
Molecular pathogenesis of Wilson disease: haplotype analysis, detection of prevalent mutations and...
9
ORIGINAL INVESTIGATION A. Gupta D. Aikath R. Neogi S. Datta K. Basu B. Maity R. Trivedi J. Ray S. K. Das P. K. Gangopadhyay K. Ray Molecular pathogenesis of Wilson disease: haplotype analysis, detection of prevalent mutations and genotype–phenotype correlation in Indian patients Received: 24 February 2005 / Accepted: 24 May 2005 / Published online: 17 August 2005 Ó Springer-Verlag 2005 Abstract Wilson disease (WD) is an autosomal recessive disorder caused by defects in the copper-transporting P- type ATPase gene (ATP7B) resulting in the accumula- tion of copper in the liver and the brain. We identified prevalent mutations in the ATP7B of Indian WD pa- tients and attempted to correlate those with the disease phenotype. Patients from 62 unrelated families and their first-degree relatives comprising 200 individuals were enrolled in this study. Three dinucleotide repeat markers flanking WD locus and a few intragenic SNPs were used to determine the genotypes and construct haplotypes of the patients. Seven recurring haplotypes accounting for 58% of the total mutant chromosomes were identified, and four underlying defects in the ATP7B representing 37% of WD chromosomes were detected. In addition, five other rare mutations were characterized. Thus a total of nine mutations including five novel changes were identified in the ATP7B of WD patients. Interestingly, homozygotes for different mutations that would be ex- pected to produce similar defective proteins showed significant disparity in terms of organ involvement and severity of the disease. We also observed WD patients with neurological symptoms with little or no manifes- tation of hepatic pathogenesis. In one WD family, the proband and a sib had remarkably different phenotypes despite sharing the same pair of mutant chromosomes. These findings suggest a potential role for yet unidenti- fied modifying loci for the observed phenotypic hetero- geneity among the WD patients. Introduction Wilson disease (WD, MIM# 277900) is an autosomal recessive disorder, which manifests as hepatolenticular degeneration that results in accumulation of copper in the brain, liver, kidney and cornea (Wilson 1912). The disease is diagnosed on the basis of typical symptoms and conventional biochemical indicators, which include low serum concentrations of ceruloplasmin, and elevated excretion of urinary copper (Sternlieb 1990). The disease has a worldwide frequency of 1/5,000 to 1/30,000 live births (Thomas et al. 1995a) and a carrier frequency of 1 in 90 (Figus et al. 1995). The gene, a P-type ATPase (ATP7B), causally associated with the disease has six metal binding regions and other conserved domains consistent with other metal transporters (Bull et al. 1993; Lutsenko et al. 2002). Studies on molecular genetics and biochemistry of WD have been described in recent re- views (Suzuki and Gitlin 1999; Lutsenko and Petris 2002; Ray et al. 2001). Mutation screening has revealed defects covering al- most the entire ATP7B gene. Prevalent mutations in ATP7B have been identified in different populations. From the haplotype analyses, Petrukhin et al. (1993) predicted that approximately half of all WD mutations would be rare in the American and Russian populations. Using this strategy Figus et al. (1995) analyzed different mutations and defined the chromosomal haplotype in 127 patients of Mediterranean descent affected by WD. There were five common haplotypes in Sardinians, three in Italians, and two in Turks, which accounted for 85, A. Gupta D. Aikath R. Neogi S. Datta K. Basu K. Ray (&) Human Genetics and Genomics Division, Indian Institute of Chemical Biology, 4 Raja S. C. Mullick Road, Jadavpur, Kolkata, 700 032, India E-mail: thisiskr@rediffmail.com Tel.: +91-33-24730350 Fax: +91-33-24735197 B. Maity R. Trivedi Central Forensic Science Laboratory, Kolkata, India J. Ray S. N. Pradhan Center for Neurosciences, University College of Medicine, University of Calcutta, Kolkata, India S. K. Das P. K. Gangopadhyay Bangur Institute of Neurology, Kolkata, India Hum Genet (2005) 118: 49–57 DOI 10.1007/s00439-005-0007-y
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Molecular pathogenesis of Wilson disease: haplotype analysis, detection of prevalent mutations and...

ORIGINAL INVESTIGATION
A. Gupta Æ D. Aikath Æ R. Neogi Æ S. Datta Æ K. BasuB. Maity Æ R. Trivedi Æ J. Ray Æ S. K. Das
P. K. Gangopadhyay Æ K. Ray
Molecular pathogenesis of Wilson disease: haplotype analysis, detectionof prevalent mutations and genotype–phenotype correlation in Indianpatients
Received: 24 February 2005 / Accepted: 24 May 2005 / Published online: 17 August 2005� Springer-Verlag 2005
Abstract Wilson disease (WD) is an autosomal recessivedisorder caused by defects in the copper-transporting P-type ATPase gene (ATP7B) resulting in the accumula-tion of copper in the liver and the brain. We identifiedprevalent mutations in the ATP7B of Indian WD pa-tients and attempted to correlate those with the diseasephenotype. Patients from 62 unrelated families and theirfirst-degree relatives comprising 200 individuals wereenrolled in this study. Three dinucleotide repeat markersflanking WD locus and a few intragenic SNPs were usedto determine the genotypes and construct haplotypes ofthe patients. Seven recurring haplotypes accounting for58% of the total mutant chromosomes were identified,and four underlying defects in the ATP7B representing37% of WD chromosomes were detected. In addition,five other rare mutations were characterized. Thus atotal of nine mutations including five novel changes wereidentified in the ATP7B of WD patients. Interestingly,homozygotes for different mutations that would be ex-pected to produce similar defective proteins showedsignificant disparity in terms of organ involvement andseverity of the disease. We also observed WD patientswith neurological symptoms with little or no manifes-
tation of hepatic pathogenesis. In one WD family, theproband and a sib had remarkably different phenotypesdespite sharing the same pair of mutant chromosomes.These findings suggest a potential role for yet unidenti-fied modifying loci for the observed phenotypic hetero-geneity among the WD patients.
