
Superconductivity near phase separation in models of correlated electrons
Upload
independentCategory
view
0download
0
0022-4766/14/5505-0799 © 2014 by Pleiades Publishing, Ltd. 799
Journal of Structural Chemistry. Vol. 55, No. 5, pp. 799-808, 2014.
Original Russian Text © 2014 A. V. Luzanov.
MEASURES OF UNPAIRED ELECTRONS
FOR LARGE CONJUGATED SYSTEMS
A. V. Luzanov UDC 519.19
It is shown that the numerical measure of effectively unpaired electrons Neff, proposed by Head-Gordon in
2003, is a particle-hole index: Neff is reduced to the average occupation of virtual particle-hole pairs.
Specific π calculations of the Neff index provide a reasonable interpretation of the radicaloid character of the
singlet ground state in complex conjugated systems. The results of the accurate π model (based on the total
configuration interaction) and approximate approaches are compared. The coupled cluster method and
various Hartree–Fock schemes (UHF and Löwdin EHF scheme) demonstrate an acceptable quantitative
description of unpairing effects, UHF being favorable in simplicity of obtaining the measure of unpairing in
megamolecules, such as graphene structures. The application of the known Yamaguchi index results in a
rougher representation of the radical character of a system.
DOI: 10.1134/S0022476614050011
Keywords: electron correlation, number of effectively unpaired electrons, π shell, polycyclic aromatic
hydrocarbons, megamolecules, graphene, nanopores.
INTRODUCTION
In modern chemistry interest in applied problems of the molecular design of carbon-based megamolecular structures
does not vanish. Indeed, unique properties of polymer systems such as carbon nanotubes and graphene along with the
microelectronics problems have additionally stimulated the chemistry of conjugated systems [1-6]. In turn, new type
problems have appeared in quantum chemistry. One of them relates to the interpretation of the electronic structure of large
molecules where the π system containing hundreds of electrons plays a special functional role. In particular, in the recent
years numerous works have been published devoted to the effects of electron unpairing and radical states in huge
polyaromatic and related systems. The first work of this type performed at the ab initio level was [7]. Within semi-empirical
schemes the related electronic problems seem to be studied for the first time in [8]. However, it is worth mentioning some not
sufficiently correct formulations in [8], where, for example, a non-physical conception of the spin-mixed singlet term was
introduced. Among the latest works let us note the analysis of the biradicaloid character of polycyclic aromatic hydrocarbons
in the context of the nonlinear optical properties [9], and also sophisticated ab initio studies of the unpairing for graphene
molecules in [10, 11], which contain a sufficiently full bibliography.
In most works on the theory of effectively unpaired electrons [12-22] quantitative estimations of some average
number of unpaired electrons are proposed. These indices are of special interest for the ground singlet state in strongly
Institute for Single Crystals, National academy of Sciences of Ukraine, Kharkov, Ukraine; [email protected]. Translated from Zhurnal Strukturnoi Khimii, Vol. 55, No. 5 pp. 845-854, September-October, 2014. Original article submitted September 10, 2013; revised October 29, 2013.

800
correlated many-electron systems. The electron unpairing index was first proposed as far back as in the 70’s as the index of
the odd, i.e. unpaired, electron [12]. It was clearly shown that the unpaired electrons were implicitly present even in a singlet
molecule [13], although total spin was zero here. The characteristic from [12] we call the Yamaguchi index. Later new
schemes appeared which are also considered below. As it became clear, unpaired electrons are always present to some extent
due to the electron correlation that is especially significant for large molecules. One of the aims of this work is to analyze the
theory of unpairing as applied to π shells, whereas another aim is to distinguish among the available methods those best
suited for the analysis of large electronic problems.
The employment of the π scheme for polyconjugated systems is motivated in this work as follows. In large
conjugated systems it is the π shell that undergoes the greatest changes, including the unpairing. Electron correlation first of
all destructs the most outer closed subshell, and this subshell is mainly formed by π orbitals. Moreover, in the theory of
conjugated polymers most physical models are still based on one or another variant of the π approximation (most often in the
form of a many-electron Hubbard model, see reviews [23]). The accent on π-subsystems is indirectly made in a number of
non-empirical works (e.g., [10, 11]), where DMRG or CASSCF techniques are practically applied for the π electron
subsystem.
