
GENETIC AND BIOCHEMICAL-CHARACTERIZATION OF B-SUBTILIS COMPETENCE MUTANTS
Upload
independentCategory
view
0download
0
Mode of action of AraR, the key regulator of L-arabinosemetabolism in Bacillus subtilis
LuõÂs Jaime Mota,1 Paulo Tavares2² and
Isabel SaÂ-Nogueira1,3*1Instituto de Tecnologia QuõÂmica e BioloÂgica,
Universidade Nova de Lisboa, Apartado 127, 2781-901
Oeiras, Portugal.2Max-Planck Institut fuÈr Molekulare Genetik, Ihnestraûe
73, D-14195 Berlin, Germany.3Faculdade de CieÃncias e Tecnologia, Universidade Nova
de Lisboa, Quinta da Torre, 2825 Monte de Caparica,
Portugal.
Summary
The AraR protein is a negative regulator involved in
L-arabinose-inducible expression of the Bacillus sub-
tilis araABDLMNPQ-abfA metabolic operon and of the
araE/araR genes that are organized as a divergent
transcriptional unit. The two ara gene clusters are
found at different positions in the bacterial chromo-
some. AraR was overproduced in Escherichia coli
and puri®ed to more than 95% homogeneity. AraR
binds speci®cally to DNA fragments carrying the pro-
moter region of the ara genes. DNase I protection
assays showed that AraR binds to two sequences
within the promoters of the araABDLMNPQ-abfA
operon and the araE gene, and to one sequence in
the araR promoter. The AraR target sequences are
palindromic and share high identity, de®ning a 16 bp
AraR consensus operator sequence showing half-
symmetry, ATTTGTAC. Binding of AraR to DNA was
inhibited by L-arabinose but not by other sugars.
The two operator sites within the araABDLMNPQ-
abfA operon and araE promoters are located on the
same side of the DNA helix, and a pattern of enhanced
and diminished DNase I cleavage was observed
between them, but not in the araR promoter. Quantita-
tive DNase I footprinting in DNA templates containing
one, two or three AraR binding sites showed that the
repressor binds cooperatively to the two operator
sites within the metabolic operon and araE promoters
but not to the site located in the araR promoter. These
results are consistent with two modes for AraR
transcriptional repression that might correlate with
different physiological requirements: a high level of
repression is achieved by DNA bending requiring
two in-phase operator sequences (metabolic operon
and araE transport gene), whereas binding to a single
operator, which autoregulates araR expression, is
10-fold less effective.
Introduction
Bacillus subtilis is able to grow on L-arabinose as the sole
carbon and energy source. Soil bacteria, such as B. sub-
tilis, participate in the early stages of plant material
decomposition, and the L-arabinose pentose sugar is pre-
sent in various hemicellulosic and pectic plant polysaccha-
rides, such as heteroglycanes (e.g. arabinoxylan and
arabinogalactan) and the homoglycane L-arabinan. The
ability to utilize L-arabinose is dependent on three intra-
cellular enzymes encoded by the araA (L-arabinose
isomerase), araB (L-ribulokinase) and araD (L-ribulose
5-phosphate 4-epimerase) genes (SaÂ-Nogueira and
Lencastre, 1989), which sequentially convert L-arabinose
to L-ribulose, L-ribulose 5-phosphate and D-xylulose
5-phosphate respectively (Lepesant and Dedonder, 1967).
D-Xylulose 5-phosphate is further catabolized through
the pentose phosphate pathway. The B. subtilis genes
involved in the utilization of L-arabinose (ara genes)
characterized so far are those belonging to the metabolic
araABDLMNPQ-abfA operon (SaÂ-Nogueira et al., 1997)
located at about 2518 in the B. subtilis chromosome
(Kunst et al., 1997) and the divergently arranged araE/
araR genes located at about 2988 (Kunst et al., 1997),
encoding the main transporter for L-arabinose and a regu-
latory protein respectively (SaÂ-Nogueira and Mota, 1997;
SaÂ-Nogueira and Ramos, 1997).
The transcriptional control of gene expression in the
L-arabinose pathways of Escherichia coli and B. subtilis
offers a valuable insight into the evolutionary differences
between Gram-negative and Gram-positive organisms.
The catabolic pathway in the two organisms is identical,
and it has been demonstrated that the three metabolic
genes of B. subtilis, araA, araB and araD, complement
de®ciencies in their E. coli counterparts (SaÂ-Nogueira
and Lencastre, 1989). In both organisms, however, tran-
scription is regulated by non-homologous DNA-binding
proteins, AraC in E. coli (Schleif, 1996, and references
therein) and AraR in B. subtilis (SaÂ-Nogueira and Mota,
Molecular Microbiology (1999) 33(3), 476±489
Q 1999 Blackwell Science Ltd
Received 5 October, 1998; revised 26 April, 1999; accepted 28 April,1999. ²Present address: Unite de PathogeÂnie Microbienne MoleÂcul-aire, Institut Pasteur 28, Rue du Dr Roux, F-75724 Paris Cedex 15,France. *For correspondence. E-mail [email protected]; Tel.(�351) 1 446 9524; Fax (�351) 1 441 1277.
1997). These proteins exert their regulatory role in a
distinct way, showing that there was no conservation of
regulatory interactions between the two organisms. In E.
coli, the AraC protein plays a dual role in regulation. In
the absence of L-arabinose, AraC represses its own tran-
scription via a DNA-looping mechanism in the divergently
transcribed araC/araBAD promoter region. In the pre-
sence of L-arabinose, it activates the araBAD metabolic
operon and the araFGH and araE transport genes
(Schleif, 1996, and references therein). In B. subtilis, the
results obtained so far indicate that AraR acts only as a
repressor.
The predicted amino acid sequence of AraR reveals a
mosaic structure. AraR exhibits signi®cant similarity to
the E. coli GalR and LacI family of bacterial regulators
(Weickert and Adhya, 1992). This similarity does not
extend, however, to the N-terminal region of AraR, which
is related to the helix±turn±helix (HTH) consensus signa-
ture sequence of the GntR family of regulatory proteins. In
this family of bacterial regulators, the amino acid sequ-
ence similarity is con®ned to the N-terminal domain,
which binds to speci®c DNA sequences (Haydon and
Guest, 1991). This chimeric organization suggests a
two-domain structure for AraR (SaÂ-Nogueira and Mota,
1997). The model proposed for the action of AraR is
that, in the absence of L-arabinose, AraR binds to operator
site(s) within the araABDLMNPQ-abfA operon promoter
and within the araE/araR promoters region, thus prevent-
ing transcription. The presence of L-arabinose induces a
conformational change in AraR such that recognition and
binding to DNA is no longer possible, thus allowing expres-
sion of the ara genes (SaÂ-Nogueira and Mota, 1997;
SaÂ-Nogueira and Ramos, 1997).
Here, we report the puri®cation and characterization of
the AraR native protein. AraR is shown to bind to similar
palindromic sequences in the araABDLMNPQ-abfA operon
and araE/araR promoter regions. The presence of L-arabi-
nose speci®cally inhibits the binding of AraR to these sequ-
ences, indicating the existence of an interaction between
AraR and L-arabinose that converts the protein into a
form that no longer binds DNA. In addition, transcriptional
repression of the araABDLMNPQ-abfA operon and of the
araE gene by AraR occurs by a mechanism different from
the autorepression of araR expression.
Results
Overproduction and puri®cation of AraR
The AraR repressor was initially puri®ed as a fusion to the
maltose-binding protein (MBP2*) of E. coli (see Experi-
mental procedures). The puri®ed fusion protein (AraR±
MBP2*) was used to obtain rabbit polyclonal antibodies
that recognize AraR and MBP2*. The speci®city of the
AraR±MBP2* antiserum to detect AraR was con®rmed
in Western blot analysis of crude extracts from wild-type
and AraR null mutant B. subtilis cells (data not shown).
The AraR±MBP2* antiserum was used to identify unam-
biguously the native protein during the ®rst attempts at
puri®cation.
Native AraR was overproduced in E. coli BL21 (DE3)
(LysS) (Studier et al., 1990) bearing plasmid pLM14,
which carries the ribosome binding site and the entire
coding sequence of araR under the control of an IPTG-
inducible T7 promoter. Figure 1 documents the puri®cation
procedure. Following lysis of the E. coli overproducing cells
in a high ionic strength buffer, AraR was found essentially
in the soluble fraction of a high-speed centrifugation.
