, 938–947 (2007).
160. Lim, S. W. et al. Cyclosporine-induced renal injury induces toll-like receptor and maturation of dendritic cells. Transplantation 80, 691–699 (2005).
161. Ozaki, K. S. et al. The loss of renal dendritic cells and activation of host adaptive immunity are long-term effects of ischemia/reperfusion injury following syngeneic kidney transplantation. Kidney Int. 81, 1015–1025 (2012).
162. Jose, M. D., Ikezumi, Y., van Rooijen, N., Atkins, R. C. & Chadban, S. J. Macrophages act as effectors of tissue damage in acute renal allograft rejection. Transplantation 76, 1015–1022 (2003).
163. Qi, F. et al. Depletion of cells of monocyte lineage prevents loss of renal microvasculature in murine kidney transplantation. Transplantation 86, 1267–1274 (2008).
164. Jose, M. D., Le Meur, Y., Atkins, R. C. & Chadban, S. J. Blockade of macrophage colony-stimulating factor reduces macrophage proliferation and accumulation in renal allograft rejection. Am. J. Transplant. 3, 294–300 (2003).
165. Ma, F. Y., Woodman, N., Mulley, W. R., Kanellis, J. & Nikolic-Paterson, D. J. Macrophages contribute to cellular but not humoral mechanisms of acute rejection in rat renal allografts. Transplantation 96, 949–957 (2013).
166. Shimizu, A., Yamada, K., Sachs, D. H. & Colvin, R. B. Mechanisms of chronic renal allograft rejection. II. Progressive allograft glomerulopathy in miniature swine. Lab. Invest. 82, 673–686 (2002).
167. Azuma, H., Nadeau, K. C., Ishibashi, M. & Tilney, N. L. Prevention of functional, structural, and molecular changes of chronic rejection of rat renal allografts by a specific macrophage inhibitor. Transplantation 60, 1577–1582 (1995).
168. Yang, J. et al. Targeting of macrophage activity by adenovirus-mediated intragraft overexpression of TNFRp55-Ig, IL-12p40, and vIL-10 ameliorates adenovirus-mediated chronic graft injury, whereas stimulation of macrophages by overexpression of IFN-γ accelerates chronic graft injury in a rat renal allograft model. J. Am. Soc. Nephrol. 14, 214–225 (2003).
169. Woltman, A. M. et al. Quantification of dendritic cell subsets in human renal tissue under normal and pathological conditions. Kidney Int. 71, 1001–1008 (2007).
170. Mayer, V. et al. Expression of the chemokine receptor CCR1 in human renal allografts. Nephrol. Dial. Transplant. 22, 1720–1729 (2007).
171. Fahim, T. et al. The cellular lesion of humoral rejection: predominant recruitment of monocytes to peritubular and glomerular capillaries. Am. J. Transplant. 7, 385–393 (2007).
172. Sund, S., Reisaeter, A. V., Scott, H., Mollnes, T. E. & Hovig, T. Glomerular monocyte/macrophage influx correlates strongly with complement
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 19
activation in 1-week protocol kidney allograft biopsies. Clin. Nephrol. 62, 121–130 (2004).
173. Grimm, P. C. et al. Clinical rejection is distinguished from subclinical rejection by increased infiltration by a population of activated macrophages. J. Am. Soc. Nephrol. 10, 1582–1589 (1999).
174. Pilmore, H. L., Painter, D. M., Bishop, G. A., McCaughan, G. W. & Eris, J. M. Early up-regulation of macrophages and myofibroblasts: a new marker for development of chronic renal allograft rejection. Transplantation 69, 2658–2662 (2000).
175. Bunnag, S. et al. Molecular correlates of renal function in kidney transplant biopsies. J. Am. Soc. Nephrol. 20, 1149–1160 (2009).
176. Scian, M. J. et al. Gene expression changes are associated with loss of kidney graft function and interstitial fibrosis and tubular atrophy: diagnosis versus prediction. Transplantation 91, 657–665 (2011).
177. Mengel, M. et al. The molecular phenotype of 6-week protocol biopsies from human renal allografts: reflections of prior injury but not future course. Am. J. Transplant. 11, 708–718 (2011).
178. Papadimitriou, J. C. et al. Glomerular inflammation in renal allografts biopsies after the first year: cell types and relationship with antibody-mediated rejection and graft outcome. Transplantation 90, 1478–1485 (2010).
179. Ozdemir, B. H., Demirhan, B. & Gungen, Y. The presence and prognostic importance of glomerular macrophage infiltration in renal allografts. Nephron 90, 442–446 (2002).
