
Monosaccharides bound to hemoglobin in normal and diabetic individuals

www.elsevier.com/locate/ybbrc
Biochemical and Biophysical Research Communications 355 (2007) 409–418
Monosaccharides modulate HCV E2 protein-derived peptidebiological properties
Javier E. Garcia a,b, Ricardo Fierro b, Alvaro Puentes a, Jimena Cortes a,Adriana Bermudez a, Gladys Cifuentes a, Magnolia Vanegas a, Manuel E. Patarroyo a,b,*
a Fundacion Instituto de Immunologıa de Colombia, Carrera 50 # 26-00, Bogota, Colombiab Universidad Nacional de Colombia, Colombia
Received 19 January 2007Available online 8 February 2007
Abstract
A hepatitis C virus E2 protein-derived sequence was selected for studying the effect of N-glycosylation on the peptide chain’s confor-mational structure. The results suggested that the 534TDVF537 motif contained in peptide 33402 (529WGENDTDVFVLNNTRY544) hada type III b-turn, relevant in antigen recognition of polyclonal antibodies, binding to human cells, and binding to HLA DRB1*0401 mol-ecules. N-Glycopeptides derived from this sequence contained monosaccharides in Asn532. N-Glycopeptides presented differences in pep-tide chain structure compared to non-glycosylated peptides. Peptide 33402 specifically bound to human cells, specificity becoming lostwhen it was N-glycosylated. N-Glycosylation decreased antigen recognition of mouse polyclonal sera against this sequence. N-Glycopep-tide binding to HLA DRB1*0401 molecules was similar to that presented by non-glycosylated peptide, indicating that N-glycosylationdid not affect binding to HLA DRB1*0401 molecules. N-Glycosylation induced changes at structural and functional level which could berelevant for modulating human cell binding properties and antibody recognition.� 2007 Elsevier Inc. All rights reserved.
Keywords: N-Glycopeptide; N-Glycosylation; HCV; HLA-DR molecules
At least 170 million people are infected by the hep-atitis C virus (HCV) throughout the world; it alsoaffects 25% of people infected by the human immuno-deficiency virus (HIV) [1]. The structural proteins arecore (p23), E1 (33 kDa), and E2 (70 kDa) envelopeproteins [2]. HCV envelope proteins specifically bindto L-SIGN and DC-SIGN receptor and E2 proteininteraction with DC-SIGN is mediated by mannose res-idues present in viral glycoprotein [3]. HCV protein N-oligosaccharides could be involved in humoral immuneresponse evasion mechanisms developed by the host,thereby masking viral protein regions which could beimmune system targets [4]. The genomic variability ofglycoproteins contrasts with potential N-glycosylation
0006-291X/$ - see front matter � 2007 Elsevier Inc. All rights reserved.
doi:10.1016/j.bbrc.2007.01.167
* Corresponding author. Address: Fundacion Instituto de Immunologıade Colombia, Carrera 50 # 26-00, Bogota, Colombia. Fax: +57 1 4815269.
E-mail address: [email protected] (M.E. Patarroyo).
sites’ high degree of conservation [5]. The importanceof HCV envelope protein glycosylation in virus–cellinteraction has been little studied; the role of oligosac-charides in the virus’ binding and invasion ability alsoremains little known.
Carbohydrates serve as cell receptors, immune responsemodulators, and pathogen ligands during infection [6].Some studies have indicated that recognition is specific,depending mainly on the carbohydrate’s peptide bindingsite, carbohydrate oligosaccharide chain length, the bind-ing site’s position in the peptide chain, and carbohydratenature and density [7].
HCV is a virus infecting different human cells, suggestingthat the virus possesses different binding and invasion mech-anisms [8]. E2 protein regions have been reported as beinginvolved in binding to target cells and recognised by serafrom patients infected by the virus. The HCV E2 protein ami-no acid sequence 523APTYSWGENDTDVFVLNNTR544

410 J.E. Garcia et al. / Biochemical and Biophysical Research Communications 355 (2007) 409–418
(1a genotype, [9]) contains a sequence which is recognised bysera from patients infected by the virus; some studies havesuggested that this region possesses B-epitopes [10–12]. Thissequence is localised in HepG2 cell binding regions, possess-es two highly conserved potential N-glycosylation sites andprediction studies have suggested that the 532Asn site maybe exposed whilst 540Asn seems to be buried within the pro-tein’s structure [5,13,14]. Studies with E2 protein recombi-nant fragments have indicated that the regions betweenresidues 532–534 and 540–542 are glycosylated in native pro-tein [14].
N-Glycopeptides were synthesised using peptide 33402(529WGENDTDVFVLNNTRY544) as an experimentalmodel for studying the effect of N-glycosylation on thepeptide chain at structural and functional level. The resultsindicated that the region (underlined and in bold) localisedbetween N-glycosylation sites Asn532 and Asn540 wasstructured, forming a type III b-turn. Our results have sug-gested that this sequence is relevant in binding to HepG2cells and RBC, possesses epitopes which are specificallyrecognised by polyclonal antibodies induced against thisregion and that Phe537 is the relevant for specific bindingto HLA DRB1*0401 molecules. N-Glycosylation inducesconformational changes in the peptide chain, specificallyin residues neighbouring N-glycosylation sites. N-Glyco-sylation reduced antigen recognition of polyclonal anti-bodies and induced a loss of specificity in binding tohuman cells.
Materials and methods
Glycopeptides, peptides, and radiolabelling. Peptide 32900 (523CAPTYSWGENDTDVFVLNNTR543), 32901 (CAPTYSWGENDTDVFVLNNTRGC), 33402 (529WGENDTDVFVLNNTRY544), and peptide 33402-Gly-analogues were synthesised by solid-phase peptide synthesis (SPPS) usingt-Boc strategy. N-Glycopeptides 33402-(Man)Asn532 (529WGEN(Man)DTDVFVLNNTRY544), 33402-Gal(Asn)532 (529WGEN(Gal)DTDVFVLNNTRY544), and 33402-(Gluc)Asn532 (529WGEN(Gluc)DTDVFVLNNTRY544) were synthesised by SPPS using Fmoc strategy and building-blockmethodology. The assays were also done with jumbled-peptide 28(CTNFTERVNYVDWANTDLSPG; a peptide having the same aminoacid composition as peptide 33402 but having random sequence) as con-trol. Peptides and N-glycopeptides were radiolabelled according to themethodology reported by Garcia et al. [12,15].