After precipitation with 75% ammonium sulphate, AraR
was puri®ed by af®nity chromatography on a heparin±
Sepharose column followed by ion-exchange chromato-
graphy on a Hi-Load SP-Sepharose column. Approxi-
mately one-quarter of the AraR precipitated during
desalting before loading onto the SP-Sepharose column.
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 33, 476±489
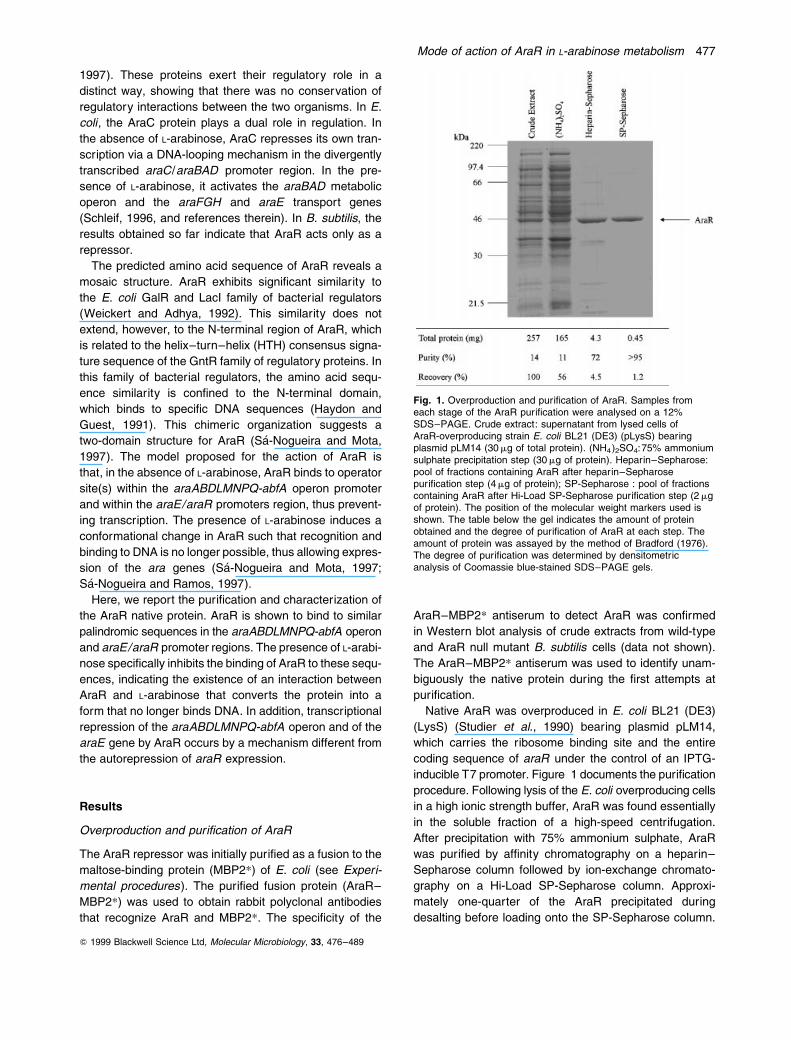
Fig. 1. Overproduction and puri®cation of AraR. Samples fromeach stage of the AraR puri®cation were analysed on a 12%SDS±PAGE. Crude extract: supernatant from lysed cells ofAraR-overproducing strain E. coli BL21 (DE3) (pLysS) bearingplasmid pLM14 (30 mg of total protein). (NH4)2SO4:75% ammoniumsulphate precipitation step (30 mg of protein). Heparin±Sepharose:pool of fractions containing AraR after heparin±Sepharosepuri®cation step (4 mg of protein); SP-Sepharose : pool of fractionscontaining AraR after Hi-Load SP-Sepharose puri®cation step (2 mgof protein). The position of the molecular weight markers used isshown. The table below the gel indicates the amount of proteinobtained and the degree of puri®cation of AraR at each step. Theamount of protein was assayed by the method of Bradford (1976).The degree of puri®cation was determined by densitometricanalysis of Coomassie blue-stained SDS±PAGE gels.
Mode of action of AraR in L-arabinose metabolism 477
Only the soluble fraction was applied to the column. With
this puri®cation procedure, about 400 mg of AraR, exceed-
ing 95% purity (Fig. 1), was obtained from 257 mg of total
protein. Recovery was greatly compromised because only
the fractions most enriched in AraR were pooled after
passing through the heparin±Sepharose column, and
also because of precipitation of the partially puri®ed
AraR in low salt. The puri®ed AraR preparation was
used in all in vitro protein±DNA interaction assays
described below. The amino-terminal microsequence of
AraR (MLPKYAQVKE) and its electrophoretic mobility in
sodium dodecyl sulphate polyacrylamide gel electrophore-
sis (SDS±PAGE) (42.7 kDa; Fig. 1) correlated well with
the properties predicted from the analysis of its nucleotide
sequence (SaÂ-Nogueira and Mota, 1997). On size-exclu-
sion chromatography, the native AraR protein had the
hydrodynamic behaviour expected of a globular protein
with an apparent molecular mass of 60 kDa, i.e. a value
intermediate between that expected for a monomer
(40.4 kDa) and that expected for a dimer (data not shown).
Binding of AraR to the promoter region of the
araABDLMNPQ-abfA operon and araE/araR genes
Previous work with araA8-lacZ, araE8-lacZ and araR8-lacZ
transcriptional fusions in the wild-type and araR null
mutant strains established that expression of the ara
genes is negatively regulated by AraR (SaÂ-Nogueira and
Mota, 1997; SaÂ-Nogueira and Ramos, 1997). By analogy
with other bacterial repressors, AraR is believed to bind
to the promoter region of the ara genes, thus preventing
transcription. Regions of dyad symmetry detected in both
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 33, 476±489
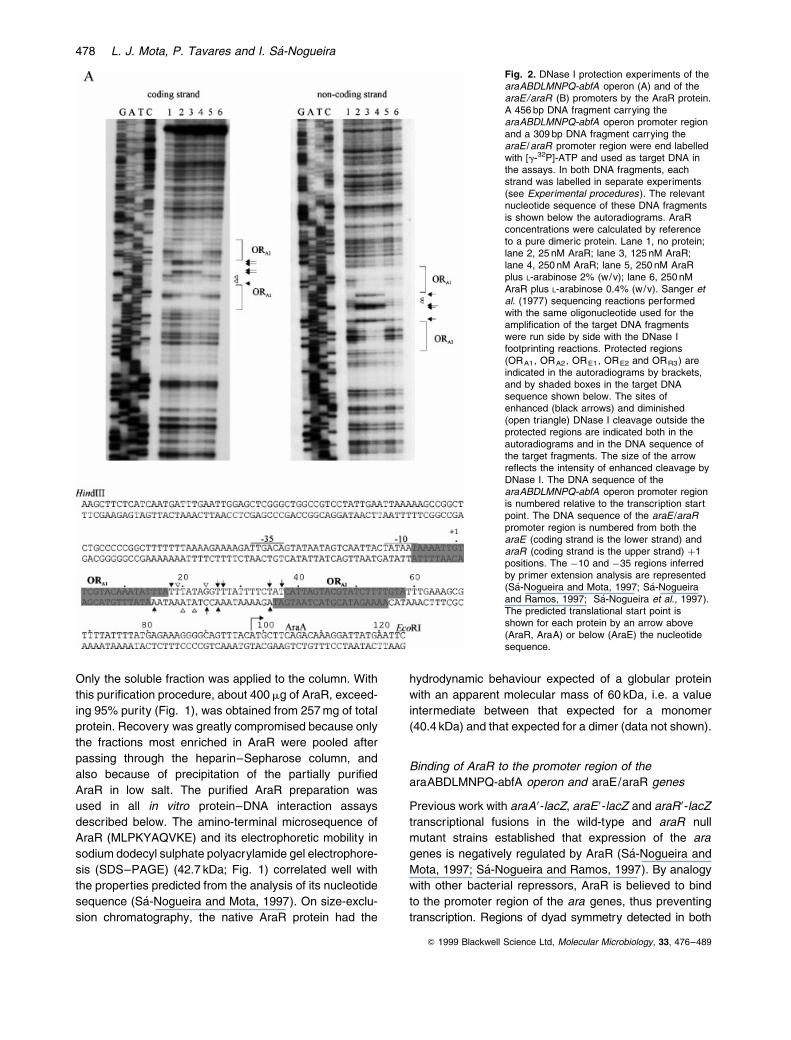
Fig. 2. DNase I protection experiments of thearaABDLMNPQ-abfA operon (A) and of thearaE/araR (B) promoters by the AraR protein.A 456 bp DNA fragment carrying thearaABDLMNPQ-abfA operon promoter regionand a 309 bp DNA fragment carrying thearaE /araR promoter region were end labelledwith [g-32P]-ATP and used as target DNA inthe assays. In both DNA fragments, eachstrand was labelled in separate experiments(see Experimental procedures). The relevantnucleotide sequence of these DNA fragmentsis shown below the autoradiograms. AraRconcentrations were calculated by referenceto a pure dimeric protein. Lane 1, no protein;lane 2, 25 nM AraR; lane 3, 125 nM AraR;lane 4, 250 nM AraR; lane 5, 250 nM AraRplus L-arabinose 2% (w/v); lane 6, 250 nMAraR plus L-arabinose 0.4% (w/v). Sanger etal. (1977) sequencing reactions performedwith the same oligonucleotide used for theampli®cation of the target DNA fragmentswere run side by side with the DNase Ifootprinting reactions. Protected regions(ORA1, ORA2, ORE1, ORE2 and ORR3) areindicated in the autoradiograms by brackets,and by shaded boxes in the target DNAsequence shown below. The sites ofenhanced (black arrows) and diminished(open triangle) DNase I cleavage outside theprotected regions are indicated both in theautoradiograms and in the DNA sequence ofthe target fragments. The size of the arrowre¯ects the intensity of enhanced cleavage byDNase I. The DNA sequence of thearaABDLMNPQ-abfA operon promoter regionis numbered relative to the transcription startpoint. The DNA sequence of the araE /araRpromoter region is numbered from both thearaE (coding strand is the lower strand) andaraR (coding strand is the upper strand) �1positions. The ÿ10 and ÿ35 regions inferredby primer extension analysis are represented(SaÂ-Nogueira and Mota, 1997; SaÂ-Nogueiraand Ramos, 1997; SaÂ-Nogueira et al., 1997).The predicted translational start point isshown for each protein by an arrow above(AraR, AraA) or below (AraE) the nucleotidesequence.