Introduction
Wilson disease (WD, MIM# 277900) is an autosomalrecessive disorder, which manifests as hepatolenticulardegeneration that results in accumulation of copper inthe brain, liver, kidney and cornea (Wilson 1912). Thedisease is diagnosed on the basis of typical symptomsand conventional biochemical indicators, which includelow serum concentrations of ceruloplasmin, and elevatedexcretion of urinary copper (Sternlieb 1990). The diseasehas a worldwide frequency of 1/5,000 to 1/30,000 livebirths (Thomas et al. 1995a) and a carrier frequency of 1in 90 (Figus et al. 1995). The gene, a P-type ATPase(ATP7B), causally associated with the disease has sixmetal binding regions and other conserved domainsconsistent with other metal transporters (Bull et al. 1993;Lutsenko et al. 2002). Studies on molecular genetics andbiochemistry of WD have been described in recent re-views (Suzuki and Gitlin 1999; Lutsenko and Petris2002; Ray et al. 2001).
Mutation screening has revealed defects covering al-most the entire ATP7B gene. Prevalent mutations inATP7B have been identified in different populations.From the haplotype analyses, Petrukhin et al. (1993)predicted that approximately half of all WD mutationswould be rare in the American and Russian populations.Using this strategy Figus et al. (1995) analyzed differentmutations and defined the chromosomal haplotype in127 patients of Mediterranean descent affected by WD.There were five common haplotypes in Sardinians, threein Italians, and two in Turks, which accounted for 85,
A. Gupta Æ D. Aikath Æ R. Neogi Æ S. Datta Æ K. BasuK. Ray (&)Human Genetics and Genomics Division,Indian Institute of Chemical Biology,4 Raja S. C. Mullick Road, Jadavpur,Kolkata, 700 032, IndiaE-mail: [email protected].: +91-33-24730350Fax: +91-33-24735197
B. Maity Æ R. TrivediCentral Forensic Science Laboratory,Kolkata, India
J. RayS. N. Pradhan Center for Neurosciences,University College of Medicine, University of Calcutta,Kolkata, India
S. K. Das Æ P. K. GangopadhyayBangur Institute of Neurology, Kolkata, India
Hum Genet (2005) 118: 49–57DOI 10.1007/s00439-005-0007-y

32, and 30% of the WD chromosomes, respectively.Loudianos et al. (1999) characterized the WD mutation,a 15-nucleotide deletion, underlying the most commonhaplotype among Sardinians. With the addition of thismutation, 92% of the WD chromosomes among Sardi-nians were accounted for. Among Europeans, twoATP7B defects, His1069Gln and Gly1267Arg, togetheraccount for 38% of the mutations characterized (Tho-mas et al. 1995b). Kim et al. (1998) identified three novelmutations in the ATP7B in Korean patients with WD.One of these, Arg778Leu was found in six chromosomesof eight unrelated patients, giving an allele frequency of37.5%. The spectrum of mutations in the ATP7B isavailable in the HGMD database (http://uwcmml1-s.uwcm.ac.uk/uwcm/mg/search/120494.html) and theHUGO mutation database (http://www.uofa-medical-genetics.org/wilson/index.php).
Very little information is available regarding theincidence and spectrum of mutations causing WDamong Indians (Kumar et al. 2005). The Indian popu-lation being old and multi-ethnic, with a high degree ofpopulation mixing and outbreeding, is expected to havehigh heterogeneity in its mutation profile. At the sametime, since WD is a comparatively rare disorder with alow frequency of de novo mutations, it is expected that afew prevalent mutations would be present in the popu-lation due to founder effect. Towards that goal, weanalyzed three di-nucleotide repeat markers (D13S316,D13S133, and D13S314) flanking the ATP7B for hap-lotype analysis (Thomas et al. 1994), and collated similarhaplotypes to a few major groups. These haplogroupscoalesced to a few distinct single nucleotide polymor-phism (SNP)-based haplotypes. The underlying muta-tions were characterized and an attempt was made todecipher the genotype–phenotype relationship in thepatients with the prevalent mutations.
Materials and methods
Patients
Wilson disease patients, mostly with neurological prob-lems, were examined at the Bangur Institute of Neu-rology, Kolkata, with referrals being made to theRegional Institute of Ophthalmology, Kolkata and theGastroenterology and Paediatrics Departments, SSKMHospital, Kolkata, for ophthalmologic and hepaticcases, respectively. The diagnosis was based—(a) clini-cally, on the presence of neurological features like dys-tonia, tremor, rigidity and bradykinesia and/or hepaticfeatures like signs and symptoms of acute or chronicliver failure, etc., and Kayser-Fleischer (K-F) ring in thecornea; and (b) biochemically, on low serum cerulo-plasmin (<20 mg/dl) and high 24-h urinary copperexcretion (>100 lg) (Sternlieb 1990), and on CT &MRI scanning where applicable. Diagnosis was basedon the entire profile, and presence or absence of any onefeature did not contribute towards either diagnosis or
exclusion thereof. Other relevant tests were conductedaccording to the need of the patients and for the diag-nosis of the disease. We collected 200 blood samplesfrom 62 families that included at least one patient ineach family. The internal review committee on researchusing human subjects cleared the project after properreview as per the regulations of the Indian Council ofMedical Research.
Collection of blood samples and GenomicDNA preparation
Approximately 10.0 ml peripheral blood samples werecollected with informed consent of WD patients andtheir family members. Genomic DNA was preparedfrom fresh whole blood using the conventional phenol–chloroform method, followed by ethanol precipitationafter which the DNA was dissolved in TE (10 mM Tris–HCl, 0.1 mM EDTA, pH 8.0) (Sambrook and Russell2001).
