
Mesocosms of Aquatic Bacterial Communities from the Cuatro Cienegas Basin (Mexico): A Tool to Test...
13
MICROBIOLOGY OF AQUATIC SYSTEMS Mesocosms of Aquatic Bacterial Communities from the Cuatro Cienegas Basin (Mexico): A Tool to Test Bacterial Community Response to Environmental Stress Silvia Pajares & German Bonilla-Rosso & Michael Travisano & Luis E. Eguiarte & Valeria Souza Received: 5 December 2011 / Accepted: 8 March 2012 / Published online: 30 March 2012 # Springer Science+Business Media, LLC 2012 Abstract Microbial communities are responsible for impor- tant ecosystem processes, and their activities are regulated by environmental factors such as temperature and solar ultraviolet radiation. Here we investigate changes in aquatic microbial community structure, diversity, and evenness in response to changes in temperature and UV radiation. For this purpose, 15 mesocosms were seeded with both micro- bial mat communities and plankton from natural pools with- in the Cuatro Cienegas Basin (Mexico). Clone libraries (16S rRNA) were obtained from water samples at the beginning and at the end of the experiment (40 days). Phylogenetic analysis indicated substantial changes in aquatic community composition and structure in response to temperature and UV radiation. Extreme treatments with elevation in temper- ature or UV radiation reduced diversity in relation to the Control treatments, causing a reduction in richness and increase in dominance, with a proliferation of a few resistant operational taxonomic units. Each phylum was affected dif- ferentially by the new conditions, which translates in a differ- ential modification of ecosystem functioning. This suggests that the impact of environmental stress, at least at short term, will reshape the aquatic bacterial communities of this unique ecosystem. This work also demonstrates the possibility of designing manageable synthetic microbial community eco- systems where controlled environmental variables can be manipulated. Therefore, microbial model systems offer a com- plementary approach to field and laboratory studies of global research problems associated with the environment. Introduction The existence of microbial communities resembling modern microbial mats and calcified stromatolites has been regis- tered in the fossil record since the Archaean, starting from small localized patches to large areas covering most of the shallow oceans in the Proterozoic. Microbial mats are the oldest ecosystems known to date and have proven to be successful ecological assemblages since they have survived billions of years of environmental change due to their stable but adaptable structural properties and their capabilities for biogeochemical transformations and energy flows [22, 26, 47]. Habitats that contain microbial mats provide windows to an understanding of how life persisted in the past and potentially how life will function in the future [56]. Cuatro Cienegas Basin (CCB) is an enclosed evaporitic basin in the Mexican Chihuahua Desert. It consists of a diverse range of pools dominated by living microbial mats and stromatolites that have evolved in relative isolation and under biogeochemical constraints [9, 54]. Despite the ex- treme oligotrophy of the ecosystem, particularly for Electronic supplementary material The online version of this article (doi:10.1007/s00248-012-0045-7) contains supplementary material, Microb Ecol (2012) 64:346–358 DOI 10.1007/s00248-012-0045-7 which is available to authorized users. S. Pajares : G. Bonilla-Rosso : L. E. Eguiarte : V. Souza (*) Departamento de Ecología Evolutiva, Instituto de Ecología, Universidad Nacional Autónoma de México (UNAM), AP 70-275, CU, Coyoacán, 04510 Mexico City, Mexico e-mail: [email protected] V. Souza e-mail: [email protected] S. Pajares Consejo Superior de Investigaciones Científicas (IRNA-CSIC), Salamanca, Spain M. Travisano Department of Ecology, Evolution and Behavior, University of Minnesota, Saint Paul, MN 55108, USA
Transcript of Mesocosms of Aquatic Bacterial Communities from the Cuatro Cienegas Basin (Mexico): A Tool to Test...

MICROBIOLOGY OF AQUATIC SYSTEMS
Mesocosms of Aquatic Bacterial Communitiesfrom the Cuatro Cienegas Basin (Mexico): A Tool to TestBacterial Community Response to Environmental Stress
Silvia Pajares & German Bonilla-Rosso &
Michael Travisano & Luis E. Eguiarte & Valeria Souza
Received: 5 December 2011 /Accepted: 8 March 2012 /Published online: 30 March 2012# Springer Science+Business Media, LLC 2012
Abstract Microbial communities are responsible for impor-tant ecosystem processes, and their activities are regulatedby environmental factors such as temperature and solarultraviolet radiation. Here we investigate changes in aquaticmicrobial community structure, diversity, and evenness inresponse to changes in temperature and UV radiation. Forthis purpose, 15 mesocosms were seeded with both micro-bial mat communities and plankton from natural pools with-in the Cuatro Cienegas Basin (Mexico). Clone libraries (16SrRNA) were obtained from water samples at the beginningand at the end of the experiment (40 days). Phylogeneticanalysis indicated substantial changes in aquatic communitycomposition and structure in response to temperature andUV radiation. Extreme treatments with elevation in temper-ature or UV radiation reduced diversity in relation to theControl treatments, causing a reduction in richness and
increase in dominance, with a proliferation of a few resistantoperational taxonomic units. Each phylum was affected dif-ferentially by the new conditions, which translates in a differ-ential modification of ecosystem functioning. This suggeststhat the impact of environmental stress, at least at short term,will reshape the aquatic bacterial communities of this uniqueecosystem. This work also demonstrates the possibility ofdesigning manageable synthetic microbial community eco-systems where controlled environmental variables can bemanipulated. Therefore, microbial model systems offer a com-plementary approach to field and laboratory studies of globalresearch problems associated with the environment.
Introduction
The existence of microbial communities resembling modernmicrobial mats and calcified stromatolites has been regis-tered in the fossil record since the Archaean, starting fromsmall localized patches to large areas covering most of theshallow oceans in the Proterozoic. Microbial mats are theoldest ecosystems known to date and have proven to besuccessful ecological assemblages since they have survivedbillions of years of environmental change due to their stablebut adaptable structural properties and their capabilities forbiogeochemical transformations and energy flows [22, 26,47]. Habitats that contain microbial mats provide windowsto an understanding of how life persisted in the past andpotentially how life will function in the future [56].
Cuatro Cienegas Basin (CCB) is an enclosed evaporiticbasin in the Mexican Chihuahua Desert. It consists of adiverse range of pools dominated by living microbial matsand stromatolites that have evolved in relative isolation andunder biogeochemical constraints [9, 54]. Despite the ex-treme oligotrophy of the ecosystem, particularly for
Electronic supplementary material The online version of this article(doi:10.1007/s00248-012-0045-7) contains supplementary material,
Microb Ecol (2012) 64:346–358DOI 10.1007/s00248-012-0045-7
which is available to authorized users.
S. Pajares :G. Bonilla-Rosso : L. E. Eguiarte :V. Souza (*)Departamento de Ecología Evolutiva, Instituto de Ecología,Universidad Nacional Autónoma de México (UNAM),AP 70-275, CU, Coyoacán,04510 Mexico City, Mexicoe-mail: [email protected]
V. Souzae-mail: [email protected]
S. PajaresConsejo Superior de Investigaciones Científicas (IRNA-CSIC),Salamanca, Spain
M. TravisanoDepartment of Ecology, Evolution and Behavior,University of Minnesota,Saint Paul, MN 55108, USA

phosphorus (P), CCB has a high level of diversity and speciesendemism at both the macro- and microscopic scales [40, 54,55, 58]. The aquatic microbiota of the pools exhibits ancestralmarine affinities [2, 15, 41], and the composition of thisbacterial community is dominated by Proteobacteria (mainlyGammaproteobacteria, Betaproteobacteria, and Alphaproteo-bacteria subdivisions), followed in abundance by Bacteroi-detes and Actinobacteria [12, 16, 55]. This is in agreementwith 16S rRNA-based studies in continental freshwater lakes,where the Actinobacteria lineage acI appears to be the mostcommon and persistent bacterioplankton lineage [42, 60, 64].
