
Mixed-Valent Polynuclear Cobalt Complexes Incorporating Tetradentate Phenoxyamine Ligands
6
RESEARCH FRONT CSIRO PUBLISHING Full Paper Aust. J. Chem. 2009, 62, 1124–1129 www.publish.csiro.au/journals/ajc Mixed-Valent Polynuclear Cobalt Complexes Incorporating Tetradentate Phenoxyamine Ligands Joo Chuan Ang, A Yanyan Mulyana, A Chris Ritchie, A Rodolphe Clérac, B,C and Colette Boskovic A,D A School of Chemistry, University of Melbourne,Vic. 3010, Australia. B Centre National De La Recherche Scientifique (CNRS), UPR8641, Centre de Recherche Paul Pascal (CRPP), Equipe ‘Matériaux Moléculaires Magnétiques’, 115 avenue du Dr Albert Schweitzer, Pessac, F-33600, France. C Université de Bordeaux, UPR 8641, Pessac, F-33600, France. D Corresponding author. Email: [email protected] The new potentially tetradentate ligand precursor 2-[(bis(2-hydroxyethyl)amino)methyl]-4,6-bis-tert -butylphenol (H 3 L) has been synthesized and characterized. The reaction of H 3 L with cobalt(ii) acetate has afforded the novel mixed-valent tetra- and pentanuclear cobalt complexes [Co II 2 Co III 2 (OAc) 2 (L) 2 (HL)] (1) and [Co II Co III 4 (OAc) 2 (L) 4 ](2). Single-crystal X-ray diffraction studies of these complexes indicate different coordination geometries for the divalent cobalt centres in each complex, with distorted trigonal bipyramidal and distorted tetrahedral coordination evident in 1 and 2, respectively. The variable temperature magnetic susceptibility data for complex 1 reveal behaviour dominated by antiferromagnetic coupling between the two cobalt(ii) centres. Their approximate trigonal bipyramidal coordination geometries give rise to a 4 A 2 ground term, allowing a spin-only treatment assuming local spin quantum numbers of S i = 3/2. Fitting the data to the Heisenberg exchange Hamiltonian ( ˆ H ex =−2J S 1 ·S 2 ) results in J =−6.9(1) cm −1 and g = 2.30(5). Manuscript received: 29 April 2009. Manuscript accepted: 2 June 2009. Introduction The synthesis of new polynuclear complexes of paramagnetic metal centres is driven in part by their potential to display novel magnetic properties that may ultimately give rise to new molecular magnetic materials. [1–3] The development of such spin clusters relies on the exploration of new families of organic ligands that can bridge the metal centres, or act as capping ligands that prevent agglomeration beyond a certain size by encapsulating the metal-containing core. An ongoing focus of our research is the exploration of organic ligands that can simul- taneously chelate and bridge metal centres for the preparation of new spin clusters. In previous work, we have investigated the use of phenoxide-containing Schiff base ligands for the syn- thesis of new polynuclear Mn, Fe, Co, and Ni complexes. [4] We have separately explored the use of a tripodal bridging and chelating ligands with an (N,O,O)-donor set for the formation of polynuclear Fe, Co, Ni, and mixed metal Cu–Ln (Ln = Gd, Tb, and Dy) complexes. [5] The obvious extension of this was to combine the two functionalities in the one ligand, exploit- ing their differing coordination tendencies to obtain polynuclear complexes with novel structural features and resulting prop- erties. Herein we report the synthesis and characterization of the new proligand 2-[(bis(2-hydroxyethyl)amino)methyl]-4,6- bis-tert -butylphenol (H 3 L, Scheme 1), which contains both phenol and tripodal amine-alcohol functionalities, and its use in the synthesis of two novel mixed-valent polynuclear cobalt complexes. H 3 L 4 H 3 L (D OH) HL 1 (D OMe) HL 2 (D NEt 2 ) HL 3 (D CH 2 NMe 2 ) HO N t Bu t Bu D D HO N NO 2 OH OH Scheme 1. Proligands. Although the proligand H 3 L has not been reported previ- ously, ligands similar to H 3 L but bearing amine or ether donors on the pendant arms of the ligand (Scheme 1: HL 1 , HL 2 , and HL 3 ), in place of alcohol groups, have been synthesized, together with their mononuclear Mg, Ca, Al, Zn, Cr(iii), Co(ii), and Zr complexes. [6,7] A dinuclear dichloride-bridged Fe(ii) complex of the ligand (L 1 ) − has been structurally characterized, as has a dinuclear K complex with the ligand (L 2 ) − . In the latter com- plex, the phenoxide oxygen atoms of the ligand serve to bridge the K centres. Tertiary amine and ether donors are unlikely to be able to bridge metal centres; thus their replacement with alco- hol groups in the present work was pursued in order to obtain © CSIRO 2009 10.1071/CH09256 0004-9425/09/091124
Transcript of Mixed-Valent Polynuclear Cobalt Complexes Incorporating Tetradentate Phenoxyamine Ligands

RESEARCH FRONT
CSIRO PUBLISHINGFull Paper
Aust. J. Chem. 2009, 62, 1124–1129 www.publish.csiro.au/journals/ajc
Mixed-Valent Polynuclear Cobalt Complexes IncorporatingTetradentate Phenoxyamine Ligands
Joo Chuan Ang,A Yanyan Mulyana,A Chris Ritchie,A Rodolphe Clérac,B,C
and Colette BoskovicA,D
ASchool of Chemistry, University of Melbourne, Vic. 3010, Australia.BCentre National De La Recherche Scientifique (CNRS), UPR 8641, Centre de Recherche Paul Pascal(CRPP), Equipe ‘Matériaux Moléculaires Magnétiques’, 115 avenue du Dr Albert Schweitzer,Pessac, F-33600, France.
