
Avaliação do óleo essencial folhas de Cymbopogon citratus (DC.) Stapf após o processo de secagem

i
UNIVERSIDADE FEDERAL DO RIO GRANDE DO SUL
ESCOLA DE ENGENHARIA
DEPARTAMENTO DE ENGENHARIA QUÍMICA
TRABALHO DE CONCLUSÃO EM ENGENHARIA QUÍMICA
MODELAGEM DA CINÉTICA DE TRANSESTERIFICAÇÃO DO ÓLEO DE SOJA EM ETANOL PRÓXIMO AO PONTO CRÍTICO
Aluno: Renato Corrêa Arrieche
Cartão: 00113529
Professor Orientador: Carlos Alberto Krahl
Porto Alegre, Junho de 2009

ii
Trabalho de Conclusão apresentado como requisito parcial para a obtenção do grau de
Engenheiro Químico
Prof. Carlos Alberto Krahl Orientador
Prof. Dr. Rafael de Pelegrini Soares
Co-orientador
UNIVERSIDADE FEDERAL DO RIO GRANDE DO SUL Reitor: Prof. Carlos Alexandre Netto
Vice-reitor: Prof. Rui Vicente Oppermann Pró-Reitora Adjunta de Graduação: Profa. Valquiria Linck Bassani
Diretor do Escola de Engenharia: Prof. Nilson Romeu Marcílio

iii
Coordenador do Curso de Engenharia Química: Prof. Oscar William Perez Lopez
AGRADECIMENTOS
Agradeço à minha família, por todo o apoio, amor e carinho que me deram durante o curso e durante toda a minha vida. Em especial à minha mãe, por todo o amor e carinho dedicados nos últimos 27 anos da minha vida.
Agradeço ao professor Carlos Alberto Krahl e ao professor Rafael Pelegrini Soares pela orientação que estes grandes mestres me deram ao longo deste trabalho.
Agradeço à colaboração dos químicos Luiz Antônio Mazzini e Ana Mello pelas análises efetuadas.
Agradeço à CIENTEC, em especial ao DEPROC, pela oportunidade de realizar este trabalho.
Agradeço ao Departamento de Engenharia Química pela excelência do conhecimento que me foi transmitido durante o curso.
Agradeço a Deus por, mesmo eu sendo ateu, me dar força, paz interior e muita sorte. Muita sorte.
SUMÁRIO
1. INTRODUÇÃO....................................................................................... 1
1.1. O Biodiesel............................................................................... 1
1.2. A CIENTEC e o DEPROC ........................................................ 2
2. REVISÃO BIBLIOGRÁFICA ............. ............. ............. ............. .......... 3
2.1. A Transesterificação de óleos vegetais.................................... 3
2.2. Principais rotas de produção..................................................... 4
2.3. Vantagens e desvantagens do processo sem catalisador ....... 5
2.4. Efeito das condições de processo ............. ............. ............... 6
2.5. O uso do etanol ........................................................................ 8
2.6. Observações sobre a cinética no supercrítico ............. ........... 9
3. REVISÃO TEÓRICA E SOFTWARES UTILIZADOS ............................. 10

iv
3.1. Termodinâmica ....................................................................... 10
3.2. Cinética ..................................................................................... 11
3.3. Balanço material .................................................................. 12
3.4. Estimação das propriedades dos fluidos ................................... 12
3.5. O Simulador EMSO .................................................................... 12
3.6. O pacote termodinâmico VRTherm ............................................ 13
3.7. Conceitos gerais em modelagem .............................................. 14
4. MATERIAIS E MÉTODOS ......................................................................... 15
4.1. O Reator .................................................................................... 15
4.2. O óleo de soja ............................................................................. 15
4.3. O etanol ................................................................................... 15
4.4. Propriedades estimadas ........................................................... 15
4.5. Dados experimentais ............. ................................................... 16
5. MODELAGEM DO SISTEMA ............. .................................................... 18
5.1. Simplificações............................................................................ 18
5.2. Modelagem do reator ............................................................... 19
5.3. Modelagem da reação............................................................... 19
5.4. Sistema de equações................................................................ 20
5.5. Estimação de parâmetros..................................................... 21
5.6. Considerações sobre o modelo.................................................. 21
6. ANÁLISE E DISCUSSÃO DOS RESULTADOS ............. ............. ......... 23
6.1. Análise dos experimentos ……………………………………….. 23
6.2. Discussão dos resultados ……………………………………….. 26
7. CONCLUSÕES ............. ............. ............. ............. ............. ............. 31
8. BIBLIOGRAFIA............. ............. ............. ............. ............. ............. . 34
ANEXO …………………………………………………………………………
LISTA DE SIGLAS E ABREVIATURAS

v
CIENTEC Fundação de Ciência e Tecnologia
DEPROC Departamento de Processos Químicos
R1, R2, R3,R4 Diferentes radicais
DG Diglicerídeo
MG Monoglicerídeo
TG Triglicerídeo
A Álcool; etanol
B Triglicerídio; Óleo De Soja; Trioleína
C Biodíesel; éster do óleo de soja
D Glicerina
L ácido linoleico; líquido
O oxigênio; ácido oleico
P ácido palmítico;
St ácido esteárico
LLL trilinoleína, triglicerídeo do ácido linoleico
V vapor
SC supercrítico
LV líquido-vapor
LVE equilíbrio líquido-vapor (o mesmo que LV)
exp experimental
calc calculado
CNTP condições normais de temperatura e pressão (0 °C, 1 atm)
LISTA DE SÍMBOLOS
RAB : razão molar álcool-óleo (adimensional)
T temperatura
P pressão
volume do reator
N número de moles
R constante dos gases
Z fator de compressibilidade

vi
ω fator acêntrico
i componente qualquer
fugacidade do componente i na solução
R constante dos gases
fração do componente i na solução
constante de distribuição do componente i
fugacidade do componente i na solução
A, B, C, D, E constantes da equação de Arrhenius completa
taxa da reação;
avanço da reação
Ci concentração do componente i na mistura
k constante cinética aparente
KE constante de equilíbrio aparente
Pv pressão de vapor
LISTA DE FIGURAS
Figura 1: Reação de transesterificação…3
Figura 2: Concentração molar dos glicerídeos em função do tempo do tempo…4
Figura 3: Efeito da temperatura da reação sobre a conversão de óleo … 6
Figura 4: constante de equilíbrio versus temperatura: gráfico de Arrhenius … 7
Figura 5: Efeito da pressão sobre a conversão. … 7
Figura 6: Efeito da razão álcool-óleo sobre a conversão de óleo … 8
Figura 7: o simulador EMSO … 13
Figura 8: interface gráfica do VRTherm ..14
Figura 9: Temperatura e pressão verusus tempo total de reação e pressão versus
temperatura … 17
Figura 10: Dados expermentais da queda de pressão versus tempo de reação… 24
Figura 11: constante cinética versus tempo: dados experimentais e reta ajustada…24
Figura 12: Constante aparente de equilíbrio versus temperatura….25

vii
Figura 13: influência da pressão inicial sobre Ke, J e ΔPe… 25
Figura 14: corrida 58… 27
Figura 15: Corrida 59… 27
Figura 16: Corrida 61…. 27
Figura 17: Corrida 62 … 28
Figura 18: Corrida 65 … 28
Figura 19: Corrida 67 … 28
Figura 20: Erro relativo de todas as corridas … 29
Figura 21: Comparação do erro relativo em relação à faixa de temperatura. … 29
Figura 22: Comparação da conversão estimada pelo modelo … 32
LISTA DE TABELAS
Tabela 1: Comparação entre as diferentes rotas de produção de biodiesel… 4
Tabela 2: Comparação entre a rota alcalina e a rota supercrítica…ç 5
Tabela 3: Comparação entre o etanol e o etanol para a produção de biodiesel … 9
Tabela 4: Propriedades estimadas do biodiesel (≈ etil linoleato) e óleo de soja (≈
trilinoleína)….. 16
Tabela 5: Condições gerais das corridas realizadas … 16
Tabela 6: Comparação geral dos valores da constante cinética e dos valores
experimentais para cada corrida…. 23

