
Machine learning in computational docking
17
1 Mohamed A. Khamis, Walid Gomaa, Walaa F. Ahmed, Machine learning in computational docking, Artificial Intelligence In Medicine (2015), http://dx.doi.org/10.1016/j.artmed.2015.02.002 http://dx.doi.org/10.1016/j.artmed.2015.02.002
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Machine learning in computational docking

1
Mohamed A. Khamis, Walid Gomaa, Walaa F. Ahmed,
Machine learning in computational docking, Artificial Intelligence In Medicine (2015),
http://dx.doi.org/10.1016/j.artmed.2015.02.002
http://dx.doi.org/10.1016/j.artmed.2015.02.002

Objective
http://dx.doi.org/10.1016/j.artmed.2015.02.002 2
The objective of this paper is to highlight the state-of-the-art machine learning (ML) techniques in computational docking.
The use of smart computational methods in the life cycle of drug design is relatively a recent development that has gained much popularity and interest over the last few years.
Computational docking is the process of predicting the best pose (orientation + conformation) of a small molecule (drug candidate) when bound to a target larger receptor molecule (protein) in order to form a stable complex molecule.

Background
3
• Background for protein-ligand interactions: Physical, chemical, and biological
• Molecular data formats: e.g., .mol, .pdb, .sdf, etc.
• Docking software programs: e.g., AutoDock, eHiTS, iDock, etc.
• Molecular databases: Containing data of proteins with their possible ligands e.g., PDB, PDBbind, Binding DB, DUD etc.
http://dx.doi.org/10.1016/j.artmed.2015.02.002
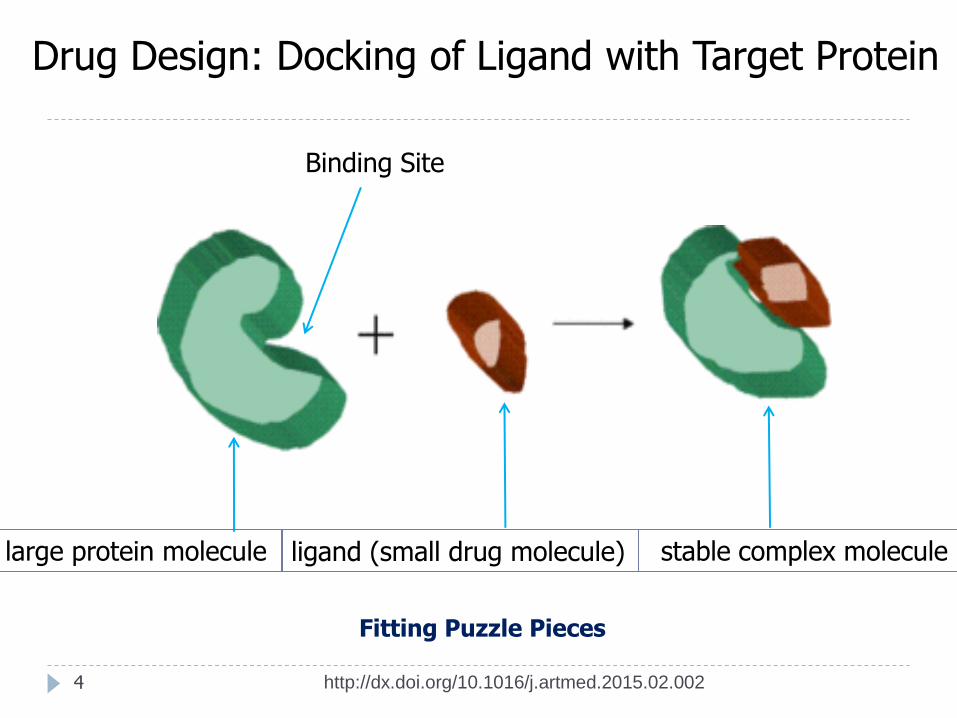
4
ligand (small drug molecule) large protein molecule stable complex molecule
Fitting Puzzle Pieces
Drug Design: Docking of Ligand with Target Protein
Binding Site
http://dx.doi.org/10.1016/j.artmed.2015.02.002

