
Primary Culture and Characterization of Human Renal Inner Medullary Collecting Duct Epithelial Cells
Upload
independentCategory
view
1download
0
THYROID CANCER AND NODULES
Molecular Analysis of the RET Proto-Oncogene Key Exonsin Patients with Medullary Thyroid Carcinoma:
A Comprehensive Study of the Iranian Population
Ehsan Alvandi,1 Seyed Mohammad Akrami,1 Mohsen Chiani,2,* Mehdi Hedayati,3 Babak Noori Nayer,2,{
Mohammad Reza Mohajeri Tehrani,4 Manouchehr Nakhjavani,5 and Mehrdad Pedram1
Background: Germ-line mutations of RET proto-oncogene are the known cause of hereditary medullary thyroidcarcinoma (MTC), which account for approximately 25% of all MTC cases and occur as multiple endocrineneoplasia type 2 syndromes. Here, we present the first comprehensive genetic screening and analysis of MTCamong Iranian families.Methods: A total of 55 patients with MTC (male to female ratio¼ 1:1.6; average age of disease onset¼ 33� 13years) from 53 independent families participated in this study. All of the patients had undergone total thy-roidectomy between 1999 and 2006, and 51 of them were clinically characterized as apparently sporadic cases.Genomic DNA samples were obtained and following highly-specific polymerase chain reaction amplification ofthe 6 RET key exons (10, 11, 13, 14, 15, and 16) were subjected to direct DNA sequencing without a requirementfor a purification step.Results: Sequence analysis revealed that 9 (17.6%) of the apparently sporadic cases (from 8 kindreds) carried an RETgerm-line mutation. Of the seven different mutations identified among all of the families studied, five were in thecysteine codons, with Cys634Arg having the highest prevalence (45.5%) among the afflicted families. Mutationcarriers have an earlier age of onset (21� 6) versus the sporadic cases (37� 12).Conclusions: This is the first comprehensive genetic screening and analysis of MTC among Iranian families. Theresults further confirm the need and advantages of DNA sequencing for identification of hereditary MTC cases.There does not seem to be a meaningful correlation between single nucleotide polymorphism patterns and theaverage age of disease onset. Geographical distribution of the sporadic cases, however, shows a significantconcentration toward the Northern regions of the country, noticeably the provinces situated directly to the southof the Caspian Sea.
Introduction
Medullary thyroid carcinoma (MTC), which ac-counts for 4%–10% of all thyroid cancers, stems from
parafollicular calcitonin-producing (C) cells. About 25% ofall MTC cases are hereditary and occur as multiple endo-crine neoplasia type 2 (MEN2) syndromes with an auto-somal dominant mode of inheritance (1,2). Based on theinvolvement of the type of tissues and clinical symptoms,MEN2 syndromes are classified into three major variants:
MEN2B, which is the most aggressive type (characterizedby MTC and pheochromocytoma along with develop-mental disorders such as marfanoid habitus, mucosal andintestinal ganglioneuromatosis) but accounts for only 5%of cases; MEN2A, which accounts for the vast majority(85%) of cases and is characterized by MTC, pheochro-mocytoma, and hyperparathyroidism; and familial MTC(FMTC) that could be considered as a ‘‘clinical variant’’ oran extremely mild version of MEN2A characterized byMTC alone (1–3).
1Department of Medical Genetics, School of Medicine, Tehran University of Medical Sciences (TUMS), Tehran, Iran.2Research Center for Gastroenterology and Liver Diseases and 3Research Institute for Endocrine Sciences, Shahid Beheshti University
of Medical Sciences, Tehran, Iran.4Endocrinology and Metabolism Research Center, TUMS, Tehran, Iran.5Endocrinology and Metabolism Research Center, Vali-Asr Hospital, Tehran, Iran.*Present address: Pilot Biotechnology Department, Pasteur Institute of Iran, Tehran, Iran.{Present address: Department of Gastrology and Hepatology, Mehrad General Hospital, Tehran, Iran.
THYROIDVolume 00, Number 00, 2011ª Mary Ann Liebert, Inc.DOI: 10.1089/thy.2010.0267
1

Germ-line missense mutations of RET proto-oncogene arethe known cause of all hereditary MEN2 variants (3,4). RETgene (RET), which is located on chromosome 10 near thecentromere (10q11.2), encodes a transmemebrane receptortyrosine kinase protein that is primarily expressed in neuralcrest and urogenital precursor cells. RET molecule contains anextracelluar, a transmembrane, and two intracellular tyrosinekinase domains. Traditionally, elevated serum calcitoninlevels, both basal and following pentagastrin stimulation,were used as an indicator for C-cell hyperfunction andMEN2 carrier status. However, molecular genetic studieshave provided a reliable method for identification of RETmutations and established a strong correlation between thegenotypes and MEN2 phenotypes (2,5–7). Mutations in ex-ons 10 and 11 (e.g., Cys620Tyr and Cys634Arg), affecting thecysteine-residue encoding codons within RET extra-cellulardomain, play a major role in causing MEN2A. These muta-tions often lead to a gain-of-function in RET tyrosine kinasesignaling as a result of auto-dimerization of two RET mole-cules. Some other mutations in exons 10 and 11 as well asmutations in exons 13, 14, and 15 (which encode the RETintra-cellular domain) could lead to FMTC. By contrast,MEN2B is almost exclusively related to Met918Thr mutationin exon 16 that also leads to a gain-of-function as a result ofsubstrate alteration (8–13). Therefore, analysis of the 6 RETkey exons (no. 10, 11, 13, 14, 15, and 16) is a very reliablemethod in order to differentiate hereditary MTC cases fromsporadic ones (14).
Before the present work, the available published data onthe rate, distribution, and mutation analysis of MTC amongthe Iranian population were very limited. In a retrospectivestudy, Larijani and colleagues analyzed the medical records offive major tertiary referral centers in the Iranian capital,Tehran. The statistical analysis of 1,177 thyroid carcinomacases, from a 14-year period (1980–1994), showed that only 42patients (3.6%) were diagnosed as MTC, with a female to maleratio of 1:1.1 (15). The molecular study of patients with MTCin Iran has been limited to a report by Hedayati et al., in whichexamination of RET exons 10 and 11 in 57 MTC cases (with afemale to male ratio of 1.2:1) by polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) led toidentification of only four germ-line mutation carriers (16).
Here, we present the first comprehensive molecular anal-ysis of patients with MTC among the Iranian population.After a thorough genetic counseling phase, a total of 55 pa-tients with MTC from 53 independent kindreds were exam-ined by direct DNA sequencing of all 6 RET key exons:10, 11,13, 14, 15, and 16. This led to a reliable strategy for identifi-cation of MTC germ-line mutation carriers, exact genotypingof all index patients, and a prelude to molecular screening oftheir first-degree relatives. Molecular characterization of theasymptomatic germ-line mutation carriers provided a highlysignificant preventive opportunity by their timely referral forprophylactic thyroidectomy.
