
Genome Landscape and Evolutionary Plasticity of Chromosomes in Malaria Mosquitoes

Lethal fragmentation of bacterial chromosomesmediated by DNA gyrase and quinolones
Muhammad Malik, Xilin Zhao and Karl Drlica*Public Health Research Institute, 225 Warren Street,Newark, NJ 07103, USA.
Summary
When DNA gyrase is trapped on bacterial chromo-somes by quinolone antibacterials, reversible com-plexes form that contain DNA ends constrained byprotein. Two subsequent processes lead to rapid celldeath. One requires ongoing protein synthesis; theother does not. The prototype quinolone, nalidixicacid, kills wild-type Escherichia coli only by the firstpathway; fluoroquinolones kill by both. Both lethalprocesses correlated with irreversible chromosomefragmentation, detected by sedimentation and viscos-ity of DNA from quinolone-treated cells. However,only fluoroquinolones fragmented purified nucleoidswhen incubated with gyrase purified from wild-typecells. A GyrA amino acid substitution (A67S) expectedto perturb a GyrA–GyrA dimer interface allowed nali-dixic acid to fragment chromosomes and kill cells inthe absence of protein synthesis; moreover, it made anon-inducible lexA mutant hypersusceptible to nalid-ixic acid, a property restricted to fluoroquinoloneswith wild-type cells. The GyrA variation also facili-tated immunoprecipitation of DNA fragments by GyrAantiserum following nalidixic acid treatment of cells.The ability of changes in both gyrase and quinolonestructure to enhance protein synthesis-independentlethality and chromosome fragmentation is explainedby drug-mediated destabilization of gyrase–DNAcomplexes. Instability of type II topoisomerase–DNAcomplexes may be a general phenomenon that can beexploited to kill cells.
Introduction
DNA topoisomerases are ubiquitous enzymes that play acentral role in chromosome architecture. As a normal partof their reaction mechanism, these enzymes bind to chro-mosomal DNA and form complexes that can be trappedby other proteins (Vizan et al., 1991; Bernard and Coutu-rier, 1992; Heddle et al., 2001a), by the quinolone anti-
bacterials (Heddle et al., 2000; Drlica and Malik, 2003)and by certain antitumour drugs (Liu et al., 2000; Wilster-mann and Osheroff, 2003). The ternary quinolone–enzyme–DNA complexes block DNA replication (Wentzelland Maxwell, 2000; Pohlhaus and Kreuzer, 2005), RNApolymerase movement (Willmott et al., 1994) and cellgrowth (reviewed in Drlica and Malik, 2003). While con-tinued inhibition of DNA and RNA synthesis eventuallyleads to cell death, several observations indicate thatrapid killing by quinolones involves events beyond ternarycomplex formation. For example, drug concentrationsrequired to rapidly kill cells are generally higher than thoseneeded to block DNA synthesis, a surrogate for inhibitionof growth (Chow et al., 1988; Chen et al., 1996). In addi-tion, first-generation quinolones, such as nalidixic andoxolinic acids, block DNA synthesis but do not kill cells inthe absence of protein synthesis (Deitz et al., 1966; Chenet al., 1996). More recently, a quinolone dimer was foundto block mycobacterial growth without killing cells (Maliket al., 2005). Thus, rapid killing appears to involve at leasttwo steps in which the first is formation of bacteriostaticquinolone–topoisomerase–DNA complexes. The subse-quent step(s) is poorly understood.
One hypothesis attributes a lethal step to events asso-ciated with blockage of replication fork movement (Brom-berg et al., 2003; Hong and Kreuzer, 2003; Pohlhaus andKreuzer, 2005). Early support for this idea was obtainedwhen DNA breaks were found to arise from collision ofreplication forks with eukaryotic topoisomerase I trappedon DNA by camptothecin (D’Arpa et al., 1990). However,attempts to extend the hypothesis to bacterial systemsfailed to reveal DNA breaks when replication forks orhelicases collided with quinolone–topoisomerase–DNAcomplexes (Hiasa et al., 1996; Shea and Hiasa, 1999;2000; 2003). Thus, the possibility was raised that haltingreplication fork movement facilitates endonuclease attackof DNA (Hong and Kreuzer, 2003; Pohlhaus and Kreuzer,2005). We now find that quinolone lethality is unaffectedby a pretreatment that halts replication [shift of atemperature-sensitive dnaB mutant to restrictive condi-tions (Zhao et al., 2006)]. Consequently, another explana-tion for rapid quinolone-mediated death is needed.
In principle, the ternary complexes could themselves bea source of lethal DNA breaks, because DNA in the com-plexes is broken (Gellert et al., 1977; Sugino et al., 1977;Williams and Maxwell, 1999). However, these breaks are
Accepted 6 June, 2006. *For correspondence. E-mail: [email protected]; Tel. (+1) 973 8543360; Fax: (+1) 973 8543101.
Molecular Microbiology (2006) 61(3), 810–825 doi:10.1111/j.1365-2958.2006.05275.xFirst published online 27 June 2006
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd

readily resealed (Gellert et al., 1977; Sugino et al., 1977;Pohlhaus and Kreuzer, 2005). To be rapidly lethal, thebreaks must become irreversible. That could happen if theDNA ends were released from the protein constraint exist-ing within the complexes. Evidence for end release iscurrently indirect: nucleoids, isolated from Escherichia coliin the absence of protein denaturants, cannot maintainDNA supercoils when obtained from cells treated withquinolones under lethal conditions (Chen et al., 1996). Itwas argued (Chen et al., 1996) that loss of supercoilmaintenance reflects widespread chromosome fragmen-tation, because isolated nucleoids are constrained intomany topologically independent domains that would notexhibit extensive relaxation from a few breaks (Worceland Burgi, 1972).
The present work examined the hypothesis that rapid,quinolone-mediated cell death arises from the intracellularrelease of DNA breaks from constraint in ternarycomplexes. When bacterial culture conditions, quinolonestructure, and gyrase structure were varied, cell deathcorrelated with chromosome fragmentation measured byDNA sedimentation and viscosity. For example, nalidixicacid and the fluoroquinolone gatifloxacin killed exponen-tially growing cells and fragmented chromosomes, butonly gatifloxacin did so in the absence of proteinsynthesis. These data also supported chromosome frag-mentation occurring by two pathways, one that requiresprotein synthesis and one that does not. Nalidixic acidacted only by the former. Because gatifloxacin, but notnalidixic acid, fragmented isolated nucleoids when incu-bated with gyrase purified from wild-type cells, it appearedthat the fluoroquinolone could destabilize ternary drug–gyrase–DNA complexes. Destabilization also occurred ina gyrA mutant expected to have an altered GyrA–GyrAdimer interface: the mutation allowed nalidixic acid tofragment chromosomes and rapidly kill cells even inthe absence of ongoing protein synthesis. Such drug-mediated destabilization of topoisomerase–DNA com-plexes explains stimulation of illegitimate recombinationby antitopoisomerase agents in both bacteria and eukary-otic organisms (Asami et al., 2002; Ikeda et al., 2004), andit provides new ways to evaluate potential antitumour andantimicrobial agents.
Results
Chromosome fragmentation associated with nalidixicacid treatment of wild-type cells
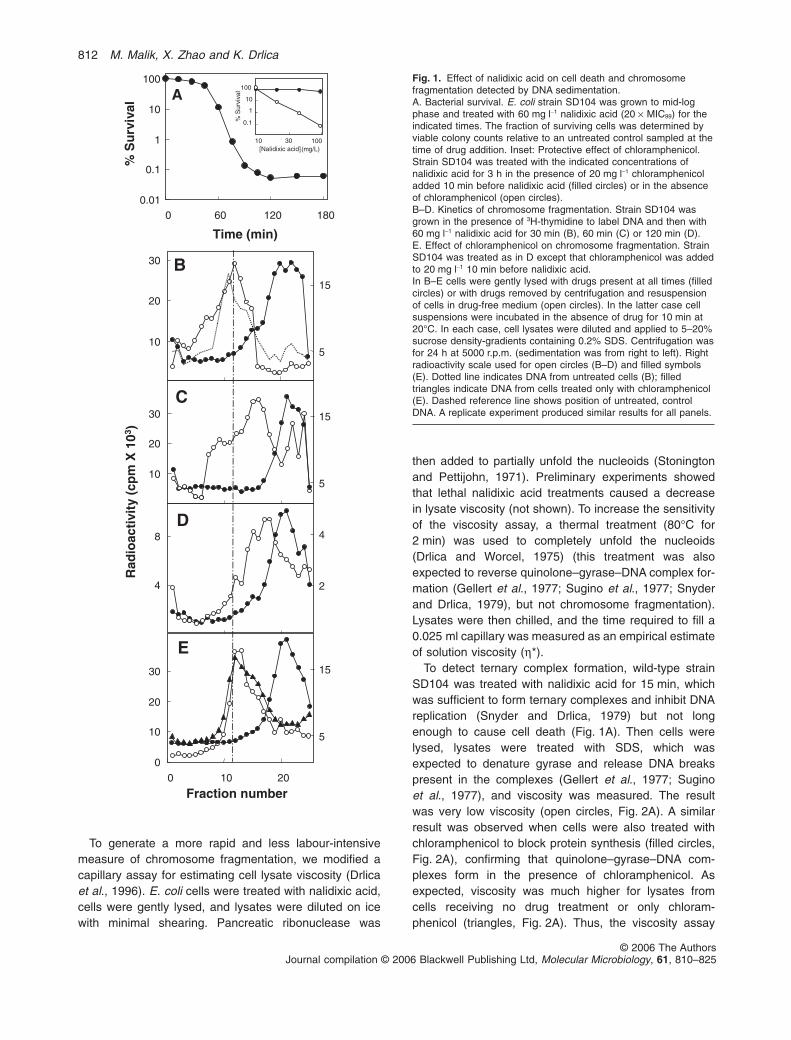
Nalidixic acid, at 20 times its minimal inhibitory concen-tration (MIC99), exhibited a 45 min lag when killing wild-type E. coli (Fig. 1A; see Table 1 for strain descriptions).This lag in cell death contrasted with inhibition of repli-cation by quinolones, which occurs within a few minutes
(Snyder and Drlica, 1979; Pohlhaus and Kreuzer, 2005).To examine quinolone-mediated DNA breakage, wetreated E. coli with nalidixic acid for various times andthen processed the cells in two ways. In one, we lysedthe cells in the presence of drug and directly applied thesamples to SDS-containing sucrose density-gradients forcentrifugation. In this procedure SDS denatures proteinand releases broken DNA from ternary complexes. Inthe other method, nalidixic acid was removed and thecells were incubated at 20°C for 10 min immediatelybefore cell lysis. This procedure allowed reversal ofternary complex formation and resealing of DNA breaksthat had not progressed to chromosome fragmentation.Then the samples were sedimented into SDS-containinggradients. Figure 1B shows that radioactively labelledDNA from untreated cells applied directly to sucrose gra-dients sedimented rapidly (dotted line), while DNA fromcells treated for 30 min with nalidixic acid sedimentedslowly (filled circles). These data confirmed the presenceof ternary complexes containing DNA breaks. Removalof nalidixic acid prior to cell lysis led to the recovery ofrapidly sedimenting DNA (open circles, Fig. 1B), indicat-ing reversal of ternary complexes and resealing of DNAbreaks. After longer, lethal incubation in nalidixic acid(60 or 120 min), removal of drug prior to cell lysis failedto fully restore the DNA to the fast-sedimenting form(Fig. 1 C and D). A longer treatment time (180 min)caused the ‘reversed’ sample to sediment almost asslowly as the sample sedimented directly into SDS-containing gradients (not shown). Thus, irreversiblerelease of DNA breaks from ternary complexes (chromo-some fragmentation) and cell death occur over thecourse of a few hours.
