
Light-assisted 1, 4-dioxane degradation
14
~ Pergamon Chemosphere, Vol. 35, No. 11, pp. 2675-2688, 1997 © 1997 Elsevier Science Ltd All rights reserved. Printed in Great Britain 0045-6535/97 $17.00+0.00 PII: S0045-6535(97)00322-6 LIGHT-ASSISTED 1,4-DIOXANE DEGRADATION • .~¢ V.Maurino, P.Calza, C.Minero, E.Pehzzettl and M.Vincenti Dipartimento di Chimica Analitica, Universitk degli Studi di Torino, via P. Giuria 5, 10125 Torino - Italy (Receieed in Switzedand 19 March 1997; accepted 12 June 1997) ABSTRACT The oxidative degradation induced by light of 1,4-dioxane been investigated in homogeneous solution in the presence of inorganic peroxides (either hydrogen peroxide or peroxydisulfate) and under heterogeneous photocatalytic conditions in the presence of titanium dioxide. The effect of photon energy spectral distribution, pH, 02 and scavengers has been examined. The degradation mechanism involves common intermediates. Ethylene glycol diformate was observed as the main intermediate. In the presence of UV light peroxydisulfate showed high degradation efficiency. A tentative chain radical oxidation mechanism is postulated. The distinctive features of heterogeneous photocatalytic treatment were the good efficiency under simulated solar spectrum and the absence of inhibition of the degradation rate by HCO3-. The presence of peroxydisulfate enhances the TiO2 photocatalytic degradation rate. © 1997 Elsevier Science Ltd INTRODUCTION 1,4-Dioxane is a synthetic organic compound with no known natural sources [1]. It is used as industrial solvent [2] and found as by-product of several chemical processes involving ethylene glycol or ethylene oxide [3]• This compound appears to be persistent to degradation in the conventional industrial biotreatment processes as well as under aerobic or anaerobic conditions occurring in the natural environment. Owing to its physical properties, 1,4-dioxane is expected to migrate rapidly into the aquifer [4,5]. There is a growing concern about 1,4-dioxane since it is a suspected carcinogen [1,6]. Among the physical technologies capable of removing 1,4-dioxane, the most effective is distillation whereas carbon adsorption and air stripping are inadequate [1]. Due to the economic costs of distillation, chemical treatments should be considered as an alternative. Nevertheless, the oxidation with chlorine or permanganate is ineffective, or could even lead to the formation of more toxic compounds [7-9]. 2675
-
Upload
independent -
Category
Documents
-
view
8 -
download
0
Transcript of Light-assisted 1, 4-dioxane degradation

~ Pergamon Chemosphere, Vol. 35, No. 11, pp. 2675-2688, 1997 © 1997 Elsevier Science Ltd
All rights reserved. Printed in Great Britain 0045-6535/97 $17.00+0.00
PII: S0045-6535(97)00322-6
LIGHT-ASSISTED 1,4-DIOXANE DEGRADATION
• .~¢ V.Maurino, P.Calza, C.Minero, E.Pehzzettl and M.Vincenti
Dipartimento di Chimica Analitica, Universitk degli Studi di Torino, via P. Giuria 5, 10125 Torino - Italy
(Receieed in Switzedand 19 March 1997; accepted 12 June 1997)
A B S T R A C T
The oxidative degradation induced by light of 1,4-dioxane been investigated in homogeneous solution
in the presence of inorganic peroxides (either hydrogen peroxide or peroxydisulfate) and under heterogeneous
photocatalytic conditions in the presence of titanium dioxide. The effect of photon energy spectral
distribution, pH, 02 and scavengers has been examined. The degradation mechanism involves common
intermediates. Ethylene glycol diformate was observed as the main intermediate. In the presence of UV light
peroxydisulfate showed high degradation efficiency. A tentative chain radical oxidation mechanism is
postulated. The distinctive features of heterogeneous photocatalytic treatment were the good efficiency under
simulated solar spectrum and the absence of inhibition of the degradation rate by HCO3-. The presence of
peroxydisulfate enhances the TiO 2 photocatalytic degradation rate. © 1997 Elsevier Science Ltd
I N T R O D U C T I O N
1,4-Dioxane is a synthetic organic compound with no known natural sources [1]. It is used as industrial
solvent [2] and found as by-product of several chemical processes involving ethylene glycol or ethylene
oxide [3]• This compound appears to be persistent to degradation in the conventional industrial biotreatment
processes as well as under aerobic or anaerobic conditions occurring in the natural environment. Owing to its
physical properties, 1,4-dioxane is expected to migrate rapidly into the aquifer [4,5]. There is a growing
concern about 1,4-dioxane since it is a suspected carcinogen [1,6].
Among the physical technologies capable of removing 1,4-dioxane, the most effective is distillation
whereas carbon adsorption and air stripping are inadequate [1]. Due to the economic costs of distillation,
chemical treatments should be considered as an alternative. Nevertheless, the oxidation with chlorine or
permanganate is ineffective, or could even lead to the formation of more toxic compounds [7-9].
2675

2676
Advanced oxidation processes (e.g. ozone, Fenton's reagent, H202/ozone ) have been evaluated
showing some degree of effectiveness. Neither hydrogen peroxide nor ozone [10, 11 ] alone readily oxidize
1,4-dioxane. Using Fenton's reagent the concentration of 1,4-dioxane and the related total organic carbon
(TOC) content was reduced within 10 h by 97% and 11%, respectively [8]. The combination H202/ozone
was found capable to oxidize 1,4-dioxane to more readily biodegradable oxidation products [11].