CUniversité de Bordeaux, UPR 8641, Pessac, F-33600, France.DCorresponding author. Email: [email protected]
The new potentially tetradentate ligand precursor 2-[(bis(2-hydroxyethyl)amino)methyl]-4,6-bis-tert-butylphenol (H3L)has been synthesized and characterized. The reaction of H3L with cobalt(ii) acetate has afforded the novel mixed-valenttetra- and pentanuclear cobalt complexes [CoII
2 CoIII2 (OAc)2(L)2(HL)] (1) and [CoIICoIII
4 (OAc)2(L)4] (2). Single-crystalX-ray diffraction studies of these complexes indicate different coordination geometries for the divalent cobalt centres ineach complex, with distorted trigonal bipyramidal and distorted tetrahedral coordination evident in 1 and 2, respectively.The variable temperature magnetic susceptibility data for complex 1 reveal behaviour dominated by antiferromagneticcoupling between the two cobalt(ii) centres. Their approximate trigonal bipyramidal coordination geometries give rise toa 4A′
2 ground term, allowing a spin-only treatment assuming local spin quantum numbers of Si = 3/2. Fitting the data tothe Heisenberg exchange Hamiltonian (Hex = −2J S1·S2) results in J = −6.9(1) cm−1 and g = 2.30(5).
Manuscript received: 29 April 2009.Manuscript accepted: 2 June 2009.
Introduction
The synthesis of new polynuclear complexes of paramagneticmetal centres is driven in part by their potential to displaynovel magnetic properties that may ultimately give rise to newmolecular magnetic materials.[1–3] The development of such spinclusters relies on the exploration of new families of organicligands that can bridge the metal centres, or act as cappingligands that prevent agglomeration beyond a certain size byencapsulating the metal-containing core. An ongoing focus ofour research is the exploration of organic ligands that can simul-taneously chelate and bridge metal centres for the preparationof new spin clusters. In previous work, we have investigated theuse of phenoxide-containing Schiff base ligands for the syn-thesis of new polynuclear Mn, Fe, Co, and Ni complexes.[4]
We have separately explored the use of a tripodal bridging andchelating ligands with an (N,O,O)-donor set for the formationof polynuclear Fe, Co, Ni, and mixed metal Cu–Ln (Ln = Gd,Tb, and Dy) complexes.[5] The obvious extension of this wasto combine the two functionalities in the one ligand, exploit-ing their differing coordination tendencies to obtain polynuclearcomplexes with novel structural features and resulting prop-erties. Herein we report the synthesis and characterization ofthe new proligand 2-[(bis(2-hydroxyethyl)amino)methyl]-4,6-bis-tert-butylphenol (H3L, Scheme 1), which contains bothphenol and tripodal amine-alcohol functionalities, and its usein the synthesis of two novel mixed-valent polynuclear cobaltcomplexes.
H3L4H3L (D � OH)
HL1 (D � OMe) HL2 (D � NEt2) HL3 (D � CH2NMe2)
HO N
tBu
tBu
D
D
HO N
NO2
OH
OH
Scheme 1. Proligands.
Although the proligand H3L has not been reported previ-ously, ligands similar to H3L but bearing amine or ether donorson the pendant arms of the ligand (Scheme 1: HL1, HL2, andHL3), in place of alcohol groups, have been synthesized, togetherwith their mononuclear Mg, Ca, Al, Zn, Cr(iii), Co(ii), and Zrcomplexes.[6,7] A dinuclear dichloride-bridged Fe(ii) complexof the ligand (L1)− has been structurally characterized, as has adinuclear K complex with the ligand (L2)−. In the latter com-plex, the phenoxide oxygen atoms of the ligand serve to bridgethe K centres. Tertiary amine and ether donors are unlikely to beable to bridge metal centres; thus their replacement with alco-hol groups in the present work was pursued in order to obtain
© CSIRO 2009 10.1071/CH09256 0004-9425/09/091124

RESEARCH FRONT
Mixed-Valent Polynuclear Cobalt Complexes 1125
HO N
tBu
tBu
OH
OH
HO
tBu
tBu
HN
OH
OH
�CH2O
MeOH
Scheme 2. Synthesis of proligand H3L.
