
Localization of neutrophil cationic proteins and loss of anionic charges in glomeruli of patients...
16
CLINICAL IMMUNOLOGY AND EMMUNOPATHOLOGY &t,299-314 (1982) Localization of Neutrophil Cationic Proteins and Loss of Anionic Charges in Glomeruli of Patients with Systemic Lupus Erythematosus Glomerulonephritisl GIOVANNI CAMUW, CIRO TETTA, GIUSEPPE SEGOLONI, RENATO CODA,* AND ANTONIO VERCELLONE Lahorutorio di Immunopatologia, Cattedra di Nejiiologia Medica, Divisione di Nefrologia e Dialisi. Ospedale Maggiore S.G. Butt&a, and *Zstituto di Anatomia ed Istologia Patologica I, Centro di Microscopia Elettronica, Turin, Ita1.v The localization of human neutrophil cationic protein (NCP) antigens in 14 patients with systemic lupus erythematosus (SLE) glomerulonephritis was investigated by immu- nofluorescence, using a rabbit anti-NCP serum. The specificity of the antiserum was demonstrated by its ability to bind exclusively to cytoplasmic structures of human poly- morphonuclear neutrophils (PMN), but not of lymphocytes, monocytes, or platelets. Furthermore, the immunofluorescence reaction was abolished by absorption of the anti- serum with purified NCP. In 7 patients with active SLE, severe proteinuria, and nephrotic syndrome, NCP were detected in glomerular capillary walls (GCW). The glomerular deposits of NCP were associated with a loss of glomerular polyanions (GPA). as revealed by colloidal iron and Alcian blue staining. The PMN, present in the peripheral blood of these patients, were markedly depleted of NCP. The depletion of NCP in circulating PMN, and the presence of NCP deposits in GCW with concomitant loss of GPA are consistent with the interpretation that NCP are released in raivo and that they directly contribute to the development of glomerular injury and proteinuria in pa- tlents with active SLE glomerulonephritis. INTRODUCTION Fixed anionic sites in glomerular capillary wall (GCW)’ are known to influence glomerular permeability to plasma proteins (1- 8). Several studies have shown the distribution and the nature of the glomerular polyanions (GPA) (5, 7-12). The glomerular perfusion with polycations induces spreading of foot processes, oblit- eration of slit pores, and reduction of GPA (13, 14). Since polyanions or neutral molecules show none of these effects, Seiler et al. (13, 14) hypothesized that anionic sites, not only condition glomerular permeability to anionic molecules such as albumin, but also maintain, by electrostatic repulsive forces, the structural organization of the foot processes. In rats injected with aminonucleoside of puromycin, the epithelial changes do not result from proteinuria, but are due to ’ This work was supported by CNR, Rome Ctr. 80.00602.04. ? Abbreviations used: GCW, glomerular capillary wall; GPA, glomerular polyanions; GBM, glomerular basement membrane; IC, immune complexes: SLE, systemic lupus erythematosus: CP. cationic proteins: PMN, polymorphonuclear neutrophils: NCP, neutrophil cationic proteins: TT-BSA. T&buffered Tyrode’s solution with bovine serum albumin. 0090- 1229/82/090299- 16$01.00/O Copyright 0 1982 by Academic Press, Inc. All rights of reproduction in any form reserved.
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Localization of neutrophil cationic proteins and loss of anionic charges in glomeruli of patients...

CLINICAL IMMUNOLOGY AND EMMUNOPATHOLOGY &t,299-314 (1982)
Localization of Neutrophil Cationic Proteins and Loss of Anionic Charges in Glomeruli of Patients with Systemic
Lupus Erythematosus Glomerulonephritisl
GIOVANNI CAMUW, CIRO TETTA, GIUSEPPE SEGOLONI, RENATO CODA,* AND ANTONIO VERCELLONE
Lahorutorio di Immunopatologia, Cattedra di Nejiiologia Medica, Divisione di Nefrologia e Dialisi. Ospedale Maggiore S.G. Butt&a, and *Zstituto di Anatomia ed Istologia Patologica I,
Centro di Microscopia Elettronica, Turin, Ita1.v
The localization of human neutrophil cationic protein (NCP) antigens in 14 patients with systemic lupus erythematosus (SLE) glomerulonephritis was investigated by immu- nofluorescence, using a rabbit anti-NCP serum. The specificity of the antiserum was demonstrated by its ability to bind exclusively to cytoplasmic structures of human poly- morphonuclear neutrophils (PMN), but not of lymphocytes, monocytes, or platelets. Furthermore, the immunofluorescence reaction was abolished by absorption of the anti- serum with purified NCP. In 7 patients with active SLE, severe proteinuria, and nephrotic syndrome, NCP were detected in glomerular capillary walls (GCW). The glomerular deposits of NCP were associated with a loss of glomerular polyanions (GPA). as revealed by colloidal iron and Alcian blue staining. The PMN, present in the peripheral blood of these patients, were markedly depleted of NCP. The depletion of NCP in circulating PMN, and the presence of NCP deposits in GCW with concomitant loss of GPA are consistent with the interpretation that NCP are released in raivo and that they directly contribute to the development of glomerular injury and proteinuria in pa- tlents with active SLE glomerulonephritis.
INTRODUCTION
Fixed anionic sites in glomerular capillary wall (GCW)’ are known to influence glomerular permeability to plasma proteins (1- 8). Several studies have shown the distribution and the nature of the glomerular polyanions (GPA) (5, 7-12). The glomerular perfusion with polycations induces spreading of foot processes, oblit- eration of slit pores, and reduction of GPA (13, 14). Since polyanions or neutral molecules show none of these effects, Seiler et al. (13, 14) hypothesized that anionic sites, not only condition glomerular permeability to anionic molecules such as albumin, but also maintain, by electrostatic repulsive forces, the structural organization of the foot processes. In rats injected with aminonucleoside of puromycin, the epithelial changes do not result from proteinuria, but are due to
’ This work was supported by CNR, Rome Ctr. 80.00602.04. ? Abbreviations used: GCW, glomerular capillary wall; GPA, glomerular polyanions; GBM,
glomerular basement membrane; IC, immune complexes: SLE, systemic lupus erythematosus: CP. cationic proteins: PMN, polymorphonuclear neutrophils: NCP, neutrophil cationic proteins: TT-BSA. T&buffered Tyrode’s solution with bovine serum albumin.
0090- 1229/82/090299- 16$01.00/O Copyright 0 1982 by Academic Press, Inc. All rights of reproduction in any form reserved.

300 CAMUSSI ET AL.
the primary loss of GPA induced by the drug (15- 17). Moreover, the chronic administration of aminonucleoside is followed by loss of GPA, collapse of GCW, plasma protein accumulation in the subendothelial part of the glomerular base- ment membrane (GBM) and in the mesangium and, finally, by glomerular sclerosis (17). In nephrotoxic glomerulonephritis, the loss of GPA precedes the onset of proteinuria (18), a finding which further supports the contention that neutraliza- tion of GPA leads to altered charge-selective permeability. Furthermore, the de- position of immune complexes (IC) in the GCW is associated with loss and/or redistribution of GPA (19, 20). In mice with systemic lupus erythematosus (SLE) nephritis and proteinuria, the passage of plasma proteins across the GCW is caused by nonuniform increases in glomerular permeability (20), related to a focal loss of GPA (19).