478 L. J. Mota, P. Tavares and I. SaÂ-Nogueira

araABDLMNPQ-abfA and araE/araR promoter regions
are likely candidates for the AraR binding site(s)
(SaÂ-Nogueira and Mota, 1997).
The localization of the AraR binding sites within the ara
promoters was determined by DNase I footprinting on both
strands of the DNA using a 100-fold excess of competitor
DNA (Fig. 2). A 456 bp fragment and a 309 bp fragment
containing all the putative operator-like sequences in the
araABDLMNPQ-abfA operon and araE/araR promoters
region, respectively, were the target DNA in the experi-
ments. These two fragments carry all the sequences
necessary for regulation of the operon and of the araE
gene by AraR, as demonstrated by transcriptional fusions
integrated into the amyE locus of the B. subtilis chromo-
some (data not shown). The DNA sequences protected
by DNase I digestion overlap the predicted binding
sites for AraR based on the analysis of the nucleotide
sequence. Two binding sites were detected in the
araABDLMNPQ-abfA operon promoter region and three
AraR binding sites in the araE/araR promoter region. In
the araABDLMNPQ-abfA operon coding strand (Fig. 2A),
AraR protects the regions between positions ÿ7 and
�17 (ORA1) and between positions �38 and �58 (ORA2).
In the araE coding strand (Fig. 2B), AraR protects the
regions between positions ÿ6 and �14 (ORE1) and
between positions �39 and �59 (ORE2), and, in the
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 33, 476±489
Fig. 2. Continued.
Mode of action of AraR in L-arabinose metabolism 479

araR coding sequence, it protects the region between
positions ÿ6 and �14 (ORR3) (Fig. 2B). The same sequ-
ences are protected in the non-coding strands. Some
dif®culty in de®ning precisely the boundaries of the
protected regions in each of the strands is caused by the
high AT content of the operator ¯anking sequences that
makes them partly resistant to DNase I attack. A pattern
of DNase I enhanced and diminished cleavage was
observed between ORA1 and ORA2, and between ORE1
and ORE2, but not between ORE1 and ORR3 (Fig. 2A
and B). These protection and hypersensibility patterns
occur in similar positions relative to the start of transcrip-
tion of the araA and araE genes respectively. AraR
binding to the two in-phase operators of the metabolic
operon and of the transport gene promoter regions thus
seems to occur in a similar way, producing in both cases
a distortion of the DNA helix. Such an effect is not
observed when AraR binds to the single operator within
the araR promoter.
L-Arabinose speci®cally inhibits binding of AraR to its
operator sequences
Induction of AraR-responsive genes occurs upon growth
of B. subtilis in minimal medium in the presence of L-arabi-
nose (SaÂ-Nogueira et al., 1997; SaÂ-Nogueira and Mota,
1997; SaÂ-Nogueira and Ramos, 1997). This has led to
the suggestion that L-arabinose interacts with AraR to
prevent binding to the DNA. We initially tested the effect
of L-arabinose in vitro using concentrations of 0.4% and
2% (w/v). Both concentrations of L-arabinose strongly
inhibited the binding of AraR to its operator regions
(Fig. 2). Further tests showed that concentrations as
low as 0.02% (w/v) L-arabinose abolished binding of
AraR to all three promoters under analysis (not shown).
This is the miminum sugar concentration at which full
induction of the araABDLMNPQ-abfA operon expression
is observed in vivo (SaÂ-Nogueira and Mota, 1997). The
effect of other carbohydrates, including two catabolized
pentoses, D-xylose and D-ribose, on AraR binding was
tested (Fig. 3). No effect on the af®nity of AraR for its
operator regions was observed with these sugars, indicat-
ing that L-arabinose is the effector that modulates AraR
binding to DNA, acting as a speci®c inducer to inhibit the
binding of a repressor protein to a control site.
Relative af®nity of AraR to the operator regions
The relative af®nity of AraR for each operator site in the
promoter region of the araABDLMNPQ-abfA operon and
for the araE/araR genes was measured by quantitative
DNase I footprinting over a wide range of repressor con-
centrations. DNA templates containing one, two or three
(in case of the araE/araR genes) AraR binding sites
were characterized (Figs 4 and 5). The apparent dissoci-
ation constant (Kapp) for each individual binding site was
determined as the repressor concentration at which
half-maximum site occupancy was observed.
With a DNA fragment from the promoter region of the
araABDLMNPQ-abfA operon containing the two AraR
binding sites, ORA1 and ORA2 were protected at approxi-
mately the same concentration of repressor (< 40 nM;
Fig. 4A). The footprinting exhibited the characteristic pat-
tern of enhanced and diminished DNase I cleavage sites
between the two operator sequences that was described
above. When binding to the individual operators, ORA1
and ORA2, was analysed in templates carrying only one
of the sequences (Fig. 4B and C), the Kapp of AraR
for each site decreased about eightfold relative to that
obtained with the two-operator fragment. Furthermore,
no DNase I hypersensitivity was detected in surrounding
sequences. These results suggested that binding to
ORA1-ORA2 is cooperative and that it promotes a distor-
tion of the DNA helix, which requires binding of AraR to
the two sites. To support this point further, we quanti®ed
AraR binding to the ORA1mut-ORA2 fragment carrying a
single nucleotide substitution in ORA1 (Fig. 4D). The sub-
stitution in a highly conserved position of the AraR target
sequence (Fig. 6A) was designed to inhibit the binding
of AraR to ORA1mut . This inhibitory effect was indeed
observed, and it reduced dramatically the apparent af®nity
of AraR for the second operator ORA2 (compare the foot-
prints in Fig. 4A and D). Cross-talk between AraR mol-
ecules binding to the two operator sequences is thus
necessary for high-af®nity binding, which binding pro-
motes distortion of the double helix. Some protection of
ORA1mut and ORA2, and the presence of faint hypersensi-
tive bands between the two operators, indicated that the
ORA1mut single mutation still allowed low-af®nity binding
of AraR and some cross-talk between the two operators
(Fig. 4D).