Chloramphenicol blocks the lethal action of nalidixicacid (Deitz et al., 1966; Fig. 1A inset). To observe theeffect of chloramphenicol on nalidixic acid-mediated chro-mosome fragmentation, cells were treated with chloram-phenicol for 10 min, and then nalidixic acid was added foran additional 2 h. When cells were lysed in the presenceof the drugs, slowly sedimenting DNA was recovered(filled circles, Fig. 1E), indicating that ternary complexescontaining broken DNA were present. The DNA in thesecomplexes resealed when the drugs were removed priorto cell lysis (open circles, Fig. 1E). As expected, chloram-phenicol alone had no effect on DNA sedimentation (filledtriangles, Fig. 1E). Similar results were obtained whennucleoids were isolated by preparative centrifugationand subsequently applied to analytical SDS-containingsucrose density-gradients (data not shown). In this caseresealing of DNA breaks in the complexes was achievedby addition of 50 mM EDTA to cell lysates prior to nucleoidisolation. Collectively these data show that chlorampheni-col protects from both cell death and chromosome frag-mentation caused by nalidixic acid.
Bacterial chromosome fragmentation 811
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 61, 810–825
To generate a more rapid and less labour-intensivemeasure of chromosome fragmentation, we modified acapillary assay for estimating cell lysate viscosity (Drlicaet al., 1996). E. coli cells were treated with nalidixic acid,cells were gently lysed, and lysates were diluted on icewith minimal shearing. Pancreatic ribonuclease was
then added to partially unfold the nucleoids (Stoningtonand Pettijohn, 1971). Preliminary experiments showedthat lethal nalidixic acid treatments caused a decreasein lysate viscosity (not shown). To increase the sensitivityof the viscosity assay, a thermal treatment (80°C for2 min) was used to completely unfold the nucleoids(Drlica and Worcel, 1975) (this treatment was alsoexpected to reverse quinolone–gyrase–DNA complex for-mation (Gellert et al., 1977; Sugino et al., 1977; Snyderand Drlica, 1979), but not chromosome fragmentation).Lysates were then chilled, and the time required to fill a0.025 ml capillary was measured as an empirical estimateof solution viscosity (h*).
To detect ternary complex formation, wild-type strainSD104 was treated with nalidixic acid for 15 min, whichwas sufficient to form ternary complexes and inhibit DNAreplication (Snyder and Drlica, 1979) but not longenough to cause cell death (Fig. 1A). Then cells werelysed, lysates were treated with SDS, which wasexpected to denature gyrase and release DNA breakspresent in the complexes (Gellert et al., 1977; Suginoet al., 1977), and viscosity was measured. The resultwas very low viscosity (open circles, Fig. 2A). A similarresult was observed when cells were also treated withchloramphenicol to block protein synthesis (filled circles,Fig. 2A), confirming that quinolone–gyrase–DNA com-plexes form in the presence of chloramphenicol. Asexpected, viscosity was much higher for lysates fromcells receiving no drug treatment or only chloram-phenicol (triangles, Fig. 2A). Thus, the viscosity assay
0.01
0.1
1
10
100
0 60 120 180
Time (min)
% S
urv
ival
A1
100
30 10010[Nalidixic acid] (mg/L)
% S
urvi
val
10
0.1
4
8
10
20
30
Rad
ioac
tivi
ty (
cpm
X 1
03 )
10
20
30
Fraction number
0
10
20
30
0 10 20
B
C
E
D
5
15
5
15
4
2
5
15
Fig. 1. Effect of nalidixic acid on cell death and chromosomefragmentation detected by DNA sedimentation.A. Bacterial survival. E. coli strain SD104 was grown to mid-logphase and treated with 60 mg l-1 nalidixic acid (20 ¥ MIC99) for theindicated times. The fraction of surviving cells was determined byviable colony counts relative to an untreated control sampled at thetime of drug addition. Inset: Protective effect of chloramphenicol.Strain SD104 was treated with the indicated concentrations ofnalidixic acid for 3 h in the presence of 20 mg l-1 chloramphenicoladded 10 min before nalidixic acid (filled circles) or in the absenceof chloramphenicol (open circles).B–D. Kinetics of chromosome fragmentation. Strain SD104 wasgrown in the presence of 3H-thymidine to label DNA and then with60 mg l-1 nalidixic acid for 30 min (B), 60 min (C) or 120 min (D).E. Effect of chloramphenicol on chromosome fragmentation. StrainSD104 was treated as in D except that chloramphenicol was addedto 20 mg l-1 10 min before nalidixic acid.In B–E cells were gently lysed with drugs present at all times (filledcircles) or with drugs removed by centrifugation and resuspensionof cells in drug-free medium (open circles). In the latter case cellsuspensions were incubated in the absence of drug for 10 min at20°C. In each case, cell lysates were diluted and applied to 5–20%sucrose density-gradients containing 0.2% SDS. Centrifugation wasfor 24 h at 5000 r.p.m. (sedimentation was from right to left). Rightradioactivity scale used for open circles (B–D) and filled symbols(E). Dotted line indicates DNA from untreated cells (B); filledtriangles indicate DNA from cells treated only with chloramphenicol(E). Dashed reference line shows position of untreated, controlDNA. A replicate experiment produced similar results for all panels.
812 M. Malik, X. Zhao and K. Drlica
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 61, 810–825

detected ternary complexes containing broken DNAwhen SDS was present to release the breaks fromprotein-mediated constraint.
In the absence of SDS, the 15 min nalidixic acid treat-ment had little effect on viscosity (open circles, Fig. 2B);the effect on viscosity was even less when chloram-phenicol was also present (filled circles, Fig. 2B).However, if cells were treated with nalidixic acid for 3 h,lysate viscosity was much lower (open circles, Fig. 2C).Addition of chloramphenicol 10 min before nalidixic acidblocked the chromosome fragmentation associated withthe 3-h nalidixic acid treatment (filled circles, Fig. 2C)without affecting the formation of ternary complexes(when SDS was added, lysates from cells treated withchloramphenicol and nalidixic acid for 15 min and 3 hhad similar, low levels of viscosity (not shown). Aspointed out above, the 3-h treatment with nalidixic acidkilled cells (Fig. 1A inset, open circles), but not whenchloramphenicol was present (Fig. 1A inset, filledcircles). Thus, viscosity measurements distinguished for-mation of DNA breaks trapped in quinolone–gyrase–DNA complexes from lethal chromosome fragmentationand showed that fragmentation correlates with celldeath. This concordance with conclusions reached fromsedimentation measurements validated the use of vis-cosity to detect chromosome fragmentation.
Chromosome fragmentation and cell death associatedwith fluoroquinolone treatment
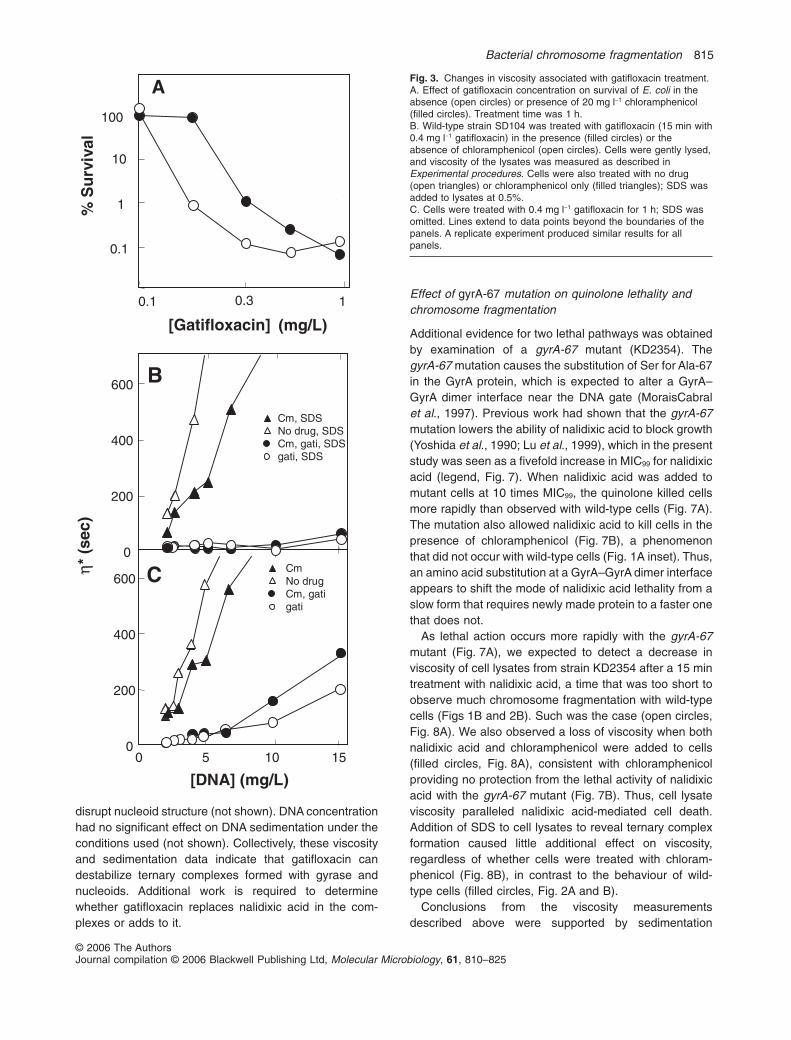
Gatifloxacin, a new, commercially available fluoroqui-nolone, killed E. coli when chloramphenicol was present,although slightly higher concentrations were required thanin the absence of chloramphenicol (Fig. 3A). As expected,gatifloxacin formed ternary complexes when added to thewild-type strain SD104, as revealed by the loss of viscos-ity after addition of SDS to cell lysates (circles, Fig. 3B). Inthe absence of SDS, viscosity was also low, even whencells were treated with both chloramphenicol and gati-floxacin (Fig. 3C). Sedimentation measurements con-firmed that a 15 min treatment with gatifloxacin at 20
times MIC99 caused chromosome fragmentation in thepresence of chloramphenicol (not shown). Thus, unlikenalidixic acid, fluoroquinolones such as gatifloxacin frag-ment chromosomes and kill E. coli even in the absence ofprotein synthesis. These data are consistent with theexistence of a second pathway leading to chromosomefragmentation and cell death.
