
Bone morphogenetic protein-4 (BMP4)- A paracrine regulator of human adrenal C19 steroid synthesis
Upload
independentCategory
view
1download
0
1
Lung Selective Gene Responses to Alveolar Hypoxia: Potential Role for the Bone
Morphogenetic Antagonist Gremlin in Pulmonary Hypertension
Christine M. Costello1#, Katherine Howell1, Edwina Cahill1, Jean McBryan2, Melanie
Konigshoff3, Oliver Eickelberg3, Sean Gaine4, Finian Martin2, Paul McLoughlin1
1School of Medicine and Medical Science, 2School of Biomolecular and Biomedical Science,
UCD Conway Institute of Biomolecular and Biomedical Sciences, University College Dublin,
Dublin 4, Ireland; 3 Dept. of Medicine II, University of Giessen Lung Center, Giessen,
Germany; 4Department of Respiratory Medicine, Mater Misericordiae University Hospital,
University College Dublin, Dublin 7, Ireland.
#Correspondence should be addressed to: [email protected]
Running title: Lung selective gene responses to alveolar hypoxia
Abbreviations: ECs: Endothelial cells, BMP: Bone morphogenetic protein, HMCEC-L:
Human microvascular endothelial cells (lung), HMCEC-C: Human microvascular endothelial
cells (cardiac), GREM1: Gremlin 1, SLM-2: Sam68-like mammalian protein 2, CXCR7:
Chemokine (C-X-C motif) receptor 7, SLC2A1: Solute carrier family 2 (facilitated glucose
transporter), member 1, VEGF-A: Vascular endothelial growth factor A.
Page 1 of 45Articles in PresS. Am J Physiol Lung Cell Mol Physiol (May 9, 2008). doi:10.1152/ajplung.00358.2007
Copyright © 2008 by the American Physiological Society.

2
ABSTRACT
Pulmonary hypoxia is a common complication of chronic lung diseases leading to the
development of pulmonary hypertension. The underlying sustained increase in vascular
resistance in hypoxia is a response unique to the lung. Thus, we hypothesised that there are
genes whose expression is altered selectively in the lung in response to alveolar hypoxia.
Using a novel subtractive array strategy, we compared gene responses to hypoxia in primary
human pulmonary microvascular endothelial cells (HMVEC-L) to those in cardiac
microvascular endothelium and identified 90 genes (forming nine clusters) differentially
regulated in the lung endothelium. From one cluster, we confirmed that the bone
morphogenetic protein (BMP) antagonist, Gremlin 1, was upregulated in the hypoxic murine
lung in vivo, but was unchanged in five systemic organs. We also demonstrated that gremlin
protein was significantly increased by hypoxia in vivo and inhibited HMVEC-L responses to
BMP stimulation in vitro. Furthermore, significant up-regulation of gremlin was measured in
lungs of patients with pulmonary hypertensive disease. From a second cluster, we showed
that CXC receptor 7, a receptor for the pro-angiogenic chemokine CXCL12, was selectively
upregulated in the hypoxic lung in vivo, confirming that our subtractive strategy had
successfully identified a second lung-selective hypoxia-responsive gene. We conclude that
hypoxia, typical of that encountered in pulmonary disease, causes lung-specific alterations in
gene expression. This gives new insights into the mechanisms of pulmonary hypertension
and vascular loss in chronic lung disease and identifies gremlin 1 as a potentially important
mediator of vascular changes in hypoxic pulmonary hypertension.
Keywords: Pulmonary endothelium; Gremlin 1; CXCR7; angiogenesis
Page 2 of 45

3
INTRODUCTION
Sustained pulmonary hypoxia is a common consequence of chronic lung diseases and
leads to the development of pulmonary hypertension as a result of increased pulmonary
vascular resistance (25, 39). When pulmonary hypertension occurs in this setting, it leads to
increased morbidity and reduced survival (reviewed in Hopkins et al (25)). The increased
pulmonary vascular resistance underlying chronic hypoxic pulmonary hypertension is
accompanied by characteristic changes in the pulmonary arterioles, which include thickening
of the pulmonary arterial wall and associated reductions in lumen diameter in vivo (47, 48).
More recently, it has been shown that persistent vasoconstriction, mediated by the small G-
protein RhoA and its downstream kinases ROCK I and II, is also a major contributor to the
hypoxia-induced increase in vascular resistance (29, 38, 53). These long-term structural and
functional changes in the pulmonary vessels in response to hypoxia are unique to this
circulation and are not observed in the other organs of the body. These observations suggest
that there are genes whose expression is selectively modulated by hypoxia in the lung while
simultaneously remaining unaffected in other organs.
The important roles of the vascular endothelium in the local control of vascular
smooth muscle tone and in modulating changes in the structure of the vessel wall are now
well recognised. Such effects are mediated through the release of specific signalling
molecules such as nitric oxide, prostacyclin and endothelin (1). The endothelial cells can also
produce extracellular matrix molecules such as laminin, fibronectin and elastin that can
contribute to the formation of the extracellular matrix of the vessel wall (5). However,
endothelial cells from different organs show considerable heterogeneity and their roles in
control of the vasculature differ from organ to organ. Ultrastructural diversity is well
recognised as endothelial structure ranges from discontinuous, through fenestrated to
continuous tight phenotypes (11, 14). Expression of cell surface molecules also varies
considerably between endothelial cells from different organs. For example, lung endothelium
Page 3 of 45

4
shows high basal expression of angiotensin converting enzyme (4, 19), thrombomodulin (54)
and membrane dipeptidase (13, 21, 40) when compared to that of systemic organs. Moreover,
these tissue specific features are maintained in culture conditions suggesting that they are
intrinsic to the mature endothelial cells and do not depend on the organ specific environment
in which they normally reside (30). Taken together, these observations suggest that the
pulmonary endothelium shows organ-specific responses to hypoxia that play an important role
in the development of the vascular changes leading to chronic hypoxic pulmonary
hypertension.
The aim of our study was to identify genes whose expression was altered in the
pulmonary endothelium in response to hypoxia while remaining unaltered (or changing in the
opposite direction) in the endothelial cells of other organs. Using a microarray approach, we
adopted a novel subtractive strategy to compare global gene responses to hypoxia in primary
human pulmonary microvascular endothelial cells to those observed in primary cardiac
microvascular endothelial cells and identified a cohort of lung-selective, hypoxia responsive
genes. We selected two exemplary clusters from amongst this cohort for further examination
in mice exposed to environmental hypoxia (10% O2). This approach confirmed that gremlin 1
(GREM1) gene expression, a bone morphogenetic protein (BMP) antagonist, was selectively
upregulated in the murine lung in response to hypoxia when compared to other organs. We
found expression of gremlin protein in the normoxic murine lung in vivo and demonstrated
that this was markedly increased by hypoxia. Gremlin expression in lung tissue from patients
with idiopathic pulmonary hypertension was also significantly elevated. Finally, we showed
that gremlin inhibited BMP stimulated human pulmonary endothelial cell wound healing in
vitro. From a second exemplary cluster, we showed that chemokine (C-X-C motif) receptor 7
(CXCR7), a G–protein coupled receptor for the pro-angiogenic chemokine CXCL–12, was
selectively upregulated in the hypoxic lung in vivo, confirming that our subtractive strategy
had successfully identified lung-selective hypoxia-responsive genes. This study gives new
Page 4 of 45

5
insights into the mechanisms of hypoxic pulmonary hypertension and, in particular, provides
the first evidence of a potentially important role for gremlin, an endogenous inhibitor of the
BMP pathways, in pulmonary hypertension.
Page 5 of 45

6
MATERIALS AND METHODS
Cell Culture and In Vitro Model of Hypoxia
Primary human microvascular endothelial cells from lung (HMVEC-L) and cardiac
tissue (HMVEC-C) were grown on sterile tissue culture dishes in Endothelial Growth
Medium (EGM-2MV; Code: CC-3202) according to the manufacturers instructions (Lonza
Bioscience formerly Cambrex, UK). At experiment start, medium was changed and dishes
were transferred to the hypoxia chamber (Coy Labs, USA) and cultured in an atmosphere of
1% O2, 5% CO2 and 94% N2 for 3hr, 24hr or 48hrs. Control conditions were achieved by
culture in 21% O2, 5% CO2 and 74% N2 in a cell-culture incubator. At experiment end, cell
medium was removed, cells washed with PBS and thoroughly lysed in RLT buffer (RNeasy
Mini kit, Qiagen, UK). All cells used in these experiments were passage 6-7 and were
routinely checked for mycoplasma contamination using the VenorGeM PCR kit (Cambio Ltd,
UK.). Six independent hypoxic time-course experiments were carried out for both lung and
cardiac cells.
RNA Extraction
Total RNA extraction (RNeasy Mini Kit, Qiagen Ltd, UK), with on-the-column
DNase treatment, was carried out as previously described (17). RNA was determined to be
free of contaminating genomic DNA by PCR amplification of GAPDH (17), quantified
spectrophotometrically and confirmed to be intact by standard gel electrophoresis. Hypoxic
conditions were confirmed by RT-PCR with two well-established hypoxia-induced genes,
VEGF-Α and SLC2A1 (formerly GLUT-1). Briefly, using standard RT-PCR conditions and a
final primer concentration of 0.2µM (Table 1), the PCR cycling profile was as follows: 2 min
at 95°C, and n cycles of 94°C for 45 sec, 60°C for 30 sec, 72°C for 90 sec, and a final
extension step of 72°C for 10 min. Samples were run on a 2% agarose-EtBr gel and
amplicons were visualised under UV light (Gene Genius BioImaging systems, Syngene).
Page 6 of 45

