Lyn but Not Fyn Kinase Controls IgG-Mediated Systemic Anaphylaxis
9
The Journal of Immunology Lyn but Not Fyn Kinase Controls IgG-Mediated Systemic Anaphylaxis Yves T. Falanga,* Natalia S. Chaimowitz, † Nicolas Charles, ‡ Fred D. Finkelman, x,{,‖ Nicholas A. Pullen,* Suzanne Barbour, † Kevin Dholaria,* Travis Faber,* Motunrayo Kolawole,* Bernice Huang, † Sandra Odom, # Juan Rivera, # Jason Carlyon, † Daniel H. Conrad, † Sarah Spiegel,** Carole A. Oskeritzian,** and John J. Ryan* Anaphylaxis is a rapid, life-threatening hypersensitivity reaction. Until recently, it was mainly attributed to histamine released by mast cells activated by allergen crosslinking (XL) of Fc«RI-bound allergen-specific IgE. However, recent reports established that anaphylaxis could also be triggered by basophil, macrophage, and neutrophil secretion of platelet-activating factor subsequent to FcgR stimulation by IgG/Ag complexes. We have investigated the contribution of Fyn and Lyn tyrosine kinases to FcgRIIb and FcgRIII signaling in the context of IgG-mediated passive systemic anaphylaxis (PSA). We found that mast cell IgG XL induced Fyn, Lyn, Akt, Erk, p38, and JNK phosphorylation. Additionally, IgG XL of mast cells, basophils, and macrophages resulted in Fyn- and Lyn-regulated mediator release in vitro. FcgR-mediated activation was enhanced in Lyn-deficient (knockout [KO]) cells, but decreased in Fyn KO cells, compared with wild-type cells. More importantly, Lyn KO mice displayed significantly exacerbated PSA features whereas no change was observed for Fyn KO mice, compared with wild-type littermates. Intriguingly, we establish that mast cells account for most serum histamine in IgG-induced PSA. Taken together, our findings establish pivotal roles for Fyn and Lyn in the regulation of PSA and highlight their unsuspected functions in IgG-mediated pathologies. The Journal of Immunology , 2012, 188: 4360–4368. T he initiation of an allergic response is commonly attributed to mast cell activation via their high-affinity IgE receptor (FcεRI) (1, 2). In some cases, the exposure to IgE-bound allergen causes rapid and exacerbated hypersensitivity reactions called anaphylaxis. Consequently, the role of mast cells in allergy, inflammatory diseases, and immune homeostasis has been most extensively studied downstream of IgE receptor signaling (3–9), although interesting studies reported that mast cells can also be activated by their IgG receptor (10). Data generated from murine models demonstrate that an anaphylactic reaction can be induced through the IgG/FcgR pathway (11–14). In this “alternative” pathway of anaphylaxis, FcgR activation of basophils, macro- phages, and neutrophils elicits anaphylaxis symptoms, including a drop in core body temperature. FcεRI activation involves a plethora of signaling molecules, including Fyn and Lyn kinases. Lyn kinase, which is expressed as 53- and 56-kDa isoforms due to alternative splicing (15–17), is expressed in nearly all immune cells, except the T cell lineage. For example, the association of Lyn kinase with the BCR (18) and the high-affinity IgE receptor, FcεRI (19), places Lyn in a critical position to control inflammation. Because of the known stimula- tory roles for Src family kinases, it was assumed that Lyn pos- sessed prostimulatory roles in signaling. Consistent with this, Lyn knockout (KO) mast cells were found to have poor calcium flux responses after IgE-mediated activation (20). However, further study showed that Lyn deficiency exacerbated IgE-mediated mast cell activation and anaphylaxis (21). These apparently contradic- tory data were explained by the ability of Lyn to recruit several inhibitory proteins, including SHIP-1, SHP-2, DOK-1, and CBP (22–25). These negative regulators reduce signaling through other Src family members, including PI3K and the Ras/MAPK cascade. Hence, Lyn appears to be a negative regulator of the mast cell response. Alternatively, Fyn is a 59-kDa Src family protein tyrosine kinase involved in mast cell degranulation and cytokine release (26, 27). Once phosphorylated, Fyn activates the adaptor protein Gab2, initiating the PI3K pathway. This enhances calcium flux through a TRPC1-mediated process leading to cortical F-actin depoly- merization (28). Additionally, Fyn enhances transcription factor activity, degranulation, and cytokine release (2, 21). Fyn activation is currently viewed as a positive regulator of IgE-mediated in- flammatory signals, similar to the well-documented Syk kinase pathway. IgG receptors (FcgR) share a common g subunit with FcεRI and share similar signaling pathways. Although the importance of Syk *Department of Biology, Virginia Commonwealth University, Richmond, VA 23284; † Department of Microbiology and Immunology, Virginia Commonwealth University School of Medicine, Richmond, VA, 23298; ‡ INSERM Unite ´ 699, “Immunopatho- logie Re ´nale, Re ´cepteurs et Inflammation,” Faculte ´ de Me ´decine Xavier Bichat– Universite ´ Paris VII Denis Diderot, 75870 Paris Cedex 18, France; x Research Service, Cincinnati Veterans Affairs Medical Center, Cincinnati, OH 45220; { Divi- sion of Immunology, University of Cincinnati College of Medicine, Cincinnati, OH 45267; ‖ Division of Immunobiology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229; # Laboratory of Molecular Immunogenetics, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, MD 20892; and **Department of Biochemistry and Molecular Biology, Virginia Commonwealth University School of Medicine, Richmond, VA 23298 Received for publication September 28, 2010. Accepted for publication February 28, 2012. This work was supported by the intramural research program of the National Insti- tute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (to J.R.), and National Institutes of Health Grants 1R01AI59638 and U19AI077435 (to J.R.), KO1AR053186 (to C.A.O.), and a Veterans Affairs Merit Grant (to F.D.F.). Address correspondence and reprint requests to Prof. John J. Ryan, Department of Biology, Virginia Commonwealth University, 1000 W. Cary Street, Richmond, VA 23284. E-mail address: [email protected] Abbreviations used in this article: BMMC, bone marrow-derived mast cell; KO, knockout; LTC4, leukotriene C 4 ; PAF, platelet-activating factor; PAFAH, platelet- activating factor acetylhydrolase; PSA, passive systemic anaphylaxis; pY, tyrosine- phosphorylated; SLE, systemic lupus erythematosus; WT, wild-type; XL, crosslink- ing. www.jimmunol.org/cgi/doi/10.4049/jimmunol.1003223
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Lyn but Not Fyn Kinase Controls IgG-Mediated Systemic Anaphylaxis

The Journal of Immunology
Lyn but Not Fyn Kinase Controls IgG-Mediated SystemicAnaphylaxis
Yves T. Falanga,* Natalia S. Chaimowitz,† Nicolas Charles,‡ Fred D. Finkelman,x,{,‖
Nicholas A. Pullen,* Suzanne Barbour,† Kevin Dholaria,* Travis Faber,*
Motunrayo Kolawole,* Bernice Huang,† Sandra Odom,# Juan Rivera,# Jason Carlyon,†
Daniel H. Conrad,† Sarah Spiegel,** Carole A. Oskeritzian,** and John J. Ryan*
Anaphylaxis is a rapid, life-threatening hypersensitivity reaction. Until recently, it was mainly attributed to histamine released by
mast cells activated by allergen crosslinking (XL) of Fc«RI-bound allergen-specific IgE. However, recent reports established that
anaphylaxis could also be triggered by basophil, macrophage, and neutrophil secretion of platelet-activating factor subsequent to
FcgR stimulation by IgG/Ag complexes. We have investigated the contribution of Fyn and Lyn tyrosine kinases to FcgRIIb and
FcgRIII signaling in the context of IgG-mediated passive systemic anaphylaxis (PSA). We found that mast cell IgG XL induced
Fyn, Lyn, Akt, Erk, p38, and JNK phosphorylation. Additionally, IgG XL of mast cells, basophils, and macrophages resulted in
Fyn- and Lyn-regulated mediator release in vitro. FcgR-mediated activation was enhanced in Lyn-deficient (knockout [KO]) cells,
but decreased in Fyn KO cells, compared with wild-type cells. More importantly, Lyn KO mice displayed significantly exacerbated
PSA features whereas no change was observed for Fyn KO mice, compared with wild-type littermates. Intriguingly, we establish
that mast cells account for most serum histamine in IgG-induced PSA. Taken together, our findings establish pivotal roles for Fyn
and Lyn in the regulation of PSA and highlight their unsuspected functions in IgG-mediated pathologies. The Journal of
Immunology, 2012, 188: 4360–4368.
