
Marked action potential prolongation as a source of injury current leading to border zone...
11
Volume 127, Number 6 American Heml Journal Kupersmith, Li, and Maldonado 13. 14. 15. 16. 17. effective in treating neurocardiogenic syncope [Abstract]. J Am Co11 Cardiol 1992;3:339A. Abboud FM. Ventricular syncope: is the heart a sensory organ? N Engl J Med 1989;320:390-2. Pongiglione G, Fish FA, Strasburger JF, Benson W. Heart rate and blood pressure response to upright tilt in young patients with unexplained syncope. J Am Co11 Cardiol 1990;16:165-70. Mark AL. The Bezold-Jarish reflex revisited: clinical implica- tions in inhibiting reflexes originating in the heart. J Am Co11 Cardiol 1983;1:90-102. Oberg B, Thoren P. Increased activity in left ventricular receptors during hemorrhage or occlusion of canal veins in the cat: a possible cause of the vaaovagal reaction. Acta Physiol Stand-1972;85:164-73. Epstein SE, Stampfer M, Beiser GD. Role of capacitance and resistance vessels in vasovagal syncope. Circulation 1968; 37:524-33. 18. Perry JC, Garson A. The child with recurrent syncope: auto- nomic function testing and beta-adrenergic hypersensitivity. J Am Co11 Cardiol 1991;17:1168-71. 19. Fitzpatrick AP, Sutton R. Tilt-induced syncope [Letter]. N Engl J Med 1989;321:31. 20. Lippman N, Lerman BB. Differential responses to therapy with beta-blockers in patients with isoproterenol-dependent and independent neurocardiac syncope [Abstract]. Circula- tion 1992$6(suppl l):I-578. 21. Thames MD. Effect of d- and 1-nronranolol on the discharge of cardiac vagal C fibers. Am J Physiol 1980;238:H465-70. - 22. Thoren P. Role of cardiac C-fibers in cardiovascular control. Rev Physiol Biochem Pharmacol 1979;86:1-94. 23. Milstein S, Buetikofer J, Dunnigan A, Benditt DG, Gornick C, Reyes WJ. Usefulness of disopyramide for prevention of up- right tilt-induced hypotension-bradycardia. Am J Cardiol 1990;65:1339-44. Marked action potential prolongation as a source of injury current leading to border zone arrhythmogenesis The objective of this study was to delineate electrophysiologic phenomena in a border zone adjacent to a zone of marked action potential prolongation. By means of a standard microelectrode technique, we studied sheep Purkinje fibers placed in a partitioned chamber and superfused with Tyrode’s solution. Ethylenediame tetraacetic acid (EDTA) was added to one chamber. Recordings were made in the abnormal segment (ABN) superfused with EDTA and at two sites in the normal segment (NL)-at the border within 0.5 mm (NL-8) and 3 to 4 mm from the partition (NL-D). Exposure of ABN to EDTA caused marked prolongation of the action potential duration (APD) and triggered activations (TAs), which were found to have the earliest recorded activation at NL-B (n = 20), at ABN (n = 8) or at both sites (n = 12). NL-B recordings displayed prolonged low-amplitude secondary plateaus, which were termed “border zone early afterdepolarizations.” These were coincident with the plateaus of the prolonged action potentials in ABN and appeared to be due to electrotonic transmission of current from ABN to NL-B. Border zone TAs arose from these low-amplitude plateaus and were either eliminated by the addition of lidocaine to NL consistent with their presumed NL site of origin or occurred after localized withdrawal of EDTA from one segment in fibers rendered quiescent at the plateau by generalized superfusion with EDTA. In conclusion, APD and membrane potential inhomogeneities lead to electrotonic transmission of injury current to border zones adjacent to zones of abnormal APD prolongation. This injury current leads to TAs originating at the border zone. These findings may be relevant to the role of injury current in clinical arrhythmias. (AM HEART J 1994;127:1543-53.) Joel Kupersmith, MD,” Zhen-Yuan Li, PhD,a and Claudio Maldonado, MSb East Lansing, Mich., and Louisville, Ky. From %he Department of Medicine, College of Human Medicine, Michigan State University; and bthe Cardiovascular Division, Department of Medi- cine, University of Louisville School of Medicine. Received for publication March 22, 1993; accepted Oct. 15, 1993. Reprint requests: Joel Kupersmith, MD, Michigan State University, De- partment of Medicine, B338 Clinical Center, East Lansing, MI 48824. Copyright @I 1994 by Mosby-Year Book, Inc. 0002-8703/94/$3.00 + 0 4/l/53801 Injury currents have long been postulated to be a mechanism of arrhythmias.le5 In these postulates it is presumed that differences in ionic potential exist be- tween contiguous zones of the heart. Current there- fore flows from one zone to another and causes elec- trophysiologic abnormalities. These abnormalities could assume various forms. In one form injury cur- 1543
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Marked action potential prolongation as a source of injury current leading to border zone...

Volume 127, Number 6
American Heml Journal Kupersmith, Li, and Maldonado
13.
14.
15.
16.
17.
effective in treating neurocardiogenic syncope [Abstract]. J Am Co11 Cardiol 1992;3:339A. Abboud FM. Ventricular syncope: is the heart a sensory organ? N Engl J Med 1989;320:390-2. Pongiglione G, Fish FA, Strasburger JF, Benson W. Heart rate and blood pressure response to upright tilt in young patients with unexplained syncope. J Am Co11 Cardiol 1990;16:165-70. Mark AL. The Bezold-Jarish reflex revisited: clinical implica- tions in inhibiting reflexes originating in the heart. J Am Co11 Cardiol 1983;1:90-102. Oberg B, Thoren P. Increased activity in left ventricular receptors during hemorrhage or occlusion of canal veins in the cat: a possible cause of the vaaovagal reaction. Acta Physiol Stand-1972;85:164-73. Epstein SE, Stampfer M, Beiser GD. Role of capacitance and resistance vessels in vasovagal syncope. Circulation 1968; 37:524-33.
