
Myelopoiesis modulation by ACE hyperfunction in kinin B1 receptor knockout mice: Relationship with...
8
Chemico-Biological Interactions 184 (2010) 388–395 Contents lists available at ScienceDirect Chemico-Biological Interactions journal homepage: www.elsevier.com/locate/chembioint Myelopoiesis modulation by ACE hyperfunction in kinin B 1 receptor knockout mice: Relationship with AcSDKP levels Carlos R. Oliveira a , Edgar J. Paredes-Gamero b , Christiano M.V. Barbosa b , Fábio D. Nascimento c , Elice C. Batista b , Felipe C.G. Reis b , Antonio H.B. Martins b , Alice T. Ferreira b , Adriana K. Carmona b , João B. Pesquero b , Ivarne L.S. Tersariol c , Ronaldo C. Araújo b,∗ , Claudia Bincoletto a,∗∗ a Departamento de Farmacologia, Escola Paulista de Medicina, Universidade Federal de São Paulo (UNIFESP), Rua 3 de maio, 100, 04040-020 São Paulo, SP, Brazil b Departamento de Biofísica, Escola Paulista de Medicina, Universidade Federal de São Paulo (UNIFESP), Rua Botucatu, 862, 04023-062 São Paulo, SP, Brazil c Departamento de Bioquímica, Universidade Federal de São Paulo, Escola Paulista de Medicina, São Paulo, SP, Brazil article info Article history: Received 14 October 2009 Received in revised form 11 January 2010 Accepted 12 January 2010 Available online 22 January 2010 Keywords: ACE hyperfunction BK Bone marrow B1KO mice GM-CFC AcSDKP levels abstract Angiotensin I-converting enzyme (ACE), a common element of renin–angiotensin system (RAS) and kallikrein–kinin system (KKS), is involved in myelopoiesis modulation, mainly by cleaving the tetrapep- tide N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP). Based on this finding and in our results showing B1 and B2 kinin receptors expression in murine bone marrow (BM) cells, we evaluated the ACE influ- ence on myelopoiesis of kinin B1 receptor knockout mice (B1KO) using long-term bone marrow cultures (LTBMCs). Captopril and AcSDKP were used as controls. Enhanced ACE activity, expressed by non- hematopoietic cells (Ter-199 − and CD45 − ), was observed in B1KO LTBMCs when compared to wild-type (WT) cells. ACE hyperfunction in B1KO cells was maintained when LTBMCs from B1KO mice were treated with captopril (1.0 M) or AcSDKP (1.0 nM). Although no alterations were observed in ACE mRNA and pro- tein levels under these culture conditions, 3.0 nM of AcSDKP increased ACE mRNA levels in WT LTBMCs. No alteration in the number of GM-CFC was seen in B1KO mice compared to WT animals, even when the former were treated with AcSDKP (10 g/kg) or captopril (100 mg/kg) for 4 consecutive days. Hemato- logical data also revealed no differences between WT and B1KO mice under basal conditions. When the animals received 4 doses of lipopolysaccharide (LPS), a decreased number of blood cells was detected in B1KO mice in relation to WT. We also found a decreased percentage of Gr1 + /Mac-1 + , Ter119 + , B220 + , CD3 + , and Lin − Sca1 + c-Kit + (LSK) cells in the BM of B1KO mice compared to WT animals. Low AcSDKP levels were observed in BM cultures from B1KO in comparison to WT cultures. We conclude that ACE hyperfunction in B1KO mice resulted in faster hydrolysis of AcSDKP peptide, which in turn decreased in BM tissues allowing HSC to enter the S stage of the cell cycle. © 2010 Elsevier Ireland Ltd. All rights reserved. 1. Introduction All blood cells can be generated from a common hematopoi- etic stem cell (HSC) through an extremely dynamic process called hematopoiesis or blood cell formation. Postnatal hematopoiesis takes place in the bone marrow (BM), where the HSCs as well as a complex mix of dividing and maturing cells of different lin- eages can be found [1,2]. Regulation of HSCs is governed by two intimately involved entities. One is the gene expression pattern in ∗ Corresponding author. ∗∗ Corresponding author. Tel.: +55 11 5576 4443: fax: +55 11 5576 4443. E-mail addresses: [email protected] (R.C. Araújo), [email protected] (C. Bincoletto). the cell and the other is the composition of external signals from the BM microenvironment. Transcription factors are internal sig- nals that regulate gene expression, while external signals from the BM microenvironment can be mediated by cell–cell interactions, cell–extracellular matrix (ECM) interactions, and soluble growth factors secreted from hematopoietic and stromal cells [3–5]. Apart from transcription factors, there are also other intrinsic factors within the HSCs that are important regulators of stem cell fate. These include effector molecules in the cell cycle machin- ery. For instance, high levels of cyclin D 2 have been detected in cycling HSCs, suggesting an important role in HSC cycling, whereas studies of knockout mice have shown that the cyclin- dependent kinase inhibitor (CDKI) p21 acts to maintain HSCs in a quiescent state [6,7]. Signals from internal and external regula- tory factors decide whether the HSCs are maintained in quiescence, 0009-2797/$ – see front matter © 2010 Elsevier Ireland Ltd. All rights reserved. doi:10.1016/j.cbi.2010.01.015
Transcript of Myelopoiesis modulation by ACE hyperfunction in kinin B1 receptor knockout mice: Relationship with...

Mm
CEJa
0b
0c
a
ARRAA
KABBBGA
1
ehtaei
c
0d
Chemico-Biological Interactions 184 (2010) 388–395
Contents lists available at ScienceDirect
Chemico-Biological Interactions
journa l homepage: www.e lsev ier .com/ locate /chembio int
yelopoiesis modulation by ACE hyperfunction in kinin B1 receptor knockoutice: Relationship with AcSDKP levels
arlos R. Oliveiraa, Edgar J. Paredes-Gamerob, Christiano M.V. Barbosab, Fábio D. Nascimentoc,lice C. Batistab, Felipe C.G. Reisb, Antonio H.B. Martinsb, Alice T. Ferreirab, Adriana K. Carmonab,oão B. Pesquerob, Ivarne L.S. Tersariol c, Ronaldo C. Araújob,∗, Claudia Bincolettoa,∗∗
Departamento de Farmacologia, Escola Paulista de Medicina, Universidade Federal de São Paulo (UNIFESP), Rua 3 de maio, 100,4040-020 São Paulo, SP, BrazilDepartamento de Biofísica, Escola Paulista de Medicina, Universidade Federal de São Paulo (UNIFESP), Rua Botucatu, 862,4023-062 São Paulo, SP, BrazilDepartamento de Bioquímica, Universidade Federal de São Paulo, Escola Paulista de Medicina, São Paulo, SP, Brazil
r t i c l e i n f o
rticle history:eceived 14 October 2009eceived in revised form 11 January 2010ccepted 12 January 2010vailable online 22 January 2010
eywords:CE hyperfunctionKone marrow1KO miceM-CFCcSDKP levels
a b s t r a c t
Angiotensin I-converting enzyme (ACE), a common element of renin–angiotensin system (RAS) andkallikrein–kinin system (KKS), is involved in myelopoiesis modulation, mainly by cleaving the tetrapep-tide N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP). Based on this finding and in our results showingB1 and B2 kinin receptors expression in murine bone marrow (BM) cells, we evaluated the ACE influ-ence on myelopoiesis of kinin B1 receptor knockout mice (B1KO) using long-term bone marrow cultures(LTBMCs). Captopril and AcSDKP were used as controls. Enhanced ACE activity, expressed by non-hematopoietic cells (Ter-199− and CD45−), was observed in B1KO LTBMCs when compared to wild-type(WT) cells. ACE hyperfunction in B1KO cells was maintained when LTBMCs from B1KO mice were treatedwith captopril (1.0 �M) or AcSDKP (1.0 nM). Although no alterations were observed in ACE mRNA and pro-tein levels under these culture conditions, 3.0 nM of AcSDKP increased ACE mRNA levels in WT LTBMCs.No alteration in the number of GM-CFC was seen in B1KO mice compared to WT animals, even when theformer were treated with AcSDKP (10 �g/kg) or captopril (100 mg/kg) for 4 consecutive days. Hemato-
logical data also revealed no differences between WT and B1KO mice under basal conditions. When theanimals received 4 doses of lipopolysaccharide (LPS), a decreased number of blood cells was detectedin B1KO mice in relation to WT. We also found a decreased percentage of Gr1+/Mac-1+, Ter119+, B220+,CD3+, and Lin−Sca1+c-Kit+ (LSK) cells in the BM of B1KO mice compared to WT animals. Low AcSDKPlevels were observed in BM cultures from B1KO in comparison to WT cultures. We conclude that ACEhyperfunction in B1KO mice resulted in faster hydrolysis of AcSDKP peptide, which in turn decreased into en
BM tissues allowing HSC. Introduction
All blood cells can be generated from a common hematopoi-tic stem cell (HSC) through an extremely dynamic process calledematopoiesis or blood cell formation. Postnatal hematopoiesis
akes place in the bone marrow (BM), where the HSCs as wells a complex mix of dividing and maturing cells of different lin-ages can be found [1,2]. Regulation of HSCs is governed by twontimately involved entities. One is the gene expression pattern in∗ Corresponding author.∗∗ Corresponding author. Tel.: +55 11 5576 4443: fax: +55 11 5576 4443.