7
GeneChip Experiments
Endothelial cells were cultured and exposed to hypoxia on six separate occasions and
total RNA extracted. Experimental samples (n=3) were formed by randomly pooling RNA
from two independent experiments. This process was repeated at each of 4 experimental
time-points yielding 12 separate arrays for lung and cardiac experiments. Affymetrix
GeneChip Human Genome U133A chips, which allow the simultaneous analysis of 22,283
gene transcripts, were used. Total RNA (5µg) was reverse transcribed to cDNA (one-cycle
cDNA synthesis kit), clean up of double stranded cDNA, biotin labelling of anti-sense cRNA
(integrity checked on the Bioanalyzer 2100, Agilent technologies, USA) and clean up and
fragmentation of biotinylated cRNA were all carried out according to the manufacturers
protocol (Affymetrix, USA). Fragmented cRNA (15µg) was hybridised to chips for 16 hrs at
45ºC in a hybridisation oven (AffyMetrix, USA) with rotation at 60rpm. Immediately
following hybridisation, the array underwent automated washing and staining protocol on the
automatic fluidics station 400, (AffyMetrix, USA) and chips were scanned (GeneChip 3000
scanner, AffyMetrix, USA). One “3hr hypoxic lung chip” failed due to uneven hybridisation
and was therefore removed from all subsequent analysis.
Affymetrix .cel files were imported and normalised using algorithms of GeneSpring
6.0 software i.e. data was normalised by applying the following data transformation criteria:
a) measurements less than 0.01 were set to 0.01, b) per chip: normalised to 50th percentile and
c) per gene: normalise to median. Genes that were absent in a majority of chips were
eliminated from further analyses yielding a list of 10,476 genes. Using this list, a condition
tree was generated by hierarchical clustering using a Pearson correlation (GeneSpring 6.0
software).
In order to discern those genes that were uniquely up-regulated in the lung we adopted
the following stringent subtractive approach. We first selected those genes that increased at
Page 7 of 45

8
least 2-fold between basal normoxic expression and any hypoxic time-point in the lung ECs.
We next subtracted from that list those genes that increased at least 1.2-fold between
normoxia and any hypoxic time-point in the heart ECs. This strategy identified those genes
that increased at least 2-fold in lung cells but decreased, remained the same or increased
marginally (<1.2-fold) in cardiac cells. A similar analysis was carried out to discern those
genes that decreased 2-fold in lung cells but increased, remained the same or decreased only
marginally (<1.2-fold) in cardiac cells. These gene lists were combined and subjected to
hierarchical clustering to identify interesting clusters for follow-on analyses (GeneSpring 6.0
software).
Real-time PCR (TaqMan)
Total RNA (1µg) was reverse-transcribed (RT) to cDNA using Superscript II RNase
H-Reverse Transcriptase kit (Invitrogen, USA). Real-time PCR was performed on 384-well
plates and each sample was measured in duplicate. An initial minus RT reaction confirmed
that all RNA samples were free of contaminating genomic DNA. The Eukaryotic 18S rRNA
(VIC/TAMRA) pre-developed assay reagent kit was used as the endogenous control gene
according to the TaqMan PCR protocol (Applied Biosystems (ABI), USA). Probes and
primers were either designed to non-redundant sequence using Primer Express software or
ordered from ABI as Assay-on-Demand Gene Expression Assays (Table 2a). Reactions were
carried out on the ABI PRISM 7900 Sequence Detection System and mRNA levels were
determined using the standard curve method (ABI Prism 7700 Sequence Detection System
User Bulletin #2).
In Vivo Animal Model of Hypoxia
Chronic hypoxic pulmonary hypertension was induced by housing adult male C57Bl6
mice (n=8) in hypoxic conditions (inspired oxygen 10% for 2 days) (20). This mouse strain
Page 8 of 45

9
demonstrates pulmonary hypertension, right ventricular hypertrophy and pulmonary vascular
remodelling similar to that is seen in other species following sustained exposure to a hypoxic
environment (26, 29). Control mice (n=8) were housed in normoxic conditions (inspired
oxygen 21%) in the same room. After 48hrs, mice were killed by cervical dislocation, and the
thymus, lungs, heart (left ventricle), kidney, liver and spleen were rapidly removed and
immediately flash frozen in liquid nitrogen. RNA extraction was carried out as described
above with the exception that tissue samples were first crushed to a fine powder using a
mortar and pestle and passed through QIA shredder columns (Qiagen Ltd, UK) to ensure
complete breakdown of tissue. All experiments were approved by the UCD Animal Research
Ethics Sub-Committee and carried out under license from the Department of Health.
Immunohistochemical Analysis
Adult male C57Bl6 mice were maintained in normoxia (n=5) or exposed to 10%
oxygen (n=5) for two days as above. Mice were euthanised (sodium pentobarbitone, 60mg/kg
i.p.), the heart and lungs were removed en-bloc and lungs were fully inflated via the trachea
with standard pressures (25 cm H2O) in fixative (4% paraformaldehyde) overnight and then
embedded in paraffin. Following fixation for 24 hours, the vertical axis of each left lung was
identified and the lung was cut perpendicular to this axis into 4 mm thick slices with a sharp
blade beginning at a position chosen by random number within the first slice. Seven µm thick
sections were cut from the surface of each slice, mounted onto polysine slides and baked
overnight at 37°C, de-waxed and hydrated. Endogenous peroxidase activity was blocked with
0.3% H2O2 in methanol for 5 min. Antigen retrieval was performed with 10mM sodium
citrate buffer (pH 6.0) for 10 min in a microwave. Slides were washed in PBS, blocked in
rabbit serum (10% diluted in PBS) for 30 min and incubated in 1:10 dilution of primary goat
polyclonal anti Drm/gremlin (R&D Systems; final concentration 10µg/ml) in PBS overnight
at 4°C. Following PBS washes, endogenous peroxidase activity was quenched with 0.3%
Page 9 of 45

10
H2O2 in methanol. Slides were next incubated in 1:150 dilution biotinylated rabbit anti-goat
secondary antibody (Vector Laboratories, USA; final concentration: 10µg/ml) for 1 hour,
washed in PBS and incubated in immunoperoxidase solution (ABC Vectastain Kit, Vector
Laboratories) for 1 hour. Negative control slides were exclusion of primary or secondary
antibody. After allowing the diaminobenzidine reaction product to develop for 20 min,
sections were washed extensively in PBS and counterstained with haematoxylin (BDH
Laboratory, UK) before being examined microscopically on a BX-61 microscope (Olympus,
UK) (x 40 objective).
Quantification of Gremlin Staining
The extent of gremlin staining was determined by quantitative stereological techniques
as previously described (8, 24) using a computer based analysis system (CAST, Visiopharm,
Denmark) combined with light microscopy (Leica, Laboratory Instruments). Random fields
of view were acquired (x 40 objective) from all sections of each lung using a systematic
random sampling strategy, digitised and displayed on screen. A point counting grid was
digitally superimposed on these images to determine the fraction of the alveolar walls that
was occupied by cells staining positively for gremlin.
Scratch Assay Protocol
At the start of the experiment, three horizontal lines were drawn on the back of 12 well
plastic plates (Greiner Bio-One Cellstar). HMVEC-L were allowed grow into a confluent
monolayer and the medium was changed. Twenty-four hours later, a single vertical scratch
was applied to each chamber using a 1-100ul pipette tip (S1120-1840, Starlab, UK). After
injury, cells were allowed to settle for 2-3 hrs and then treated with either vehicle (4mM HCl-
0.1% BSA), BMP-4 (80ng/ml final conc.), BMP4 + gremlin (2µg/ml final conc.) or BMP4 +
condition medium. The condition medium was obtained from HMVEC-L cells grown in 1%
Page 10 of 45

11
O2 for 24hrs and spun at 1,500rpm for 3 mins to remove any cellular debris. The BMP4 and
gremlin were from R & D Systems. The scratch was visualised using phase contrast (x10
objective) and the width of the wound measured at 0hr and 24 hrs (AxioVision 4.4 software).
Six fields in each well were measured by taking images along the length of the scratch at six
pre-defined positions (above and below each drawn line) and averaged. For each treatment,
six separate well analyses were carried out.
Gremlin Expression in Human Tissues
Lung tissue samples were obtained from five patients (mean age 39 ± 10 years; 3
females) with idiopathic pulmonary arterial hypertension (IPAH) and six control subjects
whose lungs were rejected from transplantation due to recipient incompatibility (organ
donors, mean age 42 ± 13 years; 3 females). Pulmonary hypertension was defined as a
pulmonary mean arterial pressure >25mmHg at rest or >30mmHg on exercise. None of the
IPAH patients exhibited known BMPR2 mutations. Total RNA and cDNA was prepared as
described above. Real-time PCR was performed using fluorogenic SYBR Green and the
Sequence Detection System Fast 7500 (PE Applied Biosystems). Human gremlin primers are
shown in table 2b. Hypoxanthine phosphoribosyltransferase (HPRT)-1, a ubiquitously and
equally expressed gene free of pseudogenes, was used as a reference gene (table 2b). PCR
was performed using the primers at a final concentration of 200 nM and relative transcript
abundance was determined using the comparative Ct method (ABI Prism 7700 Sequence
Detection System User Bulletin #2).
Statistical Analyses
All statistics were carried out using Statistica 7.0 software. For normally distributed
data, responses are reported as mean ± standard error of the mean (SEM). Comparison of
means in two group experiments was carried out using a t-test. For non-normally distributed
Page 11 of 45

12
data, responses are reported as mean ± standard deviation (SD) and statistical comparisons
made using Mann-Whitney U test. In experimental designs with three or more groups
statistical analysis was performed using the Kruskal-Wallis test followed by Mann–Whitney
U with the Bonferroni post-hoc correction to test for differences between specific groups;
values of P<0.05 were accepted as statistically significant.
Page 12 of 45

