
Molecular cloning and functional characterization ... - doi@nrct
216
MOLECULAR CLONING AND FUNCTIONAL CHARACTERIZATION OF CALRETICULIN FROM THE HUMAN LIVER FLUKE, OPISTHORCHIS VIVERRINI BY MISS WANLAPA CHAIBANGYANG A DISSERTATION SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF THE DOCTOR OF PHILOSOPHY (BIOMEDICAL SCIENCES) GRADUATE PROGRAM IN BIOMEDICAL SCIENCES FACULTY OF ALLIED HEALTH SCIENCES THAMMASAT UNIVERSITY ACADEMIC YEAR 2017 COPYRIGHT OF THAMMASAT UNIVERSITY Ref. code: 25605412330010EAN
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of Molecular cloning and functional characterization ... - doi@nrct

MOLECULAR CLONING AND FUNCTIONAL
CHARACTERIZATION OF CALRETICULIN
FROM THE HUMAN LIVER FLUKE,
OPISTHORCHIS VIVERRINI
BY
MISS WANLAPA CHAIBANGYANG
A DISSERTATION SUBMITTED IN PARTIAL FULFILLMENT
OF THE REQUIREMENTS FOR THE DEGREE OF
THE DOCTOR OF PHILOSOPHY (BIOMEDICAL SCIENCES)
GRADUATE PROGRAM IN BIOMEDICAL SCIENCES
FACULTY OF ALLIED HEALTH SCIENCES
THAMMASAT UNIVERSITY
ACADEMIC YEAR 2017
COPYRIGHT OF THAMMASAT UNIVERSITY
Ref. code: 25605412330010EAN

MOLECULAR CLONING AND FUNCTIONAL
CHARACTERIZATION OF CALRETICULIN
FROM THE HUMAN LIVER FLUKE,
OPISTHORCHIS VIVERRINI
BY
MISS WANLAPA CHAIBANGYANG
A DISSERTATION SUBMITTED IN PARTIAL FULFILLMENT
OF THE REQUIREMENTS FOR THE DEGREE OF
THE DOCTOR OF PHILOSOPHY (BIOMEDICAL SCIENCES)
GRADUATE PROGRAM IN BIOMEDICAL SCIENCES
FACULTY OF ALLIED HEALTH SCIENCES
THAMMASAT UNIVERSITY
ACADEMIC YEAR 2017
COPYRIGHT OF THAMMASAT UNIVERSITY
Ref. code: 25605412330010EAN


(1)
Dissertation Title MOLECULAR CLONING AND FUNCTIONAL
CHARACTERIZATION OF CALRETICULIN
FROM THE HUMAN LIVER FLUKE,
OPISTHORCHIS VIVERRINI
Author Miss Wanlapa Chaibangyang
Degree Doctor of Philosophy in Biomedical Sciences
Department/Faculty/University Graduate Program in Biomedical Sciences
Faculty of Allied Health Sciences
Thammasat University
Dissertation Advisor Associate Professor Hans Rudi Grams, Dr. rer. nat.
Dissertation Co‐Advisor Professor Peter Smooker, Ph.D.
Dissertation Co‐Advisor Associate Professor Smarn Tesana, Ph.D.
Dissertation Co‐Advisor Associate Professor Suksiri Vichasri Grams, Dr.
rer. nat.
Dissertation Co‐Advisor Assistant Professor Amornrat Geadkaew Krenc,
Ph.D.
Academic Year 2017
ABSTRACT
Opisthorchis viverrini (Ov), a human liver fluke endemic in parts of
Thailand, causes opisthorchiasis and is associated with cholangiocarcinoma, a serious
health problem in the country. Calreticulin (CALR) is an endoplasmic reticulum‐
resident multifunctional protein in mammals that is involved in various biological
functions inside and outside the cell including calcium homeostasis, chaperoning,
apoptotic cell clearance, cell adhesion, as well as angiogenesis. In this study, RNA
expression and protein distribution of O. viverrini calreticulin (OvCALR) were
analyzed, and its intracellular and extracellular functions were investigated. A cDNA
encoding OvCALR was cloned from total RNA of the adult parasite. The deduced
amino acid sequence of OvCALR contains various characteristics of calreticulin
including the calreticulin family signature 1 and 2, a signal peptide, an ER retention
Ref. code: 25605412330010EAN

(2)
signal, three functional and structural domains (N‐, P‐, and C‐domain), three
conserved cysteine residues, and three each of two tandem repeated motifs.
Additionally, it has very high sequence conservation at 96.6% identity to Clonorchis
sinensis calreticulin and significantly less to blood fluke calreticulin, Schistosoma
mansoni (57.5%) and S. japonicum (53.6%), and mammalian calreticulin, Mus
musculus (50.7%) and Homo sapiens (50.9%), respectively. OvCALR mRNA was
detected by reverse transcription PCR in all analyzed developmental stages from the
newly excysted juvenile to the mature parasite. Western blot analyses using mouse
anti‐rOvCALR antiserum detected native protein in soluble and insoluble crude
parasite extracts as well as in the excretory/secretory product of the adult parasite.
Moreover, rOvCALR was recognized by Ov‐infected hamster sera starting eight
weeks postinfection. Immunohistochemical detection of OvCALR in adult
O. viverrini revealed its presence in several tissues and cell types, including
tegumental cell bodies, cecal epithelium, testes, ovary, prostate gland, Mehlis’ gland,
vitelline cells, eggs, and lining of seminal vesicle. OvCALR was not detected in liver
tissue of Ov‐infected hamster. Recombinant OvCALR demonstrated in vitro
protection of citrate synthase from thermal‐induced aggregation. Recombinant
OvCALR C‐domain showed a mobility shift in native‐PAGE in the presence of
calcium. Recombinant OvCALR was shown to bind to human C1q in a dose‐
dependent manner and to suppress C1q‐mediated hemolysis of human sensitized red
blood cells. In addition, it also was shown to inhibit proliferation, migration, and
sprouting of human endothelial cells in vitro. The observed effects were similar to
those observed in rMmCALR. However, transacetylase activity of rOvCALR as
previously reported for Haemonchus contortus calreticulin could not be observed. In
conclusion, OvCALR has ER‐functions including chaperoning and calcium binding
that are comparable to human calreticulin. Moreover, OvCALR is a released protein
from the parasite with potential to modulate the host immune system and
cholangiocarcinoma development from its anti‐angiogenesis activity and C1q‐
mediated interference of the host immune system. OvCALR is an interesting molecule
that might be a target for opisthorchiasis control and diagnosis.
Ref. code: 25605412330010EAN

(3)
Keywords: Platyhelminthes, CALR, calcium-binding, chaperone, transacetylase, cell
proliferation, cell motility, endothelial sprouting, angiogenesis, complement C1q
Ref. code: 25605412330010EAN

(4)
ACKNOWLEDGEMENTS
This dissertation would not be accomplished without any supports and
collaboration of many persons and institutes. The most important person who gave me
an invaluable opportunity to do my Ph.D. is my major advisor, Associate Professor
Dr. Hans Rudi Grams. I would like to express my sincere gratitude and greatest
appreciation to him for the generous guidance and the endless support throughout of
my study in his laboratory at Thammasat University.
I am very thankful to all co‐advisors of my dissertation. I would like to
express my deep thankfulness to Associate Professor Dr. Smarn Tesana, Department
of Parasitology, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand
for his valuable advice and generous support. Equally, I would like to express my
deep gratefulness to Associate Professor Dr. Suksiri Vichasri Grams for her
encouragement, valuable suggestions and comments. Likewise, my deepest
appreciation goes to Assistant Professor Dr. Amornrat Geadkaew Krenc for her
kindness support and guidance in my laboratory work.
I would like to offer my special thanks and deepest appreciation to my
foreign co‐advisor, Professor Dr. Peter Smooker, School of Science, Biosciences and
Food Technology, RMIT University, Australia who provided the great opportunity to
conduct my research in his laboratory for his encouragement, kindness, and support
throughout the ten months of my visit. I would like to thank Dr. Paul Ramsland and
Dr. Anna Walduck, School of Science, Biosciences and Food Technology, RMIT
University, Australia for their kind support and useful suggestions and comments.
Special thanks also to all students and staff at Biotechnology Laboratory, School of
Science, RMIT University for their help, encouragement, and marvelous time in
Melbourne, Australia.
I am deeply grateful to Professor Major General Dr. Oytip Nathalang and
Assistant Professor Dr. Potjanee Srimanote for their valuable suggestions and kindly
providing materials to complete my laboratory work. I would like to give my
appreciation to Associate Professor Dr. Ratchaneewan Aunpad for kindly providing
materials and the opportunity to use her laboratory facilities. I am also thankful to
Ref. code: 25605412330010EAN

(5)
Assistant Professor Dr. Tullayakorn Plengsuriyakarn, Graduate Studies, Chulabhorn
International College of Medicine, Thammasat University for kindly providing
paraffin‐embedded hamster liver and O. viverrini‐infected hamster liver.
I would like to express my gratitude to the members of my dissertation
defense committee, Associate Professor Dr. Smarn Tesana, Professor Dr. Peter
Smooker, Associate Professor Dr. Ratchaneewan Aunpad, and Associate Professor
Dr. Poom Adisakwattana, Faculty of Tropical Medicine, Mahidol University, for their
acceptance to be my dissertation defense committee and kindness to correct my
dissertation.
My study would have not been possible without financial support from
the Thailand Research Fund (TRF) and Thammasat University through the Royal
Golden Jubilee Ph.D. Program (RGJ-PHD) under the supervision of Associate
Professor Dr. Hans Rudi Grams.
I would like to thank all friends, instructors, and staff members of the
Graduate Program in Biomedical Sciences, Faculty of Allied Health Sciences,
Thammasat University for their support, especially all members of the Molecular
Biology and Parasitology Unit.
Last but not least, I would like to express my deepest appreciation to my
beloved family for their consistent support and the greatest encouragement.
Miss Wanlapa Chaibangyang
Ref. code: 25605412330010EAN

(6)
TABLE OF CONTENTS
Page
ABSTRACT (1)
ACKNOWLEDGEMENTS (4)
TABLE OF CONTENTS (6)
LIST OF TABLES (14)
LIST OF FIGURES (15)
LIST OF ABBREVATIONS (19)
CHAPTER 1 INTRODUCTION 1
CHAPTER 2 OBJECTIVES 3
CHAPTER 3 REVIEW OF LITERATURES 4
3.1 Opisthorchis viverrini 4
3.1.1 Taxonomy and typical characterization 4
3.1.2 Life cycle 4
3.1.3 Morphology 6
3.1.3.1 Adult 6
3.1.3.2 Egg 6
3.1.3.3 Miracidium 6
3.1.3.4 Sporocyst 7
3.1.3.5 Redia 7
Ref. code: 25605412330010EAN

(7)
TABLE OF CONTENTS (Cont.)
Page
3.1.3.6 Cercaria 7
3.1.3.7 Metacercaria 7
3.1.4 Geographical distribution 8
3.1.5 Clinical manifestation and pathogenesis 10
3.1.6 Diagnosis 12
3.1.7 Treatment and control 13
3.1.8 Opisthorchis viverrini‐associated cholangiocarcinoma 14
3.2 Calreticulin (CALR) 19
3.2.1 Genes and structure of calreticulin 20
3.2.2 Biological functions of calreticulin 25
3.2.2.1 Calreticulin functions inside of the ER 25
(1) Chaperone activity 25
(2) Calcium homeostasis 27
3.2.2.2 Calreticulin functions outside of the ER 29
(1) Cell adhesion 29
(2) Focal adhesion disassembly and cell migration 29
(3) Phagocytosis 30
(4) Wound healing 30
(5) Angiogenic activity 31
3.2.3 Calreticulin of pathogenic protozoans and helminths 33
3.2.3.1 Protozoans 33
3.2.3.2 Helminths 36
(1) Nematodes 36
(2) Cestode 37
(3) Trematodes 37
CHAPTER 4 MATERIALS AND METHODS 39
Ref. code: 25605412330010EAN

(8)
TABLE OF CONTENTS (Cont.)
Page
4.1 Molecular cloning of Opisthorchis viverrini calreticulin 39
(OvCALR)
4.1.1 Preparation of total RNA 39
4.1.1.1 Isolation of adult O. viverrini total RNA 39
4.1.1.2 Separation of nucleic acids by agarose gel 39
electrophoresis
4.1.1.3 DNase I treatment of O. viverrini total RNA 40
4.1.2 Reverse transcription and PCR amplification of 40
OvCALR
4.1.3 Ligation of OvCALR cDNA into pGEM®‐T Easy 43
vector and transformation of Escherichia coli (E. coli)
cells with ligation product
4.1.4 Preparation of chemically competent E. coli cells 44
4.1.5 Transformation of E. coli cells 45
4.1.6 Isolation of plasmid DNA using alkaline extraction 46
method
4.1.7 Positive transformants screening by colony PCR 46
4.1.8 Sequence analysis 47
4.1.9 Storage of bacterial clones 48
4.2 Production of rOvCALR using a bacterial system 48
4.2.1 Construction of recombinant expression vector 48
4.2.1.1 Construction of pQE‐30‐OvCALR expression 48
vector
4.2.1.2 Construction of pET21b(+)‐OvCALR expression 49
vector
(1) DNA cloning of OvCALR 49
(2) Ligation of OvCALR into pET21b(+) vector 50
Ref. code: 25605412330010EAN

(9)
TABLE OF CONTENTS (Cont.)
Page
4.2.2 Small‐scale expression and time‐course analysis 51
4.2.3 Determination of target‐protein solubility 52
4.2.4 Large‐scale expression of rOvCALR 52
4.2.4.1 Large‐scale expression of M15‐derived 52
rOvCALR
4.2.4.2 Large‐scale expression of Rosetta‐ 52
gami(DE3)pLysS‐derived rOvCALR
4.2.5 Protein analysis by sodium dodecyl sulfate‐ 53
polyacrylamide gel electrophoresis (SDS‐PAGE)
4.2.5.1 Gel casting, sample preparation and 53
electrophoresis
4.2.5.2 Staining of protein in polyacrylamide gel 54
4.2.6 Purification of rOvCALR using Ni‐NTA affinity 54
chromatography
4.2.6.1 Purification of rOvCALR under denaturing 54
conditions
4.2.6.2 Purification of rOvCALR under native 55
conditions
4.2.7 Concentration and dialysis of rOvCALR 55
4.2.7.1 Concentration of rOvCALR using centrifugal 55
concentrator
4.2.7.2 Dialysis of rOvCALR 55
4.2.8 Measurement of rOvCALR concentration using 56
Bradford’s assay
4.3 Production of polyclonal antibody in mice 56
4.3.1 Production of polyclonal mouse anti‐rOvCALR 56
antibody
4.3.2 Production of polyclonal mouse anti‐OvES antibody 57
Ref. code: 25605412330010EAN

(10)
TABLE OF CONTENTS (Cont.)
Page
4.3.3 Determination of antibody titer by enzyme linked 57
immunosorbent assay (ELISA)
4.4 Preparation of parasite antigens 58
4.4.1 Parasite crude extracts 58
4.4.2 Excretory/secretory (ES) product 58
4.4.3 Measurement of protein concentration using BCA assay 59
4.5 Characterization of OvCALR expression 59
4.5.1 Preparation of different developmental stages of 59
O. viverrini
4.5.2 Stage‐specific reverse transcriptase polymerase chain 60
reaction (RT‐PCR)
4.5.3 Detection of native OvCALR using Western blot 61
analysis
4.5.3.1 Protein blotting by semi‐dry transfer 61
4.5.3.2 Detection of protein blotting 62
4.5.4 Immunohistochemical detection of OvCALR 63
4.6 Detection of rOvCALR by O. viverrini‐infected hamster sera 64
4.6.1 Blood collection from O. viverrini‐infected hamsters 64
4.6.2 Indirect ELISA 64
4.6.3 Western blot analysis 65
4.7 Functional analysis of rOvCALR 65
4.7.1 Production of experimental control proteins 65
4.7.1.1 Production of recombinant Mus musculus 65
calreticulin (rMmCALR)
(1) Isolation of mouse total RNA 65
(2) Molecular cloning of MmCALR 65
(3) Construction of pET21b(+)‐MmCALR 67
expression vector
Ref. code: 25605412330010EAN

(11)
TABLE OF CONTENTS (Cont.)
Page
(4) Expression and purification of rMmCALR 68
4.7.1.2 Production of recombinant Fasciola gigantica 68
calcium‐binding protein 1 (rFgCaBP1)
4.7.1.3 Production of recombinant Schistosoma japonicum 68
glutathione S‐transferase (rSjGST)
4.7.2 Thermal induced protein aggregation assay 69
4.7.3 Calcium binding assay 69
4.7.3.1 Production of rOvCALR C‐domain 69
(1) Construction of pGEX‐5X‐1‐OvCALR C‐domain 69
expression vector
(2) Expression of rSjGST‐OvCALR C‐domain 71
(3) Purification of rSjGST‐OvCALR C‐domain 72
4.7.3.2 Determination of calcium‐binding activity using 72
native‐PAGE
4.7.4 Determination of transacetylase activity 73
4.7.4.1 Indirect transacetylase activity measurement 73
using GST assay
4.7.4.2 Western blot analysis of acetylated protein 74
4.7.5 C1q binding assay 75
4.7.6 Hemolytic assay 75
4.7.7 In vitro angiogenesis assays 76
4.7.7.1 Preparation of recombinant proteins for tissue 76
culture
4.7.7.2 Cell culture 77
4.7.7.3 MTT assay 77
4.7.7.4 Scratch assay 78
4.7.7.5 Tube formation assay 78
Ref. code: 25605412330010EAN

(12)
TABLE OF CONTENTS (Cont.)
Page
CHAPTER 5 RESULTS 80
5.1 Molecular cloning of Opisthorchis viverrini calreticulin 80
(OvCALR) cDNA and its sequence analysis
5.2 Production of rOvCALR and other control proteins using a 88
bacterial system
5.2.1 Production of rOvCALR 88
5.2.1.1 Small‐scale expression of rOvCALR 88
5.2.1.2 Large‐scale expression and purification of 88
rOvCALR
5.2.2 Production of rOvCALR C‐domain 94
5.2.3 Production of recombinant Mus musculus calreticulin 95
(rMmCALR)
5.2.4 Production of recombinant Fasciola gigantica 99
calcium‐binding protein 1 (rFgCaBP1)
5.2.5 Production of recombinant Schistosoma japonicum 100
glutathione S‐transferase (rSjGST)
5.3 Production of polyclonal antibody in mice 101
5.3.1 Production of polyclonal mouse anti‐rOvCALR 101
antibody
5.3.2 Production of polyclonal mouse anti‐OvES antibody 102
5.4 Characterization of OvCALR expression in O. viverrini 103
5.4.1 Stage‐specific reverse transcriptase polymerase chain 103
reaction (RT‐PCR)
5.4.2 Western blot analyses 104
5.4.3 Immunohistochemical analyses 108
5.5 Analysis of OvCALR‐specific antibody response by 112
O. viverrini‐infected hamster sera
Ref. code: 25605412330010EAN

(13)
TABLE OF CONTENTS (Cont.)
Page
5.6 Functional analysis of rOvCALR 114
5.6.1 Chaperoning activity 114
5.6.2 Calcium‐binding activity 116
5.6.3 Transacetylase activity 117
5.6.4 Human C1q binding 120
5.6.5 Inhibition of classical complement pathway 121
5.6.6 In vitro angiogenic properties 123
5.6.6.1 Endothelial cell proliferation 124
5.6.6.2 Endothelial cell migration 125
5.6.6.3 Endothelial cell sprouting 126
CHAPTER 6 DISCUSSION 128
CHAPTER 7 CONCLUSIONS 136
REFERENCES 138
APPENDICES 165
APPENDIX A SPECIFICATIONS OF DNA VECTORS 166
APPENDIX B REAGENT PREPARATIONS 170
APPENDIX C ANIMAL ETHICS PERMISSION 183
APPENDIX D HUMAN ETHICS PERMISSION 184
BIOGRAPHY 185
Ref. code: 25605412330010EAN

(14)
LIST OF TABLES
Table Page
4.1 List of primer 42
4.2 List of E. coli strains and appropriate antibiotic concentrations 46
Ref. code: 25605412330010EAN

(15)
LIST OF FIGURES
Figure Page
3.1 Life cycle of the liver fluke, Opisthorchis viverrini 5
3.2 The prevalence of O. viverrini and C. sinensis 9
3.3 Incidence of liver and bile duct cancer in different regions 15
of Thailand during the year 2010–2012
3.4 Incidence of cholangiocarcinoma worldwide reported 16
between 1977 and 2007
3.5 Proposed pathogenesis mechanisms of O. viverrini 18
infection‐induced cholangiocarcinoma (CCA)
3.6 Genes, mRNAs, and protein of calreticulin in human 21
3.7 Structure of calreticulin 23
3.8 Comparison of amino acids sequences of calreticulin from 24
different species
3.9 Calreticulin chaperone activity in glycoprotein folding 26
control
3.10 Functions of calreticulin and its dependent pathway 28
3.11 Schematic representation of calreticulin‐mediated 31
phagocytosis
3.12 The complement pathway 34
4.1 Timeline diagram of anti‐rOvCALR antisera production in 57
Mice
5.1 Agarose gel showing the RT‐PCR product of OvCALR 82
obtained from adult O. viverrini total RNA
5.2 Agarose gel showing the OvCALR cDNA product obtained 83
from transformant‐screening methods
5.3 Nucleic acid and deduced amino acid sequences of OvCALR 84
5.4 Signal peptide prediction of OvCALR by SignalP 4.1 85
Ref. code: 25605412330010EAN

(16)
LIST OF FIGURES (Cont.)
Figure Page
5.5 Multiple alignment of the deduced amino acid sequences 86
of calreticulin from O. viverrini (UniProt: A0A1P8P1U1),
C. sinensis (UniProt: K7NB00), S. mansoni
(UniProt: Q06814), S. japonicum (UniProt: O45034),
M. musculus (UniProt: P14211), H. sapiens (UniProt: P27797)
5.6 Time‐course and target‐protein solubility analyses of E. coli 89
M15/pQE30‐OvCALR
5.7 Protein expression and target‐protein solubility analyses 90
of E. coli Rosetta‐gami(DE3)pLysS/pET21b(+)‐OvCALR
5.8 Purification of rOvCALR using Ni‐NTA affinity 91
chromatography under denaturing conditions
5.9 Purification of rOvCALR using Ni‐NTA affinity 92
chromatography under native conditions
5.10 SDS‐PAGE showing purified rOvCALR (5 µg) obtained 93
from denaturing purification
5.11 Purification of rSjGST‐OvCALR C‐domain using glutathione 94
affinity chromatography
5.12 Agarose gel showing (A) total RNA isolated from mouse 96
(B) the RT‐PCR product obtained from mouse total RNA
with specific primers for MmCALR
5.13 Protein expression and target‐protein solubility analyses 97
of E. coli Rosetta‐gami(DE3)pLysS/pET21b(+)‐MmCALR
5.14 Purification of rMmCALR using Ni‐NTA affinity 98
chromatography under native conditions
5.15 Purification of rFgCaBP1 using Ni‐NTA affinity 99
chromatography under native conditions
5.16 Purification of rSjGST using glutathione affinity 100
Chromatography
Ref. code: 25605412330010EAN

(17)
LIST OF FIGURES (Cont.)
Figure Page
5.17 Line graph showing antibody level of polyclonal 101
anti‐rOvCALR antibody (dilution 1:400) determined
by indirect ELISA
5.18 Line graph showing antibody level of polyclonal anti‐OvES 102
antibody (dilution 1:6,400) determined by indirect ELISA
5.19 Stage‐specific analysis of OvCALR from total RNA of 103
newly excysted juveniles (NEJ), 2‐, 4‐ and 8‐week‐old
parasites (2wk, 4wk and 8wk) by reverse transcription PCR
5.20 SDS‐PAGE of parasite extracts from adult O. viverrini 105
5.21 Western blot detection of OvCALR by anti‐rOvCALR 106
antisera from three mice
5.22 Western blot detection of OvCALR by anti‐rOvCALR 107
Antiserum
5.23 Immunohistochemical detection of OvCALR in adult 109
O. viverrini
5.24 Immunohistochemical detection of O. viverrini antigens 110
in infected hamster liver sections
5.25 Graph of the absorbance values (mean ± SD) of preinfection 112
and 12‐week postinfection hamster sera (n=10) against
rOvCALR detected by indirect ELISA
5.26 Indirect ELISA and immunoblot detection of rOvCALR by 113
pooled (n=10) preinfection and postinfection hamster sera
5.27 Line graph showing protective effect of OvCALR on citrate 115
synthase (CS) against thermal induced aggregation
5.28 Native PAGE showing shift in the migration of OvCALR in 116
the system presenting of calcium
5.29 Bar graph showing transacetylase activity of calreticulin 118
determined by GST activity assay
Ref. code: 25605412330010EAN

(18)
LIST OF FIGURES (Cont.)
Figure Page
5.30 Immunoblot detection of protein acetylation by 119
anti‐acetylated lysine antibody
5.31 Graph showing human C1q binding of OvCALR obtained 120
by competitive ELISA
5.32 Bar graph showing effect of rOvCALR against classical 122
complement‐mediated hemolysis
5.33 Bar graph showing effect of rOvCALR on HUVECs 123
proliferation determined by MTT assay
5.34 Effect of rOvCALR on HUVECs migration determined 125
by scratch assay
5.35 Effect of rOvCALR on HUVECs sprouting determined by 127
tube formation assay
Ref. code: 25605412330010EAN

(19)
LIST OF ABBREVIATIONS
Symbols/Abbreviations Terms
%
% (v/v)
% (w/v)
Percent
Volume/volume percent
Weigh/volume percent
×g Gravitational acceleration
°C
α
β
γ
Degree Celsius
Alpha
Beta
Gamma
µg Microgram(s)
µl Microliter(s)
µm
1st
2nd
7‐AMC
8‐oxodG
AA
ABC
AEC
ANOVA
APS
ASR
BCA
BCIP/NBT
BiP
BLAST
bp
Micrometer(s)
First
Second
7‐Acetoxy‐4‐methylcoumarin
8-Hydroxydeoxyguanosine
Amino acid
Avidin‐biotin complex
3‐amino‐9‐ethylcarbazole
Analysis of variance
Ammonium persulfate
Age‐standardized incidence rates
Bicinchoninic acid
5‐Bromo‐4‐chloro‐3‐indolyl phosphate/ Nitro
blue tetrazolium chloride
Immunoglobulin heavy‐chain binding protein
The Basic Local Alignment Search Tool
Base pair(s)
Ref. code: 25605412330010EAN

(20)
LIST OF ABBREVIATIONS (Cont.)
Symbols/Abbreviations Terms
BSA
C. sinensis
C1q
Ca
CaBP3
CALR
CAM
CCA
CD4+
CD47
CD91
cDNA
CDNB
cm2
CNX
CO2
CRP55
CRTase
CW
CYPR
DEPC
DMSO
DNA
DNase
Bovine serum albumin
Clonorchis sinensis
Complement component 1q
Calcium
Calcium binding protein 3
Calreticulin
Chick chorioallantoic membrane
Cholangiocarcinoma
T‐helper cell
Cluster of Differentiation 47 or Integrin‐
associated protein
Cluster of Differentiation 91 or Low‐density‐
lipoprotein‐related protein 1
Complementary deoxyribonucleic acid
1‐chloro‐2,4‐dinitrobenzene
Square centimeter
Calnexin
Carbon dioxide
55 kDa calcium binding protein of the ER
lumen
Calreticulin transacetylase
Crude worm extract
NADPH cytochrome c reductase
Diethyl pyrocarbonate
Dimethyl sulfoxide
Deoxyribonucleic acid
Deoxyribonuclease
Ref. code: 25605412330010EAN

(21)
LIST OF ABBREVIATIONS (Cont.)
Symbols/Abbreviations Terms
dNTPs
dT
DTT
DW
E. coli
E. dispar
E. histolytica
e.g.
EBV
EDTA
EhCALR
ELISA
EMBOSS
eNOS
ER
ERAD
ERp57
ERp60
ES
FAK
FECT
FgCaBP1
FGF
Grp78
h
H. contortus
Deoxy nucleoside triphosphate(s)
Deoxythymine
Dithiothreitol
Distilled water
Escherichia coli
Entamoeba dispar
Entamoeba histolytica
Exempli gratia (for example)
Epstein‐Barr virus
Ethylenediaminetetraacetic acid
Entamoeba histolytica calreticulin
Enzyme‐linked immunosorbent assay
European Molecular Biology Open Software
Suite
Endothelial nitric oxide synthase
Endoplasmic reticulum
ER‐associated degradation
ER protein 57
Endoplasmic reticulum resident protein 60
Excretory/secretory products
Focal adhesion kinase
Formalin ethyl‐acetate concentration technique
Fasciola gigantica calcium‐binding protein 1
Fibroblast growth factor
Immunoglobulin heavy‐chain binding protein
Hour(s)
Haemonchus contortus
Ref. code: 25605412330010EAN

(22)
LIST OF ABBREVIATIONS (Cont.)
Symbols/Abbreviations Terms
H2O2
HCC
HpCALR
HPCVE
HRP
HUVEC
IARC
IFN
Ig
IL
InsP3R
IPTG
Kb
KD
kDa
kg
L. donovani
L‐NAME
LAMP
Lao PDR
LB
LC‐MS/MS
LdCALR
LRP1
mA
MAC
MAPK
Hydrogen peroxide
Hepatocellular carcinoma
Heligmosomoides polygyrus calreticulin
Horseradish peroxidase
Human placenta chorionic villi explants
Human umbilical vein endothelial cell
International Agency for Research on Cancer
Interferon
Immunoglobulin
Interleukin
Inositol 1,4,5‐triphosphate receptor
Isopropyl‐1‐thio‐β‐D‐galactopyranoside
Kilobase(s)
Dissociation constant
Kilo Dalton
Kilogram(s)
Leishmania donovani
NG‐nitro‐L‐Arginine ethyl ester
Loop‐mediated isothermal amplification
Lao People's Democratic Republic
Luria Bertani
Liquid chromatography‐mass spectrometry
Leishmania donovani calreticulin
Low‐density‐lipoprotein‐related protein 1
Milliampere(s)
Membrane‐attack complex
Mitogen‐activated protein kinase
Ref. code: 25605412330010EAN

(23)
LIST OF ABBREVIATIONS (Cont.)
Symbols/Abbreviations
MEF
mg
MHC
MIF
min
ml
mm
mM
mRNA
n
N. americanus
NCBI
NF‐AT
Ni‐NTA
nm
NMR
NO
NOS
OD
O. felineus
OPD
Ov
O. viverrini
OvCALR
Ov‐GRN‐1
Ov‐Trx‐1
Terms
Myocyte‐enhancer factor
Milligram(s)
Major histocompatibility complex
Minute intestinal flukes
Minute(s)
Milliliter(s)
Millimeter(s)
Millimolar(s)
Messenger ribonucleic acid
Sample size
Necator americanus
National Center for Biotechnology Information
Nuclear factor of activated T‐cells
Nickle‐nitrilotriacetic acid
Nanometer(s)
Nuclear magnetic resonance
Nitric oxide
Nitric oxide synthase
Optical density
Opisthorchis felines
O‐Phenylenediamine dihydrochloride
Opisthorchis viverrini
Opisthorchis viverrini
Opisthorchis viverrini calreticulin
Opisthorchis viverrini granulin‐like growth
factor
Opisthorchis viverrini thioredoxin
Ref. code: 25605412330010EAN

(24)
LIST OF ABBREVIATIONS (Cont.)
Symbols/Abbreviations Terms
PBS
PBST
PCR
PDI
PEDF
pH
PS
r
RNA
RNase
ROI
ROS
rpm
RT‐PCR
s
S. haematobium
S. japonicum
S. mansoni
SDS
SDS‐PAGE
SERCA
ShCALR
SjGST
Phosphate buffered saline
Phosphate buffered saline with Tween® 20
Polymerase chain reaction
Protein disulfide isomerase
Pigment epithelium‐derived factor
Negative logarithm of hydrogen ion
concentration
Phosphatidylserine
Recombinant
Ribonucleic acid
Ribonuclease
Reactive oxygen intermediates
Reactive oxygen species
Round per minute
Reverse transcriptase polymerase chain
reaction
Second(s)
Schistosoma haematobium
Schistosoma japonicum
Schistosoma mansoni
Sodium dodecyl sulfate
Sodium dodecyl sulfate‐Polyacrylamide gel
electrophoresis
Sarco/endoplasmic reticulum calcium ATPase
Schistosoma haematobium calreticulin
Schistosoma japonicum glutathione S‐
transferase
Ref. code: 25605412330010EAN

(25)
LIST OF ABBREVIATIONS (Cont.)
Symbols/Abbreviations
SR
SR‐A
T. solium
TBE
TBS
TcCALR
TEMED
TGF
TM
TMB
TNF
TsCALR
TSP
UDP
UGGT
UTR
UV
V
VEGF
WHO
Terms
Sarcoplasmic reticulum
Scavenger receptor type A
Taenia solium
Tris/Borate/EDTA
Tris buffered saline
Trypanosoma cruzi calreticulin
Tetramethylethylenediamine
Transforming growth factor
Melting temperature
3,3′,5,5′‐Tetramethylbenzidine
Tumor necrosis factor
Taenia solium calreticulin
Thrombospondin
Uridine diphosphate
UDP‐glucose/glycoprotein glucosyltransferase
Untranslated region
Ultraviolet
Volt(s)
Vascular endothelial growth factor
World Health Organization
Ref. code: 25605412330010EAN

(26)
LIST OF ABBREVIATIONS (Cont.)
Amino acid codes
Full name 1-letter abbreviation 3-letter abbreviation
Alanine A Ala
Arginine R Arg
Asparagine N Asn
Aspartic acid D Asp
Cysteine C Cys
Glutamic acid E Glu
Glutamine Q Gln
Glycine G Gly
Histidine H His
Isoleucine I Ile
Leucine L Leu
Lysine K Lys
Methionine M Met
Phenylalanine F Phe
Proline P Pro
Serine S Ser
Threonine T Thr
Tryptophan W Trp
Tyrosine Y Tyr
Valine V Val
Ref. code: 25605412330010EAN

1
CHAPTER 1
INTRODUCTION
Opisthorchiasis, a parasitic infection caused by the liver fluke
Opisthorchis viverrini, is an important public health problem in Thailand, especially
in North and Northeast Thailand, and other countries in Southeast Asia including Lao
People’s Democratic Republic (Lao PRD), Vietnam and Cambodia. In Thailand, it
can cause economic loss about $120 million annually for medical care and lost wage.1
Human, who is a definitive host, can be infected by ingestion of raw or undercooked
freshwater fish that are contaminated with metacercariae. The number of globally
O. viverrini infected people is estimated to be around ten million.2 Most of the
infected persons are asymptomatic and long‐term infection leads to severe symptoms
and serious complications including cholangiocarcinoma. The principal diagnosis is
microscopic identification of parasite eggs in stool specimen. Praziquantel, the only
one drug of choice for opisthorchiasis treatment, is still effective for the treatment. In
Thailand, the liver fluke control program operation at region‐wide scale started in
19873, however, the recent prevalence of O. viverrini infection in some areas is still
high.
O. viverrini infection is carcinogenic to human and chronic infection can
result in cholangiocarcinoma development. Cholangiocarcinoma, a cancer of the
biliary system, is difficult to diagnose and has very poor prognosis, with an average 5‐
year survival rate of 5–10%.4 It is relatively resistant to chemotherapy and radiation,
therefore, surgery is the only possibility of a cure. Cholangiocarcinoma has low
incidence worldwide but not in Thailand. It is the predominant type of cancer
incidence particularly in the north and northeast Thailand, where O. viverrini is
endemic. The mechanisms of O. viverrini infection‐involved cholangiocarcinoma
development are not fully understood. O. viverrini secreted molecules have been
thought to partly contribute to the microenvironment that promotes tumorigenesis. A
parasite‐secreted growth factor, O. viverrini granulin‐like growth factor (Ov‐GRN‐
1)5,6
, has been reported that might participate in development of cholangiocarcinoma
Ref. code: 25605412330010EAN

2
in chronic O. viverrini infection. However, a potential angiogenic molecule from the
parasite has not been studied yet.
Calreticulin is a well‐researched multifunctional protein that is involved
in various biological functions including calcium homeostasis, ER‐chaperoning,
apoptotic cells clearance, cell adhesion and migration, as well as angiogenic activity.
It is present in all nucleated cells of higher organism and has been described in a wide
range of organisms, such as mammals, higher plants, helminths and protozoans.
Calreticulin not only plays an important role in physiological conditions but is also
involved in various pathological conditions including cancer.
To understand how important O. viverrini calreticulin (OvCALR) is in the
parasite and in the host‐parasite interplay, this study molecular characterized its DNA,
RNA, and protein. Protein functions including chaperoning, calcium binding, acetyl
group transferring, C1q binding, and angiogenic properties were analyzed. This study
provides a part to fulfill the basic knowledge of host‐parasite interplay during
opisthorchiasis as well as O. viverrini infection‐associated cholangiocarcinoma that
could conduce to the new target molecule on diagnosis and vaccine development.
Ref. code: 25605412330010EAN

3
CHAPTER 2
OBJECTIVES
The main objective of this study is to investigate molecular and functional
properties of O. viverrini calreticulin (OvCALR), a potential angiogenic protein from
O. viverrini. The specific objectives are as follows:
1. To molecularly clone and characterize the cDNAs encoding
calreticulin from Opisthorchis viverrini
2. To produce recombinant OvCALR (rOvCALR) and its C‐domain as
soluble proteins
3. To produce mouse polyclonal antisera against rOvCALR
4. To identify native OvCALR in parasite antigen extracts and parasite
tissue
5. To determine the immunogenicity of OvCALR in O. viverrini‐
infected hamster sera
6. To investigate biochemical properties of rOvCALR including calcium
binding, chaperoning, acetyl group transferring, and C1q binding
7. To investigate in vitro angiogenic properties of rOvCALR
Ref. code: 25605412330010EAN

4
CHAPTER 3
REVIEW OF LITERATURE
3.1 Opisthorchis viverrini
3.1.1 Taxonomy and typical characterization
O. viverrini is a food‐borne trematode that is classified into the
phylum Platyhelminthes, class Trematoda, sub‐class Digenea, order Opisthorchiida
and family Opisthorchiidae. This family also includes two other closely related
species, which are medically important pathogens: Clonorchis sinensis and
O. felineus. The characteristics of adult digenean body are dorsoventrally flat and
bilaterally symmetrical, but varying in size according to the species. The typical
characteristics of trematodes are mostly hermaphroditic, an oral and a ventral suckers
presenting but lacking of respiratory and circulatory systems. Their body is covered
with the tegument that involved in nutrient absorption, sensory functions,
osmoregulation and also protection from host immune response.7
3.1.2 Life cycle
The life cycle of O. viverrini (Figure 3.1) is similar to those of
O. felineus and C. sinensis with differences in susceptible intermediate host species.
They undergo various larval stages in three different hosts with sexual and asexual
reproductions. The adult flukes of O. viverrini reside mainly in the bile ducts of the
liver and gall bladder of the definitive host including cat, dog and human. The flukes
lay a large number of embryonated eggs, which pass into the duodenum along with
the bile and release into the environment along with the feces. After reaching
freshwater reservoirs, these eggs are ingested by Bithynia snails and then miracidia
hatch in the gastrointestinal tract of the snail by mechanical and chemical
stimulations. The miracidia penetrate to the lymph spaces and transform to
sporocysts. The sporocysts reproduce to rediae and then cercariae by asexual
reproduction. Free‐swimming cercariae leave the snail, actively seek and attach to
susceptible cyprinid fish species. They penetrate the skin of the fish and encyst in the
flesh as metacercariae, which are the infective stage. The definitive host can be
Ref. code: 25605412330010EAN
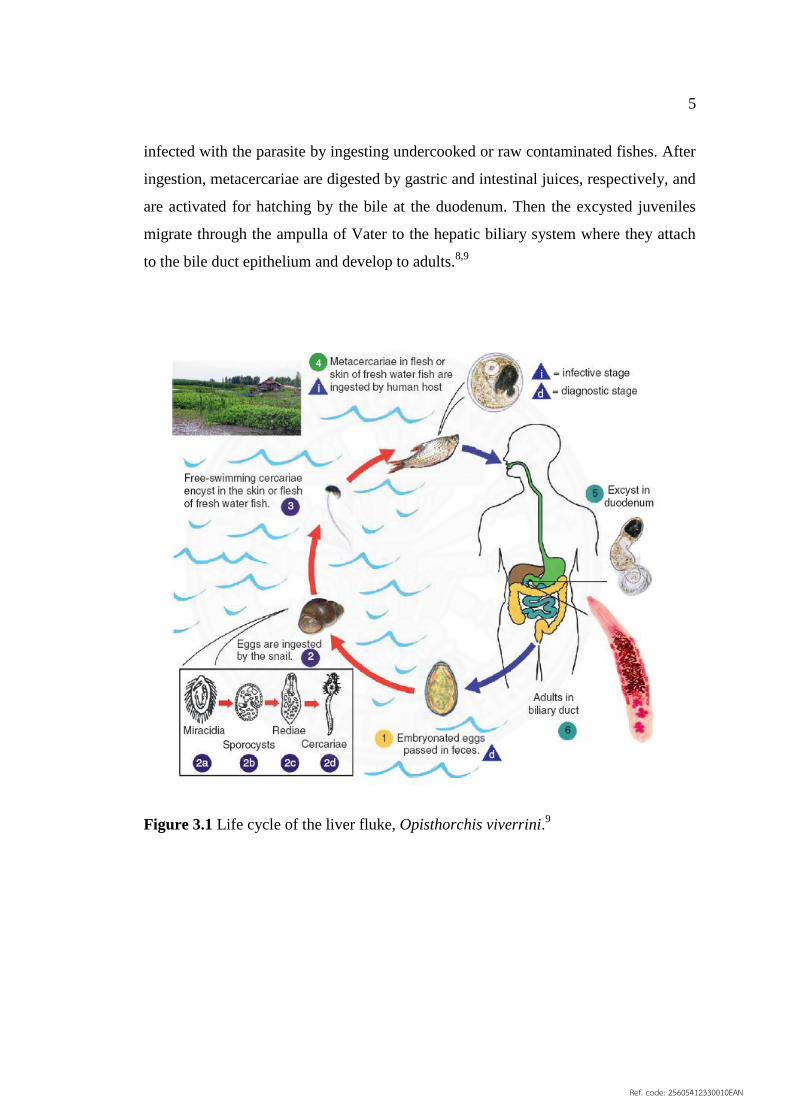
5
infected with the parasite by ingesting undercooked or raw contaminated fishes. After
ingestion, metacercariae are digested by gastric and intestinal juices, respectively, and
are activated for hatching by the bile at the duodenum. Then the excysted juveniles
migrate through the ampulla of Vater to the hepatic biliary system where they attach
to the bile duct epithelium and develop to adults.8,9
Figure 3.1 Life cycle of the liver fluke, Opisthorchis viverrini.9
Ref. code: 25605412330010EAN

6
3.1.3 Morphology
3.1.3.1 Adult
The adult worms of O. viverrini are dorsoventrally flat, leaf‐
shaped and transparent. The average size of the mature worm in human is 7 (5.4–
10.2) mm in length and 1.5 (0.8–1.9) mm in width, slightly larger than that found in
cat and dog.10
There are two suckers, the oral sucker is sub‐terminal and the ventral
sucker is located at the anterior part about one‐fifth of the body length. The two
branches of the blind ending digestive tract, which are connected at the short
esophagus, run in parallel and end at the posterior part of the body.11
Cirrus sac and
cirrus are not present. The numerous follicles of the vitellaria are in the lateral part of
the body, situated beside the intestine between ventral sucker and testes. The
multilobated ovary is located in front of the anterior testis and behind the uterus,
which originates from this site and ends with an opening at the genital pore in front of
the ventral sucker. The seminal vesicle is long, slightly coiled and opening at the
genital pore via the ejaculatory duct. The two testes are deeply lobed, diagonal
situated at the posterior part and the excretory bladder, a long S‐shaped tube, runs
between them.8
3.1.3.2 Egg
The embryos develop to maturity while the eggs move along
the uterus. The fully developed miracidia present in the eggs at the distal uterus. The
young eggs at the proximal uterus are larger than the mature eggs at the distal
uterus.12
The mature eggs laid from the adult stage of O. viverrini are oval in shape,
yellowish brown in color, and approximately 27 µm × 15 µm in size. The operculum
is surrounded with a thickened eggshell, the so‐called shoulder. The shell surface is
rough with a muskmelon‐like appearance, and an abopercular knob is present. The
morphology of O. viverrini, O. felineus, C. sinensis eggs and those of heterophyid
flukes is similar and causes problems in identification. Eggs can be found in the feces
approximately four weeks after infection.8,13
In human, adult O. viverrini can lay eggs
in a range from 2,000 to 4,200 per day.10
3.1.3.3 Miracidium
The miracidium develops in the egg and hatches after
ingested by a Bithynia snail. The study of O. viverrini eggs using electron microscopy
Ref. code: 25605412330010EAN

7
showed that the miracidium larva in the fully developed egg has a conical anterior
end, and its entire body is covered with a ciliated tegument except at the anteriormost
end. Inside the body, it contains plenty of glands: anterior apical gland, large cephalic
gland, and posterior secretory glands. At the posterior part of body, it contains a small
germinal region composing of 3–13 germ cells and a pair of flame cells.12
3.1.3.4 Sporocyst
The body of the mature sporocyst is very thin‐walled,
generally coiled, and approximately 1.1 mm × 0.65 mm sized. It contains many
developing rediae.14
3.1.3.5 Redia
The redia has a mount on the top, a thick walled pharynx and
short gut. The average size of a redia is 520 µm in length and 90 µm in width.8 Each
redia contains as many as 15 developing cercariae.14
3.1.3.6 Cercaria
The developing cercariae leave from the brood chambers of
the rediae and mature in the hemolymph space of the snail. The free‐swimming
cercaria shed by the snail is pipe‐form, oculate, pleurolophocercus, geo‐ and photo‐
tropic. The body size and tail size in average are 154 µm by 75 µm and 392 µm by 26
µm, respectively. The two eye spots are situated laterally between oral sucker and
pharynx. The body is covered with minute spines and brownish pigment scattering
bilaterally throughout the body. It has at least five pairs of penetration glands and at
least ten sensory hairs on each side of the body. The ventral sucker is inconspicuous
located slightly anterior to the roughly spherical excretory bladder.8,14
3.1.3.7 Metacercaria
The metacercaria is contained in a double‐walled cyst, which
is usually oval shaped. The outer cyst wall is about 3 to 8 µm thick, while the inner
cyst wall is very thin that can be recognized following the parasite excysted only. The
encysted and excysted metacercariae are morphologically similar and have an average
size of 201 µm by 167 µm and 558 µm by 145 µm, respectively. The oral and ventral
suckers are obvious. The appearances of the excretory bladder is oval shaped
containing a dense mass of dark granules, and of the surface body is a scatter of
brownish‐yellow pigment throughout its body.8
Ref. code: 25605412330010EAN

8
3.1.4 Geographical distribution
O. viverrini infection has been reported in Southeast Asia from
Thailand, Lao PDR, Cambodia and Vietnam (Figure 3.2).9 In 1980–1981, the highest
prevalence of O. viverrini infection in Thailand based on the region was 34.60% in
the northeast, and the rate in other regions was 6.34% in the central, 5.59% in the
north, and 0.01% in the south. The overall prevalence was 14% or approximately 7
million people infected.15
Following intensive and continuous control programs, the
prevalence of infection in 2001 declined in the northeast (15.7%), central (3.8%) and
south (0%), while in the north increased to 19.3%.3 In 2009, the overall prevalence
declined to 8.7% with an unabated prevalence in the northeast (16.6%) and north
(10%).16
However, the prevalence rate reported during 2012–2013 in some provinces
in the northeast was as high as 40.9% (Nakhon Phanom)17
, 38.68% (Yasothon)18
and
over 78% in some villages such as Ankam (Sakon Nakhon) and Kongswang (Nakhon
Phanom).17
In Lao PDR, over 2 million people are infected with O. viverrini,
mostly in the south of the country. The national scale survey of prevalence rate in
primary schoolchildren estimated during 2000–2002 from 17 provinces and Vientiane
municipality was 10.9%, and the prevalence was higher in the regions along the
Mekong River, up to 32.2%, 25.9% and 21.5% in Khammuane, Savannakhet and
Saravane Province, respectively.19
In 2006–2007, the prevalences of O. viverrini
infection in Saravane20
, Khong21
and Mounlapamok21
District, where are located in
the south of Lao PDR, were reported up to 58.5%, 92.0% and 90.9%, respectively.
The problematic identification of O. viverrini egg from those of small intestinal
trematodes resulted in prevalent report as Opisthorchis‐like eggs or
O. viverrini/minute intestinal fluke eggs in some surveys. However, several reports
confirmed O. viverrini infection by morphological and molecular analyses from
collected adult worms. They found that multiple trematodes infection is common in
Lao PDR, and Haplorchis taichui, a small intestinal trematode, usually found along
with O. viverrini.21-23
In 2002–2003, the prevalences of O. viverrini and minute
intestinal fluke (MIF) infections in inhabitant were 67.1% in Savannakhet Province23
and 81.1% in Khammouane Province22
. The prevalences in five provinces along the
Mekong River estimated during 2009–2011 as Ov/MIF revealed that the prevalence in
Ref. code: 25605412330010EAN
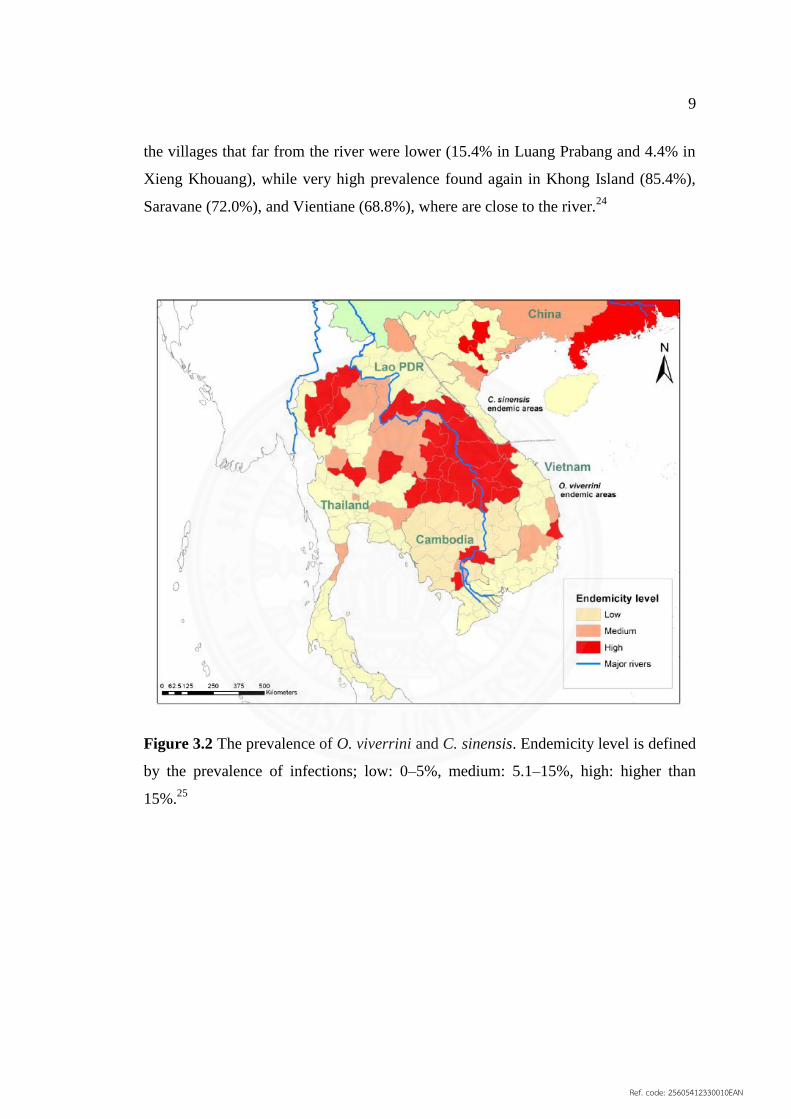
9
the villages that far from the river were lower (15.4% in Luang Prabang and 4.4% in
Xieng Khouang), while very high prevalence found again in Khong Island (85.4%),
Saravane (72.0%), and Vientiane (68.8%), where are close to the river.24
Figure 3.2 The prevalence of O. viverrini and C. sinensis. Endemicity level is defined
by the prevalence of infections; low: 0–5%, medium: 5.1–15%, high: higher than
15%.25
Ref. code: 25605412330010EAN