polynuclear complexes. The promise in this regard of the pro-ligand H3L was also inferred from the previously reported useof the related polyalcohol-containing proligand H3L4 (Scheme1) for the synthesis of di- and heptanuclear Mn complexes.[8,9]
Results and DiscussionSynthesis and CharacterizationThe ligand precursor H3L was prepared by a Mannich con-densation between 2,3-di-tert-butylphenol, diethanolamine, andparaformaldehyde in methanol (Scheme 2). The product wasobtained as a white crystalline material in reasonable yield, thepurity of which was established by elemental analysis and 1H and13C NMR spectroscopy. A similar preparation has been reportedpreviously for the related ligand precursors HL1, HL2, and HL3
(Scheme 1).[6,7]
The overnight treatment of a solution of cobalt(ii) acetate inacetonitrile with one equivalent of the proligand H3L afforded asolution from which crystals of [Co4(OAc)2(L)2(HL)]·2MeCN(1·2MeCN) were obtained on standing. A similar reactioninvolving pretreatment of the proligand with two equiva-lents of triethylamine and recrystallization of the productfrom dichloromethane/hexane afforded a crystalline sample of[Co5(OAc)2(L)4] (2). Both reactions involved partial aerial oxi-dation of the cobalt starting material and can be summarized asfollows:
4 Co(OAc)2 + 3 H3L → [CoII2 CoIII
2 (OAc)2(L)2(HL)]
+ 6 AcOH + 2 H+ + 2 e− (1)
5 Co(OAc)2 + 4 H3L → [CoIICoIII4 (OAc)2(L)4]
+ 8 AcOH + 4 H+ + 4 e− (2)
Both reactions appear to be driven by protonation of theacetate ligands of the starting material and their replacementby the incoming chelating ligands. This process is accompaniedby structural rearrangement and agglomeration, which is com-monly observed in the synthesis from acetate-containing startingmaterials of polynuclear complexes bearing ligands that simulta-neously chelate and bridge the metal centres.[1,5] It is noteworthythat addition of base to the reaction mixture afforded complex 2,with complete deprotonation of the proligand, as opposed to thepartial deprotonation observed in 1, which was synthesized with-out added base.Although both complexes could only be obtainedin modest yield, the syntheses are reproducible. The low yield islargely due to the very high solubility of both complexes in mostorganic solvents, which in turn results from the presence of thesolubilizing tert-butyl substituents on the aromatic ring.
Structure DescriptionsCompound 1·2MeCN crystallizes in the monoclinic spacegroup P21/c with one tetranuclear complex and two acetonitrile
Table 1. Selected interatomic distances [Å] and angles [◦] for 1·2MeCNand 2
Parameter 1·2MeCN 2
CoII· · ·CoII 3.427(1)CoII· · ·CoIII 3.043(1)–3.075(1) 3.188(1)CoIII· · ·CoIII 2.855(1) 2.825(1)CoII–O 1.948(3)–2.074(3) 1.988(8)–2.022(8)CoII–N 2.204(4)CoIII–O 1.874(3)–1.969(3) 1.796(9)–2.054(7)CoIII–N 1.935(4)–1.939(4) 1.924(4)CoII–O–CoII 122.8(2)CoII–O–CoIII 90.4(1)–105.0(1) 107.7(3)–112.3(3)CoIII–O–CoIII 94.6(1)–96.3(1) 92.6(3)–97.7(3)O–CoII–OA 112.1(1)–121.4(1)O–CoII–OB 167.4(1)O–CoII–NB 173.7(1)O–CoII–OC 90.8(3)–119.8(3)O–CoIII–OD 81.7(1)–94.5(1) 74.0(3)–98.0(3)O–CoIII–OE 175.2(1)–178.5(1) 169.7(2)–175.8(2)O–CoIII–NCD 85.9(1)–95.1(1) 81.8(2)–96.8(2)O–CoIII–NE 165.0(2)–166.9(2) 163.4(3)–168.5(2)
ATrigonal bipyramidal, equatorial.BTrigonal bipyramidal, axial.CTetrahedral.DOctahedral, cis.EOctahedral, trans.
