Characteristics of protein residue-residue contacts and their ...
Upload
thermofisherCategory
view
0download
0
2592-2600 Nucleic Acids Research, 1994, Vol. 22, No. 13 © 1994 Oxford University Press
Methylphosphonate mapping of phosphate contacts criticalfor RNA recognition by the human immunodeficiency virustat and rev proteins
Clare E.Pritchard + , Jane A.Grasby, Frangois HamyS, Anthony M.Zacharek11, Mohinder Singh,Jonathan Karn and Michael J.Gait*MRC Laboratory of Molecular Biology, Hills Road, Cambridge CB2 2QH, UK
Received March 22, 1994; Revised and Accepted May 25, 1994
ABSTRACT
The HIV-1 regulatory proteins tat and rev are both RNAbinding proteins which recognize sequences in duplexRNA which are close to structural distortions. Here weidentify phosphate contacts which are critical for eachbinding reaction by use of a new method. Model RNAbinding sites are constructed carrying substitutions ofindividual phosphodiesters by uncharged methylphos-phonate derivatives isolated separately as ftp and Spdiastereoisomers and tested for protein binding bycompetition assays. In the binding of tat to the trans-activation response region (TAR), three phosphates,P21 and P22 which are adjacent to the U-rich bulge andP40 on the opposite strand, are essential and in eachcase both isomers inhibit binding. Similarly, in theinteraction between the HIV-1 rev protein and the rev-responsive element (RRE) both methylphosphonateisomers at P103, P104, P124 and P125 interfere withrev binding. At P106, only the ftp methylphosphonateisomer is impaired in rev binding ability and it isproposed that the ftp oxygen is hydrogen-bonded toan uncharged amino acid or to a main chain hydrogenatom. Synthetic chemistry techniques also provideevidence for the conformations of non-Watson - CrickG106:G129 and G105:A131 base-pairs in the RRE 'bubble'structure upon rev binding. Almost all functionalgroups on the 5 bulged residues in the bubble havebeen ruled out as sites of contact with rev but, bycontrast, the AT-positions of each G residue in theflanking base-pairs are identified as sites of likelyhydrogen-bonding to rev. The results show that bothtat and rev recognize the major groove of distorted RNAhelixes and that both proteins make specific contactswith phosphates which are displaced from the sites ofbase-pair contact.
INTRODUCTION
Gene expression in human immunodeficiency virus (HIV)requires two viral proteins: the frans-activator, tat, and theregulator of virion expression, rev (1, 2). Tat stimulatestranscriptional elongation from the viral long terminal repeat(LTR), whereas rev is required for the efficient cytoplasmicexpression of mRNAs encoding the viral enzymes and structuralproteins.
Tat interacts with the fra/w-activation responsive region (TAR),a 59-residue stem-loop structure that is located at the 5'-end ofall mRNAs (Figure 1) (3 —5). Recombinant tat protein binds TARRNA stoichiometrically and with high efficiency (Kd 1 —3 nM)(6, 7). The binding site for tat surrounds a distortion in the TARRNA duplex created by a trinucleotide bulge (7—12). Residuesessential for tat recognition include the first residue in the bulge,U23, and the two base-pairs immediately above the bulge,G26:C39 and A27:U38 (7, 8, 11, 12). The two base-pairs belowthe bulge also make a contribution to tat binding affinity (11,12). The other residues in the bulge, U24 and U25, appear to actpredominantly as spacers and may be replaced by othernucleotides or even by non-nucleotide linkers (10).
Rev is also an RNA binding protein which binds directly toa second genetic element encoded by HIV, the rev-responseelement (RRE) (13-17). The complete biologically active RREis 351 nucleotides long and forms a complicated secondarystructure (18). The binding reaction is complex and involves aninitial interaction with a high affinity site followed by the co-operative addition of rev monomers to lower affinity sites (14,19-21). Initial high affinity interaction of rev with the RRE RNAoccurs at a unique 'bubble' structure (Figure 1) (22—25) whichis stabilized by cross-strand non-Watson-Crick G:G and G:Apairs (19, 24, 25). In preliminary work, it was shown that theApposition of G1M flanking the bubble is essential to revbinding, suggesting that, like tat, rev also recognizes the majorgroove of an RNA duplex at a site of a local distortion (25) [NB.
*To whom correspondence should be addressed
Present addresses: +Department of Biochemistry, University of Oxford, Oxford OX1 3QU, UK, §Ciba-Geigy AG, CH-4002 Basel, Switzerland and'Department of Biochemical Sciences, Harvard University, Cambridge, MA, USA
by guest on August 18, 2016
http://nar.oxfordjournals.org/D
ownloaded from

Nucleic Acids Research, 1994, Vol. 22, No. 13 2593
(a)
GGU GC A
CGGC
24 y
AUCG
A C G
GCAUU AU AGCGUU ACGUGCG
c U A
UAGCGCGC
5' 3'
(b)
3' 51
CGG C Stem llaU AGC
106 Stem Mbra A
CAGCGUC^ ^ a a * ^ ^ ^ ^ ^ a ^ ^ r *
u
:<B-CAGCGUC. . ^ M ™wCAGUCGCAGu
U G G 131 y 129130
AC 13°GCUACGU A Stem lieGCUAUA
UU
Figure 1. (a) Secondary structure of the rrans-activation responsive region (TAR)RNA. Highlighted residues define the region involved with tat recognition, (b)Partial secondary structure for the stem-loop II region of the rev-response element(RRE) RNA. Highlighted residues define the region involved with initial highaffinity rev recognition.
Numbers for nucleotide residues are +70 compared to ourprevious publication (25)]. At higher rev concentrations,additional rev molecules become associated with the RRE,packing around selected regions of primarily double strandedRNA. The packing of rev around the RRE correlates well withrev activity in vivo (18).
In addition to base recognition, specific contacts throughphosphate residues are often essential elements in protein-nucleicacid interactions. For example in the high resolution structuralmodel of the interaction of the Zif268 complex with DNA,interactions are seen between phosphate and serine residues aswell as between phosphate and arginine or histidine residues (26).In the interaction of helix 3 of Hin recombinase with the majorgroove of a DNA duplex, there are simultaneous contacts ofphosphate 8 with a tyrosine and an arginine and also of phosphate9 with a threonine and the main chain amide of a glycine (27).Whereas contacts to uncharged amino acids are hydrogen bonds,contacts to the side-chains of basic residues may have bothhydrogen-bonding as well as some electrostatic character, dueto the presence of the opposing charges.
The most commonly used method for mapping phosphatecontacts in nucleic acid-protein interactions is ethylationinterference, and this technique has been used to define positionswhich are important for both the tat-TAR and the rev-RREinteractions. Ethylation of either of two adjacent phosphates inTAR, between G2] and A22 (P21) or between A22 and U23
(P22), interfered with the binding of a short basic peptide (28)as well as of the amino acid arginine (29). Ethylation interferencestudies of TAR using the complete tat protein revealed additional
sites of interference at five phosphates on the opposite strand(P36-P40) (12). Ethylation interference analysis using rev anda rev-derived peptide has identified three phosphates on one sideof the bubble and three phosphates on the other strand distantfrom the bubble as important to the interaction (22).
