
Laboratory accelerated and natural weathering of styrene–ethylene–butylene–styrene (SEBS)...
7
Laboratory accelerated and natural weathering of styreneeethyleneebutyleneestyrene (SEBS) block copolymer C.C. White * , K.T. Tan * , D.L. Hunston, T. Nguyen, D.J. Benatti, D. Stanley, J.W. Chin National Institute of Standards and Technology,100 Bureau Dr. MS 8615, Gaithersburg, MD 20899-8615, USA article info Article history: Received 29 September 2010 Received in revised form 20 January 2011 Accepted 10 March 2011 Available online 23 March 2011 Keywords: Thermoplastic elastomers Aging Degradation Outdoor exposure SEBS Weathering abstract Indoor accelerated and outdoor weathering of polystyrene-b-(ethylene-co-butylene)-b-styrene (SEBS) was studied by infrared spectroscopy. Accelerated conditions involved simultaneous exposure of spec- imens to ultravioletevisible radiation between 295 nm and 450 nm and each of four temperature/ relative humidity (RH) environments, i.e., (a) 30 C 1 C at <1% RH, (b) 30 C 1 C at 80% RH, (c) 55 C 1 C at <1% RH, and (d) 55 C 1 C at 80% RH. Outdoor exposure was conducted in Gai- thersburg, MD, in two different time periods. Similar photooxidative mechanisms were operative under all conditions. In the case of indoor accelerated exposure, the rate of photooxidation was found to depend strongly on temperature. Unlike the exposure at 55 C, moisture-assisted photooxidation was insignifi- cant at 30 C. A quantitative study on the synergistic effect of environmental stressors revealed that the degrading effect of combined temperature and moisture on photooxidation was greater than the sum of the two effects exerted independently. Outdoor weathered specimens exhibited significantly slower photooxidation. Acceleration of photooxidation ranged from 2.5 to 10 times in comparison to the outdoor exposure, depending on the indoor accelerated conditions. Published by Elsevier Ltd. 1. Introduction Thermoplastic elastomers (TPEs) are commercially important materials due to their attractive combination of fluid processing characteristics, excellent chemical resistance, and desirable physical properties equivalent to vulcanized rubbers [1,2]. Since their commercial introduction, many types of TPEs have been produced. The largest fraction of the TPE market are materials based on block copolymers of polystyrene (PS) and elastomer having basic structures of poly(styreneebutadieneestyrene) (SBS) or poly(- styreneeisopreneestyrene) (SIS). These materials are often exposed to various environmental stressors, including temperature, ozone, moisture, ultraviolet (UV) radiation and oxygen, which inevitably result in some undesirable modifications to their properties. Indeed, studies have shown that weathering leads to a variety of changes ranging from surface discoloration to extensive decreases in mechanical properties [3,4]. In many instances, combinations of these stressors may be encountered, producing potential synergism that may be more aggressive in terms of environmental attack than the individual factors acting independently. For SBS and SIS, the elastomeric phase is known to be inherently more susceptible to degradation than the PS phase [5,6]. This is ascribed to the presence of reactive double bonds in, and the low glasserubber transition temperature (T g ) of, the elastomeric phase. As a result of commercial importance of these polymers [5,7e10], oxidative degradation of SBS and SIS under UV exposure has been extensively reported in the literature. Under UV radiation, these block copolymers undergo autocatalytic degradation reactions that lead to the formation of various oxygenated products and crosslinking, which result in extensive embrittlement and premature failure. A second generation of PS-elastomer block copolymers con- taining hydrogenated elastomeric mid-blocks was brought to the marketplace ten years after commercial introduction of SBS [11]. These new materials are poly(styreneeethyleneebutylenee styrene) (SEBS) copolymers and have been claimed to be more resistant to degradation than SBS and SIS copolymers [11,12]. The improved weathering resistance of SEBS is due to the chemical resistance of the saturated rubbery segments [11,12]. However, as with other unmodified organic polymers, SEBS undergo degrada- tion upon exposure to environmental extremes [13e15]. For instance, Allen et al. [15] showed that SEBS decomposes via chain scission at the linkage between PS and olefin phases at 180 C, yielding acetophenone end groups at the PS phase and carboxylic acids at the olefin chain-ends. These studies further showed that there is extensive oxidation and crosslinking associated with * Corresponding authors. E-mail addresses: [email protected] (C.C. White), [email protected] (K.T. Tan). Contents lists available at ScienceDirect Polymer Degradation and Stability journal homepage: www.elsevier.com/locate/polydegstab 0141-3910/$ e see front matter Published by Elsevier Ltd. doi:10.1016/j.polymdegradstab.2011.03.003 Polymer Degradation and Stability 96 (2011) 1104e1110
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Laboratory accelerated and natural weathering of styrene–ethylene–butylene–styrene (SEBS)...

lable at ScienceDirect
Polymer Degradation and Stability 96 (2011) 1104e1110
Contents lists avai
Polymer Degradation and Stability
journal homepage: www.elsevier .com/locate/polydegstab
Laboratory accelerated and natural weatheringof styreneeethyleneebutyleneestyrene (SEBS) block copolymer
C.C. White*, K.T. Tan*, D.L. Hunston, T. Nguyen, D.J. Benatti, D. Stanley, J.W. ChinNational Institute of Standards and Technology, 100 Bureau Dr. MS 8615, Gaithersburg, MD 20899-8615, USA
a r t i c l e i n f o
Article history:Received 29 September 2010Received in revised form20 January 2011Accepted 10 March 2011Available online 23 March 2011
Keywords:Thermoplastic elastomersAgingDegradationOutdoor exposureSEBSWeathering
* Corresponding authors.E-mail addresses: [email protected] (C
(K.T. Tan).