N1
N2
O1
O2
O3
O11O10
O4
O5
O12
O6
O7
O8O9
O13
Co1Co2
Co3
Co4
N3
Fig. 1. Structural representation of complex 1 in 1·2MeCN. Colour code:CoII (cyan), CoIII (green), O (red), N (blue).
molecules in the asymmetric unit. A summary of the impor-tant interatomic distances and angles is provided in Table 1.The asymmetric neutral tetranuclear complex 1 (Fig. 1) has a{CoII
2 CoIII2 (µ2-O)4(µ3-O)} core where the five oxygen atoms
are provided by the ethoxo arms of the single HL2− and twoL3− ligands. Bond valence sum calculations indicate that cobaltcentres Co1 and Co2 are trivalent, whereas Co3 and Co4 aredivalent.[10] The trivalent centres both display distorted octahe-dral coordination geometries. However, the divalent centres areboth five-coordinate, with distorted trigonal bipyramidal coor-dination geometries, where the axial coordination of the trigonalbipyramids is defined by O5 and O7 for Co3 and N3 and O13for Co4. A distortion parameter, τ, can be calculated for five-coordinate complexes, such that the parameter τ is 100% and
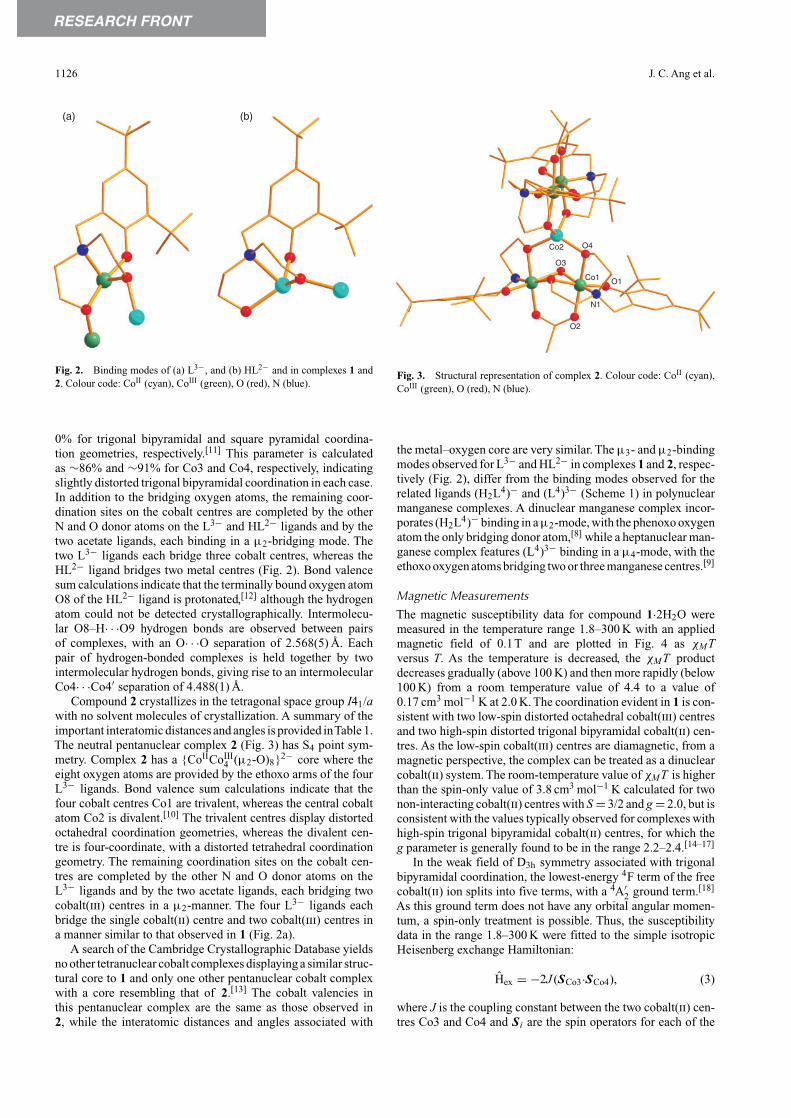
RESEARCH FRONT
1126 J. C. Ang et al.
(a) (b)
Fig. 2. Binding modes of (a) L3−, and (b) HL2− and in complexes 1 and2. Colour code: CoII (cyan), CoIII (green), O (red), N (blue).
0% for trigonal bipyramidal and square pyramidal coordina-tion geometries, respectively.[11] This parameter is calculatedas ∼86% and ∼91% for Co3 and Co4, respectively, indicatingslightly distorted trigonal bipyramidal coordination in each case.In addition to the bridging oxygen atoms, the remaining coor-dination sites on the cobalt centres are completed by the otherN and O donor atoms on the L3− and HL2− ligands and by thetwo acetate ligands, each binding in a µ2-bridging mode. Thetwo L3− ligands each bridge three cobalt centres, whereas theHL2− ligand bridges two metal centres (Fig. 2). Bond valencesum calculations indicate that the terminally bound oxygen atomO8 of the HL2− ligand is protonated,[12] although the hydrogenatom could not be detected crystallographically. Intermolecu-lar O8–H· · ·O9 hydrogen bonds are observed between pairsof complexes, with an O· · ·O separation of 2.568(5) Å. Eachpair of hydrogen-bonded complexes is held together by twointermolecular hydrogen bonds, giving rise to an intermolecularCo4· · ·Co4′ separation of 4.488(1) Å.