Although ethylation interference analysis has proved valuablefor suggesting possible phosphate contacts in both tat—TAR andrev-RRE interactions, the method has some limitations. First,the introduction of bulky ethyl groups by treatment of RNA withethylnitrosourea (ENU) is likely to cause substantial sterichindrance to interactions with a protein. Furthermore, ethylationresults in the non-stereo-specific introduction of an 0-ethyl groupto give a mixture of both Rp and Sp ethyl phosphotriesters.Because both diastereoisomers are present, the ENU interferencedata does not allow distinctions to be made between electrostaticcontacts, hydrogen-bonding contacts, or combinations of thesetwo types of contact. Thus, ENU footprints should not beregarded as definitive evidence for sites of phosphate-proteincontact.
Recently we have begun to explore an alternative approachto the study of phosphate residues in RNA important for proteinbinding. Synthetic chemistry is used to replace individualphosphodiesters in the RNA by methylphosphonates (30). In thesederivatives, a methyl group replaces one of the phosphate oxygensto give an uncharged isosteric analogue of phosphate. The smallmethyl group is much less likely to interfere with protein bindingthan a bulky O-ethyl group. Here we describe the separation ofoligonucleotides carrying the individual Rp and Spmethylphosphonate isomers and assembly into RNA modelsrepresenting TAR and RRE. The results show the effects ofsubstitution of individual oxygen atoms of phosphate residues andgive important insights into the location and nature of phosphatecontacts in both the rev —RRE and the tat—TAR interactions.
MATERIALS AND METHODS
Chemical synthesis of oligoribonucleotides
Oligoribonucleotides were synthesized by the phosphoramiditemethod on a 1 mmol scale using an ABI 380B DNA/RNASynthesizer (25, 30, 31). Ribonucleoside phosphoramidites,2'-deoxyncleoside methylphosphonoamidites (dT, dC, dG anddA), spacer phosphoramidite C3, A^-deaza-dA and A^-deaza-dGphosphoramidites were obtained from Glen Research (viaCambio). A sample of dU methylphosphonoamidite was a giftfrom Dr Nanda Sinha (Millipore Corporation). A*-Benzoyl-2'-deoxyadenosine and Apt-benzoyl-2'-deoxycytidine phosphor-amidites were obtained from Cruachem (UK) and A^-phenoxy-acety 1-2'-deoxyguanosine was obtained from Pharmacia. Otherreagents were from Applied Biosystems Ltd or Cruachem.C^-Methylguanosine phosphoramidite was obtained as prev-iously described (31). 2'-Deoxy-2'-fluoroadenosine phosphor-amidite was a gift from F.Eckstein (32). Oligonucleotides werecleaved from the support and base-deprotected with saturatedmethanolic ammonia at room temperature for 24 h (or 48 h inthe case of benzoyl removal from A^-deazaA). C^-MethylG-containing oligonucleotides were deprotected with 10% DBU inmethanol as previously described (31). 2'-O-t-Butyldimethylsilylgroups were removed by treatment with 1 M tetrabutylammoniumfluoride (Aldrich) in tetrahydrofuran (1 ml) at room temperaturefor 24-48 h. After desalting, oligonucleotides were purified bystrong anion exchange HPLC on a 1 X 25 cm Partisil-10 SAXcolumn (Whatman) as previously described (25, 30, 31) and
by guest on August 18, 2016
http://nar.oxfordjournals.org/D
ownloaded from

2594 Nucleic Acids Research, 1994, Vol. 22, No. 13
checked for purity by reversed-phase HPLC on a /t-BondapakC18 column (0.4x25 cm) (Millipore). Typical yields for 15-mersafter purification were 10/l260 units (ca 5% overall yield). Foroligonucleotides containing single 2'-deoxy-3'-methylphosphonatesubstitutions, individual diastereoisomers were resolved using ashallow gradient of acetonitrile (generally 7.5 — 17.5% in 25 min,Figure 3). The product in each peak was collected and desalted.
Analysis of oligonucleotidesNucleoside analysis was carried out by digestion of theoligonucleotides with snake venom phosphodiesterase(Boehringer) and alkaline phosphatase (Boehringer) followed byHPLC analysis on a /i-Bondapak C18 reversed-phase column(0.4x25 cm) as previously described (25). Retention times(0—15% acetonitrile gradient in 25 min) for unmodifiednucleosides were: C, 6.2; U, 8.4; G, 16.0; A, 19.4; dG, 18.2;dA, 22.5 min. Retention times for nucleosides containingmodified nucleosides were: C^-methylG, 22.9; 2'-fluoro-2'-dA,24.8; A^-deaza-2'-dA, 22.7; A^-deaza-2'-dG, 19.5 min.Oligonucleotides containing single 2'-deoxy-3'-methylphos-phonates were digested under the same conditions and theresultant methylphosphonate dimers separated by reversed-phaseHPLC using a 0—20% acetonitrile gradient. For example,digestion of the fast eluting isomer (25.1 min) of CGUGUGGdG-mprCGCAGC (methylphosphonate at P106, i-e. between G106and C107, Figure 2) gave methylphosphonate dimer (dGmprC)isomer 1 eluting at 22.8 minutes, whereas the slow eluting isomer(25.4 minutes) of the same oligonucleotide gave methylphos-phonate dimer isomer 2 eluting at 23.8 min. All other methyl-phosphonate-containing oligonucleotides were checked in thisway. In each case the slow eluting oligomer always producedthe slow eluting dimer after nuclease digestion.
The positions of 2'-deoxynucleoside substitutions were checkedby 32P end-labelling of the oligonucleotides, by treatment with[7-32P]ATP and T4 polynucleotide kinase, followed by partialalkaline hydrolysis and polyacrylamide gel electrophoresis aspreviously described (30). The absence of a radioactive bandsignified the presence of a 2'-deoxynucleoside at that position,since such residues are resistant to alkaline hydrolysis. Bycontrast, 2'-deoxy-3'-methylphosphonate-containing oligonucle-otides were cleaved at the methylphosphonate linkage underpartial alkaline hydrolysis (data not shown) and produced aradioactive band which migrated slower than the equivalent bandobtained for hydrolysis of the corresponding inter-ribonucleotidelinkage. This band was therefore diagnostic for the presence ofthe methylphosphonate. No migration differences were observedbetween individual Rp and Sp isomers.
Tat binding assaysTat protein was prepared and binding assays were carried outas previously described (12). 32P-Labelled unmodified duplexeswere formed by annealing a 1.5-fold molar excess of 17-merstrand with 14-mer strand labelled with [Y- 3 2 P]ATP and T4polynucleotide kinase. Competition binding assays (25 1̂)included 0.1% Triton X100, 50 mM Tris-HCl (pH 7.4), 20mM KC1, 0.1 M DTT, 7.5 nM 32P-labelled unmodified duplexRNA (20 000 c.p.m.), 20-30 nM tat protein and variousconcentrations of unlabelled competitor duplex RNA. Bindingreactions were incubated on ice for 30 min and non-denaturingpolyacrylamide gel electrophoresis carried out at 4°C (8% gel,20x20 cm) in 0.5 X TB (TB = 44.5 mM Tris base, 44.5 mMboric acid), 0.1 % Triton for 35 min at 12 W. Gels were dried
and complexes detected by autoradiography. Relative bindingconstants were calculated after scanning of the resultantautoradiographs by densitometry.