0141-3910/$ e see front matter Published by Elsevierdoi:10.1016/j.polymdegradstab.2011.03.003
a b s t r a c t
Indoor accelerated and outdoor weathering of polystyrene-b-(ethylene-co-butylene)-b-styrene (SEBS)was studied by infrared spectroscopy. Accelerated conditions involved simultaneous exposure of spec-imens to ultravioletevisible radiation between 295 nm and 450 nm and each of four temperature/relative humidity (RH) environments, i.e., (a) 30 �C � 1 �C at <1% RH, (b) 30 �C � 1 �C at 80% RH,(c) 55 �C � 1 �C at <1% RH, and (d) 55 �C � 1 �C at 80% RH. Outdoor exposure was conducted in Gai-thersburg, MD, in two different time periods. Similar photooxidative mechanisms were operative underall conditions. In the case of indoor accelerated exposure, the rate of photooxidation was found to dependstrongly on temperature. Unlike the exposure at 55 �C, moisture-assisted photooxidation was insignifi-cant at 30 �C. A quantitative study on the synergistic effect of environmental stressors revealed that thedegrading effect of combined temperature and moisture on photooxidation was greater than the sum ofthe two effects exerted independently. Outdoor weathered specimens exhibited significantly slowerphotooxidation. Acceleration of photooxidation ranged from 2.5 to 10 times in comparison to the outdoorexposure, depending on the indoor accelerated conditions.
Published by Elsevier Ltd.
1. Introduction
Thermoplastic elastomers (TPEs) are commercially importantmaterials due to their attractive combination of fluid processingcharacteristics, excellent chemical resistance, and desirable physicalproperties equivalent to vulcanized rubbers [1,2]. Since theircommercial introduction, many types of TPEs have been produced.The largest fraction of the TPE market are materials based onblock copolymers of polystyrene (PS) and elastomer having basicstructures of poly(styreneebutadieneestyrene) (SBS) or poly(-styreneeisopreneestyrene) (SIS). These materials are often exposedto various environmental stressors, including temperature, ozone,moisture, ultraviolet (UV) radiation and oxygen, which inevitablyresult in some undesirable modifications to their properties. Indeed,studies have shown that weathering leads to a variety of changesranging from surface discoloration to extensive decreases inmechanical properties [3,4]. Inmany instances, combinationsof thesestressors may be encountered, producing potential synergism thatmay be more aggressive in terms of environmental attack than theindividual factors acting independently.
.C. White), [email protected]
Ltd.
For SBS and SIS, the elastomeric phase is known to be inherentlymore susceptible to degradation than the PS phase [5,6]. This isascribed to the presence of reactive double bonds in, and the lowglasserubber transition temperature (Tg) of, the elastomeric phase.As a result of commercial importance of these polymers [5,7e10],oxidative degradation of SBS and SIS under UV exposure has beenextensively reported in the literature.UnderUVradiation, these blockcopolymers undergo autocatalytic degradation reactions that lead tothe formation of various oxygenated products and crosslinking,which result in extensive embrittlement and premature failure.
A second generation of PS-elastomer block copolymers con-taining hydrogenated elastomeric mid-blocks was brought to themarketplace ten years after commercial introduction of SBS[11]. These new materials are poly(styreneeethyleneebutyleneestyrene) (SEBS) copolymers and have been claimed to be moreresistant to degradation than SBS and SIS copolymers [11,12]. Theimproved weathering resistance of SEBS is due to the chemicalresistance of the saturated rubbery segments [11,12]. However, aswith other unmodified organic polymers, SEBS undergo degrada-tion upon exposure to environmental extremes [13e15]. Forinstance, Allen et al. [15] showed that SEBS decomposes via chainscission at the linkage between PS and olefin phases at 180 �C,yielding acetophenone end groups at the PS phase and carboxylicacids at the olefin chain-ends. These studies further showed thatthere is extensive oxidation and crosslinking associated with

0
1
2
3
4
5
250 350 450 550 650Wavelength (nm)
Irra
dia
nc
e (
Wm
-2
nm
-1
)
0
0.5
1
1.5
2
2.5
c
b
d
e
a
Fig. 1. Spectral distribution of output UVevisible radiation from (a) the SPHERE, andthe sunlight recorded on July 31, 2006 at a time of (b) 09:00, (c) 12:00, (d) 15:00, and(e) 18:00.
C.C. White et al. / Polymer Degradation and Stability 96 (2011) 1104e1110 1105
hydroperoxides that are formed initially in the olefinic phase but noevidence of crosslinking was found in the PS phase.
To date, the susceptibility of SEBS to a combination of temper-ature, humidity and UVevisible irradiation has not been adequatelystudied. The interaction of these environmental stressors, eitheracting independently or in combination, will affect polymer prop-erties and determine their long-term service life. Hence, anunderstanding of agingmechanisms due to various stressors wouldbe useful for development of strategies to improve materialperformance. Also, increasing demand for shorter product devel-opment cycles have led to urgent needs for meaningful acceleratedtest methodologies to generate durability data. The present study,therefore, has three major objectives. The first objective is to usea laboratory accelerated testing protocol for screening the relativeimportance of two aging stressors e temperature and humidity e
acting independently and in combination on the photooxidation ofSEBS. A second objective is to employ this test methodology toidentify the failure mechanisms that are operative during theaccelerated and outdoor tests. A third objective is to establisha linkage between accelerated tests and outdoor field exposuretests, since such a linkage is required for the accelerated test me-thodology to be meaningful.
2. Experimental1
2.1. Materials and specimen preparation
The SEBS block copolymer used was a commercial gradematerial having the chemical structure shown below:
CH2 bm
CH CH2CH2 CHCH2
CH2
CH3
co CH2CHbn m2
Specimens were prepared from solution casting using a mixtureof 20% mass fraction of the copolymer in toluene. Mixing wasperformed using a mechanical stirrer. After degassing the mixturewith a vacuum pump, the solution was spin-cast on calcium fluo-ride (CaF2) disks (19 mm diameter � 4 mm thick) at 104.7 rad s�1
for 20 s in a virtually CO2-free dry glove box. CaF2 disks wereselected as the substrate due to their excellent heat and moistureresistance properties, and their transparency to both UV andinfrared radiation [16]. The coated disks were cured at 50 �C for anhour under nitrogen to minimize oxidation. The thickness of theresulting coatings ranged from 20 mm to 30 mm.