E-mail addresses: [email protected] (R.C. Araújo),[email protected] (C. Bincoletto).
009-2797/$ – see front matter © 2010 Elsevier Ireland Ltd. All rights reserved.oi:10.1016/j.cbi.2010.01.015
ter the S stage of the cell cycle.© 2010 Elsevier Ireland Ltd. All rights reserved.
the cell and the other is the composition of external signals fromthe BM microenvironment. Transcription factors are internal sig-nals that regulate gene expression, while external signals from theBM microenvironment can be mediated by cell–cell interactions,cell–extracellular matrix (ECM) interactions, and soluble growthfactors secreted from hematopoietic and stromal cells [3–5].
Apart from transcription factors, there are also other intrinsicfactors within the HSCs that are important regulators of stem cellfate. These include effector molecules in the cell cycle machin-ery. For instance, high levels of cyclin D2 have been detected
in cycling HSCs, suggesting an important role in HSC cycling,whereas studies of knockout mice have shown that the cyclin-dependent kinase inhibitor (CDKI) p21 acts to maintain HSCs ina quiescent state [6,7]. Signals from internal and external regula-tory factors decide whether the HSCs are maintained in quiescence,
gical I
p[
ilttpaBtibpmaa[etehcHb
oatem
catb[haibt
tvaiipnoaietBt[
aualiop
C.R. Oliveira et al. / Chemico-Biolo
roliferate, undergo apoptosis, or migrate out of the BM space8].
Haznedaroglu et al. [9] proposed that other mechanisms are alsonvolved in hematopoiesis regulation. They hypothesized that aocally active, intrinsic renin–angiotensin system (RAS) exists inhe BM, where it affects production, proliferation, and differen-iation of the cells in both normal and malignant hematologicalrocesses. In accordance with this preliminary evidence, Strawn etl. [10] verified that all RAS components are detectable in normalM derived blood cells at molecular and protein levels. In line withhese studies, angiotensin-II type 1a (AT1a) receptors are presentn CD34+ hematopoietic cells and stimulate the proliferation ofoth BM and umbilical cord blood hematopoietic progenitors inhysiological or pathological states [11–16]. Part of these events isodulated by the angiotensin I-converting enzyme (ACE), which
lso cleaves the negative physiological hematopoietic regulator Ncetyl-seryl-aspartyl-lysyl-proline (AcSDKP) into its inactive form10,12,17,18]. Therefore, ACE hyperfunction may lead to the accel-ration of AcSDKP metabolism, which in turn lowers its levels inhe BM microenvironment, finally removing the antiproliferativeffects of the peptide on the hematopoietic cells and blasts. Theemoregulatory tetrapeptide AcSDKP, originally isolated from fetalalf BM [19,20], reversibly prevents the recruitment of pluripotentSCs and normal early progenitors into the S stage of the cell cycley maintaining them quiescent.
Zambidis et al. [21], in an elegant study, demonstrated the rolef renin–angiotensin axis in directly regulating human embryonicngio-hematopoietic genesis and also proved that captopril inhibi-ion of ACE activity severely inhibited human hemangioblast colonyxpansion, similarly to the effects on progenitors described forurine BM cultures [11].ACE and angiotensin-II (Ang II) receptor inhibition is also asso-
iated with improved HSC recovery after chemotherapy induction,n effect that was originally interpreted to be due to the tetrapep-ide AcSKDP [22]. Since the Ang II peptide is a direct mitogen foroth human erythroid and CD34+ hematopoietic cell progenitors23,12], and specific AGTR1 inhibition has been reported to favorematopoietic progenitor expansion through an unknown mech-nism [24], the modulator activities of ACE, AcSDKP, and Ang IIn hematopoiesis may also be complicated by the involvement ofradykinin, another important peptide cleaved by ACE into its inac-ive form.
The kallikrein–kinin system (KKS) acts via two types of recep-ors: B1 and B2. B2 receptors are widely distributed in theasculature and expressed constitutively, whereas B1 receptorsre weakly expressed under physiological conditions, but stronglynduced by pathological stimuli such as inflammation or tissuenjury [25,26]. In general, activation of the B1 receptor producesrolonged, sustained, or oscillatory responses. This persistent sig-aling appears due to lack of desensibilization and internalizationf B1 receptor [27–29]. Bradykinin (BK) is a potent inflammatorygent, which is an enzymatically produced peptide generated atnflammatory sites by the action of kallikreins [30,31]. This peptidexerts a variety of effects measurable as smooth muscle contrac-ion, increased vascular permeability, vasodilation, and pain [32].K sensitization of myeloid progenitors to prostaglandin (PGE) fur-her suggests that this peptide may act to influence myelopoiesis32].
Intrigued by the possible role of RAS and KKS in myelopoiesisnd based on the fact that ACE is a key component of both systems,sing kinin B1 receptor knockout mice (B1KO) we evaluated ACE
ctivity and the possible role of AcSDKP in murine BM cell pro-iferation. Cellular localization and BM cell populations involvedn ACE expression were also assessed. For this purpose, through-ut the experiments we used captopril and AcSDKP respectively asositive controls of ACE inhibition and ACE substrate.nteractions 184 (2010) 388–395 389
2. Materials and methods
2.1. Reagents
All reagents were purchased from Sigma Chemical Co. (St.Louis, MO, USA), unless otherwise indicated, and cell culture mediaand supplements were purchased from Gibco-BRL (Bethesda,MD, USA). ACE fluorescence resonance energy transfer substrateAbz-FRK(Dnp)P-OH was generously supplied by the BiophysicsDepartment of the Universidade Federal de São Paulo (UNIFESP,São Paulo, Brazil).