Materials and Methods
Patients
Fifty-five patients with MTC from 53 independent familieswere referred from three main specialized medical centers inthe capital city, Tehran (TUMS Endocrinology and Metabo-lism Research Center, EMRC, at Shariati Hospital; Shahid
Beheshti University’s Endocrine Research Center at TaleghaniHospital; and Endocrinology and Metabolism ResearchCenter at Vali-Asr Hospital), which deal with most MTC casesin Iran. These cases were diagnosed and treated during aneight-year period from 1999 to 2006.
MTC or C-cell hyperplasia was initially detected by ele-vated serum-calcitonin levels, followed by fine needle aspira-tion. After surgical removal of the thyroid gland, the diagnosisof MTC was further confirmed by histopathology. The initialMEN2 variant type assignments were made based on clinicalmanifestations, medical records, including biochemical dataand histopathological features, and family pedigrees.
Genetic counseling
All of the index patients were subjected to genetic coun-seling, during which background of the disease, the aim of thestudy, and the potential risk for other family members werediscussed. An informed consent form was obtained from eachpatient and approved by TUMS ethics committee in accor-dance with institutional guidelines and national regulations.
After molecular analysis, a formal report containing theresults was prepared for each family/patient, by addressingrespective endocrinologists. After identification of the fami-lies with RET germ-line mutation carrier/s, a second geneticcounseling session was held, during which arrangementswere made for collecting blood samples from the first-degreerelatives of the index patients. In case of detecting anasymptomatic germ-line carrier during the screening process,he/she was referred for prophylactic thyroidectomy.
DNA extraction
Using a modified salting-out technique, genomic DNA wasextracted from peripheral blood lymphocytes of ethylenedia-minetetraacetic acid-anticogulated samples. Briefly, 500ml ofeach blood sample was mixed with 1 mL of RBC-lysis buffer(0.32 M sucrose, 10 mM Tris-HCl, pH 8.2, 5 mM MgCl2, and 1%v/v Triton X-100), incubated on ice for 15 min, and centrifugedat 10,000 rpm for 2 min. After discarding the supernatant, thesame procedure was repeated twice before addition of 300mlof WBC-lysis buffer (0.45 M NaCl, 10 mM Tris-HCl, pH 8.2,and 2 mM ethylenediaminetetraacetic acid), 20ml of 10% so-dium dodecyl sulfate, and 10ml of proteinase K (20 mg/mL),followed by incubation at 558C for 2 h. gDNA was precipitatedby addition of 100ml of 5M NaCl and isopropanol (1v/v),fished out using a thin glass rod, washed with 70% EtOH, anddissolved in 1�TE buffer. The quality of gDNA samples andtheir quantification were determined by agarose gel electro-phoresis and UV spectrophotometry (A260/A280).
PCR amplification of RET key exons
Forward (F) and reverse (R) primers for RET exons 10, 11, 13,14, 15, and 16 were designed based on analysis of DNA se-quences as well as some previous studies (17–20): exon 10,RET10-F2 (50-CTATGCTTGCGACACCAGTTG-30) and RET10-R2 (50-CTCCTGGGTGGAGTAACAGAG-30); exon 11, RET11-F2 (50-CAGAGCATACGCAGCCTGTAC-30) and RET11-R1(50-GCCTCGTCTGCCCAGCGTTG-30); exon 13, RET13-F1 (50-AGAAGCCTCAAGCAGCATCGTC-30) and RET13-R1 (50-AGGAGCAGTAGGGAAAGGGAGA-30); exon 14, RET14-F1(50-GAAGACCCAAGCTGCCTGAC-30) and RET14-R1
2 ALVANDI ET AL.

(50-GGCTAGAGTGTGGCATGGTG-30); exon 15, RET15-F1(50-GGTCTCACCAGGCCGCTAC-30) and RET15-R2 (50-TCGGTATCTTTCCTAGGCTTC-30); exon 16, RET16-F1 (50-AGGGA-TAGGGCCTGGGCTTC-30) and RET16-R1 (50-CCAGCCATTTGCCTCACGAAC-30).
PCRs were performed in 25-mL mixtures containing50–100 ng of template gDNA, 0.2–0.4 mM of each primer,1�PCR buffer with 1–2 mM of Mg2þ, and 0.625 U of Taqpolymerase (Roche Diagnostics). The concentrations of Mg2þ
(CinnaGen Inc.), dNTPs (CinnaGen Inc.), and template gDNAwas specifically worked out for each exon. PCR amplificationwas performed with a pre-heating cycle of 948C for 3 min,followed by 35 cycles of denaturation at 948C for 30 s, anneal-ing at 608C–628C for 30 s, extension at 728C for 40 s, and a finalextension cycle at 728C for 7 min. Afterward, 5ml of each PCRwas analyzed by gel electrophoresis on 1% agarose, and aportion of each reaction was used for direct DNA sequencing.
DNA sequence analysis
DNA sequencing reactions were performed by BigDye Ter-minator Cycle Sequencing kit (Applied Biosystems) in a 20mlfinal volume, using 10 ng of each PCR product as template and3.2 pmol of the respective primer per reaction. The reactionswere run on an automated ABI Sequence Analyzer 3130xl(Applied Biosystems). The PCRs for each sample were initiallysequenced using the forward primers. In the case of germ-linemutation carriers, however, the results were also re-confirmed
by another round of PCR and DNA sequencing using the re-spective reverse primers for further assurance. The ABI chro-matograms were each carefully examined using SequenceScanner (Applied Biosystems) before subjecting them to align-ment analysis with Lasergene MegAlign software (DNASTAR).
Restriction fragment length polymorphism
In order to detect the Cys634Arg mutation (TGC?CGC)by a restriction endonuclease, the PCR products of RET exon11 (454 bp) were first purified and quantified, as mentionedearlier, and were then digested by Hha I (CinnaGen Inc.) fol-lowing the manufacturer’s instructions. Digested fragmentsalong with nondigested ones (as negative controls) were an-alyzed on 2% agarose gels.
Results
MTC patients characterization and demographics
A total of 55 patients with MTC, with a female to male ratioof 1.6:1, from 53 kindreds (i.e., unrelated families) were iden-tified by reviewing the medical records from major referralcenters in the Iranian capital city, Tehran, and enrolled for thisstudy (Table 1). The average age of disease onset was 33 (�13)years. All of these cases had undergone total thyroidectomyand were still alive during the course of study. Based on re-viewing the clinical records and information obtained duringthe genetic counseling sessions, only four patients from four
Table 1. Distribution of Patients/Kindreds with Medullary Thyroid Carcinoma
Indexed by Age of Disease Onset, Gender, and Province
Family/patient no. MTC age of onset Gender Province Family/patient No. MTC age of onset Gender Province
1 32 F Markazi 29 38 F Gilan2a 26 F E. Azarbayejan 30 48 F Tehran3 29 M Markazi 31/1 21 F Markazi4 45 M Kermanshah 31/2a 21 F Markazi5 10 M Khorasan 31/3 21 M Markazi6 31 F Khorasan 32 29 F NC7 13 F Tehran 33 37 M NC8 47 F Esfahan 34 20 F Ardebil9 50 F Lorestan 35 38 F NC
10 48 F Gilan 36 36 F NC11 27 F Gilan 37 45 F NC12 25 F Khorasan 38 43 M Mazandaran13 40 F Gilan 39 20 M NC14 37 F Mazandaran 40 38 F Kerman15 28 F Mazandaran 41 33 M NC16 39 F E. Azarbayejan 42 45 F NC17 61 M Semnan 43 42 M Khuzestan18 32 M Kerman 44 17 F Semnan19 49 M Mazandaran 45 25 M Esfahan20 34 F Gilan 46 32 M Gilan21 15 F E. Azarbayejan 47 21 M Markazi22 34 F Gilan 48 22 F Esfahan23 57 F Mazandaran 49 33 M E. Azarbayejan24 65 F Tehran 50 13 F Gilan25 43 M Zanjan 51 22 M Markazi26b 26 M Tehran 52 23 F NC27 46 M Markazi 53c 14 F Khuzestan28 57 M Mazandaran
Index patients’ phenotypes: aMEN2A; bFMTC; cMEN2B.NC, not confirmed; MTC, medullary thyroid carcinoma; MEN2A, multiple endocrine neoplasia type 2.
