
Mechanism of 4-HPR-induced apoptosis in glioma cells: evidences suggesting role of...
12
Mechanism of 4-HPR-induced apoptosis in glioma cells: evidences suggesting role of mitochondrial-mediated pathway and endoplasmic reticulum stress Meenakshi Tiwari, Ashok Kumar, Rohit Anthony Sinha, Ashutosh Shrivastava, Anil Kumar Balapure 1 , Ramesh Sharma 1 , Virendra Kumar Bajpai 2 , Kalyan Mitra 2 , Satish Babu and Madan Madhav Godbole Department of Endocrinology, Sanjay Gandhi Post Graduate Institute of Medical Sciences, Raebareli Road, Lucknow, 226 014, India and 1 Tissue Culture Laboratory and 2 Electron Microscopy Division, Central Drug Research Institute, Mahatma Gandhi Marg, Lucknow, 226 001, India To whom correspondence should be addressed. Tel: +91 522 2668700 ext. 2368; Fax: +91 522 2668017; Email: [email protected] N-(4-Hydroxyphenyl)retinamide (4-HPR), a synthetic reti- noid is under clinical evaluation as a therapeutic agent in a variety of cancers. Its mechanism(s) of action involves mul- tiple overlapping pathways that still remain unclear. In glioma cells its mechanism of action is not well elucidated. Here, we show that 4-HPR and not all-trans retinoic acid and 9-cis retinoic acid effectively induce apoptosis in glioma cells. 4-HPR-induced apoptosis is associated with hydroperoxide production and loss of mitochondrial mem- brane potential (DC m ). Ultrastructural changes further indicate 4-HPR-induced mitochondrial swelling, endoplas- mic reticulum (ER) dilation as well as close proximity of mitochondria and ER. As suggested by dilated ER, 4-HPR treatment increased the free cytosolic Ca 2+ as well as mito- chondrial Ca 2+ . Chelation of extracellular Ca 2+ by EGTA did not prevent Ca 2+ elevation, thus suggesting involve- ment of intracellular calcium stores in the release. Buffer- ing of intracellular calcium by BAPTA-AM did not prevent 4-HPR-induced apoptosis; however, blocking the release of Ca 2+ from ER by heparin inhibited apoptosis, indicating the role of depletion of Ca 2+ from ER stores in apoptosis. 4-HPR treatment also resulted in an increase in Bax levels along with its translocation to mitochondria that promote mitochondrial membrane permeabilization. 4-HPR- induced apoptosis was further associated with the release of cytochrome c and apoptosis-inducing factor (AIF) from mitochondria to cytosol and nucleus, respectively, along with caspase-3 and caspase-7 activation. However, AIF nuclear translocation, peripheral chromatin condensation and apoptosis were not completely prevented by general caspase inhibitors, thus suggesting involvement of a caspase-dependent and caspase-independent pathway in 4-HPR-induced apoptosis. Taken together, these results suggest the role of mitochondrial-mediated pathway and ER stress as a key event in 4-HPR-induced apoptosis in glioma cells. Introduction Retinoids are a class of natural and synthetic compounds with pleiotropic effects including antitumor activity. Retinoids have demonstrated their ability to inhibit growth and induce differ- entiation in a variety of malignancies; however, many reti- noids like all-trans retinoic acid (ATRA) and 9-cis retinoic acid (9-cisRA), have limited therapeutic utility because of side effects (1,2). N-(4-Hydroxyphenyl)retinamide (4-HPR) is a synthetic retinoid that was developed as retinoic acid receptor (RAR) b and RAR-g selective agonist and has emerged as a potential chemotherapeutic agent owing to its effectiveness relative to toxicity in a variety of malignancies (3–5). Its anticancer activity is attributed to its ability to inhibit tumor cell growth and apoptosis induction. 4-HPR initiates cell-type- specific responses, and the apoptotic pathways involve various overlapping pathways (5). 4-HPR may act through RAR-dependent (3), RAR- independent (6), or both the pathways (7,8). 4-HPR leads to activation of caspases in a variety of cancer cells through intrinsic (9–11) as well as extrinsic pathway (12); recent report also suggests involvement of caspase-independent pathway (13). Modulations of members of Bcl-2 family proteins (11), p53-independent (14) and ceramide-mediated pathways (8) have been implicated in 4-HPR-induced apoptosis. Various reports have suggested that 4-HPR-induced apoptosis is predominantly associated with generation of reactive oxygen species (ROS) and loss of mitochondrial membrane potential (DY m ) (5,15–19). ROS levels and DY m are known to regulate intracellular Ca 2+ homoeostasis. Disruption of cellular Ca 2+ homeostasis has been proposed to be a critical event in both apoptosis and necrosis (20,21). However, the role of Ca 2+ in 4-HPR-induced apoptosis is not determined. Mitochondria play an important role during apoptosis by releasing various apoptotic proteins that leads to apoptosis induction. In 4- HPR-induced apoptosis, mitochondria play a central role by releasing cytochrome c from intermembrane space, which thereby initiates apoptosis by activating executioner caspases (9–11). Apoptosis-inducing factor (AIF), another such protein that resides in mitochondria and under various conditions, translocates to nucleus and results in apoptosis induction in a caspase-independent manner (22). AIF-mediated apoptosis has been reported in the case of synthetic retinoid CD437 in human bronchial epithelial cells (23); its role in 4-HPR- induced apoptosis needs to be evaluated. Further, growing evidence suggests that besides mitochondria endoplasmic reticulum (ER) are also a major point of integration for damage Abbreviations: DY m , mitochondrial membrane potential; 4-HPR, N-(4- hydroxyphenyl)retinamide; 9-cis RA, 9-cis retinoic acid; AA, ascorbic acid; AIF, apoptosis-inducing factor; ATRA, all-trans retinoic acid; BAPTA-AM, 1,2-bis(O-aminophenoxy)ethane-N,N,N,N-tetraacetic acid acetomethyl ester; CsA, cyclosporin A; DCF-DA, 2 0 ,7 0 -dichlorofluorescine diacetate; DMSO, dimethyl sulfoxide; EGTA, ethyleneglycol-bis(b-aminoethyl)-N,N,N 0 ,N 0 - tetraacetic acid; EM, electron microscopy; ER, endoplasmic reticulum; GSH, reduced glutathione; KRB, Krebs–Ringer buffer; L-NAME, N G -nitro-L- arginine methyl ester; MMP, mitochondrial membrane permeablization; MN, mannitol; MPT, membrane permeability transition; PBS, phosphate-buffered saline; PI, propidium iodide; RAR, retinoic acid receptor; RPA, rat primary astrocytes; ROS, reactive oxygen species; RXR, retinoic X receptor. Carcinogenesis vol.27 no.10 pp.2047–2058, 2006 doi:10.1093/carcin/bgl051 Advance Access publication May 4, 2006 # The Author 2006. Published by Oxford University Press. All rights reserved. For Permissions, please email: [email protected] 2047 by guest on October 10, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 10, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 10, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 10, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 10, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 10, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 10, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 10, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 10, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 10, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 10, 2013 http://carcin.oxfordjournals.org/ Downloaded from by guest on October 10, 2013 http://carcin.oxfordjournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Mechanism of 4-HPR-induced apoptosis in glioma cells: evidences suggesting role of...

Mechanism of 4-HPR-induced apoptosis in glioma cells: evidences suggesting roleof mitochondrial-mediated pathway and endoplasmic reticulum stress
Meenakshi Tiwari, Ashok Kumar, Rohit Anthony Sinha,Ashutosh Shrivastava, Anil Kumar Balapure1,Ramesh Sharma1, Virendra Kumar Bajpai2,Kalyan Mitra2, Satish Babu and MadanMadhav Godbole�Department of Endocrinology, Sanjay Gandhi Post Graduate Institute ofMedical Sciences, Raebareli Road, Lucknow, 226 014, India and 1TissueCulture Laboratory and 2Electron Microscopy Division, Central DrugResearch Institute, Mahatma Gandhi Marg, Lucknow, 226 001, India
�To whom correspondence should be addressed. Tel: +91 522 2668700ext. 2368; Fax: +91 522 2668017;Email: [email protected]
N-(4-Hydroxyphenyl)retinamide (4-HPR), a synthetic reti-noid is under clinical evaluation as a therapeutic agent in avariety of cancers. Its mechanism(s) of action involves mul-tiple overlapping pathways that still remain unclear. Inglioma cells its mechanism of action is not well elucidated.Here, we show that 4-HPR and not all-trans retinoic acidand 9-cis retinoic acid effectively induce apoptosis inglioma cells. 4-HPR-induced apoptosis is associated withhydroperoxide production and loss of mitochondrial mem-brane potential (DCm). Ultrastructural changes furtherindicate 4-HPR-induced mitochondrial swelling, endoplas-mic reticulum (ER) dilation as well as close proximity ofmitochondria and ER. As suggested by dilated ER, 4-HPRtreatment increased the free cytosolic Ca2+ as well as mito-chondrial Ca2+. Chelation of extracellular Ca2+ by EGTAdid not prevent Ca2+ elevation, thus suggesting involve-ment of intracellular calcium stores in the release. Buffer-ing of intracellular calcium by BAPTA-AM did not prevent4-HPR-induced apoptosis; however, blocking the releaseof Ca2+ from ER by heparin inhibited apoptosis, indicatingthe role of depletion of Ca2+ from ER stores in apoptosis.4-HPR treatment also resulted in an increase in Bax levelsalong with its translocation to mitochondria that promotemitochondrial membrane permeabilization. 4-HPR-induced apoptosis was further associated with the releaseof cytochrome c and apoptosis-inducing factor (AIF) frommitochondria to cytosol and nucleus, respectively, alongwith caspase-3 and caspase-7 activation. However, AIFnuclear translocation, peripheral chromatin condensationand apoptosis were not completely prevented by generalcaspase inhibitors, thus suggesting involvement of a
caspase-dependent and caspase-independent pathway in4-HPR-induced apoptosis. Taken together, these resultssuggest the role of mitochondrial-mediated pathway andER stress as a key event in 4-HPR-induced apoptosis inglioma cells.