10
There are few data about O. viverrini infection in Cambodia.
However, over 600,000 people in Cambodia have been estimated to be infected with
O. viverrini.2 A small survey of O. viverrini infection in primary schoolchildren in
Kampongcham conducted in 2002 from 251 stool specimens was 4%.26
The national
scale survey conducted between 2006 and 2011 based on the Kato‐Katz thick smear
technique showed an overall prevalence of O. viverrini/MIF infection of 5.7%. The
central and southern regions, especially in Takeo and Kampongcham Provinces,
showed the highest prevalence of O. viverrini/MIF infection at 23.8–24%.27
Meanwhile, another survey in Takeo Province revealed 47.5% prevalence of
O. viverrini/MIF infection found in riverside villages of the Ang Svay Chek in the
Prey Kabas District.28
In Vietnam, both O. viverrini and C. sinensis infections have been
reported and estimated at 2 million people infected.2 Endemic areas of O. viverrini
have been reported in the central and southern regions including Nui Thanh, Mo Duc,
Phu My, Song Cau, Tuy An and Buon Don, whereas those of C. sinensis have been
reported in the northern region.29
The infection rates of O. viverrini reported from
three endemic southern provinces ranged from 15.2% to 36.9%.25
Recently, a small
survey in Phu My District from 254 stool samples revealed 11.4% of O. viverrini
infection based on the Kato‐Katz technique, and some of positive individuals were
confirmed by morphological and molecular analyses from collected adult worms.30
3.1.5 Clinical manifestation and pathogenesis
O. viverrini infection causes opisthorchiasis that often shows no
specific signs and symptoms. Only 5–10% of all infections show flatulence,
dyspepsia, fatigue, lost appetite, upper right quadrant abdominal pain and mild
hepatomegaly. Clinical symptoms are related to the intensity of infection, in severe
cases may be found with weakness, weight loss and ascites. Hematological and
biochemical features are not notable. In asymptomatic individuals, gallbladder
abnormalities including enlargement, sludge, gallstones and poor function can be
detected by ultrasonography. However, these abnormalities can be resolved after
treatment with praziquantel. Many complications can be found in opisthorchiasis
including obstructive jaundice, cholangitis, cholecystitis, intra‐abdominal mass,
gallbladder or intrahepatic stones, and cholangiocarcinoma, the most serious
Ref. code: 25605412330010EAN

11
complication. Cholangiocarcinoma is a malignant tumor of the biliary epithelium in
the intrahepatic biliary tree and may invade the sinusoids of the liver parenchyma.
The main clinical manifestation of opisthorchiasis associated cholangiocarcinoma is
jaundice that accounts for 60% of patients reported in northeast Thailand. The patients
may show obstructive jaundice alone or obstructive jaundice with either fever or acute
abdominal complications. Non‐jaundiced patients may suffer from dyspeptic pain,
anorexia, weight loss, upper right abdominal mass and distant metastasis.7,9,31
The pathological consequences of O. viverrini infection take mainly
place in the liver, extrahepatic bile ducts and gallbladder. Hepatomegaly, subcapsular
bile duct dilation and thick gallbladder are commonly found in infected animals and
humans. Histopathologic changes in opisthorchiasis include inflammation, epithelial
desquamation, epithelial and adenomatous hyperplasia, goblet cell metaplasia,
periductal fibrosis and granuloma formation. Gallstone can be found in up to 5% of
people in endemic areas.9
Pathogenesis of opisthorchiasis may be caused by mechanical or
chemical irritation from parasite and/or host immune response. The parasite’s suckers,
during feed and migration, cause mechanical injury and contribute to biliary
ulceration. The parasite eggs become entrapped through the ulcer and induce
granulomatous inflammation. The granulomata are usually found in experimental
hamster infections and occasionally found in human infection with bile duct
obstruction.32
Moreover, the metabolic products from the parasite that presented in the
excretory/secretory (ES) product can induce cell proliferation and may lead to the
biliary hyperplasia in opisthorchiasis. The host immune system responds to the
infection via inflammation that is mainly found around the bile ducts and periportal
areas. Severe inflammation can increase oxidative DNA damage and result in genetic
alteration. Furthermore, anthelmintic treatment with praziquantel leads to a
pronounced recruitment of inflammatory cells in the infected area and causes more
oxidative DNA damage. The effect of treatment combined with repeated infections
may enhance cholangiocarcinogenesis.9
Ref. code: 25605412330010EAN

12
3.1.6 Diagnosis
The gold standard for diagnosis of O. viverrini infection is
microscopic examination of eggs in feces, bile or duodenal fluid. For fecal sample,
formalin ethyl‐acetate concentration technique (FECT) is the current gold standard
method for O. viverrini detection.33
The fecal examination is widely used as routine
diagnosis. The methods for egg detection include the Kato‐Katz thick smear, Stoll’s
dilution and the quantitative formalin ethyl‐acetate concentration. However, the
sensitivity and accuracy of detection depend on the used method and the proficiency
of microscopist. Limitation of microscopic examination is morphological similarity of
trematode eggs, especially in the following families: Opisthorchiidae, Heterophyidae
and Lecithodendriidae. Hence, the diagnosis of opisthorchiasis, clonorchiasis and
minute intestinal flukes in co‐endemic areas such as Lao PDR and northern Thailand
are problematic.9
Immunodiagnostic approaches have been developed for
opisthorchiasis including immunoelectrophoresis, indirect‐hemagglutination, radio‐
immunoprecipitation, enzyme‐linked immunosorbent assay (ELISA), and dot‐
ELISA.34
Various parasite antigen extracts, including somatic crude extract, surface
tegumental extract and excretory/secretory product, have been used in antibody
detection assay. The promising one is enzyme‐linked immunosorbent assay using
partially purified antigens. Five fractions of adult O. viverrini were obtained from
chemical extraction and gel filtration chromatography: tegumental extract, somatic
extract and fraction 1 to 3. This assay showed 100% sensitivity of tegumental extract,
somatic extracts and fraction 1, whereas 94.9%, 84.0% and 57.1% specificity,
respectively, when tested against infected human sera compared to FECT method.35
Research focus then switched to specific parasite antigens for diagnosis. For example,
purified O. viverrini oval antigen was used in dot‐ELISA to detect antibodies in serum
samples. The assay showed 100% sensitivity and specificity compared to the modified
Kato‐Katz method and adult worm detection following praziquantel treatment.36
However, most antigens of this parasite are not species‐specific leading to cross‐
reactivity with sera from other parasitic infections. In addition, antibody levels show
no considerable decrease after 12 months of praziquantel treatment. For antigen
detection using copro‐ELISA, monoclonal antibodies were developed to detect the
Ref. code: 25605412330010EAN

13
parasite antigens in stool samples; however, obtained sensitivity and specificity were
unsatisfactory.34
Recently, monoclonal antibody against excretory/secretory antigen
of adult O. viverrini has been developed to detected O. viverrini urinary antigens in
urine sample using ELISA. This assay showed 81% sensitivity and 70% specificity
compared to FECT method, meanwhile the assay result showed cross reactivity with
some individuals who infected with Strongyloides stercoralis and hookworm. In
addition, the efficacy of this assay to detect the infection depended on the intensity of
infection.33
A new approach for O. viverrini detection, polymerase chain
reaction (PCR)‐based approach, has been developed to enhance the capability and
accuracy of detection. PCR‐based diagnosis focused on detection of O. viverrini egg
DNA in fecal specimens with the aim to increase sensitivity of O. viverrini detection
in light infection cases.37
Several DNA fragments from mitochondrial DNA38
and
ribosomal DNA39
of O. viverrini have been used as the target in molecular diagnosis.
Moreover, colorimetric loop‐mediated isothermal amplification (LAMP) assays have
been developed to detect O. viverrini with potentially used for point‐of‐care
diagnosis.40,41
3.1.7 Treatment and control
Praziquantel is the only drug currently recommended by World
Health Organization (WHO) for treatment of O. viverrini infection. There are two
optional recommended regimens: 3 times daily at oral dose of 25 mg/kg for 2–3
consecutive days or single oral dose of 40 mg/kg.42
In Thailand, the studies in 1980–
1983 showed that the treatment with a single oral dose of 40 mg praziquantel per 1 kg
body weight was effective for opisthorchiasis, with a cure rate as high as 91–95.5%.3
This regimen has been used in a large‐scale control program in Thailand. The adverse
effects including headache, vertigo, nausea, fatigue, vomiting, and abdominal pain are
common; however, all of these are transient and relatively minor severe.9,43
Concerning about the development of drug resistance resulted in several attempts to
find optional drugs for opisthorchiasis treatment.44
The promising one is
tribendimidine, which is effective drug for treatment of soil‐transmitted helminth
infections that approved in China. It showed very high cure and egg reduction rates in
Ref. code: 25605412330010EAN

14
opisthorchiasis treatment of clinical phase 2 trial; however, non‐inferiority compared
to praziquantel treatment was not shown.43,45
Prevention and control of O. viverrini infection comprise three main
strategies: (1) elimination of human host and animal reservoirs by treatment, (2)
interruption of disease transmission by improved sanitation, and (3) interruption of
infection by health education.3
3.1.8 Opisthorchis viverrini‐associated cholangiocarcinoma
Primary liver cancer, comprising two major types based on
histological characteristics and origin: hepatocellular carcinoma (HCC) and
cholangiocarcinoma (CCA), is a predominant type of cancer in Thailand.46
Liver
cancer causes high mortality rates, with total 27,500 deaths reported in Thailand in
2004. Moreover, liver cancer is the disease that caused the highest burden of
premature death in both sexes.47
It was the first leading cancer in men with an ASR of
33.9, and the third in women with an ASR of 12.9 during the year 2010–2012 (Figure
3.3).46
The incidence rate of liver cancer reported in 2008 as ASR from cancer
registries in Thailand ranged between 14.3 and 83.1 in men and between 5.0 and 43.2
in women in Surat Thani and Nakhon Phanom Province, respectively. Based on the
histological types, cholangiocarcinoma accounted for more than 55% in some
provinces in the north and northeast, where O. viverrini is endemic, and up to 73–82%
in Ubon Ratchathani, Khon Kaen, and Nakhon Phanom.48
On the other hand,
cholangiocarcinoma is relatively rare worldwide, hepatocellular carcinoma accounting
for 70% to 85% of the total cancer burden worldwide. In 2008, low rates of liver
cancer were found in several parts of Asia (southern, central and western) and Europe
(central, northern and eastern), with ASR < 5 in both sexes. On the other hand, top
two highest rates were found in eastern Asia (ASR = 35.5 in men and 12.7 in women)
and south eastern Asia (ASR = 21.4 in men and 9.0 in women), where are the
endemic areas of the liver flukes O. viverrini and C. sinensis.49
The incidences of
cholangiocarcinoma worldwide are shown in Figure 3.4, and the highest rate was
reported in northeastern Thailand.
Ref. code: 25605412330010EAN
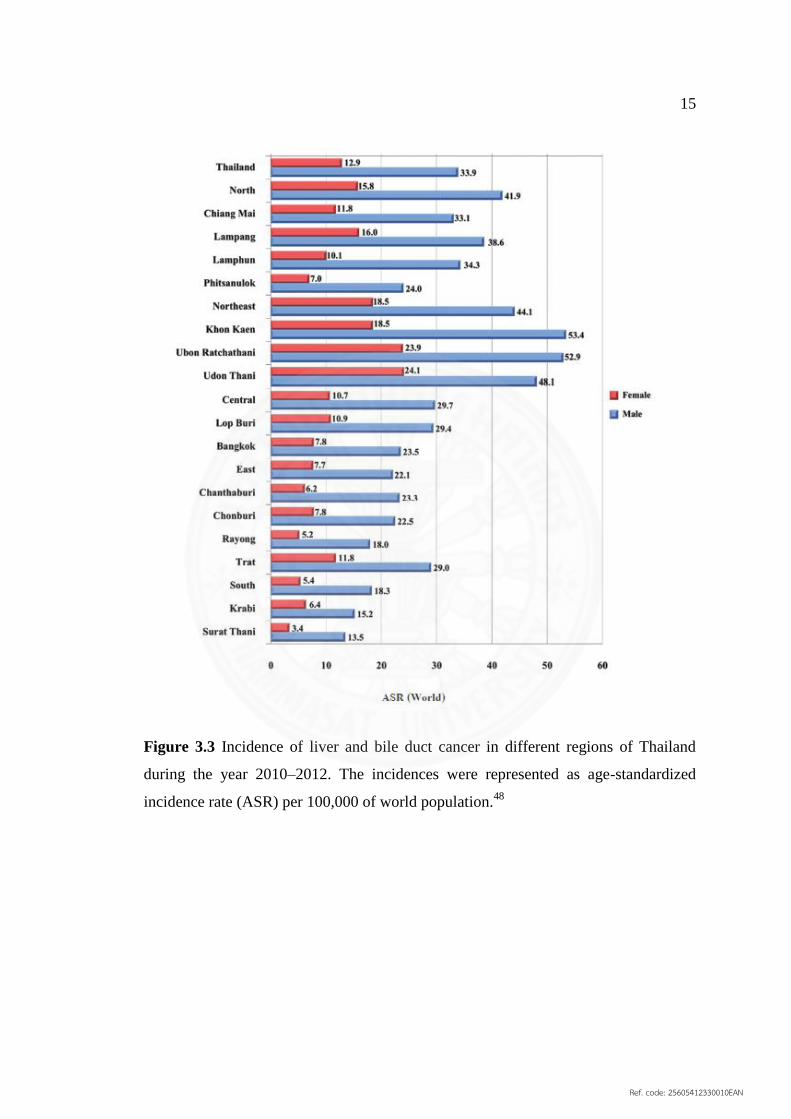
15
Figure 3.3 Incidence of liver and bile duct cancer in different regions of Thailand
during the year 2010–2012. The incidences were represented as age‐standardized
incidence rate (ASR) per 100,000 of world population.48
Ref. code: 25605412330010EAN

16
Figure 3.4 Incidence of cholangiocarcinoma worldwide reported between 1977 and
2007.50,51
In 1994, the International Agency for Research on Cancer (IARC),
a part of the World Health Organization (WHO), has classified O. viverrini as group 1
carcinogen; infection with this parasite is carcinogenic to humans.52
Until now, there
are many studies including epidemiology, human, and experimental animal that
demonstrated the positive relationship between O. viverrini infection and
cholangiocarcinoma. The data derived from cross‐sectional and case control studies
conducted in Thailand to evaluate the association between O. viverrini infection and
the risk of cholangiocarcinoma showed the range of odds ratios from 1.3–27.1,
whereas no significant association was found between O. viverrini infection and
hepatocellular carcinoma. In experimental animal studies, administration of N‐nitroso
compounds (N‐nitroso dimethylamine or N‐nitroso dihydroxy di‐n‐propylamine) in
combination with O. viverrini infection in hamsters increased the incidences of
cholangiocarcinoma. Such increase was not found in O. viverrini infection only. So
the IARC working group evaluated that chronic infection with O. viverrini causes
cholangiocarcinoma.2
Ref. code: 25605412330010EAN

17
There are three main mechanisms that have been proposed to
contribute to CCA: (1) mechanical damage to biliary epithelium caused by parasites,
(2) immunopathology due to inflammatory response to parasite infection, and (3)
toxic effects of the parasite excretory/secretory (ES) product (Figure 3.5). The
physical damage that occurs from parasite feeding and migratory activities leads to
incomplete wound repair. This constant cell division in the presence of exogenous
cofactors such as dietary nitrosamine may result in DNA damage and subsequent
oncogenesis. These carcinogenic properties were demonstrated in hamsters with
surgically ligated bile duct, followed by sub‐carcinogenic oral dose of nitrosamine
administration. The hamsters showed a significant development of biliary lesions
which was not found in controls that received either biliary ligation or nitrosamine.
Furthermore, DNA damage was revealed in biliary epithelial cells adjacent to
O. viverrini in chronically infected hamsters by staining with 8‐oxodG which is a
DNA damage marker.53
Nitric oxide (NO) is a product of the inflammatory response
to the parasite infection produced by macrophages, mast cells and other cell types.
Nitric oxide is not only cytotoxic but also genotoxic and mutagenic, resulting in the
release of endogenous nitrosamine that is carcinogenic. In a dietary nitrosamine
control study, opisthorchiasis patients had increased levels in plasma and urinary
nitrate compared to uninfected persons. The elevated nitrate level indicates a high
endogenous nitrosamine production, as nitrates are excretory metabolite of
nitrosamines. This condition can be abolished by co‐administration of ascorbic acid
with proline or treatment with praziquantel.54,55
The molecules that are released by the
parasite have become of interest and widely studied as factors facilitating to
cholangiocarcinogenesis. The results of co‐culture between adult O. viverrini and
NIH‐3T3 mouse fibroblasts in a Transwell chamber showed a marked increase in
NIH‐3T3 cell proliferation when compared to that without parasites. The O. viverrini
ES product promoted cell proliferation by stimulating the expression of cyclin D1 and
phosphorylated retinoblastoma (pRB) that resulted in driving the cell through the
G1/S transition point into the S‐phase of the cell cycle. In addition, a gene microarray
approach revealed that mRNAs encoding growth‐promoting proteins such as
Ref. code: 25605412330010EAN

18
Figure 3.5 Proposed pathogenesis mechanisms of O. viverrini infection‐induced
cholangiocarcinoma (CCA).53
Three major factors contributed by O. viverrini
infection lead to cholangiocarcinogenesis including (1) mechanical damage, (2) host
immune response, and (3) excretory/secretory (ES) product.
Ref. code: 25605412330010EAN

19
transforming growth factor were found to be overexpressed in NIH‐3T3 cells co‐
cultured with O. viverrini ES product.54
A notable growth factor that is present in the
O. viverrini ES product is a homolog of human granulin. O. viverrini granulin‐like
growth factor (Ov‐GRN‐1) is a secreted protein by the adult flukes and detected on
the biliary epithelial cells of infected hamsters.5 Recombinant Ov‐GRN‐1 promoted
proliferation of NIH‐3T3 mouse fibroblast and human CCA cell lines via the mitogen‐
activated protein kinase (MAPK).54
In addition, it promoted in vitro angiogenesis in
human endothelial cells.56
Thioredoxin is another molecule that presented in
O. viverrini ES product.57
In vitro study of the effect of O. viverrini thioredoxin (Ov‐
Trx‐1) on human cholangiocyte found that recombinant Ov‐Trx‐1 suppressed bile
duct epithelial cells from oxidative stress‐induced apoptosis by downregulating
apoptotic genes in MAPK pathway and upregulated anti‐apoptosis‐related genes.58
Novel oxysterol derivatives, bile aldehyde and bile alcohol, were found in O. viverrini
extracts using a liquid chromatography‐mass spectrometry (LC‐MS/MS) approach.
Oxysterols, which is oxygenated derivatives of cholesterol, can be mutagenic and
genotoxic. Moreover, it possesses pro‐oxidative and pro‐inflammation properties that
can promote carcinogenesis. A study of binding domains in human genes
demonstrated an association between different types of oxysterols and the
development and progression of various cancers: colon, lung, breast and bile ducts.59
All of the proposed mechanisms may act in concert to create a unique
microenvironment that stimulates cholangiocarcinogenesis.
3.2 Calreticulin (CALR)
Calreticulin was first isolated as a high affinity calcium‐binding protein
found in the rabbit skeletal muscle sarcoplasmic reticulum (SR) in 1974.60
It had been
discovered and described under various names including calregulin, CRP55, CaBP3,
ERp60 and calsequestrin‐like protein. Following an advance in molecular biology, it
was later proved that they are all the same protein, namely calreticulin, and it is not
only present in skeletal muscle sarcoplasmic reticulum but also abundant in non‐
muscle endoplasmic reticulum (ER). The non‐muscle endoplasmic reticulum lumen
contains many calcium‐binding proteins including protein disulfide isomerase (PDI),
Ref. code: 25605412330010EAN

20
immunoglobulin heavy‐chain binding protein (BiP; Grp78), endoplasmin (Grp94), but
the major one is calreticulin. The name of calreticulin, widely accepted then, is from
calcium binding protein localized to the endoplasmic/sarcoplasmic reticulum
membranes.61
Over four decades since first identified, calreticulin has been identified
extensively in diverse organisms including vertebrates (e.g., human, mouse, bovine,
dog, porcine, chicken, fish, and frog)62-68
, invertebrates (e.g., shrimp, mollusks,
roundworms, blood flukes, tapeworms, fruit fly, and protozoans)69-82
, and as well as
plants83
. Moreover, it is widely recognized that it is not restricted only in the
endoplasmic/sarcoplasmic reticulum but ubiquitous. It is also localized in
intracellular, at the cell surface, and extracellular compartments in various kinds of
cell and tissue as a multifunctional protein involved in many physiological and
pathological conditions.84
3.2.1 Genes and structure of calreticulin
Mammalian calreticulin (CALR) had been previously described as
one gene, one protein, and one mRNA, no evidence for RNA alternative splicing.85
On the other hand, at least two isoforms of calreticulin have been reported in plants.83
Recently, the second isoform of calreticulin, CALR386
, has been discovered in human
and mouse.87
However, this novel isoform of calreticulin has not been characterized
further, providing limited information. Both of human CALR and CALR3 genes are
located in the region 13 on the short arm of chromosome 19 and consist of nine exons,
with 4.2 kb and 17 kb in length, respectively (Figure 3.6A).87,88
In mouse, CALR gene
is located on chromosome 8 with a length of 4.8 kb and is organized into nine exons
similar to human calreticulin.89,90
The northern blot detection of calreticulin mRNA in
various human tissues revealed CALR mRNA at 1.9 kb approximately sized in all
tested tissues (spleen, thymus, prostate, testis, ovary, small intestine, colon, and
peripheral blood leucocyte), while CALR3 mRNA was detected only in testis at 1.3
kb approximately sized. The CALR mRNA contains an extensive 3′ UTR, about 600
base pairs, resulted in larger than CALR3 mRNA, whereas their 5′ UTR length are
comparable (Figure 3.6B).87
Human CALR3 isoform, 384 amino acids in length,
shared only about 50% homology with human CALR, 417 amino acids in length,
Ref. code: 25605412330010EAN
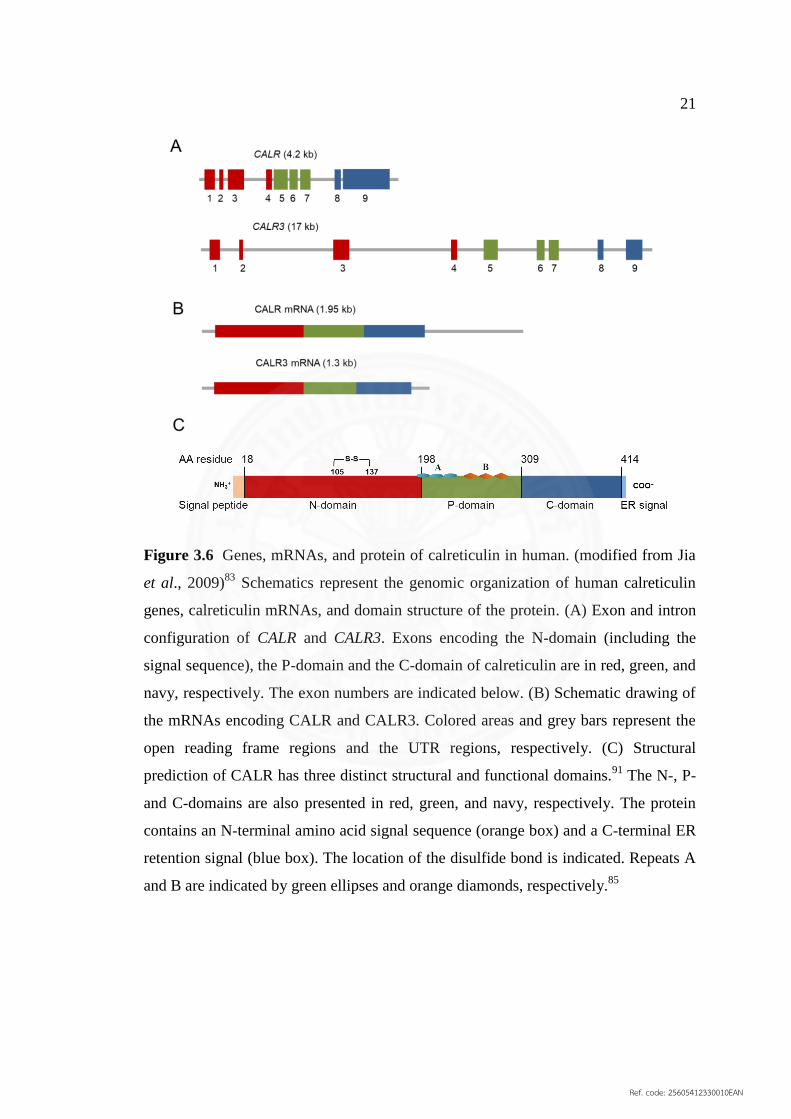
21
Figure 3.6 Genes, mRNAs, and protein of calreticulin in human. (modified from Jia
et al., 2009)83
Schematics represent the genomic organization of human calreticulin
genes, calreticulin mRNAs, and domain structure of the protein. (A) Exon and intron
configuration of CALR and CALR3. Exons encoding the N‐domain (including the
signal sequence), the P‐domain and the C‐domain of calreticulin are in red, green, and
navy, respectively. The exon numbers are indicated below. (B) Schematic drawing of
the mRNAs encoding CALR and CALR3. Colored areas and grey bars represent the
open reading frame regions and the UTR regions, respectively. (C) Structural
prediction of CALR has three distinct structural and functional domains.91
The N‐, P‐
and C‐domains are also presented in red, green, and navy, respectively. The protein
contains an N‐terminal amino acid signal sequence (orange box) and a C‐terminal ER
retention signal (blue box). The location of the disulfide bond is indicated. Repeats A
and B are indicated by green ellipses and orange diamonds, respectively.85
Ref. code: 25605412330010EAN

22
likewise in mouse calreticulin isoforms.87
At amino acid level, calreticulin has a
typical structure comprising of three distinct structural and functional domains: N‐, P‐
and C‐domains. It carries a signal sequence preceded the N‐domain and an ER
retention signal at the end of the C‐domain (Figure 3.6C).85
The N‐domain (N‐terminal domain, residues 18–197) is highly
conserved between species. It is usually contains the three‐conserved cysteine, which
a pair of them forms an intramolecular disulfide bridge that essential for the correct
folding of calreticulin (Figure 3.7).92
In addition, it contains two patterns of
calreticulin family signature motif: calreticulin family signature 1 and 2 with
consensus pattern [KRHN]‐x‐[DEQN]‐[DEQNK]‐x(3)‐C‐G‐G‐[AG]‐[FY]‐[LIVM]‐
[KN]‐[LIVMFY](2) and [LIVM](2)‐F‐G‐P‐D‐x‐C‐[AG], respectively.93
This domain
plays an important role in chaperone activity, and it can bind zinc, rubella virus RNA,
α‐integrin, protein disulfide‐isomerase (PDI), ER protein 57 (ERp57) and also
interacts with the DNA‐binding domain of the glucocorticoid receptor.85
In order to
work as chaperone, the N‐domain, providing the oligosaccharide‐ and polypeptide‐
binding regions, and the P‐domain, providing another binding site, require to work
together forming the folding unit. The amino acids residues involved with sugar
binding in the N‐domain have been identified, including Tyr109, Lys111, Asp135,
Tyr128, and as well as Asp317 in the C‐domain. The changed conformation of
calreticulin was observed in a mutation of His170 resulted in calreticulin loosed its
chaperone function.94
Moreover, the N‐domain of calreticulin, named vasostatin, was
shown to inhibit angiogenesis and suppress tumor growth.95,96
Recently,
crystallographic studies of human97
and mouse98
calreticulin uncovered a single of the
high‐affinity but low‐capacity calcium‐binding site in the globular domain that was
previously mapped in the P‐domain based on biochemical study99
.
The P‐domain (residues 198–308) is a proline rich region, which
also takes part in chaperone activity.85
Nuclear magnetic resonance (NMR) studies of
the P‐domain of calreticulin revealed an extended‐arm like structure and ERp57
binding sites in this region100
(Figure 3.7). The mutation of some amino acid residues
in the ERp57 binding sites at the tip of the P‐domain, Glu239, Asp241, Glu243 and
Trp244, leaded to binding disruption of ERp57.94
The P‐domain has two sets of three
repeats, so called repeat A (amino acid sequence PXXIXDPDAXKPEDWDE) and B
Ref. code: 25605412330010EAN

23
(amino acid sequence GXWXPPXIXNP XYX), respectively (Figure 3.6). These
repeats are crucial for the lectin‐like chaperone activity of calreticulin as well as the
calcium‐binding activity.85
Figure 3.7 Structure of calreticulin.101
Schematic representation of three‐dimensional
model of the calreticulin based on the crystallographic structure of the N‐domain, the
NMR structure of the P‐domain, and de novo model of the C‐domain of calreticulin.
The structural and functional domains of calreticulin are indicated: the globular N‐
domain (green), the extended‐arm P‐domain (yellow), and the acidic C‐domain (red).
The yellow ball represents the calcium‐binding site.
Ref. code: 25605412330010EAN

24
The C‐domain (C‐terminal domain, residues 309–417) is acidic and
high negatively charged domain, which composed of aspartic and glutamic acid
residues clusters responsible for calcium buffering activity inside the ER (Figure 3.7).
This acidic region generates calcium‐binding sites with low‐affinity but high capacity
(25 mol Ca2+
/mol protein, Kd= ~ 250 µM).102,103
Several known calcium‐binding
proteins, such as PDI, BiP, and endoplasmin, contain similar clusters of acidic amino
acid residues.61
Moreover, this domain can bind to blood‐clotting factors, including
factor IX and X, and plays a key role in the regulation of calreticulin interaction with
PDI, and ERp57 through the binding of calcium.85
In mammalian calreticulin, the C‐
domain is usually followed by the sequence KDEL, an endoplasmic reticulum
retention signal. This ER retention signal sequence has been found to be varied
depending on species between the calreticulin homologs (Figure 3.8).
Figure 3.8 Comparison of amino acids sequences of calreticulin from different
species.85
Overall comparisons of different calreticulin are highly similar. The ER
retention signal sequences of calreticulin from selected species are shown. Gaps in the
amino acid sequence are indicated by the white area, and amino acid mismatches are
indicated by short vertical line.
Ref. code: 25605412330010EAN

25
3.2.2 Biological functions of calreticulin
Beside the calcium buffering and chaperoning, the primary roles of
calreticulin in the endoplasmic reticulum, calreticulin has multiple duties not only
inside but also outside of the ER, and is involved in a variety of cellular signaling
pathways.85,102
3.2.2.1 Calreticulin functions inside of the ER
(1) Chaperone activity
The ER is an essential compartment of the cell, where is a
factory of protein folding and modifying for secretory and membrane proteins that
accounted for one third of total cellular protein and a control center for calcium
storage and balance.104,105
The main function of calreticulin in the ER is in the folding
control of newly synthesized proteins and glycoproteins to prevent misfolded protein
aggregation by identifying and banning those proteins from the ER and sending to
ubiquitin‐mediated proteolysis (Figure 3.9). This folding chaperone activity of
calreticulin occurs mainly via its lectin site in the N‐domain by interacting with
ERp57 to form a folding unit.84
The glycoprotein folding control occurs in the
calreticulin/calnexin (CNX) cycle via recognized monoglucosylated N‐linked
oligosaccharides (GlcMan9GlcNAc2) by those lectin‐like chaperones.104
The
importance of calreticulin in chaperone activity is shown in major histocompatibility
complex (MHC) class I molecule production. Calreticulin is involved in folding
control and assembly of MHC class I through its lectin and peptide‐binding sites as
well as ERp57 interaction.101
The absence of calreticulin leads to an increase of
unstable MHC class I molecules exported from the ER, a decrease of MHC class I cell
surface expression and an impairment of antigen presentation.106
In addition,
calreticulin have been shown to suppress in vitro precipitation/aggregation of non–
and glycosylated protein at either physiological temperature or higher (42–50°C) in
calcium‐dependent and independent conditions.107-109
Therefore, calreticulin
deficiency impairs protein and glycoprotein quality control, resulting in accumulation
and export of misfolded proteins.110
Ref. code: 25605412330010EAN
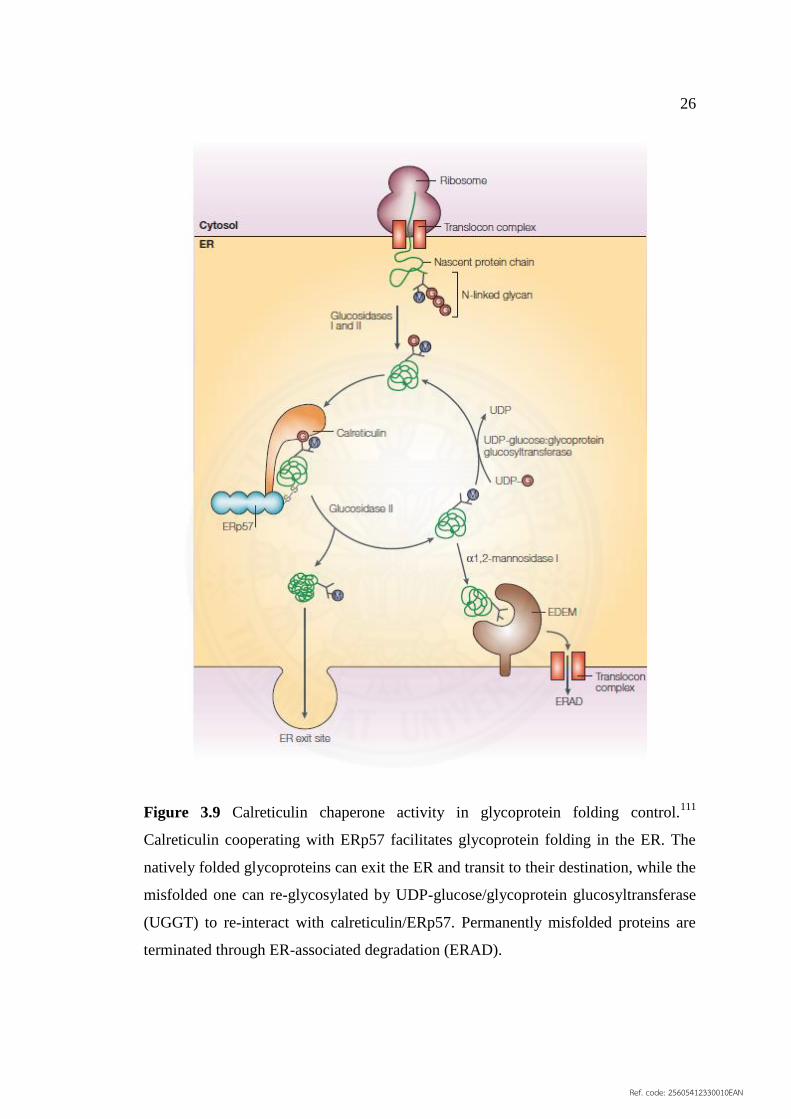
26
Figure 3.9 Calreticulin chaperone activity in glycoprotein folding control.111
Calreticulin cooperating with ERp57 facilitates glycoprotein folding in the ER. The
natively folded glycoproteins can exit the ER and transit to their destination, while the
misfolded one can re‐glycosylated by UDP‐glucose/glycoprotein glucosyltransferase
(UGGT) to re‐interact with calreticulin/ERp57. Permanently misfolded proteins are
terminated through ER‐associated degradation (ERAD).
Ref. code: 25605412330010EAN

27
(2) Calcium homeostasis
Calcium plays an important role in signal transduction
pathways that involved with diverse cellular processes, including muscle contraction,
gene expression, protein synthesis and stress signaling. The ER is the major calcium
storage site and is essential in balancing cellular calcium homeostasis. Calcium
releasing from the ER is controlled by the inositol 1,4,5‐triphosphate receptor
(InsP3R) and the ryanodine receptor, while calcium restoring is operated by the
sarco/endoplasmic reticulum calcium ATPase (SERCA) pump (Figure 3.10).102
Inside ER, there are a number of calcium‐binding proteins, which serve as calcium‐
buffering and other responsibilities in ER lumen, including calreticulin, calnexin, BiP,
GRP94, calumenin, and the reticulocalbins.104
Calreticulin is a major calcium
buffering protein, which binds to over 50% of calcium stored in the ER and regulates
calcium storage and release within the ER. Calreticulin deficient cells exhibited a
reduction of calcium capacity and free calcium concentration in the ER lumen and an
inhibition of InsP3‐dependent calcium release from the ER.94,112
There was a report that calreticulin‐deficiency in mice
results in embryonic lethality because of impaired cardiac development.113
This
embryonic lethality can be rescued by the expression of activated calcineurin,
however, the mice showed growth retardation, hypoglycemia, and high level of blood
cholesterol and triglyceride.114
The mechanism of calreticulin and calcineurin‐
dependent embryonic lethality has been revealed that calreticulin‐dependent
calcineurin activation is essential for nuclear translocation of cardiac‐specific
transcription factors, nuclear factor of activated T‐cells (NF‐AT) and myocyte‐
enhancer factor (MEF) 2C.114,115
Therefore, the role of calreticulin in calcium release
is crucial in early stage of cardiac development through calcium and calcineurin‐
dependent pathways (Figure 3.10).
The calcium buffering homeostasis of calreticulin may
involve in adipocyte differentiation. The study of adipose‐derived mesenchymal stem
cell differentiation showed that high level of extracellular calcium and ionophore, a
chemical that facilitates calcium influx, inhibited mesenchymal stem cells
differentiation and upregulated calreticulin expression.116
Ref. code: 25605412330010EAN

28
Figure 3.10 Functions of calreticulin and its dependent pathway.102
Calreticulin (CRT
used only in this figure) is involved in the calreticulin/calnexin cycle to regulate
folding of glycoproteins by cooperated with ERp57. The MHC class I loading
complex comprised of CRT, CNX, ERp57, transporter associated with antigen
processing (TAP) and tapasin. It produces a proper functioning MHC class I molecule
by assembling the class I heavy chain and 2‐microglobulin with a stabilizing
peptide. CRT maintains homeostasis of cytosolic and ER calcium levels by regulating
calcium transport through InsP3R and SERCA. Cytoplasmic calcium level is essential
for calcineurin activation that allows nuclear translocation of the transcription factors
for cardiac development.
Ref. code: 25605412330010EAN

29
3.2.2.2 Calreticulin functions outside of the ER
There are many reports concerning the function of
calreticulin outside the ER. However, the mechanism of calreticulin translocation to
cell surface and extracellular release is not fully understood. A widely accepted
hypothesis is ER‐stress‐mediated apoptosis or cell dying, possibly caused by
oxidative stress induced by reactive oxygen species (ROS), UV irradiation, ER
calcium homeostasis disruption, as well as cell infection.84,101,117,118
There are three
major mechanisms have been proposed for cell surface exposure of calreticulin:
association of cytosolic calreticulin with phosphatidylserine (PS) via a membrane flip‐
flop mechanism during apoptosis and ERp57‐dependent or ‐independent translocation
through secretory pathway.84,101
The mechanism of cytoplasmic release of calreticulin
has been described through retrotranslocation from the ER. It can escape from signal
dependent ER‐retention to secretory pathway and avoid the ubiquitylation and
degradation after retrotranslocation.119
(1) Cell adhesion
Cytoplasmic calreticulin was shown to bind to the
KxGFFFKR amino acid sequence found in the cytoplasmic domains of the α subunits
of integrin possibly to stabilize integrin‐ligand binding and activate focal adhesion
kinase (FAK).84
Calreticulin deficient mouse embryo fibroblasts showed a reduction
collagen Ⅳ binding activity and the activity was restored when presenting of
cytosolic calreticulin.119
Cell surface calreticulin was shown to bind to glycosylated
forms of laminin to mediate spreading of laminin‐adherent melanoma cells.120
In
addition, murine embryonic stem cells lacking calreticulin exhibited severely
impaired integrin function.103
(2) Focal adhesion disassembly and cell migration
Cell surface calreticulin of endothelial cells and fibroblasts
has been shown to interact with the heparin‐binding domain at the N terminal of
thrombospondin 1 (TSP1) in association with the low‐density‐lipoprotein‐related
protein 1 (LRP1/CD91) to mediate focal adhesion disassembly, which is an important
process of cell migration. The binding site that is responsible for this activity is
located in the N‐domain of calreticulin, amino‐acid residues 19–36.84
Calreticulin
deficient mouse embryo fibroblasts showed a reduction in cell migration on
Ref. code: 25605412330010EAN

30
fibronectin and laminin as well as in migration chamber when compared to wild‐type
cells.84,119
Moreover, exogenous calreticulin can stimulate keratinocyte and fibroblast
migration using migration chamber and in vitro scratch assay.121
(3) Phagocytosis
Cell surface expression of calreticulin stimulates the
phagocytic ingestion of dead, ER‐stressed and cancer cells, so called eat‐me signal.
This mechanism is mediated by the binding of calreticulin to cell surface LRP1
(CD91) of phagocyte. In normal cell, the expression of anti‐phagocytic molecule,
CD47 (integrin‐associated protein), on the cell surface is higher than calreticulin,
while the cancer cell increases the expression of CD47 to evade the phagocytosis.101
Cell surface calreticulin in association with phosphatidylserine (PS) presented on
apoptotic cell has been shown to provide the recognition signal for apoptotic cells
clearance by phagocytes (Figure 3.11).84
Moreover, exogenous recombinant
calreticulin is capable to promote phagocytosis of normal bone marrow cells by
human macrophages.122
Additionally, calreticulin also plays an important role as a
bridging molecule through LRP1(CD91) in the recognition and removal of apoptotic
cells by binding directly to apoptotic cells‐recognized molecules, including C1q,
collectin, and ficolin.84
The C1q binding site on calreticulin is located spanning the N‐
and P‐domains of amino acid residues 160–283, specifically called the S‐domain.123
(4) Wound healing
The presence of calreticulin in the extracellular matrix is
associated with wound healing processes. Topically applied calreticulin significantly
increased the rate of wound repair in pig124
and mouse125
. Moreover, the applying of
calreticulin to wounds increased the expression of transforming growth factor β3, a
crucial wound healing factor, and increased cellular proliferation and migration of
keratinocytes and fibroblasts. The quality of wound healing was improved by both the
epidermal and dermal cutaneous repair process. Therefore, extracellular calreticulin
plays an important role in the process of tissue repair and remodeling.84,102,121
Ref. code: 25605412330010EAN

31
Figure 3.11 Schematic representation of calreticulin‐mediated phagocytosis.
(modified from Raghavan et al., 2013)101
Normal live cells (a) and cancer cells in the
absence of anti‐CD47 (b) are not phagocytosed, while cancer cells in the presence of
anti‐CD47 (b), drug‐induced stress cells (c) and apoptotic cells (d) upregulate cell
surface calreticulin expression resulting in phagocytosis.
(5) Angiogenic activity
Blood vessels are essential for supply of oxygen and
nutrients, waste disposal, and immune surveillance.126
New blood vessel formation,
termed neovascularization, can occur via two mechanisms: vasculogenesis and
angiogenesis. Vasculogenesis is the de novo formation of blood vessel from precursor
cells such as angioblasts, a cell type with potency to differentiate into endothelial cells
that are derived from hemangioblasts, differentiated from mesodermal stem cells.127
This process is driven by the recruitment of undifferentiated mesodermal cells to
become endothelial cells and the assembly of the cells into blood vessels. In contrast,
angiogenesis is the formation of new blood vessel from endothelial cells of existing
vasculature, a process driven by endothelial cell proliferation.128
In the embryo, a
primitive vessel network is created by vasculogenesis and followed by the process of
angiogenesis that is sprouting, branching and stabilization. These two mechanisms of
new blood vessel formation occur throughout life, from embryonic to adult stage, and
are involved in both physiological and pathological conditions.128,129
Angiogenesis is
required for physiological processes, such as wound healing and reproduction, as well
as pathological processes, such as invasive tumor growth. Therefore, it is tightly
Ref. code: 25605412330010EAN

32
regulated by a balance between pro‐angiogenic factors, such as vascular endothelial
growth factor (VEGF) and fibroblast growth factor (FGF), and anti‐angiogenic
factors, such as thrombospondin‐1 and pigment epithelium‐derived factor (PEDF).130
Initiation of blood vessel growth needs up‐regulation of pro‐angiogenic factors and
follows by down‐regulation of anti‐angiogenic factors.131
The curious finding in athymic mice that an Epstein‐Barr
virus‐immortalized cell line is not tumorigenic and promotes regression of
experimental Burkitt lymphoma, colon carcinoma, and other human malignancies
through vasculature‐based process leading to further studies of EBV‐immortalized
cells.132,133
Factors secreted from these cells were investigated, and one factor that was
purified from the supernatant of an EBV‐immortalized cell line was identified as an
NH2‐terminal fragment of human calreticulin (amino acids 1–180), named vasostatin.
Vasostatin was shown to inhibit the proliferation of endothelial cells induced by basic
fibroblast growth factor (bFGF or FGF‐2) but not cells of other lineages such as
human foreskin fibroblasts, Burkitt lymphoma cells, lung carcinoma cells, and colon
adenocarcinoma cells, and suppress angiogenesis in vivo. When recombinant
vasostatin was inoculated into athymic mice, it significantly reduced growth of human
Burkitt lymphoma and human colon carcinoma; however, tumor cells were not
growth‐inhibited by vasostatin in vitro.95
Further studies to identify a specific region responsible for
angiogenic activity in calreticulin have been reported. Calreticulin lacking the N‐
terminal 1–120 amino acids exhibited an inhibition of endothelial cell proliferation
in vitro and an inhibition of Burkitt tumor growth in vivo comparably to vasostatin.
Similar results were observed in a calreticulin fragment encompassing amino acid
residues 120–180. The results suggested that the anti‐angiogenic activity of vasostatin
and full‐length calreticulin is located on the N‐terminal amino acid residues 120–180
of calreticulin.134
In another study, it was shown that a vasostatin fragment,
encompassing amino acid residues 135–164, inhibited proliferation of endothelial
cells induced by bFGF comparably with vasostatin (amino acids 120–180). The
inhibitory effect in human endothelial cell proliferation and neovascularization in
chick embryo chorioallantoic membrane (CAM) were stronger when vasostatin was
Ref. code: 25605412330010EAN

33
fused with platelet factor‐4 (C‐13 fragment) by a nonpolar peptide bridge of Gly‐Pro‐
Gly.135
The mechanism of vasostatin to inhibit endothelial cell
proliferation, angiogenesis, and tumor growth is still unclear. However, it has been
reported that vasostatin specifically binds to α5 and γ1 chains of laminin, the
extracellular matrix protein expressed by endothelial cells. Laminins are high‐
molecular‐weight protein comprising polypeptides of α, β, and γ chains linked by
disulfide bonds. At least five α, three β, and three γ chains have been described to
assemble into twelve identified different laminin isoforms. The endothelial cell
attachment to laminin was inhibited after the addition of vasostatin to endothelial cell
cultures resulting in a reduction of endothelial cell proliferation.136
Moreover, an
in vivo study of plasmid‐encoding vasostatin effects on human hepatocellular
carcinoma xenograft in nude mice revealed an inhibitory effect of vasostatin on the
tumor growth and metastasis and an increase of survival time. In addition, the
histological result revealed that vasostatin treatment increased apoptosis and
suppressed angiogenesis.137
On the other hand, a pro‐angiogenic activity of calreticulin
has been reported that it can promote the proliferation, migration, and tube formation
of human umbilical vein endothelial cells through nitric oxide signaling pathway.
These effects were inhibited by a specific endothelial nitric oxide synthase (eNOS)
inhibitor, NG‐nitro‐L‐Arginine ethyl ester (L‐NAME). Pro‐angiogenic activity of
calreticulin might be involved in pathological angiogenesis event in rheumatoid
arthritis that found a significant increase calreticulin level in serum and synovial fluid
compared to healthy controls and osteoarthritis patients.138
Moreover, calreticulin has
been expressed in various cancers and it might be involved in tumorigenesis.139
3.2.3 Calreticulin of pathogenic protozoans and helminths
3.2.3.1 Protozoans
Pathogenic protozoan calreticulins are likely to implicate in a
mechanism of the parasite evading the host immune response. The complement
system is a part of innate immune system of host defense against common pathogenic
infection and complement activation results in pathogen opsonization, lysis, and
Ref. code: 25605412330010EAN
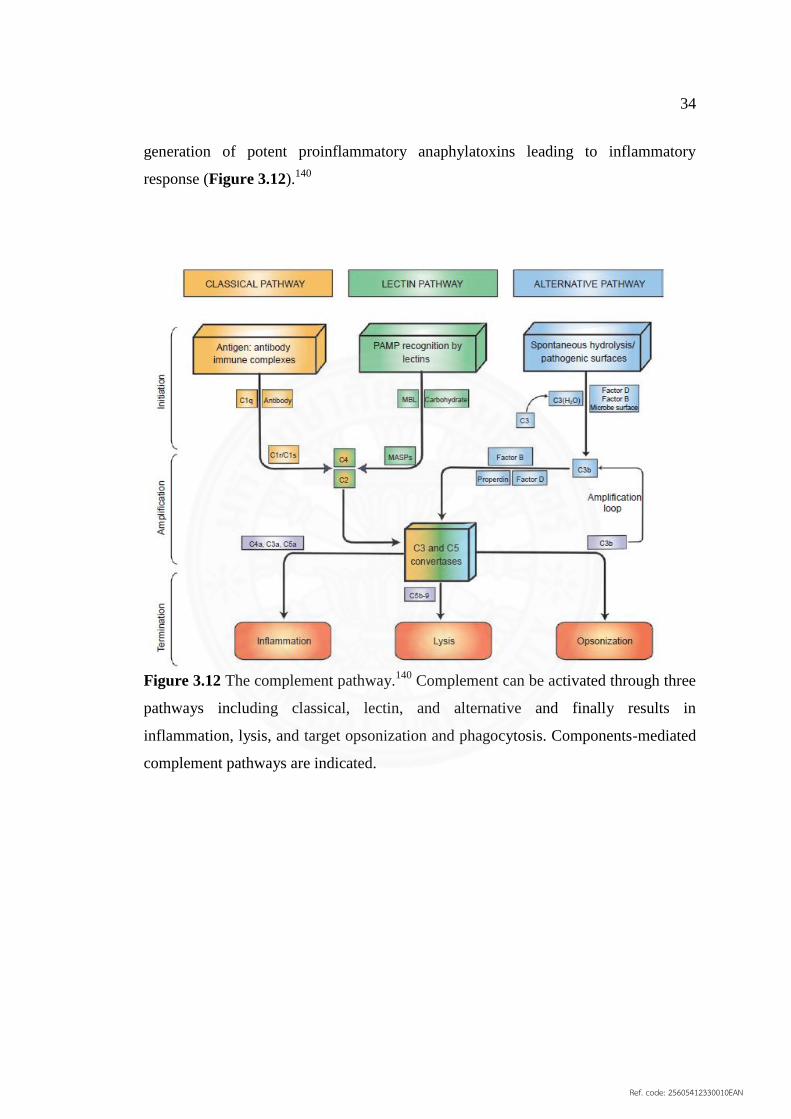
34
generation of potent proinflammatory anaphylatoxins leading to inflammatory
response (Figure 3.12).140
Figure 3.12 The complement pathway.140
Complement can be activated through three
pathways including classical, lectin, and alternative and finally results in
inflammation, lysis, and target opsonization and phagocytosis. Components‐mediated
complement pathways are indicated.
Ref. code: 25605412330010EAN