To determine whether aspects of fluoroquinolonestructure affect the two lethal pathways differently, threestructurally related fluoroquinolones, PD161144,PD161148 and PD160788 (Fig. 4A) were examined withan E. coli strain (KD2361) that contained a parCquinolone-resistance mutation to make gyrase the soledrug target. The three compounds exhibited no differ-ence when bacterial survival was measured in theabsence of chloramphenicol (Fig. 4B; in these experi-ments lethal action was expressed as a function ofMIC99 to correct for differences in factors such as druguptake and efflux). When protein synthesis was blocked,the three compounds varied in lethality (Fig. 4C). As themost active compound, PD161144, differed from theother two by single moieties, enhanced lethality wasattributed to a C-8-methoxy group (PD161144 vs.PD160788) and to the position at which an ethyl sub-stituent was attached to the C-7 piperazinyl ring(PD161144 vs. PD161148). Although additional work isrequired to understand exactly how these moieties facili-tate lethal action, the data support the existence of twomechanistically different forms of lethality.
Chromosome fragmentation due to fluoroquinolonetreatment of isolated nucleoids
To examine chromosome fragmentation in a cell-freesystem, we first prepared nucleoids from cells treated withcoumermycin to relax DNA supercoiling (Drlica andSnyder, 1978). Incubation of nucleoids with purifiedgyrase and gatifloxacin then caused viscosity to dropby 50% (P � 0.05, Table 2). Neither gyrase alone norgatifloxacin alone reduced viscosity. Omission of cou-mermycin treatment prevented the gatifloxacin-gyrase-
Table 1. Bacterial strains.
Strain Relevant genotype Reference/source
1596 C600 gyrA-1596 (NorR S83L) parC-1596 (NorR S80L) A. Khodursky (Khodursky et al., 1995)AQ8300 lexA-3(Ind–), malF-3089::Tn10 T. KogomaDG75 Wild type A. Worcel (Worcel and Burgi, 1972)KD1911 gyrA-67 zfa-3145::Tn10 (KanR) T. Lu (Lu et al., 1999)KD2354 SD104 gyrA-67 This work by transduction of gyrA-67 and zfa-3145::Tn10KanR from
KD1911KD2361 DG75 parC-1596 X. Zhao by transduction of NorR from 1596KD2424 KD2354 lexA-3 This work by transduction of lexA-3 and malF-3089::Tn10 from AQ8300KD2451 SD104 lexA-3 This work by transduction of lexA-3 and malF-3089::Tn10 from AQ8300SD104 Wild type S. DiNardo (DiNardo et al., 1984)
Bacterial chromosome fragmentation 813
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 61, 810–825

dependent reduction in viscosity (not shown), consistentwith gyrase preferentially binding to relaxed DNA. Nalid-ixic acid failed to fragment isolated nucleoids when incu-
bated with wild-type gyrase (Table 2), a result that wasconsistent with this first-generation quinolone requiringnewly synthesized protein to fragment chromosomes andto kill cells (Fig. 1). These data indicate that gyrase plusnucleoids are sufficient for chromosome fragmentation bygatifloxacin and presumably for the protein synthesis-independent pathway of fragmentation.
To determine whether gatifloxacin can use preformedternary complexes as a substrate, we treated wild-typecells (SD104) with chloramphenicol for 10 min followed byaddition of nalidixic acid for an additional 15 min, condi-tions shown in Figs 1E and 2 to trap gyrase on DNA withlittle chromosome fragmentation. Cells were lysed, andnucleoids were isolated by preparative sucrose density-gradient centrifugation. Chloramphenicol and nalidixicacid, which were present at all times, maintainedquinolone–gyrase–DNA complexes on the nucleoids, asshown by the ability of SDS to lower viscosity (compareopen triangles and open circles, Fig. 5A). When isolatednucleoids containing nalidixic acid–gyrase–DNA com-plexes were treated with gatifloxacin prior to incubation at80°C, viscosity dropped (compare circles, Fig. 5A). Omis-sion of nalidixic acid prior to cell lysis prevented gatifloxa-cin from lowering viscosity (squares, Fig. 5A), indicatingthat nalidixic acid–gyrase–DNA complexes were requiredfor this gatifloxacin-dependent chromosome fragmen-tation. EDTA blocked chromosome fragmentation whenadded to complex-containing nucleoids before, but notafter, gatifloxacin treatment (compare filled circles andfilled triangles, Fig. 5B), consistent with EDTA reversingternary complex formation but not chromosomefragmentation.
Sedimentation experiments confirmed key aspects ofthe release of DNA breaks from nalidixic acid–gyrase–DNA complexes by gatifloxacin. DNA from cells treatedwith chloramphenicol and nalidixic acid sedimentedslowly in SDS-containing sucrose density-gradients whengatifloxacin was added to cell lysates prior to EDTA [filledcircles, Fig. 6; gatifloxacin concentration was saturating,because the same result was obtained at 1/4 the concen-tration (not shown)]. DNA also sedimented slowly whenEDTA and gatifloxacin were omitted (not shown), whichallowed release of DNA breaks from nalidixic acid-containing ternary complexes by SDS present in the ana-lytical sucrose density-gradients. When EDTA was addedimmediately before gatifloxacin, DNA sedimented rapidly(open circles, Fig. 6), indicating that DNA breaks in thecomplexes were resealed. DNA also sedimented rapidlyin the absence of gatifloxacin if EDTA was present toreverse complex formation (not shown). A 2 min incuba-tion at 80°C had no effect on DNA sedimentation in SDS-containing gradients (not shown). Results similar to thosein Fig. 6 were also obtained when SDS was omitted fromthe gradients, but then the 80°C treatment was required to
0
200
400
600A
0
200
400
600
h* (
sec)
B
0
200
400
600
0 5 10 15
[DNA] (mg/L)
C
Cm, SDSNo drug, SDSCm, Nal, SDS Nal, SDS
Cm No drugCm, NalNal
Cm No drugCm, NalNal
Fig. 2. Nalidixic acid-mediated chromosome fragmentationdetected by viscosity. Wild-type E. coli strain SD104 was treatedwith nalidixic acid at 30 mg l-1 in the presence (filled circles) or theabsence of chloramphenicol (open circles). Cells were gently lysed,and viscosity of the lysates was measured as described inExperimental procedures. Cells were also treated with no drug(open triangles) or chloramphenicol only (filled triangles).A. Cells were treated for 15 min with nalidixic acid, and SDS wasadded to lysates at 0.5%.B. Cells were treated for 15 min with nalidixic acid, but SDS wasomitted.C. Cells were treated for 3 h with nalidixic acid, and SDS wasomitted. Lines extend to data points beyond the boundaries of thepanels. A replicate experiment produced similar results for allpanels.
814 M. Malik, X. Zhao and K. Drlica
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 61, 810–825
disrupt nucleoid structure (not shown). DNA concentrationhad no significant effect on DNA sedimentation under theconditions used (not shown). Collectively, these viscosityand sedimentation data indicate that gatifloxacin candestabilize ternary complexes formed with gyrase andnucleoids. Additional work is required to determinewhether gatifloxacin replaces nalidixic acid in the com-plexes or adds to it.
Effect of gyrA-67 mutation on quinolone lethality andchromosome fragmentation
Additional evidence for two lethal pathways was obtainedby examination of a gyrA-67 mutant (KD2354). ThegyrA-67 mutation causes the substitution of Ser for Ala-67in the GyrA protein, which is expected to alter a GyrA–GyrA dimer interface near the DNA gate (MoraisCabralet al., 1997). Previous work had shown that the gyrA-67mutation lowers the ability of nalidixic acid to block growth(Yoshida et al., 1990; Lu et al., 1999), which in the presentstudy was seen as a fivefold increase in MIC99 for nalidixicacid (legend, Fig. 7). When nalidixic acid was added tomutant cells at 10 times MIC99, the quinolone killed cellsmore rapidly than observed with wild-type cells (Fig. 7A).The mutation also allowed nalidixic acid to kill cells in thepresence of chloramphenicol (Fig. 7B), a phenomenonthat did not occur with wild-type cells (Fig. 1A inset). Thus,an amino acid substitution at a GyrA–GyrA dimer interfaceappears to shift the mode of nalidixic acid lethality from aslow form that requires newly made protein to a faster onethat does not.