A number of cationic proteins (CP) has been identified as being released from a variety of inflammatory cells such as platelets (21, 22), macrophages (23), and polymorphonuclear neutrophils (PMN) (24-26). Among these mediators, three CP, isolated and purified from rabbit PMN (MW 12,000), have been shown to promote an increase in vascular permeability. A fourth cationic component (MW 5000) induces degranulation of, and histamine release from, mastocytes (24).
Evidence for an active release of CP from stimulated rabbit PMN has been provided (25). More recently, Olsson and Venge purified and characterized human neutrophil cationic proteins (NCP) from leukemic leukocytes (26) with a molecu- lar weight ranging from 21,000 to 29,000 and we have shown their active release from IC-stimulated normal human PMN (27, 28).
NCP display several important biological activities as they increase vascular permeability when injected intradermally (28); they activate the complement cas- cade with generation of anaphylatoxin activity in serum (29), have a bactericidal capacity (30), cause basophil degranulation and release of mediators (28), have chemotactic activity (29), aggregate PMN in vitro, cause neutropenia in Iho, and induce the release of platelet-activating factor (PAF) from PMN (27, 31, 32).
We have previously shown an in vi\‘0 release and urinary excretion of NCP in acute phases of SLE glomerulonephritis (33). The present study describes the localization of NCP in glomeruli of patients with active SLE glomerulonephritis, the concomitant loss of GPA in GCW and of NCP in PMN of the peripheral blood. These findings support the hypothesis that NCP are released in viva and exert a direct pathogenetic effect on the glomeruli of these patients.
MATERIALS AND METHODS
Preparation of Cells
Leukocytes, obtained from peripheral blood of normal donors, were collected in siliconized plastic tubes containing 5 x 10M3 M (final concentration) of ethyl- enediaminetetraacetate (EDTA), pH 7.2. After separation by the Hysopaque- Ficoll step gradient (34), 95% pure PMN were prepared, as previously described (27), from the granulocyte-erythrocyte-containing sediment by subsequent gela- tin sedimentation, osmotic shock and finally washed three times with TT-BSA (no

NEUTROPHIL CATIONIC PROTEINS AND SLE NEPHRITIS 301
Caz+, no Mg2+)3 to avoid appreciable platelet contamination (less than 1%). Finally, PMN were resuspended in TT-BSA.
Plastic-adherent, mononuclear cells (98% pure) were obtained from the high- density mononuclear-rich fraction of the Hysopaque - Ficoll gradient by allowing cell adherence in 35mm-diameter plastic petri dishes, as previously described (32). Adherent cells were characterized as monocytes by esterase staining (35), coupled with inhibition by sodium fluoride (36). Washed human platelets were obtained as previously described (37). Briefly, human blood was collected on citric acid-citrate-dextrose (for 9 ml of blood, 1 ml of a solution of 8 g. of citric acid monohydrate, 22 g of citrate trisodium dehydrate, and 22.3 g per liter of distilled water was used). Platelets were washed in TT-BSA, no Ca’+, no Mg2+, centrifuged twice at 1OOOg for 15 min on a cushion of erythrocytes, separated by centrifugation at 90g for 15 min, and finally resuspended in TT-BSA.
Purification of Human Cationic Proteins
A. Neutrophil cationic proteins. NCP were obtained from PMN as previously described (27). Briefly, the sonicates (1 x 108/ml of PMN) were extracted with 5 vol of 0.2 M acetate buffer, pH 4, at 4” C for 3 hr. After centrifugation at 40,OOOg for 30 min, the supernatant was collected and recentrifuged under similar condi- tions. The supernatant was dialyzed for 48 hr against 0.01 M phosphate buffer, pH 8.5, then chromatographed on a 1.5 x 30-cm DEAE-Sephadex A-50 (Pharmacia Fine Chemicals, lot 3537, Uppsala, Sweden) column equilibrated with the same buffer. After elution at pH 8.5, NCP were concentrated to approximately 5 ml with an Amicon ultrafiltration unit, using a UM 2 membrane, then dialyzed against 0.1 M acetate buffer, pH 3.7. The whole procedure of purification was carried out at 4°C to avoid digestion by proteolytic enzymes. Molecular sieve filtration was performed on a 2.6 x loo-cm Sephadex G-100 (Pharmacia Fine Chemicals, lot 9069) column, equilibrated with 0.1 M acetate buffer pH 3.7. The flow rate of the column was 12 mVhr and 3-ml fractions were collected. The molecular weight of NCP was estimated according to Andrews’ method (38), using as reference sub- stances BSA (66,000) (Boehringer GmbH, lot 19602991, Mannheim, West Ger- many) labeled with ‘251, hemoglobin (64,000) (Sigma Chemical Co., lot H-2707, St. Louis, MO.) peroxidase (40,000) (Grade II, Boehringer, lot 127361), cytochrome c (12,000) (Type XY, from rabbit heart, Sigma, lot C-9137), and glucagon (3600) (crystalline, Sigma, lot 4250) labeled with 1251. The electrophoretic identification of NCP was performed on cellulose acetate strips in 0.05 M acetate buffer, pH 4.0, according to Olsson and Venge (26). NCP were also examined by polyacrylamide gel electrophoresis (gel concentration 8%)) carried out in Tris -glycine - sodium
Ii Composition of buffers was as follows. Tyrode’s: KCl, 2.6 x lo-” M: MgCl,6H,O, 1.0 x lo--” M: NaCI, 1.37 x 10-i M: NaHCO,, 1.0 X lo-* M; CaClZ6H,0, 3.0 X 10m3 M: glucose 0.1%. Tris-buffered Tyrode’s (TT): Tyrode’s with 1 x 10e3 M Tris, used instead of bicarbonate. When indicated, 0.25% bovine serum albumin (BSAl was added to TT (‘IT-BSA). When calcium and magnesium ions were
omitted in TT-BSA, the latter is designated as TT-BSA, no Ca*+ no Mg”+. Phosphate-buffered saline
(PBS): 0.01 M phosphate buffer, NaCl, 0.15 M. All solutions were buffered at pH 7.4 at room temper- ature.

302 CAMUSSI ET AL.
dodecyl sulfate (SDS) buffer, 0.025 44, pH 8.8, at room temperature for 90 min at 200 V and 30 mA (39). Proteins were stained with Coomassie brilliant blue R250, 0.2% (w/v), in methanol:acetic acid:water (9:2:9, v/v), the dye-free solvent being used as washing solution. IgG (IgG Standard, Boehringer, lot TRA-07) heavy (52,000) and light (22,000) chains, lysozyme (crystalline, from egg white, Boehringer, lot 107255), and carboxypeptidase B (Boehringer, lot 15265) were used as marker polypeptides for determination of molecular weights. The five NCP components, exhibiting on analytical electrophoresis a cathodal mobility higher than that of lysozyme, had a molecular weight ranging from 25,000 to 32,000 by the Andrews’ method (38) and ranging from 17,000 to 25,000 in four distinct bands by SDS-polyacrylamide gel electrophoresis (Fig. 1) (27). The five components displayed biological activity, based on the blueing reaction induced in the skin of rabbits injected with Evans’ blue (28) (minimal effective dose 2.5 pg protein) (27) and on in vitro basophil degranulation (28) (minimal effective dose 0.25 pg protein) (27). The antigenic inoculum used to immunize rabbits was ob- tained by pooling several homogeneous preparations of NCP.