18. Perry JC, Garson A. The child with recurrent syncope: auto- nomic function testing and beta-adrenergic hypersensitivity. J Am Co11 Cardiol 1991;17:1168-71.
19. Fitzpatrick AP, Sutton R. Tilt-induced syncope [Letter]. N Engl J Med 1989;321:31.
20. Lippman N, Lerman BB. Differential responses to therapy with beta-blockers in patients with isoproterenol-dependent and independent neurocardiac syncope [Abstract]. Circula- tion 1992$6(suppl l):I-578.
21. Thames MD. Effect of d- and 1-nronranolol on the discharge of cardiac vagal C fibers. Am J Physiol 1980;238:H465-70. -
22. Thoren P. Role of cardiac C-fibers in cardiovascular control. Rev Physiol Biochem Pharmacol 1979;86:1-94.
23. Milstein S, Buetikofer J, Dunnigan A, Benditt DG, Gornick C, Reyes WJ. Usefulness of disopyramide for prevention of up- right tilt-induced hypotension-bradycardia. Am J Cardiol 1990;65:1339-44.
Marked action potential prolongation as a source of injury current leading to border zone arrhythmogenesis
The objective of this study was to delineate electrophysiologic phenomena in a border zone adjacent to a zone of marked action potential prolongation. By means of a standard microelectrode technique, we studied sheep Purkinje fibers placed in a partitioned chamber and superfused with Tyrode’s solution. Ethylenediame tetraacetic acid (EDTA) was added to one chamber. Recordings were made in the abnormal segment (ABN) superfused with EDTA and at two sites in the normal segment (NL)-at the border within 0.5 mm (NL-8) and 3 to 4 mm from the partition (NL-D). Exposure of ABN to EDTA caused marked prolongation of the action potential duration (APD) and triggered activations (TAs), which were found to have the earliest recorded activation at NL-B (n = 20), at ABN (n = 8) or at both sites (n = 12). NL-B recordings displayed prolonged low-amplitude secondary plateaus, which were termed “border zone early afterdepolarizations.” These were coincident with the plateaus of the prolonged action potentials in ABN and appeared to be due to electrotonic transmission of current from ABN to NL-B. Border zone TAs arose from these low-amplitude plateaus and were either eliminated by the addition of lidocaine to NL consistent with their presumed NL site of origin or occurred after localized withdrawal of EDTA from one segment in fibers rendered quiescent at the plateau by generalized superfusion with EDTA. In conclusion, APD and membrane potential inhomogeneities lead to electrotonic transmission of injury current to border zones adjacent to zones of abnormal APD prolongation. This injury current leads to TAs originating at the border zone. These findings may be relevant to the role of injury current in clinical arrhythmias. (AM HEART J 1994;127:1543-53.)
Joel Kupersmith, MD,” Zhen-Yuan Li, PhD,a and Claudio Maldonado, MSb
East Lansing, Mich., and Louisville, Ky.
From %he Department of Medicine, College of Human Medicine, Michigan State University; and bthe Cardiovascular Division, Department of Medi- cine, University of Louisville School of Medicine.
Received for publication March 22, 1993; accepted Oct. 15, 1993.
Reprint requests: Joel Kupersmith, MD, Michigan State University, De- partment of Medicine, B338 Clinical Center, East Lansing, MI 48824.
Copyright @I 1994 by Mosby-Year Book, Inc. 0002-8703/94/$3.00 + 0 4/l/53801
Injury currents have long been postulated to be a mechanism of arrhythmias.le5 In these postulates it is presumed that differences in ionic potential exist be- tween contiguous zones of the heart. Current there- fore flows from one zone to another and causes elec- trophysiologic abnormalities. These abnormalities could assume various forms. In one form injury cur-
1543

June 1994 1544 Kupersmith, Li, and Maldonado American Heart Journal
rent could lead to enhanced automaticity, that is, an increased intrinsic firing rate, which would lead to arrhythmias. We have been examining these types of phenomena in in vitro microelectrode preparations of Purkinje fibers in a double-chamber bath. This bath
is designed so that localized electrophysiologic ab- normalities can be induced and interactions between normal and abnormal segments of Purkinje fibers studied. We induced marked prolongation of the ac- tion potential duration, which is known to be associ- ated with triggered activations (TAs), that is, non- driven action potentials that follow and are caused by previous action potentials.
In our double-chambered bath preparation we found evidence of another mechanism of triggered activity. The border zone between normal and ab- normal segments is highly reactive, and we found that border zones displayed action potentials with low-amplitude secondary plateaus.6-8 These plateaus corresponded to and appeared to represent electro- tonic transmission of depolarizing current from the long plateaus present in the abnormal segment. In addition, certain TAs in fact seemed to arise in the border zone and be conducted into abnormal and more distal normal segments.8 These observations are consistent with the suggestion that “injury cur- rent” arising from prolonged action potentials in the abnormal segment moves into the normal segment border zone and causes automatic activity (rapid fir- ing).g This rapid firing could translate into clinical arrhythmias. Thus the border zone is a possible site of arrhythmogenesis, a concept that has considerable clinical implications. In the present study we exam- ined this postulate.