2.2. Exposure conditions
Specimens were exposed to both indoor accelerated andoutdoor conditions. In the indoor accelerated environments, thespecimens were exposed to UVevisible radiationwith wavelengthsranging between 295 nm and 450 nm using the NIST integratingsphere-based weathering chamber (Simulated Photodegradationvia High Energy Radiant Exposure (SPHERE) [17]). The spectralemission distribution of the UVevisible lamp is shown in Fig. 1a.During UVevisible irradiation, specimens were subjected toeach of four temperature/relative humidity (RH) environments:
1 Certain commercial products or equipment are described in this paper in orderto specify adequately the experimental procedure. In no case does such identifi-cation imply recommendation or endorsement by the National Institute of Stan-dards and Technology (NIST), nor does it imply that it is necessarily the bestavailable for the purpose.
(a) 30 �C � 1 �C and <1% RH, (b) 30 �C � 1 �C and 80% RH,(c) 55 �C � 1 �C and <1% RH and (d) 55 �C � 1 �C and 80% RH.
For outdoor tests, two sets of specimens with different exposurestarting dates (May and July 2006) were exposed on the roof ofa NIST laboratory in Gaithersburg, MD. These particular startingdates were of interest because the extended duration of sunlight,high temperatures and high RHs during the summer are delete-rious and can accelerate photochemical reactions in polymers usedoutdoors. Specimens were placed in a custom-built chamber thatwas positioned facing south toward the equator at an angle of 5�
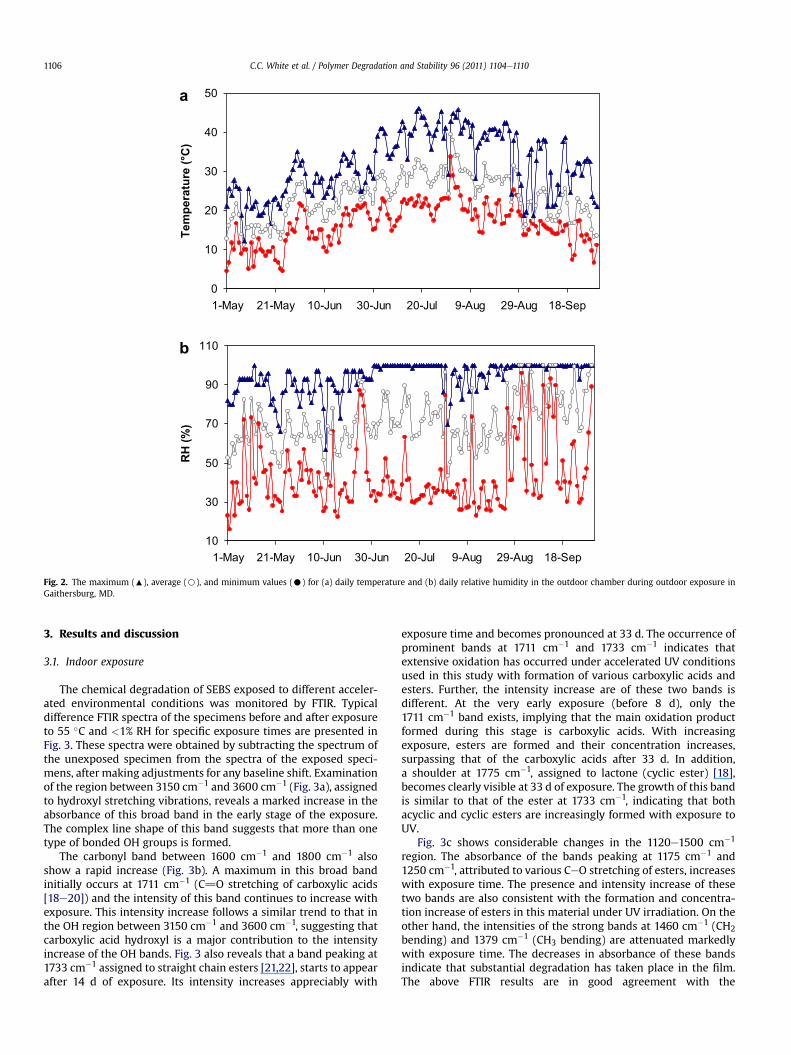
from the horizontal plane. Solar radiation reached the specimensthrough a borosilicate glass plate at the top of the chamber. Theupper surface of the bottom side of the chamber consisted ofa 3 mm thick polytetrafluoroethylene sheet, which was used tominimize heating in the chamber via reflection of the solar radia-tion; while the sides were made with a breathable cloth thatallowed transmission of water vapor, but prevented dust fromentering the chamber. The temperature, RH and total irradiance inthe chamber were monitored continuously during exposure usingthermocouples, humidity sensors, and a radiometer, respectively.Fig. 1bee show representative solar spectral irradiance distributionrecorded at several different times in a clear day at the exposuresite. It can be seen that the solar wavelength spectrum changesconstantly during a daily cycle and extends to z295 nm, which isconsistent with the cut-off wavelength range for the artificial lightsource used in the present indoor accelerated study (see Fig. 1a).Also, during this period, daily temperatures in the chamber variedbetween 7 �C and 46 �C, and RHs ranged from 23% to 100%, asshown in Fig. 2.
2.3. Spectroscopic characterization
FTIR measurements were carried out in transmission modeusing a Nexus 670 spectrometer equipped with a liquid nitrogen-cooled mercury cadmium telluride detector. All spectra werecollected over a range of 1000e4000 cm�1 at a nominal resolutionof 4 cm�1 and averaged over 128 scans. Band height was used asa measure of FTIR intensity. Five replicates were measured for eachexposure environment and the error bars in the figures representplus or minus one standard deviation, �1s, from the mean values.