Polymerase chain reaction of markers and Genotyping
Polymerase chain reaction (PCR) was carried out in atotal volume of 25.0 ll containing 20–50 ng of genomicDNA, 0.4 lM of each primer, 0.2 mM of each dNTP,2.5 mM of MgCl2, and 0.5 unit of Taq polymerase(Invitrogen, Carlsbad, California) in a thermocycler(GeneAmp-9700, PE Applied Biosystems, Foster City,CA, USA). Dinucleotide repeats (D13S133, D13S314,and D13S316) flanking the WD locus were genotyped.The repeat D13S316 is at a distance of 375 kb from theWD locus towards the telomere and D13S314 is 917 kbfrom the WD locus on the centromeric end. D13S133lies between the WD locus and D13S316 (Thomas et al.1995a). These repeats were amplified using fluorescent-labeled primers as described previously (Thomas et al.1994). Amplified PCR products were separated by 6%polyacrylamide gel analysis in an ABI 377 DNAsequencing system (Applied Biosystems Inc., FosterCity, CA, USA) using Genescan-500 TAMRA SizeStandard (Applied Biosystems Inc., Foster City, CA,USA). Genotypes were determined for each of the threedi-nucleotide repeat-markers for each member of thecore family (parents and sibs) of the index patients. Ineach case the haplotype was constructed using thegenotype data, following the Mendelian pattern ofinheritance of the markers in the family.
Amplification of exons and DNA sequencing
PCR was done to amplify the exons and flanking regionsof the WD gene from the DNA of patients using primersdescribed previously (Waldenstrom et al. 1996). Addi-tional primers were also designed to amplify regionsflanking the exons (sequences available on request).
50
PCR was carried out for each fragment in a total volumeof 25.0 ll as described above using 1.5–2.5 mM ofMgCl2 (as appropriate) with 35 cycles of denaturation at94�C (30 s), appropriate annealing temperature (varyingbetween 50�C and 60�C) for 30 s and an extensiontemperature of 72�C (30 s).
The PCR products, free of contaminating bands dueto non-specific amplification, were column purified usingQiagen PCR-purification kits (Qiagen, Hilden, Ger-many), and bi-directional sequencing was done using anABI Prism 377 DNA sequencer or an ABI 3100 AvantDNA sequencer with dye-termination chemistry.Nucleotide changes were detected by comparing the se-quence obtained in the chromatogram with the normalgene sequence (NT_024524; Homo sapiens chromosome13 genomic contig) using Pairwise BLAST (Tatusovaand Madden 1999). The nucleotide changes were con-firmed in the DNA samples from either of the parents ofthe proband.
Results
Sixty-four WD patients from 62 unrelated families andtheir first-degree relatives, a total of 200 individuals,were enrolled in this study. Genotyping was done usingthree highly polymorphic dinucleotide repeat markersD13S314 (CA repeat), D13S133 (GA14GT17 repeat), andD13S316 (CA repeat).
Marker distribution pattern in normaland WD chromosomes
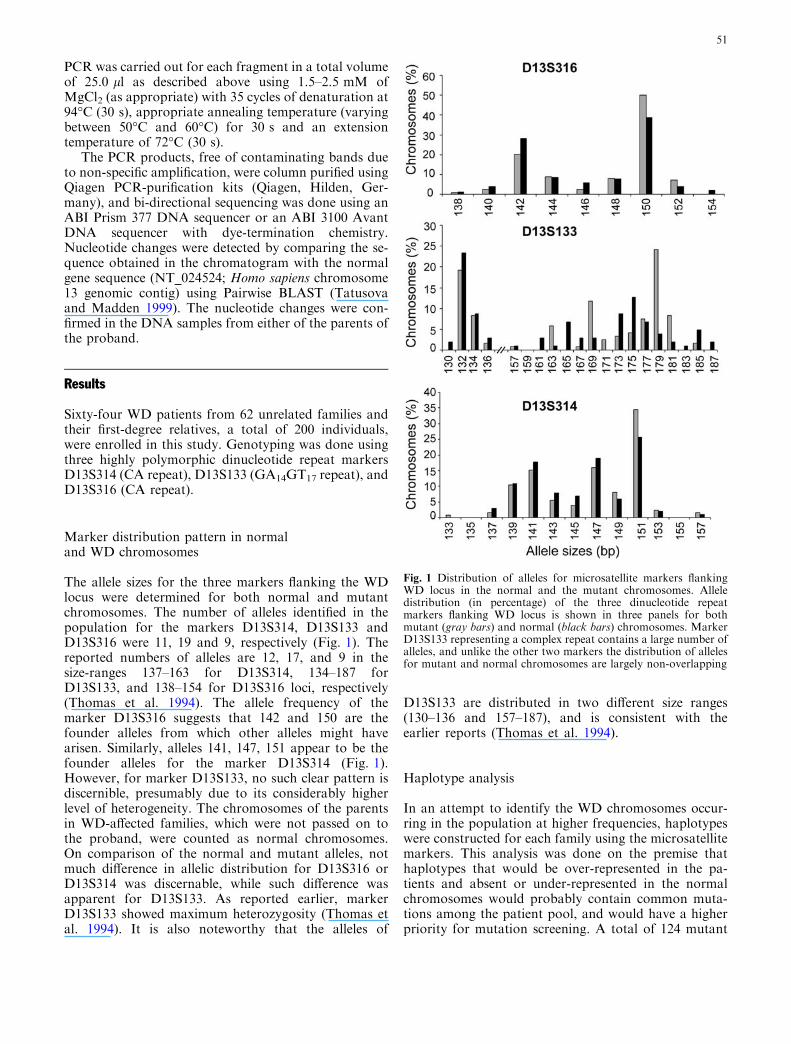
The allele sizes for the three markers flanking the WDlocus were determined for both normal and mutantchromosomes. The number of alleles identified in thepopulation for the markers D13S314, D13S133 andD13S316 were 11, 19 and 9, respectively (Fig. 1). Thereported numbers of alleles are 12, 17, and 9 in thesize-ranges 137–163 for D13S314, 134–187 forD13S133, and 138–154 for D13S316 loci, respectively(Thomas et al. 1994). The allele frequency of themarker D13S316 suggests that 142 and 150 are thefounder alleles from which other alleles might havearisen. Similarly, alleles 141, 147, 151 appear to be thefounder alleles for the marker D13S314 (Fig. 1).However, for marker D13S133, no such clear pattern isdiscernible, presumably due to its considerably higherlevel of heterogeneity. The chromosomes of the parentsin WD-affected families, which were not passed on tothe proband, were counted as normal chromosomes.On comparison of the normal and mutant alleles, notmuch difference in allelic distribution for D13S316 orD13S314 was discernable, while such difference wasapparent for D13S133. As reported earlier, markerD13S133 showed maximum heterozygosity (Thomas etal. 1994). It is also noteworthy that the alleles of
D13S133 are distributed in two different size ranges(130–136 and 157–187), and is consistent with theearlier reports (Thomas et al. 1994).