Introduction
Retinoids are a class of natural and synthetic compounds withpleiotropic effects including antitumor activity. Retinoids havedemonstrated their ability to inhibit growth and induce differ-entiation in a variety of malignancies; however, many reti-noids like all-trans retinoic acid (ATRA) and 9-cis retinoicacid (9-cisRA), have limited therapeutic utility because of sideeffects (1,2). N-(4-Hydroxyphenyl)retinamide (4-HPR) is asynthetic retinoid that was developed as retinoic acid receptor(RAR) b and RAR-g selective agonist and has emerged as apotential chemotherapeutic agent owing to its effectivenessrelative to toxicity in a variety of malignancies (3–5). Itsanticancer activity is attributed to its ability to inhibit tumorcell growth and apoptosis induction. 4-HPR initiates cell-type-specific responses, and the apoptotic pathways involve variousoverlapping pathways (5).
4-HPR may act through RAR-dependent (3), RAR-independent (6), or both the pathways (7,8). 4-HPR leads toactivation of caspases in a variety of cancer cells throughintrinsic (9–11) as well as extrinsic pathway (12); recent reportalso suggests involvement of caspase-independent pathway(13). Modulations of members of Bcl-2 family proteins(11), p53-independent (14) and ceramide-mediated pathways(8) have been implicated in 4-HPR-induced apoptosis. Variousreports have suggested that 4-HPR-induced apoptosis ispredominantly associated with generation of reactive oxygenspecies (ROS) and loss of mitochondrial membrane potential(DYm) (5,15–19). ROS levels and DYm are known to regulateintracellular Ca2+ homoeostasis. Disruption of cellular Ca2+
homeostasis has been proposed to be a critical event in bothapoptosis and necrosis (20,21). However, the role of Ca2+ in4-HPR-induced apoptosis is not determined. Mitochondriaplay an important role during apoptosis by releasing variousapoptotic proteins that leads to apoptosis induction. In 4-HPR-induced apoptosis, mitochondria play a central role byreleasing cytochrome c from intermembrane space, whichthereby initiates apoptosis by activating executioner caspases(9–11). Apoptosis-inducing factor (AIF), another such proteinthat resides in mitochondria and under various conditions,translocates to nucleus and results in apoptosis induction ina caspase-independent manner (22). AIF-mediated apoptosishas been reported in the case of synthetic retinoid CD437 inhuman bronchial epithelial cells (23); its role in 4-HPR-induced apoptosis needs to be evaluated. Further, growingevidence suggests that besides mitochondria endoplasmicreticulum (ER) are also a major point of integration for damage
Abbreviations: DYm, mitochondrial membrane potential; 4-HPR, N-(4-hydroxyphenyl)retinamide; 9-cis RA, 9-cis retinoic acid; AA, ascorbic acid;AIF, apoptosis-inducing factor; ATRA, all-trans retinoic acid; BAPTA-AM,1,2-bis(O-aminophenoxy)ethane-N,N,N,N-tetraacetic acid acetomethyl ester;CsA, cyclosporin A; DCF-DA, 20,70 -dichlorofluorescine diacetate; DMSO,dimethyl sulfoxide; EGTA, ethyleneglycol-bis(b-aminoethyl)-N,N,N0,N0-tetraacetic acid; EM, electron microscopy; ER, endoplasmic reticulum;GSH, reduced glutathione; KRB, Krebs–Ringer buffer; L-NAME, NG-nitro-L-arginine methyl ester; MMP, mitochondrial membrane permeablization; MN,mannitol; MPT, membrane permeability transition; PBS, phosphate-bufferedsaline; PI, propidium iodide; RAR, retinoic acid receptor; RPA, rat primaryastrocytes; ROS, reactive oxygen species; RXR, retinoic X receptor.
Carcinogenesis vol.27 no.10 pp.2047–2058, 2006doi:10.1093/carcin/bgl051Advance Access publication May 4, 2006
# The Author 2006. Published by Oxford University Press. All rights reserved. For Permissions, please email: [email protected] 2047
by guest on October 10, 2013
http://carcin.oxfordjournals.org/D
ownloaded from
by guest on O
ctober 10, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
by guest on October 10, 2013
http://carcin.oxfordjournals.org/D
ownloaded from
by guest on O
ctober 10, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
by guest on October 10, 2013
http://carcin.oxfordjournals.org/D
ownloaded from
by guest on O
ctober 10, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
by guest on October 10, 2013
http://carcin.oxfordjournals.org/D
ownloaded from
by guest on O
ctober 10, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
by guest on October 10, 2013
http://carcin.oxfordjournals.org/D
ownloaded from
by guest on O
ctober 10, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
by guest on October 10, 2013
http://carcin.oxfordjournals.org/D
ownloaded from
by guest on O
ctober 10, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

sensing, pro-apoptotic signaling and caspase activation (24).Thus, the underlying mechanism(s) in 4-HPR-induced apop-tosis seems to remain unclear. Therefore, the present study wasconducted to get a better understanding of some of the possiblemechanisms involved in 4-HPR-induced apoptosis requiredfor its full clinical evaluation.
Malignant gliomas are aggressively growing brain tumorsthat are resistant to treatments including chemotherapy andother adjuvant therapies. Till date, chemotherapeutic agentfor malignant gliomas still needs to be identified. Betterefficacy of 4-HPR than natural retinoids has been shown inapoptosis induction in glioma cell lines that involves down-regulation of Bcl-2 and Bcl-XL and PARP cleavage (25–27).However, the mechanisms involved in glioma cells are notwell understood.
We investigated the molecular mechanism of 4-HPR-induced apoptosis in glioma cell lines, U87MG and U373MG.4-HPR-induced apoptosis was associated with ROS genera-tion, loss of DYm and disruption of Ca2+ homeostasis alongwith mitochondrial swelling and ER dilation. 4-HPR increasedBax expression and its translocation to mitochondria, leadingto release of cytochrome c and AIF. Downstream events fur-ther involved caspase-3 and caspase-7 activation; however,apoptosis also takes place in the presence of caspase inhibitorsvia AIF nuclear translocation. Taken together, our results sug-gest that 4-HPR-induced apoptosis involves two parallel path-ways emanating from mitochondria and also leads to ERstress.
Materials and methods
Cell lines and reagents
The human glioma cell lines U87MG (p53-wild-type) and U373MG (p53-mutant) were procured from National Center for Cell Science, Pune (India). 4-HPR, 5,50,6,60-tetrachlo-1-10,3,30tetraethylbenzimidazolylcarbocyanine iodide(JC-1) and caspase inhibitors were purchased from Calbiochem (San Diego,CA). ATRA, 9-cisRA, reduced glutathione (GSH), mannitol (MN), ascorbicacid (AA), NG-nitro-L-arginine methyl ester (L-NAME), cyclosporin A (CsA),dimethyl sulfoxide (DMSO), 20,70-dichlorofluorescine diacetate (DCF-DA),ethyleneglycol-bis(b-aminoethyl)-N,N,N0,N0-tetraacetic acid (EGTA), 1,2-bis(O-aminophenoxy)ethane-N,N,N,N-tetraacetic acid acetomethyl ester(BAPTA-AM), heparin (low molecualr weight sodium salt) and propidiumiodide (PI) were purchased from Sigma Chemicals (St Louis, MO). Fluo-3AM,Hoechst 33258 and Mitotracker Red CM-H2Xros were from Molecular Probes(Eugene, OR). ApoAlert caspase-3 detection kit was from Clontech (Palo Alto,CA). Retinoids were dissolved in DMSO at a concentration of 10�2 M andwere stored in aliquots at �80�C. Other chemicals, drugs, probes and stainsused in this work were of molecular biological grade.
Cell culture and drug treatment
Rat primary astrocytes (RPA) were developed from neonatal rat cerebellumwith institutional animal ethics committee permission. The single cells wereobtained by mechanical disruption and cultured in Dulbeco’s modified Eagle’smedium with fetal calf serum (FCS) (10%). Glial origin of cells was confirmedby glial fibrillary acidic protein staining (Sigma).
U87MG and U373MG cells were cultured in Minimal Essential Medium(MEM) with non-essential amino acids, containing glutamine (4 mM), andpenicillin–streptomycin (50 IU/ml) with FCS (10%), at 37�C in a humidifiedatmosphere of 5% CO2. For experimental purposes, exponentially growingcells were seeded at the densities that allowed untreated cells to reach a nearlyconfluent state (�70–80%) at the end of the experiment. Cells were treatedwith drugs, 24 h of plating in fresh MEM. Retinoids were replenished after 48h. Untreated cells received the same amount of DMSO (0.1%) as the treatedcells and were used as control.
Cell viability assay
The cytotoxic effect of retinoids on glioma cell lines was determined bycounting the viable cells with trypan blue staining. Briefly, cells (2 · 104
cells/well) were seeded in 24-well plate. After 24 h of plating the cells
were exposed to various concentrations of retinoids for 72 h, and both attachedand non-attached cells were collected and resuspended in phosphate-bufferedsaline (PBS). Viable cells were counted using a Coulter counter after stainingwith trypan blue dye. The IC50 concentrations were determined from concen-tration response curve. For further experiments, unless specified otherwise,IC50 concentrations were used.