B. Platelet cationic proteins. CP were obtained from sonicated platelets ac- cording to the technique described by Nachman et al. (22), consisting in the separation of CP from the acid extracts by anion exchange chromatography and gel filtration using sequentially DEAE and Sephadex G-75 (Pharmacia) columns
FIG. 1. Polyacrylamide gel electrophoresis of NCP (2) (gel concentration 8%), performed in Tris-glycine-sodium dodecyl sulfate buffer, 0.025 M, pH 8.8, at room temperature for 90 min at 200 V and 30 mA. IgG heavy (52,000) and light (22,000) chains and lysozyme (14,500) were used as reference substances (1). NCP show separate bands with molecular weights ranging from 17,000 to 25,000.
NEUTROPHIL CATIONIC PROTEINS AND SLE NEPHRITIS 303
(22). Platelet CP were used to exclude immunologic cross-reactivity with serum from rabbits immunized to NCP.
Preparation of Rabbit Anti-Human NCP Serum
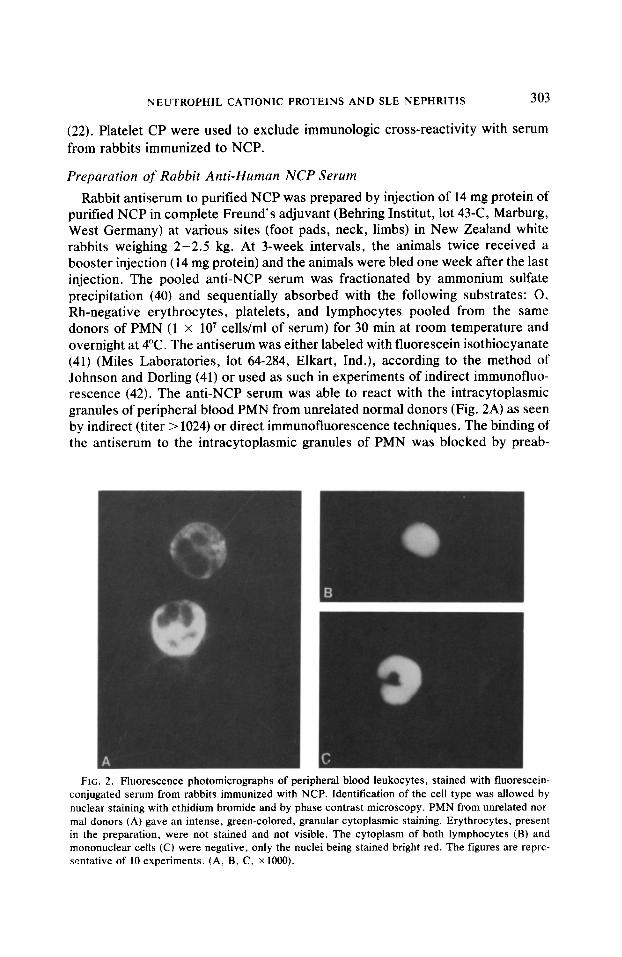
Rabbit antiserum to purified NCP was prepared by injection of 14 mg protein of purified NCP in complete Freund’s adjuvant (Behring Institut, lot 43-C, Marburg, West Germany) at various sites (foot pads, neck, limbs) in New Zealand white rabbits weighing 2-2.5 kg. At 3-week intervals, the animals twice received a booster injection (14 mg protein) and the animals were bled one week after the last injection. The pooled anti-NCP serum was fractionated by ammonium sulfate precipitation (40) and sequentially absorbed with the following substrates: 0, Rh-negative erythrocytes, platelets, and lymphocytes pooled from the same donors of PMN (1 x 10’ cells/ml of serum) for 30 min at room temperature and overnight at 4°C. The antiserum was either labeled with fluorescein isothiocyanate (41) (Miles Laboratories, lot 64-284, Elkart, Ind.), according to the method of Johnson and Dorling (41) or used as such in experiments of indirect immunofluo- rescence (42). The anti-NCP serum was able to react with the intracytoplasmic granules of peripheral blood PMN from unrelated normal donors (Fig. 2A) as seen by indirect (titer > 1024) or direct immunofluorescence techniques. The binding of the antiserum to the intracytoplasmic granules of PMN was blocked by preab-
FIG. 2. Fluorescence photomicrographs of peripheral blood leukocytes, stained with fluorescein- conjugated serum from rabbits immunized with NCP. Identification of the cell type was allowed by nuclear staining with ethidium bromide and by phase contrast microscopy. PMN from unrelated nor- mal donors (A) gave an intense, green-colored, granular cytoplasmic staining. Erythrocytes, present in the preparation, were not stained and not visible. The cytoplasm of both lymphocytes (B) and mononuclear cells (C) were negative, only the nuclei being stained bright red. The figures are repre- sentative of 10 experiments. (A, B, C, x 1000).

304 CAMUSSI ET AL.
sorption with purified NCP. No reactivity with human erythrocytes (Fig. 2A), washed or unwashed platelets, lymphocytes (Fig. 2B) or mononuclear cells in peripheral blood smears (Fig. 2C) or with purified plastic-adherent macrophages was observed. By immunofluorescence, the antiserum did not react with normal human skin or kidney tissue. Studies of immunoprecipitation by double im- munodiffusion (43) against normal human plasma, purified y globulin (Cohn Frac- tion II, Sigma, lot HG-II), and CP purified from platelets showed absence of cross-reactivity. In contrast, the anti-NCP serum reacted with purified NCP. These studies demonstrated that serum from rabbits immunized to NCP specifi- cally reacted with NCP.
Patients
Fourteen patients (13 females and 1 male), aged 19 to 65 years (mean 32), satisfying the criteria of the American Rheumatism Association for the diagnosis of SLE (44), were selected for this study. Patients with drug-induced, lupus-like or overlapping syndromes were not included in this study. The clinical activity was independently assessed at the time of the renal biopsy by two investigators on the basis of the following signs and symptoms (44): fever, active rash, serositis, ar- thritis, central nervous system involvement, and active renal disease. As de- scribed in a previous publication (37), patients were divided into three groups on the basis of various degrees of disease activity: Grade I (active) comprised pa- tients with at least three or more of the above-cited symptoms. In addition, all of them had depressed C3 and C4 serum levels, high titers of antinuclear antibodies (45) with a homogeneous pattern, high titers of anti-native DNA antibodies (46) and of circulating IC (47,48). Furthermore, these patients had a leukopenia of less than 4000 white blood cells per cubic millimeter while they were not exposed to drugs known to induce leukopenia. Grade II (moderately active) comprised pa- tients with one or two of the above-cited symptoms. Patients lacking any of these symptoms or signs of serological SLE activity were assigned to Grade III.
Eight normal human kidney specimens, obtained by open, surgical biopsy and six kidney specimens, obtained by needle biopsy from patients with Berger’s glomerulonephritis, were used as controls in the immunofluorescent and histo- logical studies.