Compound 2 crystallizes in the tetragonal space group I41/awith no solvent molecules of crystallization. A summary of theimportant interatomic distances and angles is provided inTable 1.The neutral pentanuclear complex 2 (Fig. 3) has S4 point sym-metry. Complex 2 has a {CoIICoIII
4 (µ2-O)8}2− core where theeight oxygen atoms are provided by the ethoxo arms of the fourL3− ligands. Bond valence sum calculations indicate that thefour cobalt centres Co1 are trivalent, whereas the central cobaltatom Co2 is divalent.[10] The trivalent centres display distortedoctahedral coordination geometries, whereas the divalent cen-tre is four-coordinate, with a distorted tetrahedral coordinationgeometry. The remaining coordination sites on the cobalt cen-tres are completed by the other N and O donor atoms on theL3− ligands and by the two acetate ligands, each bridging twocobalt(iii) centres in a µ2-manner. The four L3− ligands eachbridge the single cobalt(ii) centre and two cobalt(iii) centres ina manner similar to that observed in 1 (Fig. 2a).
A search of the Cambridge Crystallographic Database yieldsno other tetranuclear cobalt complexes displaying a similar struc-tural core to 1 and only one other pentanuclear cobalt complexwith a core resembling that of 2.[13] The cobalt valencies inthis pentanuclear complex are the same as those observed in2, while the interatomic distances and angles associated with
O2
O3
O1
O4
Co1
Co2
N1
Fig. 3. Structural representation of complex 2. Colour code: CoII (cyan),CoIII (green), O (red), N (blue).
the metal–oxygen core are very similar. The µ3- and µ2-bindingmodes observed for L3− and HL2− in complexes 1 and 2, respec-tively (Fig. 2), differ from the binding modes observed for therelated ligands (H2L4)− and (L4)3− (Scheme 1) in polynuclearmanganese complexes. A dinuclear manganese complex incor-porates (H2L4)− binding in a µ2-mode, with the phenoxo oxygenatom the only bridging donor atom,[8] while a heptanuclear man-ganese complex features (L4)3− binding in a µ4-mode, with theethoxo oxygen atoms bridging two or three manganese centres.[9]
Magnetic MeasurementsThe magnetic susceptibility data for compound 1·2H2O weremeasured in the temperature range 1.8–300 K with an appliedmagnetic field of 0.1T and are plotted in Fig. 4 as χMTversus T. As the temperature is decreased, the χMT productdecreases gradually (above 100 K) and then more rapidly (below100 K) from a room temperature value of 4.4 to a value of0.17 cm3 mol−1 K at 2.0 K. The coordination evident in 1 is con-sistent with two low-spin distorted octahedral cobalt(iii) centresand two high-spin distorted trigonal bipyramidal cobalt(ii) cen-tres. As the low-spin cobalt(iii) centres are diamagnetic, from amagnetic perspective, the complex can be treated as a dinuclearcobalt(ii) system. The room-temperature value of χMT is higherthan the spin-only value of 3.8 cm3 mol−1 K calculated for twonon-interacting cobalt(ii) centres with S = 3/2 and g = 2.0, but isconsistent with the values typically observed for complexes withhigh-spin trigonal bipyramidal cobalt(ii) centres, for which theg parameter is generally found to be in the range 2.2–2.4.[14–17]
In the weak field of D3h symmetry associated with trigonalbipyramidal coordination, the lowest-energy 4F term of the freecobalt(ii) ion splits into five terms, with a 4A′
2 ground term.[18]
As this ground term does not have any orbital angular momen-tum, a spin-only treatment is possible. Thus, the susceptibilitydata in the range 1.8–300 K were fitted to the simple isotropicHeisenberg exchange Hamiltonian:
Hex = −2J (SCo3·SCo4), (3)
where J is the coupling constant between the two cobalt(ii) cen-tres Co3 and Co4 and Si are the spin operators for each of the

RESEARCH FRONT
Mixed-Valent Polynuclear Cobalt Complexes 1127
00
1
2
3
4
5
50 100 150 200 250 300
χ MT
[cm
3 m
ol�
1 K
]
T [K]
Fig. 4. Plot of χMT versus T for compound 1·2H2O at 0.1 T; the solid lineis the fitted to the data as described in the text.
centres. An excellent fit (solid line in Fig. 4) was obtained withJ = −6.9(1) cm−1, g = 2.30(5), and ρ = 0.087 (ρ correspondsto the residual paramagnetism observed below 10 K, taking intoaccount the presence of monomeric S = 3/2 cobalt(ii) impurityspecies), giving rise to an S = 0 ground state.