0
10
20
30
40
50
1-May 21-May 10-Jun 30-Jun 20-Jul 9-Aug 29-Aug 18-Sep
Tem
peratu
re (°C
)
10
30
50
70
90
110
1-May 21-May 10-Jun 30-Jun 20-Jul 9-Aug 29-Aug 18-Sep
RH
(%
)
a
b
Fig. 2. The maximum (:), average (B), and minimum values (C) for (a) daily temperature and (b) daily relative humidity in the outdoor chamber during outdoor exposure inGaithersburg, MD.
C.C. White et al. / Polymer Degradation and Stability 96 (2011) 1104e11101106
3. Results and discussion
3.1. Indoor exposure
The chemical degradation of SEBS exposed to different acceler-ated environmental conditions was monitored by FTIR. Typicaldifference FTIR spectra of the specimens before and after exposureto 55 �C and <1% RH for specific exposure times are presented inFig. 3. These spectra were obtained by subtracting the spectrum ofthe unexposed specimen from the spectra of the exposed speci-mens, after making adjustments for any baseline shift. Examinationof the region between 3150 cm�1 and 3600 cm�1 (Fig. 3a), assignedto hydroxyl stretching vibrations, reveals a marked increase in theabsorbance of this broad band in the early stage of the exposure.The complex line shape of this band suggests that more than onetype of bonded OH groups is formed.
The carbonyl band between 1600 cm�1 and 1800 cm�1 alsoshow a rapid increase (Fig. 3b). A maximum in this broad bandinitially occurs at 1711 cm�1 (C]O stretching of carboxylic acids[18e20]) and the intensity of this band continues to increase withexposure. This intensity increase follows a similar trend to that inthe OH region between 3150 cm�1 and 3600 cm�1, suggesting thatcarboxylic acid hydroxyl is a major contribution to the intensityincrease of the OH bands. Fig. 3 also reveals that a band peaking at1733 cm�1 assigned to straight chain esters [21,22], starts to appearafter 14 d of exposure. Its intensity increases appreciably with
exposure time and becomes pronounced at 33 d. The occurrence ofprominent bands at 1711 cm�1 and 1733 cm�1 indicates thatextensive oxidation has occurred under accelerated UV conditionsused in this study with formation of various carboxylic acids andesters. Further, the intensity increase are of these two bands isdifferent. At the very early exposure (before 8 d), only the1711 cm�1 band exists, implying that the main oxidation productformed during this stage is carboxylic acids. With increasingexposure, esters are formed and their concentration increases,surpassing that of the carboxylic acids after 33 d. In addition,a shoulder at 1775 cm�1, assigned to lactone (cyclic ester) [18],becomes clearly visible at 33 d of exposure. The growth of this bandis similar to that of the ester at 1733 cm�1, indicating that bothacyclic and cyclic esters are increasingly formed with exposure toUV.
Fig. 3c shows considerable changes in the 1120e1500 cm�1
region. The absorbance of the bands peaking at 1175 cm�1 and1250 cm�1, attributed to various CeO stretching of esters, increaseswith exposure time. The presence and intensity increase of thesetwo bands are also consistent with the formation and concentra-tion increase of esters in this material under UV irradiation. On theother hand, the intensities of the strong bands at 1460 cm�1 (CH2bending) and 1379 cm�1 (CH3 bending) are attenuated markedlywith exposure time. The decreases in absorbance of these bandsindicate that substantial degradation has taken place in the film.The above FTIR results are in good agreement with the

310032003300340035003600
Wavenumber (cm-1
)
8 d
14 d
21 d
33 d
Ab
so
rb
an
ce
0.04 AU
16501700175018001850
Wavenumber (cm-1
)
8 d
14 d
21 d
33 d
Ab
so
rb
an
ce
17111733
1775
0.4 AU
11201200128013601440
Wavenumber (cm-1
)
11751250
1379
1460
8 d
14 d
21 d
33 d
Ab
so
rb
an
ce
0.2 AU
a b
c
Fig. 3. FTIR difference spectra for various exposure times at 55 �C and <1% RH for different spectral regions: (a) 3600e3100 cm�1, (b) 1850e1650 cm�1, and (c) 1500e1120 cm�1.Only spectra for four exposure times are shown for clarity.
C.C. White et al. / Polymer Degradation and Stability 96 (2011) 1104e1110 1107
photooxidative mechanism proposed by Allen et al. [15,18,19] forSEBS in which chain scission and oxidation primarily occurring atlinkages between styrenic and olefin phases.
Fig. 4 shows the correlation plot between the absorbance of twocharacteristic FTIR bands associated with photooxidation, i.e.,1711 cm�1 (C]O stretching of carboxylic acids [18e20]) and3350 cm�1 (bonded OH stretching) [15,18] for all four exposureconditions. All data points fall on the same line, indicating that therelative proportions of the photoproducts are independent of the
0
0.04
0.08
0.12
0.16
0 0.4 0.8 1.2 1.6 2
Absorbance of 1711 cm-1
Ab
so
rb
an
ce
o
f 3
35
0 c
m-1
Fig. 4. Correlation between absorbance of 3350 cm�1 and 1711 cm�1 for SEBS exposedto different environments: (-) 30 �C and <1% RH, (B) 30 �C and 80% RH, (6)55 �Cand <1% RH, and (:) 55 �C and 80% RH. Error bars represent �1s from the meanvalues.
exposure conditions. Basically, for different exposure conditions,when the peak ratios show a similar trend, the degradationmechanisms tend to be similar, even though the kinetics of thedegradation are quite different. Thus, the superposition of thesefour curves suggests that the primary mechanism of photooxida-tion in the SEBS specimens does not vary with temperature or RH.