Theinitiation of an allergic response is commonly attributedto mast cell activation via their high-affinity IgE receptor(FcεRI) (1, 2). In some cases, the exposure to IgE-bound
allergen causes rapid and exacerbated hypersensitivity reactionscalled anaphylaxis. Consequently, the role of mast cells in allergy,inflammatory diseases, and immune homeostasis has been mostextensively studied downstream of IgE receptor signaling (3–9),although interesting studies reported that mast cells can also beactivated by their IgG receptor (10). Data generated from murinemodels demonstrate that an anaphylactic reaction can be inducedthrough the IgG/FcgR pathway (11–14). In this “alternative”
pathway of anaphylaxis, FcgR activation of basophils, macro-phages, and neutrophils elicits anaphylaxis symptoms, includinga drop in core body temperature.FcεRI activation involves a plethora of signaling molecules,
including Fyn and Lyn kinases. Lyn kinase, which is expressed as53- and 56-kDa isoforms due to alternative splicing (15–17), isexpressed in nearly all immune cells, except the T cell lineage.For example, the association of Lyn kinase with the BCR (18) andthe high-affinity IgE receptor, FcεRI (19), places Lyn in a criticalposition to control inflammation. Because of the known stimula-tory roles for Src family kinases, it was assumed that Lyn pos-sessed prostimulatory roles in signaling. Consistent with this, Lynknockout (KO) mast cells were found to have poor calcium fluxresponses after IgE-mediated activation (20). However, furtherstudy showed that Lyn deficiency exacerbated IgE-mediated mastcell activation and anaphylaxis (21). These apparently contradic-tory data were explained by the ability of Lyn to recruit severalinhibitory proteins, including SHIP-1, SHP-2, DOK-1, and CBP(22–25). These negative regulators reduce signaling through otherSrc family members, including PI3K and the Ras/MAPK cascade.Hence, Lyn appears to be a negative regulator of the mast cellresponse.Alternatively, Fyn is a 59-kDa Src family protein tyrosine kinase
involved in mast cell degranulation and cytokine release (26, 27).Once phosphorylated, Fyn activates the adaptor protein Gab2,initiating the PI3K pathway. This enhances calcium flux througha TRPC1-mediated process leading to cortical F-actin depoly-merization (28). Additionally, Fyn enhances transcription factoractivity, degranulation, and cytokine release (2, 21). Fyn activationis currently viewed as a positive regulator of IgE-mediated in-flammatory signals, similar to the well-documented Syk kinasepathway.IgG receptors (FcgR) share a common g subunit with FcεRI and
share similar signaling pathways. Although the importance of Syk
*Department of Biology, Virginia Commonwealth University, Richmond, VA 23284;†Department of Microbiology and Immunology, Virginia Commonwealth UniversitySchool of Medicine, Richmond, VA, 23298; ‡INSERM Unite 699, “Immunopatho-logie Renale, Recepteurs et Inflammation,” Faculte de Medecine Xavier Bichat–Universite Paris VII Denis Diderot, 75870 Paris Cedex 18, France; xResearchService, Cincinnati Veterans Affairs Medical Center, Cincinnati, OH 45220; {Divi-sion of Immunology, University of Cincinnati College of Medicine, Cincinnati, OH45267; ‖Division of Immunobiology, Cincinnati Children’s Hospital Medical Center,Cincinnati, OH 45229; #Laboratory of Molecular Immunogenetics, National Instituteof Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health,Bethesda, MD 20892; and **Department of Biochemistry and Molecular Biology,Virginia Commonwealth University School of Medicine, Richmond, VA 23298
Received for publication September 28, 2010. Accepted for publication February 28,2012.
This work was supported by the intramural research program of the National Insti-tute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutesof Health (to J.R.), and National Institutes of Health Grants 1R01AI59638 andU19AI077435 (to J.R.), KO1AR053186 (to C.A.O.), and a Veterans Affairs MeritGrant (to F.D.F.).
Address correspondence and reprint requests to Prof. John J. Ryan, Department ofBiology, Virginia Commonwealth University, 1000 W. Cary Street, Richmond, VA23284. E-mail address: [email protected]
Abbreviations used in this article: BMMC, bone marrow-derived mast cell; KO,knockout; LTC4, leukotriene C4; PAF, platelet-activating factor; PAFAH, platelet-activating factor acetylhydrolase; PSA, passive systemic anaphylaxis; pY, tyrosine-phosphorylated; SLE, systemic lupus erythematosus; WT, wild-type; XL, crosslink-ing.
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1003223

kinase in IgG signaling has been demonstrated (29, 30), little isknown about Src family kinase functions. In this study we inves-tigated the importance of the Lyn and Fyn kinases in FcgR sig-naling using mast cells, basophils, and macrophages. Our findingsshow that Fyn, Lyn, Akt, Erk, p38, and JNK are activated uponFcgR stimulation, and that Fyn and Lyn regulate FcgR-mediateddegranulation and cytokine and chemokine release not only inmast cells, but also in basophils and macrophages. Furthermore, wedemonstrate that mast cells account for the total amount of circu-lating histamine during FcgR-induced passive systemic anaphy-laxis (PSA), which is regulated by Fyn and Lyn. Moreover, weshow that Lyn kinase, but not Fyn kinase, is a major regulator ofIgG-mediated PSA. These results bring new insights to the functionof Fyn and Lyn kinases downstream of FcgR stimulation.
Materials and MethodsAnimals
C57BL/63 129sv wild -type (WT), C57BL/63 129sv Fyn-deficient (KO),129svWT, and 129sv Lyn KO inbred strains were described previously (21,26). Mast cell-deficient Wsh2/2 and their C57BL/6 control mice werepurchased from The Jackson Laboratory. All mice were used at a minimumof 9 wk age, and all experiments received approval from the VirginiaCommonwealth University Institutional Animal Care and Use Committee.
Cytokines and reagents
Cytokines and ELISA assay kits were purchased from PeproTech (RockyHill, NJ). The Bio-Plex Pro cytokine assay kits were purchased from Bio-Rad (Hercules, CA). Histamine and leukotriene C4 (LTC4) ELISA kitswere purchased from Cayman Chemicals (Ann Arbor, MI). Cytokinemeasurements were performed as per the manufacturer’s directions. Absspecific for tyrosine-phosphorylated (pY) Lyn and total Lyn were pur-chased from Santa Cruz Biotechnology (Santa Cruz, CA). Abs specific forthe tyrosine-phosphorylated form of the Src kinase activation loop (pY416)and for Fyn were purchased from Cell Signaling Technologies (Danvers,MA). Anti-mouse FcεRIa was purchased from eBioscience (San Diego,CA). Rat anti-mouse FcgRII/RIII (2.4G2), purified mouse IgE, rat anti-mouse IgE, rat IgG isotype control, and rat IgG anti–c-Kit (CD117) werepurchased from BD Pharmingen (San Diego, CA).
Cells
Mouse bone marrow-derived mast cells (BMMCs) were derived by culturein complete RPMI 1640 medium (Invitrogen Life Technologies, Carlsbad,CA) containing 10% heat-inactivated FBS, 2 mM L-glutamine, 100 U/mlpenicillin, 100 mg/ml streptomycin, 1 mM sodium pyruvate, and 1 mMHEPES (all from Biofluids, Rockville, MD), supplemented with IL-3–containing supernatant from WEHI-3 cells and stem cell factor-containingsupernatant from BHK-MKL cells. The final concentration of IL-3 andstem cell factor was adjusted to 1 and 10 ng/ml, respectively, as measuredby ELISA. Mouse bone marrow-derived basophils were derived by cul-turing bone marrow cells for 6 d in complete RPMI 1640 medium sup-plemented with 5 ng/ml rIL-3 (PeproTech, Rocky Hill, NJ). At day 6, theywere analyzed and sorted by flow cytometry on the basis of FcεRI-positiveand Kit-negative characteristics. Mouse bone marrow-derived macro-phages were derived by culturing bone marrow cells for 8 d in completeRPMI supplemented with 50 ng/ml macrophage CSF (PeproTech).
Flow cytometry
Mice were bled via tail vein nick. Cells were labeled following RBC lysisand filtration through 40-mm cell strainers. Abs included unlabeled 2.4G2,FITC-conjugated anti-mouse c-Kit (2B8) and Gr-1 (RB6-6B2); PE-conjugated anti-FcεRIa (MAR-1) and Gr-1 (RB6-8C5); Alexa 647-con-jugated anti-mouse CD49b (DX5) and B220 (RA3-6B2); PE-Cy7–conju-gated anti-mouse CD3 (145-2C11) and CD11b (M1/70) from BioLegend;and PE-conjugated anti–Siglec-F (E50-2440) from BD Biosciences. Flowcytometric analysis was performed using a FACSCanto (BD Biosciences),and data analysis was conducted with FlowJo software (Tree Star).
Western blot
Cells were dissociated in radioimmunoprecipitation assay buffer andWestern blotting was performed using 50 mg total cell lysate per sample.Proteins were loaded and separated over an 8–16 or 4–20% gradient SDS-polyacrylamide gel (Bio-Rad). Proteins were transferred to nitrocellulose
membranes (Pall, Ann Arbor, MI) and blocked for 60 min in Blotto Bbuffer (Rockland Immunochemicals, Gilbertsville, PA) plus 0.1% Tween-20. Blots were incubated in a solution of TBS supplemented with 0.1%Tween-20 and 5% BSA (TBST), with the indicated Abs overnight at 4˚Cwith gentle rocking. Blots were washed six times for 10 min each in TBST,followed by incubation in Blotto B containing a 1:5000 dilution of HRP-linked anti-IgG matched to the relevant species, from Cell SignalingTechnology (Danvers, MA). Size estimates for proteins were obtainedusing molecular mass standards from Bio-Rad.
Passive systemic anaphylaxis
Mice were injected i.v. with 200 ml PBS containing 5 mg histamine, 700 ngplatelet-activating factor (PAF), or 500 mg rat anti-mouse CD16/CD32,clone 2.4G2 (13, 31). The body temperature of each animal was mea-sured using a rectal microprobe (Physitemp Instruments). Mice were theneuthanized, and blood was collected by cardiac puncture to prepare serum.
PAF acetylhydrolase activity assays
Substrate (50 mM 1-alkyl-2-acetyl-sn-glycero-3-phosphocholine with 0.05mCi hexadecyl-2-acetyl-sn-glyceryl-3-phosphocholine, 1-O-[acetyl-(N)-3H][NEN/PerkinElmer; 13.5 Ci/mmol] added as a tracer) was combined withserum and incubated for 30 min at 37˚C to determine PAF acetylhydrolase(PAFAH) enzymatic activity. PAFAH activity was expressed as releaseof [3H]acetate, using a method that we described previously (32). The1-alkyl-2-acetyl-sn-glycero-3-phosphocholine (PAF) was purchased fromAvanti Polar Lipids (Alabaster, AL).
Statistical analysis
Data are presented as the means 6 SEM of at least three independentexperiments. Comparisons were made by the two-tailed Student t test forindependent samples. A p value ,0.05 was considered statistically sig-nificant. Analysis was performed with GraphPad Prism software.