The photocatalytic behavior of saturated heterocyclic compounds has been reported [12]. However,
data concerning 1,4-dioxane are lacking. The present research examined the 1,4-dioxane degradation under
photocatalytic conditions on titanium dioxide [13-15], and under processes induced by light in the presence
of inorganic peroxides (either hydrogen peroxide or peroxydisulfate).
E X P E R I M E N T A L
Materials and Reagents
1,4-Dioxane and 1,4-dioxane-2,3-diol were obtained from Merck (p.a.) and Aldrich (98%),
respectively, and used as received. Aqueous stock solutions were prepared daily in deionized-doubly distilled
water. All other chemicals, except ethylene glycol diformate (EGDF), were commercially available (at least
analytical grade) and used without further purification. EGDF was purposely synthesized. A mixture of
ethylene glycol (EG, 1 mole), formic acid 85% (3 moles, 162.5 g) and sulfuric acid 98% (5 mL) was heated
under reflux until 3.3 moles of water were distilled. The mixture was then neutralized with solid NaHCO3,
treated with 100 mL of Na2SO 4 1.0 M and extracted with 100 mL of ethyl acetate. The organic layer was
washed with a solution of Na2SO 4 1.0 M / NaHCO 3 0.5 M. After elimination of ethyl acetate in a rotary
evaporator under reduced pressure, the residue was vacuum distilled (51 °C, 0.2 torr). The desired compound
was obtained with 95% GC purity, the remainder was ethylene glycol monoformate (EGMF). Electron
Impact Mass Spectrometry (El-MS) gave spectra for EGDF characterized by m/z 29 (48%), 31 (100%), 43
(27%), 44 (44%), 60 (34%), 72 (6%), 74 (0.3%), 103 (0.3%), 118 (0.1%); for EGMF characterized by m/z 29
(36%), 31 (100%), 44 (26%), 60 (60%), 73 (5%), 75 (0.2%); IH NMR spectra presented two singlet signals at
4.16 ppm (100%) and 7.86 ppm (50.9%) (reference CHC13 7.27 ppm). Titania TiO 2 P25 (BET area ca 55
m2/g, Degussa A.G., Frankfurt, Germany) was irradiated in aerated aqueous suspension for at least 12 hours
and washed with deionized-doubly distilled water before use.
Irradiation Experiments
The slurries were prepared by suspending in water the required amount of photocatalyst powder using
sonication. Substrate aqueous solution was then added in the required amount. The measured pH of a water
suspension containing 200 mg L -I of TiO 2 is 5.5. In the experiments carried out at pH 10 the required
adjustment was made adding drops of 1 M NaOH. The irradiation experiments were carried out in Pyrex
glass (with cut-off at 295 nm) or quartz cylindrical cells (4.0 cm diameter, 2.3 cm height), containing 5 mL of
the aqueous suspension of the photocatalyst, powder and substrate, using a 1500 watt Xenon lamp (Solarbox,
CO.FO.MEGRA, Milan Italy) equipped with a 340 nm cut off filter simulating AMI solar light.
Homogeneous degradation with the peroxide/UV systems were carried out in the same cells made from Pyrex
(L>295 nm) or quartz (lamp full spectnma), using the Xenon lamp without any cut-off filter. The irradiance
spectrum and the cells have been described elsewhere [16,17]. The total photonic fluxes in the cells were:
3.45.10 -7, 2.71.10 .7 and 1.92-10 .7 Einstein s -1 in the range 280-400 rim, 295-400 run and 340-400 rim,

2677
respectively. The cell temperature during irradiation was 60 °C. The experiments in the absence of air were
prepared by purging the closed irradiation cells containing either the TiO 2 suspension or water with He.
Then, the required volume of substrate stock solution and, when relevant, the peroxide solution were injected
in the cell with a microsyringe. During irradiation the slurries were magnetically stirred.
o 1 0
O 0.8
d ,-- 0 .6
x 0 0.4
I O.2
0
/ I
"" A
" - • . . . . 7"/ e I ~'1~},, n: (a) pH 5.5 ,
. ~ . o: (a) pH 11 " ~ , \ [ 3 ~ o -k: (b) pH 5.5 " ,
~ r \ \ ~ *: (b) He, pH 5.5 *
. . . .
0 5 10 15 20 50 250
o 1 0
O 0.8
d . - 0 .6
o 0 .4
I 0.2
0
B .. v : (a} HCOa- 0.1M
• . 4: (a) S20a 2- 0.1M \ \ ' " . . <>: (a) He. S2082 - 0.1M <~ '"V.
.%.°.
i : y . . . . , . . . . .
5 10 15 20 25
Irradiation time, minutes
Figure 1. Light assisted 1,4-dioxane degradation over TiO 2. Condition: C O = 4.0x10 -4 M, (a)
TiO 2 P25 200 mg L -l, radiation of E>340 nm; (b) TiO 2 P25 200 mg L -1, xenon lamp full
spectrum.
Analytical Determinations.
After irradiation the suspensions were filtered through a solvent resistant 0.45 I~m flter membrane
(Millex HV, Millipore). The determination of 1,4-dioxane was carried out with a Purge-and-Trap-GC-FID
system (Tekmar LSC 2000) equipped with a VOCARB 2000 trap (SUPELCO). 0.5 mL of the aqueous
sample was diluted with 4.5 mL ofNa2SO 4 2.0 M and stripped with He in the flitted purge sampler of 5-mL
capacity. GC determination was performed on a Varian STAR 3400 CX equipped with a SPB5 capillary

2678
column of 60 m length, 0.32 mm I.D. and I ~tm film thickness (SUPELCO). EGMF and EGDF were
identified with a GC-MS system (GC: Varian STAR 3400 CX. MS: Finnigan MAT 95 Q) in El mode, and
quantified on a GC-FID (Varian STAR 3400 CX) using a direct injection of the aqueous solution (1 I~L).