Changes in the FTIR band intensities related to bonded OHstretching (3350 cm�1) as a function of exposure time (Fig. 5) wereexamined to investigate the thermal and moisture effects onphotooxidation. It can be seen that the absorbance of this bandinitially increases with exposure time for all four environments. At30 �C and<1% RH or 30 �C and 80% RH, the absorbance continues toincrease within the timescale of the experiment, i.e., 130 d. Therates of absorbance increase initially remains constant for 55 �C and80% RH or 55 �C and<1% RH but then decrease after about 20 d and40 d, respectively. A decrease in absorbance at 55 �C and 80% RH isevident after prolonged exposure. Owing to limited experiments,the reasons for the falloff in absorbance in the later stages ofexposure are too speculative to warrant detailed analysis at thispoint. One possibility is due to the strong affinity of water mole-cules to the oxidation products that are highly hydrophilic. Thisresults in dissolution/washing of oxidative products by watermolecules and subsequent drainage because the specimens weremounted vertically in the chambers to promote cleaner surfaces.Therefore, the absorbance recorded after prolonged exposure at55 �C does not meaningfully reflect the primary mechanism.
The coupled effects of UVevisible irradiation and moisture ondegradation were assessed by examining the initial formation rateof bonded OH stretching at the same temperature. The initial rate isdetermined by a least-squares fitting of the initial linear region ofeach curve, and the values obtained are shown in Table 1. Relativelyhigh values of corresponding correlation coefficients (R2 > 0.98)confirm the reliability of the slopes obtained. Notably, the coupledeffect of UVevisible irradiation and moisture at 30 �C is

0
0.04
0.08
0.12
0.16
0 20 40 60 80 100 120 140
Time of exposure (days)
Ab
so
rb
an
ce
of 3
35
0 c
m-1
Fig. 5. Changes of the absorbance of 3350 cm�1 as a function of exposure time fordifferent environments: (-) 30 �C and <1% RH, (B) 30 �C and 80% RH, (6) 55 �C and<1% RH, and (:) 55 �C and 80% RH. Error bars represent �1s from the mean values.
0
0.4
0.8
1.2
1.6
0 20 40 60 80 100 120 140
Time of exposure (days)
Ab
so
rb
an
ce
o
f 1
71
1 c
m-1
Fig. 6. Changes of the absorbance of 1711 cm�1 as a function of exposure time fordifferent environments: (-) 30 �C and <1% RH, (B) 30 �C and 80% RH, (6) 55 �C and<1% RH, (:) 55 �C and 80% RH, (,) and (C) outdoor exposures started in May 2006and July 2006, respectively. Error bars represent �1s from the mean values.
C.C. White et al. / Polymer Degradation and Stability 96 (2011) 1104e11101108
insignificant as shown by the fact that at 30 �C the formation ratesof bonded OH stretching are statistically similar for both RHs. Incontrast, an increased rate of more than 30% in the formation ofbonded OH stretching at 55 �C and 80% RH compared to that at55 �C and <1% RH. Similar trends are noted with changes incarbonyl-containing compounds (1711 cm�1) as a function ofexposure time as shown in Fig. 6, further reinforcing the observa-tions made with the bonded OH stretching. It is possible that therewas some plasticization by absorbed water molecules occurring inthe specimens at 55 �C and 80% RH, and this enhanced the rate ofoxygen and radical diffusion. The degradation in a solid polymer isgenerally a diffusion-controlled process, thus the increase inoxygen and radical diffusion in the materials provide greaterpotential for degradation.
Turning now to the effect of temperature, higher temperatureleads to a faster initial photooxidation rate as shown in Fig. 5. Inparticular, the temperature-enhanced photooxidation is man-ifested in significantly higher initial rates of bonded OH stretchingthat are approximately three and four times greater than that of thelower temperature exposures at <1% RH and 80% RH, respectively.This increase in rate is attributed to the increase in average kineticenergy of polymeric chains and other reactants with increasingtemperature, increases the probability of molecular interaction toreact with each others, and thereby leads to a faster photooxida-tion. In addition, temperature contributes directly to degradationby increasing the diffusion rates of oxygen and radicals, furtherenhancing the accessibility of oxygen and radicals for the photo-oxidative process [23]. However, chemical degradation directly dueto thermal effect from 30 �C or 55 �C is insufficient to cause bondcleavage in commercial polymers that require dissociation energiesof 70e90 kcal/mol [24]. Thus, it is the combination of UVevisible
Table 1Initial formation rate of 3350 cm�1 for various exposure conditions.
Exposure conditions _AT ;RH
30 �C � 1 �C and <1% RH 1 � 10�3
30 �C � 1 �C and 80% RH 1 � 10�3
55 �C � 1 �C and <1% RH 3 � 10�3
55 �C � 1 �C and 80% RH 4 � 10�3
Outdoor tests started in May 2006 0.4 � 10�3
Outdoor tests started in July 2006 0.4 � 10�3
irradiation and elevated temperature that contributed to the majorchemical changes.
The degrading actions of the combined effect of temperatureand moisture on photooxidation are greater than the sum of
the two effects taken independently, i.e., _A55�C; 80% RH ¼ 43ð _A30�C;
80% RHþ _A55�C; <1% RH � _A30�C; <1% RHÞ, where _AT ; RH is the rate ofchange in infrared absorbance with respect to time for a giventemperature and RH. These observations strongly suggest thedeleterious effect of combining UVevisible irradiation, tempera-ture and moisture in terms of environmental attack. Such aninteraction of environmental stressors on degradation is alsoreported for the combination of cyclic fatigue, temperature andmoisture [25] in which sealant specimens are able to resist cyclicfatigue in the absence of high temperature and RH, but degradedsubstantially when exposed to combination of cyclic fatigue witha high temperature, high RH, or the combination of these threefactors. The existence of interactions among the degrading factorsraises a serious concern as to whether viewing environmentaleffect of these factors independently is meaningful, thus showingthe importance of accounting for such effect in the development ofscientifically meaningful accelerated durability tests.