35
Calreticulin of Trypanosoma cruzi (TcCALR), one of a well‐
characterized calreticulin, is a 45‐kDa protein that is mainly located in the ER but also
found in the Golgi complex, reservosomes, flagellar pocket, cell surface, cytosol,
nucleus, and kinetoplast.141
TcCALR presented on the surface of infective‐stage
trypomastigotes can bind to host C1q; the first component participated in the
complement system, leading to inhibition of the classical complement pathway and
promotion of infectivity in macrophage as well as in human placenta chorionic villi
explants (HPCVE).142-144
The conserved C1q binding activity found in TcCALR
probably caused by a conserved C1q binding region of T. cruzi calreticulin (amino
acid residues 159–281), over 70% identity to the S‐domain of human calreticulin.142
Similar results also observed in Entamoeba histolytica and E. dispar, the parasite
calreticulins can bind to human C1q and inhibit classical complement pathway‐
mediated hemolysis in vitro. Moreover, pre‐incubated‐rEhCALR trophozoites were
significantly resistant to lysis by human serum that is a virulent indicator of
E. histolytica trophozoites.145
In Leishmania donovani, overexpression of either the P‐
domain of LdCALR or full‐length LdCALR in transfected L. donovani resulted in
significant reduction of survival ability inside human macrophage at 72 h after co‐
incubation with macrophage. The better result was observed in the P‐domain
overexpression, possibly caused by a disruption of virulent proteins trafficking
through an impaired secretory pathway from chaperone activity interfering.146
Beside the conserved lectin‐dependent activity, TcCALR and
its N‐terminal vasostatin‐like domain (N‐TcCALR) were shown to inhibit a capillary
growth in the rat aortic ring assay, an in vitro morphogenesis, proliferation and
chemotaxis of human umbilical vein endothelial cells (HUVECs), an endothelial cell
migration into Matrigel subcutaneous implanted in mice as well as an angiogenesis in
CAM assay.141,147
The anti‐angiogenic activity of TcCALR in these assays show
significantly higher activity than human calreticulin at equimolar concentration.148
Moreover, TcCALR also inhibits the growth of mammary tumor cells in mice.149
In
contrary, recombinant TcCALR was reported that it increased proliferation and
migration of human fibroblasts in vitro and promoted wound healing in a rat skin
model.150
Ref. code: 25605412330010EAN

36
High immunogenicity of calreticulin has been reported in
human parasitic infection; patients with either T. cruzi infection or amoebic liver
abscess (E. histolytica infection) were shown to develop high levels of serum
antibodies against the parasite calreticulin.151,152
3.2.3.2 Helminths
(1) Nematodes
Calreticulin in nematode was first identified in
Onchocerca volvulus, a causative agent of onchocerciasis or river blindness, as RAL1
antigen that induced a production of human calreticulin autoantibody in patients with
onchocerciasis.153,154
Immunogenicity of nematode calreticulins have been studied
and reported in Heligmosomoides polygyrus and Necator americanus. H. polygyrus is
a parasitic nematode of mice that is widely used as an experimental model to study
helminth associated immune mechanisms. Calreticulin of H. polygyrus (HpCALR) is
expressed in all stages, especially in tissue invasive larvae stage (L4) as a secreted
protein. Native HpCALR stimulated CD4+ T cells production of IL‐4 and IL‐10 in
H. polygyrus infected mice. Similarly, mice immunized with recombinant HpCALR
showed significant production of IL‐4 and specific IgG1 and IgE antibodies. These
results indicate the T helper 2‐related immune response against HpCALR. Moreover,
recombinant HpCALR can bind to scavenger receptor type A (SR‐A) on dendritic
cells leading to internalization of HpCALR.75
Calreticulin of N. americanus can
stimulate the production of histamine from basophils and is a strong immunogen that
is recognized by IgE antibodies from the infected patients.73,155
Moreover, it has been
a target for vaccine development, the application of the recombinant calreticulin of
N. americanus resulted in the reduction of worm burdens by 43–49% compared to
controls.156
Pathogenic nematode calreticulins are likely to implicate in
a host immunomodulation and a parasite survival as well. The nematode
Haemonchus contortus, a gastrointestinal parasite of sheep and goat, secrets
calreticulin which displays anti‐coagulation properties by binding to calcium and
clotting factors leading to prolong plasma coagulation time in vitro. Similar nematode
calreticulins of Brugia malayi157
and N. americanus73
, it can bind to C1q and inhibit
classical complement‐mediated hemolysis. Additionally, it can bind to host C‐reactive
Ref. code: 25605412330010EAN

37
protein, which is a marker for inflammatory response that can bind to complement
C1q and acts as pro‐coagulant protein.158
These functions of calreticulin in
H. contortus may facilitate the parasite to feed continuously on host blood and survive
by modulating the host immune response.74
(2) Cestode
Taenia solium is an intestinal tapeworm that can cause
taeniasis and cysticercosis, especially neurocysticercosis, which is a serious public
health in human. Calreticulin of T. solium was widely distributed and differential
expression was found during development, particularly in germ cell and
embryogenesis.159
Contrary to other reported parasite calreticulins, TsCALR was
poorly recognized by human and pig sera with cysticercosis.78
The ability of TsCALR
as vaccine candidate has been studied in hamster by oral administration with cholera
toxin as adjuvant. The results showed that immunization with rTsCALR induced IL‐4
and IFN‐γ expression leading to goblet cell hyperplasia in the mucosa surrounding the
parasite and resulted in a reduction of worm burden in infected hamster as well as a
decrease of genotoxicity‐induced tapeworm infection.160,161
In addition, fecal specific
IgA and serum specific IgG antibodies against TsCALR were detected after
immunization, and specific IgA antibody was more pronounced after the parasite
challenge.161
Anti‐inflammatory activity of TsCALR was studied in experimental
induced‐colitis in mouse model. Orally administered recombinant TsCALR prior
colitis induction significantly reduced the inflammatory parameters including IL‐6,
TNF‐α, and IL‐1β and increased the regulatory cytokines IL‐10 and TGF‐β leading to
less clinical and histopathological severity.162
(3) Trematodes
Calreticulin of Schistosoma japonicum was molecularly
cloned but only its calcium‐binding activity was determined.77,163
Calreticulin of
S. mansoni (62 kDa) is a good T‐ and B‐cell antigen and represents a potential vaccine
candidate.164
After stimulation with S. mansoni calreticulin, 20–30% of the
schistosomiasis resistant individuals showed detectable levels of IL‐2, IL‐4, and IFN‐
γ and 59% of them possessed anti‐S. mansoni calreticulin IgM specific antibodies.165
Moreover, S. mansoni calreticulin was assessed as a marker for schistosomiasis
diagnosis; however, cross‐reacting antibody was detected in healthy controls.166
In
Ref. code: 25605412330010EAN

38
S. haematobium, differential recognition of adult parasite detected by the infected
human serum showed that ShCALR was recognized by IgE and IgG1 antibodies but
not by IgA and IgG4 antibodies.167
Ref. code: 25605412330010EAN

39
CHAPTER 4
MATERIALS AND METHODS
4.1 Molecular cloning of Opisthorchis viverrini calreticulin (OvCALR)
4.1.1 Preparation of total RNA
4.1.1.1 Isolation of adult O. viverrini total RNA
Total RNA of adult O. viverrini was isolated using TRIzol™
Reagent (Invitrogen, Carlsbad, CA, USA). The parasites were homogenized in
TRIzol™
Reagent (1 ml reagent per 50–100 mg parasite wet weight) using a tissue
homogenizer (Ultra‐Turrax T25, IKA, Staufen, Germany). The homogenate was
centrifuged at 12,000×g for 10 min at 4°C. The clear supernatant was collected and
incubated at room temperature for 5 min. Chloroform (0.2 ml per 1 ml of TRIzol™
Reagent) was added, and the tube was shaken vigorously and further incubated at
room temperature for 2–3 min. The sample was centrifuged at 12,000×g for 15 min at
4°C, and the upper aqueous phase was carefully aspirated into a new tube. To
precipitate total RNA, 100% isopropanol (0.5 ml per 1 ml of TRIzol™
Reagent) was
added, incubated at room temperature for 10 min, and centrifuged at 12,000×g for 10
min at 4°C. The pellet was collected and subsequently washed with 75% (v/v) ethanol
(1 ml per 1 ml of TRIzol™
Reagent). The tube was centrifuged afterward at 7,500×g
for 5 min at 4°C, and the pellet was completely dried by air‐drying. Finally, the RNA
pellet was dissolved in RNase‐free water and stored at −80°C. The quality of isolated
total RNA was determined by 1% (w/v) agarose gel electrophoresis (see Section
4.1.1.2).
4.1.1.2 Separation of nucleic acids by agarose gel electrophoresis
Agarose gel was prepared by melting agarose (Ultrapure™
Agarose, Invitrogen, Carlsbad, CA, USA) in 0.5X TBE buffer (45 mM Tris‐base, 45
mM boric acid, 1 mM EDTA, pH 8.3), 0.5 µg/ml ethidium bromide using a
microwave oven. The agarose gel has been allowed to cool down before poured into
an assembled gel tray. After the gel was solidified, the comb was removed and the gel
was transferred into electrophoresis tank (Horizon®, Gibco‐BRL, Piscatawan, NJ,
Ref. code: 25605412330010EAN

40
USA). Agarose gel electrophoresis buffer (0.5X TBE buffer, 0.5 µg/ml ethidium
bromide) was poured into the tank until cover the gel. The samples were prepared by
mixing with sample loading solution (5% [v/v] glycerol, 0.025% [w/v] bromophenol
blue, 0.025% [w/v] xylene cyanol FF) and loaded into each well. The electrophoresis
condition was set at 80 V for 1 h, and then the gel was visualized under UV light
using gel documentation system.
4.1.1.3 DNase I treatment of O. viverrini total RNA
Contaminating DNA in the total RNA was eliminated by
DNase I treatment. The reaction mixture was prepared as shown below.
Components Volume (µl)
Total RNA
DNase 10X Reaction Buffer
RNase‐free water
RNase‐free DNase (1U/µl) (Promega, Fitchburg, WI, USA)
x (1 µg RNA)
2
17−x
1
Total volume 20
The reaction mixture was incubated at 37°C for 30 min, and
then 1 µl DNase Stop Solution (Promega, Fitchburg, WI, USA) was added to
terminate the reaction. Finally, the reaction tube was incubated at 65°C for 10 min to
inactivate the DNase.
4.1.2 Reverse transcription and PCR amplification of OvCALR
OvCALR cDNA was generated by reverse transcriptase PCR (RT‐
PCR) from total RNA of adult O. viverrini. One microgram of DNase‐treated RNA
was reverse transcribed using RevertAid Reverse Transcriptase (Thermo Scientific,
Vilnius, Lithuania) with Oligo(dT)20 primer. The reaction mixture was prepared as
shown on the next page.
Ref. code: 25605412330010EAN

41
Components Volume (µl)
Total RNA
DEPC‐treated ultrapure water
Oligo(dT)20 primer (100 µM)
5X Reaction Buffer
Thermo Scientific™ RiboLock RNase Inhibitor
Mixed dNTP (10 mM each)
RevertAid Reverse Transcriptase (200U/µl)
X
11.5−X
1
4
0.5
2
1
Total volume 20
Firstly, one microgram of DNase‐treated RNA was mixed with
Oligo(dT)20 primer, incubated at 70°C for 5 min and then the reaction tube was
chilled on ice. Subsequently, other components except RevertAid Reverse
Transcriptase (Thermo Scientific, Vilnius, Lithuania) were added into the reaction
tube and incubated at 37°C for 5 min. Reverse Transcriptase was added into the
reaction tube, followed by briefly spin and then incubated at 42°C for 1 h and 70°C
for 10 min, respectively. The reverse transcription reaction product was stored at
−20°C if not immediately used.
The first‐strand cDNA products were used as template for PCR
amplification of cDNA containing coding sequence of OvCALR. Specific primers
were designed based on an expressed sequence tag (Ov_Contig3944) of the encoding
gene (HRG303 forward primer and HRG304 reverse primer, see Table 4.1). These
primers introduced recognition sites for PaeI (SphI) and PstI restriction endonucleases
at 5′ and 3′ end, respectively, to facilitate the cloning into expression plasmid. The
PCR reaction mixtures were prepared as shown on the next page. The thermal cycling
conditions were set as follows: initial denaturation at 94°C for 3 min, 30 cycles of
denaturation at 95°C, annealing at 55°C, and extension at 72°C for 1 min each, and
final extension at 72°C for 10 min.
Ref. code: 25605412330010EAN
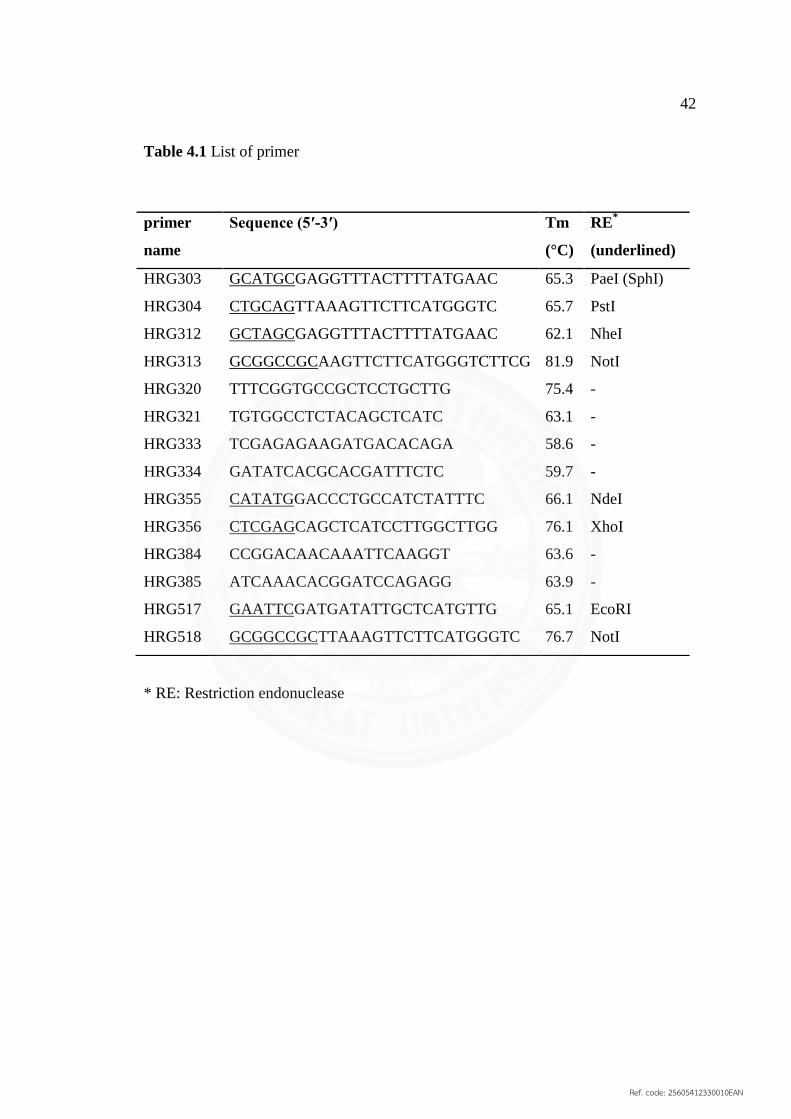
42
Table 4.1 List of primer
* RE: Restriction endonuclease
primer
name
Sequence (5′‐3′) Tm
(°C)
RE*
(underlined)
HRG303 GCATGCGAGGTTTACTTTTATGAAC 65.3 PaeI (SphI)
HRG304 CTGCAGTTAAAGTTCTTCATGGGTC 65.7 PstI
HRG312 GCTAGCGAGGTTTACTTTTATGAAC 62.1 NheI
HRG313
HRG320
HRG321
GCGGCCGCAAGTTCTTCATGGGTCTTCG
TTTCGGTGCCGCTCCTGCTTG
TGTGGCCTCTACAGCTCATC
81.9
75.4
63.1
NotI
-
-
HRG333 TCGAGAGAAGATGACACAGA 58.6 -
HRG334
HRG355
HRG356
GATATCACGCACGATTTCTC
CATATGGACCCTGCCATCTATTTC
CTCGAGCAGCTCATCCTTGGCTTGG
59.7
66.1
76.1
-
NdeI
XhoI
HRG384 CCGGACAACAAATTCAAGGT 63.6 -
HRG385
HRG517
HRG518
ATCAAACACGGATCCAGAGG
GAATTCGATGATATTGCTCATGTTG
GCGGCCGCTTAAAGTTCTTCATGGGTC
63.9
65.1
76.7
-
EcoRI
NotI
Ref. code: 25605412330010EAN

43
Components Volume (µl)
First‐strand cDNA products
HRG303 forward primer (10 µM)
HRG304 reverse primer (10 µM)
10X Taq Buffer with (NH4)2SO4
25 mM MgCl2
Mixed dNTP (10 mM each)
Taq DNA Polymerase (Thermo Scientific, Vilnius, Lithuania)
Sterile ultrapure water
2
1
1
5
3
1
0.5
36.5
Total volume 50
The PCR products was analyzed by 1% (w/v) agarose gel
electrophoresis as described in Section 4.1.1.2. The agarose gel containing expected
band was excised and extracted using the QIAquick Gel Extraction Kit (Qiagen,
Hilden, Germany) following the manufacturer’s instruction.
4.1.3 Ligation of OvCALR cDNA into pGEM®‐T Easy vector and
transformation of Escherichia coli (E. coli) cells with ligation
product
Purified OvCALR DNA fragments from previous section was
ligated into the pGEM®
‐T Easy vector (Promega, Fitchburg, WI, USA) using the
following ligation reaction.
Components Volume (µl)
2X Rapid Ligation Buffer, T4 DNA Ligase
Purified OvCALR DNA fragments
pGEM®‐T Easy vector
T4 DNA Ligase (3 Weiss units/µl)
5
3
1
1
Total volume 10
The reaction was incubated at room temperature for 1 h and
subsequently introduced into E. coli XL1‐Blue competent cells using a chemical
Ref. code: 25605412330010EAN

44
transformation method as described in Section 4.1.5. The transformants carrying
recombinant plasmid were identified by colony PCR as described in Section 4.1.7.
The plasmid DNA was isolated from positive clones using the AxyPrep Plasmid
Miniprep Kit (Axygen Biosciences, Union City, CA, USA) following manufacturer’s
instruction. The purified recombinant plasmid DNA was double‐digested with PaeI
(SphI) and PstI restriction enzymes (Thermo Scientific, Vilnius, Lithuania) to confirm
the insertion of target fragment. The double digestion reaction was prepared as shown
below.
Components Volume (µl)
10X Buffer B
Purified recombinant plasmid DNA
PaeI (SphI) (10 U/µl)
PstI (10 U/µl)
2
16.5
0.5
1
Total volume 20
The reaction was incubated at 37°C for 2 h and then inactivated at
80°C for 20 min. The digested and undigested recombinant plasmid DNA were
analyzed in parallel by agarose gel electrophoresis (see Section 4.1.1.2). The pGEM®‐
T Easy carrying OvCALR was sent to a commercial DNA sequencing service (1st
Base Asia, Singapore) to verify the nucleic acid sequence of OvCALR cDNA. A
verified plasmid DNA harboring the correct nucleic acid sequence of OvCALR
cDNA was used in the further experiments.
4.1.4 Preparation of chemically competent E. coli cells
The competent cells were prepared from the following E. coli
strains: XL1‐Blue, M15, BL21, and Rosetta‐gami(DE3)pLysS. These E. coli strains
were freshly grown on Luria‐Bertani (LB) agar plate containing appropriate antibiotic
as shown in Table 4.2. A single colony of each E. coli strain was grown at 37°C with
250 rpm shaking for 16–20 h in 5 ml LB broth containing appropriate antibiotic (see
Table 4.2). On the next day, 200 µl of fresh overnight culture was inoculated in 100
ml LB broth in 1 liter Erlenmeyer flask and incubated at 37°C with 250 rpm shaking.
A bacterial growing was monitored periodically by spectrophotometer at 600 nm.
Ref. code: 25605412330010EAN

45
When OD600 of 0.5 was reached, the culture was harvested by centrifugation at 6,000
rpm for 8 min at 4°C. The pellet was collected, resuspended in 20 ml of ice‐cold 0.1
M MgCl2, and then centrifuged at 6,000 rpm for 8 min at 4°C. The pellet was
collected and resuspended in 20 ml of ice‐cold 0.1 M CaCl2. After incubation on ice
for 20 min, the cell suspension was centrifuged at 6,000 rpm for 8 min at 4°C. The
pellet was collected and gently resuspended in 4.3 ml of 0.1 M CaCl2 and 0.7 ml of
glycerol. The competent cells were pipetted into 100 µl aliquots and then flash‐frozen
in liquid nitrogen. The cells were stored at −80°C until used in the experiment.
4.1.5 Transformation of E. coli cells
Either 10 µl of the ligation reaction or 1 µl of recombinant plasmid
DNA suspension was pipetted into a pre‐chilled 15 ml conical tube. Subsequently,
100 µl of competent cells that had been thawed on ice was transferred into the tube
and mixed gently. The tube was incubated on ice for 20 min and then heat‐shocked at
42°C for 45 s in a water bath. The tube was incubated on ice for 2 min afterward.
Nine hundred microliter of LB broth was added into the tube and incubated at 37°C
with shaking for 30 min. The transformation culture (100–200 µl) was plated on LB
agar plate containing appropriate antibiotics as follows: 100 µg/ml ampicillin for
XL1‐Blue and BL21, 100 µg/ml ampicillin and 25 µg/ml kanamycin for M15, and
100 µg/ml ampicillin, 34 µg/ml chloramphenicol, 12.5 µg/ml kanamycin and 3.125
µg/ml tetracycline for Rosetta‐gami(DE3)pLysS. The plates were incubated overnight
at 37°C.
Ref. code: 25605412330010EAN

46
Table 4.2 List of E. coli strains and appropriate antibiotic concentrations
E. coli strain Antibiotic Final concentration
(µg/ml)
XL1‐Blue tetracycline 12.5 (agar plate only)
M15 kanamycin 25
BL21 - -
Rosetta‐gami(DE3)pLysS kanamycin
tetracycline
chloramphenicol
12.5
3.125
34
4.1.6 Isolation of plasmid DNA using alkaline extraction method
A single colony of plasmid‐carrying E. coli was picked into 5 ml
LB broth and grown overnight in a 15 ml conical tube at 37°C with 250 rpm shaking.
The culture was harvested by centrifugation at 4,000 rpm for 5 min. The bacterial
pellet was collected, resuspended in 200 µl solution I (25 mM Tris pH 8.0, 50 mM
glucose, 10 mM EDTA), and transferred into a 1.5 ml microcentrifuge tube. Four
hundred microliters of solution Ⅱ (0.1 N NaOH, 1% [w/v] SDS) were added to the
tube, mixed by inverting, and incubated on ice for 5 min. The previous step was
repeated by adding 300 µl solution Ⅲ (2.7 M potassium acetate, pH 4.8). After
incubation, the tube was centrifuged at 12,000×g for 5 min at room temperature, and
the supernatant was collected to a new tube. To precipitate plasmid DNA, 0.6 volume
of isopropanol was added to the tube, mixed by inverting, and centrifuged at
12,000×g for 5 min at room temperature. The pellet was collected, washed with 70%
(v/v) ethanol, and centrifuged at 12,000×g for 5 min at room temperature. Finally, the
pellet was collected, air‐dried, and dissolved in 50 µl ultrapure water. Isolated‐
plasmid suspension was treated with RNAse A (Thermo Scientific, Vilnius,
Lithuania) as manufacturer’s instruction to eliminate RNA contamination before
restriction enzyme digestion.
Ref. code: 25605412330010EAN

47
4.1.7 Positive transformants screening by colony PCR
Single bacterial colonies growing on selective LB agar plate were
picked and each suspended in 50 µl sterile DW. These bacterial suspensions were
used as PCR template for PCR amplification using specific primers. The PCR
reaction mixtures were prepared as shown below. The thermal cycling conditions
were programmed with optimal conditions based on previous results. The PCR
products were analyzed by 1% (w/v) agarose gel electrophoresis as described in
Section 4.1.1.2.
Components Volume (µl)
Bacterial suspension
HRG299 forward primer (10 µM)
HRG300 reverse primer (10 µM)
10X Taq Buffer with (NH4)2SO4
25 mM MgCl2
Mixed dNTP (10 mM each)
Taq DNA Polymerase (Thermo Scientific, Vilnius, Lithuania)
Sterile ultrapure water
5
0.5
0.5
2.5
1.5
0.5
0.25
14.25
Total volume 25
4.1.8 Sequence analysis
Obtained DNA sequencing results were analyzed using
bioinformatics tools. The EMBOSS program168
was used to analyze nucleic acid and
amino acid sequences. A signal peptide was predicted by SignalP 4.1 server169
(http://www.cbs.dtu.dk/services/SignalP/). Motifs in deduced amino acid sequence
were screened by ScanProsite tool170
. Jemboss, pepstats (Jemboss version 1.5;
http://emboss.sf.net/Jemboss/) was used to calculate molecular weight and isoelectric
point of protein. BLAST® (https://blast.ncbi.nlm.nih.gov/Blast.cgi), which is a
powerful alignment search tool, was used to search for homologous nucleotide and
protein sequences. Multiple sequence alignment was performed by Jalview171
(Jalview version 2.10.3; http://www.jalview.org/Download) and SeaView172
(SeaView version 4.6.3; http://doua.prabi.fr/software/seaview). Jemboss, needle
Ref. code: 25605412330010EAN

48
(Jemboss version 1.5; http://emboss.sf.net/Jemboss/) was used to analyze pairwise
sequence conservation.
4.1.9 Storage of bacterial clones
To prepare bacterial glycerol stock for long‐term storage, an
overnight culture in liquid medium was grown. Seven hundred and fifty microliter of
the overnight culture was mixed thoroughly with 250 microliter of 60% (v/v) glycerol
in a 1.5 ml microcentrifuge tube. The tube was frozen in liquid nitrogen and stored at
−80°C.
4.2 Production of rOvCALR using a bacterial system
4.2.1 Construction of recombinant expression vector
4.2.1.1 Construction of pQE‐30‐OvCALR expression vector
The verified pGEM®‐T Easy‐OvCALR plasmid (from
previous section) and pQE‐30 expression vector (Qiagen, Hilden, Germany) were
double‐digested with PaeI (SphI) and PstI restriction enzymes (Thermo Scientific,
Vilnius, Lithuania) to create sticky‐end DNA products. The digestion reactions and
conditions were prepared and processed as described in Section 4.1.3. The digestion
products were analyzed by 1% (w/v) agarose gel electrophoresis (see Section 4.1.1.2).
After that, the expected bands were excised from the agarose gel and extracted using
the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany) following the
manufacturer’s instruction. The OvCALR DNA fragment and pQE‐30 vector were
joined by T4 DNA ligase (Thermo Scientific, Vilnius, Lithuania) using the ligation
reaction as shown on the next page.
Ref. code: 25605412330010EAN

49
Components Volume (µl)
10X T4 DNA Ligase Buffer
Purified OvCALR DNA fragments
Digested pQE‐30
T4 DNA Ligase (5 Weiss U/µl)
Sterile ultrapure water
1
3
1
1
4
Total volume 10
The ligation proceeded overnight at 4°C and the product was
then introduced into E. coli XL1‐Blue competent cells by chemical transformation
(see Section 4.1.5). Colony PCR and restriction enzyme digestion were performed to
confirm the clone harboring pQE‐30‐OvCALR recombinant plasmid as described in
Section 4.1.7 and Section 4.1.3, respectively. After getting a verified clone, the
recombinant plasmid DNA was isolated from positive clones using alkaline extraction
method (see Section 4.1.6) and introduced into the expression host, E. coli M15 (see
Section 4.1.5). Colony PCR was performed to identify the positive clones as
described in Section 4.1.7.
4.2.1.2 Construction of pET21b(+)‐OvCALR expression vector
(1) DNA cloning of OvCALR
The verified OvCALR (from previous section) was used as a
template for PCR amplification. The forward and reverse primers (HRG312 forward
primer and HRG313 reverse primer, see Table 4.1) introduced recognition sites for
NheI and NotI restriction endonucleases, respectively, to facilitate the cloning into
pET21b(+) vector. The PCR reaction was prepared as shown on the next page.
Ref. code: 25605412330010EAN

50
Components Volume (µl)
Purified OvCALR DNA fragments
HRG312 forward primer (10 µM)
HRG313 reverse primer (10 µM)
10X Taq Buffer with (NH4)2SO4
25 mM MgCl2
Mixed dNTP (10 mM each)
Taq DNA Polymerase (Thermo Scientific, Vilnius, Lithuania)
Sterile ultrapure water
2
1
1
5
3
1
0.5
36.5
Total volume 50
The thermal cycling conditions were set as follows: initial
denaturation at 95°C for 5 min, 30 cycles of denaturation at 95°C, annealing at 55°C,
and extension at 72°C for 1 min each, and final extension at 72°C for 10 min. The
PCR products were analyzed by 1% (w/v) agarose gel electrophoresis (see Section
4.1.1.2), and the agarose gel containing expected band was excised and extracted
using the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany) following the
manufacturer’s instruction. The purified OvCALR DNA fragment was inserted into
the pGEM®‐T Easy vector (Promega, Fitchburg, WI, USA), introduced into E. coli
XL1‐Blue, screened for positive clones, and the DNA sequence verified as described
in Section 4.1.3.
(2) Ligation of OvCALR into pET21b(+) vector
After getting the verified recombinant plasmid, pGEM®
‐T
Easy‐OvCALR and pET21b(+) expression vector (Novagen, Darmstadt, Germany)
were double‐digested with NheI and NotI restriction enzymes (Thermo Scientific,
Vilnius, Lithuania). The double digestion reaction was prepared into two steps as
shown on the next page.
Ref. code: 25605412330010EAN

51
Step Components Volume (µl)
1
10X Tango Buffer
pGEM®‐T‐OvCALR or pET21b(+) vector
NheI (10 U/µl)
2
17.5
0.5
2 10X Tango Buffer
NotI (10 U/µl)
Sterile ultrapure water
6
4
10
Total volume 40
All components in the first step were incubated at 37°C for 2
h. After that, the components in the second step were added and further incubated at
37°C for 2 h. The reaction was inactivated by incubation at 65°C for 20 min and
analyzed by 1% (w/v) agarose gel electrophoresis (see Section 4.1.1.2). The expected
bands was excised from the agarose gel and extracted using the QIAquick Gel
Extraction Kit (Qiagen, Hilden, Germany). The purified OvCALR DNA fragment and
digested‐pET‐21b(+) were ligated and further processed as described above for pQE‐
30‐OvCALR expression vector construction. After getting a verified clone, the
recombinant plasmid DNA was isolated from positive clones using alkaline extraction
method (see Section 4.1.6) and introduced into the expression host, E. coli Rosetta‐
gami(DE3)pLysS (see Section 4.1.5). Colony PCR was performed to identify the
positive clones as described in Section 4.1.7.
4.2.2 Small‐scale expression and time‐course analysis
Three positive colonies were randomly selected to determine the
time‐course expression. A single colony was inoculated in 5 ml LB broth containing
100 µg/ml ampicillin and other appropriate antibiotics (see Table 4.2), and incubated
overnight at 37°C with 250 rpm shaking. On the next day, 20 ml LB broth containing
appropriate antibiotics as previously mentioned was inoculated with 1 ml of the
overnight culture, and incubated at 37°C with 250 rpm shaking until the OD600 of 0.6
was reached. One milliliter of the culture was collected as non‐induced sample, and
then Isopropyl‐β‐D‐1‐thiogalactopyranoside (IPTG) (Fermentas, Vilnius, Lithuania)
was added into the culture to a final concentration of 1 mM. For M15/pQE‐30‐
OvCALR, the cultures were further grown in the previous conditions for 4 h, and 1 ml
Ref. code: 25605412330010EAN

52
of the culture was collected every hour following induction as induced samples. For
Rosetta‐gami(DE3)pLysS/pET‐21b(+)‐OvCALR, the cultures were further grown
overnight at 28°C with 250 rpm shaking, and then 1 ml of each culture was collected
on the next day as induced sample. Additional 1‐ml samples were hourly collected for
protein solubility analysis. All samples were centrifuged at 12,000×g for 1 min. The
pellet was resuspended in 50 µl of denaturing lysis buffer (100 mM NaH2PO4, 10 mM
Tris‐HCl, 8 M Urea, pH 8.0) and then analyzed by SDS‐PAGE (see Section 4.2.5).
4.2.3 Determination of target‐protein solubility
Additional collected samples at pre‐ and post‐induction were
centrifuged at 12,000×g for 1 min. The supernatant was discarded, and the pellet was
resuspended in 100 µl of native lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10
mM imidazole, pH 8.0). The bacterial cells were lysed by sonication (six 9.9 s bursts
with 9.9 s pauses) at 27% amplitude. The lysate was centrifuged at 12,000×g for 20
min at 4°C. The supernatant was collected as soluble fraction, and the pellet was
resuspended in 50 µl of denaturing lysis buffer as insoluble fraction. Then soluble and
insoluble fractions of each time point were analyzed by SDS‐PAGE as described in
Section 4.2.5.
4.2.4 Large‐scale expression of rOvCALR
4.2.4.1 Large‐scale expression of M15‐derived rOvCALR
A single positive colony verified by small‐scale screening
was cultured overnight at 37°C with shaking in 5 ml LB broth containing 100 µg/ml
ampicillin, 25 µg/ml kanamycin. On the next day, 100 ml LB broth containing
appropriate antibiotics as previously mentioned was inoculated with 5 ml of the
overnight culture. The culture was grown at 37°C, 250 rpm shaking until the OD600 of
0.6 was reached, and then the expression was induced by IPTG at a final
concentration of 1 mM. The culture was further incubated at 37°C, 250 rpm shaking
for 4 h. The bacterial cells were harvested by centrifugation at 4,000×g for 20 min at
4°C and stored at −20°C.
4.2.4.2 Large‐scale expression of Rosetta‐gami(DE3)pLysS‐
derived rOvCALR
A single positive colony verified by small‐scale screening
was cultured overnight at 37°C with shaking in 5 ml LB broth containing 100 µg/ml
Ref. code: 25605412330010EAN

53
ampicillin, 34 µg/ml chloramphenicol, 12.5 µg/ml kanamycin and 3.125 µg/ml
tetracycline. On the next day, 100 ml LB broth containing appropriate antibiotics as
mentioned above was inoculated with 5 ml of the overnight culture. The culture was
grown at 37°C, 250 rpm shaking until the OD600 of 0.6 was reached, and then the
expression was induced by IPTG at a final concentration of 1 mM. The culture was
further incubated overnight at 28°C, 250 rpm shaking. On the next day, the culture
was harvested by centrifugation at 4,000×g for 20 min at 4°C and stored at −20°C.
4.2.5 Protein analysis by sodium dodecyl sulfate‐polyacrylamide gel
electrophoresis (SDS‐PAGE)
4.2.5.1 Gel casting, sample preparation and electrophoresis
The discontinuous Tris‐glycine SDS‐PAGE system was used
for protein electrophoresis. Gel casting components of Hoefer SE260 Mini‐gel
System (Amersham biosciences, GE Healthcare, Piscatawan, NJ, USA) were
assembled following the manufacturer’s manual. A single gel of 12.5% resolving gel
was prepared by pipetting the following solutions into a 50 ml conical tube: 2.5 ml
30% (w/v) polyacrylamide/Bis solution 29:1 (Bio‐Rad, Shanghai, China), 1.5 ml 1.5
M Tris‐HCl pH 8.8, 60 µl 10% (w/v) SDS, 1.88 ml ultrapure water, 60 µl 10% (w/v)
ammonium persulfate (APS), and 2.5 µl TEMED. All components were mixed by
gently swirling and immediately poured into between a glass plate and an alumina
plate up to 2/3 of the glass plate. Distilled water was overlaid on the top of the
resolving gel, and the gel was allowed to polymerize for 1 h. After the gel
polymerizing, the water was discarded and totally removed using paper towel. To
prepare a single 4% stacking gel, the following components were mixed by gently
swirling in a 50 ml conical tube: 270 µl 30% (w/v) polyacrylamide/Bis solution 29:1
(Bio‐Rad, Shanghai, China), 500 µl 0.5 M Tris‐HCl pH6.8, 20 µl 10% (w/v) SDS,
1.18 ml ultrapure water, 20 µl 10% (w/v) APS, 2 µl TEMED. The mixed solution was
topped on the resolving gel by pipetting, and a comb was inserted promptly. The gel
was allowed to polymerize for 1 h. After that, the comb had been removed, and the
wells were washed with ultrapure water. The gel apparatus was assembled following
the manual, and electrophoresis buffer (25 mM Tris‐base, 202 mM glycine, 0.1%
[w/v] SDS) was filled into the upper and lower chamber.
Ref. code: 25605412330010EAN

54
The samples were prepared by mixing with 2X sample
electrophoresis buffer (0.125 M Tris‐HCl pH 6.8, 0.2 M DTT, 0.02% [w/v]
bromophenol blue, 4% [w/v] SDS, 20% [v/v] sterile glycerol), heating at 95°C for 5
min, putting on ice for 1 min, and briefly spinning. The Broad Range Molecular
Weight Standards (Bio‐Rad Laboratories, Hercules, CA, USA) and the samples were
loaded into each well, and a power was applied at 20 mA/gel. The run was finished
when the tracking dye reached the bottom of the gel. The gel was carefully removed
from the gel cassette, and the stacking gel was discarded. The remaining gel was
washed several times in distilled water afterward.
4.2.5.2 Staining of protein in polyacrylamide gel
The gel (resolving gel) was stained with Coomassie dye
staining solution (0.025% [w/v] Coomassie Blue R‐250, 40% [v/v] methanol, and 7%
[v/v] glacial acetic acid) by soaking the gel overnight with gently shaking. After that,
the gel was destained by soaking in high‐methanol destaining solution (40% [v/v]
methanol, 7% [v/v] glacial acetic acid) and low‐methanol destaining solution (5%
[v/v] methanol, 7% [v/v] glacial acetic acid), respectively. The protein bands in the
gel were visualized and photographed using a scanner (Canon CanoScan LiDE 700F).
For long‐term storage, the gel was laid between two sheets of cellophane membrane
on a glass plate and allowed to dry at room temperature for 2 days.
4.2.6 Purification of rOvCALR using Ni‐NTA affinity
chromatography
4.2.6.1 Purification of rOvCALR under denaturing conditions
The cell pellet was resuspended in 5 ml denaturing lysis
buffer (100 mM NaH2PO4, 10 mM Tris‐HCl, 8 M urea, pH 8.0) and incubated on a
rotary mixer at room temperature for 1 h. The cell lysate was centrifuged at 10,000×g
for 30 min at 4°C, and the supernatant was collected. The Ni‐NTA slurry (QIAGEN,
Hilden, Germany) was mixed with the clear supernatant on a rotary mixer at 4°C for 1
h before loading into a polypropylene column. The resin was allowed to settle for 15
min. The column’s bottom cap was removed afterward, and the flow‐through was
collected. The column was washed twice with 4 ml wash buffer (100 mM NaH2PO4,
10 mM Tris‐HCl, 8 M urea, pH 6.3) and eluted four times each with 500 µl elution
buffer at pH 5.9 and pH 4.5, respectively (100 mM NaH2PO4, 10 mM Tris‐HCl, 8 M
Ref. code: 25605412330010EAN

55
urea, pH 5.9 or pH 4.5). All fractions were collected and analyzed by SDS‐PAGE as
described in Section 4.2.5.
4.2.6.2 Purification of rOvCALR under native conditions
The cell pellet was resuspended in native lysis buffer (50
mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0) using 5 ml per gram wet
weight. After that, the bacterial cells were broken by ultrasonication (Sonics vibra
cell™ VC 750, Sonics and Materials, Inc., Newtown, CT, USA) using 6×9.9 s with
9.9 s pauses at 27% amplitude. The total cell lysate was centrifuged at 10,000×g for
30 min at 4°C, and supernatant was collected as cell lysate. One milliliter of Ni‐NTA
slurry (QIAGEN, Hilden, Germany) was mixed with the cell lysate on a rotary mixer
at 4°C for 1 h. Afterward, the mixture was loaded into a polypropylene column, and
the resin was allowed to settle for 15 min. The column’s bottom cap was removed,
and the flow‐through was collected. The column was washed twice with 4 ml of wash
buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0) and eluted four
times with 500 µl of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM
imidazole, pH 8.0). All fractions were collected and analyzed by SDS‐PAGE as
described in Section 4.2.5.
4.2.7 Concentration and dialysis of rOvCALR
4.2.7.1 Concentration of rOvCALR using centrifugal
concentrator
The eluates containing rOvCALR (from Section 4.2.6) were
pooled and concentrated using Amicon Ultra‐15 Centrifugal Filter Units (Merck
Millipore, County Cork, Ireland). The pooled sample was filled into the concentrator
and then centrifuged at 5,000×g for 30 min at 4°C using Sorvall Legend RT+
centrifuge (Thermo Scientific, Osterode, Germany). This centrifugation step was
repeated several times until the volume of sample reached the desired volume.
4.2.7.2 Dialysis of rOvCALR
To remove some chemicals which interfere the subsequent
assays, the sample was dialyzed against 10 mM PBS pH 7.2 using 3 Spectra/Por®
dialysis membrane tubing (Spectrumlabs, Rancho Dominguez, CA, USA). The
dialysis membrane was prepared by soaking in distilled water for 30 min and then
rinsing thoroughly with distilled water. The dialysis tubing was tightly closed at the
Ref. code: 25605412330010EAN

56
bottom, and the sample was loaded into the dialysis bag. Subsequently, the bag was
tightly closed at the top and submerged in dialysate (10 mM PBS pH 7.2). The
volume of dialysate was 1,000 times of the sample volume. The dialysis was
performed with stirring at 4°C, and the dialysate was changed at 2 h and 16 h
following the dialysis start. The sample was collected following 24 h of dialysis.
The quality and quantity of rOvCALR were determined by
SDS‐PAGE and Bradford’s assay as described in Section 4.2.5 and Section 4.2.8,
respectively. After that, the quantified rOvCALR was aliquoted and stored at −20°C.
4.2.8 Measurement of rOvCALR concentration using Bradford’s
assay
The recombinant protein concentration was measured by a
Bradford’s assay (Bio‐Rad, Hercules, CA, USA). Bovine serum albumin (BSA)
(Sigma Aldrich, St. Louis, MO, USA) was used as protein standard at following
concentrations: 0.1, 0.2, 0.3, 0.4 and 0.5 mg/ml. The dye reagent was prepared by
dilution of one part of dye reagent concentrate with four parts of ultrapure water. Ten
microliters of each protein standard and sample were pipetted into separate microtiter
plate wells (NUNC, Jiangsu, China) in duplicate. The protein diluent such as 10 mM
PBS, pH 7.2 was used as a blank. Two hundred microliters of diluted dye reagent
were added into each well, and the plate was mixed by tapping and then incubated at
room temperature for 5 min. The absorbance was measured at 595 nm using a
microplate reader. A standard protein curve of BSA was plotted, and a linear equation
was used to calculate the concentration of sample.
4.3 Production of polyclonal antibody in mice
4.3.1 Production of polyclonal mouse anti‐rOvCALR antibody
Three female 6–8 week‐old ICR mice were immunized with E. coli
M15‐derived rOvCALR to produce polyclonal antisera. The pre‐immune sera were
collected one week before immunization as control sera. The amount of antigen for
immunization was prepared at 30 µg/mouse for priming and at 20 µg/mouse for
boosting. The antigen preparation was processed as follows: resolving rOvCALR in
12.5% polyacrylamide gel (see Section 4.2.5), staining the gel with 0.5% (w/v)
Ref. code: 25605412330010EAN

57
Coomassie Blue G‐250 (USB Corporation, Cleveland, OH, USA), destaining with
distilled water, excising the target band, and grinding the gel with 10 mM PBS pH 7.2
by glass homogenizer. The mice were intraperitoneally injected with 100 µl prepared
antigen, three times every 3‐week intervals: priming, first and second boosting. The
blood was collected from their tail using heparinized capillary tubes every 2 weeks
after immunization. The capillary tubes were centrifuged at 10,000×g for 5 min at
room temperature, and the serum was kept at −20°C. The timeline diagram of anti‐
rOvCALR antisera production is shown in Figure 4.1.
Figure 4.1 Timeline diagram of anti‐rOvCALR antisera production in mice. Black
arrowheads indicate blood collecting time points, and white arrowheads indicate
immunization time points.
4.3.2 Production of polyclonal mouse anti‐OvES antibody
Two female 6–8 weeks‐old BALB/c mice were immunized with 100
µl prepared antigen at 20 µg/mouse for priming and at 10 µg/mouse for boosting. The
antigen was prepared by mix equal volume of O. viverrini ES product and TiterMax
Gold Adjuvant (Sigma Aldrich, St. Louis, MO, USA) using 1‐ml insulin syringe with
21G needle. Mice were immunized and blood collected as described in Section 4.3.1.
4.3.3 Determination of antibody titer by enzyme linked
immunosorbent assay (ELISA)
To prepare the rOvCALR‐coated microtiter plate, rOvCALR was
prepared in carbonate buffer, pH9.6 (30 mM Na2CO3, 75 mM NaHCO3) and pipetted
into each well of a NUNC MaxiSorp 96‐well plate (NUNC, Roskide, Denmark) (150
ng/100 µl/well) and incubated overnight at 4°C. The wells were washed three times
with distilled water. To block unspecific protein binding, the wells were incubated
with 0.25% (w/v) BSA (Sigma Aldrich, St. Louis, MO, USA) in coating buffer at
Ref. code: 25605412330010EAN

58
room temperature for 30 min. The plate was washed afterward three times with
distilled water. Pre‐immune, priming, first boosting, and second boosting sera were
stepwise 2‐fold diluted in an antibody diluent (0.25% [w/v] BSA in 10 mM PBS, pH
7.2) starting from 1:100 to 1:204,800 and pipetted into each well in duplicate. The
plate was incubated at 37°C for 1 h and washed three times as before. Goat anti‐
mouse HRP conjugated antibody (Invitrogen, Frederick, MD, USA) at dilution
1:4,000 was added and incubated at 37°C for 1 h. The wells were washed afterward,
and 100 µl of o‐Phenylenediamine dihydrochloride (OPD) substrate (Sigma Aldrich,
St. Louis, MO, USA) was added, incubated in the dark at room temperature for 30
min. The reaction was stopped by adding 50 µl of 2 M H2SO4, and the absorbance
was measured at 492 nm.
4.4 Preparation of parasite antigens
4.4.1 Parasite crude extracts
Adult parasites (from Section 4.5.1) were homogenized on ice using
a tissue homogenizer (Ultra‐Turrax T25, IKA, Staufen, Germany) in homogenization
buffer (10 mM PBS, pH 7.2, 150 mM NaCl, 0.5% [v/v] Triton X‐100, 1 mM PMSF, 1
mM EDTA). The homogenate was centrifuged at 12,000×g for 15 min at 4°C to
remove insoluble material. The supernatant was collected as soluble crude worm
extract, and the pellet was further extracted as insoluble crude worm extract. The
pellet was solubilized in 50 mM Tris‐HCl, pH 8.0, 3% (w/v) SDS and incubated at
37°C for 1 h. After that, the sample was centrifuged at 12,000×g for 15 min, and the
supernatant was collected as insoluble crude worm extract. The parasite extracts were
determined quantity and quality using a bicinchoninic acid (BCA) assay (see Section
4.4.3) and SDS‐PAGE (see Section 4.2.5), respectively. The parasite crude extracts
were aliquoted and stored at −80°C.
4.4.2 Excretory/secretory (ES) product
Adult O. viverrini was freshly collected from experimentally
infected hamsters (from Section 4.5.1). Alive parasites were washed twice with
physiological saline solution (0.85% [w/v] sodium chloride) and washed once with 10
mM PBS, pH 7.2. The buffer was changed to fresh 10 mM PBS, pH 7.2 and the
Ref. code: 25605412330010EAN

59
parasites were incubated in CO2 incubator (Forma™ Steri‐Cycle™ CO2 Incubator,
Thermo Fisher Scientific, Marietta, OH, USA) at 37°C, 5% CO2 for 4 h. Then the
buffer was collected and centrifuged at 5,000×g for 20 min at 4°C to remove insoluble
materials and eggs. The supernatant was collected and concentrated using a
centrifugal concentrator with 3kDa cut‐off (GE Healthcare, Buckinghamshire, UK).
The concentrated ES product was determined quantity and quality using Bradford’s
assay (see Section 4.2.8) and SDS‐PAGE (see Section 4.2.5), respectively. The
concentrated OvES product was aliquoted and stored at −80°C.
4.4.3 Measurement of protein concentration using BCA assay
BCA protein assay is a detergent‐compatible quantitative protein
measurement. PierceTM
BCA Protein Assay Kit (Thermo Scientific, Rockford, IL,
USA) was used to determine protein concentration of parasite crude extracts using
microplate procedure. BSA (Sigma Aldrich, St. Louis, MO, USA) was used as protein
standard at following concentrations: 0.125, 0.25, 0.5, 0.75, 1.0, 1.5 and 2.0 mg/ml.
The BCA working reagent was prepared by mixing 50 parts of BCA reagent A with 1
part of BCA reagent B. Ten microliters of each protein standard and sample were
pipetted into separate microtiter plate wells (NUNC, Jiangsu, China) in duplicate. The
protein diluent such as 10 mM PBS, pH 7.2 was used as a blank. Two hundred
microliters of BCA working reagent were added into each well, and the plate was
mixed by tapping and then incubated at 37°C for 30 min. The absorbance was
measured at 562 nm using a microplate reader. A standard protein curve of BSA was
plotted, and a linear equation was used to calculate the concentration of sample.
4.5 Characterization of OvCALR expression
4.5.1 Preparation of different developmental stages of O. viverrini
O. viverrini metacercariae were isolated from naturally infected
cyprinoid fish in the northeast of Thailand. Newly excysted juveniles were obtained
from mechanically excysted metacercariae using a disposal needle. Other
developmental stages of O. viverrini were obtained from the liver of experimentally
infected hamsters. Syrian golden hamsters were infected with 50 metacercariae by
intragastric intubation. The juveniles and adults O. viverrini were harvested from
Ref. code: 25605412330010EAN

60
livers and bile ducts of infected hamsters at 2‐, 4‐ and 8‐week postinfection,
respectively. The parasites were washed in physiological saline solution (0.85% [w/v]
sodium chloride) and kept in liquid nitrogen for further experiments.
4.5.2 Stage‐specific reverse transcriptase polymerase chain reaction
(RT‐PCR)
Total RNA of newly excysted juvenile O. viverrini was isolated
using TRIzol™
Reagent with PureLink™ RNA Mini Kit (Ambion, Foster City, CA,
USA) following the manufacturer’s user manual. Total RNA of 2‐week juvenile, 4‐
week juvenile and adult O. viverrini (8‐week‐old) was isolated using TRIzol™
Reagent (Invitrogen, Carlsbad, CA, USA) as described in Section 4.1.1.1. The quality
of isolated total RNA was determined by 1% (w/v) agarose gel electrophoresis (see
Section 4.1.1.2). The concentration of isolated total RNA from each stage was
measured using a spectrophotometer, and followed by DNase I treatment as described
in Section 4.1.1.3.
To verify the expression of OvCALR from each developmental
stage of the parasite, 100 ng of DNase‐treated total RNA from each stage was used as
a template for reverse transcription. Two reverse specific primers were used to
generate cDNA in the same reaction: HRG385 reverse primer for OvCALR and
HRG334 reverse primer for OvActin as internal control (see Table 4.1). The reaction
mixture was prepared as shown below.
Components Volume (µl)
Total RNA (100 ng)
DEPC‐treated ultrapure water
HRG334 reverse primer (10 µM)
HRG385 reverse primer (10 µM)
5X Reaction Buffer
Thermo Scientific™ RiboLock RNase Inhibitor
Mixed dNTP (10 mM each)
RevertAid Reverse Transcriptase (200U/µl)
X
12.5−X
1
1
6
0.5
3
1
Total volume 30
Ref. code: 25605412330010EAN

61
The cDNA generation was processed as described in Section 4.1.2.
After that, the reverse transcription product was amplified by separate PCR reactions.
The PCR reactions were prepared as shown on the next page. The thermal cycling
conditions were set as follows: 5 min initial denaturation at 95°C, 34 cycles of
denaturation at 95°C, annealing at 55°C, and extension at 68°C for 1 min each, and 5
min final extension at 68°C. The PCR products were analyzed by 1% (w/v) agarose
gel electrophoresis (see Section 4.1.2.2).
Components Volume
(µl)
First‐strand cDNA products
HRG303 (OvActin) or HRG384 (OvCALR) forward primer (10 µM)
HRG304 (OvActin) or HRG385 (OvCALR) reverse primer (10 µM)
10X Taq Buffer with (NH4)2SO4
25 mM MgCl2
Mixed dNTP (10 mM each)
Taq DNA Polymerase (Thermo Scientific, Vilnius, Lithuania)
Sterile ultrapure water
2
0.5
0.5
2.5
1.5
0.5
0.2
17.3
Total volume 25
4.5.3 Detection of native OvCALR using Western blot analysis
4.5.3.1 Protein blotting by semi‐dry transfer
The parasite antigens (from Section 4.4.1 and Section 4.4.2),
10 µg each of insoluble, soluble crude worm extracts and ES product, and 100 ng of
rOvCALR were size‐separated by 12.5% SDS‐PAGE as described in Section 4.2.5.1.
The resolving gel was washed in distilled water and subsequently equilibrated in
semi‐dry transfer buffer (50 mM Tris, 40 mM glycine, 0.04% [w/v] SDS, 20% [v/v]
methanol) for 5 min. The nitrocellulose membrane (Immobilon‐NC Transfer
Membrane, Merck Millipore, Darmstadt, Germany) and filter paper (3 MM Chr filter
paper, Whatman®, GE healthcare, Buckinghamshire, UK) were cut to 8 × 8.5 cm
2 (1
sheet for membrane and 12 sheets for filter paper). Before blotting, the membrane was
equilibrated in semi‐dry transfer buffer for 15 min, and the filter paper was soaked in
Ref. code: 25605412330010EAN