METHODS Experimental preparation. The hearts of 26 sheep were
removed within 2 to 3 minutes after the animals were killed and quickly rinsed in chilled Tyrode’s solution with the following composition (in mmol/L): NaCI, 137; NaHCOa, 24; NaHzPOb, 1.8; MgC12, 0.5; CaC12, 1.8; KCl, 4.0; and dextrose, 5.3 and a pH of 7.3 to 7.4. Free-running false ten- dons containing Purkinje fibers and muscle from both ventricles were removed, placed in chilled Tyrode’s solu- tion, given a gas mixture of 95 % oxygen and 5 % carbon di- oxide, and quickly transported to the electrophysiology laboratory. Preparations were then immediately immersed in a lucite tissue bath superfused with Tyrode’s solution at 5 to 10 ml/min and maintained at 35’ to 37’ C by means of a glass heat exchanger.
The preparations were longitudinally separated into two segments by use of a Plexiglas partition with a 2 mm semi- circular opening at the bottom to allow for passage of the fiber. Silicone jelly was applied around the opening and along the edges of the partition to completely isolate the two chambers. Each chamber had its own inflow and out-
flow to allow for independent superfusion. Each chamber was also individually grounded. Absence of interchamber leakage was established after recording stimuli and ground- ing electrodes were set up at each end of the preparations. While recording with both microelectrodes, we confirmed the absence of leakage by removing a grounding electrode from one of the chambers. If the noise level increased con- siderably, we considered the two chambers to be well iso- lated. In addition, when ethylenediame tetraacetic acid (EDTA) was added to one chamber, the configuration of the stimulated action potential from the opposite chamber was observed. Lack of change in the configuration indi- cated a good seal of the partition.
Recording and stimulating techniques. After allowing 1 hour for stabilization of the preparation, standard mi- croelectrode techniques were used to record transmem- brane potential characteristics. Simultaneous recordings were obtained from the preparation on both sides of the partition by means of glass microelectrodes (Narashige model PE-2, Narashige Co., Ltd., Tokyo, Japan) filled with 3 mol/L potassium chloride and with a tip resistance of 10 to 30 mega Q. Signals were fed into an amplifier (WP Instruments M707) with high-input impedance (1014 Q) and input capacity neutralization. Recordings were dis- played on an oscilloscope (Tektronix, Inc., Beaverton, Ore.) and recorded on a Gould 2200 brush recorder (Gould, Inc., Valley View, Ohio). Data were also recorded, digitized, and analyzed by Computer Scope ISC-67, a hardware- software system (R. C. Electronics, Santa Barbara, Calif.). This information was then displayed in analogue form at high speed for precise timing of events. Bipolar Teflon- coated silver-silver electrodes were placed near the free ends of each segment to allow for stimulation of the prep- aration on either side. Pacing when performed was achieved with a WP Instruments stimulator delivering rectangular stimuli with a pulse width of 2 msec and an amplitude of 5 to 7 mA.
Experimental protocol. Purkinje fibers were impaled by microelectrodes in the following three locations: (1) 3 to 4 mm from the partition in one chamber, (2) within 0.5 mm from the partition, and (3) 3 to 4 mm from the partition in the other chamber (Fig. 1). After baseline recordings were obtained, EDTA (3.3 mmol/L) was added to the chamber with the single microelectrode (Fig. 1) while continuing to superfuse the other chamber with unmodified Tyrode’s so- lution. Continuous recordings were made during selective superfusion of EDTA to this “abnormal side” (ABN) and to the normal side (NL) at the border within 0.5 mm from the partition (NL-B) and at the more distal site 3 to 4 mm
from the partition (NL-D). A total of 40 experiments were performed (Table I). In
certain experiments the protocol was varied as follows: lidocaine (10 mg/L or 21.3 mmol/L) was selectively added to NL (n = lo), and superfusion of both chambers with EDTA (3.3 mmol/L) was followed by selective withdrawal of EDTA from NL (n = 7). All values were reported as means ? standard error. The investigation conformed to the “Guide for the care and use of laboratory animals,” published by the National Institutes of Health.

Volume 127, Number 6
American Heart Journal Kupersmith, Li, and Maldonado 1545
ABN NL
4 3 2 1 1234 mm mm
Fig. 1. Schematic diagram of two-chambered tissue bath. In each experiment three microelectrodes were impaled in Purkinje fiber (PF), one in the abnormal segment (ABN) 3 to 4 mm from the partition (P), one in the normal segment (NL) border zone (NL-B) up to 0.5 mm from the partition, and another at the more distal site (NL-D) 3 to 4 mm from the partition.
Terminology. Terminology concerning early afterdepo- larizations (EADs), triggered activity, and abnormal action potential prolongation has been somewhat variable. For the purposes of this study we used the following terminol- ogy, which we believe conforms to that of Cranefield and Aronsonl” with some modifications based on our findings (we recognize that it represents somewhat of a departure from previous terminology inasmuch as these are newly described phenomena).
Early afterdepolarizations. These are afterdepolariza- tions that interrupt or delay phase 3 of the action poten- tial and prolong it. lo In this study we were concerned with the following two types of EADs: those originating in ABN and those observed at NL-B sites. EADs at NL-B sites ap- peared to be associated with electrotonic transmission of current from the plateaus of abnormally prolonged action potentials in ABN (described later in this article). We termed the latter “border zone EADs.”
Triggered activations. These nondriven activations oc- cur in the presence of EADs (see also under Discussion- activation originating in the border zone).
Maximum diastolic potential and take-off potential. In general, maximum diastolic potential refers to the maxi- mum negative potential at the end of phase 3 of the action potential. However, in the preparations we studied there were multiple activations before full repolarization in ABN and NL-B, that is, the definition of diastole represented somewhat of a problem. The maximum diastolic potential after these activations was often relevant to our discussion. We used the term “maximum postactivation potential” (MPAP) to define the maximum potential that occurred in a sequence before full repolarization. We used the term “take-off potential” in the conventional sense, that is, ac- tivation voltage.