13
RESULTS
Global Gene Expression in Hypoxia
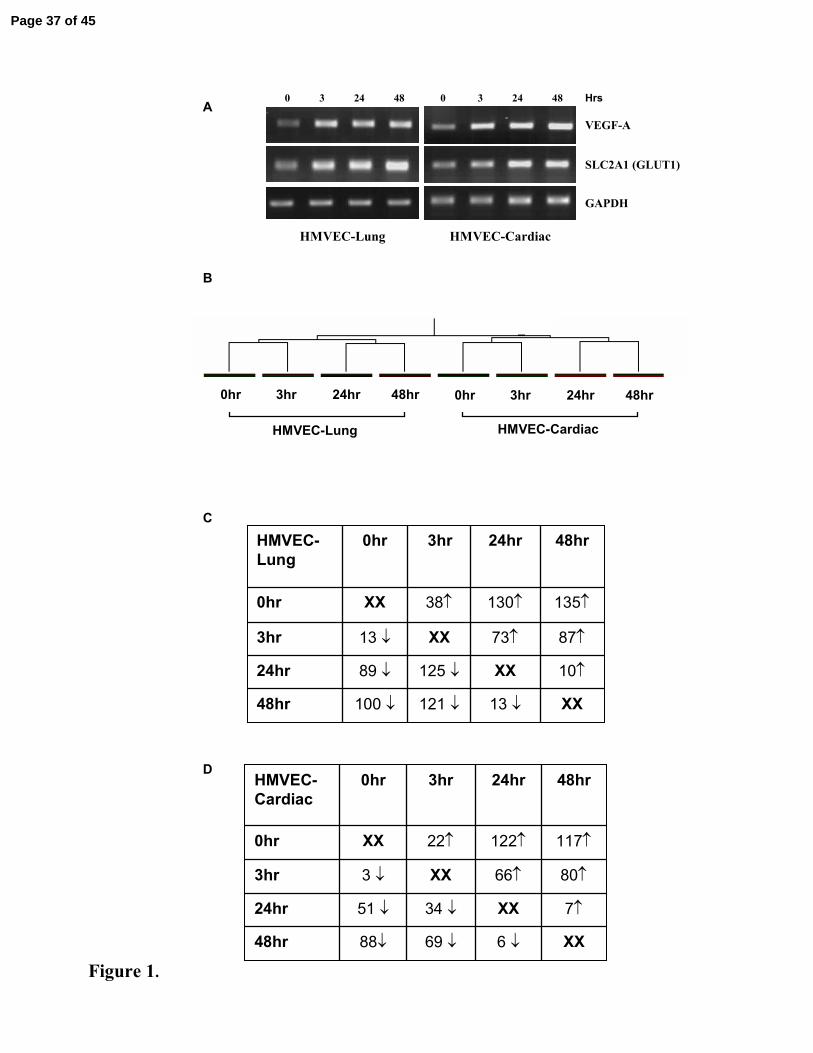
We confirmed that primary human microvascular ECs in our in vitro model were
hypoxic by showing that, as expected, the expression of VEGF-A and SLC2A1 (GLUT1) was
upregulated as early as 3hrs exposure to hypoxia and further increased with longer exposure
to 1% O2 (Figure 1A). Using AffyMetrix U133A chips (~22,283 gene transcripts), we next
investigated the global expression profile induced in cells in response to increasing periods of
hypoxia. Hierarchical clustering of the arrays based on the genes whose expression was
reliable (10,746 genes) clustered cells from the lung or cells from the heart together, forming
clearly separate cell-specific sub-trees (Figure 1B). This finding supported our hypothesis
that there were genes whose expression was modulated by hypoxia in the pulmonary
endothelium in a manner that was specific to that cell type and was different from the pattern
of gene response observed in cardiac endothelial cells. In the lung cells 428 genes were
differentially up or down-regulated at some time point during hypoxia while in the cardiac
cells the corresponding number was 299 genes. A broad-overview of the time course of these
changes is shown in Figure 1C and 1D.
Identification of Lung Endothelium Selective Hypoxic Responsive Genes
Our subtractive approach revealed 51 genes that were up-regulated (Table 3a) and 39
genes that were down-regulated in the lung cells > 2-fold (Table 3b) but had a different
pattern of expression in cardiac cells. These genes are presented in broad functional
categories based on Gene Ontologies (Table 3). Hierarchical clustering of these 90
differentially regulated genes identified distinct cohorts of genes with shared expression
profiles over a time course of hypoxia (Figure 2). One very striking finding from this analysis
was a cluster of seven genes (outlined in yellow in Figure 2A) which showed a basal gene
expression that was greatly elevated (>3-10 fold) in lung cells compared to cardiac cells
Page 13 of 45

14
(Figure 2B). Furthermore, this expression was upregulated >2-fold in response to hypoxia (at
least one time-point) only in the lung cells (Figure 2C). In light of these characteristics, we
chose to further examine this cluster in more detail. Within this cluster was the secreted BMP
antagonist, gremlin 1 (GREM1) and the, as yet, unknown protein, LIM and calponin
homology domains 1 (LIMCH1), both of which were represented by two independent probes.
This cluster also included VEGF165 RNA processing protein, Sam68-like phosphotyrosine
protein (SLM2), the extracellular matrix protein Spondin 1 (SPON1) and the potent tumour
angiogenic factor ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2) (12, 16, 51).
The remaining two genes in this cluster represented genes of unknown specific function,
namely protein kinase Y-linked (PRKY) and macrophage expressed gene 1 (MPEG1).
In Vitro Transcriptional Changes Confirmed by Real-time PCR analysis
We confirmed that basal expression of the majority of genes (6 of 7) in the gremlin
cluster was significantly elevated in lung when compared to cardiac endothelial cells in each
of six separate hypoxia experiments at 48 hrs (Figure 3). Indeed, extremely low gene
expression of GREM1, SPON1, ENPP2 and PRKY was observed in cardiac endothelium.
Although mean LIMCH1 basal levels were higher in lung cells, this difference was not
significant (p=0.08). Finally, we confirmed that GREM1 expression was significantly
increased in response to hypoxia (48 hrs) only in the pulmonary microvascular endothelium
(Figure 3). Analysis of all genes, at all experimental time-points, confirmed that gremlin was
the only significantly differentially regulated gene in hypoxia in the cell culture model (data
not shown). Because vascular remodelling occurs most prominently in smaller vessels in the
lung, we asked if GREM1 expression was different in endothelial cells from large pulmonary
vessels. We therefore examined GREM1 expression in endothelial cells isolated from human
pulmonary artery (HPAEC - Lonza Bioscience formerly Cambrex, UK) in a separate series of
experiments. We found low basal expression (similar to that measured in cardiac cells) in
Page 14 of 45

15
these cells, which was not altered with hypoxia (data not shown), suggesting that GREM1
expression is specific to the pulmonary microvascular endothelium.
Lung Selective Genes Confirmed in an In Vivo Animal Model of Hypoxia
To examine whether the effects observed in the cell culture model were relevant to the
in vivo situation, we examined gene expression in organs from mice exposed to 10%
environmental oxygen for 2 days. In a similar manner to that seen in vitro, basal expression
of the majority (5 of 7) of genes in this cluster were significantly elevated in the whole lung
compared to the heart. Exceptions were SLM2, which was significantly higher in the heart
(Figure 4A) and PRKY, which remained unchanged in the two tissue types. We also
observed a significant up-regulation of GREM1 and SLM2 expression in lung tissue in
response to the two day hypoxic challenge. In order to establish whether this finding was
selective to the lung, we extended our gene expression analysis to include the kidney, liver,
spleen and thymus. Expression analysis confirmed that GREM1 (>3-fold) and SLM2 (~2-
fold) were only up-regulated in the lung in response to hypoxia (Figure 4B & C).
Gremlin 1 Protein is Higher in Hypoxic Lung Tissue
Immunohistochemical staining showed that gremlin protein was expressed basally in
the normoxic lung (Figures 5A and 5B) and increased substantially after two days in hypoxia
(Figures 5C and 5D). Gremlin immunoreactivity was clearly observed in the tissue
surrounding the alveolar spaces. Gremlin staining was absent when primary antibody was
omitted from normoxic (Figure 5E) or hypoxic (Figure 5F) tissue or when secondary antibody
was omitted (data not shown). Images shown are representative of findings in five control
animals and five hypoxic animals. Quantification of the volume of gremlin stained tissue per
unit volume of alveolar wall in the lung showed that gremlin protein was significantly higher
in the hypoxic lungs (n=5) than in normoxic lungs (n=4) (Figure 5G).
Page 15 of 45