GENETIC ANALYSIS OF MTC IN IRAN 3

families were characterized with MEN2 phenotypes: no. 2 and31/2, MEN2A; no. 26, FMTC; and no. 53, MEN2B. The re-maining 51 patients, representing 49 independent families,were considered as apparently sporadic cases.
Molecular analysis of the RET proto-oncogenekey exons
PCR set-ups. One of the goals of the present work was todevise an efficient, time-saving, and cost-effective strategy for
amplification and sequencing of the 6 RET key exons (10, 11,13, 14, 15, and 16). This was achieved by working out a de-tailed PCR-setup for highly specific amplification of eachexon, as well as efficient primer usage, without the need foreither a gel- or PCR-purification step before DNA sequencing(Table 2). An example of this approach, how the specificconditions were worked out for RET exon 11, is shown inFigure 1 (also see Materials and Methods for details ongDNAs quality and sequencing reactions). It should be notedthat except for Lane 1, which was the first amplification
Table 2. Polymerase Chain Reaction Conditions and Oligonucleotide Primers Used for Efficient
Amplification of RET Key Exons for the Purpose of Direct DNA Sequencing
ExonPrimer
sequenceSize of PCR
productMg2þ
(mM)dNTPs(mM)a
DNA(ng)
Annealingtemperature (8C)
10 10-F2: 50-CTATGCTTGCGACACCAGTTG-30 392 bp 1.5 0.4 100 6010-R2: 50-CTCCTGGGTGGAGTAACAGAG-30
11 11-F2: 50-CAGAGCATACGCAGCCTGTAC-30 454 bp 2 0.4 100 6011-R1: 50-GCCTCGTCTGCCCAGCGTTG-30
13 13-F1: 50-AGAAGCCTCAAGCAGCATCGTC-30 348 bp 1.5 0.4 100 6213-R1: 50-AGGAGCAGTAGGGAAAGGGAGA-30
14 14-F1: 50-GAAGACCCAAGCTGCCTGAC-30 385 bp 1.5 0.4 50 6014-R1: 50-GGCTAGAGTGTGGCATGGTG-30
15 15-F1: 50-GGTCTCACCAGGCCGCTAC-30 332 bp 2 0.2 100 6015-R2: 50-TCGGTATCTTTCCTAGGCTTC-30
16 16-F1: 50-AGGGATAGGGCCTGGGCTTC-30 351 bp 1 0.2 100 6016-R1: 50-CCAGCCATTTGCCTCACGAAC-30
Reaction volume: 50ml.aConcentration of each of dNTP.F, forward primer; R, reverse primer; PCR, polymerase chain reaction.
FIG. 1. RET exon 11 polymerase chain reaction (PCR) set-up. (A) The PCR conditions for various reactions are listed belowthe lanes. Each lane is a representative of a series of reactions aimed at optimization of the final product. Lane 7 shows thefinal set-up for exon-11 amplification, with no visible non-specific products and an efficient primer usage. (B) Application ofPCR conditions listed for lane 7 in panel A to various gDNA samples. A portion of each reaction, without going through apurification step, was sent out for direct DNA sequencing. Lanes: M, 100-bp DNA ladder; 1–7, representatives of various PCRexperiments; 33–42, index patients; C, no-template negative control. Arrow indicates the 454-bp specific band for exon 11.
4 ALVANDI ET AL.

reaction from the 6-exon PCR set, each lane is a representativeof a set of reactions focusing on a specific PCR condition, suchas the amount of template gDNA, annealing temperature, andMg2þ or dNTPs concentrations (Fig. 1).
Characterization and distributions of RET muta-tions. Among the 53 kindreds studied here, 11 (20.7%)were identified as carrying RET germ-line mutation carriers(Table 3). The 4 families (no. 2, 26, 31, and 53) with earlierMEN2 phenotype assignments matched their predictive ge-notypes. There were 13 patients who were index mutation-carriers with an average age of disease onset of 20.5 years old(�6), ranging from 10 to 31 years. Each of these identifiedmutation carriers harbored only a single germ-line missensepoint-mutation. Although one cannot rule out the possibilityof changes within other RET exons, we did not find any evi-dence of double mutations or deletion/insertions within the 6
key exons examined here. Overall, a total of 7 different het-erozygote missense point-mutations were identified in RETexons 10, 11, 14, and 16 (Fig. 2). There were no mutations inexons 13 and 15. The most affected part of the RET moleculecame from exons 11 (seven families, 63.6%) and 10 (twofamilies, 18.2%), which encode the cysteine-rich regions of theextra-cellular domain, followed by exons 14 and 16 (onefamily each, 9.1%), which encode the tyrosine kinase regionsof the intra-cellular domain. In fact, 9 of the 11 afflicted fam-ilies carry germ-line mutations in cysteine codons.
The most prevalent mutation, Cys634Arg (TGC?CGC),which is indicative of MEN2A, was seen in 7 carriers(53.9%) from 5 (45.5%) kindreds. In fact, apart from caseswith assigned MEN2 phenotypes, the Cys634Arg mutationwas found in 4 out of 49 (i.e., 8.2%) apparently sporadicMTC cases. The remaining six mutations (Cys611Tyr,Cys618Arg, Cys630Arg, Cys634Tyr, Val804Met, andMet918Thr) each represented a single index patient fromone (9.1%) mutation-carrier kindred. Surprisingly, themutation carrier with the youngest age of disease onset(patient no. 5, 10 years) was genotyped as Val804Met(GTG?ATG), which is typically classified as level 1 (i.e.,the lowest) risk level.
Single nucleotide polymorphism mapping and geographi-cal distribution of MTC cases. In addition to characteriza-tion of germ-line mutations, DNA sequencing provided acomplete and detailed genotype for each index MTC patient.Table 4 shows the distribution of known single nucleotidepolymorphism (SNPs) among all 53 patients with MTC. Onlyone of the observed SNPs, codon 691 from exon 11, results inan amino-acid change: GGT?AGT; Gly691Ser. In addition tothe SNPs listed on the table, we detected a potentially newSNP (AAG?AAA; Lys887Lys) at codon 887 from exon 15 infour members of kindred number 31, including two indexpatients: 31/1 and 31/2 (Fig. 3).