In the proposed analysis of the reliability of various procedures a key role is played by the full configuration
interaction (FCI) method. As is known, an accurate solution of the many-electron problem in the finite-dimensional
representation by atomic orbitals corresponds to the FCI method. A direct comparison with FCI provides the evaluation of the
results of substantially simplified electronic models while the latter, unlike FCI, are applicable also to very large systems. In
this work, within the semi-empirical Pariser–Parr–Pople π scheme for FCI we show that as a rule, the calculations yield quite
reliable results even with the help of a simple unrestricted Hartree–Fock (UHF) method. Here the Head–Gordon index is
taken as the main index of unpairing [16].
FORMALISM OF UNPAIRING INDICES
The above mentioned Yamaguchi index [12] is denoted as Nodd. It estimates the degree of electron unpairing by
means of the nonlinear measure of the deviation of eigenvalue spectrum {λk} of the spin-free one-electron density matrix D1
from its analogue in the restricted Hartree–Fock (RHF) method. Without going into details (see, e.g., [12, 14]), let us write
the working expression for Nodd through eigenvalues λk (natural occupation numbers)
odd
(2 ) .k k
k
N = − λ λ∑ (1)
In practice, Nodd usually gives overestimated values of unpaired electrons [16] (often two times higher [18]). Head-Gordon
proposed a better definition of the index of unpairing: the index
eff
min[ , 2 ].k k
k
N = λ − λ∑ (2)
In this work, this index is the main one. The basis for this choice are not only particular numerical results, but a less formal
representation of index (2) itself. In a more transparent form the index of unpairing was derived in our work [17] and it was
based on an interesting Kutzelnigg conception about a measure of the electron shell openness [24]. Further it turned out that a
similar construction appears again in the analysis of particle-hole components of the one-electron density matrix [18]. For
“normal” molecules (without pathology in the distribution of occupation numbers {λk}, see in detail the Supplement) index
Neff (2) exactly coincides with the average occupation of particles and holes, i.e. with index Neff[h – p] introduced in [18].
Therefore (2) proves to be a direct particle-hole characteristic of the electron unpairing. It can be calculated as a doubled sum
of the occupation numbers of all virtual natural orbitals [25]
eff
2 .a
a n
N
>
= λ∑ (3)

801
Here n = N/2 is the maximum possible number of electron pairs in a singlet molecule with N electrons. In record (3) it is
assumed that the eigenvalues {λk} are arranged in decreasing order. An explicit proof and the equivalence condition of
expressions (2) and (3) have not been presented previously. The necessary analysis is given in the Supplement section for
convenience.
Note also that a precursor of index (3) is the qualitative measure of the biradicaloid character in [13]. In this work,
the biradicaloid character was estimated by the closeness of occupation numbers λk to 1. Another essential moment is the
application of indices Nodd and Neff in the UHF scheme. The UHF method is most easily implemented for large molecules and
in quantum chemistry of conjugated polymers it was applied already in [26]. Indices (1) and (2) are calculated very simply in
UHF because in this case
1
,D α β= ρ + ρ (4)
where ρα and ρβ are the standard density matrices of α- and β-shells. However, the application of the index Nodd within UHF
causes a difficulty in the interpretation of the idea of electron unpairing itself. In [14] it was shown that for the singlet states
Nodd equaled the double ⟨S2⟩ value (the average value of the spin square). In other words, in the approach [12, 14] the measure
of the electron unpairing is identified with the measure of the method defectiveness: spin contamination differing UHF from
more correct (pure with respect from spin) methods. This approach does not seem so attractive (see also [16]). Nonetheless,
this approach is applied in [27] as very available (it is not necessary to know the spectrum of density matrices). In the
illustrations we present the values of the corresponding index, denoting it as UHF
odd.N For the singlet states this index is
calculated by the relation [14]
UHF 2
odd UHF2 2Tr( ),N N α β= ⟨ ⟩ = − ρ ρS (5)
which is equivalent to the formula UHF
odd2Tr ( ).N N β α β= − ρ ρ ρ
As argued above, we prefer (2) and (3) to the unpairing measure Neff, further denoting the calculated data for Neff in
each model through FCI
eff,N UHF
odd,N and so on. If we use the known results [28] for the spectrum of matrix (4), then with
regard to definition (3) the index UHF
effN can also be represented in the explicit matrix form
UHF 1/2
eff2Tr[( ) ].N N β α β= − ρ ρ ρ (6)
Since the singular numbers of the nonsymmetric matrix ραρβ (i.e., if eigenvalues of the symmetric matrix
[(ραρβ)+(ραρβ)]
1/2 ≡ (ρβραρβ)1/2) are below unit, then from (5) and (6) it follows that UHF UHF
eff odd.N N≤
Along with UHF we employ a rarely used but sufficiently meaningful model of different orbitals for different spins
in the correct Löwdin formulation [32]. It is usually called the extended Hartree–Fock (EHF) method. In due time, many
works have been devoted to this model (see reviews [30, 31]). In our calculations a convenient algorithm is used [32].