Both system were equipped with a capillary column of 30 m length, 0.32 mm I.D., coated with PAG of 0.25
~m film thickness (SUPELCO).
/=,
1 .................. <> A 0.8
0.6 n : (c) " : : :
1 't,', \ , ,: (d) -,, 0 0.4 -] ,\~x,. ~ v: (c), He _ "",,,
2- 0 0 2 4 6 20 60
o 1 0
tO 0.8
,- 0.6
x o 0.4
I 0.2 ,¢
0
i ¢
'~, ~ ' ~ ~ : (e), HCO 3- 0.1M %
0 10 20 30 40 50 60 200 400
Irradiation time, minutes
Figure 2. Light assisted 1,4-dioxane degradation in the presence of peroxides. Condition: C O =
4.0x10 -4 M, (c) $2082- 0.01 M, full xenon lamp spectrum; (d) $2082- 0.1 M, full xenon lamp
spectrum; (e) H202 0.1 M, radiation of L>295 nm; (f) H202 0.01 M, full xenon lamp spectrum.
A: $2082-/UV; B: H202/UV.
The temporal variation of formaldehyde, glycolaldehyd¢ and glyoxal in the degradation experiments
was monitored by HPLC-UV (Hitachi model L6200 pump with I.A200 UV-Vis detector) at Z=360 nm after
precolumn derivatization with 2,4-dinitrophenylhydrazine [18,19] using a Lichrocart Cl$ column (Merck,
125 mm length, 4 mm I.D., 5 ~m packing). The determination of glyoxal required a modification to the
original procedure, consisting in the r~placernent of CH3CN with tetrahydrofuran both in the HPLC eluant

2679
and in the derivatizing reagent. Formic, oxalic and glycolic acids were measured by suppressed ion
chromatography employing a Biotronik IC 5000 apparatus equipped with a Dionex AS 4A separation column
and an alkaline buffer eluant containing K2CO 3 (1.5 mmol L -l) and KHCO 3 (1.5 mmol L -l) at a flow rate of
1.5 mL min d. Total Carbon (TC) and Inorganic Carbon (IC) determinations were carded out on a Shimadzu
model TOC-5000 total carbon analyzer.
Table 1. Pseudo-first order kinetic data for the light-assisted 1,4-dioxane degradation.
# TiO 2 (mg L -1)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
200 (pH 5.5)
200 (PH 11)
200 (pH 5.5)
200 (pH 8.0)
200 (pH 5.5)
200 (pH 5.5)
200 (pH 5.5)
Peroxide
S2082- S2Os 2"
H202 H202
H202
H202
H202
$2082- S2082- $2082- $2082" S2082"
Concentration ! (M)
0.1
0.1
0.01
0.1
0.1
0.1
0.01
0.1
0.01
0.01
0.01
0.I
Radiation
>340nm
>340rim
full spectrum
>340rma
>340nm
>340nm
>340nm
>295nm
>295nm
>295nm
>295nm
full spectrum
>295nm
full spectnlm
full spectrum
full spectrum
full spectrum
Scavenger
HCO 3- (0.1 M)
]HCO 3- (0.1 M)
HCO 3- (0.1 M)
gaseous phase
air air
air
air
air
He
He
air
air
He
air
air
air
air
He
air
air
Kinetic constant ~min -I)
0.17
0.20
0.29
0.18
0.27
0.50
0.0028
0.018
0.040
0.040
0.0047
0.12
no reaction
0.44
0.86
0.060
1.57
RESULTS AND DISCUSSION
Light assisted degradation of 1,4-dioxane was carried out in the presence of either hydrogen peroxide or
sodium peroxydisulfate, and under photocatalytic conditions in aqueous suspensions of TiO 2 powder. Typical
concentration profiles are shown in Figure I for TiO 2/hv, and in Figure 2 for S2082-/UV and H202/UV
treatments, respectively. The degradation processes follow quite closely (pseudo)first order kinetics over at
least two half-lives. Table 1 summarizes all the experimental conditions explored along with the kinetic
constants calculated using a best fit of the experimental data. Measurements of peroxo species concentration
showed that the peroxide depletion is less than 10% of its initial concentration after substrate disappearance.