As lethal action occurs more rapidly with the gyrA-67mutant (Fig. 7A), we expected to detect a decrease inviscosity of cell lysates from strain KD2354 after a 15 mintreatment with nalidixic acid, a time that was too short toobserve much chromosome fragmentation with wild-typecells (Figs 1B and 2B). Such was the case (open circles,Fig. 8A). We also observed a loss of viscosity when bothnalidixic acid and chloramphenicol were added to cells(filled circles, Fig. 8A), consistent with chloramphenicolproviding no protection from the lethal activity of nalidixicacid with the gyrA-67 mutant (Fig. 7B). Thus, cell lysateviscosity paralleled nalidixic acid-mediated cell death.Addition of SDS to cell lysates to reveal ternary complexformation caused little additional effect on viscosity,regardless of whether cells were treated with chloram-phenicol (Fig. 8B), in contrast to the behaviour of wild-type cells (filled circles, Fig. 2A and B).
Conclusions from the viscosity measurementsdescribed above were supported by sedimentation
1
100
A
1
[Gatifloxacin] (mg/L)
10
0.1
% S
urv
ival
0.1 0.3
0
200
400
600 B
0
200
400
600 C
0 5 10 15
[DNA] (mg/L)
Cm, SDSNo drug, SDSCm, gati, SDSgati, SDS
CmNo drugCm, gatigati
η* (
sec)
Fig. 3. Changes in viscosity associated with gatifloxacin treatment.A. Effect of gatifloxacin concentration on survival of E. coli in theabsence (open circles) or presence of 20 mg l-1 chloramphenicol(filled circles). Treatment time was 1 h.B. Wild-type strain SD104 was treated with gatifloxacin (15 min with0.4 mg l-1 gatifloxacin) in the presence (filled circles) or theabsence of chloramphenicol (open circles). Cells were gently lysed,and viscosity of the lysates was measured as described inExperimental procedures. Cells were also treated with no drug(open triangles) or chloramphenicol only (filled triangles); SDS wasadded to lysates at 0.5%.C. Cells were treated with 0.4 mg l-1 gatifloxacin for 1 h; SDS wasomitted. Lines extend to data points beyond the boundaries of thepanels. A replicate experiment produced similar results for allpanels.
Bacterial chromosome fragmentation 815
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 61, 810–825

analysis. When nucleoids were obtained from EDTA-treated mutant cell lysates and DNA was sedimented intoSDS-containing sucrose density-gradients, DNA fromuntreated cells sedimented rapidly (Fig. 9A), while DNAfrom the gyrA-67 mutant, treated with nalidixic acid (Fig.9B) or chloramphenicol and nalidixic acid (Fig. 9C), sedi-mented slowly. These data were consistent with EDTAtreatment being unable to reverse ternary complex forma-tion and reseal DNA breaks with mutant gyrase. Asexpected, DNA from mutant cells treated only withchloramphenicol sedimented rapidly (not shown). Weconclude that the gyrA-67 mutation allows nalidixic acid tofragment chromosomes and kill cells in the absence ofprotein synthesis.
The gyrA-67 mutant also provided a way to determinewhether intracellular processes distinguish the two lethalpathways. The lexA-3 (Ind–) allele, which blocks inductionof the SOS response, is reported to increase killing byfluoroquinolones but to have no effect on the lethality ofnalidixic acid (Lewin et al., 1989; Howard et al., 1993;Chen et al., 1996). We confirmed that the lexA-3 allele has
1
10
100
% S
urv
ival
B
1
10
100
1 10
[Fluoroquinolone] (x MIC99)
C
PD161144
PD161148
PD160788
PD161148 PD160788
A
PD161144
N
O O
OHF
N
N O
C3H
N
O O
OHF
N
N OC3H
N
O O
OHF
N
N
Fig. 4. Effect of fluoroquinolone structure on lethal action in theabsence of protein synthesis.A. Structures of fluoroquinolones tested. Arrows point to positionsat which PD161144 differs from the two other compounds.B. Lethal action with exponentially growing cells. E. coli strainKD2361 was treated with PD161144 (filled circles), PD161148(open circles), or PD160788 (filled triangles) for 1 h. At the end ofthe incubation, cells were diluted, and the fraction of surviving cellswas determined.C. Lethal action in the absence of protein synthesis. Strain KD2361was treated with chloramphenicol (20 mg l-1) for 10 min beforeaddition of fluoroquinolone (symbols are the same as in B).Fluoroquinolone concentrations are expressed as multiples ofMIC99, which were 0.08, 0.10 and 0.025 mg l-1, respectively, forPD161144, PD161148 and PD160788. A replicate experimentproduced similar results for both panels.
Table 2. Chromosome fragmentation mediated by purified gyrase.
Gyraseaddeda
Quinolonetreatmentb
Viscosityc
(% ± SD)
No None 100Yes None 97 ± 8No Gatifloxacin 90 ± 10Yes Gatifloxacin 48 ± 9No Nalidixic acid 103 ± 3Yes Nalidixic acid 102 ± 11Yes (GyrA-67)d Nalidixic acid 50 ± 3Yes (GyrA-67)d Gatifloxacin 32 ± 10
a. Gyrase (10 unit mixture of GyrA and GyrB protein) was addedalong with 100 mM ATP and isolated nucleoids, as described inExperimental procedures.b. Gatifloxacin concentration was 0.1 mg ml-1; nalidixic acid concen-tration was 1 mg ml-1.c. Viscosity was determined as described in Experimental proce-dures after a brief incubation at 80°C to unfold nucleoids (15 mg l-1
DNA). No treatment with gyrase or quinolone was taken as 100%.d. GyrA protein was obtained from a plasmid-borne gyrA-67 mutantgene (see Experimental procedures).
816 M. Malik, X. Zhao and K. Drlica
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 61, 810–825

no effect on nalidixic acid lethality with gyrA+ cells (Fig.10A). In contrast, the gyrA-67 mutation rendered a lexA-3mutant hypersusceptible to nalidixic acid (Fig. 10B).These data indicate that the wild-type SOS regulonpartially protects cells from the protein synthesis-independent mode of quinolone-mediated cell death, butnot from the mode that requires ongoing proteinsynthesis. Reduced lethality at high concentration (Fig.10), commonly called the quinolone paradox (Crumplinand Smith, 1975), is currently unexplained.
If the effects of the gyrA-67 mutation arise from desta-bilization of a GyrA–GyrA dimer interaction, purifiedGyrA-67 protein, when combined with GyrB, should facili-tate cleavage of nucleoids by quinolones. Gatifloxacinfragmented isolated nucleoids slightly more when incu-bated with mutant GyrA than with the wild-type enzyme(Table 2), even though ternary complex formation wasexpected to be lower (mutant MIC99 for gatifloxacin wastwo times higher than wild-type MIC99, not shown). Nalid-ixic acid also exhibited activity with mutant gyrase(Table 2).
Chromosome fragmentation due to destabilization ofquinolone–gyrase–DNA complexes was expected toleave GyrA protein covalently attached to DNA (Mizuuchiet al., 1980; Morrison et al., 1980). To seek residualgyrase on DNA, the gyrA-67 mutant (strain KD2354)was labelled with 3H-thymidine, and cells were treated
0
200
400
600
0 5 10 15
[DNA] (mg/L)
B0
200
400
600
ANal, Nal, gati Nal, SDS gati,SDSgati No drug
EDTA then no gati EDTA then gatiNo gati then EDTA gati then EDTA
h* (
sec)
Fig. 5. Changes in viscosity associated with gatifloxacin treatmentof nucleoids in vitro following isolation from E. coli treated withnalidixic acid and chloramphenicol.A. Gatifloxacin-mediated DNA cleavage. Wild-type strain SD104was treated with chloramphenicol (20 mg l-1) followed by nalidixicacid (6 mg l-1) for 15 min. Cells were lysed, and nucleoids wereobtained as a band sedimenting in a preparative sucrosedensity-gradient (see Experimental procedures). Nucleoids werediluted in 0.02 M Tris-HCl (pH 8) containing gatifloxacin at100 mg l-1 (final concentration; filled circles) or lacking gatifloxacin(open circles). Samples were incubated at 37°C for 10 min andthen at 30°C for 20 min before being assayed for viscosity asdescribed in Experimental procedures. An additional sample wastreated with 0.5% SDS after incubation without gatifloxacin (opentriangles). Nucleoids were also isolated from cells that were nottreated with nalidixic acid and then incubated at 37°C for 10 minfollowed by 30°C for 20 min with (filled squares) or without (opensquares) gatifloxacin. One sample from cells lacking nalidixic acidtreatment was treated in vitro with gatifloxacin and also treated with0.5% SDS (filled triangles).B. Effect of EDTA on gatifloxacin-mediated cleavage. Strain SD104was treated with chloramphenicol and nalidixic acid as in A,nucleoids were isolated and treated with (filled symbols) or without(open symbols) gatifloxacin at 100 mg l-1. Viscosity was measuredafter EDTA was added to 50 mM either before incubation (triangles)with or without gatifloxacin or after incubation (circles) with orwithout gatifloxacin (incubation was as in A). Nucleoids were thentreated and analysed for viscosity as in Experimental procedures.Three replicate experiments produced similar results.
Rad
ioac
tivi
ty (
cpm
X 1
03 )
0
2
4
6
0 10 20
Fraction number
1
2
Fig. 6. Changes in DNA sedimentation associated with gatifloxacintreatment of lysates from E. coli incubated with chloramphenicoland nalidixic acid. Wild-type strain SD104 was grown in thepresence of 3H-thymidine and then treated with chloramphenicol(20 mg l-1) for 10 min followed by nalidixic acid (6 mg l-1) for 15 min.Cells were lysed and diluted in 0.02 M Tris-HCl pH 8 containinggatifloxacin at a final concentration of 100 mg l-1. EDTA (50 mM)was added to lysates before (open circles, right radioactivity scale)or after (filled circles, left radioactivity scale) addition of gatifloxacin.Following incubation, DNA samples were diluted 50-fold in 0.02 MTris-HCl pH 8, layered onto 5–20% sucrose density-gradients, andsedimented for 19 h at 6300 r.p.m. Sedimentation was from right toleft. Replicate experiments produced similar results.