16
Gremlin Inhibits BMP4 Induced Endothelial Wound Healing
Given the known role of gremlin as a potent antagonist of BMP induced proliferation
and migration, we next examined its ability to inhibit wound healing. As shown in Figure 6A,
the scratch applied to lung endothelial cells treated with vehicle alone (Panel 1) was still
visible after 24hr, which was in sharp contrast to that seen in cells treated with BMP4
(80ng/ml), where the scratch width was substantially reduced (Panel 2). Conversely, blocking
BMP4 activity with the addition of gremlin (2µg/ml) resulted in a scratch width similar to
control (Panel 3). Furthermore, treatment of the cells with condition medium from hypoxic
cells also blocked wound healing (panel 4). Figure 6B shows the mean scratch width from 6
separate wells. This showed BMP4 significantly induced wound healing and treatment with
gremlin or hypoxic condition medium significantly blocked this effect.
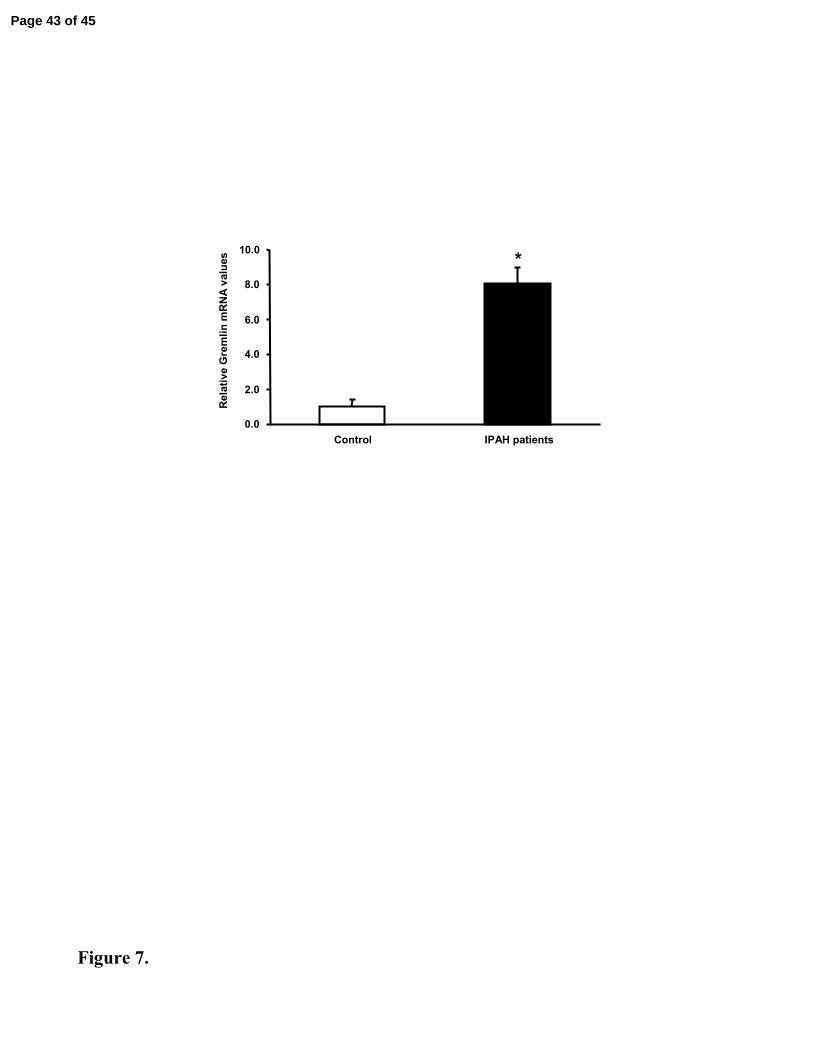
Gremlin Expression is Higher in Patients with IPAH
In patients with idiopathic pulmonary arterial hypertension expression of gremlin was
significantly greater than in that observed in non-diseased lungs (Figure 7).
Second Cluster of Genes Examined in In Vivo Animal Model of Hypoxia
In order to demonstrate the robustness of our lung selective gene findings, we next
focussed on a second smaller cluster of four genes, where hierarchical clustering showed a
marked up-regulation at 24hr and 48hr hypoxia in lung ECs but remained virtually unchanged
in the cardiac cells in vitro (Figure 8A). This cluster was comprised of TM2D1 domain
containing 1 protein (also annotated as beta-amyloid binding protein (BBP)), the pro-
angiogenic CXCL12 binding G-protein coupled receptor CXCR7, the motor protein Myosin
IB (MYO1B) and a newly described mediator of angiogenesis, brain-derived neurotrophic
factor (BDNF) (7, 27, 31, 43). Two genes were confirmed as hypoxia responsive in vivo,
Page 16 of 45

17
namely CXCR7 and BDNF. However, CXCR7 was the only one of these two that was
hypoxia responsive in the lung but not in the heart, as confirmed by real-time PCR (Figure
8B).
CXCR7 has only very recently been shown to bind the pro-angiogenic chemokine
CXCL12. To investigate this pathway further, we examined gene expression in vivo.
CXCL12 increased >2-fold in hypoxic lung tissue, with more modest changes observed in
heart and spleen tissue (Figure 9A). CXCR7 expression was markedly elevated after hypoxic
stress, a response that was unique to the lung (Figure 9B), implying that the CXCL12/CXCR7
axis may have a special role in hypoxic responses in lung.
Page 17 of 45

18
DISCUSSION
Chronic lung diseases, including asthma, chronic obstructive pulmonary disease,
emphysema, cystic fibrosis and occupational lung diseases are frequently complicated by the
development of pulmonary hypertension caused by a sustained elevation of pulmonary
vascular resistance. This complication causes right ventricular failure, increases disability and
reduces life expectancy (25). One major stimulus that contributes to pulmonary hypertension
is hypoxia caused by the underlying lung disease. In the present study, we detected genes
whose expression was selectively altered in the lung in response to levels of hypoxia
encountered in pulmonary disease.
A number of the 90 genes identified in our initial array studies were already linked to
pathways known to be involved in the development of pulmonary hypertension e.g.
peroxisome proliferative activated receptor and arginase (18, 44, 57). This observation
provided support for our subtractive strategy; however, we did not pursue these well-explored
pathways further.
We first focused our attention on a cluster of seven genes which showed high basal
expression in the pulmonary endothelial cells and hypoxic responsiveness (figure 2 and 3). In
vivo, five of these genes demonstrated very high basal expression values when compared to
the heart (Figure 4A). In view of our recent demonstration that angiogenesis is a prominent
feature of the pulmonary vascular responses to hypoxia (26, 29), it was of interest to note that
the majority of the genes in this cluster had documented roles in angiogenesis, either
inhibiting (51) or promoting it (9, 15). Two of the genes in the cluster demonstrated lung-
selective hypoxia responses in vivo, although one of these, SLM2, was the one gene in the
cluster that had significantly higher basal expression in the heart. The second of these two
genes, gremlin, which showed both high basal expression in the lung and lung selective
hypoxia responses in vivo, was of particular interest because it is a soluble inhibitor of the
BMP signaling axis and inhibition of this signaling pathway has recently been identified as
Page 18 of 45

19
playing a central role in the development of hypoxic pulmonary hypertension (22, 49, 50, 56).
Inhibition of BMP signalling also plays a crucial role in the development of idiopathic
pulmonary arterial hypertension (IPAH) (23). Heterozygous non-functional BMPR2
mutations leading to haploinsufficiency strongly predispose to the development of IPAH but
the precise pathways linking the BMPR2 mutations to the vascular lesions observed have not
been elucidated (28, 33, 35). Moreover, BMPs together with their antagonists, including
gremlin, are key mediators of lung development in the embryo (2, 10, 36, 46, 52, 55).
Given that background, we were interested to further explore changes in gremlin
expression in the lung in vivo. We found that gremlin protein was present under normoxic
conditions and significantly more extensively expressed in the tissue surrounding the alveolar
spaces in hypoxia, a pattern of increase similar to that of its mRNA (Figure 5). The extensive
staining observed in hypoxia suggests that other cells in addition to endothelial cells may
express gremlin including epithelial cells, myofibroblats and macrophages (32, 37).
In systemic organs, BMPs have been linked to endothelial angiogenesis but an action
on the pulmonary endothelium has not previously been documented (6, 34, 41, 42, 45). We
report for the first time that BMP-4 promotes pulmonary endothelial wound healing responses
and that gremlin can block this action (Figure 6). Since gremlin is a secreted BMP antagonist,
we postulated that the hypoxia-induced upregulation of this protein in pulmonary endothelial
cells would exert a blocking effect on BMP. Our finding that conditioned medium from
hypoxic endothelial cells blocked BMP4 stimulation of endothelial wound healing supports
this hypothesis. In contrast, Stabile et al (45) recently reported that gremlin was pro-
angiogenic and can play a BMP-independent role in the angiogenic process by directly
binding the endothelial cell surface via as yet uncharacterised cell-surface heparan-sulfate
proteoglycans. These potentially divergent results most likely reflect the different cell types
used in the two studies (immortalised cell lines or systemic cells in the latter study and
Page 19 of 45

20
pulmonary ECs in our study) but clearly, further experiments are required to decipher the
complex interactions between gremlin, BMPs, and their cognate receptors.
Since alterations in BMP signaling play a central role in the development of
pulmonary arterial hypertension, we examined gremlin expression in the explanted lungs of
patients who were undergoing transplantation and found that its expression was markedly
increased compared to controls (Figure 7). This suggests that the underlying pathogenetic
mechanisms may involve a previously unsuspected role of this endogenous BMP antagonist.
Furthermore, the finding of high basal expression of gremlin in the lung may provide an
explanation for the previously unexplained observation that the pulmonary circulation is the
only vascular bed that suffers in patients with BMPR2 mutations although the mutation is
somatic; further experiments are needed to test this possibility.
In order to confirm that that our subtractive approach had revealed other lung selective
hypoxia responsive genes, we examined a second cluster in vivo, the “CXCR7 cluster”. This
cluster showed initial low expression levels (when compared to the gremlin cluster) but
marked hypoxic responses. Of the four genes identified using array data, two, CXCR7 and
BDNF, were upregulated by hypoxia in the lung. Of these two, BDNF was also upregulated
in the heart demonstrating that it was not lung specific and so we did not pursue this further.
CXCR7, formerly the orphan receptor CMKOR1, has recently been identified as a
receptor for the pro-angiogenic chemokine CXCL12 (3, 7). We examined the responses of
both these genes to hypoxia in vivo in our extended panel of organs and found that CXCL12
was increased in the hypoxic lung but that this increase was not limited to the lung (Figure 8).
This is in good agreement with our original microarray data, where CXCL12 was >2-fold
upregulated in lung cells, but also increased in the heart (~1.7-fold) and so had not been
identified as lung selective (data not shown). However, it is worth noting that both gene array
and real-time PCR showed that basal levels of CXCL12 in lung endothelial cells were > 15-
fold higher than in cardiac endothelial cells (data not shown). However, CXCR7 was
Page 20 of 45

21
upregulated in the lung alone at levels of hypoxia found in lung disease, suggesting a potential
novel mechanism whereby CXCL12 could show lung specific pro-angiogenic actions (Figure
9). Further experiments are required to substantiate this hypothesis. Nonetheless, the
demonstration that a further gene from a different cluster demonstrates lung selective hypoxic
response suggests that our array-based approach has identified a cohort of such pulmonary
selective genes.
In conclusion, we have shown that lung selective changes in gene expression are
caused by alveolar hypoxia typical of that found in disease in vivo. One pathway identified
was selective upregulation of the endogenous BMP antagonist gremlin, which is of specific
interest because of the key role that inhibition of BMP signalling is known to play in the
development of pulmonary hypertension. We confirmed that a further gene, CXCR7, from a
separate cluster identified in our cell model, showed lung selective hypoxic responses in vivo.
Thus, our cohort of lung selective hypoxia responsive genes gives new insights into potential
mechanisms underlying hypoxic pulmonary hypertension.
Page 21 of 45

22
ACKNOWLEDEGEMENTS
Grants: This work was funded by grants from the Health Research Board and the Higher
Education Authority (Programme for Research in Third-Level Institutions) and an
unrestricted research grant from Actelion. The funders had no role in study design, data
collection and analysis, decision to publish, or preparation of the manuscript.
Supporting Information: The expert technical assistance of Emilie Duval is gratefully
acknowledged.
Page 22 of 45