RBC binding assays. These assays were performed according to themethodology reported by Garcia et al. [15]. 125I-peptide or 125I-N-glyco-peptide concentrations were between 0 and 1200 nM and unlabelledpeptide or N-glycopeptide concentration was 12 lM.
HepG2 binding assays. These assays were performed according toGarcia et al. [12,15]. 125I-peptide or 125I-N-glycopeptides concentrationswere between 0 and 1200 nM and unlabelled peptide or N-glycopeptideconcentration was 12 lM. 1 · 106 HepG2 cells were used.
125I-N-glycopeptide human cell binding inhibition assays. A fixedconcentration of 125I-N-glycopeptide was added to 1 · 106 cells (HepG2cells or RBCs) in the presence of different non-radiolabelled inhibitorconcentrations. Cell-associated radioactivity was quantified as in thespecific binding assays. N-Glycopeptides, peptide 33402, D-mannose, D-galactose, and D-glucose were used as inhibitors (0.32, 0.21, 0.11, and0.02 lmol); 125I-N-glycopeptide concentration ranged from 0.15 to0.20 pmol.
Peptide 33402 HepG2 cell competition binding assays. The methodologyused was that reported by Garcia et al. [12,15]. 125I-peptide 33402
(350 nM) was incubated with 1 · 106 HepG2 cells in the presence orabsence of unlabelled original or analogue peptide (400, 4, 0.4, and0.016 lM) in 110 ll final volume; the methodology was that used in spe-cific binding assays.
CD assays. CD spectra were recorded according to the methodologyreported by Garcia et al. [15]. Each spectrum was obtained (0.1 mMpeptides in aqueous 30% TFE solutions) from averaging three scans takenat 20 nm/min scan-rate with 1 nm spectral bandwidth, corrected forbaseline.
NMR N-glycopeptide characterization. Peptide or N-glycopeptide wasdissolved in DMSO and a Bruker DRX-6001H was used for obtainingspectra. Double-quantum filter COSY (DQF-COSY) experiments wereused for spectrum assignment and (TOCSY) pulse sequence; phase-sen-sitive NOESY experiments determined NOEs between protons. Thesedata were processed on an Indy computer (Silicon Graphics) equippedwith updated XWINNMR software (Bruker). A 4.75 ppm water signalwas used as reference for proton chemical displacement. All spectra wererun at 295 K.
Structural calculations. NOESY spectra recorded at different mixingtimes (150–600 ms) were used for ensuring that signals obtained werenot due to a spin diffusion effect at 295 K. All NOE intensities weredivided into three distance ranges (1.8–2.7 (strong), 1.8–3.3 (medium),and 1.8–5.0 �A
0(weak)). Structure calculations were made using X-PLOR
1993 with Topa-nilges, Pro- and Para-nilges, Pro-topology, andparameter sets. Secondary structure calculations used CNS protocolsfor direct NOE intensity refinement using a full-relaxation matrixapproach.
Structure prediction N-glycopeptide 33402-(Gal)Asn532 using
molecular dynamics. Discover software (Insight II) molecular dynamicstechnique was used for identifying structures presenting minimumpotential energy. Such structures were NOE-restricted (i, i + 4interaction) as identified in NMR experiments for N-glycopeptide33402-(Gal)Asn532. Molecular dynamics’ calculations, followed byenergy minimisation, considered that the molecule had an aqueousenvironment (5 A H2O layer).
Obtaining polyclonal antibodies. The antibodies were obtainedaccording to Garcia et al. [15] and ELISAs were performed using con-ventional methodology.
Peptide-binding competition assays. Peptide HLA-DR molecule bind-ing assays were done according to Vargas et al. [16]. The following bio-tinylated peptides were used: (i) peptide 33402, (ii) haemagglutininprotein-derived peptide 307PKYVKQNTLKLAT318 binding to HLADRB*0101, DRB*0301, and DRB*0401 molecules, (iii) tetanic toxoidprotein-derived peptide 830TTQYIKANSKFIGIIE844 binding to HLADRB*0701 molecules, and (iv) peptide GFKAAAAAAA binding to HLADRB*1101 molecules.
Results and discussion
The HCV E2 protein-derived peptide 32900 (CAPTYS
WGENDTDVFVLNNTR) is localised in the bindingregions of cells susceptible to infection and recognisedby sera from patients infected with the virus. This regionhas two potential N-glycosylation motifs. Peptide 32900was selected for inducing polyclonal antibodies againstthis sequence in BALB/c mice. Shorter peptides weresynthesised from the peptide 32900 sequence to enableN-glycopeptide synthesis. Peptide 33402 (WGENDTDVFVLNNTRY) was selected to be glycosylated it had tohave the same antigenic properties as peptide 32900 (present-ing equal recognition of polyclonal antibodies anti-peptide32900); epitope-prediction showed that this sequencebound to MHC molecules (http://www.syfpeithi.de/Scripts/MHCServer.dll/EpitopePrediction.htm).