RESULTS
Fig. 2 shows experiments in which TAs arose in ABN and were conducted to NL. Fig. 2, A shows baseline recordings when both segments were super-
Table I. Experiment protocol and results
Border Abnormal zone zone
Intervention Total TAs TAs Both
All experiments 40 20 8 12 Lidocaine to normal zone* 10 10 Selective EDTA washout 7 7 -
*Lidocaine was added in experiments with border zone TAs only.
fused in unmodified Tyrode’s solution. Fig. 2, B and C show recordings 20 and 23 minutes after exposure of ABN to EDTA (3.3 mmol/L). Action potentials in ABN were markedly prolonged and recordings dis- played TAs, whereas NL-B and NL-D recordings displayed activity corresponding to TAs. Fig. 2, D shows an analogue printout of a high-speed recording of an example of TA. Note that the earliest recorded activation was in ABN. Fig. 2, E shows a return to baseline after washout of EDTA from ABN.
Fig. 3 shows an experiment in which the earliest recorded TAs were in NL-B rather than ABN. Fig. 3, A shows baseline recordings during superfusion of both segments with unmodified Tyrode’s solution. Fig. 3, B and C show results of selective exposure of ABN to EDTA. Action potential in ABN became prolonged, and activations occurred in ABN (Fig. 3, B and C) with corresponding activations in NL-B and NL-D. However, as the example of TA in Fig. 3, D shows, in contrast to that in Fig. 2, D, the earliest re- corded timing of these activations was at NL-B with later timing at ABN and NL-D. We will refer to this type of TA as a border zone TA. Fig. 3, E shows a re- turn to baseline after washout of EDTA from ABN.
These border zone TAs were of particular interest.
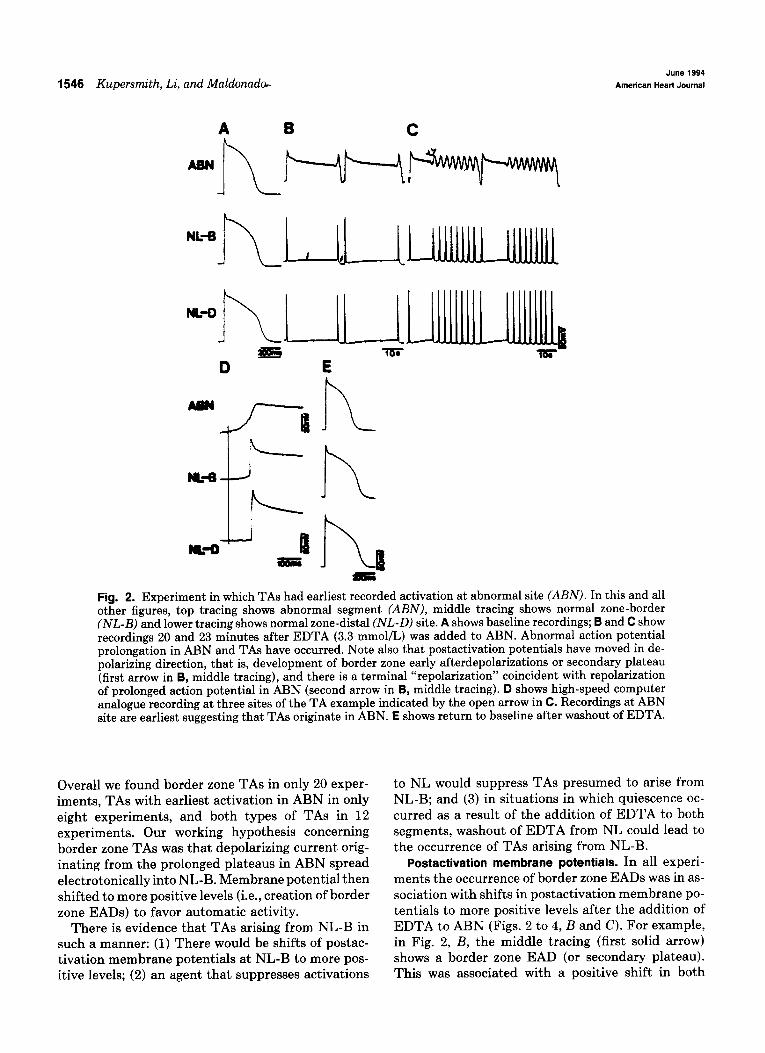
1546 Kupersmith, Li, and Maldonador June 1994
American Heart Journal
h
Fig. 2. Experiment in which TAs had earliest recorded activation at abnormal site (ABN). In this and all other figures, top tracing shows abnormal segment (ABN), middle tracing shows normal zone-border (NL-B) and lower tracing shows normal zone-distal (NL-D) site. A shows baseline recordings; B and C show recordings 20 and 23 minutes after EDTA (3.3 mmol/L) was added to ABN. Abnormal action potential prolongation in ABN and TAs have occurred. Note also that postactivation potentials have moved in de- polarizing direction, that is, development of border zone early afterdepolarizations or secondary plateau (first arrow in B, middle tracing), and there is a terminal “repolarization” coincident with repolarization of prolonged action potential in ABN (second arrow in B, middle tracing). D shows high-speed computer analogue recording at three sites of the TA example indicated by the open arrow in C. Recordings at ABN site are earliest suggesting that TAs originate in ABN. E shows return to baseline after washout of EDTA.
Overall we found border zone TAs in only 20 exper- iments, TAs with earliest activation in ABN in only eight experiments, and both types of TAs in 12 experiments. Our working hypothesis concerning border zone TAs was that depolarizing current orig- inating from the prolonged plateaus in ABN spread electrotonically into NL-B. Membrane potential then shifted to more positive levels (i.e., creation of border zone EADs) to favor automatic activity.