As mentioned above, the g value of 2.3 derived for 1·2H2Ocompares well with the values obtained for other complexeswith high-spin cobalt(ii) centres in trigonal bipyramidal coor-dination geometries. This deviation from the free-ion valuege can be attributed to a second-order Zeeman effect associ-ated with admixture of a higher-energy 4E′′ term with orbitalangular momentum.[16] The two cobalt(ii) centres in complex2 are bridged by an alkoxo and an acetate ligand, with the Jparameter reflecting net antiferromagnetic superexchange alongthese pathways.Although relatively few examples of structurallyanalogous complexes are available in the literature for compar-ison, the J value obtained for 1·2H2O is consistent with thevalue of −8.5 cm−1 obtained for a dinuclear cobalt(ii) com-plex, where two alkoxo ligands bridge two trigonal bipyramidalcobalt(ii) centres.[14] The alkoxo bridge is likely the dominantsuperexchange pathway in 1, with a CoII–O–CoII bridging angleof 123◦. Although magnetostructural correlations between thecoupling constant and the M–O–M bridging angle are not wellestablished for cobalt, as they are for copper and nickel,[19,20]
it is consistent with the Goodenough–Kanemori approach thata Co–O–Co bridging angle larger than 100◦ should give riseto an antiferromagnetic coupling.[21] Given the distorted trigo-nal bipyramidal coordination geometries of Co3 and Co4, thesingly occupied magnetic orbitals on each cobalt centre are thedxy, dx2–y2 , and dz2 orbitals. The bridging alkoxo ligand providesa superexchange pathway for overlap of the dz2 orbital on Co3with the dx2–y2 or dxy (degenerate for a strict trigonal bipyramid)orbitals on Co4.
Conclusion
A new tetradentate phenoxyamine ligand and two new mixed-valent polynuclear cobalt complexes bearing this ligand havebeen synthesized and characterized. The phenoxyamine ligandsimultaneously chelates and bridges the metal centres, dis-playing µ3- and µ2-bridging modes in its triply and doubly
deprotonated forms, respectively. The coordinative flexibility ofthis ligand suggests that it and its derivatives have considerablepromise for the synthesis of new spin clusters.The divalent cobaltcentres in the reported complexes manifest relatively uncommontrigonal bipyramidal and tetrahedral coordination geometries.An investigation of the magnetic properties of the tetradentatecomplex reveals a moderate antiferromagnetic coupling betweenthe divalent cobalt centres.
ExperimentalMaterialsAll manipulations were performed under aerobic conditions,using materials as received.
MeasurementsInfrared spectra (KBr disk) were recorded on a BioRad 175Fourier-transform (FT)-IR spectrometer. 1H and 13C NMR spec-tra were recorded on a Varian Unity+ 400 MHz spectrometer.Electrospray ionization (ESI) mass spectra were performed ona Micromass Quattro II mass spectrometer. Elemental analy-ses were performed by Chemical and Microanalytical Services,Belmont, Australia, and Campbell Microanalytical Laboratory,Dunedin, New Zealand. Thermogravimetric analyses (TGA)were performed on a Mettler Toledo thermal analyzer.
Syntheses2-[(bis(2-Hydroxyethyl)amino)methyl]-4,6-bis-tert-butylphenol (H3L)Paraformaldehyde (0.700 g, 23.2 mmol) and diethanolamine
(2.44 g, 23.2 mmol) were refluxed in MeOH (50 mL) for 90 min,followed by the addition of a solution of 2,4-di-tert-butylphenol(4.79 g, 23.2 mmol) in MeOH (50 mL) and refluxed for a further24 h. After cooling to room temperature, the resulting solutionwas evaporated to dryness and the residue was dissolved inCH2Cl2 and washed with water (3 × 30 mL). The CH2Cl2 solu-tion was dried with magnesium sulfate and n-hexane was addedto crystallize the product as white needles, yield 43%. (Anal.calc. for H3L, C19H33NO3: C 70.55, H 10.28, N 4.33%. FoundC 70.83, H 9.23, N 4.30.) Selected νmax/cm−1 3294, 2961, 2926,2870, 2830, 1482, 1390, 1385, 1289, 1235, 1164, 1125, 1073,996, 757, 723. δH (400 MHz, CDCl3) 1.26 (s, 9H, C(CH3)3),1.39 (s, 9H, C(CH3)3), 2.78 (t, 4H, CH2CH2OH), 3.74 (t, 4H,CH2CH2OH), 3.84 (s, 2H, ArCH2N), 6.83 (s, 1H, ArH), 7.21(s, 1H, ArH). δC (13C{1H}, 100 MHz, CDCl3) 29.6, 31.7, 34.2,34.9, 56.5, 60.3, 60.4, 121.5, 123.3, 123.6, 136.0, 141.1, 153.7.m/z (ESI MS, CH2Cl2) 324.4 [M + H]+.