Tab
le1
EL
ISA
sw
ith
sera
fro
mB
AL
B/c
mo
use
imm
un
ised
wit
hp
epti
de
3290
1
Pep
tid
esre
cogn
itio
nfo
rse
ra(P
-III
)d
eter
min
edfo
rE
LIS
As
An
tib
od
yti
ters
agai
nst
pep
tid
e33
402
and
N-g
lyco
pep
tid
es
Ser
um
Ino
cula
ted
pep
tid
eE
valu
ated
pep
tid
eA
nti
bo
dy
tite
rsag
ain
stp
epti
de
3340
233
402-
(Man
)Asn
532
3340
2-(G
al)A
sn532
3340
2-(G
luc)
Asn
532
Pep
tid
ean
alo
gue
Asn
532
3290
032
901
2833
402
Pre
-im
mu
ne
sera
P-I
IIse
raP
-III
sera
P-I
IIse
raP
-III
sera
P-I
IIse
ra
132
901
++
�+
01:
128,
000
1:80
001:
8000
1:16
,000
1:16
,000
232
901
��
��
ND
ND
ND
ND
ND
ND
332
901
��
��
ND
ND
ND
ND
ND
ND
432
901
++
�+
01:
128,
000
ND
ND
ND
ND
532
901
++
�+
01:
>1,
024,
000
1:16
,000
1:80
001:
16,0
001:
64,0
006
3290
1+
+�
+0
1:32
,000
ND
ND
ND
ND
732
901
++
�+
01:
128,
000
ND
ND
ND
ND
832
901
++
�+
01:
>1,
024,
000
1:16
,000
1:80
001:
32,0
001:
64,0
009
3290
1+
+�
+0
1:40
00N
DN
DN
DN
D10
3290
1+
+�
+0
1:16
,000
ND
ND
ND
ND
1128
��
��
ND
ND
ND
ND
ND
ND
1228
��
��
ND
ND
ND
ND
ND
ND
1328
��
��
ND
ND
ND
ND
ND
ND
1428
��
��
ND
ND
ND
ND
ND
ND
1528
��
��
ND
ND
ND
ND
ND
ND
16C
on
tro
lgr
ou
p�
��
�0
00
00
0
J.E. Garcia et al. / Biochemical and Biophysical Research Communications 355 (2007) 409–418 411
Antigenic recognition of peptides 32900 and 33402 by
polyclonal antibodies
ELISAs were done with polyclonal antibodies obtainedfrom BALB/c mice immunised with polymer peptide32901(GCAPTYSWGENDTDVFVLNNTRGC). ELISAsshowed that 8/10 sera (P-III) specifically recognised peptides32900 (CAPTYSWGENDTDVFVLNNTR) and 33402(WGENDTDVFVLNNTRY). None of the sera specificallyrecognised jumbled-peptide 28 (CTNFTERVNYVDWANTDLSPG), nor immune sera from mice immunisedwith jumbled-peptide 28 recognise peptides 32900, 32901,and 33402. Sera specifically recognising peptide 33402 weretitred by ELISA for determining the minimum concentra-tion at which these sera could specifically recognise peptide33402. Results (Table 1) showed that most sera had high lev-els of polyclonal antibodies specific against peptide 33402.Sera 5 and 8 presented the highest antibody levels(1:>1,024,000), whilst sera 9 and 10 presented the lowest ones(Table 1). The results indicated that polymerised peptide32901 (GCAPTYSWGENDTDVFVL NNTRGC) inducedhigh polyclonal antibody production in BALB/c mice andthat these antibodies specifically recognised peptide 32900C-terminal and central regions, suggesting that epitopeswere recognised in this region (underlined and in bold above)(Table 1). These results agreed with studies reporting that theregion between residues 528 and 546 possesses B-epitopesand is recognised by sera from patients infected with HCV.This region is recognised by monoclonal antibodies fromBALB/c mice immunised with HCV E2 protein recombinantfragments [11].
ELISAs with eight sera at 1:100 revealed that all N-glyco-peptides were equally and specifically recognised by thesesera (data not shown). ELISAs established the minimumconcentration at which sera 1, 5, and 8 could specifically rec-ognise the three N-glycopeptides and analogue 33402-Asn532