Staining of Intracytoplasmic Cationic Proteins in Peripheral Blood Smears
In order to assess the intracytoplasmic CP content of PMN from SLE patients in respect to controls, direct immunofluorescence using anti-NCP serum was performed on peripheral blood smears. The intensity of the immunofluorescence staining of the intracytoplasmic PMN granules was graded from 0 to 3 by two independent observers unaware of the pathologic findings. A minimum of 50 PMN per slide was evaluated.
Histological and Pathological Study of Renal Tissue
All 14 patients were submitted to percutaneous renal biopsy. The renal tissue was divided into two parts, one used for light and the other for immunofluores-

NEUTROPHIL CATIONIC PROTEINS AND SLE NEPHRITIS 305
cence microscopy. Tissue for light microscopy was fixed in Serra’s solution (60% ethyl alcohol, 12% phormol, and 10% glacial acetic acid) for 2 hr, postfixed in 95% alcohol for 2 hr, and embedded in paraffin. The sections were stained with hema- toxylin and eosin and periodic acid-Schiff (PAS) reaction and the histological findings were classified according to Appel er al. (49) in five classes as indicated herein: Class I, normal kidneys; Class II, mesangial changes; Class III, focal and segmental glomerulonephritis; Class IV, diffuse proliferative glomerulonephritis: Class V, membranous glomerulonephritis. Immunofluorescence was performed on renal tissue snap-frozen in liquid nitrogen. Sections, 2-4 pm thick, were obtained in a cryostat. After washing in PBS, the sections were incubated with fluoresceinated rabbit antisera specific for human IgG, IgM, IgA, C3, Clq, C4, and fibrinogen (Behringwerke). In order to amplify the fluorescence staining reaction for NCP, an indirect immunofluorescence test was performed using goat anti- rabbit IgG (Behringwerke), labeled with fluorescein isothiocyanate. In prelimi- nary experiments, the goat anti-rabbit IgG serum, preabsorbed with human y globulins, was unable to stain the renal sections from normal as well as from SLE individuals. The renal sections were examined with a Zeiss microscope equipped with a HBO-200 high-pressure mercury vapor lamp, using a GB 12 exciter filter and a KV 490 barrier filter. A minimun of four glomeruli per section (range 4-23) was examined. The intensity of the fluorescent staining was evaluated indepen- dently by two observers and the intensity and extent were graded from 0 to 3. When staining for NCP was present, serial sections were incubated with antise- rum previously absorbed with purified NCP to assess the specificity of the reac- tion. PMN present in the glomeruli were quantitated independently by two ob- servers. The PMN index was evaluated by counting the total number of intra- glomerular PMN divided by the number of glomeruli, and expressed the number of PMN per glomerulus.
Staining of Glomerular Polyanions
The presence of GPA was evaluated by the colloidal iron (50) and the Alcian blue (14) staining techniques. Staining was performed on 4-pm-frozen or paraffin embedded sections of renal tissue. For the colloidal iron staining, after rinsing (30 set) in 12% glacial acetic acid aqueous solution, the sections were stained for 1 hr and 30 min in the colloidal iron solution, followed by four (3 min each) rinses in 12% glacial acetic acid. The pH of both the staining and the rinsing solutions was between 1.1 and 1.3 to insure selective staining of acidic polysaccharides without coloration of nucleic acids. The Prussian blue color was developed for 20 min in a freshly prepared mixture of equal parts of 2% hydrochloric acid and 2% potassium ferrocyanide. Carballumen (15 min) was used as counterstain. The amount of staining was evaluated independently by two examiners who were unaware of the pathologic findings in the renal sections. The intensity and extent of GPA staining were semiquantitatively evaluated and expressed as the number of glomeruli which exhibited segmental or total loss of GPA (Table 1). Specimens from eight normal subjects and from six patients with Berger’s glomerulonephritis served as controls. Furthermore, duplicate sections from each specimen were not exposed

306 CAMUSSI ET AL.
to colloidal iron but were processed with the HCl-potassium ferrocyanide part of the colloidal iron method in order to rule out nonspecific staining due to naturally occurring hemosiderin. In nine patients, GPA were also evaluated by the Alcian blue staining technique, a more selective but weaker stain (50) that invariably provided results comparable to those obtained by the colloidal iron technique.
RESULTS
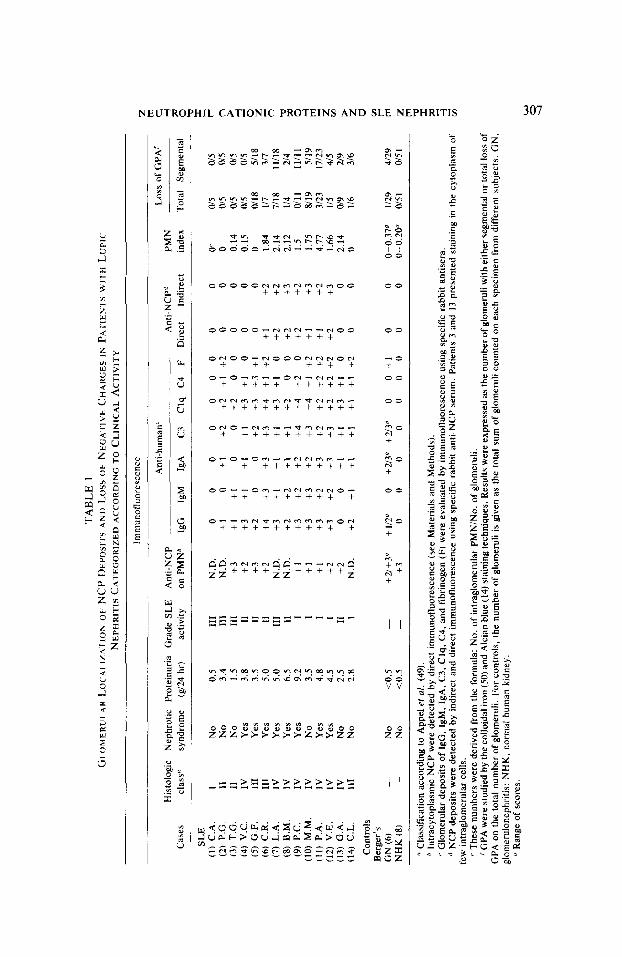
As shown in Table 1, the study by direct and indirect immunofluorescence showed deposits of NCP in glomeruli of seven out of fourteen patients with SLE. The specificity of the immunofluorescence staining was confirmed by the loss of the reaction when the antiserum was absorbed with purified NCP. The deposits of NCP were localized in a linear, segmental manner along the peripheral part of the GCW (Figs. 3B, D, F) and markedly differed from the granular or patchy deposits of immunoglobulins (Figs. 3A, C, E) or complement. In contrast, aside from the segmental pattern, the deposits of NCP resembled those of polyanions detected by colloidal iron or Alcian blue stainings in normal human glomeruli (Figs. 4A, B).
Both the intensity and extent of NCP deposits were greater in glomeruli of patients with grade I of SLE activity. The glomeruli of these patients also had the greater loss in GPA (Figs. 4C-F), a loss that was especially marked in patients with severe proteinuria and nephrotic syndrome (Table 1). The loss in GPA, however, appeared more widespread than the deposits of NCP.