Rev binding assaysRev protein was prepared as described (19, 34). Competitionbinding reactions (20 /tl) shown in Figure 5 contained 0.5%glycerol, 0.05% Triton X100, 40 mM Tris-HCl (pH 7.9), 20mM KC1, 3.5 nM 32P-labelled unmodified duplex RNA, 28 nMrev protein and various concentrations of unlabelled competitorduplex RNA. Other competition binding reactions (20 ftl) (Table3) were carried out similarly except that 25 nM 32P-labelledduplex RNA and 70 nM rev protein concentrations were used.Non-denaturing gel electrophoresis was carried out with 4%(Figure 5, Table 2) or 7.5% (Table 3) polyacrylamide gels andrunning buffer 0.5 x TBE (TBE = 44.5 mM Tris base, 44.5mM boric acid, 1 mM EDTA), 0.1% Triton, 1% glycerol.
RESULTS
Synthetic RNA models for TAR RNA and RRE RNAcontaining methylphosphonate diastereomer substitutionsBoth the binding site for tat on TAR and the high affinity sitefor rev on the RRE can be mimicked by short synthetic RNAduplexes (Figure 2) (10, 25, 30). Models of this type lendthemselves to studies where modified bases are incorporated intospecific sites. This approach has been used to identify the//'-positions of both G26 and A27 and the AP-H and/orOppositions of U23 as crucial to tat binding (10, 30). Similarly,functional group substitution experiments at the high affinity revbinding site have suggested specific base-pairing schemes for thenon-Watson—Crick base-pairs in the 'bubble' and indicated thatrev interacts with the N1 of Gl04 (25).
Single 2'-deoxy-3'-methylphosphonate linkages wereincorporated into these model RNA duplexes by chemicalsynthesis. Because methylphosphonate linkages 3' to ribose unitsare highly unstable, a ribonucleoside phosphoramidite wasreplaced by the corresponding 2'-deoxy-3'-methylphosphono-amidite at die appropriate coupling step. Oligonucleotides havingthe same sequence but with the corresponding 2'-deoxynucleotideincorporated at the equivalent site were synthesized as controlsfor the possible effects of the sugar substitution on proteinbinding.
Oligonucleotides containing single methylphosphonate isomerswere resolved by careful high performance liquid chromatography(HPLC) using a reversed-phase column and shallow gradientsof acetonitrile eluents. A typical example of diastereoisomerseparation by HPLC is shown in Figure 3.
It has been shown recently that for all 16 pairs ofdi-2'-deoxynucleoside 3'—5'-methylphosphonates isomers the Rpisomer elutes faster and the Sp isomer slower on reversed-phaseHPLC (35). Therefore in order to distinguish the stereochemistryat the methylphosphonate for the synthesized oligonucleotides,we digested each pair of the separated oligonucleotides with snakevenom phosphodiesterase followed by alkaline phosphatase. Thisyields the four nucleosides and a dinucleoside 3'—5'-methylphos-phonate. These products were analysed by reversed-phase HPLC(data not shown). As expected, in each case the dinucleoside3' —5'-methylphosphonate released from the fast eluting oligo-nucleotide product was eluted faster than the correspondingdinucleoside 3' —5'-methylphosphonate obtained from the slow
by guest on August 18, 2016
http://nar.oxfordjournals.org/D
ownloaded from
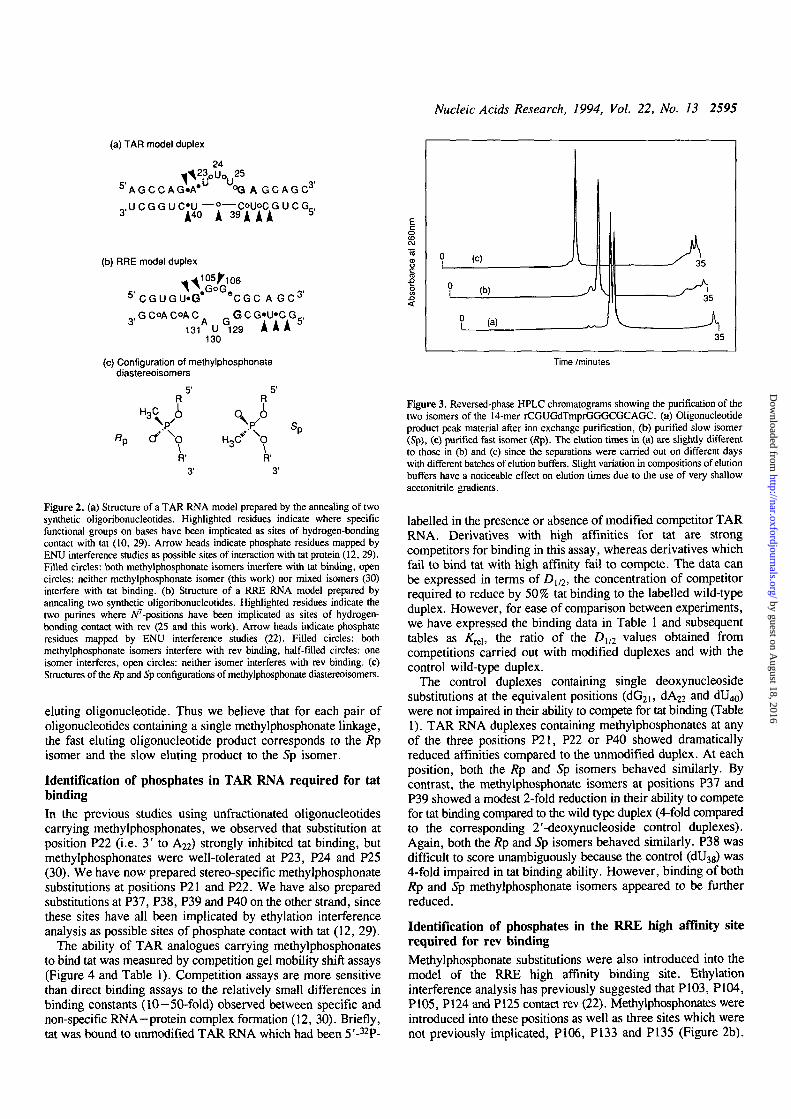
Nucleic Acids Research, 1994, Vol. 22, No. 13 2595
(a) TAR model duplex
24
5'AGCCAG»A* °GAGCAGC3'
(b) RRE model duplex
5'CGUGU«G* e C G C A G C 3 '
., GCoACoAC. -GCG'U'CG,,3 A , , fj 1 i k 0
131 U 129 A * *130
(c) Configuration of methylphosphonatediastereoisomers
51 5'
1 1H3>.< V sp
" p Cf O H3C* OR' R'
Figure 2. (a) Structure of a TAR RNA model prepared by the annealing of twosynthetic oligoribonucleotides. Highlighted residues indicate where specificfunctional groups on bases have been implicated as sites of hydrogen-bondingcontact with tat (10, 29). Arrow heads indicate phosphate residues mapped byENU interference studies as possible sites of interaction with tat protein (12, 29).Filled circles: both methylphosphonate isomers interfere with tat binding, opencircles: neither methylphosphonate isomer (this work) nor mixed isomers (30)interfere with tat binding, (b) Structure of a RRE RNA model prepared byannealing two synthetic oligoribonucleotides. Highlighted residues indicate thetwo purines where Appositions have been implicated as sites of hydrogen-bonding contact with rev (25 and this work). Arrow heads indicate phosphateresidues mapped by ENU interference studies (22). Filled circles: bothmethylphosphonate isomers interfere with rev binding, half-filled circles: oneisomer interferes, open circles: neither isomer interferes with rev binding, (c)Structures of the Rp and Sp configurations of methylphosphonate diastereoisomers.
eluting oligonucleotide. Thus we believe that for each pair ofoligonucleotides containing a single methylphosphonate linkage,the fast eluting oligonucleotide product corresponds to the Rpisomer and the slow eluting product to the Sp isomer.
