
Magic Polyicosahedral Core-Shell Clusters
15
Magic polyicosahedral core-shell clusters Giulia Rossi, 1 Arnaldo Rapallo, 2 Christine Mottet, 3 Francesca Baletto, 4 Alessandro Fortunelli, 5 Riccardo Ferrando 1* 1 INFM and IMEM/CNR, Dipartimento di Fisica dell’Universit` a di Genova Via Dodecaneso 33, Genova, I16146, Italy 2 ISMAC/CNR, Via Bassini 15, Milano, I20133, Italy 3 CRMCN/CNRS, Campus de Luminy, Marseille, F13288, France 4 ICTP, Strada Costiera 2, Trieste, I34014, Italy 5 IPCF/CNR, Via Alfieri 1, Ghezzano, I56010, Italy * To whom correspondence should be addressed; E-mail: [email protected] A new family of magic cluster structures is found by combining genetic global optimization and density functional calculations. These clusters are Ag-Ni and Ag-Cu nanoparticles with an inner Ni or Cu core and an Ag external shell, as experimentally observed for Ag-Ni, and present polyicosahedral charac- ter. The interplay of the core-shell chemical ordering with the polyicosahedral structural arrangement gives high-symmetry clusters of remarkable struc- tural, thermodynamic and electronic stability, which can have high melting points (they melt higher than pure clusters of the same size), large energy gaps and (in the case of Ag-Ni) non-zero magnetic moments. Bimetallic nanoclusters have received recently a great attention (1) both from the point of view of basic science, because of their peculiar properties, which can be very different from those of the pure nanoclusters of their constituents (2,3), and for a variety of applications, rang- 1
Transcript of Magic Polyicosahedral Core-Shell Clusters

Magic polyicosahedral core-shell clusters
Giulia Rossi,1 Arnaldo Rapallo,2 Christine Mottet,3 Francesca Baletto,4
Alessandro Fortunelli,5 Riccardo Ferrando1∗
1INFM and IMEM/CNR, Dipartimento di Fisica dell’Universita di Genova
Via Dodecaneso 33, Genova, I16146, Italy2ISMAC/CNR, Via Bassini 15, Milano, I20133, Italy
3CRMCN/CNRS, Campus de Luminy, Marseille, F13288, France4ICTP, Strada Costiera 2, Trieste, I34014, Italy
5IPCF/CNR, Via Alfieri 1, Ghezzano, I56010, Italy
∗To whom correspondence should be addressed; E-mail: [email protected]
A new family of magic cluster structures is found by combining genetic global
optimization and density functional calculations. These clusters are Ag-Ni and
Ag-Cu nanoparticles with an inner Ni or Cu core and an Ag external shell,
as experimentally observed for Ag-Ni, and present polyicosahedral charac-
ter. The interplay of the core-shell chemical ordering with the polyicosahedral
structural arrangement gives high-symmetry clusters of remarkable struc-
tural, thermodynamic and electronic stability, which can have high melting
points (they melt higher than pure clusters of the same size), large energy gaps
and (in the case of Ag-Ni) non-zero magnetic moments.
Bimetallic nanoclusters have received recently a great attention (1) both from the point of
view of basic science, because of their peculiar properties, which can be very different from
those of the pure nanoclusters of their constituents (2,3), and for a variety of applications, rang-
1

ing from catalysis to optics (4, 5). What renders bimetallic nanoclusters very attractive is that
their properties can vary dramatically not only with size, as happens in pure nanoclusters, but
also with composition. Thus, a precise control of their structure and chemical ordering can be
a very promising way to prepare the building blocks for specifically tailored cluster-assembled
materials (6). For this application, a key step is to single out magic cluster structures, namely
those structures presenting special structural, and hence electronic and thermodynamic, stabil-
ity. In recent experiments (7), it has been shown that silver-nickel nanoclusters adopt a core-
shell configuration, where a well-defined outer silver shell embeds a nickel core. The analysis
of the low-energy ion scattering data has shown that the silver shell is often of monoatomic
thickness, but a more precise determination of their structure is not available yet.