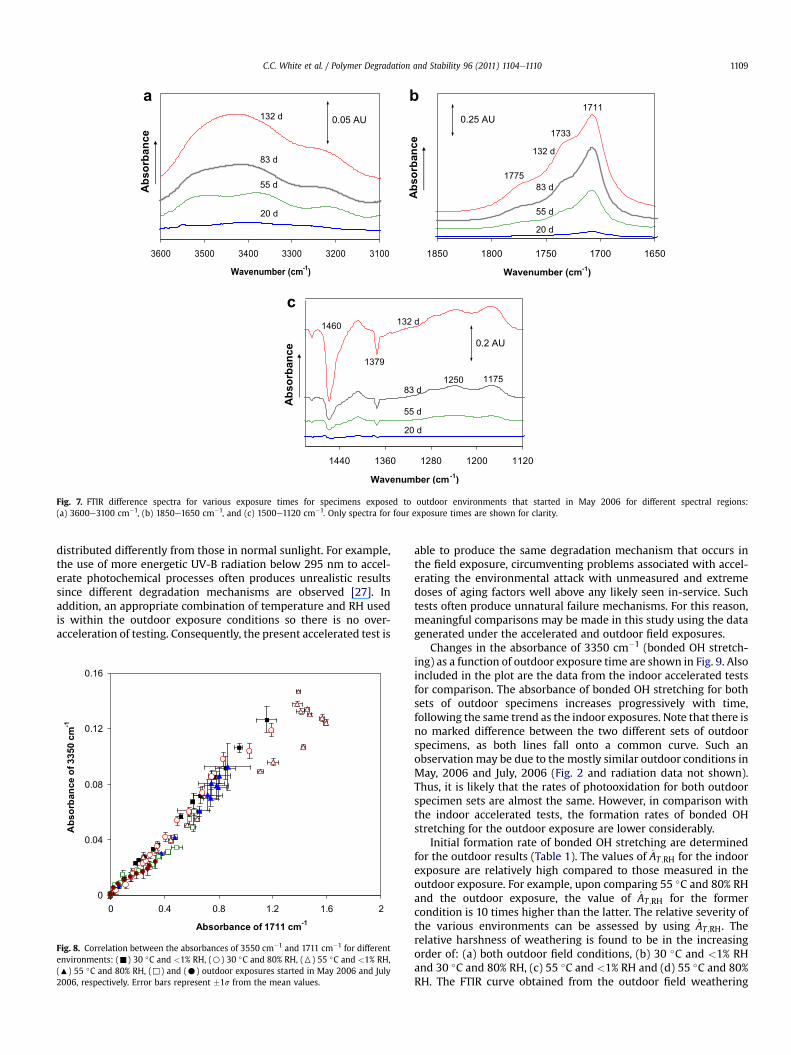
3.2. Outdoor exposure
Fig. 7 shows the representative FTIR difference spectra for theexposure started in May, 2006. The degradation products formedduring the outdoor exposure are consistent with those described inthe indoor accelerated exposure (compare to Fig. 3). Further, thereis a good correlation between outdoor and indoor exposures for the3350 cm�1 vs. 1711 cm�1 curves (compare Figs. 4 and 8). Therefore,the photooxidative mechanism operating in the outdoor andindoor accelerated conditions appears to be the same. Sucha correlation between the two different exposures is not surprisingsince the UV radiation between 295 nm and 400 nm is known to bephotolytically active in degrading polymeric materials [26,27], andthe artificial light source used consists of radiation that is rich in thesame wavelength range. It is indeed known that one difficulty inrelating field and laboratory results has been attributed to the factthat the wavelengths of the accelerated indoor light source are
310032003300340035003600
Wavenumber (cm-1
)
20 d
55 d
83 d
132 d
Ab
so
rb
an
ce
0.05 AU
16501700175018001850
Wavenumber (cm-1
)
20 d
55 d
83 d
132 d
Ab
so
rb
an
ce
1711
1733
1775
0.25 AU
11201200128013601440
Wavenumber (cm-1
)
11751250
1379
1460
20 d
55 d
83 d
132 d
Ab
so
rb
an
ce
0.2 AU
a b
c
Fig. 7. FTIR difference spectra for various exposure times for specimens exposed to outdoor environments that started in May 2006 for different spectral regions:(a) 3600e3100 cm�1, (b) 1850e1650 cm�1, and (c) 1500e1120 cm�1. Only spectra for four exposure times are shown for clarity.
C.C. White et al. / Polymer Degradation and Stability 96 (2011) 1104e1110 1109
distributed differently from those in normal sunlight. For example,the use of more energetic UV-B radiation below 295 nm to accel-erate photochemical processes often produces unrealistic resultssince different degradation mechanisms are observed [27]. Inaddition, an appropriate combination of temperature and RH usedis within the outdoor exposure conditions so there is no over-acceleration of testing. Consequently, the present accelerated test is
0
0.04
0.08
0.12
0.16
0 0.4 0.8 1.2 1.6 2
Absorbance of 1711 cm-1
Ab
so
rb
an
ce
of 3
35
0 c
m-1
Fig. 8. Correlation between the absorbances of 3550 cm�1 and 1711 cm�1 for differentenvironments: (-) 30 �C and <1% RH, (B) 30 �C and 80% RH, (6) 55 �C and <1% RH,(:) 55 �C and 80% RH, (,) and (C) outdoor exposures started in May 2006 and July2006, respectively. Error bars represent �1s from the mean values.
able to produce the same degradation mechanism that occurs inthe field exposure, circumventing problems associated with accel-erating the environmental attack with unmeasured and extremedoses of aging factors well above any likely seen in-service. Suchtests often produce unnatural failure mechanisms. For this reason,meaningful comparisons may be made in this study using the datagenerated under the accelerated and outdoor field exposures.