[CoII2CoIII
2 (OAc)2(L)2(HL)] 1Co(OAc)2·4H2O (0.623 g, 2.5 mmol) and H3L (0.808 g,
2.5 mmol) were dissolved in MeCN (30 mL) and the solutionstirred overnight. A small amount of precipitate was removed byfiltration and the filtrate was loosely covered and left to stand.Purple diamond-shaped crystals formed after 4 days. A sam-ple for crystallography was maintained in contact with motherliquor to prevent the loss of interstitial solvent. The crystals wereisolated by filtration and washed with MeCN; yield 5%. The air-dried sample appears to be hygroscopic. (Anal. calc. for 1·2H2O,C61H101N3Co4O15: C 54.18, H 7.53, N 3.11%. Found C 53.57,H 7.31, N 3.43.)The degree of hydration was confirmed byTGA.Selected νmax/cm−1 3436, 2952, 2868, 1567, 1440, 1360, 1240,1077, 1002, 535.

RESEARCH FRONT
1128 J. C. Ang et al.
Table 2. Crystallographic data for 1·2MeCN and 2
1·2MeCN 2
Formula C65H102Co4N5O13 C80H126Co5N4O16
Formula weight 1397.24 1694.50Space group P21/c I41/aa [Å] 14.603(4) 22.116(2)b [Å] 18.007(5) 22.116(2)c [Å] 27.995(7) 16.765(2)β [◦] 102.709(5) 90V [Å3] 7181(3) 8200(1)Z 4 4T [K] 130(2) 130(2)ρcalc [g cm−3] 1.292 1.373µ [mm−1] 0.968 1.056Reflections measured 43962 25653Unique reflections 16312 4711Data/restraints/parameters 16312/1/762 4711/28/224Rint 0.1429 0.1806R1(I > 2σ(I )) 0.0648 0.0709wR2 (all data) 0.1665 0.1857Goodness-of-fit on F2 0.917 0.993�ρmax,min [e Å−3] 1.128, −0.839 1.076, −0.577
[CoIICoIII4 (OAc)2(L)4] 2
Co(OAc)2·4H2O (0.498 g, 2.00 mmol) was added to a solu-tion of NEt3 (0.404 g, 4 mmol) and H3L (0.646 g, 2.00 mmol) inMeCN (30 mL) and the resulting solution was stirred overnightand filtered. The filtrate was evaporated to dryness on a rotaryevaporator and subjected to high vacuum overnight. A concen-trated solution of the residue in CH2Cl2 was filtered throughCelite and cotton wool and layered with two volumes of n-hexane. Dark purple diamond-shaped crystals formed after aweek, together with an amorphous purple solid. The sample forcrystallography was maintained in contact with mother liquorto prevent the loss of interstitial solvent. Although it was pos-sible to hand-pick individual crystals for single crystal X-raydiffraction studies, it was not possible to separate the crystallineand amorphous materials and obtain a pure sample for bulkmeasurements.
X-Ray CrystallographyThe data for compounds 1·2MeCN and 2 (Table 2) were col-lected on a BrukerApex I Diffractometer at 130 K using graphitemonochromatic Mo-Kα radiation (0.71073 Å). The structures of1 and 2 were solved by direct methods and refined by full-matrixleast-squares method on F2 using the SHELXTL-97 crystallo-graphic package.[22] During the refinement of compounds 1 and2, the tert-butyl groups of the HL2− and L3− ligands and themethyl groups of the bound acetate ligands were found to bedisordered. Significant disorder of the ethoxo arms of the L3−ligands was also observed for compound 2. The disorder hasbeen refined using free variables.
CCDC 728620 (1·2MeCN) and 728621 (2) contain the sup-plementary crystallographic data for the present paper. Thesedata can be obtained free of charge fromThe Cambridge Crystal-lographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Magnetic MeasurementsVariable-temperature magnetic susceptibility and magnetizationmeasurements were obtained using a Quantum Design SQUID
magnetometer MPMS-XL. This magnetometer works between1.8 and 400 K for applied DC fields ranging from −7 to 7T.Measurements were performed on a polycrystalline sample ofcomplex 1·2H2O weighing 10.82 mg. The sample bag was pre-pared in glove box and then immediately put into the SQUID formeasurement. The magnetic data were corrected for the sampleholder and the diamagnetic contribution.
AcknowledgementsThe present work was supported by the Australian Research Council, theUniversity of Melbourne, the University of Bordeaux, the CNRS, the RegionAquitaine and MAGMANet (NMP3-CT-2005–515767). We thank RobertGable for helpful discussions.
References[1] R. E. P. Winpenny, Compr. Coord. Chem. II 2004, 7, 125.
doi:10.1016/B0-08-043748-6/06199-5[2] D. Gatteschi, R. Sessoli, J. Villain, Molecular Nanomagnets 2007
(Oxford University Press: Oxford).[3] E. Coronado, K. R. Dunbar, Inorg. Chem. 2009, 48, 3293.