23
REFERENCES
1. Aaronson PI, Robertson TP, and Ward JP. Endothelium-derived mediators and hypoxic pulmonary vasoconstriction. Respir Physiol Neurobiol 132: 107-120., 2002.2. Adams D, Larman B, and Oxburgh L. Developmental expression of mouse Follistatin-like 1 (Fstl1): Dynamic regulation during organogenesis of the kidney and lung. Gene Expr Patterns 7: 491-500, 2007.3. Balabanian K, Lagane B, Infantino S, Chow KY, Harriague J, Moepps B, Arenzana-Seisdedos F, Thelen M, and Bachelerie F. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem 17, 2005.4. Balyasnikova IV, Yeomans DC, McDonald TB, and Danilov SM. Antibody-mediated lung endothelium targeting: in vivo model on primates. Gene Ther 9: 282-290., 2002.5. Botto L, Beretta E, Daffara R, Miserocchi G, and Palestini P. Biochemical and morphological changes in endothelial cells in response to hypoxic interstitial edema. Respir Res 7:7.: 45:50, 2006.6. Boyd NL, Dhara SK, Rekaya R, Godbey EA, Hasneen K, Rao RR, West FD, 3rd, Gerwe BA, and Stice SL. BMP4 promotes formation of primitive vascular networks in human embryonic stem cell-derived embryoid bodies. Exp Biol Med (Maywood) 232: 833-843, 2007.7. Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, Penfold MET, Sunshine MJ, Littman DR, Kuo CJ, Wei K, McMaster BE, Wright K, Howard MC, and Schall TJ. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med 203: 2201-2213, 2006.8. Cadogan E, Hopkins N, Giles S, Bannigan JG, Moynihan J, and McLoughlin P.Enhanced expression of inducible nitric oxide synthase without vasodilator effect in chronically infected lungs. Am J Physiol 277: L616-627, 1999.9. Canalis E, Economides AN, and Gazzerro E. Bone morphogenetic proteins, their antagonists, and the skeleton. Endocr Rev 24: 218-235., 2003.10. Cardoso WV. Lung morphogenesis revisited: old facts, current ideas. Dev Dyn 219: 121-130, 2000.11. Carmeliet P. Cardiovascular biology. Creating unique blood vessels. Nature 412: 868-869., 2001.12. Chen M and O'Connor KL. Integrin alpha6beta4 promotes expression of autotaxin/ENPP2 autocrine motility factor in breast carcinoma cells. Oncogene 24: 5125-5130, 2005.13. Christofidou-Solomidou M, Kennel S, Scherpereel A, Wiewrodt R, Solomides CC, Pietra GG, Murciano JC, Shah SA, Ischiropoulos H, Albelda SM, and Muzykantov VR. Vascular immunotargeting of glucose oxidase to the endothelial antigens induces distinct forms of oxidant acute lung injury: targeting to thrombomodulin, but not to PECAM-1, causes pulmonary thrombosis and neutrophil transmigration. Am J Pathol 160: 1155-1169, 2002.14. Cleaver O and Melton DA. Endothelial signaling during development. Nat Med 9: 661-668., 2003.15. Cohen CD, Doran PP, Blattner SM, Merkle M, Wang GQ, Schmid H, Mathieson PW, Saleem MA, Henger A, Rastaldi MP, and Kretzler M. Sam68-like mammalian protein 2, identified by digital differential display as expressed by podocytes, is induced in proteinuria and involved in splice site selection of vascular endothelial growth factor. J Am Soc Nephrol 16: 1958-1965., 2005.16. Cohen CD, Doran PP, Blattner SM, Merkle M, Wang GQ, Schmid H, Mathieson PW, Saleem MA, Henger A, Rastaldi MP, and Kretzler M. Sam68-like mammalian
Page 23 of 45

24
protein 2, identified by digital differential display as expressed by podocytes, is induced in proteinuria and involved in splice site selection of vascular endothelial growth factor. J Am Soc Nephrol 16: 1958-1965, 2005.17. Costello CM, Mah N, Hasler R, Rosenstiel P, Waetzig GH, Hahn A, Lu T, Gurbuz Y, Nikolaus S, Albrecht M, Hampe J, Lucius R, Kloppel G, Eickhoff H, Lehrach H, Lengauer T, and Schreiber S. Dissection of the inflammatory bowel disease transcriptome using genome-wide cDNA microarrays. PLoS Med 2: e199., 2005.18. Crossno JT, Jr., Garat CV, Reusch JE, Morris KG, Dempsey EC, McMurtry IF, Stenmark KR, and Klemm DJ. Rosiglitazone attenuates hypoxia-induced pulmonary arterial remodeling. Am J Physiol Lung Cell Mol Physiol 292: L885-897, 2007.19. Danilov SM, Gavrilyuk VD, Franke FE, Pauls K, Harshaw DW, McDonald TD, Miletich DJ, and Muzykantov VR. Lung uptake of antibodies to endothelial antigens: key determinants of vascular immunotargeting. Am J Physiol Lung Cell Mol Physiol 280: L1335-1347, 2001.20. Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, and McMurtry IF.Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol 287: L656-664, 2004.21. Ford VA, Stringer C, and Kennel SJ. Thrombomodulin is preferentially expressed in Balb/c lung microvessels. J Biol Chem 267: 5446-5450, 1992.22. Frank DB, Abtahi A, Yamaguchi DJ, Manning S, Shyr Y, Pozzi A, Baldwin HS, Johnson JE, and de Caestecker MP. Bone morphogenetic protein 4 promotes pulmonary vascular remodeling in hypoxic pulmonary hypertension. Circ Res 97: 496-504, 2005.23. Gaine SP and Rubin LJ. Primary pulmonary hypertension. Lancet 352: 719-725, 1998.24. Hopkins N, Cadogan E, Giles S, and McLoughlin P. Chronic airway infection leads to angiogenesis in the pulmonary circulation. J Appl Physiol 91: 919-928, 2001.25. Hopkins N and McLoughlin P. The structural basis of pulmonary hypertension in chronic lung disease: remodelling, rarefaction or angiogenesis? J Anat 201: 335-348., 2002.26. Howell K, Preston RJ, and McLoughlin P. Chronic hypoxia causes angiogenesis in addition to remodelling in the adult rat pulmonary circulation. J Physiol 547: 133-145., 2003.27. Hu Y, Wang YD, Guo T, Wei WN, Sun CY, Zhang L, and Huang J. Identification of brain-derived neurotrophic factor as a novel angiogenic protein in multiple myeloma. Cancer Genet Cytogenet 178: 1-10, 2007.28. Humbert M, Deng Z, Simonneau G, Barst RJ, Sitbon O, Wolf M, Cuervo N, Moore KJ, Hodge SE, Knowles JA, and Morse JH. BMPR2 germline mutations in pulmonary hypertension associated with fenfluramine derivatives. Eur Respir J 20: 518-523, 2002.29. Hyvelin JM, Howell K, Nichol A, Costello CM, Preston RJ, and McLoughlin P.Inhibition of Rho-kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Circ Res 97: 185-191., 2005.30. Kieda C, Paprocka M, Krawczenko A, Zalecki P, Dupuis P, Monsigny M, Radzikowski C, and Dus D. New human microvascular endothelial cell lines with specific adhesion molecules phenotypes. Endothelium 9: 247-261., 2002.31. Kirfel G, Borm B, Rigort A, and Herzog V. The secretory beta-amyloid precursor protein is a motogen for human epidermal keratinocytes. Eur J Cell Biol 81: 664-676, 2002.32. Koli K, Myllarniemi M, Vuorinen K, Salmenkivi K, Ryynanen MJ, Kinnula VL, and Keski-Oja J. Bone morphogenetic protein-4 inhibitor gremlin is overexpressed in idiopathic pulmonary fibrosis. Am J Pathol 169: 61-71, 2006.33. Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, Nichols WC, and Trembath RC. Heterozygous germline mutations in BMPR2, encoding a
Page 24 of 45

25
TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet 26: 81-84, 2000.34. Langenfeld EM and Langenfeld J. Bone morphogenetic protein-2 stimulates angiogenesis in developing tumors. Mol Cancer Res 2: 141-149., 2004.35. Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, Phillips JA, 3rd, Newman J, Williams D, Galie N, Manes A, McNeil K, Yacoub M, Mikhail G, Rogers P, Corris P, Humbert M, Donnai D, Martensson G, Tranebjaerg L, Loyd JE, Trembath RC, and Nichols WC. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet 68: 92-102, 2001.36. Michos O, Panman L, Vintersten K, Beier K, Zeller R, and Zuniga A. Gremlin-mediated BMP antagonism induces the epithelial-mesenchymal feedback signaling controlling metanephric kidney and limb organogenesis. Development 131: 3401-3410., 2004.37. Myllarniemi M, Lindholm P, Ryynanen MJ, Kliment CR, Salmenkivi K, Keski-Oja J, Kinnula VL, Oury TD, and Koli K. Gremlin-mediated decrease in bone morphogenetic protein signaling promotes pulmonary fibrosis. Am J Respir Crit Care Med177: 321-329, 2008.38. Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa H, McMurtry IF, and Oka M. Inhaled Rho Kinase Inhibitors are Potent and Selective Vasodilators in Rat Pulmonary Hypertension. Am J Respir Crit Care Med 24, 2004.39. Penaloza D and Arias-Stella J. The heart and pulmonary circulation at high altitudes: healthy highlanders and chronic mountain sickness. Circulation 115: 1132-1146., 2007.40. Rajotte D and Ruoslahti E. Membrane dipeptidase is the receptor for a lung-targeting peptide identified by in vivo phage display. J Biol Chem 274: 11593-11598, 1999.41. Ramoshebi LN and Ripamonti U. Osteogenic protein-1, a bone morphogenetic protein, induces angiogenesis in the chick chorioallantoic membrane and synergizes with basic fibroblast growth factor and transforming growth factor-beta1. Anat Rec 259: 97-107, 2000.42. Rothhammer T, Bataille F, Spruss T, Eissner G, and Bosserhoff AK. Functional implication of BMP4 expression on angiogenesis in malignant melanoma. Oncogene 26: 4158-4170, 2007.43. Salas-Cortes L, Ye F, Tenza D, Wilhelm C, Theos A, Louvard D, Raposo G, and Coudrier E. Myosin Ib modulates the morphology and the protein transport within multi-vesicular sorting endosomes. J Cell Sci 118: 4823-4832, 2005.44. Sasaki A, Doi S, Mizutani S, and Azuma H. Roles of accumulated endogenous nitric oxide synthase inhibitors, enhanced arginase activity, and attenuated nitric oxide synthase activity in endothelial cells for pulmonary hypertension in rats. Am J Physiol Lung Cell Mol Physiol 292: L1480-1487, 2007.45. Stabile H, Mitola S, Moroni E, Belleri M, Nicoli S, Coltrini D, Peri F, Pessi A, Orsatti L, Talamo F, Castronovo V, Waltregny D, Cotelli F, Ribatti D, and Presta M.Bone morphogenic protein antagonist Drm/gremlin is a novel proangiogenic factor. Blood109: 1834-1840, 2007.46. Stanley EG, Biben C, Allison J, Hartley L, Wicks IP, Campbell IK, McKinley M, Barnett L, Koentgen F, Robb L, and Harvey RP. Targeted insertion of a lacZ reporter gene into the mouse Cer1 locus reveals complex and dynamic expression during embryogenesis. Genesis 26: 259-264, 2000.47. Stenmark KR, Fagan KA, and Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99: 675-691., 2006.48. Stenmark KR and McMurtry IF. Vascular remodeling versus vasoconstriction in chronic hypoxic pulmonary hypertension: a time for reappraisal? Circ Res 97: 95-98., 2005.
Page 25 of 45