Microbial communities play a central role in the cyclingof nutrients in aquatic ecosystems, and they constitute afundamental link in the carbon transfer process [5]. There-fore, the study of factors, such as solar ultraviolet radiationand temperature, which directly affect the bacterial compo-sition in the communities is of primary importance. UVradiation could be particularly deleterious for bacteria be-cause these organisms have simple haploid genomes withlittle or no functional redundancy, and they are small, whichprecludes effective cellular protective pigmentation [19].Damage caused by UV in bacterial systems in aquatic envi-ronments eventually affects the whole community, havingan impact on photosynthesis, biomass production, and thecommunity composition [4, 62]. Temperature also plays akey role in community dynamics, since it has been shownthat microbial activity and turnover increase with tempera-ture, favoring thermotolerant organisms with fast growthrates [1, 48]. Few studies have linked environmental stressto bacterial diversity in freshwater environments, given theinherent difficulties of working with microbial communitiesin the field over extended periods of time [8, 57]. In the caseof the microbial communities of CCB, we have a uniqueopportunity to do so, since in their aquatic habitats we haveobserved a strong cohesivity and redundancy [9, 47].
On the other hand, global scale environmental problemsare rarely regarded as amenable to traditional scientificexperiment. We argue here that it is possible to conductreplicated experiments on model systems, with the ease ofexperimentation increasing as the scale of the model systemgets smaller. Such simplicity enables high degrees of exper-imental Control and replication that are necessary to addressmany questions that are inaccessible through field observa-tion or experimentation [32]. Herein we studied the responseof CCB aquatic ecosystem, focusing on its microbial com-munities, to new environments (changes of temperature andUV radiation) by using a mesocosms approach that includes“synthetic” microbial mats, bacterioplankton, and water fromCCB pools as starting point. Furthermore, by considering eachmesocosm as autosustainable closed system, we could assessthe change in the bacterial community structure in response toenvironmental stresses. This was performed using 16S rRNAclone libraries and supported by observations with optical
microscopy, finding that environmental perturbations have adifferential impact on the community biodiversity, even at ashort time scale in experimental mesocosms.
Material and Methods
Experimental Design
In order to measure the response of aquatic microbial com-munity to environmental stress, we constructed 15 meso-cosms in standard 40-L fish tanks, which included microbialmats, bacterioplankton, and composite water sample fromCCB pools. To do this, in November 2007, nine apparatusconsisting of three acrylic trays with 12 glass slides eachwere placed into three pools (P1, P2, and P3) in PozasAzules Ranch of CCB (Supplementary Fig. S1). The poolswere chosen because they are stable ecosystems from thesame hydrological system located in close proximity to oneanother and are no more than 20 m in diameter and no lessthan 3 m in depth. These pools represent well-mixedsprings, rich in calcium carbonates and maintain the growthof microbial mat communities.
In July 2009 three water samples were collected fromeach pool for the purpose of nutrient analyses (Table S1).Physicochemical variables (pH, dissolved O2, electrical con-ductivity, and salinity) were obtained in situ using aYSI600QS instrument equipped with a Hydrolab YSI600R(YSI Inc., Yellow Springs, OH). These measurements sug-gest that the water within these pools was pH neutral, highlysaturated with oxygen and slightly saline. The ratio of C:N:Pin each pool was P1 460:7:1, P2 370:9:1, and P3 200:7:1,respectively, suggesting in all of them a slight N limitation(F. García-Oliva, personal communication).
Given the similarity of the physicochemical parameterswithin each pool, 600 L of water was retrieved from P1, P2,and P3 and was subsequently mixed into a composite sam-ple in order to homogenize the community at the beginningof the experiment (T0). Composite water sample was addedto each mesocosm in addition to one acrylic tray with 12randomly selected glass slides from the pools (Fig. S1).
The mesocosms systems were exposed to five treatments,each replicated three times (for a total of 15 mesocosms): (a)Control—similar temperature (30 °C) and UV intensity tothe original natural pools, (b) Fluct—fluctuating tempera-ture (temperature range in summer was 25–45 °C) andsimilar UV intensity to the pools, (c) 40C—increase to aconstant temperature of 40 °C and similar UV intensity tothe pools, (d) UVplus—artificial increase in UV radiationduring 12 h per day and similar temperature to the pools,and (e) UVmin—reduction in UV light using acrylic filtersand similar temperature to the pools. The regulation of watertemperature and aerated conditions were maintained using a
Bacterial Community Response to Stress 347

BOYU Chiller L-075. In the Fluct treatment, no chiller wasadded, allowing the formation of an oxygen gradient wheretemperature fluctuates according to the ambient. A HOBOPendant Temperature/Light Data Logger was placed intoeach tank to monitor variations in light and temperature.For the UVplus treatment, a UV light lamp was added to thefish tank (BioPro underwater light 110–120 V). In theUVmin treatment, UV sun light was blocked by an acrylicfilter covering the mesocosms. The tanks were refilled withsterile distilled water when their water volume decreased byevaporation (between 0.5 and 1 L every week). The meso-cosms were placed at random on a patio of a house in thetown of CCB for 40 days, approximately 17 km from thecollection sites.
DNA Isolation and Sequencing of 16S rRNA CloneLibraries
To measure the response of the community to each treatment,16S rRNA clone libraries were obtained from water samplesat the beginning (T0) and at the end of the experiment (40 dayslater). Water samples of 3 L were collected from each meso-cosm and were immediately filtered using 0.45-μm filters anda field vacuum pump. Filters were placed into sterile Falcontubes and stored at −20 °C. The filters were subsequentlystored at −80 °C prior to DNA extraction.
Genomic DNA was extracted from two filtered samplesper mesocosm using an UltraClean Water DNA Kit (MoBio, Carlsbad, CA, USA), according to the manufacturer’sinstructions. A bead-beating step is incorporated as part ofthis protocol in order to disrupt the gram-positive cells aswell as the hardy cyanobacteria. Two different PCR ampli-fications were performed on a MJ Research thermocycler bytargeting 16S rRNA gene fragments of the bacterial domain[34] and the phylum Cyanobacteria [43]. Each PCR reactionfor bacterial 16S rRNA fragments contained 20–30 ng ofDNA per 50 μl reaction volume, 1× High Fidelity PCRbuffer, 1.5 mM MgSO4, 0.4 mM of each deoxyribonucleo-tide, 300 mM of the universal primers 27F (5′-AGA GTTTGA TCC TGG CTC AG-3′) and 1492R (5′-GGT TACCTT GTT ACG ACT T-3′), 1 mg mL−1 bovine serumalbumin, 5 % DMSO, and 1 U of Platinum Taq DNApolymerase High Fidelity (Invitrogen, CA, USA). PCR am-plification was performed according to the following cycle:initial denaturation at 94 °C for 3 min, 30 cycles of dena-turation at 94 °C for 1 min, annealing at 52 °C for 1 min,and extension at 72 °C for 1.2 min, followed by a finalextension period of 3 min at 72 °C. Each PCR reaction forcyanobacterial 16S rRNA fragments contained 20–30 ng ofDNA per 50 μl, 1× of PCR buffer, 2.5 mM MgCl2, 0.2 mMof each deoxyribonucleotide, 500 mM of the cyanobacterialprimers CYA359F (5′-GGG GAA TYT TCC GCA ATGGG-3′) and CYA781bR (5′-GAC TAC AGG GGT ATC
TAA TCC CTT T-3′), and 2.5 U of Taq DNA polymerase(Facultad de Veterinaria, UNAM, Mexico). PCR amplifica-tion was performed according to the following cycle: 94 °Cfor 1 min, 60 °C for 1 min, and 72 °C for 1 min, repeatedover 35 cycles. The PCR products of three replicate reactionmixtures from each sample were mixed and separated by1 % agarose gel electrophoresis and purified with a QIA-quick PCR purification kit (Qiagen Inc., the Netherlands).Purified PCR products were cloned using Topo TA Cloningkit with pCR2.1 vectors and transformed into Escherichiacoli TOP10 cells (Invitrogen, CA, USA), following themanufacturer’s instructions. Cloned inserts were extractedand isolated for sequencing using the Plasmid Miniprep96kit (Millipore Corp.). Plasmids were shipped to the DNAlaboratory of the University of Washington (Seattle, USA)for sequencing. The bacterial partial 16S rRNA gene frag-ments were sequenced using the primer 27F, and the cyano-bacterial region was sequenced with the primer M13R.