Protein HIV-1 protease (hsg1.pdb)
5
Ligand (Drug) Indinavir (ind.pdb)
Formula: C36H47N5O4
Indinavir (IDV; trade name Crixivan, manufactured by Merck) is inhibitor used
to treat HIV infection and AIDS.
http://dx.doi.org/10.1016/j.artmed.2015.02.002
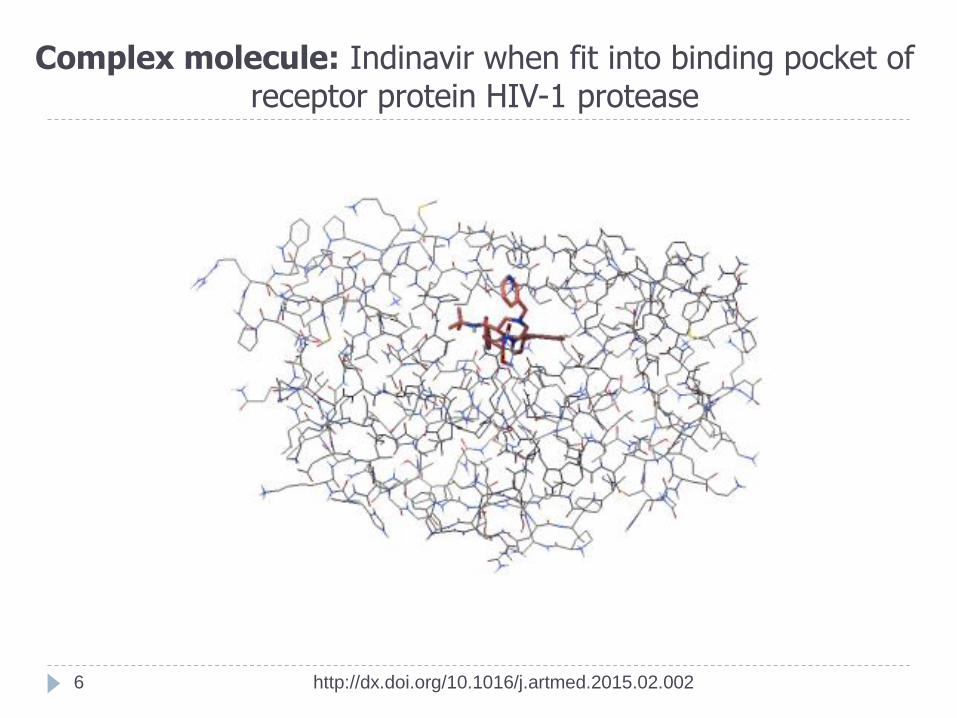
Complex molecule: Indinavir when fit into binding pocket of receptor protein HIV-1 protease
6 http://dx.doi.org/10.1016/j.artmed.2015.02.002

Traditional Drug Design Methods
7
• Traditional drug design techniques - such as random screening and chance discovery are essentially trial and error methods.
• And so they are very time consuming (10-15 years), very expensive ($300M), with extremely low yield.
• For instance, over last 50 years, 500,000 compounds have been tested for anti-cancer; Only 25 are in wide use today [1].
• On other hand, CADD is target specific, structure-based,
automatic, fast, and very low cost with high success rate.
1. Denny, William A., New Zealand Institute of Chemistry, The Design and development of anti-cancer drugs. Available at http://nzic.org.nz/ChemProcesses/biotech/12J.pdf.
http://dx.doi.org/10.1016/j.artmed.2015.02.002