Here we show theoretically that there is a whole family of new magic Ag-Ni and Ag-Cu
nanoclusters, which are characterized by the common structural properties of being perfect
core-shell clusters and polyicosahedra (pIh). Perfect core-shell clusters have all Ag atoms on
the surface and all Ni or Cu atoms inside. Polyicosahedra are clusters built by packing up
elementary icosahedra of 13 atoms (Ih13), as seen in Fig. 1. We demonstrate that the interplay
between the polyicosahedral structural ordering and the core-shell chemical ordering gives a
net driving force for building up clusters of remarkable energetic stability. Among this family
of core-shell pIh we single out a few high-symmetry clusters presenting special thermodynamic
and electronic stability, with high melting points and large HOMO-LUMO gaps. Some of these
structures are also magnetic in their ground state.
Our computational procedure consists of several steps: (i) we model the clusters by realistic
many-body atom-atom potentials (8), and search for the global energy minimum by genetic
algorithm optimization (9), at some selected sizes and variable composition ; (ii) at a given
size, we look for the compositions corresponding to the most stable structures; (iii) we optimize
the most significant structures locally by Density Functional Theory (DFT) calculations (10) to
2

confirm the trends obtained in (ii) and single out the clusters with high electronic stability; (iv)
we check the thermodynamic stability of the magic structures by making Molecular Dynamics
melting simulations and calculating the temperature-dependent occupation probabilities of the
global minima by harmonic thermodynamics (11).
Why core-shell pIh can be magic structures? A driving force for the formation of high-
stability clusters is the maximization of the number of nearest-neighbor bonds among the atoms.
This favors compact quasi-spherical structures, such as the the Ih13 and other icosahedra. How-
ever, interatomic distances in icosahedra are different from their ideal value in the bulk crystal,
and this produces an internal strain. The competition between the maximization of the bond
number and the accumulation of strain determines the most favorable structures in pure systems
described by pair potentials (12), such as Lennard-Jones (LJ) and Morse. For example, small LJ
clusters prefer the icosahedral shape over the decahedral and the cuboctahedral ones (the latter
being a piece of fcc bulk crystal) (13). Adding further atoms, a series of magic polycosahedral
LJ clusters is produced at sizesN = 19, 23, 26, 29. However, aboveN = 29, the gain in the
bond number (which scales roughly as the cluster surface area) does not compensate any more
the internal strain (which scales as the cluster volume), so that LJ pIh become unfavorable. In
the case of noble and transition metal pure clusters the situation is more complicated, due to
the many-body character of the atom-atom interaction, which tends to make icosahedra less
favorable compared to the case of pair potentials. In fact, the many-body part of the metallic
interaction induces a significantbond order - bond length correlation(14), which means that
the optimal bond lengthincreases with the coordinationof the atoms. This is exactly the op-
posite of what happens in the icosahedral geometry, where internal bonds, pertaining to highly
coordinated atoms, are on the average shorter than surface bonds, that join low-coordination
atoms. For example, in the Ih13, internal bonds are 5% shorter than surface bonds. Compact
polyicosahedral clusters suffer seriously from the same problem, since they have even more
3

compressed internal distances, thus a huge internal strain. Therefore they are not expected to
exhibit special stability in pure transition metal clusters (15).
However the situation can change drastically in binary systems. In fact, if we substitute the
internal atoms in a pure polyicosahedral cluster withsmaller atoms, the optimal bond lengths
of both internal and surface atoms may be recovered, and the strain strongly decreased. This
leads naturally to the idea ofcore-shell polyicosahedral clusters, which both maximize the bond
number and minimize the strain. Other important factors favoring such clusters are that the large
atoms must have a strong tendency to segregate at the surface with respect to the small atoms,
and possibly the two elements should have a weak tendency to mixing or alloying in the bulk
phase. All these features are present in the Ag-Ni and Ag-Cu systems. Ni and Cu atoms are16%
and13% smaller than Ag atoms; Ag has a strong tendency to surface segregation with respect
to both Ni and Cu (16); Ag-Ni and Ag-Cu bulk systems present very extended miscibility
gaps (17). The results reported here demonstrate that the Ag-Ni and the Ag-Cu systems are
really suitable for building up magic core-shell polyicosahedra. For these systems we show first
that, given the size, the most stable structures are obtained at the compositions where perfect
core-shell pIh can be formed. Then, among these perfect core-shell pIh, we single out those
with the most remarkable stability features. These are also very nicely symmetric structures.