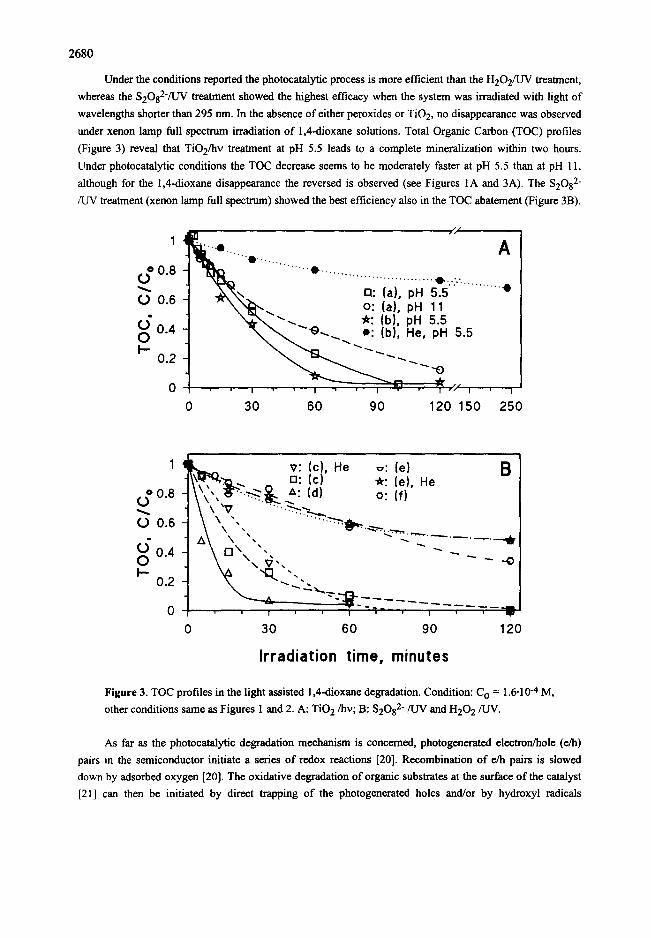
2680
Under the conditions reported the photocatalytic process is more efficient than the H202/UV treatment,
whereas the $2082-/UV treatment showed the highest efficacy when the system was irradiated with light of
wavelengths shorter than 295 nm. In the absence of either peroxides or Tie2, no disappearance was observed
under xenon lamp full spectrum irradiation of 1,4-dioxan¢ solutions. Total Organic Carbon (TOC) profiles
(Figure 3) reveal that TiO2/hv treatment at pH 5.5 leads to a complete mineralization within two hours.
Under photocatalytic conditions the TOC decrease seems to be moderately faster at pH 5.5 than at pH I l,
although for the 1,4-dioxane disappearance the reversed is observed (see Figures 1A and 3A). The $2082-
/UV treatment (xenon lamp full spectrum) showed the best efficiency also in the TOC abatement (Figure 3B).
f,3 0.8
0 0.6
(-)" 0 . 4 0 I---
0 .2
• o "; ' A " 'O"'.,. , .
~ i O'" ....... • ............................ O . . ' . ' . .
I::1: ( a ) , pH 5.5 . . . . . . . . •
" \'N;I-, o" (a}, pH 11 X~,,'~'--..~ ~: (b), pH 5.5
' ~ " "0 . . . . *: (b), He, pH 5.5
, , , , , , , , i r . , , q'~ - ; " i , i
0 30 60 90 120 150 250
t j ° 0.8
O 0.6
O" 0.4 0 I.-
0 .2
0
,•,:•.,. V : ( c } , H e ,~: ( e ) B n: {c) -g':. (e), He
(d) o: of) \ \ "v \ \ ", ...... " ~ " t ~
0 30 60 90 120
Irradiation time, minutes
Figure 3. TOC profiles in the light assisted 1,4-dioxanc degradation. Condition: C O = 1.6.10 -4 M,
other conditions same as Figures 1 and 2. A: T ie 2/he; B: $2082-/UV and H202/UV.
As far as the photocatalytic degradation mechanism is concerned, photogenerated electron/hole (e/h)
pairs in the semiconductor initiate a series of redox reactions [20]• Recombination of e/h pairs is slowed
down by adsorbed oxygen [20]. The oxidative degradation of organic substrates at the surface of the catalyst
[21] can then be initiated by direct trapping of the photogcnerated holes and/or by hydroxyl radicals

2681
generated at the TiO 2 surface (either free or bound). The oxidative degradation of organic substrates with the
H202/UV treatment is initiated by hydroxyl radicals generated through the photodeoomposition of hydrogen
peroxide [22]. UV inadiation of solutions containing $2082- induces a photolytie decomposition generating
the radical anion SO4 °-, which reacts primarily as electron abstractor. In aqueous solution this radical is in
equilibrium with the hydroxyl radical [23].
0 . 4
E
c" 0.3 0
o 2 e,-
o 0.1 e,- o
0 0
o: EGDF n: Glycolic acid v: Formic acid
. . . ~ . - . . ~ , ~ A: Formaldehyde . " " • . . . . . : . . . . . . i . , . . . . .
m l = T - , L - I I 1 I 1 I
0 10 20 30 40 50 60
Irradiation time, minutes
F i g u r e 4. Intermediates evolution in the photocatalytic degradation of 1,4-dioxane. Conditions:
initial substrate concentration 4.0.10 -4 M, TiO 2 P25 200 mg L -l, pH 5.5. The dotted line shows
the calculated concentration vs. time profile for EGDF. Calculations were carried out by using a
model consisting of two consecutive pseudo-first order processes. See text for details.
Independent of the nature of the oxidizing species (OH* free radical, for H202/UV, hole and]or trapped
OH*, for TiO2/hv, SO4"- radical, for S2082-/UV), the early steps of the degradation process (TiO2/hv pH 5.5
and peroxides/UV) lead to the formation of substantial amounts of EGDF, indicating an oxidative ring
opening mechanism. It is worth noting that the maximum concentration of EGDF is about 75% of the initial
dioxane concentration. Synthesis of EGDF by controlled oxidation of 1,4-dioxane in the presence of air and
cobalt acetate has been reported [24]. The EGDF formation could require some intermediate steps before the
ring fission. 1,4-Dioxanol was observed in the anodic oxidation of 1,4-dioxane in acidic medium [25]. In the
present case 1,4-dioxanol was not detected, probably owing to its little stability toward further oxidation
leading to very low steady-state concentrations (under 0.1 mg L-I). A kinetic model consisting of two
consecutive pseudo-first order processes leads to the calculated evolution of EGDF concentration showed as
dotted line in figure 4. The comparison with the experimental profiles recorded for EGDF (Figure 4) shows
that the formation of EGDF is slightly delayed with respect to the calculated profile, but the concentration
maximum is fairly well reproduced. The kinetic constants used in the calculation are 0.17 and 0.026 min-I for
the first and second process, respectively (see Table 1 entry 1 and the discussion below about the
photocatalytic degradation of EGDF).