ResultsFcgR stimulation induces rapid Fyn, Lyn, Akt, Erk, p38, andJNK activation in BMMCs
To compare the effect of the IgE versus IgG receptor stimulationon the phosphorylation state of Fyn kinase, BMMCs were stim-ulated in vitro with IgE plus Ag or 2.4G2 plus anti-IgG (referred toas IgE or 2.4G2 crosslinking [XL], respectively). Fyn kinase wasthen immunoprecipitated and proteins were separated by SDS-PAGE. The phosphorylated form of Fyn was detected by immu-noblotting for the activated loop of the Src kinase family on residue416 (pY416) (Fig. 1A). Our results show that Fyn kinase (59 kDa)is rapidly activated downstream of both the IgE and the IgGreceptors. Quantification of Western blot signal intensity by den-sitometry after adjusting for loading showed that FcgR and FcεRIactivate Fyn to a similar extent.Similar to Fyn, we found that both Lyn kinase isoforms (53
and 56 kDa) (17) were also activated following 2.4G2 XL (Fig.1B). FcgR-mediated Lyn activation was nearly as strong as IgE-mediated effects as attested by densitometry analysis of band in-tensity.Intracellular signaling events occurring in BMMCs after FcεRI
activation of mast cells has been intensively studied and is wellestablished. Because our results showed that comparable activa-tion levels of Fyn and Lyn can be achieved through FcgR or FcεRIstimulation, we next decided to investigate the kinetics of phos-phorylation events subsequent to Fyn and Lyn activation. Wefound that Fyn activation after 2.4G2 XL occurred rapidly, withmaximal phosphorylation observed within 3 min (Fig. 1C). Ad-ditionally, FcgR activation of mast cells triggered the activation ofAkt, Erk, p38, and JNK (Fig. 1D). A representative densitometryanalysis of band intensity shows the phosphorylation kinetics ofthese signaling molecules (Fig. 1E), with ERK phosphorylationbeing the strongest, reaching ∼70-fold increase in less thana minute after the initiation of 2.4G2 XL. Our results showedparallel kinetics for Fyn, Akt, and Erk during the early time points
The Journal of Immunology 4361
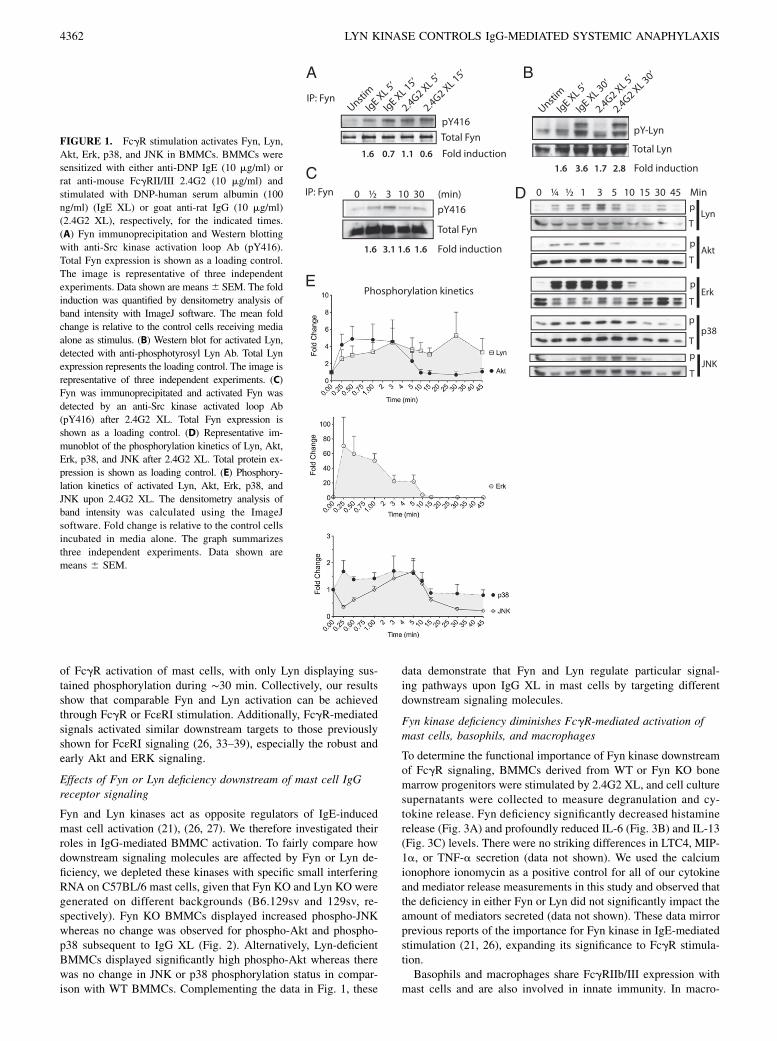
of FcgR activation of mast cells, with only Lyn displaying sus-tained phosphorylation during ∼30 min. Collectively, our resultsshow that comparable Fyn and Lyn activation can be achievedthrough FcgR or FcεRI stimulation. Additionally, FcgR-mediatedsignals activated similar downstream targets to those previouslyshown for FcεRI signaling (26, 33–39), especially the robust andearly Akt and ERK signaling.
Effects of Fyn or Lyn deficiency downstream of mast cell IgGreceptor signaling
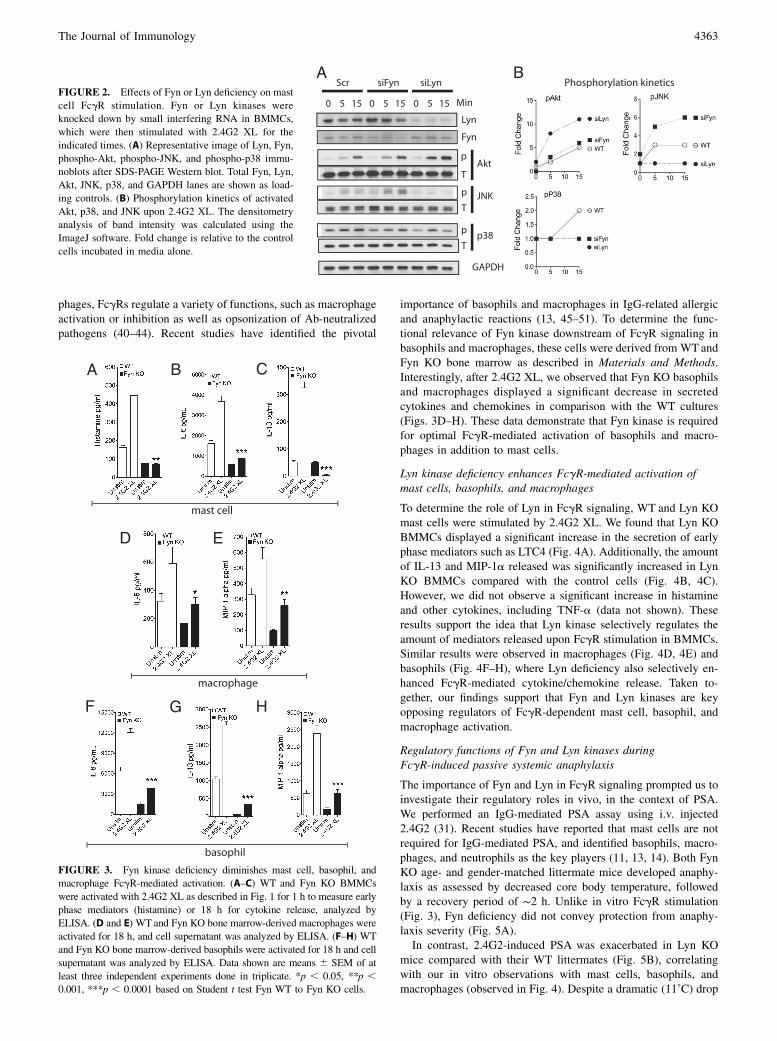
Fyn and Lyn kinases act as opposite regulators of IgE-inducedmast cell activation (21), (26, 27). We therefore investigated theirroles in IgG-mediated BMMC activation. To fairly compare howdownstream signaling molecules are affected by Fyn or Lyn de-ficiency, we depleted these kinases with specific small interferingRNA on C57BL/6 mast cells, given that Fyn KO and Lyn KO weregenerated on different backgrounds (B6.129sv and 129sv, re-spectively). Fyn KO BMMCs displayed increased phospho-JNKwhereas no change was observed for phospho-Akt and phospho-p38 subsequent to IgG XL (Fig. 2). Alternatively, Lyn-deficientBMMCs displayed significantly high phospho-Akt whereas therewas no change in JNK or p38 phosphorylation status in compar-ison with WT BMMCs. Complementing the data in Fig. 1, these
data demonstrate that Fyn and Lyn regulate particular signal-ing pathways upon IgG XL in mast cells by targeting differentdownstream signaling molecules.
Fyn kinase deficiency diminishes FcgR-mediated activation ofmast cells, basophils, and macrophages
To determine the functional importance of Fyn kinase downstreamof FcgR signaling, BMMCs derived from WT or Fyn KO bonemarrow progenitors were stimulated by 2.4G2 XL, and cell culturesupernatants were collected to measure degranulation and cy-tokine release. Fyn deficiency significantly decreased histaminerelease (Fig. 3A) and profoundly reduced IL-6 (Fig. 3B) and IL-13(Fig. 3C) levels. There were no striking differences in LTC4, MIP-1a, or TNF-a secretion (data not shown). We used the calciumionophore ionomycin as a positive control for all of our cytokineand mediator release measurements in this study and observed thatthe deficiency in either Fyn or Lyn did not significantly impact theamount of mediators secreted (data not shown). These data mirrorprevious reports of the importance for Fyn kinase in IgE-mediatedstimulation (21, 26), expanding its significance to FcgR stimula-tion.Basophils and macrophages share FcgRIIb/III expression with
mast cells and are also involved in innate immunity. In macro-
FIGURE 1. FcgR stimulation activates Fyn, Lyn,
Akt, Erk, p38, and JNK in BMMCs. BMMCs were
sensitized with either anti-DNP IgE (10 mg/ml) or
rat anti-mouse FcgRII/III 2.4G2 (10 mg/ml) and
stimulated with DNP-human serum albumin (100
ng/ml) (IgE XL) or goat anti-rat IgG (10 mg/ml)
(2.4G2 XL), respectively, for the indicated times.