To compare the relative stability of clusters of different sizes and compositions, we monitor
the quantities∆ and∆2 (18,19). These are routinely used in the case of pure clusters; here we
adapt them to the analysis of binary clusters.∆ is the excess energy of the cluster with respect
to N bulk atoms, divided byN2/3 (which scales roughly as the number of surface atoms):
∆ =EGM(N, N1)−N1ε
coh1 −N2ε
coh2
N2/3, (1)
whereN1 andεcoh1 are the number and the bulk cohesive energy of Ag atoms, whereasN2 =
N − N1 andεcoh2 are the same quantities of either Ni or Cu, andEGM(N,N1) is the global-
4

minimum energy at the given size and composition. Stable structures are identified by low∆
values.∆2 is the energy second difference
∆2(N, N1) = EGM(N,N1 + 1) + EGM(N, N1 − 1)− 2EGM(N, N1). (2)
Maxima of∆2 indicate structures of special relative stability compared to those of the same size
and nearby compositions.
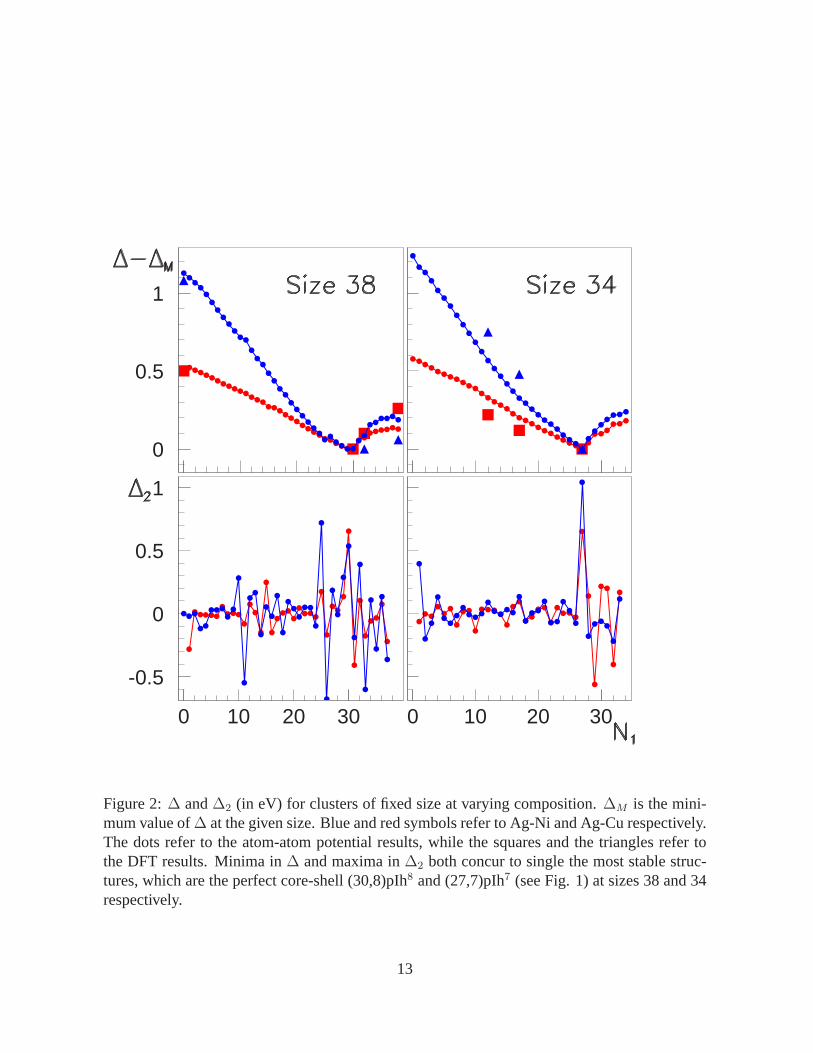
We start our analysis at a specific size,N = 38, and look for the global energy minima at
varying composition. Size 38 is magic for the regular truncated octahedron, a high-symmetry
piece of fcc bulk lattice, thus is expected to correspond to pure clusters of good stability. Plotting
∆ and∆2 vs. N1, for both Ag-Ni and Ag-Cu, the same conclusion follows (see Fig. 2): the
minimum∆ and a high peak in∆2 correspond to the perfect core-shell structure which includes
the maximum number of small atoms inside, namely 30 external Ag atoms and 8 inner Ni or Cu
atoms. This structure (see Fig. 1) is a pIh made up of 8 interpenetrating Ih13. In the following,
a pIh made up ofm Ih13 and containingN1, N2 atoms, is denoted as(N1, N2)pIhm, so that
the above cluster is the(30, 8)pIh8. Other perfect core-shell pIh are the(31, 7)pIh7 and the