Despite the absence of the size consistency in the EHF model, it has an important advantage. Unlike UHF, non-trivial
solutions of EHF equations (not reduced to the RHF solution) necessarily exist [31]. Therefore in EHF there is always
electron unpairing, including for weakly correlated electrons. The corresponding measure calculated by (3) is denoted as EHF
eff.N
Finally, let us briefly consider an approximate CCD scheme from the CC theory in which pair electron correlations
are taken into account in a size-consistent manner way (see, e.g., book [33]). The size consistency is especially important for
large systems. In the CCD method a certain basis molecular spin-orbitals is selected and a matrix of the two-electron
excitation operator t2 = ||tab,ij|| is constructed, where indices i and j belong to the occupied orbitals of the reference
determinant, and indices a and b belong to unoccupied orbitals. A rather complex nonlinear equation for tab,ij provides the
possibility to correctly calculate the correlation energy and also to estimate the corresponding density matrix CCD
1D for

802
systems far from degeneration [33, Ch. 11] (recall that CCD is not a variational scheme). The simplest way of the
approximate calculation of CCD
1D seems to be described in [19]. First, from t2 a “renormalized” operator is constructed
1/2
2 2 2 2( ) .C t I t t
+ −
= + (7)
This operator is identified with the analogue of the excitation operator generating doubly excited configurations in the CID
method (variational taking into account of all doubly excited configurations). Then it is easy to use relatively simple formulas
of the density matrix theory obtained previously in [34]. This makes it possible to directly apply formula (2), thus giving the
size-consistent CCD estimate CCD
effN of the number of effectively unpaired electrons.
For not strongly degenerate electronic systems the CCD method is sufficiently effective, especially if the matrix t2 is
constructed in the basis set of so-called Brueckner or similar orbitals [35-37]. In practice we apply a quite simple and
effective variant of the CCD scheme in the basis set of natural UHF orbitals, i.e. eigenvectors of matrix (4). The latter differ
from the RHF orbitals only at a sufficiently substantial electron correlation, more precisely, under the condition of the so-
called triplet Hartree-Fock instability [31]. In fact, all conjugated systems studied here are of this type (otherwise UHF UHF
odd eff0).N N= = The use of natural UHF orbitals for the problems of the configurational interaction was proposed in [38].
CASE STUDIES
The data in Tables 1-3 were obtained using the standard π parameters. The resonance integral of the π bond and the
one-center integral of Coulomb repulsion were taken to be –2.4 eV and 11.13 eV respectively, and the two-center repulsion
integrals were estimated by the Ohno formula. The geometry of the π core was set by regular polygons with the С–С bond
length of 1.4 Å. All systems from Tables 1 and 2 or their derivatives are known in the experiment. In particular, the synthesis
of the indacenodiphenalene molecule (system 15 in Table 2) is described in [39].
In Tables 1-3 the structural formula reflect also the atomic distribution eff
{ }Dµ
of effectively unpaired electrons. In
each table the best approximation (FCI for Table 1, CCD for Table 2, and UHF for Table 3) was used for the atomic
distributions. The distribution eff{ }D
µ was normalized to the corresponding Neff value FCI
eff(N for Table 1, CCD
effN for Table 2,
and UHF
effN for Table 3). The atomic distribution eff
{ }Dµ
was obtained in the form of a weighted sum of the squared LCAO
coefficients 2
kcµ
of the corresponding virtual orbitals: eff 22
k k
k n
D cµ µ
>
= λ∑ (see formula (3)). This partition automatically
provides the normalization eff
eff.D N
µ
µ
=∑
Let us consider some specific results. Indices presented in Table 1 make it possible to directly estimate the quality of
different approaches in comparison with FCI. It is obvious that the CCD method is on average closest to the FCI results. The
largest deviations from FCI
effN is observed in the index UHF
odd,N which is not surprising in the light of the critics of the
Yamaguchi index in [16, 18]. Nonetheless, for strongly localized unpaired electrons, as it occurs in biradicals 9 and 10 from
Table 1, the UHF
oddN index also yields quite adequate values. Still within the UHF approximation the UHF
effN characteristic
seems to be more acceptable, i.e. particle-hole index (3) for density matrix (4). This comparative estimation of UHF
oddN and
UHF
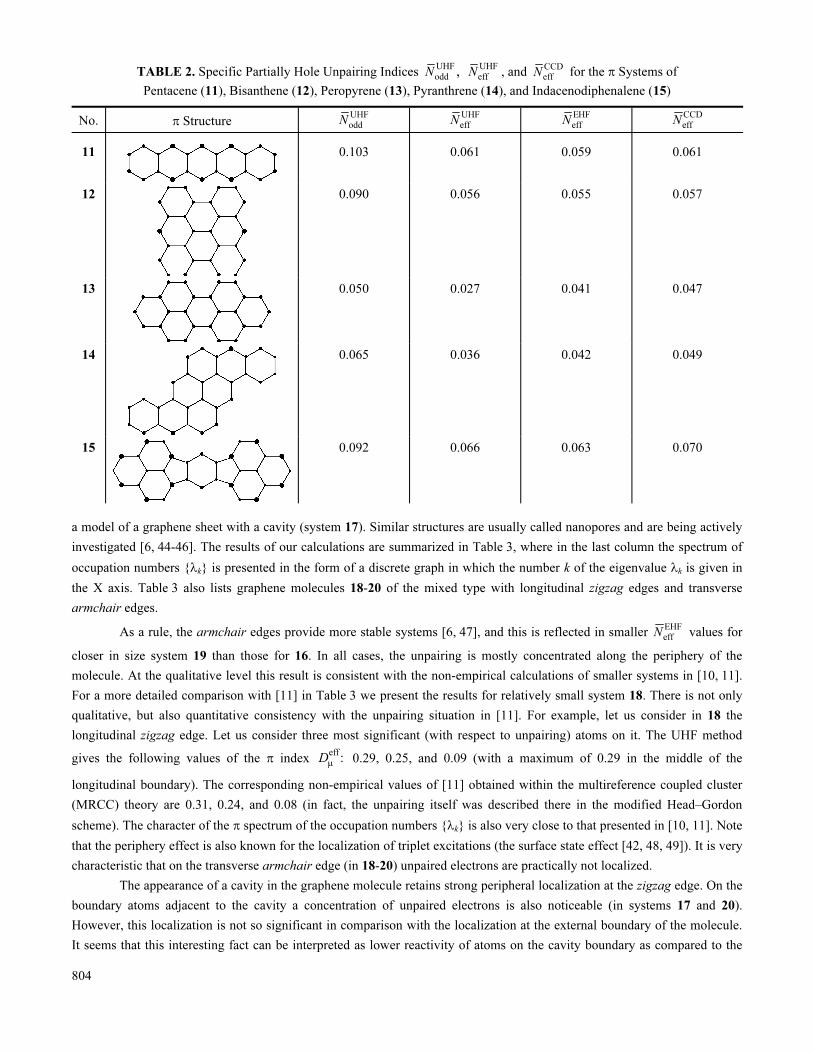
effN is confirmed by Table 2 for medium-sized π systems that already cannot be studied by FCI.
In this case, in the estimation of the quality of UHF we are based on the CCD method as the most reliable (of
available) dimensionally correlated scheme of the calculation of Neff. Table 2 lists the specific values: the numbers
eff eff/ ,N N N= where N is the total number of valence electrons (the number of π electrons in this case). The corresponding
CCD
effN values and others in Table 2 can be compared with similar results for related molecules in the FCI method. In

803
particular, in anthratcene not mentioned in Table 1, the calculations give FCI
effN = 0.052, with EHF
effN = 0.046 and CCD
effN = 0.049
(for anthracene the UHF and RHF results are practically identical, i.e. UHF
effN ≈ 0). From Table 2 we see that apart from a few
exceptions, UHF
effN corresponds to FCI
effN practically at the quantitative level, and hence, the UHF method is indeed quite
acceptable for the approximate description of the radical character in sufficiently large systems with strong electron
correlation. From Table 2 it follows that the EHF Löwdin approximation and the standard UHF scheme most often yield
close results. At the same time, for weakly correlated systems such as 13 (peropyrene), EHF yields noticeably better results.
However, we should not forget the absence of size consistency in EHF.
Data listed in Tables 1 and 2 somehow extenuate the application of the UHF scheme in the previous works [27, 40],
where the Yamaguchi index UHF
oddN or values close to it were applied. However, as seen from Tables 1 and 2, the UHF
oddN index
most often gives a too rough description while the particle-hole index UHF
effN leads to a more adequate (moderate) situation of
the electron unpairing.
Consequently, a simple procedure of the analysis of unpairing based on the UHF
effN index can be extended to
megamolecular conjugated systems. The latter have been intensively studied in the current works on physics and chemistry of
graphene-like structures, e.g. in [41-43]. In particular, in [41, 42] the spin states of hexagonal graphene molecules with the
general formula C6m2H6m were theoretically studied. The latter have zigzag edges. We consider a special case of this system
for the value m = 6: the singlet C216H36 molecule (system 16 in Table 3). Moreover, based on the same structure, we consider
TABLE 1. Comparison of the Particle-Hole Unpairing index FCI
effN (complete configurational interaction)
with Approximate Estimates UHF
odd,N UHF
effN , and CCD
effN for Different Type π Systems
No. π Structure UHF
oddN UHF
effN EHF
effN
CCD
effN FCI
effN
1
1.54 0.97 0.71 0.78 0.97
2
1.38 0.81 0.73 0.90 0.92
3
0.96 0.53 0.65 0.67 0.98
4
3.64 2.40 1.96 1.72 1.68
5
1.46 0.88 0.84 1.06 1.13
6
1.80 1.18 0.94 1.04 1.09
7
1.86 1.03 0.97 0.89 1.09
8
2.49 1.97 1.90 2.30 1.93
9
2.14 2.07 2.23 2.41 2.48
10
2.52 2.25 2.54 2.51 2.57
804
TABLE 2. Specific Partially Hole Unpairing Indices UHF
odd,N UHF
effN , and CCD
effN for the π Systems of
Pentacene (11), Bisanthene (12), Peropyrene (13), Pyranthrene (14), and Indacenodiphenalene (15)
No. π Structure UHF
oddN UHF
effN EHF
effN CCD
effN
11
0.103 0.061 0.059 0.061
12
0.090 0.056 0.055 0.057
13
0.050 0.027 0.041 0.047
14
0.065 0.036 0.042 0.049
15
0.092 0.066 0.063 0.070
a model of a graphene sheet with a cavity (system 17). Similar structures are usually called nanopores and are being actively
investigated [6, 44-46]. The results of our calculations are summarized in Table 3, where in the last column the spectrum of
occupation numbers {λk} is presented in the form of a discrete graph in which the number k of the eigenvalue λk is given in
the X axis. Table 3 also lists graphene molecules 18-20 of the mixed type with longitudinal zigzag edges and transverse
armchair edges.
As a rule, the armchair edges provide more stable systems [6, 47], and this is reflected in smaller EHF
effN values for
closer in size system 19 than those for 16. In all cases, the unpairing is mostly concentrated along the periphery of the
molecule. At the qualitative level this result is consistent with the non-empirical calculations of smaller systems in [10, 11].
For a more detailed comparison with [11] in Table 3 we present the results for relatively small system 18. There is not only
qualitative, but also quantitative consistency with the unpairing situation in [11]. For example, let us consider in 18 the
longitudinal zigzag edge. Let us consider three most significant (with respect to unpairing) atoms on it. The UHF method
gives the following values of the π index eff:D
µ 0.29, 0.25, and 0.09 (with a maximum of 0.29 in the middle of the
longitudinal boundary). The corresponding non-empirical values of [11] obtained within the multireference coupled cluster
(MRCC) theory are 0.31, 0.24, and 0.08 (in fact, the unpairing itself was described there in the modified Head–Gordon
scheme). The character of the π spectrum of the occupation numbers {λk} is also very close to that presented in [10, 11]. Note
that the periphery effect is also known for the localization of triplet excitations (the surface state effect [42, 48, 49]). It is very
characteristic that on the transverse armchair edge (in 18-20) unpaired electrons are practically not localized.
The appearance of a cavity in the graphene molecule retains strong peripheral localization at the zigzag edge. On the
boundary atoms adjacent to the cavity a concentration of unpaired electrons is also noticeable (in systems 17 and 20).
However, this localization is not so significant in comparison with the localization at the external boundary of the molecule.