Cooperativity of binding was also observed for the two
AraR binding sites, ORE1 and ORE2, located in the pro-
moter region of the araE/araR genes. The Kapp of AraR
binding to ORE1 and ORE2 present in the same DNA frag-
ment (Fig. 5B) was 3- and 2.3-fold higher, respectively,
than the af®nities found with fragments carrying the single
operator sequences (Fig. 5D and E). The appearance of
DNase I hypersensitive sites was only observed in foot-
printings with fragments carrying both ORE1 and ORE2.
These features are similar to the ones found for AraR
binding to the ORA1ORA2 operators sequence (see
above). The presence of ORR3 in the araE/araR promoter
regions seems to promote binding to the ORE1ORE2 sites
(Fig. 5A and B). In the absence of ORE2, the af®nity of
AraR for ORE1 increases 2.3-fold when ORR3 is present
(Fig. 5C). This apparent cross-talk between ORE1 and
ORR3 is not associated with DNase I hypersensitivity in
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 33, 476±489
480 L. J. Mota, P. Tavares and I. SaÂ-Nogueira

the region between the two operators. In contrast to the
cooperative effects described above, the af®nity of AraR
for ORR3, located in the araR promoter, was about the
same irrespective of the presence of ORE1 or ORE1 and
ORE2 (Fig. 5F, C and A respectively). AraR binds ORR3
with a Kapp of 40 nM, an af®nity signi®cantly higher than
that found for any other single operator sequence. In the
presence of 0.02% (w/v) L-arabinose, the binding of the
repressor to all individual operator sequences was almost
abolished (Kapp 250 $ nM; Figs 4 and 5).
De®ning the AraR consensus operator sequence
The AraR binding sequences identi®ed by DNase I foot-
printing are palindromic with the symmetry centre in a
CG pair of nucleotides (Fig. 2). Based on the alignment
of all ®ve operator sequences, it is possible to deduce a
16 bp AraR consensus operator sequence showing half-
symmetry, ATTTGTAC (Fig. 6A). This operator motif
shows uninterrupted dyad symmetry like most of the
sites for other members of the GntR family of bacterial
repressors (Fig. 6B). FadR and FarR are the exception,
showing no obvious symmetry (Quail et al., 1994; Clark
and Cronan, 1996). The operator sequences revealed
highly conserved sequences, positions 1 and 4 ±7, but
more information regarding speci®c amino acid basepair
contacts between these regulators and their operator
sites will be required to evaluate the signi®cance of this
conservation.
In spite of the high similarity among the AraR target sites
(Fig. 6A), the repressor binds to individual operator sequ-
ences with af®nities ranging from 40 nM (ORR3) to
>250 nM (ORA2). The apparent af®nities for individual
operators follows the order ORR3>ORE1>ORE2>
ORA1 $ ORA2 (Figs 4 and 5). Correlation of these values
with the operator DNA sequence and the symmetry of
each site (Fig. 6A) suggests that sequences outside the
16 bp inverted repeats in¯uence the intrinsic af®nity of
the repressor for the operators.
Discussion
AraR is the key regulator of the genes involved in L-arabi-
nose utilization in B. subtilis. In this study, we puri®ed the
native AraR and showed by DNase I footprinting experi-
ments that AraR binds to the araABDLMNPQ-abfA
operon promoter and to the araE/araR promoter region.
Binding is sequence speci®c and is strongly inhibited by
L-arabinose. The results con®rm our proposed model
(SaÂ-Nogueira and Mota, 1997) of the molecular mechan-
ism of how AraR regulates gene expression by direct bind-
ing to DNA and inhibition of this interaction by the effector
L-arabinose. L-Arabinose acts as a typical inducer that
inhibits binding of a repressor protein to its control sites.
Based on the alignment of all the AraR binding sequ-
ences, it was possible to deduce a 16 bp consensus opera-
tor sequence, ATTTGTACGTACAAAT, which shows
uninterrupted dyad symmetry, like most of the sites for
other members of GntR family of bacterial regulators
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 33, 476±489
Fig. 3. Effect of different carbohydrates on DNase I protection ofthe araE /araR promoters by the AraR protein. The 309 bp fragmentcorresponding to the araE coding strand (Fig. 2B) was used astarget DNA. The AraR protein was used at a concentration of125 nM. AraR concentration was calculated by reference to a puredimeric protein. The different carbohydrates were used at a ®nalconcentration of 0.02% (w/v). The presence (�) or absence (ÿ) ofthe different carbohydrates and of the AraR repressor is shown in thetop panel. The brackets indicate the extent of the protected regions.The black arrows indicate sites of enhanced cleavage by DNase I.
Mode of action of AraR in L-arabinose metabolism 481

(Fig. 6). We could not draw any conclusions about the
oligomeric state of AraR in solution. However, the dyad
symmetry of its target sequence and the probability that
the protein±DNA interaction is mediated by a putative
HTH domain identi®ed in the amino terminus of AraR sug-
gest that the repressor is in a multimeric form when bound
to DNA. Both features are commonly observed in bacterial
transcriptional repressors that bind DNA as homodimers
or homotetramers (Pabo and Sauer, 1992). These pro-
teins may be stable as homomultimers in solution at
physiological concentrations or, alternatively, dimerization
may depend on stable binding to DNA. The hypothesis
that AraR multimerization occurs is further supported by
the alignment of its primary structure with proteins belong-
ing to the LacI-GalR family of repressors, which shows
amino acid identity in some of the regions implicated in
dimerization of the E. coli members of this family
(SaÂ-Nogueira and Mota, 1997).
The binding patterns of AraR to the araABDLMNPQ-
abfA operon promoter and to the araE promoter are simi-
lar but differ from that of AraR binding to its own promoter.
A pattern of DNase I enhanced and diminished cleavage
was observed between ORA1 and ORA2, and between
ORE1 and ORE2, but not between ORE1 and ORR3
(Fig. 2). The distance between the DNase I hypersensitive
sites ranges from 8 to 11 bp, corresponding to about one
helix turn (10.5 bp per helix turn for B-DNA in vitro and
11 bp per helix turn for E. coli DNA in vivo ; Lee and Schleif,
1989), consistent with a distortion of the DNA helix that
alters the width of the minor groove (Drew and Travers,
1984). Loop formation between the in-phase operator sequ-
ences is a possibility. DNA looping in well-characterized
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 33, 476±489
Fig. 4. Quantitative DNase I footprinting of different DNA fragments from the promoter region of the araABDLMNPQ-abfA operon. DNAfragments carrying one or two AraR binding sites or a single point mutation in ORA1 (ORA1mut) were used.A. ORA1±ORA2 (228 bp).B. ORA2 (173 bp).C. ORA1 (218 bp).D. ORA1mut±ORA2 (254 bp).A schematic representation of the DNA fragments used in the experiments is shown above the autoradiograms: AraR binding sites arerepresented in grey, DNA from the araABDLMNPQ-abfA operon promoter in white and heterologous DNA (from plasmid pSN32 ± seeExperimental procedures) as dotted boxes. Labelling of the footprinting autoradiogram follows the symbology used in Fig. 2. Total repressorconcentration at which half-maximal site occupancy is achieved (a value that represents Kapp, the apparent af®nity of AraR for each site) isindicated within parenthesis for each operator in the different DNA fragments. For ORA2 alone, or ORA1mut and ORA2 in fragmentORA1mut±ORA2, the sites protection was less than 50% at the highest concentration of AraR tested. The results shown in the ®gure werecalculated for one single experiment. The assays with DNA fragments ORA1±ORA2, ORA1 and ORA2 were performed at least twice and theKapp values showed a deviation of less than 25%. In the case of DNA fragments ORA1±ORA2, ORA1mut±ORA2 and ORA2, assays were alsoperformed on the opposite strand and similar results were obtained. Lane 1, no protein; lane 2, 25 nM AraR; lane 3, 50 nM AraR; lane 4,100 nM AraR; lane 5, 150 nM AraR; lane 6, 200 nM AraR; lane 7, 250 nM AraR; lane 8, 250 nm AraR plus L-arabinose 0.02% (w/v). AraRconcentrations were calculated by reference to a pure dimeric protein. The araA coding strand of the templates was end labelled with [g-32P]-ATP and used as a target in the assays shown.