(32, 6)pIh6. The latter is a highly symmetric (group D6h) pancakestructure; the six Cu or Ni
atoms inside do not form a compact aggregate, but are placed on a perfect hexagonal ring. The
structure at composition (29,9) is an incomplete pIh: adding a single Ag atom on its surface we
obtain the very stable global minimum(30, 9)pIh9.
At size 34, the predominance of the core-shell polyicosahedral structure is even clearer.
This is the right size for forming a high-symmetry (group D5h) pIh made of 7 Ih13. This struc-
ture underlies most of the minima at varying composition, and attains its maximum stability
when the perfect core-shell arrangement is formed. This happens at composition (27,7): the
(27,7)pIh7 contains a compact decahedral nucleus of Ni or Cu, and the all Ag atoms are placed
in an external anti-Mackay (20) shell. The special energetic stability of perfect core-shell pIh
5

at sizes 34 and 38 is nicely confirmed by our DFT calculations, whose results are reported in
Fig. 2 and Table I. In passing, we note that the atom-atom potential geometries are only slightly
modified by the DFT relaxation, thus validating the accuracy of the semi-empirical approach
for these elements.
The core-shell pIh of highest symmetry is at larger size: the anti-Mackay icosahedron of
45 atoms. This cluster is obtained by covering an Ih13 of Ni or Cu by a complete anti-Mackay
shell of Ag atoms, and is made of 12 interpenetrating Ih13 sharing the central atom as a common
vertex (being thus classified as (32,13)pIh12).
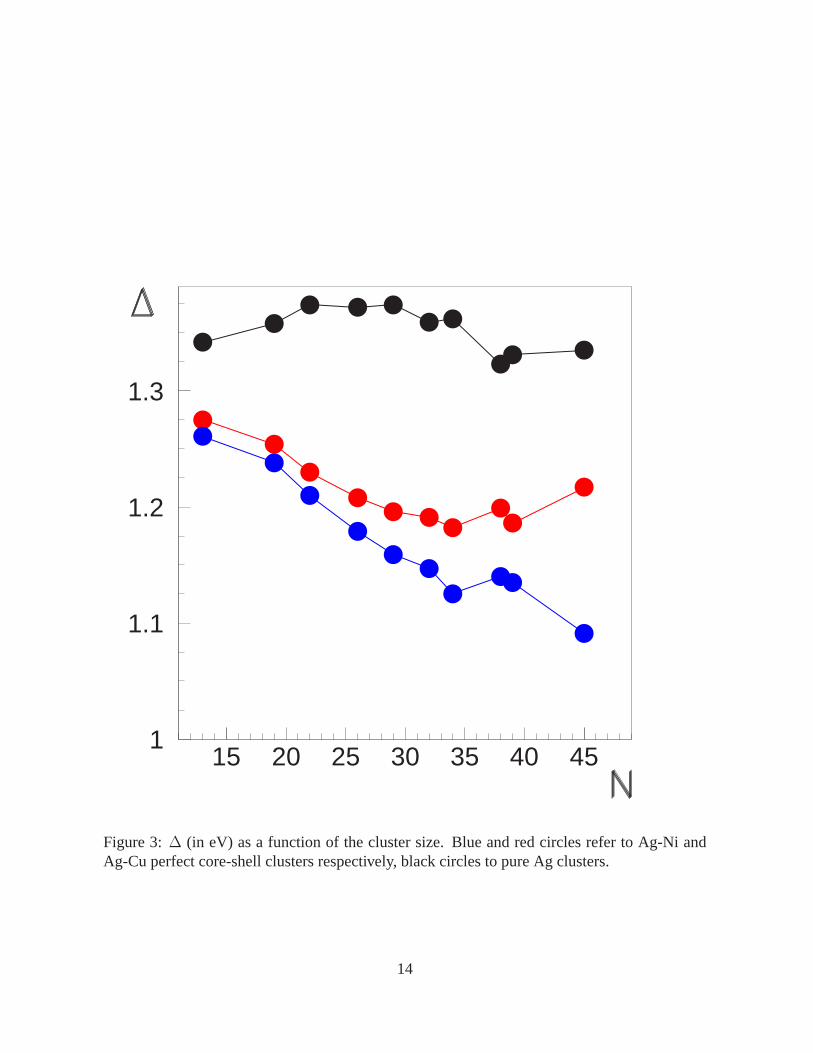
High-stability core-shell pIh constitute a rich family, being found at many sizes below 45,
with ∆ and∆2 behaving qualitatively as atN = 34 and 38. For example, there is a series of