Bacterial chromosome fragmentation 817
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 61, 810–825

for 15 min with nalidixic acid (10 ¥ MIC) plus chloram-phenicol at 20 mg l-1, conditions expected to fragmentchromosomes. Cells were lysed, and lysates were treatedwith 50 mM EDTA to reverse ternary complexes, if any,that were intact. Nucleoids were then isolated by sedi-mentation into preparative sucrose density-gradients(nascent RNA entanglements maintain a compact nucle-oid structure even when DNA breaks are present). Mutantnucleoids, which in control experiments were precipitatedby GyrA antiserum-protein A sepharose treatment but notby pre-immune serum, were gently diluted without
mechanical DNA breakage and then sedimented intoSDS-containing sucrose density-gradients. Fractionationof the gradients and precipitation with GyrA antiserumshowed that GyrA cosedimented with bulk DNA fragments(Fig. 11). The slightly slower sedimentation of GyrA-bound DNA was expected because shorter fragmentswould have a higher concentration of ends and presum-ably GyrA protein. GyrA antiserum did not precipitatenucleoids from wild-type cells isolated by preparativesucrose gradient centrifugation following treatment withEDTA to reverse complex formation (not shown); conse-quently, wild-type DNA was not examined by resedimen-tation into SDS-containing gradients. Collectively thesedata indicate that DNA fragments generated by the
[Nalidixic acid] (mg/L)
Cfu
(%
of
un
trea
ted
co
ntr
ol)
0.1
1
10
100
0 2 4Time (hrs)
A
1
10
100
10 100
B
Fig. 7. Effect of gyrA-67 mutation on lethal action of nalidixic acid.A. Rate of killing by nalidixic acid. Cells of strain KD2354 (gyrA-67,filled circles) or SD104 (wild-type, open circles) were incubated forthe indicated times with nalidixic acid at 10 ¥ MIC99, after whichcultures were diluted and the fraction of surviving cells wasmeasured (MIC99 for nalidixic acid was 3 and 16 mg l-1,respectively, with SD104 and KD2354).B. Effect of chloramphenicol on the lethal action of nalidixic acid. E.coli cells (strain KD2354) were treated with (filled circles) or without(open circles) chloramphenicol (20 mg l-1) for 10 min, and thennalidixic acid was added at the indicated concentrations for 3 h.Cells were diluted, and the fraction of surviving cells wasdetermined. A replicate experiment produced similar results for bothpanels.
0
200
400
600 B
0 5 10 15
[DNA] (mg/L)
0
200
400
600 A
Cm, SDSNo drug, SDSCm, Nal, SDSNal, SDS
CmNo drugCm, NalNal
h* (
sec)
Fig. 8. Changes in cell lysate viscosity associated with nalidixicacid treatment of a gyrA-67 mutant. An E. coli GyrA A67S variant(strain KD2354) was treated with nalidixic acid (160 mg l-1; 10 ¥MIC99) for 15 min in the presence (filled circles) or the absence ofchloramphenicol (20 mg l-1, open circles). Cells were gently lysed,and viscosity of the lysates was measured as described inExperimental procedures. Cells were also treated with no drug(open triangles) or chloramphenicol only (filled triangles).A. SDS omitted.B. SDS added to lysates at 0.5%. Lines extend to data pointsbeyond the boundaries of the panels. Three replicate experimentsproduced similar results for both panels.
818 M. Malik, X. Zhao and K. Drlica
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 61, 810–825

protein synthesis-independent pathway are linked to GyrAprotein.
Discussion
DNA topoisomerases break DNA and bind covalently toDNA ends as a normal part of their reaction mechanism
(Heddle et al., 2000); quinolones trap the covalentprotein–DNA complexes by preventing DNA religation(Anderson et al., 1999; 2000). Quinolone-mediated trap-ping of gyrase–DNA complexes is depicted schemati-cally in Fig. 12, step b. The ternary complexes, whichcontain a pair of single-stranded DNA breaks staggeredby 4 bp, block DNA synthesis and cell growth. However,complex formation can be reversed by removal of thequinolone (Pohlhaus and Kreuzer, 2005). Complex for-
0 5 10 15 20 25
Fraction number
0
10
20
30
40
0
5
10
15
20
10
20
30
Rad
ioac
tivi
ty (
cpm
X 1
02 )
C
B
A
Fig. 9. Changes in DNA sedimentation associated with nalidixicacid treatment of a gyrA-67 mutant. E. coli strain KD2354 (gyrA-67)was grown in 3H-thymidine to label DNA and then was treated withno additional agent (A), with nalidixic acid (160 mg l-1; 10 ¥ MIC99)for 15 min in the presence of chloramphenicol (20 mg l-1, B), or withnalidixic acid (160 mg l-1) in the absence of chloramphenicol (C).Cells were gently lysed, and EDTA was added to 50 mM. Nucleoidswere isolated by preparative sucrose density-gradientcentrifugation, and peak fractions were diluted and recentrifuged in5–20% sucrose density-gradients containing 0.2% SDS for 16 h at4800 r.p.m. Sedimentation was from right to left. Replicateexperiments produced similar results.
1
100
Cfu
(%
of
un
trea
ted
co
ntr
ol)
A
1
100
1 10 100
[Nalidixic acid] (mg/L)
B
Fig. 10. Effect of lexA-3 mutation on nalidixic acid-mediated celldeath.A. Lethal action with gyrA+ cells. Wild-type cells (strain SD104,open circles) or an isogenic lexA-3 mutant (strain KD2451, filledcircles) were treated with nalidixic acid at the indicatedconcentrations for 3 h. At the end of the incubation, cells werediluted, and the number of colony-forming units (cfu) wasdetermined and expressed as a percent of cfu at the time of drugaddition.B. Lethal action with gyrA-67 cells. GyrA A67S variant (strainKD2354, open circles) and a gyrA-67 lexA-3 double mutant (strainKD2424, filled circles) were treated as in A. Replicate experimentsproduced similar results.
Bacterial chromosome fragmentation 819
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 61, 810–825
mation can also be reversed in vitro and the breaksresealed by addition of EDTA or a mild thermal treat-ment (Gellert et al., 1977; Sugino et al., 1977; Snyderand Drlica, 1979). If ternary complexes are allowed toremain on the chromosome, treatment of cell lysates ornucleoids with a protein denaturant, such as SDS, dis-sociates the complexes and releases DNA breaks(Fig. 12, step e). Previous study of quinolone–gyrase–DNA complexes has almost always involved proteindenaturation prior to analysis of DNA size; such analy-ses reflect formation of ternary complexes (also calledcleavage, cleavable, or cleaved complexes) rather thanchromosome fragmentation. Chromosome fragmentationis seen as (i) the inability to maintain chromosomal DNAsupercoiling (Chen et al., 1996); (ii) a decrease in DNAsize when monitored in the absence of protein denatur-ation (Fig. 2); and (iii) DNA cleavage that is not reversedby drug removal, heat, or EDTA treatment prior to addi-tion of protein denaturant (Fig. 1 and Pohlhaus andKreuzer, 2005). These assays revealed that rapid qui-nolone lethality correlates with chromosome fragmenta-tion when quinolone incubation time was varied andwhen the protective effect of chloramphenicol wasmanipulated using quinolone and gyrase variants(Figs 1–3 and 7–9).
Several lines of evidence indicate that chromosomefragmentation and cell death occur in two ways (Fig. 12,
steps c and d). First, fragmentation and death caused byfirst-generation quinolones (nalidixic acid) required ongo-ing protein synthesis (Figs 1 and 2), while gatifloxacin-mediated fragmentation and death did not (Fig. 3).Second, lethal activity, normalized to bacteriostatic activ-ity to correct for drug uptake, efflux and ternary complexformation, was enhanced by the C-8-methoxy group andC-7-ring structure only when protein synthesis wasblocked (Fig. 4). Third, gatifloxacin, but not nalidixic acid,fragmented isolated nucleoids when incubated with wild-type gyrase (Table 2). Gatifloxacin also fragmentednucleoids on which gyrase had been trapped by pre-treatment of cells with a combination of chloramphenicoland nalidixic acid (Figs 5 and 6). With these in vitrosystems, detection of quinolone action is suboptimal,because it requires higher quinolone concentrations thanintracellular treatments; a comparable concentration
400
800
0 5 10
Fraction number
Rad
ioac
tivi
ty (
cpm
)
Fig. 11. Immunoprecipitation of chromosomal DNA by GyrAantiserum. DNA in E. coli strain KD2354 (gyrA-67) wasradioactively labelled by growth in 3H-thymidine, and cells werethen treated chloramphenicol (20 mg l-1) for 10 min after whichnalidixic acid (10 times MIC99) was added to cultures for 15 min.Cells were lysed, treated with 50 mM EDTA, and nucleoids wereisolated by preparative centrifugation (see Experimentalprocedures). Fractions were assayed for acid-precipitableradioactivity; nucleoids from the peak fraction were diluted fivefoldin 0.02 M Tris-HCl pH 8 buffer and recentrifuged in SDS-containingsucrose gradients at 6300 r.p.m. in a Beckman SW50.1 rotor for 19h at 20°C. Then acid-precipitable radioactivity (open circles) andradioactivity immunoprecipitated with GyrA antiserum (filled circles)was determined. Sedimentation was from right to left. Similarresults were obtained in a replicate experiment.