viii
RESUMO
A transesterificação de óleos vegetais tornou-se um importante método de
produção de um combustível denominado biodiesel, semelhante ao óleo diesel usado
nos motores Diesel, com a vantagem de ser proveniente de fontes renováveis. O
biodiesel tem como matéria prima o etanol e óleos vegetais, entre os quais se destaca
o óleo de soja, usado neste trabalho.
A produção de biodiesel por transesterificação é feita neste trabalho sem
catalisador em condições de elevadas temperaturas e pressões. A não utilização de
catalisadores químicos torna mais fácil a separação dos produtos
dessa reação em relação aos métodos convencionais (catálise ácida e catálise
alcalina), porém apresenta algumas desvantagens devido à severidade das condições,
além de grandes quantidades de álcool.Ainda são necessárias, para efetiva aplicação
industrial, investigações mais profundas do processo.
No presente trabalho foi avaliada a cinética da transesterificação na faixa de
temperaturas e pressões próximas ao ponto crítico do etanol. Foram realizadas
estimações dos parâmetros cinéticos e termodinâmicos a partir de dados de pressão e
temperatura, ambos em função do tempo da reação.A estimativa da cinética foi feita
considerando a queda de pressão proporcional ao avanço da reação. A constante de
equilíbrio foi calculada a partir dos dados da conversão final do óleo de soja em
ésteres e em glicerina. As propriedades físicas do óleo e do biodiesel foram estimadas
tanto por métodos de contribuição de grupo como por correlações empíricas. Para fins
de validação, os dados de pressão calculados pelos modelos propostos são
comparados com os dados experimentais.
As reações de transesterificação foram simplificadas em uma única reação
global e um modelo cinético de primeira ordem em relação a cada reagente foi
proposto. O equilíbrio de fases foi ignorado na estimativa cinética.

1
1) INTRODUÇÃO
1.1) O Biodiesel
O interesse por combustíveis a partir de fontes renováveis cresce em todo
planeta. Uma alternativa é o biodiesel. O biodiesel tem potencial para ser a resposta
de dois grandes problemas da humanidade: a futura escassez de combustíveis
fósseis e o aquecimento global causado pela emissão de CO2 para a atmosfera.
O biodiesel de cadeia longa é um combustível composto de mono-alquil-éster
derivado de ácidos graxos proveniente de uma fonte lipídica renovável como óleo
vegetal ou gordura animal. É tipicamente produzido através da reação de óleos
vegetais ou gorduras animais com álcool, sob a ação de catalisadores. Tipicamente o
álcool é o metanol ou o etanol. O subproduto da reação é, idealmente, a glicerina.
De uma forma geral, pode-se afirmar que os monoalquil-ésteres de ácidos
graxos podem ser produzidos a partir de qualquer tipo de óleo vegetal, mas nem todo
óleo vegetal pode (deve) ser utilizado como matéria-prima para produção de biodiesel.
Isto porque alguns óleos vegetais apresentam propriedades não ideais, como alta
viscosidade ou alto número de iodo, que são transferidas para o biocombustível e que
o tornam inadequado para o uso direto em motores do ciclo diesel. No Brasil, têm-se
dezenas de alternativas de matérias primas para produção de biodiesel (oleaginosas);
tais como: mamona, dendê, soja, girassol, pinhão manso, caroço de algodão, nabo
forrageiro, amendoim, babaçu, entre outros. Porém, dada a dimensão do agronegócio
da soja no mercado brasileiro (90% da produção de óleo vegetal no Brasil), é
relativamente fácil reconhecer que essa oleaginosa apresenta o maior potencial para
servir de modelo ao desenvolvimento de um programa nacional de biodiesel. Devido a
estes fatos, o óleo de soja foi o escolhido para este trabalho. (Revista Biodíesel,
fascículo 1 apud Santos, 2008)
Para produção convencional de biodiesel são utilizados catalisadores alcalinos,
tais como NaOH, KOH ou seus derivados metoxidos e etoxidos, tendo algumas
vantagens, tais como menor investimento inicial, condições mais brandas e tecnologia
mais desenvolvida. Porém, apresenta inconveniências, tais como produzir sabões,
consumo do catalisador e forte sensibilidade à água. (Revista Biodíesel, fascículo 1
apud Santos, 2008)
A obtenção de ésteres etílicos com o uso de fluidos supercríticos tem como
vantagem a não utilização de catalisadores químicos que torna mais fácil a separação

2
dos produtos dessa reação. A reação no supercrítico tem também a vantagem de
poder utilizar qualquer tipo de óleo ou gordura, não necessitando de uma etapa de
purificação da matéria prima. Mesmo sendo a altas pressões, o tratamento cinético no
supercrítico (SC) é simples devido a, principalmente, a homogeneidade do sistema.
Porém, em torno do ponto crítico, há equilíbrio entre as fases líquida e vapor, sendo
que este não é matematicamente trivial.
A modelagem cinética em função da pressão pode vir a baratear, simplificar e
acelerar posteriores investigações no processo, visto que a pressão é uma variável
muito mais fácil de medir e mais acessível que a composição do sistema. Em
modelagens cinéticas, são necessárias medições em função do tempo de reação, o
que torna necessário que se tenha várias medidas de composição ao longo do tempo.
Porém, são necessários muitos experimentos e modelos que descrevem
suficientemente o sistema, usando para isso a termodinâmica. A modelagem
termodinâmica a altas pressões, porém, é muito mais complexa que para situações de
pressão normal e ainda constitui um vasto campo a ser explorado.
1.2) A CIENTEC e o DEPROC
Os dados experimentais foram obtidos no Departamento de Processos
Químicos (DEPROC), pertencente à Fundação Estadual de Ciência e Tecnologia
(CIENTEC). (SANTOS, 2008)
Fundação de Ciência e Tecnologia - CIENTEC, é uma entidade pública de
direito privado, vinculada a Secretaria da Ciência e Tecnologia do Estado do Rio
Grande do Sul. Foi criada pela Lei n.º 6.370, de 6 de junho de 1972, como sucessora
do Instituto Tecnológico do Estado do Rio Grande do Sul - ITERS, criado em 11 de
dezembro de 1942.
A CIENTEC atua no mercado realizando prestação de serviços tecnológicos
para empresas públicas e privadas, órgãos públicos, associações, entidades e
pessoas físicas, através da realização de ensaio, calibração, consultoria, inspeção,
pesquisa , desenvolvimento, extensão tecnológica e informação tecnológica. As
atividades desenvolvidas concentram-se nas seguintes áreas: Alimentos; Engenharia
de Edificações; Materiais de Construção Civil; Engenharia Eletro-Eletrônica;
Tecnologia Metal-Mecânica; Engenharia de Processos; Química; Geotecnia.
Também conta com uma incubadora, a qual proporciona um espaço para
colocar em prática ações inovadoras de base tecnológica.

3
2) REVISÃO BIBLIOGRÁFICA
2.1) A Transesterificação de óleos vegetais
A reação de transesterificação para a produção de biodiesel é a formação de
um éster a partir de glicerídeos e álcoois. Tipicamente ela é feita na presença de
catalisadores, com metanol ou etanol como álcool e óleos vegetais como fonte de
triglicerídeos, como mostra a figura abaixo: (DAMBISKI, 2007)
Figura 1: Reação de transesterificação (DAMBISKI, 2007)
Como pode ser visto na figura acima, a reação consiste em:
álcool + triglicerídeo = éster + glicerina
Mais especificamente:
onde A representa o álcool, C é o éster, TG, DG e MG são, respectivamente,
triglicerídeo, diglicerídeo e monoglicerídeo. A glicerina formada está representada por
D.