Haplotype analysis
In an attempt to identify the WD chromosomes occur-ring in the population at higher frequencies, haplotypeswere constructed for each family using the microsatellitemarkers. This analysis was done on the premise thathaplotypes that would be over-represented in the pa-tients and absent or under-represented in the normalchromosomes would probably contain common muta-tions among the patient pool, and would have a higherpriority for mutation screening. A total of 124 mutant
Fig. 1 Distribution of alleles for microsatellite markers flankingWD locus in the normal and the mutant chromosomes. Alleledistribution (in percentage) of the three dinucleotide repeatmarkers flanking WD locus is shown in three panels for bothmutant (gray bars) and normal (black bars) chromosomes. MarkerD13S133 representing a complex repeat contains a large number ofalleles, and unlike the other two markers the distribution of allelesfor mutant and normal chromosomes are largely non-overlapping
51

and 103 normal unrelated chromosomes were accountedfor in a total of 200 individuals based on haplotypeanalysis. The distribution of haplotypes between normaland WD patients was largely non-overlapping—that is,only 11 haplotypes (totaling 16 chromosomes) wereshared between the two groups (data not shown).Though clustering of haplotypes in small discrete groupswas evident, the data set was mostly scattered. Hence,haplotypes differing in 1–3 repeat units in one of thethree loci were collated to haplogroups (Table 1) on thebasis that such groups would harbor single mutationsacquired by founder effect, which possibly acquired newalleles at the marker loci due to slippage during DNAreplication in subsequent generations (Levinson andGutman 1987). In each cluster of haplotypes, majordifference in allele size was noted for the D13S133 locus.
Haplogroup construction
Among 124 mutant chromosomes, a total of 69 haplo-types were pooled in four haplogroups. Haplogroup 1–4represented 28, 23, 11 and 7 mutant chromosomes,respectively (Table 1). While sequencing was done in afew samples from each haplogroup to identify the
underlying ATP7B mutation, SNPs were identifiedwhich were used to create SNP-based haplotypes. It wasthus observed that a larger number of microsatellitemarker-based haplotypes in each haplogroup coalescedto 1–3 SNP-based haplotype(s) only (Table 1). Based onthe numbers of mutant chromosomes in seven commonhaplotypes, we conclude that approximately 58% of themutations in the test population resulted from only afew founder defects in the ATP7B, while the rest of theWD chromosomes were most likely to harbor indepen-dent mutations.
Nucleotide variants in the ATP7B gene
ATP7B gene of 2–3 patients from each haplogroup wasscanned for mutation by sequencing. Once the muta-tion at a specific location on the gene was identified inthose representative samples, all other individuals ofthe same haplogroup were sequenced at that specificlocation to confirm the presence of that mutation. Inaddition, screening for rare mutation in samples notrepresented in any haplogroup was done as part ofongoing study.
Table 1 Haplotypes andATP7B gene defectsrepresenting prevalentmutations in Indian population
a These microsatellite-based h-aplotypes do not belong to therelevant haplogroup but sharethe same haplotype based onSNP. The disparity might bedue to recombination betweenthe microsatellite loci located atrelatively larger distance (375–917 kb) from the ATP7B geneba= c.447G>C (Exon 2);
b = c.1216T>G (Exon 2);c = c.1544-53A>C (Intron 3);d= c.2145C>T (Exon 8);e = c.2484C>T (Exon 10);f = c.2495A>G (Exon 10)c
Exons 2–17 have been scannedfor nucleotide variants. Screen-ing of the remaining exons ofATP7B and the promoter re-gion is in progress
Microsatellite-basedhaplotype D13S 316-133-314(number of chromosomes)
Haplogroups SNP-basedhaplotype a-b-c-d-e-fb
(number of chromosomes)
Mutation(number ofchromosomes)
150-177-151 (3)150-179-151 (13)150-179-149 (4) G-T-A-C-T-A (23) Cys271Stop (23)150-179-147 (1) Common haplotype#1152-179-151 (1)150-132-141 (1)a
1152-181-147 (1) G-T-A-C-C-A (4) Tyr187Stop (4)150-179-147 (1) Common haplotype#2152-181-141 (2)a
150-181-151 (3) G-T-A-T-C-A (4) Not determinedc
150-181-147 (1) Common haplotype#3
142-132-143 (2)142-132-137 (2)142-132-139 (4) G-G-C-C-C-G (12) c.1708-1G>C (12)144-132-139 (2) Common haplotype#4144-132-141 (1)144-132-137 (1)
2142-132-139 (1)142-132-141 (1)142-134-141 (6) G-G-C-C-C-G (11) Not determinedc
142-134-139 (1) Common haplotype#5144-134-139 (1)144-134-141 (1)
150-169-147 (3) G-T-C-C-C-A (11) Not determinedc
150-169-151 (6) 3 Common haplotype#6150-169-149 (2)
148-163-147 (3) C-T-C-C-C-A (7) c.448_452delGAGGG (7)148-163-145 (1) 4 Common haplotype#7148-163-151(2)150-163-151(1)
52

Prevalent mutations Two mutations c.813C>A(Cys271Stop) and c.1708-1G>C were found to beassociated with 23 and 12 WD chromosomes, respec-tively. While Cys271Stop mutation was reported previ-ously in the Caucasian population (Curtis et al. 1999),c.1708-1G>C was reported in two individuals of Indianorigin (Thomas et al. 1995b). Two novel mutations,c.561T>A (Tyr187Stop) and c.448_452delGAGGG,were detected in 4 and 7 WD chromosomes, respectively.Polyacrylamide gel electrophoresis of the PCR amplifiedexon 2 in the WD with the deletion mutationc.448_452delGAGGG clearly showed evidence of het-eroduplex formation (data not shown). We have not yetdetected mutations underlying three haplotypesaccounting for a total of 26 WD chromosomes. For thesechromosomes, exons 2–17 have been scanned for nucle-otide variants and screening of the remaining exons ofATP7B and the promoter region is in progress. Together,seven haplotypes accounted for 72 mutant chromosomesin a total of 62 patients representing 58% of the under-lying mutations in the test population (Table 1).