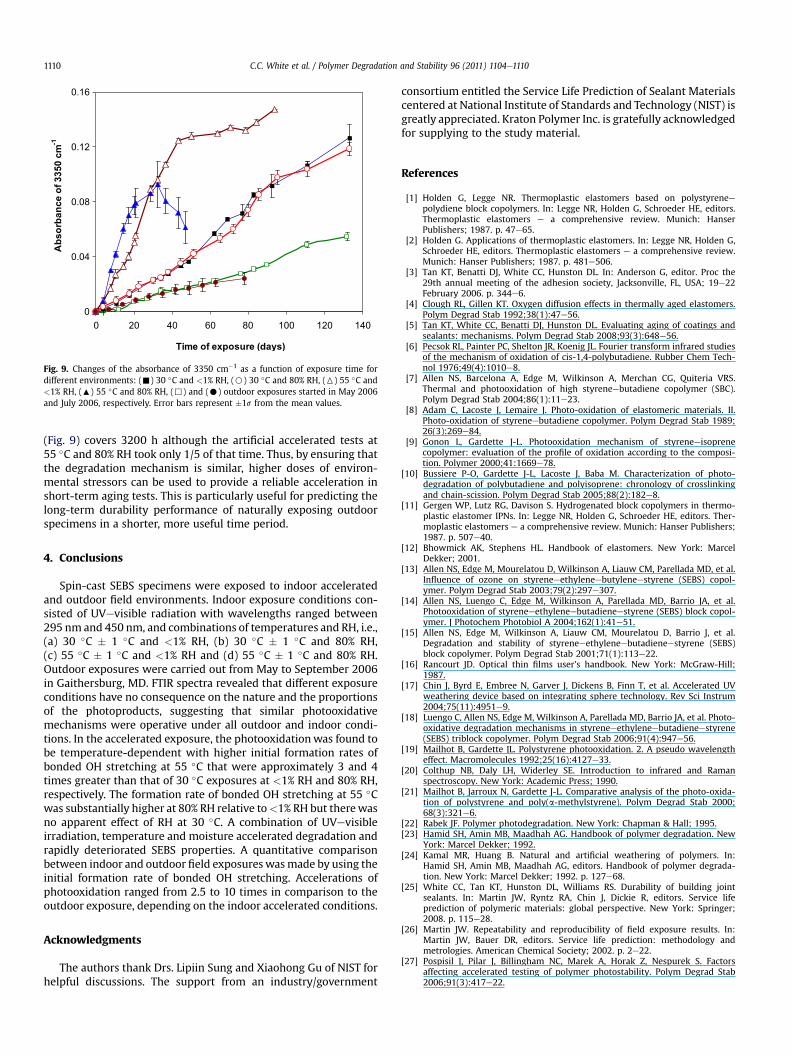
Changes in the absorbance of 3350 cm�1 (bonded OH stretch-ing) as a function of outdoor exposure time are shown in Fig. 9. Alsoincluded in the plot are the data from the indoor accelerated testsfor comparison. The absorbance of bonded OH stretching for bothsets of outdoor specimens increases progressively with time,following the same trend as the indoor exposures. Note that there isno marked difference between the two different sets of outdoorspecimens, as both lines fall onto a common curve. Such anobservation may be due to the mostly similar outdoor conditions inMay, 2006 and July, 2006 (Fig. 2 and radiation data not shown).Thus, it is likely that the rates of photooxidation for both outdoorspecimen sets are almost the same. However, in comparison withthe indoor accelerated tests, the formation rates of bonded OHstretching for the outdoor exposure are lower considerably.
Initial formation rate of bonded OH stretching are determinedfor the outdoor results (Table 1). The values of _AT ;RH for the indoorexposure are relatively high compared to those measured in theoutdoor exposure. For example, upon comparing 55 �C and 80% RHand the outdoor exposure, the value of _AT ;RH for the formercondition is 10 times higher than the latter. The relative severity ofthe various environments can be assessed by using _AT ;RH. Therelative harshness of weathering is found to be in the increasingorder of: (a) both outdoor field conditions, (b) 30 �C and <1% RHand 30 �C and 80% RH, (c) 55 �C and <1% RH and (d) 55 �C and 80%RH. The FTIR curve obtained from the outdoor field weathering
0
0.04
0.08
0.12
0.16
0 20 40 60 80 100 120 140
Time of exposure (days)
Ab
so
rb
an
ce
of 3
35
0 c
m-1
Fig. 9. Changes of the absorbance of 3350 cm�1 as a function of exposure time fordifferent environments: (-) 30 �C and <1% RH, (B) 30 �C and 80% RH, (6) 55 �C and<1% RH, (:) 55 �C and 80% RH, (,) and (C) outdoor exposures started in May 2006and July 2006, respectively. Error bars represent �1s from the mean values.
C.C. White et al. / Polymer Degradation and Stability 96 (2011) 1104e11101110
(Fig. 9) covers 3200 h although the artificial accelerated tests at55 �C and 80% RH took only 1/5 of that time. Thus, by ensuring thatthe degradation mechanism is similar, higher doses of environ-mental stressors can be used to provide a reliable acceleration inshort-term aging tests. This is particularly useful for predicting thelong-term durability performance of naturally exposing outdoorspecimens in a shorter, more useful time period.
4. Conclusions
Spin-cast SEBS specimens were exposed to indoor acceleratedand outdoor field environments. Indoor exposure conditions con-sisted of UVevisible radiation with wavelengths ranged between295 nm and 450 nm, and combinations of temperatures and RH, i.e.,(a) 30 �C � 1 �C and <1% RH, (b) 30 �C � 1 �C and 80% RH,(c) 55 �C � 1 �C and <1% RH and (d) 55 �C � 1 �C and 80% RH.Outdoor exposures were carried out from May to September 2006in Gaithersburg, MD. FTIR spectra revealed that different exposureconditions have no consequence on the nature and the proportionsof the photoproducts, suggesting that similar photooxidativemechanisms were operative under all outdoor and indoor condi-tions. In the accelerated exposure, the photooxidationwas found tobe temperature-dependent with higher initial formation rates ofbonded OH stretching at 55 �C that were approximately 3 and 4times greater than that of 30 �C exposures at <1% RH and 80% RH,respectively. The formation rate of bonded OH stretching at 55 �Cwas substantially higher at 80% RH relative to<1% RH but therewasno apparent effect of RH at 30 �C. A combination of UVevisibleirradiation, temperature and moisture accelerated degradation andrapidly deteriorated SEBS properties. A quantitative comparisonbetween indoor and outdoor field exposures wasmade by using theinitial formation rate of bonded OH stretching. Accelerations ofphotooxidation ranged from 2.5 to 10 times in comparison to theoutdoor exposure, depending on the indoor accelerated conditions.