Scoring Function
8
Is mathematical predictive model that produces a score that represents binding free energy and hence stability of resulting complex molecule.
Generally, such function should produce set of credible ligands ranked according to their binding stability along with their binding poses
X-Score: Wang R, Lai L, Wang S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J Computer-Aided Molecular Design 2002;16:11–26.
http://dx.doi.org/10.1016/j.artmed.2015.02.002
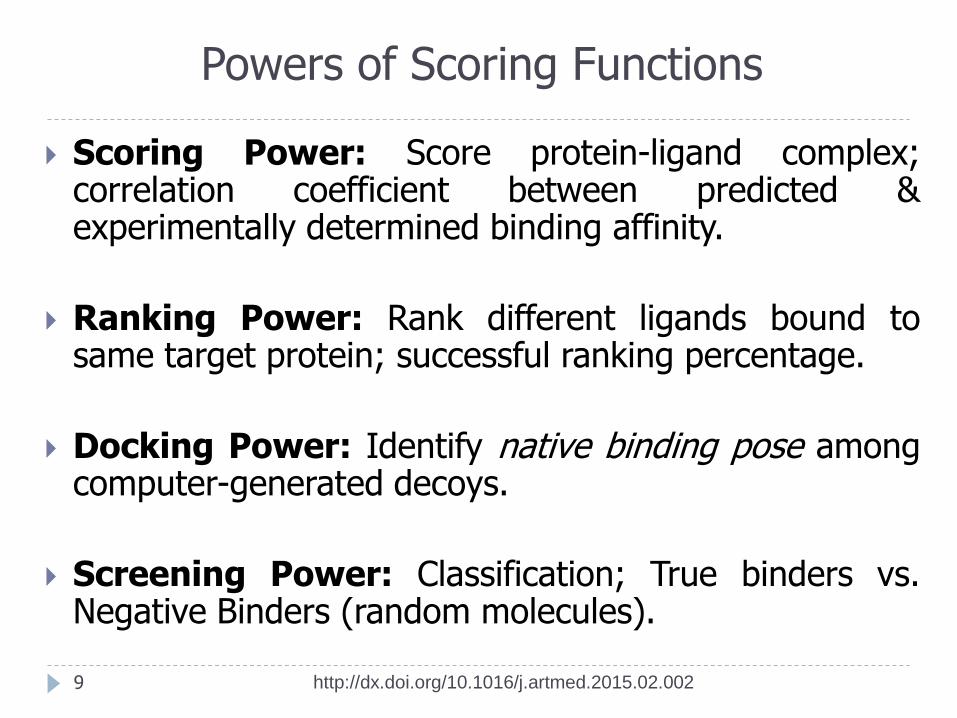
Powers of Scoring Functions
9
Scoring Power: Score protein-ligand complex; correlation coefficient between predicted & experimentally determined binding affinity.
Ranking Power: Rank different ligands bound to same target protein; successful ranking percentage.
Docking Power: Identify native binding pose among computer-generated decoys.
Screening Power: Classification; True binders vs. Negative Binders (random molecules).
http://dx.doi.org/10.1016/j.artmed.2015.02.002