From the above arguments it follows that oxidation pathways involving dioxanediols with the hydroxyl
groups on non-adjacent carbon atoms (e.g. 1,4-dioxane-2,5-diol and 1,4-dioxane-2,6-diol) are of limited
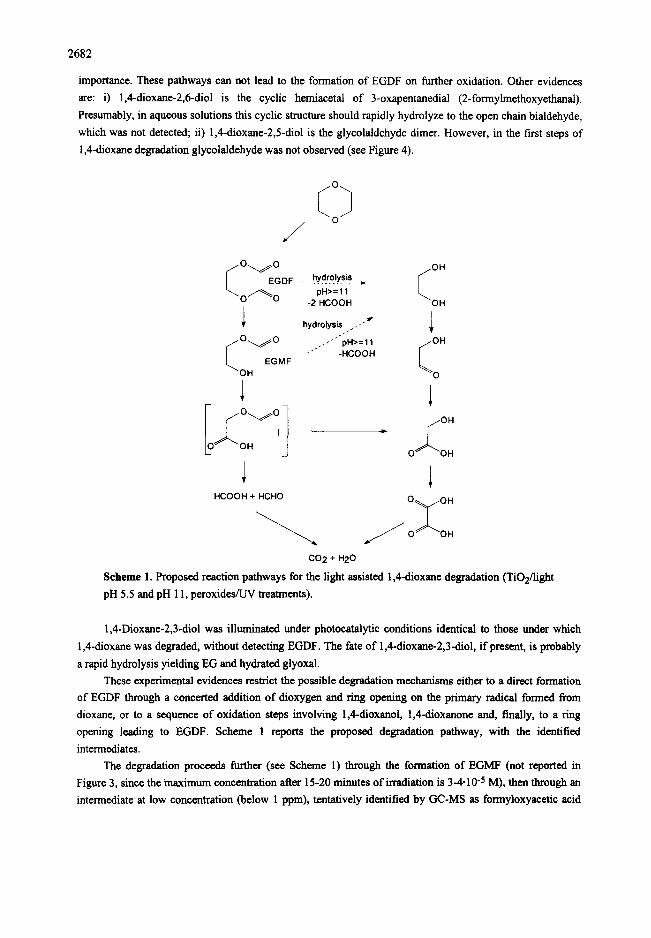
2682
importance. These pathways can not lead to the formation of EGDF on further oxidation. Other evidences
are: i) 1,4-dioxane-2,6-diol is the cyclic hemiacetal of 3-oxapentanedial (2-formylmethoxyethanal).
Presumably, in aqueous solutions this cyclic structure should rapidly hydrolyze to the open chain bialdehyde,
which was not detected; ii) 1,4-dioxane-2,5-diol is the glycolaldehyde dimer. However, in the first steps of
1,Cdioxane degradation glycolaldehyde was not observed (see Figure 4).
OWO OH
EG DF _ .h?!!.?‘~?.i?. +
0-o pH>=ll
i
-2 HCOOH OH
hydrolysis ,,e**
(
OWO ,_*’ /$+=,,
i OH
-HCOOH EGMF
OH c 0
I I
O-0
!n II
OH
0 OH * 1 0 OH
1 i HCOOH + HCHO
\ /XI:
CO2 + Hz0
Scheme 1. Proposed reaction pathways for the light assisted 1,Cdioxane degradation (TiOl/light
pH 5.5 and pH 11, peroxides/W treatments).
1,4-Dioxane-2,3-diol was illuminated under photocatalytic conditions identical to those under which
1,4-dioxane was degraded, without detecting EGDF. The fate of 1,4-dioxane-2,3-diol, if present, is probably
a rapid hydrolysis yielding EG and hydrated glyoxal.
These experimental evidences restrict the possible degradation mechanisms either to a direct formation
of EGDF through a concerted addition of dioxygen and ring opening on the primary radical formed from
dioxane, or to a sequence of oxidation steps involving 1,Cdioxanol, 1,Cdioxanone and, finally, to a ring
opening leading to EGDF. Scheme 1 reports the proposed degradation pathway, with the identified
intermediates.
The degradation proceeds further (see Scheme 1) through the formation of EGMF (not reported in
Figure 3, since the maximum concentration after 15-20 minutes of irradiation is 3-4-10-5 M), then through an
intermediate at low concentration (below 1 ppm), tentatively identified by GC-MS as formyloxyacetic acid

2683
(I), which in turn transforms either to gly¢olic acid and subsequently to oxalic acid by deformylation, or to
formic acid and formaldehyde by decarboxylation. Apart from the time scale, the intermediates observed in
the degradation process with TiO2/light and with peroxidesRYV are very similar (Figure 5).
~ 0 . 4 "~
0.3
~ 0.2
~ 0.1
8 o:
o: EGDF A .. ...................... v: Formic acid
V " 'v ............ A: Formaldehyde j° ' " . . . . . . .
10 20 30
o.4 1 o: EGDF B 0.3 -I : Formaldehyde
'~, .~ o: Oxalic Acid
~ 0.1
8oi . . . . . . . . . . . . . . . . . . . . .