to determine whether N-glycosylation affected peptide 33402antigen recognition. Results showed that when Asn532 waschanged for Gly, sera 1, 5, and 8, peptide analogue (analogueAsn532) recognition became significantly reduced (Table 1).A similar pattern appeared when the same assay was repeat-ed with N-glycosylated peptides. When Asn532 peptideanalogue antigen recognition was compared to that of N-gly-copeptides, less N-glycopeptide antigen recognition was seen(Table 1), indicating that peptide 33402 N-glycosylation atAsn532 interfered with antibody recognition and did notdepend on bound monosaccharide. These results suggestedthat the carbohydrates possibly induced conformationalchanges in the peptide and/or interacted with the side chains,thereby increasing steric impediment, significantly affectingantibody recognition.
Human cell binding studies using peptides and
N-glycopeptides
Human cell binding assays determined peptide and N-glycopeptide specific binding activity and whether N-glyco-
RBCs Inhibition assays
HepG2 cells Inhibition assays
% B
indi
ng I
nhib
itio
n
added inhibitor (umol)
% B
indi
ng I
nhib
itio
n
added inhibitor (umol)
N-glycopeptide 125I-33402-(Man)Asn532
0
25
50
75
100
0 0.25 0.5
0 0.25 0.5
HBS
33402-(Man)Asn532
33402-(Gal)Asn532
33402-(Gluc)Asn532
D-mannose
D-galactose
D-glucose
N-glycopeptide 125I-33402-(Man)Asn532
0
25
50
75
100
HBS33402-(Man)Asn53233402-(Gal)Asn53233402-(Gluc)Asn532D-mannoseD-galactoseD-glucose
B
A
125I-Peptide added (nM)
125 I-
Pep
tide
bou
nd (p
mol
)
Binding assays to HepG2 cells
Peptide 33402
0
0.04
0.08
0 400 800
Peptide 33402
0
0.008
0.016
0 400 800
N-glycopeptide 33402-(Man)Asn532
0
0.008
0.016
0 300 600
N-glycopeptide 33402-(Man)Asn532
0
0.04
0.08
0 300 600
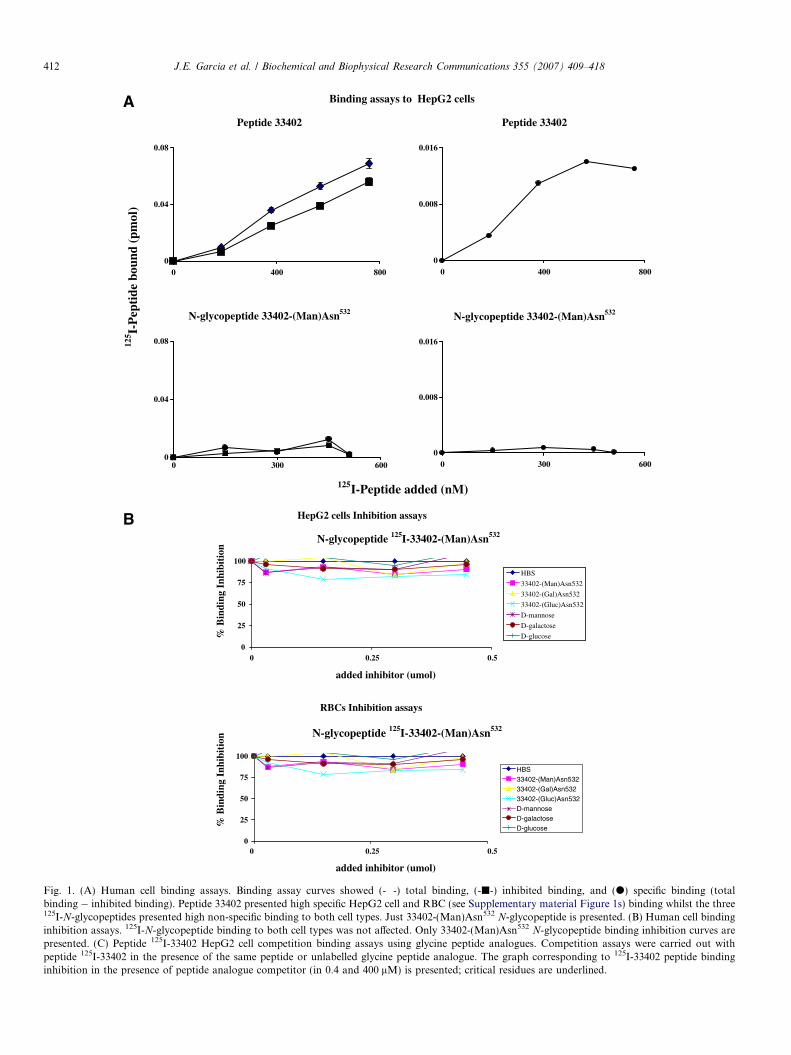
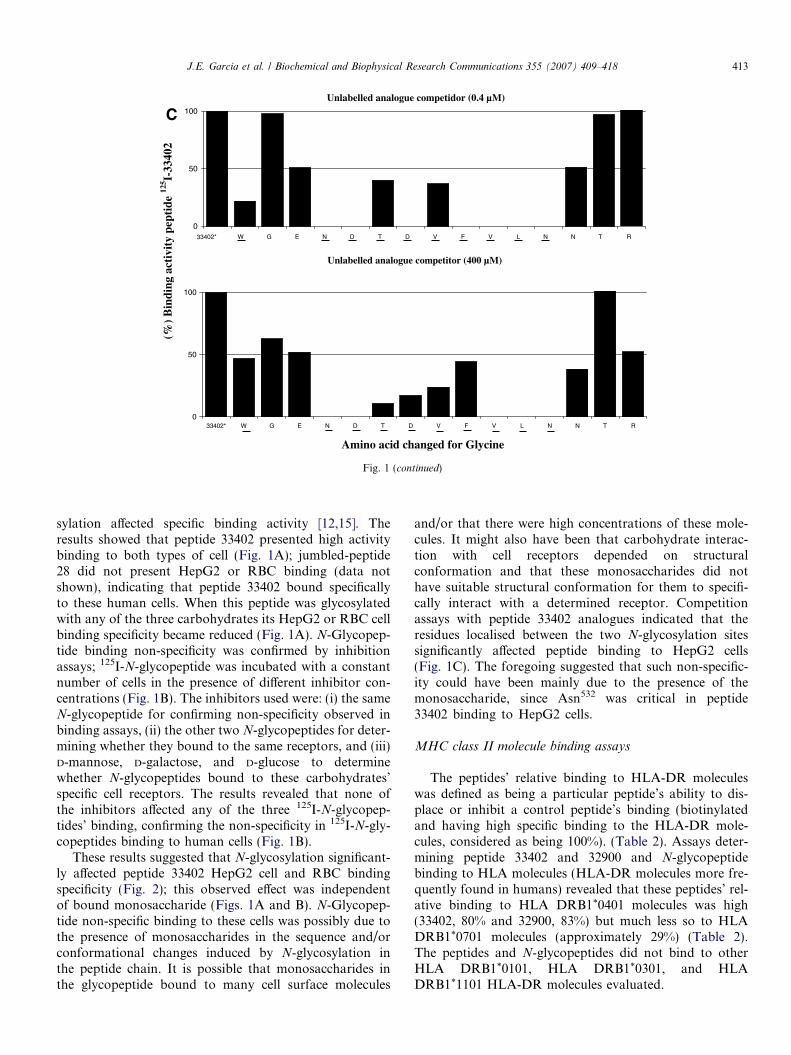
Fig. 1. (A) Human cell binding assays. Binding assay curves showed (-�-) total binding, (-j-) inhibited binding, and (d) specific binding (totalbinding � inhibited binding). Peptide 33402 presented high specific HepG2 cell and RBC (see Supplementary material Figure 1s) binding whilst the three125I-N-glycopeptides presented high non-specific binding to both cell types. Just 33402-(Man)Asn532 N-glycopeptide is presented. (B) Human cell bindinginhibition assays. 125I-N-glycopeptide binding to both cell types was not affected. Only 33402-(Man)Asn532 N-glycopeptide binding inhibition curves arepresented. (C) Peptide 125I-33402 HepG2 cell competition binding assays using glycine peptide analogues. Competition assays were carried out withpeptide 125I-33402 in the presence of the same peptide or unlabelled glycine peptide analogue. The graph corresponding to 125I-33402 peptide bindinginhibition in the presence of peptide analogue competitor (in 0.4 and 400 lM) is presented; critical residues are underlined.