62
semi‐dry transfer buffer for 2 min. A blotting tower was constructed by sequentially
placing down the following on the anode side of the blotting instrument (Fastblot
B33, Whatman Biometra, Goettingen, Germany): six sheets of filter paper,
nitrocellulose membrane, gel, and remaining six sheets of filter paper. The air bubble
between the blotting stack was removed by rolling a glass rod over the stack of filter
paper. The lid of the blotting instrument had been placed properly, and the power was
applied at 0.8 mA/cm2 for 50 min. After that, the membrane was stained with 0.1%
(w/v) Ponceau S (Sigma Aldrich, Steinheim, Germany), 5% (v/v) acetic acid for 3
min and then washed with distilled water until the protein bands were observed. The
membrane was allowed to dry and directly processed for immunodetection or be
stored at −20°C.
4.5.3.2 Detection of protein blotting
The nitrocellulose membrane was washed in distilled water
for 10 min to remove the remaining Ponceau S on the membrane. Afterward, the
membrane was equilibrated in TBS, pH 7.5 (20 mM Tris‐HCl, 150 mM NaCl) for 5
min. Unspecific binding sites on the membrane were blocked in 5% (w/v) skim milk
(Oxoid, Hants, UK) in TBS, pH 7.5 at room temperature for 1 h with gentle shaking.
The membrane‐bound antigens were probed with either mouse anti‐rOvCALR
antiserum or mouse pre‐immune serum at dilution 1:3,000 in antibody diluent (1%
[w/v] skim milk in TBS, pH 7.5) by incubation overnight at 4°C with gentle shaking.
On the following day, the membranes had been washed three times for 5 min each in
washing buffer (TBS, pH 7.5, 0.05% [v/v] Tween®
20) and followed by incubation
with goat anti‐mouse IgG (whole molecule) ‐alkaline phosphatase (Sigma Aldrich, St.
Louis, MO, USA) in antibody diluent (dilution 1:30,000) for 1 h at room temperature
with gentle shaking. The membranes were washed in washing buffer as before and
subsequently equilibrated in detection buffer (0.1 M Tris‐HCl, pH 9.5, 0.1 M NaCl,
50 mM MgCl2) for 5 min. The chromogenic substrate, 5‐bromo‐4‐chloro‐3‐indolyl
phosphate/nitro blue tetrazolium (BCIP/NBT) (Amresco, Solon, OH, USA), was
added to develop the signals as dark blue band by incubation in the dark. After
adequate signal strength had been observed, the developed membranes were washed
several times in distilled water to stop the reaction.
Ref. code: 25605412330010EAN

63
4.5.4 Immunohistochemical detection of OvCALR
Freshly collected O. viverrini adults (from Section 4.5.1) were
cleaned in physiological saline solution, fixed in methacarn solution, and embedded in
paraffin (Q Path Paraffin Normal, LABONORD SAS, Templemars, France). The
paraffin embedded parasite tissue was sectioned at 8 µm thickness on a microtome
(LEICA RM 2235, Nussloch, Germany) and mounted on gelatin‐coated microscope
slides. The slides were placed on a heating plate at 42°C overnight. The parasite
sections were deparaffinized twice in xylene for 10 min each and rehydrated in a
series of decreasing alcohol concentrations (absolute ethanol to 70% [v/v] ethanol) for
5 min each. Epitope retrieval was performed by heating parasite sections in sodium
citrate buffer (10 mM Sodium citrate, 0.05% [v/v] Tween®
20, pH 6.0) for 5 min in a
microwave oven. The parasite sections had been allowed to cool down before rinsed
in washing buffer (10 mM PBS, pH 7.2, 0.1% [v/v] Tween® 20). After that,
unspecific binding sites were blocked by incubation in 1% (w/v) glycine, 10 mM
PBS, pH 7.2 and then in 4% (w/v) BSA, 10 mM PBS, pH 7.2 for 30 min each at room
temperature. After blocking, the parasite sections were incubated overnight at 4°C
with either mouse anti‐rOvCALR antiserum or pre‐immune serum at dilution 1:2,000
in 1% (w/v) BSA in 10 mM PBS, pH 7.2. On the following day, the parasite sections
were washed three times in washing buffer for 5 min each with gentle shaking, and
the endogenous peroxidase activity was blocked by incubation of the sections in 3%
(v/v) hydrogen peroxide (Merck, Hohenbrunn, Germany) twice at room temperature
for 10 min each in the dark. After washing three times with washing buffer, the
parasite sections were incubated in polyclonal rabbit anti‐mouse immunoglobulin
biotinylated (dilution 1:200) (Dako, Glostrup, Denmark) at 37°C for 1 h. The parasite
sections were washed as before and subsequently incubated with avidin‐biotin
complex (ABC) peroxidase (ABC Peroxidase Standard Staining Kit, Thermo
Scientific, Rockford, IL, USA) at room temperature for 30 min. The parasite sections
were washed again, the chromogenic substrate, 3‐amino‐9‐ethylcarbazole (AEC)
(Vector Laboratories, Burlingame, CA, USA), was added and incubated at room
temperature in the dark until a signal developed. The reaction was stopped by washing
several times in PBST (10 mM PBS, pH 7.2, 0.1% [v/v] Tween® 20). The tissue
sections were mounted in mounting medium (10% (v/v) glycerol in 10 mM PBS, pH
Ref. code: 25605412330010EAN

64
7.2) and photographed using a microscopy camera (Olympus BX51, Japan
[microscope], PixeLINK, Ottawa, ON, Canada [digital camera]).
To detect OvCALR in O. viverrini‐infected hamster liver tissue, the
liver tissue embedded paraffin was sectioned at 5 µm thickness on a microtome
(LEICA RM 2235, Nussloch, Germany), mounted on gelatin‐coated microscope
slides and processed as described above. Antigen retrieval was performed by heating
liver sections in Tris‐EDTA buffer (10mM Tris‐Base, 1mM EDTA, 0.05% [v/v]
Tween® 20, pH 9.0). The tissue sections were counterstained with hematoxylin before
mounting. Normal hamster liver tissue was processed as above and used as negative
control for immunostaining at identical conditions.
4.6 Detection of rOvCALR by O. viverrini‐infected hamster sera
4.6.1 Blood collection from O. viverrini‐infected hamsters
Syrian golden hamsters (n=10) were experimentally infected with
50 metacercariae by intragastric intubation. Blood samples were collected by retro
orbital blood collection at preinfection and 2, 4, 8 and 12 weeks postinfection. The
blood samples were allowed to clot at room temperature for 2 h and then centrifuged
at 5,000×g for 10 min at room temperature. The hamster sera were collected,
aliquoted and kept at −20°C.
4.6.2 Indirect ELISA
An indirect ELISA technique was used to detect rOvCALR by
O. viverrini‐infected hamster sera as described in Section 4.3.2. Briefly, 100 ng of
rOvCALR was coated in each well of 96‐well plate (NUNC MaxiSorp, NUNC,
Roskide, Denmark). In washing step, the plate was stringently washed three times
with PBST (10 mM PBS, pH 7.2, 0.05% [v/v] Tween® 20). Unspecific protein
binding to the wells was blocked by incubation with 0.5% (w/v) skim milk (Oxoid,
Hants, UK) in 10 mM PBS, pH 7.2. Single serum of pre‐ and post‐infection (12
weeks) hamster sera, and pooled serum of pre‐ and post‐infection hamster sera (2, 4, 8
and 12 weeks) were diluted to 1:200 in 0.5% (w/v) skim milk in 10 mM PBS, pH 7.2
and assayed in duplicate. Goat anti‐hamster IgG (H+L) HRP antibody (Invitrogen,
Frederick, MD, USA) at dilution 1:3,000 was used as secondary antibody.
Ref. code: 25605412330010EAN

65
4.6.3 Western blot analysis
Western blot was used to detect rOvCALR by O. viverrini‐infected
hamster sera as described in Section 4.5.3. Briefly, 200 ng of rOvCALR per lane was
resolved in 12.5% SDS‐PAGE and transferred onto nitrocellulose membrane
(Immobilon‐NC Transfer Membrane, Merck Millipore, Darmstadt, Germany) by
semi‐dry blotting. Pooled preinfection hamster sera and pooled O. viverrini‐infected
sera at 2, 4, 8 and 12 weeks were prepared by dilution to 1:100 in antibody diluent.
The membrane was cut into strips, and each strip was submerged in each antibody
solution and incubated overnight at 4°C. Goat anti‐hamster IgG (H+L) HRP antibody
(Invitrogen, Frederick, MD, USA) at dilution 1:1,000 was used as secondary
antibody. AEC (Vector Laboratories, Burlingame, CA, USA) was used as
chromogenic substrate.
4.7 Functional analysis of rOvCALR
4.7.1 Production of experimental control proteins
4.7.1.1 Production of recombinant Mus musculus calreticulin
(rMmCALR)
(1) Isolation of mouse total RNA
Heart, lung and kidney were collected from an ICR mouse.
The organs were chopped into small pieces, weighed, and immediately immersed in
liquid nitrogen. TRIzol™
Reagent (Invitrogen, Carlsbad, CA, USA) was added
depending on their weight (1 ml per 50–100 mg tissue), and the tissue was
homogenized by a tissue homogenizer (Ultra‐Turrax T25, IKA, Staufen, Germany).
The RNA extraction was performed as described in Section 4.1.1.
(2) Molecular cloning of MmCALR
MmCALR cDNA was generated by AMV Reverse
Transcriptase (Fermentas, Vilnius, Lithuania) from heart‐isolated total RNA using
designed specific primers based on GenBank accession NM_007591. The reaction
mixture was prepared as shown on the next page. The reaction was incubated at 50°C
for 1 h.
Ref. code: 25605412330010EAN

66
Components Volume (µl)
DNase‐treated total RNA
DEPC‐treated ultrapure water
HRG321 reverse specific primer (10 µM)
5X AMV RT Buffer
Thermo Scientific™ RiboLock RNase Inhibitor
Mixed dNTP (10 mM each)
AMV Reverse Transcriptase (20 U/µl)
5
6.5
1
4
0.5
2
1
Total volume 20
The first‐strand cDNA products were used as template for
PCR amplification using the following primers: HRG320 forward primer and
HRG321 reverse primer (see Table 4.1). The conventional PCR mixture was prepared
as shown below.
Components Volume (µl)
First‐strand cDNA products
HRG320 forward primer (10 µM)
HRG321 reverse primer (10 µM)
10X Taq Buffer with (NH4)2SO4
25 mM MgCl2
Mixed dNTP (10 mM each)
Taq DNA Polymerase (Thermo Scientific, Vilnius, Lithuania)
Sterile ultrapure water
2
1
1
5
3
1
0.5
36.5
Total volume 50
The thermal cycling conditions were set as follows: initial
denaturation at 95°C for 5 min, 30 cycles of denaturation at 95°C, annealing at 60°C
and extension at 72°C for 1 min each, and final extension at 72°C for 5 min. The PCR
product was re‐amplified using HRG355 forward primer and HRG356 reverse primer
(see Table 4.1) that were introduced the recognition sites of NdeI and XhoI restriction
Ref. code: 25605412330010EAN

67
endonuclease, respectively, to facilitate the cloning into pET21b(+) vector. The PCR
mixture and conditions were set as shown above, except the annealing step at 55°C.
The PCR products were analyzed by 1% (w/v) agarose gel electrophoresis (see
Section 4.1.1.2). The agarose gel containing expected band was excised and extracted
using the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany) following the
manufacturer’s protocol.
The purified MmCALR cDNA was inserted into the
pGEM®‐T Easy vector (Promega, Fitchburg, WI, USA) and then introduced into
E. coli XL1‐Blue competent cells (see Section 4.1.3). Positive transformants were
identified by colony PCR (see Section 4.1.7) and restriction enzyme digestion. The
DNA sequence was verified by commercial DNA sequencing services (1st Base Asia,
Singapore).
(3) Construction of pET21b(+)‐MmCALR expression vector
After getting the verified recombinant plasmid, pGEM®
‐T
Easy‐MmCALR and pET21b(+) expression vector (Novagen, Darmstadt, Germany)
were double‐digested with NdeI and XhoI restriction enzymes (Thermo Scientific,
Vilnius, Lithuania) to create sticky‐ends DNA products. The double digestion reaction
was prepared as shown below.
Components Volume (µl)
10X Buffer O
Plasmid DNA
NdeI (10 U/µl)
XhoI (10 U/µl)
2
16.5
0.5
1
Total volume 20
The reaction was incubated at 37°C for 2 h, inactivated at
80°C for 20 min, and then analyzed by 1% (w/v) agarose gel electrophoresis (see
Section 4.1.1.2). The expected bands were excised from the agarose gel and extracted
using the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). The purified
MmCALR DNA fragments and digested pET‐21b(+) were jointed and introduced into
E. coli XL1‐Blue (see Section 4.2.1.2). After getting a verified clone, the recombinant
Ref. code: 25605412330010EAN

68
plasmid DNA, pET‐21b(+)‐MmCALR, was isolated from positive clones using
alkaline extraction method (see Section 4.1.6) and introduced into the expression
host, E. coli Rosetta‐gami(DE3)pLysS (see Section 4.1.5). Colony PCR was
performed to identify the positive clones carrying pET‐21b(+)‐MmCALR as
described in Section 4.1.7.
(4) Expression and purification of rMmCALR
Small‐scale expression to determine time‐course expression
(see Section 4.2.2) and target‐protein solubility (see Section 4.2.3) were performed to
find out the optimal conditions. After getting the conditions, rMmCALR was
expressed at large‐scale and purified using Ni‐NTA affinity chromatography under
native conditions as described in Section 4.2.4.2 and Section 4.2.6.2.
Purified rMmCALR was concentrated and dialyzed against
10 mM PBS, pH 7.2 (see Section 4.2.7). Its quality and quantity were determined
using SDS‐PAGE (see Section 4.2.5) and Bradford’s assay (see Section 4.2.8),
respectively. The qualified protein was aliquoted and stored at −20°C until used.
4.7.1.2 Production of recombinant Fasciola gigantica calcium‐
binding protein 1 (rFgCaBP1)
The constructed expression plasmid, pQE30‐FgCaBP1173
was introduced into E. coli M15 competent cells (see Section 4.1.5) and checked for
its expression (see Section 4.2.2 and Section 4.2.3). Recombinant FgCaBP1 was
purified using Ni‐NTA affinity chromatography under native conditions (see Section
4.2.6.2) and dialyzed against 10 mM PBS, pH 7.2 (see Section 4.2.7.2). Its quality
and quantity were determined using SDS‐PAGE (see Section 4.2.5) and Bradford’s
assay (see Section 4.2.8), respectively. The qualified protein was aliquoted and stored
at −20°C until used.
4.7.1.3 Production of recombinant Schistosoma japonicum
glutathione S‐transferase (rSjGST)
S. japonicum GST serves as a fusion protein of pGEX
vectors in the GST gene fusion system. To produce rSjGST, the pGEX‐5X‐1 vector
(GE Healthcare, Little Chalfont, UK) was introduced into E. coli BL21 competent
cells (see Section 4.1.5) and checked for its expression (see Section 4.2.2 and Section
4.2.3) to find out the optimal conditions. Recombinant SjGST was expressed at large‐
Ref. code: 25605412330010EAN

69
scale with 0.5 mM IPTG induction and purified using glutathione affinity
chromatography as described in Section 4.7.3.1 (2) and Section 4.7.3.1 (3),
respectively. After that, the protein was concentrated and dialyzed against 10 mM
PBS, pH 7.2 (see Section 4.2.7.2). Its quality and quantity were determined using
SDS‐PAGE (see Section 4.2.5) and Bradford’s assay (see Section 4.2.8),
respectively. The qualified protein was aliquoted and stored at −20°C until used.
4.7.2 Thermal induced protein aggregation assay
Citrate synthase, a non‐glycosylated protein, was used to
demonstrate the chaperoning activity of calreticulin174
. Citrate synthase (Sigma
Aldrich, St. Louis, MO, USA) was dissolved in 10 mM PBS, pH 7.2 at final
concentration of 1 mg/ml. Fixed molar concentration of citrate synthase (1 µM) was
mixed with various molar concentrations of rOvCALR (0.25–1 µM) in 200 µl total
volume of 10 mM PBS, pH 7.2. Recombinant MmCALR (1 µM) and BSA (Sigma
Aldrich, St. Louis, MO, USA) (1 µM) were used as positive and negative control,
respectively. All samples were pipetted into each well of 96‐well UV transparent
microplate (Thermo Scientific, Roskilde, Denmark) in duplicate. The plate was
incubated at 45°C, and the light scattering at 360 nm was measured every 10 min for
1 h using Varioskan Flash Spectral Scanning Multimode Reader (Thermo Scientific,
Waltham, MA, USA) to monitor citrate synthase aggregation.
4.7.3 Calcium binding assay
4.7.3.1 Production of rOvCALR C‐domain
(1) Construction of pGEX‐5X‐1‐OvCALR C‐domain
expression vector
The acidic OvCALR C‐domain was used to study the
calcium binding activity in native PAGE. Recombinant OvCALR C‐domain was
produced in E. coli in fusion with SjGST using pGEX‐5X‐1 vector (GE Healthcare,
Little Chalfont, UK). The verified OvCALR was used as a template for PCR
amplification. The HRG517 forward primer and HRG518 reverse primer (see Table
4.1) were designed to amplify the 321 bp (included stop codon) OvCALR C‐domain
fragment, and the recognition sites of EcoRI and NotI restriction endonuclease were
introduced into the forward and reverse primer, respectively, to facilitate the cloning
into pGEX‐5X‐1 vector. The PCR reaction was prepared as shown on the next page.
Ref. code: 25605412330010EAN

70
Components Volume (µl)
Purified OvCALR
HRG517 forward primer (10 µM)
HRG518 reverse primer (10 µM)
10X Taq Buffer with (NH4)2SO4
25 mM MgCl2
Mixed dNTP (10 mM each)
Taq DNA Polymerase (Thermo Scientific, Vilnius, Lithuania)
Sterile ultrapure water
2
1
1
5
3
1
0.5
36.5
Total volume 50
The thermal cycling conditions were set as follows: initial
denaturation at 95°C for 5 min, 30 cycles of 1 min denaturation at 95°C, 30 s
annealing at 55°C, and 45 s extension at 72°C, and final extension at 72°C for 2 min.
The PCR products were analyzed by 2% (w/v) agarose gel electrophoresis as
described in Section 4.1.1.2, except the voltage applied at 60 V for 2 h. The agarose
gel containing expected band was excised and extracted using the GeneJET Gel
Extraction Kit (Thermo Scientific, Vilnius, Lithuania) following the manufacturer’s
instruction. The purified OvCALR C‐domain DNA fragments were inserted into the
pGEM®‐T Easy vector (Promega, Fitchburg, WI, USA) and then introduced into
E. coli XL1‐Blue (see Section 4.1.3). The positive transformants were identified by
colony PCR (see Section 4.1.7) and restriction enzyme digestion. DNA sequence of
OvCALR C‐domain was verified by commercial DNA sequencing services (1st Base
Asia, Singapore).
After getting the verified recombinant plasmid, pGEM®
‐T
Easy‐OvCALR C‐domain and pGEX‐5X‐1 expression vector (GE Healthcare, Little
Chalfont, UK) were double‐digested with EcoRI and NotI restriction enzymes
(Thermo Scientific, Vilnius, Lithuania) to create sticky‐ends DNA products. The
double digestion reaction was prepared as shown on the next page.
Ref. code: 25605412330010EAN

71
Components Volume (µl)
10X Buffer O
Plasmid DNA
EcoRI (10 U/µl)
NotI (10 U/µl)
2
17
0.5
0.5
Total volume 20
The reaction was incubated at 37°C for 2 h, inactivated at
65°C for 20 min, and then analyzed by 2% (w/v) agarose gel electrophoresis at 60 V
for 2 h (see Section 4.1.1.2). The expected bands was excised from the agarose gel
and extracted using the GeneJET Gel Extraction Kit (Thermo Scientific, Vilnius,
Lithuania). The purified OvCALR C‐domain DNA fragment and digested pGEX‐5X‐
1 were ligated by T4 DNA ligase and introduced into E. coli XL1‐Blue as described
in Section 4.2.1.1. After getting a verified clone, the recombinant plasmid DNA,
pGEX‐5X‐1‐OvCALR C‐domain, was isolated from a positive clone using alkaline
extraction method (see Section 4.1.6) and introduced into the expression host, E. coli
BL21 (see Section 4.1.5). Colony PCR was performed to identify the positive clones
as described in Section 4.1.7.
(2) Expression of rSjGST‐OvCALR C‐domain
Small‐scale expression to determine time‐course expression
(see Section 4.2.2) and target‐protein solubility (see Section 4.2.3) were performed to
find out the optimal conditions.
To express in large‐scale, a single positive colony verified
by small‐scale screening was cultured overnight at 37°C with shaking in 5 ml LB
broth containing 100 µg/ml ampicillin. On the next day, 200 ml LB broth containing
100 µg/ml ampicillin was inoculated with 4 ml of the overnight culture and incubated
at 37°C with 250 rpm shaking. The expression was induced with 0.2 mM IPTG at
final concentration when OD600 of the culture was ~ 0.6. The culture was further
incubated for 4 h at 28°C, 250 rpm shaking. After that, the culture was harvested by
centrifugation at 4,000×g for 20 min at 4°C, and the bacterial pellet was stored at
−20°C.
Ref. code: 25605412330010EAN

72
(3) Purification of rSjGST‐OvCALR C‐domain
The GST‐fusion protein was purified by glutathione affinity
chromatography using the Glutathione Purification Kit (Clontech Laboratories,
Fitchburg, WI, USA). The cell pellet was resuspended in precooled extraction/loading
buffer (10 mM NaH2PO4, 1.8 mM KH2PO4, 140 mM NaCl, pH 7.5) at 4 ml per gram
wet weight. Then the bacterial cells were broken by ultrasonication (Sonics vibra
cell™ VC 750, Sonics and Materials, Inc., Newtown, CT, USA) using 6×9.9 s with
9.9 s pauses at 27% amplitude. The total cell lysate was centrifuged at 12,000×g for
20 min at 4°C, and supernatant was collected as cell lysate and kept on ice. The
following steps were performed in a refrigerator at 4°C. One milliliter of Glutathione‐
Superflow Resin (Clontech Laboratories, Fitchburg, WI, USA) was thoroughly mixed
with the cell lysate by pipetting and then transferred into a polypropylene column.
The resin was allowed to settle for 20 min. The column’s bottom cap was removed,
and the flow‐through was collected. The column was washed three times with 4 ml of
precooled extraction/loading buffer. After that, the column was eluted six times with 1
ml of elution buffer (33 mM reduced glutathione, 50 mM Tris‐HCl, pH 8.0). All
fractions were collected and analyzed by SDS‐PAGE as described in Section 4.2.5.
Purified rSjGST‐OvCALR C‐domain was dialyzed against
10 mM PBS, pH 7.2 (see Section 4.2.7). Its quality and quantity were determined
using SDS‐PAGE (see Section 4.2.5) and Bradford’s assay (see Section 4.2.8),
respectively. The qualified protein was aliquoted and stored at −20°C until used.
4.7.3.2 Determination of calcium‐binding activity using native‐
PAGE
In this assay, sample buffer, polyacrylamide gel, and
electrophoresis running buffer were added either 1 mM CaCl2 or EDTA. Gel casting
components of Hoefer SE260 Mini‐gel System (Amersham biosciences, GE
Healthcare, Piscatawan, NJ, USA) were assembled following the manufacturer’s
manual. The 8.5% native polyacrylamide gel was prepared by mixing the following
solutions: 1.98 ml 30% (w/v) polyacrylamide/Bis solution 29:1 (Bio‐Rad, Shanghai,
China), 1.75 ml 1.5 M Tris‐HCl pH 8.8, 3.181 ml ultrapure water, 70 µl 10% (w/v)
ammonium persulfate (APS), 5 µl TEMED, and 14 µl of either 0.5 mM CaCl2 or 0.5
mM EDTA. The mixed solution was poured between a glass plate and an alumina
Ref. code: 25605412330010EAN

73
plate by pipetting up to the top of the alumina plate, and a comb was inserted
immediately. The gel was allowed to polymerize for 1 h. After that, the comb was
removed, and the wells were washed with ultrapure water. The gel apparatus was
assembled following the manual, and electrophoresis buffer (25 mM Tris‐base, 202
mM glycine, 1 mM CaCl2 or 1 mM EDTA) was filled into the upper and lower
chamber.
The samples including rSjGST‐OvCALR C‐domain,
rFgCaBP1 (from Section 4.7.1.2) as positive control, and rSjGST (from Section
4.7.1.3) as negative control were prepared by incubating 5 µg each with either 1 mM
CaCl2 or 1 mM EDTA on ice for 20 min. After incubation, the samples were mixed
with 2X sample buffer (120 mM Tris‐HCl, pH 6.8, 20% [v/v] glycerol, 0.05% [w/v]
Bromophenol Blue, 01% [w/v] DTT, 2 mM CaCl2 or 2 mM EDTA) and loaded into
each well of 8.5% polyacrylamide gel. Power was applied at 20 mA/gel, 200 V, 90
min. Finally, the gel was removed carefully from the gel cassette and stained with
staining solution as described in Section 4.2.5.2.
4.7.4 Determination of transacetylase activity
4.7.4.1 Indirect transacetylase activity measurement using GST
assay
Transacetylase activity of CALR was indirectly determined
by inhibition of GST in the presence of acetyl derivative of 4‐methylcoumarins.175,176
To measure transacetylase activity of OvCALR, the assay was performed as
previously described with some modifications. The following solutions were freshly
prepared: 1 mM 7‐Acetoxy‐4‐methylcoumarin (7‐AMC) (Sigma Aldrich, Buchs,
Switzerland) in DMSO Hybri‐Max® (Sigma Aldrich, St. Louis, MO, USA), 100 mM
1‐chloro‐2,4‐dinitrobenzene (CDNB) (Sigma, Dorset, UK) in absolute ethanol, 200
mM reduced glutathione (Serva, Heidelberg, Germany). Samples were prepared by
mixing 0.5 µg rSjGST with 2 µg either rOvCALR or rMmCALR and 250 µM 7‐AMC
in 0.25 M potassium phosphate buffer pH 6.5, 1 mM EDTA at 20 µl of total volume.
These mixtures were incubated at 37°C in a Thermomixer (Eppendorf, Hamburg,
Germany) and then GST activity was measured in triplicate at 15, 30 and 45 min of
incubation. The GST activity was measured by pipetting 20 µl of the samples into UV
96‐well plate (Thermo Scientific, Roskilde, Denmark) and followed by adding 180 µl
Ref. code: 25605412330010EAN

74
of substrate solution (1 mM CDNB, 2 mM reduced glutathione in 0.25 M potassium
phosphate buffer, pH 6.5). The absorbance at 340 nm was read immediately and every
60 sec thereafter for 10 min on a Varioskan Flash Spectral Scanning Multimode
Reader (Thermo Scientific, Waltham, MA, USA). Meanwhile the sample containing
only DMSO (mock solution) and substrate solution were used to determine
background values at each time point, and the control samples containing either
rSjGST alone or rSjGST plus other components (7‐AMC, rOvCALR, rMmCALR)
were used to determine calreticulin transacetylase activity and the effect of those
components on GST activity. Linear regression was plotted between time and
background‐corrected absorbance values using Prism 6 (GraphPad Software, Inc.,
CA, USA). Relative GST activity of samples was calculated by comparison of slope
value against untreated‐rSjGST at each time point.
4.7.4.2 Western blot analysis of acetylated protein
The samples were prepared by mixing 0.5 µg rSjGST with or
without 2 µg rOvCALR in the presence of 7‐AMC (50–250 µM in 5 µL of DMSO) in
0.25 M potassium phosphate buffer pH 6.5, 1mM EDTA at 20 µL of total volume.
These mixtures were incubated at 37°C using Thermomixer (Eppendorf, Hamburg,
Germany) for 20 and 40 min. These samples were size‐separated by 12.5 % SDS‐
PAGE (see Section 4.2.5.1) and transferred onto a nitrocellulose membrane (Bio‐Rad,
Goettingen, Germany) by semi‐dry blotting (see Section 4.5.3.1). Western blot
detection was performed as described in Section 4.5.3.2. Polyclonal rabbit anti‐
acetylated lysine antibody (Cell signaling, Danvers, MA, USA) at dilution 1:1,000 in
5% (w/v) BSA in TBS pH 7.5, 0.1% (v/v) Tween®
20 was used as primary antibody.
Alkaline phosphatase conjugated goat anti rabbit IgG (Dako, Glostrup, Denmark) at
dilution 1:1,000 in 1% (w/v) BSA in TBS pH 7.5, 0.05% (v/v) Tween® 20 was used
as secondary antibody.
Acetylation of protein by 7‐AMC was also tested with BSA
and rFgCaBP1 by incubation with 250 µM 7‐AMC for 30 min at 37°C and analysis
by western blot as described above.
Ref. code: 25605412330010EAN

75
4.7.5 C1q binding assay
Competitive ELISA was used to determine the binding of
rOvCALR to human C1q. Each well of a 96‐well plate (NUNC MaxiSorp, NUNC,
Roskide, Denmark) was coated with 20 ng native human C1q protein (Abcam,
Cambridge, UK) in 100 µl carbonate buffer, pH 9.6 (30 mM Na2CO3, 75 mM
NaHCO3) by incubation overnight at 4°C. The wells were washed three times with
washing buffer (0.05% [v/v] Tween® 20 in 10 mM PBS, pH7.4). Unspecific binding
to the wells was blocked by incubation in 200 µl blocking solution (1% [w/v] skim
milk in 10 mM PBS, pH 7.4, 0.05% [v/v] Tween® 20) at room temperature for 90
min. After washing the wells as before, either rOvCARL (0–2 µg) alone or 1 µg
rOvCALR mixed with rMmCALR (0–2 µg) or 1 µg rOvCALR mixed with BSA
(Sigma Aldrich, St. Louis, MO, USA) (0–2 µg) in protein diluent (1:3 of 10 mM PBS,
pH 7.4 in ultrapure water) was added to the wells. The plate was incubated for 1 h at
37°C, and the wells were washed as before. Mouse anti‐rOvCALR antiserum diluted
at 1:10,000 in antibody diluent (1% [w/v] skim milk in 10 mM PBS, pH 7.4, 0.05%
[v/v] Tween®
20) was added into the wells and incubated for 90 min at 37°C. The
wells were washed as before, and sheep anti‐mouse IgG HRP conjugated (GE
Healthcare, Little Chalfont, UK) diluted at 1:3,000 in antibody diluent was added into
each well. The plate was incubated for 1 h at 37°C and the wells were washed again.
TMB substrate solution (Life Technologies, Carlsbad, CA, USA) was used at 100
µl/well, and the plate was incubated in the dark for 30 min at room temperature. The
reaction was stopped by adding 100 µl of 1 N HCl. The absorbance at 450 nm was
read on a POLARstar Omega plate reader (BMG Labtech, Ortenberg, Germany). This
assay was performed two times in duplicate. Curves fitted between mean of
absorbance values and amounts of protein were plotted in DataGraph 4.2 (Visual Data
Tools, Inc.)
4.7.6 Hemolytic assay
Hemolytic assay of classical pathway complement activation was
performed using sensitized human red blood cells. Human EDTA blood and serum of
blood group AB were used in this experiment. Red blood cells were washed twice in
gelatin veronal buffer (0.1% [w/v] gelatin in Calcium‐Magnesium‐Veronal buffer
[bioMérieux, Charbonnières les Bains, France]) by centrifugation at 1,000×g for 5
Ref. code: 25605412330010EAN

76
min. Red blood cells were sensitized with Anti‐B reagent (CE‐Immundiagnostika
GmbH, Eschelbronn, Germany) at dilution 1:50 in gelatin veronal buffer for 5 min at
4°C. The sensitized red blood cells were washed as before and prepared as 10%
sensitized red blood cells in gelatin veronal buffer. Normal human serum (dilution
1:10) alone or mixed with 10–50 µg of either rOvCALR, rMmCALR (positive
control), or BSA (negative control) in a total volume of 300 µl gelatin veronal buffer
were pre‐incubated at 37°C for 30 min. After that, 20 µl of sensitized red blood cell
suspension was added into each tube and further incubated at 37°C for 60 min. The
tubes were centrifuged at 1,500×g for 5min and supernatants were pipetted into each
well of a 96‐well plate. The hemolytic activity was determined by measuring the
absorbance at 540 nm on a Varioskan Flash spectral scanning multimode reader
(Thermo Scientific, MA, USA). Spontaneous hemolysis in the tube containing 10 mM
PBS, pH 7.2 in gelatin veronal buffer at identical conditions was measured and used
as background. This assay was performed two times in duplicate. The hemolytic
activity was expressed as percentage of relative hemolysis. Statistical analysis using
ANOVA with Dunnett’s multiple comparison test was accomplished by SPSS 16.0
(SPSS, Inc.).
4.7.7 In vitro angiogenesis assays
4.7.7.1 Preparation of recombinant proteins for tissue culture
Recombinant OvCALR, MmCALR and SjGST were
prepared as described previously, concentrated and dialyzed against 10 mM PBS, pH
7.4. Bacterial endotoxin was removed twice using a Pierce® High Capacity Endotoxin
Removal Spin Column (Thermo Scientific, Rockford, IL, USA). To equilibrate the
resin, the spin column had been equilibrated to room temperature, and the spin
column was inserted into a collection tube and centrifuged at 500×g for 1 min to
remove the storage solution. The resin was regenerated by mixing with 8 ml of 0.2 N
NaOH in a 15 ml conical tube on a rotary mixer, overnight at room temperature. On
the following day, the resin suspension was transferred into the spin column, and the
column was inserted into a collection tube and centrifuged at 500×g for 1 min to
remove the solution. Then the resin was mixed thoroughly with 8 ml of 2 M NaCl and
centrifuged as before. The resin was washed by mixing with 8 ml ultrapure water and
then centrifuged as before to remove the water. The resin was equilibrated in 10 mM
Ref. code: 25605412330010EAN

77
PBS, pH 7.4 by mixing the resin with 8 ml of 10 mM PBS, pH 7.4 and removing the
solution as before. This equilibrating step was repeated three times. The sample was
mixed with the resin in a 15 conical tube on a rotary mixer at 4°C overnight. On the
next day, the sample was collected by transferring the resin suspension into the spin
column and centrifugation at 500×g for 1 min. All endotoxin removal steps were
repeated with the collected sample to minimize the bacterial endotoxin in the sample.
4.7.7.2 Cell culture
Pre‐screened human umbilical vein endothelial cells
(HUVECs) were purchased from Lonza (Clonetics™ Cells, Lonza, Basel,
Switzerland) and grown in endothelial growth medium (EGM™‐2) (Lonza, Basel,
Switzerland). Cell passaging was done using ReagentPack™ Subculture Reagents
(Lonza, Basel, Switzerland). The instruction of Clonetics™ Endothelial Cell System
was followed for cell culture, cell maintenance, subculturing, and cell storage. In vitro
angiogenesis assays were performed using HUVECs passage number 3 to 6 with cell
viability greater than 80%. Cell viability was determined using Countess™
Automated Cell Counter (Invitrogen, Carlsbad, CA, USA) with trypan blue staining.
4.7.7.3 MTT assay
HUVECs were seeded at 1×104 cells/well into 96‐well cell
culture plate (Corning, Pittston, PA, USA). The plate was incubated in a humidified
CO2 incubator (37°C, 5% CO2) for 4 h to allow the cells to attach. One hundred
microliter of EGM™‐2 (Lonza, Basel, Switzerland) containing either 10 mM PBS pH
7.4, rOvCARL, rMmCALR (from Section 4.7.1.1) or rSjGST (from Section 4.7.1.3)
at 0.1, 1, 10 and 20 µM concentration were prepared and added into the wells. The
plate was incubated for 48 h in the humidified CO2 incubator. Afterward, 12 mM
Thiazolyl Blue Tetrazolium Bromide (MTT) (Sigma Aldrich, St. Louis, MO, USA)
was added at 20 µl/well, and then the plate was incubated in the incubator for 4 h.
Finally, the medium was removed, and 100 µl of solubilizing solution (0.1 N HCl in
isopropanol) was added to each well to dissolve formazan crystals. The absorbance at
570 nm was read with background wavelength at 690 nm on a POLARstar Omega
plate reader (BMG Labtech, Ortenberg, Germany). This assay was performed two
times in triplicate. A bar graph with mean and standard deviation values of relative
viability was plotted by comparison to untreated HUVECs (10 mM PBS, pH 7.4).
Ref. code: 25605412330010EAN

78
Statistical analysis using two‐way ANOVA with Tukey’s multiple comparison test
was accomplished by SPSS 16.0 (SPSS, Inc.).
4.7.7.4 Scratch assay
Scratch assay177
was performed with some modifications.
HUVEC cells were seeded into a 24‐well cell culture plate (Corning, Pittston, PA,
USA) and grown until confluence. Two straight‐line scratches were made
perpendicularly with a 200 µl pipette tip. Debris cells were removed, and the wells
were washed with 10 mM PBS, pH 7.4. Four hundred microliter of EGM™‐2 (Lonza,
Basel, Switzerland) containing either 10 mM PBS pH 7.4, rOvCARL, rMmCALR
(from Section 4.7.1.1) or rSjGST (from Section 4.7.1.3) at concentrations of 20 and
50 µM were prepared and replaced into the wells. Markings were made on the plate as
a reference point to get the same field during image captures. Pre‐incubation images
were captured using an inverted microscope (Nikon Eclipse TS100 [microscope],
Infinity3 Lumenera [digital camera]). The plate was incubated in the humidified CO2
incubator (37°C, 5% CO2) for 24 h, and post‐incubation images were captured by
matching with the reference point. A fixed area occupied by HUVECs was analyzed
by using ImageJ software (ImageJ version 1.51h, National Institutes of Health, USA;
http://imagej.nih.gov/ij/). This assay was performed four times. A bar graph with
mean and standard deviation values of relative migration area was plotted by
comparison to untreated HUVECs (10 mM PBS, pH 7.4). Statistical analysis using
two‐way ANOVA with Tukey’s multiple comparison test was accomplished by SPSS
16.0 (SPSS, Inc.).
4.7.7.5 Tube formation assay
Tube formation assay178
was performed with some
modifications. Matrigel Growth Factor Reduced (Corning, NY, USA) was thawed
overnight on ice at 4°C. In this experiment, pipette tips and 96‐well plate were
precooled before used and they were maintained on ice during the coating process. A
Matrigel‐coated plate was prepared by pipetting 60 µl of the Matrigel to each well of
a 96‐well cell culture plate (Corning, Pittston, PA, USA). The plate was incubated at
room temperature for 10 min and then moved into the humidified CO2 incubator for
30 min. Seventy‐five microliter of EGM™‐2 medium containing either 10 mM PBS
pH 7.4, rOvCARL, rMmCALR (from Section 4.7.1.1) or rSjGST (from Section
Ref. code: 25605412330010EAN

79
4.7.1.3) at two times concentrations of 20 and 50 µM were prepared and added into
the Matrigel pre‐coated wells. Freshly prepared HUVECs suspension was seeded into
each well at 2.5 × 103 cells/well in 75 µl of EGM™‐2 medium. The plate was
incubated in the incubator (37°C, 5% CO2) for 18 h. Formation of tube‐like structure
by HUVECs was observed under an inverted microscope (Nikon Eclipse TS100
[microscope], Infinity3 Lumenera [digital camera]). The tube length was measured by
ImageJ software with angiogenesis analyzer plug‐in (ImageJ version 1.51h, National
Institutes of Health, USA; http://imagej.nih.gov/ij/). This assay was performed three
times in duplicate. A bar graph with mean and standard deviation values of relative
viability was plotted by comparison to untreated HUVECs (10 mM PBS, pH 7.4).
Statistical analysis using two‐way ANOVA with Tukey’s multiple comparison test
was accomplished by SPSS 16.0 (SPSS, Inc.).
Ref. code: 25605412330010EAN

80
CHAPTER 5
RESULTS
5.1 Molecular cloning of Opisthorchis viverrini calreticulin (OvCALR) cDNA
and its sequence analysis
Based on the transcriptome data of O. viverrini179
, specific
oligonucleotide primers were designed for amplification of the uncharacterized
OvCALR cDNA not including the protein’s predicted signal peptide sequence. This
cDNA was reverse transcribed and amplified using PCR from total RNA of adult
O. viverrini as described in Section 4.1.2. The PCR products were resolved in 1%
(w/v) agarose gel, and the expected product showed as a major band at about 1,200 bp
(Figure 5.1A). This product was purified from the agarose gel, ligated into the
pGEM®‐T Easy vector and introduced into E. coli XL1‐Blue. Transformants were
screened by colony PCR (Figure 5.2A) and restriction enzyme double digestion
(Figure 5.2B). The recombinant plasmid was isolated from positive‐screened clones
and then the nucleic acid sequence of the insert determined using a commercial DNA
sequencing service (1st Base Asia, Singapore).
Comparison of the verified OvCALR cDNA sequence and the
transcriptome data of O. viverrini179
revealed the 1,248 bp open reading frame of
OvCALR cDNA. The complete coding sequence of OvCALR was submitted into the
NCBI database and is available under the GenBank accession number KX722539.
The sequence of the OvCALR cDNA was used to deduce the amino acid sequence of
OvCALR and revealed a protein of 415 amino acid residues of pre‐mature
O. viverrini calreticulin (Figure 5.3). The mature OvCALR protein (399 amino acids)
has a calculated molecular weight of 46.23 kDa and a theoretical isoelectric point of
4.3. Signal peptide prediction indicated a 16 amino acid residues signal peptide
(Figure 5.4) at the N terminus, and ScanProsite, a motifs scanning tool, indicated an
ER retention signal (HEEL) at the C terminus of OvCALR (Figure 5.3). Moreover,
ScanProsite analysis of the deduced amino acid sequence of OvCALR showed that
OvCALR has the conserved calreticulin family signature 1 at residues 99–114
Ref. code: 25605412330010EAN

81
(KYEQSVSCGGAYLKLL) (Figure 5.3). The calreticulin family signature 2
(consensus pattern: [LIVM](2)‐F‐G‐P‐D‐x‐C‐[AG], ProSite documentation
PDOC00636) is not found in OvCALR because of the first amino acid changing from
isoleucine in mammalian and blood fluke calreticulin to phenylalanine in OvCALR
and CsCALR (Figure 5.5). The other characteristics of calreticulin including three
functional domains (N‐, P‐, and C‐domain), a conserved disulfide bond, and three
each of two tandem repeated motifs (repeat A: PXXIXDPDAXKPEDWDE, repeat B:
GXWXPPXIXNPXYX) are present. So OvCALR can be classified as a novel
member of the calreticulin family. The other homologs of calreticulin were searched
through the NCBI database for multiple sequence alignment (Figure 5.5). The
deduced amino acid sequence of OvCALR has very high sequence conservation at
96.6% identity to C. sinensis calreticulin. The sequence conservation is significantly
less when compared to blood fluke calreticulin, S. mansoni (57.5%) and S. japonicum
(53.6%), and mammalian calreticulin, M. musculus (50.7%) and H. sapiens (50.9%),
respectively. Less conserved regions can be observed at the N‐ and C‐termini of these
sequences.
Ref. code: 25605412330010EAN

82
Figure 5.1 Agarose gel showing the RT‐PCR product of OvCALR obtained from
adult O. viverrini total RNA. Lane M: GeneRuler™ 100 bp Plus DNA Ladder
(Thermo Scientific, Vilnius Lithuania).
Ref. code: 25605412330010EAN

83
Figure 5.2 Agarose gel showing the OvCALR cDNA product obtained from
transformant‐screening methods. (A) Colony PCR presenting the 1,200 bp product of
OvCALR from bacterial clones 1–5 (lanes 1–5) and no product from negative control
(lane 6); lane M: GeneRuler™ 100 bp Plus DNA Ladder (Thermo Scientific, Vilnius
Lithuania). (B) Restriction enzyme analysis of pGEM®‐T Easy carrying the OvCALR
cDNA; lane M: GeneRuler™ 1 kb DNA Ladder (Thermo Scientific, Vilnius
Lithuania), lane 1: undigested pGEM®‐T Easy carrying the OvCALR cDNA, lane 2:
pGEM®‐T Easy carrying the OvCALR cDNA after PaeI (SphI) and PstI double‐
digestion. An arrowhead indicates the location of OvCALR.
A B
Ref. code: 25605412330010EAN

84
Figure 5.3 Nucleic acid and deduced amino acid sequences of OvCALR. Sequence
analysis and graphic were created with EMBOSS showorf. The boxed sequence
indicates the signal peptide and the underlined sequence indicates the ER retention
signal. The calreticulin family signature 1 is indicated by double‐underlined sequence.
Ref. code: 25605412330010EAN
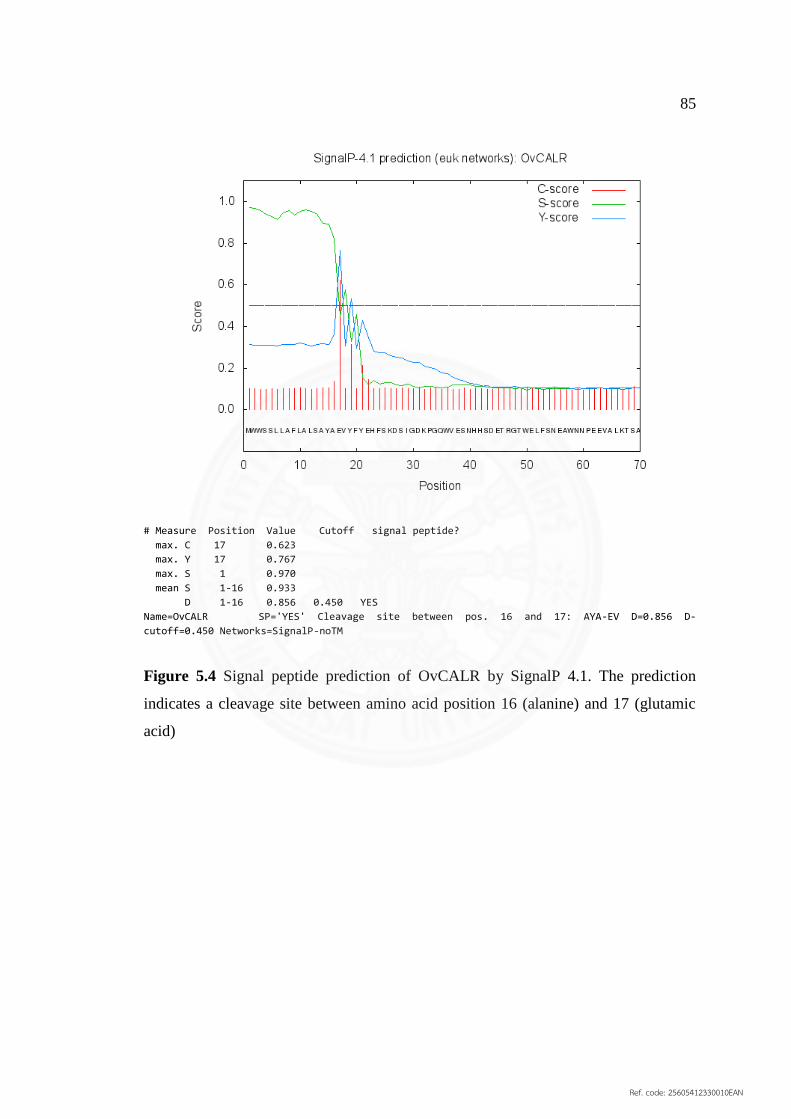
85
# Measure Position Value Cutoff signal peptide?
max. C 17 0.623
max. Y 17 0.767
max. S 1 0.970
mean S 1-16 0.933
D 1-16 0.856 0.450 YES
Name=OvCALR SP='YES' Cleavage site between pos. 16 and 17: AYA-EV D=0.856 D-
cutoff=0.450 Networks=SignalP-noTM
Figure 5.4 Signal peptide prediction of OvCALR by SignalP 4.1. The prediction
indicates a cleavage site between amino acid position 16 (alanine) and 17 (glutamic
acid)
Ref. code: 25605412330010EAN
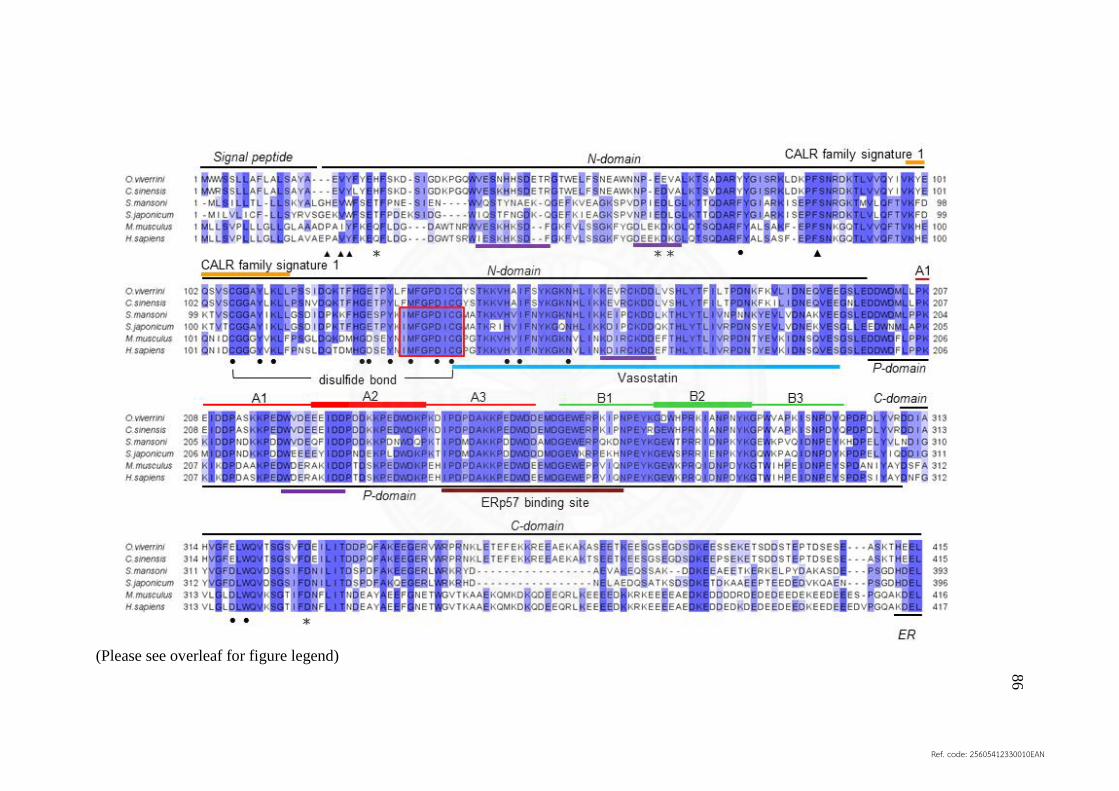
(Please see overleaf for figure legend)
86
Ref. code: 25605412330010EAN

Figure 5.5 Multiple alignment of the deduced amino acid sequences of calreticulin from O. viverrini (UniProt: A0A1P8P1U1),
C. sinensis (UniProt: K7NB00), S. mansoni (UniProt: Q06814), S. japonicum (UniProt: O45034), M. musculus (UniProt: P14211),
H. sapiens (UniProt: P27797). Characteristic N-, P-, C-domains, signal peptide, ER retention signal, repeat motifs A and B, and family
signatures 1 and 2 (red box) of calreticulin are indicated. Cysteine residues forming disulfide bond are indicated, and residues forming
high‐affinity calcium‐binding site based on mouse calreticulin98
are indicated by asterisks. Residues participate in carbohydrate‐
binding97,98
and polypeptide‐binding180
based on human and mouse calreticulin are indicated by black dots and black arrowheads,
respectively. The angiogenic active region as peptide vasostatin134
and region involved with oxidoreductase ERp57 interaction181
are
indicated. The C1q binding sites identified in human calreticulin182
are indicated by purple lines.
87
Ref. code: 25605412330010EAN