doi:10.1021/IC900218F[4] (a) C. Boskovic, R. Bircher, P. L. W. Tregenna-Piggott, H. U. Güdel,
C. Paulsen, W. Wernsdorfer, A. L. Barra, E. Khatsko, A. Neels,H. Stoeckli-Evans, J. Am. Chem. Soc. 2003, 125, 14046. doi:10.1021/JA0367086(b) C. Boskovic, A. Sieber, G. Chaboussant, H. U. Güdel, J. Ensling,W. Wernsdorfer, A. Neels, G. Labat, H. Stoeckli-Evans, S. Janssen,Inorg. Chem. 2004, 43, 5053. doi:10.1021/IC049600F(c)A. Sieber, C. Boskovic, R. Bircher, O.Waldmann, S.T. Ochsenbein,H. U. Güdel, N. Kirchner, J. van Slageren, W. Wernsdorfer, A. Neels,H. Stoeckli-Evans, S. Janssen, F. Juranyi, H. Mutka, Inorg. Chem.2005, 44, 4315. doi:10.1021/IC050134J
[5] (a) K. G. Alley, R. Bircher, O. Waldmann, S. T. Ochsenbein,H. U. Güdel, B. Moubaraki, K. S. Murray, F. Fernandez-Alonso,B. F. Abrahams, C. Boskovic, Inorg. Chem. 2006, 45, 8950.doi:10.1021/IC060938E(b) K. G. Alley, A. Mukherjee, R. Clérac, C. Boskovic, Dalton Trans.2008, 59. doi:10.1039/B710755B
[6] S. Groysman, E. Sergeeva, I. Goldberg, M. Kol, Inorg. Chem. 2005,44, 8188. doi:10.1021/IC051400W
[7] A. R. F. Cox, V. C. Gibson, E. L. Marshall, A. J. P. White, D. Yeldon,Dalton Trans. 2006, 5014. doi:10.1039/B607753F
[8] S. Koizumi, M. Nihei, H. Oshio, Chem. Lett. 2004, 33, 896.doi:10.1246/CL.2004.896
[9] S. Koizumi, M. Nihei, T. Shiga, M. Nakano, H. Nojiri, R. Bircher,O. Waldmann, S. T. Ochsenbein, H. U. Güdel, F. Fernandez-Alonso,H. Oshio, Chem. Eur. J. 2007, 13, 8445. doi:10.1002/CHEM.200700714
[10] N. E. Brese, M. O’Keefe, Acta Crystallogr. 1991, B47, 192.[11] A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn, G. C. Verschoor, J.
Chem. Soc., Dalton Trans. 1984, 1349. doi:10.1039/DT9840001349[12] C. Hormillosa, S. Healy, T. Stephen, I. D. Brown, Bond Valence Cal-
culator Version 2.00, 1993 (distributed by I. D. Brown, Institute forMaterials Research, McMaster University, Ontario, Canada).
[13] A. Ferguson, A. Parkin, M. Murrie, Dalton Trans. 2006, 3627.doi:10.1039/B603622H
[14] L. Banci, A. Dei, Inorg. Chim. Acta 1980, 39, 35. doi:10.1016/S0020-1693(00)93630-2
[15] C. Benelli, D. Gatteschi, G. Speroni, Inorg. Chim. Acta 1984, 90, 179.doi:10.1016/S0020-1693(00)80743-4
[16] M. J. Hossain, H. Sakiyama, Inorg. Chim. Acta 2002, 338, 255.doi:10.1016/S0020-1693(02)01053-8
[17] L. F. Jones, C. A. Kilner, M. A. Halcrow, N. J. Chem. 2007, 31, 1530.doi:10.1039/B700987A
[18] J. S. Wood, Inorg. Chem. 1968, 7, 852. doi:10.1021/IC50063A002

RESEARCH FRONT
Mixed-Valent Polynuclear Cobalt Complexes 1129
[19] (a) J. Reim, K. Griesar, W. Haase, B. Krebs, J. Chem. Soc., DaltonTrans. 1995, 2649. doi:10.1039/DT9950002649(b) L. K. Thompson, S. K. Mandal, S. S. Tendon, J. N. Bridson,M. K. Park, Inorg. Chem. 1996, 35, 3117. doi:10.1021/IC9514197
[20] (a) M.A. Halcrow, J.-S. Sun, J. C. Huffman, G. Christou, Inorg. Chem.1995, 34, 4167. doi:10.1021/IC00120A022(b) S. Mukherjee, T. Weyhermüller, E. Bothe, K. Wieghardt,P. Chaudhuri, Eur. J. Inorg. Chem. 2003, 863. doi:10.1002/EJIC.200390116
[21] (a) J. B. Goodenough, Phys. Rev. 1955, 100, 564. doi:10.1103/PHYSREV.100.564(b) J. B. Goodenough, J. Phys. Chem. Solids 1958, 6, 287.doi:10.1016/0022-3697(58)90107-0(c) J. Kanamori, J. Phys. Chem. Solids 1959, 10, 87. doi:10.1016/0022-3697(59)90061-7
[22] G. M. Sheldrick, SHELXS97 and SHELXTL-97 1997 (University ofGöttingen: Germany).




