Identification of phosphates in TAR RNA required for tatbindingIn the previous studies using unfractionated oligonucleotidescarrying methylphosphonates, we observed that substitution atposition P22 (i.e. 3' to A22) strongly inhibited tat binding, butmethylphosphonates were well-tolerated at P23, P24 and P25(30). We have now prepared stereo-specific methylphosphonatesubstitutions at positions P21 and P22. We have also preparedsubstitutions at P37, P38, P39 and P40 on the other strand, sincethese sites have all been implicated by ethylation interferenceanalysis as possible sites of phosphate contact with tat (12, 29).
The ability of TAR analogues carrying methylphosphonatesto bind tat was measured by competition gel mobility shift assays(Figure 4 and Table 1). Competition assays are more sensitivethan direct binding assays to the relatively small differences inbinding constants (10-50-fold) observed between specific andnon-specific RNA—protein complex formation (12, 30). Briefly,tat was bound to unmodified TAR RNA which had been 5'-32P-
Time /minutes
Figure 3. Reversed-phase HPLC chromatograms showing the purification of thetwo isomers of the 14-mer rCGUGdTmprGGGCGCAGC. (a) Oligonucleotideproduct peak material after ion exchange purification, (b) purified slow isomer(Sp), (c) purified fast isomer (Rp). The elution times in (a) are slightly differentto those in (b) and (c) since the separations were carried out on different dayswith different batches of elution buffers. Slight variation in compositions of elutionbuffers have a noticeable effect on elution times due to the use of very shallowacetonitrile gradients.
labelled in the presence or absence of modified competitor TARRNA. Derivatives with high affinities for tat are strongcompetitors for binding in this assay, whereas derivatives whichfail to bind tat with high affinity fail to compete. The data canbe expressed in terms of Z)1/2, the concentration of competitorrequired to reduce by 50% tat binding to the labelled wild-typeduplex. However, for ease of comparison between experiments,we have expressed the binding data in Table 1 and subsequenttables as KTeb the ratio of the Dl/2 values obtained fromcompetitions carried out with modified duplexes and with thecontrol wild-type duplex.
The control duplexes containing single deoxynucleosidesubstitutions at the equivalent positions (dG2i, dA22 and dU^)were not impaired in their ability to compete for tat binding (Table1). TAR RNA duplexes containing methylphosphonates at anyof the three positions P21, P22 or P40 showed dramaticallyreduced affinities compared to the unmodified duplex. At eachposition, both the Rp and Sp isomers behaved similarly. Bycontrast, the methylphosphonate isomers at positions P37 andP39 showed a modest 2-fold reduction in their ability to competefor tat binding compared to the wild type duplex (4-fold comparedto the corresponding 2'-deoxynucleoside control duplexes).Again, both the Rp and Sp isomers behaved similarly. P38 wasdifficult to score unambiguously because the control (dU38) was4-fold impaired in tat binding ability. However, binding of bothRp and Sp methylphosphonate isomers appeared to be furtherreduced.
Identification of phosphates in the RRE high affinity siterequired for rev bindingMethylphosphonate substitutions were also introduced into themodel of the RRE high affinity binding site. Ethylationinterference analysis has previously suggested that PI03, PI04,P105, P124 and P125 contact rev (22). Methylphosphonates wereintroduced into these positions as well as three sites which werenot previously implicated, P106, P133 and P135 (Figure 2b).
by guest on August 18, 2016
http://nar.oxfordjournals.org/D
ownloaded from

2596 Nucleic Acids Research, 1994, Vol. 22, No. 13
Competitor
CompetitorRNA conc/nM
dG2imp Sp
Oo 10
0200
400
800
dG2imp Rp
o o 100
200
400 008
wt TAR duplex
o o 100
200
400
1800
Table 1. Competition tat binding for methylphosphonate-modified TAR RNAduplexes
complex - ^
Free RNA -
Competitor
CompetitorRNA conc/nM
Sp
8 8
Rp
© 8 8 8
wt TAR duplex
8 8
Complex- —>*••-
Free RNA -
Figure 4. Competition band-shift assay for a t binding. Each lane contains 7.5nM 32P-labelled wild-type TAR duplex RNA and 20-30 nM tat protein.Concentrations of unlabelled competitor RNA are as shown: top panel, dG21mpSp, dG21mp Rp and wild-type RNA duplex; bottom panel, dC37mp Sp, dC37mpRp and wild-type RNA duplex.
In each case the Rp and Sp diastereoisomers were readily resolvedby use of reversed-phase HPLC.
The effect of the methylphosphonates on rev binding wasmeasured in competition gel-mobility shift assays similar to thosecarried out to study the tat-TAR interaction (Figure 5 and Table2). At three of the positions mapped by ethylation interference,PI03 and PI04 on the top strand and PI25 on the bottom strand,both methylphosphonate isomers strongly inhibited rev bindingmore than 8-fold, whilst the corresponding 2'-deoxynucleoside-containing control oligonucleotides were hardly affected. Atpositions PI03 and PI25 the methylphosphonate substitutionswere carried out using commercially available dT-methylphos-phonoamidite derivative. However, in both cases the control dT-containing duplexes were unaffected in rev binding ability. Bycontrast, at position PI24 there was a significant effect of2'-deoxy substitution (dC^) and, although the methylphos-phonate appeared to additionally reduce binding, it is difficultto score this position unambiguously.
At PI05, which had previously been shown to inhibit revbinding in ethylation interference experiments, neithermethylphosphonate isomer nor the control dGios was impairedin their ability to compete for rev binding. Methylphosphonatesubstitution at either position P133 or P135, which are eachlocated well away from the bubble, also had no effect oncompetitive rev binding ability. The corresponding 2'-deoxy-nucleotide-containing controls (dA133 and dA135) were able tocompete normally for rev binding.
An example of stereo-specific inhibition by methylphosphonatesis provided by Pi06, which is located within the 'bubble'. The
Wildtype
Duplex RNA
unmodified
dG2l
dA22
dC37
dU38
dC39
dU40
Krela
1.0
1.5
6.4
4.3
0.6
2.2
Methylphosphonate
Duplex RNA
dG2imp(/?p)
dG2imp(Sp)
dA22mp (Rp)
dA22mp(Sp)
dC37mp (Rp)
dC37mp (Sp)
dU38mp (Rp)
dU3gmp (Sp)
dC39mp (Rp)
dC39mp (Sp)
dU40mp (Rp)
dU4Qmp (Sp)
Krel
5.6
6.4
>g
>8
1.6
1.8
>8
>8
'1.1
2.X
>8
>8
*Krel is the concentration (nM) of mutant competitor duplex required to reduceby 50% tat binding to 32P-labelled wild-type duplex divided by the concentration(nM) of wild-type unmodified duplex required to reduce by 50% tat binding to32P-labelled wild-type duplex.
Rp methylphosphonate isomer competed poorly for rev bindingwhereas the Sp isomer competed as well as the control RNAduplex and the d G ^ control.