perfect core-shell pIh which is made of fragments of the anti-Mackay pIh12. This series passes
through the (27,7)pIh7 and the (30,9) pIh9, and comprises several smaller clusters [(17,2)pIh2,
(20,3)pIh3, (22,4)pIh4 (of tetrahedral symmetry), (24,5)pIh5, (26,6)pIh7], which are obtained
one from the other by adding a single interpenetrating Ih13 at a time.
All the perfect core-shell polyicosaedra are well separated in energy from their homo-
tops (21) because of the tendency to surface segregation of Ag. Therefore, the structures being
eventually in energetic competition with the global minima are other core-shell clusters of dif-
ferent symmetry. The situation is different for those clusters that are not perfect core-shell
particles. For example, at size 34, we obtain the D5h pIh7 also at compositions (12,22) and
(17,17), but these structures have several quasi-degenerate homotops.
Among this series of core-shell pIh, now we focus on the structure which has the most
intriguing structural and electronic properties, which, from the results of Table I, is the (27,7)-
pIh7. In the case of Ag-Cu, this cluster has a large gap (0.82 eV), indicating a strong electronic
stability (22), and no magnetic moment. In the case of Ag-Ni, the gap is still rather large
(0.81 and 0.46 eV in the majority and minority spin, respectively), and the ground state has a
6

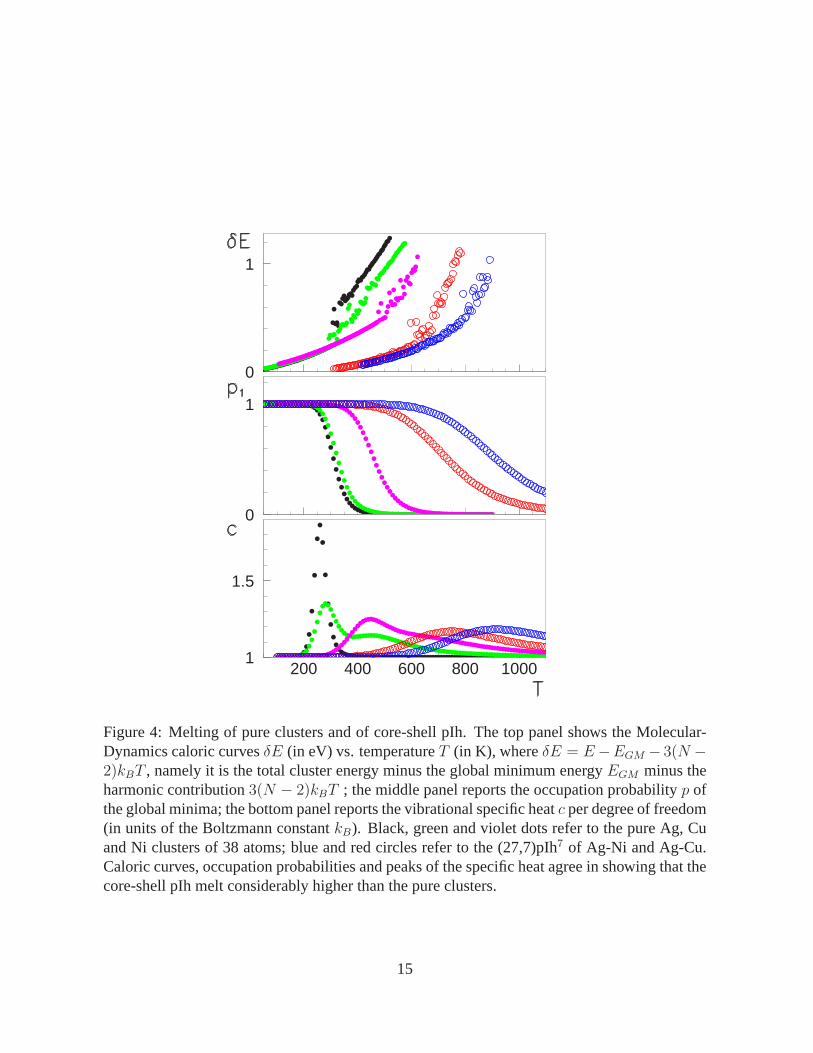
non-negligible magnetic moment. In both cases, the (27,7)-pIh7 is also of remarkable thermo-
dynamic stability, as follows from the analysis of its melting behavior. In fact (see Fig. 4),
this core-shell cluster melts considerablyhigher than the pure Ag, Cu, and Ni clusters in the
same size range. This is at variance with the behavior of Ag-Cu and Ag-Ni bulk systems, where