We found no apparent significant correlation between ob-served SNPs and patient gender or age of disease onset. Bycontrast, verified geographical distribution of the patientswith MTC shows a significant concentration of sporadic cases
Table 3. Characterization of RET Germ-Line Mutation Carriers
RET mutation
Family/patient No. MTC age of onset Gender Exon Codon Nucleotide/amino acid Phenotype
2 26 F 11 634 TGC?TAC (Cys634Tyr) MEN2A5 10 M 14 804 GTG?ATG (Val804Met) Sporadic6 31 F 10 611 TGC?TAC (Cys611Tyr) Sporadic7 13 F 11 634 TGC?CGC (Cys634Arg) Sporadic
11 27 F 10 618 TGC?CGC (Cys618Arg) Sporadic21 15 F 11 634 TGC?CGC (Cys634Arg) Sporadic26 26 M 11 630 TGC?CGC (Cys630Arg) FMTC31/1 21 F 11 634 TGC?CGC (Cys634Arg) Sporadic (?)31/2 21 F 11 634 TGC?CGC (Cys634Arg) MEN2A31/3 21 M 11 634 TGC?CGC (Cys634Arg) Sporadic (?)39 20 M 11 634 TGC?CGC (Cys634Arg) Sporadic51 22 M 11 634 TGC?CGC (Cys634Arg) Sporadic53 14 F 16 918 ATG?ACG (Met918Thr) MEN2B
Notice that phenotypes assignments were done before molecular characterization. Question marks for patients 31/1 and 31/3 indicatesuspicion for being a mutation carrier based on their family history.
FMTC, familial medullary thyroid carcinoma.
FIG. 2. Distribution of RET germ-line mutations. Thebrackets show the affected codons and resulting amino acidchanges, followed by the number of kindreds identified. Colorimages available online at www.liebertonline.com/thy.
GENETIC ANALYSIS OF MTC IN IRAN 5
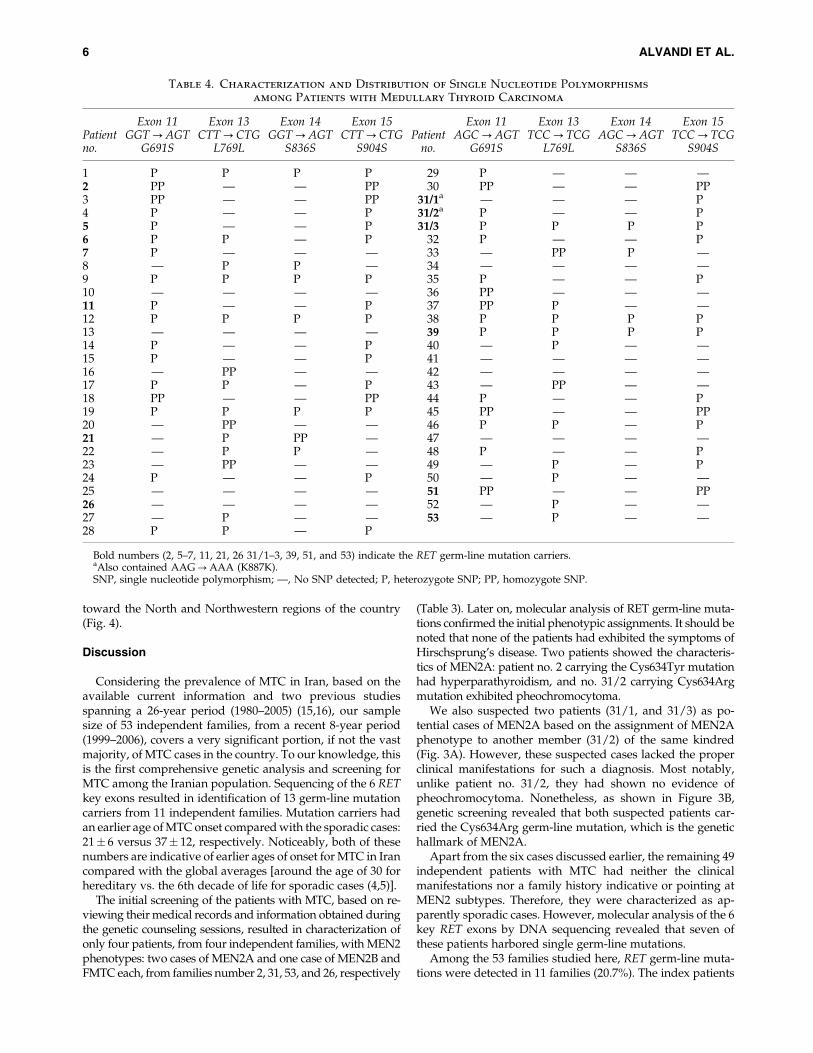
toward the North and Northwestern regions of the country(Fig. 4).
Discussion
Considering the prevalence of MTC in Iran, based on theavailable current information and two previous studiesspanning a 26-year period (1980–2005) (15,16), our samplesize of 53 independent families, from a recent 8-year period(1999–2006), covers a very significant portion, if not the vastmajority, of MTC cases in the country. To our knowledge, thisis the first comprehensive genetic analysis and screening forMTC among the Iranian population. Sequencing of the 6 RETkey exons resulted in identification of 13 germ-line mutationcarriers from 11 independent families. Mutation carriers hadan earlier age of MTC onset compared with the sporadic cases:21� 6 versus 37� 12, respectively. Noticeably, both of thesenumbers are indicative of earlier ages of onset for MTC in Irancompared with the global averages [around the age of 30 forhereditary vs. the 6th decade of life for sporadic cases (4,5)].
The initial screening of the patients with MTC, based on re-viewing their medical records and information obtained duringthe genetic counseling sessions, resulted in characterization ofonly four patients, from four independent families, with MEN2phenotypes: two cases of MEN2A and one case of MEN2B andFMTC each, from families number 2, 31, 53, and 26, respectively
(Table 3). Later on, molecular analysis of RET germ-line muta-tions confirmed the initial phenotypic assignments. It should benoted that none of the patients had exhibited the symptoms ofHirschsprung’s disease. Two patients showed the characteris-tics of MEN2A: patient no. 2 carrying the Cys634Tyr mutationhad hyperparathyroidism, and no. 31/2 carrying Cys634Argmutation exhibited pheochromocytoma.
We also suspected two patients (31/1, and 31/3) as po-tential cases of MEN2A based on the assignment of MEN2Aphenotype to another member (31/2) of the same kindred(Fig. 3A). However, these suspected cases lacked the properclinical manifestations for such a diagnosis. Most notably,unlike patient no. 31/2, they had shown no evidence ofpheochromocytoma. Nonetheless, as shown in Figure 3B,genetic screening revealed that both suspected patients car-ried the Cys634Arg germ-line mutation, which is the genetichallmark of MEN2A.
Apart from the six cases discussed earlier, the remaining 49independent patients with MTC had neither the clinicalmanifestations nor a family history indicative or pointing atMEN2 subtypes. Therefore, they were characterized as ap-parently sporadic cases. However, molecular analysis of the 6key RET exons by DNA sequencing revealed that seven ofthese patients harbored single germ-line mutations.