Sequence and Statistical Analysis
After trimming and editing with the Bioedit program,sequences of average length of 760 bp for universal primerproducts and 420 bp for cyanobacterial primer productswere aligned using the NAST aligner against the Green-Genes database ([14]; http://greengenes.lbl.gov/). Align-ments were corrected manually, and all sequences thatwere identified as putative chimeras in Bellerophon programwere discarded from the dataset. Two neighbor joiningphylogenetic trees of representative 16S rRNA gene sequen-ces, one for the general data set and another for the cyano-bacterial region, were constructed using Quicktree programwith 10,000 bootstrap replicates. The resulting phylogenetictrees were edited with iTOL program ([36]; http://itol.embl.de/index.shtml). Sequences were deposited in GenBank withaccession numbers JF412835-JF414016 for bacterial andJF423322-JF423470 for cyanobacterial 16S rRNA genesequences.
Operational taxonomic units (OTUs) were defined andidentified using the Mothur v1.12 software package [52]. Athreshold of 97 % sequence similarity was used to defineeach OTU. For the cyanobacterial sequences, the thresholdwas set to 99 % due to the shorter length of the products.Alpha diversity indices, richness estimates, and rarefactioncurves based on the rank abundance of OTUs defined byclones sharing >97 % sequence similarity were calculatedwith the Mothur program. Pooled data per treatment servedas the experimental units for microbial diversity indices,since the pooled data can give a better estimate of overalldiversity in each treatment. The Shannon (H′), Simpson(1/D), and Berger–Parker (BP) indices were used to estab-lish relative rankings of diversity levels among treatments.Chao1 was used to estimate the community’s species
348 S. Pajares et al.

richness [28, 39]. Rarefaction curves provide a measure ofsample coverage and are a good comparative method tostandardize and compare species richness computed fromsamples of different sizes [27]. To evaluate relationshipsbetween treatments based on the shared relative abundanceof OTUs (beta diversity), we performed pair wise compar-isons between similarity indices [13, 39]. Similarity indicesSorensen (S), Sorensen-chao1 (S-chao1), Morisita Horn(Mo), and Yue and Clayton’s theta (θ) were also calculatedwith Mothur.
In order to estimate the relationship between dominanceand diversity, Rényi’s entropy profiles [50] for ten scales(α00, 0.25, 0.5, 1, 2, 4, 8, 16, 32, infinite) were calculatedfor each treatment with R program. This profile provides aparametric measure of the uncertainty of predicting therelative abundance of species at different scales betweenthe different treatments.
Additional statistical analyses were performed with SPSS16.0 program. Physicochemical data from the pools weretested for normal distribution and subjected to single classi-fication ANOVA (p<0.05). As the number of clones obtainedin each replicate was highly variable, due to the cloningprocedure, we also performed an ANOVA of the relativefrequencies of 16S rRNA OTUs (97 %) between treatmentsto identify possible differences and to verify the variabilityamong replicates of each treatment.
Results
Mesocosms Communities
The primary goal of this study was to investigate the short-term effects of environmental stress on aquatic microbialcommunities. Community structure, diversity, and evennessindices were compared at initial (T0) and final (Tf, 40 dayslater) time points. We recovered 27 OTUs out of 224sequences using a 97 % cutoff at T0 (Table S2). At timeT0, the communities were largely dominated by Actinobac-teria (71 %), followed by Alphaproteobacteria (8 %) andCyanobacteria (7.1 %; Figs. 1 and 2). Evenness was low dueto the dominance of a single OTU from family Microbacter-iaceae (Fig. 1 and Table S3). Alphaproteobacteria membersbelonged to orders Rhizobiales, Rhodobacterales, Sphingo-monadales, and Caulobacterales, while the 16S rRNAsequences from Cyanobacteria belonged to order Chroococ-cales and phylum Rhodophyta (chloroplast). At Tf, 168unique phylotypes from 11 major lineages of Eubacteriawere recovered out of 959 sequences using the 97 % cutoff,suggesting a general increase in diversity across all treat-ments. Mesocosm communities were not sampled to ex-haustion, as indicated by the rarefaction analyses where nocurve reached an asymptote (Fig. 3).
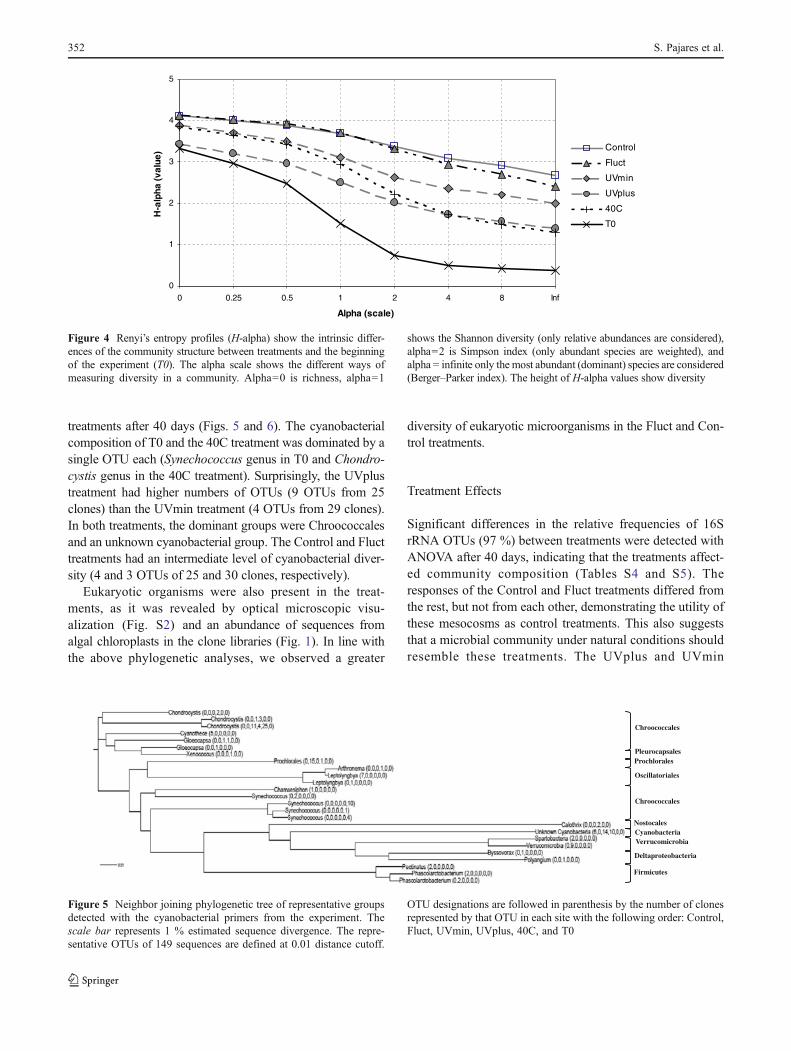
Control and Fluct mesocosms had the higher richness anddiversity (Fig. 4 and Table S3). In the Control treatment, atotal of 58 OTUs were recovered out of 173 16S rRNAsequences. The Control treatment was dominated byAlphaproteobacteria from orders Caulobacterales, Sphingo-monadales, Rhizobiales, and Rhodospirillales. The phyla Cya-nobacteria (orders Chroococcales and a fraction ofuncharacterized Cyanobacteria), Betaproteobacteria (ordersBurkholderiales and Rhodocyclales) and Gammaproteobacte-ria (orders Legionellales, Xanthomonadales, and Pseudomo-nadales) were also numerous. In the Fluct treatment, 62 OTUswere recovered out of 168 sequences in ten phyla, displayingthe highest diversity in all treatments. Alphaproteobacteriadominated this treatment (orders Caulobacterales, Rhizo-biales, Sphingomonadales, and Rhodospirillales), with partic-ular dominance of OTUs from Methylocystaceae,Caulobacteraceae, and Brevundimonas. This was followed inabundance by phylum Planctomycetes and a single OTUbelonging to family Microbacteriaceae (Actinobacteria).