4
Figura 2: Concentração molar dos glicerídeos em função do tempo do tempo.
(Carvalho et al, simulado no EMSO)
Esta é uma reação de equilíbrio e em tres etapas . Por motivos práticos, as três
reações podem ser representadas em uma única reação global, dessa forma:
onde a letra B agora representa o triglicerídeo.
2.2) Principais rotas de produção
Diferentes tipos de reação (rotas de produção) podem ser usados na produção
de biodiesel em escala industrial(REFERÊNCIAS, PELO MENOS 2), sendo estas
rotas identificadas principalmente pelo catalisador utilizado. SãO elas:
- Homogênea (catálise ácida ou alcalina)
- Heterogênea (catálise por óxidos ou carbonatos)
- Enzimática (lipases atuam como catalisador)
- Supercrítica (sem catalisador)
Os diferentes métodos são comparados na tabela abaixo:

5
Tabela 1: Comparação entre as diferentes rotas de produção de biodiesel
2.3) Vantagens e desvantagens do processo sem catalisador
2.3.1) Sem catalisador – visão geral
De modo geral, o processo sem catalisador apresenta as seguintes vantagens
e desvantagens: (Kusdiana & Saka, 2000; Silva et al, 2007; Kusdiana & Saka, 2003;
Kiwjaroun et al,2008)
• Vantagens
- A presença de água melhora o processo, pois ajuda na remoção da glicerina.
A possibilidade da presença de água é a grande vantagem da rota supercrítica.
(Kusdiana & Saka, 2000). A presença de água pode ainda beneficiar a cinética
através da hidrólide do TG em ácidos graxos, mais reativos que o próprio TG.
(Minami & Saka, 2004; Kusdiana &Saka, 2003)
- Pode ser usado qualquer tipo de óleo (inclusive óleo de fritura) ou gordura
animal (sobras do abate de gado, por exemplo).
- Ácidos graxos livres contribuem positivamente para a reação (Minami & Saka,
2004).
- Alta pureza da glicerina como subproduto.
2.3.2) Comparação com a rota alcalina
A transesterificação em meio homogêneo alcalino utiliza bases fortes como
catalisador. É o principal meio de produção industrial de biodiesel, sendo útil uma
comparação com a rota supercrítica utilizada neste trabalho.
6
Tabela 2: Comparação entre a rota alcalina e a rota supercrítica
2.4) Efeito das condições de processo
Dambiski (2007) comparou divesras condições de processo(temperatura,
pressão e razão álcool-óleo) utilizadas em diversos trabalhos, bem como os resultados
importantes (tempo de reação, conversão do óleo em biodiesel) dos mesmos.
Analisando o efeito de cada condição em particular:
• Temperatura
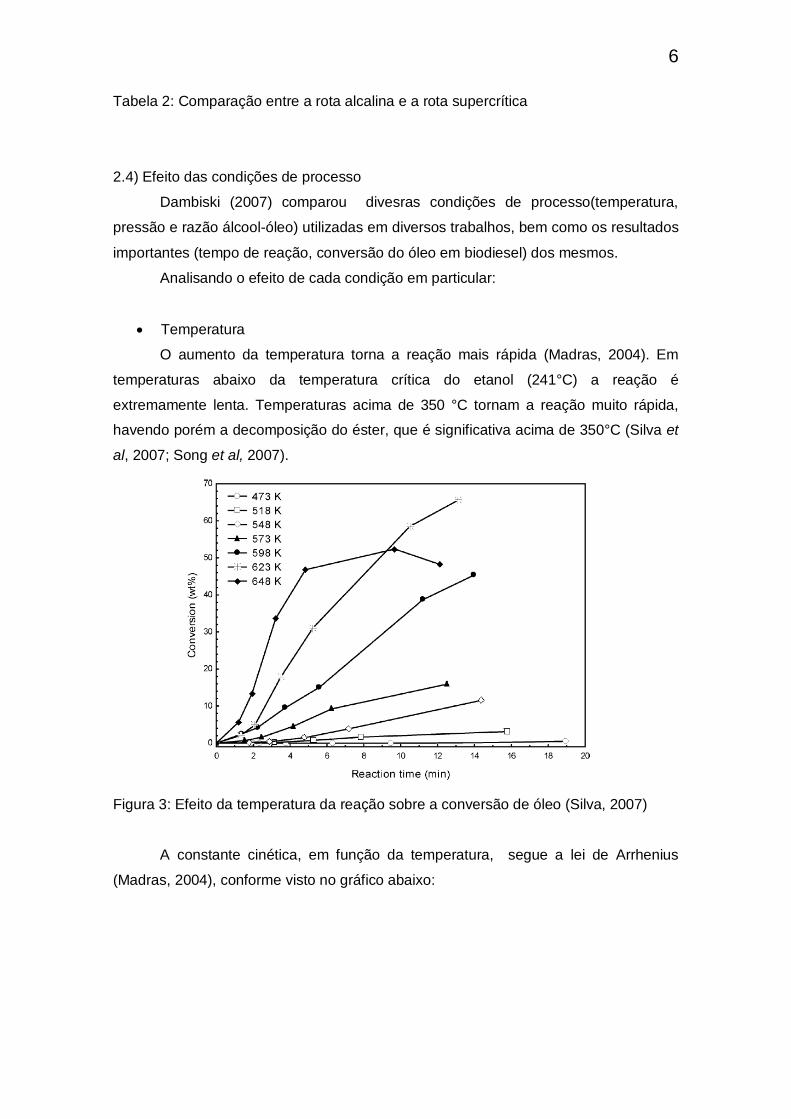
O aumento da temperatura torna a reação mais rápida (Madras, 2004). Em
temperaturas abaixo da temperatura crítica do etanol (241°C) a reação é
extremamente lenta. Temperaturas acima de 350 °C tornam a reação muito rápida,
havendo porém a decomposição do éster, que é significativa acima de 350°C (Silva et
al, 2007; Song et al, 2007).
Figura 3: Efeito da temperatura da reação sobre a conversão de óleo (Silva, 2007)
A constante cinética, em função da temperatura, segue a lei de Arrhenius
(Madras, 2004), conforme visto no gráfico abaixo:

7
Figura 4: constante de equilíbrio versus temperatura: gráfico de Arrhenius (Madras,
2004)
• Efeito da Pressão
Silva et al (2007) analisaram o efeito da pressão:
Figura 5: Efeito da pressão sobre a conversão. (Silva et al, 2007).
Conforme visto no gráfico acima, o aumento da pressão aumenta a conversão
final. Porém, a reação se torna um pouco mais lenta em pressões muito elevadas,
devido principalmente a efeitos de transporte (difusão) das moléculas no meio
reacional (Silva et al, 2007).
O volume molar, em estados próximos ao ponto crítico, é muito afetado pela
pressão (Dambiski, 2007). Visto que a velocidade da reação é afetada pela densidade
do meio (Madras, 2004), pode-se concluir que, próximo ao ponto crítico do álcool, a
pressão exerce forte influência sobre a taxa da reação. Sendo assim, um rigoroso
controle de pressão se faz necessário a fim de se ter um bom controle da cinética da
reação.

8
• Efeito da rezão molar álcool/óleo (RAB)
O aumento da proporção entre álcool e óleo, no início da reação, desloca o
equilíbrio na direção da formação de biodiesel (Madras, 2004; Song et al, 2007;
Kusdiana & Saka, 2000).
Noureddini & Zhu (1997) apud Carvalho et al. utilizaram um modelo que ilustra
bem o efeito da RAB na conversão da reação, junto com seus intermediários.
Figura 6: Efeito da razão álcool-óleo sobre a conversão de óleo (Carvalho et al,
simulado no EMSO)
A razão álcool-óleo é mais significativa, em termos cinéticos, no início da
reação, sendo que a velocidade é maior para maiores razões molares (Kusdiana &
Saka, 2000; Madras,2004). a RAB também aumenta a superfície de contato entre as
fases (Kusdiana & Saka, 2000; Dambiski, 2007).
Um efeito negativo de se usar uma alta RAB é a dificulade de remoção da
glicerina após a reação, devido à miscibilidade parcial do sistema álcool-éster-
glicerina. O álcool dissolve a glicerina que dissolve o éster. O triglicerídeo, porém,
forma uma única fase com o éster (Song et al, 2007).
2.5) O uso do etanol
O uso do etanol na produção de biodiesel confere ao combustível o rótulo
“verde”, se o etanol tiver origem da cana de açúcar. (Shimoyama et al, 2007; Melo,
2007 apud Santos, 2008; Dambiski, 2007)
Para fins de comparação, relacionou-se alguns dados na produção do biodiesel
pela via metílica em comparação com a etílica, como mostra a tabela abaixo: (Melo,
2007 apud Santos, 2008; Dambiski, 2007)