Uuo acts as a spacerWe have previously reported that U130 in the bubble may bereplaced by dU, 5-bromodU or by AP-methylU (25). Wetherefore suggested that this nucleoside is not likely to be involvedin cross-strand base-pairing. However, use of these analoguesdid not address the issue of potential recognition of U130 by rev(e.g. at CP- or at O*). We therefore replaced U130 by a C3-spacerand observed the effect on rev binding. The C3-spacer is a propyllinker that maintains the correct number and type of atomsbetween two neighbouring phosphate residues in oneoligonucleotide strand, yet the sugar and base is entirely absent.C-3 spacers have previously been used in assessing the role ofU residues in TAR (11). The results showed that there was noeffect of the C-3 spacer on rev binding (Table 3). Thus U130
appears to play no part either in cross-strand hydrogen-bondingor in rev recognition.
Configuration of non-Watson-Crick base-pairs in the RREbubblePrevious studies have suggested that the bubble in the RRE isstabilized by non-Watson-Crick base-pairs between Gl06 andG[29 and between G105 and A131 (19, 24, 25). We have nowcompleted chemical substitution experiments (Table 3) which,taken together with our previous studies (25), provide strongevidence for the conformations of the proposed base-pairs shownin Figure 6. The data also suggest that the non-Watson —Crickbase-pairs are unlikely to form specific contacts to rev, but insteadthey are essential to form a unique structural element for revbinding.
by guest on August 18, 2016
http://nar.oxfordjournals.org/D
ownloaded from
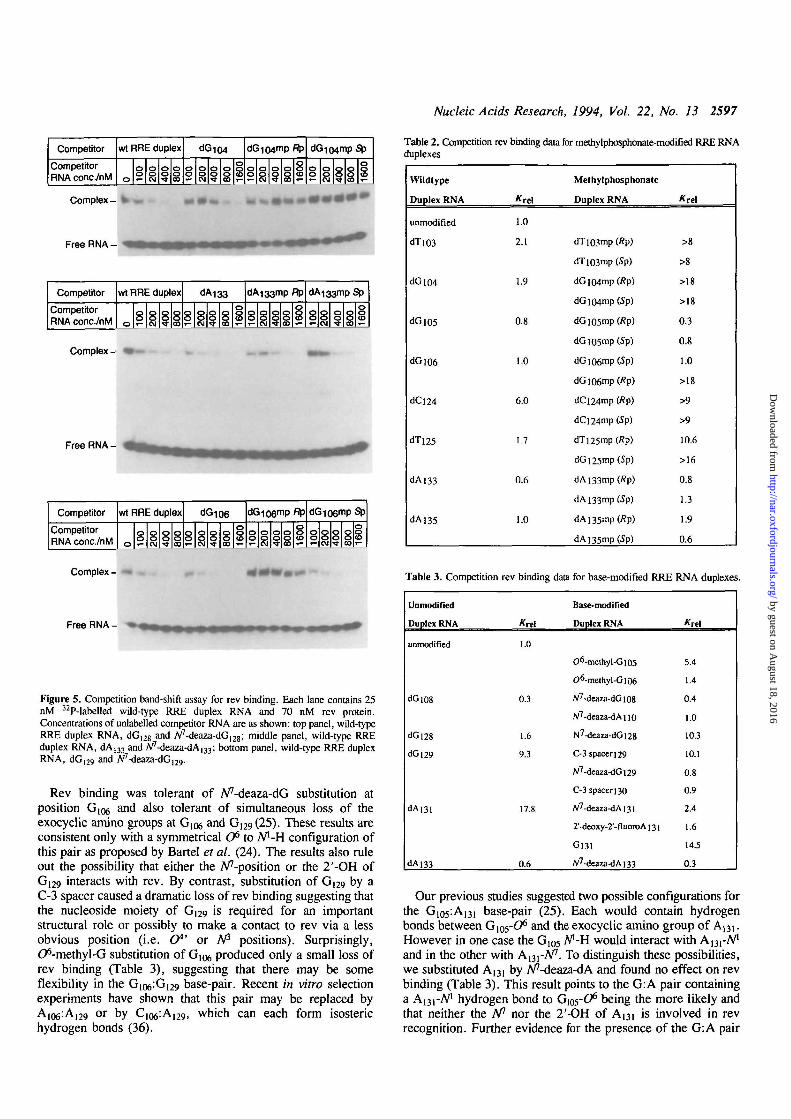
Nucleic Acids Research, 1994, Vol. 22, No. 13 2597
Competitor
CompetitorRNA conc/nM
wt RRE duplex (JG104
8
"P
8 \°8
Complex- » —
Free RNA - , ,
Competitor wt RRE duplex
CompetitorRNA conc/nM S 8
Ftp
8 8 8
Complex -
Free RNA -
Competitor
CompetitorRNA conc./nM
wt RRE duplex
8 888 8
Complex - »
Free RNA-
Figure 5. Competition band-shift assay for rev binding. Each lane contains 25nM 32P-labelled wild-type RRE duplex RNA and 70 nM rev protein.Concentrations of unlabelled competitor RNA are as shown: top panel, wild-typeRRE duplex RNA, dG12g and A^-deaza-dG^g; middle panel, wild-type RREduplex RNA, dA133 and A^-deaza-dA^; bottom panel, wild-type RRE duplexRNA, dG|29 and N'
Rev binding was tolerant of A/'-deaza-dG substitution atposition G106 and also tolerant of simultaneous loss of theexocyclic amino groups at G106 and G129(25). These results areconsistent only with a symmetrical O6 to A/'-H configuration ofthis pair as proposed by Bartel et al. (24). The results also ruleout the possibility that either the Apposition or the 2'-OH ofG129 interacts with rev. By contrast, substitution of G(29 by aC-3 spacer caused a dramatic loss of rev binding suggesting thatthe nucleoside moiety of G129 is required for an importantstructural role or possibly to make a contact to rev via a lessobvious position (i.e. (7*' or A/3 positions). Surprisingly,C^-methyl-G substitution of G106 produced only a small loss ofrev binding (Table 3), suggesting that there may be someflexibility in the Gio^G^g base-pair. Recent in vitro selectionexperiments have shown that this pair may be replaced byA106:Ai29 or by C I Q ^ A ^ , which can each form isosterichydrogen bonds (36).
Table 2. Competition rev binding data for methylphosphonate-modified RRE RNAduplexes
Wildtype
Duplex RNA
unmodified
<H"1O3
dGlO4
dGios
dGiO6
dCl24
dTi25
dAi33
dAl35
*rel
1.0
2.1
13
0.8
1.0
6.0
1.7
0.6
1,0
Methylphosphonate
Duplex RNA
dT]03mp (fip)
dTi03mp (Sp)
dGlO4mp(/?p)
dGlO4mp(5p)
dG i05mp (Rp)
dGio5mp(5p)
dGiO6mp(Sp)
dGl06mp(Rp)
dCi24mp (Rp)
dCl24mp(Sp)
dTi25mp (Rp)
dGi25mp(Sp)
dAi33mp(Rp)
dAi33mp(5p)
dA 135mp (Rp)
dA]35mp(Sp)
*rel
>8
>8
>18
>18
0.3
0.8
1.0
>18
>9
>9
10.6
>16
0.8
1.3
1.9
0.6
Table 3. Competition
Unmodified
Duplex RNA
unmodified
dGl08
dGl28
dGl29
dAm
dA]33
rev binding data
Krel
1.0
0.3
1.6
9.3
17.8
0.6
for base-modified RRE
Base-modified
Duplex RNA
O6-methyI-Gl05
O6-me[hyl-Gl06
A/7-deaza-dGi08
W7-deaza-dAno
N7-deaza-dGl28
C-3 spaceri29
W7-deaza-dGi29
C-3 spacer 130
A/7-deaza-dAi3i
2'-deoxy-2'-fluoroA 131
G131
/V7-deaza-dAi33
RNA duplexes
Krel
5.4
1.4
0.4
1.0
10.3
10.1
0.8
0.9
2.4
1.6
14.5
0.3
Our previous studies suggested two possible configurations forthe G105:Ai31 base-pair (25). Each would contain hydrogenbonds between Gios-O6 and the exocyclic amino group of A131.However in one case the G105 A"-H would interact with A^-A/1
and in the other with A^-N1. To distinguish these possibilities,we substituted A131 by A^-deaza-dA and found no effect on revbinding (Table 3). This result points to the G: A pair containinga A^i-A71 hydrogen bond to G105-O6 being the more likely andthat neither the A" nor the 2'-OH of A13i is involved in revrecognition. Further evidence for the presence of the G: A pair
by guest on August 18, 2016
http://nar.oxfordjournals.org/D
ownloaded from

2598 Nucleic Acids Research, 1994, Vol. 22, No. 13
W7-deaza-dG
A/6-Me-dA W?-deaza-dA
dl \ \*=\
A/7-deaza-dG \ ...-•
G105M31
dl
Figure 6. Proposed non-Watson—Crick base-pairing schemes for G ^ G ^ anc*G[05:A|31 showing functional group substitutions that are either unaffected in revbinding (bold) or significantly reduced in rev binding (outlined).