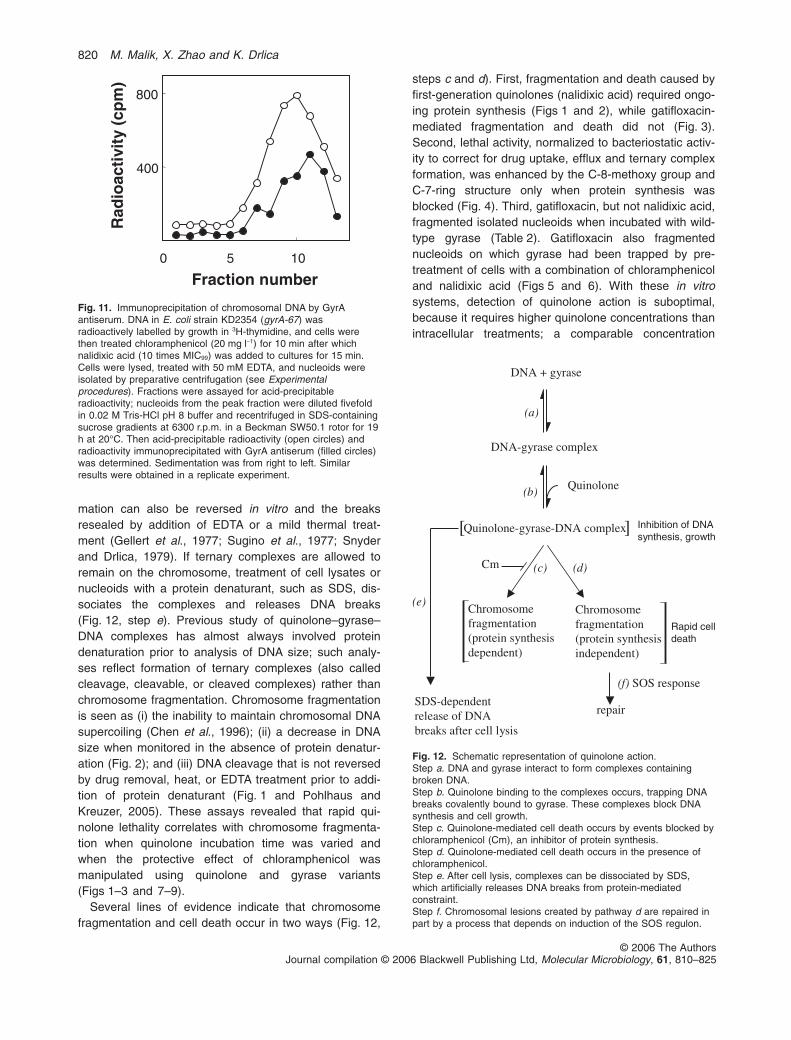
Quinolone
DNA + gyrase
(c) (d)
(a)
(b)
Cm
(f) SOS response
repair
DNA-gyrase complex
Quinolone-gyrase-DNA complex
Chromosome fragmentation(protein synthesisdependent)
Chromosome fragmentation(protein synthesisindependent)
SDS-dependentrelease of DNA breaks after cell lysis
(e)
][ Inhibition of DNAsynthesis, growth
Rapid celldeath
Fig. 12. Schematic representation of quinolone action.Step a. DNA and gyrase interact to form complexes containingbroken DNA.Step b. Quinolone binding to the complexes occurs, trapping DNAbreaks covalently bound to gyrase. These complexes block DNAsynthesis and cell growth.Step c. Quinolone-mediated cell death occurs by events blocked bychloramphenicol (Cm), an inhibitor of protein synthesis.Step d. Quinolone-mediated cell death occurs in the presence ofchloramphenicol.Step e. After cell lysis, complexes can be dissociated by SDS,which artificially releases DNA breaks from protein-mediatedconstraint.Step f. Chromosomal lesions created by pathway d are repaired inpart by a process that depends on induction of the SOS regulon.
820 M. Malik, X. Zhao and K. Drlica
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 61, 810–825

effect has been noted with plasmid DNA substrates(Sato et al., 1986).
Other observations also fit with the existence of twolethal pathways. For example, the lethality of fluoroquino-lones, but not nalidixic acid, is enhanced by the lexA-3mutation (Lewin et al., 1989), suggesting that the SOSresponse preferentially repairs damage caused bypathway d. Moreover, shifting E. coli to anaerobic condi-tions has a similar effect to chloramphenicol: both nalidixicacid and the fluoroquinolone PD161144 block anaerobicgrowth, but nalidixic acid kills cells only when growingaerobically (M. Malik, S. Hussain and K. Drlica, unpubl.obs.). PD161144 kills cells under both aerobic andanaerobic conditions. Third, earlier work showed that inhi-bition of protein synthesis blocks the lethal action and thesupercoil-eliminating activity of first-generation quinolo-nes but not that of fluoroquinolones (Chen et al., 1996).We conclude that one type of lethal chromosome frag-mentation requires newly made protein (Fig. 12, step c)while the other does not (Fig. 12, step d).
The induced protein(s) responsible for Fig. 12 (step c)has not been identified, although mutants can be obtainedby cycles of enrichment that allow norfloxacin and severalother quinolones to block growth with little lethal effect(Wolfson et al., 1989). We have obtained comparablemutants that show only bacteriostatic susceptibility to nali-dixic acid (Zhao et al., unpubl. obs.). Mapping the respon-sible mutations has proven difficult, suggesting thatmultiple genes may be involved. Little is known about theputative suicide factor except that it is likely to turn overrapidly: chloramphenicol or anaerobic treatment, admin-istered after nalidixic acid, quickly reverse quinolonelethality (Crumplin and Smith, 1975) (M. Malik, S. Hussainand K. Drlica, unpubl. obs.).
The present work focused on the protein synthesis-independent pathway (Fig. 12, step d) by examining prop-erties of the gyrA-67 mutation. This mutation, whichconfers modest protection from quinolone action (Yoshidaet al., 1990; Lu et al., 1999), increased the rate of killing(Fig. 7A), eliminated the ability of chloramphenicol toblock nalidixic acid-mediated cell death (Fig. 7B) or chro-mosome fragmentation (Figs 8A and 9), and rendered alexA-3 mutant hypersusceptible to killing by nalidixic acid(Fig. 10). Collectively these results are consistent with thegyrA-67 mutation shifting the lethal action of nalidixic acidfrom a pathway requiring ongoing protein synthesis(Fig. 12, step c) to one that does not (Fig. 12, step d).
The gyrA-67 mutation may destabilize ternaryquinolone–gyrase–DNA complexes by weakening aGyrA–GyrA interaction at an interface near the DNA gate(MoraisCabral et al., 1997). Such an idea fits with Ikeda’sobservation that quinolones stimulate a form of illegiti-mate recombination that can be explained by gyrasesubunit dissociation–reassociation (Ikeda, 1986; Ikeda
et al., 2004). It also fits with our observation that mutantgyrase was bound to DNA fragments following nalidixicacid treatment of cells (Fig. 11). Whether DNA fragmentsfrom wild-type cells treated with nalidixic acid under lethalconditions (Fig. 12, step c) are also bound to GyrA proteinremains to be determined.
A prediction of the destabilization hypothesis is that theGyrA-67 protein forms unstable ternary complexes. Whenthe protein was incubated with nucleoids and gatifloxacin,viscosity dropped more than with wild-type gyrase(Table 2). This result encourages more detailed biochemi-cal experiments using plasmid DNA rather than isolatednucleoids as a substrate. At present we do not knowwhether proteins other than gyrase are involved in therelease of DNA breaks from protein constraint, becauseisolated nucleoids contain many different proteins (Por-talier and Worcel, 1976). Another expectation of the dis-sociation hypothesis is that chromosome fragmentationand cell death occur quickly with the mutant, because newprotein need not be synthesized. Such was the case(Figs 7A and 8).
In previous work we reported that the gyrB-225 muta-tion also enhances lethal action of first-generation quino-lones when chloramphenicol is present (Heddle et al.,2001b). When this mutation was introduced by transduc-tion into a strain carrying the gyrA-67 mutation, the result-ing double mutant was killed more extensively by nalidixicacid in the presence of chloramphenicol than either singlemutant, particularly at low quinolone concentration (notshown). Thus, the two mutations appear to destabilizedifferent aspects of gyrase structure, consistent with theirlocation in different genes.
Viscosity and sedimentation assays for chromosomefragmentation have different attributes. Viscosity is moresensitive to a small number of breaks, but the shearforces required for the measurement orient large DNAmolecules (for example, see Drlica and Worcel, 1975).Thus, the assay is empirical. Moreover, it provides littleinformation about DNA size heterogeneity. In contrast,sedimentation methods display the size distribution ofDNA molecules. In the present study both methodsrevealed large changes associated with quinolone treat-ment, as expected for the chromosome being cut intomany pieces (Snyder and Drlica, 1979). Because onedouble-stranded break is potentially lethal (Krasin andHutchinson, 1977) and because many are likely to be farfrom replication forks, collision of replication forks withternary complexes is unlikely to account for the DNAbreaks. However, our data do not exclude the occurrenceof chromosome lesions comparable to those detectedwith plasmid DNA when replication is blocked by a 6 mintreatment with norfloxacin (Pohlhaus and Kreuzer, 2005).It will be interesting to determine whether these plasmidlesions increase with longer incubation times and
Bacterial chromosome fragmentation 821
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 61, 810–825

disappear when norfloxacin lethality is blocked bychloramphenicol.
Chromosome fragmentation arising from the destabili-zation of gyrase–DNA complexes (Fig. 12, step d) has atleast two potential applications. First, it may contribute tothe development of new assays for inhibitors of type IIeukaryotic topoisomerases, because prokaryotic andeukaryotic type II topoisomerases probably act by similarmechanisms (Hong and Kreuzer, 2003) and becausetopoisomerase-mediated illegitimate recombination isobserved with both cell types (Asami et al., 2002; Ikedaet al., 2004). The destabilization hypothesis also providesa framework for quinolone action and the design of moreactive compounds. For example, pathway d occurs underanaerobic conditions and in the absence of DNA, RNAand protein synthesis. We recently found with mycobac-teria that chloramphenicol interferes with the lethal activityof all fluoroquinolones tested except moxifloxacin (Maliket al., 2005). Thus, moxifloxacin may become a lead com-pound for the development of new fluoroquinolones thatkill Mycobacterium tuberculosis during growth arrest.