2.2. Animals
This study was approved by an Institutional Ethics Committeefrom Mogi das Cruzes University (CEMEA/UMC), Mogi das Cruzes,SP, Brazil. All experiments reported were carried out as stated inthe National Institutes of Health Guide for the Care and Use ofLaboratory Animals Committee. The experiments were carried outwith kinin B1 receptor knockout mice (B1KO) and C57Bl/6 wild-type (WT) animals were used as controls. All the animals wereobtained from the Centro de Desenvolvimento de Modelos Exper-imentais para a Medicina e Biologia at the UNIFESP and housed inplastic cages, in groups of five animals, in air-conditioned roomsat 22 ± 2 ◦C, with a 12-h light–dark natural cycle and allowed adlibitum access to food and tap water. The animals were euthanizedby cervical dislocation, their femurs and spleens were dissectedout, and the femur cellularity was approximately 1 × 107 nucle-ated cells. For in vivo studies, WT and B1KO mice (n = 5) weredaily treated intraperitonially (ip) with captopril (100 mg/kg) orAcSDKP (10 �g/kg) [33] for 4 consecutive days and 24 h afterthe last dose, BM cells were removed by flushing to be used inthe granulocyte-macrophage colony-forming cell (GM-CFC) assay,which was conducted as described in Section 2.4. Spleen weight ofB1KO and WT mice was evaluated as well.
To carry out hematological studies, WT and B1KO mice (n = 7)were injected (i.p.) with lipopolysaccharide (LPS E. coli, 0111 : B4)for 4 consecutive days (3 days with the dose of 400 �g/kg andone last dose of 14 mg/kg). or saline and leucocyte, erythrocyte,hemoglobin, hematocrit, and platelet levels were counted by stan-dard methods 24 h after the last dose of LPS administration. Giemsastained blood smears and a total of 100 cells were counted.
2.3. Establishment of long-term bone marrow cultures (LTBMCs)from WT and B1KO mice
Femurs excised from WT and B1KO mice were flushed withIscove’s Modified Dulbecco’s Medium (IMDM) supplemented with20% horse serum (HS), 50 international units (IU) benzyl penicillin,50 �g/mL streptomycin, 2.0 mM l-glutamine, and 10−6 M hydro-cortisone into 6-well tissue culture plates. Half of the mediumin each 6-well plate was replaced weekly with an equal amountof fresh medium. At the end of the fourth week, after stromaformation, new BMs from other mice were collected in supple-mented IMDM and cultured (2 h) in tissue culture flasks (75 cm2).Non-adherent cells were collected by removing the medium and106 cells/well were added to pre-cultured stroma. The cultureswere submitted to in vitro treatment receiving captopril (1.0 �M),or AcSDKP (1.0 nM), or DBK (1.0 �M), or Lys-bradykinin (LBK)(1.0 �M) at the beginning of the culture.
2.4. Granulocyte-macrophage colony-forming cell (GM-CFC)
assayCells from BM of mice treated with captopril or AcSDKP(105 cells/mL) for 4 consecutive days were suspended in Dul-becco’s medium supplemented with 20% HS, 50 IU benzyl penicillin,

3 gical
5acosaoc
2
(w0sPgf6mn4afisw
2
MChcmwctwwQp
2W
f(fp4awiw
2
(fb[A5
90 C.R. Oliveira et al. / Chemico-Biolo
0 �g/mL streptomycin, and 2.0 mM l-glutamine with 0.3% meltedgar (Bacto Agar, Difco Labs, Detroit, MI, USA). After that, 1 mL ofell suspension was plated in 35-mm Petri dishes in the presencef 40 U/mL recombinant murine granulocyte-macrophage colonytimulating factor (rmGM-CSF), incubated in a fully humidifiedtmosphere in 10% CO2 incubator at 37 ◦C for 7 days. At the endf the incubation period, we counted the colonies of more than 50ells using a dark field microscope.
.5. Confocal microscopy
BM cells from WT mice were seeded onto glass cover slips13 mm), subsequently fixed with 2% formaldehyde for 30 min,ashed with 0.1 M glycine, followed by permeabilization with
.01% saponin for 15 min, and washed with phosphate bufferedaline (PBS) before incubation with anti-ACE antibody diluted inBS with 1% albumin. Anti-mouse IgG Alexa Fluor 488 conju-ated (Invitrogen/Molecular Probes, Eugene, OR, USA) was usedor 40 min as a secondary antibody. Nuclei were stained with 4′--diamidino-2-phenylindole (DAPI) (20 �g/mL) for 20 min. Lighticroscopy analyses were performed using a confocal laser scan-
ing microscope (Zeiss, LSM 510 META, Jena, Germany). Alexa Fluor88 fluorophores were excited by a 488-nm spectral line of anrgon-ion laser and the light emission was detected using a bypasslter (�ex = 500–550 nm). DAPI was excited by a titanium-dopedapphire multiphoton laser (�ex = 750 nm) and the light emissionas detected using a bypass filter (�ex = 350–410 nm).
.6. Immunolabeling
We used antibodies anti-Ter-119 (erythroid lineage), anti-ac-1, anti-Gr-1 (myeloid lineage), B220 (lymphoid B), an
D3 (lymphoid T) at appropriate concentrations to recognizeematopoietic stem cell populations present in WT and B1KO BMells. All antibodies were diluted in PBS with 1% bovine serum albu-in (BSA) and 0.05% sodium azide. Antibody–antigen incubationsere performed on ice for 20 min. To identify murine progenitor
ells, we applied a biotin-conjugated lineage (Lin) antibody cock-ail (anti-Gr-1, Mac-1, CD3�, TER-119, and B220), and after labelingith streptavidin-PerCP, Sca-1-PE, and c-Kit-APC, data analysesere performed using a FACSCalibur flow cytometer with Celluest (Becton Dickinson, San Jose, CA, USA). All antibodies wereurchased from Becton Dickinson Biosciences (San Diego, CA, USA).
.7. ACE expression in BM hematopoietic and stromal cells fromT and B1KO mice
To determine the cell population expressing ACE in the BM,resh cells from WT and B1KO mice were obtained by flushing106 cells), fixing with 2% paraformaldehyde, and treating themor 15 min with 0.01% saponin for permeabilization. Detection waserformed using anti-mouse IgG Alexa Fluor 488 conjugated for0 min. Next, the cells were incubated with mouse anti-ACE IgGntibody in 1% BSA dissolved in PBS for 2 h, washed, and labeledith Ter-119 and CD45 for 20 min. CD45+/Ter-119+ population was
dentified as hematopoietic cells; and CD45−/Ter-119− populationas identified as non-hematopoietic fraction.
.8. ACE activity assay
After 4 weeks of treatment with captopril (1.0 �M), or AcSDKP1.0 nM), or DBK (1.0 �M), or LBK (1.0 �M), the non-adherent cells
rom the LTBMCs were collected and the ACE activity was measuredy using Abz-FRK(Dnp)P-OH as substrate as described previously34]. In this assays, 200 �L of lysate were incubated with 10 �Mbz-FRK(Dnp)P-OH at 37 ◦C in 0.1 M Tris–HCl, pH 7.0, containing0 mM NaCl and 10 �M ZnCl2 in a final volume of 1.0 mL. EnzymaticInteractions 184 (2010) 388–395
activity was continuously monitored in a Hitachi F-2000 fluorime-ter, measuring the fluorescence at �em = 420 nm and �ex = 320 nm.