482 L. J. Mota, P. Tavares and I. SaÂ-Nogueira

nucleoprotein complexes (e.g. l repressor; Hochschild
and Ptashne, 1986) was found to cause a pattern of
enhanced DNase I bands in the intervening sequence
between target sites that need to be separated by an
integer number of helix turns. The CG centres of ORA1
and ORA2 are indeed separated by 42 bp and, in the
case of ORE1 and ORE2, by 43 bp, which corresponds to
4.0 and 4.09 helix turns respectively, placing the AraR
molecules on the same side of the DNA helix (Fig. 7). In
contrast, ORR3 in the araR promoter is 7.7 helix turns
from ORE1, indicating that they are not in phase. These
results suggest that the binding of AraR molecules to the
operator sites ORA1 and ORA2 and to ORE1 and ORE2 pro-
motes DNA looping, whereas occupation of ORR3 does
not cause a detectable distortion of the double-helix.
Loop formation most probably involves cooperative inter-
actions. AraR does, in fact, bind cooperatively to the two
operator sites within the araABDLMNPQ-abfA metabolic
operon and araE promoters, but not to the site located in
the araR promoter (Figs 4 and 5). Negative regulation of
metabolic and transport genes involving the formation of
a DNA loop has been described in several E. coli systems,
such as lac, ara, gal and nag (reviewed in Schleif, 1992),
and proposed for negative regulation of the arg
(Czaplewski et al., 1992) and xyl (GoÈsseringer et al.,
1997) genes of B. subtilis. The present study makes it
very likely that loop formation is important for the regu-
lation of ara gene transcription, a mechanism not reported
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 33, 476±489
Fig. 5. Quantitative DNase I footprinting of different DNA fragments from the promoter region of the araE/araR genes by AraR. DNAfragments carrying one, two or three AraR binding sites were used:A. ORR3±ORE1±ORE2 (292 bp).B. ORE1±ORE2 (227 bp).C. ORR3±ORE1 (236 bp).D. ORE2 (180 bp).E. ORE1 (223 bp).F. ORR3 (203 bp).Schematic representations, labelling of the autoradiograms, determination of the Kapp values and the AraR concentrations used are as inFig. 4. The protection of ORE1 on fragment ORR3±ORE1±ORE2 was more than 50% at the lowest concentration of AraR used. The resultsshown in the ®gure were calculated for one single experiment. Assays with the DNA fragments ORR3±ORE1±ORE2 and ORE2 were performedat least twice and the Kapp values showed a deviation of less than 25%. The araE coding strand of the templates was end-labelled with[g-32P]-ATP and used as target in all assays shown.
Mode of action of AraR in L-arabinose metabolism 483

yet for the members of the GntR family of bacterial
regulators.
Transcription repression is less effective when opera-
tors overlap the ÿ10 and �1 sites but leave the ÿ35 region
open for productive interaction with RNA polymerase and
competition with the repressor (Lanzer and Bujard, 1988).
This is the case for the AraR binding sites in the promoter
region of the genes regulated by AraR (Fig. 7). However,
cooperative interactions between AraR molecules bound
to the two operator sites in the araABDLMNPQ-abfA
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 33, 476±489
Fig. 6. A. AraR consensus operator sequence. Alignment of the ®ve AraR boxes identi®ed by DNase I footprinting. DNA sequences showncorrespond to the coding strand of each ara gene. Shaded boxes indicate the nucleotides conserved in the ®ve operator sequences. At leastthree identical nucleotides were used as criteria to de®ne the 16 bp palindromic consensus sequence. The relative intrinsic af®nity (Kapp) ofAraR for each individual operator determined from the footprintings of Figs 4 and 5 is shown. The single point mutation introduced into ORA1
(G to T at position 9) to obtain the fragment ORA1mut ±ORA2 (Fig. 4) is also shown above the sequence.B. Comparison of the operator sequences from proteins belonging to the GntR family of bacterial regulators with the AraR consensus operatorsequence (numbered). FadR from E. coli (Clark and Cronan, 1996); GntR from B. subtilis (Fujita and Miwa, 1989); PhdR from E. coli (Quailand Guest, 1995); FarR from E. coli (Quail et al., 1994); HutC from Pseudomonas putida (Haydon and Guest, 1991); and TreR from B. subtilis(SchoÈck and Dahl, 1996). Shaded boxes identify the existence of at least four identical nucleotides between the sequences.
Fig. 7. Negative regulation of the L-arabinose-inducible promoters by AraR. The repressionof the ara promoters by AraR that occurs inthe absence of L-arabinose is schematicallyrepresented. The araABDLMNPQ-abfAoperon and araE/araR promoters region isdrawn to scale, with the AraR binding sitesand the distance between its CG centresoutlined. Arrows pointing at AraR binding sitesrepresent the mode action of AraR and thelevel of repression of the AraR-responsivegenes, as estimated by transcriptional fusions(SaÂ-Nogueira and Mota, 1997; SaÂ-Nogueiraand Ramos, 1997; SaÂ-Nogueira et al., 1997),is indicated. Transcriptional start points (�1)and the `±10' and `±35' positions are alsoshown.
484 L. J. Mota, P. Tavares and I. SaÂ-Nogueira

operon promoter and in the araE promoter, concomitant
with DNA bending in both cases, may maintain the DNA
in a conformation unsuitable for transcription initiation,
thus allowing a higher level of repression of the metabolic
operon and transport gene expression. This mechanism
would not be operative in the case of araR (see above),
implying that the divergent transcriptional units encoding
araR and araE are regulated asymmetrically. Considering
that the af®nity of AraR for all the operator sequences
under analysis is relatively similar (Figs 4A and 5A), it
seems most likely that alteration of the DNA conformation
in the araABDLMNPQ-abfA and araE promoter regions is
an essential feature underlying the 12.5-fold higher level
of transcription repression of araABDLMNPQ-abfA and
araE compared with araR (Fig. 7). Induction by L-arabi-
nose has been shown to increase the expression of the
metabolic operon and araE gene about 50-fold, whereas
the expression of araR increases about 4-fold (SaÂ-Nogueira
and Mota, 1997; SaÂ-Nogueira and Ramos, 1997;
SaÂ-Nogueira et al., 1997; Fig. 7). These differences
re¯ect distinct physiological requirements. In the absence
of L-arabinose, the weak repression of araR allows for a
low level of basal transcription, which keeps the system
repressed. Tight regulation of the metabolic and trans-
port genes, on the other hand, ensures expression
uniquely when the L-arabinose substrate is present.
The presence of an inducer leads to a moderate
increase in the cytoplasmic pool of AraR, which is an
effective strategy to promote the fast shut-off of the
metabolic genes' expression upon depletion of
L-arabinose.
The AraE permease, the main transporter for L-arabi-
nose in B. subtilis (SaÂ-Nogueira and Ramos, 1997), is
also responsible for the transport of the pentose D-xylose
and the hexose D-galactose into the cell (Krispin and
Allmansberg, 1998). The fact that three structurally differ-
ent substrates are subjected to AraR regulation suggests
that AraR may also control other genes and operons and
may represent a global regulator of carbohydrate meta-
bolism. A search of the B. subtilis database (SubtiList;
Moszer et al., 1995) for the AraR consensus operator
sequence, allowing a maximum of three mismatches,
identi®ed several sequences, which are currently being
investigated. Among these potential AraR binding sites,
two were located in the promoter region of the abnA and
asd genes, coding for an arabinase and an arabinosidase
(Wipat et al., 1996) respectively. These genes are involved
in the utilization of arabinan, a L-arabinofuranose homo-
glycan found in the hemicellulose and pectin of plant cell
walls. The ongoing characterization of the role of AraR in
the regulation of these genes' expression will extend our
view of how genetic regulation circuitry and L-arabinose
metabolism interplay to yield an economic and ef®cient
way to metabolize this sugar.
Experimental procedures
Microbiological methods
The E. coli strains DH5a (Gibco BRL) or XL1 blue (Strata-gene) were used as hosts for all plasmids. E. coli BL21(DE3) (pLysS) (Studier et al., 1990) was used for overproduc-tion of AraR. E. coli strains were grown on LB (Luria±Bertanimedium; Miller, 1972). Ampicillin (100 mg mlÿ1), chlorampheni-col (30 mg mlÿ1), X-Gal (5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside; 40 mg mlÿ1), and IPTG (isopropyl-b-D-galactopyranoside; 1 mM) were added as appropriate. Solidmedium was made with LB containing 1.5% (w/v) agar. E.coli transformations were carried out according to standardmethods (Sambrook et al., 1989).