(A) Fyn immunoprecipitation and Western blotting
with anti-Src kinase activation loop Ab (pY416).
Total Fyn expression is shown as a loading control.
The image is representative of three independent
experiments. Data shown are means6 SEM. The fold
induction was quantified by densitometry analysis of
band intensity with ImageJ software. The mean fold
change is relative to the control cells receiving media
alone as stimulus. (B) Western blot for activated Lyn,
detected with anti-phosphotyrosyl Lyn Ab. Total Lyn
expression represents the loading control. The image is
representative of three independent experiments. (C)
Fyn was immunoprecipitated and activated Fyn was
detected by an anti-Src kinase activated loop Ab
(pY416) after 2.4G2 XL. Total Fyn expression is
shown as a loading control. (D) Representative im-
munoblot of the phosphorylation kinetics of Lyn, Akt,
Erk, p38, and JNK after 2.4G2 XL. Total protein ex-
pression is shown as loading control. (E) Phosphory-
lation kinetics of activated Lyn, Akt, Erk, p38, and
JNK upon 2.4G2 XL. The densitometry analysis of
band intensity was calculated using the ImageJ
software. Fold change is relative to the control cells
incubated in media alone. The graph summarizes
three independent experiments. Data shown are
means 6 SEM.
4362 LYN KINASE CONTROLS IgG-MEDIATED SYSTEMIC ANAPHYLAXIS
phages, FcgRs regulate a variety of functions, such as macrophageactivation or inhibition as well as opsonization of Ab-neutralizedpathogens (40–44). Recent studies have identified the pivotal
importance of basophils and macrophages in IgG-related allergicand anaphylactic reactions (13, 45–51). To determine the func-tional relevance of Fyn kinase downstream of FcgR signaling inbasophils and macrophages, these cells were derived from WT andFyn KO bone marrow as described in Materials and Methods.Interestingly, after 2.4G2 XL, we observed that Fyn KO basophilsand macrophages displayed a significant decrease in secretedcytokines and chemokines in comparison with the WT cultures(Figs. 3D–H). These data demonstrate that Fyn kinase is requiredfor optimal FcgR-mediated activation of basophils and macro-phages in addition to mast cells.
Lyn kinase deficiency enhances FcgR-mediated activation ofmast cells, basophils, and macrophages
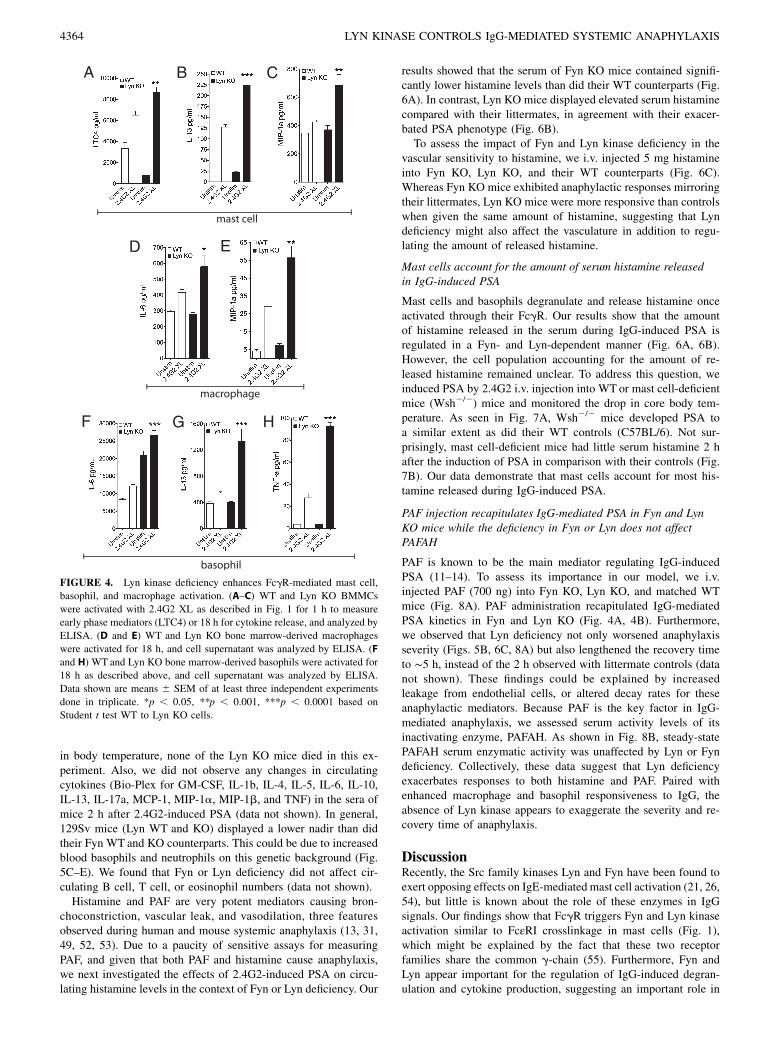
To determine the role of Lyn in FcgR signaling, WT and Lyn KOmast cells were stimulated by 2.4G2 XL. We found that Lyn KOBMMCs displayed a significant increase in the secretion of earlyphase mediators such as LTC4 (Fig. 4A). Additionally, the amountof IL-13 and MIP-1a released was significantly increased in LynKO BMMCs compared with the control cells (Fig. 4B, 4C).However, we did not observe a significant increase in histamineand other cytokines, including TNF-a (data not shown). Theseresults support the idea that Lyn kinase selectively regulates theamount of mediators released upon FcgR stimulation in BMMCs.Similar results were observed in macrophages (Fig. 4D, 4E) andbasophils (Fig. 4F–H), where Lyn deficiency also selectively en-hanced FcgR-mediated cytokine/chemokine release. Taken to-gether, our findings support that Fyn and Lyn kinases are keyopposing regulators of FcgR-dependent mast cell, basophil, andmacrophage activation.
Regulatory functions of Fyn and Lyn kinases duringFcgR-induced passive systemic anaphylaxis
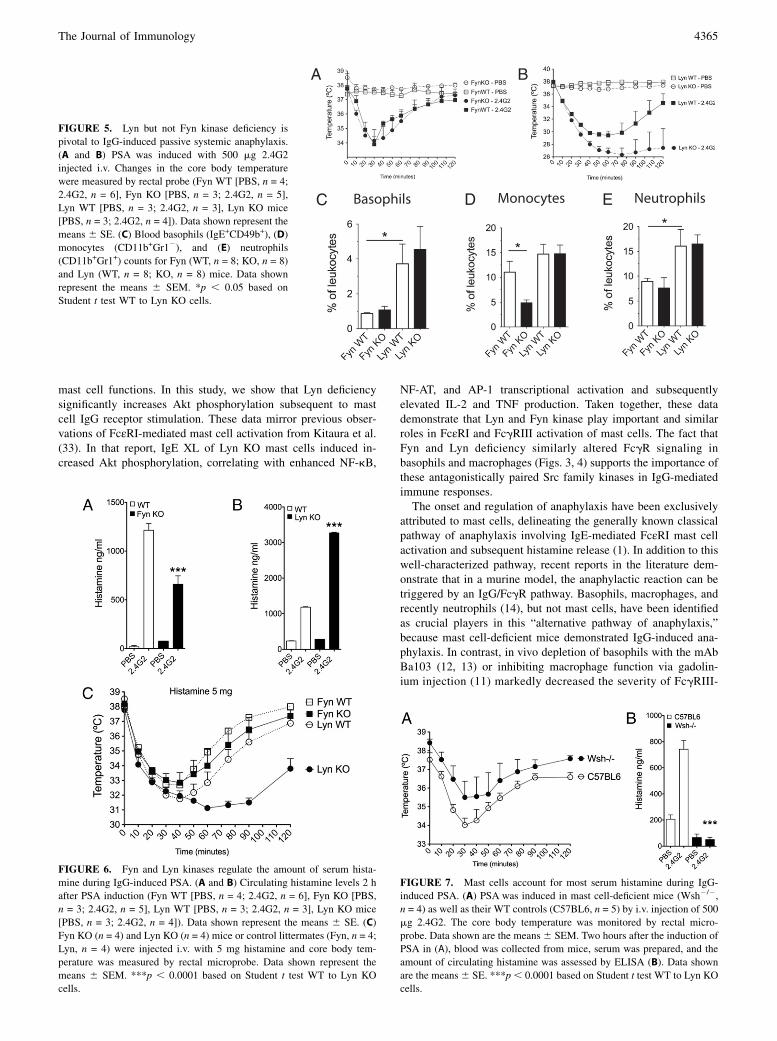
The importance of Fyn and Lyn in FcgR signaling prompted us toinvestigate their regulatory roles in vivo, in the context of PSA.We performed an IgG-mediated PSA assay using i.v. injected2.4G2 (31). Recent studies have reported that mast cells are notrequired for IgG-mediated PSA, and identified basophils, macro-phages, and neutrophils as the key players (11, 13, 14). Both FynKO age- and gender-matched littermate mice developed anaphy-laxis as assessed by decreased core body temperature, followedby a recovery period of ∼2 h. Unlike in vitro FcgR stimulation(Fig. 3), Fyn deficiency did not convey protection from anaphy-laxis severity (Fig. 5A).In contrast, 2.4G2-induced PSA was exacerbated in Lyn KO
mice compared with their WT littermates (Fig. 5B), correlatingwith our in vitro observations with mast cells, basophils, andmacrophages (observed in Fig. 4). Despite a dramatic (11˚C) drop
FIGURE 2. Effects of Fyn or Lyn deficiency on mast
cell FcgR stimulation. Fyn or Lyn kinases were
knocked down by small interfering RNA in BMMCs,
which were then stimulated with 2.4G2 XL for the
indicated times. (A) Representative image of Lyn, Fyn,
phospho-Akt, phospho-JNK, and phospho-p38 immu-
noblots after SDS-PAGE Western blot. Total Fyn, Lyn,
Akt, JNK, p38, and GAPDH lanes are shown as load-
ing controls. (B) Phosphorylation kinetics of activated
Akt, p38, and JNK upon 2.4G2 XL. The densitometry
analysis of band intensity was calculated using the
ImageJ software. Fold change is relative to the control
cells incubated in media alone.