412 J.E. Garcia et al. / Biochemical and Biophysical Research Communications 355 (2007) 409–418
Amino acid changed for Glycine
(%)
Bin
ding
act
ivit
y pe
ptid
e 12
5 I-33
402
Unlabelled analogue competitor (400 µM)
0
50
100
33402* W G E N D T D V F V L N N T R
Unlabelled analogue competidor (0.4 µM)
0
50
100
33402* W G E N D T D V F V L N N T R
C
Fig. 1 (continued)
J.E. Garcia et al. / Biochemical and Biophysical Research Communications 355 (2007) 409–418 413
sylation affected specific binding activity [12,15]. Theresults showed that peptide 33402 presented high activitybinding to both types of cell (Fig. 1A); jumbled-peptide28 did not present HepG2 or RBC binding (data notshown), indicating that peptide 33402 bound specificallyto these human cells. When this peptide was glycosylatedwith any of the three carbohydrates its HepG2 or RBC cellbinding specificity became reduced (Fig. 1A). N-Glycopep-tide binding non-specificity was confirmed by inhibitionassays; 125I-N-glycopeptide was incubated with a constantnumber of cells in the presence of different inhibitor con-centrations (Fig. 1B). The inhibitors used were: (i) the sameN-glycopeptide for confirming non-specificity observed inbinding assays, (ii) the other two N-glycopeptides for deter-mining whether they bound to the same receptors, and (iii)D-mannose, D-galactose, and D-glucose to determinewhether N-glycopeptides bound to these carbohydrates’specific cell receptors. The results revealed that none ofthe inhibitors affected any of the three 125I-N-glycopep-tides’ binding, confirming the non-specificity in 125I-N-gly-copeptides binding to human cells (Fig. 1B).
These results suggested that N-glycosylation significant-ly affected peptide 33402 HepG2 cell and RBC bindingspecificity (Fig. 2); this observed effect was independentof bound monosaccharide (Figs. 1A and B). N-Glycopep-tide non-specific binding to these cells was possibly due tothe presence of monosaccharides in the sequence and/orconformational changes induced by N-glycosylation inthe peptide chain. It is possible that monosaccharides inthe glycopeptide bound to many cell surface molecules
and/or that there were high concentrations of these mole-cules. It might also have been that carbohydrate interac-tion with cell receptors depended on structuralconformation and that these monosaccharides did nothave suitable structural conformation for them to specifi-cally interact with a determined receptor. Competitionassays with peptide 33402 analogues indicated that theresidues localised between the two N-glycosylation sitessignificantly affected peptide binding to HepG2 cells(Fig. 1C). The foregoing suggested that such non-specific-ity could have been mainly due to the presence of themonosaccharide, since Asn532 was critical in peptide33402 binding to HepG2 cells.
MHC class II molecule binding assays
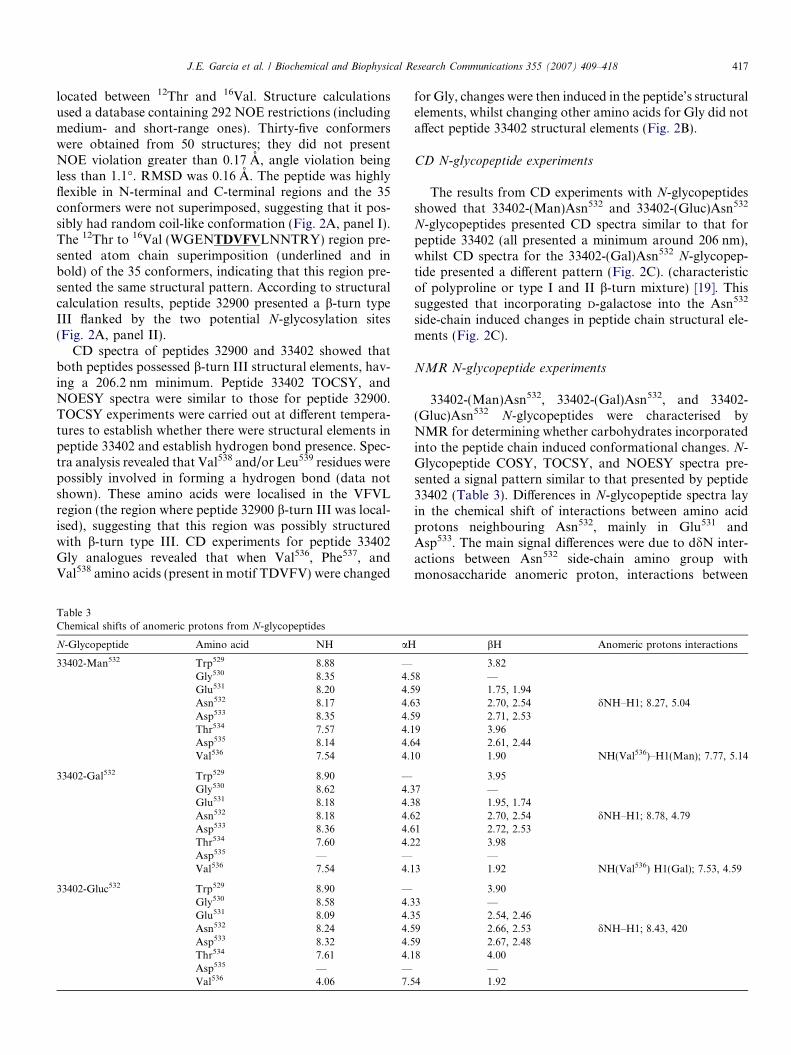
The peptides’ relative binding to HLA-DR moleculeswas defined as being a particular peptide’s ability to dis-place or inhibit a control peptide’s binding (biotinylatedand having high specific binding to the HLA-DR mole-cules, considered as being 100%). (Table 2). Assays deter-mining peptide 33402 and 32900 and N-glycopeptidebinding to HLA molecules (HLA-DR molecules more fre-quently found in humans) revealed that these peptides’ rel-ative binding to HLA DRB1*0401 molecules was high(33402, 80% and 32900, 83%) but much less so to HLADRB1*0701 molecules (approximately 29%) (Table 2).The peptides and N-glycopeptides did not bind to otherHLA DRB1*0101, HLA DRB1*0301, and HLADRB1*1101 HLA-DR molecules evaluated.
414 J.E. Garcia et al. / Biochemical and Biophysical Research Communications 355 (2007) 409–418
The results also showed that N-glycopeptides boundto HLA DRB1*0401 and HLA DRB1*0701 moleculessimilar to peptide 33402 binding, suggesting that peptideN-glycosylation did not affect peptide-HLA complex for-mation. These results also indicated that this proteinregion could contain B-epitopes which could be present-ed within MHC Class II for inducing an immuneresponse against the virus. It must also be taken intoaccount that 30% of women infected with HCV whohave the HLA DRB1*0401 allele present clearance of
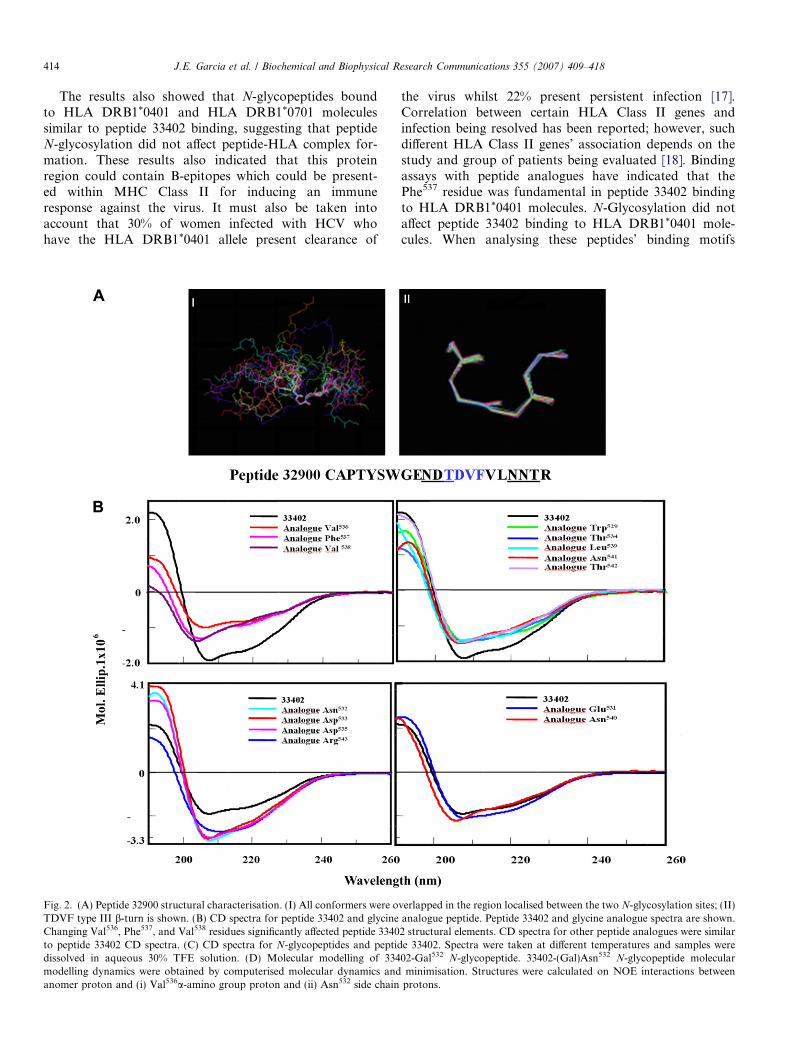
Fig. 2. (A) Peptide 32900 structural characterisation. (I) All conformers were oTDVF type III b-turn is shown. (B) CD spectra for peptide 33402 and glycineChanging Val536, Phe537, and Val538 residues significantly affected peptide 3340to peptide 33402 CD spectra. (C) CD spectra for N-glycopeptides and peptiddissolved in aqueous 30% TFE solution. (D) Molecular modelling of 334modelling dynamics were obtained by computerised molecular dynamics andanomer proton and (i) Val536a-amino group proton and (ii) Asn532 side chain
the virus whilst 22% present persistent infection [17].Correlation between certain HLA Class II genes andinfection being resolved has been reported; however, suchdifferent HLA Class II genes’ association depends on thestudy and group of patients being evaluated [18]. Bindingassays with peptide analogues have indicated that thePhe537 residue was fundamental in peptide 33402 bindingto HLA DRB1*0401 molecules. N-Glycosylation did notaffect peptide 33402 binding to HLA DRB1*0401 mole-cules. When analysing these peptides’ binding motifs
verlapped in the region localised between the two N-glycosylation sites; (II)analogue peptide. Peptide 33402 and glycine analogue spectra are shown.2 structural elements. CD spectra for other peptide analogues were similare 33402. Spectra were taken at different temperatures and samples were02-Gal532 N-glycopeptide. 33402-(Gal)Asn532 N-glycopeptide molecularminimisation. Structures were calculated on NOE interactions between
protons.

Fig. 2 (continued)
J.E. Garcia et al. / Biochemical and Biophysical Research Communications 355 (2007) 409–418 415
and registers it was found that both contained an 8-mersequence containing the critical 537Phe residue to fit intothese HLA-DR molecules’ Pocket 1, 540Asn to fit intoPocket 4 and 542Thr to fit into Pocket 6, missing residueGly in the monomer form but present in the polymerimmunising molecule.
Structural characterisation of peptides by CD and NMR
Peptide 32900 was characterised by NMR; experimentswere carried out in DMSO and daN, dNN, and daN(i,i + 2) interactions were obtained for this peptide. Low tem-perature coefficient indicated that the hydrogen bond was

Table 2Thirty three thousand four hundred and two derived peptide and N-glycopeptide relative binding to HLA-DR molecules
*Glycosilated (Asn532) residue with the carbohydrate.
416J
.E.
Ga
rciaet
al.