DNA manipulation, sequencing and polymerase chain
reaction ampli®cation
DNA manipulations were carried out as described in Sam-brook et al. (1989). Restriction enzymes were used accordingto the manufacturer's instructions. DNA was eluted from aga-rose gels using the QIAquick gel extraction kit (Qiagen). DNAsequencing was performed by the method of Sanger et al.(1977) with a Sequenase kit (T7 DNA polymerase; UnitedStates Biochemical Corporation). All polymerase chain reac-tion (PCR) ampli®cations were done using proofreadingUltma DNA polymerase (Perkin Elmer) or native Pfu DNApolymerase (Stratagene). PCR products were puri®ed usinga QIAquick PCR puri®cation kit (Qiagen).
Plasmids and vectors
Construction of plasmid pLM3 was as described bySaÂ-Nogueira and Mota (1997). An Acc I (®lled-in)±EcoRI1.2 kb fragment from pLM3 containing the araR ribosomebinding site, entire coding sequence and putative transcrip-tional terminator was subcloned in pBluescript II KS (ÿ)(Stratagene) restricted with Sma I±EcoRI. In the resultingplasmid, pLM14, araR is placed downstream from a T7 pro-moter, making it the only gene in the chimeric plasmid to beexpressed from this promoter. The araR coding sequencewas ampli®ed from pLM14 using oligonucleotides 70141 and60815AZ (Table 1). The PCR product was restricted withEcoRI±HindIII and ligated to plasmid pMal-c2 (New EnglandBiolabs). The resulting plasmid, pLM28, contains a fusion ofthe AraR N-terminal domain to the C-terminal domain of theE. coli maltose-binding protein (MBP2*).
Plasmid pLM9 was constructed by subcloning a 249 bpHindIII±EcoRI (®lled-in) DNA fragment from pSNL9(SaÂ-Nogueira et al., 1997), containing the promoter regionof the araABDLMNPQ-abfA operon, at the unique Sma I siteof pBluescript II SK (�) (Stratagene). The Sma I site in theamyE backsequence of pDH32 (Perego, 1993) was elimi-nated by insertion of the 2.7 kb Nru I±BamHI DNA fragmentfrom pAC5 (Martin-Verstraete et al., 1992) between theNru I and BamHI sites of pDH32. The resulting plasmid,pSN32, was used as the cloning vector for constructs bearingDNA fragments with one, two or three AraR binding sites.
A DNA fragment from the araABDLMNPQ-abfA operonpromoter region carrying ORA1 and ORA2 was obtainedby PCR ampli®cation from pLM9 with oligonucleotides
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 33, 476±489
Mode of action of AraR in L-arabinose metabolism 485

B1554F09 and B1554F12 (Table 1). To construct pLM32 andpLS13, the PCR product was restricted with BamHI±EcoRIand BamHI±DraI, yielding a 204 bp and a 170 bp DNA frag-ment, respectively, which contain ORA1 and ORA2. Thesefragments were inserted between the BamHI and EcoRIand between the BamHI and Sma I sites of pSN32 respec-tively. By PCR ampli®cation from pLM9 with oligonucleotidesB1554F09 and B1554F10 (Table 1), a 117 bp DNA fragment,bearing ORA1 alone, was obtained after digestion with Bgl II±EcoRI. Its insertion into pSN32 restricted with BamHI±EcoRIyielded plasmid pLM34. Plasmid pLM51 was obtained by sub-cloning a 204 bp BamHI±EcoRI DNA fragment from pLM32between the BamHI and EcoRI sites of pBluescript II KS(�) (Stratagene). To construct plasmid pLM71, a 105 bpBamHI±Ssp I DNA fragment from pLM51, containing ORA2
alone, was subcloned between the BamHI and Sma I sitesof pSN32.
Plasmid pLM3, bearing the araE /araR promoter region,was used as a template to amplify, by PCR, different DNAfragments with speci®c pairs of oligonucleotides: 220 bp,161 bp, 170 bp, 137 bp and 111 bp DNA fragments wereobtained with oligonucleotides B1553F01 and B1553F03,B1553F01 and B1554F08, B1554F07 and B1553F03,B1553F02 and B1553F03, and B1554F07 and B1554F08respectively (Table 1). These PCR products were restrictedwith BamHI±EcoRV, BamHI±ScaI or BamHI±DraI andinserted into the BamHI and Sma I sites of pSN32. The result-ing plasmids, pLS8, pLS11, pLS5, pLS10 and pLS4, contain,respectively, ORE1, ORE2 and ORR3; ORE1 and ORE2; ORE1
and ORR3; ORR3 alone; and ORE1 alone. A 156 bp DNA frag-ment bearing ORE2 alone was obtained by PCR ampli®cationfrom chromosomal DNA of B. subtilis 168T� wild-type strainwith oligonucleotides B0603H11 and B5460B02 (Table 1)and restricted with EcoRI. Insertion of this DNA fragmentinto pSN32 digested with Sma I and EcoRI yielded pLM72.
Plasmid pLM51 was used as the target DNA for site-directed
mutagenesis with oligonucleotides B4406G11 and B4406G12(Table 1) using a QuikChange site-directed mutagenesis kit(Stratagene). The resulting plasmid, pLM54, bears a singlepoint mutation in ORA1: AAAATTGTTCTTACAAATATTTA(the single change G to T indicated in bold). To construct plas-mid pLM56, a 204 bp BamHI±EcoRI DNA fragment fromplasmid pLM54 was subcloned in pSN32 restricted withBamHI±EcoRI. The sequence of all inserts obtained by PCRwas con®rmed by DNA sequencing of the constructs.
Protein expression and puri®cation
The fusion protein AraR±MBP2* was puri®ed from an extractof E. coli XL1 Blue (Stratagene) bearing plasmid pLM28 usingan amylose resin column (New England Biolabs) according tothe manufacturer's instructions. Rabbit polyclonal serumagainst AraR and MBP2* was produced by immunizationwith puri®ed AraR±MBP2*. E. coli BL21 (DE3) (pLysS)(Studier et al., 1990) transformed with pLM14 was used forthe overproduction and puri®cation of native AraR. The stab-ility of the biological system and the overproduction of a40 kDa protein were checked in a small-scale procedure,which was then scaled up. The AraR-overproducing strainwas grown in 10 l of LB medium at 378C in a fermentor. T7RNA polymerase production was induced for 30 min by theaddition of 2 mM IPTG at an optical density, at 550 nm(OD550), of about 0.6 (1±5 ´ 108 cfu mlÿ1). The E. coli nativeRNA polymerase was then inhibited by the addition of150 mg mlÿ1 rifampicin, and the growth continued for an addi-tional 2 h and 30 min. All subsequent steps were carried out at0±48C. All puri®cation steps were done with the columns con-nected to a fast-protein liquid chromatography (FPLC) system(Pharmacia). Cells were harvested by centrifugation (40 min,12 000 g) and washed in ice-cold medium containing 10%(v/v) glycerol. The cell pellets were frozen in liquid nitrogenand stored at ÿ808C until use. The pellet, corresponding to
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 33, 476±489
Table 1. Oligonucleotides used in this work.
Oligonucleotide Sequences (58!38)a Complementary sequence
B1554F09 ÿ87CCTATTGAATTCAA AAGCCGG ÿ67 araABDLMNPQ-abfAB4406G11 ÿ13CTATAATAAAATTGTTCTTACAAATATTTATTTATAGG�25 araABDLMNPQ ± abfAB4406G12 �25CCTATAAATAAATATTTGTAAGAACAATTTTATTATAGÿ 3 araABDLMNPQ ± abfAB1554F10 �47CGTACTAAAGATCTAAAATAAACC�24 araABDLMNPQ ± abfAB1554F12 �124GAATTCAGGATCCTTTGTCTGAAGC�100 araABDLMNPQ ± abfAB5460H02 �5GTACGAATTCAATGTAATTTTCG�27 araEB1553F02 �8AGTAAGTACTAATATAGAATAATCTTGTTGCÿ 23 araEB1554F07 �47GTACTTATTTTAAAATTTCG�26 araEB1553F01 �92CCCGGATATCGAGTAAAGCGTTTTCATTTAAACC�59 araE81666 �137CATTTGGTTCTAATTGAGTTGG�115 araEB0603H11 �165CCCATTGAATGGCTTCTTGTTACAGG�140 araE70141 TTTGGGGATCCGGAATTC�28ATGTTACCAAAATACGCGC�46 araRB1554F08 �17CCAAAATGAATTCGGATCCATTTCAGÿ 9 araRB1553F03 �67GAACTGGATCCTTCTTTGAATTCCGCGTATTTTGG�34 araR82737 �100GATCGGGCAGTATTTTGCC�82 araR60815AZ �1133CCCAAAGCTTGCTGAATTTATTCATTCAGTTTTCGTGC�1096 araRB3326E12 ÿ39TAAGGGTAACTATTGCCGÿ22 pSN32B5120E10 �35AGTGTATCAACAAGCTGG�17 pSN32B5460B01 �77CTTCCACAGTAGTTCACC�60 pSN32
a. The number within the primers refers to the position of the sequence in the araABDLMNPQ-abfA operon, araE, and araR, relative to the tran-scription start point of each gene, or in pSN32 relative to the EcoRI site (�1) in the multiple cloning site. Restriction sites used are underlined in theoligonucleotide sequences, BamHI (GGATCC), Bgl II (AGATCT), EcoRI (GAATTC), EcoRV (GATATC), Dra I (TTTAAA), HindIII (AAGCTT) andSca I (GATATC).