FIGURE 3. Fyn kinase deficiency diminishes mast cell, basophil, and
macrophage FcgR-mediated activation. (A–C) WT and Fyn KO BMMCs
were activated with 2.4G2 XL as described in Fig. 1 for 1 h to measure early
phase mediators (histamine) or 18 h for cytokine release, analyzed by
ELISA. (D and E) WT and Fyn KO bone marrow-derived macrophages were
activated for 18 h, and cell supernatant was analyzed by ELISA. (F–H) WT
and Fyn KO bone marrow-derived basophils were activated for 18 h and cell
supernatant was analyzed by ELISA. Data shown are means 6 SEM of at
least three independent experiments done in triplicate. *p , 0.05, **p ,0.001, ***p , 0.0001 based on Student t test Fyn WT to Fyn KO cells.
The Journal of Immunology 4363
in body temperature, none of the Lyn KO mice died in this ex-periment. Also, we did not observe any changes in circulatingcytokines (Bio-Plex for GM-CSF, IL-1b, IL-4, IL-5, IL-6, IL-10,IL-13, IL-17a, MCP-1, MIP-1a, MIP-1b, and TNF) in the sera ofmice 2 h after 2.4G2-induced PSA (data not shown). In general,129Sv mice (Lyn WT and KO) displayed a lower nadir than didtheir Fyn WT and KO counterparts. This could be due to increasedblood basophils and neutrophils on this genetic background (Fig.5C–E). We found that Fyn or Lyn deficiency did not affect cir-culating B cell, T cell, or eosinophil numbers (data not shown).Histamine and PAF are very potent mediators causing bron-
choconstriction, vascular leak, and vasodilation, three featuresobserved during human and mouse systemic anaphylaxis (13, 31,49, 52, 53). Due to a paucity of sensitive assays for measuringPAF, and given that both PAF and histamine cause anaphylaxis,we next investigated the effects of 2.4G2-induced PSA on circu-lating histamine levels in the context of Fyn or Lyn deficiency. Our
results showed that the serum of Fyn KO mice contained signifi-cantly lower histamine levels than did their WT counterparts (Fig.6A). In contrast, Lyn KO mice displayed elevated serum histaminecompared with their littermates, in agreement with their exacer-bated PSA phenotype (Fig. 6B).To assess the impact of Fyn and Lyn kinase deficiency in the
vascular sensitivity to histamine, we i.v. injected 5 mg histamineinto Fyn KO, Lyn KO, and their WT counterparts (Fig. 6C).Whereas Fyn KO mice exhibited anaphylactic responses mirroringtheir littermates, Lyn KO mice were more responsive than controlswhen given the same amount of histamine, suggesting that Lyndeficiency might also affect the vasculature in addition to regu-lating the amount of released histamine.
Mast cells account for the amount of serum histamine releasedin IgG-induced PSA
Mast cells and basophils degranulate and release histamine onceactivated through their FcgR. Our results show that the amountof histamine released in the serum during IgG-induced PSA isregulated in a Fyn- and Lyn-dependent manner (Fig. 6A, 6B).However, the cell population accounting for the amount of re-leased histamine remained unclear. To address this question, weinduced PSA by 2.4G2 i.v. injection into WTor mast cell-deficientmice (Wsh2/2) mice and monitored the drop in core body tem-perature. As seen in Fig. 7A, Wsh2/2 mice developed PSA toa similar extent as did their WT controls (C57BL/6). Not sur-prisingly, mast cell-deficient mice had little serum histamine 2 hafter the induction of PSA in comparison with their controls (Fig.7B). Our data demonstrate that mast cells account for most his-tamine released during IgG-induced PSA.
PAF injection recapitulates IgG-mediated PSA in Fyn and LynKO mice while the deficiency in Fyn or Lyn does not affectPAFAH
PAF is known to be the main mediator regulating IgG-inducedPSA (11–14). To assess its importance in our model, we i.v.injected PAF (700 ng) into Fyn KO, Lyn KO, and matched WTmice (Fig. 8A). PAF administration recapitulated IgG-mediatedPSA kinetics in Fyn and Lyn KO (Fig. 4A, 4B). Furthermore,we observed that Lyn deficiency not only worsened anaphylaxisseverity (Figs. 5B, 6C, 8A) but also lengthened the recovery timeto ∼5 h, instead of the 2 h observed with littermate controls (datanot shown). These findings could be explained by increasedleakage from endothelial cells, or altered decay rates for theseanaphylactic mediators. Because PAF is the key factor in IgG-mediated anaphylaxis, we assessed serum activity levels of itsinactivating enzyme, PAFAH. As shown in Fig. 8B, steady-statePAFAH serum enzymatic activity was unaffected by Lyn or Fyndeficiency. Collectively, these data suggest that Lyn deficiencyexacerbates responses to both histamine and PAF. Paired withenhanced macrophage and basophil responsiveness to IgG, theabsence of Lyn kinase appears to exaggerate the severity and re-covery time of anaphylaxis.
DiscussionRecently, the Src family kinases Lyn and Fyn have been found toexert opposing effects on IgE-mediated mast cell activation (21, 26,54), but little is known about the role of these enzymes in IgGsignals. Our findings show that FcgR triggers Fyn and Lyn kinaseactivation similar to FcεRI crosslinkage in mast cells (Fig. 1),which might be explained by the fact that these two receptorfamilies share the common g-chain (55). Furthermore, Fyn andLyn appear important for the regulation of IgG-induced degran-ulation and cytokine production, suggesting an important role in
FIGURE 4. Lyn kinase deficiency enhances FcgR-mediated mast cell,
basophil, and macrophage activation. (A–C) WT and Lyn KO BMMCs
were activated with 2.4G2 XL as described in Fig. 1 for 1 h to measure
early phase mediators (LTC4) or 18 h for cytokine release, and analyzed by
ELISA. (D and E) WT and Lyn KO bone marrow-derived macrophages
were activated for 18 h, and cell supernatant was analyzed by ELISA. (F
and H) WT and Lyn KO bone marrow-derived basophils were activated for
18 h as described above, and cell supernatant was analyzed by ELISA.
Data shown are means 6 SEM of at least three independent experiments
done in triplicate. *p , 0.05, **p , 0.001, ***p , 0.0001 based on
Student t test WT to Lyn KO cells.
4364 LYN KINASE CONTROLS IgG-MEDIATED SYSTEMIC ANAPHYLAXIS
mast cell functions. In this study, we show that Lyn deficiencysignificantly increases Akt phosphorylation subsequent to mastcell IgG receptor stimulation. These data mirror previous obser-vations of FcεRI-mediated mast cell activation from Kitaura et al.(33). In that report, IgE XL of Lyn KO mast cells induced in-creased Akt phosphorylation, correlating with enhanced NF-kB,
NF-AT, and AP-1 transcriptional activation and subsequentlyelevated IL-2 and TNF production. Taken together, these datademonstrate that Lyn and Fyn kinase play important and similarroles in FcεRI and FcgRIII activation of mast cells. The fact thatFyn and Lyn deficiency similarly altered FcgR signaling inbasophils and macrophages (Figs. 3, 4) supports the importance ofthese antagonistically paired Src family kinases in IgG-mediatedimmune responses.The onset and regulation of anaphylaxis have been exclusively
attributed to mast cells, delineating the generally known classicalpathway of anaphylaxis involving IgE-mediated FcεRI mast cellactivation and subsequent histamine release (1). In addition to thiswell-characterized pathway, recent reports in the literature dem-onstrate that in a murine model, the anaphylactic reaction can betriggered by an IgG/FcgR pathway. Basophils, macrophages, andrecently neutrophils (14), but not mast cells, have been identifiedas crucial players in this “alternative pathway of anaphylaxis,”because mast cell-deficient mice demonstrated IgG-induced ana-phylaxis. In contrast, in vivo depletion of basophils with the mAbBa103 (12, 13) or inhibiting macrophage function via gadolin-ium injection (11) markedly decreased the severity of FcgRIII-
FIGURE 5. Lyn but not Fyn kinase deficiency is
pivotal to IgG-induced passive systemic anaphylaxis.
(A and B) PSA was induced with 500 mg 2.4G2
injected i.v. Changes in the core body temperature
were measured by rectal probe (Fyn WT [PBS, n = 4;
2.4G2, n = 6], Fyn KO [PBS, n = 3; 2.4G2, n = 5],
Lyn WT [PBS, n = 3; 2.4G2, n = 3], Lyn KO mice
[PBS, n = 3; 2.4G2, n = 4]). Data shown represent the
means 6 SE. (C) Blood basophils (IgE+CD49b+), (D)
monocytes (CD11b+Gr12), and (E) neutrophils
(CD11b+Gr1+) counts for Fyn (WT, n = 8; KO, n = 8)
and Lyn (WT, n = 8; KO, n = 8) mice. Data shown
represent the means 6 SEM. *p , 0.05 based on
Student t test WT to Lyn KO cells.