2.9. Detection of ACE mRNAs by real-time polymerase chainreaction
RNA was isolated from LTBMC cells previously treated for 4weeks with AcSDKP (1.0, 3.0, and 5.0 nM), or captopril (1.0 �M),or DBK (1.0 �M), or LBK (1.0 �M) using Trizol reagent (Gibco-BRL,Bethesda, MD, USA). This evaluation was also performed with freshBM cells obtained from WT, B1KO, and B2KO mice. For these exper-iments, the RNAs were processed and analyzed separately. TheRNA was incubated with DNase (Promega Corporation, Madison,WI, USA) to eliminate any residual DNA that would amplify dur-ing the polymerase chain reaction (PCR). Approximately 200 ng ofcDNA was amplified (in triplicate) by real-time (RT) quantitativePCR (Taq-Man, ABI Prism 7700 Sequence Detection System, AppliedBiosystems, Foster City, CA, USA). Amplification reactions included900 nM primers and 250 nM probe (FAM dye-labeled TaqMan-MGBprobe) in TaqMan Universal PCR master mix (Applied Biosystems)and were performed at universal thermal cycling conditions (a firststep of 5 min at 50 ◦C to activate AmpErase-UNG enzyme, a secondstep of 10 min at 95 ◦C to activate AmpliTaq polymerase, followedby 40 cycles of 15 s at 95 ◦C, and 60 s at 60 ◦C) according to themanufacturer’s instructions.
The following TaqMan assays were used for RT-PCR at afinal concentration of 250 nM TaqMan probe and 900 nM ofeach primer: B1 receptor: forward 5′-CCATACAAAACCCCAGCT-GAA-3′; reverse 5′-CTTTGGTTAGAAGGCTGTAGCTTCA-3′; probe 5′-CACAGGAACCCAGACAG-3′. B2 receptor: forward 5′-CCCTTCC-TCTGGGTCCTCTT-3′; reverse 5′-GAAGAACACGCTGAGGACAAAGA-3′; probe 5′-CCGCCACTGGAGAAC-3′. ACE: forward 5′-GGAGACGA-CTTACAGTGTAGCC-3′; reverse 5′-CTCCATGTTCACAGAGGTACACT-3′; probe 5′-CTCAGCCTGGGACTTCTACAAC-3′. As internal con-trol for normalization, we amplified the �-actin gene (for-ward 5′-CTGGCCTCAACTGTCCACCTT-3′; reverse 5′-CGGACTCAT-CGTACTCCTGCTT-3′; probe 5′-CTGATCCACATCTGCT-3′).
All amplification batches included controls without templateand cDNA aliquots from two samples that served as positivecontrols and were repeatedly quantified to assess inter-assay vari-ability. ACE mRNA levels were quantified using the delta thresholdcycle (CT) method, based on the relative quantification of ACEexpression normalized to the housekeeping gene �-actin afterdetermining the first cycle of fluorescence detection (thresholdcycle) and calculating the differences of these threshold cycles.Samples with CT values ≥36 were excluded from analysis becausethey were outside the proven linear dynamic range of the assays.
2.10. AcSDKP levels in LTBMC from WT and B1KO mice treatedwith captopril
After establishing the LTBMCs from WT and B1KO cells, cul-tures were treated with captopril (1.0 �M) for 4 weeks. The AcSDKPlevels were measured by a competitive enzyme immunoassay(EIA) as previously described by Pradelles et al. [35]. Briefly, wellsof microtitre plates were coated with mouse monoclonal anti-rabbit immunoglobulins (ALPCO Diagnostics, Windham, NH, USA).Enzyme-labelled competitor (AcSDKP conjugated with acetyl-cholinesterase), diluted anti-AcSDKP antiserum, and standards orsamples (each diluted with 50 mL of EIA buffer) were added toeach well. After 18 h of incubation at 4 ◦C, the plates were washed
(10−2 mol/L phosphate buffer containing 0.05% Tween 20, pH 7.4)and 200 mL of enzymatic substrate were added. After 12 h of incu-bation in a dark chamber, the optical density was measured at405 nm, and the results expressed in pmol of AcSDKP per mL ofsupernatant.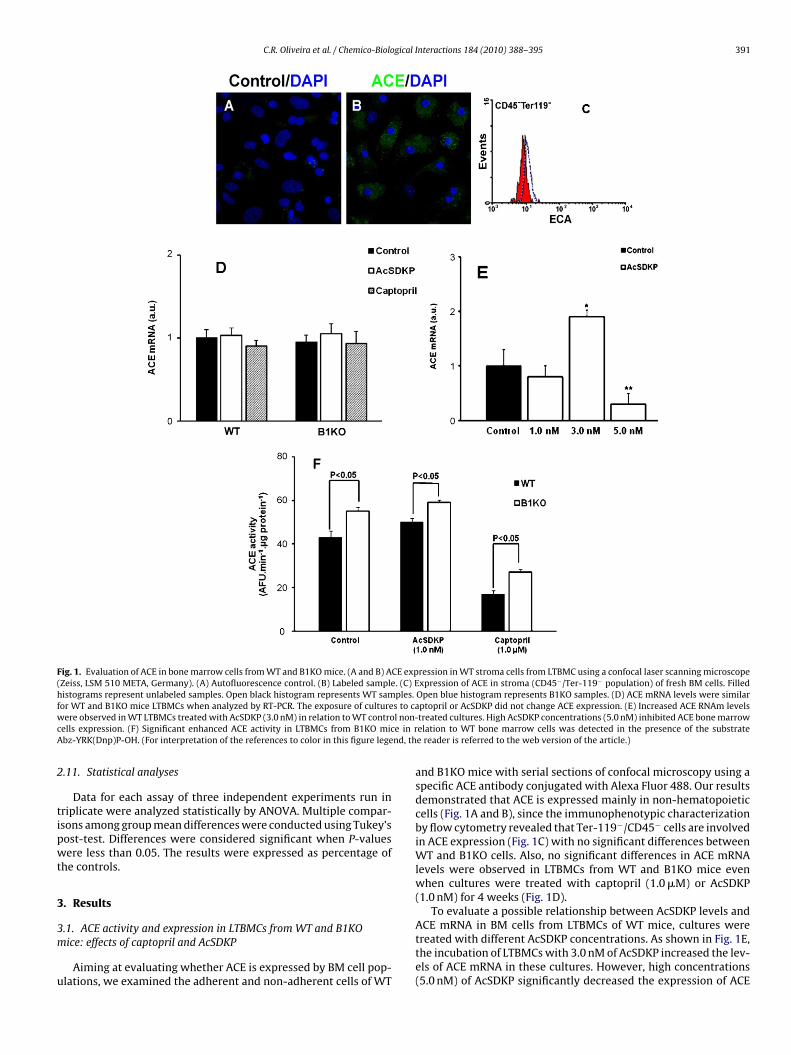
C.R. Oliveira et al. / Chemico-Biological Interactions 184 (2010) 388–395 391
Fig. 1. Evaluation of ACE in bone marrow cells from WT and B1KO mice. (A and B) ACE expression in WT stroma cells from LTBMC using a confocal laser scanning microscope(Zeiss, LSM 510 META, Germany). (A) Autofluorescence control. (B) Labeled sample. (C) Expression of ACE in stroma (CD45−/Ter-119− population) of fresh BM cells. Filledhistograms represent unlabeled samples. Open black histogram represents WT samples. Open blue histogram represents B1KO samples. (D) ACE mRNA levels were similarf s to caw l non-c e in rA nd, th
2
tipwt
3
3m
u
or WT and B1KO mice LTBMCs when analyzed by RT-PCR. The exposure of cultureere observed in WT LTBMCs treated with AcSDKP (3.0 nM) in relation to WT contro
ells expression. (F) Significant enhanced ACE activity in LTBMCs from B1KO micbz-YRK(Dnp)P-OH. (For interpretation of the references to color in this figure lege
.11. Statistical analyses
Data for each assay of three independent experiments run inriplicate were analyzed statistically by ANOVA. Multiple compar-sons among group mean differences were conducted using Tukey’sost-test. Differences were considered significant when P-valuesere less than 0.05. The results were expressed as percentage of
he controls.