Diversity in the more extreme treatments (UVmin, UVplus,and 40C) was not evenly distributed. The greater diversity inUVmin was due to a combination of lower dominance (Figs. 3and 4) and a richness equivalent to the 40C treatment. Thelowest richness was observed in the UVplus treatment, andalthough its most abundant OTU was at a larger frequencythan its equivalent in the 40C treatment, overall dominancewas also larger in the UVplus treatment.
In the UVmin treatment, 46 OTUs were recovered out of227 sequences. Alphaproteobacteria are diverse, but abun-dant OTUs are Novosphingobium, Inquilinus, bacteriumEllin333, Devosia, Starkeya, and Xanthobacteraceae. It alsohad a large abundance of a single OTU from family Micro-bacteriaceae with close affinity toMicrocella or Salinibacterwithin the Actinobacteria. Betaproteobacteria mostly belongto a single OTU with close affinity to genus Limnobacter.
The greatest dominance of Alphaproteobacteria wasfound in the 40C treatment (45 OTUs recovered out of220 sequences), where most sequences belonged to a singleOTU closely related to Phaeospirillum (Rhodospirillales)and several other OTUs from orders Sphingomonadales,Caulobacterales, and Rhizobiales. No Actinobacteria werefound in this treatment, and all Cyanobacteria belonged toorder Chroococcales.
The UVplus treatment had the lowest number of OTUs (30OTUs recovered out of 171 sequences) and a substantially
Figure 1 Neighbor joining phylogenetic tree of representative 16SrRNA gene sequences from the mesocosms experiment. The scale barrepresents 1 % estimated sequence divergence. The representativeOTUs of 1183 sequences are defined at 0.03 distance cutoff. OTUdesignations are followed in parenthesis by the number of clonesrepresented by that OTU in each site with the following order: Control,Fluct, UVmin, UVplus, 40C, and T0
b
Bacterial Community Response to Stress 349

Alphaproteobacteria
Gammaproteobacteria
Betaproteobacteria
Deltaproteobacteria
Epsilonproteobacteria
Verrucomicrobia
Planctomycete
Firmicutes
Cyanobacteria
Bacteroidete
Actinobacteria
350 S. Pajares et al.

reduced proportion of Cyanobacteria in the community (usingthe 16S rRNA universal primers). Alphaproteobacteriabelonged mostly to the same Phaeospirillum OTU as in the40C treatment. A large abundance ofPorphyrobacterwas alsodetected. Actinobacteria was the second most abundant phy-lum, with sequences clustered in three OTUs from familyMicrobacteriaceae. Only four OTUs were found in alltreatments: two OTUs from order Rhizobiales (families Xan-thobacteraceae and Methylocystaceae), one from Sphin-gobium (Sphingomonadales) and one from Limnobacter(Betaproteobacteria).
From the cyanobacterial clone libraries, we obtained atotal of 149 sequences that contained 25 unique phylotypes(OTUs at 99 % cutoff; Figs. 5 and 6). Of these, 18 OTUsbelonged to the Cyanobacteria phylum (87.2 % of the
clones), 3 OTUs to Firmicutes (4 % of the clones), 2 OTUsto Verrucomicrobia (7.4 % of the clones), and 2 OTUs toDeltaproteobacteria (1.3 % of the clones), indicating thatthese primers are not exclusively specific to the Cyanobac-teria phylum. Within the cyanobacterial phylum, Chroococ-cales was the most abundant group (48.3 % of the clonesand 11 OTUs), followed by an unknown group of Cyano-bacteria (20.1 % of the clones constituted this OTU), Pro-chlorales (10.7 % of the clones within 1 OTU), Oscillatoriales(6 % of the clones and 3 OTUs), Nostocales (1.3 % of theclones and 1 OTU), and Pleurocapsales (0.7 % of the clonesand 1 OTU).
In line with the remaining bacterial diversity, there wasan increase in the composition within the Cyanobacterialphylum recovered with these primers in almost all the
Alphaproteobacteria
Actinobacteria
Cyanobacteria
Betaproteobacteria
Bacteroidete
Gammaproteobacteria
Verrucomicrobia
Planctomycete
Firmicutes
Deltaproteobacteria
Epsilonproteobacteria
0
10
20
30
40
50
60
70
80
90
100
% C
lone
s
Control N=177 Fluct N=165 UVmin N=226 UVplus N=171 40C N=220 T0 N=224
Treatments
Figure 2 Relative abundance (in percent) of bacterial taxonomic groups from the clone library data of the different treatments and at the beginning ofthe experiment (T0). Error bars indicate standard deviation, and the number of clones obtained from each site is indicated below each one as N
0
10
20
30
40
50
60
70
0 25 50 75 100 125 150 175 200 225 250
Nº clones
Nº
OTU
s
Control
lci
hci
Fluct
lci
hci
UVmin
lci
hci
UVplus
lci
hci
40C
lci
hci
T0
lci
hci
UVmin
Control Fluct
40C
UVplus T0
Figure 3 Rarefaction curves of16S rRNA gene sequences fromthe beginning of the experiment(T0) and each treatment,displaying number of OTUsdetected (at 97 % cutoff) versusnumber of sequences analyzedin each treatment
Bacterial Community Response to Stress 351
treatments after 40 days (Figs. 5 and 6). The cyanobacterialcomposition of T0 and the 40C treatment was dominated by asingle OTU each (Synechococcus genus in T0 and Chondro-cystis genus in the 40C treatment). Surprisingly, the UVplustreatment had higher numbers of OTUs (9 OTUs from 25clones) than the UVmin treatment (4 OTUs from 29 clones).In both treatments, the dominant groups were Chroococcalesand an unknown cyanobacterial group. The Control and Flucttreatments had an intermediate level of cyanobacterial diver-sity (4 and 3 OTUs of 25 and 30 clones, respectively).
Eukaryotic organisms were also present in the treat-ments, as it was revealed by optical microscopic visu-alization (Fig. S2) and an abundance of sequences fromalgal chloroplasts in the clone libraries (Fig. 1). In line withthe above phylogenetic analyses, we observed a greater
diversity of eukaryotic microorganisms in the Fluct and Con-trol treatments.