9
Tabela 3: Comparação entre o etanol e o etanol para a produção de biodiesel
2.6) Observações sobre a cinética no supercrítico
Na reação utilizando álcool supercrítico, a cinética é favorecida por 2 motivos:
- Formação de única fase: A constante dielétrica do álcool diminui em
temperatura maior que sua respectiva temperatura crítica, tornando-o miscível com os
compostos apolares (Song et al, 2007). Caso contrário, reação ocorre em 2 fases
líquidas e há resistência à transferência de massa entre as fases, o que leva à
diminuição da conversão e da taxa de reação (Kusdiana & Saka, 2000).
- Enfraquecimento das ligações de hidrogênio: No supercrítico (SC), o
álcool se comporta como monômero livre, pois as ligações de hidrogênio são mais
fracas em comparação com as condições subcríticas. Kusdiana et al, (2004) apud
Dambiski (2007) mostra que o número de ligações de hidrogênio cai de 1,93 (a 25°C,
1 atm) para 0,7 (250 °C, 20 Mpa).
Outro fenômeno interessante da cinética no SC é a autocatálise dos ácidos
graxos livres (free fatty acids, FFA) na presença de água: Minami & Saka (2006),
mostraram que, com um teor de 10% de FFA no óleo, a velocidade e o rendimento da
reação são fortemente beneficiados nos primeiros minutos da reação. Porém, a
velocidade reacional é diminuída com o aumento da razão álcool-óleo, o contrário do
que acontece com baixos teores de FFA. Minami & Saka (2006) também compararam
a cinética de 1 passo (alcoólise, normalmente empregada) com a de 2 passos
(hidrólise + alcoólise) e concluiram que a cinética desta exige condições menos
severas que as da primeira (alcoólise).

10
3 TEORIA E MÉTODOS COMPUTACIONAIS
Neste trabalho, os conceitos básicos de termodinâmica, cinética, estmação de
propriedades e modelagem matemática serão brevemente revistos. Também haverá
uma breve descrição dos softwares utilizados.
3.1) Termodinâmica
• Equilíbrio de fases
Para o equilíbrio líquido-vapor, a condição de equilíbrio de fases é dada por:
onde e são as fugacidades (Pa) do vapor e do líquido, respectivamente,
estando o componente i na solução.
A fugacidade do componente i é definida como:
onde (fase) pode ser líquido ou vapor, e é substituído por se for fase líquida e por
caso a fase seja vapor. Dessa forma, igualando as fugacidades tem-se que:
onde é a constante de distribuição do componente i entre o vapor e o líquido.
• Equilíbrio químico.
- Condição de equilíbrio químico
A condição termodinâmica de equilíbrio químico é que a energia livre
de Gibbs atinge um mínimo quando uma dada reação atinge o equilíbrio.
- Cálculo da constante de equilíbrio
A constante de equilíbrio é dada por:
onde é a atividade do componente i na reação. A atividade, por sua vez, é
definida por:

11
Para fins de simplificação, a constante de equilíbrio aparente é deduzida, a
partir da equação anterior, como
onde é a concentração (kmol/m3) do componente i.
3.2) Cinética
• Estequiometria:
O coeficiente estequimetrico ( )de uma reação é a proporção na qual cada
componente participa da mesma.
• Equação da taxa reacional:
A velocidade de reação é dada por:
onde (kmol/m3/s)é a taxa da reação, N (kmol) é o vetor do número de moles do
sistema, V (m^3)o volume reacional e é a constante cinética cujas unidades
dependem da forma com que se expressa a taxa da reação. Para uma taxa na forma
da Lei das Potências, a expressão (666) se torna:
onde é a concentração (kmol/m3) do componente i e u é a ordem da reação.
Dessa forma, a dimensão da constante cinética é s-1.(m3/kmol)(u-1).
• Conversão e avanço da reação
Existem várias maneiras de expressar o quanto uma reação avança ao longo
do tempo. Uma delas é o avanço da reação, dado por
é o número de mols de i no início da reação, enquanto é o número de
moles do componente i num dado instante.
• Constantes cinéticas em função da temperatura.
As constantes cinéticas seguem, em geral, a relação de Arrhenius, que é

12
(J) é a energia de ativação da reação. O parâmetro A é o fator pré exponencial da
equação.
Uma forma mais geral desta equação é:
onde A, B, C e D são parâmetros ajustáveis.
3.3) Balanço material
O blanço material é caracterizado pela seguinte relação:
Entra – Sai + Gerado = Acúmulo (eq.12)
3.4) Estimação das propriedades dos fluidos
As seguintes propriedades termodinâmicas foram estimadas: fator acêntrico
( ), pressão de vapor (Pvap ), pressão crítica (PC), temperatura crítica (TC),
temperatura normal de ebulição (TB) e volume molar na CNTP (Vm).
Para estimar PC , TC , TB e Vm , o método utilizado é descrito por Constantinou
& Gani (2008). Este método utiliza a contribuição de cada grupo prsente na molécula,
cujo valor (do grupo) é tabelado para cada propriedade que se quer estimar. A
contriguição de cada grupo presente na molécula é somada e entra no cálculo da
propriedade estimada da seguinte maneira:
onde é uma função da propriedade a ser estimada, Ni é o número de vezes que o
grupo i (primeira ordem) ou j (segunda ordem) aparecem na molécula, Cx é a
contribuição do grupo (i ou j) tabelada para cada grupo funcional.
Para o cálculo do fator acêntrico, a seguinte equação é utilizada (Reid):
onde Pc está em atm e θ = TB / TC .
3.5) O Software EMSO
Criado por Rafael de Pelegrini Soares e desenvolvido pelo projeto ALSOC, o
EMSO (Enviroment for Modeling, Simulation and Optimization) é o simulador baseado
em equações utilizado neste trabalho. O EMSO utiliza uma linguagem de modelagem
própria, a fim de descrever qualquer modelo utilizado.
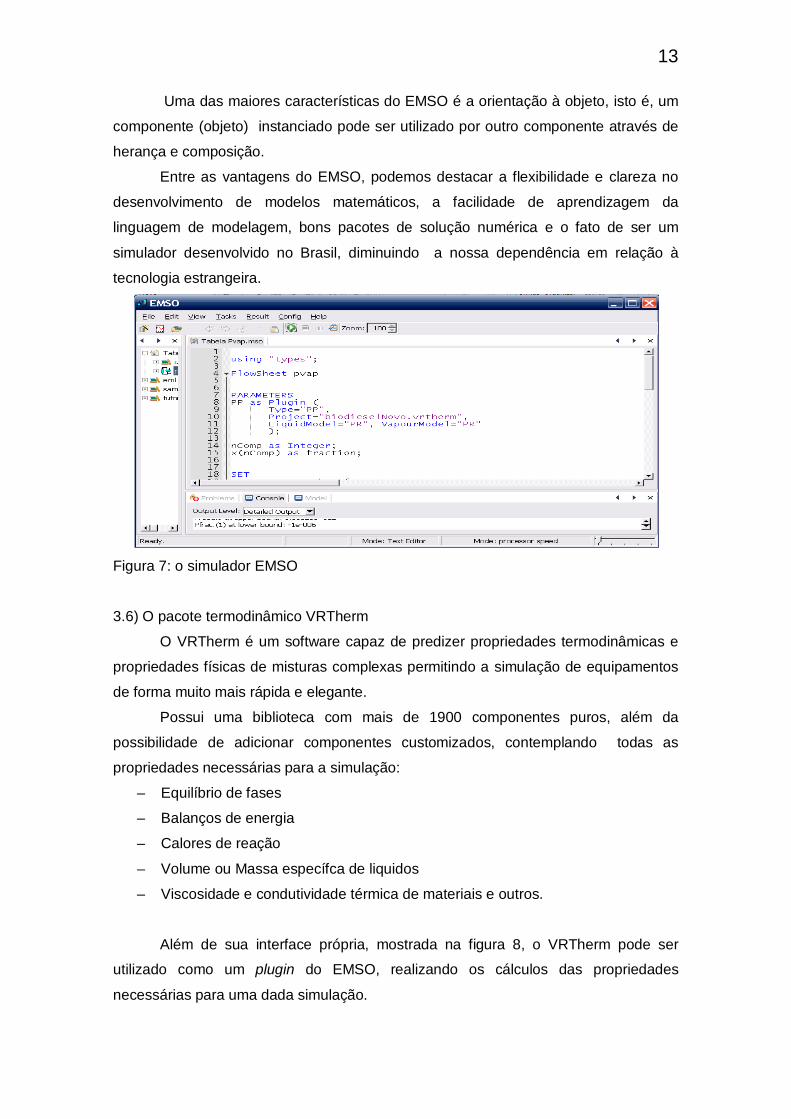
13
Uma das maiores características do EMSO é a orientação à objeto, isto é, um
componente (objeto) instanciado pode ser utilizado por outro componente através de
herança e composição.
Entre as vantagens do EMSO, podemos destacar a flexibilidade e clareza no
desenvolvimento de modelos matemáticos, a facilidade de aprendizagem da
linguagem de modelagem, bons pacotes de solução numérica e o fato de ser um
simulador desenvolvido no Brasil, diminuindo a nossa dependência em relação à
tecnologia estrangeira.
Figura 7: o simulador EMSO
3.6) O pacote termodinâmico VRTherm
O VRTherm é um software capaz de predizer propriedades termodinâmicas e
propriedades físicas de misturas complexas permitindo a simulação de equipamentos
de forma muito mais rápida e elegante.
Possui uma biblioteca com mais de 1900 componentes puros, além da
possibilidade de adicionar componentes customizados, contemplando todas as
propriedades necessárias para a simulação:
– Equilíbrio de fases
– Balanços de energia
– Calores de reação
– Volume ou Massa específca de liquidos
– Viscosidade e condutividade térmica de materiais e outros.
Além de sua interface própria, mostrada na figura 8, o VRTherm pode ser
utilizado como um plugin do EMSO, realizando os cálculos das propriedades
necessárias para uma dada simulação.