88
5.2 Production of rOvCALR and other control proteins using a bacterial system
5.2.1 Production of rOvCALR
5.2.1.1 Small‐scale expression of rOvCALR
The confirmed OvCALR cDNA was inserted into the pQE30
expression vector using the restriction endonuclease sites of PaeI and PstI. The
constructed recombinant plasmid pQE30‐OvCALR was introduced into the
expression host E. coli M15, and transformants were screened by direct colony PCR.
The positive screened colonies that carried the pQE30‐OvCALR plasmid were
checked for protein expression by small‐scale expression. Time‐course and target‐
protein solubility analyses were performed as described in Section 4.2.2 and Section
4.2.3 to find out the optimal expression conditions. Recombinant OvCALR in E. coli
M15 was already observed after the first hour and its amount increased over the
period of induction, however, it was mainly found in the insoluble fraction (Figure
5.6). To obtain soluble OvCALR, the expression vector pET21b(+) and host strain
E. coli Rosetta‐gami(DE3)pLysS were tried next. The OvCALR cDNA was
subcloned into the pET21b(+) expression vector using the restriction endonuclease
sites of NheI and NotI. Positive screened transformants were checked for time‐course
expression and target‐protein solubility. Expression of rOvCALR in E. coli Rosetta‐
gami(DE3)pLysS showed that rOvCALR existed in soluble fraction after overnight
induction at 28°C (Figure 5.7).
5.2.1.2 Large‐scale expression and purification of rOvCALR
Based on small‐scale expression, the optimal conditions for
rOvCALR expression in E. coli M15 were 1 mM IPTG induction at 37°C for 4 h
(Figure 5.6) and for rOvCALR expression in E. coli Rosetta‐gami(DE3)pLysS were 1
mM IPTG overnight induction at 28°C (Figure 5.7). These optimal conditions were
applied to large‐scale expression of rOvCALR as described in Section 4.2.4.
Recombinant OvCALR expressed in E. coli M15 was purified using Ni‐NTA affinity
chromatography under denaturing conditions (Figure 5.8), whereas rOvCALR
expressed in E. coli Rosetta‐gami(DE3)pLysS was purified under native conditions
(Figure 5.9). Eluates containing rOvCALR from the two purification conditions were
pooled separately and dialyzed against 10 mM PBS, pH 7.2 (Figure 5.10).
Ref. code: 25605412330010EAN

89
Figure 5.6 Time‐course and target‐protein solubility analyses of E. coli M15/pQE30‐
OvCALR. The culture was grown at 37°C until OD600 reached 0.6 and then the
expression was induced with 1 mM IPTG at final concentration. The culture was
further grown at 37°C for 4 h. Lane M: Broad Range Molecular Weight Standards
(Bio‐Rad, Hercules, CA, USA), lane 1: non‐induced total bacterial lysate, lanes 2–5:
IPTG‐induced total bacterial lysate at 1 h, 2 h, 3 h and 4 h, respectively, lane 6:
soluble fraction of the induced bacterial culture at 3 h, lane 7: insoluble fraction of the
induced bacterial culture at 3 h. The location of rOvCALR is indicated by an
arrowhead.
Ref. code: 25605412330010EAN
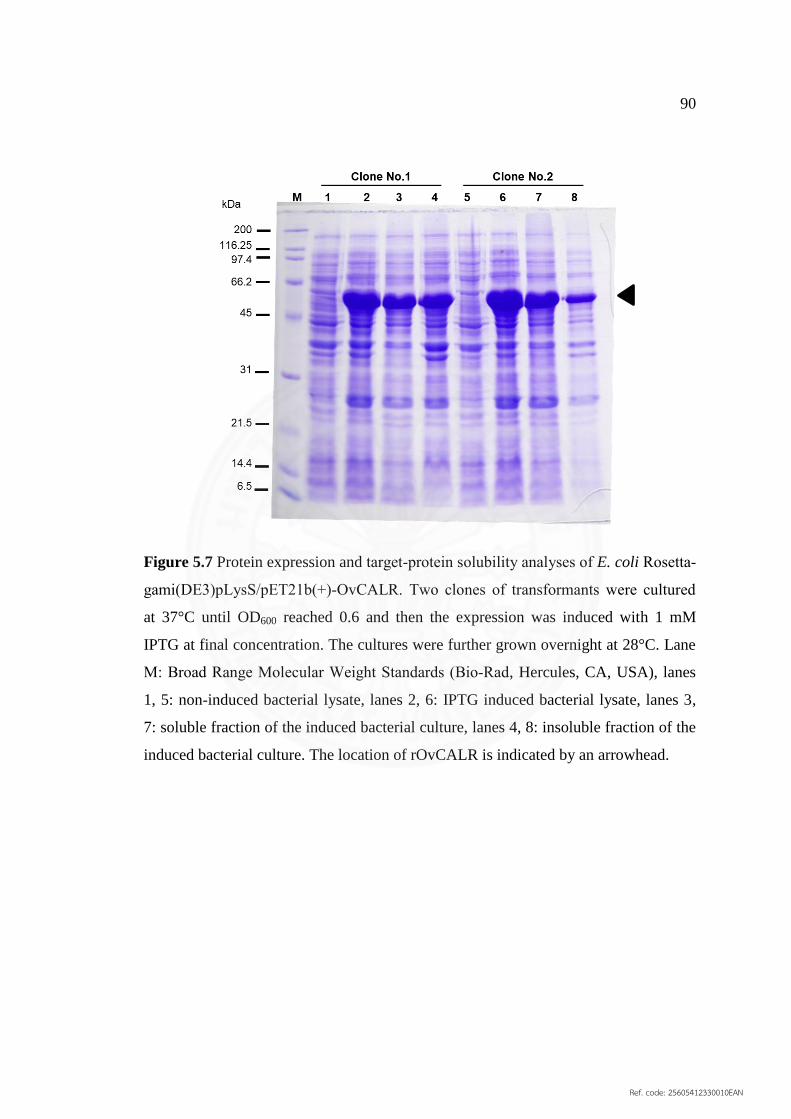
90
Figure 5.7 Protein expression and target‐protein solubility analyses of E. coli Rosetta‐
gami(DE3)pLysS/pET21b(+)‐OvCALR. Two clones of transformants were cultured
at 37°C until OD600 reached 0.6 and then the expression was induced with 1 mM
IPTG at final concentration. The cultures were further grown overnight at 28°C. Lane
M: Broad Range Molecular Weight Standards (Bio‐Rad, Hercules, CA, USA), lanes
1, 5: non‐induced bacterial lysate, lanes 2, 6: IPTG induced bacterial lysate, lanes 3,
7: soluble fraction of the induced bacterial culture, lanes 4, 8: insoluble fraction of the
induced bacterial culture. The location of rOvCALR is indicated by an arrowhead.
Ref. code: 25605412330010EAN
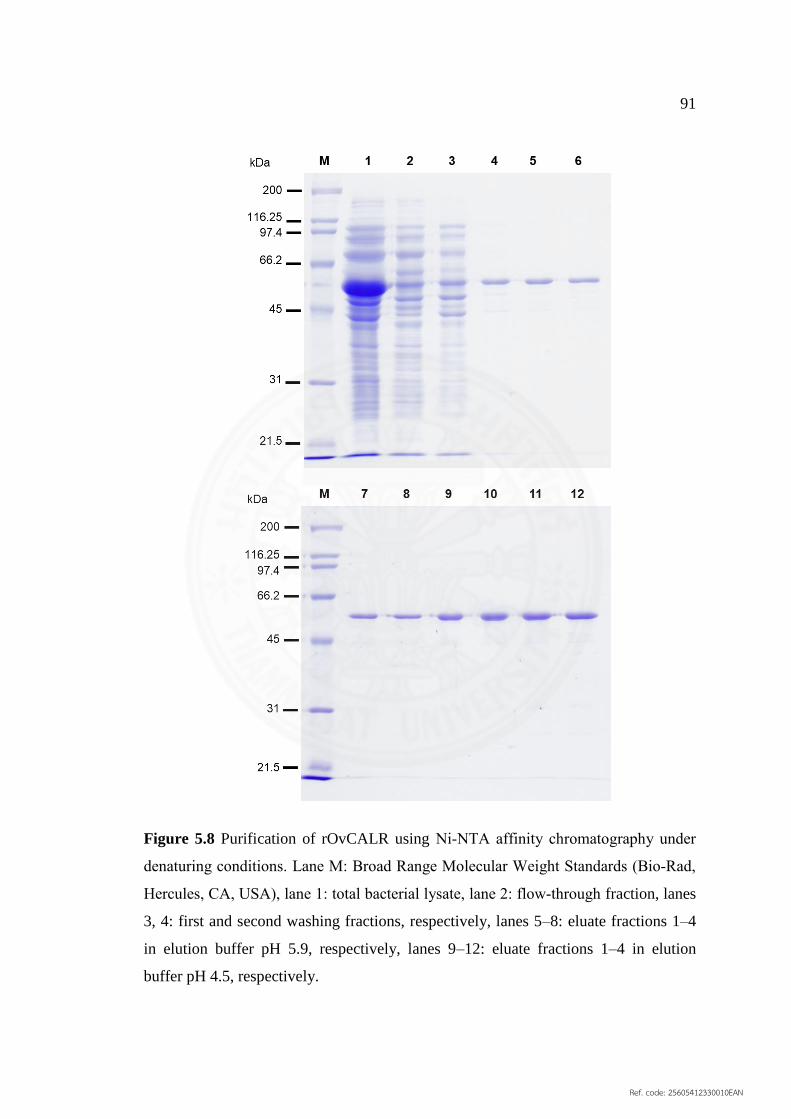
91
Figure 5.8 Purification of rOvCALR using Ni‐NTA affinity chromatography under
denaturing conditions. Lane M: Broad Range Molecular Weight Standards (Bio‐Rad,
Hercules, CA, USA), lane 1: total bacterial lysate, lane 2: flow‐through fraction, lanes
3, 4: first and second washing fractions, respectively, lanes 5–8: eluate fractions 1–4
in elution buffer pH 5.9, respectively, lanes 9–12: eluate fractions 1–4 in elution
buffer pH 4.5, respectively.
Ref. code: 25605412330010EAN

92
Figure 5.9 Purification of rOvCALR using Ni‐NTA affinity chromatography under
native conditions. Lane M: Broad Range Molecular Weight Standards (Bio‐Rad,
Hercules, CA, USA), lane 1: total bacterial lysate, lane 2: flow‐through fraction, lanes
3, 4: first and second washing fractions, respectively, lanes 5–8: eluate fractions 1–4,
respectively.
Ref. code: 25605412330010EAN

93
Figure 5.10 SDS‐PAGE showing purified rOvCALR (5 µg) obtained from denaturing
purification. Lane M: Broad Range Molecular Weight Standards (Bio‐Rad, Hercules,
CA, USA)
Ref. code: 25605412330010EAN
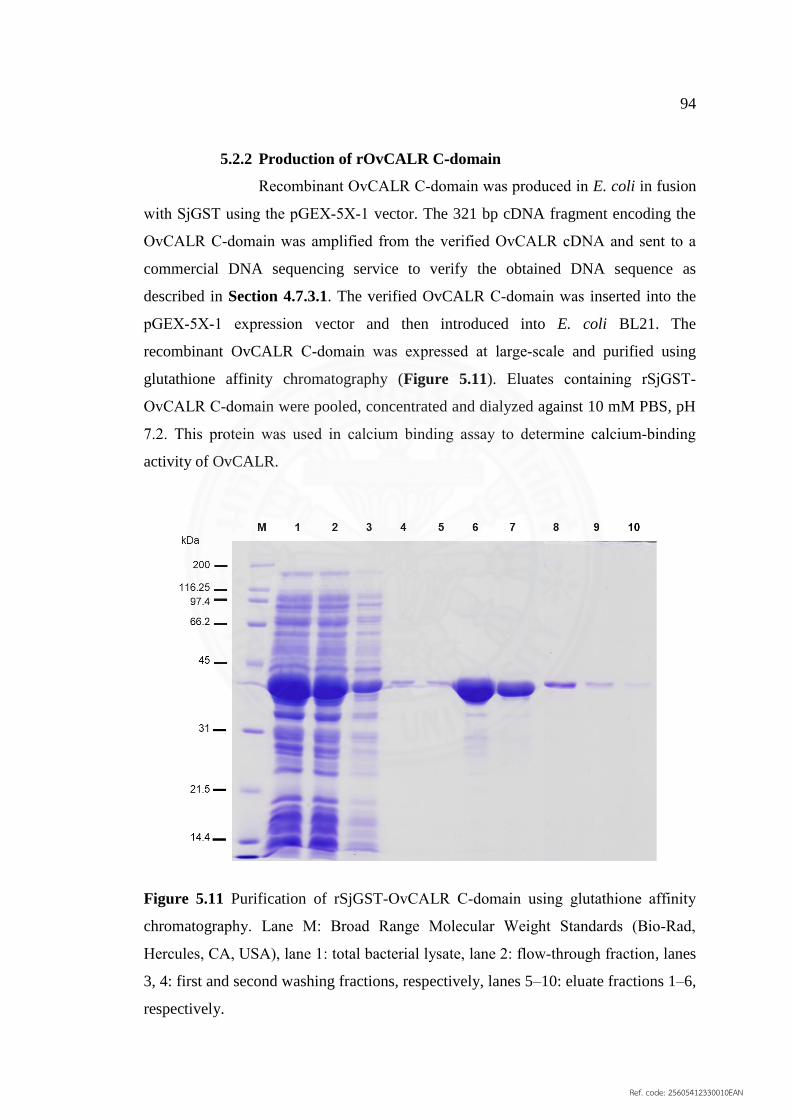
94
5.2.2 Production of rOvCALR C‐domain
Recombinant OvCALR C‐domain was produced in E. coli in fusion
with SjGST using the pGEX‐5X‐1 vector. The 321 bp cDNA fragment encoding the
OvCALR C‐domain was amplified from the verified OvCALR cDNA and sent to a
commercial DNA sequencing service to verify the obtained DNA sequence as
described in Section 4.7.3.1. The verified OvCALR C‐domain was inserted into the
pGEX‐5X‐1 expression vector and then introduced into E. coli BL21. The
recombinant OvCALR C‐domain was expressed at large‐scale and purified using
glutathione affinity chromatography (Figure 5.11). Eluates containing rSjGST‐
OvCALR C‐domain were pooled, concentrated and dialyzed against 10 mM PBS, pH
7.2. This protein was used in calcium binding assay to determine calcium‐binding
activity of OvCALR.
Figure 5.11 Purification of rSjGST‐OvCALR C‐domain using glutathione affinity
chromatography. Lane M: Broad Range Molecular Weight Standards (Bio‐Rad,
Hercules, CA, USA), lane 1: total bacterial lysate, lane 2: flow‐through fraction, lanes
3, 4: first and second washing fractions, respectively, lanes 5–10: eluate fractions 1–6,
respectively.
Ref. code: 25605412330010EAN

95
5.2.3 Production of recombinant Mus musculus calreticulin
(rMmCALR)
Total RNA isolated from mouse tissue was used as a template for
cloning of MmCALR (Figure 5.12A). The coding sequence of MmCALR was
amplified without the region encoding the predicted signal peptide using specific
primers as described in Section 4.7.1.1. The RT‐PCR products were analyzed by 1%
(w/v) agarose gel electrophoresis, and the expected product was observed at about
1,200 bp (Figure 5.12B). This product was purified from the agarose gel, ligated into
pGEM®‐T Easy vector and introduced into E. coli XL1‐Blue. The recombinant
plasmid DNA was extracted from positive screened transformants and sent to a
commercial DNA sequencing service (1st Base Asia, Singapore) to verify the obtained
cDNA sequence. The verified MmCALR cDNA was inserted into pET21b(+)
expression vector and then introduced into E. coli Rosetta‐gami(DE3)pLysS. The
positive screened colonies that carried pET21b(+)‐MmCALR plasmid were checked
for optimal protein expression by small‐scale expression. The result showed that
rMmCALR can be expressed in E. coli Rosetta‐gami(DE3)pLysS as soluble protein
with following induction conditions: 1 mM IPTG at final concentration, overnight at
28°C (Figure 5.13). Recombinant MmCALR was expressed at large‐scale and
purified using Ni‐NTA affinity chromatography under native conditions (Figure
5.14). Eluates containing rMmCALR were pooled, concentrated and dialyzed against
10 mM PBS, pH 7.2.
Ref. code: 25605412330010EAN
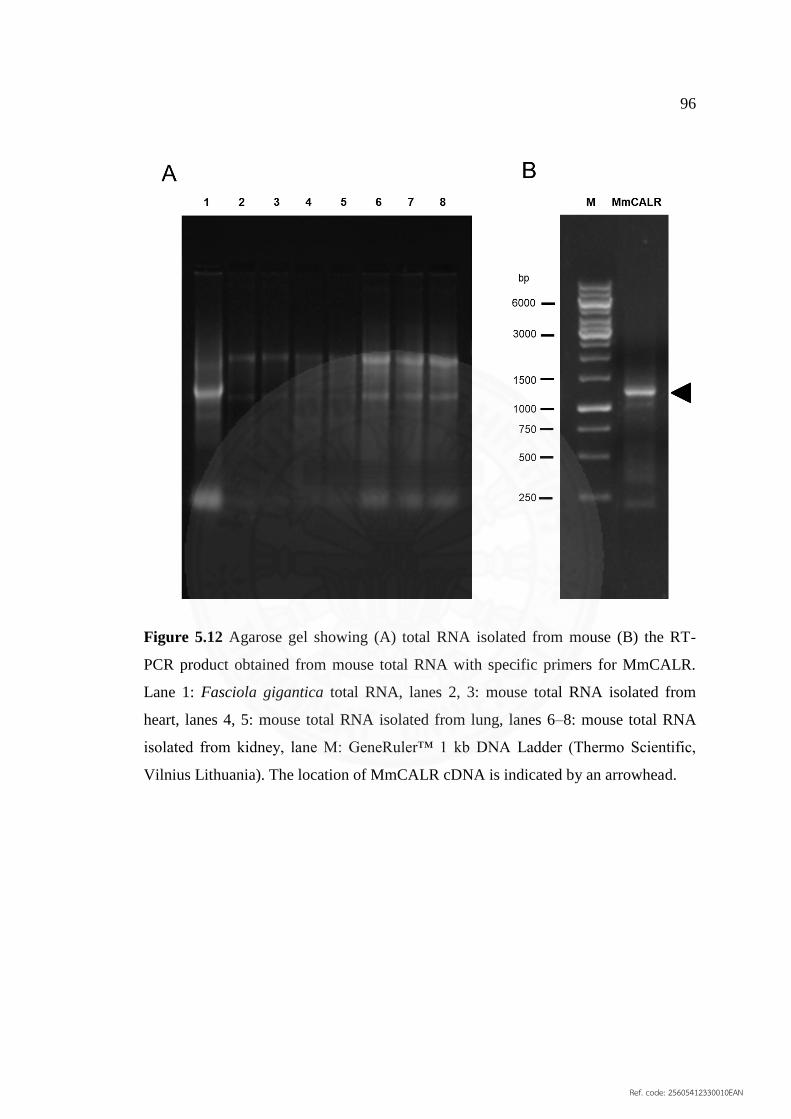
96
Figure 5.12 Agarose gel showing (A) total RNA isolated from mouse (B) the RT‐
PCR product obtained from mouse total RNA with specific primers for MmCALR.
Lane 1: Fasciola gigantica total RNA, lanes 2, 3: mouse total RNA isolated from
heart, lanes 4, 5: mouse total RNA isolated from lung, lanes 6–8: mouse total RNA
isolated from kidney, lane M: GeneRuler™ 1 kb DNA Ladder (Thermo Scientific,
Vilnius Lithuania). The location of MmCALR cDNA is indicated by an arrowhead.
Ref. code: 25605412330010EAN

97
Figure 5.13 Protein expression and target‐protein solubility analyses of E. coli
Rosetta‐gami(DE3)pLysS/pET21b(+)‐MmCALR. Two transformant clones were
cultured at 37°C until OD600 reached 0.6 and then the expression was induced with 1
mM IPTG at final concentration. The cultures were further grown overnight at 28°C.
(A) SDS‐PAGE of rMmCALR expression in E. coli Rosetta‐gami(DE3)pLysS; lane
M: Broad Range Molecular Weight Standards (Bio‐Rad, Hercules, CA, USA), lanes
1, 3: non‐induced bacterial lysate, lanes 2, 4: IPTG induced bacterial lysate from
clone number 1 and 2, respectively. (B) SDS‐PAGE of rMmCALR solubility
analysis; lane M: Broad Range Molecular Weight Standards (Bio‐Rad, Hercules, CA,
USA), lanes 1, 3: soluble fraction of the induced bacterial culture, lanes 2, 4: insoluble
fraction of the induced bacterial culture from clone number 1 and 2, respectively.
Arrowheads indicate the locations of rMmCALR.
Ref. code: 25605412330010EAN
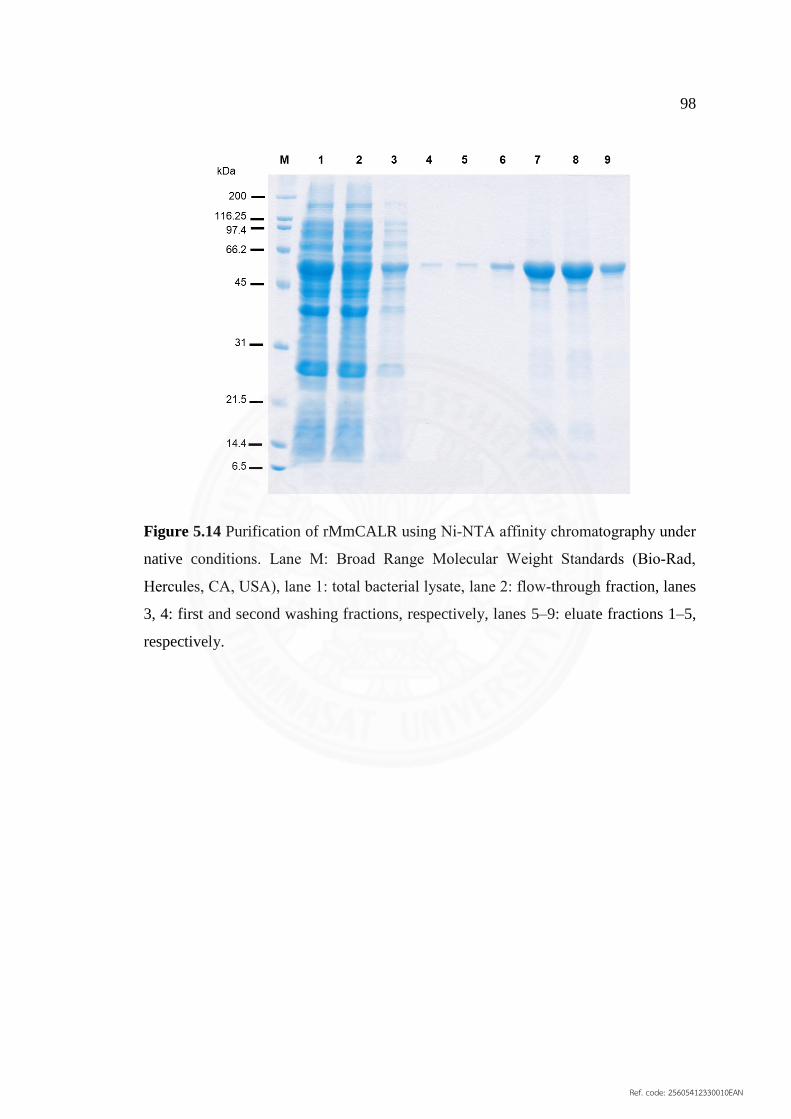
98
Figure 5.14 Purification of rMmCALR using Ni‐NTA affinity chromatography under
native conditions. Lane M: Broad Range Molecular Weight Standards (Bio‐Rad,
Hercules, CA, USA), lane 1: total bacterial lysate, lane 2: flow‐through fraction, lanes
3, 4: first and second washing fractions, respectively, lanes 5–9: eluate fractions 1–5,
respectively.
Ref. code: 25605412330010EAN
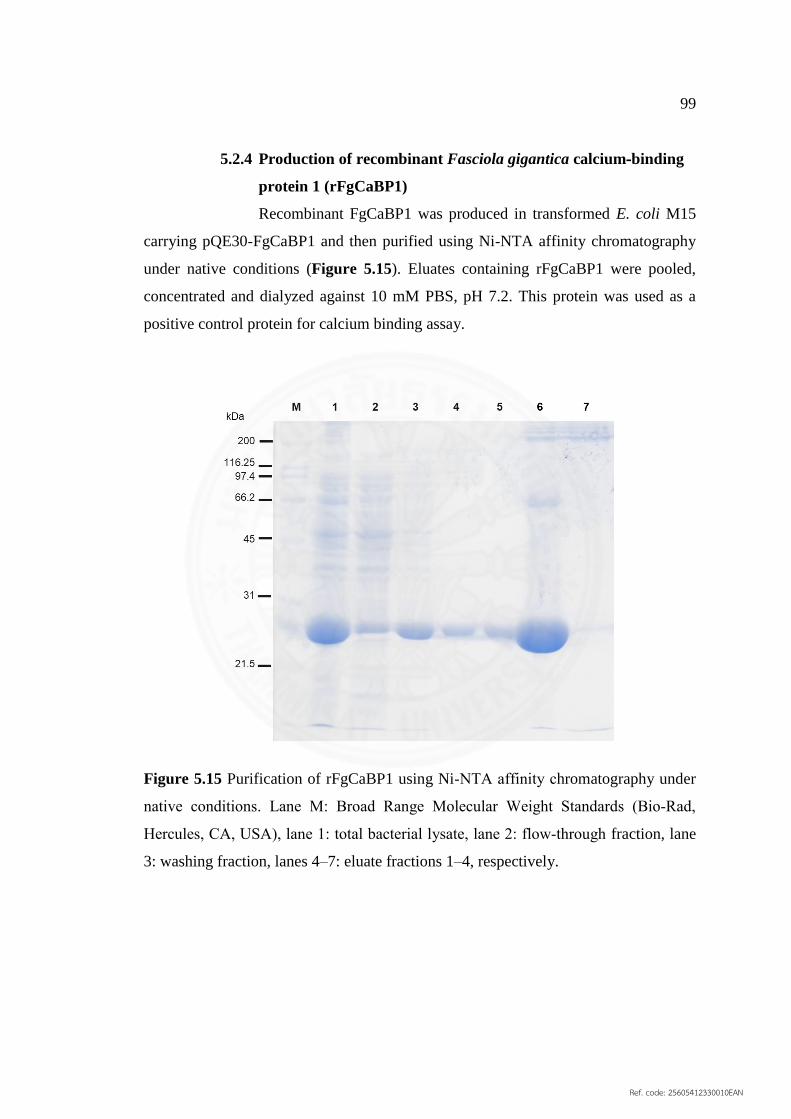
99
5.2.4 Production of recombinant Fasciola gigantica calcium‐binding
protein 1 (rFgCaBP1)
Recombinant FgCaBP1 was produced in transformed E. coli M15
carrying pQE30‐FgCaBP1 and then purified using Ni‐NTA affinity chromatography
under native conditions (Figure 5.15). Eluates containing rFgCaBP1 were pooled,
concentrated and dialyzed against 10 mM PBS, pH 7.2. This protein was used as a
positive control protein for calcium binding assay.
Figure 5.15 Purification of rFgCaBP1 using Ni‐NTA affinity chromatography under
native conditions. Lane M: Broad Range Molecular Weight Standards (Bio‐Rad,
Hercules, CA, USA), lane 1: total bacterial lysate, lane 2: flow‐through fraction, lane
3: washing fraction, lanes 4–7: eluate fractions 1–4, respectively.
Ref. code: 25605412330010EAN
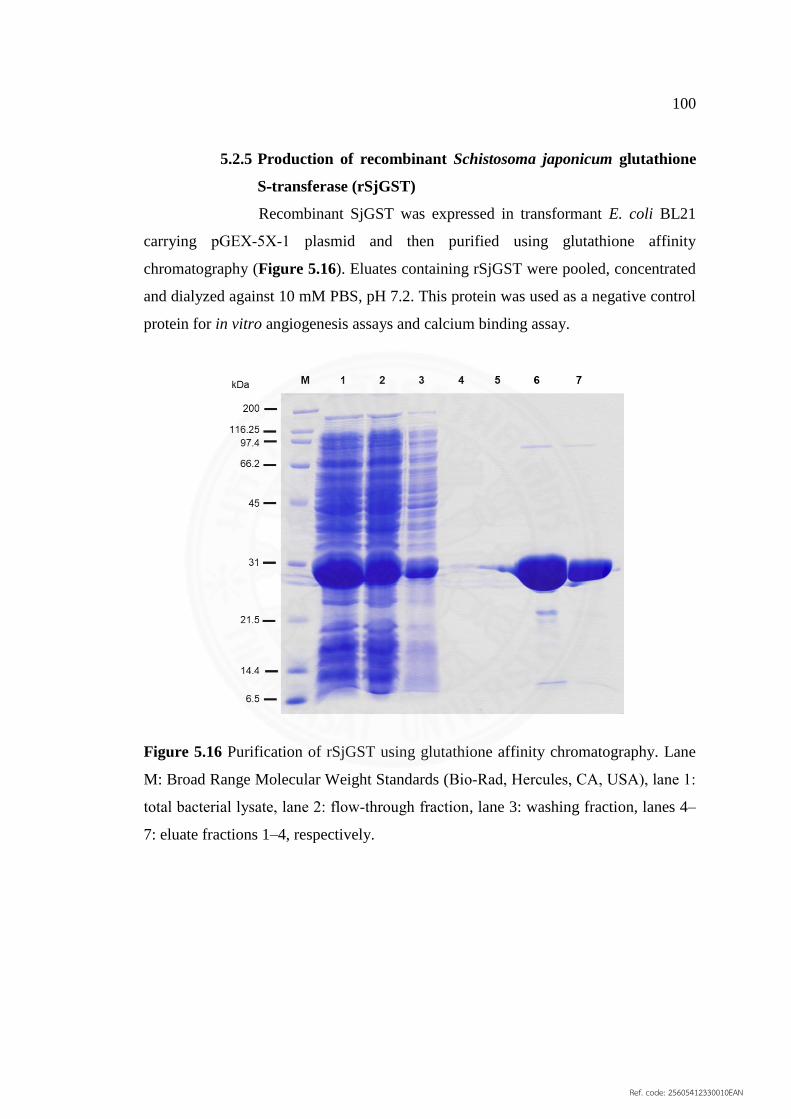
100
5.2.5 Production of recombinant Schistosoma japonicum glutathione
S‐transferase (rSjGST)
Recombinant SjGST was expressed in transformant E. coli BL21
carrying pGEX‐5X‐1 plasmid and then purified using glutathione affinity
chromatography (Figure 5.16). Eluates containing rSjGST were pooled, concentrated
and dialyzed against 10 mM PBS, pH 7.2. This protein was used as a negative control
protein for in vitro angiogenesis assays and calcium binding assay.
Figure 5.16 Purification of rSjGST using glutathione affinity chromatography. Lane
M: Broad Range Molecular Weight Standards (Bio‐Rad, Hercules, CA, USA), lane 1:
total bacterial lysate, lane 2: flow‐through fraction, lane 3: washing fraction, lanes 4–
7: eluate fractions 1–4, respectively.
Ref. code: 25605412330010EAN
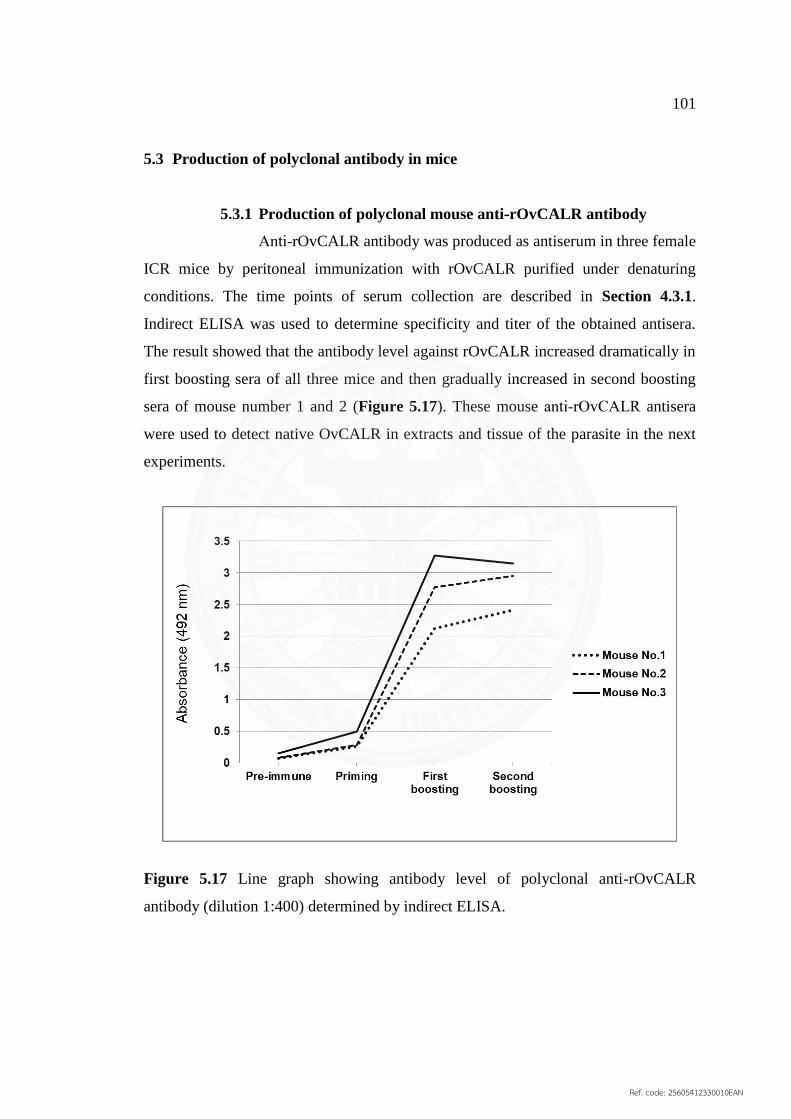
101
5.3 Production of polyclonal antibody in mice
5.3.1 Production of polyclonal mouse anti‐rOvCALR antibody
Anti‐rOvCALR antibody was produced as antiserum in three female
ICR mice by peritoneal immunization with rOvCALR purified under denaturing
conditions. The time points of serum collection are described in Section 4.3.1.
Indirect ELISA was used to determine specificity and titer of the obtained antisera.
The result showed that the antibody level against rOvCALR increased dramatically in
first boosting sera of all three mice and then gradually increased in second boosting
sera of mouse number 1 and 2 (Figure 5.17). These mouse anti‐rOvCALR antisera
were used to detect native OvCALR in extracts and tissue of the parasite in the next
experiments.
Figure 5.17 Line graph showing antibody level of polyclonal anti‐rOvCALR
antibody (dilution 1:400) determined by indirect ELISA.
Ref. code: 25605412330010EAN
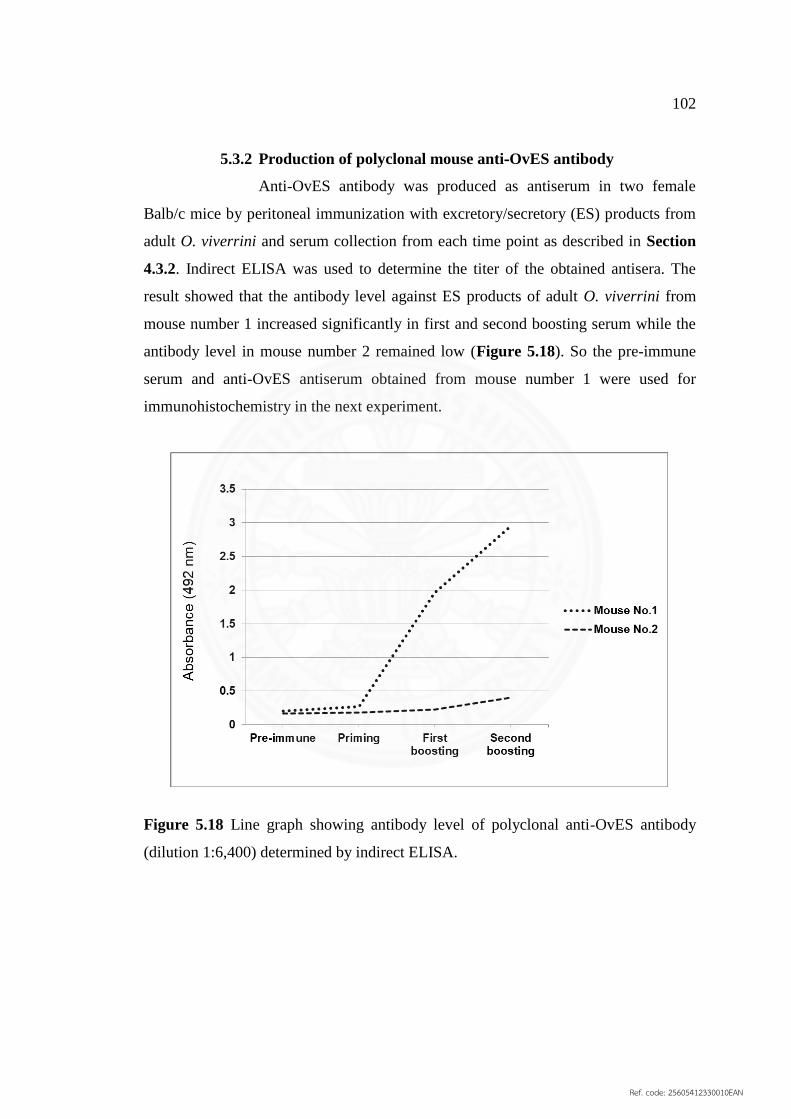
102
5.3.2 Production of polyclonal mouse anti‐OvES antibody
Anti‐OvES antibody was produced as antiserum in two female
Balb/c mice by peritoneal immunization with excretory/secretory (ES) products from
adult O. viverrini and serum collection from each time point as described in Section
4.3.2. Indirect ELISA was used to determine the titer of the obtained antisera. The
result showed that the antibody level against ES products of adult O. viverrini from
mouse number 1 increased significantly in first and second boosting serum while the
antibody level in mouse number 2 remained low (Figure 5.18). So the pre‐immune
serum and anti‐OvES antiserum obtained from mouse number 1 were used for
immunohistochemistry in the next experiment.
Figure 5.18 Line graph showing antibody level of polyclonal anti‐OvES antibody
(dilution 1:6,400) determined by indirect ELISA.
Ref. code: 25605412330010EAN
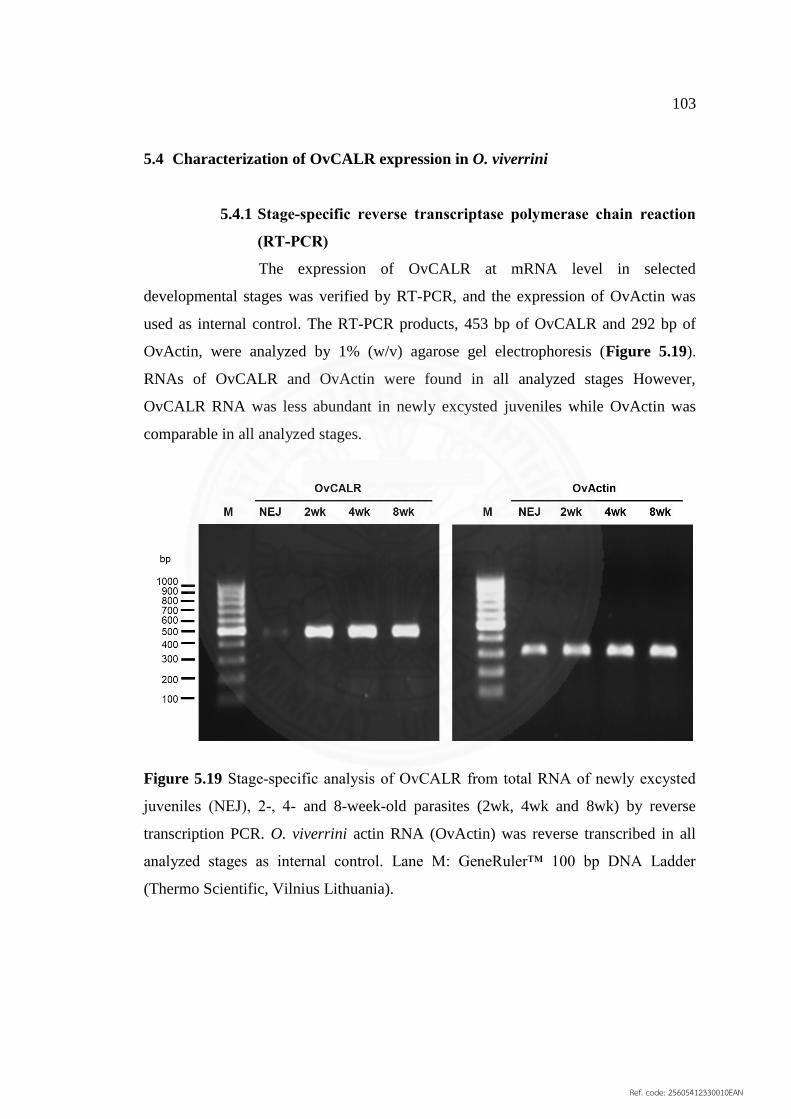
103
5.4 Characterization of OvCALR expression in O. viverrini
5.4.1 Stage‐specific reverse transcriptase polymerase chain reaction
(RT‐PCR)
The expression of OvCALR at mRNA level in selected
developmental stages was verified by RT‐PCR, and the expression of OvActin was
used as internal control. The RT‐PCR products, 453 bp of OvCALR and 292 bp of
OvActin, were analyzed by 1% (w/v) agarose gel electrophoresis (Figure 5.19).
RNAs of OvCALR and OvActin were found in all analyzed stages However,
OvCALR RNA was less abundant in newly excysted juveniles while OvActin was
comparable in all analyzed stages.
Figure 5.19 Stage‐specific analysis of OvCALR from total RNA of newly excysted
juveniles (NEJ), 2‐, 4‐ and 8‐week‐old parasites (2wk, 4wk and 8wk) by reverse
transcription PCR. O. viverrini actin RNA (OvActin) was reverse transcribed in all
analyzed stages as internal control. Lane M: GeneRuler™ 100 bp DNA Ladder
(Thermo Scientific, Vilnius Lithuania).
Ref. code: 25605412330010EAN

104
5.4.2 Western blot analyses
Parasite antigens including excretory/secretory product (ES),
soluble and insoluble crude worm extracts were prepared as described in Section 4.4
(Figure 5.20). The parasite extracts were used to analyze spatial expression of
OvCALR using immunoblot with anti‐rOvCALR antisera. Pre‐immune sera and anti‐
rOvCALR antisera from three mice were used to detect OvCALR on the
nitrocellulose membranes. These mouse antisera could detect rOvCALR and native
OvCALR in soluble crude worm extract and excretory/secretory product while the
pre‐immune sera did not show any reactivity to those proteins (Figure 5.21). All
mouse antisera comparably detected OvCALR at dilution 1:3,000. However, only
antiserum from mouse number 1 was used for immunodetection of OvCALR in
further experiments. Anti‐rOvCALR antiserum was used to determine specificity and
cross‐reactivity to rMmCALR and rOvCALR C‐domain as well. The antiserum did
not show cross‐reactivity to rMmCALR while it could detect rOvCALR C‐domain in
fusion with GST and native OvCALR in both soluble and insoluble crude worm
extracts (Figure 5.22). The estimated molecular weight of OvCALR in SDS‐PAGE
was higher than the predicted size of 46 kDa and likewise found in rMmCALR (MW:
46.35 kDa).
Ref. code: 25605412330010EAN

105
Figure 5.20 SDS‐PAGE of parasite extracts from adult O. viverrini. Ten micrograms
each of parasite extracts were resolved by 12.5% SDS‐PAGE: (A) excretory/secretory
(ES) product, (B) soluble crude worm extract (CW), and (C) insoluble crude worm
extract. Lane M: Broad Range Molecular Weight Standards (Bio‐Rad, Hercules, CA,
USA).
Ref. code: 25605412330010EAN
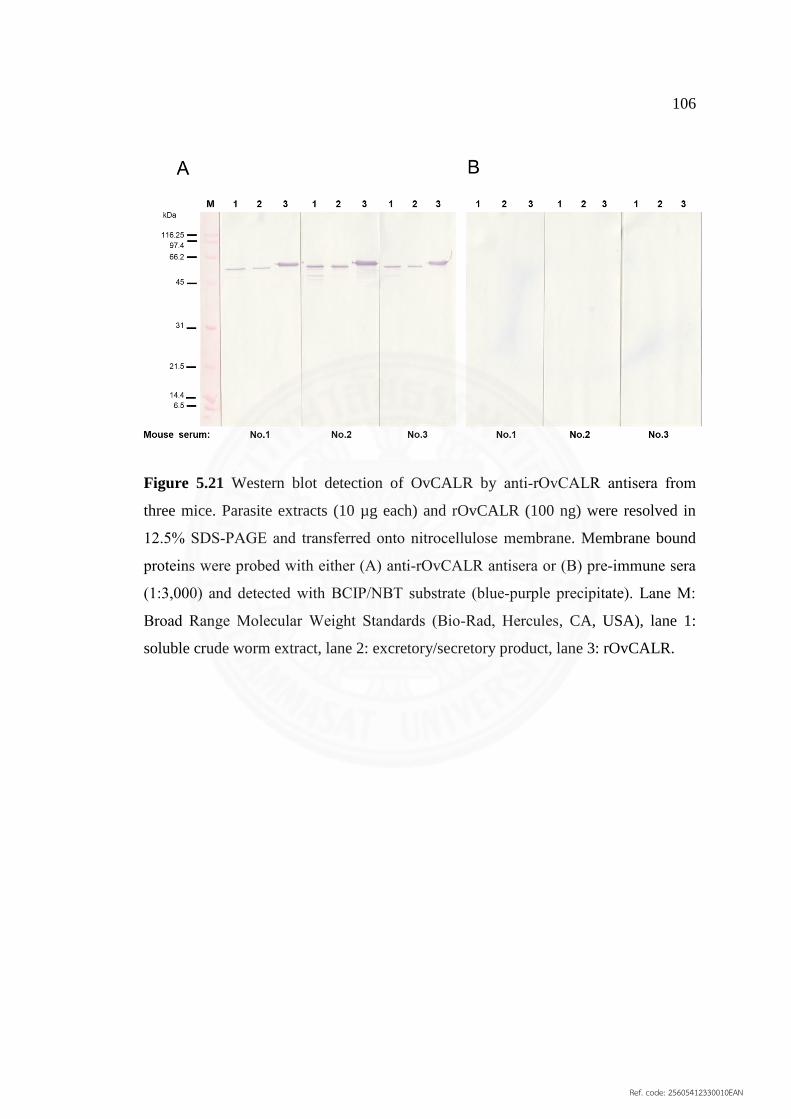
106
Figure 5.21 Western blot detection of OvCALR by anti‐rOvCALR antisera from
three mice. Parasite extracts (10 µg each) and rOvCALR (100 ng) were resolved in
12.5% SDS‐PAGE and transferred onto nitrocellulose membrane. Membrane bound
proteins were probed with either (A) anti‐rOvCALR antisera or (B) pre‐immune sera
(1:3,000) and detected with BCIP/NBT substrate (blue‐purple precipitate). Lane M:
Broad Range Molecular Weight Standards (Bio‐Rad, Hercules, CA, USA), lane 1:
soluble crude worm extract, lane 2: excretory/secretory product, lane 3: rOvCALR.
Ref. code: 25605412330010EAN
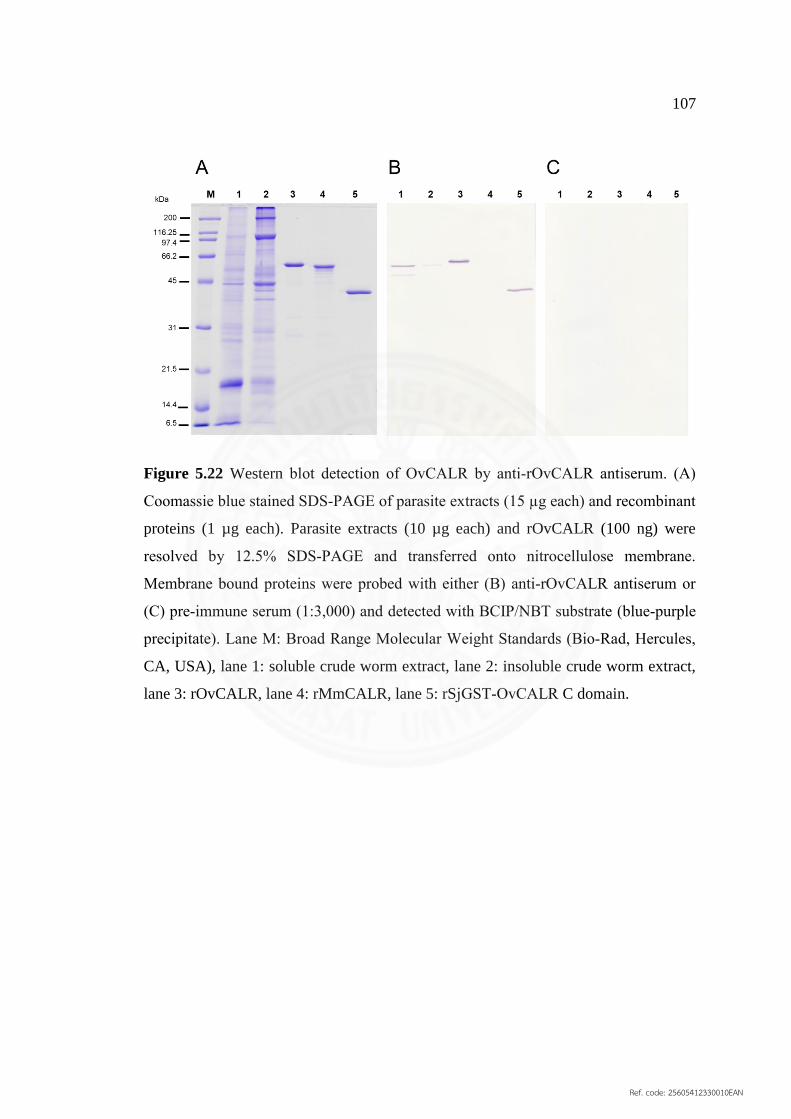
107
Figure 5.22 Western blot detection of OvCALR by anti‐rOvCALR antiserum. (A)
Coomassie blue stained SDS‐PAGE of parasite extracts (15 µg each) and recombinant
proteins (1 µg each). Parasite extracts (10 µg each) and rOvCALR (100 ng) were
resolved by 12.5% SDS‐PAGE and transferred onto nitrocellulose membrane.
Membrane bound proteins were probed with either (B) anti‐rOvCALR antiserum or
(C) pre‐immune serum (1:3,000) and detected with BCIP/NBT substrate (blue‐purple
precipitate). Lane M: Broad Range Molecular Weight Standards (Bio‐Rad, Hercules,
CA, USA), lane 1: soluble crude worm extract, lane 2: insoluble crude worm extract,
lane 3: rOvCALR, lane 4: rMmCALR, lane 5: rSjGST‐OvCALR C domain.
Ref. code: 25605412330010EAN