Treatment Effects
Significant differences in the relative frequencies of 16SrRNA OTUs (97 %) between treatments were detected withANOVA after 40 days, indicating that the treatments affect-ed community composition (Tables S4 and S5). Theresponses of the Control and Fluct treatments differed fromthe rest, but not from each other, demonstrating the utility ofthese mesocosms as control treatments. This also suggeststhat a microbial community under natural conditions shouldresemble these treatments. The UVplus and UVmin
0
1
2
3
4
5
0 0.25 0.5 1 2 4 8 Inf
Alpha (scale)
H-a
lpha
(val
ue) Control
Fluct
UVmin
UVplus
40C
T0
Figure 4 Renyi’s entropy profiles (H-alpha) show the intrinsic differ-ences of the community structure between treatments and the beginningof the experiment (T0). The alpha scale shows the different ways ofmeasuring diversity in a community. Alpha00 is richness, alpha01
shows the Shannon diversity (only relative abundances are considered),alpha02 is Simpson index (only abundant species are weighted), andalpha 0 infinite only the most abundant (dominant) species are considered(Berger–Parker index). The height of H-alpha values show diversity
Firmicutes
Verrucomicrobia
Deltaproteobacteria
Pleurocapsales
Oscillatoriales
Chroococcales
Chroococcales
Nostocales
Prochlorales
Cyanobacteria
Figure 5 Neighbor joining phylogenetic tree of representative groupsdetected with the cyanobacterial primers from the experiment. Thescale bar represents 1 % estimated sequence divergence. The repre-sentative OTUs of 149 sequences are defined at 0.01 distance cutoff.
OTU designations are followed in parenthesis by the number of clonesrepresented by that OTU in each site with the following order: Control,Fluct, UVmin, UVplus, 40C, and T0
352 S. Pajares et al.

treatments significantly differed from one to another, butneither could be distinguished from the 40C treatment.
A reduction in diversity was observed in the extremetreatments. The largest number of shared OTUs betweencommunities was found between the Control and Fluct treat-ments. The lowest shared OTUs were found between theFluct and 40C treatments. The largest Mo similarities weredetected between 40C–UVplus and Control–UVmin treat-ments, meaning that temperature and UV have a similareffect on structure and that UVmin is very little affected inits structure. All similarity values (S, S-chao1, Mo, and θ)were less than 0.5, which indicates that the treatmentsreached dissimilar community compositions (Table 1), withthe exception of Mo between 40C–UVplus (0.661) andControl–UVmin (0.528). Altogether, this suggests that tem-perature fluxes are the most important factor affecting com-munity composition. It also suggests that changes in UVregimes mostly affect community structure, and that a com-bination of changes to both temperature and UV modify thecommunity most drastically.
Discussion
We used a mesocosms experiment to determine the influ-ence of UV radiation and temperature on the compositionand diversity of the bacterioplankton community. Even in ashort time, we observed dramatic effects on microbial di-versity caused by the extreme treatments, with little diver-gence of planktonic microbiota between replicates withineach treatment. We conclude that short-term community as-sembly was driven primarily by direct responses to environ-mental stress and less by stochastic factors.
Microbial diversity in the initial community (T0) includ-ed many typical freshwater taxa, but lacked some frequentlyobserved species. The most abundant phylum was Actino-bacteria, which was dominated by a single MicrobacteriaceaeOTU. Actinobacteria is one of the most abundant, diverse, andwidespread phyla in continental freshwater lakes [20, 60],with members of the dominant clades Luna and acI [42].The freshwater Actinobacteria are free living, open water de-fense specialists, with possible photothrophic and heterotrophic
0%
20%
40%
60%
80%
100%
Control N=25 Fluct N=30 UVmin N=29 UVplus N=25 40C N=25 T0 N=15
Treatments
% C
lone
s
Deltaproteobacteria
Firmicutes
Verrucomicrobia
Pleurocapsales
Nostocales
Oscillatoriales
Prochlorales
Unknown Cyanobacteria
Chroococcales
Figure 6 Distribution andrelative abundance (in percent)of groups detected with theCyanobacteria primers of eachtreatment and at the beginningof the experiment (T0). Eachbar refers to a differenttreatment, and the number ofclones obtained from each siteis indicated below each oneas N
Table 1 Shared diversity indi-ces of 16S rRNA sequences be-tween treatments (OTUs at97 % cutoff)
S Sorensen; S-chao1 Sorensen-chao1; Mo Morisita; θ Theta
Comparison OTUs shared between Abundance based Structure based
Sharedsobs Sharedchao1 Sorensen Sorensen-chao1 Morisita Theta
40C Control 18 22.9 0.330 0.219 0.325 0.194
40C Fluct 8 11.5 0.145 0.123 0.055 0.028
40C UVplus 10 22.0 0.253 0.325 0.661 0.494
40C UVmin 19 25.7 0.396 0.345 0.223 0.125
Control Fluct 19 36.3 0.304 0.290 0.313 0.185
Control UVplus 16 29.0 0.340 0.291 0.232 0.132
Control UVmin 19 25.3 0.342 0.237 0.528 0.359
Fluct UVplus 11 14.8 0.232 0.167 0.217 0.122
Fluct UVmin 9 14.3 0.161 0.150 0.319 0.190
UVplus UVmin 10 13.0 0.247 0.187 0.422 0.267
Bacterial Community Response to Stress 353

energy generation lifestyles [42, 49]. The dominant OTU at T0belonged to the Luna/acII/Aquiluna clade, while another OTUbelonged to the uncultured actinobacterial clade acI. Actino-bacteria has already been reported as an abundant anddiverse phylum in CCB, with members of Microbacter-iaceae (Cellulomonas, Agrococcus, and Micrococcus) wellrepresented in aquatic communities [12]. An abundance ofChroococcales (Cyanobacteria) as well as members of genusBrevundimonas (Caulobacterales) and order Sphingomona-dales was observed in CCB, and is also commonly abundantin freshwater lakes [42].
The abundance of Sphingomonadales can be explained asthis group is favored in P-limited environments, since theyproduce sphingolipids instead of lipopolysaccharides intheir outer membrane [63]. A notable difference with otherfreshwater lakes was the low abundance of Bacteroidetes,even though the identified sequences belonged to the Chiti-nophagaceae (bacI) and Saprospiraceae (bacIV) families,the dominant groups in freshwater lakes [42], suggestingthat CCB is an anomalous site, a kind of hybrid between itscontinental present and its marine past.
No community in any of the mesocosms retained theinitial species composition, and all mesocosms shifted indominance from Actinobacteria to Alphaproteobacteria. It isevident that the inoculation of samples into mesocosms andthe subsequent mesocosm maintenance modified the com-position and structure of the communities, even for theControl treatment. We used the Control and Fluct treatmentsas contrasts to the other treatments as they were most similarto the natural conditions, with ambient UV exposure andeither constant (Control) or ambient (Fluct) temperature.These two treatments were statistically indistinguishable intheir diversity and had high taxonomic similarity as well as asimilar community structure. The variability in temperaturein the Fluct treatment had small effects relative to the Con-trol treatment, and only a few groups were affected by thesefluctuations and were present in Control and not in Flucttreatment mesocosms, most notably Phaeospirillum andother Rhodospirillales.
The Control and Fluct mesocosms had the largest diver-sity both in terms of richness and evenness. More diversecommunities are thought to contribute to ecosystem func-tions to a greater extent than those that are less diverse: eachspecies uses slightly different resources and is more highlyniche specific, thus providing more services to the ecosys-tem [6, 37]. Since the initial community (T0) showed amarked dominance and no community was sampled toexhaustion (as demonstrated by rarefaction analysis), it islikely that confining the community in a mesocosm caused aperturbation that strongly affected the most dominant organ-isms in T0, allowing for the proliferation of other taxawhose abundances were formerly below detection levels.This is supported by a higher evenness observed in Control
and Fluct treatments. Furthermore, we should take intoaccount that T0 represented the microbial composition ofthe pools at the time that they were sampled, while theControl treatment showed the potential composition of themicrobial community, because this treatment simulated thepotential environmental conditions of these pools. On theother hand, the ability to sample the mesocosms to satura-tion using high-throughput next generation sequencingmethods, such as 454 pyrosequencing, will be an importantstep in developing a thorough understanding of the adapta-tion and evolution of the microbial communities in responseto different environmental conditions. In addition, the abilityto temporally sample the mesocosms’ microbial communitywill provide valuable insights into the temporal changes inthe bacterial composition and structure.