/B
ioch
emica
la
nd
Bio
ph
ysica
lR
esearch
Co
mm
un
icatio
ns
35
5(
20
07
)4
09
–4
18
J.E. Garcia et al. / Biochemical and Biophysical Research Communications 355 (2007) 409–418 417
located between 12Thr and 16Val. Structure calculationsused a database containing 292 NOE restrictions (includingmedium- and short-range ones). Thirty-five conformerswere obtained from 50 structures; they did not presentNOE violation greater than 0.17 A, angle violation beingless than 1.1�. RMSD was 0.16 A. The peptide was highlyflexible in N-terminal and C-terminal regions and the 35conformers were not superimposed, suggesting that it pos-sibly had random coil-like conformation (Fig. 2A, panel I).The 12Thr to 16Val (WGENTDVFVLNNTRY) region pre-sented atom chain superimposition (underlined and inbold) of the 35 conformers, indicating that this region pre-sented the same structural pattern. According to structuralcalculation results, peptide 32900 presented a b-turn typeIII flanked by the two potential N-glycosylation sites(Fig. 2A, panel II).
CD spectra of peptides 32900 and 33402 showed thatboth peptides possessed b-turn III structural elements, hav-ing a 206.2 nm minimum. Peptide 33402 TOCSY, andNOESY spectra were similar to those for peptide 32900.TOCSY experiments were carried out at different tempera-tures to establish whether there were structural elements inpeptide 33402 and establish hydrogen bond presence. Spec-tra analysis revealed that Val538 and/or Leu539 residues werepossibly involved in forming a hydrogen bond (data notshown). These amino acids were localised in the VFVLregion (the region where peptide 32900 b-turn III was local-ised), suggesting that this region was possibly structuredwith b-turn type III. CD experiments for peptide 33402Gly analogues revealed that when Val536, Phe537, andVal538 amino acids (present in motif TDVFV) were changed
Table 3Chemical shifts of anomeric protons from N-glycopeptides
N-Glycopeptide Amino acid NH aH
33402-Man532 Trp529 8.88 —Gly530 8.35 4.5Glu531 8.20 4.5Asn532 8.17 4.6Asp533 8.35 4.5Thr534 7.57 4.1Asp535 8.14 4.6Val536 7.54 4.1
33402-Gal532 Trp529 8.90 —Gly530 8.62 4.3Glu531 8.18 4.3Asn532 8.18 4.6Asp533 8.36 4.6Thr534 7.60 4.2Asp535 — —Val536 7.54 4.1
33402-Gluc532 Trp529 8.90 —Gly530 8.58 4.3Glu531 8.09 4.3Asn532 8.24 4.5Asp533 8.32 4.5Thr534 7.61 4.1Asp535 — —Val536 4.06 7.5
for Gly, changes were then induced in the peptide’s structuralelements, whilst changing other amino acids for Gly did notaffect peptide 33402 structural elements (Fig. 2B).
CD N-glycopeptide experiments
The results from CD experiments with N-glycopeptidesshowed that 33402-(Man)Asn532 and 33402-(Gluc)Asn532
N-glycopeptides presented CD spectra similar to that forpeptide 33402 (all presented a minimum around 206 nm),whilst CD spectra for the 33402-(Gal)Asn532 N-glycopep-tide presented a different pattern (Fig. 2C). (characteristicof polyproline or type I and II b-turn mixture) [19]. Thissuggested that incorporating D-galactose into the Asn532
side-chain induced changes in peptide chain structural ele-ments (Fig. 2C).
NMR N-glycopeptide experiments
33402-(Man)Asn532, 33402-(Gal)Asn532, and 33402-(Gluc)Asn532 N-glycopeptides were characterised byNMR for determining whether carbohydrates incorporatedinto the peptide chain induced conformational changes. N-Glycopeptide COSY, TOCSY, and NOESY spectra pre-sented a signal pattern similar to that presented by peptide33402 (Table 3). Differences in N-glycopeptide spectra layin the chemical shift of interactions between amino acidprotons neighbouring Asn532, mainly in Glu531 andAsp533. The main signal differences were due to ddN inter-actions between Asn532 side-chain amino group withmonosaccharide anomeric proton, interactions between
bH Anomeric protons interactions
3.828 —9 1.75, 1.943 2.70, 2.54 dNH–H1; 8.27, 5.049 2.71, 2.539 3.964 2.61, 2.440 1.90 NH(Val536)–H1(Man); 7.77, 5.14
3.957 —8 1.95, 1.742 2.70, 2.54 dNH–H1; 8.78, 4.791 2.72, 2.532 3.98
—3 1.92 NH(Val536) H1(Gal); 7.53, 4.59
3.903 —5 2.54, 2.469 2.66, 2.53 dNH–H1; 8.43, 4209 2.67, 2.488 4.00
—4 1.92

418 J.E. Garcia et al. / Biochemical and Biophysical Research Communications 355 (2007) 409–418
Asn532 side-chain amino group protons and Asp533 side-chain protons and chemical shifts of Glu531 protons(depending on carbohydrate bound to Asn532) (Table 3).N-Glycopeptide 33402-(Gal)Asn532 (strong interaction)and 33402-Man(Asn)532 (weak interaction) NOESY exper-iments revealed signal d(H-1)dN (i, i + 4) interactionbetween monosaccharide anomer proton and Val536 protonamino group (Table 3).
Molecular modelling
Molecular dynamics’ calculations were based on model-ling the 33402-(Gal)Asn532 N-glycopeptide using the identi-fied d(H-1)dN (i, i + 4) interaction and assuming that themolecule was in an aqueous microenvironment. A set ofseven minimum energy structures had the best superimpo-sition between Asn532 and Val536 (0.44 A average RMSD)(shown in blue in Fig. 2D). It can also be seen that the Cand N-terminal regions were not superimposed (brown).The Asn532 side chain (red) and D-galactose (fuchsia) wereorientated in the opposite direction to that of the turn pres-ent in the superimposition (Fig. 2D). phi (/) and psi (w)angles were measured, but no values corresponded to anyspecific conformation. Such predicted structure suggestedthat the carbohydrate incorporated into the sequence couldhave affected peptide conformation, possibly due tohydroxyl group interaction with the side chains of thoseamino acids neighbouring N-glycosylation motifs.
The results obtained here suggested that N-glycosylationof the peptide chain with D-mannose, D-glucose, and D-galac-tose monosaccharides induced very specific conformationalchanges (glycosylated site-neighbouring residues) and struc-tural changes did not need to be induced in the whole peptidechain for modulating peptide 33402 biological properties.Results obtained could also have been due to the carbohy-drate’s intrinsic biological properties modulating the peptidechain’s physicochemical and biological properties.