26
49. Takahashi H, Goto N, Kojima Y, Tsuda Y, Morio Y, Muramatsu M, and Fukuchi Y. Downregulation of type II bone morphogenetic protein receptor in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 290: L450-458, 2006.50. Takahashi K, Kogaki S, Matsushita T, Nasuno S, Kurotobi S, and Ozono K.Hypoxia induces alteration of bone morphogenetic protein receptor signaling in pulmonary artery endothelial cell. Pediatr Res 61: 392-397, 2007.51. Terai Y, Abe M, Miyamoto K, Koike M, Yamasaki M, Ueda M, Ueki M, and Sato Y. Vascular smooth muscle cell growth-promoting factor/F-spondin inhibits angiogenesis via the blockade of integrin alphavbeta3 on vascular endothelial cells. J Cell Physiol 188: 394-402, 2001.52. Tsuchida K, Nakatani M, Matsuzaki T, Yamakawa N, Liu Z, Bao Y, Arai KY, Murakami T, Takehara Y, Kurisaki A, and Sugino H. Novel factors in regulation of activin signaling. Mol Cell Endocrinol 225: 1-8, 2004.53. Wang J, Weigand LA, Foxson J, Shimoda LA, and Sylvester JT. Ca2+ Signaling in Hypoxic Pulmonary Vasoconstriction: Effects of Myosin Light Chain and Rho Kinase Antagonists. Am J Physiol Lung Cell Mol Physiol, 2007.54. Wang J, Yao A, Wang JY, Sung CC, Fink LM, Hardin JW, and Hauer-Jensen M. cDNA cloning and sequencing, gene expression, and immunolocalization of thrombomodulin in the Sprague-Dawley rat. DNA Res 6: 57-62., 1999.55. Weaver M, Yingling JM, Dunn NR, Bellusci S, and Hogan BL. Bmp signaling regulates proximal-distal differentiation of endoderm in mouse lung development. Development 126: 4005-4015, 1999.56. Wu X, Chang MS, Mitsialis SA, and Kourembanas S. Hypoxia regulates bone morphogenetic protein signaling through C-terminal-binding protein 1. Circ Res 99: 240-247, 2006.57. Xu W, Kaneko FT, Zheng S, Comhair SA, Janocha AJ, Goggans T, Thunnissen FB, Farver C, Hazen SL, Jennings C, Dweik RA, Arroliga AC, and Erzurum SC.Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. Faseb J 18: 1746-1748, 2004.
Page 26 of 45

27
FIGURE LEGENDS
Figure 1. Hypoxia Modulated Gene Expression in Pulmonary and Cardiac
Microvascular Endothelial Cells
A. Human microvascular endothelial cells from lung (HMVEC-L) and cardiac (HMVEC-C)
cells under normoxic (0hr – 21% O2) and hypoxic conditions (1% O2) at various time-points
(3hr, 24h and 48hr). An incremental increase in the expression of two well-characterised
hypoxia responsive marker genes, namely vascular endothelial growth factor (VEGFA) and
solute carrier family 2 member 1 (SLC2A1 (previously called GLUT1)), was measured by
RT-PCR. GAPDH levels confirm equal sample loading. B. Hierarchical clustering of arrays
into a condition tree based on reliably expressed genes (10,476 genes). C. Temporal profile
of the number of genes up or down-regulated (>2-fold), revealed by comparison between
experimental time-points in HMVEC-L (428 genes) and D. HMVEC-C (299 genes).
Figure 2. Hierarchical Clustering of Lung Selective Hypoxia Responsive Genes
(A) Hierarchical clustering of lung selective genes identified a cluster (highlighted in yellow
and termed the Gremlin cluster) where (B) basal gene expression was uniquely elevated in the
lung cells (>3-10 fold) (represented as mean fluorescence values expressed in arbitrary units),
and (C) was further upregulated in response to hypoxia (represented as fold over time 0hr i.e.
non-hypoxic cell values). Each gene is represented by a single row of coloured boxes and
each time point is represented by a single column. The colour scale is red for high expression
values and green for low expression values.
Figure 3. Real-Time PCR Confirmation of Gremlin Cluster Genes in In Vitro Cell
Experiments
Page 27 of 45

28
Real-time PCR was used to confirm array results in unpooled RNA (n=6) from normoxic and
48hr hypoxic in in vitro experiments. Values are normalised to 18S rRNA and are presented
as mean±SD. † indicates significant differences between normoxic lung cells and normoxic
heart cells (p<0.05); * indicates significant differences between normoxic lung cells compared
to hypoxic lung cells (p<0.05).
Figure 4. In Vivo Real-Time PCR Analysis of Gremlin Cluster Genes in a Hypoxic
Animal Model
Mice (n=8) were maintained in normoxic or hypoxic conditions (inspired oxygen 10%) for
two days. A. Real-time PCR analysis of all seven genes in the gremlin cluster in whole lung
and cardiac tissue from normoxic mice. Values are normalised to 18S rRNA and data
presented as mean ±SEM. B. Gremlin 1 and C. SLM2 expression in the heart, kidney, liver,
lungs, spleen and thymus of normoxic and hypoxic mice. Values were normalised to 18S
rRNA and expressed as fold-change relative to the normoxic control for each organ. †
indicates significant differences between normoxic lung and normoxic heart (p<0.05); **
indicates significant differences between normoxic compared to hypoxic lung (p<0.001).
Figure 5. Immunohistochemical Localisation of Gremlin in Normoxic and Hypoxic
lungs
A. Immunohistochemical staining (brown colour) indicated that gremlin protein was
expressed basally in normoxic lung tissue (panels A & B) and increased with exposure to
hypoxia (panels C & D). Gremlin staining was not observed when primary antibody was
omitted from normoxic (panel E) or hypoxic (panel F) tissue or when secondary antibody was
omitted (data not shown). Two separate control and hypoxic animals are shown. Using
stereology, the volume of cells within the alveolar wall that were gremlin positive was
expressed as a fraction of total alveolar wall volume (volume fraction) and was significantly
Page 28 of 45

29
higher in hypoxic lungs (panel G). Data are presented as mean ±SEM. * indicates significant
differences between normoxic and hypoxic lung values (p<0.05).
Figure 6. Scratch Wound Assay
A. Human pulmonary microvascular endothelial cells were grown to confluence and a
vertical wound was applied to the monolayer. Cells were either exposed to 1. vehicle, 2.
BMP4 (80ng/ml), 3. BMP4 & Gremlin (2ug/ml) or 4. BMP4 & hypoxic condition medium
(CM) for 24hrs. Representative images are shown. The arrow indicates the position of the
original scratch. B. Mean wound width was measured for six independent wounds. Results
are expressed as % wound closure after 24hrs. Horizontal bars indicate median values. #
indicates significant difference from all other groups (p<0.05).
Figure 7. In Vivo Real-Time PCR Analysis of Gremlin mRNA in IPAH patients
Real-time PCR analysis of gremlin in control and IPAH lung tissue samples showed a
significant increase in mRNA levels in the patient group. Values were normalised to HPRT
and expressed as fold-change relative to the mean normoxic control value. *indicates
significant differences between groups (p<0.05).
Figure 8. Examination of CXCR7 Gene Cluster In Vivo
A. Hierarchical clustering of CXCR7 genes, where basal levels of gene expression increased
with hypoxia in the lung but remained unchanged in the cardiac endothelial cells. B. Real-
time PCR analysis of all four genes in the CXCR7 cluster in whole lung and cardiac tissue
from normoxic and hypoxic mice showed that the expression levels of CXCR7 and BDNF
were significantly elevated in the hypoxic lung. Expression of BDNF was also significantly
upregulated in the hypoxic heart. Values are normalised to 18S rRNA endogenous control
Page 29 of 45

30
and data are presented as mean ±SEM. * indicates significant difference between normoxic
and hypoxic tissues (p<0.05).
Figure 9. In Vivo Expression Profile of CXCL12 and CXCR7 in a Panel of Murine
Tissues
Expression of (a) CXCL12 and (b) CXCR7 in tissues from mice maintained in normoxic
(white columns) or hypoxic (black columns) conditions (inspired oxygen 10% for 48hrs).
CXCL12 expression increased >2-fold in hypoxic lung tissue, with more modest changes
observed in heart and spleen tissue. CXCR7 was significantly increased (>2fold) only in lung
tissue after hypoxic stress. Values are normalised to 18S rRNA and expressed as fold-change
relative to the normoxic control for each organ. Asterisk/s indicate significant difference
between normoxic and hypoxic tissues (* = p<0.05; ** = p<0.0001).
Page 30 of 45