Our mesocosms follow a “seed bank” model [46], wherethe most abundant taxa (“core taxa”) are responsible forcarbon and energy flow throughout an ecosystem, whereasthe long tail of rare taxa (“seed bank”) are at tolerancelimits. If environmental conditions change and become ad-equate for the growth, rare taxa originating from this “seedbank” can become abundant. This is corroborated by lowvalues of beta diversity between treatments, indicating thatthe microbial community responded both rapidly and differ-ently to changes in environmental conditions, probably dueto the increase of rare taxa under particular conditions.
On the other hand, the elimination of UV radiation in theUVmin treatment allowed the proliferation of organismsthat are negatively affected by UV, increasing the evennessof the community. However, it is possible that this alsoincreased competition, reducing species richness. The lattercan also be explained by a reduction in the number ofphotosynthetic organisms and a concomitant reduction inprimary production [3]. In particular, a proliferation ofAlphaproteobacteria was observed, and even some groupsin this phylum (Starkeya, Stappia) were not present in theother samples.
The high temperature in the 40C treatment increased thedominance in the community. A group of heat-tolerant bac-teria proliferated in the community, as other competitorsfound their metabolic capacities affected. The most affectedorganisms were Actinobacteria, to the point where nosequences from this phylum were recovered. This was sur-prising, since a previous work has reported numerous cul-turable thermostable Actinobacteria in CCB [12]. It may bethat Actinobacteria are sensitive to increased temperatures,forming spores, which would readily germinate when platedin culture media. This is also consistent with the hypothesisthat the widespread distribution of Actinobacteria in fresh-water lakes is a result of their resistance structures beingblown and dispersed by the wind [42]. In addition, weobserved that while numerous cyanobacterial sequenceswere recovered in this treatment, diversity within this phylum
354 S. Pajares et al.

was strongly reduced to only members of Chroococcales.Hence, the higher temperature could impair carbon fixationand primary production.
As expected, the most drastic reduction in diversity wasobserved in the UVplus mesocosms, leading to dominanceof two phyla that together comprise more than 90 % of thecommunity. Most notably, no Cyanobacteria were detectedwith 16S rRNA universal primers, indicating a dramaticreduction of highly efficient oxygenic photoautotrophs. Alarge proportion of Actinobacteria was observed in thistreatment. A correlation between the actinobacterial propor-tion in freshwater lakes communities and UV transparency[61] suggests that Actinobacteria may be favored in highUV environments. This could be explained due to: (1) alower rate of TT dimer formation because of their highGC content [7, 38], (2) cytoplasmic protection to UV bymeans of a thicker gram-positive cell wall [31], and (3)pigment production protection in many actinobacterialstrains [23, 24]. Our results support this hypothesis, sincethe UVplus treatment had the highest proportion of Actino-bacteria. Nevertheless, the widespread distribution of Acti-nobacteria in freshwater lakes might also be explained bythe formation of spores for dispersal [42]; their ability tocompete via inhibitory secondary products or other environ-mental variables correlated with UV incidence [60]. Moreover,the existence of abundant and widespread actinobacterialrhodopsins [53] has led to the suggestion that these organ-isms may be involved in metabolisms tightly connected tolight, and perhaps even novel photosynthetic metabolisms[21, 53]. This is also supported by our results, since a modi-fication of illumination regimes strongly affected the propor-tion of Actinobacteria.
Porphyrobacter is a freshwater Alphaproteobacteriagroup that increased its abundance in UVplus treatment. Itis a chemo-organotrophic genus that synthesizes bacterio-chlorophyll under aerobic conditions and produces reddish-orange pigmented colonies [18]. The pigmentation wouldprotect these bacteria from UV light damage by antioxidantactivity [44].
Two other major changes in the UVplus mesocosms, andshared with 40C mesocosms, were an increase in the abun-dance of Phaeospirillum (Rhodospirillales) and Sphingo-pyxis (Sphingomonadales). Both Sphingopyxis andPhaeospirillum assimilate ammonia via the NADH-dependent glutamine synthetase/glutamate synthase pathway,suggesting that rapid ammonia incorporation is a factor pro-moting their dominance under stressed conditions. Sphingo-pyxis is a genus of ultramicrobacteria with manyadaptations to survive under oligotrophic conditions [35,45] and stress [11], including an elevated resistance to UV[33] and the capacity to degrade complex organic compounds[29]. The optimal growth temperature for Phaeospirillum is38 °C, although this genus may survive temperatures of up to
42 °C [25], therefore Phaeospirillum may have adapted tothe temperature increase of 40 °C. The dominance ofPhaeospirillum in treatments UVplus and 40C is of greatimportance since it is the most abundant known photosyn-thetic organism in those mesocosms, especially in theUVplus treatment in which Cyanobacteria were observed tobe at low abundance.
The photosynthetic pigments in Phaeospirillum, bacte-riochlorophyll a and the carotenoid spirilloxanthin [30],were probably important in the UVplus treatment meso-cosms. Bacteriochlorophyll a absorbs larger wavelengthsthan chlorophyll near the infrared (830–890 nm), while thecarotenoid spirilloxanthin has two absorption peaks near theultraviolet (380 and 494 nm) [17], thus protecting the cellfrom UV while allowing it to continue energetic metabo-lism. Most primary production in UVplus mesocosms wasthen likely depended on this group of anaerobic or micro-aerophillic purple bacteria. Thus, a simultaneous increase inboth temperature and UV caused a major shift in primaryproductivity from a very efficient oxygenic photosynthesisdriven by Cyanobacteria towards a slower, less efficient pho-toheterotrophic anoxygenic production mostly driven by pur-ple bacteria.
Cyanobacteria have traditionally been recognized as afundamental component of freshwater communities andplay a key part in nutrient cycling in lakes [10]. Here, theinitial community (T0) was dominated by Chroococcales,the most abundant order in freshwater lakes [42]. Thisgroup, which provides most of the primary production inall aquatic ecosystems, was the most affected by the envi-ronmental changes explored in this study. While the Controltreatment had an even abundance of Chroococcales, Oscil-latoriales, and uncharacterized Cyanobacteria, the Flucttreatment shifted abundances towards Prochlorales. UnderUVmin conditions, the cyanobacterial community was dom-inated by Chroococcales and uncharacterized Cyanobacte-ria. The high temperature treatment dramatically eliminateddiversity and only Chroococcales persist. The cyanobacte-rial community was the most impacted under UVplus treat-ment, since the relative proportion of these organismsrecovered with the 16S rRNA universal primers (that givea measure of the global proportion of the microbial commu-nity) almost disappeared in the community. This suggeststhat there was insufficient time for these organisms to accli-mate to an increase in UV radiation. According to Ross andVincent [51], the speed of this acclimatization depends onbiosynthetic rates and is also affected by temperature. Cya-nobacterial primers recovered some sequences from Chroo-coccales and an unknown cyanobacterial group, whichmeans that we were able to detect these groups using par-ticular primers for Cyanobacteria, but they were actuallyrare in this treatment. In any case, the few remaining Cya-nobacteria in UVplus mesocosms were most likely resistant
Bacterial Community Response to Stress 355

to the damaging effects of UV radiation throughout diversemechanisms [59]: escape through migration, screening byUV-absorbing pigments, quenching of reactive oxygen spe-cies (i.e., by carotenoids, peroxidases, catalases), and dam-age repair mechanisms.