FIGURE 6. Fyn and Lyn kinases regulate the amount of serum hista-
mine during IgG-induced PSA. (A and B) Circulating histamine levels 2 h
after PSA induction (Fyn WT [PBS, n = 4; 2.4G2, n = 6], Fyn KO [PBS,
n = 3; 2.4G2, n = 5], Lyn WT [PBS, n = 3; 2.4G2, n = 3], Lyn KO mice
[PBS, n = 3; 2.4G2, n = 4]). Data shown represent the means 6 SE. (C)
Fyn KO (n = 4) and Lyn KO (n = 4) mice or control littermates (Fyn, n = 4;
Lyn, n = 4) were injected i.v. with 5 mg histamine and core body tem-
perature was measured by rectal microprobe. Data shown represent the
means 6 SEM. ***p , 0.0001 based on Student t test WT to Lyn KO
cells.
FIGURE 7. Mast cells account for most serum histamine during IgG-
induced PSA. (A) PSA was induced in mast cell-deficient mice (Wsh2/2,
n = 4) as well as their WT controls (C57BL6, n = 5) by i.v. injection of 500
mg 2.4G2. The core body temperature was monitored by rectal micro-
probe. Data shown are the means6 SEM. Two hours after the induction of
PSA in (A), blood was collected from mice, serum was prepared, and the
amount of circulating histamine was assessed by ELISA (B). Data shown
are the means6 SE. ***p, 0.0001 based on Student t test WT to Lyn KO
cells.
The Journal of Immunology 4365

mediated anaphylaxis. A recent report further showed thatFcgRIV-neutrophil–induced anaphylaxis also occurs, as mice de-ficient in FcgRI/FcgRIIB/FcgRIIIA/FcεRI/FcεRII2/2 (5KO mice)still developed IgG-mediated anaphylaxis (14). In this alternativeform of anaphylaxis, PAF, rather than histamine, has been indi-cated as a pivotal mediator. Although PAF serum levels are quitechallenging to measure, the in vivo administration of a PAF an-tagonist protected mice from developing IgG- but not IgE-mediated anaphylaxis (13). Thus, it is thought that IgG-inducedanaphylaxis operates through FcgR-mediated activation of baso-phils, neutrophils, and macrophages, with subsequent PAF releasetriggering vasodilation, vascular fluid leak, and loss of core bodytemperature (11, 13, 14, 48).Our data support and extend the understanding of IgG-mediated
anaphylaxis at the molecular signaling level. First, we found thatFyn or Lyn deletion has opposing effects on histamine release andcytokine secretion during IgG activation of mast cells, basophils,and macrophages in vitro. Interestingly, although the effects onhistamine release were consistent in vivo, they did not predict theseverity of FcgR-induced PSA. Whereas Lyn KO mice showedincreased histamine and worsened hypothermia, Fyn KO mice haddiminished histamine but no change in hypothermia versus re-spective WT mice. Additionally, although histamine and PAF areresponsible for anaphylaxis, we found that mast cells were mainlyresponsible for the amount of circulating histamine present in theserum during the course of FcgR-induced PSA (Fig. 7), extendingthe involvement of mast cells in IgG-related pathologies. Inter-estingly, a recent study found histamine produced by non-mastcell sources in a model of contact hypersensitivity (56), suggest-ing that in vivo histamine production varies with the elicitingstimulus. Based on previous reports (11–14), the drop in bodytemperature observed in mast cell-deficient animals over thecourse of anaphylaxis should be attributed to PAF released bybasophils, macrophages, and neutrophils after FcgRIII activation.Furthermore, we did not observe any significant difference inperivascular cell infiltration and pulmonary edema between FynKO mice, Lyn KO mice, and their controls subsequent to 2.4G2-induced PSA (data not shown).The exacerbated response of Lyn KO mice could be ascribed to
many factors: increased PAF secretion, decreased PAFmetabolism,or enhanced PAF signaling in vascular endothelium. We found nochanges in PAFAH activity, suggesting that reduced PAF catabo-lism is not an explanation. Furthermore, we found no overt changes
in circulating basophils, monocytes, or neutrophils in Lyn KOmice. It is also possible that Lyn KO endothelial cells, just like mastcells, basophils, and macrophages, are hyperresponsive to anystimuli involving Lyn kinase. In fact, Kuruvilla et al. (57) havedemonstrated that Fyn and Lyn are phosphorylated upon PAFreceptor activation, leading to phosphorylation of the p85 regu-latory subunit of PI3K. Additionally, Yu et al. (58) showed thatphosphotyrosine activated Lyn kinase colocalized with PI3K inthe lipid body fraction of PMN leukocytes subsequent to PAFreceptor stimulation. Taken together, and in line with our find-ings, we therefore speculate that Lyn KO endothelial cells arehyperresponsive to PAF stimulation, triggering a longer and moresevere vasodilation episode in comparison with Lyn-sufficient cells.Based on our data, we can hypothesize that patients with altered
Lyn function or expression could be hyperresponsive to IgE- andIgG-mediated pathology. There is precedence for this, as mostsystemic lupus erythematosus (SLE) patients have been shownto have reduced Lyn expression in two clinical studies (59, 60).Although SLE is regarded as an IgG immune complex-mediateddisease, Charles et al. (61) recently showed that basophils acti-vated by IgE may participate in the onset or the exacerbation oflupus-like symptoms in a mouse model. In the same report, thisgroup demonstrated that aged Lyn KO mice displayed increasedbasophil numbers in the lymph nodes, blood, and spleen. Fur-thermore, Lyn KO mice display significantly elevated circulatingautoantibodies, which, in conjunction with many other factors,lead to the development of SLE-like symptoms and contribute tothe elevated mortality in this population (62–64). Combined withour data, Lyn deficiency is postulated to worsen either IgG- orIgE-mediated inflammation, which could contribute to the devel-opment of SLE in aged mice.Several studies have reported the presence of allergen-specific
IgG in allergic individuals. The involvement of this Ig isotype inthe onset and the development of the allergic reaction remainspoorly understood (65, 66). However, Bandukwala et al. (67) useda murine model to demonstrate the importance of FcgRI andFcgRIII in airway inflammation and hyperresponsiveness. Thisstudy showed that C57BL/6 mice that were sensitized with non-infectious parasitic Schistosoma mansoni eggs and challengedwith soluble egg Ag displayed airway inflammation, includingeosinophil infiltration and severe peribronchial and perivascularinflammation. This response was greatly decreased in FcgRI2/2 orFcgRIII2/2 mice. This group also demonstrated that deficiency ofFcgRIII, but not FcgRI, reduced lung resistance upon methacho-line challenge, compared with littermate controls. Thus, in addi-tion to the well-established role of IgE/FcεRI in the onset and thedevelopment of allergy and hypersensitivity, this study providesevidence that FcgRIII activation can participate in airway diseasepathogenesis in a murine model. Combined with our data, thesefindings support the theory that the exacerbated 2.4G2-mediatedPSA we noted in Lyn KO mice is elicited by FcgRIII, as previ-ously supported by Daeron and colleagues (68) and by Ravetchand colleagues (69–71).The high-affinity (FcgRI) and low-affinity (FcgRIII) IgG re-
ceptors are known to be important in the activation of numerouscell types of the immune system and in the phagocytosis ofopsonized microbes. In contrast, one of the members of the FcgRfamily, FcgRIIb, has emerged as an inhibitory receptor (31, 72,73). Using a mouse model of allergic asthma, Dharajiya et al. (74)demonstrated that in addition to upregulating the expression ofFcgRIIb in CD14+MHC class II+ mononuclear as well as inCD11b+ cells in the lungs, ragweed extract challenge also ledto signs of severe allergic asthma-like symptoms in FcgRIIb-deficient mice, delineated by increased airway eosinophilia, mu-
FIGURE 8. Intravenous administration of PAF recapitulates IgG-in-
duced PSA kinetics while the deficiency in Fyn or Lyn does not affect
PAFAH. (A) PSA was induced by i.v. delivery of 700 ng PAF in Fyn KO
(n = 3) and Lyn KO (n = 4) as well as their littermate controls (Fyn, n = 4;
Lyn, n = 4). Core body temperature was measured by rectal microprobe.
Data shown represent the means 6 SE. (B) Enzymatic activity of serum
levels of PAFAH in Fyn and Lyn KO mice and their control littermates
(n $ 9/group). Data shown represent the means 6 SEM.
4366 LYN KINASE CONTROLS IgG-MEDIATED SYSTEMIC ANAPHYLAXIS

cin production, and allergen-specific IgE. Furthermore, a tremen-dous increase in macrophage, eosinophil, and lymphocyte re-cruitment was observed in the bronchoalveolar lavage fluid ofragweed extract-challenged FcgRIIb KO mice in comparison withWT controls. These data, as well as many others, consolidate theidea that IgG and Fcg receptors play important roles in the reg-ulation of immune system homeostasis. Our data support the hy-pothesis that Fyn and Lyn kinases are pivotal antagonistic reg-ulators of this paired IgG receptor system.The data in this study demonstrate that Fyn and Lyn kinases are
activated during IgG-mediated signaling and have opposing reg-ulatory functions in mast cells, basophils, and macrophages. Inaddition to Fyn and Lyn activation, we found that FcgR stimulationalso led to the phosphorylation of Akt, Erk, p38, and JNK. Fur-thermore, we also uncover the unsuspected contribution of mastcells as major producers of serum histamine during IgG-inducedPSA, regulated in a Fyn- and Lyn-dependent manner. More im-portantly, we show that overall Lyn but not Fyn kinase regulatesthe severity of IgG-induced passive systemic anaphylaxis, byenhancing the amount of vasoactive mediators secreted and ex-acerbating endothelial cell responsiveness to PAF and histamine.In line with previous studies, our findings extend the under-standing of IgG-related pathologies and demonstrate the pivotalrole of Lyn kinase as the key regulator of IgG-mediated inflam-mation.
DisclosuresThe authors have no financial conflicts of interest.
References1. Galli, S. J., S. Nakae, and M. Tsai. 2005. Mast cells in the development of
adaptive immune responses. Nat. Immunol. 6: 135–142.2. Kalesnikoff, J., and S. J. Galli. 2008. New developments in mast cell biology.