Acknowledgments
The authors thank Drs. Lipiin Sung and Xiaohong Gu of NIST forhelpful discussions. The support from an industry/government
consortium entitled the Service Life Prediction of Sealant Materialscentered at National Institute of Standards and Technology (NIST) isgreatly appreciated. Kraton Polymer Inc. is gratefully acknowledgedfor supplying to the study material.
References
[1] Holden G, Legge NR. Thermoplastic elastomers based on polystyreneepolydiene block copolymers. In: Legge NR, Holden G, Schroeder HE, editors.Thermoplastic elastomers e a comprehensive review. Munich: HanserPublishers; 1987. p. 47e65.
[2] Holden G. Applications of thermoplastic elastomers. In: Legge NR, Holden G,Schroeder HE, editors. Thermoplastic elastomers e a comprehensive review.Munich: Hanser Publishers; 1987. p. 481e506.
[3] Tan KT, Benatti DJ, White CC, Hunston DL. In: Anderson G, editor. Proc the29th annual meeting of the adhesion society, Jacksonville, FL, USA; 19e22February 2006. p. 344e6.
[4] Clough RL, Gillen KT. Oxygen diffusion effects in thermally aged elastomers.Polym Degrad Stab 1992;38(1):47e56.
[5] Tan KT, White CC, Benatti DJ, Hunston DL. Evaluating aging of coatings andsealants: mechanisms. Polym Degrad Stab 2008;93(3):648e56.
[6] Pecsok RL, Painter PC, Shelton JR, Koenig JL. Fourier transform infrared studiesof the mechanism of oxidation of cis-1,4-polybutadiene. Rubber Chem Tech-nol 1976;49(4):1010e8.
[7] Allen NS, Barcelona A, Edge M, Wilkinson A, Merchan CG, Quiteria VRS.Thermal and photooxidation of high styreneebutadiene copolymer (SBC).Polym Degrad Stab 2004;86(1):11e23.
[8] Adam C, Lacoste J, Lemaire J. Photo-oxidation of elastomeric materials. II.Photo-oxidation of styreneebutadiene copolymer. Polym Degrad Stab 1989;26(3):269e84.
[9] Gonon L, Gardette J-L. Photooxidation mechanism of styreneeisoprenecopolymer: evaluation of the profile of oxidation according to the composi-tion. Polymer 2000;41:1669e78.
[10] Bussiere P-O, Gardette J-L, Lacoste J, Baba M. Characterization of photo-degradation of polybutadiene and polyisoprene: chronology of crosslinkingand chain-scission. Polym Degrad Stab 2005;88(2):182e8.
[11] Gergen WP, Lutz RG, Davison S. Hydrogenated block copolymers in thermo-plastic elastomer IPNs. In: Legge NR, Holden G, Schroeder HE, editors. Ther-moplastic elastomers e a comprehensive review. Munich: Hanser Publishers;1987. p. 507e40.
[12] Bhowmick AK, Stephens HL. Handbook of elastomers. New York: MarcelDekker; 2001.
[13] Allen NS, Edge M, Mourelatou D, Wilkinson A, Liauw CM, Parellada MD, et al.Influence of ozone on styreneeethyleneebutyleneestyrene (SEBS) copol-ymer. Polym Degrad Stab 2003;79(2):297e307.
[14] Allen NS, Luengo C, Edge M, Wilkinson A, Parellada MD, Barrio JA, et al.Photooxidation of styreneeethyleneebutadieneestyrene (SEBS) block copol-ymer. J Photochem Photobiol A 2004;162(1):41e51.
[15] Allen NS, Edge M, Wilkinson A, Liauw CM, Mourelatou D, Barrio J, et al.Degradation and stability of styreneeethyleneebutadieneestyrene (SEBS)block copolymer. Polym Degrad Stab 2001;71(1):113e22.
[16] Rancourt JD. Optical thin films user’s handbook. New York: McGraw-Hill;1987.
[17] Chin J, Byrd E, Embree N, Garver J, Dickens B, Finn T, et al. Accelerated UVweathering device based on integrating sphere technology. Rev Sci Instrum2004;75(11):4951e9.
[18] Luengo C, Allen NS, Edge M, Wilkinson A, Parellada MD, Barrio JA, et al. Photo-oxidative degradation mechanisms in styreneeethyleneebutadieneestyrene(SEBS) triblock copolymer. Polym Degrad Stab 2006;91(4):947e56.
[19] Mailhot B, Gardette JL. Polystyrene photooxidation. 2. A pseudo wavelengtheffect. Macromolecules 1992;25(16):4127e33.
[20] Colthup NB, Daly LH, Widerley SE. Introduction to infrared and Ramanspectroscopy. New York: Academic Press; 1990.
[21] Mailhot B, Jarroux N, Gardette J-L. Comparative analysis of the photo-oxida-tion of polystyrene and poly(a-methylstyrene). Polym Degrad Stab 2000;68(3):321e6.
[22] Rabek JF. Polymer photodegradation. New York: Chapman & Hall; 1995.[23] Hamid SH, Amin MB, Maadhah AG. Handbook of polymer degradation. New
York: Marcel Dekker; 1992.[24] Kamal MR, Huang B. Natural and artificial weathering of polymers. In:
Hamid SH, Amin MB, Maadhah AG, editors. Handbook of polymer degrada-tion. New York: Marcel Dekker; 1992. p. 127e68.
[25] White CC, Tan KT, