
Muscle ring finger 1 mediates cardiac atrophy in vivo
10
Muscle ring finger 1 mediates cardiac atrophy in vivo Monte S. Willis, 1,2 Mauricio Rojas, 1 Luge Li, 2 Craig H. Selzman, 6 Ru-Hang Tang, 3 William E. Stansfield, 3 Jessica E. Rodriguez, 2 David J. Glass, 4 and Cam Patterson 1,5 1 Carolina Cardiovascular Biology Center, 2 Department of Pathology and Laboratory Medicine, and 3 Department of Surgery, University of North Carolina, Chapel Hill, North Carolina; 4 Novartis Institutes for Biomedical Research Incorporated, Cambridge, Massachusetts; 5 Departments of Medicine, Pharmacology, and Cell and Developmental Biology, University of North Carolina, Chapel Hill, North Carolina; 6 Division of Cardiothoracic Surgery, University of Utah, Salt Lake City, Utah Submitted 23 June 2008; accepted in final form 21 January 2009 Willis MS, Rojas M, Li L, Selzman CH, Tang R, Stansfield WE, Rodriguez JE, Glass DJ, Patterson C. Muscle ring finger 1 mediates cardiac atrophy in vivo. Am J Physiol Heart Circ Phys- iol 296: H997–H1006, 2009. First published January 23, 2009; doi:10.1152/ajpheart.00660.2008.—Pathological cardiac hypertro- phy, induced by various etiologies such as high blood pressure and aortic stenosis, develops in response to increased afterload and rep- resents a common intermediary in the development of heart failure. Understandably then, the reversal of pathological cardiac hypertrophy is associated with a significant reduction in cardiovascular event risk and represents an important, yet underdeveloped, target of therapeutic research. Recently, we determined that muscle ring finger-1 (MuRF1), a muscle-specific protein, inhibits the development of experimentally induced pathological; cardiac hypertrophy. We now demonstrate that therapeutic cardiac atrophy induced in patients after left ventricular assist device placement is associated with an increase in cardiac MuRF1 expression. This prompted us to investigate the role of MuRF1 in two independent mouse models of cardiac atrophy: 1) cardiac hypertrophy regression after reversal of transaortic con- striction (TAC) reversal and 2) dexamethasone-induced atrophy. Us- ing echocardiographic, histological, and gene expression analyses, we found that upon TAC release, cardiac mass and cardiomyocyte cross- sectional areas in MuRF1 / mice decreased 70% less than in wild type mice in the 4 wk after release. This was in striking contrast to wild-type mice, who returned to baseline cardiac mass and cardiomy- ocyte size within 4 days of TAC release. Despite these differences in atrophic remodeling, the transcriptional activation of cardiac hyper- trophy measured by -myosin heavy chain, smooth muscle actin, and brain natriuretic peptide was attenuated similarly in both MuRF1 / and wild-type hearts after TAC release. In the second model, MuRF1 / mice also displayed resistance to dexamethasone-induced cardiac atrophy, as determined by echocardiographic analysis. This study demonstrates, for the first time, that MuRF1 is essential for cardiac atrophy in vivo, both in the setting of therapeutic regression of cardiac hypertrophy and dexamethasone-induced atrophy. ubiquitin ligase; cardiac hypertrophy; cardiac atrophy; left ventricular assist device PATHOLOGICAL CARDIAC HYPERTROPHY develops in response to increases in afterload and represents a common intermediary in the development of heart failure, a leading cause of mortality in the United States. Left ventricular (LV) hypertrophy is an independent risk factor for several adverse outcomes, including cardiac mortality, arrhythmias, and myocardial infarction (18, 25, 28, 29). Results from numerous studies have suggested that reducing heart mass in patients with pathological cardiac hy- pertrophy may reduce morbidity and mortality and improves patient outcomes (6, 10, 26, 35, 36, 44, 45). However, the only treatments currently proven to reverse both structural and functional cardiac abnormalities associated with pathological cardiac hypertrophy are antihypertensive therapies and aortic valve replacement (for aortic stenosis), both of which have a limited success rate (14, 39). Understanding the underlying processes regulating the plasticity of the heart will allow us to identify specific pathways against which to target new thera- pies and may improve the long-term outcomes of patients with pathological cardiac hypertrophy. Muscle ring finger (MuRF) family proteins are striated muscle-specific proteins involved in cardiomyocyte develop- ment and the regulation of muscle mass (3, 32). MuRF1 localizes specifically to the M line, a central structure of the sarcomere thick filament that has recently been recognized as a center of mechanical sensing (21). The ring finger domain of MuRF1 has ubiquitin ligase capabilities (34), targeting sarco- meric proteins such as troponin I and -/slow myosin heavy chain (MHC) for degradation (13, 24). This degradation occurs through the coordinated placement of polyubiquitin chains on recognized substrates, which are subsequently degraded by the proteasome (50). MuRF1 also interacts with and inhibits serum response factor (SRF) activity, a transcription factor critical to the development of cardiac hypertrophy (49). MuRF1’s local- ization in the sarcomere places it in a unique position to both recognize the mechanical stresses that induce cardiac hyper- trophy and regulate its subsequent development by controlling the degradation of targeted sarcomeric proteins. Indeed, results from studies using animal models where the expression of MuRF1 has been altered have implicated MuRF1 in the regu- lation of the development of cardiac hypertrophy: increasing MuRF1 in cardiomyocytes inhibits the development of hyper- trophy (2), whereas the complete lack of MuRF1 results in the development of an exaggerated cardiac hypertrophy in re- sponse to pressure overload (49). The present study was prompted by our observation that human cardiac tissue samples obtained from patients after placement of a LV assist device (LVAD) expressed signifi- cantly higher levels of MuRF1 protein than samples taken before the device was implanted. The unloading of the heart by a LVAD device leads to a decrease in the workload of the Address for reprint requests and other correspondence: M. S. Willis, Dept. of Pathology and Laboratory Medicine, Carolina Cardiovascular Biology Center, Univ. of North Carolina, Medical Biomolecular Research Bldg., Rm. 2336, 103 Mason Farm Rd., Chapel Hill, NC 27599-7525 (e-mail: monte_ [email protected]). The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. Am J Physiol Heart Circ Physiol 296: H997–H1006, 2009. First published January 23, 2009; doi:10.1152/ajpheart.00660.2008. 0363-6135/09 $8.00 Copyright © 2009 the American Physiological Society http://www.ajpheart.org H997 by 10.220.33.5 on August 19, 2016 http://ajpheart.physiology.org/ Downloaded from
-
Upload
uniatlantico -
Category
Documents
-
view
0 -
download
0
Transcript of Muscle ring finger 1 mediates cardiac atrophy in vivo

Muscle ring finger 1 mediates cardiac atrophy in vivo
Monte S. Willis,1,2 Mauricio Rojas,1 Luge Li,2 Craig H. Selzman,6 Ru-Hang Tang,3 William E. Stansfield,3
Jessica E. Rodriguez,2 David J. Glass,4 and Cam Patterson1,5
1Carolina Cardiovascular Biology Center, 2Department of Pathology and Laboratory Medicine, and 3Department of Surgery,University of North Carolina, Chapel Hill, North Carolina; 4Novartis Institutes for Biomedical Research Incorporated,Cambridge, Massachusetts; 5Departments of Medicine, Pharmacology, and Cell and Developmental Biology,University of North Carolina, Chapel Hill, North Carolina; 6Division of Cardiothoracic Surgery, University of Utah,Salt Lake City, Utah
Submitted 23 June 2008; accepted in final form 21 January 2009
Willis MS, Rojas M, Li L, Selzman CH, Tang R, StansfieldWE, Rodriguez JE, Glass DJ, Patterson C. Muscle ring finger 1mediates cardiac atrophy in vivo. Am J Physiol Heart Circ Phys-iol 296: H997–H1006, 2009. First published January 23, 2009;doi:10.1152/ajpheart.00660.2008.—Pathological cardiac hypertro-phy, induced by various etiologies such as high blood pressure andaortic stenosis, develops in response to increased afterload and rep-resents a common intermediary in the development of heart failure.Understandably then, the reversal of pathological cardiac hypertrophyis associated with a significant reduction in cardiovascular event riskand represents an important, yet underdeveloped, target of therapeuticresearch. Recently, we determined that muscle ring finger-1 (MuRF1),a muscle-specific protein, inhibits the development of experimentallyinduced pathological; cardiac hypertrophy. We now demonstrate thattherapeutic cardiac atrophy induced in patients after left ventricularassist device placement is associated with an increase in cardiacMuRF1 expression. This prompted us to investigate the role ofMuRF1 in two independent mouse models of cardiac atrophy:1) cardiac hypertrophy regression after reversal of transaortic con-striction (TAC) reversal and 2) dexamethasone-induced atrophy. Us-ing echocardiographic, histological, and gene expression analyses, wefound that upon TAC release, cardiac mass and cardiomyocyte cross-sectional areas in MuRF1�/� mice decreased �70% less than in wildtype mice in the 4 wk after release. This was in striking contrast towild-type mice, who returned to baseline cardiac mass and cardiomy-ocyte size within 4 days of TAC release. Despite these differences inatrophic remodeling, the transcriptional activation of cardiac hyper-trophy measured by �-myosin heavy chain, smooth muscle actin, andbrain natriuretic peptide was attenuated similarly in both MuRF1�/�
and wild-type hearts after TAC release. In the second model,MuRF1�/� mice also displayed resistance to dexamethasone-inducedcardiac atrophy, as determined by echocardiographic analysis. Thisstudy demonstrates, for the first time, that MuRF1 is essential forcardiac atrophy in vivo, both in the setting of therapeutic regression ofcardiac hypertrophy and dexamethasone-induced atrophy.
ubiquitin ligase; cardiac hypertrophy; cardiac atrophy; left ventricularassist device
PATHOLOGICAL CARDIAC HYPERTROPHY develops in response toincreases in afterload and represents a common intermediary inthe development of heart failure, a leading cause of mortality inthe United States. Left ventricular (LV) hypertrophy is anindependent risk factor for several adverse outcomes, includingcardiac mortality, arrhythmias, and myocardial infarction (18,
25, 28, 29). Results from numerous studies have suggested thatreducing heart mass in patients with pathological cardiac hy-pertrophy may reduce morbidity and mortality and improvespatient outcomes (6, 10, 26, 35, 36, 44, 45). However, the onlytreatments currently proven to reverse both structural andfunctional cardiac abnormalities associated with pathologicalcardiac hypertrophy are antihypertensive therapies and aorticvalve replacement (for aortic stenosis), both of which have alimited success rate (14, 39). Understanding the underlyingprocesses regulating the plasticity of the heart will allow us toidentify specific pathways against which to target new thera-pies and may improve the long-term outcomes of patients withpathological cardiac hypertrophy.
Muscle ring finger (MuRF) family proteins are striatedmuscle-specific proteins involved in cardiomyocyte develop-ment and the regulation of muscle mass (3, 32). MuRF1localizes specifically to the M line, a central structure of thesarcomere thick filament that has recently been recognized asa center of mechanical sensing (21). The ring finger domain ofMuRF1 has ubiquitin ligase capabilities (34), targeting sarco-meric proteins such as troponin I and �-/slow myosin heavychain (MHC) for degradation (13, 24). This degradation occursthrough the coordinated placement of polyubiquitin chains onrecognized substrates, which are subsequently degraded by theproteasome (50). MuRF1 also interacts with and inhibits serumresponse factor (SRF) activity, a transcription factor critical tothe development of cardiac hypertrophy (49). MuRF1’s local-ization in the sarcomere places it in a unique position to bothrecognize the mechanical stresses that induce cardiac hyper-trophy and regulate its subsequent development by controllingthe degradation of targeted sarcomeric proteins. Indeed, resultsfrom studies using animal models where the expression ofMuRF1 has been altered have implicated MuRF1 in the regu-lation of the development of cardiac hypertrophy: increasingMuRF1 in cardiomyocytes inhibits the development of hyper-trophy (2), whereas the complete lack of MuRF1 results in thedevelopment of an exaggerated cardiac hypertrophy in re-sponse to pressure overload (49).
The present study was prompted by our observation thathuman cardiac tissue samples obtained from patients afterplacement of a LV assist device (LVAD) expressed signifi-cantly higher levels of MuRF1 protein than samples takenbefore the device was implanted. The unloading of the heart bya LVAD device leads to a decrease in the workload of the
Address for reprint requests and other correspondence: M. S. Willis, Dept.of Pathology and Laboratory Medicine, Carolina Cardiovascular BiologyCenter, Univ. of North Carolina, Medical Biomolecular Research Bldg., Rm.2336, 103 Mason Farm Rd., Chapel Hill, NC 27599-7525 (e-mail: [email protected]).
The costs of publication of this article were defrayed in part by the paymentof page charges. The article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Am J Physiol Heart Circ Physiol 296: H997–H1006, 2009.First published January 23, 2009; doi:10.1152/ajpheart.00660.2008.
0363-6135/09 $8.00 Copyright © 2009 the American Physiological Societyhttp://www.ajpheart.org H997
by 10.220.33.5 on August 19, 2016
http://ajpheart.physiology.org/D
ownloaded from

failing heart and a decrease in LV mass. This form of cardiacatrophy is beneficial to the patient as it decreases the stress onthe heart. Although previous studies have identified thatMuRF1 is essential for the inhibition of the development ofpathological cardiac hypertrophy, there is no evidence linkingMuRF1 to the reversal of hypertrophy (i.e., cardiac atrophy). Inthis report, we used two independent models to demonstratethat MuRF1 is a necessary and significant mediator in theregulation of cardiac atrophy in vivo.
MATERIALS AND METHODS
Animals. The MuRF1�/� mice used in these experiments havepreviously been described (3, 49).
Human cardiac LVAD samples. MuRF1 protein levels were deter-mined from heart samples from patients undergoing cardiac unloadingby the placement of a LVAD. Heart samples were collected frompatients with end-stage ischemic heart disease who received a LVADfor decompensated heart failure as a bridge to transplantation aspreviously described (40, 53). During the placement of the LVAD, acore of tissue is removed to prepare the LV for LVAD inflow. Thisspecimen was collected at the time of LVAD placement (pre-LVADsample), snap frozen in liquid nitrogen, and stored at �80°C. Whenpatients returned for a heart transplant, ventricular samples wereexcised just adjacent to the LVAD placement site (post-LVAD sam-ple), snap frozen, and stored at �80°C. Use of the human tissue usedin this study was approved by the University of North CarolinaInstitution Review Board (no. 03-1359).
Mouse cardiac hypertrophy reversal. Twelve to fifteen-week-oldMuRF1�/� and wild-type (WT) mice were subjected to the reversible(slipknot) transaortic constriction (TAC) procedure recently describedby our laboratory (42). All experiments used �50% male and 50%female MuRF1�/� mice and WT littermate controls. All animalprotocols were reviewed by the University of North Carolina Institu-tional Animal Care Advisory Committee and were in compliance withthe rules governing animal use published by the National Institutes ofHealth.
Dexamethasone model of cardiac atrophy. MuRF1�/� and WTlittermates were maintained on a standard diet with unlimited access towater. Dexamethasone (5 mg �kg�1 �day�1) or saline vehicle was givenby a daily subcutaneous injection for 2 wk as previously described (1,16). In additional experiments, dexamethasone (1 mg �kg�1 �day�1) orDMSO vehicle was continuously infused by a dorsally implanted osmoticminipump (model 2002, Alzet, Palo Alto, CA) for 2 wk. Echocardiog-raphy was performed at baseline and after 2 wk of dexamethasonetreatment.
Echocardiography. Echocardiography on mice was performed on aVisualSonics Vevo 660 ultrasound biomicroscopy system as previ-ously described (49).
Hemodynamic assessment of aorta flow. To assess the aorticconstriction after TAC and TAC reversal, right and left carotid arteryflow were assessed using a 20-MHz probe driven by a high-frequencypulsed Doppler signal processing workstation (Induc Instruments,Houston, TX) as previously described (42).
Total RNA isolation/real-time PCR determination of mRNA expres-sion. Total RNA was isolated from cardiac ventricular tissue aspreviously described (49). mRNA expression was determined using atwo-step reaction. cDNA was made using a High Capacity cDNAArchive kit (Applied Biosystems, Foster City, CA). PCR productswere amplified on an ABI Prism 7900HT Sequence Detection Systemusing cDNA and either 1) the TaqMan probe set in TaqMan UniversalPCR Master Mix or 2) unlabeled primers in Power CYBR GreenMaster Mix. The TaqMan probes used in these experiments includedbrain natriuretic peptide (BNP; Mm00435304_g1), smooth muscle�-actin (Mm00808218_g1), �-MHC (Mm00600555_m1), MuRF1(Mm01188690_m1), MuRF2 (Mm01292963_g1), atrogin-1/ muscle
atrophy F box/F box only protein 32 (Mm00499518_m1), and 18S(Hs99999901_s1) (Applied Biosystems). The unlabeled primers formouse tissue inhibitor of metalloproteinase (TIMP)-1, TIMP-2, ma-trix metalloproteinase (MMP)-2, pro-collagen type I (ColI), pro-collagen type III (ColIII), laminin B (Lamb), and GAPDH were aspreviously published (43). Samples were run in triplicate, and relativemRNA expression was determined using 18S (TaqMan Probes) orGAPDH (unlabeled primers) as internal endogenous controls.
Histology and lectin staining. Hearts were perfused, processed forhistology, and stained with hematoxylin and eosin, trichrome, orTriticum vulgaris lectin TRITC conjugate as previously described(49). Myocyte area was determined using NIH ImageJ (version1.38X) based on photomicrographs of a standard graticle ruler.
Western immunoblots. Human ventricular (50 �g) samples wereprepared in denaturing sample loading buffer, separated by 8% SDS-PAGE, and transferred to a polyvinylidene difluoride membrane. Themembrane was incubated overnight at 4°C in 5% milk and Tris-buffered saline-Tween with polyclonal goat anti-MuRF1 antibody(NB100-2406, Novus Biologicals, Littleton, CO). The membrane waswashed, incubated with horseradish peroxidase (HRP)-conjugatedanti-goat antibody (sc-2768, Santa Cruz Biotechnology, Santa Cruz,CA), and washed again. The HRP signal was then detected using theECL Plus Western Blotting system (RPN2132, GE Healthcare) ac-cording to the manufacturer’s protocol.
Statistical analysis. One-way ANOVA or Student’s t-test wasperformed using Sigma Stat 3.5 (Systat Software, San Jose, CA) andbasic statistics on Microsoft Excel 2007 (Microsoft, Seattle, WA).Results are expressed as means � SE, with statistical significancedefined as P � 0.05.
RESULTS
Cardiac MuRF1 increases after cardiac unloading. Theplacement of a LVAD in patients waiting for a heart transplanthelps improve cardiac output by taking over the pumping ofblood, effectively unloading the work the heart has to do. Anumber of studies have reported that LVAD-induced un-loading results in beneficial cardiac atrophy, which is evi-denced by a reduction in LV mass (4, 31, 40, 53). In thisstudy, we collected samples from six patients before andafter the placement of a LVAD, including two patients withmatched consecutive samples (Fig. 1A). Post-LVAD levelsof cardiac MuRF1 protein were significantly elevated(�60%) compared with MuRF1 protein levels found insamples taken pre-LVAD (Fig. 1B). In the two matchedsamples, post-LVAD cardiac MuRF1 levels increased 52%and 39% from pre-LVAD levels taken from adjacent tissue(Fig. 1A). This suggested that MuRF1 was intrinsicallyinvolved in the cardiac atrophy resulting from LVAD-induced cardiac unloading. This result led us to propose thehypothesis that MuRF1 is involved in the regulation ofcardiac atrophy. To test this hypothesis, we challengedMuRF1�/� mice to two independent models of cardiacatrophy: 1) cardiac hypertrophy regression after reversal ofTAC and 2) dexamethasone-induced atrophy.
MuRF1 is necessary for the reversal of pathological cardiachypertrophy in vivo. We (49) have recently identified thathearts taken from adult MuRF1�/� mice are indistinguishablefrom WT littermate controls. We (49) also discovered thatMuRF1�/� mice develop an exaggerated cardiac hypertrophyafter the induction of pressure overload by TAC, suggestingthat MuRF1 antagonizes the development of pathological car-diac hypertrophy. In the present study, we investigated the rolethat MuRF1 plays in the cardiac atrophy that is seen with the
H998 MURF1 MEDIATES CARDIAC ATROPHY IN VIVO
AJP-Heart Circ Physiol • VOL 296 • APRIL 2009 • www.ajpheart.org
by 10.220.33.5 on August 19, 2016
http://ajpheart.physiology.org/D
ownloaded from

reversal of experimentally induced pathological cardiac hyper-trophy. MuRF1�/� and WT control mice underwent reversibleTAC, which was released 4 wk later. The ability of MuRF1�/�
mice to decrease cardiac mass after TAC release was thendetermined by echocardiography, histology, and gene expres-
sion analyses. By calculating the LV mass index by echocar-diography, we discovered that 4 wk after TAC, the increase incardiac mass in MuRF1�/� mice was �2.4-fold higher than theincrease seen in WT mice (71.8% vs. 29.4% from baseline,respectively; Fig. 2A). After TAC release [the success of which
Fig. 1. Expression of cardiac muscle ringfinger-1 (MuRF1) after unloading of humanand mouse hearts. A: cardiac MuRF1 proteinlevels increased after mechanical unloadingwith a left ventricular (LV) assist device(LVAD). n � 6 (with 2 of the 4 samplesmatched), all run on the same gel. LV massdetermination from an echocardiography M-mode analysis taken from patient 2 and pa-tient 4 demonstrated a change of 56.3 g(226.8 g pre-LVAD and 170.5 g post-LVAD) and 26.4 g (187.3 g pre-LVAD and160.9 g post-LVAD), respectively. B: densi-tometric analysis of MuRF1 immunoblots(IB). Student’s t-test was performed to com-pare patient groups. *P � 0.009.
Fig. 2. Histological and mass analysis ofMuRF1�/� and wild-type (WT) mice after 4wk of transaortic constriction (TAC) and 4wk after TAC reversal (Rev/R). A: echocar-diographic determination of LV mass. BL,baseline; Max, maximum. n � 3–25 mice/group. B: actual determination of heartweight/body weight. n � 3–7 mice/group.Arrows indicate the reversal of TAC. C andD: representative histological cross sectionsat low power (C; hematoxylin and eosinstained; magnification: 0.7) and highpower (D; Masson’s trichrome stained; mag-nification: 20). In C, the histological anal-ysis was representative of 2–3 individualmice/group.
H999MURF1 MEDIATES CARDIAC ATROPHY IN VIVO
AJP-Heart Circ Physiol • VOL 296 • APRIL 2009 • www.ajpheart.org
by 10.220.33.5 on August 19, 2016
http://ajpheart.physiology.org/D
ownloaded from

was confirmed by Doppler flow experiments (Table 1) andhistological analysis (Supplemental Fig. 1)],1 the LV massindex of WT mice regressed to baseline within 4 days. Asignificant decrease in cardiac mass was also seen inMuRF1�/� mice 4 days after TAC release; however, this rateof cardiac atrophy was not maintained. Four weeks after TACrelease, the cardiac mass of MuRF1�/� mice remained 37.6%higher than the pre-TAC baseline. The inability of MuRF1�/�
mice to fully reverse the TAC-induced hypertrophy was con-firmed by the determination of the actual whole heart mass inrepresentative mice (Fig. 2B). As with the LV mass index,MuRF1�/� hearts exhibited an exaggerated total cardiac massafter 4 wk of TAC (60.4%) compared with WT mice (35.6%;Fig. 2B). However, whereas the total cardiac mass returned tobaseline levels in WT mice by 4 days after TAC release,MuRF1�/� mice decreased their mass by only �50% (60.4%vs. 35.7%) and remained at 32.5% greater mass than WT heartsafter 4 wk of TAC release (Fig. 2B). This response to TAC andTAC release was also seen by histological analyses (Fig. 2, Cand D).
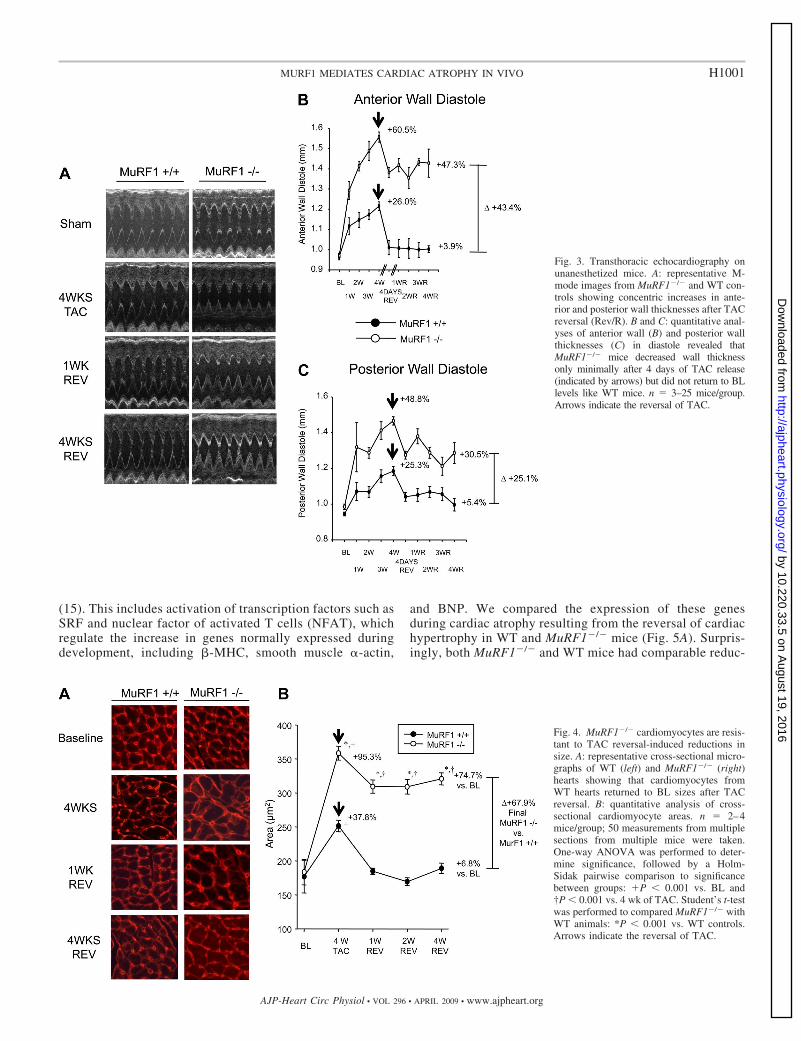
MuRF1�/� mice retain increases in wall thicknesses andcardiomyocyte size after TAC release. To ascertain if thesustained increase in the LV mass index seen in MuRF1�/�
mice after TAC release correlated with a lack of cardiac wallatrophy, we analyzed serial echocardiographic images of an-terior and posterior ventricular walls in WT and MuRF1�/�
mice before and after TAC release (Fig. 3). M-mode imagingidentified an increase in both anterior and posterior wall thick-nesses in WT and MuRF1�/� mice after 4 wk of TAC (Fig.3A). Quantitative analysis of wall thickness in diastole dem-onstrated that MuRF1�/� mice increased anterior wall thick-ness 60.5% from baseline, which was �2.3-fold higher thanthe increase identified in WT mice (26.0%; Fig. 3B). Similarly,posterior wall thickness in MuRF1�/� mice increased 48.8%
from baseline values, which was 1.9-fold higher than theincrease identified in WT mice (25.3%; Fig. 3C). Consistentwith our LV mass index results, anterior and posterior wallthicknesses in WT mice returned to baseline levels 4 days afterTAC release. However, in MuRF1�/� mice, decreases of47.3% and 30.5% (anterior and posterior wall thicknesses indiastole, respectively) occurred after 4 days post-TAC releasebut did not decrease further in the ensuing 4 wk. Surprisingly,during the TAC release time course, heart rate, percent frac-tional shortening, percent ejection fraction, and LV interven-tricular distance did not differ between MuRF1�/� and WTmice (Supplemental Fig. 2), indicating that neither MuRF1�/�
nor WT heart function deteriorated during cardiac hypertrophyinduction or after TAC release.
To determine whether the maintained cardiac wall thicknessand mass seen in MuRF1�/� mice after TAC release repre-sented a sustained increased in cardiomyocyte size, we nextanalyzed the cross-sectional area of cardiomyocytes from WTand MuRF1�/� hearts (Fig. 4). Representative cross-sectionalareas demonstrated that hearts lacking MuRF1 had an exag-gerated increase in cardiomyocyte size compared with WTmice after TAC (Fig. 4A, 4 wk). Quantitative analysis of thecross-sectional areas revealed that MuRF1�/� cardiomyocytesincreased their cross-sectional areas 95.3%, �2.5 times theincrease in cross-sectional areas of WT mice at 4 wk of TAC(37.8%; Fig. 4B). In WT mice, the individual cardiomyocytecross-sectional area decreased to baseline levels by 1 wk afterTAC release, whereas at the same time point, MuRF1�/�
cardiomyocyte cross-sectional area had decreased by only20.6% (Fig. 4B). No further decrease in cardiomyocyte cross-sectional area was seen in MuRF1�/� hearts for the remainderof the TAC release period (Fig. 4A, 1 wk Rev and 4 wkRev). A confounding issue with these findings is thatMuRF1�/� hearts have an exaggerated cardiac hypertrophy,which proportionally decreases to the same extent as WTmice (�30%; Fig. 2A). By several measures, including heartweight (Fig. 2) and wall thickness (Fig. 3), MuRF1�/� micehypertrophy to nearly the same extent after 1 wk of TAC asWT mice do after 4 wk of TAC. To more clearly delineatethe role of MuRF1 in pathological cardiac hypertrophyreversal, MuRF1�/� mice underwent TAC for 1 wk toachieve comparable cardiac hypertrophy to WT mice after 4wk. The TAC was then released, and the degree of cardiacwall thickness was followed by echocardiography and his-tology (Supplemental Fig. 3 and Supplemental Table 1).Surprisingly, little if any decrease in anterior and posteriorwall thicknesses was detected by echocardiography (Sup-plemental Fig. 3, A and B). Histological analysis of cardio-myocyte cross-sectional areas demonstrated a 9.6% de-crease in size 7 days after the release of TAC (SupplementalFig. 3, C and D). This contrasts to the 100% decrease incardiac hypertrophy identified in WT mice 7 days afterrelease of TAC and comparable cardiac hypertrophy (Figs.2– 4). These results demonstrate that MuRF1 is involved inregulating the decrease in cardiomyocyte mass during car-diac atrophy associated with hypertrophy regression.
Lack of MuRF1 does not affect the suppression of hypertro-phy-associated transcriptional activity after debanding. Thedevelopment of pressure overload-induced pathological car-diac hypertrophy is associated with signaling processes thatresult in the activation of distinct transcriptional programs
1 Supplemental material for this article is available online at the AmericanJournal of Physiology-Heart and Circulatory Physiology website.
Table 1. Physiological assessment of aortic constrictionat baseline and after transaortic constriction reversalby right and left carotid blood flow determination bypulse-Doppler analysis
Peak RightCarotid ArteryVelocity, m/s
Peak Left CarotidArtery Velocity,
m/s
Peak Right/LeftCarotid Artery
Velocity
Prebanding baselineWT 42.1�6.3 40.8�5.5 1.0�0.5MuRF1�/� 50.5�6.2 48.1�2.4 1.0�0.4
Postbanding baselineWT 21.7�9.3 91.0�8.9 4.2�0.6*MuRF1�/� 25.7�13.7 98.4�6.6 3.8�1.2*
4 days postdebandingWT 63.9�8.5 31.2�9.6 2.6�0.4†MuRF1�/� 75.4�7.3 80.6�5.6 1.2�0.5
1 wk postdebandingWT 57.8�6.8 58.3�3.0 0.9�0.8MuRF1�/� 67.2�6.6 61.8�5.5 1.1�0.5
Values are means � SE; n � 3 mice/group. Carotid Doppler velocities wereperformed at baseline and 4 and 7 days after mice had been debanded. WT, wildtype; MuRF1, muscle ring finger-1. *P � 0.001 compared with baseline; †P �0.001 compared with postdebanded MuRF1�/� and baseline WT peak velocities.
H1000 MURF1 MEDIATES CARDIAC ATROPHY IN VIVO
AJP-Heart Circ Physiol • VOL 296 • APRIL 2009 • www.ajpheart.org
by 10.220.33.5 on August 19, 2016
http://ajpheart.physiology.org/D
ownloaded from
(15). This includes activation of transcription factors such asSRF and nuclear factor of activated T cells (NFAT), whichregulate the increase in genes normally expressed duringdevelopment, including �-MHC, smooth muscle �-actin,
and BNP. We compared the expression of these genesduring cardiac atrophy resulting from the reversal of cardiachypertrophy in WT and MuRF1�/� mice (Fig. 5A). Surpris-ingly, both MuRF1�/� and WT mice had comparable reduc-
Fig. 3. Transthoracic echocardiography onunanesthetized mice. A: representative M-mode images from MuRF1�/� and WT con-trols showing concentric increases in ante-rior and posterior wall thicknesses after TACreversal (Rev/R). B and C: quantitative anal-yses of anterior wall (B) and posterior wallthicknesses (C) in diastole revealed thatMuRF1�/� mice decreased wall thicknessonly minimally after 4 days of TAC release(indicated by arrows) but did not return to BLlevels like WT mice. n � 3–25 mice/group.Arrows indicate the reversal of TAC.
Fig. 4. MuRF1�/� cardiomyocytes are resis-tant to TAC reversal-induced reductions insize. A: representative cross-sectional micro-graphs of WT (left) and MuRF1�/� (right)hearts showing that cardiomyocytes fromWT hearts returned to BL sizes after TACreversal. B: quantitative analysis of cross-sectional cardiomyocyte areas. n � 2–4mice/group; 50 measurements from multiplesections from multiple mice were taken.One-way ANOVA was performed to deter-mine significance, followed by a Holm-Sidak pairwise comparison to significancebetween groups: P � 0.001 vs. BL and†P � 0.001 vs. 4 wk of TAC. Student’s t-testwas performed to compared MuRF1�/� withWT animals: *P � 0.001 vs. WT controls.Arrows indicate the reversal of TAC.
H1001MURF1 MEDIATES CARDIAC ATROPHY IN VIVO
AJP-Heart Circ Physiol • VOL 296 • APRIL 2009 • www.ajpheart.org
by 10.220.33.5 on August 19, 2016
http://ajpheart.physiology.org/D
ownloaded from

tions in all three fetal genes examined after TAC release,indicating that the genes associated with pathological car-diac hypertrophy were similarly inactivated in bothMuRF1�/� and WT hearts after unloading of the heart (Fig.5A). This result suggests that the sustained cardiac hyper-trophy seen in MuRF1�/� mice after TAC release is not dueto continued prohypertrophic transcriptional programs andmay instead be linked to impairments in mechanisms asso-ciated with cardiac muscle mass reduction.
Genes associated with cardiac remodeling do not increasein MuRF1�/� mice after TAC release. A clinical study (47) hasdemonstrated that the development of pathological cardiac hyper-trophy and subsequent therapeutic atrophy is accompanied bychanges in the cardiac extracellular matrix (ECM). The ECM is anetwork of collagens that sustains myocyte structure and function.
The balance of collagen turnover is controlled by specific MMPsand inhibitors of MMPS called TIMPs. During LV atrophyassociated with surgical repair of aortic stenosis, increases inMMP-2, TIMP-1, and TIMP-2 have been reported (47). In thepresent study, we investigated the remodeling mechanism inMuRF1-/- hearts by determining the mRNA levels of proteinsassociated with regulation of the cardiac ECM, including pro-collagen I, procollagen III, and laminin. We also investigated themRNA levels of enzymes that degrade these proteins, includingMMP-2, MMP-13, TIMP-1, and TIMP-2 (Fig. 5B). At 4 daysafter TAC release, WT mice expressed higher levels of cardiacTIMP-1, TIMP-2, MMP-2, and ColI compared with MuRF1�/�
mice. We did not identify increases (or differences) in ColIII,Lamb, or MMP-13 during atrophy 4 or 7 days after TAC releasein MuRF1�/� or WT mice (data not shown). These findings
Fig. 5. Quantitative real-time PCR analysis of mRNA at baseline, after 4 wk of TAC, and 4 and 7 days after TAC reversal. A: expression of genes involved incardiac hypertrophy [�-myosin heavy chain (MHC), smooth muscle �-actin, and brain natriuretic peptide (BNP)]. B: expression of genes associated with cardiacremodeling [tissue inhibitor of metalloproteinase (TIMP)-1, TIMP-2, matrix metalloproteinase (MMP)-2, and pro-collagen I]. One-way ANOVA was performedto determine significance, followed by a Holm-Sidak pairwise comparison to significance between groups: §P � 0.05 vs. WT mice at 4 wk of TAC; †P � 0.05vs. WT and MuRF1�/� at BL, 4 days of TAC reversal, and 7 days TAC reversal; and *P � 0.001 vs. all other groups. Arrows indicate the reversal of TAC.
H1002 MURF1 MEDIATES CARDIAC ATROPHY IN VIVO
AJP-Heart Circ Physiol • VOL 296 • APRIL 2009 • www.ajpheart.org
by 10.220.33.5 on August 19, 2016
http://ajpheart.physiology.org/D
ownloaded from

indicate that the expression of some genes associated with theECM remodeling that accompanies atrophy associated withpathological cardiac hypertrophy regression (i.e., TIMP-1 andTIMP-2) is attenuated in mice lacking MuRF1, which may beassociated with the apparent inability of MuRF1�/� cardiomyo-cytes to decrease in size after TAC release.
MuRF1 mediates the cardiac atrophy induced by dexametha-sone treatment. The experiments described above point to acritical role of MuRF1 in mediating cardiac atrophy associatedwith the regression of pathological hypertrophy. However,there has been some debate as to whether or not the process ofhypertrophy regression involves the same cellular mechanicsas pure muscle atrophy, that is, a decrease in muscle mass froma steady-state level. To test whether or not the effects ofMuRF1 described above are specific to pathological hypertro-phic regression, we also tested whether or not MuRF1 isinvolved in the cardiac atrophy induced by chronic dexametha-sone treatment. A previous study (7) has already demonstratedthat MuRF1 plays a significant role in mediating dexametha-
sone-induced skeletal muscle atrophy. We tested the effect ofchronic dexamethasone treatment on cardiac wall thickness inWT and MuRF1�/� mice. Daily dexamethasone injectionswere given to mice for 2 wk and followed by echocardiographyand histological analysis. When sham mice (saline injectionsonly) were compared with WT mice receiving dexamethasonetreatment, significant decreases in anterior and posterior wallthicknesses were identified (Fig. 6, A–C), with parallel de-creases in cardiomyocyte cross-sectional areas (Fig. 6, D andE). However, MuRF1�/� animals were resistant to dexameth-asone-induced cardiac atrophy and appeared to trend towardincreased cardiac mass by heart weight/body weight measures(Table 2). In parallel experiments, osmotic pumps that releaseddexamethasone were implanted dorsally and left in place for 2wk (7), followed by cardiac wall thickness assessment byechocardiography. When sham mice (osmotic pumps releasingvehicle only) were compared with WT mice undergoing dexa-methasone treatment, significant decreases in anterior andposterior wall thicknesses in dexamethasone-treated animals
Fig. 6. Histological and echocardiographic analysis of MuRF1�/� mice after daily treatment with subcutaneous dexamethasone (Dex) treatment.A: representative histological cross sections at low power (hematoxylin and eosin stained; magnification: 0.7). B: quantitative analysis of anterior and posteriorwall thicknesses in diastole revealed that MuRF1�/� mice were resistant to Dex-induced wall thinning. C: representative M-mode images of mouse hearts after2 wk of sham or Dex treatment. D: quantitative analysis of cross-sectional cardiomyocyte areas; 100 measurements from multiple sections from multiple micewere taken with representative histological sections shown in E. Histological analysis was representative of 2–3 individual mice/group. One-way ANOVA wasperformed to determine significance, followed by a Holm-Sidak pairwise comparison to significance between groups: *P � 0.05 vs. all other groups.
H1003MURF1 MEDIATES CARDIAC ATROPHY IN VIVO
AJP-Heart Circ Physiol • VOL 296 • APRIL 2009 • www.ajpheart.org
by 10.220.33.5 on August 19, 2016
http://ajpheart.physiology.org/D
ownloaded from

were identified, as anticipated (Supplemental Fig. 4), alongwith cardiac dilation (Supplemental Table 2). In contrast,MuRF1�/� mice were resistant to dexamethasone-induced car-diac atrophy, as indicated by little or no changes in anterior andposterior wall thicknesses in diastole (Supplemental Fig. 4) orchamber dilation (Supplemental Table 2). Although it wasnoted that the osmotic pump experiments demonstrated agreater overall atrophy than the daily dexamethasone injec-tions, technical issues with wound dehiscence (likely due to thedexamethasone treatment) made further analysis of this obser-vation impractical. These findings suggest that MuRF1 medi-ates atrophy in the dexamethasome model in the same mannerthat it does in the atrophy associated with pathological cardiachypertrophy regression. MuRF1 may therefore play a moregeneralized role in decreasing cardiac muscle mass in a varietyof clinical scenarios.
DISCUSSION
The development of pathological cardiac hypertrophy is acommon precursor to heart failure and heightens the risk ofheart failure and arrhythmias. Not surprisingly, the reversal ofpathological cardiac hypertrophy reduces these risks and istherefore an attractive process against which to target potentialtherapies. In the present study, we used two models of cardiacatrophy to investigate the role of MuRF1 in decreasing cardiacmuscle mass in vivo. The concept of cardiac atrophy in thepresent study has been expanded from muscle mass loss frombaseline levels to the decrease in muscle mass seen in thetherapeutic regression of hypertrophic states. Our results dem-onstrate that MuRF1 is an essential mediator of the cardiacatrophy associated with both regression of TAC-inducedpathological hypertrophy as well as the atrophy resulting fromchronic dexamethasone treatment. Together, these findingsdemonstrate, for the first time, a major role for MuRF1 in theprocess of reducing cardiac muscle mass in vivo. As preclinicaltrials have demonstrated the value of blunting hypertrophicgrowth without compromising cardiac performance, the poten-tial for antihypertrophy therapy has been suggested (19, 20).This strongly supports the notion of exploring MuRF1 as a
useful therapeutic target in the quest to improve clinical out-comes and prevent heart failure in a wide range of patients.
While the reversal of pathological cardiac hypertrophy thatoccurs after the removal of pressure overload appears toinvolve a reduction in cardiomyocyte size, it is only one aspectof a broader process that involves the restoration of diastolicfunction and remodeling of the ECM. In clinical scenarioswhere high blood pressure is adequately treated or aorticstenosis is surgically repaired, reversal of pathological cardiachypertrophy occurs in patients. In this situation, several studieshave reported that regression of pathological cardiac hypertro-phy parallels improvements in diastolic dysfunction (12, 22,30, 46). A balance of degradation by MMPs and TIMPs in theECM is vital to the remodeling process that occurs duringhypertrophy regression. In failing hearts, alterations in thebalance of MMPs and their endogenous inhibitors (TIMPs)have been reported. The regulation of MMPs and TIMPsduring the regression of pathological cardiac hypertrophy,which occurs in as little as 4 days in the present study, has notbeen previously reported. We identified that WT mice had anexpected transient increase in MMP-2, TIMP-1, TIMP-2, andColI mRNA levels 4 days after TAC release. In contrast, thesedid not change in MuRF1�/� mice. While we don’t necessarilybelieve that MuRF1 has a direct effect on ECM regulation,MuRF1’s effect on cardiomyocyte size might allow it toindirectly affect the ECM.
In the present study, we identified, for the first time, thatdexamethasone treatment in adult mice leads to a reduction incardiac mass. This should be contrasted to the effects that dexa-methasone has on the heart in both human and experimentalneonates. Dexamethasone therapy in neonates for bronchpulmo-nary dysplasia, premature birth, and chronic lung disease has beenreported to be associated with increased cardiac mass repeatedly(23, 38, 41, 48, 52). In a randomized clinical trial, a common sideeffect of dexamethasone therapy is cardiac hypertrophy aftertherapy for as little as 7 days, resulting in clinically significantsymptoms (54). Therefore, the effects of dexamethasone-inducedcardiac atrophy reported in the present study are observationslikely confined to adult mouse hearts. The role of MuRF1 in this
Table 2. Transthoracic echocardiography on unanesthetized MuRF1�/� and WT control mice after 2 wk of dailysubcutaneous saline or dexamethasone treatment
Saline Treatment Dexamethasone Treatment
WT MuRF1�/� WT MuRF1�/�
n 9 9 8 6Heart rate, beats/min 581.3�22.5 614.9�12.1 585.8�15.7 596.8�13.1Anterior wall at diastole, mm 1.200.02 1.260.02 1.03�0.02* 1.26�0.04Posterior wall at diastole, mm 1.180.04 1.190.01 0.97�0.01* 1.18�0.01Anterior wall at systole, mm 1.830.04 1.960.05 1.43�0.03 1.86�0.02Posterior wall at systole, mm 1.700.04 1.580.03 1.44�0.03* 1.67�0.03LVEDD 2.80.4 3.40.3 3.00.1 3.20.1LVESD 1.30.2 1.50.2 1.40.1 1.50.1LV mass/body weight, mg/g 3.990.19 4.040.19 3.32�0.12* 5.47�0.18*LV mass/tibia length, mg/mm 6.880.34 7.430.40 5.87�0.22* 9.06�0.41*Heart weight/body weight, mg/g 5.200.28 5.740.25 4.18�0.07* 6.01�0.25Fractional shortening, % 54.51.0 55.00.9 52.8�1.8 54.8�0.8Ejection fraction, % 86.50.90 86.50.7 84.7�1.5 86.4�0.6
Values are means � SE; n, no. of mice/group. LVEDD, left ventricular (LV) end-diastolic dimension; LVESD, LV end-systolic dimension. LV mass wascalculated as follows: (external LV diameter in diastole3 � LVEDD3) 1.055. Fractional shortening was calculated as follows: (LVEDD � LVESD)/LVEDD 100. The ejection fraction was calculated as follows: (end Simpson’s diastolic volume � end Simpson’s systolic volume)/end Simpson’s diastolic volume 100.*P � 0.05 vs. all other groups.
H1004 MURF1 MEDIATES CARDIAC ATROPHY IN VIVO
AJP-Heart Circ Physiol • VOL 296 • APRIL 2009 • www.ajpheart.org
by 10.220.33.5 on August 19, 2016
http://ajpheart.physiology.org/D
ownloaded from

age-dependent effect in cardiac mass is tantalizing, however,beyond the scope of the present study.
MuRF1 is found exclusively in skeletal muscle and the heartand has been localized to the M-line of the sarcomere (3, 5).Structurally, MuRF1 contains a ring finger domain, a motifknown to have ubiquitin ligase activity. Ubiquitin ligases interactwith ubiquitin-activating and ubiquitin-conjugating enzymes toplace ubiquitin chains on substrates to be recognized and degradedby the 26S proteasome (50). Our laboratory was the first toidentify that MuRF1 is a bona fide ubiquitin ligase capableof interacting with cardiac troponin I and tagging it for degrada-tion in a proteasome-dependent manner (24). Other investigatorshave identified that MuRF1 interacts with additional sarcomericproteins including telethonin and myotilin (51) as well as �-MHC,which is degraded in vivo by MuRF1 (13). In the present study,mice deficient for MuRF1 exhibited a significant attenuation incardiac atrophy after TAC release, implicating MuRF1 as a keyplayer in the sarcomeric degradation that occurs after cardiacunloading. The fact that MuRF1�/� hearts displayed some degreeof cardiac atrophy after TAC release suggests that MuRF1 is notthe only ubiquitin ligase operating in this process. A previousstudy (3) has demonstrated that the muscle-specific ubiquitinligase atrogin-1, in addition to MuRF1, is capable of regulatingthe decrease in skeletal muscle mass that occurs during atrophy.When we examined the expression of both MuRF1 and atrogin-1 inWT mice in our model system, we found that both proteinswere increased after the induction of TAC (Supplemental Fig.5, A and B). The increase in atrogin-1 expression was compa-rable between MuRF1�/� and WT mice during cardiac atrophyafter TAC release; however, the levels of MuRF2, a relatedMuRF family member, did not significantly change in eitherMuRF1�/� or WT mice after pathological cardiac hypertrophy(Supplemental Fig. 5C). The robust increase in atrogin-1 dur-ing cardiac atrophy associated with hypertrophy regression inMuRF1�/� mice could account for the small amount of hyper-trophic regression seen in these mice during the first week afterTAC release.
In the heart, MuRF1 was initially identified as a protein thatinhibits the development of pathological cardiac hypertrophy byblocking PKC-ε signaling and degrading cardiac troponin I (2,24). Consistent with this described role, we (49) have previouslydemonstrated that MuRF1�/� mice exhibit an amplified patho-logical cardiac hypertrophic response to TAC-induced pressureoverload and that this effect persists for up to 2 wk after TACinduction. The development of pathological cardiac hypertrophyinvolves enhanced protein synthesis by individual myocytes (8,33, 37). Recent studies (9, 11) have identified that, in addition toincreased protein synthesis, proteasomal activity and protein turn-over are also enhanced during the development of pathologicalcardiac hypertrophy. Given that the ubiquitin-proteasome systemdegrades as much as 30% of newly synthesized cellular proteins(17), it stands to reason that mice deficient in MuRF1, a ubiquitinligase known to interact with and degrade multiple sarcomericproteins, exhibit enhanced cardiac hypertrophy in response toTAC. Without MuRF1, a key component of the protein qualitycontrol system is missing in these mice, allowing for the extrav-agant build up of cardiac muscle with little or no protein degra-dation against which to balance.
We propose that one of MuRF1’s key roles in the heart is toregulate the development and maintenance of pathologicalcardiac hypertrophy, perhaps by actively degrading worn sar-
comeric proteins as part of a broader process of protein qualitycontrol. This would parallel MuRF1’s proposed role in theprotein quality control of creatine kinase, whereby oxidizedforms of creatine kinase are preferentially ubiquitinated andtargeted for degradation (27, 55). In addition to the role thatMuRF1 appears to play in preventing pathological cardiachypertrophy, we have also uncovered evidence in this studythat MuRF1 is critical to the process of cardiac atrophy,paralleling the results of another study (3) in which skeletalmuscle atrophy was induced in MuRF1�/� mice by limbdenervation. In that study (3), MuRF1�/� mice had a 36%sparing of muscle mass loss after denervation, indicating thatMuRF1 plays an active role in skeletal muscle atrophy.MuRF1’s ability to degrade specific sarcomeric proteins islikely the underlying process that mediates both cardiac andskeletal muscle atrophy in these studies.
ACKNOWLEDGMENTS
The authors acknowledge the assistance of Janice Weaver in the AnimalHistopathology Laboratory at the University of North Carolina for assistancein preparing histological specimens.
All work was performed at the University of North Carolina-Chapel Hill.
GRANTS
This work was supported by the University of North Carolina ResearchCouncil, the R. J. Reynolds Faculty Development Award from the Universityof North Carolina Foundation, an American Heart Association Scientist De-velopment Grant, the Children’s Cardiomyopathy Foundation (to M. S. Wil-lis), and National Heart, Lung, and Blood Institute Grant R01-HL-065619 (toC. Patterson).
REFERENCES
1. Agbenyega ET, Wareham AC. Effect of clenbuterol on skeletal muscleatrophy in mice induced by the glucocorticoid dexamethasone. CompBiochem Physiol Comp Physiol 102: 141–145, 1992.
2. Arya R, Kedar V, Hwang JR, McDonough H, Li HH, Taylor J,Patterson C. Muscle ring finger protein-1 inhibits PKCε activation andprevents cardiomyocyte hypertrophy. J Cell Biol 167: 1147–1159, 2004.
3. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA,Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valen-zuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ.Identification of ubiquitin ligases required for skeletal muscle atrophy.Science 294: 1704–1708, 2001.
4. Burkhoff D, Klotz S, Mancini DM. LVAD-induced reverse remodeling:basic and clinical implications for myocardial recovery. J Card Fail 12:227–239, 2006.
5. Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, BangML, Trombitas K, Granzier H, Gregorio CC, Sorimachi H, Labeit S.Identification of muscle specific ring finger proteins as potential regulatorsof the titin kinase domain. J Mol Biol 306: 717–726, 2001.
6. Cipriano C, Gosse P, Bemurat L, Mas D, Lemetayer P, N’Tela G,Clementy J. Prognostic value of left ventricular mass and its evolutionduring treatment in the Bordeaux cohort of hypertensive patients. Am JHypertens 14: 524–529, 2001.
7. Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, BurovaE, Rakhilin SV, Stitt TN, Patterson C, Latres E, Glass DJ. The E3ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab 6: 376–385, 2007.
8. Coleman PS, Parmacek MS, Lesch M, Samarel AM. Protein synthesisand degradation during regression of thyroxine-induced cardiac hypertro-phy. J Mol Cell Cardiol 21: 911–925, 1989.
9. Depre C, Wang Q, Yan L, Hedhli N, Peter P, Chen L, Hong C,Hittinger L, Ghaleh B, Sadoshima J, Vatner DE, Vatner SF, MaduraK. Activation of the cardiac proteasome during pressure overload pro-motes ventricular hypertrophy. Circulation 114: 1821–1828, 2006.
10. Devereux RB, Wachtell K, Gerdts E, Boman K, Nieminen MS, Papa-demetriou V, Rokkedal J, Harris K, Aurup P, Dahlof B. Prognosticsignificance of left ventricular mass change during treatment of hyperten-sion. JAMA 292: 2350–2356, 2004.
H1005MURF1 MEDIATES CARDIAC ATROPHY IN VIVO
AJP-Heart Circ Physiol • VOL 296 • APRIL 2009 • www.ajpheart.org
by 10.220.33.5 on August 19, 2016
http://ajpheart.physiology.org/D
ownloaded from

11. Dickhout JG, Austin RC. Proteasomal regulation of cardiac hypertrophy:is demolition necessary for building? Circulation 114: 1796–1798, 2006.
12. Dyadyk AI, Bagriy AE, Lebed IA, Yarovaya NF, Schukina EV,Taradin GG. ACE inhibitors captopril and enalapril induce regression ofleft ventricular hypertrophy in hypertensive patients with chronic renalfailure. Nephrol Dial Transplant 12: 945–951, 1997.
13. Fielitz J, Kim MS, Shelton JM, Latif S, Spencer JA, Glass DJ,Richardson JA, Bassel-Duby R, Olson EN. Myosin accumulation andstriated muscle myopathy result from the loss of muscle RING finger 1 and3. J Clin Invest 117: 2486–2495, 2007.
14. Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a newtherapeutic target? Circulation 109: 1580–1589, 2004.
15. Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly.Annu Rev Physiol 65: 45–79, 2003.
16. Gilson H, Schakman O, Combaret L, Lause P, Grobet L, Attaix D,Ketelslegers JM, Thissen JP. Myostatin gene deletion prevents glucocor-ticoid-induced muscle atrophy. Endocrinology 148: 452–460, 2007.
17. Goldberg AL. Protein degradation and protection against misfolded ordamaged proteins. Nature 426: 895–899, 2003.
18. Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventric-ular mass and hypertrophy are associated with increased risk for suddendeath. J Am Coll Cardiol 32: 1454–1459, 1998.
19. Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, WangZ, Kerber RE, Weiss RM. Cardiac hypertrophy is not a requiredcompensatory response to short-term pressure overload. Circulation 101:2863–2869, 2000.
20. Hill JA, Olson EN. Cardiac plasticity. N Engl J Med 358: 1370–1380, 2008.21. Hoshijima M. Mechanical stress-strain sensors embedded in cardiac
cytoskeleton: Z disk, titin, and associated structures. Am J Physiol HeartCirc Physiol 290: H1313–H1325, 2006.
22. Ikonomidis I, Tsoukas A, Parthenakis F, Gournizakis A, KassimatisA, Rallidis L, Nihoyannopoulos P. Four year follow up of aortic valvereplacement for isolated aortic stenosis: a link between reduction inpressure overload, regression of left ventricular hypertrophy, and diastolicfunction. Heart 86: 309–316, 2001.
23. Israel BA, Sherman FS, Guthrie RD. Hypertrophic cardiomyopathyassociated with dexamethasone therapy for chronic lung disease in preterminfants. Am J Perinatol 10: 307–310, 1993.
24. Kedar V, McDonough H, Arya R, Li HH, Rockman HA, Patterson C.Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degradescardiac troponin I. Proc Natl Acad Sci USA 101: 18135–18140, 2004.
25. Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH. Relation ofleft ventricular mass and geometry to morbidity and mortality in uncompli-cated essential hypertension. Ann Intern Med 114: 345–352, 1991.
26. Koren MJ, Ulin RJ, Koren AT, Laragh JH, Devereux RB. Leftventricular mass change during treatment and outcome in patients withessential hypertension. Am J Hypertens 15: 1021–1028, 2002.
27. Koyama S, Hata S, Witt CC, Ono Y, Lerche S, Ojima K, Chiba T, DoiN, Kitamura F, Tanaka K, Abe K, Witt SH, Rybin V, Gasch A, FranzT, Labeit S, Sorimachi H. Muscle RING-finger protein-1 (MuRF1) as aconnector of muscle energy metabolism and protein synthesis. J Mol Biol376: 1224–1236, 2008.
28. Krumholz HM, Larson M, Levy D. Prognosis of left ventricular geo-metric patterns in the Framingham Heart Study. J Am Coll Cardiol 25:879–884, 1995.
29. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognosticimplications of echocardiographically determined left ventricular mass inthe Framingham Heart Study. N Engl J Med 322: 1561–1566, 1990.
30. Loureiro J, Smith S, Fonfara S, Swift S, James R, Dukes-McEwan J. Caninedynamic left ventricular outflow tract obstruction: assessment of myocardialfunction and clinical outcome. J Small Anim Pract 49: 578–586, 2008.
31. Maybaum S, Mancini D, Xydas S, Starling RC, Aaronson K, PaganiFD, Miller LW, Margulies K, McRee S, Frazier OH, Torre-Amione G.Cardiac improvement during mechanical circulatory support: a prospec-tive multicenter study of the LVAD Working Group. Circulation 115:2497–2505, 2007.
32. McElhinny AS, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC.Muscle-specific RING finger-1 interacts with titin to regulate sarcomericM-line and thick filament structure and may have nuclear functions via itsinteraction with glucocorticoid modulatory element binding protein-1.J Cell Biol 157: 125–136, 2002.
33. Morgan HE, Kira Y, Gordon EE. Aortic pressure, substrate utilizationand protein synthesis. Eur Heart J 5, Suppl F: 141–146, 1984.
34. Mrosek M, Labeit D, Witt S, Heerklotz H, von Castelmur E, Labeit S,Mayans O. Molecular determinants for the recruitment of the ubiquitin-ligase MuRF-1 onto M-line titin. FASEB J 21: 1383–1392, 2007.
35. Muiesan ML, Salvetti M, Rizzoni D, Castellano M, Donato F, Agabiti-Rosei E. Association of change in left ventricular mass with prognosis duringlong-term antihypertensive treatment. J Hypertens 13: 1091–1095, 1995.
36. Okin PM, Devereux RB, Jern S, Kjeldsen SE, Julius S, Nieminen MS,Snapinn S, Harris KE, Aurup P, Edelman JM, Wedel H, LindholmLH, Dahlof B. Regression of electrocardiographic left ventricular hyper-trophy during antihypertensive treatment and the prediction of majorcardiovascular events. JAMA 292: 2343–2349, 2004.
37. Parmacek MS, Magid NM, Lesch M, Decker RS, Samarel AM. Cardiacprotein synthesis and degradation during thyroxine-induced left ventricularhypertrophy. Am J Physiol Cell Physiol 251: C727–C736, 1986.
38. Riede FT, Schulze E, Vogt L, Schramm D. Irreversible cardiac changesafter dexamethasone treatment for bronchopulmonary dysplasia. PediatrCardiol 22: 363–364, 2001.
39. Schillaci G. Pharmacogenomics of left ventricular hypertrophy reversal:beyond the “one size fits all” approach to antihypertensive therapy.J Hypertens 22: 2273–2275, 2004.
40. Selzman CH, Sheridan BC. Off-pump insertion of continuous flow leftventricular assist devices. J Card Surg 22: 320–322, 2007.
41. Skelton R, Gill AB, Parsons JM. Cardiac effects of short course dexa-methasone in preterm infants. Arch Dis Child Fetal Neonatal Ed 78:F133–F137, 1998.
42. Stansfield WE, Rojas M, Corn D, Willis M, Patterson C, Smyth SS,Selzman CH. Characterization of a model to independently study regres-sion of ventricular hypertrophy. J Surg Res 142: 387–393, 2007.
43. Ueberham E, Low R, Ueberham U, Schonig K, Bujard H, GebhardtR. Conditional tetracycline-regulated expression of TGF-beta1 in liver oftransgenic mice leads to reversible intermediary fibrosis. Hepatology 37:1067–1078, 2003.
44. Verdecchia P, Angeli F, Borgioni C, Gattobigio R, de Simone G,Devereux RB, Porcellati C. Changes in cardiovascular risk by reductionof left ventricular mass in hypertension: a meta-analysis. Am J Hypertens16: 895–899, 2003.
45. Verdecchia P, Schillaci G, Borgioni C, Ciucci A, Gattobigio R, Zampi I,Reboldi G, Porcellati C. Prognostic significance of serial changes in leftventricular mass in essential hypertension. Circulation 97: 48–54, 1998.
46. Villari B, Vassalli G, Monrad ES, Chiariello M, Turina M, Hess OM.Normalization of diastolic dysfunction in aortic stenosis late after valvereplacement. Circulation 91: 2353–2358, 1995.
47. Walther T, Schubert A, Falk V, Binner C, Kanev A, Bleiziffer S,Walther C, Doll N, Autschbach R, Mohr FW. Regression of leftventricular hypertrophy after surgical therapy for aortic stenosis is asso-ciated with changes in extracellular matrix gene expression. Circulation104: I54–58, 2001.
48. Werner JC, Sicard RE, Hansen TW, Solomon E, Cowett RM, Oh W.Hypertrophic cardiomyopathy associated with dexamethasone therapy forbronchopulmonary dysplasia. J Pediatr 120: 286–291, 1992.
49. Willis MS, Ike C, Li L, Wang DZ, Glass DJ, Patterson C. Muscle ringfinger 1, but not muscle ring finger 2, regulates cardiac hypertrophy invivo. Circ Res 100: 456–459, 2007.
50. Willis MS, Schisler JC, Patterson C. Appetite for destruction: E3 ubiquitin-ligase protection in cardiac disease. Future Cardiol 4: 65–75, 2008.
51. Witt SH, Granzier H, Witt CC, Labeit S. MURF-1 and MURF-2 target aspecific subset of myofibrillar proteins redundantly: towards understandingMURF-dependent muscle ubiquitination. J Mol Biol 350: 713–722, 2005.
52. Yeh TF, Lin YJ, Hsieh WS, Lin HC, Lin CH, Chen JY, Kao HA,Chien CH. Early postnatal dexamethasone therapy for the prevention ofchronic lung disease in preterm infants with respiratory distress syndrome:a multicenter clinical trial. Pediatrics 100: E3, 1997.
53. Zafeiridis A, Jeevanandam V, Houser SR, Margulies KB. Regressionof cellular hypertrophy after left ventricular assist device support. Circu-lation 98: 656–662, 1998.
54. Zecca E, Papacci P, Maggio L, Gallini F, Elia S, De Rosa G, RomagnoliC. Cardiac adverse effects of early dexamethasone treatment in preterminfants: a randomized clinical trial. J Clin Pharmacol 41: 1075–1081, 2001.
55. Zhao TJ, Yan YB, Liu Y, Zhou HM. The generation of the oxidizedform of creatine kinase is a negative regulation on muscle creatine kinase.J Biol Chem 282: 12022–12029, 2007.
H1006 MURF1 MEDIATES CARDIAC ATROPHY IN VIVO
AJP-Heart Circ Physiol • VOL 296 • APRIL 2009 • www.ajpheart.org
by 10.220.33.5 on August 19, 2016
http://ajpheart.physiology.org/D
ownloaded from




