comes from the finding that substitution of Gi05 by d^-methyl-G resulted in a moderate loss of rev binding ability (Table 3).Severe loss of binding ability resulted when A,31 was replacedby G (Table 3) or by dl (25). The same structure for the G:Apair was also suggested from recent chemical probing studieson an in vitro selected RRE model RNA (37).
It is interesting to note that substitution by the corresponding2'-deoxynucleoside at either G129 and A131 inhibited rev binding,whereas substitution by the corresponding A^-deaza-2'-deoxy-nucleoside had no effect on rev binding. One interpretation ofthese results is that rev binding is greatly influenced by theconformations of the sugars at these two positions and that theseconformations are affected in turn by the identities of thecorresponding bases. Thus perhaps the 2'-deoxynucleosidesugars, which prefer a C2'-endo configuration, force alternativebase-pairing schemes less favourable for rev binding, whereasfor the corresponding iV'-deaza^'-deoxynucleosides, alternativebase-pairing is disallowed and the sugars are forced into theC3'-endo configuration. To address this question, we substitutedA131 by 2'-deoxy-2'-fluoroA and found no loss of competitionfor rev binding (Table 3). Since it has been shown that a2'-deoxy-2'-fluoronucleoside adopts predominantly the C3'-endoconfiguration commonly found for ribonucleosides (32), thisresult suggests that a C3'-endo configuration at A m is essentialfor the maintenance of the correct bubble structure to ensure goodrev binding.
Rev recognition in the major groove of the RREIn previous studies it had been found that A^-deaza-dG-substitution of G104, located in a base-pair flanking the bubblein the model duplex, resulted in a severe loss of rev binding (25)suggesting that rev may interact in the major groove of the RNAduplex. 2'-Deoxy-A/7-deaza-substitutions were examined at G108,Ano> G12g and Ai33 (Figure 2b). Competition band shift analysis
(Table 3) showed that 2'-deoxy-Aff-deaza substitution at G,2gimmediately flanking the bubble dramatically reduced rev bindingwhereas the 2'-dG substitution alone had no effect. By contrast,none of the 2'-deoxy-A'7-deaza-substitutions in the three purinesfurther away from the bubble, G(08, An0 and A]33, had anyeffect on rev binding.
The results show that the function of the bubble structureprincipally seems to be to create a local distortion in the RNAduplex allowing accessibility of the major groove at each of theflanking base-pairs. Major groove recognition of the RNA duplexoccurs in both purines flanking the bubble but this does not extendto more distant purines. Instead, rev seems to interact with theA^-positions of each of the guanines in the flanking base-pairs(G1(M and Gi28), data which are consistent with carboxy-ethylation interference data for rev binding (22, 37).
DISCUSSION
Methylphosphonate mapping methodStereo-specific methylphosphonates were first introduced intooligodeoxynucleotides by Noble et al. (38) for the study of theinteraction of a DNA duplex with lac repressor, but the techniquehas not previously been applied to the study of RNA-proteininteractions. The results of our methylphosphonate mapping onthe TAR binding site and the RRE high affinity site suggest thatthe method has several advantages over ethylation interferencemapping. In particular, because there is less risk of steric clashand because stereochemically pure substitution can be obtained,more precise mapping of phosphate contacts as well asinformation on the directionality of the interaction can beobtained.
In our studies of tat and rev binding, we have obtainedexamples of both stereo-specific and non-specific interference bymethylphosphonates. Where both methylphosphonate isomersinterfere with binding, we propose that this phosphate interactswith a charged partner (e.g. Arg or Lys), since in addition tothe interference with hydrogen-bonding, the removal of thephosphate charge would be expected to be disruptive of theelectrostatic component of the interaction independent of thestereochemistry of the modified phosphate. An alternative butperhaps less likely explanation for non-stereo-specific interferenceby methylphosphonates is that each oxygen atom is hydrogen-bonded to a different partner. When only one methylphosphonateisomer interferes, as in the case of P106 in RRE, we suggestthat there is a single hydrogen bond to an uncharged amino acidside chain or to the main chain. The hydrogen bond musttherefore be to the oxygen atom affected by methylphosphonatesubstitution (i.e. pro-/? for Rp substitution). We have also seenexamples of sites which were identified as potential sites ofphosphate contacts to protein by ethylation interference mappingbut where methylphosphonate substitutions are well tolerated.The most likely explanation for these divergent results is thatthe ethylation interfered with binding through a steric clash, ratherthan through removal of a hydrogen-bonding contact.
The tat-TAR complexRecent NMR data has suggested that free TAR RNA containsa continuous A-form helix which extends throughout the bulgeregion (39, 40). However, the molecule is highly flexible andcan be stabilized in an 'open' configuration with a widened majorgroove after binding to basic peptides, or even to the amino acid
by guest on August 18, 2016
http://nar.oxfordjournals.org/D
ownloaded from

Nucleic Acids Research, 1994, Vol. 22, No. 13 2599
derivative argininamide. On the basis of these observations,Puglisi et al. (39, 40) suggested that a base triple is formedbetween U23 and A27:U38, which creates a binding pocket andallows arginine to insert into the major groove by forming twohydrogen bonds with G26 as well as contacts to P2i and P22.Although this work has demonstrated the feasibility of NMRstudies of the tat-TAR interaction, there is still insufficient NMRdata to unambiguously position the argininamide in the complex,to define the width of the major groove, or to directly demonstratethe existence of the base triple.
Preliminary NMR work in this laboratory (G.Varani, personalcommunication) has focussed on studies of complexes betweena model peptide carrying both the 'core' and basic regions oftat and TAR RNA. This 37-residue peptide (ADP-1) more closelyresembles tat in its binding specificity (12) than do the short basicpeptides and argininamide studied by Puglisi et al. (39, 40). Inagreement with Puglisi et al., we have found that TAR isrearranged after peptide binding and that there are NOEs linkingone of the arginines in the peptide to the base moieties of U23and the adjacent A22. However, we have also found that ADP-1makes a number of additional contacts to TAR RNA below theTAR bulge.