Acknowledgments
This research has been supported by the Fundacion parael desarrollo de la ciencia y la tecnologıa ‘‘Francisco Josede Caldas’’ (COLCIENCIAS) contract RC 060/2006. Weespecially thank Jason Garry for translating the manu-script and Dr Johan Hoebeke for his collaboration andsuggestions.
Appendix A. Supplementary data
Supplementary data associated with this article can befound, in the online version, at doi:10.1016/j.bbrc.2007.01.167.
References
[1] F. Chisari, Unscrambling hepatitis C virus-host interactions, Nature436 (2005) 930–932.
[2] M. Flint, J. McKeating, The role of the hepatitis C virus glycopro-teins in infection, Rev. Med. Virol. 10 (2000) 101–117.
[3] P.Y. Lozach, H. Lortat-Jacob, A. de Lacroix de Lavalette, I.Staropoli, S. Foung, S. Amara, C. Houles, F. Fieschi, O. Schwartz,J.L. Virelizier, F. Arenzana-Seisdedos, R. Altmeyer, DC-SIGN andL-SIGN are high affinity binding receptors for hepatitis C virus E2glyco protein, J. Biol. Chem. 278 (2003) 20358–20366.
[4] A. Fournillier, C. Wychowski, D. Boucreux, T. Baumert, J. Meunier,D. Jacobs, S. Muguet, E. Depla, G. Inchauspe, Induction of HepatitisC Virus E1 envelope protein-specific immune response can beenhanced by mutation of N-glycosylation sites, J. Virol. 75 (2001)12088–12097.
[5] A. Goffard, J. Dubuisson, Glycosylation of hepatitis C virus envelopeproteins, Biochimie 85 (2003) 295–301.
[6] K. Yarema, C.R. Bertozzi, Chemical approaches to glycobiology andemerging carbohydrate-based therapeutic agents, Curr. Opin. Chem.Biol. 2 (1998) 49–61.
[7] P. Chong, N. Chan, A. Kandil, B. Tripet, O. James, Y.P. Yang, S.P.Shi, M. Klein, A strategy for rational design of fully syntheticglycopeptide conjugate vaccines, Infect. Immun. 65 (1997) 4918–4925.
[8] R. Bartenschlager, V. Lohmann, Replication of hepatitis C virus, J.Gen. virol. 81 (2000) 1631–1648.
[9] Q.L. Choo, K. Richman, H. Han, K. Berger, C. Lee, C. Dong, C.Gallegos, D. Coit, A. Medona-Selby, P.J. Barr, A.J. Weiner, D.W.Bradley, G. Kuo, M. Houghton, Genetic organization and diversityof the Hepatitis C virus, Proc. Natl. Acad. Sci. USA 88 (1991) 2451–2455.
[10] M.A. Mink, S. Benichou, P. Madaule, P. Tiollais, A.M. Prince, G.Inchauspe, Characterization and mapping of a B-cell immunogenicdomain in hepatitis C virus E2 glycoprotein using a yeast peptidelibrary, Virology 200 (1994) 246–255.
[11] J.W. Lee, K. Kim, S.H. Jung, K.J. Lee, E.C. Choi, Y.C. Sung, C.Y.Kang, Identification of a domain containing B-cell epitopes inhepatitis C virus E2 glycoprotein by using mouse monoclonalantibodies, J. Virol. 73 (1999) 11–18.
[12] J.E. Garcia, A. Puentes, J. Suarez, R. Lopez, R. Vera, L.E.Rodriguez, M. Ocampo, H. Curtidor, F. Guzman, M. Urquiza,M.E. Patarroyo, Hepatitis C virus (HCV) E1 and E2 proteinregions that specifically bind to HepG2 cells, J. Hepatol. 36 (2002)254–262.
[13] A.T. Yagnik, A. Lahm, A. Meola, R.M. Roccasecca, B.B. Ercole, A.Nicosia, A. Tramontano, A model for the hepatitis C virus envelopeglyco E2 protein, Proteins 40 (2000) 355–366.
[14] T. Slater-handshy, D. Droll, X. Fan, A. Bisceglie, T.J. Chambers,HCV E2 glycoprotein: mutagenesis of N-linked glycosylation sitesand its effects on E2 expression and processing, Virology 319 (2004)36–48.
[15] J.E. Garcia, A. Puentes, H. Curtidor, R. Vera, L.E. Rodriguez, J.Valbuena, R. Lopez, M. Ocampo, J. Cortes, M. Vanegas, J.E. Rosas,C. Reyes, M.E. Patarroyo, Peptides from the Plasmodium falciparum
STEVOR putative protein bind with high affinity to normal humanred blood cells, Peptides 26 (2005) 1133–1143.
[16] L.E. Vargas, C.A. Parra, L.M. Salazar, F. Guzman, M. Pinto, M.E.Patarroyo, MHC allele-specific binding of a malaria peptide makes itbecome promiscuous on fitting a glycine residue into pocket 6,Biochem. Biophys. Res. Commun. 307 (2003) 148–156.
[17] L.J. Fanning, J. Levis, E. Kenny-Walsh, F. Wynne, M. Whelton, F.Shanahan, Viral clearance in hepatitis C (1b) infection: relationshipwith human leukocyte antigen Class II in a homogeneous population,Hepatology 31 (2000) 1334–1337.
[18] D.D. Eckels, H. Wang, T.H. Bian, N. Tabatabai, J.C. Gill,Immunobiology of hepatitis C virus (HCV) infection: the role ofCD4 T cells in HCV, Infect. Immunol. Rev. 174 (2000) 90–97.
[19] A. Percze1, M. Hollbsi, B.M. Foxman, G.D. Fasman, Conforma-tional analysis of pseudocyclic hexapeptides based on quantitativecircular dichroism (CD), NOE, and X-ray data. The pure CD spectraof type i and type I1 b-turns, J. Am. Chem. Soc. 113 (1991) 9184–9772.
Copyright © 2022 FDOKUMEN




