There is evidence that TAs arising from NL-B in such a manner: (1) There would be shifts of postac- tivation membrane potentials at NL-B to more pos- itive levels; (2) an agent that suppresses activations
to NL would suppress TAs presumed to arise from NL-B; and (3) in situations in which quiescence oc- curred as a result of the addition of EDTA to both segments, washout of EDTA from NL could lead to the occurrence of TAs arising from NL-B.
Postactivation membrane potentials. In all experi- ments the occurrence of border zone EADs was in as- sociation with shifts in postactivation membrane po- tentials to more positive levels after the addition of EDTA to ABN (Figs. 2 to 4, B and C). For example, in Fig. 2, B, the middle tracing (first solid arrow) shows a border zone EAD (or secondary plateau). This was associated with a positive shift in both

Volume 127, Number 6
American Heart Journal Kupersmith, Li, and Maldonado 1547
Fig. 3. Example of experiment in which earliest recorded activations were timed at NL-B. A shows base- line recordings. B and C show recordings 16 and 18 minutes after addition of EDTA (3.3 mmol/L) to ABN. Note again development of border zone EADs and TAs. D shows high-speed computer analogue printout of TA example indicated by open arrow in C. NL-B recording is earliest suggesting that TAs arise in this region. E shows return to baseline after washout of EDTA.
MPAP and take-off potential. Such border zone EADs have been reported previously under similar conditions.g The second solid arrow in Fig. 2, B shows repolarization of the secondary plateau (or border zone EAD). This repolarization was coincident with repolarization of the action potential in ABN. The same phenomenon was seen in Fig. 3, B and C, and occurred in all experiments.
Fig. 4 shows the mean membrane potentials in NL-B before and after the addition of EDTA to ABN in 10 experiments in which values could be obtained for a wide range of cycle lengths including the longer intervals. Mean MPAPs of the first border zone ac- tivation and take-off potentials of the subsequent activation are shown before (Fig. 4, A) and after (Fig. 4, B) the selective addition of EDTA to ABN. All cy- cle lengths shown were obtained during pacing. In all
10 of these experiments TAs arose entirely in the border zone. Both MPAP and TAs shifted in a pos- itive direction at all cycle lengths.
Effects of lidocaine. Our postulate is that injury current transmitted into NL enhances automaticity leading to TAs and that TAs therefore arise in NL rather than ABN. If this postulate is correct, the se- lective addition of an agent to depress automaticity in NL would inhibit TAs. Lidocaine is known to sup- press automaticity among its other actions. Fig. 5 shows the effects of the selective addition of lidocaine to NL in an experiment in which border zone TAs had occurred. After selective exposure of ABN to EDTA (Fig. 5, B), TAs have occurred with the earliest recorded activation in NL-B (example in Fig. 5, C). After EDTA, changes in MPAP and take-off poten- tial of TAs were similar to those in other experiments.

1548 Kupersmith, Li, and Maldonudo June 1994
American Heart Journal
A -60
T
-100 * 0.0 0.1 0.2 0.3 0.4 0.6 0.6 0.7 0.6 6.9 1.0
HZ
-60 T
- 1004 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.6 0.9 1.0
HZ
Fig. 4. Plot of transmembrane potentials at NL-B sites in 10 experiments. In each of these experiments, values could be obtained over a wide range of cycle lengths including rather long intervals. Horizontal axis is cycle length in Herz (Hz), and vertical axis is transmembrane potential in millivolts (mV). Solid line represents MPAP and dotted line indicates takeoff potential. A represents values at baseline, and B rep- resents values obtained after addition of EDTA (3.3 mmol/L) to ABN. All cycle lengths were paced. Note that both MPAP and takeoff potentials shift in positive direction in B.

Volume 127, Number 6
American Heart Journal Kupersmith, Li, and Maldonado 1549
Jar Mr Fig. 5. Effects of lidocaine. A shows baseline recordings. B shows recordings made after addition of EDTA to ABN. There is a 4-minute gap in 8. Abnormal action potential prolongation and TAs have occurred. Open arrow shows TA example occurred in C. As shown in C, the TAs have earliest recorded activation at NL-B site. D shows recordings made 10 minutes after addition of lidocaine (21.3 mmol/L+ 10 mg/L) selectively to NL. TAs were suppressed and fiber became quiescent. E shows recordings made 8 minutes after washout of lidocaine in NL. TAs returned.
After the selective addition of lidocaine to NL (Fig. 5, D), border zone TAs were abolished. After washout of lidocaine (Fig. 5, E), fibers returned to the post- EDTA state. This experiment shows that lidocaine selectively added to NL abolished border zone TAs. This finding is consistent with these activations in fact having their site of origin at NL-B, similar results were obtained in all 10 experiments in which lidocaine was added in this manner.
Reversal of quiescence at plateau. EDTA caused prolongation of the APD, and this state frequently progressed to arrest at the plateau level of the mem- brane potential, that is, the action potential never repolarized. In seven experiments EDTA was first added to both chambers of the bath, leading to a state in which there was arrest at the plateau level in both chambers, that is, the fiber was quiescent (Table I). Our postulate was that withdrawal of EDTA from one chamber would lead to an electrophysiologic im-
balance and inhomogeneities favoring flow of injury current to cause the type of abnormalities previously described.