Conclusion
This mesocosms experiment depicts how microbial commu-nities respond to environmental stress in a short time period.The aquatic microbial communities in this experimentshowed a definite response to environmental stress by shiftsin community composition and structure. Diversity wasreduced in the extreme treatments, meaning that increasesin both temperature and UV incidence can translate to areduction of richness and increased dominance. This couldbe caused because the acclimatization time to the newenvironments for some groups of organisms was insuffi-cient, as could be the case with Cyanobacteria. Interestingly,the UV treatment showed resistant bacterial strains to UVradiation. Physiological traits, such as carotenoids produc-tion and other “sunscreen” molecules, and polysaccharidescould be factors determining the resistance of bacteria to UVradiation. Our data highlight the necessity to investigate thephysiology and ecology of various uncultured, and dominantand responsive aquatic community members of this uniqueecosystem. This type of experiment will help us to understandthe resilience and response of aquatic ecosystems to human-induced environmental changes, and underscores the utility ofmicrobial model systems for answering ecological questionsthat can be difficult to address using field systems.
Acknowledgments This work was done with the grants to VS: SEPCONACyT 50705, SEMARNAT 0023459. and WWF-Alianza CarlosSlim OL039. SP had a postdoctoral scholarship from CSIC (Spain). VSand LEE worked on the manuscript during their sabbatical leave with aDGAPA support for VS and LEE and UC-Mexus/Conacyt support forLEE. We thank particularly R. González Chauvet, M. Rodríguez, C.Alonso, and A. Uscanga, as well as people from the Molecular andExperimental Evolution Laboratory (UNAM) for the technical assis-tance in the field, L. Espinosa Asuar for the technical assistance in thelaboratory, L. Falcón for the methodological advise, and C. Rooks forreading the manuscript. We specially thank PRONATURA Noreste forthe access to the Pozas Azules ranch, the office of APFF of CuatroCienegas for their constant support, and the Hotel Marielena forproviding long-term housing and support for the experiment.
References
1. Adams HE, Crump BC, Kling GW (2010) Temperature controls onaquatic bacterial production and community dynamics in arcticlakes and streams. Environ Microbiol 12:1319–1333
2. Alcaraz LD, Olmedo G, Bonilla G, Cerritos R, Hernández G(2008) The genome of Bacillus coahuilensis reveals adaptationsessential for survival in the relic of an ancient marine environment.Proc Natl Acad Sci U S A 105:5803–5808
3. Alonso-Sáez L, Gasol JM, Lefort T, Hofer J, Sommaruga R (2006)Effect of natural sunlight on bacterial activity and differentialsensitivity of natural bacterioplankton groups in northwesternMediterranean coastal waters. Appl Environ Microbiol 72:5806–5813
4. Arrieta JM, Weinbauer MG, Herndl G (2000) Interspecific vari-ability in sensitivity to UV radiation and subsequent recovery inselected isolates of marine bacteria. Appl EnvironMicrobiol 66:1468–1473
5. Azam F, Malfatti F (2007) Microbial structuring of marine ecosys-tems. Nat Rev Microbiol 5:782–791
6. Bell T, Newman JA, Silverman BW, Turner SL, Lilley AK (2005)The contribution of species richness and composition to bacterialservices. Nature 436:1157–1160
7. Bentley SD, Parkhill J (2004) Comparative genomic structure ofprokaryotes. Annu Rev Genet 38:771–791
8. Bertilsson S, Eiler A, Nordqvist A, Jørgensen NOG (2007) Linksbetween bacterial production, amino-acid utilization and commu-nity composition in productive lakes. ISME J 1:532–544
9. Breitbart M, Hoare A, Nitti A et al (2009) Metagenomic and stableisotopic analyses of modern freshwater microbialites in CuatroCienegas, Mexico. Environ Microbiol 11:16–34
10. Callieri C (2007) Picophytoplankton in freshwater ecosystems: theimportance of small-sized phototrophs. Freshwat Rev 1:1–28
11. Cavicchioli R, Ostrowski M, Fegatella F, Goodchild A, Guixa-Boixereu N (2003) Life under nutrient limitation in oligotrophicmarine environments: an eco/physiological perspective of Sphin-gopyxis alaskensis (formerly Sphingomonas alaskensis). MicrobEcol 45:203–217
12. Cerritos R, Eguiarte LE, Avitia M et al (2011) Diversity of cultur-able thermo-resistant aquatic bacteria along an environmental gradientin Cuatro Cienegas, Coahuila, Mexico. Antonie Van Leeuwenhoek99:303–318
13. Chao A, Chazdon RL, Colwell RK, Shen TJ (2005) A new statis-tical approach for assessing compositional similarity based onincidence and abundance data. Ecol Lett 8:148–159
14. DeSantis TZ, Dubosarskiy I, Murray SR, Andersen GL (2003)Comprehensive aligned sequence construction for automated de-sign of effective probes (CASCADE-P) using 16S rDNA. Bioin-formatics 19:1461–1468
15. Desnues C, Rodriguez-Brito B, Rayhawk S et al (2008) Biodiver-sity and biogeography of phages in modern stromatolites andthrombolites. Nature 452:340–343
16. Escalante AE, Eguiarte LE, Espinosa L et al (2008) Diversity ofaquatic prokaryotic communities in the Cuatro Cienegas basin. FEMSMicrobiol Ecol 65:50–60
17. Fuchs BM, Spring S, Teeling H et al (2007) Characterization of amarine gammaproteobacterium capable of aerobic anoxygenicphotosynthesis. Proc Natl Acad Sci U S A 104:2891
18. Fuerst JA, Hawkins JA, Holmes A et al (1993) Porphyrobacterneustonensis gen. nov., sp. nov., an aerobic bacteriochlorophyll-synthesizing budding bacterium from fresh water. Int J Syst Bacteriol43:125–134
19. García-Pichel F (1994) A model for the internal self-shading inplanktonic organisms and its implications for the usefulness ofultraviolet sunscreens. Limnol Oceanogr 39:1704–1717
20. Glöckner FO, Zaichikov E, Belkova N et al (2000) Comparative16S rRNA analysis of lake bacterioplankton reveals globally dis-tributed phylogenetic clusters including an abundant group ofactinobacteria. Appl Environ Microbiol 66:5053–5065
21. González JM, Fernández-Gómez B, Fernández-Guerra A et al(2008) Genome analysis of the proteorhodospin-containing marine