Among the 53 families studied here, RET germ-line muta-tions were detected in 11 families (20.7%). The index patients
Table 4. Characterization and Distribution of Single Nucleotide Polymorphisms
among Patients with Medullary Thyroid Carcinoma
Exon 11 Exon 13 Exon 14 Exon 15 Exon 11 Exon 13 Exon 14 Exon 15Patientno.
GGT?AGT CTT?CTG GGT?AGT CTT?CTG Patientno.
AGC?AGT TCC?TCG AGC?AGT TCC?TCGG691S L769L S836S S904S G691S L769L S836S S904S
1 P P P P 29 P — — —2 PP — — PP 30 PP — — PP3 PP — — PP 31/1a — — — P4 P — — P 31/2a P — — P5 P — — P 31/3 P P P P6 P P — P 32 P — — P7 P — — — 33 — PP P —8 — P P — 34 — — — —9 P P P P 35 P — — P10 — — — — 36 PP — — —11 P — — P 37 PP P — —12 P P P P 38 P P P P13 — — — — 39 P P P P14 P — — P 40 — P — —15 P — — P 41 — — — —16 — PP — — 42 — — — —17 P P — P 43 — PP — —18 PP — — PP 44 P — — P19 P P P P 45 PP — — PP20 — PP — — 46 P P — P21 — P PP — 47 — — — —22 — P P — 48 P — — P23 — PP — — 49 — P — P24 P — — P 50 — P — —25 — — — — 51 PP — — PP26 — — — — 52 — P — —27 — P — — 53 — P — —28 P P — P
Bold numbers (2, 5–7, 11, 21, 26 31/1–3, 39, 51, and 53) indicate the RET germ-line mutation carriers.aAlso contained AAG?AAA (K887K).SNP, single nucleotide polymorphism; —, No SNP detected; P, heterozygote SNP; PP, homozygote SNP.
6 ALVANDI ET AL.
in six (54.5%) of these families harbored a mutation in codon634, with Cys634Arg (TGC?CGC) being the most frequentone. The Cys634Arg is the most frequent, and exclusive,mutation associated with MEN2A. There is also generally astrong correlation between mutations of codon 634 and theoccurrence of pheochromocytoma and/or hyperparathy-roidism as well as an increasing probability of developingthese phenotypes with age (1–3). Therefore, we concludedand recommended that all of the Cys634Arg carriers gothrough regular clinical follow-ups.
Despite the exact criteria of International RET MutationConsortium for classification of FMTC (i.e., families withmore than 10 carriers and affected members over the age of50) (14), we classified one family (no. 26) as FMTC. Based onthe clinical data, only four members of this family had de-veloped MTC, and they all lacked other MEN2-associatedmanifestations. However, this is in line with a less rigid def-inition of FMTC and the recent trend advocated by theAmerican Thyroid Association (2). Moreover, random geneticscreening of two of the four members revealed that they both
FIG. 3. Pedigree analysis and sequencing results for kindred no. 31. (A) As the pedigree shows, three members of the family(II-2, �4, and �7) had been diagnosed with medullary thyroid carcinoma (MTC), but only patient II-4 (indicated by thearrow) exhibited the clinical manifestation of multiple endocrine neoplasia type 2 phenotype. (B) DNA sequencing chro-matograms. Molecular analysis of the three patients with MTC revealed that they all carried the genetic hallmark of multipleendocrine neoplasia type 2 (i.e., Cys634Arg; TGC?CGC). Later on, screening of asymptomatic first-degree relatives alsorevealed that members I-2 and III-2 were RET germ-line mutation carriers as well. The rectangle on each sequencing diagram,except for the last one (codon 887), indicates the codon 634. M shows the position of the heterozygote point mutation. The lastdiagram shows a potentially new single nucleotide polymorphisms (SNP) (AAG?AAA) identified in codon 887 of fourfamily members. Black circles and squares in panel A indicate patients with MTC or the proband. The gray squares show themember identified as mutation carriers. The strikethrough circle marks a deceased female member. Color images availableonline at www.liebertonline.com/thy.
GENETIC ANALYSIS OF MTC IN IRAN 7
carried the rare germ-line mutation TGC?CGC (Cys630Arg)in exon 11, which is in agreement with FMTC characterization(21,22).
We detected a Val804Met mutation in RET exon 14 of a 10-year-old patient (family no. 5). In some reports, mutations incodon 804 have been reported to be associated with late age ofMTC onset and low tumor aggressiveness (4,5,23). By con-trast, another study reported a Val804Met carrier showing the
symptoms of the disease at the age of six, who died 6 yearslater due to widespread metastases (24). Finally, the presenceof pheochromocytoma and/or hyperparathyroidism inVal804Met carriers have been reported in a number of studies(25–27). Patient no. 5 did not exhibit any other endocrine-related phenotypes. However, the presence of MTC at the ageof 10 supports the previously proposed wide range for the ageof disease onset among Val804Met carriers (28).
In approximately 95% of cases, MEN2B is associated with amethionine-to-threonine substitution in the intra-cellular do-main of RET as a result of mutation in codon 918 of exon 16(Met918Thr) (10–12,29). We found this germ-line mutation inpatient no. 53 (age of MTC onset: 14) who had already beencharacterized as MEN2B based on her medical records. In-terestingly, both parents, who were over the age of 35, ap-peared healthy. Although we did not succeed in arranging forgenetic screening of the parents, considering their ages, it islikely that the index patient is a de novo germ-line mutationcarrier. The presence of de novo germ-line Met918Thr muta-tion has been reported by other investigators as well and, infact, accounts for more than 50% of the cases (2,30,31).