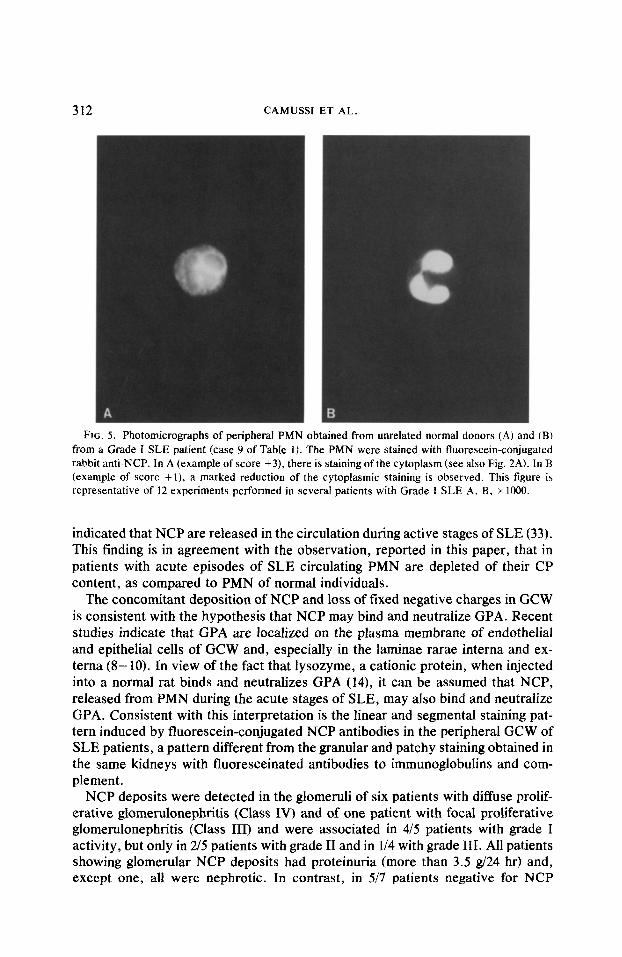
The study by light microscopy of glomeruli with NCP deposits revealed no correlation with local accumulation of PMN. Only in two patients (cases 3 and 13, Table l), with absence of NCP in GCW, were CP detected in the cytoplasm of PMN present in the glomeruli. In patients with grade I of SLE activity and with deposits of NCP in GCW (cases 6- 12, of Table l), the PMN present in glomeruli (PMN index: 2.45 t 1.48, mean + 1 SEM) failed to stain for NCP. Likewise, the PMN present in the peripheral blood of these patients appeared markedly depleted of NCP (Fig. 5B), as compared with PMN from normal individuals (Fig. SA).
Deposits of NCP were never detected in glomeruli from normal subjects or from patients with Berger’s glomerulonephritis. These glomeruli always showed normal amounts of polyanions, as detected by the colloidal iron or Alcian biue techniques (Table 1).
DISCUSSION
The results of the present study show that deposits of NCP and loss of GPA occur concomitantly in the GCW of patients with active SLE glomerulonephritis, proteinuria, and nephrotic syndrome. These findings are associated with depletion of NCP in circulating PMN.
SLE glomerulonephritis is a prototype of IC disease (51). In this condition, PMN are attracted and stimulated by IC to release lysosomal enzymes (52) and PAF (31, 32). These mediators may contribute to a local inflammatory reaction and to increase glomerular permeability. It has been shown that in SLE the in- teraction between IC and PMN occurs during the active phases of the disease (49) and results in neutropenia, a sign of disease activity (44). Our previous work
TABL
E 1
GI~O
MER
~.TI
AR
Lo
ch1
IZAT
ION
OF
NCP
DEPO
SITS
AN
D Lo
ss
OF
NEG
ATIV
E CH
ARGE
S IN
PA
TIEN
TS
WIT
H LU
PIC
NEPH
RITI
S CA
TEGO
RIZE
D AC
CORD
ING
TO
CLIN
ICAL
AC
TIVI
TY
Imm
unof
luor
esce
nce
Case
s
Anti-
hum
an’
Hist
olog
ic Ne
phro
tic
Prot
einur
ia Gr
ade
SLE
Anti-
NCP
Anti-
NCP”
cla
ss”
synd
rom
e (g
/24
hr)
activ
ity
on
PMN”
kG
kM
lg
.4
C3
Clq
C4
F Di
rect
In
dire
ct
SLE
(11
C.A.
(2
) P.
G.
(31
T.G
. (4
) V.
C.
(5)
G.F.
(6
) C.
R.
(7)
L.A.
(8
) B.
M.
(9)
P.C.
(1
0)
M.M.
(1
1)
P.A.
(1
2)
V.E.
(1
3)
G.A.
(1
4)
C.L.
Cont
rols
Berg
er’s
GN
(6)
NHK
(8)
I 11
II IV
III
III
IV
IV
IV
IV
IV
IV
IV
III - -
No
0.5
No
3.4
No
1.5
Yes
3.8
Yes
3.5
Yes
5.0
Yes
5.0
Yes
6.5
Yes
9.2
No
3.5
Yes
4.8
Yes
4.5
No
2.5
No
2.8
No
No
<OS
co.5
III
111
III II II II III II I I I I II I
N.D.
0
0 N.
D.
+1
0 +3
+1
+1
t2
+3
+1
+3
+2
0
t2
+4
13
N.D.
f3
fl
N.D.
+2
+2
+1
+3
t2
fl
+3
+3
+1
t3
t2
+2
+3
t2
+2
0 0
N.D.
+2
t1
+2/+
3g
t 11
20
0 -
+3
0 0
0 +1
0 +I
0 +3
+1
+1
t2
t2
+3
+3
+I
3-I
t 213
0 0
0 00
0 +2
+2
+1
i-2
0
+2
0 0
+1
+3
t1
0 t2
t3
13
+I
+3
t4
t1
+2
fl
+3
+1
0 +1
t2
0
0 t4
f4
+2
0
t3
t4
t1
+2
+2
t2
t2
t2
t3
i-2
t2
t2
+1
t3
+I
0 t1
+1
+1
1-
2
t2/3
g 0
0 +1
0
000
0 0
0 0
0 0
0 0
0 0
+1
+2
t2
t2
t2
t3
t2
+2
t1
t3
+I
t2
t2
t3
0 0
0 0
0 0
0 0
PMN
index
0’
O/5
015
0 O/
S 01
5 0.
14
O/5
O/5
0.15
01
5 O/
5 0
O/l8
51
18
1.84
I/7
31
7 2.
14
7118
11
/18
2.12
11
4 2/
4 1.
5 O
il1
1111
1 1.
75
8119
51
19
4.77
31
23
17/2
3 1.
66
l/S
415
2.14
o/
9 21
9 0
116
3/6
o-o.
379
1129
41
29
O-0.
208
0151
o/
5 I
Loss
of
G
PA’
Tota
l Se
gmen
tal
r( Cl
assif
icatio
n ac
cord
ing
to
Appe
I ef
nl.