14
Figura 8: interface gráfica do VRTherm
3.7) Estimação de parâmetros
A estimação de parâmetros, através de dados experimentais, é feita pela
minimização de uma função objetivo que relaciona dados experimentais com dados
previstos pelo modelo. Para o caso em que esta função objetivo é o somatório do
quadrado da diferença entre um dados experimentais e dados calculados, a estimação
é conhecida por Mínimos Quadrados e é dada por:
onde V é a variável a ser comparada.

15
4) MATERIAIS E MÉTODOS
4.1) O reator
Santos (2008) descreve o reator utilizado nos experimentos. O material é aço
inox, possui controle de temperatura, agitação magnética e indicador de pressão. O
volume total é de 974 cm3 mas, devido á dilatação do material, seu volume foi
considerado como 985 cm3 na temperatura da reação. (Santos, 2008)
4.2) Óleo de soja:
Vem da Oleoplan, de Veranópolis. É composto por mais de 95% de
tracilgliceróis.
A composição média do óleo de soja é:
• Ácidos graxos (insaturações; %massa; símbolo): linoleico (18:2 ; 53%; L),
oleico (18:1 ;23%; O), palmítico(16:0; 11%, P), linolênico (18:3; 8%; Ln),
esteárico (18:0; 4%; St).
• Principais triacilgliceróis: LLL (17,6%), LLO (15,3%), LLP (10,2%), LLLn
(7,9%), LLSt(4,2%) e outros
Por simplificação, o óleo de soja é composto por somente LLL (uma
trilinoleína), e o biodiesel (éster) derivado é o etil linoleato.
4.3) O etanol: Anidro, da marca Vertec. Suas propriedades são conhecidas, estão no
banco de dados do Vrtech e não serão descritas aqui.
4.4) Propriedades estimadas
As propriedades fisico-químicas do óleo de soja e do biodiesel foram
estimadas pelos métodos de grupos funcionais descrito no capitulo 3 (Constantinou &
Gani, 1994; Reid et al) e são mostrados no quadro a seguir:

16
Tabela 4: Propriedades estimadas do biodiesel (≈ etil linoleato) e óleo de soja (≈
trilinoleína).
4.5) Dados experimentais
Santos (2008) realizou 5 dos 6 experimentos utilizados neste trabalho. O
último foi realizado posteriormente na CIENTEC.
Os experimentos estão registrado com os nomes originais das corridas
(experimento em função do tempo). A seguir, segue uma tabela geral dos
experimenos, suas condições e massas de etanol e óleo adicionadas.
Tabela 5: Condições gerais das corridas realizadas
A conversão da corrida 67 tinha uma incerteza experimental de ±9,8 % e seu
valo, mesmo podendo estar muito errado, será considerado nos cálculos.
• Gráficos das corridas
Os gráficos totais das corridas, desde o início do aquecimento até o fim do
experimento, são mostrados a seguir:

17
Figura 9: Temperatura e pressão verusus
tempo total de reação e pressão versus
temperatura
A despeito do tempo de aquecimento, considera-se que a reação inicia no
momento em que o set point de temperatura é alcançado. Logo, o período de
aquecimento do reator (cerca de 3,3 h em média) será desconsiderado das análises.

18
5 . MODELAGEM DO SISTEMA
5.1) Simplificações
As seguintes simplificações foram feitas sobre o sistema:
• Componentes:
- O óleo de soja é composto somente por triglicerídeos, do tipo LLL, isto é,
somente triglicerídeo de ácido linoleico. Não há ácidos graxos livres no óleo.
- O biodiesel formado é o etil linoleato.
• Reação
- A reação é de equilíbrio, de primeira ordem em relação aos reagentes e não
há intermediários na reação.
- A reação interfacial e a transferência de massa entre as fases foram
desprezadas.
- O volume da reação é o volume do reator (constante).
- A constante cinética depende apenas da temperatura.
- A constante de equilíbrio depende apenas da composição final do sistema e
varia apenas com a temperatura.
- A reação avança até o equilíbrio, sendo que o mesmo é atingido até o tempo
especificado (medição final) . As conversões e a queda de pressão total,
medidas no fim da reação, são as respectivas variáveis de equilíbrio.
- A queda de pressão no reator é proporcional ao avanço da reação, sendo que
o etanol, por ser mais volátil e por estar em excesso, é o único responsável
pela pressão do sistema.
- A reação só começa a ocorrer depois que o set point da temperatura é
atingido.
- A constante de proporcionalidade é a razão entre a queda de pressão no
equilíbrio e o avanço total de equilíbrio.
• Reator
- O reator é tratado, a princípio, como uma caixa preta, sendo ignorados todos
os fenômenos que ocorrem dentro do mesmo, exceto a reação química e um
possível equilíbrio de fases.

19
- Qualquer dinâmica que não seja a cinética (temperatura, pressão e trocas de
calor, por exemplo) é desprezível.
- O set point de temperatura é atingido instantaneamente e a pressão é medida
a partir deste ponto.
• Equilíbrio de fases
- Quando houver equilíbrio líquido-vapor, haverá apenas etanol na fase vapor.
- Quando houver equilíbrio líquido-vapor, a reação acontecerá apenas na fase
líquida.
- O volume do vapor é desprezível frente ao do líquido.
- Qualquer possível estado de equilíbrio é atingido instantaneamente.
5.2) Modelagem do reator
Do balanço de massa mostrado na seção 666, temos, para o componente i:
{entra} – {sai} + {gerado} = {acúmulo}
Substituindo o número de moles do componente i pela diferença entre o
número de moles inicial e o avanço estequiométrico de i, temos:
Para o cálculo da queda de pressão do reator, temos:
A constante J (Pa/kmol) é a razão entre a queda de pressão total e o avanço
total:
A pressão do sistema é caculada, então, como sendo:
onde (Pa) é a pressão inicial do sistema, medida experimentalmente.
• Observação: A presença da constante de proporcionalidade no modelo lembra
uma espécie de “ajuste de curva”, a fim de se trabalhar diretamente com os
dados experimentais. A origem de toda esta modelagem é a constante J, cuja
dedução está em anexo.
5.3) Modelagem da reação

20
A reação de transesterificação é dada pela seguinte equação:
3 C2H5O + 1 C57H98O6 3 C20H36O2 + C3H8O3
Por comodidade, os nomes das substâncias e suas fórmulas serão substituídos
pelas letras A, B, C e D na ordem em que aparecem na reação. A relação
estequiométrica será relativa ao etanol (A), visto que este é o componente mais
importante da modelagem da pressão. A reação fica escrita, então, como:
O vetor número de moles fica calculado na equação acima , reduzindo o
sistema de 4 equações diferenciais para apenas uma, em função do avanço da reação
( ).
A constante de equilíbrio, simplificada para o caso em que o equilíbrio depende
apenas da concentração molar no sistema em equilíbrio, é:
A taxa da reação então fica:
Duas variáveis importantes para a visualização dos resultados são a fração
molar total, e a conversão molar de óleo em éster.
Por comodidade, as massas iniciais do sistema serão fornecidas como
especificação de entrada, a fim de calcular o número de moles no início da reação.
onde é a massa molar (kg/kmol) e é a massa de cada componente no início
da reação.
5.4) Sistema de equações
Juntando tudo o que foi visto acima, temos o sistema completo de equações
que será implementado no software EMSO.