Experimental procedures
Bacterial strains, culture conditions and quinolones
Relevant features of the E. coli strains used are described inTable 1. Cells were grown at 37°C as liquid cultures in Luria–Bertani (LB) medium (Miller, 1972); colonies were grown onLB agar plates. Nalidixic acid was obtained from SigmaChemical (St. Louis, MO). Investigational fluoroquinolonesPD160788, PD161144 and PD161148 were provided by DrJohn Domagala, Pfizer Global Research and Development,Ann Arbor, MI. Gatifloxacin was obtained from Bristol-Myers-Squibb (Wallingford, CT). Quinolones were dissolved in0.1 ml of 1 M NaOH (1/10 of the final volume), and thensterile water was added to obtain a final concentration of10 mg ml-1. This stock solution was divided into aliquots forstorage at -80°C. Dilutions were prepared with sterile distilledwater; solutions were kept at -20°C for several weeks duringexperiments.
Measurement of fluoroquinolone susceptibility
The MIC for 99% of the cells (MIC99) was determined graphi-cally from plots of quinolone concentration in agar versus thefraction of colony-forming units recovered. Lethal activity ofquinolones was determined by growing cells in LB liquidmedium to mid-log phase at 37°C with shaking (250 r.p.m.)and then incubating 2 ml aliquots with quinolone at variousconcentrations for various times. Shaking was continuedduring treatment. Cells were diluted, applied to LB agarplates lacking drug, and incubated overnight to allow colonyformation. All calculations of percent survival were relative toa no-drug control sampled at the time of quinolone addition.In selected experiments chloramphenicol was added to20 mg l-1 10 min prior to addition of quinolone and maintained
throughout treatment. Control experiments showed no effectfrom drug carryover to agar plates.
Cell lysis and preparation of bacterial nucleoids
Escherichia coli cell lysates were obtained by incubation withegg white lysozyme and non-ionic detergents at 20°C for2–3 min as described previously for isolation of membrane-free bacterial nucleoids (Snyder and Drlica, 1979). Nucleoidswere obtained as a rapidly sedimenting band during prepara-tive sucrose-density gradient centrifugation using the spermi-dine method for obtaining membrane-attached nucleoids(Kornberg et al., 1974). For viscosity experiments usingnucleoids (described below), cell lysates were large enoughfor the nucleoid band to be visible in preparative sucrosegradients. Preparative sucrose density-gradients contained10–30% sucrose, 10 mM Tris-HCl pH 8, 5 mM MgCl2 and50 mM NaCl. Centrifugation was for 18 min at 10 000 r.p.m.at 4°C with a Beckman SW50.1 rotor. When necessary, frac-tions were collected from the bottom of polyallomer centrifugetubes (Seton Scientific, Los Gatos, CA). For sedimentationstudies (described below), DNA was labelled by growth ofcells in 3H-thymidine (25 mCi ml-1 culture) for about one gen-eration prior to lysis or treatment with chloramphenicol and/orquinolone.
Detection of chromosome fragmentation by viscositymeasurement
Cell lysates were divided into aliquots (0.2 ml), and twofoldserial dilutions were distributed to 10 ¥ 75 mm glass tubes.Pancreatic ribonuclease was added to 20 mg l-1, andsamples were incubated at 80°C for 2 min to unfold chromo-somal DNA. Samples were chilled on ice and brought to 20°Cin a water bath. A 0.025 ml glass microcapillary pipet (KimbleGlass, Cat. no. 71900–25) was placed in each tube, and thetime required to fill the capillary, less the time for buffer alone,was taken as an empirical measure of viscosity. Nucleoids,obtained from preparative sucrose gradients as describedabove, were treated in a similar way except that they wereincubated at 37°C for 10 min, then at 30°C for 20 min prior tothe ribonuclease and 80°C steps to allow for treatment withgatifloxacin or other agents.
Detection of chromosome fragmentation bysedimentation measurement
3H-labelled DNA in lysates containing quinolone and chloram-phenicol when appropriate were diluted 10-fold in 0.02 MTris-HCl (pH 8) on ice with gentle mixing. Aliquots (0.02–0.04 ml) were then applied, with minimal shearing, to the topsurface of preformed 5–20% sucrose density-gradients con-taining 0.2% SDS, 0.1 M NaCl and 0.05 M sodium phosphatebuffer, pH 6.8. An additional 0.01 ml aliquot of 2% SDS wasthen layered on the top of the gradient. For resealing of DNAbreaks present in ternary complexes, cells were washed ingrowth medium prior to cell lysis to remove drugs and thenincubated for 10 min at 20°C after resuspension in 1 M NaCl,0.01 M Tris-HCl (pH 8). For radioactive nucleoids obtained by
822 M. Malik, X. Zhao and K. Drlica
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 61, 810–825

preparative sucrose density-gradient centrifugation, aliquotswere diluted 5- to 20-fold in 0.02 M Tris-HCl (pH 8) andapplied to the 5–20% sucrose gradients containing 0.2%SDS, NaCl and phosphate buffer. Resealing of DNA internary complexes for nucleoid experiments was achieved byadding 0.05 M EDTA to cell lysates or to nucleoidpreparations. Centrifugation was performed with a BeckmanSW50.1 or SW 55 Ti rotor at 20°C for speeds and timesindicated in figure legends. Fractions were collected ontoWhatman #2 filters (4.25 cm diameter) from the bottom of thecentrifuge tubes, and acid-precipitable radioactivity wasdetermined with a liquid scintillation spectrometer.
Preparation of gyrase and measurement ofchromosome fragmentation in vitro
DNA gyrase subunits A and B were prepared from E. colicontaining recombinant plasmids as described (Hallett et al.,1990; Maxwell and Howells, 1999). GyrA protein with anAla-67 to Ser substitution was prepared by site-directedmutagenesis with the wild-type GyrA overexpression plasmidpPH3 (Hallett et al., 1990) using a Stratagene (La Jolla, CA)Quickchange kit according to the manufacturer’s instructionsfollowed by overexpression and purification as described(Maxwell and Howells, 1999). Membrane-attached nucleoidswere prepared from preparative sucrose gradients asdescribed above following treatment of the wild-type strainSD104 with 30 mg l-1 coumermycin A1 for 15 min to relaxDNA supercoiling (Drlica and Snyder, 1978). Sucrose wasremoved by dialysis in gyrase-binding buffer (Hiasa andMarians, 1996). Gyrase (10 units), ATP (100 mM) and quino-lones were added followed by incubation at 30°C for 30 min.The preparation was chilled on ice, and viscosity was mea-sured as described above following treatment with RNaseand incubation at 80°C for 2 min.
Preparation of GyrA antiserum and assay forgyrase-bound DNA
Anti-GyrA antiserum was prepared commercially (LampireBiological Laboratories, Pipersville, PA) by immunizing arabbit with GyrA protein purified from E. coli. GyrA (0.4 mg in0.5 ml) was mixed with 0.5 ml of Incomplete Freund’s Adju-vant and injected. Booster doses using the same amount ofprotein were delivered at days 21, 42, 56 and 84. 3H-labellednucleoids, obtained from preparative sucrose density-gradients as described above, or DNA from 5 to 20% sucrosedensity-gradients were diluted fourfold in 0.02 M Tris-HCl(pH 8) and incubated overnight with a 1:60 dilution of rabbitantiserum at 4°C. Protein A-Sepharose (Pharmacia Biotech,Sweden) was added as a 1:10 dilution followed by incubationat room temperature for 2 h and then collection by low-speedcentrifugation. Beads were washed twice with 0.02 M Tris-HCl pH 6.8. Radioactivity associated with the beads wasdetermined by liquid scintillation counting.
Acknowledgements
We thank Gunda Schlaeffer for initiating the project, AnthonyMaxwell and Alison Howells for providing guidance and facili-
ties for the purification of DNA gyrase, and the following forcritical comments and discussions: John Domagala, MarilaGennaro, Sam Kayman, Anthony Maxwell, Will Parks,Richard Pine and Brian Quinn. The work was supported byNIH Grants AI 35257 and AI 063431.
References
Anderson, V.E., Zaniewski, R.P., Kaczmarek, F.S., Gootz,T.D., and Osheroff, N. (1999) Quinolones inhibit DNA reli-gation mediated by Staphylococcus aureus topoisomeraseIV. J Biol Chem 274: 35927–35932.
Anderson, V., Zaniewski, R., Kaczmarek, F., Gootz, T., andOsheroff, N. (2000) Action of quinolones against Staphy-lococcus aureus topoisomerase IV: basis for DNA cleavageenhancement. Biochem 39: 2726–2732.
Asami, Y., Jia, D., Tatebayashi, K., Yamagata, K., Tanokura,M., and Ikeda, H. (2002) Effect of the DNA topoisomeraseII inhibitor VP-16 on illegitimate recombination in yeastchromosomes. Gene 291: 251–257.
Bernard, P., and Couturier, M. (1992) Cell killing by the Fplasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes. J Mol Biol 226: 735–745.
Bromberg, K.D., Burgin, A.B., and Osheroff, N. (2003)Quinolone action against human topoisomerase IIalpha:stimulation of enzyme-mediated double-stranded DNAcleavage. Biochemistry 42: 3393–3398.
Chen, C.-R., Malik, M., Snyder, M., and Drlica, K. (1996) DNAgyrase and topoisomerase IV on the bacterial chromo-some: quinolone-induced DNA cleavage. J Mol Biol 258:627–637.
Chow, R., Dougherty, T., Fraimow, H., Bellin, E., and Miller,M. (1988) Association between early inhibition of DNAsynthesis and the MICs and MBCs of carboxyquinoloneantimicrobial agents for wild-type and mutant [gyrA nfxB(ompF) acrA] Escherichia coli K-12. Antimicrob AgentsChemother 32: 1113–1118.
Crumplin, G.C., and Smith, J.T. (1975) Nalidixic acid: anantibacterial paradox. Antimicrob Agents Chemother 8:251–261.
D’Arpa, P., Beardmore, C., and Liu, L.F. (1990) Involvementof nucleic acid synthesis in cell killing mechanisms of topoi-somerase poisons. Cancer Res 50: 6919–6924.
Deitz, W.H., Cook, T.M., and Goss, W.A. (1966) Mechanismof action of nalidixic acid on Escherichia coli. III. Conditionsrequired for lethality. J Bacteriol 91: 768–773.
DiNardo, S., Voelkel, K., and Sternglanz, R. (1984) DNAtopoisomerase II mutant of Saccharomyces cerevisiae:topoisomerase II is required for segregation of daughtermolecules at the termination of DNA replication. Proc NatlAcad Sci USA 81: 2616–2620.