(49)
. ’
Intra
cyto
plas
mic
NCP
were
de
tect
ed
by
dire
ct
imm
unof
luor
esce
nce
(see
M
ater
ials
and
Met
hods
). ’
Glom
erula
r de
posit
s of
Ig
G,
IgM,
IgA,
C3
, Cl
q,
C4,
and
fibrin
ogen
(F
l we
re
evalu
ated
by
im
mun
oflu
ores
cenc
e us
iug
spec
ific
rabb
it an
tiser
a.
d NC
P de
posit
s we
re
dete
cted
by
in
dire
ct
and
dire
ct
imm
unof
luor
esce
nce
usin
g sp
ecifi
c ra
bbit
anti-
NCP
seru
m.
Patie
nts
3 an
d I3
pr
esen
ted
stai
ning
in
the
cy
topl
asm
of
fe
w int
raglo
mer
ular
cells
. ’
Thes
e nu
mbe
rs
were
de
rived
fro
m
the
form
ula:
No.
of
intra
glom
erula
r PM
NINo
. of
gl
omer
uli.
’ GP
A we
re
stud
ied
by t
he
collo
idal
iro
n (5
01 a
nd
Alcia
n bl
ue
(14)
st
aini
ng
tech
niqu
es.
Resu
lts
were
ex
pres
sed
as th
e nu
mbe
r of
glo
mer
uli
with
eit
her
segm
enta
l or
to
tal
loss
of
GPA
on
the
tota
l nu
mbe
r of
gl
omer
uli.
For
cont
rols,
th
e nu
mbe
r of
glo
mer
uli
is g
iven
as t
he
tota
l su
m
of
glom
erul
i co
unte
d on
ea
ch
spec
imen
fro
m
diffe
rent
su
bjec
ts.
GN.
glom
erul
onep
hritis
: NH
K.
norm
al hu
man
kid
ney.
” Ra
nge
of
scor
es.
- -
.- ,..
-
.- -
- --
-
- -
1-1
-
-.
_ .I
I
_ -,
I.
-
.*
- ._
. .I
.-
-

308 CAMUSSI ET AL.
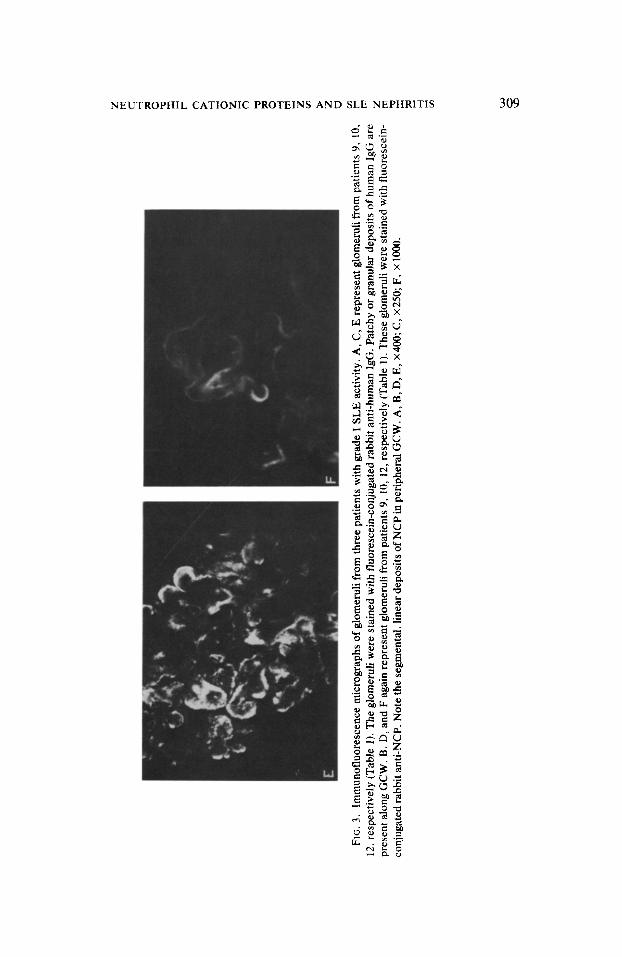
FIG
. 3.
Im
mun
oflu
ores
cenc
e m
icrog
raph
s of
glo
mer
uli
from
thr
ee
patie
nts
with
gr
ade
I SL
E ac
tivity
. A,
C,
E re
pres
ent
glom
erul
i fro
m
patie
nts
9, 1
0,
i 12
. res
pect
ivel
y (T
able
1). T
he g
lom
erul
i we
re
stai
ned
with
flu
ores
cein
-con
juga
ted
rabb
it an
ti-hu
man
Ig
G.
Patc
hy o
r gr
anul
ar
depo
sits
of h
uman
Ig
G a
re
pres
ent
alon
g G
CW
. B,
D,
and
F ag
ain
repr
esen
t gl
omer
uli
from
pat
ient
s 9,
10,
12,
resp
ectiv
ely
(Tab
le 1)
. The
se
glom
erul
i we
re
stai
ned
with
flu
ores
cein
- 3
conj
ugat
ed
rabb
it an
ti-N
CP.
N
ote
the
segm
enta
l, lin
ear
depo
sits
of N
CP
in p
erip
hera
l G
CW
. A,
B,
D,
E, X
400;
C
, X2
50;
F, X
100
0.