21
- Parâmetros: , , , , , total: 4 + 2.i
- Variáveis: , , , , , , , , , total: 7 + 4.i
- Equações: 5 + 3.i
- Especificações: , , total: 2 + i
- Graus de liberdade: 0
- Equações diferenciais: 1
- Condições iniciais: total: 1
- Graus de liberdade dinâmicos: 0
A simulação está pronta para rodar. Mas é interessante observar que e
mesmo sendo funções da temperatura, são considerados parâmetros, pois a
temperatura é considerada constante para cada corrida. Os valores de e são
calculados separadamente, a partir dos dados experimentais, enquanto será
estimado por mínimos quadrados não lineares, usando os dados de pressão.
O código EMSO deste modelo se encontra em anexo.
5.5) Estimação de parâmetros
O parâmetro é estimado pelo método de Mínimos Quadrados, isto é, a
minimização da seguinte função:
, onde é o dado experimental de pressão em um determinado tempo, é o
dado equivalente calculado pelo modelo e é o tempo total no qual a reação atinge o
equilíbrio (tempo final).
5.6) Considerações sobre o modelo:

22
• Erro aleatório do medidor de pressão
A medição da pressão foi feita em um indicador de pressão acoplado ao reator.
Tal indicador apresentava grandes erros aleatórios, chegando ao ponto de, em alguns
casos, apresentar pressão absoluta negativa no começo da reação.
• Efeito das altas pressões sobre o sistema
Muitas das simplificações comumente empregadas na literatura levam em
consideração que a pressão não influencia significativamente o sistema,
principalmente no caso de misturas líquidas. Porém, altas pressões causam grandes
desvios de idealidades próximos ao ponto crítico de qualquer um dos componentes.
As altas pressões influenciam este modelo em 2 casos:
- Constante de equilíbrio: conforme visto na equação 4, a constante de
equilíbrio depende das atividades de cada componente. Estas são função da
fugacidade de cada componente que, por sua vez, é função da pressão do sistema. A
simplificação da constante de equilíbrio para sua dependência exclusiva da
composição do sistema pode levar a grandes erros de modelagem. (Smith et al)
- Equação cúbica de estado: equações cúbicas apresentam , em altas
pressões, um falso ponto crítico (geralmente em temperaturas menores que o
verdadeiro ponto crítico) e falsas raízes na região de equilíbrio líquido-vapor (LVE).
(Reid et al)
• Tempo de aquecimento – conversão inicial
A reação foi considerada como nula durante o tempo de aquecimento do reator
(> 3h). Na prática, em 3 horas de reação, a conversão pode chegar em até 10% em
peso de óleo, na maior temperatura trabalhada no experimento (265°C = 538 K)
(Madras, 2004).
• Propriedades críticas estimadas
As propriedades críticas do óleo e do biodiesel, mostradas no capítulo 4, foram
obtidas por métodos de regressão numérica a partir dos grupos constituintes de cada
molécula. No entanto, estas propriedades são totalmente teóricas, visto que não
podem ser verificadas experimentalmente devido ao fato de que as moléculas se
decompõem em temperaturas muito inferiores às temperaturas críticas em questão. E
como as equações de estado dependem desses parâmetros críticos, há uma grande
incerteza quanto à validade dos resultados gerados pelas equações de estado.

23
6) ANÁLISE E DISCUSSÃO DOS RESULTADOS
6.1) Análise dos experimentos
6.1.1) Tabela dos experimentos (corridas)
Um resumo de todas as corridas realizadas encontra-se abaixo:
Tabela 6: Comparação geral dos valores da constante cinética e dos valores
experimentais para cada corrida.
Por praticidade, os nomes originais das corridas foram mantidos.
6.1.2) Tendências gerais
Juntando as informações globais de todos os experimentos,pode-se perceber
algumas tendências gerais a partir dos gráficos abaixo:
• Queda de pressão vesus tempo: todas as corridas

24
Figura 10: Dados expermentais da queda de pressão versus tempo de reação.
• Constante cinética versus temperatura (gráfico de Arrhenius):
Figura 11: constante cinética versus tempo: dados experimentais e reta ajustada.
Pelo que se pode perceber do gráfico abaixo, a constante cinética não
apresentou uma tendência verdadeiramente linear, apesar de que os pontos acima da
do etanol tem valor maior que os abaixo da temperatura crítica. O ponto mais à
esquerda foi o da corrida 61, cuja temperatura (265°C) foi a maior de todas. No
entanto, sua constante cinética foi menor que as corridas 58 e 59, de 250°C e 263°C
respectivamente. Possivelmente a influência da densidade do sistema foi
predominante e mais corridas nessa temperatura deveriam ser feitas.
A energia de ativação calculada foi de 0,6 kJ/mol, inferior ao valor de 2 kJ/mol
encontrado por Madras (2004).

25
• Constante de equilíbrio versus temperatura
Figura 12: Constante aparente de equilíbrio versus temperatura.
Nenhuma informação significativa pode ser tirada deste gráfico devido ao fato
de haver poucos experimentos em cada condição.
• Influência da pressão inicial
A constante de equilíbrio, o fator de ajuste da pressão e a queda de pressão
total parecem ser influênciados pela pressão total do sistema, como mostram os
gráficos a seguir:

26
Figura 13: influência da pressão inicial sobre Ke, J e ΔPe
De acordo com os gráficos acima, a pressão inicial do sistema, com o seu
aumento, faz diminuir a constante de equilíbrio, mas aumenta a queda de pressão total
e o fator de ajuste. É interessante notar que o único ponto que ficou fora das
tendências gerais foi a corrida 59, que apresenta o maior valor de pressão inicial.
6.2) Discussão dos resultados
6.2.1) Gráficos das corridas
Ao comparar os dados experimentais com os valores calculados, é possível
visualizar a confirmação (ou não) da hipótese de que a queda de pressão é
proporcional ao consumo de etanol. Para isso, dois gráficos serão de grande auxílio
nesta visualização: a pressão do sistema versus o tempo da reação e a pressão do
sistema versus a conversão de óleo. O segundo gráfico é mais indicado para
visualização da corrida, enquanto o primeiro é o que realmente descreve a realidade,
visto que a conversão também é uma variável calculada pelo modelo.
As corridas 58, 59 e 61 representam um sistema (provavelmente) homogêneo;
a corrida 62 tem líquido subresfriado (comprimido) no reator e as corridas 65 e 67
estão na condição de LVE. Os gráficos de cada um destes conjuntos são mostrados
abaixo:
Podemos observar, pelas figuras acima, que na situação de temperatura
superior à crítica do etanol a queda de pressão variou quase que linearmente com a
pressão, confirmando a hipótese principal do trabalho. É interessante notar que, na
corrida 58 (T > TC, P < PC), o erro relativo é maior no último ponto devido a um
possível equilíbrio líquido-vapor do etanol no fim da reação, sendo que este muito
provavelmente se deve à formação de glicerina que é polar e parcialmente miscível
com o biodiesel.