Classical Scoring functions
10
Classical scoring functions e.g., X-Score rely only on fixed set of molecular features (e.g., energy terms)
Summed in linear weighted manner that fails to model non-linear relationships among individual energy terms.
In addition, weights of those individual energy terms are calibrated based on specific protein family (using linear regression),
Hence, classical scoring functions are more prone to over-fitting.
http://dx.doi.org/10.1016/j.artmed.2015.02.002
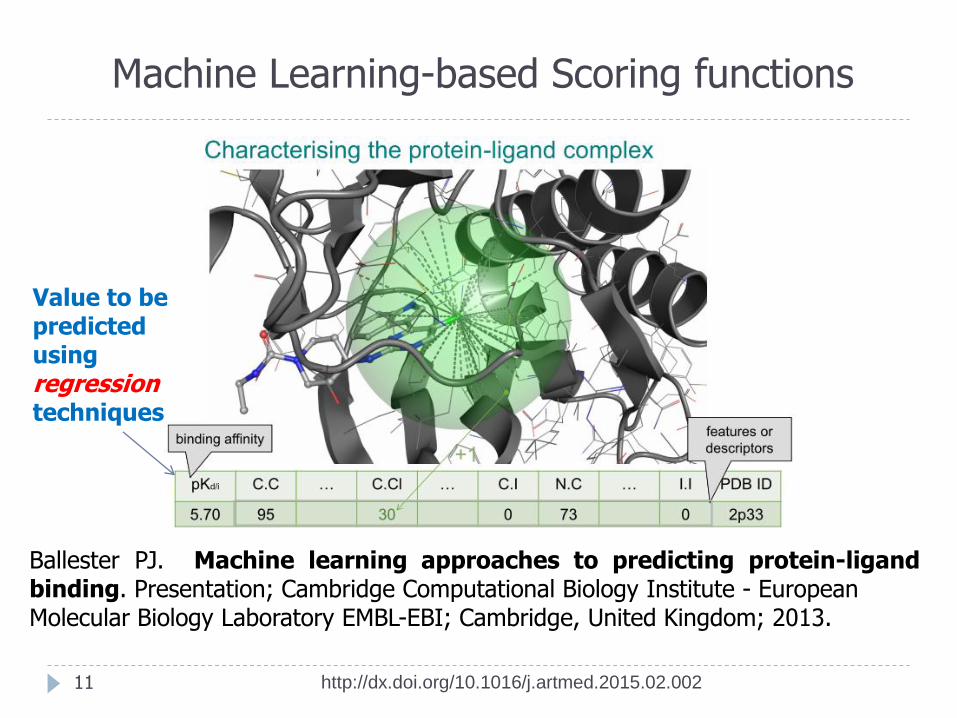
Machine Learning-based Scoring functions
11
Ballester PJ. Machine learning approaches to predicting protein-ligand binding. Presentation; Cambridge Computational Biology Institute - European Molecular Biology Laboratory EMBL-EBI; Cambridge, United Kingdom; 2013.
Value to be predicted using regression techniques
http://dx.doi.org/10.1016/j.artmed.2015.02.002

Training & Testing sets of PDBbind v. 2007
12
Ballester PJ. Machine learning approaches to predicting protein-ligand binding. Presentation; Cambridge Computational Biology Institute - European Molecular Biology Laboratory EMBL-EBI; Cambridge, United Kingdom; 2013.
http://dx.doi.org/10.1016/j.artmed.2015.02.002

Results
http://dx.doi.org/10.1016/j.artmed.2015.02.002 13
We survey this paradigm shift elaborating on the main building components of ML approaches used in molecular docking.
For instance, the best random forest (RF)-based scoring function (Li, 2014) on PDBbind v2007 achieves a Pearson correlation coefficient between the predicted and experimentally determined binding affinities of 0.803 while the best classical scoring function achieves 0.644 (Cheng, 2009).
The best RF-based ranking power (Ashtawy, 2012) ranks the ligands correctly based on their experimentally determined binding affinities with accuracy 62.5% and identifies the top binding ligand with accuracy 78.1%.

Conclusion
14
Machine Learning techniques give ability to utilize as many relevant molecular features (e.g., geometric features, pharmacophore features, etc.) as possible.
Particularly, ensemble-based machine learning approaches (e.g., random forest, boosted regression trees, etc.) are resilient to over fitting.
Yield good results not only on training complexes but on any testing complexes as well.
http://dx.doi.org/10.1016/j.artmed.2015.02.002

Acknowledgement
15
This work is supported:
Mainly by Information Technology Industry Development Agency (ITIDA) under ITAC Program grant number CFP#58
In part by E-JUST Research Fellowship
http://dx.doi.org/10.1016/j.artmed.2015.02.002

Publications
16
Mohamed A. Khamis, Walid Gomaa, 2015, Comparative Assessment of Scoring and Ranking Powers of Machine-Learning-Based Scoring Functions on an Updated Benchmark PDBbind 2013, Engineering Applications of Artificial Intelligence, Elsevier. (submitted)
Mohamed A. Khamis, Walid Gomaa, Basem Galal, 2015, Deep Learning Competes Random Forest in Computational Docking, IEEE/ACM Transactions on Computational Biology and Bioinformatics. (submitted)
http://dx.doi.org/10.1016/j.artmed.2015.02.002

Questions
http://dx.doi.org/10.1016/j.artmed.2015.02.002 17
E-mail:




