. Results
.1. ACE activity and expression in LTBMCs from WT and B1KOice: effects of captopril and AcSDKP
Aiming at evaluating whether ACE is expressed by BM cell pop-lations, we examined the adherent and non-adherent cells of WT
ptopril or AcSDKP did not change ACE expression. (E) Increased ACE RNAm levelstreated cultures. High AcSDKP concentrations (5.0 nM) inhibited ACE bone marrowelation to WT bone marrow cells was detected in the presence of the substratee reader is referred to the web version of the article.)
and B1KO mice with serial sections of confocal microscopy using aspecific ACE antibody conjugated with Alexa Fluor 488. Our resultsdemonstrated that ACE is expressed mainly in non-hematopoieticcells (Fig. 1A and B), since the immunophenotypic characterizationby flow cytometry revealed that Ter-119−/CD45− cells are involvedin ACE expression (Fig. 1C) with no significant differences betweenWT and B1KO cells. Also, no significant differences in ACE mRNAlevels were observed in LTBMCs from WT and B1KO mice evenwhen cultures were treated with captopril (1.0 �M) or AcSDKP(1.0 nM) for 4 weeks (Fig. 1D).
To evaluate a possible relationship between AcSDKP levels and
ACE mRNA in BM cells from LTBMCs of WT mice, cultures weretreated with different AcSDKP concentrations. As shown in Fig. 1E,the incubation of LTBMCs with 3.0 nM of AcSDKP increased the lev-els of ACE mRNA in these cultures. However, high concentrations(5.0 nM) of AcSDKP significantly decreased the expression of ACE
392 C.R. Oliveira et al. / Chemico-Biological Interactions 184 (2010) 388–395
Fait
ics
FcimwtWNtc
3c
B4bmmApd
3
ah
3c
wTmrbc
(ELISPOT) assay. The results showed an increase in AcSDKP levelsin the supernatant of LTBMCs from WT-captopril-treated cul-tures (1.0 �M) when compared to B1KO-captopril-treated cultures(Fig. 4). Decreased AcSDKP concentrations (35%) were also observed
ig. 2. Effects of captopril (100 mg/kg) and AcSDKP (10 �g/kg) on GM-CFC of WTnd B1KO-treated mice for 4 consecutive days (n = 5). No significant differencesn GM-CFC numbers were observed in B1KO mice compared to WT animals in allreatments. Values are means + SEM of three separate experiments.
n relation to control non-treated cultures (Fig. 1E). These resultslearly show a relationship between AcSDKP levels and ACE expres-ion in BM cells from WT mice.
Besides using a selective fluorescence substrate for ACE [Abz-RK(Dnp)P-OH] in WT and B1KO LTBMCs, we also evaluatedaptopril- and AcSDKP-treated LTBMCs for ACE activity. Interest-ngly, we observed an enhanced ACE activity in cultures from B1KO
ice in relation to WT BM cells (Fig. 1F), which was maintainedhen AcSDKP was added to the LTBMCs for 4 weeks. As expected,
he specific ACE inhibitor captopril decreased ACE activity inT and B1KO LTBMCs compared to control non-treated cultures.
onetheless, a significant decrease was observed in captopril-reated LTBMCs from WT in comparison to those from B1KOultures (Fig. 1F).
.2. Number of GM-CFC in WT and B1KO mice treated withaptopril or AcSDKP
The number of GM-CFC was determined after treating WT and1KO mice daily with 100 mg/kg captopril and 10 �g/kg AcSDKP forconsecutive days. No differences were observed in GM-CFC num-ers between B1KO, B1KO-captopril-, and B1KO-AcSDKP-treatedice compared to WT, WT-captopril, and WT-AcSDKP-treatedice, respectively (Fig. 2). These data suggest that, althoughCE hyperfunction is present in BM cells from B1KO mice, thishenomenon did not modify murine myelopoiesis under basal con-itions.
.3. Spleen weight
B1KO mice (18 months old) presented massive splenomegalynd their average spleen weight (90.1 ± 4.2 mg) was significantlyigher than that of WT animals (70.1 ± 6.3 mg).
.4. Cell populations in BM from WT and B1KO mice by flowytometry
To asses whether ACE modulates a specific BM cell population,e quantified BM cells from WT and B1KO mice by flow cytometry.
+
he results showed a decrease in erythroid (Ter119 ), granulocytic-acrophage (Gr-1+/Mac-1+), B220, CD3+, and LSK cells in B1KO inelation to WT mice (Figs. 3A and 4B). However, increased num-ers of CD45−/Ter-119− cells were observed in BM from B1KO miceompared with WT BM cells (Fig. 3A).
Fig. 3. BM cell populations from WT and B1KO mice. (A) Ter119+ and Gr1+/Mac1+.*Significant in relation to WT mice. (B) LSK population detected in the BM from WTand B1KO mice. *Significant in relation to WT cultures.
3.5. Effects of captopril on LTBMCs AcSDKP supernatant levelsfrom WT and B1KO mice
In order to evaluate whether captopril enhances the levels ofACE substrate AcSDKP in LTBMCs, we quantified AcSDKP levelsin the supernatant of LTBMC by the enzyme-linked immunospot
Fig. 4. AcSDKP levels in the supernatant of LTBMCs from WT and B1KO mice treatedfor 4 weeks with captopril (1.0 �M). Half of the medium in each flask was replacedweekly with an equal amount of fresh medium. Note the increased levels of AcSDKPin WT LTBMC treated cells in relation to B1KO-captopril-treated cells. *Significantin relation to WT cultures.
C.R. Oliveira et al. / Chemico-Biological Interactions 184 (2010) 388–395 393
Table 1Leucocyte, erythrocyte, hemoglobin, hematocrit, and platelet levels in WT and B1KO mice (n = 7) under basal conditions and 24 h after LPS administration.
Animal Leucocytes (103/�L) Erithrocytes (×106/�L) Hemoglobin (mg/dL) Hematocrit (%) Platelets (×103/�L)
Basal conditionsWT 7.9 ± 1.0 11.0 ± 0.3 17.6 ± 0.5 55.2 ± 0.8 865 ± 160B1KO 8.2 ± 1.0 10.0 ± 0.2 15.7 ± 0.4 49.0 ± 0.4 1069 ± 79
iac
3a
fBwrTdrDb
3e
WPg
F(A
After LPS administrationWT 8.4 ± 3.0 9.4 ± 0.3B1KO 3.3 ± 0.7* 8.7 ± 0.3*
* P < 0.05 in relation to WT LPS-treated animals.
n B1KO LTBMCs in relation to WT cultures. These results are inccordance with ACE hyperfunction described herein for B1KO BMells.
.6. Total peripheral blood cells in WT and B1KO mice after LPSdministration
Table 1 shows that under basal conditions there are no dif-erences in total number of peripheral blood cells from WT and1KO mice, similarly to the results observed for CFU-GM. However,hen WT and B1KO animals received LPS (14 mg/kg), we detected a
eduction in blood cells from B1KO mice in relation to WT animals.hese data strongly suggest that the alteration in BM compartmentescribed in this study for B1KO mice contributed to their defectiveesponse to infections, and consequently that in addition to AcS-KP, kinin B1 receptor also plays an important role in myelopoiesisalance.
.7. Influence of kinin receptor agonists (DBK and LBK) on ACExpression in LTBMCs
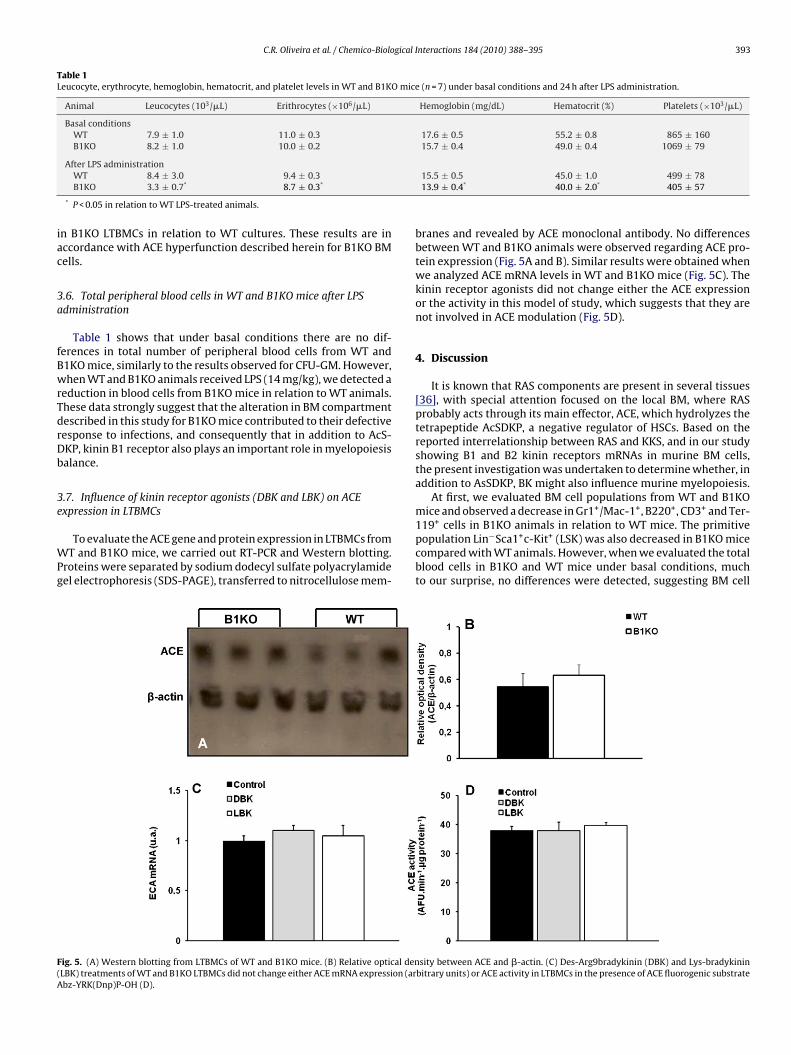
To evaluate the ACE gene and protein expression in LTBMCs fromT and B1KO mice, we carried out RT-PCR and Western blotting.
roteins were separated by sodium dodecyl sulfate polyacrylamideel electrophoresis (SDS-PAGE), transferred to nitrocellulose mem-
ig. 5. (A) Western blotting from LTBMCs of WT and B1KO mice. (B) Relative optical denLBK) treatments of WT and B1KO LTBMCs did not change either ACE mRNA expression (arbz-YRK(Dnp)P-OH (D).
15.5 ± 0.5 45.0 ± 1.0 499 ± 7813.9 ± 0.4* 40.0 ± 2.0* 405 ± 57
branes and revealed by ACE monoclonal antibody. No differencesbetween WT and B1KO animals were observed regarding ACE pro-tein expression (Fig. 5A and B). Similar results were obtained whenwe analyzed ACE mRNA levels in WT and B1KO mice (Fig. 5C). Thekinin receptor agonists did not change either the ACE expressionor the activity in this model of study, which suggests that they arenot involved in ACE modulation (Fig. 5D).
4. Discussion
It is known that RAS components are present in several tissues[36], with special attention focused on the local BM, where RASprobably acts through its main effector, ACE, which hydrolyzes thetetrapeptide AcSDKP, a negative regulator of HSCs. Based on thereported interrelationship between RAS and KKS, and in our studyshowing B1 and B2 kinin receptors mRNAs in murine BM cells,the present investigation was undertaken to determine whether, inaddition to AsSDKP, BK might also influence murine myelopoiesis.
At first, we evaluated BM cell populations from WT and B1KOmice and observed a decrease in Gr1+/Mac-1+, B220+, CD3+ and Ter-
119+ cells in B1KO animals in relation to WT mice. The primitivepopulation Lin−Sca1+c-Kit+ (LSK) was also decreased in B1KO micecompared with WT animals. However, when we evaluated the totalblood cells in B1KO and WT mice under basal conditions, muchto our surprise, no differences were detected, suggesting BM cellsity between ACE and �-actin. (C) Des-Arg9bradykinin (DBK) and Lys-bradykininbitrary units) or ACE activity in LTBMCs in the presence of ACE fluorogenic substrate

3 gical
mtu
taiasOotmBltdvpa
tcsetiv
ipncin
wnBApcamafiobsa
oAsLitec
sftenf
94 C.R. Oliveira et al. / Chemico-Biolo
igration to other compartments. In line with this hypothesis, inhe present study we found significant splenomegaly in B1KO micender basal conditions.
AcSDKP, a physiological negative regulator of hematopoiesishat acts by blocking the cell cycle entry of quiescent stem cellsnd progenitors [15], is probably involved with our results show-ng decreased BM progenitor cells in B1KO mice in relation to WTnimals, since the levels of this tetrapeptide, together with otherignals observed in BM compartment, regulates HSC proliferation.f note, we detected decreased levels of AcSDKP in the supernatantf B1KO LTBMCs and B1KO-captopril-treated cultures comparedo WT and WT-captopril-treated cultures, respectively. This result
ight partly explain the decreased progenitor cells we found inM from B1KO mice in relation to WT animals, because decreased
evels of AcSDKP allow BM progenitor cells to enter the S stage ofhe cell cycle. In accordance with these results, Kanasaki et al. [37]emonstrated that AcSDKP suppresses mesangial cell proliferationia upregulation of cell cycle modulators, such as p53, p27kip1, and21cip1. Moreover, increased stability of p53 protein by AcSDKP waslso reported by the authors.
When cleaved by angiotensin converting enzyme (ACE), theetrapeptide AcSDKP releases the bone precursor cells to the cellycle indirectly by blocking the effect of an unknown stem celltimulator of proliferation [14] or by interfering with the periph-ral production or degradation of other cytokines that may reachhe BM. This peptide, whose precursor could be thymosin-�4 [38],s known to be secreted by BM [39] and ubiquitously distributed inivo [40].
During phase-specific anticancer drug treatment, admin-stration of exogenous AcSDKP selectively maintains normalrogenitors in the quiescent state. In vivo, administration of exoge-ous AcSDKP increases survival in mice treated with lethal doses ofytosine arabinoside [41] and doxorubicin [42] or with sub-lethalrradiation [43]. In humans, AcSDKP may be useful for protectingormal HSCs against damage due to cytotoxic therapy [44].
The importance of kinin B1 receptor for myelopoiesis balanceas also demonstrated by our results, showing that although theumbers of GM-CFC and total blood cells were similar for both1KO and WT animals even when these animals were treated withcSDKP or captopril, the response of B1KO mice to LPS was com-romised. This was evidenced by the decreased numbers of bloodells observed in B1KO mice exposed to LPS compared with WTnimals under the same conditions, suggesting that the former areore susceptible to infections, possibly due to the BM impairment
lready described in this study for B1KO animals. Based on thesendings, we suggest that in addition to the influence of AcSDKPn myelopoiesis, kinins are also involved and may contribute to itsalance, since many parameters of this process evaluated in ourtudy presented different results for B1KO mice compared to WTnimals.
Given that the relationship between kinins and AcSDKP is ofur main interest and with the purpose of investigating whetherCE was involved in the results described above, we used the ACEubstrate Abz-FRK(Dnp)P-OH and observed an increase in B1KOTBMC ACE activity in relation to WT cultures. The results obtainedn this assay could partly explain our findings, since ACE hyperfunc-ion might inactivate AcSDKP more efficiently allowing the HSC tonter the S stage of the cell cycle and resulting in the decreased BMell populations as observed in B1KO mice.
Since hematopoietic cells are smaller and circular in shape andtromal cells are bigger with variable shapes, these morphological
eatures were used in their identification. Our results demonstratedhat stromal cells from LTBMCs might be mainly responsible for ACExpression in BM microenvironment because Ter-119− and CD45−egative cells were responsible for ACE expression in the LTBMCrom WT cells.
[
[
Interactions 184 (2010) 388–395
Of interest, after treatment with 1.0 �M of captopril or 1.0 nMof AcSDKP for 4 weeks, ACE protein levels and mRNA expres-sion did not change and were similar in both B1KO and WT cells.Nevertheless, when WT LTBMCs were treated with higher AcS-DKP concentrations (3.0 nM), increased ACE mRNA levels wereobserved, which also suggests the occurrence of a dose-responserelationship between ACE expression and AcSDKP concentrationsin BM cells.
Using kinin agonists, we detected no alterations in ACE mRNA orprotein levels in BM cells from B1KO and WT mice, suggesting thatthere might be another phenomenon that could account for ourresults showing ACE hyperfunction and myelopoiesis modulationin B1KO cells. Our findings could be explained, at least partially, bythe work of Chen et al. [7], in which the authors have successfullyshown heterodimer formation between ACE and B2 receptor andco-localization of both proteins at the plasma membrane. Althoughboth the underlying mechanisms involved in receptor oligomer-ization and the exact role this phenomenon plays in G-coupledreceptor function remain largely unknown, it is possible that thishappening was present in our experiment and explains, at least inpart, the enhanced ACE activity we found in B1KO mice. Using Chi-nese hamster ovary cell monolayer co-expressing the somatic formof the enzyme and B2 coding region, Sabatini et al. [45] demon-strated an augmentation in ACE catalytic activity, which in turnexplains part of the results presented herein.
Further studies of clinical relevance to provide a mechanisticrelationship for ACE, AcSDKP, and BK in normal BM as well as inBM-related pathologies are being carried out in our laboratory.Moreover, the relationship between AcSDKP levels and ACE expres-sion is of great interest and deserves special attention.
Conflict of interest
None.
Acknowledgements
This work was supported by grants from the Fundacão deAmparo à Pesquisa do Estado de São Paulo (Process no. 06/57253-4)and Conselho Nacional de Desenvolvimento Científico e Tec-nológico (CNPq).
References
[1] M. Ogawa, Differentiation and proliferation of hematopoietic stem cells, Blood81 (1993) 2844–2853.
[2] S.H. Orkin, Diversification of haematopoietic stem cells to specific lineages, Nat.Rev. Genet. 1 (2000) 57–64.
[3] M. Tavian, L. Coulombel, D. Luton, H.S. Clemente, F. Dieterlen-Lièvre, B. Péault,Aorta-associated CD34+ hematopoietic cells in the early human embryo, Blood87 (1996) 67–72.
[4] T. North, T.L. Gu, T. Stacy, Q. Wang, L. Howard, M. Binder, M. Marín-Padilla,N.A. Speck, Cbfa2 is required for the formation of intra-aortic hematopoieticclusters, Development 126 (1999) 2563–2575.
[5] S.H. Orkin, L.I. Zon, Hematopoiesis and stem cells: plasticity versus develop-mental heterogeneity, Nat. Immunol. 3 (2002) 323–328.
[6] S.H. Cheshier, S.J. Morrison, X. Liao, I.L. Weissman, In vivo proliferation and cellcycle kinetics of long-term self-renewing hematopoietic stem cells, Proc. Natl.Acad. Sci. U.S.A. 96 (1999) 3120–3125.
[7] T. Cheng, N. Rodrigues, H. Shen, Y. Yang, D. Dombkowski, M. Sykes, D.T. Scadden,Hematopoietic stem cell quiescence maintained by p21cip1/waf1, Science 287(2000) 1804–1808.
[8] J. Larsson, S. Karlsson, The role of Smad signaling in hematopoiesis, Oncogene24 (2005) 5676–5692.
[9] I.C. Haznedaroglu, S. Tuncer, M. Gürsoy, A local renin–angiotensin system in
the bone marrow, Med. Hypotheses 46 (1996) 507–510.10] W.B. Strawn, R.S. Richmond, E.A. Tallant, P.E. Gallagher, C.M. Ferrario,Renin–angiotensin system expression in rat bone marrow haematopoietic andstromal cells, Br. J. Haematol. 126 (2004) 120–126.
11] A. Okamura, H. Raukugi, M. Ohishi, Y. Yanagitani, S. Takiuchi, K. Moriguchi,P.A. Fennessy, J. Higaki, T. Ogihara, Upregulation of renin–angiotensin system

gical I
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
550.
C.R. Oliveira et al. / Chemico-Biolo
during differentiation of monocytes to macrophages, J. Hypertens. 17 (1999)537–545.
12] J.E. Chisi, J. Wdzieczak-Bakala, J. Thierry, C.V. Briscoe, A.C. Riches, Captoprilinhibits the proliferation of hematopoietic stem and progenitor cells in murinelong-term bone marrow cultures, Stem Cells 17 (1999) 339–344.
13] K.E. Rodgers, S. Xiong, R. Steer, G.S. diZerega, Effect of angiotensin II onhematopoietic progenitor cell proliferation, Stem Cells 18 (2000) 287–294.
14] K.J. Rieger, N. Saez-Servent, M.P. Papet, J. Wdzieczak-Bakala, J.L. Morgat, J.Thierry, W. Voelter, M. Lenfant, Involvement of human plasma angiotensinI-converting enzyme in the degradation of the haemoregulatory peptide N-acetyl-seryl-aspartyl-lysyl-proline, Biochem. J. 296 (1993) 373–378.
15] S. Robinson, M. Lenfant, J. Wdzieczak-Bakala, J. Melville, A. Riches, The mech-anism of action of the tetrapeptide acetyl-N-Ser-Asp-Lys-Pro (AcSDKP) inthe control of haematopoietic stem cell proliferation, Cell Prolif. 25 (1992)623–632.
16] J. Cole, D. Ertoy, H. Lin, R.L. Sutliff, E. Ezan, T.T. Guyene, M. Capecchi, P. Corvol,K.E. Bernstein, Lack of angiotensin II-facilitated erythropoiesis causes anemiain angiotensin-converting enzyme-deficient mice, J. Clin. Invest. 106 (2000)1391–1398.
17] L. Comte, V. Lorgeot, L. Volkov, A. Allegraud, J.C. Aldigier, V. Praloran, Effects ofthe angiotensin-converting enzyme inhibitor enalapril on blood haematopoi-etic progenitors and Acetyl-N-Ser-Asp-Lys-Pro concentrations, Eur. J. Clin.Invest. 27 (1997) 788–790.
18] H. Abali, I.C. Haznedaroglu, H. Goker, I. Celik, D. Özatli, Z. Koray, M. Caglar,Circulating and local bone marrow renin–angiotensin system in leukemichematopoiesis: preliminary evidences, Hematology 7 (2002) 75–82.
19] E. Frindel, M. Guigon, Inhibition of CFU entry into cycle by a bone marrowextract, Exp. Hematol. 5 (1977) 74–76.
20] M. Lenfant, J. Wdzieczak-Bakala, E. Guittet, J.C. Prome, D. Sotty, E. Frindel,Inhibitor of hematopoietic pluripotent stem cell proliferation: purification anddetermination of its structure, Proc. Natl. Acad. Sci. U.S.A. 86 (1989) 779–782.
21] E.T. Zambidis, T.S. Park, W. Yu, A. Tam, M. Levine, X. Yuan, M. Pryzhkova, B.Péault, Expression of angiotensin-converting enzyme (CD143) identifies andregulates primitive hemangioblasts derived from human pluripotent stemcells, Blood 112 (2008) 3601–3614.