the melting point is depressed at mixed compositions (17). The special thermodynamic stabil-
ity of the (27,7)pIh7 follows from two factors. First, this cluster is well separated from higher
isomers: at least by 0.3 eV for Ag-Cu and 0.45 eV for Ag-Ni according to the atom-atom poten-
tial, but the DFT calculations give a much larger separation, of more than 0.8 eV. Second, our
harmonic thermodynamics calculations show that the entropic contributions to the free energy
are favorable to the (27,7)pIh7 structure, in analogy with previous results on pure strontium
clusters (23).
The electronic and magnetic properties of this new family of magic clusters can be of great
interest, and are currently under investigation. For example, the large gap and the stability of
the Ag-Cu (27,7)pIh7 make this cluster potentially suitable for applications in opto-electronic
and single-electron tunnelling devices. On the other hand, the same cluster is a promising
candidate for single-molecule magnetism in the case of Ag-Ni. Finally, since the requirements
for these structures are of quite general character (size mismatch, large atoms segregating at the
surface with respect to small atoms, possibly a weak tendency to mix in the bulk phase), we
expect that magic core-shell polyicosahedra are likely to be encountered in a large variety of
binary metallic systems, for example silver-cobalt, gold-nickel, gold-cobalt, and (with a weaker
tendency) silver-palladium and gold-copper.
References and Notes
1. J. Jellinek, E. B. Krissinel,Theory of Atomic and Molecular Clusters, J. Jellinek, ed.
(Springer, Berlin, 1999), pp. 277–308.
7

2. M. P. Andrews, S. C. OBrien,J. Phys. Chem.96, 8233 (1992).
3. S. Darby, T. V. Mortimer-Jones, R. L. Johnston, C. Roberts,J. Chem. Phys.116, 1536
(2002).