Rare mutations We detected five other mutations eachfound in single patients in heterozygous condition—onemissense (c.2128G>A; Gly710Ser) and four in/del types(c.172_173insC, c.892delC, c.2298_2299insC andc.2495_2496insG). The novel change c.172_173insCabolishes restriction digestion sites for the enzyme EaeI.The location of these mutations on the ATP7B, thenumber of chromosomes harboring these mutations andthe predicted implication of the gene defect in thetranslated mutant protein are described in Table 2.Chromatograms for the novel mutations are shown inFig. 2.
Single nucleotide polymorphism We detected ten singlenucleotide variants which include seven innocuouschanges in the coding sequence that would translate tosilent and conservative changes in aminoacids—c.447G>C (Val149Val), c.540C>T (As-n180Asn), c.1216T>G (Ser406Ala), c.2145C>T(Tyr715Tyr), c.2484C>T (Gly828Gly), c.2495A>G(Lys832Arg), c.2855 A>G (Lys952Arg)—and three
Table 2 Nucleotide changes detected in the ATP7B gene of WD patients
Nucleotide changes Location Predicted change in protein/number of chromosomes (percentage) Reference
Nonsense mutationc.561 TAT>TAA Exon 2 Tyr187Stop; Nonfunctional protein
with only the first 2 Cu bindingdomains/4 (3.2)
Novel
c.813 TGC>TGAb Exon 2 Cys271Stop; Nonfunctional proteinwith complete first 2 and an incomplete3rd Cu binding domains/23 (18.5)
Curtis et al. 1999
Splice-junction mutationc.1708-1G>Cb Intron 4 Loss of 6th Cu binding domain;
Protein without the catalytic activity/12 (9.6)Thomas et al. 1995b
Frameshift mutationc.172_173insC Exon 2 Ala58Ala-fs; Truncated protein
without any of its conserved domains/1 (0.8)Novel
c.448_452delGAGGG Exon 2 Glu150His-fs; Truncated protein withonly the 1st Cu binding domains/7 (5.6)
Novel
c.892delC Exon 2 Gln298Lys-fs; Truncated protein withonly the 1st 3 Cu binding domains/1 (0.8)
Novel
c.2298_2299insCb Exon 8 Pro767Pro-fs; Truncated protein havingall 6 Cu binding domains but no otherfunctional domains/1 (0.8)
Thomas et al. 1995b
c.2495_2496insG Exon 10 Lys832Lys-fs; Truncated protein havingall 6 Cu binding domains but noother functional domains/1 (0.8)
Novel
Missense mutationc.2128GGT>AGTb Exon 8 Gly710Ser; Affects 2nd transmembrane
domain/1 (0.8)Waldenstrom et al. 1996
SNPc.447 GTG>GTC Exon 2 Val149Val/7 (5.6) Novelc.1216TCT>GCT Exon 2 Ser406Ala/23 (18.5) Curtis et al. 1999c.540 AAC>AAT Exon 2 Asn180Asn/1 (0.8) Novelc.1544-53A>C Intron 3 No change/62 (50.0) rs2147363a
c.1870-23A>C Intron 5 No change/1 (0.8) Novelc.2145 TAC>TAT Exon 8 Tyr715Tyr/4 (3.2) Novelc.2484 GGC>GGT Exon 10 Gly828Gly/23 (18.5) Curtis et al. 1999c.2495 AAG>AGGb Exon 10 Lys832Arg/23 (18.5) Figus et al. 1995c.2855 AAA>AGA Exon 12 Lys952Arg/11 (8.8) Figus et al. 1995c.2866-105G>A Intron 12 No change/ 11 (8.8) Novel
Total number of chromosomes examined in each case is 124a rs2147363: dbSNP IDb Reported nucleotide changes are enlisted in the WD database: http://www.uofa-medical-genetics.org/wilson/index.php
53

changes (c.1544-53A>C, c.1870-23A>C, and c.2866-105G>A) in the intervening sequence of the gene.Among these SNPs two variants (c.1216T>G andc.2495A>G) were observed to be in linkage disequilib-rium. As shown in Table 1, six of the SNPs were used tobuild haplotypes that collated the microsatellite-basedhaplogroups to fewer haplotypes. The relative locationof the mutations and the SNPs on the ATP7B are shownin Fig. 3.