108
5.4.3 Immunohistochemical analyses
Immunohistochemistry was used to investigate the distribution of
OvCALR in parasite tissue using anti‐rOvCALR antiserum. Pre‐immune serum did
not show any staining on adult stage O. viverrini tissue, whereas anti‐rOvCALR
antiserum led to staining of many parts of the parasite especially in the reproductive
system (Figure 5.23). Punctate staining was found in the parenchyma and intense
staining in tegumental cell bodies, cecal epithelium, gametocytes in testes and ovary,
prostate gland, Mehlis’ gland, vitelline cells, and lining of seminal vesicle (Figure
5.23 and Figure 5.24). Eggs in the proximal uterus were strongly stained and the
staining intensity decreased toward the distal uterus.
The O. viverrini‐infected hamster liver section detected with anti‐
rOvCALR antiserum showed only staining of parasite tissues but not of host tissues,
whereas anti‐OvES antiserum detected parasite antigens in the parasite tissue with
intense staining and in the host tissues surrounding the parasite (Figure 5.24). This
result suggests that some parasite antigens can be trapped in the bile duct epithelium
and connective tissue surrounding the bile duct of the host. Although OvCALR
presented in excretory/secretory products of the parasite, it could not be detected in
the host tissues with anti‐rOvCALR antiserum at appropriate conditions. The selected
dilution of the mouse antisera did not cause any staining in the normal hamster liver
sections at identical conditions (Figure 5.24).
Ref. code: 25605412330010EAN

Figure 5.23 Immunohistochemical detection of OvCALR in adult O. viverrini. Sagittal sections of the parasite were probed by either
mouse anti‐rOvCALR antiserum (A and B) or pre‐immune serum (C) at dilution 1:2,000 and detected with AEC substrate (red staining).
Tg: tegumental cell bodie (arrowheads), Sv: seminal vesicle, Pg: prostate gland cells, Eg: eggs, Mg: Mehlis’s gland, Ov: ovary, Sr:
seminal receptacle, Te: testis.
Anterior Posterior
Ventral
Dorsal
200 µm
A
B
C
109
Ref. code: 25605412330010EAN
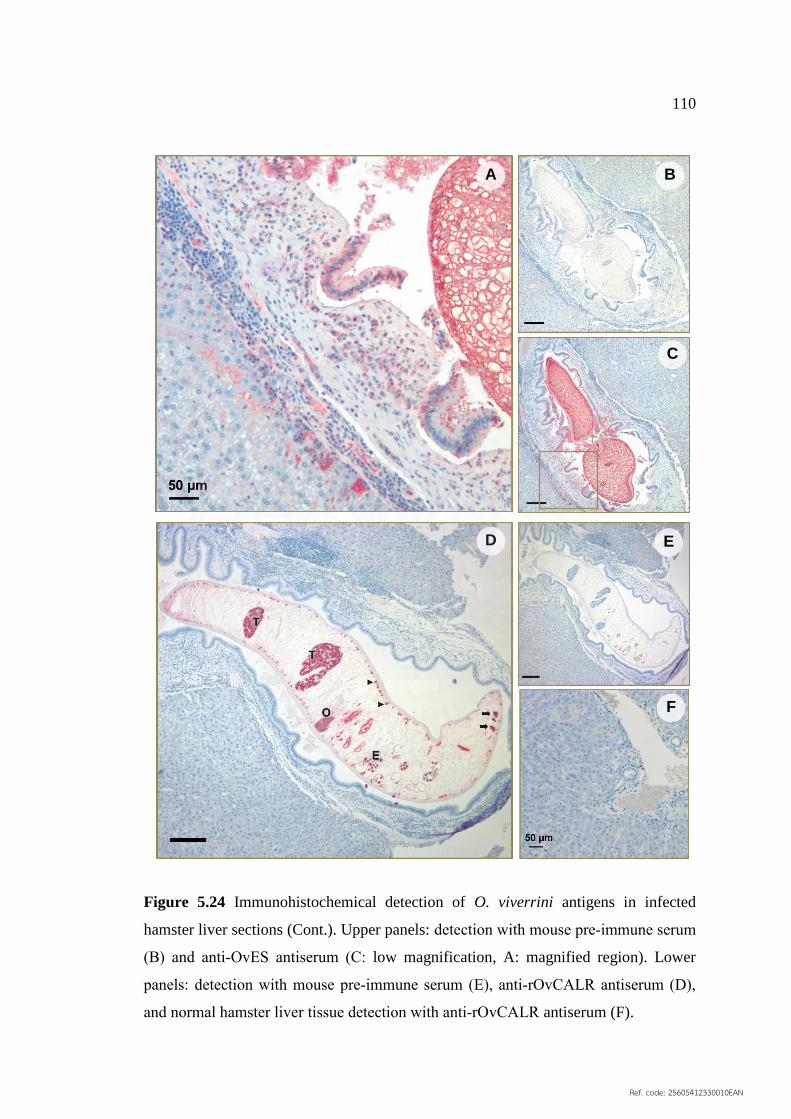
110
Figure 5.24 Immunohistochemical detection of O. viverrini antigens in infected
hamster liver sections (Cont.). Upper panels: detection with mouse pre‐immune serum
(B) and anti‐OvES antiserum (C: low magnification, A: magnified region). Lower
panels: detection with mouse pre‐immune serum (E), anti‐rOvCALR antiserum (D),
and normal hamster liver tissue detection with anti‐rOvCALR antiserum (F).
A
D
C
B
F
E
Ref. code: 25605412330010EAN
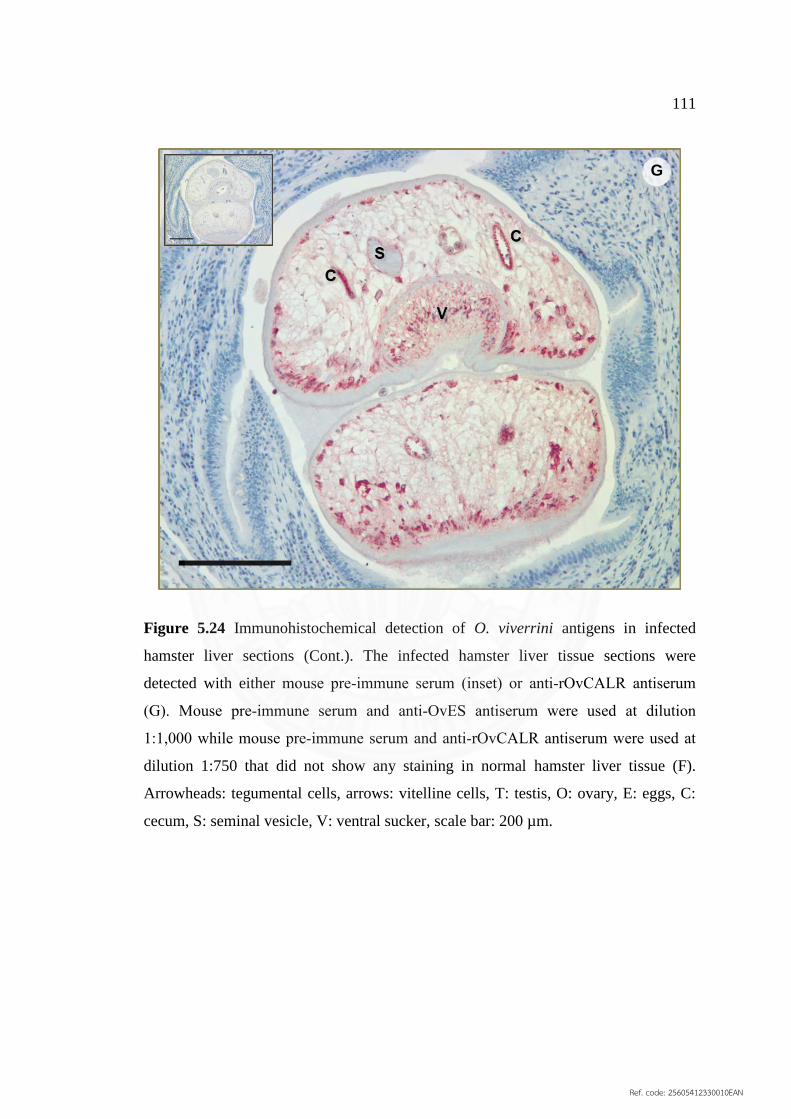
111
Figure 5.24 Immunohistochemical detection of O. viverrini antigens in infected
hamster liver sections (Cont.). The infected hamster liver tissue sections were
detected with either mouse pre‐immune serum (inset) or anti‐rOvCALR antiserum
(G). Mouse pre‐immune serum and anti‐OvES antiserum were used at dilution
1:1,000 while mouse pre‐immune serum and anti‐rOvCALR antiserum were used at
dilution 1:750 that did not show any staining in normal hamster liver tissue (F).
Arrowheads: tegumental cells, arrows: vitelline cells, T: testis, O: ovary, E: eggs, C:
cecum, S: seminal vesicle, V: ventral sucker, scale bar: 200 µm.
G
Ref. code: 25605412330010EAN
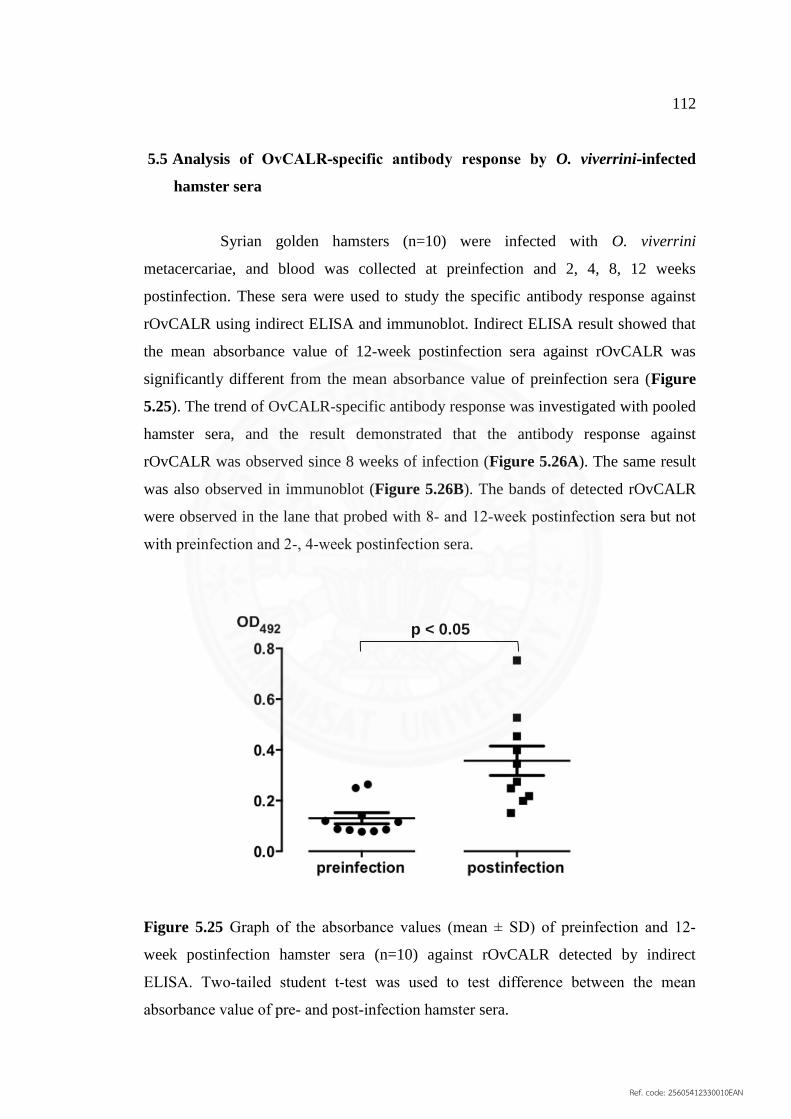
112
5.5 Analysis of OvCALR‐specific antibody response by O. viverrini‐infected
hamster sera
Syrian golden hamsters (n=10) were infected with O. viverrini
metacercariae, and blood was collected at preinfection and 2, 4, 8, 12 weeks
postinfection. These sera were used to study the specific antibody response against
rOvCALR using indirect ELISA and immunoblot. Indirect ELISA result showed that
the mean absorbance value of 12‐week postinfection sera against rOvCALR was
significantly different from the mean absorbance value of preinfection sera (Figure
5.25). The trend of OvCALR‐specific antibody response was investigated with pooled
hamster sera, and the result demonstrated that the antibody response against
rOvCALR was observed since 8 weeks of infection (Figure 5.26A). The same result
was also observed in immunoblot (Figure 5.26B). The bands of detected rOvCALR
were observed in the lane that probed with 8‐ and 12‐week postinfection sera but not
with preinfection and 2‐, 4‐week postinfection sera.
Figure 5.25 Graph of the absorbance values (mean ± SD) of preinfection and 12‐
week postinfection hamster sera (n=10) against rOvCALR detected by indirect
ELISA. Two‐tailed student t‐test was used to test difference between the mean
absorbance value of pre‐ and post‐infection hamster sera.
p < 0.05
Ref. code: 25605412330010EAN
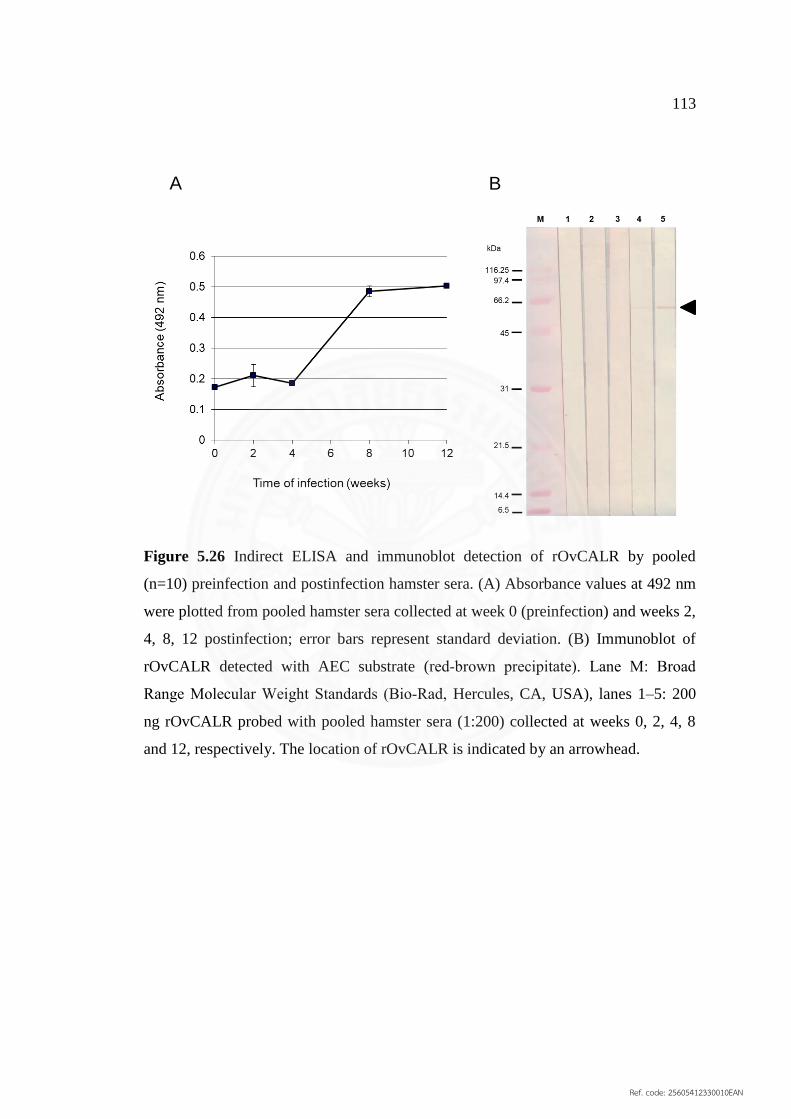
113
Figure 5.26 Indirect ELISA and immunoblot detection of rOvCALR by pooled
(n=10) preinfection and postinfection hamster sera. (A) Absorbance values at 492 nm
were plotted from pooled hamster sera collected at week 0 (preinfection) and weeks 2,
4, 8, 12 postinfection; error bars represent standard deviation. (B) Immunoblot of
rOvCALR detected with AEC substrate (red‐brown precipitate). Lane M: Broad
Range Molecular Weight Standards (Bio‐Rad, Hercules, CA, USA), lanes 1–5: 200
ng rOvCALR probed with pooled hamster sera (1:200) collected at weeks 0, 2, 4, 8
and 12, respectively. The location of rOvCALR is indicated by an arrowhead.
A B
Ref. code: 25605412330010EAN

114
5.6 Functional analysis of rOvCALR
5.6.1 Chaperoning activity
Citrate synthase, a non‐glycosylated protein, was used to
demonstrate the chaperoning activity of calreticulin determined by a thermal induced
protein aggregation assay as described in Section 4.7.2. The relative light scattering at
360 nm of each test and control protein was plotted against incubation time for total 1
hour (Figure 5.27). The result showed that the light scattering in the conditions
containing citrate synthase alone and BSA/citrate synthase (molar ratio 1:1) increased
over time. On the other hand, the light scattering in the conditions containing
rOvCALR /citrate synthase molar ratio of 1:1 did not change over time, likewise in
the rMmCALR/citrate synthase that was used as positive control. The light scattering
slightly increased over time when the molar ratio of rOvCALR/ citrate synthase was
changed to 1:2 and 1:4, respectively. This result suggested that rOvCALR suppressed
citrate synthase aggregation at high temperature similar to rMmCALR and suppressed
aggregation completely up to 1 hour at molar ratio of 1:1.
Ref. code: 25605412330010EAN
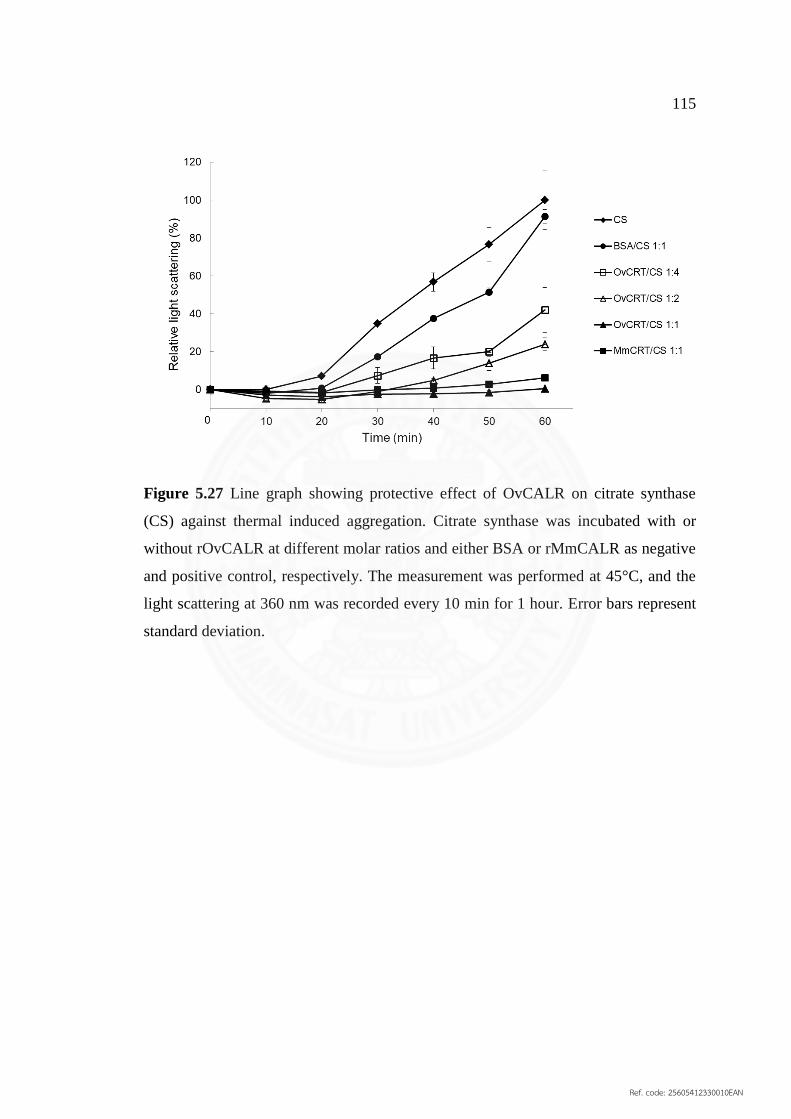
115
Figure 5.27 Line graph showing protective effect of OvCALR on citrate synthase
(CS) against thermal induced aggregation. Citrate synthase was incubated with or
without rOvCALR at different molar ratios and either BSA or rMmCALR as negative
and positive control, respectively. The measurement was performed at 45°C, and the
light scattering at 360 nm was recorded every 10 min for 1 hour. Error bars represent
standard deviation.
Ref. code: 25605412330010EAN

116
5.6.2 Calcium‐binding activity
Recombinant OvCALR C‐domain in fusion with S. japonicum
glutathione S‐transferase (rSjGST‐OvCALR C‐domain) was used to study calcium‐
binding activity using native PAGE in the presence of either calcium or EDTA
(Figure 5.28). Recombinant F. gigantica calcium‐binding protein 1 (rFgCaBP1) that
carries two calcium‐binding EF‐hands and rSjGST were used as positive and negative
control, respectively. The result showed that a shift in the migration of protein in
native PAGE containing calcium in the system was observed with rOvCALR C‐
domain and readily with rFgCaBP1 that served as positive control, but not with
rSjGST (Figure 5.28).
Figure 5.28 Native PAGE showing shift in the migration of OvCALR in the system
presenting of calcium. Lane 1: rSjGST‐OvCALR C‐domain, Lane 2: positive control
rFgCaBP1, Lane 3: negative control rSjGST. Arrowheads and lines indicate the
location of rSjGST.
Ref. code: 25605412330010EAN

117
5.6.3 Transacetylase activity
Transacetylase activity of calreticulin was indirectly determined by
inhibition of GST in the presence of 7‐Acetoxy‐4‐methylcoumarin (7‐AMC) as a
substrate. The result showed that GST activity was suppressed in the presence of
either rOvCALR or rMmCALR without 7‐AMC and slightly suppressed in the
presence of 7‐AMC alone (Figure 5.29). However, the suppression of GST activity
was enhanced in the combination of calreticulin and 7‐AMC. Further investigation by
immunoblot with antibody against acetylated‐lysine revealed that acetylation of GST
by 7‐AMC occurred without the presence of calreticulin (Figure 5.30A), and GST
without 7‐AMC treatment did not show any detected band (Figure 5.30B). In
addition, direct acetylation by 7‐AMC was observed for BSA and recombinant
F. gigantica calcium‐binding protein 1 (Figure 5.30B).
Ref. code: 25605412330010EAN
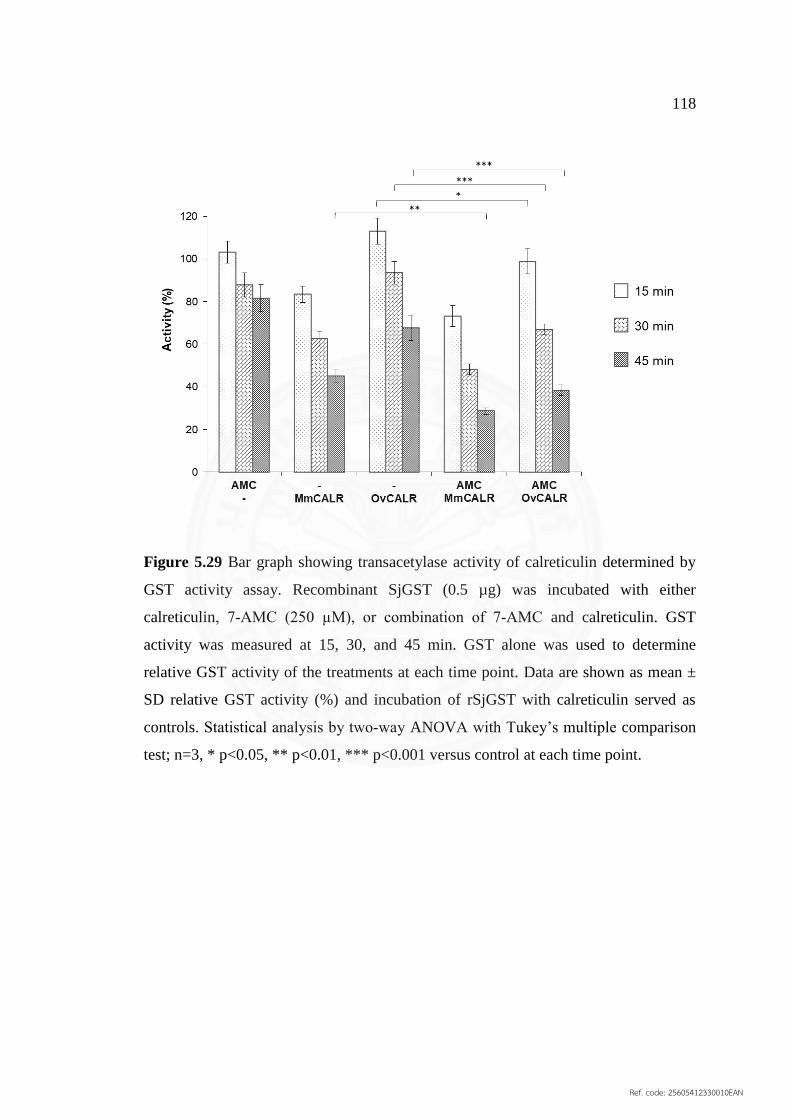
118
Figure 5.29 Bar graph showing transacetylase activity of calreticulin determined by
GST activity assay. Recombinant SjGST (0.5 µg) was incubated with either
calreticulin, 7‐AMC (250 µM), or combination of 7‐AMC and calreticulin. GST
activity was measured at 15, 30, and 45 min. GST alone was used to determine
relative GST activity of the treatments at each time point. Data are shown as mean ±
SD relative GST activity (%) and incubation of rSjGST with calreticulin served as
controls. Statistical analysis by two‐way ANOVA with Tukey’s multiple comparison
test; n=3, * p<0.05, ** p<0.01, *** p<0.001 versus control at each time point.
Ref. code: 25605412330010EAN
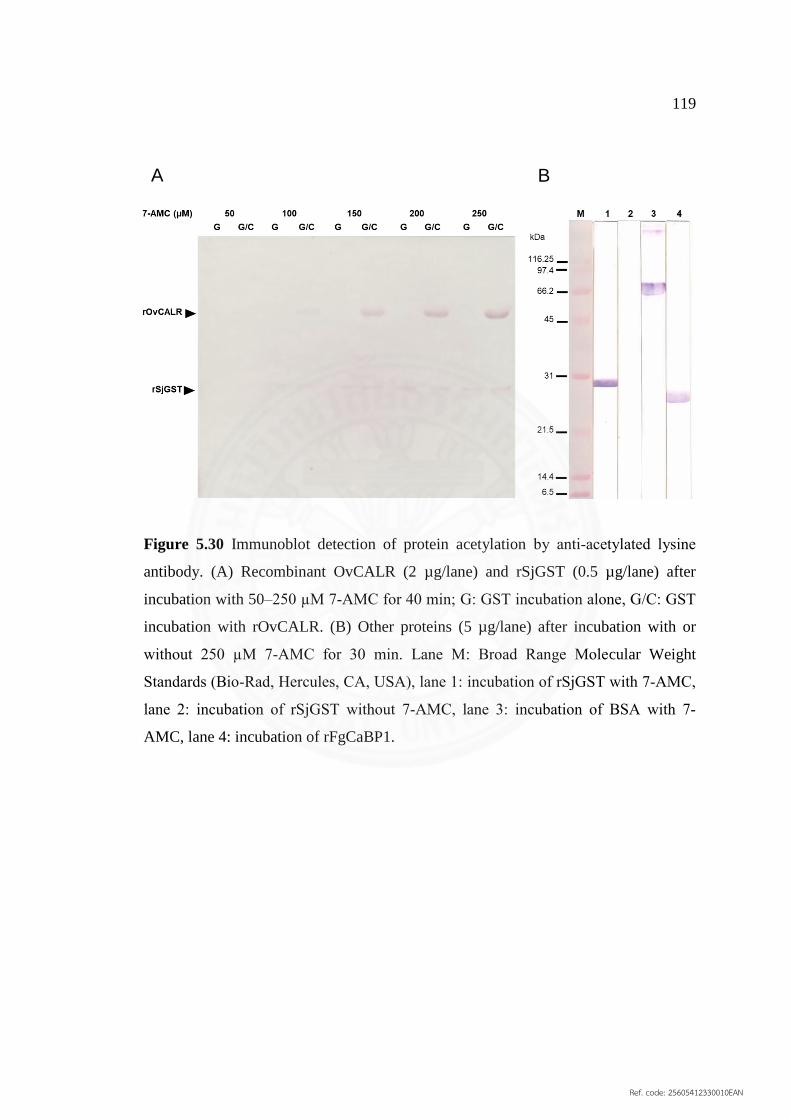
119
Figure 5.30 Immunoblot detection of protein acetylation by anti‐acetylated lysine
antibody. (A) Recombinant OvCALR (2 µg/lane) and rSjGST (0.5 µg/lane) after
incubation with 50–250 µM 7‐AMC for 40 min; G: GST incubation alone, G/C: GST
incubation with rOvCALR. (B) Other proteins (5 µg/lane) after incubation with or
without 250 µM 7‐AMC for 30 min. Lane M: Broad Range Molecular Weight
Standards (Bio‐Rad, Hercules, CA, USA), lane 1: incubation of rSjGST with 7‐AMC,
lane 2: incubation of rSjGST without 7‐AMC, lane 3: incubation of BSA with 7‐
AMC, lane 4: incubation of rFgCaBP1.
A B
Ref. code: 25605412330010EAN

120
5.6.4 Human C1q binding
Competitive ELISA was used to determine the interaction of
rOvCALR and human C1q. In this assay, mouse calreticulin and BSA were used as
positive and negative competitor, respectively. Binding of rOvCALR was measured
by indirect ELISA using anti‐rOvCALR antibody (Figure 5.31). The result
demonstrated that rOvCALR bound to human C1q in a dose‐dependent manner and
this specific binding was confirmed by rMmCALR, a mammalian calreticulin. Mouse
calreticulin inhibited the binding of rOvCALR to human C1q by competing for the
binding sites on C1q, whereas BSA did not significantly affect the binding of
rOvCALR to human C1q (Figure 5.31).
Figure 5.31 Graph showing human C1q binding of OvCALR obtained by competitive
ELISA. Mean and standard deviation values with fitted curves are shown for 20 ng
C1q incubated with either rOvCALR (0–2 µg), rOvCALR (1 µg)/rMmCALR (0–
2 µg) or rOvCALR (1 µg)/BSA (0–2 µg). Binding of rOvCALR was detected by a
mouse anti‐rOvCALR antiserum.
Ref. code: 25605412330010EAN

121
5.6.5 Inhibition of classical complement pathway
Hemolytic assay of sensitized human red blood cells was used to
determine the effect of rOvCALR against classical complement activation. In this
assay, mouse calreticulin and BSA were used as positive and negative control,
respectively. Pre‐incubated normal human serum with/without tested proteins was
mixed with sensitized red blood cells and observed for hemolysis. Classical
complement‐mediated hemolysis was determined by measuring hemoglobin release at
540 nm. All absorbance values were subtracted with spontaneous hemolysis
absorbance value, and pre‐incubated normal human serum alone was used to calculate
the relative hemolysis of treatments (Figure 5.32). The result showed significant
decrease in relative hemolysis of sensitized red blood cells in the conditions of either
rOvCALR or rMmCALR pre‐incubated normal human serum at 30 and 50 µg amount
of protein. BSA pre‐incubated normal human serum did not show significant decrease
in relative hemolysis at the same concentrations. This result indicated that rOvCALR
suppressed the classical complement‐mediated hemolysis in a dose‐dependent
manner.
Ref. code: 25605412330010EAN
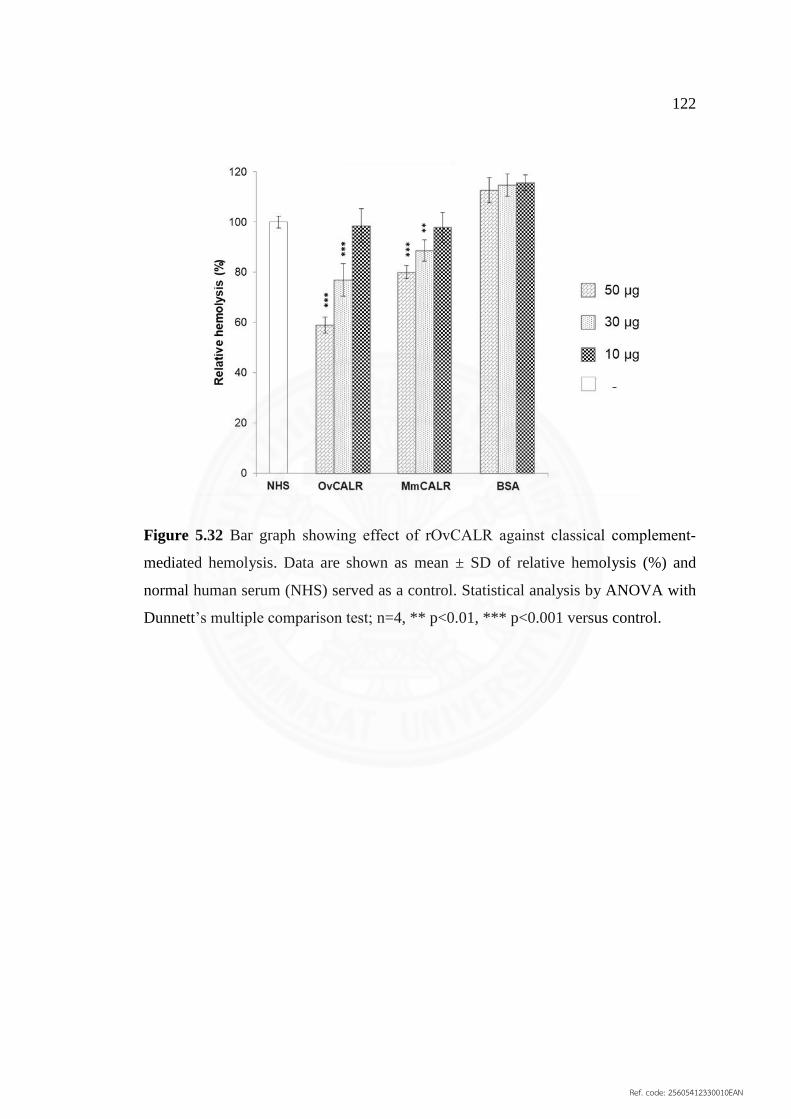
122
Figure 5.32 Bar graph showing effect of rOvCALR against classical complement‐
mediated hemolysis. Data are shown as mean ± SD of relative hemolysis (%) and
normal human serum (NHS) served as a control. Statistical analysis by ANOVA with
Dunnett’s multiple comparison test; n=4, ** p<0.01, *** p<0.001 versus control.
Ref. code: 25605412330010EAN
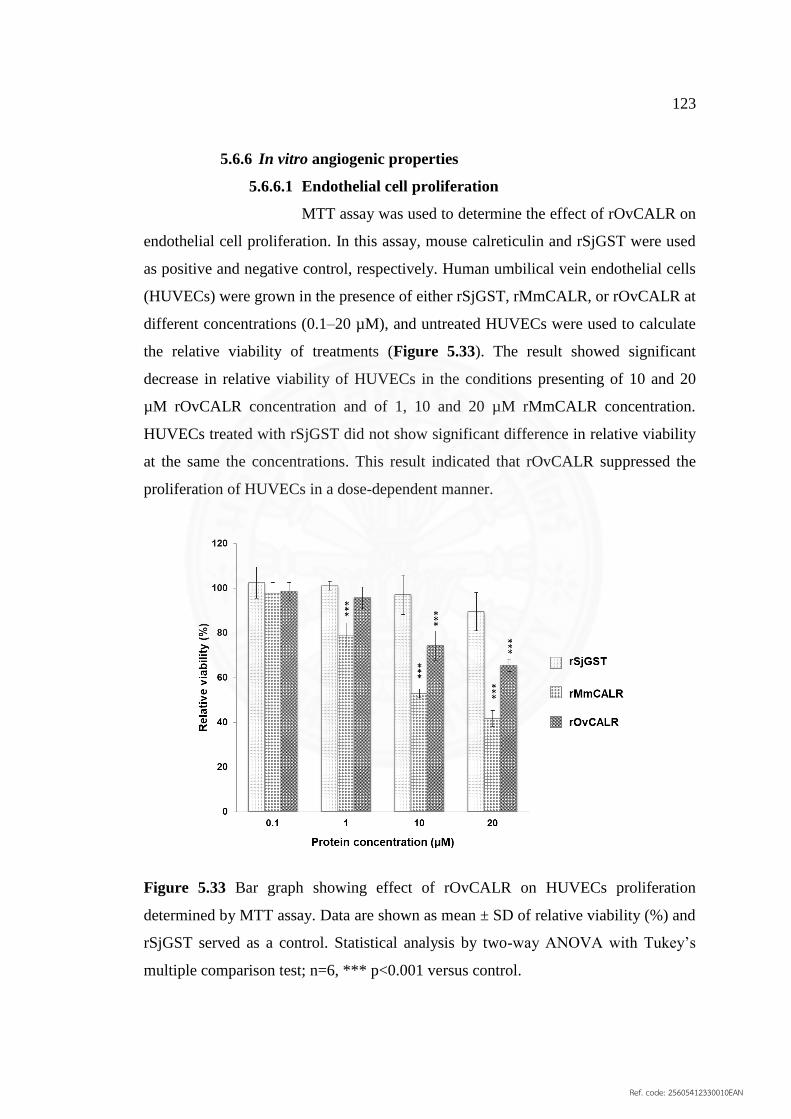
123
5.6.6 In vitro angiogenic properties
5.6.6.1 Endothelial cell proliferation
MTT assay was used to determine the effect of rOvCALR on
endothelial cell proliferation. In this assay, mouse calreticulin and rSjGST were used
as positive and negative control, respectively. Human umbilical vein endothelial cells
(HUVECs) were grown in the presence of either rSjGST, rMmCALR, or rOvCALR at
different concentrations (0.1–20 µM), and untreated HUVECs were used to calculate
the relative viability of treatments (Figure 5.33). The result showed significant
decrease in relative viability of HUVECs in the conditions presenting of 10 and 20
µM rOvCALR concentration and of 1, 10 and 20 µM rMmCALR concentration.
HUVECs treated with rSjGST did not show significant difference in relative viability
at the same the concentrations. This result indicated that rOvCALR suppressed the
proliferation of HUVECs in a dose‐dependent manner.
Figure 5.33 Bar graph showing effect of rOvCALR on HUVECs proliferation
determined by MTT assay. Data are shown as mean ± SD of relative viability (%) and
rSjGST served as a control. Statistical analysis by two‐way ANOVA with Tukey’s
multiple comparison test; n=6, *** p<0.001 versus control.
Ref. code: 25605412330010EAN

124
5.6.6.2 Endothelial cell migration
Scratch assay, a simple method to measure migration of cells,
was used to study the effect of rOvCALR on HUVECs migration. The scratch was
made and the image was captured before and after incubation with proteins at
different concentrations. Figure 5.34A shows the images of scratches at 0 hour and
24 hour after incubation with either rOvCALR, rMmCALR (positive control), or
rSjGST (negative control). The migration area was measured, and the migration area
of untreated HUVECs was used to calculate relative migration area following
treatment (Figure 5.34B). The result showed significant decrease in relative migration
area of HUVECs in the conditions containing rOvCALR and rMmCALR at 20 and 50
µM concentration. HUVECs treated with rSjGST did not show significant difference
in relative migration area between the concentrations. This result indicated that
rOvCALR suppressed the migration of HUVECs in a dose‐dependent manner.
Ref. code: 25605412330010EAN
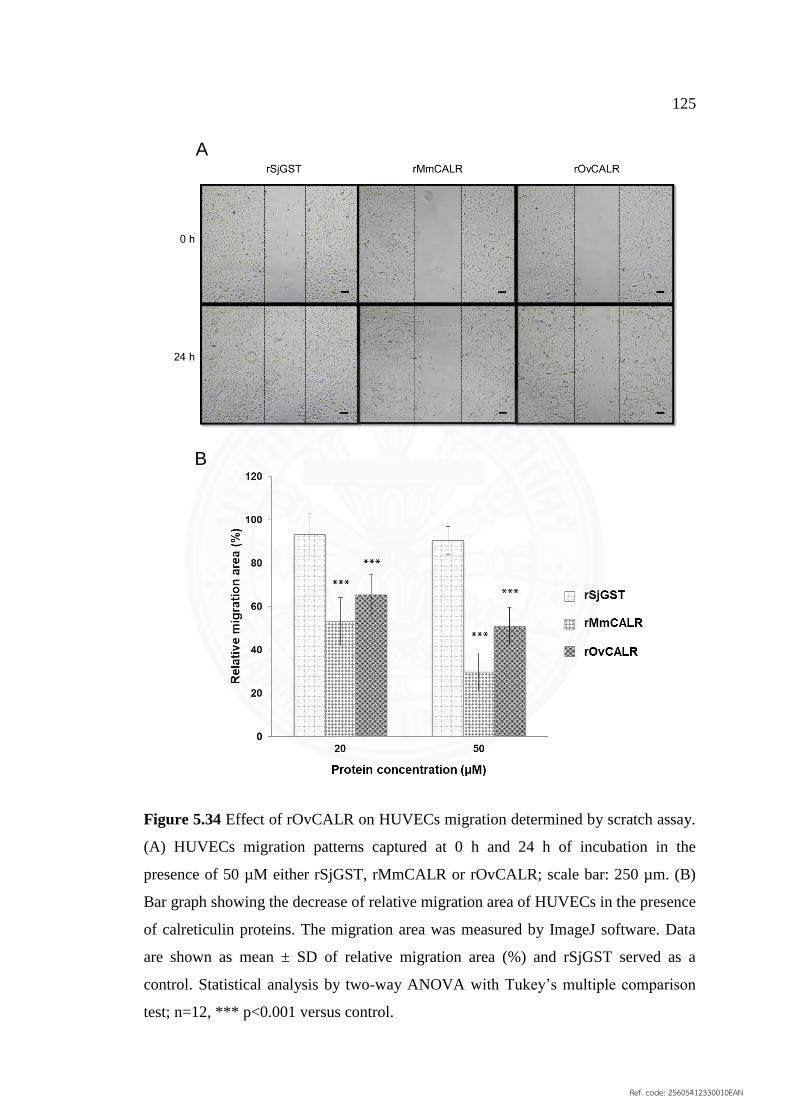
125
Figure 5.34 Effect of rOvCALR on HUVECs migration determined by scratch assay.
(A) HUVECs migration patterns captured at 0 h and 24 h of incubation in the
presence of 50 µM either rSjGST, rMmCALR or rOvCALR; scale bar: 250 µm. (B)
Bar graph showing the decrease of relative migration area of HUVECs in the presence
of calreticulin proteins. The migration area was measured by ImageJ software. Data
are shown as mean ± SD of relative migration area (%) and rSjGST served as a
control. Statistical analysis by two‐way ANOVA with Tukey’s multiple comparison
test; n=12, *** p<0.001 versus control.
A
B
Ref. code: 25605412330010EAN

126
5.6.6.3 Endothelial cell sprouting
Tube formation assay using Matrigel as an extracellular
basement membrane matrix for endothelial cell sprouting was used to study the effect
of rOvCALR on the sprouting of HUVECs. Figure 5.35A shows the tube‐like
structures formed by HUVECs following incubation with either PBS, rOvCALR,
rMmCALR (positive control), or rSjGST (negative control). HUVECs treated with
rSjGST showed sprouting comparable to untreated HUVECs (PBS), whereas
HUVECs treated with calreticulin showed less sprouting particularly at 50 µM
concentration. The tube length was measured by ImageJ angiogenesis analyzer, and
the tube length of untreated HUVECs (PBS) was used to calculate relative tube length
of treatments (Figure 5.35B). The result showed significant decrease in relative tube
length of HUVECs‐formed tube‐like structure in the conditions containing rOvCALR
and rMmCALR at 20 and 50 µM concentration. HUVECs treated with rSjGST did
not show significant difference in relative tube length between the concentrations.
This result indicated that rOvCALR suppressed the sprouting of HUVECs in a dose‐
dependent manner.
Ref. code: 25605412330010EAN

127
Figure 5.35 Effect of rOvCALR on HUVECs sprouting determined by tube formation
assay. (A) Tube‐like structure formation by HUVECs after incubated with proteins for
18 hour; scale bar: 250 µm. (B) Bar graph showing the decrease of relative tube
length of HUVECs‐formed tube‐like structure in the presence of calreticulin proteins.
Data are shown as mean ± SD of relative tube length (%) and rSjGST served as a
control. Statistical analysis by two‐way ANOVA with Tukey’s multiple comparison
test; n=6, * p<0.05, ** p<0.01, *** p<0.001 versus control.
A
B
Ref. code: 25605412330010EAN

128
CHAPTER 6
DISCUSSION
The present study elucidates molecular sequence, expression, distribution,
and functional roles of O. viverrini calreticulin to demonstrate how important it is
inside and outside the parasite, particularly in the host‐parasite interplay. This study
started with molecular cloning of OvCALR from adult parasite total RNA using
reverse transcription PCR that revealed the 1,248 bp open reading frame of the
OvCALR cDNA. The deduced amino acid sequence of OvCALR carries the
conserved calreticulin family signature 1 as well as the signature 2 with one amino
acid miss‐match, isoleucine in mammalian CALR is replaced by phenylalanine in
O. viverrini (Figure 5.5). This miss‐match is also found in the closely related species,
C. sinensis, however, phenylalanine has a hydrophobic sidechain like isoleucine. In
addition, the presence of other calreticulin characteristics in the OvCALR amino acid
sequence indicates that OvCALR is a novel member of the calreticulin family. These
characteristics are a N-terminal signal peptide, a C-terminal ER retention signal, three
structural domains (N‐, P‐, and C‐domain), three conserved cysteine residues, two of
which are used for disulfide bridge forming, and three each of two tandem repeated
motifs. At least two isoforms of calreticulin have been reported in higher plants83
and
animals, while only one isoform has been reported in lower animals87
. The published
O. viverrini genome data contains only one calreticulin gene.183
Recombinant OvCALR was expressed using bacterial systems and used
for functional studies and antibody production. At first, rOvCALR was expressed in
E. coli M15, it was already present since the first hour of IPTG induction but was
insoluble (Figure 5.6), whereas recombinant MmCALR, the selected positive control
for functional studies, could not be expressed at all at identical conditions (data not
shown). Moreover, the bacterial growth as observed by OD600 stopped after IPTG
induction, indicating that expression of MmCALR was toxic for E. coli M15.
Eventually, rOvCALR and rMmCALR were expressed as soluble proteins in E. coli
Rosetta‐gami(DE3)pLysS and purified by affinity chromatography. The purified
rOvCALR was used to produce polyclonal anti‐rOvCALR antibodies in mice, and the
Ref. code: 25605412330010EAN

129
obtained antisera were evaluated for specificity and titer against rOvCALR by indirect
ELISA. Because calreticulin is an immunodominant antigen184,185
, the obtained
antisera from three mice showed high specific antibody levels after first boosting by
immunization with crushed polyacrylamide gel as adjuvant (Figure 5.17).
The expression level of OvCALR RNA during development in the
experimental mammalian host (Mesocricetus auratus) was comparable high in 2‐, 4‐,
and 8‐week‐old parasites but RNA was less abundant in newly excysted juveniles
(Figure 5.19). The western analyses of parasite antigen extracts using anti‐rOvCALR
antisera revealed the presence of OvCALR in soluble and insoluble crude worm
extracts as well as excretory/secretory (ES) product of adult parasites (Figures 5.21
and 5.22) demonstrating that calreticulin is not only an intracellular but also an
extracellular protein. Calreticulin has been detected as soluble molecule in sera of
patients with autoimmune diseases186-188
and in parasite ES product189,190
.
Interestingly, the intensity of detected OvCALR was readily comparable in soluble
crude worm extract and ES product, while it was faint in insoluble crude worm extract
at the same total amount of protein. This result suggests that OvCALR is mainly
present as soluble protein with functions in- and outside of the parasite. Anomalous
migration of native OvCALR, rOvCALR, and rMmCALR in SDS‐PAGE, which was
slower than expected from their calculated molecular mass, had previously been
observed for mammalian calreticulin and was explained by its highly disorder in the
P‐domain and highly acidic character of the C‐domain leading to poor SDS-binding
and retaining of its tertiary structure.61,191
The cross‐reactivity between human anti‐
Onchocerca volvulus calreticulin in sera from patients with onchocerciasis and
autoantigen153,154
as well as between the sera of patients with systemic lupus
erythematosus and recombinant S. mansoni calreticulin192
has been reported. In this
study, the raised mouse anti‐rOvCALR antiserum specifically recognized native
OvCALR and rOvCALR C‐domain in fusion with GST but not rMmCALR,
indicating the lacking of cross‐reactivity between OvCALR and MmCALR that
shares 50% identity in their amino acid sequences. The immunohistochemical staining
by anti‐rOvCALR antiserum showed that OvCALR was widely distributed in adult
parasite tissue including tegumental cell bodies, cecal epithelium and particularly in
the genital organs (Figure 5.23). Similar findings were also observed in the tissue
Ref. code: 25605412330010EAN

130
distribution of calreticulin in S. mansoni76
and T. solium159
. The staining of
gametocytes in the gonads suggests a role of OvCALR in spermatogenesis and
oogenesis as well as in eggshell formation12
since the staining was also found in the
Mehlis’ gland, and vitelline cells. Interestingly, spermatogenic cells in the testes
showed intense staining, while the staining was absent in the spermatozoa found in
seminal vesicles and seminal receptacle (Figure 5.23). This finding might correlate
with the lack of an endoplasmic reticulum in mature spermatozoa.193
Moreover,
differential expression of OvCALR in eggs in different regions of the uterus was
found to correlate with embryogenesis12
. The developing miracidia found in the eggs
of the middle uterus showed high OvCALR expression while OvCALR was absent in
the fully mature miracidia found in the eggs of the distal uterus. This result suggests a
role of OvCALR during embryogenesis in O. viverrini. Overall, intense
immunostaining was found in highly active cells either undergoing rapid cell
divisions or with high protein output for example, gametocyte during gametogenesis,
fertilized egg during embryogenesis, and secretory cells. This might be correlated to
the conserved major role of calreticulin in protein/glycoprotein chaperoning in the
ER. Continuous tegumental membrane reparation194
, protease secretion from cecal
epithelium195-197
, and massive secretion from accessory glands to support the
unremitting reproductive process12
can explain the high expression of OvCALR in
these tissues. The low abundance of OvCALR RNA in the newly excysted juvenile
can be explained with the absence of developed reproductive organs198
(Figure 5.19).
This result corresponds to those findings in 18 h schistosomula and cercariae of
S. mansoni.76
Although OvCALR was detected in the ES product of the parasite and
rOvCALR recognized by O. viverrini‐infected hamster sera (Figures 5.25 and 5.26),
it was not detected in the liver tissue of O. viverrini‐infected hamster by anti‐
rOvCALR antiserum (Figures 5.24 and 5.25). Immunostaining of the host tissue at
least at the bile duct epithelium has been observed for other O. viverrini proteins, e.g.
granulin‐like growth factor5, tetraspanins
199, and cathepsin F cysteine protease
197. The
mechanism of internalization of parasite antigens by cholangiocytes is still not
clear.200
However, immunostaining of host tissue using an antiserum against
O. viverrini ES product demonstrated that released parasite antigens infiltrate
neighboring host tissue (Figure 5.25). Recently, the internalization of extracellular
Ref. code: 25605412330010EAN

131
vesicles secreted by O. viverrini and promoting proliferation of cholangiocytes has
been reported. The vesicles were found to contain various proteins, e.g. tetraspanins,
calcium‐binding proteins, and cathepsin D.201
OvCALR was not reported and it can be
assumed that it is released through an unrelated pathway. Unlike the antibody
responses against ES and somatic antigens of O. viverrini that were readily observed
after four weeks of infection202
, the response against rOvCALR was first detected in
hamster sera eight weeks after infection (Figure 5.26) indicating release of OvCALR
from mature parasites. The protein might be released from the reproductive system of
adult parasites.
Calreticulin plays a major role as soluble ER chaperone by binding to
monoglucosylated N‐linked oligosaccharides via its lectin site.104
However, it has
been reported that it can interact with non‐glycosylated proteins via its overlapping
polypeptide‐binding and lectin sites.97,180
In vitro protein aggregation assay, which has
been used to demonstrate the protective effect of calreticulin from various organisms
against thermal induced protein/glycoprotein aggregation174,203
was used to determine
chaperoning activity of rOvCALR. Recombinant OvCALR can protect citrate
synthase, a non‐glycosylated protein, from aggregation at high temperature similar to
rMmCALR (Figure 5.27) suggesting the capability of OvCALR as ER chaperone.
Mutagenesis180
and deletion‐analysis204
of calreticulin suppressing protein
aggregation have been used to map the polypeptide‐binding site on the globular lectin
domain including the N‐domain and proximal region of the C‐domains indicating the
correct folding of rOvCALR in the required globular domain. Additionally, multiple
alignment of calreticulin sequences revealed high sequence conservation of residues
participating in carbohydrate‐ and polypeptide‐binding as well as ERp57‐binding,
which is a co‐chaperone (Figure 5.5)97,180,181
, supporting the conserved chaperoning
property in OvCALR and other trematode orthologs.
The binding of calcium to maintain calcium homeostasis inside the ER is
another important role of calreticulin. Calreticulin contains a single high‐affinity, low‐
capacity calcium‐binding site within the globular domain and multiple low‐affinity,
high‐capacity calcium‐binding sites within the C‐domain.97,99
The low‐affinity, high‐
capacity calcium‐binding C‐domain of OvCALR in fusion with S. japonicum
glutathione S‐transferase (SjGST) was demonstrated to bind calcium resulting in
Ref. code: 25605412330010EAN