It seems that this interesting fact can be interpreted as lower reactivity of atoms on the cavity boundary as compared to the

805
TABLE 3. Specific Particle-Hole Unpairing Indices UHF
oddN and
UHF
effN and Natural Occupation Numbers
{λk} for Graphene Megamolecules with Zigzag Edges (16), (17) and Armchair-Zigzag Edges (18)-(20)
No. Megamolecule UHF
oddN
UHF
effN {λk}
1 2 3 4 5
16
0.075 0.047
17
0.094 0.058
18
0.086 0.060
19
0.056 0.042

806
TABLE 3. (Cont.)
1 2 3 4 5
20
0.063 0.049
outer boundary atoms. It is also interesting to note an obvious increase in the specific unpairing index on passing to a
structure with a cavity due to the boundary expansion, i.e. an increase in the number of surface atoms. The relationship,
although ambiguous, between the distribution of effectively unpaired electrons with reactivity is discussed in the literature
(see, e.g., [27, 40]).
CONCLUSIONS
From the results of the study two main conclusions can be drawn. The first belongs to the choice of the most
appropriate definition of the quantitative measure of electron unpairing. Index Neff (2) introduced by Head-Gordon [16] is
equivalent to the average occupation of virtual particle–hole electron pairs (3), i.e. Neff is in fact a particle-hole characteristic
of the unpairing. This fact proved in this work gives the clear physical meaning to the index Neff. This is the difference of Neff
from a number of others, somewhat more artificial definitions of the electron unpairing. And it seems to be not occasional
that Yamaguchi index Nodd (2) as well as our collectivity index from [17] (not presented here) leads to too rough results,
especially when Hartree–Fock models of the UHF type are applied. The second conclusion relates to practical
recommendations: which approximate quantum chemical model can be applied for large conjugated systems with hundreds
of atoms. For small systems a comparison of UHF results with the results of higher level models (FСI, ССD) shows that in
the UHF method a quite acceptable description level is reached, but with the necessary condition of using particle-hole index UHF
effN (6). It is also important that where a direct comparison is possible, the results of the proposed calculative π scheme are
well consistent with a significantly more difficult non-empirical approach in [11].
Further it should be noted that for very large systems with thousands of atoms it difficult to apply even the UHF π
model. The electronic problems are quite relevant for such systems in the physics of graphene systems with their often
intricate external boundary, cavities (holes) of various shapes, and so on [6, 44-46]. Such objects without the translational
symmetry cannot be studied within the band structure theory. At present, we see one possible way to solve the mentioned
problem. It consists in the application of even simpler one-electron scheme than that in UHF. For the alternant conjugated π
systems such a model was proposed in [50]. A preliminary investigation shows that the generalized Hückel model [50] can
roughly reproduce the Table 3 results and qualitatively reflect the electron unpairing at the boundaries of the graphene
molecule, including a megamolecule with a cavity defect. Such approaches reflecting the molecular topology seem to be
useful as a first step in the study of strongly correlated π megasystems that are too difficult for rigorous theory.
SUPPLEMENT.
EQUIVALENCE OF PARTICLE-HOLE INDEX (3) TO HEAD-GORDON INDEX (2)
For simplicity let us limit the proof by the most important for us singlet ground state. Then n = N/2 equals the
number of electron pairs. For the ground Hartree–Fock (RHF) state the first n of natural occupation numbers {λi}1 ≤ i ≤ n are

807
identical and amount to the maximum possible value of 2. Although, even at strong unpairing (at strong electron correlation)
the mentioned occupation numbers decrease, they remain, as a rule, to be limited from below by a value of 1, i.e. 1 < λi for all
i ≤ n. This behavior of the upper part of the {λi}1 ≤ i ≤ n spectrum is typical of electronic states without pathology. In this case,
during the complete decay of the ith Hartree–Fock electron pair the λi number deviates from the value of 2 an approaches 1
from above. Thus, in conventional problems the condition 1 < λi is maintained for all first n of the spectrum numbers.
In exclusive cases that we call pathological, the condition 1 < λi turns out to be valid for a smaller number of
eigenvalues: i ≤ n′, where a fortiori n′ < n. This is possible, for example, for superexcited states. The study of such
pathological cases is also possible in principle. It is necessary for them to replace n by the dimensionality n′ that we define as
the amount of occupation numbers with the condition {λi} > 1. In these cases, n′ becomes an essential characteristic of the
electronic state. Usually, n′ = n, and below we analyze only the normal case n′ = n.