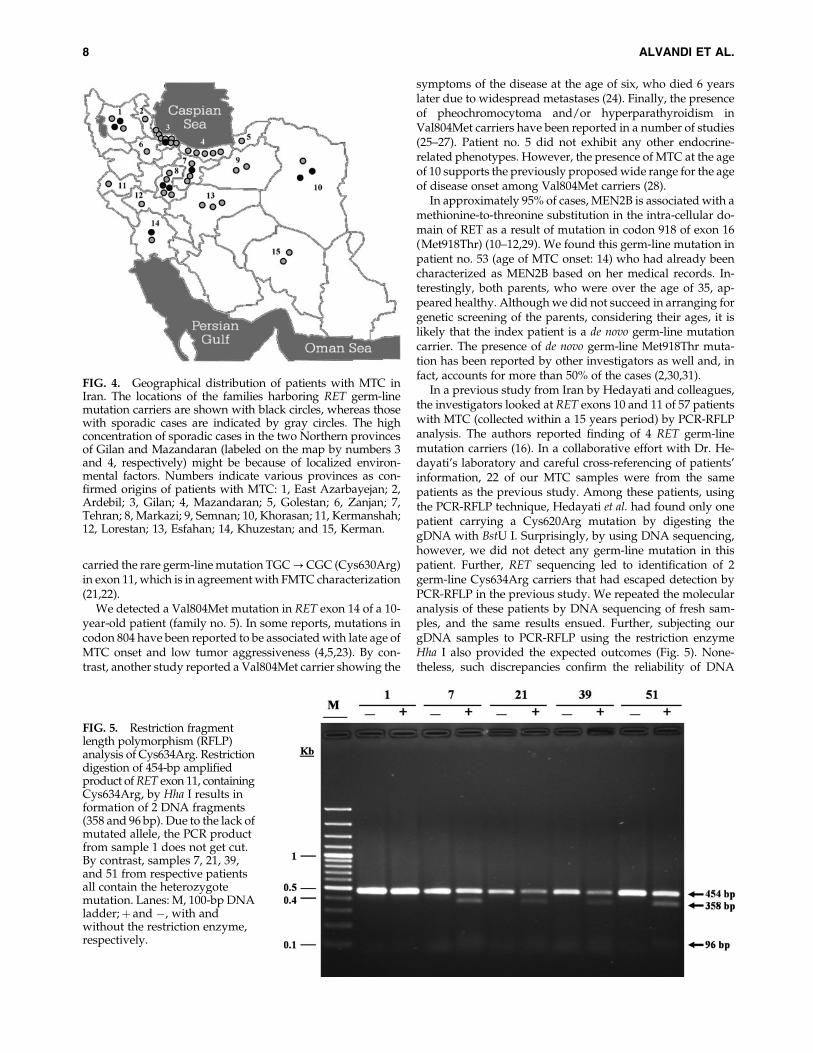
In a previous study from Iran by Hedayati and colleagues,the investigators looked at RET exons 10 and 11 of 57 patientswith MTC (collected within a 15 years period) by PCR-RFLPanalysis. The authors reported finding of 4 RET germ-linemutation carriers (16). In a collaborative effort with Dr. He-dayati’s laboratory and careful cross-referencing of patients’information, 22 of our MTC samples were from the samepatients as the previous study. Among these patients, usingthe PCR-RFLP technique, Hedayati et al. had found only onepatient carrying a Cys620Arg mutation by digesting thegDNA with BstU I. Surprisingly, by using DNA sequencing,however, we did not detect any germ-line mutation in thispatient. Further, RET sequencing led to identification of 2germ-line Cys634Arg carriers that had escaped detection byPCR-RFLP in the previous study. We repeated the molecularanalysis of these patients by DNA sequencing of fresh sam-ples, and the same results ensued. Further, subjecting ourgDNA samples to PCR-RFLP using the restriction enzymeHha I also provided the expected outcomes (Fig. 5). None-theless, such discrepancies confirm the reliability of DNA
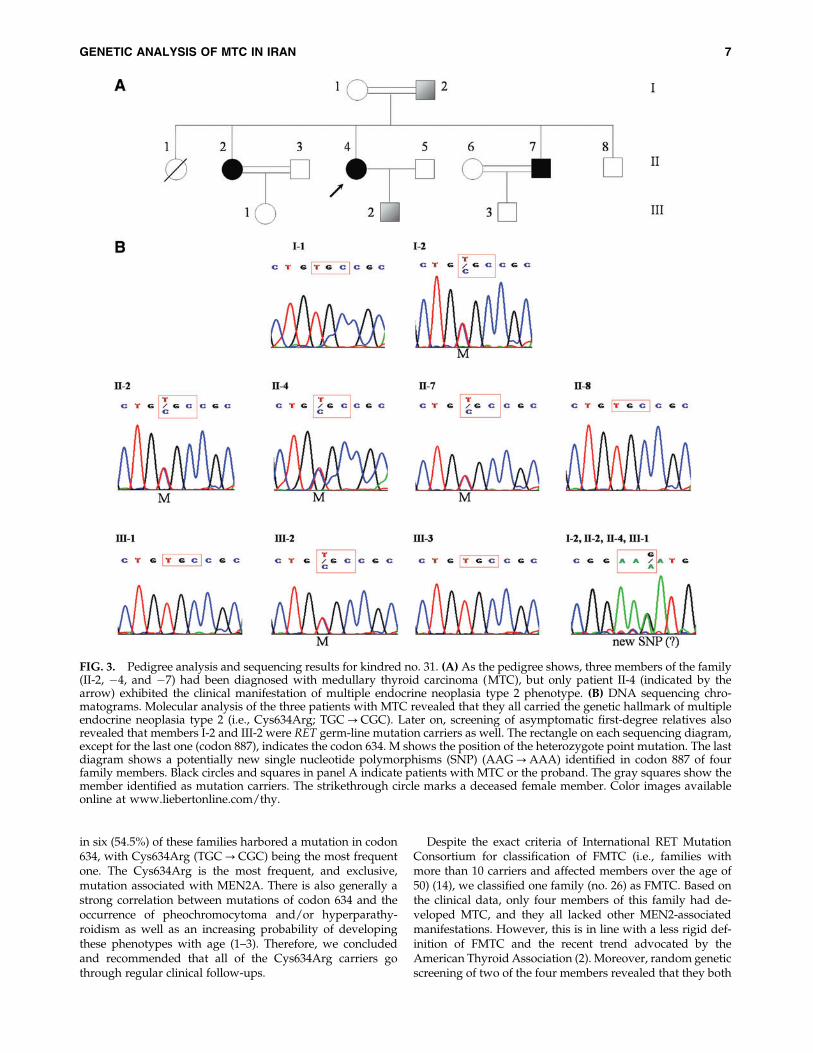
FIG. 4. Geographical distribution of patients with MTC inIran. The locations of the families harboring RET germ-linemutation carriers are shown with black circles, whereas thosewith sporadic cases are indicated by gray circles. The highconcentration of sporadic cases in the two Northern provincesof Gilan and Mazandaran (labeled on the map by numbers 3and 4, respectively) might be because of localized environ-mental factors. Numbers indicate various provinces as con-firmed origins of patients with MTC: 1, East Azarbayejan; 2,Ardebil; 3, Gilan; 4, Mazandaran; 5, Golestan; 6, Zanjan; 7,Tehran; 8, Markazi; 9, Semnan; 10, Khorasan; 11, Kermanshah;12, Lorestan; 13, Esfahan; 14, Khuzestan; and 15, Kerman.
FIG. 5. Restriction fragmentlength polymorphism (RFLP)analysis of Cys634Arg. Restrictiondigestion of 454-bp amplifiedproduct of RET exon 11, containingCys634Arg, by Hha I results information of 2 DNA fragments(358 and 96 bp). Due to the lack ofmutated allele, the PCR productfrom sample 1 does not get cut.By contrast, samples 7, 21, 39,and 51 from respective patientsall contain the heterozygotemutation. Lanes: M, 100-bp DNAladder;þ and �, with andwithout the restriction enzyme,respectively.
8 ALVANDI ET AL.

sequencing and its preference over RFLP in detection of RETgerm-line mutations as also stated by the International RETMutation Consortium (14).
In addition to identifying the germ-line mutations in anunbiased manner, another advantage of RET sequencing isthat it provides a clear genotype, which includes an SNP mapas well. We have obtained clear and complete genotypic pro-files of the 6 RET key exons for our patients with MTC. In-terestingly, we also detected a potentially new SNP, withoutamino acid substitution, in exon 15 (AAG?AAA; Lys887Lys)of four members of one family (Fig. 3). Whether this nucleotidechange has any effect on MTC progression requires furtherinvestigation. There have been some contradictory results indifferent studies regarding the association of SNPs with agerm-line mutation and/or mean age of the disease onset (32–40). Given our limited sample size, we did not find a statisti-cally significant correlation between the SNP maps and patientgender and/or age of disease onset within our samples.