180. Hoffmann, U. et al. Impact of chemokine receptor CX3CR1 in human renal allograft rejection. Transpl. Immunol. 23, 204–208 (2010).
181. Copin, M. C. et al. Diagnostic and predictive value of an immunohistochemical profile in asymptomatic acute rejection of renal allografts. Transpl. Immunol. 3, 229–239 (1995).
182. Yoshimoto, K. et al. CD68 and MCP-1/CCR2 expression of initial biopsies reflect the outcomes of membranous nephropathy. Nephron Clin. Pract. 98, c25–c34 (2004).
183. Eardley, K. S. et al. The relationship between albuminuria, MCP-1/CCL2, and interstitial macrophages in chronic kidney disease. Kidney Int. 69, 1189–1197 (2006).
184. Eardley, K. S. et al. The role of capillary density, macrophage infiltration and interstitial scarring in the pathogenesis of human chronic kidney disease. Kidney Int. 74, 495–504 (2008).
185. Cuzic, S., Ritz, E. & Waldherr, R. Dendritic cells in glomerulonephritis. Virchows Arch. B Cell Pathol. Incl Mol. Pathol. 62, 357–363 (1992).
186. De Palma, G. et al. The possible role of ChemR23/Chemerin axis in the recruitment of dendritic cells in lupus nephritis. Kidney Int. 79, 1228–1235 (2011).
187. Segerer, S. et al. Compartment specific expression of dendritic cell markers in human glomerulonephritis. Kidney Int. 74, 37–46 (2008).
188. Noessner, E., Lindenmeyer, M., Nelson, P. J. & Segerer, S. Dendritic cells in human renal inflammation—Part II. Nephron Exp. Nephrol. 119, e91–e98 (2011).
189. Kassianos, A. J. et al. Increased tubulointerstitial recruitment of human CD141hi CLEC9A+ and CD1c+ myeloid dendritic cell subsets in renal fibrosis and chronic kidney disease. Am. J. Physiol. Renal Physiol. 305, F1391–F1401 (2013).
190. Tucci, M., Ciavarella, S., Strippoli, S., Dammacco, F. & Silvestris, F. Oversecretion of cytokines and chemokines in lupus nephritis is regulated by intraparenchymal dendritic cells: a review. Ann. N. Y Acad. Sci. 1173, 449–457 (2009).
191. Tucci, M. et al. Glomerular accumulation of plasmacytoid dendritic cells in active lupus nephritis: role of interleukin-18. Arthritis Rheum. 58, 251–262 (2008).
192. Loverre, A. et al. Ischemia-reperfusion injury-induced abnormal dendritic cell traffic in the transplanted kidney with delayed graft function. Kidney Int. 72, 994–1003 (2007).
193. Wilde, B. et al. Dendritic cells in renal biopsies of patients with ANCA-associated vasculitis. Nephrol. Dial. Transplant. 24, 2151–2156 (2009).
194. Steinmetz, O. M. et al. Analysis and classification of B-cell infiltrates in lupus and ANCA-associated nephritis. Kidney Int. 74, 448–457 (2008).
195. Kerjaschki, D. et al. Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J. Am. Soc. Nephrol. 15, 603–612 (2004).
196. Loike, J. D. et al. CD11c/CD18 on neutrophils recognizes a domain at the N. terminus of the A α chain of fibrinogen. Proc. Natl Acad. Sci. USA 88, 1044–1048 (1991).
197. Fancke, B., Suter, M., Hochrein, H. & O’Keeffe, M. M-CSF: a novel plasmacytoid and conventional dendritic cell poietin. Blood 111, 150–159 (2008).
198. Fleetwood, A. J., Lawrence, T., Hamilton, J. A. & Cook, A. D. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J. Immunol. 178, 5245–5252 (2007).
199. Hume, D. A. Macrophages as APC and the dendritic cell myth. J. Immunol. 181, 5829–5835 (2008).
200. Desmedt, M., Rottiers, P., Dooms, H., Fiers, W. & Grooten, J. Macrophages induce cellular immunity by activating Th1 cell responses and suppressing Th2 cell responses. J. Immunol. 160, 5300–5308 (1998).
201. Jung, S. et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell Biol. 20, 4106–4114 (2000).
202. MacDonald, K. P. et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood 116, 3955–3963 (2010).
203. Sapoznikov, A. et al. Organ-dependent in vivo priming of naive CD4+, but not CD8+, T cells by plasmacytoid dendritic cells. J. Exp. Med. 204, 1923–1933 (2007).
204. Laouar, Y., Sutterwala, F. S., Gorelik, L. & Flavell, R. A. Transforming growth factor-β controls T helper type 1 cell development through regulation of natural killer cell interferon-γ. Nat. Immunol. 6, 600–607 (2005).