Fig. 6 shows an example of this phenomenon. Here EDTA was initially superfused into both chambers and was then selectively withdrawn from NL. Expo- sure of both chambers to EDTA at first (10 to 15 minutes) caused TAs to occur. These recordings are not shown. Later, action potentials would arrest at plateau level with a homogeneous membrane poten- tial throughout the fiber (Fig. 6, B). The gap in this figure was 10 minutes during which time EDTA was selectively washed out of NL. Subsequently the fiber repolarized in NL-B and NL-D, creating an inhomo- geneity of membrane potential, and this was followed by a period during which TAs occurred. The earliest recorded activation of these TAs was in NL-B (ex- ample in Fig. 6, C). After washout of EDTA from ABN, the preceding changes reverted to baseline

1660 Kupersmith, Li, and Maldonado June 1994
American Hearl Journal
Fig. 6. Generalized superfusion of fiber with EDTA followed by selective withdrawal of EDTA from one side. A shows baseline recordings. EDTA was then added to both chambers. Initially TAs occurred (not shown), but then action potential would stop at plateau level as shown in 6, which was recorded after ad- dition of EDTA (3.3 mmol/L) to both chambers. Gap in B was 10 minutes, during which time EDTA was selectively washed out of NL. Fiber on NL side repolarized and then TAs occurred. C shows that earliest recorded activations in latter part of B (example indicated by open arrow) were timed at NL-B site. D and E show washout of EDTA from ABN. D shows recordings while EDTA was being washed out with reversal of automatic activity. Solid arrow shows one aced heat. E shows final washout at 25 minutes.
(Fig. 6, D and E). Thus selective washout of EDTA from one segment of fiber that was quiescent at the plateau led to the initiation of border zone TAs in association with inhomogeneity of the APD and membrane potential.
DISCUSSION
The most interesting observations in the present study were the events in the border zone. We ob- served border zone EADs and TAs. Border zone EADs appeared to be due to electrotonic spread of depolarizing “injury” current from the prolonged ac- tion potentials in ABN. Fig. 7 shows a schematic di- agram of our interpretation of the data. As shown in Fig. 7, A there is marked inhomogeneity of the APD. As shown in Fig. 7, B this leads to movement of cur- rent from the abnormal zone with a longer action po- tential to the normal zone causing enhanced auto- maticity, which in turn causes TAs.
Border zone TAs and pacemaker activity appear to be related to the fact that EADs induced by injury current moved membrane potential at this site in a positive direction (Fig. 4), presumably up to the threshold for inward Na+ current. This injury cur- rent was derived from the APD inhomogeneities cre- ated by the selective addition of EDTA to ABN. Ev- idence favoring this interpretation regarding border zone TAs includes the following: (1) depolarizing movement of postactivation membrane potential in the border zone, (2) observation of earliest recorded TAs in the border zone, (3) temporal coincidence of border zone EADs with phases 2 and 3 of the abnor- mally prolonged action potentials in ABN, (4) the observation that lidocaine added selectively to NL abolished border zone TAs (this presumably was due to lidocaine-induced suppression of automaticity in the region), and (5) induction of border zone TAs by selective local withdrawal of EDTA in fibers previ-

Volume 127, Number 6
American Heart Journal Kupersmith, Li, and Maldonado 1551
B
ABN
BZ
I%- n!!JL
Fig. 7. Schematic diagram of border zone automaticity based on findings in this study. A, Abnormally prolonged (ABN) and normal border zone (BZ) action potential. Open arrow shows movement of injury current from ABN to BZ. B, Injury current leads to enhanced automaticity, which in turn causes border zone TA. This TA then conducts back to ABN (solid arrow).
ously rendered quiescent at the plateau by general- ized superfusion with EDTA.
hjury currents. Injury currents have been a long- standing concept of arrhythmia formation. They have been thought to lead to automatic activity,ll* l2 focal reexcitation,il-l3 or reentry via slowed conduc- tion.12s l4 However, the amount of experimental evi- dence in favor of these concepts has been limited. It is known that direct application of prolonged depo- larizing current to Purkinje fibers in diastole en- hances pacemaker activity. Trautwein and Kasse- baum15 showed that depolarizing current applied in diastole leads to repetitive activations or prolonged damped oscillations not reaching threshold (these resembled our border zone EADs) (Fig. 2 in reference 16). Spread of depolarizing injury current as a mech- anism of enhanced automaticity was observed by Sano and Sawanoborig adjacent to a zone in which membrane potential was depolarized via EDTA (ca- nine false tendon) and by Katzung et a1.16 adjacent to a zone superfused with high-K+ solution (guinea pig papillary muscle).
Injury currents similar to those in our experiments may also arise from disparities in the timing of action potential repolarization as a result of conduction block. In a porcine model of ischemia, Janse et a1.17 attributed arrhythmia to this mechanism. They found evidence that injury currents arising in ischemic zones induced pacemaker activity in adjacent normal zones, although they also attributed ischemic ar- rhythmias to reentrant wavelets. Because delayed repolarization was considered necessary to explain the injury currents in their preparation, delayed
conduction to ischemic tissue causing delayed repo- larization and 2:l block causing prolonged APD were invoked as explanations. I8 Computer simulations were consistent with this hypothesis in showing that injury currents arising because of conduction-in- duced delays in repolarization could spread to neigh- boring zones and enhance automaticity.i8 These findings were also considered consistent with the findings of Harris and Rojaslg that ectopic impulses after coronary occlusion appeared to originate in the border zone. Also of interest were observations of prolonged action potentials in dog Purkinje fibers isolated from l-day-old infarcted hearts.20l 21
Electrotonic current from the plateaus of action potentials has also been observed as a mechanism of reentry via reflection. 22-25 In this model a three- chamber tissue bath with an inexcitable or “ischem- ic” gap in the center was used. The gap created a block, but it also allowed electrotonic current to pass through. Action potentials on one side of this gap, appropriately timed with diastole on the other side, led to enhanced automatic activity. With critical timing of stimulated action potentials, the electro- tonic current resulted in trains of reflected reentrant beats or sequences resembling parasystole or other arrhythmias.