2 r, CiJ
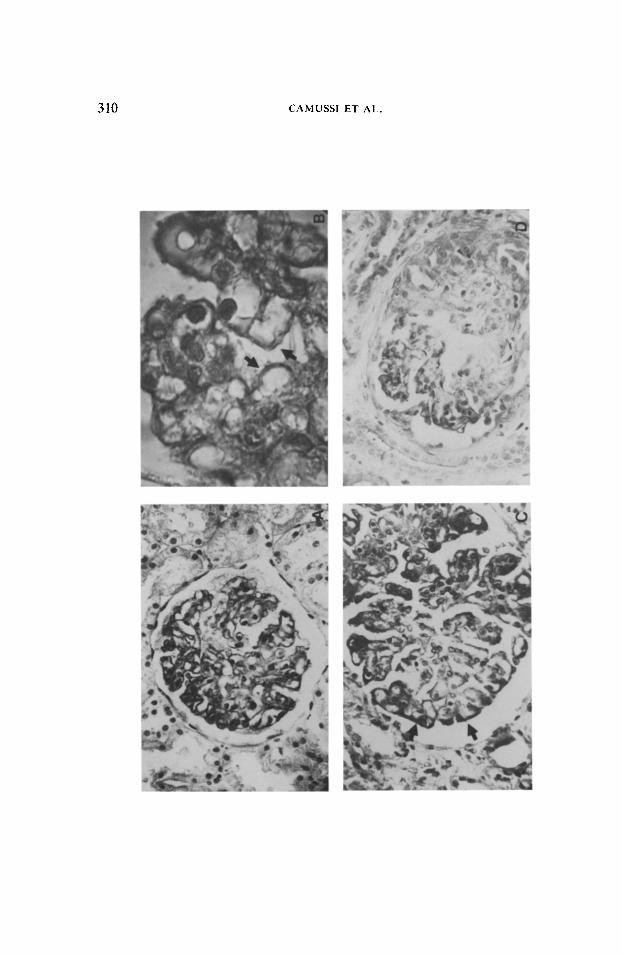
310 CAMUSSI ET AL.
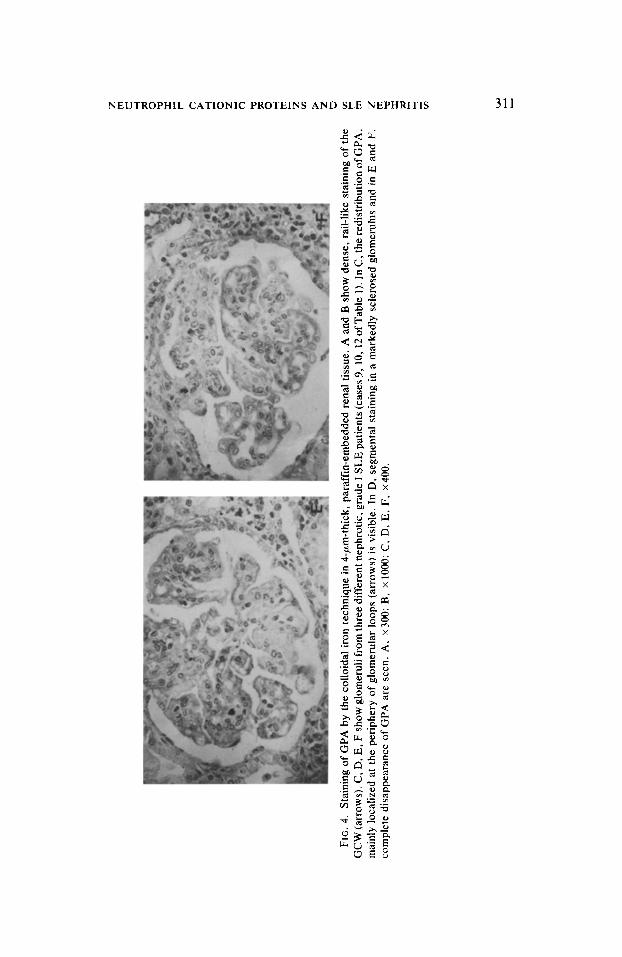
FIG
. 4.
St
aini
ng
of G
PA
by t
he c
ollo
idal
iro
n te
chni
que
in 4
-pm
-thic
k,
para
ffin-
embe
dded
re
nal
tissu
e.
A an
d B
show
de
nse,
ra
il-lik
e st
aini
ng
of t
he
F G
CW
(ar
rows
). C
, D
, E,
F s
how
glom
erul
i fro
m t
hree
diff
eren
t ne
phro
tic,
grad
e I
SLE
patie
nts
(cas
es 9
, 10,
12
of T
able
1). I
n C
, th
e re
dist
ribut
ion
of G
PA,
m
mai
nly
loca
lized
at
the
per
iphe
ry
of g
lom
erul
ar
loop
s (a
rrows
) is
vis
ible
. In
D
, se
gmen
tal
stai
ning
in
a m
arke
dly
scle
rose
d gl
omer
ulus
an
d in
E a
nd
F.
mz
com
plet
e di
sapp
eara
nce
of C
PA
are
seen
. A,
X3
00:
B,
x 10
00:
C.
D,
E, F
, x4
00.
i =i
;;
312 CAMUSSI ET AL.
FIG. 5. Photomicrographs of peripheral PMN obtained from unrelated normal donors (A) and (B) from a Grade I SLE patient (case 9 of Table I). The PMN were stained with fluorescein-conjugated rabbit anti-NCP. In A (example of score +3), there is staining of the cytoplasm (see also Fig. 2A). In B (example of score +l), a marked reduction of the cytoplasmic staining is observed. This figure is representative of 12 experiments performed in several patients with Grade I SLE A, B. x 1000.
indicated that NCP are released in the circulation during active stages of SLE (33). This finding is in agreement with the observation, reported in this paper, that in patients with acute episodes of SLE circulating PMN are depleted of their CP content, as compared to PMN of normal individuals.
The concomitant deposition of NCP and loss of fixed negative charges in GCW is consistent with the hypothesis that NCP may bind and neutralize GPA. Recent studies indicate that GPA are localized on the plasma membrane of endothelial and epithelial cells of GCW and, especially in the laminae rarae interna and ex- terna (8- 10). In view of the fact that lysozyme, a cationic protein, when injected into a normal rat binds and neutralizes GPA (14), it can be assumed that NCP, released from PMN during the acute stages of SLE, may also bind and neutralize GPA. Consistent with this interpretation is the linear and segmental staining pat- tern induced by fluorescein-conjugated NCP antibodies in the peripheral GCW of SLE patients, a pattern different from the granular and patchy staining obtained in the same kidneys with fluoresceinated antibodies to immunoglobulins and com- plement.
NCP deposits were detected in the glomeruli of six patients with diffuse prolif- erative glomerulonephritis (Class IV) and of one patient with focal proliferative glomerulonephritis (Class III) and were associated in 4/5 patients with grade I activity, but only in 2/5 patients with grade II and in l/4 with grade III. All patients showing glomerular NCP deposits had proteinuria (more than 3.5 g/24 hr) and, except one, all were nephrotic. In contrast, in 97 patients negative for NCP

NEUTROPHIL CATIONIC PROTEINS AND SLE NEPHRITIS 313
deposits, proteinuria was lower than 3.5 g/24 hr and no nephrotic syndrome oc- curred. The association between the deposition of NCP and active SLE, pro- teinuria as well as nephrotic syndrome suggests a pathogenetic relation between these events. However, while NCP binding was invariably associated with the loss of GPA (7/7 patients), this appeared more widespread than the NCP deposits. It is possible that the amount of NCP, sufficient to neutralize GPA, may be too low to be detected by immunofluorescence. It is also conceivable that cationic mediators other than NCP may have been released in the circulation and fixed by GCW. The most likely candidates are CP released from platelets. Platelet aggre- gation can be induced by IC (53) or by PAF (54, 55). Platelet could initiate the glomerular inflammatory (56) injury or amplify an inflammatory reaction initiated by NCP. Further studies are needed to clarify this issue.
The depletion of CP in circulating PMN, the presence of NCP deposits in GCW with the concomitant loss of GPA seems to suggest that NCP are released in V~VO from stimulated PMN and contribute to the pathogenesis of glomerular injury and proteinuria in patients with active SLE glomerulonephritis.
ACKNOWLEDGMENT
The authors wish to thank Professor G. A. Andres for his help.
REFERENCES I. Bennett, C. M., Glassock, R. J., Chang, R. L. S., Deen, W. M.. Robertson, C. R., and Brenner.
B. M., J. Clin. Invest. 57, 1287, 1976. 2. Boher, M. P., Bay& C., Humes, H. D., Glassock, R. J., Robertson, C. R., and Brenner. B. M..
.I. C’lin. Invest. 61, 72, 1978. 3. Brenner, B. M., Bohrer, M. P., Baylis, C., and Deen, W. M., Kidney Int. 12, 229, 1977. 4. Brenner, B. M., Hostetter, T. H., and Humes, H. D., N. EngI. J. Med. 298, 826, 1978. 5. Rennke, H. G., Cotran, R. S., and Venkatachalam, M. A., J. Cell Biol. 67, 638, 197.5. 6. Rennke. H. G., Pate], Y., and Venkatachalam, M. A., Kidney fnt. 13, 324. 1978.