0 60 120 180 240
Irradiation time, minutes
Figure 5. Intermediates evolution in the light-assisted degradation of 1,4-dioxane: A) in the
presence of $2Os2- 1.0.10 -2 M and with UV irradiation (full spectrum); B) in the presence of
H202 1.0.10 .2 and UV irradiation (k>295 nm)
Figure 6 shows the decrease of EGDF concentration together with its TOC profile in the photocatalytic
degradation at pH 5.5. It is worth to note that the kinetic constant of EGDF degradation (0.026 rain q) is
substantially lower than that of 1,4-dioxane. Also in this case an almost complete mineralization is reached
within two hours, but the EGDF and TOC profiles are much more close together than those for 1,4-dioxane.
This observation suggests that i) the intermediates produced from EGDF degradation are very quickly
mineralized; ii) EGDF oxidation is the slow step of 1,4-dioxane degradation. Point i) was recently discussed
[26] for pentachlorophenol by considering a sequence of consecutive reactions. If successive kinetic constants
are much lower than that of the original substrate degradation or have all the same value, CO 2 should be

2684
produced at times much longer than that of original substrate disappearance (depending on the number of
intermediate stages), showing a sigmoidal behavior [27]. Alternatively, if all kinetic constants are much
larger than that of the original substrate degradation, the normalized rate of TOC abatement equals the rate of
original substrate abatement. In this case the sigmoidal function of TOC vs. time is replaced by an
exponential decay behavior [26], as it is experimentally observed here.
o
o. 0 I -
Ll.
W
& 1 O: TOC 0.8 t " ~ \ \ 13: EGDF
0.6 - "~ ,~ \ \ \
0.4 - \-..
0
0 30 60 90 120
Irradiation time, minutes
Figure 6, Photocatalytic degradation of EGMF. Conditions: initial substrate concentration 4.0.10-
4 M, TiO 2 P25 200 mg L -l, pH 5.5.
In basic media under photocatalytic conditions the reaction pathways undergo a change. As noted
earlier the (pseudo)first order constant (0.20 min -1) is fairly larger than in neutral/slightly acidic media. In
contrast the TOC decrease is slower than in acidic media. Organic intermediates exhibiting a negative charge
at pH 11 (e.g. carboxylic acids) are formed during the degradation, and it is well-known that their further
degradation could be inhibited at pH > pHzp c of the catalyst [28]. In addition, also reductive (and
competitive) pathways can play a role. This leads to a steady state condition between the C 1 and C 2
compounds. The steady state is due to redox cycling of these carbon species over the photoeatalyst [29]. At
pH 11, in the early degradation steps, EGDF and EGMF formation were not observed (at least above the
estimated detection limits of 0.1 mg L-I). Yet, it was observed the appearance of relatively small quantities of
EG, and relatively large amounts of formaldehyde, glycolaldehyde, formic and glycolic acid (Figure 7).
Separated experiments showed that EGDF is quickly hydrolyzed to EG and formate in dilute aqueous
solutions at pH > 11 (>95% in 90 seconds). Once formed under photocatalytic conditions, EGDF is
quantitatively hydrolyzed to EG and formate. Scheme I suggests a reaction pathway able to justify these
observations.
As far as the peroxide treatment is concerned, it is worth to note that the kinetic behavior of the 82082-
/UV system is quite different from that of H202/UV. In the first case no reaction was detected when the
solution (in the presence of air) was exposed to radiation of wavelengths longer than 295 nm. However the
$2082-/UV system is the most efficient when irradiated at wavelengths shorter than 295 nm. This evidence
could not be explained in terms of the reactivity of the radical anion SO4 °- toward organic substrates. The

2685
second order kinetic constants for the reaction of S04"- with alcohols and ethers are two order of magnitude
lower than the kinetic constants for the reaction between hydroxyl radicals and the same substrates [30]. For
1,4-dioxane in water the reported kinetic constants are 1.8.109 M-Is -1 and 1.6.107 M-Is -I for OH" and SO4"-,
respectively [31].
0.5
~ 0 . 4 g "~ 0.3
0.2
0.1 o (9 0
o: EG e: Glycolaldehyde El: Glycolic acid O: Oxalic acid A: Formaldheyde
.V.."V'.... v: Formic acid
0 30 60 90 120
Irradiation time, minutes
Figure 7. Intermediates evolution in the photocatalytic degradation of 1,4-dioxane at pH 11.
Conditions: initial substrate concentration 4.0.10 .4 M, TiO 2 P25 200 mg L "1 .
In the presence of S2Os2"/UV, dioxygen showed a quite noticeable inhibition on the degradation
process (Figure 2A and Table 1, entry 14 and 15). The H202/UV treatment was scarcely affected by the
presence of oxygen (Figure 2B and Table 1, entry 9 and 10), whereas it was necessary for the TiO2/hv
treatment (Figure 1 and Table 1, entry 1 and 7). As already pointed out, dioxygen act as electron scavenger in
TiO 2 mediated photocatalytic degradation, slowing down electron-hole recombination. However, the addition
of peroxydisulfate to the TiO2/hv treatment caused a substantial increase in the rate of degradation, and the
presence of oxygen showed an inhibitory effect (Figure 1A and Table 1, entry 5 and 6). Extinction
coefficients and quantum yields for the photodissociation of peroxydisulfate and hydrogen peroxide are quite
similar [32,33]. Even if the SO4"- converts very rapidly to OH" reacting with water or OH-, one could expect
an upper limit for the degradation rate with $2082-/UV equal to the rate observed with the H202/UV
treatment. Yet, we observed a ratio between the degradation rates for $2082-/UV and H202R.IV treatments of
3.7 and 7.2 in the presence and in the absence of O2, respectively (see Table I, entry 12, 14 and 15).