4. A. M. Molenbroek, S. Haukka, B. S. Clausen,J. Phys. Chem. B102, 10680 (1998).
5. U. Kreibig, M. Vollmer,Optical Properties of Metal Clusters(Springer, Berlin, 1995).
6. P. Jensen,Rev. Mod. Phys.71, 1695 (1999).
7. M. Gaudry,et al., Phys. Rev. B67, 155409 (2003).
8. The many body atom-atom potential is developed in the framework of the second moment
approximation to the tight-binding model. Form and parameters are given in (24,25).
9. Our genetic algorithm evolves in parallel 10 subpopulations of 50 individuals each; indi-
viduals are subjected to mutation and mating operations. The exchange mutation between
atoms of different species, which has been proven useful in optimizing binary clusters (3)
is included.
10. The Density Functional calculations are carried out with the DFT module of the NWChem
package (26), and use the Becke functional (27) for exchange and the Perdew-Wang
functional (28) for correlation. (7s6p6d)/(5s3p2d) Gaussian-type-orbital basis sets and ef-
fective core potentials are used for all elements, derived from ref. (29, 30) for Ag and
ref. (31) for Cu and Ni, and modified combining the suggestions in ftp://ftp.chemie.uni-
karlsruhe.de/pub/basen and ref. (32). A charge density fitting basis was used to compute
the Coulomb potential (courtesy of Dr. Florian Weigend, Karlsruhe, Germany). More de-
tails on the numerical procedure can be found in ref. (32).
11. J. P. K. Doye, F. Calvo,Phys. Rev. Lett.86, 3170 (2001).
8

12. J. P. K. Doye, D. J. Wales, R. S. Berry,J. Chem. Phys.103, 4234 (1995).
13. See the Cambridge Cluster Database, http://www-wales.ch.cam.ac.uk/CCD.html.
14. L. Pauling,The Nature of the Chemical Bond(Cornell University Press, New York, 1960).
15. In the case of aluminum, recent Embedded Atom Method calculations (33) have shown that
polyicosahedral structures are global minima up toN = 34 and 38, due to a specific feature
of the Al-Al bonding. However, the global minima at 34 an 38 are not of special stability.
16. J.-M. Roussel, A. Saul, G. Treglia, B. Legrand,Phys. Rev. B55, 10931 (1997).
17. M. Hansen,Constitution of Binary Alloys(Mc Graw - Hill, New York, 1958).
18. C. L. Cleveland, U. Landman,J. Chem. Phys.94, 7376 (1991).
19. K. Clemenger,Phys. Rev. B32, 1359 (1985).
20. J. A. Northby,J. Chem. Phys.87, 6166 (1987).
21. Following (34), homotops are defined as clusters of the same size, structure and composi-
tion, differing only in the chemical ordering, namely by exchages between the positions of
atoms of different species.
22. The HOMO-LUMO gap is a good indicator of the chemical hardness of the system.
23. In (35) it was shown that the global minimum of Sr34 is not the pIh7, but an entropy driven
solid-solid transition makes it the most probable structure above 130 K.
24. F. Baletto, C. Mottet, R. Ferrando,Phys. Rev. B66, 155420 (2002).
25. F. Baletto, C. Mottet, R. Ferrando,Phys. Rev. B90, 135504 (2003).
26. R. A. Kendall,et al., Comp. Phys. Comm.128, 260 (2000).
9

27. A. D. Becke,Phys. Rev. A38, 3098 (1988).
28. J. P. Perdew,et al., Phys. Rev. B46, 6671 (1992).
29. A. Schaefer, C. Huber, R. Ahlrichs,J. Chem. Phys.100, 5289 (1994).
30. D. Andrae, U. Haeussermann, M. Dolg, H. Stoll, H. Preuss,Theor. Chim. Acta77, 123
(1990).