It is convenient to divide the complete spectrum {λj} into two spectral sets: the set {λi}1 ≤ i ≤ n with the condition
1 < λi and the set {λa}a > n with the condition λa ≤ 1. Here the first set is associated with the occupied orbitals of the reference
determinant from natural orbitals and the second is associated with a set similar to virtual MOs. Now we may turn to formula
(2). Let us take into account the validity of two equalities: min[λi,2 – λi] = 2 – λi (for the first spectral set) and min[λa,2 –
λa] = λa (for the second spectral set). From here we have eff
2 .i a
i a
N n= − λ + λ∑ ∑ Let us take into account the spectrum
normalization to the number of electrons, i.e. the equality i a
i a
λ + λ =∑ ∑ 2n. Substituting it into the previous relationship,
we obtain formula (3) and so on. In the pathological case, instead of (3) we obtain eff
2 2( ).a
a n
N n n
′>
′= λ − −∑ This formula
already does not allow a simple interpretation in terms of the occupations of conventional particle-hole pairs. For the ground
state the anomalous case n′ < n is far from the practice. The general conclusion from the analysis performed consists in that
for real problems it is possible to apply indices (2) and (3) as equivalent.
REFERENCES
1. Y. Geerts, G. Klärner, and K. Müllen, Electronic Materials: The Oligomer Approach, Wiley-VCH, Weinheim (1998);
K. P. Loh, Q. Bao, P. K. Ang, and J. Yang, J. Mater. Chem., 20, No. 12, 2277-2289 (2010).
2. K. Müllen and U. Scherf (eds.),in: Organic Light Emitting Devices, Synthesis, Properties and Applications, Wiley-VCH,
Weinheim (2006).
3. K. Mullen and J. P. Rabe, Acc. Chem. Res., 41, No. 4, 511-520 (2008).
4. W. Choi, I. Lahiri, R. Seelaboyina, and Y. S. Kang, Crit. Rev. Solid State, 35, No. 1, 52-71 (2010).
5. K. M. Kadish and F. D’Souza (eds.), in: Handbook of Carbon Nano Materials. Volume 1. Synthesis and
Supramolecular Systems, World Scientific Publishing, Singapore (2011).
6. A. L. Ivanovskii, Usp. Khim., 81, No. 7, 571-605 (2012).
7. J. Hachmann, J. J. Dorando, M. Aviles, and G. K.-L. Chan, J. Chem. Phys., 127, No. 13, 134309-1-134309-9 (2007).
8. E. F. Sheka, Int. J. Quant. Chem., 100, No. 4, 375-387 (2004).
9. K. Yoneda, M. Nakano, K. Fukuda, and B. Champagne, J. Phys. Chem. Lett., 3, No. 22, 3338-3342 (2012).
10. W. Mizukami, Y. Kurashige, and T. Yanai, J. Chem. Theor. Comp., 9, No. 1, 401-407 (2012).
11. F. Plasser, H. Pašalic, M. H. Gerzabek, et al., Angew. Chem. Int. Ed., 52, No. 9, 2581-2584 (2013).
12. K. Takatsuka, T. Fueno, and K. Yamaguchi, Theor. Chim. Acta, 48, No. 3, 175-183 (1978).
13. D. Döhnert and J. Koutecký, J. Am. Chem. Soc., 102, No. 6, 1789-1796 (1980).
14. V. N. Staroverov and E. R. Davidson, Chem. Phys. Lett., 330, Nos. 1-2, 161-168 (2000).
15. R. C. Bochicchio, L. Lain, and A. Torre, Chem. Phys. Lett., 374, Nos. 5-6, 567-571 (2003); L. Lain, A. Torre,
D. R. Alcoba, and R. C. Bochicchio, Chem. Phys. Lett., 476, Nos. 1-3, 101-103 (2009).

808
16. M. Head-Gordon, Chem. Phys. Lett., 380, Nos. 3-4, 488/489 (2003).
17. A. V . Luzanov and O. A. Zhikol, Int. J. Quant. Chem., 104, No. 2, 167-180 (2005).
18. A. V. Luzanov and O. V. Prezhdo, J. Chem. Phys., 124, No. 22, 224109-1-224109-16 (2006).
19. A. V. Luzanov and O. V. Prezhdo, J. Chem. Phys., 125, No. 15, 154106-1-154106-14 (2006).
20. C. Lambert, Angew. Chem. Int. Ed. Eng., 50, No. 8, 1756-1758 (2011).
21. P. Karafiloglou and K. Kyriakidou, Int. J. Quant. Chem., 113, No. 13, 1775-1786 (2013).
22. J. R. Dias, Mol. Phys., 111, No. 6, 735-751 (2013).
23. A. A. Ovchinnikov, I. I. Ukrainskii, and G. V. Kventsel, Usp. Fiz. Nauk, 108, No. 1, 81-111 (1972); O. V. Yazyev, Acc.