Genotype–phenotype correlation
To correlate the phenotypes of the patients with theirrespective genotypes, we initially examined patients whowere homozygous for the ATP7B mutation as opposedto compound heterozygotes, which would produce twokinds of defective proteins. Generally, it was observedthat the clinical feature and age of disease onset weresimilar among the three unrelated homozygotes for
Fig. 2 Chromatograms showing the novel mutations in ATP7B.The sequences containing the novel mutations and correspondingregions of wild type are shown in bottom and top location,respectively in each panel. Panel A depicts a homozygous mutation(c.561TAT>TAA) resulting in an amino acid alteration (Tyr187-Stop). The mutation has been identified by direct sequencing inthree unrelated patients. Panel B depicts a single base heterozygousdeletion (c.892delC) in a patient resulting in overlapping sequencedownstream to the deletion. This rare mutation has been confirmedby the sequencing of a second sample aliquot of the patient.Chromatogram in Panel C shows the heterozygous mutationc.2495_2496insG (in reverse complement sequence) depicting singlebase insertion with heterozygous signal thereafter. This rare
mutation also has been confirmed by sequencing of a secondsample aliquot of the patient. Chromatogram in the Panel D showsa heterozygous insertion mutation (c.172_173insC). In addition tosequencing the sample in second aliquot, restriction analysis usingEaeI (loss of site on mutation) confirmed the sequence alteration.Panel E depicts homozygous deletion of five bases (c.448_452del-GAGGG), which has been marked by a box in the wild-typesequence. The mutation has been identified in multiple unrelatedpatients by sequencing. In addition, polyacrylamide gel electro-phoresis of the PCR amplified ATP7B exon 2 of the parents(obligate carrier) of the patients clearly showed evidence ofheteroduplex formation (data not shown). A polymorphism(c.447GTG>GTC), associated with this deletion has been indi-cated in the mutant strand
54

Cys271Stop mutation as well as the three unrelatedhomozygotes for c.448_452delGAGGG mutation.However, as shown in Table 3, there were clear differ-ences in the clinical profile between the homozygotes forthe above two mutations as well as with the singlehomozygote for the Tyr187Stop mutation. It wasinteresting to note that although the Cys271Stop ho-mozygotes produced truncated, functionless proteins,they presented with less severe phenotype. On the con-trary, three c.448_452delGAGGG homozygotes, with asimilar fate of the protein, showed more severe pheno-type with an earlier age of onset (8–10 years).
As Cys271Stop is the most prevalent mutationamong the patient pool, we identified a few hetero-zygotes for it, whose second mutation was also char-acterized. The clinical features of these compound
heterozygotes were compared to record the phenotypicdifferences resulting from the second heterogeneousmutation. As described in Table 4 the difference wasquite remarkable.
We observed interesting clinical differences betweentwo affected sibs with same genotype at the WD locus(c.448_452delGAGGG and a common haplotype forwhich underlying mutation has not been characterized).Both girls had a predominantly hepatic disease, in theform of heterogeneous echotexture and a shrunken li-ver size, splenomegaly and free fluid in the peritonealcavity—all indicating cirrhotic changes. Unlike otherWD patients bearing the c.448_452delGAGGG muta-tion, neither of these two WD-affected sibs had anyextrapyramidal symptoms (dystonic movements,bradykinesia or rigidity) or other neurological features.However, the age of onset of the elder sister was16 years while that of the younger sister was 14. Theyounger sister, in addition to the above hepatologicalfeatures, had psychological manifestations of WD inthe form of masked face, drooling of saliva, silly smileand impairment of memory and learning abilities, andalso had arthralgia, which persisted even after chelationtherapy.
Fig. 3 Location of the mutations in ATP7B. The ATP7B gene,consisting of 21 exons, is shown schematically. The copper bindingdomains (Cu1–6), transmembrane domains (Tm1–7), a transduc-tion domain (Td) that derives energy from ATP for metaltransport, the transmembrane cation channel/phosphorylationdomain (ChPh), and the ATP binding domain (ATP loop) areillustrated (Thomas et al. 1995b). All the mutations (boxed) andSNPs with high frequency are shown. Novel mutations and SNPsare italicized
Table 3 Comparison of clinical features between different homozygous mutants
Clinical parameters Cys271Stop (n= 3) Tyr187Stop (n= 1) c.448_452delGAGGG (n= 3)
Age of onset 17–19 years 11 years 8–10 yearsLiver involvement Not evident Hepatomegaly Not evidentExtrapyramidalfeatures
Coarse resting tremor,bradykinesia, rigidity,freezing in motion andpostural instability
Dystonic posturingand rigidity of limbs,shuffling gait,postural instability(L>R), tremor of lipsand perioral muscles(Rabbit syndrome)
No bradykinesia,tremor, chorea/athetosisor any dystonic movement.Only dystonic posturingand gait evident
Psychiatric symptoms Lability of mood andaffect, impairment ofcognition, withschizophrenia-likesymptoms
Mild, with alterationof behavior with respectto mood and affect,withdrawn nature andrefusal to talk
Cognitive functionimpaired in all caseswith no alteration ofmood and affect
Liver function tests Reduced Albumin:Globulin (1–1.2),Other liver and urinary function tests within normal limits
All liver function testswithin normal limits
All the patients listed had predominantly neurological disease, with no history of seizure
55

Discussion
ATP7B, the causal gene for WD, is fairly large in size(�80 kb) and mutations have been reported encom-passing the entire gene, which is interrupted by 21intervening sequences. Thus, mutation screening in WDpatients is an arduous job. In any population it would bedesirable to identify the prevalent mutations first, sincedownstream benefit in terms of the diagnostics as well asgenetic counseling would be available to a larger seg-ment of affected people. Towards this goal we hadundertaken haplotyping of the nuclear members of theaffected families to identify the mutant chromosomepresent in patient samples in larger numbers and char-acterize underlying common mutations with priority.