We found no specific geographical concentration of RETgerm-line mutation carriers in Iran. By contrast, geographicaldistribution of sporadic cases shows a high concentrationwithin the Northern regions, especially in the provinces ofGilan and Mazandaran, which are located by the Caspian Sea(Fig. 4). Although these regions are densely populated, con-sidering the fact that they make up only about 8% of the entirepopulation of the country (41), this might be suggestive ofinvolvement of some environmental factors.
This study is the first comprehensive survey of patients withMTC in Iran. Despite the poor system of assorting data of pa-tients with MTC, along with very low frequency of the disease,through concerted effort and teamwork, we managed to collectdata from three major endocrinology and surgery departmentsin the country. We also set up a reliable step-by-step strategyfor molecular analysis of RET proto-oncogene for patients withMTC in Iran. During the course of this study, by screening thefirst-degree relatives of identified hereditary cases among threeof the 11 families containing index germ-line carriers, we weresuccessful in detecting four asymptomatic germ-line mutationcarriers and referred them for prophylactic thyroidectomy.Further follow-up of families carrying germ-line mutation isneeded to improve their health state.
Acknowledgment
This work was supported in part by TUMS grant number3320.
Author Disclosure Statement
No competing financial interests exist.
References
1. Leboulleux S, Baudin E, Travagli JP, Schlumberger M 2004Medullary thyroid carcinoma. Clin Endocrinol (Oxf) 61:299–310.
2. Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, Gharib H,Moley JF, Pacini F, Ringel MD, Schlumberger M, Wells SA Jr.2009 Medullary thyroid cancer: management guidelines ofthe American Thyroid Association. Thyroid 19:565–612.
3. Hansford JR, Mulligan LM 2000 Multiple endocrine neo-plasia type 2 and RET: from neoplasia to neurogenesis. JMed Genet 37:817–827.
4. Eng C 1999 RET proto-oncogene in the development ofhuman cancer. J Clin Oncol 17:380–393.
5. Machens A, Gimm O, Hinze R, Hoppner W, Boehm BO,Dralle H 2001 Genotype-phenotype correlations in heredi-tary medullary thyroid carcinoma: oncological features andbiochemical properties. J Clin Endocrinol Metab 86:1104–1109.
6. Yip L, Cote GJ, Shapiro SE, Ayers GD, Herzog CE, Sellin RV,Sherman SI, Gagel RF, Lee JE, Evans DB 2003 Multiple en-docrine neoplasia type 2: evaluation of the genotype-phenotype relationship. Arch Surg 138:409–416; discussion 416.
7. Kouvaraki MA, Shapiro SE, Perrier ND, Cote GJ, Gagel RF,Hoff AO, Sherman SI, Lee JE, Evans DB 2005 RET proto-oncogene: a review and update of genotype-phenotypecorrelations in hereditary medullary thyroid cancer and as-sociated endocrine tumors. Thyroid 15:531–544.
8. Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K,Lairmore TC, Howe JR, Moley JF, Goodfellow P, Wells SA Jr.1993 Mutations in the RET proto-oncogene are associatedwith MEN 2A and FMTC. Hum Mol Genet 2:851–856.
9. Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C,Gardner E, Love DR, Mole SE, Moore JK, Papi L, PonderMA, Telenius H, Tunnacliffe A, Ponder BAJ 1993 Germ-linemutations of the RET proto-oncogene in multiple endocrineneoplasia type 2A. Nature 363:458–460.
10. Carlson KM, Dou S, Chi D, Scavarda N, Toshima K, JacksonCE, Wells SA Jr., Goodfellow PJ, Donis-Keller H 1994 Singlemissense mutation in the tyrosine kinase catalytic domain ofthe RET protooncogene is associated with multiple endo-crine neoplasia type 2B. Proc Natl Acad Sci USA 91:1579–1583.
11. Eng C, Smith DP, Mulligan LM, Nagai MA, Healey CS,Ponder MA, Gardner E, Scheumann GF, Jackson CE, Tun-nacliffe A, Ponder BAJ 1994 Point mutation within the ty-rosine kinase domain of the RET proto-oncogene in multipleendocrine neoplasia type 2B and related sporadic tumours.Hum Mol Genet 3:237–241.
12. Hofstra RM, Landsvater RM, Ceccherini I, Stulp RP, Stel-wagen T, Luo Y, Pasini B, Hoppener JW, van Amstel HK,Romeo G, Lips CJ, Buys CH 1994 A mutation in the RETproto-oncogene associated with multiple endocrine neopla-sia type 2B and sporadic medullary thyroid carcinoma.Nature 367:375–376.
13. Bolino A, Schuffenecker I, Luo Y, Seri M, Silengo M, Tocco T,Chabrier G, Houdent C, Murat A, Schlumberger M, Tour-niaire J, Lenoir GM, Romeo G 1995 RET mutations in exons13 and 14 of FMTC patients. Oncogene 10:2415–2419.
14. Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-PeccozP, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG, LibroiaA, Lips CJ, Lombardi G, Mannelli M, Pacini F, Ponder BA,Raue F, Skogseid B, Tamburrano G, Thakker RV, ThompsonNW, Tomassetti P, Tonelli F, Wells SA Jr., Marx SJ 2001Guidelines for diagnosis and therapy of MEN type 1 andtype 2. J Clin Endocrinol Metab 86:5658–5671.
15. Larijani B, Aghakhani S, Khajeh-Dini H, Baradar-Jalili R2003 Clinico-pathological features of thyroid cancer as ob-served in five referral hospitals in Iran—a review of 1177cases. Acta Oncol 42:334–337.
16. Hedayati M, Nabipour I, Rezaei-Ghaleh N, Azizi F 2006Germline RET mutations in exons 10 and 11: an Iraniansurvey of 57 medullary thyroid carcinoma cases. Med JMalaysia 61:564–569.
17. Berndt I, Reuter M, Saller B, Frank-Raue K, Groth P, Grus-sendorf M, Raue F, Ritter MM, Hoppner W 1998 A new hot
GENETIC ANALYSIS OF MTC IN IRAN 9

spot for mutations in the ret protooncogene causing familialmedullary thyroid carcinoma and multiple endocrine neo-plasia type 2A. J Clin Endocrinol Metab 83:770–774.
18. Chung YJ, Kim HH, Kim HJ, Min YK, Lee MS, Lee MK, KimKW, Ki CS, Kim JW, Chung JH 2004 RET proto-oncogenemutations are restricted to codon 634 and 618 in Koreanfamilies with multiple endocrine neoplasia 2A. Thyroid14:813–818.
19. Jindrichova S, Vcelak J, Vlcek P, Neradilova M, Nemec J,Bendlova B 2004 Screening of six risk exons of the RETproto-oncogene in families with medullary thyroid carci-noma in the Czech Republic. J Endocrinol 183:257–265.