Methylphosphonate substitution mapping of the TAR RNAstructure is of particular value since current NMR techniquesgive only limited and indirect information as tophosphate-protein contacts. The model of Puglisi et al. (39, 40)includes direct hydrogen bonds from argininamide to both P21and P22 inferred from ethylation interference studies (28, 29).Here we report that both the Rp and Sp isomers ofmethylphosphonate substituted at either P21 or P22 interfere withtat binding. The absence of stereo-specificity for these sites isconsistent with interaction of each of these phosphate residueswith a charged residue, such as arginine. However, the data doesnot distinguish between a single contact, as postulated by Puglisiet al. for the argininamide complex (39, 40), or a model for thetat—TAR complex in which there are separate contacts to thetwo phosphates made by two different charged amino acid sidechains.
In addition to P21 and P22, phosphates P36 to P40 from theopposite strand of TAR RNA have been implicated by ENUinterference as sites of tat interaction (12). Peptides which bindTAR RNA specifically (such as ADP-1, residues 37-72) showa strong ENU footprint involving phosphates P36—P40, whereaspeptides which bind with lower specificity (such as ADP-3,residues 48 — 72) do not interact strongly with these phosphates(12). In agreement with these observations, the insertion of eitherRp or Sp methylphosphonate isomers at P40 strongly interferedwith tat binding to TAR RNA and it seems likely that P40 istherefore a site of interaction with a charged amino acid side chainof tat. By contrast, the methylphosphonate isomers at P37, P38and P39 have a weaker effect on tat binding (2- to 4-fold),suggesting that none of these phosphates forms strong hydrogen-bonding contacts with tat. It is probable that tat protein closelyapproaches these residues on this side of the RNA duplex, withthe result that ethylation of P36-P40 phosphates producessubstantial steric hindrance. Electrostatic contacts have also beensuggested by Tao and Frankel to be of importance for the P23,P24, and P25 phosphates located in the TAR RNA bulge (41),although it should be noted that methylphosphonate substitutionsare well-tolerated at these positions (30).
The picture of the tat—TAR complex which is emerging fromboth chemical and NMR studies is that there is a conformational
change in TAR RNA following binding by tat. However, theprecise nature of the contacts made between tat and TAR RNA,and the structure of TAR RNA in the bound configuration, remainto be unequivocally determined.
The rev-RRE high affinity complexThe high affinity binding site for rev, like the binding site fortat, is also characterized by a local distortion of an RNA duplex.In the TAR structure, the distortion is produced by the bulgeduridines, whereas in the RRE structure, the distortion is due tothe presence of a single bulged uridine (U130) and to two non-Watson-Crick base-pairs (G106:G129 and G105:A131). NMRstudies suggest that the rev binding site, like the tat binding siteon TAR, is flexible and can be stabilized by binding to smallligands such as peptides (42). The precise structure of the bindingsite has not yet been determined, but model building studies basedon the structures of a series of variant rev binding sites (aptamers)have suggested that the major groove is substantially widenedby the presence of the non-Watson-Crick base-pairs (43).
Our functional group substitution experiments have allowedus to define the likely configuration of the G ^ G ^ andGio5;Ai3i base-pairs. The G106:G129 pair appears to be in asymmetrical O6 to A -̂H configuration, whereas in the G1O5:A131
base-pair, G^yO6 hydrogen bonds with the exocyclic aminogroup of A131, and Gi05 Nl-H interacts with A131-W. Thesepairing schemes are consistent with NMR data obtained fromstudies of RRE—peptide complexes (42). Assuming bond anglesfor the sugars and phosphates normally observed in duplex RNA,G105, A131 and G106 would be expected to be in andconfigurations and Gl29 in a syn configuration (25, 43).However recent NMR studies predict an unusual andconformation for G129 rather than the expected syn configurationfor this base (42). Although our chemical substitution experimentsdo not directly address whether the G129 base is in a syn or andconformation, our finding that C^-methylG substitution at GIQ6was not harmful to rev binding suggests that the G106:G129 pairmay be rather flexible, even in the rev-bound state. Thereforeit is possible that Gl2g may be able to adopt either syn or andconformations depending on the ligand bound to the RRE RNA.
The methylphosphonate substitution experiments reported herehave established that rev makes important contacts to the highaffinity RRE binding site at PI03 and PI04 on one side of thebubble and at PI24 and PI25 on the other strand, somewhatdistant to the bubble. Evidence that PI03, PI04, PI24 and PI25play a critical role in rev recognition was also obtained inethylation interference studies of a RRE high affinity sitegenerated by in vitro selection (37). At each of the four sites ofmethylphosphonate interference both isomers are impaired in revbinding ability. These results are consistent with these phosphatesbeing involved in hydrogen-bonding interactions with chargedamino acids, or with two different ligands. Tan et al. haveproposed that the basic region of rev takes up an a-helicalstructure and that 6 amino acids (Thr-34, Arg-35, Arg-38,Arg-39, Asn-40 and Arg-44) make specific contacts with theRNA (43). Our results are consistent with this proposal sincean a-helix could easily bridge the distance between P103/P104and P124/P125 and some of these identified arginines couldindeed be responsible for such contacts.
The discovery that a single methylphosphonate isomer {Rp)at position P106 was impaired in rev binding ability is particularlyinformative. We interpret this result to indicate that the pro-/?oxygen atom of this phosphate makes a hydrogen bond with rev
by guest on August 18, 2016
http://nar.oxfordjournals.org/D
ownloaded from

2600 Nucleic Acids Research, 1994, Vol. 22, No. 13
either to an uncharged amino acid side chain (e.g. Thr-34 orAsn-40) or alternatively to a main chain amide functionality. Thisphosphate was not previously predicted by ethylation interferencemapping as a site of rev interaction.
Surprisingly, we have not obtained any evidence for directcontacts between rev and the five residues in the 'bubble'. Eachof the functional groups tested on these bases is either non-essential or appears to be involved in base-pairing. By contrast,major groove recognition of the RNA duplex occurs at theA^-positions of both purines flanking the bubble ( G ^ and G^s),but this does not extend to more distant purines. Thus, manyof the specific contacts made by rev to the high affinity bindingsite appear to be with phosphates rather than to the bases.
CONCLUSION
Studies of the interactions of tat and rev with RNA have providednew insights into the chemistry of nucleic acid recognition. Thereare several examples of RNA binding proteins that recognizebases displayed in single-stranded loop and bulge structures (45).By contrast, born tat and rev associate primarily with functionalgroups on Watson -Crick base-pairs that are exposed within themajor groove of a distorted double-stranded RNA (9, 12, 20).In addition to base-specific contacts, our methylphosphonatemapping shows that both proteins make specific phosphatecontacts on both strands of the flanking RNA duplexes. The dataon both tat binding to TAR and rev binding to the RRE suggestthat there are numerous contacts made between the amino acidside chains of tat and rev and functional groups displayed in theRNA binding sites. In the absence of high-resolution structuraldata, chemical modification studies, such as those described hereare providing our most precise images of the structures of thesetwo important RNA—protein complexes.
ACKNOWLEDGEMENTS
We diank Terry Smith, Jan Fogg and Richard Grenfell for helpwith synthesis of oligoribonucleotides, Christina Lamont andSheila Green for help in preparation of tat protein, Anne Kelleyfor provision of rev protein and Gabriele Varani and Jo Butlerfor helpful discussions. We are very grateful to Professor FritzEckstein (Max-Planck-Institiit fur Experimentelle Medizin,Gottingen) for provision of 2'-deoxy-2'-fluoroadenosine and itscorresponding phosphoramidite and to Dr Nanda Sinha (MilliporeCorporation, Bedford, Massachusetts) for 2'-deoxuridinemethylphosphonoamidite. This work was supported by a grantfrom the MRC AIDS Directed Programme.