7. Rennke, H. G., and Venkatachalam, M. A., Kidney Int. 11, 44, 1977. 8. Kamowsky, M. J., Amer. Rev. Med. 30, 213, 1979. 9. Kanwar, Y. S., and Farquahar, M. G., J. Cell Biol. 81, 137, 1979.
10. Kanwar, Y. S., and Farquahar, M. G., Proc. Nut. Acad. Sci. I/.$A 76, 1303, 1979.
11. Caulfield, J. P., and Farquhar, M. G., Proc. Nut. Acad. Sci. USA 73, 1646, 1976. 12. Nevins, T. E., and Michael, A. F., Kidney Int. 19, 553, 1981. 13. Seiler, M. W., Venkatachalam, M, A., and Cotran, R. S.. Science 189, 390, 1975. 14. Seiler, M. W., Rennke, H. G., Venkatachalam, M. A., and Cotran, R. S., L&. Invest. 36, 48,
1975. 1.5. Michael, A. F., Blau, E., and Vernier, R. L., Lab. invest. 23, 649, 1970. 16. Caulfield, J. P., and Farquahar, M. G., Lab. Invest. 39, 505, 1978. 17. Velosa, J. A., Glasser, R. J., Mevius, T. E., and Michael, A. F., Lab. Invest. 36, 527. 1977. 18. Kreisberg, J. I., Wayne, D. B.. and Kamowsky, M. J.. Kidne.v Int. 16, 290, 1979. 19. Kelley, V. E., and Cavallo, T., Lab. Imvest. 42, 59, 1980. 20. Kelley, V. E., and Cavallo, T., Lab. Invest. 37, 265, 1977. 21. Nachman, R. L., Weksler, B., and Ferris, B.,J. C/in. Invest. 51, 549, 1972. 22. Nachman, R. L., Weksler, B., and Ferris, B., J. Clin. Int*esr. 49, 274, 1970. 23. Patterson-Delafteld, .I., Martinez, R. J., and Lehrer, R. J., Infect. immun. 30, 180, 1980. 24. Ranadive, N. S., and Cochrane, C. G., J. Exp. Med. 128,605, 1966. 25. Ranadive, N. S., Sajnani, A. H., Alihuerka. K., and Movat, H. Z., Inr. Arch. Allergy &p/.
Immunol. 45, 880, 1973. 26. Glsson, I., and Venge, P., &and. J. Haematol. 9, 204. 1972.

314 CAMUSSI ET AL.
27. Camussi, G., Tetta, C., Bussolino, F., Caligaris-Cappio, F., Coda, R., Masera, C., and Segoloni, G., Int. Arch. Allergy Appl. Immunol. 62, 1, 1980.
28. Camussi, G., Mencia-Huerta, J. M,, and Benveniste, J., Immunology 33, 523, 1977. 29. Venge, I., and Olsson, P.. J. Inr~rrunol. 115, 1505, 1975. 30. Odeberg. H., and Olsson, P., J. C&n. int*r.sr. 56, 1 f 18, 1975. 31. Camussi, G., Tetta, C., Bussolino, F., Caligaris-Cappio, F., Coda, R., Masera, C., and Segoloni,
G., Int. Arch. Allergy Appl. Immunol. 64, 25, 1980. 32. Camussi, G., Aglietta, M., Coda, R., Bussolino, F., Piacibello. W., and Tetta, C., Immunology
42, 191, 1981. 33. Camussi. G., Segoloni, G., Stratta, P., Mencia-Huerta, J. M.. Ragni. R.. Piccoli. G.. and Vercel-
lone, A., Pror. Eur. Diul. Transplant. Assoc. 478, 1977. 34. Boyum, A., Stand. J. C/it!. Lab. Irrvest., Srrppl. 97, 21, 1968. 35. Yam, L. T., Li. C. Y., and Crosby, W. H., Amer. J. Pathol. 55, 283, 1971. 36. Loeffler, H., n “Nuentes Freib rg Symposium” (H. Merker. Ed.) p. 275. Springer-Verlag,
Berlin/Heidelberg/New York, 1963. 37. Camussi. G., Tetta, C., Coda, R., and Benveniste, J., Lab. lnvest. 3, 241, 1981. 38. Andrews, P., Biochem. J. 96, 595, 196.5. 39. Weber, K., and Osborne, M.. J. Biol. Cl7en-l. 244, 4406, 1969. 40. Stelos. P.. It7 “Handbook of Experimental Immunology” (D. M. Weir, Ed.), pp. 4-6, Blackwell,
Oxford, 1967. 41. Johnson, G. D., and Dorling, J., In “Techniques in Clinical Immunology” (R. A. Thompson,
Ed.), pp. 85-115, Blackwell, Oxford, 1977. 42. Coons. A. H., and Kaplan, M. H., J. Exp. Med. 91, 1, 1950. 43. Ouchterlony. 8., Acta Pu~hol. Miuwbiol. Stand. 26, 507, 1949. 44. Cohen, A. S., Reynolds. W. E., Franklin. E. C., Kutka, I. P.. Ropes, M. W., Schulman. L. E.,
and Wallance, S. L., Bull. Rheum. Dis. 21, 643, 1971. 45. Rothfield, N. F., Frangione, B., and Franklin, E. C., J. C/in. Inresr. 44, 62. 1965. 46. Aarden, L. A., De Groot, E. R., and Feltkamp, T. E. W.. Ann. N. Y. Acad. Sri. 254, 505, 1975.
47. Levinsky, R. J., and Soothill, J. F., C/in. Exp. Immunol. 29, 428, 1977. 48. Camussi, G., Caligaris-Cappio. F., Messina, M.. Coppo, R.. Stratta, P., and Vercellone, A., C/in.
Nephrol. 6, 280, 1980. 49. Appel. G. B., Silva. F. G.. Pirani, C. L., Meltzer. J. I., and Estes. D.. Medicine 57, 371. 1978. 50. Mowry, R. W., Lab. Invest. 7, 566, 1958. 51. Koffler, D.. Schur. P. H., and Kunkel, H. J., J. Exp. Med. 126, 607, 1967. 52. Weissman, G., Korchok, H. M., Perez, D., Smolen, J. E.. and Hoffstein, S. T., hr “Advances in
Inflammation Research” (G. Weissman et (II.. Eds.), p. 95, Raven Press, New York, 1979. 53. Mueller-Eckradt, C., and Luscher, E. F., Thromb. Diath. Haemorrh. 20, 821, 1968. 54. Benveniste, J., Henson. P. M., and Cochrane, C. G., J. E.x~. Med. 136, 1356, 1972. 55. Pinckard. R. N., McManus, L. M., Demopoulos, C. A., Halonen. M., Clark, P. O., Shaw, J. O.,
Kniker. W. T., and Hanahan, D. J., J. Reticuloendothel. Sot. 28 (Suppl.), 95. 1980. 56. Nachman, R. L.. and Polley, N., It7 “Advances in Inflammation Research” (G. Weissman et a/. ,
Eds.), p. 169, Raven Press, New York, 1979.
Received December 8. 1981: accepted with revisions March 13. 1982.




