20. Ceccherini I, Hofstra RM, Luo Y, Stulp RP, Barone V, Stel-wagen T, Bocciardi R, Nijveen H, Bolino A, Seri M 1994DNA polymorphisms and conditions for SSCP analysis ofthe 20 exons of the ret proto-oncogene. Oncogene 9:3025–3029.
21. Dourisboure RJ, Belli S, Domenichini E, Podesta EJ, Eng C,Solano AR 2005 Penetrance and clinical manifestations ofnon-hotspot germline RET mutation, C630R, in a familywith medullary thyroid carcinoma. Thyroid 15:668–671.
22. Machens A, Schneyer U, Holzhausen HJ, Raue F, Dralle H2004 Emergence of medullary thyroid carcinoma in a familywith the Cys630Arg RET germline mutation. Surgery136:1083–1087.
23. Lombardo F, Baudin E, Chiefari E, Arturi F, Bardet S, Cail-lou B, Conte C, Dallapiccola B, Giuffrida D, Bidart JM,Schlumberger M, Filetti S 2002 Familial medullary thyroidcarcinoma: clinical variability and low aggressiveness asso-ciated with RET mutation at codon 804. J Clin EndocrinolMetab 87:1674–1680.
24. Frohnauer MK, Decker RA 2000 Update on the MEN 2Ac804 RET mutation: is prophylactic thyroidectomy indicat-ed? Surgery 128:1052–1057; discussion 1057–1058.
25. Gibelin H, Bezieau S, Misso C, Bouin-Pineau MH, Mar-echaud R, Kraimps JL 2004 Germline RET V804M mutationassociated with multiple endocrine neoplasia type 2A. Br JSurg 91:1458–1459.
26. Pinna G, Orgiana G, Riola A, Ghiani M, Lai ML, Carcassi C,Mariotti S 2007 RET proto-oncogene in Sardinia: V804M isthe most frequent mutation and may be associated withFMTC/MEN-2A phenotype. Thyroid 17:101–104.
27. Recasens M, Oriola J, Fernandez-Real JM, Roig J, Rodriguez-Hermosa JI, Font JA, Galofre P, Lopez-Bermejo A, Ricart W2007 Asymptomatic bilateral adrenal pheochromocytoma ina patient with a germline V804M mutation in the RET proto-oncogene. Clin Endocrinol (Oxf) 67:29–33.
28. Feldman GL, Edmonds MW, Ainsworth PJ, Schuffenecker I,Lenoir GM, Saxe AW, Talpos GB, Roberson J, Petrucelli N,Jackson CE 2000 Variable expressivity of familial medul-lary thyroid carcinoma (FMTC) due to a RET V804M(GTG?ATG) mutation. Surgery 128:93–98.
29. Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, GagelRF, Ploos van Amstel HK, Lips CJM, Nishisho I, Takai S-I,Marsh DJ, Robinson BG, Frank-Raue F, Xue F, Noll WW,Romeo G, Pacini F, Fink M, Niederle B, Zedenius J, Nor-denskjold M, Komminoth P, Hendy GN, Gharib H, Thibo-deau SN, Lacroix A, Frilling A, Ponder BAJ, Mulligan LM1996 The relationship between specific RET proto-oncogenemutations and disease phenotype in multiple endocrineneoplasia type 2. International RET mutation consortiumanalysis. JAMA 276:1575–1579.
30. Carlson KM, Bracamontes J, Jackson CE, Clark R, Lacroix A,Wells SA Jr., Goodfellow PJ 1994 Parent-of-origin effects in
multiple endocrine neoplasia type 2B. Am J Hum Genet55:1076–1082.
31. Kitamura Y, Scavarda N, Wells SA Jr., Jackson CE, Good-fellow PJ 1995 Two maternally derived missense mutationsin the tyrosine kinase domain of the RET protooncogene in apatient with de novo MEN 2B. Hum Mol Genet 4:1987–1988.
32. Gimm O, Neuberg DS, Marsh DJ, Dahia PL, Hoang-Vu C,Raue F, Hinze R, Dralle H, Eng C 1999 Over-representationof a germline RET sequence variant in patients with sporadicmedullary thyroid carcinoma and somatic RET codon 918mutation. Oncogene 18:1369–1373.
33. Robledo M, Gil L, Pollan M, Cebrian A, Ruiz S, Azanedo M,Benitez J, Menarguez J, Rojas JM 2003 PolymorphismsG691S/S904S of RET as genetic modifiers of MEN 2A.Cancer Res 63:1814–1817.
34. Fernandez RM, Pecina A, Antinolo G, Navarro E, Borrego S2006 Analysis of RET polymorphisms and haplotypes in thecontext of sporadic medullary thyroid carcinoma. Thyroid16:411–417.
35. Lesueur F, Cebrian A, Robledo M, Niccoli-Sire P, SvenssonKA, Pinson S, Leyland J, Whittaker J, Pharoah PD, PonderBA 2006 Polymorphisms in RET and its coreceptors and li-gands as genetic modifiers of multiple endocrine neoplasiatype 2A. Cancer Res 66:1177–1180.
36. Baumgartner-Parzer SM, Lang R, Wagner L, Heinze G,Niederle B, Kaserer K, Waldhausl W, Vierhapper H 2005Polymorphisms in exon 13 and intron 14 of the RET proto-oncogene: genetic modifiers of medullary thyroid carcino-ma? J Clin Endocrinol Metab 90:6232–6236.
37. Wiench M, Wloch J, Wygoda Z, Gubala E, Oczko M, Paw-laczek A, Kula D, Lange D, Jarzab B 2004 RET polymor-phisms in codons 769 and 836 are not associated withpredisposition to medullary thyroid carcinoma. Cancer De-tect Prev 28:231–236.
38. Elisei R, Cosci B, Romei C, Bottici V, Sculli M, Lari R, BaraleR, Pacini F, Pinchera A 2004 RET exon 11 (G691S) poly-morphism is significantly more frequent in sporadic med-ullary thyroid carcinoma than in the general population. JClin Endocrinol Metab 89:3579–3584.
39. Cardot-Bauters C, Leteurtre E, Leclerc L, Vantyghem MC,Do Cao C, Wemeau JL, d’Herbomez M, Carnaille B, BarbuV, Pinson S, Pigny P 2008 Does the RET variant G691S in-fluence the features of sporadic medullary thyroid carcino-ma? Clin Endocrinol (Oxf) 69:506–510.
40. Weinhaeusel A, Scheuba C, Lauss M, Kriegner A, Kaserer K,Vierlinger K, Haas OA, Niederle B 2008 The influence ofgender, age, and RET polymorphisms on C-cell hyperplasiaand medullary thyroid carcinoma. Thyroid 18:1269–1276.
41. Islamic Republic of Iran Census Figures. Statistical Centre ofIran. Available at www.sci.org.ir. Accessed August 03, 2010.www.sci.org.ir).
Address correspondence to:Mehrdad Pedram, Ph.D.
Department of Medical GeneticsSchool of Medicine
Tehran University of Medical Sciences100 Poursina Ave.
Keshavarz Blvd.Tehran 14176-1351
Iran
E-mail: [email protected]
10 ALVANDI ET AL.
Copyright © 2022 FDOKUMEN




