These experimental evidences could be rationalized assuming that in the $2082-/UV system some
organic radicals react with peroxydisulfate regenerating the SO4"- radical, according to reactions (1-3).
hv S2082- ~ 2 SO4"- (1) SO4"- + RH 2 > SO42- + H + + RH" (2)
RH" + $2082- ~ R + SO42" + H + + SO4"- (3)

2686
The net result is a photoinitiated radical chain reaction between the peroxydisulfate and the organic
substrate. Taking into account the above mentioned values for the second order kinetic constants of the
reaction of SO4 °- and OH" with 1,4-dioxane, chain lengths of 810 and 410 propagation steps (in the absence
and in the presence of oxygen, respectively) follow from the observed ratios between the degradation rates
for the $2082-/UV and H202/UV systems. According to this hypothesis 02 is inhibitory due to its ability to
terminate the radical chain reactions.
The higher efficiency ofTiO 2 with respect to H202, for both initial compound and TOC disappearance,
could be attributed to: i) the presence of an electron scavenger rises the steady-state concentration of holes,
and related bound OH" radicals. However, their concentration could not be greater than the number of
incident photons and likely equal to [OH'] or [SO4 °-] formed in homogeneous solutions containing peroxide.
This imply that the degradation is driven not only by primary species (bound OH" or hole), but also by
secondary oxidants like the superoxide ion, generated through electron scavenging by 02 [34]; ii) the wider
light spectrum absorbed by TiO 2. The band gap absorption edge of the Ti02 powder (390 nm) is located at a
wavelength ca 70 nm higher than the appearance of the H202 absorption band.
The efficacy of these light-assisted degradation processes in the presence of hydrogen carbonate, a
common anion in natural and waste water, was also tested (Figure 1 and table 1, entry 4, 1! and 16). When
the TiO2/hv treatment is considered, the kinetic of the primary degradation process is slightly affected,
indicating a good efficiency of the process also in these media. We observed a strong inhibition of the
H202/UV and S2082-/UV treatments when HCO 3- was present (Figure 1B and C). In these oxidation
processes the oxidizing species are scavenged by HCO 3- in homogeneous solution, leading to the formation
of CO3 °" [35]. Second order kinetic constants for the reaction of HCO 3- with SO4 °- and OH" are,
respectively, 9.1"106 and 8.5"106 M-ls -1 [30]. The rate decrease can be explained on the basis of the reduced
reactivity of these radicals toward organic substrates according to reaction (4-6), where RH 2 is an organic
substrate of structure related to the presently investigated substances (primary and intermediates).
CO3 °- + RH 2 ) HCO 3- + RI--I ° (4)
SO4"- + RH 2 ) SO42- + H + + RH ° (5)
OH" + RH 2 ) H20 + RH" (6)
Second order kinetic constants for the reactions 4, 5 and 6 are of the order of 105, 107 and 109 M-Is -1
respectively [30]. In the case of TiO 2 the photocatalytic degradation occurs at the surface where HCO 3- is
excluded because of the negatively charged surface (pHpz c = 6.3 for TiO2) [28].
Acknowledgment. The authors are very grateful for the financial support from CIEMAT, MURST and
CNR.
1.
REFERENCES
p-Dioxane, In Reviews of Environmental Contamination and Toxicology (Edited by G.W. Ware), vol.
106, pp. 113-121. Springer Verlag, New York (1988).

2687
2. D.E. Keeley, Ethers, In Kirk-Othmer Encyclopedia of Chemical Technology (3rd Edn., Edited by
G.T. Seaborg, H.F. Mark, C.G. Overberger and D.F. Othmer), vol. 9, p. 381. J.Wiley and Sons, New
York (1979).
3. A.V. Popoola, Mechanism of reaction involving the formation of dioxane byproduct during the
production ofpoly(ethylene terephthalate), J. Appl. Polym. Sci. 43, 1875-1877 (1992).
4. F.B. DeWaUe, B. Foppe and E.S.K. Chian, Detection of trace organics in well water near a solid
waste landfill, J. Am. Water Works Assoc. 73, 206-210 (1981).
5. S. Lesage, R.E. Jackson, M.W. Priddle and P.G. Riemann, Occurrence and fate of organic solvent
residues in anoxie groundwater at the Gloucester landfill, Canada, Environ. Sci. Technol. 24, 559-566
(1990).
6. R. Hartung, Health and Environmental Effects Assesment for 1,4-Dioxane, Gelrnan Sciences, Ann
Arbor, Mi (1989).
7. M.J. McGuire, I.H, Suffet, J.V. Radziul, Assessment of unit processes for the removal of trace
organic compounds from drinking water, 3". Am. Water Works Assoc. 10, 565-572 (1978).
8. G.M. Kl~cka and S.J. Gonsior, Removal of 1,4-dioxane from waste water, J. Hazard. Mater. 13, 161-
168 (1986).
9, Y.-T. Woo, B.J. Neuburger, J.C. Arcos, M.F. Argus, K. Nishiyama, G.W. Griffin, Enhancement of
toxicity and enzyme-repressing activity ofp-dioxane by chlorination. Stereoselective effects, Toxicol.
Lett. 5, 69-75 (1980).
10. J. Hoign~, H. Bader, Rate constants of reaction of ozone with organic and inorganic compounds in
water I. Non dissociating organic compounds, Water Res. 17, 173-183 (1983).
11. C.D. Adams, P.A. Scanlan, and N.D. Secrist, Oxidation and biodegradability enhancement of 1,4-
dioxane using hydrogen peroxide and ozone, Environ. Sci. Technol. 28, 1812-1818 (1994).
12. S. Doherty, C. Guillard, P. Pichat, Kinetics and products of the photocatalytic degradation of
morpholine (tetrahydro-2H-l,4-oxazine) in TiO 2 aqueous suspension, J. Chem. Soc. Faraday Trans.