132
migration shifting on native‐PAGE (Figure 5.28). Calreticulin C-domain lacks high‐
affinity calcium‐binding EF‐hand motifs that are found in F. gigantica calcium‐
binding protein 1 (FgCaBP1) and binds calcium through its acidic amino acid
cluster.99
Hence, rOvCALR C‐domain required the presence of calcium in the
electrophoresis running buffer to show calcium-binding (data not shown), which
supports the low affinity binding reported in mammalian calreticulin.99,205
The C‐
domains of human calreticulin (HsCALR) and OvCALR have only 33% sequence
identity and OvCALR carries a smaller number of acidic residues (33.0%) than
HsCALR (42.6%). The high‐affinity, low‐capacity calcium‐binding property of
OvCALR was not analyzed in this study. However, multiple alignment of calreticulin
sequences showed conservation of the involved residues Gln26, Lys62, Lys64 that
provide backbone carbons and high conservation of Asp328 that provides the side
chain to form high‐affinity calcium‐binding site (Figure 5.5)98
, suggesting that high
affinity calcium‐binding is conserved in trematode calreticulins.
Calreticulin transacetylase (CRTase), an activity to transfer acetyl group
from polyphenolic acetates to certain receptor molecule, is a novel characterized
function of calreticulin mapped in the P‐domain.206,207
The possible receptor proteins
including GST, nitric oxide synthase (NOS), NADPH cytochrome c reductase
(CYPR), cytochrome P450 and various substrates including acetoxycoumarins and
acetyl coenzyme A have been identified for CRTase.208
Calreticulin‐mediated GST
acetylation resulting in reduction of GST activity has been developed as an indirect
measurement of CRTase activity.209
Transacetylase activity of rOvCALR was
analyzed by the same assay using 7‐Acetoxy‐4‐methylcoumarin (7‐AMC) as
substrate210-212
. Unlike calreticulin of H. contortus with its previously demonstrated
CRTase activity through inhibition of GST activity176,207
, both of rOvCALR and
rMmCALR demonstrated suppression of GST activity without the presence of 7‐
AMC (Figure 5.30A) and direct acetylation by 7‐AMC was observed in various
tested proteins (Figure 5.30B). The loss of GST activity might be explained by the
direct acetylation of GST by 7‐AMC and the direct interaction between GST and
calreticulin by its chaperoning function.
The complement system is a part of the innate immune system of the host
defense against common pathogenic infection. C1q is the complement component that
Ref. code: 25605412330010EAN

133
recognizes antigen‐bound antibodies and initiates classical complement pathway
activation. Recombinant OvCALR demonstrated human C1q binding as shown by
competitive ELISA with rMmCALR (Figure 5.31). In addition, rOvCALR inhibited
C1q‐mediated hemolysis of sensitized red blood cells (Figure 5.32) as previously
shown for human calreticulin.213
This is the first time to demonstrate the suppression
of human C1q‐mediated hemolysis of sensitized human red blood cells by a trematode
calreticulin. Interestingly, the inhibition effect of rOvCALR was significant stronger
than that of rMmCALR. C1q binding region has been mapped in the N‐ and P‐
domains of calreticulin213
, and the specific binding sites have been identified in
human calreticulin including IESKHKSDF, DEEKDFG, KDIRCKDD and
WDERAKID.182
Some motifs of HsCALR C1q binding sequence are absent or altered
in OvCALR and other trematode orthologs (Figure 5.5), however, the most conserved
motif is KEVRCKDD, which is KDIRCKDD in HsCALR that has been shown to
bind excellently to the globular head of C1q and inhibit complement‐mediated
hemolytic activity.182
Similarly, the absence of some HsCALR C1q binding motifs is
also observed in the H. contortus calreticulin sequence; nevertheless, two additional
binding sites mapped in the N‐domain of HcCALR have been identified.214
In case of
OvCALR, it might carry other unidentified C1q binding sites different from HsCALR
resulting in the stronger hemolytic inhibition. However, a difference in folding quality
of the recombinant proteins cannot be excluded but then again a significant difference
in chaperoning activity was not observed between rOvCALR and rMmCALR (Figure
5.27). Parasite interaction with the host complement is a double‐edged sword; killing
of parasites by complement system has been shown in various parasites including
O. viverrini215,216
but on the other side, mechanisms for host immune evasion and
infectivity have been reported as well.217
Accordingly, human C1q binding and
suppression of classical complement activation through OvCALR might support
parasite survival and decrease the consequences of complement activation including
the membrane‐attack complex (MAC) formation. The ability of parasite calreticulin to
bind to host C1q and inhibit C1q‐mediated hemolysis are found in various parasites
including T. cruzi142
, N. americanus73
, Trichinella spiralis218
indicating a conserved
function for host immune evasion and survival. The binding of calreticulin is not
limited only to the collagen‐like fragments but also takes place at the globular heads
Ref. code: 25605412330010EAN

134
of C1q.219
Hence, the binding of parasite calreticulin to the globular region of C1q
might affect to Fc receptor‐mediated phagocytosis220
as well as apoptotic cell
clearance221,222
of the host. Concerning the effect of C1q on tumor growth, there are
both positive and negative effects on tumor growth that have been reported depending
on the studied type of tumor. However, the presence of C1q in the microenvironment
of solid tumors growth has been reported with independent‐classical pathway
activation.223
This finding supports the upregulation of C1q during O. viverrini
infection induced CCA in hamster.224
The relationship between OvCALR and C1q‐
mediated tumor growth needs to be further investigated as well as any direct effect of
OvCALR on CCA development. Nevertheless, the angiogenic properties of OvCALR
have been investigated in this study. Recombinant OvCALR significantly suppressed
in vitro proliferation, migration, and sprouting of human endothelial cells similar to
rMmCALR but at lesser potency (Figures 5.33, 5.34, and 5.35) suggesting a role of
OvCALR as angiogenesis inhibitor. This finding is supported by the highly conserved
sequence of the peptide vasostatin134
present in the OvCALR sequence as well as in
other trematode orthologs (Figure 5.5).
Calreticulin is a well‐researched multifunctional protein involved in
various physiological and pathological conditions and has been highlighted since it
was found in a wide variety of cells and organisms. This study of calreticulin of
O. viverrini demonstrated the conservation of the basic properties of calreticulin
including chaperoning and calcium‐binding activities. OvCALR was detected in all
developmental stages in the host and found to be widely distributed in the mature
parasite especially in the reproductive organs indicating an important role in parasite
development and reproduction. It could be anticipated that suppression of OvCALR
will affect to parasite development and survival. However, calreticulin deficiency
observed in C. elegans demonstrated only a defect in reproduction in adult worms but
not embryonic lethality203
as reported in calreticulin‐deficient mouse.225
In addition,
compensation by upregulation of other proteins including heat shock protein‐70
family members (hsp70) and disulfide isomerase (PDI) as alternative chaperons was
found in calreticulin-deficient C. elegans.226
Similarly, the embryonic lethality found
in calreticulin‐deficient mouse was caused by cardiac development failure but other
embryonic organs were found to be normal developed suggesting that the lethal effect
Ref. code: 25605412330010EAN

135
resulted from calcium buffering but not chaperoning activity of calreticulin.94,225
The
additional investigation in calreticulin-deficient O. viverrini can benefit our
understanding on its importance for the parasite and eventually provide a new parasite
control strategy. The presence of OvCALR in the parasite ES product indicates that
the role of calreticulin is not limited to the ER as soluble calcium‐binding chaperone.
OvCALR is a parasite‐secreted antigen with potential to modulate the host immune
system through its C1q binding property as well as to intervene host angiogenesis via
its anti‐angiogenic properties. These host‐modulating properties found in OvCALR
might facilitate host immune evasion for their survival and indirectly affect to CCA
development, suppressing tumor growth via angiogenesis inhibition and C1q‐
mediated tumor growth.
Ref. code: 25605412330010EAN

136
CHAPTER 7
CONCLUSIONS
This section concludes molecular, biochemical, and biological properties
of the analyzed Opisthorchis viverrini calreticulin (OvCALR). The major findings are
summarized as follows:
1. In this study, OvCALR has been characterized. The complete coding
sequence of the OvCALR cDNA comprises 1,248 bp and encodes the 415 amino
acids pre‐mature OvCALR. The 399 amino acids mature OvCALR has a calculated
molecular weight of 46.23 kDa and a theoretical isoelectric point of 4.3.
2. The deduced amino acid sequence of OvCALR contains many
characteristics of calreticulin including the conserved calreticulin family signature 1
and 2, a 16‐amino‐acid‐residue signal peptide, an His‐Glu‐Glu‐Leu (HEEL) ER
retention signal, three functional and structural domains (N‐, P‐, and C‐domain), three
conserved cysteine residues, and three each of two tandem repeated motifs.
3. The deduced amino acid sequence of OvCALR has very high
sequence conservation at 96.6% identity to C. sinensis calreticulin and significantly
less to blood fluke calreticulin, S. mansoni (57.5%) and S. japonicum (53.6%), and
mammalian calreticulin, M. musculus (50.7%) and H. sapiens (50.9%), respectively.
4. A multiple amino acid sequence alignment of OvCALR and
calreticulin from other species showed high conservation in the P‐domain and less
conservation at the beginning of the N‐domain and almost all of the C‐domain. The
carbohydrate‐, peptide‐, ERp57‐binding sites involved in chaperoning activity, the
high‐affinity calcium‐binding site, and anti‐angiogenic vasostatin sequence based on
mammalian calreticulin were found highly conserved in the OvCALR sequence.
5. Soluble recombinant OvCALR and MmCALR were produced using a
bacterial system comprising expression vector pET21b(+) and host strain E. coli
Rosetta‐gami(DE3)pLysS and purified by Ni‐NTA affinity chromatography.
6. Mouse polyclonal anti‐rOvCALR antisera were produced and the
obtained antisera showed high antibody level and high specificity against rOvCALR
Ref. code: 25605412330010EAN

137
and rSjGST‐OvCALR C‐domain. Additionally, the antisera did not show cross‐
reactivity with rMmCALR.
7. OvCALR RNA was found in all analyzed developmental stages
including newly excysted, 2‐week‐old, 4‐week‐old juveniles and adult O. viverrini but
at lower abundance in newly excysted juveniles.
8. Native OvCALR was mainly detected as soluble protein and as a
released protein in the excretory/secretory product of the adult parasite.
9. Native OvCALR was found widely distributed in the adult parasite
including tegumental cell bodies, cecal epithelium, gametocytes in testes and ovary,
prostate gland, Mehlis’ gland, vitelline cells, eggs, and lining of seminal vesicle. It
was not detected infiltrating liver tissue of O. viverrini‐infected hamster.
10. Recombinant OvCALR was recognized by O. viverrini‐infected
hamster sera indicating the antigenic property of the protein.
11. Recombinant OvCALR protected citrate synthase from in vitro
thermal‐induced aggregation similar to rMmCALR indicating its chaperoning
property.
12. Recombinant OvCALR C‐domain in fusion with SjGST showed a
mobility shift in native‐PAGE in the presence of calcium indicating its calcium‐
binding property.
13. Recombinant OvCALR did not show transacetylase activity by
indirect GST inhibition with 7‐Acetoxy‐4‐methylcoumarin (7‐AMC) as substrate. In
addition, western blot analysis using anti‐acetylated lysine antibody demonstrated
direct acetylation by 7‐AMC in various proteins.
14. Recombinant OvCALR bound to human C1q in a dose‐dependent
manner and inhibited classical complement pathway‐mediated hemolysis of
sensitized‐human red blood cells similar to rMmCALR.
15. Recombinant OvCALR suppressed proliferation, migration, and
sprouting of human endothelial cells in vitro indicating its anti‐angiogenic properties.
Ref. code: 25605412330010EAN

138
REFERENCES
1. Kaewpitoon N, Kootanavanichpong N, Kompor P, Chavenkun W, Kujapun J,
Norkaew J, et al. Review and current status of Opisthorchis viverrini infection at the
community level in Thailand. Asian Pac J Cancer Prev. 2015;16(16):6825-30. doi:
10.7314/APJCP.2015.16.16.6825. PubMed PMID: 26514452.
2. WHO, International Agency for Research on Cancer Monograph Working
Group. A review of human carcinogens Part B: Biological agents [internet]. IARC:
Lyon (France); 2012 (cited 2018 Mar 13). Available from:
http://monographs.iarc.fr/ENG/Monographs/vol100B/mono100B-13.pdf.
3. Jongsuksuntigul P, Imsomboon T. Opisthorchiasis control in Thailand. Acta
Trop. 2003;88(3):229-32. doi: 10.1016/j.actatropica.2003.01.002. PubMed PMID:
14611877.
4. Anderson CD, Pinson CW, Berlin J, Chari RS. Diagnosis and treatment of
cholangiocarcinoma. Oncologist. 2004;9(1):43-57. doi: 10.1634/theoncologist.9-1-43
PubMed PMID: 14755014.
5. Smout MJ, Laha T, Mulvenna J, Sripa B, Suttiprapa S, Jones A, et al. A
granulin-like growth factor secreted by the carcinogenic liver fluke, Opisthorchis
viverrini, promotes proliferation of host cells. PLoS Pathog. 2009;5(10):e1000611.
doi: 10.1371/journal.ppat.1000611. PubMed PMID: 19816559.
6. Papatpremsiri A, Smout MJ, Loukas A, Brindley PJ, Sripa B, Laha T.
Suppression of Ov-grn-1 encoding granulin of Opisthorchis viverrini inhibits
proliferation of biliary epithelial cells. Exp Parasitol. 2015;148:17-23. doi:
10.1016/j.exppara.2014.11.004. PubMed PMID: 25450776.
7. Keiser J, Utzinger J. Food-borne trematodiases. Clin Microbiol Rev.
2009;22(3):466-83. doi: 10.1128/CMR.00012-09. PubMed PMID: 19597009.
8. Kaewkes S. Taxonomy and biology of liver flukes. Acta Trop.
2003;88(3):177-86. doi: 10.1016/j.actatropica.2003.05.001. PubMed PMID:
14611872.
9. Sripa B, Kaewkes S, Intapan PM, Maleewong W, Brindley PJ. Food-borne
trematodiases in Southeast Asia epidemiology, pathology, clinical manifestation and
Ref. code: 25605412330010EAN

139
control. In: Rollinson D, Stothard JR, editors. Advances in Parasitology. Important
helminth infections in Southeast Asia: diversity and potential for control and
elimination, part A. 72. London: Academic Press; 2010. p. 305-50.
10. Upatham ES. A review of experimental and field research on the human liver
fluke, Opisthorchis viverrini. JSciSocThailand. 1988;14:245-62.
11. Schuster RK. Opisthorchiidosis--a review. Infect Disord Drug Targets.
2010;10(5):402-15. doi: 10.2174/187152610793180902. PubMed PMID: 20701569.
12. Khampoosa P, Jones MK, Lovas EM, Srisawangwong T, Laha T, Piratae S, et
al. Light and electron microscopy observations of embryogenesis and egg
development in the human liver fluke, Opisthorchis viverrini (Platyhelminthes,
Digenea). Parasitol Res. 2012;110(2):799-808. doi: 10.1007/s00436-011-2557-3.
PubMed PMID: 21786067.
13. King S, Scholz T. Trematodes of the family Opisthorchiidae: a minireview.
Korean J Parasitol. 2001;39(3):209-21. doi: 10.3347/kjp.2001.39.3.209. PubMed
PMID: 11590910.
14. Wykoff DE, Harinasuta C, Juttijudata P, Winn MM. Opisthorchis viverrini in
Thailand: the life cycle and comparison with O. felineus. J Parasitol. 1965;51:207-14.
doi: 10.2307/3276083 PubMed PMID: 14275209.
15. Harinasuta C, Harinasuta T. Opisthorchis viverrini: life cycle, intermediate
hosts, transmission to man and geographical distribution in Thailand.
Arzneimittelforschung. 1984;34(9B):1164-7. PubMed PMID: 6542383.
16. Wongsaroja T, Nithikathkulb C, Rojkitikulc W, Nakaia W, Royal L,
Rammasutc P. National survey of helminthiasis in Thailand. Asian Biomedicine.
2014;8(6):779-83. doi: 10.5372/1905-7415.0806.357.
17. Thaewnongiew K, Singthong S, Kutchamart S, Tangsawad S, Promthet S,
Sailugkum S, et al. Prevalence and risk factors for Opisthorchis viverrini infections in
upper Northeast Thailand. Asian Pac J Cancer Prev. 2014;15(16):6609-12. doi:
10.7314/APJCP.2014.15.16.6609. PubMed PMID: 25169496.
18. Saengsawang P, Promthet S, Bradshaw P. Prevalence of OV infection in
Yasothon Province, Northeast Thailand. Asian Pac J Cancer Prev. 2012;13(7):3399-
402. doi: 10.7314/APJCP.2012.13.7.3399. PubMed PMID: 22994767.
Ref. code: 25605412330010EAN

140
19. Rim HJ, Chai JY, Min DY, Cho SY, Eom KS, Hong SJ, et al. Prevalence of
intestinal parasite infections on a national scale among primary schoolchildren in
Laos. Parasitol Res. 2003;91(4):267-72. doi: 10.1007/s00436-003-0963-x. PubMed
PMID: 14574555.
20. Sayasone S, Odermatt P, Phoumindr N, Vongsaravane X, Sensombath V,
Phetsouvanh R, et al. Epidemiology of Opisthorchis viverrini in a rural district of
southern Lao PDR. Trans R Soc Trop Med Hyg. 2007;101(1):40-7. doi:
10.1016/j.trstmh.2006.02.018. PubMed PMID: 16828134.
21. Sayasone S, Mak TK, Vanmany M, Rasphone O, Vounatsou P, Utzinger J, et
al. Helminth and intestinal protozoa infections, multiparasitism and risk factors in
Champasack province, Lao People's Democratic Republic. PLoS Negl Trop Dis.
2011;5(4):e1037. doi: 10.1371/journal.pntd.0001037. PubMed PMID: 21532735.
22. Chai JY, Han ET, Shin EH, Sohn WM, Yong TS, Eom KS, et al. High
prevalence of Haplorchis taichui, Phaneropsolus molenkampi, and other helminth
infections among people in Khammouane province, Lao PDR. Korean J Parasitol.
2009;47(3):243-7. doi: 10.3347/kjp.2009.47.3.243. PubMed PMID: 19724697.
23. Chai JY, Han ET, Guk SM, Shin EH, Sohn WM, Yong TS, et al. High
prevalence of liver and intestinal fluke infections among residents of Savannakhet
Province in Laos. Korean J Parasitol. 2007;45(3):213-8. doi:
10.3347/kjp.2007.45.3.213. PubMed PMID: 17876167.
24. Yoon HJ, Ki M, Eom K, Yong TS, Chai JY, Min DY, et al. Risk factors for
Opisthorchis viverrini and minute intestinal fluke infections in Lao PDR, 2009-2011.
Am J Trop Med Hyg. 2014;91(2):384-8. doi: 10.4269/ajtmh.13-0596. PubMed PMID:
24980495.
25. Sithithaworn P, Andrews RH, Nguyen VD, Wongsaroj T, Sinuon M, Odermatt
P, et al. The current status of opisthorchiasis and clonorchiasis in the Mekong Basin.
Parasitol Int. 2012;61(1):10-6. doi: 10.1016/j.parint.2011.08.014. PubMed PMID:
21893213.
26. Lee KJ, Bae YT, Kim DH, Deung YK, Ryang YS, Kim HJ, et al. Status of
intestinal parasites infection among primary school children in Kampongcham,
Cambodia. Korean J Parasitol. 2002;40(3):153-5. doi: 10.3347/kjp.2002.40.3.153.
PubMed PMID: 12325445.
Ref. code: 25605412330010EAN

141
27. Yong TS, Chai JY, Sohn WM, Eom KS, Jeoung HG, Hoang EH, et al.
Prevalence of intestinal helminths among inhabitants of Cambodia (2006-2011).
Korean J Parasitol. 2014;52(6):661-6. doi: 10.3347/kjp.2014.52.6.661. PubMed
PMID: 25548418.
28. Yong TS, Shin EH, Chai JY, Sohn WM, Eom KS, Lee DM, et al. High
prevalence of Opisthorchis viverrini infection in a riparian population in Takeo
Province, Cambodia. Korean J Parasitol. 2012;50(2):173-6. doi:
10.3347/kjp.2012.50.2.173. PubMed PMID: 22711932.
29. WHO. Review on the epidemiological profile of helminthiases and their
control in the Western Pacific region, 1997-2008 [Internet]. MVP/WPRO, Western
Pacific Regional Office, World Health Organization. 2008 [cited 2018 Mar 13].
Available from: http://www.wpro.who.int/mvp/documents/docs/Helminths10Year
Review_reformattedv2_.pdf?ua=1.
30. Dao TT, Bui TV, Abatih EN, Gabriel S, Nguyen TT, Huynh QH, et al.
Opisthorchis viverrini infections and associated risk factors in a lowland area of Binh
Dinh Province, Central Vietnam. Acta Trop. 2016;157:151-7. doi:
10.1016/j.actatropica.2016.01.029. PubMed PMID: 26872984.
31. Mairiang E, Mairiang P. Clinical manifestation of opisthorchiasis and
treatment. Acta Trop. 2003;88(3):221-7. doi: 10.1016/j.actatropica.2003.03.001.
PubMed PMID: 14611876.
32. Sripa B, Kaewkes S, Sithithaworn P, Mairiang E, Laha T, Smout M, et al.
Liver fluke induces cholangiocarcinoma. PLoS Med. 2007;4(7):e201. doi:
10.1371/journal.pmed.0040201. PubMed PMID: 17622191.
33. Worasith C, Kamamia C, Yakovleva A, Duenngai K, Wangboon C,
Sithithaworn J, et al. Advances in the diagnosis of human opisthorchiasis:
development of Opisthorchis viverrini antigen detection in urine. PLoS Negl Trop
Dis. 2015;9(10):e0004157. doi: 10.1371/journal.pntd.0004157. PubMed PMID:
26485024.
34. Wongratanacheewin S, Sermswan RW, Sirisinha S. Immunology and
molecular biology of Opisthorchis viverrini infection. Acta Trop. 2003;88(3):195-
207. doi: 10.1016/j.actatropica.2003.02.002. PubMed PMID: 14611874.
Ref. code: 25605412330010EAN

142
35. Poopyruchpong N, Viyanant V, Upatham ES, Srivatanakul P. Diagnosis of
opisthorchiasis by enzyme-linked immunosorbent assay using partially purified
antigens. Asian Pac J Allergy Immunol. 1990;8(1):27-31. PubMed PMID: 2393450.
36. Wongsaroj T, Sakolvaree Y, Chaicumpa W, Maleewong W, Kitikoon V,
Tapchaisri P, et al. Affinity purified oval antigen for diagnosis of Opisthorchiasis
viverrini. Asian Pac J Allergy Immunol. 2001;19(4):245-58. PubMed PMID:
12009074.
37. Duenngai K, Sithithaworn P, Rudrappa UK, Iddya K, Laha T, Stensvold CR,
et al. Improvement of PCR for detection of Opisthorchis viverrini DNA in human
stool samples. J Clin Microbiol. 2008;46(1):366-8. doi: 10.1128/JCM.01323-07.
PubMed PMID: 18003810.
38. Le TH, Van De N, Blair D, Sithithaworn P, McManus DP. Clonorchis sinensis
and Opisthorchis viverrini: development of a mitochondrial-based multiplex PCR for
their identification and discrimination. Exp Parasitol. 2006;112(2):109-14. doi:
10.1016/j.exppara.2005.09.012. PubMed PMID: 16310774.
39. Sato M, Thaenkham U, Dekumyoy P, Waikagul J. Discrimination of O.
viverrini, C. sinensis, H. pumilio and H. taichui using nuclear DNA-based PCR
targeting ribosomal DNA ITS regions. Acta Trop. 2009;109(1):81-3. doi:
10.1016/j.actatropica.2008.09.015. PubMed PMID: 18952037.
40. Arimatsu Y, Kaewkes S, Laha T, Hong SJ, Sripa B. Rapid detection of
Opisthorchis viverrini copro-DNA using loop-mediated isothermal amplification
(LAMP). Parasitol Int. 2012;61(1):178-82. doi: 10.1016/j.parint.2011.08.009.
PubMed PMID: 21871581.
41. Arimatsu Y, Kaewkes S, Laha T, Sripa B. Specific diagnosis of Opisthorchis
viverrini using loop-mediated isothermal amplification (LAMP) targeting parasite
microsatellites. Acta Trop. 2015;141(Pt B):368-71. doi:
10.1016/j.actatropica.2014.09.012. PubMed PMID: 25268466.
42. WHO. Foodborne trematode infections Opisthorchiasis [Internet]. WHO. 2018
[cited 2018 Mar 16]. Available from:
www.who.int/foodborne_trematode_infections/opisthorchiasis/Opisthorchiasis_viverr
ini/en/.
Ref. code: 25605412330010EAN

143
43. Sayasone S, Keiser J, Meister I, Vonghachack Y, Xayavong S, Senggnam K,
et al. Efficacy and safety of tribendimidine versus praziquantel against Opisthorchis
viverrini in Laos: an open-label, randomised, non-inferiority, phase 2 trial. Lancet
Infect Dis. 2018;18(2):155-61. doi: 10.1016/S1473-3099(17)30624-2. PubMed
PMID: 29153938.
44. Soukhathammavong P, Odermatt P, Sayasone S, Vonghachack Y, Vounatsou
P, Hatz C, et al. Efficacy and safety of mefloquine, artesunate, mefloquine-artesunate,
tribendimidine, and praziquantel in patients with Opisthorchis viverrini: a
randomised, exploratory, open-label, phase 2 trial. Lancet Infect Dis. 2011;11(2):110-
8. doi: 10.1016/S1473-3099(10)70250-4. PubMed PMID: 21111681.
45. Duthaler U, Sayasone S, Vanobbergen F, Penny MA, Odermatt P, Huwyler J,
et al. Single-ascending-dose pharmacokinetic study of tribendimidine in Opisthorchis
viverrini-infected patients. Antimicrob Agents Chemother. 2016;60(10):5705-15. doi:
10.1128/AAC.00992-16. PubMed PMID: 27431234.
46. Pongnikorn D, Suwanrungruang K, Buasom R. Cancer incidence in Thailand.
In: Imsamran W, Chaiwerawattana A, Wiangnon S, Pongnikorn D, Suwanrungrung
K, Sangrajrang S, et al., editors. Cancer in Thailand VIII, 2010-2012. 8. Bangkok:
New Thammada Press (Thailand) Co., Ltd.; 2015. p. 5-72.
47. Bundhamcharoen K, Odton P, Phulkerd S, Tangcharoensathien V. Burden of
disease in Thailand: changes in health gap between 1999 and 2004. BMC Public
Health. 2011;11:53. doi: 10.1186/1471-2458-11-53. PubMed PMID: 21266087.
48. Attasara P, Sriplung H. Cancer incidence in Thailand. In: Khuhaprema T,
Attasara P, Sriplung H, Wiangnon S, Sangrajrang S, editors. Cancer in Thailand
Volume VII, 2007-2009. Bangkok: Ministry of Public Health; 2013. p. 8-73.
49. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer
statistics. CA Cancer J Clin. 2011;61(2):69-90. doi: 10.3322/caac.20107. PubMed
PMID: 21296855.
50. Bragazzi MC, Cardinale V, Carpino G, Venere R, Semeraro R, Gentile R, et
al. Cholangiocarcinoma: Epidemiology and risk factors. Transl Gastrointest Cancer.
2012;1:21-32. doi: 10.3978/j.issn.2224-4778.2011.11.04. PubMed PMID:
51. Bridgewater J, Galle PR, Khan SA, Llovet JM, Park JW, Patel T, et al.
Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J
Ref. code: 25605412330010EAN

144
Hepatol. 2014;60(6):1268-89. doi: 10.1016/j.jhep.2014.01.021. PubMed PMID:
24681130.
52. IARC. Schistosomes, liver flukes and Helicobacter pylori. IARC Working
Group on the Evaluation of Carcinogenic Risks to Humans. Lyon: The Agency; 1994.
53. Sripa B, Brindley PJ, Mulvenna J, Laha T, Smout MJ, Mairiang E, et al. The
tumorigenic liver fluke Opisthorchis viverrini--multiple pathways to cancer. Trends
Parasitol. 2012;28(10):395-407. doi: 10.1016/j.pt.2012.07.006. PubMed PMID:
22947297.
54. Smout MJ, Sripa B, Laha T, Mulvenna J, Gasser RB, Young ND, et al.
Infection with the carcinogenic human liver fluke, Opisthorchis viverrini. Mol
Biosyst. 2011;7(5):1367-75. doi: 10.1039/c0mb00295j. PubMed PMID: 21311794.
55. Watanapa P, Watanapa WB. Liver fluke-associated cholangiocarcinoma. Br J
Surg. 2002;89(8):962-70. doi: 10.1046/j.1365-2168.2002.02143.x. PubMed PMID:
12153620.
56. Haugen B, Karinshak SE, Mann VH, Popratiloff A, Loukas A, Brindley PJ, et
al. Granulin secreted by the food-borne liver fluke Opisthorchis viverrini promotes
angiogenesis in human endothelial cells. Front Med (Lausanne). 2018;5:30. doi:
10.3389/fmed.2018.00030. PubMed PMID: 29503819.
57. Suttiprapa S, Matchimakul P, Loukas A, Laha T, Wongkham S, Kaewkes S, et
al. Molecular expression and enzymatic characterization of thioredoxin from the
carcinogenic human liver fluke Opisthorchis viverrini. Parasitol Int. 2012;61(1):101-
6. doi: 10.1016/j.parint.2011.06.018. PubMed PMID: 21740981.
58. Matchimakul P, Rinaldi G, Suttiprapa S, Mann VH, Popratiloff A, Laha T, et
al. Apoptosis of cholangiocytes modulated by thioredoxin of carcinogenic liver fluke.
Int J Biochem Cell Biol. 2015;65:72-80. doi: 10.1016/j.biocel.2015.05.014. PubMed
PMID: 26007234.
59. Correia da Costa JM, Vale N, Gouveia MJ, Botelho MC, Sripa B, Santos LL,
et al. Schistosome and liver fluke derived catechol-estrogens and helminth associated
cancers. Front Genet. 2014;5:444. doi: 10.3389/fgene.2014.00444. PubMed PMID:
25566326.
Ref. code: 25605412330010EAN

145
60. Ostwald TJ, MacLennan DH. Isolation of a high affinity calcium-binding
protein from sarcoplasmic reticulum. J Biol Chem. 1974;249(3):974-9. PubMed
PMID: 4272851.
61. Michalak M, Milner RE, Burns K, Opas M. Calreticulin. Biochem J. 1992;285
( Pt 3):681-92. PubMed PMID: 1497605.
62. McCauliffe DP, Lux FA, Lieu TS, Sanz I, Hanke J, Newkirk MM, et al.
Molecular cloning, expression, and chromosome 19 localization of a human Ro/SS-A
autoantigen. J Clin Invest. 1990;85(5):1379-91. doi: 10.1172/JCI114582. PubMed
PMID: 2332496.
63. Smith MJ, Koch GL. Multiple zones in the sequence of calreticulin (CRP55,
calregulin, HACBP), a major calcium binding ER/SR protein. EMBO J.
1989;8(12):3581-6. PubMed PMID: 2583110.
64. Milner RE, Baksh S, Shemanko C, Carpenter MR, Smillie L, Vance JE, et al.
Calreticulin, and not calsequestrin, is the major calcium binding protein of smooth
muscle sarcoplasmic reticulum and liver endoplasmic reticulum. J Biol Chem.
1991;266(11):7155-65. PubMed PMID: 2016321.
65. Collins JH, Xi ZJ, Alderson-Lang BH, Treves S, Volpe P. Sequence homology
of a canine brain calcium-binding protein with calregulin and the human Ro/SS-A
antigen. Biochem Biophys Res Commun. 1989;164(1):575-9. doi: 10.1016/0006-
291X(89)91758-0. PubMed PMID: 2803321.
66. Khanna NC, Tokuda M, Waisman DM. Comparison of calregulins from
vertebrate livers. Biochem J. 1987;242(1):245-51. PubMed PMID: 3593237.
67. Kales S, Fujiki K, Dixon B. Molecular cloning and characterization of
calreticulin from rainbow trout (Oncorhynchus mykiss). Immunogenetics.
2004;55(10):717-23. doi: 10.1007/s00251-003-0631-4. PubMed PMID: 14669059.
68. Treveso S, Zorzato F, Chiozzi P, Melandri P, Volpe P, Pozzan T. Frog brain
expresses a 60 KDa Ca2+ binding protein similar to mammalian calreticulin.
Biochem Biophys Res Commun. 1991;175(2):444-50. doi: 10.1016/0006-
291X(91)91584-Y. PubMed PMID: 2018493.
69. Luana W, Li F, Wang B, Zhang X, Liu Y, Xiang J. Molecular characteristics
and expression analysis of calreticulin in Chinese shrimp Fenneropenaeus chinensis.
Ref. code: 25605412330010EAN

146
Comp Biochem Physiol B Biochem Mol Biol. 2007;147(3):482-91. doi:
10.1016/j.cbpb.2007.03.001. PubMed PMID: 17449312.
70. Kawabe S, Yokoyama Y. Molecular cloning of calnexin and calreticulin in the
Pacific oyster Crassostrea gigas and its expression in response to air exposure. Mar
Genomics. 2010;3(1):19-27. doi: 10.1016/j.margen.2010.01.002. PubMed PMID:
21798193.
71. Wang G, Jiang Z, Zhang M, Yang N, Zhu D. Identification of a new
calreticulin homolog from Yesso scallop (Patinopecten yessoensis) and its role in
innate immunity. Fish Shellfish Immunol. 2016;58:108-15. doi:
10.1016/j.fsi.2016.09.019. PubMed PMID: 27633681.
72. Smith MJ. A C. elegans gene encodes a protein homologous to mammalian
calreticulin. DNA Seq. 1992;2(4):235-40. doi: 10.3109/10425179209020808 PubMed
PMID: 1627827.
73. Kasper G, Brown A, Eberl M, Vallar L, Kieffer N, Berry C, et al. A
calreticulin-like molecule from the human hookworm Necator americanus interacts
with C1q and the cytoplasmic signalling domains of some integrins. Parasite
Immunol. 2001;23(3):141-52. doi: 10.1046/j.1365-3024.2001.00366.x PubMed
PMID: 11240905.
74. Suchitra S, Joshi P. Characterization of Haemonchus contortus calreticulin
suggests its role in feeding and immune evasion by the parasite. Biochim Biophys
Acta. 2005;1722(3):293-303. doi: 10.1016/j.bbagen.2004.12.020. PubMed PMID:
15716049.
75. Rzepecka J, Rausch S, Klotz C, Schnoller C, Kornprobst T, Hagen J, et al.
Calreticulin from the intestinal nematode Heligmosomoides polygyrus is a Th2-
skewing protein and interacts with murine scavenger receptor-A. Mol Immunol.
2009;46(6):1109-19. doi: 10.1016/j.molimm.2008.10.032. PubMed PMID: 19108896.
76. Khalife J, Liu JL, Pierce R, Porchet E, Godin C, Capron A. Characterization
and localization of Schistosoma mansoni calreticulin Sm58. Parasitology. 1994;108 (
Pt 5):527-32. doi: 10.1017/S0031182000077398. PubMed PMID: 8052508.
77. Scott JC, McManus DP. Molecular cloning and functional expression of a
cDNA encoding the major endoplasmic reticulum-associated calcium-binding protein,
Ref. code: 25605412330010EAN

147
calreticulin, from Philippine strain Schistosoma japonicum. Parasitol Int.
1999;48(1):35-46. doi: 10.1016/S1383-5769(98)00039-7. PubMed PMID: 11269324.
78. Mendlovic F, Ostoa-Saloma P, Solis CF, Martinez-Ocana J, Flisser A, Laclette
JP. Cloning, characterization, and functional expression of Taenia solium calreticulin.
J Parasitol. 2004;90(4):891-3. doi: 10.1645/GE-3325RN. PubMed PMID: 15357095.
79. Cabezon C, Cabrera G, Paredes R, Ferreira A, Galanti N. Echinococcus
granulosus calreticulin: molecular characterization and hydatid cyst localization. Mol
Immunol. 2008;45(5):1431-8. doi: 10.1016/j.molimm.2007.08.022. PubMed PMID:
17936905.
80. Smith MJ. Nucleotide sequence of a Drosophila melanogaster gene encoding a
calreticulin homologue. DNA Seq. 1992;3(4):247-50. doi:
10.3109/10425179209034025 PubMed PMID: 1296819.
81. Joshi M, Pogue GP, Duncan RC, Lee NS, Singh NK, Atreya CD, et al.
Isolation and characterization of Leishmania donovani calreticulin gene and its
conservation of the RNA binding activity. Mol Biochem Parasitol. 1996;81(1):53-64.
doi: 10.1016/0166-6851(96)02676-X. PubMed PMID: 8892305.
82. Labriola C, Cazzulo JJ, Parodi AJ. Trypanosoma cruzi calreticulin is a lectin
that binds monoglucosylated oligosaccharides but not protein moieties of
glycoproteins. Mol Biol Cell. 1999;10(5):1381-94. doi: 10.1091/mbc.10.5.1381.
PubMed PMID: 10233151.
83. Jia XY, He LH, Jing RL, Li RZ. Calreticulin: conserved protein and diverse
functions in plants. Physiol Plant. 2009;136(2):127-38. doi: 10.1111/j.1399-
3054.2009.1223.x. PubMed PMID: 19453510.
84. Gold LI, Eggleton P, Sweetwyne MT, Van Duyn LB, Greives MR, Naylor
SM, et al. Calreticulin: non-endoplasmic reticulum functions in physiology and
disease. FASEB J. 2010;24(3):665-83. doi: 10.1096/fj.09-145482. PubMed PMID:
19940256.
85. Michalak M, Corbett EF, Mesaeli N, Nakamura K, Opas M. Calreticulin: one
protein, one gene, many functions. Biochem J. 1999;344 Pt 2:281-92. PubMed PMID:
10567207.
Ref. code: 25605412330010EAN

148
86. NEXTPROT. CALR3-Function: SIB Swiss Institute of Bioinformatics; 2011-
2018 [updated 17 January 2018. Available from:
https://www.nextprot.org/entry/NX_Q96L12/.
87. Persson S, Rosenquist M, Sommarin M. Identification of a novel calreticulin
isoform (Crt2) in human and mouse. Gene. 2002;297(1-2):151-8. doi: 10.1016/S0378-
1119(02)00880-6. PubMed PMID: 12384296.
88. McCauliffe DP, Yang YS, Wilson J, Sontheimer RD, Capra JD. The 5'-
flanking region of the human calreticulin gene shares homology with the human
GRP78, GRP94, and protein disulfide isomerase promoters. J Biol Chem.
1992;267(4):2557-62. PubMed PMID: 1733953.
89. Rooke K, Briquet-Laugier V, Xia YR, Lusis AJ, Doolittle MH. Mapping of
the gene for calreticulin (Calr) to mouse chromosome 8. Mamm Genome.
1997;8(11):870-1. doi: 10.1007/s003359900599. PubMed PMID: 9337407.
90. Waser M, Mesaeli N, Spencer C, Michalak M. Regulation of calreticulin gene
expression by calcium. J Cell Biol. 1997;138(3):547-57. PubMed PMID: 9245785.
91. NEXTPROT. CALR Sequence 2011-2018 [updated 17 January 2018.
Available from: https://www.nextprot.org/entry/NX_P27797/sequence.
92. Matsuoka K, Seta K, Yamakawa Y, Okuyama T, Shinoda T, Isobe T. Covalent
structure of bovine brain calreticulin. Biochem J. 1994;298 ( Pt 2):435-42. doi:
10.1042/bj2980435. PubMed PMID: 8135753.
93. PROSITE. Calreticulin family signatures: SIB Swiss Institute Bioinformatics;
2018 [updated December 2004. Available from:
https://prosite.expasy.org/PDOC00636.
94. Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M. Calreticulin, a multi-
process calcium-buffering chaperone of the endoplasmic reticulum. Biochem J.
2009;417(3):651-66. doi: 10.1042/BJ20081847. PubMed PMID: 19133842.
95. Pike SE, Yao L, Jones KD, Cherney B, Appella E, Sakaguchi K, et al.
Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor
growth. J Exp Med. 1998;188(12):2349-56. PubMed PMID: 9858521.
96. Cai KX, Tse LY, Leung C, Tam PK, Xu R, Sham MH. Suppression of lung
tumor growth and metastasis in mice by adeno-associated virus-mediated expression
Ref. code: 25605412330010EAN

149
of vasostatin. Clin Cancer Res. 2008;14(3):939-49. doi: 10.1158/1078-0432.CCR-07-
1930. PubMed PMID: 18245558.
97. Chouquet A, Paidassi H, Ling WL, Frachet P, Houen G, Arlaud GJ, et al. X-
ray structure of the human calreticulin globular domain reveals a peptide-binding area
and suggests a multi-molecular mechanism. PLoS One. 2011;6(3):e17886. doi:
10.1371/journal.pone.0017886. PubMed PMID: 21423620.
98. Kozlov G, Pocanschi CL, Rosenauer A, Bastos-Aristizabal S, Gorelik A,
Williams DB, et al. Structural basis of carbohydrate recognition by calreticulin. J Biol
Chem. 2010;285(49):38612-20. doi: 10.1074/jbc.M110.168294. PubMed PMID:
20880849.
99. Baksh S, Michalak M. Expression of calreticulin in Escherichia coli and
identification of its Ca2+
binding domains. J Biol Chem. 1991;266(32):21458-65. doi:
PubMed PMID: 1939178.
100. Ellgaard L, Riek R, Herrmann T, Guntert P, Braun D, Helenius A, et al. NMR
structure of the calreticulin P-domain. Proc Natl Acad Sci U S A. 2001;98(6):3133-8.
doi: 10.1073/pnas.051630098. PubMed PMID: 11248044.
101. Raghavan M, Wijeyesakere SJ, Peters LR, Del Cid N. Calreticulin in the
immune system: ins and outs. Trends Immunol. 2013;34(1):13-21. doi:
10.1016/j.it.2012.08.002. PubMed PMID: 22959412.
102. Wang WA, Groenendyk J, Michalak M. Calreticulin signaling in health and
disease. Int J Biochem Cell Biol. 2012;44(6):842-6. doi:
10.1016/j.biocel.2012.02.009.
103. Coppolino MG, Dedhar S. Calreticulin. Int J Biochem Cell Biol.
1998;30(5):553-8. PubMed PMID: 9693955.
104. Araki K, Nagata K. Protein folding and quality control in the ER. Cold Spring
Harb Perspect Biol. 2011;3(11):a007526. doi: 10.1101/cshperspect.a007526. PubMed
PMID: 21875985.
105. Hegde RS, Ploegh HL. Quality and quantity control at the endoplasmic
reticulum. Curr Opin Cell Biol. 2010;22(4):437-46. doi: 10.1016/j.ceb.2010.05.005.
PubMed PMID: 20570125.
106. Gao B, Adhikari R, Howarth M, Nakamura K, Gold MC, Hill AB, et al.
Assembly and antigen-presenting function of MHC class I molecules in cells lacking
Ref. code: 25605412330010EAN

150
the ER chaperone calreticulin. Immunity. 2002;16(1):99-109. doi: 10.1016/S1074-
7613(01)00260-6. PubMed PMID: 11825569.
107. Nauseef WM, McCormick SJ, Clark RA. Calreticulin functions as a molecular
chaperone in the biosynthesis of myeloperoxidase. J Biol Chem. 1995;270(9):4741-7.
doi: 10.1074/jbc.270.9.4741 PubMed PMID: 7876246.
108. Jeffery E, Peters LR, Raghavan M. The polypeptide binding conformation of
calreticulin facilitates its cell-surface expression under conditions of endoplasmic
reticulum stress. J Biol Chem. 2011;286(4):2402-15. doi: 10.1074/jbc.M110.180877.
PubMed PMID: 21075854.
109. Pocanschi CL, Kozlov G, Brockmeier U, Brockmeier A, Williams DB,
Gehring K. Structural and functional relationships between the lectin and arm
domains of calreticulin. J Biol Chem. 2011;286(31):27266-77. doi:
10.1074/jbc.M111.258467. PubMed PMID: 21652723.
110. Molinari M, Eriksson KK, Calanca V, Galli C, Cresswell P, Michalak M, et al.
Contrasting functions of calreticulin and calnexin in glycoprotein folding and ER
quality control. Mol Cell. 2004;13(1):125-35. doi: 10.1016/S1097-2765(03)00494-5.
PubMed PMID: 14731400.
111. Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev
Mol Cell Biol. 2003;4(3):181-91. doi: 10.1038/nrm1052. PubMed PMID: 12612637.
112. Nakamura K, Zuppini A, Arnaudeau S, Lynch J, Ahsan I, Krause R, et al.
Functional specialization of calreticulin domains. J Cell Biol. 2001;154(5):961-72.
doi: 10.1083/jcb.200102073. PubMed PMID: 11524434.
113. Lynch J, Michalak M. Calreticulin is an upstream regulator of calcineurin.
Biochem Biophys Res Commun. 2003;311(4):1173-9. doi:
10.1016/j.bbrc.2003.08.040. PubMed PMID: 14623303.
114. Guo L, Nakamura K, Lynch J, Opas M, Olson EN, Agellon LB, et al. Cardiac-
specific expression of calcineurin reverses embryonic lethality in calreticulin-deficient
mouse. J Biol Chem. 2002;277(52):50776-9. doi: 10.1074/jbc.M209900200. PubMed
PMID: 12377773.
115. Lynch J, Guo L, Gelebart P, Chilibeck K, Xu J, Molkentin JD, et al.
Calreticulin signals upstream of calcineurin and MEF2C in a critical Ca(2+)-
Ref. code: 25605412330010EAN

151
dependent signaling cascade. J Cell Biol. 2005;170(1):37-47. doi:
10.1083/jcb.200412156. PubMed PMID: 15998798.
116. Goudarzi F, Mohammadalipour A, Khodadadi I, Karimi S, Mostoli R,
Bahabadi M, et al. The role of calcium in differentiation of human adipose-derived
stem cells to adipocytes. Mol Biotechnol. 2018;60(4):279-89. doi: 10.1007/s12033-
018-0071-x. PubMed PMID: 29488128.
117. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-
Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic
cells through trans-activation of LRP on the phagocyte. Cell. 2005;123(2):321-34.
doi: 10.1016/j.cell.2005.08.032. PubMed PMID: 16239148.
118. Jo SH, Choi JA, Lim YJ, Lee J, Cho SN, Oh SM, et al. Calreticulin modulates
the intracellular survival of mycobacteria by regulating ER-stress-mediated apoptosis.
Oncotarget. 2017;8(35):58686-98. doi: 10.18632/oncotarget.17419. PubMed PMID:
28938588.
119. Afshar N, Black BE, Paschal BM. Retrotranslocation of the chaperone
calreticulin from the endoplasmic reticulum lumen to the cytosol. Mol Cell Biol.
2005;25(20):8844-53. doi: 10.1128/MCB.25.20.8844-8853.2005. PubMed PMID:
16199864.
120. White TK, Zhu Q, Tanzer ML. Cell surface calreticulin is a putative
mannoside lectin which triggers mouse melanoma cell spreading. J Biol Chem.
1995;270(27):15926-9. doi: 10.1074/jbc.270.27.15926 PubMed PMID: 7608143.
121. Gold LI, Rahman M, Blechman KM, Greives MR, Churgin S, Michaels J, et
al. Overview of the role for calreticulin in the enhancement of wound healing through
multiple biological effects. J Investig Dermatol Symp Proc. 2006;11(1):57-65. doi:
10.1038/sj.jidsymp.5650011. PubMed PMID: 17069011.
122. Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ,
Volkmer J, et al. Calreticulin is the dominant pro-phagocytic signal on multiple
human cancers and is counterbalanced by CD47. Sci Transl Med. 2010;2(63):63ra94.
doi: 10.1126/scitranslmed.3001375. PubMed PMID: 21178137.
123. Stuart GR, Lynch NJ, Day AJ, Schwaeble WJ, Sim RB. The C1q and collectin
binding site within C1q receptor (cell surface calreticulin). Immunopharmacology.
1997;38(1-2):73-80. doi: 10.1016/S0162-3109(97)00076-3. PubMed PMID: 9476117.
Ref. code: 25605412330010EAN

152
124. Nanney LB, Woodrell CD, Greives MR, Cardwell NL, Pollins AC, Bancroft
TA, et al. Calreticulin enhances porcine wound repair by diverse biological effects.
Am J Pathol. 2008;173(3):610-30. doi: 10.2353/ajpath.2008.071027. PubMed PMID:
18753412.
125. Greives MR, Samra F, Pavlides SC, Blechman KM, Naylor SM, Woodrell
CD, et al. Exogenous calreticulin improves diabetic wound healing. Wound Repair
Regen. 2012;20(5):715-30. doi: 10.1111/j.1524-475X.2012.00822.x. PubMed PMID:
22985041.
126. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of
angiogenesis. Nature. 2011;473(7347):298-307. doi: 10.1038/nature10144. PubMed
PMID: 21593862.
127. Adair TH, Montani JP. Angiogenesis. San Rafael (CA): Morgan & Claypool
Life Sciences; 2010 [cited 2018 Mar 13]. Available from:
https://www.ncbi.nlm.nih.gov/books/NBK53242/.
128. Drake CJ. Embryonic and adult vasculogenesis. Birth Defects Res C Embryo
Today. 2003;69(1):73-82. doi: 10.1002/bdrc.10003. PubMed PMID: 12768659.
129. Clapp C, Thebault S, Jeziorski MC, Martinez De La Escalera G. Peptide
hormone regulation of angiogenesis. Physiol Rev. 2009;89(4):1177-215. doi:
10.1152/physrev.00024.2009. PubMed PMID: 19789380.
130. Nyberg P, Xie L, Kalluri R. Endogenous inhibitors of angiogenesis. Cancer
Res. 2005;65(10):3967-79. doi: 10.1158/0008-5472.CAN-04-2427. PubMed PMID:
15899784.
131. Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer.
Vasc Health Risk Manag. 2006;2(3):213-9. doi: 10.2147/vhrm.2006.2.3.21. PubMed
PMID: 17326328.
132. Tosato G, Sgadari C, Taga K, Jones KD, Pike SE, Rosenberg A, et al.
Regression of experimental Burkitt's lymphoma induced by Epstein-Barr virus-
immortalized human B cells. Blood. 1994;83(3):776-84. doi:
10.3109/10428199509107897 PubMed PMID: 8298139.
133. Angiolillo AL, Sgadari C, Sheikh N, Reaman GH, Tosato G. Regression of
experimental human leukemias and solid tumors induced by Epstein-Barr virus-
Ref. code: 25605412330010EAN