31
TABLES
Table 1. RT-PCR primer sequences for house-keeping and hypoxia induced-marker genes
Forward (5’ to 3’) Reverse (5’ to 3’) Amplicon Cycle
VEGFA CTGCTCTACCTCCACCATGC CTGCATTCACATTTGTTGTGC 350bp 30
SLC2A1 TCACTGTGCTCCTGGTTCTG CCTGTGCTCCTGAGAGATCC 230bp 27
GAPDH TCCATGACAACTTTGGTATCGTGG GACGCCTGCTTCACCACTTCT 306bp 22
VEGFA: Vascular endothelial growth factor A, SLC2A1 (or GLUT1): Solute carrier family 2 (facilitated glucose transporter), member 1, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase,
Page 31 of 45

32
Table 2a. Real-time PCR Assays-on-Demand
Gene Assay ID (Human) Assay ID (Murine)
GREM1 Hs00171951_m1 Mm00488615_s1
SLM-2 Hs00198065_m1 Mm01200515_m1
SPON1 Hs00391830_m1 Mm00524006_m1
ENPP2/Autotaxin Hs00196470_m1 Mm00516578_m1
PRKY Hs00863863_gH Mm01312555_m1
MPEG1 Hs00909102_s1 Mm01222137_g1
LIMCH1 Hs00974323_m1 Mm01211763_g1
TM2D1 Mm00731838_m1
MYO1B Mm00501753_m1
BDNF Mm00432069_m1
CXCR7 Mm00432610_m1
CXCL12 Mm00445552_m1
Table 2b. Human Real-time PCR Assays
Gene Accession Sequences (5´ � 3´) Length Amplicon
for aag gac ccc acg aag tgt tg 20bphuHPRT NM000194
rev ggc ttt gta ttt tgc ttt tcc a 22bp157bp
for tat gag ccg cac agc cta ca 20bphuGrem1 NM013372
rev gca cct tgg gac cct ttc tt 20bp96bp
Page 32 of 45
33
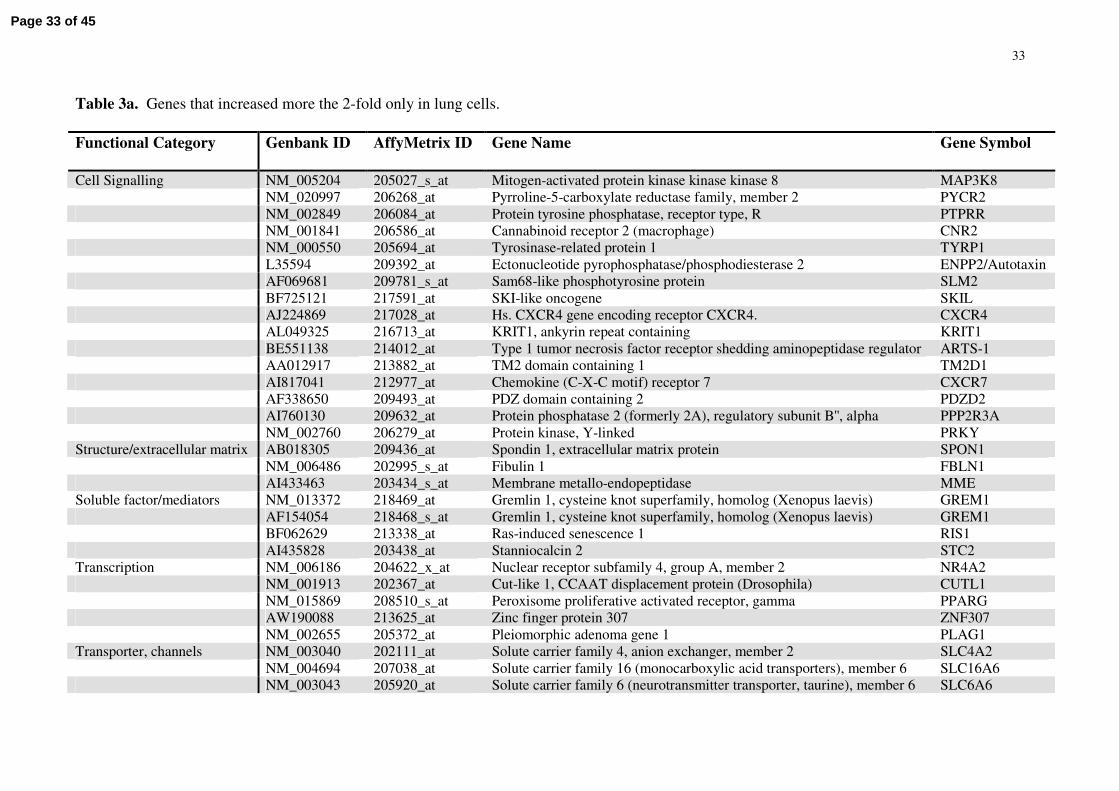
Table 3a. Genes that increased more the 2-fold only in lung cells.
Functional Category Genbank ID AffyMetrix ID Gene Name Gene Symbol
Cell Signalling NM_005204 205027_s_at Mitogen-activated protein kinase kinase kinase 8 MAP3K8NM_020997 206268_at Pyrroline-5-carboxylate reductase family, member 2 PYCR2NM_002849 206084_at Protein tyrosine phosphatase, receptor type, R PTPRRNM_001841 206586_at Cannabinoid receptor 2 (macrophage) CNR2NM_000550 205694_at Tyrosinase-related protein 1 TYRP1L35594 209392_at Ectonucleotide pyrophosphatase/phosphodiesterase 2 ENPP2/AutotaxinAF069681 209781_s_at Sam68-like phosphotyrosine protein SLM2BF725121 217591_at SKI-like oncogene SKILAJ224869 217028_at Hs. CXCR4 gene encoding receptor CXCR4. CXCR4AL049325 216713_at KRIT1, ankyrin repeat containing KRIT1BE551138 214012_at Type 1 tumor necrosis factor receptor shedding aminopeptidase regulator ARTS-1AA012917 213882_at TM2 domain containing 1 TM2D1AI817041 212977_at Chemokine (C-X-C motif) receptor 7 CXCR7AF338650 209493_at PDZ domain containing 2 PDZD2AI760130 209632_at Protein phosphatase 2 (formerly 2A), regulatory subunit B'', alpha PPP2R3ANM_002760 206279_at Protein kinase, Y-linked PRKY
Structure/extracellular matrix AB018305 209436_at Spondin 1, extracellular matrix protein SPON1NM_006486 202995_s_at Fibulin 1 FBLN1AI433463 203434_s_at Membrane metallo-endopeptidase MME
Soluble factor/mediators NM_013372 218469_at Gremlin 1, cysteine knot superfamily, homolog (Xenopus laevis) GREM1AF154054 218468_s_at Gremlin 1, cysteine knot superfamily, homolog (Xenopus laevis) GREM1BF062629 213338_at Ras-induced senescence 1 RIS1AI435828 203438_at Stanniocalcin 2 STC2
Transcription NM_006186 204622_x_at Nuclear receptor subfamily 4, group A, member 2 NR4A2NM_001913 202367_at Cut-like 1, CCAAT displacement protein (Drosophila) CUTL1NM_015869 208510_s_at Peroxisome proliferative activated receptor, gamma PPARGAW190088 213625_at Zinc finger protein 307 ZNF307NM_002655 205372_at Pleiomorphic adenoma gene 1 PLAG1
Transporter, channels NM_003040 202111_at Solute carrier family 4, anion exchanger, member 2 SLC4A2NM_004694 207038_at Solute carrier family 16 (monocarboxylic acid transporters), member 6 SLC16A6NM_003043 205920_at Solute carrier family 6 (neurotransmitter transporter, taurine), member 6 SLC6A6
Page 33 of 45