31. M. Dolg, U. Wedig, H. Stoll, H. Preuss,J. Chem. Phys.86, 866 (1987).
32. E. Apra, A. Fortunelli,J. Chem. Phys.107, 2934 (2003).
33. J. P. K. Doye,J. Chem. Phys.119, 1136 (2003).
34. J. Jellinek, E. Krissinel,Chem. Phys. Lett.258, 283 (1996).
35. G. M. Wang, E. Blastein-Barojas, A. E. Roitberg,J. Chem. Phys.115, 3640 (2001).
36. A. F. and R. F. acknowledge financial support from the Italian CNR for the project “(Supra-
)Self-Assemblies of Transition Metal Nanoclusters” within the framework of the ESF EU-
ROCORES SONS. A. F. acknowledges the Italian INSTM for a grant at the CINECA super-
computing center.
10

Table 1: DFT calculations results. Values of spin (S), and HOMO-LUMO energy difference(Gap, in eV) for alpha/beta electrons. The symbol ”JT” after the Gap value signals a Jahn-Teller system, with a degenerate HOMO (symmetry-breaking is not allowed during the DFTrelaxation in these cases).
System Size Cluster S GapAg 38 Truncated Octahedron 1 0.39/0.35 JTCu 38 Truncated Octahedron 1 0.61/0.56 JTNi 38 Truncated Octahedron 16 0.21/0.03 JTAg-Cu 38 (32,6)pIh6 0 0.34/0.34
38 (30,8)pIh8 0 0.26/0.2634 (27,7)pIh7 0 0.82/0.8234 (17,17)pIh7 0 0.94/0.9434 (12,22)pIh7 0 0.88/0.8845 (32,13)pIh12 2.5 0.49/1.00
Ag-Ni 38 (32,6)pIh6 0 0.09/0.0938 (30,8)pIh8
34 (27,7)pIh7 3.5 0.81/0.4634 (17,17)pIh7 6.5 0.19/0.0734 (12,22)pIh7 7 0.26/0.07 JT45 (32,13)pIh12 16 0.21/0.03 JT
11

Ih13
.
(27,7) (32,6) (30,8) (32,13)pIh7 pIh6 pIh8 pIh12
Figure 1: Polyicosahedral core-shell clusters. They are built by packing up elementary Ih13
clusters, as schematically shown in the top row. Each cluster is shown in four different views,in two of them the external Ag atoms are represented by small points to show the arrangementof the core atoms. Pictures of other pIh are available in the online supporting material.
12
0
0.5
1
0 10 20 30
0
0.5
1
0 10 20 30
-0.5
0
0.5
1
0 10 20 30
-0.5
0
0.5
1
0 10 20 30
Figure 2:∆ and∆2 (in eV) for clusters of fixed size at varying composition.∆M is the mini-mum value of∆ at the given size. Blue and red symbols refer to Ag-Ni and Ag-Cu respectively.The dots refer to the atom-atom potential results, while the squares and the triangles refer tothe DFT results. Minima in∆ and maxima in∆2 both concur to single the most stable struc-tures, which are the perfect core-shell (30,8)pIh8 and (27,7)pIh7 (see Fig. 1) at sizes 38 and 34respectively.
13
1
1.1
1.2
1.3
15 20 25 30 35 40 45
Figure 3:∆ (in eV) as a function of the cluster size. Blue and red circles refer to Ag-Ni andAg-Cu perfect core-shell clusters respectively, black circles to pure Ag clusters.
14
0
1
200 400 600 800 1000
0
1
200 400 600 800 1000
1
1.5
200 400 600 800 1000
Figure 4: Melting of pure clusters and of core-shell pIh. The top panel shows the Molecular-Dynamics caloric curvesδE (in eV) vs. temperatureT (in K), whereδE = E−EGM − 3(N −2)kBT , namely it is the total cluster energy minus the global minimum energyEGM minus theharmonic contribution3(N − 2)kBT ; the middle panel reports the occupation probabilityp ofthe global minima; the bottom panel reports the vibrational specific heatc per degree of freedom(in units of the Boltzmann constantkB). Black, green and violet dots refer to the pure Ag, Cuand Ni clusters of 38 atoms; blue and red circles refer to the (27,7)pIh7 of Ag-Ni and Ag-Cu.Caloric curves, occupation probabilities and peaks of the specific heat agree in showing that thecore-shell pIh melt considerably higher than the pure clusters.
15




