Among the prevalent mutations Cys271Stop, whichaccounts for �19% of the total mutations, has beendetected earlier in the European and Turkish popula-tion, but not as a prevalent one (Curtis et al. 1999).Interestingly, the Cys271Stop and c.1708-1G>C, an-other prevalent mutation, have not been reported fromMongoloid populations (Chinese, Japanese, Taiwaneseand Korean), although there is ample proof of Mon-goloid admixture with the Indian population. Also, wedid not find the prevalent mutation of the Mongoloidpopulation, i.e., Arg778Leu in our studied population(Chuang et al. 1996; Nanji et al. 1997; Kim et al. 1998;Liu et al. 2004). On the other hand, His1069Gln ac-counts for 20–40% of the WD mutations in differentCaucasian population groups (Thomas et al. 1995b;Shah et al. 1997). This mutation was not detected in ourstudy population although admixture of Caucasian andIndian blood has been reported in history before andduring the British colonization of India.
While screening for mutations in the Indian popula-tion it would be important to screen exon 2 first, wherewe observed largest number of nucleotide variantsincluding three prevalent mutations. Most of the de-tected mutations were found to be present in heterozy-gous condition in the patients that suggested that WD in
the Indian population results from a limited number ofrelatively frequent and a large number of rare mutations.To date comprehensive haplotyping data using SNPs inWD studies were not available. PCR-based assay usingnewly identified SNPs could be used to detect pre-symptomatic sib of WD patient instead of previouslyreported dinucleotide repeats flanking ATP7B which areprone to recombination (Gupta et al. 2003).
Most of our neurological patients are young (age ofonset <12 years), and the majority of them have nohepatic symptoms or signs on presentation even after1 year of follow-up. Lack of hepatic features at thisyoung age is noteworthy in the context of previous re-ports (Singh et al. 1989; Oder et al. 1991; Kalra et al.2000) suggesting that most early-onset cases of WDpresent with hepatic or mixed disease, and neurologicalpatients present at late second or third decade of life. Itis worthwhile to mention that the homozygotes for nulland frameshift mutations discussed previously, in spiteof earlier onset and severe phenotype, had respondedremarkably well to therapy comprising initial de-cop-pering with d-penicillamine and subsequent use of zincsulphate to prevent intestinal copper absorption.
Patients homozygous for the Cys271Stop mutationhad Parkinson-like features and their profile exactlycorrespond with one of the three subgroups based on theclinical and neuroimaging findings described by Oder etal. (1993). A reduced albumin–globulin ratio (1–1.2)with normal total protein points towards reduced pro-duction of albumin, due to chronic liver disease, in ab-sence of causes of loss of albumin from body (renalpathologies) and decreased protein intake (malnutrition,protein-losing enteropathies).
The patient heterozygous for Cys271Stop/Tyr187-Stop mutations would be expected to share similarphenotypic features with patients homozygous forCys271Stop mutation since in both the cases four ofthe Cu-binding domain of ATP7B would be lost.However, they exhibited substantial difference. Also,two young sibs dwelling in the same house exposed tosimilar environmental factors and sharing same two
Table 4 Comparison of clinical features of patients with Cys271Stop mutation in homozygous and heterozygous conditions
Clinical parameters Features exhibited byhomozygotes (n= 3)
HeterozygoteCys271Stop/Gly710Ser
HeterozygoteCys271Stop/c.1708-1G>C
HeterozygoteCys271Stop/Tyr187Stop
Age of onset 17–19 years 10 years 10 years 9 yearsExtrapyramidalsymptoms, in additionto dystonic posturingand rigidity— which iscommon to all patients
Coarse resting tremor,bradykinesia, rigidity,freezing in motion andpostural instability
Positive palmomentalreflex
No additional feature Chorea and action tremor
Higher mental functionsSignificant impairmentof cognitive functions
Bed-bound due tostuporous condition
Higher mental functions were preserved in both cases
Psychiatric features Lability of mood andaffect, with schizophrenia-likesymptoms
Impulsive and antisocialbehavior, anger outburstsshown previously
No significant feature was observed in both cases
Serum albumin:globulin 1.2:1 1.1:1 1:1 All results withinnormal limits
In all the cases the disease was predominantly neurological and there was no history of seizures
56

mutant alleles at WD locus show markedly differentphenotypes. These observations suggest the influence ofadditional modifier loci that modulate the diseasephenotype.
Acknowledgements The authors are thankful to the patients forparticipating in the study. The study is supported by a grant fromIndian Council of Medical Research (ICMR), Government of In-dia. AG is supported by a pre-doctoral fellowship from Council ofScientific and Industrial Research, India. DA is supported by afellowship from Indian Council of Medical Research (ICMR),India.