Nat. Immunol. 9: 1215–1223.3. Martin, T. R., S. J. Galli, I. M. Katona, and J. M. Drazen. 1989. Role of mast
cells in anaphylaxis: evidence for the importance of mast cells in the cardio-pulmonary alterations and death induced by anti-IgE in mice. J. Clin. Invest. 83:1375–1383.
4. Zabel, B. A., S. Nakae, L. Zuniga, J. Y. Kim, T. Ohyama, C. Alt, J. Pan, H. Suto,D. Soler, S. J. Allen, et al. 2008. Mast cell-expressed orphan receptor CCRL2binds chemerin and is required for optimal induction of IgE-mediated passivecutaneous anaphylaxis. J. Exp. Med. 205: 2207–2220.
5. Ryan, J. J., and J. F. Fernando. 2009. Mast cell modulation of the immune re-sponse. Curr. Allergy Asthma Rep. 9: 353–359.
6. Gilfillan, A. M., and J. Rivera. 2009. The tyrosine kinase network regulatingmast cell activation. Immunol. Rev. 228: 149–169.
7. Rivera, J., and A. Olivera. 2008. A current understanding of FcεRI-dependentmast cell activation. Curr. Allergy Asthma Rep. 8: 14–20.
8. Rivera, J., N. A. Fierro, A. Olivera, and R. Suzuki. 2008. New insights on mastcell activation via the high affinity receptor for IgE. Adv. Immunol. 98: 85–120.
9. Galli, S. J., M. Grimbaldeston, and M. Tsai. 2008. Immunomodulatory mastcells: negative, as well as positive, regulators of immunity. Nat. Rev. Immunol. 8:478–486.
10. Daeron, M., C. Bonnerot, S. Latour, and W. H. Fridman. 1992. Murine re-combinant FcgRIII, but not FcgRII, trigger serotonin release in rat basophilicleukemia cells. J. Immunol. 149: 1365–1373.
11. Strait, R. T., S. C. Morris, M. Yang, X. W. Qu, and F. D. Finkelman. 2002.Pathways of anaphylaxis in the mouse. J. Allergy Clin. Immunol. 109: 658–668.
12. Obata, K., K. Mukai, Y. Tsujimura, K. Ishiwata, Y. Kawano, Y. Minegishi,N. Watanabe, and H. Karasuyama. 2007. Basophils are essential initiators ofa novel type of chronic allergic inflammation. Blood 110: 913–920.
13. Tsujimura, Y., K. Obata, K. Mukai, H. Shindou, M. Yoshida, H. Nishikado,Y. Kawano, Y. Minegishi, T. Shimizu, and H. Karasuyama. 2008. Basophilsplay a pivotal role in immunoglobulin-G-mediated but not immunoglobulin-E-mediated systemic anaphylaxis. Immunity 28: 581–589.
14. Jonsson, F., D. A. Mancardi, Y. Kita, H. Karasuyama, B. Iannascoli, N. VanRooijen, T. Shimizu, M. Daeron, and P. Bruhns. 2011. Mouse and human neu-trophils induce anaphylaxis. J. Clin. Invest. 121: 1484–1496.
15. Stanley, E., S. Ralph, S. McEwen, I. Boulet, D. A. Holtzman, P. Lock, andA. R. Dunn. 1991. Alternatively spliced murine lyn mRNAs encode distinctproteins. Mol. Cell. Biol. 11: 3399–3406.
16. Yi, T. L., J. B. Bolen, and J. N. Ihle. 1991. Hematopoietic cells express twoforms of lyn kinase differing by 21 amino acids in the amino terminus.Mol. Cell.Biol. 11: 2391–2398.
17. Alvarez-Errico, D., Y. Yamashita, R. Suzuki, S. Odom, Y. Furumoto,T. Yamashita, and J. Rivera. 2010. Functional analysis of Lyn kinase A and Bisoforms reveals redundant and distinct roles in FcεRI-dependent mast cell ac-tivation. J. Immunol. 184: 5000–5008.
18. Yamanashi, Y., T. Kakiuchi, J. Mizuguchi, T. Yamamoto, and K. Toyoshima.1991. Association of B cell antigen receptor with protein tyrosine kinase Lyn.Science 251: 192–194.
19. Eiseman, E., and J. B. Bolen. 1992. Engagement of the high-affinity IgE receptoractivates src protein-related tyrosine kinases. Nature 355: 78–80.
20. Kawakami, Y., J. Kitaura, A. B. Satterthwaite, R. M. Kato, K. Asai,S. E. Hartman, M. Maeda-Yamamoto, C. A. Lowell, D. J. Rawlings, O. N. Witte,and T. Kawakami. 2000. Redundant and opposing functions of two tyrosinekinases, Btk and Lyn, in mast cell activation. J. Immunol. 165: 1210–1219.
21. Odom, S., G. Gomez, M. Kovarova, Y. Furumoto, J. J. Ryan, H. V. Wright,C. Gonzalez-Espinosa, M. L. Hibbs, K. W. Harder, and J. Rivera. 2004. Negativeregulation of immunoglobulin E-dependent allergic responses by Lyn kinase. J.Exp. Med. 199: 1491–1502.
22. Satterthwaite, A. B., C. A. Lowell, W. N. Khan, P. Sideras, F. W. Alt, andO. N. Witte. 1998. Independent and opposing roles for Btk and lyn in B andmyeloid signaling pathways. J. Exp. Med. 188: 833–844.
23. Takeshita, H., I. Taniuchi, J. Kato, and T. Watanabe. 1998. Abrogation of au-toimmune disease in Lyn-deficient mice by the mutation of the Btk gene. Int.Immunol. 10: 435–444.
24. Xu, Y., K. W. Harder, N. D. Huntington, M. L. Hibbs, and D. M. Tarlinton. 2005.Lyn tyrosine kinase: accentuating the positive and the negative. Immunity 22:9–18.
25. Hibbs, M. L., K. W. Harder, J. Armes, N. Kountouri, C. Quilici, F. Casagranda,A. R. Dunn, and D. M. Tarlinton. 2002. Sustained activation of Lyn tyrosinekinase in vivo leads to autoimmunity. J. Exp. Med. 196: 1593–1604.
26. Parravicini, V., M. Gadina, M. Kovarova, S. Odom, C. Gonzalez-Espinosa,Y. Furumoto, S. Saitoh, L. E. Samelson, J. J. O’Shea, and J. Rivera. 2002. Fynkinase initiates complementary signals required for IgE-dependent mast celldegranulation. Nat. Immunol. 3: 741–748.
27. Gomez, G., C. Gonzalez-Espinosa, S. Odom, G. Baez, M. E. Cid, J. J. Ryan, andJ. Rivera. 2005. Impaired FcεRI-dependent gene expression and defective ei-cosanoid and cytokine production as a consequence of Fyn deficiency in mastcells. J. Immunol. 175: 7602–7610.
28. Suzuki, R., X. Liu, A. Olivera, L. Aguiniga, Y. Yamashita, U. Blank,I. Ambudkar, and J. Rivera. 2010. Loss of TRPC1-mediated Ca2+ influx con-tributes to impaired degranulation in Fyn-deficient mouse bone marrow-derivedmast cells. J. Leukoc. Biol. 88: 863–875.
29. Crowley, M. T., P. S. Costello, C. J. Fitzer-Attas, M. Turner, F. Meng, C. Lowell,V. L. Tybulewicz, and A. L. DeFranco. 1997. A critical role for Syk in signaltransduction and phagocytosis mediated by Fcε receptors on macrophages. J.Exp. Med. 186: 1027–1039.
30. Lach-Trifilieff, E., K. Menear, E. Schweighoffer, V. L. Tybulewicz, andC. Walker. 2000. Syk-deficient eosinophils show normal interleukin-5-mediateddifferentiation, maturation, and survival but no longer respond to FcgammaRactivation. Blood 96: 2506–2510.
31. Finkelman, F. D. 2007. Anaphylaxis: lessons from mouse models. J. AllergyClin. Immunol. 120: 506–515.
32. Al-Darmaki, S., H. A. Schenkein, J. G. Tew, and S. E. Barbour. 2003. Differ-ential expression of platelet-activating factor acetylhydrolase in macrophagesand monocyte-derived dendritic cells. J. Immunol. 170: 167–173.
33. Kitaura, J., K. Asai, M. Maeda-Yamamoto, Y. Kawakami, U. Kikkawa, andT. Kawakami. 2000. Akt-dependent cytokine production in mast cells. J. Exp.Med. 192: 729–740.
34. Zhu, M., Y. Liu, S. Koonpaew, O. Granillo, and W. Zhang. 2004. Positive andnegative regulation of FcεRI-mediated signaling by the adaptor protein LAB/NTAL. J. Exp. Med. 200: 991–1000.
35. Simon, M., L. Vanes, R. L. Geahlen, and V. L. Tybulewicz. 2005. Distinct rolesfor the linker region tyrosines of Syk in FcεRI signaling in primary mast cells. J.Biol. Chem. 280: 4510–4517.
36. Xiao, W., H. Nishimoto, H. Hong, J. Kitaura, S. Nunomura, M. Maeda-Yamamoto, Y. Kawakami, C. A. Lowell, C. Ra, and T. Kawakami. 2005. Positiveand negative regulation of mast cell activation by Lyn via the FcεRI. J. Immunol.175: 6885–6892.
37. Barbu, E. A., J. Zhang, and R. P. Siraganian. 2010. The limited contribution ofFyn and Gab2 to the high affinity IgE receptor signaling in mast cells. J. Biol.Chem. 285: 15761–15768.
38. Olenchock, B. A., R. Guo, M. A. Silverman, J. N. Wu, J. H. Carpenter,G. A. Koretzky, and X. P. Zhong. 2006. Impaired degranulation but enhancedcytokine production after FcεRI stimulation of diacylglycerol kinase z-deficientmast cells. J. Exp. Med. 203: 1471–1480.
39. Liu, Y., M. Zhu, K. Nishida, T. Hirano, and W. Zhang. 2007. An essential role forRasGRP1 in mast cell function and IgE-mediated allergic response. J. Exp. Med.204: 93–103.