In all of these observations diffusion barriers are important. An important mechanism in creating dif- fusion barriers in acute ischemia may be related to osmotic changes.26-30 In ischemia, metabolism of gly- cogen and high-energy phosphate causes hyperosm- ilarity, which in turn leads to water retention, cellu- lar swelling, and edema of the extracellular space, as

June 1994 7552 Kupersmith, Li, and Maldonado American Heart Journal
well as increased tissue K+ levels. Reperfusion in- creases this water load. The edema occurs rapidly and persists for 24 hours. Among its consequences are mechanical stress on the membrane, capillary com- pression, and the no-refill phenomenon.26-30 Ische- mia-induced edema could also cause changes in elec- trical resistivity and isotropy31 (both related to increased extracellular resistance as a result of cellu- lar swelling per se and increased intracellular K+.). The increased intracellular resistance could cause an increase in coupling resistance. These changes in cel- lular hydration are thus a possible mechanism by which an abrupt electrical barrier could occur related to tissue ischemia.
The preceding ischemia and reflection models are consistent with our findings of enhanced automatic- ity as a result of spread of injury current arising from disparities in action potential plateaus. However, in both there were areas of blockage that created the conduction delays necessary for the disparities. Blockage was not necessary for this purpose in our experiments, because the current sources were action potentials that were very greatly lengthened. On the other hand, it has also been thought that such an in- tervening site of high resistance was necessary to maintain APD inhomogeneities because low-resis- tance connections between cells tend to equalize APD.32 Our observations were not consistent with this suggestion at least insofar as action potentials of greatly varying duration are concerned.
Experimental considerations. One consideration is that with electrodes in only three loci we did not map the entire fiber externally. It was also not possible to map the fiber internally. Therefore we may not have precisely localized the pacemaker cells in the border zone. However, the lidocaine data (Fig. 5) clearly es- tablished that the site of origin of the border zone TAs was in NL. High-speed recordings (e.g., Fig. 5, D) established a sequence of activation that appeared to place our border zone electrodes in reasonably close proximity (at least temporally) to the origin of these TAs. Presumably border zone TAs would originate in cells close to the partition, where border zone EADs were most prominent and membrane potential most depolarized. They would not be likely to arise at the more distant NL-D sites. Because there was a phys- ical partition between segments, there might have been some encroachment on the fiber to create blocking of impulses. However, there was no evidence of blockage across the partition to induce any of the phenomena we noted.
Inasmuch as there was a concentration gradient of EDTA across the partition, there may have been
some extracellular movement of EDTA within the fiber from ABN into NL. Although this movement could have caused border zone abnormalities, it is not a likely explanation for our findings. Bath flow should wash out most of the EDTA that arrives in NL. In addition, findings in NL would be those related to a low concentration of EDTA (e.g., minimal action po- tential lengthening) rather than the unusual action potentials we recorded with border zone EADs, which were related to electrotonic interaction. Action po- tentials with this configuration are not those seen with EDTA alone.
Our experimental setting may differ in a number of ways from the setting of arrhythmias in situ. EDTA is of course not a clinical cause of arrhythmias; how- ever, we used this agent only as a “pharmacologic current source” via APD prolongation. Another dif- ference is the fact that the demarcation between ABN and NL in our experiments was abrupt and regular. In disease states this demarcation may not be abrupt, although it probably is in many arrhythmic states such as chronic infarction and fibrosis (this is less clear for acute ischemia). On the other hand, the demarcations between diseased and normal tissue are almost certainly irregular, but this condition does not alter the implications of our findings. Such irreg- ularities in fact make for interesting possibilities, for example, the creation of peninsulas of normal or ab- normal tissue in which certain kinds of summation may occur.
Clinical relevance. Our observations of a role for in- jury current may have clinical relevance in many sit- uations. Although we should stress that EDTA is not associated with any clinical arrhythmias, APD or membrane potential inhomogeneities can occur in infarction, ischemia33p 34 (in part related to osmotic shifts),26-30 chronic fibrotic myocardiopathies, and those torsade de pointes associated with focal rather than generalized APD prolongation. These inhomo- geneities can lead to injury current, consequent TAs, and therefore tachyarrhythmias by the mechanisms shown in Fig. 7. One possible type of arrhythmia would be accelerated idioventricular rhythm. Inter- estingly there may also be relevance for ischemic reperfusion arrhythmias. Fig. 6 shows initial quies- cence at depolarized plateau membrane potentials (perhaps analogous to ischemia) and then border zone TAs arising when localized reversal of abnor- malities caused APD and membrane potential in- homogeneities (perhaps analogous to reperfusion).
In addition, if our model has clinical relevance there are innovative possibilities for treatment of ar- rhythmia. A wide variety of factors may influence ar-

volume 127, Number 6
American Heart Journal Kupersmith, Li, and Maldonado 1553
rhythmogenesis in this model, for example, initial membrane potential levels in both ABN and NL, postactivation slopes in NL-B, and APDs in both ABN and NL. In addition, cable properties at the border, conduction, thresholds, and geometry will all influence the spread of electrotonus and its conse- quences. In particular, changes in intra- and extra- cellular fluid may play a role.26-30 Arrhythmias may thus be enhanced or suppressed by influences on a wide variety of currents and membrane properties.
We thank Dr. Ralph Lazzara for his helpful comments and crit-
icism.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
Il.
12.
13.
14.
15.