34
Unknown AB029025 212328_at LIM and calponin homology domains 1 LIMCH1AK026815 212327_at LIM and calponin homology domains 1 LIMCH1NM_022136 220330_s_at SAM domain, SH3 domain and nuclear localisation signals, 1 SAMSN1NM_018043 218804_at Transmembrane protein 16A TMEM16AAV728526 212611_at Macrophage expressed gene 1 MPEG1AL135926 216556_x_at SUMO1 pseudogene 2 SUMO1P2AI093572 214238_at Clone DT1P1B6 mRNA, CAG repeat regionAI934469 213349_at KIAA0779 protein KIAA0779NM_014704 204075_s_at KIAA0562 protein KIAA0562
Other NM_014893 207703_at Neuroligin 4 NLGN4NM_004572 207717_s_at Plakophilin 2 PKP2U75667 203946_s_at Arginase, type II ARG2BF575213 221477_s_at Superoxide dismutase 2, mitochondrial SOD2AF288390 210121_at UDP-Gal:betaglcnac beta 1,3-galactosyltransferase, polypeptide 2 B3GALT2NM_001709 206382_s_at Brain-derived neurotrophic factor BDNFNM_000585 205992_s_at Interleukin 15 IL15NM_013352 218854_at Squamous cell carcinoma antigen recognized by T cells 2 SART2NM_000963 204748_at Prostaglandin-endoperoxide synthase 2 PTGS2BF432550 212364_at Myosin IB MYO1BAW026491 200951_s_at Cyclin D2 CCND2
List comprised of genes that increased more the 2-fold between control sample and any time-point of hypoxia in lung cells, but decreased, remained the same or increased marginally (<1.2-fold) in cardiac cells.
Page 34 of 45
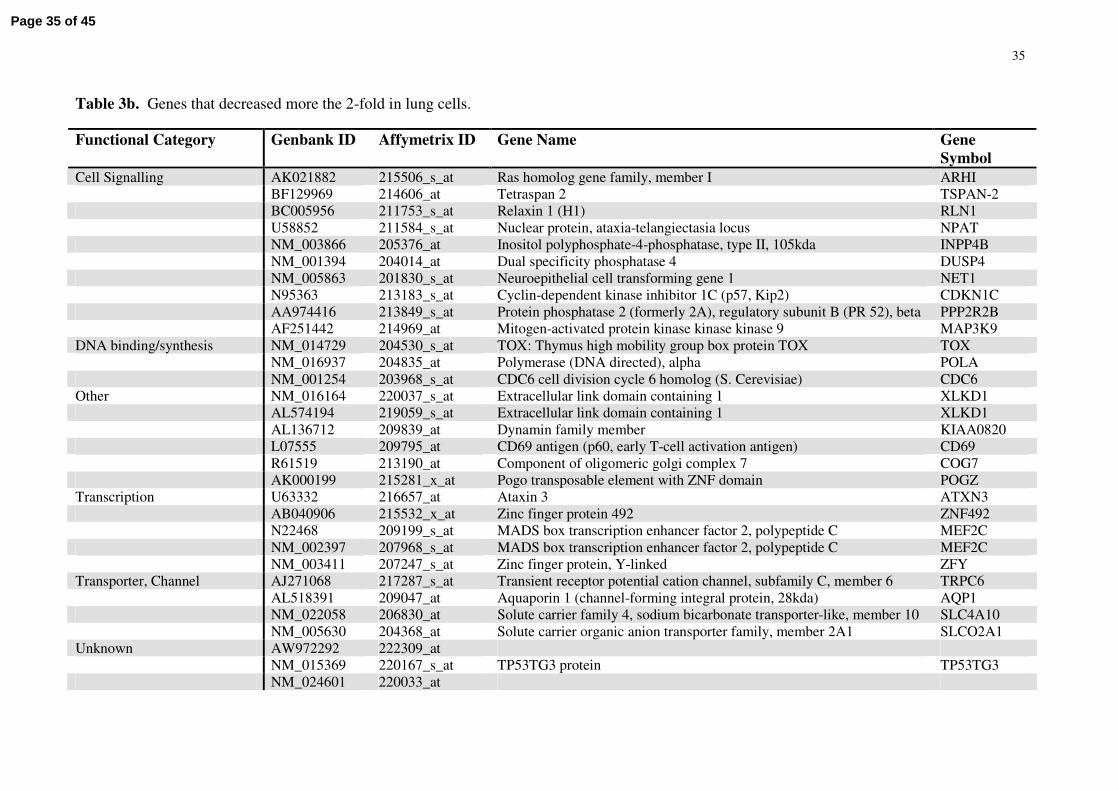
35
Table 3b. Genes that decreased more the 2-fold in lung cells.
Functional Category Genbank ID Affymetrix ID Gene Name Gene Symbol
Cell Signalling AK021882 215506_s_at Ras homolog gene family, member I ARHIBF129969 214606_at Tetraspan 2 TSPAN-2BC005956 211753_s_at Relaxin 1 (H1) RLN1U58852 211584_s_at Nuclear protein, ataxia-telangiectasia locus NPATNM_003866 205376_at Inositol polyphosphate-4-phosphatase, type II, 105kda INPP4BNM_001394 204014_at Dual specificity phosphatase 4 DUSP4NM_005863 201830_s_at Neuroepithelial cell transforming gene 1 NET1N95363 213183_s_at Cyclin-dependent kinase inhibitor 1C (p57, Kip2) CDKN1CAA974416 213849_s_at Protein phosphatase 2 (formerly 2A), regulatory subunit B (PR 52), beta PPP2R2BAF251442 214969_at Mitogen-activated protein kinase kinase kinase 9 MAP3K9
DNA binding/synthesis NM_014729 204530_s_at TOX: Thymus high mobility group box protein TOX TOXNM_016937 204835_at Polymerase (DNA directed), alpha POLANM_001254 203968_s_at CDC6 cell division cycle 6 homolog (S. Cerevisiae) CDC6
Other NM_016164 220037_s_at Extracellular link domain containing 1 XLKD1AL574194 219059_s_at Extracellular link domain containing 1 XLKD1AL136712 209839_at Dynamin family member KIAA0820L07555 209795_at CD69 antigen (p60, early T-cell activation antigen) CD69R61519 213190_at Component of oligomeric golgi complex 7 COG7AK000199 215281_x_at Pogo transposable element with ZNF domain POGZ
Transcription U63332 216657_at Ataxin 3 ATXN3AB040906 215532_x_at Zinc finger protein 492 ZNF492N22468 209199_s_at MADS box transcription enhancer factor 2, polypeptide C MEF2CNM_002397 207968_s_at MADS box transcription enhancer factor 2, polypeptide C MEF2CNM_003411 207247_s_at Zinc finger protein, Y-linked ZFY
Transporter, Channel AJ271068 217287_s_at Transient receptor potential cation channel, subfamily C, member 6 TRPC6AL518391 209047_at Aquaporin 1 (channel-forming integral protein, 28kda) AQP1NM_022058 206830_at Solute carrier family 4, sodium bicarbonate transporter-like, member 10 SLC4A10NM_005630 204368_at Solute carrier organic anion transporter family, member 2A1 SLCO2A1
Unknown AW972292 222309_atNM_015369 220167_s_at TP53TG3 protein TP53TG3NM_024601 220033_at
Page 35 of 45

36
NM_004765 219072_at B-cell CLL/lymphoma 7C BCL7CAW301305 214052_x_at Hbxag transactivated protein 2 XTP2AL050204 213929_at mRNA; cDNA dkfzp586f1223 (from clone dkfzp586f1223)AI809870 212922_s_at SET and MYND domain containing 2 SMYD2AB024518 209821_at Chromosome 9 open reading frame 26 C9orf26NM_015376 205801_s_atNM_006829 203571_s_at Adipose specific 2Chromosome 10 open reading frame 116 C10orf116Y15014 217452_s_at Hypothetical gene supported by Y15014
The list is comprised of genes that decreased more the 2-fold between control sample and any time-point of hypoxia in lung cells, but increased, remained the same or decreased marginally (<1.2-fold) in cardiac cells.
Page 36 of 45
0 3 24 48
GAPDH
SLC2A1 (GLUT1)
VEGF-A
HMVEC-Lung HMVEC-Cardiac
Hrs0 3 24 48
Figure 1.
48hr
24hr
3hr
0hr
HMVEC-Cardiac
XX6 ↓69 ↓88↓
7↑XX34 ↓51 ↓
80↑66↑XX3 ↓
117↑122↑22↑XX
48hr24hr3hr0hr
48hr
24hr
3hr
0hr
HMVEC-Lung
XX13 ↓121 ↓100 ↓
10↑XX125 ↓89 ↓
87↑73↑XX13 ↓
135↑130↑38↑XX
48hr24hr0hr 3hr
A
B
C
D
HMVEC-Lung
0hr 3hr 24hr 48hr 0hr 3hr 24hr 48hr
HMVEC-Cardiac
Page 37 of 45

Figure 2.
GREM1
GREM1
SLM2
ENPP2
PRKY
MPEG1
LIMCH1
LIMCH1
SPON1
0 3 24 48 0 3 24 48
Lung Cardiac
0 3 24 48 0 3 24 48
Lung cells Cardiac cellsLOW HIGH expression value
C. Fold over control values
B. Mean expression values
GREM1
GREM1
SLM2
ENPP2
PRKY
MPEG1
LIMCH1
LIMCH1
SPON1
0 3 24 48 0 3 24 48
Lung Cardiac
A.
Page 38 of 45

0.0
1.0
2.0
3.0
4.0
GREM1 SLM2 SPON1 ENPP2 PRKY MPEG1 LIMCH1
Nor
mal
sied
mR
NA
valu
es
HML Normoxic - 0hr
HML Hypoxic - 48hr
HMC Normoxic - 0hr
HMC Hypoxic - 48hr
†
*
†
††
†
†
Figure 3.
Page 39 of 45

0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
GREM 1 SLM 2 SPON1 ENPP2 PRKY M PEG1 LIM CH1
Nor
mal
ised
mR
NA
valu
es
Normoxic M urine LungNormoxic M urine Heart
0.0
0.5
1.01.5
2.0
2.5
3.03.5
4.0
4.5
Heart Kidney Liver Lung Spleen Thymus
Rel
ativ
em
RN
Ava
lues
GREM 1 - Contro lGREM 1 - Hypoxic
0.0
0.5
1.01.5
2.0
2.5
3.03.5
4.0
4.5
Heart Kidney Liver Lung Spleen Thymus
Rel
ativ
em
RN
Ava
lues SLM 2 - Contro l
SLM 2 - Hypoxic
Figure 4.
A
B
C
††
†
†
†
†
**
**
Page 40 of 45

Figure 5.
C. D.
50µm 50µm
A. B.
50µm50µm
E. F.
50µm 50µm
G.
0.0
0.1
0.2
0.3
Normoxic lungs Hypoxic Lungs
Volu
me
frac
tion
ofG
rem
lin *
Page 41 of 45

Figure 6.
A
2.1. 3. 4.
#
0
20
40
60
80
%w
ound
clos
ure
at24
hrs
Vehicle BMP4 BMP4 + Gremlin
BMP4 + Hypoxic CM
B
Page 42 of 45
*
Figure 7.
0.0
2.0
4.0
6.0
8.0
10.0
Control IPAH patients
Rel
ativ
eG
rem
linm
RN
Ava
lues
Page 43 of 45

0.0
3.0
6.0
9.0
TM2D1 CXCR7 MYO1B BDNF
Nor
mal
ised
mR
NA
leve
ls
Normoxic Murine Lung
Hypoxic Murine Lung
Normoxic Murine Heart
Hypoxic Murine Heart
Figure 8.
B
A
0 3 24 48 0 3 24 48
TM2D1/BBP
CXCR7
MYO1B
BDNF
Lung ECs Cardiac ECs
**
*
Page 44 of 45

A
Figure 9.
B
Rel
ativ
em
RN
Ava
lues **
0.0
1.0
2.0
3.0
Heart Kidney Liver Lung Spleen Thymus
CXCR7 - Control
CXCR7 - Hypoxic
**
*
*
0.0
1.0
2.0
3.0
Heart Kidney Liver Lung Spleen Thymus
Rel
ativ
em
RN
Ava
lues
CXCL12 - Control
CXCL12 - Hypoxic
Page 45 of 45
Copyright © 2022 FDOKUMEN




