40. Bohnsack, J. F., H. K. Kleinman, T. Takahashi, J. J. O’Shea, and E. J. Brown.1985. Connective tissue proteins and phagocytic cell function: laminin enhancescomplement and Fc-mediated phagocytosis by cultured human macrophages. J.Exp. Med. 161: 912–923.
41. Aderem, A. A., S. D. Wright, S. C. Silverstein, and Z. A. Cohn. 1985. Ligatedcomplement receptors do not activate the arachidonic acid cascade in residentperitoneal macrophages. J. Exp. Med. 161: 617–622.
42. Underhill, D. M., A. Ozinsky, A. M. Hajjar, A. Stevens, C. B. Wilson,M. Bassetti, and A. Aderem. 1999. The Toll-like receptor 2 is recruited to
The Journal of Immunology 4367

macrophage phagosomes and discriminates between pathogens. Nature 401:811–815.
43. Aderem, A., and D. M. Underhill. 1999. Mechanisms of phagocytosis in mac-rophages. Annu. Rev. Immunol. 17: 593–623.
44. Gao, J. J., V. Diesl, T. Wittmann, D. C. Morrison, J. L. Ryan, S. N. Vogel, andM. T. Follettie. 2002. Regulation of gene expression in mouse macrophagesstimulated with bacterial CpG-DNA and lipopolysaccharide. J. Leukoc. Biol. 72:1234–1245.
45. Kawakami, T. 2008. Basophils now enhance memory. Nat. Immunol. 9: 720–721.
46. Galli, S. J., and C. B. Franco. 2008. Basophils are back! Immunity 28: 495–497.47. Mukai, K., K. Matsuoka, C. Taya, H. Suzuki, H. Yokozeki, K. Nishioka,
K. Hirokawa, M. Etori, M. Yamashita, T. Kubota, et al. 2005. Basophils playa critical role in the development of IgE-mediated chronic allergic inflammationindependently of T cells and mast cells. Immunity 23: 191–202.
48. Mukai, K., K. Obata, Y. Tsujimura, and H. Karasuyama. 2009. New insights intothe roles for basophils in acute and chronic allergy. Allergol. Int. 58: 11–19.
49. Karasuyama, H., K. Mukai, Y. Tsujimura, and K. Obata. 2009. Newly discoveredroles for basophils: a neglected minority gains new respect. Nat. Rev. Immunol.9: 9–13.
50. Hida, S., S. Yamasaki, Y. Sakamoto, M. Takamoto, K. Obata, T. Takai,H. Karasuyama, K. Sugane, T. Saito, and S. Taki. 2009. Fc receptor g-chain,a constitutive component of the IL-3 receptor, is required for IL-3-induced IL-4production in basophils. Nat. Immunol. 10: 214–222.
51. Charles, N., W. T. Watford, H. L. Ramos, L. Hellman, H. C. Oettgen, G. Gomez,J. J. Ryan, J. J. O’Shea, and J. Rivera. 2009. Lyn kinase controls basophil GATA-3 transcription factor expression and induction of Th2 cell differentiation. Im-munity 30: 533–543.
52. Galli, S. J., M. Tsai, and A. M. Piliponsky. 2008. The development of allergicinflammation. Nature 454: 445–454.
53. Khodoun, M., R. Strait, T. Orekov, S. Hogan, H. Karasuyama, D. R. Herbert,J. Kohl, and F. D. Finkelman. 2009. Peanuts can contribute to anaphylactic shockby activating complement. J. Allergy Clin. Immunol. 123: 342–351.
54. Hernandez-Hansen, V., A. J. Smith, Z. Surviladze, A. Chigaev, T. Mazel,J. Kalesnikoff, C. A. Lowell, G. Krystal, L. A. Sklar, B. S. Wilson, andJ. M. Oliver. 2004. Dysregulated FcεRI signaling and altered Fyn and SHIPactivities in Lyn-deficient mast cells. J. Immunol. 173: 100–112.
55. Nimmerjahn, F., and J. V. Ravetch. 2008. Fcg receptors as regulators of immuneresponses. Nat. Rev. Immunol. 8: 34–47.
56. Dudeck, A., J. Dudeck, J. Scholten, A. Petzold, S. Surianarayanan, A. Kohler,K. Peschke, D. Vohringer, C. Waskow, T. Krieg, et al. 2011. Mast cells are keypromoters of contact allergy that mediate the adjuvant effects of haptens. Im-munity 34: 973–984.
57. Kuruvilla, A., C. Pielop, and W. T. Shearer. 1994. Platelet-activating factorinduces the tyrosine phosphorylation and activation of phospholipase C-g1, Fynand Lyn kinases, and phosphatidylinositol 3-kinase in a human B cell line. J.Immunol. 153: 5433–5442.
58. Yu, W., J. Cassara, and P. F. Weller. 2000. Phosphatidylinositide 3-kinaselocalizes to cytoplasmic lipid bodies in human polymorphonuclear leukocytesand other myeloid-derived cells. Blood 95: 1078–1085.
59. Liossis, S. N., E. E. Solomou, M. A. Dimopoulos, P. Panayiotidis,M. M. Mavrikakis, and P. P. Sfikakis. 2001. B-cell kinase lyn deficiency inpatients with systemic lupus erythematosus. J. Investig. Med. 49: 157–165.
60. Flores-Borja, F., P. S. Kabouridis, E. C. Jury, D. A. Isenberg, and R. A. Mageed.2005. Decreased Lyn expression and translocation to lipid raft signaling domainsin B lymphocytes from patients with systemic lupus erythematosus. ArthritisRheum. 52: 3955–3965.
61. Charles, N., D. Hardwick, E. Daugas, G. G. Illei, and J. Rivera. 2010. Basophilsand the T helper 2 environment can promote the development of lupus nephritis.Nat. Med. 16: 701–707.
62. Hibbs, M. L., D. M. Tarlinton, J. Armes, D. Grail, G. Hodgson, R. Maglitto,S. A. Stacker, and A. R. Dunn. 1995. Multiple defects in the immune system ofLyn-deficient mice, culminating in autoimmune disease. Cell 83: 301–311.
63. Nishizumi, H., K. Horikawa, I. Mlinaric-Rascan, and T. Yamamoto. 1998. Adouble-edged kinase Lyn: a positive and negative regulator for antigen receptor-mediated signals. J. Exp. Med. 187: 1343–1348.
64. Yu, C. C., T. S. Yen, C. A. Lowell, and A. L. DeFranco. 2001. Lupus-like kidneydisease in mice deficient in the Src family tyrosine kinases Lyn and Fyn. Curr.Biol. 11: 34–38.
65. Pepys, J., W. E. Parish, B. Stenius-Aarniala, and L. Wide. 1979. Clinical cor-relations between long-term (IgE) and short-term (IgG S-TS) anaphylacticantibodies in atopic and ‘non-atopic’ subjects with respiratory allergic disease.Clin. Allergy 9: 645–658.
66. Out, T. A., E. A. van de Graaf, N. J. van den Berg, and H. M. Jansen. 1991. IgGsubclasses in bronchoalveolar lavage fluid from patients with asthma. Scand. J.Immunol. 33: 719–727.
67. Bandukwala, H. S., B. S. Clay, J. Tong, P. D. Mody, J. L. Cannon, R. A. Shilling,J. S. Verbeek, J. V. Weinstock, J. Solway, and A. I. Sperling. 2007. Signalingthrough FcgRIII is required for optimal T helper type (Th)2 responses and Th2-mediated airway inflammation. J. Exp. Med. 204: 1875–1889.
68. Hazenbos, W. L., J. E. Gessner, F. M. Hofhuis, H. Kuipers, D. Meyer,I. A. Heijnen, R. E. Schmidt, M. Sandor, P. J. Capel, M. Daeron, et al. 1996.Impaired IgG-dependent anaphylaxis and Arthus reaction in FcgRIII (CD16)deficient mice. Immunity 5: 181–188.
69. Dombrowicz, D., V. Flamand, I. Miyajima, J. V. Ravetch, S. J. Galli, andJ. P. Kinet. 1997. Absence of FcεRI a chain results in upregulation of FcgRIII-dependent mast cell degranulation and anaphylaxis: evidence of competitionbetween FcεRI and FcgRIII for limiting amounts of FcR b and g chains. J. Clin.Invest. 99: 915–925.
70. Miyajima, I., D. Dombrowicz, T. R. Martin, J. V. Ravetch, J. P. Kinet, andS. J. Galli. 1997. Systemic anaphylaxis in the mouse can be mediated largelythrough IgG1 and FcgRIII: assessment of the cardiopulmonary changes, mastcell degranulation, and death associated with active or IgE- or IgG1-dependentpassive anaphylaxis. J. Clin. Invest. 99: 901–914.
71. Ujike, A., Y. Ishikawa, M. Ono, T. Yuasa, T. Yoshino, M. Fukumoto,J. V. Ravetch, and T. Takai. 1999. Modulation of immunoglobulin (Ig)E-mediated systemic anaphylaxis by low-affinity Fc receptors for IgG. J. Exp.Med. 189: 1573–1579.
72. Abraham, S. N., and A. L. St John. 2010. Mast cell-orchestrated immunity topathogens. Nat. Rev. Immunol. 10: 440–452.
73. Sarmay, G., G. Koncz, and J. Gergely. 1996. Human type II Fcg receptors inhibitB cell activation by interacting with the p21(ras)-dependent pathway. J. Biol.Chem. 271: 30499–30504.
74. Dharajiya, N., S. V. Vaidya, H. Murai, V. Cardenas, A. Kurosky, I. Boldogh, andS. A. Sur. 2010. FcgRIIb inhibits allergic lung inflammation in a murine modelof allergic asthma. PLoS ONE 5: e9337.
4368 LYN KINASE CONTROLS IgG-MEDIATED SYSTEMIC ANAPHYLAXIS