REFERENCES
1. Kam, J. (1991) Current Opinion in Immunol., 3, 526-536.2. Gait, M.J. and Kara, J. (1993) TIBS, 18, 255-259.3. Cullen, B.R. (1986) Cell, 46, 973-982.4. Rosen, C.A., Sodroski, J.G. and Haseltine, W.A. (1985) Cell, 41, 813-823.5. Muesing, M.A., Smith, D.H. and Capon, D.J. (1987) Cell, 48, 691 -701 .6. Dingwall, C , Ernberg, I., Gait, M.J., Green, S.M., Heaphy, S., Karn,
J., Lowe, A.D., Singh, M., Skinner, M.A. and Valerio, R. (1989) Proc.Natl. Acad. Sci. USA, 86, 6925-6929.
7. Dingwall, C , Ernberg, I., Gait, M.J., Green, S.M., Heaphy, S., Karn,J., Lowe, A.D., Singh, M. and Skinner, M.A. (1990) EMBO J., 9,4145-4153.
8. Roy, S., Delling, U., Chen, C.-H., Rosen, C.A. and Sonenberg, N. (1990)Genes & Devel., 4, 1365-1373.
9. Weeks, K.M. and Crothers, D.M. (1991) Cell, 66, 577-588.
10. Sumner-Smith, M., Roy, S., Bamett, R., Reid, L.S., Kuperman, R., Delling,U. and Sonenberg, N. (1991) J, Virol., 65, 5196-5202.
11. Delling, U., Reid, L.S., Bamett, R.W., Ma, M.Y.-X., Climie, S., Sumner-Smith, M. and Sonenberg, N. (1992) J. Virol., 66, 3018-3025.
12. Churcher, M., Lamont, C , Dingwall, C , Green, S.M., Lowe, A.D., Butler,P.J.G., Gait, M.J. and Karn, J. (1993) J. Mol. Bio/., 230, 90-110.
13. Daly, T.J., Cook, K.S., Gary, G.S., Maione, T.E. and Rusche, J.R. (1989)Nature, 342, 816-819.
14. Heaphy, S., Dingwall, C , Ernberg, I., Gait, M.J., Green, S.M., Karn,J., Lowe, A.D., Singh, M. and Skinner, M.A. (1990) Cell, 60, 685-693.
15. Malim, M.H., Hauber, J., Le, S.-Y., Maizel, J.V. and Cullen, B.R. (1989)Nature, 338, 254-257.
16. Malim, M.H., Tiley, L.S., McCarn, D.F., Rusche, J.R., Hauber, J. andCullen, B.R. (1990) Cell, 60, 675-683.
17. Zapp, M.L. and Green, M.R. (1989) Nature, 342, 714-716.18. Mann, D.A., Mikaelian, I., Zemmel, R., Green, S.M., Lowe, A.D., Kimura,
T., Singh, M., Butler, P.J.G., Gait, M.J. and Karn, J. (1994) J. Mol. Biol.,in press.
19. Heaphy, S., Finch, J.T., Gait, M.J., Karn, J., and Singh, M. (1991) Proc.Natl. Acad. Sci. USA, 88, 7366-7370.
20. Kjems, J., Brown, M., Chang, D.D. and Sharp, P.A. (1991) Proc. Natl.Acad. Sci. USA, 88, 683-687.
21. Malim, M.H. and Cullen, B.R. (1991) Cell, 65, 241-248.22. Kjems, J., Calnan, B.J., Frankel, A.D. and Sharp, P.A. (1992) EMBO].,
11, 1119-1129.23. Tiley, L.S., Malim, M.H., Tewary, H.K., Stockley, P.G. and Cullen B.R.
(1992) Proc. Natl. Acad. Sci. USA, 89, 758-762.24. Bartel, D.P., Zapp, M.L., Green, M.R. and Szostak, J.W. (1991) Cell, 67,
529-536.25. Iwai, S., Pritchard, C , Mann, D.A., Karn, J. and Gait, M.J. (1992) Nucl.
Acids Res., 20, 6465-6472.26. Pavlevitch, N.P. and Pabo, CO. (1991) Science, 252, 809-817.27. Feng, J.-A., Johnson, R.C. and Dickerson, R.E. (1994) Science, 263,
348-355.28. Calnan, B.J., Tidor, B., Biancalana, S., Hudson, D. and Frankel, A.D. (1991)
Science, 252, 1167-1171.29. Tao, J. and Frankel, A.D. (1992) Proc. Natl. Acad. Sci, USA, 89,
2723-2726.30. Hamy, F., Asseline, U., Grasby, J., Iwai, S., Pritchard, C , Slim, G., Butler,
P.J.G. Karn, J. and Gait, M.J. (1993) J. Mol. Biol., 230, 111-123.31. Grasby, J.A., Butler, P.J.G. and Gait, M.J. (1993) Nucl. Acids Res., 21,
4444-4450.32. Pieken, W.A., Olsen, D.B., Benseler, F., Aurup, H. and Eckstein, F. (1991)
Science, 253, 314-317.33. Grasby, J.A., Pritchard, C.E., Mersmann, K., Seela, F. and Gait, M.J. (1993)
Coll. Czech. Chem. Commun., 58, 154-157.34. Karn, J., Churcher, M.J., Rittner, K., Kelley, A.C., Butler, P.J.G. , Mann,
D.A. and Gait, M.J. (1994) In HIV: A Practical Approach, J. Karn (ed.),Oxford University Press, Oxford, in press.
35. Lebedev, A.V., Frauendorf, A., Vyazovkina, E.V. and Engels, J.W. (1993)Tetrahedron, 49, 1043-1052.
36. Giver, L., Bartel, D., Zapp, M., Pawul, A., Green, M. and Ellington, A.D.(1993) Nucl. Acids Res. 21, 5509-5516.
37. Jensen, K.B., Green, L., MacDougal-Waugh, S. and Tuerk, C. (1994) J.Mol. Biol., 234, 235-247.
38. Noble, S.A., Fisher, E.F. and Caruthers, M.H. (1984) Nucl. Acids Res.12, 3387-3404.
39. Puglisi, J.D., Tan, R., Calnan, B.J., Frankel, A.D. and Williamson, J.R.(1992) Science, 257, 76-80.
40. Puglisi, J.D., Chen, L., Frankel, A.D. and Williamson, J.R. (1993) Proc.Natl. Acad. Sci. USA, 90, 3680-3684.
41. Tao, J. and Frankel, A.D. (1993) Proc. Natl. Acad. Sci. USA, 90,1571-1575.
42. Battiste, J.L., Tan, R., Frankel, A.D. and Williamson, J.R. (1994)Biochemistry, 33, 2741-2747.
43. Leclerc, F., Cedergren, R. and Ellington, A.D. (1994) Nature StructuralBiol., 1, 293-300.
44. Tan, R., Chen, L., Buettner, J.A., Hudson, D. and Frankel, A.D. (1993)Cell, 73, 1031-1040.
45. Nagai, K. (1992) Curr. Opinion Struct. Biol., 2, 131-137.
by guest on August 18, 2016
http://nar.oxfordjournals.org/D
ownloaded from
Copyright © 2022 FDOKUMEN