Drlica, K., and Malik, M. (2003) Fluoroquinolones: action andresistance. Current Topics Med Chem 3: 1349–1364.
Drlica, K., and Snyder, M. (1978) Superhelical Escherichiacoli DNA: relaxation by coumermycin. J Mol Biol 120: 145–154.
Drlica, K., and Worcel, A. (1975) Conformational transitions inthe Escherichia coli chromosome: analysis by viscometryand sedimentation. J Mol Biol 98: 393–411.
Drlica, K., Xu, C., Wang, J.-Y., Burger, R.M., and Malik, M.(1996) Fluoroquinolone action in mycobacteria: similarity
Bacterial chromosome fragmentation 823
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 61, 810–825

with effects in Escherichia coli and detection by cell lysateviscosity. Antimicrob Agents Chemother 40: 1594–1599.
Gellert, M., Mizuuchi, K., O’Dea, M.H., Itoh, T., and Tomi-zawa, J.-L. (1977) Nalidixic acid resistance: a secondgenetic character involved in DNA gyrase activity. ProcNatl Acad Sci USA 74: 4772–4776.
Hallett, P., Grimshaw, A., Wigley, D., and Maxwell, A. (1990)Cloning of the DNA gyrase genes under tac promotercontrol: overexpression of the gyrase A and B proteins.Gene 93: 139–142.
Heddle, J.G., Barnard, F., Wentzell, L., and Maxwell, A.(2000) The interaction of drugs with DNA gyrase: a modelfor the molecular basis of quinolone action. NucleosidesNucleotides Nucl Acids 19: 1249–1264.
Heddle, J., Blance, S., Zamble, D., hollfelder, F., Miller, D.,Wentzell, L., et al. (2001a) The antibiotic microcin B17 is agyrase poison: characterization of the mode of inhibition. JMol Biol 307: 1223–1234.
Heddle, J., Lu, T., Zhao, X., Drlica, K., and Maxwell, A.(2001b) gyrB-225, a mutation of DNA gyrase that compen-sates for topoisomerase I deficiency: investigation of its lowactivity and quinolone hypersensitivity. J Mol Biol 309:1219–1231.
Hiasa, H., and Marians, K.J. (1996) Two distinct modes ofstrand unlinking during theta-type DNA replication. J BiolChem 271: 21529–21535.
Hiasa, H., Yousef, D., and Marians, K. (1996) DNA strandcleavage is required for replication fork arrest by a frozentopoisomerase-quinolone-DNA ternary complex. J BiolChem 271: 26424–26429.
Hong, G., and Kreuzer, K. (2003) Endonuclease cleavage ofblocked replication forks: an indirect pathway of DNAdamage from antitumor drug-topoisomerase complexes.Proc Natl Acad Sci USA 100: 5046–5051.
Howard, B.M., Pinney, R.J., and Smith, J.T. (1993) Functionof the SOS process in repair of DNA damage induced bymodern 4-quinolones. J Pharm Pharmacol 45: 658–662.
Ikeda, H. (1986) Illegitimate recombination: role of type IIDNA topoisomerase. Adv Biophys 21: 149–160.
Ikeda, H., Shiraishi, K., and Ogata, Y. (2004) Illegitimaterecombination mediated by double-strand break and end-joining in Escherichia coli. Adv Biophys 38: 3–20.
Khodursky, A.B., Zechiedrich, E.L., and Cozzarelli, N.R.(1995) Topoisomerase IV is a target of quinolones inEscherichia coli. Proc Nat Acad Sci USA 92: 11801–11805.
Kornberg, T., Lockwood, A., and Worcel, A. (1974) Replica-tion of the Escherichia coli chromosome with a solubleenzyme system. Proc Natl Acad Sci USA 71: 3189–3193.
Krasin, F., and Hutchinson, F. (1977) Repair of DNA double-strand breaks in Escherichia coli, which requires recA func-tion and the presence of a duplicate genome. J Mol Biol116: 81–98.
Lewin, C., Howard, B., Ratcliffe, N., and Smith, J. (1989)4-Quinolones and the SOS response. J Med Microbiol 29:139–144.
Liu, L.F., Desai, S., Li, T., Mao, Y., Sun, M., and Sim, S.(2000) Mechanism of action of camptothecin. Ann N YAcad Sci 922: 1–10.
Lu, T., Zhao, X., and Drlica, K. (1999) Gatifloxacin activityagainst quinolone-resistant gyrase: allele-specific enhance-ment of bacteriostatic and bactericidal activity by the C-8-
methoxy group. Antimicrob Agents Chemother 43: 2969–2974.
Malik, M., Lu, T., Zhao, X., Singh, A., Hattan, C., Domagala,J., et al. (2005) Lethality of quinolones agains Mycobacte-rium smegmatis in the presence or absence ofchloramphenicol. Antimicrob Agents Chemother 49: 2008–2014.
Maxwell, A., and Howells, A. (1999) Overexpression andpurification of bacterial DNA gyrase. Meth Mol Biol 94:135–144.
Miller, J. (1972) Experiments in Molecular Genetics. ColdSpring Harbor, NY: Cold Spring Harbor Laboratory Press.
Mizuuchi, K., Fisher, L.M., O’Dea, M., and Gellert, M. (1980)DNA gyrase action involves the introduction of transientdouble-strand breaks into DNA. Proc Natl Acad Sci USA77: 1847–1851.
MoraisCabral, J.H., Jackson, A.P., Smith, C.V., Shikotra, N.,Maxwell, A., and Liddington, R.C. (1997) Crystal structureof the breakage-reunion domain of DNA gyrase. Nature388: 903–906.
Morrison, A., Higgins, N.P., and Cozzarelli, N.R. (1980) Inter-action between DNA gyrase and its cleavage site on DNA.J Biol Chem 255: 2211–2219.
Pohlhaus, J., and Kreuzer, K.N. (2005) Norfloxacin-inducedDNA gyrase cleavage complexes block Escherichia colireplication forks, causing double-stranded breaks in vivo.Mol Microbiol 56: 1416–1429.
Portalier, R., and Worcel, A. (1976) Association of the foldedchromosome with the cell envelope of E. coli: character-ization of the proteins at the DNA-membrane attachmentsite. Cell 8: 245–255.
Sato, K., Inoue, Y., Fuji, T., Aoyama, H., Inoue, M., andMitsuhashi, S. (1986) Purification and properties of DNAgyrase from a fluoroquinolone-resistant strain of Escheri-chia coli. Antimicrob Agents Chemother 30: 777–780.
Shea, M., and Hiasa, H. (1999) Interactions between DNAhelicases and frozen topoisomerase IV-quinolone-DNAternary complexes. J Biol Chem 274: 22747–22754.
Shea, M., and Hiasa, H. (2000) Distinct effects of the UvrDhelicase on topoisomerase-quinolone-DNA ternary com-plexes. J Biol Chem 275: 14649–14658.
Shea, M., and Hiasa, H. (2003) The RuvAB branch migrationcomplex can displace topoisomerase IV-quinolone-DNAternary complexes. J Biol Chem 278: 48485–48490.
Snyder, M., and Drlica, K. (1979) DNA gyrase on the bacterialchromosome: DNA cleavage induced by oxolinic acid. JMol Biol 131: 287–302.
Stonington, O.G., and Pettijohn, D.E. (1971) The foldedgenome of Escherichia coli isolated in a protein–DNA–RNA complex. Proc Natl Acad Sci USA 68: 6–9.
Sugino, A., Peebles, C., Kruezer, K., and Cozzarelli, N.(1977) Mechanism of action of nalidixic acid: purification ofEscherichia coli nalA gene product and its relationship toDNA gyrase and a novel nicking-closing enzyme. Proc NatlAcad Sci USA 74: 4767–4771.
Vizan, J.L., Hernandez-Chico, C.I., Castillo, I., and Moreno,F. (1991) The peptide antibiotic microcin B17 inducesdouble-strand cleavage of DNA mediated by E. coli DNAgyrase. EMBO J 10: 467–476.
Wentzell, L., and Maxwell, A. (2000) The complex of DNAgyrase and quinolone drugs on DNA forms a barrier to the
824 M. Malik, X. Zhao and K. Drlica
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 61, 810–825

T7 DNA polymerase replication complex. J Mol Biol 304:779–791.
Williams, N.L., and Maxwell, A. (1999) Locking the DNA gateof DNA gyrase: investigating the effects on DNA cleavageand ATP hydrolysis. Biochemistry 38: 14157–14164.
Willmott, C.J.R., Critchlow, S.E., Eperon, I.C., and Maxwell,A. (1994) The complex of DNA gyrase and quinolone drugswith DNA forms a barrier to transcription by RNApolymerase. J Mol Biol 242: 351–363.
Wilstermann, A., and Osheroff, N. (2003) Stabilization ofeukaryotic topoisomerase II-DNA cleavage complexes.Curr Topics Med Chem 3: 321–338.
Wolfson, J.S., Hooper, D.C., Shih, D.J., McHugh, G.L., andSwartz, M.N. (1989) Isolation and characterization of an
Escherichia coli strain exhibiting partial tolerance toquinolones. Antimicrob Agents Chemother 33: 705–709.
Worcel, A., and Burgi, E. (1972) On the structure of thefolded chromosome of Escherichia coli. J Mol Biol 71:127–147.
Yoshida, H., Bogaki, M., Nakamura, M., and Nakamura, S.(1990) Quinolone resistance-determining region in theDNA gyrase gyrA gene of Escherichia coli. AntimicrobAgents Chemother 34: 1271–1272.
Zhao, X., Malik, M., Chan, N., Drlica-Wagner, A., Wang, J.-Y.,Li, X., and Drlica, K. (2006) Lethal action of quinolones witha temperature-sensitive dnaB replication mutant ofEscherichia coli. Antimicrob Agents Chemother 50: 362–364.
Bacterial chromosome fragmentation 825
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 61, 810–825
Copyright © 2022 FDOKUMEN




