91, 1853-1859 (1995).
13. Photocatalysis. Fundamental and Applications (Edited by N. Serpone and E. Pelizzetti), J.Wiley and
Sons, New York, (1989).
14. D. Bahneman, J. Cunningham, M.A. Fox, E. Pelizzetti, P. Pichat and N. Serpone, Photocatalytic
treatment of waters, In Aquatic and Surface Photochemistry (Edited by G.R. Helz, R.G. Zepp and
D.G. Crosby), pp. 261-316. Lewis Publ., Boca Raton (1994).
15. M.R. Hoffmann, S.T. Martin, W. Choi and D.W. Bahnemarm, Environmental application of
semiconductor photocatalysis, Chem. Rev. 95, 69-96 (1995).
16. E. Pelizzetti, C. Minero, V. Maurino, A. Sclafani, H. Hidaka and N. Serpone, Photocatalytic
degradation ofnonylphenol ethoxylated surfactants, Environ. Sci. Technol. 23, 1380-1384 (1989).
17. C. Minero, E. Pelizzetti, S. Malato and J. Blanco, Large solar plant photocatalytic water
decontamination: degradation ofpentachlorophenol, Chemosphere 26, 2103-2119 (1993).
18. A. Larsen, N.A. Jentott and T. Greibrokk, Determination of ppb levels of formaldehyde in air, Sci,
Total Environ. 120131, 261-269 (1992).
19. S. Steinberg and I.R. Kaplan, The determination of low molecular weight aldehydes in rain, fog and
mist by reversed phase liquid chromatography of the 2,4-dinitrophenylhydrazone derivatives, Int. J.
Environ. Anal. Chem. 18, 253-266 (1984).

2688
20.
21.
22.
23.
E. Pelizzetti and C. Minero, Mechanism of the photooxidative degradation of organic pollutants over
titanium dioxide particles, Electrochim. Acta 38, 47-55 (1993).
C. Minero, F. Catozzo and E. Pelizzetti, Role of adsorption in photocatalyzed reactions of organic
molecules in aqueous TiO 2 suspensions, Langmuir 8, 481-486 (1992).
O. Legrini, E. Oliveros and A.M. Braun, Photochemical processes for water treatment, Chem. Rev.
93, 671-698 (1993).
M.S. Tsao and W.K. Wilmarth, The aqueous chemistry of inorganic free radicals. I. The mechanism
of the photolytic decomposition of aqueous persulfate ion and evidence regarding the sulfate-
hydroxyl radical interconversion equilibrium, J. Phys. Chem. 63, 346-352 (1959).
24. H.W. Fleming (to Phillips Pert. Co.), U.S. 2,471,570 (May 31, 1949), Ethylene diformate from
dioxane, Chem.Abstr. 1949, 7503.
25. B. Wermeckes and F. Beck, Anodische oxidation cycloaliphatischer Mono- und Diether in wtlBrigen
Elektrolyten, Chem. Ber. 120, 1679-1682 (1987).
26. C. Minero, E. Pelizzetti, S. Malato and J. Blanco, Large solar plant photocatalytic water
decontamination: degradation of atrazine, Solar Energy 56, 411-419 (1996).
27. C.S. Turchi and D.F. Ollis, Mixed reactant photocatalysis: intermediates and mutual rate inhibition, J.
Catal. 119, 483-496 (1989).
28. C. Kormann, D.W. Bahnemann and M.R. Hoffmann, Photolysis of chloroform and other organic
molecules in aqueous TiO 2 suspensions, Environ. Sci. Technol. 25, 494-509 (1991).
29. (a) P. Calza, C. Minero and E. Pelizzetti, Photocatalytically assisted hydrolysis of chlorinated
methanes under anaerobic conditions, Environ. Sci. Technol. in press (1997). (b) C. Minero, P.
Piccinini, P. Calza and E. Pelizzetti, Photocatalytic reduction/oxidation processes occurring at the
carbon and nitrogen of tetranitromethane, New J. Chem. 20, 1159-1164 (1996).
30. G.V. Buxton, C.L. Greenstock, W.P. Helman and A.B. Ross, Critical review of rate constants for
reactions hydrated electrons, hydrogen atoms and hydroxyl radicals in aqueous solutions, J.. Phys.
Chem. Ref. Data 17, 513-886 (1988).
31. H. Elbenberger, S. Steenken, P. O'Neill and D. Schulte-Frohlinde, Pulse radiolysis and electron spin
resonance studies concerning the reaction of SO4"- with alcohols and ethers in aqueous solution,
Phys. Chem. 82, 749-750 (1978).
32. M.C.R. Symons, Photolysis of hydrogen peroxide in fluid and rigid media, In Peroxide Reaction
Mechanisms (Edited by J.O. Edward), pp. 137-151. John Wiley & Sons, New York (1962).
33. L.J. Heidt, J.B. Mann and H.R. Schneider, The photolysis of persulfate. II The quantum yield in
water and the effect of sodium chloride in dilute alkaline solutions, J. Am. Chem. Soc. 70, 3011-3015
(1948).
34. P. Pichat, G. Guillard, L. Amalri¢, A.C. Renard and O. Plaidy, Assessment of the importance of the
role of H202 and 02- in the photocatalytic degradation of 1,2-dimethoxybenzene, Sol. Energy Mat.
Sol. Cells 38, 391-399 (1995).
35. M. Dor~, Chimie des Oxydants au Traitment des Eaux., p 296. Lavoisier, Paris (1989).




