486 L. J. Mota, P. Tavares and I. SaÂ-Nogueira

about 4 l of cells, was thawed on ice in 80 ml of HS buffer[20 mM Hepes-KOH, pH 7.6, 10 mM EDTA, pH 8.0, 500 mMKCl, 1 mM DTE, 10% (v/v) glycerol]. The resuspended cellswere lysed by passing twice through a French press in thepresence of 1 mM phenylmethylsulphonyl ¯uoride (PMSF).The sample was centrifuged for 1 h at 15 000 g and the pro-teins were precipitated with ammonium sulphate to 75%saturation. The precipitated proteins were resuspended inLS buffer [20 mM Hepes-KOH, pH 7.6, 10 mM EDTA,pH 8.0, 50 mM KCl, 1 mM DTE, 10% (v/v) glycerol] and theammonium sulphate removed by gel ®ltration on PD10columns (Pharmacia) equilibrated with LS buffer. The samplewas applied to a pre-equilibrated XK16 column packed with20 ml of heparin±Sepharose (Pharmacia). After washingwith 20 ml of the starting buffer, proteins were eluted with a20 ml programmed KCl gradient of 50±1000 mM in 20 mMHepes-KOH, pH 7.6, 10 mM EDTA, pH 8.0, 1 mM DTE, 10%(v/v) glycerol. AraR eluted between 600 and 1000 mM KCl.Fractions enriched in AraR were pooled and the salt removedby gel ®ltration in a pre-equilibrated PD10 column (Phar-macia). Particulate material was pelletized by centrifugationfor 15 min at 15 000 g. The soluble fraction was applied to apre-equilibrated Hi-Load SP-Sepharose HP (16/10) column(Pharmacia). After washing with 20 ml of the starting buffer,the sample was eluted with a 100 ml programmed KCl gradi-ent of 50±1000 mM in 20 mM Hepes-KOH, pH 7.6, 10 mMEDTA, pH 8.0, 1 mM DTE, 10% (v/v) glycerol. AraR elutedbetween 200 and 350 mM. AraR-containing fractions werepooled and concentrated by ultra®ltration with a Centricon-30(Amicon). Protein was aliquoted, frozen in liquid nitrogen,kept at ÿ808C and used within 6 months with no detectableloss in activity. We have used lysates of AraR ÿ cells[(BL21)(DE3)(LysS)(KS-)], which were subject to the samechromatographic steps as the overproducing strain (AraR�),to identify AraR in the initial attempts of puri®cation. The pre-sence of AraR in fractions from AraR ÿ and AraR� E. coli cellswas checked by Western blot analysis using anti-AraR±MBP2*.
The native molecular weight of AraR was estimated bysize exclusion chromatography in a Superose 12 column(Pharmacia) equilibrated with buffer HS and calibrated withBio-Rad molecular weight markers [thyroglobulin (670 kDa),g-globulin (158 kDa), ovalbumin (44 kDa), myoglubin (17 kDa),vitamin B12 (1.35 kDa)]. The puri®ed protein was subjectedto N-terminal microsequencing on an ABI 494 PROCISEsequencer. Protein concentrations were determined by themethod of Bradford (1976) using bovine serum albumin(BSA) as a standard.
Synthesis and labelling of the DNA fragments
The target DNA fragments used were generated by PCR. Inthe reaction mixtures, one of the oligonucleotides has beenend labelled with [g-32P]-ATP using polynucleotide kinase.The PCR products were separated in a native polyacrylamide6% (w/v) gel, and the 58 end-labelled DNA fragments puri®edfrom the polyacrylamide gel slices as described by Ausubel etal. (1992). In the case of the araABDLMNPQ-abfA promoter,pLM9 was used as a template with M13 (ÿ20) and M13reverse oligonucleotides (Stratagene), yielding a 456 bp frag-ment. In the case of the araE/araR promoter, pLM3 plasmidDNA was used as a template with oligonucleotides 81666
and 82737 (Table 1), yielding a 309 bp fragment. These frag-ments contain all the potential regulatory sequences detectedin the ara promoters region from the analysis of the nucleotidesequence.
To obtain the DNA fragments with one, two or three AraRbinding sites used in the quantitative DNase I footprintingexperiments, pSN32 derivatives (described above) wereused as templates with oligonucleotides homologous toDNA sequences ¯anking the pSN32 multiple cloning site. Inthe case of the araABDLMNPQ-abfA promoter, DNA fromplasmids pLS13, pLM34, pLM71 and pLM56 was ampli®edwith oligonucelotides B3326E12 and B5120E10, B3326E12and B5460B01, B3326E12 and B5120E10, and B3326E12and B5120E10 respectively (Table 1), yielding the followingDNA fragments: ORA1±ORA2 (228 bp), ORA1 (218 bp), ORA2
(173 bp) and ORA1mut±ORA2 (254 bp) respectively. In thecase of the araE /araR promoter, the DNA fragmentsORR3±ORE1±ORE2 (292 bp), ORE1±ORE2 (227 bp), ORR3±ORE1 (236 bp), ORE1 (180 bp), ORE2 (223 bp) and ORR3
(203 bp) were obtained by ampli®cation of DNA from plasmidspLS8, pLS11, pLS5, pLS4, pLM72 and pLS10, respectively,with oligonucleotides B3326E12 and B5120E10, B3326E12and B5120E10, B3326E12 and B5120E10, B3326E12 andB5120E10, B3326E12 and B5120E10, and B3326E12 andB5120E10 respectively (Table 1).
DNase I footprinting
For DNase I footprinting, labelled target DNA fragments(between 1 and 2 nM) were incubated in the presence of dif-ferent amounts of AraR in 50 ml of binding buffer [20 mMHepes-KOH, pH 7.6, 4 mM EDTA, 60 mM KCl, 10 mM MgCl2,1 mM DTE, 10% (v/v) glycerol, 0.1% (w/v) BSA] with a100-fold excess of competitor DNA (polydI±dC). After 30 minequilibration at room temperature, 50 ml of cofactor mixture(10 mM MgCl2, 5 mM CaCl2) was added to each tube.DNase I (Boehringer Mannheim) was added at a ®nal concen-tration of 0.0002 U mlÿ1. Digestion was allowed for 6 min 30 son ice and quenched by the addition of 100 ml of reactionstop solution [1% (w/v) SDS, 200 mM NaCl, 20 mM EDTApH 8.0, 4 mg mlÿ1 tRNA]. After phenol±chloroform extraction,DNA fragments were precipitated with ethanol and dissolvedin 10 mM Tris-HCl, pH 8.0, 20 mM EDTA, pH 8.0, 0.05%(w/v) bromophenol blue, 0.05% (w/v) xylene cyanol, 95%(v/v) formamide. They were then denatured at 958C for2 min and fractionated in a polyacrylamide±urea 6% (w/v)gel. Gels were dried onto Whatman 3 MM paper and exposedto Hyper®lm MP (Amersham Life Science). Autoradiogramswere scanned in a computing densitometer (MolecularDynamics), and fractional protection at each binding sitewas calculated as described by Brenowitz et al. (1988)using ImageQuant software (Molecular Dynamics). The appa-rent dissociation constant (Kapp) for the different operatorswas determined as the total concentration of AraR requiredfor half-maximal site protection. Concentration of the DNAfragments was estimated to be less than 4% of the apparentdissociation constant (Kapp) for each template.