22] A. Rousseau, A. Michaud, M.T. Chauvet, M. Lenfant, P. Corvol, The homoregula-tory peptide N-acetyl-Ser-Asp-Lys-Pro is a natural and specific substrate of theN-terminal active site of human angiotensin-converting enzyme, J. Biol. Chem.270 (1995) 3656–3661.
23] M. Mrug, T. Stopka, B.A. Julian, J.F. Prchal, J.T. Prchal, Angiotensin II stimulatesproliferation of normal early erythroid progenitors, J. Clin. Invest. 100 (1997)2310–2314.
24] S. Charrier, A. Michaud, S. Badaoui, S. Giroux, E. Ezan, F. Sainteny, P. Corvol, W.Vainchenker, Inhibition of angiotensin I-converting enzyme induces radiopro-tection by preserving murine hematopoietic short-term reconstituting cells,Blood 104 (2004) 978–985.
25] M.E. Moreau, N. Garbacki, G. Molinaro, N.J. Brown, F. Marceau, A. Adam, Thekallikrein–kinin system: current and future pharmacological targets, J. Phar-macol. Sci. 99 (2005) 6–38.
26] F. Marceau, J.F. Hess, D.R. Bachvarov, The B1 receptors for kinins, Pharmacol.Rev. 50 (1998) 357–386.
27] D.B. Hara, D.F.P. Leite, E.S. Fernandes, G.F. Passos, A.O. Guimarães, J.B. Pesquero,M.M. Campos, J.B. Calixto, The relevance of kinin B1 receptor upregulation in amouse model of colitis, Br. J. Pharmacol. 154 (2008) 1276–1286.
28] C.E. Austin, A. Faussner, H.E. Robinson, S. Chakravarty, D.J. Kyle, J.M. Bathon, D.
Proud, Stable expression of the human kinin B1 receptor in Chinese hamsterovary cells. Characterization of ligand binding and effector pathways, J. Biol.Chem. 272 (1997) 11420–11425.29] S.A. Mathis, N.L. Criscimagna, L.M. Leeb-Lundberg, B1 and B2 kinin receptorsmediate distinct patterns of intracellular Ca2+ signaling in single cultured vas-cular smooth muscle cells, Mol. Pharmacol. 50 (1996) 128–139.
[
nteractions 184 (2010) 388–395 395
30] D. Regoli, J. Barabé, Pharmacology of bradykinin and related kinins, Pharmacol.Rev. 32 (1980) 1–46.
31] M. Schachter, Kallikreins (kininogenases)—a group of serine proteases withbioregulatory actions, Pharmacol. Rev. 31 (1979) 1–17.
32] E.M. Duma, J.S. Foster, R.N. Moore, Bradykinin sensitization of CSF-1-responsivemurine mononuclear phagocyte precursors to prostaglandin E, J. Immunol. 141(1988) 3186–3189.
33] J.E. Chisi, C.V. Briscoe, E. Ezan, R. Genet, A.C. Riches, J. Wdzieczak-Bakala, Cap-topril inhibits in vitro and in vivo the proliferation of primitive haematopoieticcells induced into cell cycle by cytotoxic drug administration or irradiationbut has no effect on myeloid leukaemia cell proliferation, Br. J. Haematol. 109(2000) 563–570.
34] M.C. Araujo, R.L. Melo, M.H. Cesari, M.A. Juliano, L. Juliano, A.K. Carmona,Peptidase specificity characterization of C- and N-terminal catalytic sites ofangiotensin I-converting enzyme, Biochemistry 39 (2000) 8519–8525.
35] P. Pradelles, Y. Frobert, C. Créminon, H. Ivonine, E. Frindel, Distribution ofa negative regulator of haematopoietic stem cell proliferation (AcSDKP) andthymosin beta 4 in mouse tissues, FEBS Lett. 289 (1991) 171–175.
36] D. Crisan, J. Carr, Angiotensin I-converting enzyme: genotype and disease asso-ciations, J. Mol. Diagn. 2 (2000) 105–115.
37] K. Kanasaki, H. Haneda, T. Sugimoto, K. Shibuya, M. Isono, K. Isshiki, S. Araki, T.Uzu, A. Kashiwagi, D. Koya, N-Acetyl-seryl-aspartyl-lysyl-proline inhibits DNAsynthesis in human mesangial cells via up-regulation of cell cycle modulators,Biochem. Biophys. Res. Commun. 342 (2006) 758–765.
38] I. Fleming, Signaling by the angiotensin-converting enzyme, Circ. Res. 98 (2006)887–896.
39] W. Leeanansaksiri, S.K. DeSimone, T. Huff, E. Hannappel, T.F. Huff, Thymosinbeta 4 and its N-terminal tetrapeptide, AcSDKP, inhibit proliferation, andinduce dysplastic, non-apoptotic nuclei and degranulation of mast cells, Chem.Biodivers. 1 (2004) 1091–1100.
40] J. Wdzieczak-Bakala, M.P. Fache, M. Lenfant, E. Frindel, F. Sainteny, AcSDKP,an inhibitor of CFU-S proliferation, is synthesized in mice under steady-stateconditions and secreted by bone marrow in long-term culture, Leukemia 4(1990) 235–237.
41] A.E. Bogden, P. Carde, E.D. de Paillette, J.P. Moreau, M. Tubiana, E. Frindel, Ame-lioration of chemotherapy-induced toxicity by cotreatment with AcSDKP, atetrapeptide inhibitor of hematopoietic stem cell proliferation, Ann. N. Y. Acad.Sci. 628 (1991) 126–139.
42] A. Massé, L.H. Ramirez, G. Bindoula, C. Grillon, J. Wdzieczak-Bakala, K. Rad-dassi, E. Deschamps de Paillette, J.M. Mencia-Huerta, S. Koscielny, P. Potier,F. Sainteny, P. Carde, The tetrapeptide acetyl-N-Ser-Asp-Lys-Pro (Goralatide)protects from doxorubicin-induced toxicity: improvement in mice survivaland protection of bone marrow stem cells and progenitors, Blood 91 (1998)441–449.
43] T. Watanabe, G.S. Brown, L.S. Kelsey, Y. Yan, J.D. Jackson, C. Ewel, A. Kessinger,J.E. Talmadge, In vivo protective effects of tetrapeptide AcSDKP, with or with-out granulocyte colony-stimulation factor, on murine progenitor cells aftersublethal irradiation, Exp. Hematol. 24 (1996) 713–721.
44] P. Carde, C. Chastang, E. Goncalves, N. Mathieu-Tubiana, E. Vuillemin, V.Delwail, O. Corbion, A. Vekhoff, F. Isnard, J.M. Ferrero, E. Garcia-Giralt, J.F.Gimonet, A.M. Stoppa, E. Léger-Picherit, E. Fadel, J.P. Monpezat, J.N. Munck,C. Domenge, D. Khayat, F. Guilhot, Séraspénide (acétyl SDKP): étude enphases I–II d’un inhibiteur de l’hématopoièse la protégeant de la toxicité demonochimiothérapies aracytine et ifosfamide, C. R. Acad. Sci. 315 (1992) 545–
45] R.A. Sabatini, P.B. Guimarães, L. Fernandes, F.C. Reis, P.A. Bersanetti, M.A.Mori, A. Navarro, A.M. Hilzendeger, E.L. Santos, M.C. Andrade, J.R. Cha-gas, J.L. Pesquero, D.E. Casarini, M. Bader, A.K. Carmona, J.B. Pesquero, ACEactivity is modulated by kinin B2 receptor, Hypertension 51 (2008) 689–695.




