27
Figura 14: corrida 58
Figura 15: Corrida 59
Figura 16: Corrida 61

28
Figura 17: Corrida 62
Figura 18: Corrida 65
Figura 19: Corrida 67
Para as corridas com temperatura abaixo da temperatura crítica, nota-se
grandes desvios dos dados experimentais em relação ao modelo. Porém, na corrida
62 os desvios são menores que até mesmo ás corridas no supercrítico. Isto se deve
ao fato de que, na corrida 62, tem-se líquido subresfriado, enquanto nas corridas 65 e
67 tem-se LVE e este influencia fortemente a pressão do sistema, que está no ponto
de bolha nestes casos. Observa-se que o desvio é maior quanto menor for a
temperatura e a razão álcool-óleo utilizada (corrida 67).
O erro relativo do modelo em relação aos dados experimentais srá analisado
na seção a seguir.
29
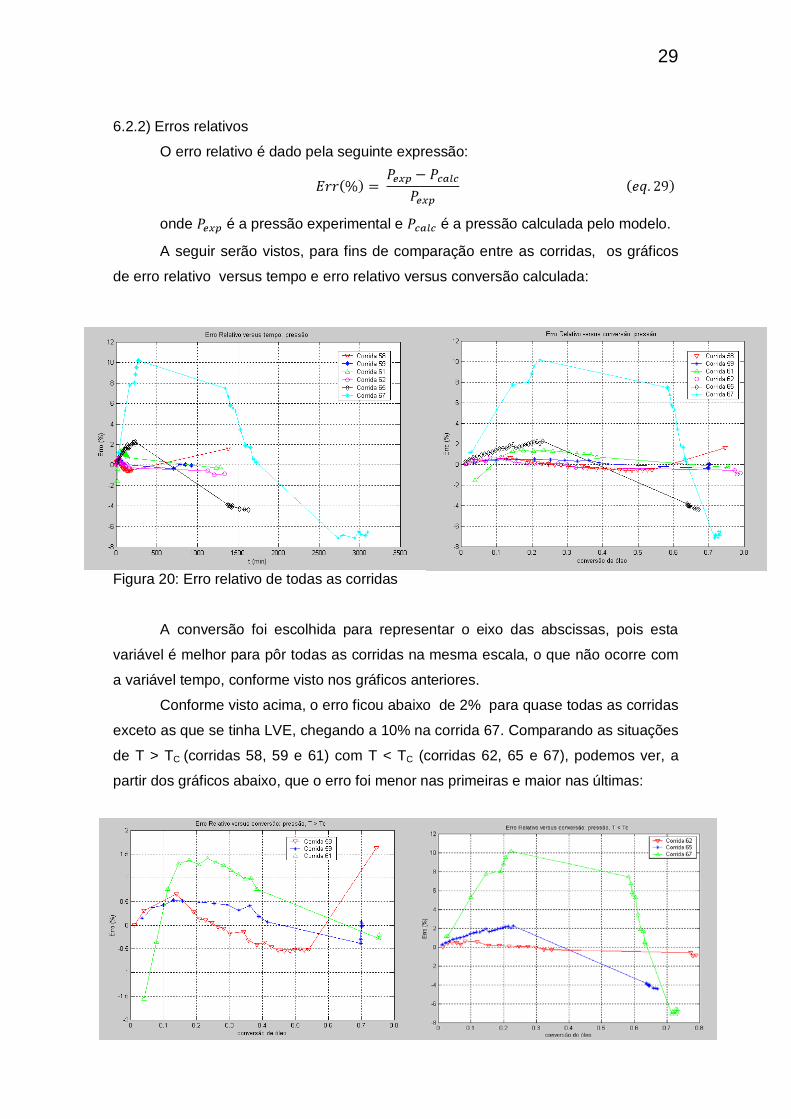
6.2.2) Erros relativos
O erro relativo é dado pela seguinte expressão:
onde é a pressão experimental e é a pressão calculada pelo modelo.
A seguir serão vistos, para fins de comparação entre as corridas, os gráficos
de erro relativo versus tempo e erro relativo versus conversão calculada:
Figura 20: Erro relativo de todas as corridas
A conversão foi escolhida para representar o eixo das abscissas, pois esta
variável é melhor para pôr todas as corridas na mesma escala, o que não ocorre com
a variável tempo, conforme visto nos gráficos anteriores.
Conforme visto acima, o erro ficou abaixo de 2% para quase todas as corridas
exceto as que se tinha LVE, chegando a 10% na corrida 67. Comparando as situações
de T > TC (corridas 58, 59 e 61) com T < TC (corridas 62, 65 e 67), podemos ver, a
partir dos gráficos abaixo, que o erro foi menor nas primeiras e maior nas últimas:

30
Figura 21: Comparação do erro relativo em relação à faixa de temperatura.
É importante salientar que a corrida 62, por se constituir de líquido sub-
resfriado, apresentou um erro semelhante aos das corridas supercríticas. Isto é devido
à homogeneidade do sistema, fato este que torna a pressão dependente apenas da
quantidade de etanol e não às suas relações de equilíbrio. Nas corridas 65 e 67, tem-
se LVE e a pressão depende tanto da quantidade de etanol como da sua relação de
equilíbrio com a fase líquida.
Outro fato digno de nota é a ocorrência de máximos e mínimos nos erros
relativos. Os máximos (Pexp > Pcalc ) ocorrem devido à formação dos intermediários DG
e MG, desconsiderados pelo modelo. Já os pontos de mínimo (Pexp < Pcalc) ocorrem
devido à interação do etanol com a glicerina formada (ambos compostos polares). Na
corrida 58 há uma possível formação de heterogeneidade no fim do experimento,
levando ao grande erro relativo ao final do mesmo.
Por último é necessário salientar que, na corrida 59, o aquecimento do reator
foi desligado devido ao fato de que a pressão do sistema foi maior que a pressão
máxima de operação segura do reator e, a qualquer momento, o selo de segurança
seria rompido. Isto levou à alguns erros experimentais, mas estes erros não foram
grandes o suficiente para se descartar o experimento.

31
7) CONCLUSÕES
Diante de tudo o que foi exposto neste trabalho, as seguintes conclusões
podem ser tiradas:
• Os experimentos são insuficientes e pouco adequados
O número de experimentos, bem como seu planejamento, foram insuficientes.
Para se ter uma avaliação adequada da cinética em função da temperatura, seria
necessário a realização de, pelo menos, 3 experimentos variando apenas a
temperatura, com a massa total e a razão álcool-óleo fixadas. Para se ter uma idéia da
influência da razão álcool-óleo, repetir-se-ia as corridas com a mesma faixa de
temperatura, porém variando a razão álcool-óleo. Quanto à massa adicionada no
reator, esta exerce uma forte influência na pressão inicial e esta, conforme visto no
capítulo 6, influencia a constante de equilíbrio, a conversão de equilíbrio, o fator de
pressão e a queda de pressão total no reator. Um dos experimentos (corrida 59) ficou
fora da tendência da presão inicial, o que necessitaria maior investigação para
determinar se foi um erro experimental ou se realmente é uma tendência da
termodinâmica do sistema.
A maior incerteza dos experimenos é, sem dúvidas, o fato da constante de
equilíbrio calculada para a corrida 61 (T = 265°C, RAB = 54) ser menor que para a
corrida 58 (T = 250 °C, RAB = 18) e a corrida 59 (T = 263 °C, RAB = 18). Vários autores
(Madras, 2004; Diasakou et al, 1998; Silva et al, 2007; Dambiski, 2007) confirmam que
a constante cinética aumenta tanto com o aumento da razão álcool-óleo como com o
aumento da temperatura: as duas situações ocorridas no experimento e que levaram a
uma constante cinética menor. Devido principalmente a este fato que mais
experimentos deveriam ter sido feitos e não o foram.
Para tornar as corridas menos sujeitas a erros tendenciosos, seria bom tornar o
tempo de aquecimento o menor possível, de preferência menor que meia hora. Os
altos tempos de aquecimento podem levar a altas incertezas sobre a conversão inicial,
prejudicando a análise dos resultados.
Outro fator de erro foi a falta de um registrador de pressão. Dados entre as 16h
de um dia e as 9h do próximo dia não puderam ser medidos, pois não havia
operadores e verbas suficientes para cumprir esta faixa de horário, levando a uma
grande incerteza sobre os fenômenos que poderiam estar acontecendo dentro do
reator.