Aronson RS. Afterpotentials and triggered activity in hyper- trophied myocardium from rats with renal hypertension. Circ Res 1981;48:720-7. Brachmann J, Scherlag BJ, Rosenshtraukh LV, Lazzara R. Bradycardia-dependent triggered activity: relevance to drug- induced multiform ventricular tachvcardia. Circulation 1983: 68~846-56. Damiano BP, Rosen MR. Effects of pacing on triggered activ- ity induced by early afterdepolarizations. Circulation 1984; 69:1013-25. Schechter E, Freeman CC, Lazzara R. Afterdepolarizations as a mechanism for the long QT syndrome: electrophysiologic studies of a case. J Am Co11 Cardiol 1984,3:1556-61. Kupersmith J, Hoff P. Occurrence and transmission of repo- larization abnormalities in vitro. J Am Co11 Cardiol1985;6:152- 60. Li ZY, Maldonado C, Zee-Cheng C, Kupersmith J. Purkinje fiber-papillary muscle interaction in the genesis of triggered activity in a guinea pig model. Cardiovasc Res 1992;6:543-8. Li ZY, Zee-Cheng C, Maldonado C, Kupersmith J. Early afterdepolarizations in sheep Purkinje fiber-transmission and suppressive effects of magnesium [Abstract]. Clin Res 1988;36:824A. Li ZY, Maldonado C, Hiromasa S, Zee-Cheng C; Kupersmith J. Conduction of early afterdepolarizations in sheep Purkinje fibers and ventricular muscle. An in vitro arrhythmia model. J Electrocardiol 1992;25:119-27. Sano T, Sawanobori T. Abnormal automaticity in canine Purkinje fibers focally subjected to low external concentra- tions of calcium. Circ Res 1972;31:158-64. Cranefield PF, Aronson RS. Cardiac arrhythmias: The role of triggered activity and other mechanisms. New York: Futura Publishing, 1988:12-8. Hoffman BF. The genesis of cardiac arrhythmias. Prog Car- diovasc Dis 1966;8:319-29. Singer D, Ten Eick RE. Pharmacology of cardiac arrhythmias. Prog Cardiovasc Dis 1969;11:488-514. Trautwein W. Generation and conduction of impulses in the heart as affected by drugs. Pharmacol Rev 1963;15:277-332. Duo GS, Hoff P, Kupersmith J. Repolarization interactions between cardiac segments of varying action potential dura- tion. AM HEART J 1989;117:854-61. Trautwein W, Kassebaum DG. On the mechanism of sponta- neous impulse generation in the pacemaker of the heart. J Gen Physiol 1961;45:317-30.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
Katzung BG, Hondeghem LM, Grant AO. Cardiac ventricular automaticity induced by current of injury. Pfliigers Arch 1975;360:193-7. Janse MJ, van Capelle JL, Morsink H, Morsink H, Kleber AG, Wilms-Schopman F, Cardinal R, d’Alnoncourt CN, Durrer D. Flow of “injury” current and patterns of excitation during early ventricular arrhythmias in acute regional myocardial is- chemia in isolated porcine and canine hearts. Evidence for two different arrhythmogenic mechanisms. Circ Res 1980;47:151- 65. Janse MJ, van Capelle FJL. Electrotonic interactions across an inexcitable region as a cause of ectopic activity in acute re- gional myocardial ischemia. A study in intact porcine and ca- nine hearts and computer models. Circ Res 1982;50:527-37. Harris AS, Rojas AG. The initiation of ventricular fibrillation due to coronary occlusion. Proc Sot Exp Med Surg 1943;1:105- 22. Friedman PL, Stewart JR, Fenoglio JJ Jr, Wit AL. Survival of subendocardial Purkinje fibers after extensive myocardial in- farction in dogs. Circ Res 1973;33:597-611. Lazzara R, El-Sherif N, Scherlag BJ. Electrophysiological properties of canine Purkinje cells in one-day-old myocardial infarction. Circ Res 1973;33:722-34. Antzelevitch C. Moe GK. Electrotonicallv mediated delaved conduction and reentry in relation to “slow responses” in mammalian ventricular conducting tissue. Circ Res 1981; 491129-39. Jalife J, Moe G. Effect of electrotonic potentials on pacemaker activity of canine Purkinje fibers in relation to parasystole. Circ Res 1976;39:801-8. Jalife J, Moe GK. Excitation, conduction, and reflection of impulses in isolated bovine and canine cardiac Purkinje fibers. Circ Res 1981;49:233-47. Rozanski GJ, Jalife J, Moe GK. Reflected reentry in nonho- mogeneous ventricular muscle as a mechanism of cardiac ar- rhythmias. Circulation 1984;69:163-73. Powers ER, DiBona DR, Powell WJ. Myocardial cell volume and coronary resistance during diminished coronary perfu- sion. Am J Physiol 1984;H467-76. Jennings RB, Schaper J, Hill ML. Effect of reperfusion late in the phase of reversible ischemic injury. Circ Res 1985;56:262- 78. Jennings RB, Murry CE, Steenbergen C, Reimer KA. Devel- opment of cell injury in sustained acute ischemia. Circulation 1990;82(suppl):II2-12. Tranum-Jensen J, Janse MJ, Fiolet JWT, Krieger WJ, D’Al- noncourt CN, Durrer D. Tissue osmolality, cell swelling, and reperfusion in acute regional myocardial ischemia in the iso- lated porcine heart. Circ Res 1981;49:364-81. Steenbergen C, Hill ML, Jennings RB. Volume regulation and plasma membrane injury in aerobic, anaerobic and ischemic myocardium in vitro. Circ Res 1985;57:864-75. Wojtczak J. Contractures and increase in internal longitudinal resistance of cow ventricular muscle induced by hypoxia. Circ Res 1979;44&3-95. Moe GK, Mendez C. Physiologic basis of premature beats and sustained tachycardias..N En&J Med 1&3;288:250-4. Janse MJ, Cinca J. Morena H. Fiolet JW. Kleber AG. de-Vries GP, Becker AE, Durrer D. The “border’zone” inmyocardial ischemia. An electrophysiological, metabolic, and histochem- ical correlation in the pig heart. Circ Res 1979;44:576-88. Kupersmith J, Shiang H, Litwak RS, Herman MV. Electro- physiological and antiarrhythmic effects of propranolol in ca- nine acute myocardial ischemia. Circ Res 1976;38:302-7.




