205. Tittel, A. P. et al. Functionally relevant neutrophilia in CD11c diphtheria toxin receptor transgenic mice. Nat. Methods 9, 385–390 (2012).
206. Duffield, J. S. et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest. 115, 56–65 (2005).
207. Tipping, P. G. & Holdsworth, S. R. T cells in crescentic glomerulonephritis. J. Am. Soc. Nephrol. 17, 1253–1263 (2006).
208. Schatzmann, U., Haas, C. & Le Hir, M. A Th1 response is essential for induction of crescentic glomerulonephritis in mice. Kidney Blood Press. Res. 22, 135–139 (1999).
209. Shirai, T., Hirose, S., Okada, T. & Nishimura, H. in Immunological Disorders in Mice (eds Rihova, B. & Vetvicka, V.) 95–136 (CRC Press Inc., 1991).
210. Dubois, E. L., Horowitz, R. E., Demopoulos, H. B. & Teplitz, R. NZB/NZW mice as a model of systemic lupus erythematosus. JAMA 195, 285–289 (1966).
211. Rigby, R. J. et al. New loci from New Zealand black and New Zealand white mice on chromosomes 4 and 12 contribute to lupus-like disease in the context of BALB/c. J. Immunol. 172, 4609–4617 (2004).
212. Morel, L. et al. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc. Natl Acad. Sci. USA 97, 6670–6675 (2000).
213. Colonna, L. et al. Abnormal costimulatory phenotype and function of dendritic cells before and after the onset of severe murine lupus. Arthritis Res. Ther. 8, R49 (2006).
214. Rudofsky, U. H., Evans, B. D., Balaban, S. L., Mottironi, V. D. & Gabrielsen, A. E. Differences in expression of lupus nephritis in New Zealand mixed H-2z homozygous inbred strains of mice derived from New Zealand black and New Zealand white mice. Origins and initial characterization. Lab. Invest. 68, 419–426 (1993).
215. Gu, L. et al. Genetic determinants of autoimmune disease and coronary vasculitis in the MRL-lpr/lpr mouse model of systemic lupus erythematosus. J. Immunol. 161, 6999–7006 (1998).
216. Anders, H. J. et al. Activation of toll-like receptor-9 induces progression of renal disease in MRL-Faslpr mice. FASEB J. 18, 534–536 (2004).
217. Zheng, D. et al. Lipopolysaccharide-pretreated plasmacytoid dendritic cells ameliorate experimental chronic kidney disease. Kidney Int. 81, 892–902 (2012).
218. Huang, X. R. et al. Mechanisms of T cell-induced glomerular injury in anti-glomerular basement membrane (GBM) glomerulonephritis in rats. Clin. Exp. Immunol. 109, 134–142 (1997).
219. Triantafyllopoulou, A. et al. Proliferative lesions and metalloproteinase activity in murine lupus nephritis mediated by type I interferons and macrophages. Proc. Natl Acad. Sci. USA 107, 3012–3017 (2010).
220. Bethunaickan, R. et al. A unique hybrid renal mononuclear phagocyte activation phenotype in murine systemic lupus erythematosus nephritis. J. Immunol. 186, 4994–5003 (2011).
221. Menke, J. et al. Circulating CSF-1 promotes monocyte and macrophage phenotypes that enhance lupus nephritis. J. Am. Soc. Nephrol. 20, 2581–2592 (2009).
222. Teichmann, L. L. et al. Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity 33, 967–978 (2010).
AcknowledgementsThe work of the authors is supported by the NIH grants R01HL108954 and R01HL112914 (J.S.I.), and R01AI67541 and U01AI51698 (A.W.T.); the University of Pittsburgh O’Brien Kidney Pilot Award (J.S.I.); the Cunningham Trust, the Mrs A. E. Hogg Charitable Trust, Kidney Research UK and the UK Medical Research Council (J.H.); an American Heart Association Postdoctoral Award (13POST14520003) and an American Society of Transplantation/Pfizer Basic Science Fellowship (N.M.R.); and a Wellcome Trust Intermediate fellowship (D.A.F.). The work of J.S.I. is also supported by the Institute for Transfusion Medicine, the Haemophilia Center of Western Pennsylvania, and the Vascular Medicine Institute.
Author contributionsJ.S.I. contributed substantially to discussion of content and reviewed/edited the manuscript before submission. All other authors made substantial contributions to all stages of the preparation of the manuscript for submission. A.W.T and J.H. contributed equally as senior co-authors.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved




