32
• Reação lenta e conversão baixa.
Madras (2004) verificou que a conversão final é muito baixa, bem como a taxa
reacional é muito lenta para temperaturas abaixo da temperatura crítica do álcool.
Silva (2007) notou uma conversão final baixa e reação demasiado lenta para
temperaturas abaixo de 325°C. Santos (2008), utilizando os mesmos experimentos
que este trabalho, concluiu que a transesterificação do óleo de soja, nas condições de
temperatura, pressão e meio reacional empregadas, não é viável industrialmente. Os
gráficos a seguir ilustram esta conclusão:
Figura 22: Comparação da conversão estimada pelo modelo
Conforme a figura acima, a corrida de maior taxa (59) alcança conversão molar
de cerca de 0,6 em 3 horas de ração, segundo o modelo proposto. Já na corrida de
maior conversão final (67), essa tem valor máximo de 0,8, segundo os experimentos.
Mas este valor só é atingido em mais de 24h, o que torna a utilização industrial destas
condições (T=200°C) economicamente inviável (Santos, 2008).
• Necessidade de refinamento do modelo:
Previsão através de equação de estado, ao invés de se utilizar apenas dados
externos e ajustes de curva. Esta é uma opção mais nobre em termos de modelagem,
visto que esta opção se fundamenta em princípios básicos ao invés de ajustar
matematicamente os modelos, cuja abordagem acaba por ser puramente matemática.
• Sugestões para trabalhos futuros:
- Analisar a cinética utilizando co-solventes (CO2, éteres, etc) que tornem as
condições de processo mais brandas (menores T e P).

33
- Analisar a adição de água subcrítica, sob o ponto de vista da hidrólise dos
triglicerídeos (reação em duas etapas).
- Comparar dados de conversão do óleo, calculados pelo modelo, com os
obtidos experimentalmente por análise química.
• Conclusão Final: O método, apesar de suas limitações, merece ser
investigado.
– Só pode ser usado em reator tipo batelada
– Altas não-linearidades da pressão
– k61 < k58, sendo que T58<T61 e RAB58<RAB61
– Porém, pode ser útil para comparação rápida entre bateladas de
mesma carga.

34
8) BIBLIOGRAFIA
CARVALHO, S.C.O; OLIVEIRA, M.E.C.; FRANÇA, L.F. Modelagem e Simulação da
Cinética de Transesterificação de Óleos Vegetais para Produção de Biodiesel,
Universidade Federal do Pará, 7 p.
CONSTANTINOU, L.; GANI, R. New group contribution method for estimating
properties of pure compound. AIChE Journal, v . 40, n . 10, p . 1697 – 1710. Out.
1994.
DAMBISKI, L. Síntese de biodiesel de óleo de nabo forrageiro empregando metanol
supercrítico, Dissertação de Mestrado, Curitiba, UTFPR, Dez. 2007, 79 p.
DEMIBRAS, A., Biodiesel production via non-catalytic SCF method
and biodiesel fuel characteristics. Journal of Energy Conversion and Management, v .
47, p . 2271 – 2282, Nov. 2005.
DIASAKOU, M.; LOULOUDI, A.; PAPAYANNAKOS, N. Kinetics of non catalytic
transesterification of sybean oil. Fuel, v.77, n . 12, p. 1297-1302, Jan. 1998.
GUNSTONE, F.D. The chemistry of oils and fats, Oxford, Blackwell Publishing, 2004,
288 p.
KIWJAROUN, C. ; TUBTIMDEE, C.;PIUMSOMBOON, P. LCA studies comparing
biodiesel synthesized by conventional and supercritical methanol methods. Journal of
Cleaner Production, v . 17, p . 143 – 153, Mar. 2008.
KUSDIANA, D.; SAKA, S.; Kinetics of transesteri®cation in rapeseed oil to biodiesel
fuel as treated in supercritical methanol. Fuel, v . 80, p . 693 – 698, Ago. 2000.
Kusdiana, D.; Saka, Effects of water on biodiesel fuel production by supercritical
methanol treatment. Bioresource Technology, v . 91, p . 289 – 295, Jun. 2003
MADRAS, G. ; KOLLURU, C. ; KUMAR, R. Synthesis of biodiesel in supercritical fluids.
Fuel, v . 83, p . 2029 – 2033, Mar. 2004.

35
MELO, J. C. Otimização da Produção de Biodiesel. 2007. Dissertação (Mestrado em
Engenharia Química)- Programa de Pós-Graduação em Engenharia Química, UFPE,
Recife.
MINAMI, E.; SAKA, S. Kinetics of hydrolysis and methyl esterification for biodiesel
production in two-step supercritical methanol process. Fuel, v . 85, p . 2479 – 2483,
Abr. 2006.
NOUREDDINI, H.; ZHU, D. Kinetics of Trasesterification of Saybean Oil. JAOCS, v.74,
n.11, p. 1457-1463, 1997.
REID, R.C. ; PRAUSNITZ, J. M.; PÓLING, B. E. The properties of gases and liquids. 4
ed, Nova Iorque, Ed McGraw – Hill, 1987, 741 p.
SANTOS, G.G.D., Revisão bibliográfica e testes de produção de biodiesel sem o uso
de catalisador, Trabalho de Conclusão, UFRGS, Nov. 2008, 30 p.
SHIMOYAMA, Y.; IWAI, Y.;ABETA, T.; ARAI, Y. Measurement and correlation of
vapor–liquid equilibria for ethanol + ethyl laurate and ethanol + ethyl myristate systems
near critical temperature of ethanol Fluid Phase Equilibria, v . 264, p . 228 – 234, Nov.
2007.
SILVA et al, Continuous Production of Fatty Acid Ethyl Esters from Soybean Oil in
Compressed Ethanol. Ind. Eng. Chem. Res., Erechim, v.46, p. 5304-5309, 2007.
SMITH, J.M.; VAN NESS, H.C.; ABBOTT, M.M. Introduction to chemical engineering
thermodynamics, 5 Ed, Nova Iorque, Ed McGraw – Hill, 1996, 763 p.
SONG, E. S.; LIM, J.; LEE,H.S.; LEE, Y.W. Transesterification of RBD palm oil using
supercritical methanol. Journal of Supercritical Fluids, v . 44, p . 356 – 363, Set. 2007.
SOARES, R.P. EMSO manual, Ago. 2007.
VRTherm, Manual do Usuário, VRTech Tecnologias Industriais Ltda. Jan. 2008.

I
ANEXO: ORIGEM DO MODELO
A modelagem cinética utilizando dados de pressão tem como pressuposto que
o número total de moles da reação em fase gasosa sofrerá uma variação global,
conforme visto nos exemplos abaixo:
No primeiro exemplo, tem-se que a variação estequiométrica do número de
moles é -1, isto é, para cada 1 mol de C formado, o sistema em si perdeu 1 mol geral,
havendo queda de pressão ao longo do avanço da reação. No exemplo 2 tem-se o
contrário: há aumento da pressão ao longo do caminho da reação. Já no terceiro
exemplo tem-se que o número de moles não varia ao longo da reação. Porém, seria
muito precipitado afirmar que não há variação da pressão, visto que isto só ocorreria
se os gases fossem ideais.
A variação da pressão ao longo da reação, para gases ideais, seria dada por:
Para o caso da transesterificação em fluido supercrítico, o meio é homogêneo e
a temperatura costuma ser maior que 300 °C. Porém , a variação líquida do número de
moles é nula. No entanto, observa-se que o etanol, agente pressurizador, está em
excesso e é muito mais volátil que os demais. Logo, a pressão do sistema é dada
simplesmente pela fração de etanol presente no meio reacional. Assim, temos que:
Onde N é o número total de moles presentes no meio reacional e j é uma constante de
proporcionalidade.
Sabendo-se que e , podemos rearranjar a
equação acima como:

II
Subtraindo-se as duas últimas equações, temos:
Eis a origem do modelo utilizado neste trabalho.
Mas é importante questionar o significado do fator J: este nada mais é que um
fator de ajuste para transformar unidades de avanço de reação (mol) para unidades
de pressão (bar). O fator J, dessa forma, é dado por:
Onde e são a queda de pressão no equilíbrio e o avanço total no equilíbrio,
ambos medidos no fim da reação (tempo suficientemente longo).
Copyright © 2022 FDOKUMEN



















![El mito indio del diluvio en su relación con los cuentos clásicos y próximo-orientales [The Indian Deluge Myth in its Relationship with Classical and Near-Eastern Narratives]](https://static.fdokumen.com/doc/165x107/63136b2785333559270c3b3e/el-mito-indio-del-diluvio-en-su-relacion-con-los-cuentos-clasicos-y-proximo-orientales.jpg)