356 S. Pajares et al.

bacterium Polaribacter sp. Proc Natl Acad Sci U S A 105:8724–8729
22. Green SJ, Jahnke LL (2010) Molecular investigations and exper-imental manipulations of microbial mats: a view to paleomicrobialecosystems. In: Seckbach J, Oren A (eds) Microbial mats: modernand ancient microorganisms in stratified systems. Springer, Berlin,pp 185–208
23. Hahn MW (2009) Description of seven candidate species affiliatedwith the phylum Actinobacteria, representing planktonic freshwa-ter bacteria. IJSEM 59:112–117
24. Hahn MW, Lünsdorf H, Wu Q et al (2003) Isolation of novelultramicrobacteria classified as Actinobacteria from five freshwa-ter habitats in Europe and Asia. Appl Environ Microbiol 69:1442–1451
25. Hisada T, Okamura K, Hiraishi A (2007) Isolation and character-ization of phototrophic purple nonsulfur bacteria from Chloro-flexus and cyanobacterial mats in hot springs. Microbes Environ22:405–411
26. Hoehler TM, Bebout BM, Des Marais DJ (2001) The role ofmicrobial mats in the production of reduced gases on the early Earth.Nature 412:324–327
27. Hughes JB, Hellmann JJ (2005) The application of rarefactiontechniques to molecular inventories of microbial diversity. MethEnzymol 397:292–308
28. Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJ (2001)Counting the uncountable: statistical approaches to estimatingmicrobial diversity. Appl Environ Microbiol 67:4399–4406
29. Hutalle-Schmelzer KM, Zwirnmann E, Krüger A, Grossart HP(2010) Enrichment and cultivation of pelagic bacteria from ahumic lake using phenol and humic matter additions. FEMSMicrobiol Ecol 72:58–73
30. Imhoff JF (2005) The Proteobacteria, part C. The Alphaproteobac-teria family I. Rhodospirillaceae. In: Brenner DJ, Kreig NR, StaleyJT, Garrity GM (eds) Bergey’s manual of systematic bacteriology,2nd edn. Springer, New York, p 32
31. Jagger J (1983) Physiological effects of near-ultraviolet radiationon bacteria. Photochem Photobiol Rev 7:1–75
32. Jessup CM, Kassen R, Forde SE et al (2004) Big questions, smallworlds: microbial model systems in ecology. Trends Ecol Evol19:189–197
33. Joux F, Jeffrey WH, Lebaron P, Mitchell DL (1999) Marine bac-terial isolates display diverse responses to UVB radiation. ApplEnviron Microbiol 65:3820–3827
34. Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackenbrandt E,Goodfellow M (eds) Nucleic acid techniques in bacterial system-atics. Wiley, Chichester, pp 115–157
35. Lauro FM, McDougald D, Thomas T et al (2009) The genomicbasis of thropic strategy in marine bacteria. Proc Natl Acad Sci U SA 106:15527–15533
36. Letunic I, Bork P (2007) Interactive Tree Of Life (iTOL): an onlinetool for phylogenetic tree display and annotation. Bioinform23:127–128
37. Loreau M, Hector A (2001) Partitioning selection and complemen-tarity in biodiversity experiments. Nature 412:72–76
38. Mackenzie C, Chidambaram M, Sodergren EJ, Kaplan S, WeinstockGM (1995) DNA repair mutants of Rhodobacter sphaeroides. JBacteriol 177:3027–3035
39. Magurran AE (2004) Measuring biological diversity. Blackwell,Oxford, 256 p
40. MinckleyW (1969) Environments of the Bolson of Cuatro Cienegas,Cuahuila, Mexico, with special reference to the aquatic biota. Uni-versity of Texas, El Paso. Science Series 2:1–65
41. Moreno-Letelier A, Olmedo G, Eguiarte LE, Martínez-Castilla L,Souza V (2011) Parallel evolution and horizontal gene transfer ofthe pst operon in Bacillus from oligotrophic environments. Int JEvol Biol. doi:10.4061/2011/781642
42. Newton RJ, Jones SE, Eiler A, McMahon KD, Bertilsson S (2011)A guide to the natural history of freshwater lake bacteria. Micro-biol Mol Biol Rev 75:14–49
43. Nübel U, García-Pichel F, Muyzer G (1997) PCR primers to amplify16S rRNA genes from cyanobacteria. Appl Environ Microbiol63:3327–3332
44. Ordoñez OF, Flores MR, Dib JR, Paz A, Farías ME (2009)Extremophile culture collection from Andean lakes: extremepristine environments that host a wide diversity of microor-ganisms with tolerance to UV radiation. Microb Ecol 58:461–473
45. Ostrowski M, Cavicchioli R, Blaauw M, Gottschal JC (2001)Specific growth rate plays a critical role in hydrogen peroxideresistance of the marine oligotrophic ultramicrobacterium Sphin-gomonas alaskensis strain RB2256. Appl Environ Microbiol67:1292–1299
46. Pedrós-Alió C (2006) Marine microbial diversity: can it be deter-mined? Trends Microbiol 14:257–263
47. Peimbert M, Alcaraz LD, Hernández I et al (2012) Extreme Red-field ratios, metagenomic and microbial diversity analyses of aseasonal shallow red pool in Cuatro Cienegas Coahuila, Mexico.Astrobiology (in press)
48. Pettersson M, Baath E (2003) The rate of change of a soil bacterialcommunity after liming as a function of temperature. Microb Ecol46:177–186
49. Philosof A, Sabehi G, Béjà O (2009) Comparative analyses ofactinobacterial genomic fragments from Lake Kinneret. EnvironMicrobiol 11:3189–3200
50. Rényi A (1961) On measures of information and entropy. In: Proc4th Berkeley Symp on Math, Statist Prob. Univ California, pp547–561
51. Ross JC, Vincent WF (1998) Temperature dependence of UVradiation effects on antarctic cyanobacteria. J Phycol 34:118–125
52. Schloss PD, Westcott SL, Ryabin T et al (2009) Introducingmothur: open-source, platform-independent, community-supported software for describing and comparing microbial com-munities. Appl Environ Microbiol 75:7537–7541
53. Sharma AK, Sommerfeld K, Bullerjahn GS et al (2009) Acti-norhodopsin genes discovered in diverse freshwater habitatsand among cultivated freshwater Actinobacteria. ISME J 3:726–737
54. Souza V, Eguiarte LE, Siefert J, Elser JJ (2008) Microbial ende-mism: does phosphorus limitation enhance speciation? Nat RevMicrobiol 6:559–564
55. Souza V, Espinosa-Asuar L, Escalante AE et al (2006) An endan-gered oasis of aquatic microbial biodiversity in the Chihuahuandesert. Proc Natl Acad Sci U S A 103:6565–6570
56. Souza V, Siefert J, Escalante AE, Elser JJ, Eguiarte LE (2012) TheCuatro Cienegas Bolson in Coahuila, Mexico: an astrobiologicalPrecambrian park. Astrobiology (in press)
57. Szabo KE, Itor POB, Bertilsson S, Tranvik L, Eiler A (2007)Importance of rare and abundant populations for the structureand functional potential of freshwater bacterial communities.Aquat Microb Ecol 47:1–10
58. Tobler M, Carson EW (2010) Environmental variation, hybridiza-tion, and phenotypic diversification in Cuatro Cienegas pupfishes.J Evol Biol 23:1475–1489
59. Vincent WF, Quesada A (1997) Microbial niches in the polarenvironment and the escape from UV radiation in non-marinehabitats. In: Battaglia B, Valencia J, Walton D (eds) Antarcticcommunities: species, structure and survival. Cambridge Univer-sity Press, Cambridge, pp 388–395
60. Warnecke F, Amann R, Pernthaler J (2004) Actinobacterial 16SrRNA genes from freshwater habitats cluster in four distinct line-ages. Environ Microbiol 6:242–253
Bacterial Community Response to Stress 357

61. Warnecke F, Sommaruga R, Sekar R, Hofer JS, Pernthaler J (2005)Abundances, identity, and growth state of Actinobacteria in moun-tain lakes of different UV transparency. Appl Environ Microbiol71:5551–5559
62. Winter C, Moeseneder MM, Herndl GJ (2001) Impact of UVradiation on bacterioplankton community composition. Appl En-viron Microbiol 67:665–672
63. Yabuuchi E, Kosako Y (2005) Order IV. Sphingomonadales ord.nov. In: Brenner DJ, Kreig NR, Staley JT, Garrity GM (eds) Bergey’smanual of systematic bacteriology, vol 2, 2nd edn. Springer, NewYork, pp 230–233
64. ZwartG,CrumpBC,AgterveldMPKW,HagenF,HanSK (2002)Typicalfreshwater bacteria: an analysis of available 16S rRNA gene sequen-ces from plankton of lakes and rivers. Aquat Microb Ecol 28:141–155
358 S. Pajares et al.




