References
Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW (1993)The Wilson disease gene is a putative copper transportingP-type ATPase similar to the Menkes gene. Nat Genet 5:327–337
Chuang L, Wu H, Jang M, Wang T, Sue W, Lin B, Cox D, Tai T(1996) High frequency of two mutations in codon 778 in exon 8of the ATP7B gene in Taiwanese families with Wilson disease.J Med Genet 33:521–523
Curtis D, Durkie M, Balac (Morris) P, Sheard D, Goodeve A,Peake I, Quarrell O, Tanner S (1999) A study of Wilson diseasemutations in Britain. Hum Mutat 14:304–311
Figus A, Angius A, Loudianos G, Bertini C, Dessi V, Loi A,Deiana M, Lovicu M, Olla N, Sole G, De Virgiliis S, Lilliu F,Farci A, Nurchi A, Giacchino R, Barabino A, Marazzi M,Zancan L, Greggio N, Marcellini M, Solinas A, Deplano A,Barbera C, Devoto M, Ozsoylu S, Kocak N, Akar N, Karay-alcin S, Mokini V, Cullufi P, Balestrieri A, Cao A, Pirastu M(1995) Molecular pathology and haplotype analysis of Wilsondisease in Mediterranean populations. Am J Hum Genet57:1318–1324
Gupta A, Neogi R, Mukherjea M, Mukhopadhyay A, Roy-choudhury S, Senapati A, Gangopadhyay P, Ray K (2003)DNA linkage based diagnosis of Wilson disease in asymptom-atic siblings. Indian J Med Res 118:208–214
Kalra V, Khurana D, Mittal R (2000) Wilson’s disease—earlyonset and lessons from a pediatric cohort in India. Indian Pe-diatr 37:595–601
Kim EK, Yoo OJ, Song KY, Yoo HW, Choi SY, Cho SW, HahnSH (1998) Identification of three novel mutations and a highfrequency of the Arg778Leu mutation in Korean patients withWilson disease. Hum Mutat 11:275–278
Kumar S, Thapa BR, Kaur G, Prasad R (2005) Identification andmolecular characterization of 18 novel mutations in the ATP7Bgene from Indian Wilson disease patients: genotype. Clin Genet67:443–445
Levinson G, Gutman G (1987) Slipped-strand mispairing: a majormechanism for DNA sequence evolution. Mol Biol Evol 4:203–221
Liu X, Zhang Y, Liu T, Hsiao K, Zhang J, Gu X, Bao K, Yu L,Wang M (2004) Correlation of ATP7B genotype with pheno-type in Chinese patients with Wilson disease. World J Gastro-enterol 10:590–593
Loudianos G, Dessi V, Lovicu M, Angius A, Figus A, Lilliu F, DeVirgiliis S, Nurchi AM, Deplano A, Moi P, Pirastu M, Cao A(1999) Molecular characterization of wilson disease in theSardinian population—evidence of a founder effect. HumMutat 14:294–303
Lutsenko S, Petris M (2002) Function and regulation of themammalian copper transporting ATPases: insights from bio-chemical and cell biological approaches. J Memb Biol 191:1–12
Lutsenko S, Efremov R, Tsivkovskii R, Walker J (2002) Humancopper-transporting ATPase ATP7B (the Wilson’s diseaseprotein): biochemical properties and regulation. J BioenergBiomembr 34:351–362
Nanji MS, Nguyen VT, Kawasoe JH, Inui K, Endo F, Nakajima T,Anezaki T, Cox DW (1997) Haplotype and mutation analysis inJapanese patients with Wilson disease. Am J Hum Genet60:1423–1429
Oder W, Grimm G, Kollegger H, Ferenci P, Schneider B, Deecke L(1991) Neurological and neuropsychiatric spectrum of Wilson’sdisease: a prospective study of 45 cases. J Neurol 238:281–287
Oder W, Prayer L, Grimm G, Spatt J, Ferenci P, Kollegger H,Schneider B, Gangl A, Deecke L (1993) Wilson’s disease: evi-dence of subgroups derived from clinical findings and brainlesions. Neurology 43:120–124
Petrukhin K, Fischer S, Pirastu M, Tanzi R, Chernov I, Devoto M,Brzustowicz L, Cayanis E, Vitale E, Russo J, Matseoane D,Boukhgalter B, Wasco W, Figus A, Loudianos J, Cao A,Sternlieb I, Evgrafov O, Parano E, Pavone L, Warburton D,Ott J, Penchaszadeh G, Scheinberg I, Gilliam T (1993) Map-ping, cloning and genetic characterization of the region con-taining the Wilson disease gene. Nat Genet 5:338–343
Ray K, Roychowdhury S, Senapati A, Basu N, Gangopadhyay PK(2001) Recent advances in Wilson’s Disease. In: Sinha KK,Chandra P, Jha DK (eds) Advances in clinical neurosciences.East Zone Neuro CME Ranchi, India, pp 247–258
Sambrook J, Russell DW (2001) Molecular cloning: a laboratorymanual. Cold Spring Harbor press, Cold Spring Harbor, NY
Shah AB, Chernov I, Zhang HT, Ross BM, Das K, Lutsenko S,Parano E, Pavone L, Evgrafov O, Ivanova-Smolenskaya IA,Anneren G, Westermark K, Urrutia FH, Penchaszadeh GK,Sternlieb I, Scheinberg IH, Gilliam TC, Petrukhin K (1997)Identification and analysis of mutations in the Wilson diseasegene (ATP7B): population frequencies, genotype–phenotypecorrelation, and functional analyses. Am J Hum Genet 61:317–328
Singh S, Dilawari JB, Chawla Y, Walia BN (1989) Wilson’s diseasein young children from northern India. Trop Gastroenterol10:46–50
Sternlieb I (1990) Perspectives on Wilson’s disease. Hepatology12:1234–1239
Suzuki M, Gitlin JD (1999) Intracellular localization of the Menkesand Wilson’s disease proteins and their role in intracellularcopper transport. Pediatr Int 41:436–442
Tatusova T, Madden T (1999) BLAST 2 Sequences, a new tool forcomparing protein and nucleotide sequences. FEMS MicrobiolLett 174:247–250
Thomas GR, Bull PC, Roberts EA, Walshe JM, Cox DW (1994)Haplotype studies in Wilson disease. Am J Hum Genet 54:71–78
Thomas GR, Roberts EA, Walshe JM, Cox DW (1995a) Haplo-types and mutations in Wilson disease. Am J Hum Genet56:1315–1319
Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW(1995b) The Wilson disease gene: spectrum of mutations andtheir consequences. Nat Genet 9:210–217
Waldenstrom E, Lagerkvist A, Dahlman T, Westermark K,Landegren U (1996) Efficient detection of mutations in Wilsondisease by manifold sequencing. Genomics 37:303–309
Wilson S (1912) Progressive lenticular degeneration: a familialnervous disease associated with cirrhosis of liver. Brain 34:295–509
57




