Flow cytometry for quantification of apoptosis
Cells were harvested and fixed in 70% ethanol at �20�C. Pelleted cells werestained for 1 h in 0.5 ml of staining solution (40 mg/ml PI, 0.5% Triton X-100and 0.1 mg/ml RNase-A in PBS). PI fluorescence was detected in FL-2 mode ina FACScan (Becton Dickinson, San Jose, CA). Approximately 10 000 events(cells) were evaluated from each sample. The sub-G1 fraction (apoptotic) wasestimated by gating hypodiploid cells in the DNA histogram using non-apoptotic population (cells treated with DMSO) as a reference to comparewith treated cells. Data were analyzed as single parameter frequency histogramusing cell Quest Alias software.
DNA fragmentation
Cells (�106) were pelleted and re-suspended in hypotonic buffer [25 mM Tris–HCl (pH 7.4), 25 mM EDTA and 0.5% Triton X-100] for 30 min on ice. Aftercentrifugation at 10 000 g, 0.1 mg/ml RNase-A was added to supernatant for2 h followed by 0.1 mg/ml of proteinase-K for 4 h at 37�C. DNA was extractedusing phenol–chloroform and then precipitated with ethanol and sodiumacetate. Pelleted DNA was dissolved in Tris–EDTA (pH 8.0) and elec-trophoresed on a 1.8% agarose gel in the presence of ethidium bromide(0.5 mg/ml).
Measurement of ROS and Dcm
Intracellular ROS generation and DYm were measured using DCF-DA andJC-1 (28), respectively. Briefly, cells were plated in a six-well plate. One hourbefore the cells were harvested, DCF-DA (10 mM), or 15 min before, JC-1(5 mM), was added directly to the culture medium at 37�C in dark. Then, thecells were harvested in PBS and analyzed immediately by FACScan equippedwith a 488 nm laser. Forward and side scatter were used to gate the viablepopulation of cells. DCF-DA emit at 530 nm (FL-1) channel, whereas JC-1emit at two wavelengths: JC-1 monomer at 527 nm (FL-1 channel), indicatingcells with low DYm, and J-aggregate emit at 590 nm (FL-2 channel), indicatingcells with high DYm.
Assay for measuring free cytosolic calcium [Ca2+]c
Free cytosolic calcium [Ca2+]c was measured using cell permeant Ca2+-sensitive fluorescent dye Fluo-3AM (29). Wherever indicated, EGTA(1 mM), BAPTA-AM (10 mM) and heparin (5 mg/ml) were added to theculture medium for 24, 4 and 12 h, respectively, before the loading withFluo-3AM. The medium was removed from the tissue culture plates andreplaced with 4 mM Fluo-3AM diluted in Krebs–Ringer buffer (KRB)[10 mM D-glucose, 120 mM NaCl, 4.5 mM KCl, 0.7 mM Na2HPO4,1.5 mM NaH2PO4 and 0.5 mM MgCl2 (pH 7.4 at 37�C)] for 20 min. Thedishes were washed once with 5 ml KRB to remove the residual dye. Thecultures were then treated for the indicated times with 4-HPR or the vehiclediluted in KRB including 2 mM CaCl2. The cells were then harvested bytrypsinization, washed with 5 ml of Ca2+-free PBS at 37�C and analyzedimmediately for Fluo-3AM fluorescence intensity at 530 nm (FL-1) byflow cytometry. Forward and side scatter were used to gate the viablepopulation of cells.
To localize mitochondria Ca2+, Fluo-3AM-loaded live cells were incubatedwith Mitotracker Red (200 nM) for 45 min and visualized under UV-fluorescence microscope.
Loading of heparin inside the cells to inhibit IP3R-mediated Ca2+ releasewas performed by cytoplasmic hypo-osmotic pinocytic lysis procedure (30).Briefly, cells were incubated in hypertonic solution polyethylene glycol 1000(10%, v/v), 0.5 M sucrose and heparin (5 mg/ml) for 5 min in MEM to a finalvolume of 1 ml to form heparin-containing pinocytic vesicles. Cells wereexposed to hypo-osmotic media (six parts MEM to four parts water) for2 min to promote pinocytic lysis and the distribution of heparin throughoutthe cytoplasm. Cells were further rinsed and cultured in normal MEM. Thisprocedure resulted in the loading of �90% cells as determined by cytoplasmiclocalization of fluorescent fluorescein isothiocyanate-dextran, and did notcontribute to cell toxicity (data not shown)
Immunofluorescence
Cells were grown on coverslips, were fixed in 4% paraformaldehyde for 30 minat 4�C and permeabilized with pre-chilled 0.2% Triton X-100/PBS. Primaryantibodies: mouse monoclonal against N-terminal of Bax YTH-6A7 (Trevigen,Gaithersburg, MD) and goat polyclonal anti-AIF E-1 (Santa Cruz Biotech-nology, Santa Cruz, CA). Before labeling with anti-Bax, cells were
M.Tiwari et al.
2048

permeabilized for 5 min with 0.0125% CHAPS/PBS to prevent artificialactivation of Bax. Secondary antibodies: FITC-conjugated immunoglobulins(DAKO, Denmark). Nuclei were counterstained with Hoechst-33258 (10 mg/ml) or PI (25 mg/ml), after incubation in 0.5% mg/ml RNase-A (Sigma). Cellswere photographed under conventional UV-fluorescence microscope.
Transmission electron microscopy
Cells were fixed in a mixture of 4% glutaraldehyde/2% parformaldehyde in0.1 M cacodylate buffer, pH 7.4, for 4 h at 4�C. Cells were washed and thesamples were post-fixed in 2% osmium tetroxide for 2 h. Cells were dehy-drated, embedded in epon/plastic mixture, sectioned using ultramicrotome(Leica ULTRACUT UCT) and stained using standard electron microscopy(EM) procedures. Specimens were photographed in an FET Tecnai-12 Twinelectron microscope at 80 kV.
Colorimetric assay for caspase-3 activation
Caspase-3-like activity was quantified by colorimetric assay according to themanufacturer’s protocol (Clontech, Palo Alto, CA). Briefly, 1 · 106 cells werelysed in 50 ml of lysis buffer, and supernatant was collected after centrifugationat 10 000 g for 10 min. Fifty microliter of the 2· reaction buffer/DTT mix wasadded to 50 ml of the cell lysate. Then, 5 ml of 1 mM of caspase-3 substrate(DEVD-pNA) was added to each sample and incubated at 37�C for 1 h.A parallel control reaction was set up that did not contain conjugated substrate.The samples were read at 405 nm in a microplate reader.
Western blot analysis
The cells were harvested after 24 and 48 h of treatment. We did not performwestern blotting of 72 h treated cells, as after 72 h majority of the cells weredetached and entered secondary necrosis and the cellular contents were lostfrom the cells. After washing twice with PBS, whole cell lysate was preparedby lysing cells by freeze-thaw in 50 mM phosphate buffer with proteaseinhibitor cocktail. For cytosolic fractions, cells were homogenized in buffer(20 mM HEPES–KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM sodium-EGTA and 1 mM dithiothretol, 250 mM sucrose and protease inhibitor cock-tail) using pyrex homogenizer type A pestle, 140 strokes. Supernatant wascollected by pelleting at 17 000 g for 20 min at 4�C. Assay of lactate dehy-drogenase and cytochrome oxidase confirmed the supernatant as cytosolicfraction and the pellet as mitochondrial fraction. Proteins were resolved on15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) (30 mg of cytochrome c) or 12% SDS–PAGE (50 mg for rest of theproteins) and transferred on nitrocellulose membrane in electrotransblot appar-atus (Amersham Biosciences, Buckinghamshire, UK). Membranes were incu-bated with primary antibody. Rabbit polyclonal anti-caspase-3, Bax, Bcl-xL,b-actin and mouse monoclonal anti-Bcl-2 antibodies were from Santa CruzBiotechnology; and rabbit polyclonal anti-capase-7 and mouse monoclonalanti-cytochrome c (7H8.2C12) antibodies were kind gifts from Dr Xiao MiuSung (University of Leicester, UK) and Dr R Jemmerson (University ofMinnesota, USA), respectively. Antibodies for RAR-b (No. 112), RAR-g(No. 453) and retinoic X receptor (RXR) a (No. 444) were kindly giftedby Dr P Chambon (IGBMC, De Strasbourg, France). The bands were detectedby using HRP-conjugated secondary antibodies to primary immunoglobulinusing enhanced chemiluminescence system (Amersham Biosciences).
Statistical analysis
Differences in mean were calculated using Student’s t-tests. P-value < 0.05was considered as statistically significant.
Results
Enhanced cytotoxic effect of 4-HPR over ATRA and 9-cisRAin glioma cell lines
The comparative cytotoxic effects of retinoids were studied onhigh-grade glioma cell lines U87MG and U373MG in com-parison with normal astrocytes; RPA developed from neonatalrat cerebellum.
We found a better cytotoxic potential of 4-HPR than ATRAand 9-CisRA in glioma cell lines as determined by cell via-bility assay (Figure 1A). The IC50 of 4-HPR in U87MG andU373MG cells was 10 and 15 mM, respectively. In contrast,ATRA and 9-cisRA showed a weak cytostatic effect and evenat higher concentration (50 mM) failed to induce a growthinhibitory effect on U87MG and U373MG cells similar to4-HPR. On RPA all the retinoids exerted weak cytostatic effect
(Figure 1A) with no cell death observed even at higher con-centration of 50 mM (data not shown), suggesting that gliomacells were more sensitive to retinoid-induced growth inhibitoryeffects in comparison with normal astrocytes.
At higher concentrations, that is, IC50 and above, 4-HPRinduced cell death by apoptosis in both the glioma cell linesbut not in normal astrocytes as determined by microscopicobservation of Hoechst-33258 nuclear staining. Typical fea-tures of apoptosis like condensed and fragmented nuclei wereobserved (data not shown). Further, to quantify the extent ofapoptosis, hypodiploid peak of PI-stained cells was measuredand DNA laddering pattern was also analyzed (Figure 1B andC). 4-HPR induced apoptosis at IC50 concentration in >50% ofthe glioma cells after 72 h of treatment along with a 180 bpladdering pattern. A similar extent of apoptosis was notachieved by ATRA and 9-cisRA even at higher concentration(50 mM).
4-HPR-induced apoptosis is associated with ROS generationand loss of DYm in glioma cell lines
Several characteristic early apoptotic events involve enhancedROS production and loss of DYm (31). One way by which4-HPR triggers apoptosis is by hydroperoxide generation andloss of DYm (15–19). To determine the association of ROS/hydroperoxide generation with apoptosis in glioma cells, theoxidation of DCF was monitored by flow cytometry(Figure 2A–C). To determine change in DYm, flow cytometricanalysis was performed using JC-1. An increase in JC-1 mono-mers (FL-1 intensity) indicating low DYm was expressed as thepercentage of cells with low DYm. The effect of 4-HPR aloneor in combination with antioxidants and CsA, a classicalinhibitor of mitochondrial membrane permeability transition(MPT), was studied. 4-HPR induces hydroperoxide productionin both the glioma cell lines at apoptosis inducing concentra-tion (Figure 2A). Since U87MG cells were more sensitive to4-HPR treatment than U373MG, time-course analysis ofhydroperoxide production was performed in U87MG cells(Figure 2B). 4-HPR significantly increased levels of hydroper-oxide (1.4-fold) within 60 min, which further increased withtime (3.2-fold after 6 h). Hydroperoxide production was notassociated with ATRA and 9-cisRA. In U87MG, pretreatment(12 h) with different antioxidants, namely GSH, MN, AA andL-NAME, effectively inhibited 4-HPR-induced hydroperoxideproduction (Figure 2C) and apoptosis (Figure 2D), suggestinginvolvement of hydroperoxide production in apoptosisinduction.
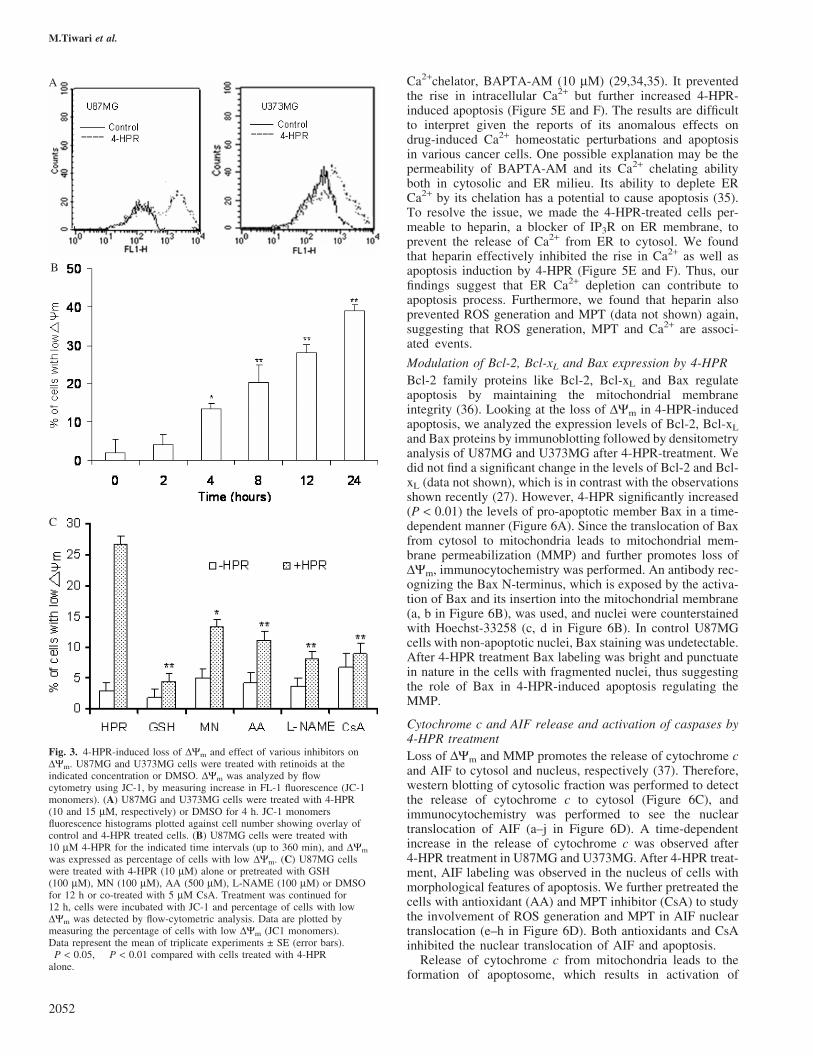
Since ROS production can be a cause and a consequence ofchange in DYm (32), effect of 4-HPR on loss of DYm wasassessed (Figure 3). 4-HPR induced loss of DYm in U87MGand U373MG cells (Figure 3A) as indicated by an increase inJC-1 monomer intensity (FL-H), suggesting loss of DYm. Inorder to study the onset of MPT, time-course analysis ofU87MG cells was performed by measuring the percentageof cells with low DYm (JC-1 monomer). We found that asignificant loss of DYm (�13.4% cells with low DYm) wasobserved only after 4 h of 4-HPR treatment, which furtherdiminished with time (Figure 3B). Since significant ROS gen-eration was observed within 1 h of 4-HPR treatment, loss ofDYm appears as a subsequent event in 4-HPR action.
We then examined the association between ROS generationand loss of DYm. U87MG cells were either pretreated (for 12 h)with various antioxidants or co-treated with CsA. Antioxidantseffectively prevented loss of DYm (Figure 3C), while CsA
Mechanism of 4-HPR-induced apoptosis in glioma cells
2049

effectively prevented ROS generation (Figure 2C) and subse-quent apoptosis (Figure 2D), suggesting that 4-HPR-inducedhydroperoxide generation and loss of DYm are related eventsand associated with apoptosis induction.
4-HPR promotes distinct cellular and subcellular alterationsin glioma cells
Since loss of DYm was associated with 4-HPR-induced apop-tosis, we were interested in the ultrastructural changes in mito-chondria and also other subcellular organelles like ER thatwould possibly provide additional clues to the underlyingstress mechanism. Thus, we performed EM of U87MG cellsexposed to 4-HPR for 48 h, the time when the cells are inexecution phase. Cells with representative characteristics arepresented in Figure 4. The control U87MG cells displayed awell-defined plasma membrane and nuclear envelope contain-ing nucleoli (Figure 4A). The mitochondria of these cells were
healthy with well-defined cristae (Figure 4C). Rough ER couldbe seen through the cytoplasm of the cell. A 48 h exposure to4-HPR led to cytoplasmic and nuclear shrinkage (Figure 4B).The nucleoli were absent and chromatin condensation wasevident along with clusters of vacuolization in the cytoplasm.A higher magnification confirmed that the larger vacuoleswere the remnants of the mitochondria, which appeared abnor-mally enlarged or swollen, along with accumulation ofelectron-dense material (Figure 4D) compared with thoseobserved in the control cells. The cristae were absent, andthe disruption of outer membrane integrity was evident.These mitochondrial changes appeared to be consistent withMPT induction. The ER was dilated and could also be iden-tified in close proximity to the mitochondria. The smallervacuoles surrounding the mitochondria presumably repre-sented what remained of the ER. Furthermore, in additionto chromatin condensation, the dilation of the ER appearedto be a common subcellular feature in 4-HPR exposed
B
A
C
Fig. 1. Effects of retinoids on cell viability and apoptosis induction in RPA and glioma cell lines U87MG and U373MG. (A) RPA, U87MG cells andU373MG cells were plated in six-well plates, cultured for 24 h, and then treated with indicated concentration of retinoids, 4-HPR, ATRA, 9-cisRA or vehicle(DMSO) for 72 h. Cell viability was determined by trypan blue staining. The data are expressed as % control cell viability, control denoting cells treatedwith DMSO alone. Average of triplicate samples are presented as ±SE (error bars). (B) For apoptosis assay, U87MG and U373MG cells were treated with4-HPR at toxic concentrations (10 mM for U87MG and 15 mM for U373MG), ATRA (50 mM), 9-cisRA (50 mM) or DMSO (control) for 72 h. Cells werestained with PI, and apoptotic fractions (sub-G1, hypodiploid peak) were quantified by flow cytometry using Cell-quest-alias Software. Data are expressed asaverage of % hypodiploid peak in quadruplicate experiments ± SE (error bars). �P � 0.05, ��P � 0.01; survival fractions of control versus treated cells.(C) Internucleosomal DNA laddering pattern was determined by agarose gel analysis. ATRA (lanes 2 and 6), 9-cisRA (lanes 3 and 7), 4-HPR (lanes 4and 8) or DMSO (Control, lanes 1 and 5), Lane M: 1 kb ladder.
M.Tiwari et al.
2050

cells, implying that ER stress was associated with apoptosisinduction.
Role of intracellular Ca2+ in 4-HPR-induced apoptosis
EM studies suggested the involvement of mitochondrial swel-ling and ER stress in 4-HPR-induced apoptosis. In cells, theER and mitochondria are the two major sites that are directlyinvolved in the storage and regulation of intracellular Ca2+
homeostasis (33). Although enhanced ROS generation andMPT are well associated with 4-HPR-induced apoptosis, analteration in Ca2+ homeostasis, which is a related event, has notbeen studied yet. Thus, we investigated whether 4-HPR couldincrease [Ca2+]c as a surrogate indicator of ER stress using theCa2+-sensitive dye Fluo-3AM by flow cytometry. 4-HPRincreased the Fluo-3-AM intensity in U87MG and U373MGcells (Figure 5A). Time-course analysis in U87MG cellsshowed a significantly increased [Ca2+]c within initial hoursof 4-HPR treatment, which further increased with time(Figure 5B), and was sustained with the DNA fragmentation(data not shown). We also determined the effect of antioxi-dants and MPT inhibitor on the alterations of [Ca2+]c
(Figure 5C). Both antioxidants and CsA could prevent therise in [Ca2+]c, suggesting the role of ROS and MPT in regu-lating intracellular Ca2+ levels (Figure 5C).
We further examined the Fluo-3AM-loaded cells that werecounter-stained with Mitotracker Red under fluorescencemicroscope, to investigate whether 4-HPR also increases themitochondrial Ca2+ levels. Results indicate that 4-HPR treat-ment not only increases [Ca2+]c but also increases mitochon-drial calcium uptake observed as yellowing of themitochondria due to overlay of green and red (Figure 5D).Further, the cells were permeabilized by exposure to 10 mMdigitonin added to the KRB to yield a free [Ca2+]c of 100 nMso as to measure mitochondrial calcium by flow cytometry,which suggested a delayed increase (�2-fold after 12 h) inmitochondrial Ca2+ after 4-HPR treatment (data not shown).
Effect of calcium chelators in 4-HPR-induced cell death
In order to determine the source of increased Ca2+, we pre-treated the cells with EGTA at a subtoxic concentration of1 mM to chelate the extracellular Ca2+ that brings extracellularCa2+ concentration in micromolar range (34). Chelation ofextracellular Ca2+ did not prevent the increase in [Ca2+]c
and subsequent apoptosis after 4-HPR treatment (Figure 5Eand F), suggesting involvement of intracellular Ca2+ stores for[Ca2+]c elevation. To test whether increase in [Ca2+]c isinvolved in progression of apoptosis, we used a cell permeant
A
B
C
D
Fig. 2. HPR-induced ROS generation and effect of various antioxidants on ROS generation and apoptosis. Glioma cells were treated with 4-HPR at theindicated concentration, retinoids or DMSO for indicated times. Then, 10 000 events were evaluated for ROS production by flow cytometry using DCF-DA.(A) U87MG and U373MG cells were treated with 4-HPR (10 and 15 mM, respectively) or DMSO for 4 h. DCF-fluorescence histograms showing overlay ofcontrol and 4-HPR treated cells. (B) Time-course analysis of U87MG cells treated with 4-HPR (10 mM), ATRA (50 mM), 9-cisRA (50 mM) or DMSO forthe indicated time intervals (up to 360 min). DCF fluorescence expressed as fold increase as compared with control. (C and D) U87MG cells were treatedwith 4-HPR (10 mM) alone or pretreated with GSH (100 mM), MN (100 mM), AA (500 mM), L-NAME (100 mM) or DMSO for 12 h or co-treated with5 mM CsA. (C) Treatment was continued for 6 h; cells were incubated with DCF-DA 1 h before harvestation. ROS generation was measured by flowcytometry. Data are expressed as fold increase in DCF levels obtained from three individual experiments ± SE (error bars). (D) Apoptosis was detected aspercentage of hypodiploid cells by flow cytometry after 72 h of treatment. Data represent the mean of triplicate experiments ± SE (error bars). �P < 0.05,��P < 0.01 compared with cells treated with 4-HPR alone.
Mechanism of 4-HPR-induced apoptosis in glioma cells
2051
Ca2+chelator, BAPTA-AM (10 mM) (29,34,35). It preventedthe rise in intracellular Ca2+ but further increased 4-HPR-induced apoptosis (Figure 5E and F). The results are difficultto interpret given the reports of its anomalous effects ondrug-induced Ca2+ homeostatic perturbations and apoptosisin various cancer cells. One possible explanation may be thepermeability of BAPTA-AM and its Ca2+ chelating abilityboth in cytosolic and ER milieu. Its ability to deplete ERCa2+ by its chelation has a potential to cause apoptosis (35).To resolve the issue, we made the 4-HPR-treated cells per-meable to heparin, a blocker of IP3R on ER membrane, toprevent the release of Ca2+ from ER to cytosol. We foundthat heparin effectively inhibited the rise in Ca2+ as well asapoptosis induction by 4-HPR (Figure 5E and F). Thus, ourfindings suggest that ER Ca2+ depletion can contribute toapoptosis process. Furthermore, we found that heparin alsoprevented ROS generation and MPT (data not shown) again,suggesting that ROS generation, MPT and Ca2+ are associ-ated events.
Modulation of Bcl-2, Bcl-xL and Bax expression by 4-HPR
Bcl-2 family proteins like Bcl-2, Bcl-xL and Bax regulateapoptosis by maintaining the mitochondrial membraneintegrity (36). Looking at the loss of DYm in 4-HPR-inducedapoptosis, we analyzed the expression levels of Bcl-2, Bcl-xL
and Bax proteins by immunoblotting followed by densitometryanalysis of U87MG and U373MG after 4-HPR-treatment. Wedid not find a significant change in the levels of Bcl-2 and Bcl-xL (data not shown), which is in contrast with the observationsshown recently (27). However, 4-HPR significantly increased(P < 0.01) the levels of pro-apoptotic member Bax in a time-dependent manner (Figure 6A). Since the translocation of Baxfrom cytosol to mitochondria leads to mitochondrial mem-brane permeabilization (MMP) and further promotes loss ofDYm, immunocytochemistry was performed. An antibody rec-ognizing the Bax N-terminus, which is exposed by the activa-tion of Bax and its insertion into the mitochondrial membrane(a, b in Figure 6B), was used, and nuclei were counterstainedwith Hoechst-33258 (c, d in Figure 6B). In control U87MGcells with non-apoptotic nuclei, Bax staining was undetectable.After 4-HPR treatment Bax labeling was bright and punctuatein nature in the cells with fragmented nuclei, thus suggestingthe role of Bax in 4-HPR-induced apoptosis regulating theMMP.
Cytochrome c and AIF release and activation of caspases by4-HPR treatment
Loss of DYm and MMP promotes the release of cytochrome cand AIF to cytosol and nucleus, respectively (37). Therefore,western blotting of cytosolic fraction was performed to detectthe release of cytochrome c to cytosol (Figure 6C), andimmunocytochemistry was performed to see the nucleartranslocation of AIF (a–j in Figure 6D). A time-dependentincrease in the release of cytochrome c was observed after4-HPR treatment in U87MG and U373MG. After 4-HPR treat-ment, AIF labeling was observed in the nucleus of cells withmorphological features of apoptosis. We further pretreated thecells with antioxidant (AA) and MPT inhibitor (CsA) to studythe involvement of ROS generation and MPT in AIF nucleartranslocation (e–h in Figure 6D). Both antioxidants and CsAinhibited the nuclear translocation of AIF and apoptosis.
Release of cytochrome c from mitochondria leads to theformation of apoptosome, which results in activation of
A
B
C
Fig. 3. 4-HPR-induced loss of DYm and effect of various inhibitors onDYm. U87MG and U373MG cells were treated with retinoids at theindicated concentration or DMSO. DYm was analyzed by flowcytometry using JC-1, by measuring increase in FL-1 fluorescence (JC-1monomers). (A) U87MG and U373MG cells were treated with 4-HPR(10 and 15 mM, respectively) or DMSO for 4 h. JC-1 monomersfluorescence histograms plotted against cell number showing overlay ofcontrol and 4-HPR treated cells. (B) U87MG cells were treated with10 mM 4-HPR for the indicated time intervals (up to 360 min), and DYm
was expressed as percentage of cells with low DYm. (C) U87MG cellswere treated with 4-HPR (10 mM) alone or pretreated with GSH(100 mM), MN (100 mM), AA (500 mM), L-NAME (100 mM) or DMSOfor 12 h or co-treated with 5 mM CsA. Treatment was continued for12 h, cells were incubated with JC-1 and percentage of cells with lowDYm was detected by flow-cytometric analysis. Data are plotted bymeasuring the percentage of cells with low DYm (JC1 monomers).Data represent the mean of triplicate experiments ± SE (error bars).�P < 0.05, ��P < 0.01 compared with cells treated with 4-HPRalone.
M.Tiwari et al.
2052

executioner caspases, namely caspase-3 and caspase-7 (38).Therefore, activation of caspase-3 and caspase-7 was assessedby colorimetric assay for caspase-3-like activity and westernblotting in U87MG and U373MG cells. 4-HPR treatmentinduced significant increase in caspase-3-like activity in atime-dependent manner (Figure 7A). A decrease in the pro-caspase-3 levels were also detected (a in Figure 7B), whichindicates an increase in its activated form and corroborateswith the increasing caspase-3-like activity. Further, an increasein the active caspase-7 subunit was detected after 4-HPR treat-ment (b in Figure 7B). The general and caspase-3-specificinhibitor, but not caspase-8 inhibitor, prevented the cellsfrom 4-HPR-induced intranucleosomal fragmentation; how-ever, peripheral chromatin condensation still took place in�40% of the cells, as assessed by nuclear morphology ofPI-stained cells (Figure 7C).
Presently, we also found that 4-HPR treatment leads tonuclear translocation of AIF (c, d in Figure 6D), and AIF isknown to induce apoptosis independent of caspases activity(22). Thus, we performed immunocytochemistry of AIF in thepresence of general caspase inhibitor. Results indicate that AIFtranslocation was not inhibited after treatment with generalcaspase inhibitors (i, j in Figure 6D); however, the nuclearapoptotic pattern differed. In the presence of general caspaseinhibitor, which prevented the generation of active caspase-3
subunit (data not shown), peripheral chromatin condensationin the absence of fragmentation was observed, thus indicatingthe involvement of both caspase-dependent and caspase-independent pathway 4-HPR-induced apoptosis in glioma cell.
4-HPR increased RAR-g and RXR-a expression
4-HPR shows a selective retinoid receptor activation profilethat induces transcriptional activation preferentially via RAR-g and to some extent by RAR-b, but not RAR-a, and inducesthe receptor expression (3). To determine if 4-HPR-mediatedsignals in glioma cultures resulted in increased expression ofRAR-b and RAR-g and its receptor partner, RXR-a, as adownstream effect, we assessed the expression of the receptorby western blotting (Figure 7D). Both the cell lines showed abasal level expression of all these receptors that significantlyincreased for RAR-g as well as for RXR-a in a time-dependentmanner at the same drug concentration at which apoptosiswas induced. The levels of RAR-b remained unchangedafter 4-HPR treatment.
Discussion
4-HPR has shown its chemotherapeutic potential in clinicaltrials in a variety of cancers (5). Although much interestand effort have been focused on the apoptotic potential of
Fig. 4. Assessment of 4-HPR-induced cellular and subcellular alterations by EM. Electron micrographs of U87MG cells (A and C), control cells and(B and D) U87MG cells exposed for 48 h to 10 mM 4-HPR are shown. The nucleus is identified by N, the mitochondria are indicated with the open arrowsand the ER is designated by the diamond arrows. The clusters of cytoplasmic vacuolization in the 4-HPR-treated U87MG cells (B) is circled. The scale barsin A and B equal 2 microns, and the scale bars in C and D equal 500 nm.
Mechanism of 4-HPR-induced apoptosis in glioma cells
2053

4-HPR as a means of eliminating cancer cells, the mechanismsunderlying 4-HPR-induced cell death are complex and stillremain elusive.
Gliomas are predominantly aggressively growing malignantbrain tumors and are associated with poor prognosis. Majorityof chemotherapeutic agents have shown negative outcomes inmalignant glioma (39). To improve the glioma treatment, a
better understanding of the molecular mechanism involved inthe drug action is required (40). The sensitivity or resistance ofcancer cells to various drugs depends on the activation ofvarious routes to apoptosis involving different signaling path-ways. The present study was performed to get a better insightinto the molecular pathway involved in 4HPR-induced apop-tosis using glioma cell lines U87MG and U373MG.
A B
C
E F
D
Fig. 5. HPR induced changes in calcium homeostasis in glioma cells. Glioma cells were treated as indicated, and levels of [Ca2+]c were determined by flowcytometry using Fluo-3AM. (A) U87MG and U373MG cells were treated with 4-HPR (10 and 15 mM, respectively) or DMSO for 4 h. Fluo-3AMfluorescence histograms showing overlay of control and 4-HPR-treated cells. (B) U87MG cells were treated with 4-HPR (10 mM), ATRA (50 mM), 9-cisRA(50 mM) or DMSO (control) for the indicated times, and then levels of [Ca2+]c were determined by flow cytometry. (C) U87MG cells were treated with4-HPR (10 mM) alone or pretreated with GSH (100 mM), MN (100 mM), AA (500 mM), L-NAME (100 mM) or DMSO for 12 h or co-treated with 5 mMCsA. Levels of [Ca2+]c were determined by flow cytometry. Data are expressed as fold increase in fluo-3AM intensity. (D) Fluorescent microscopy images ofU87MG cells treated with 4-HPR for 24 h were loaded with Fluo-3 (green fluorescence) and Miottracker Red (red fluorescence) and were excited at 488 nm.Images show the overlay of the two individual images. Green fluorescence shows increased [Ca2+]c, red localizes mitochondria, and colocalization of Ca2+
and mitochondria is represented in shades of yellow owing to overlay of green fluorescence with red. (E and F) U87MG cells were pretreated with DMSO(control), 1 mM EGTA for 24 h or 10 mM BAPTA-AM for 4 h or 5 mg/ml heparin for 12 h and then treated with 10 mM 4-HPR for indicated times in thepresence or absence of 1 mM EGTA or 10 mM BAPTA-AM, 5 mg/ml heparin. (E) Free [Ca2+]c was measured after 6 h of treatment by flow cytometry andexpressed as fold increase. (F) Apoptosis (hypodiploid peak) using PI staining after 72 h, by flow cytometry. Data are expressed as mean of triplicateexperiments ± SD (error bars). �P � 0.05 compared with cells treated with 4-HPR alone.
M.Tiwari et al.
2054

Here, we demonstrate that 4-HPR induced apoptosis inglioma cells, but not in normal astrocytes, suggesting its cancercell-specific action as shown previously (10). Results furtherdemonstrate that 4-HPR induced apoptosis through a similar
mechanism in both the glioma cell lines U87MG (p53-wild-type) and U373MG (p53-mutant), which differed in their p53status, thus supporting earlier findings (14) showing involve-ment of a p53-independent pathway.
4-HPR, which is a synthetic derivative of ATRA, inducesapoptosis in a wide variety of cancer cells as a major effectermechanism; however, ATRA and 9-cisRA are known toinduce differentiation and mediate distinct biological out-comes. 4-HPR seems to surmount the problems associatedwith natural retinoid-induced differentiation and increasedresistance to chemotherapy, owing to its apoptosis-inducingcapacity (5). In glioma cells, in comparison with 4-HPR, apop-tosis was not induced with ATRA and 9-cisRA, even at higherconcentration at which they showed growth inhibitory action.Our results further suggested that 4-HPR acts by a distinctmechanism from the one ascribed to ATRA and 9-cisRA byinvolving ROS generation, loss of DYm and elevation of Ca2+.
4-HPR has a known pro-oxidant capacity, even better thannewer synthetic retinoid, like SHetA2 (41). In glioma cells alsothe pro-oxidant capacity of 4-HPR was sustained andhydroperoxide production was associated with apoptosisinduction as suggested previously in a variety of cell types(9,10,17–19). 4-HPR also led to a loss of DYm as a subsequentevent to ROS generation, and both were associated events thatlead to apoptosis in glioma cells as shown earlier in metastaticsquamous cell carcinoma cells (15) and in human cervicalcarcinoma C33A cells (9).
During various conditions, ROS and MPT are often asso-ciated with Ca2+ homeostasis, and all three are cohort events.Any alteration therein plays an important role in apoptosisinduction (20,21). Chemotherapeutic drugs like triterpenoid-CDDO have been shown to act via a mechanism involvingmodulation of Ca2+ homeostasis leading to caspase activationand apoptosis (29). In the present study, we also determinedthe role of alteration in Ca2+ homeostasis in 4-HPR-inducedapoptosis. Here, we provide an illustration of a novel mecha-nism for the possible anticancer activity of 4-HPR. This is thefirst report indicating that 4-HPR induced rise in [Ca2+]c inglioma cells, which came from intracellular Ca2+ stores(Figure 5). We also found an association between Ca2+
increase, ROS generation and MPT. In the eukaryotic cells,the ER and mitochondria are the primary sites that areutilized for Ca2+ storage and the regulation of intracellular
A
B
C
D
Fig. 6. 4-HPR induces Bax translocation to mitochondria and releasescytochrome c and AIF from mitochondria to cytosol and nucleus,respectively. U87MG and U373 cells were treated with 4-HPR 10 mM and15 mM concentration, respectively, for the indicated times. Whole celllysate were prepared and the proteins were separated by SDS–PAGE forwestern blotting as described in Materials and methods for (A) Bax andb-actin showing loading control; (B) immunocytochemistry showing Baxactivation induced by 4-HPR treatment (shown in gray scale). Cells weretreated with 10 mM of 4-HPR for 48 h. Immunodetection of Bax with theprimary antibody directed against the N-terminal domain exposed after itsactivation (a, b) and Hoechst-33258 staining of nuclei (c, d) analyzed byUV-fluorescence microscopy: Bax is not visualized in cells with normalnuclei. Bright punctuate labeling of Bax indicative of activation andtranslocation in mitochondrial membranes is observed in cells withfragmented nucleus associated with apoptosis. (C) Cytochrome c release,determined in cytosolic fractions (see Materials and methods). (D)Immunolabeling of AIF after 48 h. AIF (green) is shown in the first panel(a, c, e, g, i); second panel (b, d, f, h, j) shows nuclei stained with PI (red).In control cells, AIF is present in the cytoplasm (in the mitochondria),whereas 4-HPR (10 mM) induced the translocation of AIF frommitochondria to the nucleus. The release of AIF from the mitochondriawas inhibited by AA (500 mM) and CsA (5 mM) and not by z-VAD FMK(50 mM) in cells with condensed nucleus.
Mechanism of 4-HPR-induced apoptosis in glioma cells
2055

Ca2+ homeostasis (21). The sustained Ca2+ release from the ERcan trigger Ca2+ uptake by the mitochondria, which possess anexceptional capacity to sequester Ca2+ and buffer local varia-tions in [Ca2+]c levels (21,37). Indeed, 4-HPR treatmentresulted in an increase in the mitochondria Ca2+ levels inglioma cells (Figure 5D). Furthermore, the ultrastructuralchanges also indicated the close proximity of the mitochondria
to the ER (Figure 4D). It has been suggested that close contactsexist between mitochondria and the sites of ER Ca2+ release,such that ER Ca2+ release leads to rapid Ca2+ accumulation inmitochondria, which may promote the loss of mitochondrialpermeability and further play an important role in apopto-sis induction (37). We further found that 4-HPR-inducedapoptosis in glioma cells was inhibited by heparin, which
0
A B
C
D
Fig. 7. Activation of caspases and RARs by 4-HPR. (A) Caspase-3-like activity in whole cell lysates, measured by using ApoAlert caspase-3 detection kit(clontech). Data are expressed as mean of triplicate experiments ± SE (error bars). (B) Pro-caspase-3 or caspase-7 (pro and active form) by western blotting.(C) U87MG cells and U373MG cells were pretreated with Z-VAD (50 mM), a general caspase inhibitor; Z-DEVD-fmk (50 mM), inhibitor for caspase-3; orZ-IETD-fmk (50 mM), caspase-8 inhibitor, for 4 h followed by treatment with 4-HPR (IC50) or were left untreated for 72 h. Percentage apoptosis wasdetermined by counting the apoptotic nuclei of PI-stained cells under microscope. Data are expressed as mean of triplicate experiments ± SD (error bars).�P � 0.05, ��P � 0.01 compared with cells treated with 4-HPR alone. (D) Western blotting for RAR-b, RAR-g , RXR-a and b-actin showing loadingcontrol. Experiment was repeated three times independently. Level of significance was drawn from densitometric analysis of the blots.
M.Tiwari et al.
2056

blocks the release of Ca2+ from ER, but not BAPTA-AM,which prevents the rise of [Ca2+]c. Thus, our results suggestan important role of ER Ca2+ pool depletion and its traffickingto cytosol and to mitochondria in 4-HPR-induced apoptosis.Furthermore, 4-HPR treatment resulted in ER vacuolization(Figure 4B). It is suggested that the ER Ca2+ pool depletionmay compromise its structural and functional integrity thatmay result in its vacuolization. As apoptosis is accompaniedby blebbing, it is likely that loss of ER integrity may direct thebleb formation through cytoskeleton rearrangement (42). Fur-thermore, the disruption of intracellular calcium homeostasisand/or ER stress can also promote the activation of caspase-12,which has been implicated in activation of caspases 8, 9 and 3(43). However, in the present study we did not study the role ofcaspase-12 activation that needs to be investigated.
Loss of DYm and MMP are early events in apoptosis (32).4-HPR induced loss of DYm, which is associated with apop-tosis in glioma cells, consistent with earlier studies showingthat 4-HPR acts directly on mitochondria to induce apoptosis(9,11,15,16). The opening of the mitochondrial ‘permeabilitytransition pore’ is regulated by endogenous effectors, includ-ing members of the Bcl-2 family proteins such as Bax (36).The pro-apoptotic Bax is normally present in the cytosol,which promotes programmed cell death by its insertion intomitochondrial membrane (44). In glioma cells 4-HPR upregu-lated expression as well as mitochondrial translocation of Bax.In mitochondria, Bax has pore-forming properties (44) that canpromote the loss of DYm and release cytochrome c and AIFfrom mitochondria (32,45). Indeed, 4-HPR leads to release ofcytochrome c and AIF in glioma cells. The release ofcytochrome c from the mitochondria and its interactionwith cytosolic factors including Apaf-1 and dATP can activatecaspase-9 and then caspases 3 and 7 (46). Caspases areinvolved in 4-HPR-induced apoptosis, and the activation ofcaspase-3 subsequent to cytochrome c release has been pre-viously assessed (5,9–11). We also found 4-HPR-inducedactivation of caspase-3 and caspase-7 in glioma cells (25).However, we found that in the presence of pan-caspaseinhibitor oligonucleosomal fragmentation was prevented butperipheral chromatin condition still took place, suggesting that4-HPR may also trigger the caspase-independent pathway.Recent reports have also suggested involvement of caspase-independent apoptosis by 4-HPR in human B lymphoma cells(13). Another mechanism for apoptosis induction, independentof caspase activity, is through AIF, a mitochondrial proteinthat is translocated into the nucleus and leads to apoptosisinduction (22). It is also suggested that the nuclear transloca-tion of AIF can be a caspase-dependent event (47). Within thenucleus, AIF, in cooperation with a mitochondrial proteinEndo G, induces peripheral chromatin condensation andlarge-scale DNA fragmentation (22). We found that 4-HPRinduced AIF nuclear translocation in the presence of caspaseinhibitors, which leads to peripheral chromatin condensationin the absence of nuclear fragmentation. AIF nuclear translo-cation was inhibited by antioxidants and CsA, suggesting roleof ROS generation and MPT in AIF-mediated apoptosis. Thus,the results suggest involvement of parallel pathways of chro-matin processing in which nuclear fragmentation is governedby caspases and peripheral chromatin condensation by AIF assuggested earlier, where role of AIF has been shown in the firststage of apoptosis and caspases in the second stage (48). Thereare growing amount of evidences that suggest involvement ofcaspase-independent pathways in synthetic retinoid-induced
apoptosis (13,23), which may provide additional advantageto overcome the resistance due to caspase inactivation incancers.
In the present study, use of antioxidants, MPT as well ascaspases inhibitors did not completely prevent 4-HPR-inducedapoptosis, suggesting role of additional mechanisms involvingceramide (8) or RAR-mediated pathway (3). Presently, we didnot study the role of ceramide pathway, but the levels of RAR-b, RAR-g and RXR-a in 4-HPR-induced apoptosis were deter-mined. 4-HPR led to upregulation of RAR-g and RXR-a levelsin glioma cells. It has been suggested that RAR-g and itsreceptor partner RXR-a activation can lead to apoptosis induc-tion. The increased RAR-g expression may further enhanceapoptosis (3). Thus, the receptor-dependent effect of 4-HPRseems to be relevant in glioma cells as shown previously inother cell types (3,5–7). The ability of 4-HPR to induce apop-tosis in retinoid-resistant tumor cells (6,49) suggested that4-HPR can mediate its effects in a receptor-independent man-ner. However, in the present study, it is not clear whether theupregulation of receptor is associated with apoptosis, as wecould not use the RAR-antagonists that are needed for furtherevaluation.
In conclusion, the present study brings insight into some ofthe unrevealed pathways of 4-HPR-induced apoptosis. Look-ing at its ability to target multiple sites of apoptotic pathways,further studies are warranted using 4-HPR as a single or com-binational agent to eliminate cancer cells.
Acknowledgements
We thank Dr Xiao Miu Sung (University of Leicester, UK) for caspase-7antibody, R. Jemmerson (University of Minnesota, MN) for cytochrome cantibody (7H8.2C12) and Dr P. Chambon (IGBMC, De Strasbourg, France)for antibodies against RAR-b (No. 112), RAR-g (No. 453) and RXR-a(No. 444). This study was supported by an intramural grant PGI/DIR/RC/681/2004 to M.M.G. and by a fellowship (No.9/590(33)/2001-EMR-I) toM.T. from the Council for Scientific Industrial Research, New Delhi, India.This work constitutes the PhD thesis of M.T.
References
1.Fontana,J.A. and Rishi,A.K. (2002) Classical and novel retinoids: theirtargets in cancer therapy. Leukemia, 16, 463–472.
2.David,M., Hodak,E. and Lowe,N.J. (1998) Adverse effect of retinoids.Med. Toxicol. Adverse Drug. Exp., 3, 273–288.
3.Fanjul,A.N., Delia,D., Pierotti,M.A., Rideout,D., Qiu,J. and Pfahl,M.(1996) 4-Hydroxyphenyl retinamide is a highly selective activator ofretinoid receptors. J. Biol. Chem., 271, 22441–22446.
4.Parchment,R.E., Jasti,B.R., Kocarek,T.A. et al. (1999) Pharmacologicissues for fenretinide chemotherapy (4HPR, NSC-374551). Clin. CancerRes., Suppl 5: 38008, (abstr 350).
5.Wu,J.M., DiPietrantonio,A.M. and Hsieh,T.C. (2001) Mechanism offenretinide (4-HPR)-induced cell death. Apoptosis, 6, 377–388.
6.Delia,D., Aiello,A., Lombardi,L. et al. (1993) N-(4-Hydroxyphenyl)retinamide induces apoptosis of malignant hemopoietic cell linesincluding those unresponsive to retinoic acid. Cancer Res., 53, 6036–6041.
7.Clifford,J.L., Menter,D.G., Wang,M., Lotan,R. and Lippman,S.M. (1999)Retinoid receptor-dependent and -independent effects of N-(4-hydroxyphenyl)retinamide in F9 embryonal carcinoma cells. CancerRes., 59, 14–18.
8.Lovat,P.E., Ranalli,M., Annichiarrico-Petruzzelli,M., Bernassola,F.,Piacentini,M., Malcolm,A.J., Pearson,A.D., Melino,G. and Redfern,C.P.(2000) Effector mechanisms of fenretinide-induced apoptosis inneuroblastoma. Exp. Cell Res., 260, 50–60.
9.Suzuki,S., Higuchi,M., Proske,R.J., Oridate,N., Hong,W.K. and Lotan,R.(1999) Implication of mitochondria-derived reactive oxygen species,cytochrome C and caspase-3 in N-(4-hydroxyphenyl)retinamide-inducedapoptosis in cervical carcinoma cells. Oncogene, 18, 6380–6387.
Mechanism of 4-HPR-induced apoptosis in glioma cells
2057

10.Asumendi,A., Morales,M.C., Alvarez,A., Arechaga,J. and Perez-Yarza,G.(2002) Implication of mitochondria-derived ROS and cardiolipinperoxidation in N-(4-hydroxyphenyl)retinamide-induced apoptosis. Br. J.Cancer, 86, 1951–1956.
11.Boya,P., Morales,M.C., Gonzalez-Polo,R.A. et al. (2003) Thechemopreventive agent N-(4-hydroxyphenyl)retinamide inducesapoptosis through a mitochondrial pathway regulated by proteins fromthe Bcl-2 family. Oncogene, 22, 6220–6230.
12.Kalli,K.R., Devine,K.E., Cabot,M.C., Arnt,C.R., Heldebrant,M.P.,Svingen,P.A., Erlichman,C., Hartmann,L.C., Conover,C.A. andKaufmann,S.H. (2003) Heterogeneous role of caspase-8 in fenretinide-induced apoptosis in epithelial ovarian carcinoma cell lines. Mol.Pharmacol., 64, 1434–1443.
13.Barna,G., Sebestyen,A., Weischede,S., Petak,I., Mihalik,R., Formelli,F.and Kopper,L. (2005) Different ways to induce apoptosis by fenretinideand all-trans-retinoic acid in human B lymphoma cells. Anticancer Res.,25, 4179–4185.
14.Corazzari,M., Lovat,P.E., Oliverio,S., Di-Sano,F., Donnorso,R.P.,Redfern,C.P. and Piacentini,M. (2005) Fenretinide: a p53-independentway to kill cancer cells. Biochem. Biophys. Res. Commun., 331, 810–815.
15.Hail,N., Jr. and Lotan,R. (2000) Mitochondrial permeability transition is acentral coordinating event in N-(4-hydroxyphenyl) retinamide-inducedapoptosis. Cancer Epidemiol. Biomarkers. Prev., 9, 1293–1301.
16.Hail,N., Jr. and Lotan,R. (2001) Mitochondrial respiration is uniquelyassociated with the prooxidant and apoptotic effects of N-(4-hydroxyphenyl)retinamide. J. Biol. Chem., 276, 45614–45621.
17.Goto,H., Takahashi,H., Fujii,H., Ikuta,K. and Yokota,S. (2003) N-(4-Hydroxyphenyl)retinamide (4-HPR) induces leukemia cell death viageneration of reactive oxygen species. Int. J. Hematol., 78, 219–225.
18.Liu,S., Brown,C.W., Berlin,K.D., Dhar,A., Guruswamy,S., Brown,D.,Gardner,G.J., Birrer,M.J. and Benbrook,D.M. (2004) Synthesis offlexible sulfur-containing heteroarotinoids that induce apoptosis andreactive oxygen species with discrimination between malignant andbenign cells. J. Med. Chem., 47, 999–1007.
19.Osone,S., Hosoi,H., Kuwahara,Y., Matsumoto,Y., Iehara,T. andSugimoto,T. (2004) Fenretinide induces sustained-activation of JNK/p38MAPK and apoptosis in a reactive oxygen species-dependent manner inneuroblastoma cells. Int. J. Cancer, 112, 219–224.
20.Duchen,M.R. (1999) Contributions of mitochondria to animal physiology:from homeostatic sensor to calcium signaling and cell death. J. Physiol.,516, 1–17.
21.Gunter,T.E. and Gunter,K.K. (2001) Uptake of calcium by mitochondria:transport and possible function. IUBMB Life, 52, 197–204.
22.Cand,C., Cohen,I., Daugas,E., Ravagnan,L., Larochette,N., Zamzami,N.and Kroemer,G. (2002) Apoptosis-inducing factor (AIF): a novelcaspase-independent death effecter released from mitochondria.Biochimie, 84, 215–222.
23.Boisvieux,E., Ulrich,T., Sourdeval,M. and Marano,F. (2005) CD437, asynthetic retinoid, induces apoptosis in human respiratory epithelialcells via caspase-independent mitochondrial and caspase-8-dependentpathways both up-regulated by JNK signaling pathway. Exp. Cell Res.,307, 76–90.
24.Rao,R.V., Ellerby,H.M. and Bredesen,D.E. (2004) Coupling endoplasmicreticulum stress to the cell death program. Cell Death Differ., 11, 372–380.
25.Puduvalli,V.K., Saito,Y., Xu,R., Kouraklis,G.P., Levin,V.A. andKyritsis,A.P. (1999) Fenretinide activates caspases and inducesapoptosis in gliomas. Clin. Cancer Res., 5, 2230–2235.
26.Saitoh,Y., Goto,T., Puduvalli,V.K., Murakami,M., Kochi,M., Levin,V.A.,Kyritsis,A.P. and Ushio,Y. (1999) Induction of apoptosis by N-(4-hydroxyphenyl) retinamide in glioma cells. Int. J. Oncol., 15, 499–504.
27.Lytle,R.A., Jiang,Z, Zheng,X., Higashikubo,R. and Rich,K.M. (2005)Retinamide-induced apoptosis in glioblastomas is associated with down-regulation of Bcl-xL and Bcl-2 proteins. J. Neurooncol., 74, 225–232.
28.Salvioli,S., Ardizzoni,A., Franceschi,C. and Cossarizza,A. (1997) JC-1, butnot DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess
delta psi changes in intact cells: implications for studies on mitochondrialfunctionality during apoptosis. FEBS. Lett., 411, 77–82.
29.Hail,N.,Jr., Konopleva,M., Sporn,M., Lotan,R. and Andreeff,M. (2004)Evidence supporting a role for calcium in apoptosis induction by thesynthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid(CDDO). J. Biol. Chem., 279, 11179–11187.
30.Okada,C.Y. and Rechsteiner,M. (1982) Introduction of macromoleculesinto cultured mammalian cells by osmotic lysis of pinocytic vesicles.Cell, 29, 33–41.
31.Kroemer,G., Zamzami,N. and Susin,S.A (1997) Mitochondrial control ofapoptosis. Immunol. Today, 18, 44–51.
32.Green,D.R. and Reed,J.C. (1998) Mitochondria and apoptosis. Science,281, 1309–1311.
33.Hajnoczky,G., Davies,E. and Madesh,M. (2003) Calcium signaling andapoptosis. Biochem. Biophys. Res. Commun., 304, 445–454.
34. Jacobson,J. and Duchen,M.R. (2002) Mitochondrial oxidative stress andcell death in astrocytes—requirement for stored Ca2+ and sustained openingof the permeability transition pore. J. Cell Sci., 115, 1175–1188.
35.Pu,Y. and Chang,D.C. (2001) Cytosolic Ca(2+) signal is involved inregulating UV-induced apoptosis in hela cells. Biochem. Biophys. Res.Commun., 282, 84–89.
36.Adams,J.M. and Cory,S. (1998) The Bcl-2 protein family: arbiters of cellsurvival. Science, 281, 1322–1326.
37.Kim,R., Emi,M. and Tanabe,K. (2005) Role of mitochondria as the gardensof cell death. Cancer. Chemother. Pharmacol., 21, 1–9.
38.Wang,Z.B., Liu,Y.Q. and Cui,Y.F. (2005) Pathways to caspase activation.Cell. Biol. Int., 29, 489–496.
39.Carpentier,A.F. (2005) New therapeutic approaches in glioblastomas.Bull. Cancer, 92, 355–359.
40.Steinbach,J.P. and Weller,M. (2004) Apoptosis in gliomas:molecular mechanisms and therapeutic implications. J. Neurooncol., 70,245–254.
41.Chun,K.H., Benbrook,D.M., Berlin,K.D., Hong,W.K. and Lotan,R. (2003)The synthetic heteroarotinoid SHetA2 induces apoptosis in squamouscarcinoma cells through a receptor-independent and mitochondria-dependent pathway. Cancer Res., 63, 3826–3832.
42.Cerella,C., D’Alessio,M., De Nicola,M., Magrini,A., Bergamaschi,A. andGhibelli,L. (2003) Cytosolic and endoplasmic reticulum Ca2+
concentrations determine the extent and the morphological type ofapoptosis, respectively. Ann. N. Y. Acad. Sci., 1010, 74–77.
43. Jimbo,A., Fujita,E., Kouroku,Y., Ohnishi,J., Inohara,N., Kuida,K.,Sakamaki,K., Yonehara,S. and Momoi,T. (2003) ER stress inducescaspase-8 activation, stimulating cytochrome c release and caspase-9activation. Exp. Cell Res., 283, 156–166.
44.Goping,I.S., Gross,A., Lavoie,J.N., Nguyen,M., Jemmerson,R., Roth,K.,Korsmeyer,S.J. and Shore,G.C. (1998) Regulated targeting of Bax tomitochondria. J. Cell Biol., 143, 207–215.
45.Bras,M., Queenan,B. and Susin,S.A. (2005) Programmed cell death viamitochondria: different modes of dying. Biochemistry, 70, 231–239.
46.Wang,Z.B., Liu,Y.Q. and Cui,Y.F. (2005) Pathways to caspase activation.Cell Biol. Int., 29, 489–96.
47.Damien,A., Brigitte,G., Mariusz,K., Juanita,C.S., Francesco,C. andRichard,J.Y. (2003) Mitochondrial release of AIF and Endo G requirescaspase activation downstream of Bax/Bak-mediated permeabilization.EMBO J., 22, 4385–4399.
48.Susin,S.A., Daugas,E., Ravagnan,L. et al. (2000) Two distinct pathwaysleading to nuclear apoptosis. J. Exp. Med., 192, 571–580.
49.Sheikh,M.S., Shao,Z.M., Li,X.S. et al. (1995) N-(4-Hydroxyphenyl)retinamide (4-HPR)-mediated biological actions involve retinoidreceptor-independent pathways in human breast carcinoma.Carcinogenesis, 16, 2477–2486.
Received February 10, 2006; revised April 10, 2006;accepted April 13, 2006
M.Tiwari et al.
2058




