Acknowledgements
We thank T. A. Trautner, H. de Lencastre and A. O. Henriquesfor their constant interest in this work and helpful discussions,
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 33, 476±489
Mode of action of AraR in L-arabinose metabolism 487

S. Schaper for critical reading of the manuscript, H. Gaengefor protein microsequencing and L. M. Sarmento for con-structing some plasmids. This work was supported by grantno. PRAXIS XXI Bio/0033/96 from FundacËaÄo para a CieÃnciae Tecnologia (FCT). L.J.M. was the recipient of fellowship no.PRAXIS XXI BD/5689/95 from FCT and of an EMBO short-term fellowship.
References
Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seid-man, J.G., Smith, J.A., and Struhl, K. (1992) Short Proto-cols in Molecular Biology, 2nd edn. Harvard MedicalSchool, NY: John Wiley and Sons.
Bradford, M.M. (1976) A rapid and sensitive method for thequanti®cation of microgram quantities of protein utilizingthe principle of protein dye-binding. Anal Biochem 72:248±253.
Brenowitz, M., Senear, D.F., and Ackers, G.K. (1988) Quan-titative DNaseI footprint titration: a method for studyingprotein±DNA interactions. Methods Enzymol 130: 132±181.
Clark, D.P., Cronan, J.R., and Cronan, J.E. (1996) Two-carbon compounds and fatty acids as carbon sources. InEscherichia coli and Salmonella: Cellular and MolecularBiology, 2nd edn. Neidhart, F.C., Curtiss III, R., Ingraham,J.L., Lin, E.C.C., Low, K., Maganasik, B., et al. (eds).Washington, D.C.: American Society for Microbiology,pp. 343±357.
Czaplewski, L.G., North, A.K., Smith, M.C., Baumberg, S.,and Stockley, P.G. (1992) Puri®cation and initial character-ization of AhrC: the regulator of arginine metabolism genesin Bacillus subtilis. Mol Microbiol 6: 267±275.
Drew, H.R., and Travers, A.A. (1984) DNA structural vari-ations in the E. coli tyrT promoter. Cell 37: 491±502.
Fujita, Y., and Miwa, Y. (1989) Identi®cation of an operatorsequence for the Bacillus subtilis gnt operon. J BiolChem 264: 4201±4206.
GoÈsseringer, R., Kuster, E., Galinier, A., Deutscher, J., andHillen, W. (1997) Cooperative and non-cooperative DNAbinding modes of catabolite control protein CcpA fromBacillus megaterium result from sensing two differentsignals. J Mol Biol 266: 665±676.
Haydon, D.J., and Guest, J.R. (1991) A new family ofbacterial regulatory proteins. FEMS Microbiol Lett 79:291±296.
Hochschild, A., and Ptashne, M. (1986) Cooperative bindingof l repressors to sites separated by integral turns of theDNA helix. Cell 44: 681±687.
Krispin, O., and Allmansberg, R. (1998) The Bacillus subtilisAraE protein displays a broad substrate speci®city forseveral different sugars. J Bacteriol 180: 3250±3252.
Kunst, F., Ogasawara, N., Moszer, I., Albertini, A.M., Alloni,G., Azevedo, V. et al. (1997) The complete genome sequ-ence of the Gram-positive bacterium Bacillus subtilis.Nature 390: 249±256.
Lanzer, M., and Bujard, H. (1988) Promoters largely determinethe ef®ciency of repressor action. Proc Natl Acad Sci USA85: 8973±8977.
Lee, D.H., and Schleif, R.F. (1989) In vivo DNA loops inaraCBAD : size limits and helical repeat. Proc Natl AcadSci USA 86: 476±480.
Lepesant, J.A., and Dedonder, R. (1967) Metabolism duL-arabinose chez Bacillus subtilis Marburg Ind 168. C RAcad Sci Ser D 264: 2683±2686.
Martin-Verstraete, I., DeÂbarbouille , M., Klier, A., and Rapo-port, G. (1992) Mutagenesis of the Bacillus subtilis ``ÿ12,ÿ24'' promoter of the levanase operon and evidence ofan upstream activating sequence. J Mol Biol 226: 85±99.
Miller, J.H. (1972) Experiments in Molecular Genetics ColdSpring Harbor, NY: Cold Spring, Harbor Laboratory Press.
Moszer, I., Glaser, P., and Danchin, A. (1995) SubtiList: arelational database for the Bacillus subtilis genome. Micro-biology 141: 261±268.
Pabo, C.O., and Sauer, R.T. (1992) Transcription factors:structural families and principles of DNA recognition.Annu Rev Biochem 61: 1053±1095.
Perego, M. (1993) Integrational vectors for genetic manipula-tion in Bacillus subtilis. In Bacillus subtilis and Other GramPositive Bacteria: Biochemistry, Physiology, and MolecularGenetics. Sonenshein, A.L., Hoch, J.A., and Losick, R.(eds). Washington, D.C.: American Society for Micro-biology, pp. 615±624.
Quail, M.A., Dempsey, C.E., and Guest, J.R. (1994) Identi®-cation of a fatty acyl responsive regulator (FarR) inEscherichia coli. FEBS Lett 356: 183±187.
Quail, M.A., and Guest, J.R. (1995) Puri®cation, characteriz-ation and mode of action of PdhR, the transcriptionalrepressor of the pdhR-aceEF-lpd operon of Escherichiacoli. Mol Microbiol 15: 519±529.
Sambrook, J., Fritsch, E.F., and Maniatis, T. (1989) Molecu-lar Cloning: a Laboratory Manual, 2nd edn. Cold SpringHarbor, NY: Cold Spring Harbor Laboratory Press.
Sanger, F., Nicklen, S., and Coulson, A.R. (1977) DNA sequ-encing with chain-terminating inhibition. Proc Natl Acad SciUSA 74: 140±144.
SaÂ-Nogueira, I., and Lencastre, H. (1989) Cloning and char-acterization of araA, araB, and araD, the structural genesfor L-arabinose utilization in Bacillus subtilis. J Bacteriol171: 4088±4091.
SaÂ-Nogueira, I., and Mota, L.J. (1997) Negative regulation ofL-arabinose metabolism in Bacillus subtilis: characteriz-ation of the araR (araC ) gene. J Bacteriol 179: 1598±1608.
SaÂ-Nogueira, I., Nogueira, T.V., Soares, S., and Lencastre,H. (1997) The L-arabinose (ara) operon of Bacillus subtilis:nucleotide sequence, genetic organization and expression.Microbiology 143: 957±969.
SaÂ-Nogueira, I., and Ramos, S. (1997) Cloning, functionalanalysis, and transcriptional regulation of the Bacillussubtilis araE gene involved in L-arabinose utilization. JBacteriol 179: 7705±7711.
Schleif, R. (1992) DNA looping. Annu Rev Biochem 61: 199±223.
Schleif, R. (1996) Two positively regulated systems, ara andmal. In Escherichia coli and Salmonella: Cellular and Mol-ecular Biology, 2nd edn. Neidhart, F.C., Curtiss III, R.,Ingraham, J.L., Lin, E.C.C., Low, K., Maganasik, B., et al.,(eds). Washington, D.C.: American Society for Microbiol-ogy, pp. 1300±1309.
SchoÈck, F., and Dahl, M.K. (1996) Expression of the treoperon of Bacillus subtilis 168 is regulated by the repressorTreR. J Bacteriol 178: 4576±4581.
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 33, 476±489
488 L. J. Mota, P. Tavares and I. SaÂ-Nogueira

Studier, F.W., Rosenberg, A.H., Dunn, J.J., and Dubendorff,J.W. (1990) Use of T7 polymerase to direct expression ofcloned genes. Methods Enzymol 185: 60±89.
Weickert, M.J., and Adhya, S. (1992) A family of bacterialregulators homologous to Gal and Lac repressors. J BiolChem 267: 15869±15874.
Wipat, A., Carter, N., Brignell, S.C., Guy, B.J., Piper, K.,Sanders, J., et al. (1996) The dnaB±pheA (256 degrees±240 degrees) region of the Bacillus subtilis chromosomecontaining genes responsible for stress responses, theutilization of plant cell walls and primary metabolism.Microbiology 142: 3067±3078.
Q 1999 Blackwell Science Ltd, Molecular Microbiology, 33, 476±489
Mode of action of AraR in L-arabinose metabolism 489
Copyright © 2022 FDOKUMEN




