Chem. Res., 46, No. 10, 2319-2328 (2013).
24. W. Kutzelnigg and V. H. Smith, Int. J. Quant. Chem., 2, No. 4, 531-552 (1968).
25. A. V. Luzanov, Int. J. Quant. Chem., 113, No. 23, 2489-2505 (2013).
26. I. A. Misurkin and A. A. Ovchinnikov, Pis’ma v ZHETF, 4, No. 7, 248-252 (1966).
27. E. F. Sheka and L. A. Chernozatonskii, J. Phys. Chem. C, 111, No. 29, 10771-10779 (2007); E. F. Sheka, Int. J. Quant.
Chem., 112, No. 18, 3076-3090 (2012).
28. A. T. Amos and G. G. Hall, Proc. R. Soc. (London) A, 263, No. 1315, 483-493 (1961).
29. P.-O. Löwdin, Phys. Rev., 97, No. 12, 1509-1520 (1955).
30. I. Mayer, Adv. Quant. Chem., 12, 189-262 (1980).
31. M. M. Mestechkin, G. E. Vaiman, V. Climo, and I. Tin’o, Extended Hartree–Fock Method and its Application to
Molecules [in Russian], Naukova dumka, Kiev (1983).
32. A. V. Luzanov and V. V. Ivanov, Theor. Èkserim. Khim., 26, No. 4, 385-396 (1990).
33. I. Shavitt and R. J. Bartlett, Many-Body Methods in Chemistry and Physics: MBPT and Coupled-Cluster Theory,
Cambridge University Press, Cambridge (2009).
34. A. V. Luzanov and Yu. F. Pedash, Theor. Èkserim. Khim., 21, No. 4, 385-391 (1985).
35. D. Crawford and H. F. Schaefer, Rev. Comput. Chem., 14, 133-136 (2000).
36. C. D. Sherrill, A. I. Krylov, E. F. C. Byrd, and M. Head-Gordon, J. Chem. Phys., 109, No. 11, 4171-4181 (1998).
37. K. Yankowski and K. Rubiniec, Mol. Phys., 100, No. 11, 1741-1754 (2002).
38. P. Pulay and T. P. Hamilton, J. Chem. Phys., 88, No. 8, 4926-4933 (1988).
39. I. Murata, Pure & Appl. Chem., 65, No. 1, 97-103 (1993).
40. V. N. Staroverov and E. R . Davidson, J. Am. Chem. Soc., 122, No. 1, 186/187 (2000).
41. J. Fernández-Rossier and J. J. Palacios, Phys. Rev. Lett., 99, No. 17, 177204-1-177204-4 (2007).
42. M. R. Philpott and Y. Kawazoe, J. Chem. Phys., 134, No. 12, 124706-1-124706-9 (2011).
43. A. S . Barnard and I. K. Snook, Modelling Simul. Mater. Sci. Eng., 19, No. 5, 054001 (2011).
44. X. Jia, J. Campos-Delgado, M. Terrones, V. Meunier, et al., Nanoscale, 3, No. 1, 86-95 (2011).
45. M. Acik and Y. J. Chabal, J. Appl. Phys., 50, 070101-1-070101-16 (2011).
46. R. Zan, Q. M. Ramasse, U. Bangert, and K. S. Novoselov, Nano Lett., 12, No. 8, 3936-3940 (2012).
47. Y. Liu, A. Dobrinsky, and B. I. Yakobson, Phys. Rev. Lett., 105, No. 23, 235502-1-235502-4 (2010).
48. A. V. Luzanov, J. Struct. Chem., 54, No. 1, 1-9 (2013).
49. P. Szakács, Á. Szabados, and P. R . Surján, Chem. Phys. Lett., 498, Nos. 4-6, 292-295 (2010).
50. V. V. Ivanov, I. P. Kisil, and A. V. Luzanov, J. Sruct. Chem., 37, No. 4, 537-543 (1996).
Copyright © 2022 FDOKUMEN




