153
immortalized B cells. Leuk Lymphoma. 1995;19(3-4):267-76. doi:
10.3109/10428199509107897. PubMed PMID: 8535218.
134. Pike SE, Yao L, Setsuda J, Jones KD, Cherney B, Appella E, et al.
Calreticulin and calreticulin fragments are endothelial cell inhibitors that suppress
tumor growth. Blood. 1999;94(7):2461-8. doi: PubMed PMID: 10498619.
135. Li X, Jiang L, Wang Y, Xiao Y, Huang Y, Yao Q, et al. Inhibition of
angiogenesis by a novel small peptide consisting of the active fragments of platelet
factor-4 and vasostatin. Cancer Lett. 2007;256(1):29-32. doi:
10.1016/j.canlet.2007.05.002. PubMed PMID: 17574330.
136. Yao L, Pike SE, Tosato G. Laminin binding to the calreticulin fragment
vasostatin regulates endothelial cell function. J Leukoc Biol. 2002;71(1):47-53. doi:
10.1189/jlb.71.1.47 PubMed PMID: 11781379.
137. Peng XC, Wang M, Chen XX, Liu J, Xiao GH, Liao HL. Plasmid-encoding
vasostatin inhibited the growth and metastasis of human hepatocellular carcinoma
cells. Mol Cell Biochem. 2014;395(1-2):265-72. doi: 10.1007/s11010-014-2135-y.
PubMed PMID: 24997628.
138. Ding H, Hong C, Wang Y, Liu J, Zhang N, Shen C, et al. Calreticulin
promotes angiogenesis via activating nitric oxide signalling pathway in rheumatoid
arthritis. Clin Exp Immunol. 2014;178(2):236-44. doi: 10.1111/cei.12411. PubMed
PMID: 24988887.
139. Zamanian M, Veerakumarasivam A, Abdullah S, Rosli R. Calreticulin and
cancer. Pathol Oncol Res. 2013;19(2):149-54. doi: 10.1007/s12253-012-9600-2.
PubMed PMID: 23392843.
140. Dunkelberger JR, Song WC. Complement and its role in innate and adaptive
immune responses. Cell Res. 2010;20(1):34-50. doi: 10.1038/cr.2009.139. PubMed
PMID: 20010915.
141. Ramirez G, Valck C, Aguilar L, Kemmerling U, Lopez-Munoz R, Cabrera G,
et al. Roles of Trypanosoma cruzi calreticulin in parasite-host interactions and in
tumor growth. Mol Immunol. 2012;52(3-4):133-40. doi:
10.1016/j.molimm.2012.05.006. PubMed PMID: 22673211.
142. Ferreira V, Valck C, Sanchez G, Gingras A, Tzima S, Molina MC, et al. The
classical activation pathway of the human complement system is specifically inhibited
Ref. code: 25605412330010EAN

154
by calreticulin from Trypanosoma cruzi. J Immunol. 2004;172(5):3042-50. doi:
PubMed PMID: 14978109.
143. Ramirez G, Valck C, Molina MC, Ribeiro CH, Lopez N, Sanchez G, et al.
Trypanosoma cruzi calreticulin: a novel virulence factor that binds complement C1 on
the parasite surface and promotes infectivity. Immunobiology. 2011;216(1-2):265-73.
doi: 10.1016/j.imbio.2010.04.001. PubMed PMID: 20472323.
144. Castillo C, Ramirez G, Valck C, Aguilar L, Maldonado I, Rosas C, et al. The
interaction of classical complement component C1 with parasite and host calreticulin
mediates Trypanosoma cruzi infection of human placenta. PLoS Negl Trop Dis.
2013;7(8):e2376. doi: 10.1371/journal.pntd.0002376. PubMed PMID: 23991234.
145. Ximenez C, Gonzalez E, Nieves ME, Silva-Olivares A, Shibayama M,
Galindo-Gomez S, et al. Entamoeba histolytica and E. dispar calreticulin: inhibition
of classical complement pathway and differences in the level of expression in
amoebic liver abscess. Biomed Res Int. 2014;2014:127453. doi:
10.1155/2014/127453. PubMed PMID: 24860808.
146. Debrabant A, Lee N, Pogue GP, Dwyer DM, Nakhasi HL. Expression of
calreticulin P-domain results in impairment of secretory pathway in Leishmania
donovani and reduced parasite survival in macrophages. Int J Parasitol.
2002;32(11):1423-34. doi: 10.1016/S0020-7519(02)00134-0. PubMed PMID:
12350377.
147. Molina MC, Ferreira V, Valck C, Aguilar L, Orellana J, Rojas A, et al. An in
vivo role for Trypanosoma cruzi calreticulin in antiangiogenesis. Mol Biochem
Parasitol. 2005;140(2):133-40. doi: 10.1016/j.molbiopara.2004.12.014. PubMed
PMID: 15760653.
148. Toledo V, Ramirez G, Valck C, Lopez N, Ribeiro CH, Maldonado I, et al.
Comparative in vivo antiangiogenic effects of calreticulin from Trypanosoma cruzi
and Homo sapiens sapiens. Biol Res. 2010;43(3):287-9. doi: /S0716-
97602010000300004. PubMed PMID: 21249299.
149. Lopez NC, Valck C, Ramirez G, Rodriguez M, Ribeiro C, Orellana J, et al.
Antiangiogenic and antitumor effects of Trypanosoma cruzi Calreticulin. PLoS Negl
Trop Dis. 2010;4(7):e730. doi: 10.1371/journal.pntd.0000730. PubMed PMID:
20625551.
Ref. code: 25605412330010EAN

155
150. Ignacio Arias J, Sepulveda C, Bravo P, Hamilton-West C, Maldonado I,
Ferreira A. Comparative effect of human and Trypanosoma cruzi calreticulin in
wound healing. J Tissue Eng Regen Med. 2015;9(1):41-54. doi: 10.1002/term.1613.
PubMed PMID: 23109509.
151. Marcelain K, Colombo A, Molina MC, Ferreira L, Lorca M, Aguillon JC, et
al. Development of an immunoenzymatic assay for the detection of human antibodies
against Trypanosoma cruzi calreticulin, an immunodominant antigen. Acta Trop.
2000;75(3):291-300. doi: 10.1016/S0001-706X(00)00062-0. PubMed PMID:
10838212.
152. Gonzalez E, Rico G, Mendoza G, Ramos F, Garcia G, Moran P, et al.
Calreticulin-like molecule in trophozoites of Entamoeba histolytica HM1:IMSS
(Swissprot: accession P83003). Am J Trop Med Hyg. 2002;67(6):636-9. doi:
10.4269/ajtmh.2002.67.636. PubMed PMID: 12518855.
153. Lux FA, McCauliffe DP, Buttner DW, Lucius R, Capra JD, Sontheimer RD, et
al. Serological cross-reactivity between a human Ro/SS-A autoantigen (calreticulin)
and the lambda Ral-1 antigen of Onchocerca volvulus. J Clin Invest.
1992;89(6):1945-51. doi: 10.1172/JCI115801. PubMed PMID: 1602002.
154. Rokeach LA, Zimmerman PA, Unnasch TR. Epitopes of the Onchocerca
volvulus RAL1 antigen, a member of the calreticulin family of proteins, recognized by
sera from patients with onchocerciasis. Infect Immun. 1994;62(9):3696-704. PubMed
PMID: 7520419.
155. Pritchard DI, Hooi DS, Brown A, Bockarie MJ, Caddick R, Quinnell RJ.
Basophil competence during hookworm (Necator americanus) infection. Am J Trop
Med Hyg. 2007;77(5):860-5. doi: 10.4269/ajtmh.2007.77.860 PubMed PMID:
17984343.
156. Winter JA, Davies OR, Brown AP, Garnett MC, Stolnik S, Pritchard DI. The
assessment of hookworm calreticulin as a potential vaccine for necatoriasis. Parasite
Immunol. 2005;27(4):139-46. doi: 10.1111/j.1365-3024.2005.00756.x. PubMed
PMID: 15910422.
157. Yadav S, Gupta S, Selvaraj C, Doharey PK, Verma A, Singh SK, et al. In
silico and in vitro studies on the protein-protein interactions between Brugia malayi
Ref. code: 25605412330010EAN

156
immunomodulatory protein calreticulin and human C1q. PLoS One.
2014;9(9):e106413. doi: 10.1371/journal.pone.0106413. PubMed PMID: 25184227.
158. Suchitra S, Anbu KA, Rathore DK, Mahawar M, Singh BP, Joshi P.
Haemonchus contortus calreticulin binds to C-reactive protein of its host, a novel
survival strategy of the parasite. Parasite Immunol. 2008;30(6-7):371-4. doi:
10.1111/j.1365-3024.2008.01028.x. PubMed PMID: 18422872.
159. Mendlovic F, Carrillo-Farga J, Torres J, Laclette JP, Flisser A. Differential
expression of calreticulin in developmental stages of Taenia solium. J Parasitol.
2006;92(4):789-95. doi: 10.1645/GE-724R1.1. PubMed PMID: 16995397.
160. Salazar AM, Mendlovic F, Cruz-Rivera M, Chavez-Talavera O, Sordo M,
Avila G, et al. Genotoxicity induced by Taenia solium and its reduction by
immunization with calreticulin in a hamster model of taeniosis. Environ Mol
Mutagen. 2013;54(5):347-53. doi: 10.1002/em.21782. PubMed PMID: 23704053.
161. Leon-Cabrera S, Cruz-Rivera M, Mendlovic F, Romero-Valdovinos M,
Vaughan G, Salazar AM, et al. Immunological mechanisms involved in the protection
against intestinal taeniosis elicited by oral immunization with Taenia solium
calreticulin. Exp Parasitol. 2012;132(3):334-40. doi: 10.1016/j.exppara.2012.08.006.
PubMed PMID: 22921496.
162. Mendlovic F, Cruz-Rivera M, Diaz-Gandarilla JA, Flores-Torres MA, Avila
G, Perfiliev M, et al. Orally administered Taenia solium Calreticulin prevents
experimental intestinal inflammation and is associated with a type 2 immune
response. PLoS One. 2017;12(10):e0186510. doi: 10.1371/journal.pone.0186510.
PubMed PMID: 29036211.
163. Huggins MC, Gibbs J, Moloney NA. Cloning of a Schistosoma japonicum
gene encoding an antigen with homology to calreticulin. Mol Biochem Parasitol.
1995;71(1):81-7. doi: 10.1016/0166-6851(95)00038-3. PubMed PMID: 7630385.
164. El Gengehi N, El Ridi R, Tawab NA, El Demellawy M, Mangold BL. A
Schistosoma mansoni 62-kDa band is identified as an irradiated vaccine T-cell antigen
and characterized as calreticulin. J Parasitol. 2000;86(5):993-1000. doi:
10.1645/0022-3395(2000)086[0993:ASMKBI]2.0.CO;2. PubMed PMID: 11128523.
Ref. code: 25605412330010EAN

157
165. El Naglaa G, Zada S, El Demellawy MA. Immune response to Schistosoma
mansoni calreticulin in schistosomiasis resistant individuals. Egypt J Immunol.
2004;11(2):37-45. PubMed PMID: 16734116.
166. El Aswad Bel D, Doenhoff MJ, El Hadidi AS, Schwaeble WJ, Lynch NJ. Use
of recombinant calreticulin and cercarial transformation fluid (CTF) in the
serodiagnosis of Schistosoma mansoni. Immunobiology. 2011;216(3):379-85. doi:
10.1016/j.imbio.2010.06.014. PubMed PMID: 20691496.
167. Mutapi F, Bourke C, Harcus Y, Midzi N, Mduluza T, Turner CM, et al.
Differential recognition patterns of Schistosoma haematobium adult worm antigens by
the human antibodies IgA, IgE, IgG1 and IgG4. Parasite Immunol. 2011;33(3):181-
92. doi: 10.1111/j.1365-3024.2010.01270.x. PubMed PMID: 21204849.
168. Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology
Open Software Suite. Trends Genet. 2000;16(6):276-7. doi: 10.1016/S0168-
9525(00)02024-2. PubMed PMID: 10827456.
169. Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating
signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785-6. doi:
10.1038/nmeth.1701. PubMed PMID: 21959131.
170. de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS,
Gasteiger E, et al. ScanProsite: detection of PROSITE signature matches and
ProRule-associated functional and structural residues in proteins. Nucleic Acids Res.
2006;34(Web Server issue):W362-5. doi: 10.1093/nar/gkl124. PubMed PMID:
16845026.
171. Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview
Version 2--a multiple sequence alignment editor and analysis workbench.
Bioinformatics. 2009;25(9):1189-91. doi: 10.1093/bioinformatics/btp033. PubMed
PMID: 19151095.
172. Gouy M, Guindon S, Gascuel O. SeaView version 4: A multiplatform
graphical user interface for sequence alignment and phylogenetic tree building. Mol
Biol Evol. 2010;27(2):221-4. doi: 10.1093/molbev/msp259. PubMed PMID:
19854763.
173. Vichasri-Grams S, Subpipattana P, Sobhon P, Viyanant V, Grams R. An
analysis of the calcium-binding protein 1 of Fasciola gigantica with a comparison to
Ref. code: 25605412330010EAN

158
its homologs in the phylum Platyhelminthes. Mol Biochem Parasitol. 2006;146(1):10-
23. doi: 10.1016/j.molbiopara.2005.10.012. PubMed PMID: 16297461.
174. Saito Y, Ihara Y, Leach MR, Cohen-Doyle MF, Williams DB. Calreticulin
functions in vitro as a molecular chaperone for both glycosylated and non-
glycosylated proteins. EMBO J. 1999;18(23):6718-29. doi:
10.1093/emboj/18.23.6718. PubMed PMID: 10581245.
175. Kumar A, Ponnan P, Raj HG, Parmar VS, Saso L. Comparative specificities of
Calreticulin Transacetylase to O-acetyl, N-acetyl and S-acetyl derivative of 4-
methylcoumarins and their inhibitory effect on AFB1-induced genotoxicity in vitro
and in vivo. Food Chem Toxicol. 2013;52:216-24. doi: 10.1016/j.fct.2012.11.018.
PubMed PMID: 23182742.
176. Singh P, Ponnan P, Krishnan S, Tyagi TK, Priya N, Bansal S, et al. Protein
acyltransferase function of purified calreticulin. Part 1: characterization of
propionylation of protein utilizing propoxycoumarin as the propionyl group donor. J
Biochem. 2010;147(5):625-32. doi: 10.1093/jb/mvq002. PubMed PMID: 20071373.
177. Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and
inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2(2):329-
33. doi: 10.1038/nprot.2007.30. PubMed PMID: 17406593.
178. Arnaoutova I, Kleinman HK. In vitro angiogenesis: endothelial cell tube
formation on gelled basement membrane extract. Nat Protoc. 2010;5(4):628-35. doi:
10.1038/nprot.2010.6. PubMed PMID: 20224563.
179. Young ND, Campbell BE, Hall RS, Jex AR, Cantacessi C, Laha T, et al.
Unlocking the transcriptomes of two carcinogenic parasites, Clonorchis sinensis and
Opisthorchis viverrini. PLoS Negl Trop Dis. 2010;4(6):e719. doi:
10.1371/journal.pntd.0000719. PubMed PMID: 20582164.
180. Lum R, Ahmad S, Hong SJ, Chapman DC, Kozlov G, Williams DB.
Contributions of the lectin and polypeptide binding sites of calreticulin to its
chaperone functions in vitro and in cells. J Biol Chem. 2016;291(37):19631-41. doi:
10.1074/jbc.M116.746321. PubMed PMID: 27413183.
181. Frickel EM, Riek R, Jelesarov I, Helenius A, Wuthrich K, Ellgaard L.
TROSY-NMR reveals interaction between ERp57 and the tip of the calreticulin P-
Ref. code: 25605412330010EAN

159
domain. Proc Natl Acad Sci U S A. 2002;99(4):1954-9. doi:
10.1073/pnas.042699099. PubMed PMID: 11842220.
182. Kovacs H, Campbell ID, Strong P, Johnson S, Ward FJ, Reid KB, et al.
Evidence that C1q binds specifically to CH2-like immunoglobulin gamma motifs
present in the autoantigen calreticulin and interferes with complement activation.
Biochemistry. 1998;37(51):17865-74. doi: 10.1021/bi973197p. PubMed PMID:
9922153.
183. Young ND, Nagarajan N, Lin SJ, Korhonen PK, Jex AR, Hall RS, et al. The
Opisthorchis viverrini genome provides insights into life in the bile duct. Nat
Commun. 2014;5:4378. doi: 10.1038/ncomms5378. PubMed PMID: 25007141.
184. Pekarikova A, Sanchez D, Palova-Jelinkova L, Simsova M, Benes Z,
Hoffmanova I, et al. Calreticulin is a B cell molecular target in some gastrointestinal
malignancies. Clin Exp Immunol. 2010;160(2):215-22. doi: 10.1111/j.1365-
2249.2009.04085.x. PubMed PMID: 20030668.
185. Nakhasi HL, Pogue GP, Duncan RC, Joshi M, Atreya CD, Lee NS, et al.
Implications of calreticulin function in parasite biology. Parasitol Today.
1998;14(4):157-60. doi: 10.1016/S0169-4758(97)01180-0. PubMed PMID:
17040734.
186. Sontheimer RD, Lieu TS, McCauliffe DP. Molecular characterization of the
Ro/SS-A autoimmune response. Semin Dermatol. 1991;10(3):199-205. doi:
10.1038/jid.1993.27. PubMed PMID: 1931569.
187. Kishore U, Sontheimer RD, Sastry KN, Zappi EG, Hughes GR, Khamashta
MA, et al. The systemic lupus erythematosus (SLE) disease autoantigen-calreticulin
can inhibit C1q association with immune complexes. Clin Exp Immunol.
1997;108(2):181-90. doi: 10.1046/j.1365-2249.1997.3761273.x PubMed PMID:
9158084.
188. Jorgensen CS, Hansen KB, Jacobsen S, Halberg P, Ullman S, Hansen D, et al.
Absence of high-affinity calreticulin autoantibodies in patients with systemic
rheumatic diseases and coeliac disease. Scand J Clin Lab Invest. 2005;65(5):403-12.
doi: 10.1080/00365510510013857. PubMed PMID: 16081363.
189. Guillou F, Roger E, Mone Y, Rognon A, Grunau C, Theron A, et al.
Excretory-secretory proteome of larval Schistosoma mansoni and Echinostoma
Ref. code: 25605412330010EAN

160
caproni, two parasites of Biomphalaria glabrata. Mol Biochem Parasitol.
2007;155(1):45-56. doi: 10.1016/j.molbiopara.2007.05.009. PubMed PMID:
17606306.
190. Mendlovic F, Cruz-Rivera M, Avila G, Vaughan G, Flisser A. Cytokine,
antibody and proliferative cellular responses elicited by Taenia solium calreticulin
upon experimental infection in hamsters. PLoS One. 2015;10(3):e0121321. doi:
10.1371/journal.pone.0121321. PubMed PMID: 25811778.
191. Varricchio L, Falchi M, Dall'Ora M, De Benedittis C, Ruggeri A, Uversky
VN, et al. Calreticulin: challenges posed by the intrinsically disordered nature of
calreticulin to the study of its function. Front Cell Dev Biol. 2017;5:96. doi:
10.3389/fcell.2017.00096. PubMed PMID: 29218307.
192. Khalife J, Trottein F, Schacht AM, Godin C, Pierce RJ, Capron A. Cloning of
the gene encoding a Schistosoma mansoni antigen homologous to human Ro/SS-A
autoantigen. Mol Biochem Parasitol. 1993;57(2):193-202. doi: 10.1016/0166-
6851(93)90195-4. PubMed PMID: 8433712.
193. Miquel J, Swiderski Z, Sripa B, Ribas A. Ultrastructural characters of the
spermatozoon of the liver fluke Opisthorchis viverrini (Poirier, 1886)
(Opisthorchiidae). Parasitol Res. 2017;116(9):2499-506. doi: 10.1007/s00436-017-
5559-y. PubMed PMID: 28725936.
194. Apinhasmit W, Sobhon P, Saitongdee P, Menayotin S, Upatham ES.
Opisthorchis viverrini: ultrastructure of the tegument of the first-week juveniles and
adult flukes. Int J Parasitol. 1994;24(5):613-21. doi: 10.1016/0020-7519(94)90113-9.
PubMed PMID: 7928062.
195. Kaewpitoon N, Laha T, Kaewkes S, Yongvanit P, Brindley PJ, Loukas A, et
al. Characterization of cysteine proteases from the carcinogenic liver fluke,
Opisthorchis viverrini. Parasitol Res. 2008;102(4):757-64. doi: 10.1007/s00436-007-
0831-1. PubMed PMID: 18092178.
196. Suttiprapa S, Mulvenna J, Huong NT, Pearson MS, Brindley PJ, Laha T, et al.
Ov-APR-1, an aspartic protease from the carcinogenic liver fluke, Opisthorchis
viverrini: functional expression, immunolocalization and subsite specificity. Int J
Biochem Cell Biol. 2009;41(5):1148-56. doi: 10.1016/j.biocel.2008.10.013. PubMed
PMID: 18996218.
Ref. code: 25605412330010EAN

161
197. Pinlaor P, Kaewpitoon N, Laha T, Sripa B, Kaewkes S, Morales ME, et al.
Cathepsin F cysteine protease of the human liver fluke, Opisthorchis viverrini. PLoS
Negl Trop Dis. 2009;3(3):e398. doi: 10.1371/journal.pntd.0000398. PubMed PMID:
19308250.
198. Nithikathkul C, Tesana S, Sithithaworn P, Balakanich S. Early stage biliary
and intrahepatic migration of Opisthorchis viverrini in the golden hamster. J
Helminthol. 2007;81(1):39-41. doi: 10.1017/S0022149X07212106. PubMed PMID:
17381865.
199. Chaiyadet S, Krueajampa W, Hipkaeo W, Plosan Y, Piratae S, Sotillo J, et al.
Suppression of mRNAs encoding CD63 family tetraspanins from the carcinogenic
liver fluke Opisthorchis viverrini results in distinct tegument phenotypes. Sci Rep.
2017;7(1):14342. doi: 10.1038/s41598-017-13527-5. PubMed PMID: 29084967.
200. Sripa B, Kaewkes S. Localisation of parasite antigens and inflammatory
responses in experimental opisthorchiasis. Int J Parasitol. 2000;30(6):735-40. doi:
10.1016/S0020-7519(00)00054-0. PubMed PMID: 10856508.
201. Chaiyadet S, Sotillo J, Smout M, Cantacessi C, Jones MK, Johnson MS, et al.
Carcinogenic liver fluke secretes extracellular vesicles that promote cholangiocytes to
adopt a tumorigenic phenotype. J Infect Dis. 2015;212(10):1636-45. doi:
10.1093/infdis/jiv291. PubMed PMID: 25985904.
202. Sripa B, Kaewkes S. Relationship between parasite-specific antibody
responses and intensity of Opisthorchis viverrini infection in hamsters. Parasite
Immunol. 2000;22(3):139-45. doi: 10.1046/j.1365-3024.2000.00286.x PubMed
PMID: 10672195.
203. Park BJ, Lee DG, Yu JR, Jung SK, Choi K, Lee J, et al. Calreticulin, a
calcium-binding molecular chaperone, is required for stress response and fertility in
Caenorhabditis elegans. Mol Biol Cell. 2001;12(9):2835-45. doi:
10.1091/mbc.12.9.2835. PubMed PMID: 11553721.
204. Leach MR, Cohen-Doyle MF, Thomas DY, Williams DB. Localization of the
lectin, ERp57 binding, and polypeptide binding sites of calnexin and calreticulin. J
Biol Chem. 2002;277(33):29686-97. doi: 10.1074/jbc.M202405200. PubMed PMID:
12052826.
Ref. code: 25605412330010EAN

162
205. Wijeyesakere SJ, Gafni AA, Raghavan M. Calreticulin is a thermostable
protein with distinct structural responses to different divalent cation environments. J
Biol Chem. 2011;286(11):8771-85. doi: 10.1074/jbc.M110.169193. PubMed PMID:
21177861.
206. Raj HG, Kumari R, Seema, Gupta G, Kumar R, Saluja D, et al. Novel function
of calreticulin: characterization of calreticulin as a transacetylase-mediating protein
acetylator independent of acetyl CoA using polyphenolic acetates. Pure Appl Chem.
2006;78(5):985-92. doi: 10.1351/pac200678050985.
207. Singh P, Ponnan P, Priya N, Tyagi TK, Gaspari M, Krishnan S, et al. Protein
acyltransferase function of purified calreticulin: the exclusive role of P-domain in
mediating protein acylation utilizing acyloxycoumarins and acetyl CoA as the acyl
group donors. Protein Pept Lett. 2011;18(5):507-17. doi:
10.2174/092986611794927938. PubMed PMID: 21235489.
208. Kumari R, Bansal S, Gupta G, Arora S, Kumar A, Goel S, et al. Calreticulin
transacylase: genesis, mechanism of action and biological applications. Biochimie.
2010;92(9):1173-9. doi: 10.1016/j.biochi.2010.01.016. PubMed PMID: 20109516.
209. Raj HG, Parmar VS, Jain SC, Kohli E, Ahmad N, Goel S, et al. Mechanism of
biochemical action of substituted 4-methylbenzopyran-2-ones. Part 7: Assay and
characterization of 7,8-diacetoxy-4-methylcoumarin:protein transacetylase from rat
liver microsomes based on the irreversible inhibition of cytosolic glutathione S-
transferase. Bioorg Med Chem. 2000;8(7):1707-12. doi: 10.1016/S0968-
0896(00)00104-8. PubMed PMID: 10976517.
210. Raj HG, Kohli E, Goswami R, Goel S, Rastogi RC, Jain SC, et al. Mechanism
of biochemical action of substituted benzopyran-2-ones. Part 8: Acetoxycoumarin:
protein transacetylase specificity for aromatic nuclear acetoxy groups in proximity to
the oxygen heteroatom. Bioorg Med Chem. 2001;9(5):1085-9. doi: 10.1016/S0968-
0896(00)00328-X. PubMed PMID: 11377166.
211. Arora S, Vohra P, Kumar A, Tyagi YK, Raj HG, Dawarkanath BS, et al.
Calreticulin transacetylase catalyzed activation of rat tracheal smooth muscle cell
nitric oxide synthase by acetoxycoumarins. Biol Pharm Bull. 2008;31(4):709-13. doi:
10.1248/bpb.31.709 PubMed PMID: 18379067.
Ref. code: 25605412330010EAN

163
212. Singh U, Kumar A, Sinha R, Manral S, Arora S, Ram S, et al. Calreticulin
transacetylase catalyzed modification of the TNF-alpha mediated pathway in the
human peripheral blood mononuclear cells by polyphenolic acetates. Chem Biol
Interact. 2010;185(3):263-70. doi: 10.1016/j.cbi.2010.02.025. PubMed PMID:
20188080.
213. Kishore U, Sontheimer RD, Sastry KN, Zaner KS, Zappi EG, Hughes GR, et
al. Release of calreticulin from neutrophils may alter C1q-mediated immune
functions. Biochem J. 1997;322 ( Pt 2):543-50. PubMed PMID: 9065775.
214. Naresha S, Suryawanshi A, Agarwal M, Singh BP, Joshi P. Mapping the
complement C1q binding site in Haemonchus contortus calreticulin. Mol Biochem
Parasitol. 2009;166(1):42-6. doi: 10.1016/j.molbiopara.2009.02.007. PubMed PMID:
19428671.
215. Sirisinha S, Rattanasiriwilai W, Puengtomwatanakul S, Sobhon P, Saitongdee
P, Koonchornboon T. Complement-mediated killing of Opisthorchis viverrini via
activation of the alternative pathway. Int J Parasitol. 1986;16(4):341-6. doi:
10.1016/0020-7519(86)90112-8. PubMed PMID: 3744673.
216. Marikovsky M, Levi-Schaffer F, Arnon R, Fishelson Z. Schistosoma mansoni:
killing of transformed schistosomula by the alternative pathway of human
complement. Exp Parasitol. 1986;61(1):86-94. doi: 10.1016/0014-4894(86)90138-4.
PubMed PMID: 3943595.
217. Goto H, Sanchez MCA. Does the complement system work for or against the
host during parasite infections. Int Trends Immun. 2013;1(2):11-23. PubMed PMID:
218. Zhao L, Shao S, Chen Y, Sun X, Sun R, Huang J, et al. Trichinella spiralis
Calreticulin Binds Human Complement C1q As an Immune Evasion Strategy. Front
Immunol. 2017;8:636. doi: 10.3389/fimmu.2017.00636. PubMed PMID: 28620388.
219. Paidassi H, Tacnet-Delorme P, Verneret M, Gaboriaud C, Houen G, Duus K,
et al. Investigations on the C1q-calreticulin-phosphatidylserine interactions yield new
insights into apoptotic cell recognition. J Mol Biol. 2011;408(2):277-90. doi:
10.1016/j.jmb.2011.02.029. PubMed PMID: 21352829.
220. Bobak DA, Gaither TA, Frank MM, Tenner AJ. Modulation of FcR function
by complement: subcomponent C1q enhances the phagocytosis of IgG-opsonized
Ref. code: 25605412330010EAN

164
targets by human monocytes and culture-derived macrophages. J Immunol.
1987;138(4):1150-6. PubMed PMID: 3492544.
221. Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B,
Fadok VA, et al. C1q and mannose binding lectin engagement of cell surface
calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp
Med. 2001;194(6):781-95. doi: 10.1084/jem.194.6.781. PubMed PMID: 11560994.
222. Navratil JS, Watkins SC, Wisnieski JJ, Ahearn JM. The globular heads of C1q
specifically recognize surface blebs of apoptotic vascular endothelial cells. J
Immunol. 2001;166(5):3231-9. doi: 10.4049/jimmunol.166.5.3231 PubMed PMID:
11207277.
223. Thielens NM, Tedesco F, Bohlson SS, Gaboriaud C, Tenner AJ. C1q: A fresh
look upon an old molecule. Mol Immunol. 2017;89:73-83. doi:
10.1016/j.molimm.2017.05.025. PubMed PMID: 28601358.
224. Wu Z, Boonmars T, Boonjaraspinyo S, Nagano I, Pinlaor S, Puapairoj A, et al.
Candidate genes involving in tumorigenesis of cholangiocarcinoma induced by
Opisthorchis viverrini infection. Parasitol Res. 2011;109(3):657-73. doi:
10.1007/s00436-011-2298-3. PubMed PMID: 21380578.
225. Mesaeli N, Nakamura K, Zvaritch E, Dickie P, Dziak E, Krause KH, et al.
Calreticulin is essential for cardiac development. J Cell Biol. 1999;144(5):857-68.
doi: 10.1083/jcb.144.5.857 PubMed PMID: 10085286.
226. Lee W, Kim KR, Singaravelu G, Park BJ, Kim DH, Ahnn J, et al. Alternative
chaperone machinery may compensate for calreticulin/calnexin deficiency in
Caenorhabditis elegans. Proteomics. 2006;6(4):1329-39. doi:
10.1002/pmic.200500320. PubMed PMID: 16404716.
Ref. code: 25605412330010EAN

165
APPENDICES
Ref. code: 25605412330010EAN

166
APPENDIX A
SPECIFICATIONS OF DNA VECTORS
1. pGEM®‐T Easy
Ref. code: 25605412330010EAN

167
2. pQE‐30 expression vector
Ref. code: 25605412330010EAN
168
3. pET21b(+) expression vector
Ref. code: 25605412330010EAN

169
Ref. code: 25605412330010EAN

170
APPENDIX B
REAGENT PREPARATIONS
1. Antibiotics stock solution
1.1 Ampicillin stock solution (100 mg/ml)
Dissolve 1 g of ampicillin in 10 ml distilled water, sterilize by
filtration through a 0.22 μm filter, aliquot into 1.5 ml microcentrifuge tubes, and store
at −20°C.
1.2 Chloramphenicol (34 mg/ml)
Dissolve 340 mg of chloramphenicol in 10 ml absolute ethanol,
aliquot into 1.5 ml microcentrifuge tubes, and store at −20°C.
1.3 Kanamycin stock solution (25 mg/ml)
Dissolve 250 mg of kanamycin in 10 ml distilled water, sterilize by
filtration through a 0.22 μm filter, aliquot into 1.5 ml microcentrifuge tubes, and store
at −20°C.
1.4 Tetracycline stock solution (12.5 mg/ml)
Dissolve 125 mg of tetracycline hydrochloride in 10 ml distilled
water, sterilize by filtration through a 0.22 μm filter, aliquot into 1.5 ml
microcentrifuge tubes, and store at −20°C.
2. Media for E. coli bacterial culture
2.1 Luria Bertani (LB) Broth (1,000 ml)
Bacto peptone 10 g
Bacto yeast extract 5 g
NaCl 5 g
Dissolve all components in 1,000 ml distilled water, sterilize the
broth by autoclaving, and store at room temperature.
Ref. code: 25605412330010EAN

171
2.2 LB agar plates
Bacto peptone 10 g
Bacto yeast extract 5 g
NaCl 5 g
Agar 15 g
Dissolve all components in 1,000 ml DW, sterilize the agar by
autoclaving, allow the media to cool down to about 60°C, pour into petri dishes, allow
to harden for several hours at room temperature, and store the plates at 4°C.
3. General buffers/stock solutions
3.1 1 M CaCl2
Dissolve 11 g CaCl2 in ultrapure water to a final volume of 100 ml,
sterilize by autoclaving, and store at room temperature.
3.2 DEPC-treated water
Add 1 ml of DEPC solution to 1000 ml ultrapure water, shake well,
remove CO2 by placing the uncapped bottle in a fume hood for overnight, sterilize the
solution by autoclaving, and store at room temperature.
3.3 0.5 M EDTA
Dissolve 18.6 g EDTA·Na2·2H2O in ultrapure water, adjust the pH
to 8.0 with NaOH, add ultrapure water to complete the volume, sterilize the solution
by autoclaving, and store at room temperature.
3.4 Ethidium bromide
Dissolve 1 g ethidium bromide in 100 ml sterile ultrapure water,
mix vigorously and carefully, and store at room temperature in a dark container.
3.5 2 M Glucose
Dissolve 18 g glucose in 100 ml ultrapure water, sterilize by
filtration through a 0.22 μm membrane filter, aliquot and store at −20°C.
3.6 60% (v/v) Glycerol
Mix 60 ml of glycerol with 40 ml ultrapure water, sterilize the
solution by autoclaving, and store at room temperature.
Ref. code: 25605412330010EAN

172
3.7 1 M IPTG
Dissolve 2.38 g IPTG in 10 ml ultrapure water, sterilize by filtration
through a 0.22 μm membrane filter, aliquot and store at −20°C.
3.8 1 M MgCl2
Dissolve 9.5 g MgCl2 in ultrapure water to a final volume of 100 ml,
sterilize by filtration through a 0.22 μm membrane filter, and store at room
temperature.
3.9 0.85% (w/v) NaCl
Dissolve 8.5 g NaCl in 1,000 ml distilled water, sterilize the
solution by autoclaving, and store at room temperature.
3.10 5 M NaCl
Dissolve 29.2 g NaCl in 100 ml ultrapure water, sterilize the
solution by autoclaving, and store at room temperature.
3.11 5 N NaOH
Dissolve 20 g NaOH pellets in 100 ml sterile ultrapure water, and
store at room temperature.
3.12 10 mM Phosphate buffered saline (PBS) (10X)
Per liter:
Na2HPO4 14.4 g
KH2PO4 2.4 g
NaCl 80 g
KCl 2 g
Dissolve all components in 800 ml ultrapure water, adjust the pH to
7.2–7.4 by adding HCl, adjust the volume to 1,000 ml with ultrapure water, sterilize
the solution by autoclaving, and store at room temperature.
3.13 20% (w/v) SDS
Dissolve 20 g SDS in 100 ml sterile ultrapure water, and store at
room temperature.
3.14 Tris-buffered saline (10X TBS)
0.2 M Tris-base
1.5 M NaCl
Ref. code: 25605412330010EAN

173
Dissolve all components in ultrapure water, adjust the pH to 7.5
with HCl, sterilize the solution by autoclaving, and store at room temperature.
3.15 1 M Tris-HCl
Dissolve 12.114 g Tris-base in 80 ml ultrapure water, adjust the pH
as required with HCl, adjust volume to 100 ml, sterilize the solution by autoclaving,
and store at room temperature.
4. Reagents for agarose gel electrophoresis
4.1 DNA/RNA agarose gel loading buffer (10X)
50% (v/v) Glycerol
0.25% (w/v) Bromophenol blue
0.25% (w/v) Xylene cyanol FF
Mix all components in sterilized ultrapure water, aliquot into 1.5 ml
microcentrifuge tubes, and store at 4°C. In case of RNA, prepare by using DEPC‐
treated water.
4.2 Tris‐Boric EDTA (TBE) buffer stock (5X)
Per liter:
Tris-base 52 g
Boric acid 27.5 g
Disodium EDTA·2H2O 4.56 g
Dissolve all components in 800 ml distilled water, adjust the pH to
8.0 by adding HCl, adjust the volume to 1,000 ml, and store at room temperature.
4.3 Tris-Boric EDTA (TBE) running buffer (0.5X)
Dilute 100 ml of 5X TBE buffer with 900 ml distilled water, add 50
μl ethidium bromide stock solution, mix well, and store at room temperature.
Ref. code: 25605412330010EAN

174
5. Reagents for ELISA
5.1 Antibody diluents
5.1.1 0.25% (w/v) BSA in 10 mM PBS, pH 7.2
Dissolve 25 mg BSA in 10 ml of 10 mM PBS,pH 7.2, and
store at 4°C.
5.1.2 0.5 % (w/v) skim milk in 10 mM PBS, pH 7.2
Dissolve 50 mg BSA in 10 ml of 10 mM PBS,pH 7.2, and
store at 4°C.
5.2 Blocking solutions
5.2.1 0.25% (w/v) BSA in carbonate coating buffer, pH 9.6
Dissolve 50 mg BSA in 20 ml of carbonate coating buffer,
and keep at 4°C until use.
5.2.2 0.5 % (w/v) skim milk in 10 mM PBS, pH 7.2
Dissolve 100 mg skim milk in 20 ml of 10 mM PBS, pH 7.2,
stir until homogenous, and keep at 4°C until use.
Ref. code: 25605412330010EAN

175
5.3 Carbonate coating buffer
30 mM Na2CO3
75 mM NaHCO3
Dissolve all components in ultrapure water, adjust the pH to 9.6,
sterilize the solution by autoclaving, and store at 4°C.
5.4 Washing buffer (prepare fresh)
10 mM PBS, pH 7.2 999.5 ml
Tween® 20 0.5 ml
6. Reagents for GST‐tagged protein purification
6.1 Extraction/loading buffer pH 7.5
10 mM NaH2PO4
1.8 mM KH2PO4
140 mM NaCl
Dissolve all components in ultrapure water, adjust the pH to 7.5,
sterilize the solution by autoclaving, and store at room temperature.
6.2 Elution buffer pH 8.0 (prepare fresh)
33 mM reduced glutathione
50 mM Tris‐HCl
Dissolve 100 mg of reduced glutathione in 10 ml of 50 mM Tris-
Base, pH 10.23, adjust the pH to 8.0, and store at 4°C or on ice.
7. Reagents for immunohistochemistry
7.1 Antibody diluent (1% BSA in PBS)
Dissolve 100 mg BSA in 10 ml of 10 mM PBS, pH 7.2, and store at
4°C.
7.2 Blocking solution (4% BSA in PBS)
Dissolve 400 mg BSA in 10 ml of 10 mM PBS, pH 7.2, and store at
4°C.
Ref. code: 25605412330010EAN

176
7.3 Epitope retrieval buffers
7.3.1 Sodium citrate buffer
10 mM sodium citrate
0.05% (v/v) Tween® 20
Dissolve sodium citrate in sterile ultrapure water, adjust the
pH to 6.0, add Tween® 20, and mix well.
7.3.2 Tris‐EDTA buffer
10mM Tris‐Base
1mM EDTA
0.05% (v/v) Tween® 20
Dissolve Tris‐Base in sterile ultrapure water, add EDTA
(from 0.5 M EDTA, pH 8.0 stock solution), adjust the pH to 9.0, add Tween® 20, and
mix well.
7.4 Fixing solution (4% paraformaldehyde)
Dissolve 4 g of paraformaldehyde in 10 ml 0.1 M PBS, pH 7.2 and
70 ml ultrapure water by adding 1 drop of 2 N NaOH and heating at 65°C, adjust
volume to 100 ml by adding ultrapure water, and store at 4°C.
7.5 Gelatin solution (glass‐slide coating solution)
0.3% (w/v) Gelatin
0.05% (w/v) Chromium potassium sulfate
Add all components to sterile ultrapure water, stir and heat the
solution at 60°C until all components are dissolved, and cool down to room
temperature before use.
7.6 Glycine blocking solution (0.1% glycine in PBS)
Dissolve 100 mg glycine in 100 ml of 10 mM PBS, pH 7.2, and
store at room temperature.
7.7 Mounting medium (10% glycerol in PBS)
Glycerol 10 ml
10X PBS, pH 7.2 10 ml
Distilled water 80 ml
Mix all above solutions, sterile the solution by autoclaving, and
store at room temperature.
Ref. code: 25605412330010EAN

177
7.8 Washing buffer (prepare fresh)
10 mM PBS, pH 7.2 999 ml
Tween® 20 1 ml
8. Reagents for Native‐PAGE
8.1 Native electrophoresis buffer
25 mM Tris‐base
202 mM glycine
Dissolve all components in sterile distilled water, and store at room
temperature.
8.2 Native sample electrophoresis buffer (2X)
120 mM Tris‐HCl, pH 6.8
0.05% (w/v) Bromophenol Blue
01% (w/v) DTT
20% (v/v) glycerol
Dissolve all components in sterile ultrapure water, add glycerol as
the last component, aliquot into 1.5 ml microcentrifuge tubes, and store at −20°C.
9. Reagents for crude worm extraction
9.1 Soluble crude worm extraction buffer
10 mM PBS, pH 7.2
150 mM NaCl
1 mM EDTA
1 mM PMSF
0.5% (v/v) Triton X-100
Mix the first three components as stock solutions in sterile ultrapure
water, mix well, and add PMSF solution and Triton X-100 before use.
Ref. code: 25605412330010EAN

178
9.2 Insoluble crude worm extraction buffer
50 mM Tris‐HCl, pH 8.0
3% (w/v) SDS
Dissolve Tris‐Base in sterile ultrapure water, adjust the pH to 8.0,
add SDS solution (from 20% [w/v] SDS stock solution) to a final concentration of
3%, and mix well.
10. Reagents for plasmid DNA isolation (quick preparation)
10.1 Solution I
25 mM Tris-base
50 mM glucose
10 mM EDTA
Dissolve all components in ultrapure water, adjust the pH to 8.0,
sterilize the solution by autoclaving, and store at room temperature.
10.2 Solution II (prepare fresh before use)
0.1 N NaOH
1% (w/v) SDS
Dilute NaOH from stock solution (5 N NaOH) to a final
concentration of 0.1 N with sterile ultrapure water, and then add SDS from stock
solution (20% [w/v] SDS) stock solution to a final concentration of 1%.
10.3 Solution III
2.7 M potassium acetate, pH 4.8
Dissolve potassium acetate in ultrapure water, adjust the pH to 4.8
by glacial acetic acid, sterilize the solution by autoclaving, and store at room
temperature.
11. Reagents for protein purification under denaturing conditions
11.1 Denaturing lysis buffer
100 mM NaH2PO4
10 mM Tris-base
Ref. code: 25605412330010EAN

179
8 M Urea
Dissolve all components in ultrapure water, adjust the pH to 8.0,
and store at room temperature.
11.2 Lysis buffer
100 mM NaH2PO4
10 mM Tris-base
8 M Urea
Dissolve all components in sterile ultrapure water, adjust the pH to
8.0 by using NaOH, and store at room temperature.
11.3 Washing buffer
100 mM NaH2PO4
10 mM Tris-base
8 M Urea
Dissolve all components in sterile ultrapure water, adjust the pH to
6.3 by using NaOH, and store at room temperature.
11.4 Elution buffer I
100 mM NaH2PO4
10 mM Tris-base
8 M Urea
Dissolve all components in sterile ultrapure water, adjust the pH to
5.9 by using NaOH, and store at room temperature.
11.5 Elution buffer II
100 mM NaH2PO4
10 mM Tris-base
8 M Urea
Dissolve all components in sterile ultrapure water, adjust the pH to
4.5 by using HCl, and store at room temperature.
12. Reagent for protein purification under native conditions
12.1 Native lysis buffer
50 mM NaH2PO4
Ref. code: 25605412330010EAN

180
300 mM NaCl
10 mM Imidazole
Dissolve all components in sterile ultrapure water, adjust the pH to
8.0, and store at room temperature.
12.2 Washing buffer
50 mM NaH2PO4
300 mM NaCl
20 mM Imidazole
Dissolve all components in sterile ultrapure water, adjust the pH to
8.0, and store at room temperature.
12.3 Elution buffer
50 mM NaH2PO4
300 mM NaCl
250 mM Imidazole
Dissolve all components in sterile ultrapure water, adjust the pH to
8.0, and store at room temperature.
13. Reagents for SDS-PAGE
13.1 10%(w/v) Ammonium persulfate
Dissolve 10 g ammonium persulfate in a total volume of 100 ml
sterile ultrapure water, aliquot into 1.5 ml microcentrifuge tubes, and store at −20°C.
13.2 Coomassie blue staining solution
0.025% (w/v) Coomassie Blue R-250
40% (v/v) Methanol
7% (v/v) Acetic acid
Dissolve Coomassie Blue R-250 dye in distilled water and
methanol, finally add acetic acid, store at room temperature.
13.3 Electrophoresis buffer
25 mM Tris‐base
202 mM glycine
0.1% (w/v) SDS
Ref. code: 25605412330010EAN

181
Dissolve all components in sterile distilled water, and store at room
temperature.
13.4 High‐methanol destaining solution
40% (v/v) Methanol
7% (v/v) Acetic acid
Dilute absolute methanol in distilled water, finally add acetic acid,
and store at room temperature.
13.5 Low‐methanol destaining solution
5% (v/v) Methanol
7% (v/v) Acetic acid
Dilute absolute methanol in distilled water, finally add acetic acid,
and store at room temperature.
13.6 Sample electrophoresis buffer (2X)
0.125 M Tris-HCl, pH 6.8
4% (w/v) SDS
0.2 M DTT
0.02% (w/v) Bromophenol blue
20% (v/v) Glycerol
Dissolve all components in sterile ultrapure water, add glycerol as
the last component, aliquot into 1.5 ml microcentrifuge tubes, and store at −20°C.
14. Reagent for transacetylase activity
14.1 0.25 M Potassium phosphate buffer, pH 6.5
KH2PO4 6.315 g
K2HPO4 2.795 g
Dissolve all the components in 500 ml ultrapure water, adjust the
pH to 6.5, sterilize the solution by autoclaving, and store at room temperature.
Ref. code: 25605412330010EAN

182
15. Reagents for Western blot analysis
15.1 Antibody diluent (1% skim milk in TBS)
Dissolve 100 mg skim milk in 10 ml TBS, pH 7.5, stir the solution
until homogenous, and keep at 4°C until use.
15.2 Blocking solution (5% skim milk in TBS)
Dissolve 500 mg skim milk in 10 ml TBS, pH 7.5, stir the solution
until homogenous, and keep at 4°C until use.
15.3 Detection buffer, pH 9.5
0.1 M Tris‐HCl
0.1 M NaCl
50 mM MgCl2
Mix all above stock solutions in sterilized ultrapure water, adjust
the pH to 9.5 with NaOH, store at room temperature.
15.4 Ponceau S dye solution
0.1% (w/v) Ponceau S dye
5% (v/v) Acetic acid
Dissolve Ponceau S dye in sterile distilled water, add acetic acid to
complete the volume, and store at room temperature.
15.5 Semi-dry transfer buffer
50 mM Tris-base
40 mM Glycine
0.04% (w/v) SDS
20% (v/v) Methanol
Dissolve all powder components in sterile ultrapure water, add
methanol to complete the volume, and store at 4°C in a tight container.
15.6 Washing buffer (prepare fresh)
10 mM PBS, pH 7.2 999.5 ml
Tween® 20 0.5 ml
Ref. code: 25605412330010EAN

183
APPENDIX C
ANIMAL ETHICAL PERMISSION
Ref. code: 25605412330010EAN

184
APPENDIX D
HUMAN ETHICAL PERMISSION
Ref. code: 25605412330010EAN

185
BIOGRAPHY
Name Miss Wanlapa Chaibangyang
Date of birth October 28, 1984
Educational attainment
2003–2006 Bachelor of Science (B.Sc.) in Medical Technology
with Second class honors, Faculty of Allied Health
Sciences, Thammasat University
2011–2017 Doctor of Philosophy (Ph.D.) in Biomedical Sciences,
Graduate Program in Biomedical Sciences,
Faculty of Allied Health Sciences,
Thammasat University
Scholarship The Royal Golden Jubilee Ph.D. Scholarship (13th
)
under the supervision of Assoc. Prof. Dr. Hans Rudi
Grams from the Thailand Research Fund and
Thammasat University
Publication
1. Chaibangyang W, Geadkaew-Krenc A, Vichasri-Grams S, Tesana S, Grams R.
Molecular and Biochemical Characterization of Opisthorchis viverrini Calreticulin.
Korean J Parasitol. 2017;55(6):643-52. doi: 10.3347/kjp.2017.55.6.643. PubMed
PMID: 29320819.
Work Experience 2007–2011: Medical Technologist, Mahachai
Hospital, Samutsakhon, Thailand
License and Training attended
1. Undergraduate research trainee at the Protein‐Ligand Engineering and Molecular
Biology Research Laboratory, National Center for Genetic Engineering and
Ref. code: 25605412330010EAN

186
Biotechnology (BIOTEC), National Science and Technology Development Agency
(NSTDA), Pathumthani, Thailand (2006)
2. Student trainee (Medical Technology) in diagnostic laboratory at Thammasat
Hospital, Pathumthani, Thailand (2006)
3. Student trainee (Medical Technology) in diagnostic laboratory at Vajira Hospital,
Bangkok, Thailand (2007)
4. License on Medical Technologist from The Medical Technology Council of
Thailand (2007–present)
5. Human Ethics Training, March 21st, 2014, Faculty of Allied Health Sciences,
Thammasat University, Pathumthani, Thailand
6. Animal Ethics Training, June 9th
, 2014, Faculty of Allied Health Sciences,
Thammasat University, Pathumthani, Thailand
7. Training in Ethical Principles and Guidelines for the Use of Animals for
Scientific Purposes, January 20th
, 2015, Thammasat University, Pathumthani,
Thailand
8. Biosafety training, July 15th
, 2015, Thammasat University, Pathumthani,
Thailand
9. Genomic Epidemiology Workshop in Asian Institute in Statistical Genetics and
Genomics (1st Asian Workshop), December 18
th, 2017, Faculty of Allied Health
Sciences, Thammasat University, Pathumthani, Thailand
10. Informatics for RNA‐seq analysis Workshop in Asian Institute in Statistical
Genetics and Genomics (1st Asian Workshop), December 19
th, 2017, Faculty of Allied
Health Sciences, Thammasat University, Pathumthani, Thailand
Seminar and Conferences attended
1. The 3rd
ASEAN Federation of Hematology Congress (AFH 2014), October 23rd
–
25th
, 2014, the Centara Grand and Bangkok Convention Centre at Centralworld,
Bangkok, Thailand
2. The 1st International Allied Health Sciences Conference, November 4
th–6
th, 2014,
Rama Gardens Hotel, Bangkok, Thailand
Ref. code: 25605412330010EAN

187
3. The 2nd
Joint Symposium of BK21‐PLUS of CUK and Thammasat University,
January 21st–23
rd, 2015, Thammasat University, Pathumthani, Thailand
4. The Joint Symposium of Universiti Kebangsaan Malaysia and Thammasat
University 2015, May 27th
–28th
, 2015, Universiti Kebangsaan Malaysia, Kuala
Lumpur, Malaysia
5. RMIT Animal Ethics Seminar, October 28th
, 2016, RMIT University (Bundoora
Campus), Victoria, Australia
6. The Royal Golden Jubilee‐Ph.D. Congress 18, June 8th
–10th
, 2017, Richmond
Stylish Convention Hotel, Nonthaburi, Thailand
Poster Presentation
1. Chaibangyang W., Geadkaew A.,Grams R., Molecular Cloning of Calreticulin
from the Human Liver Fluke Opisthorchis viverrini, November 4th
–6th
, 2014, The 1st
International Allied Health Sciences Conference, Rama Gardens Hotel, Bangkok,
Thailand
Oral Presentations
1. Chaibangyang W., Geadkaew A., Grams R., Characterization of calreticulin from
the human liver fluke Opisthorchis viverrini, The Joint Symposium of Universiti
Kebangsaan Malaysia and Thammasat University 2015, May 27th
–28th
, 2015,UKM
,Kuala Lumpur, Malaysia
2. Chaibangyang W., Geadkaew A., Grams R., Molecular and biochemical
characterization of calreticulin from the human liver fluke, Opisthorchis viverrini,
The Royal Golden Jubilee‐Ph.D. Congress 18, June 8th
–10th
, 2017, Richmond Stylish
Convention Hotel, Nonthaburi, Thailand
Ref. code: 25605412330010EAN




















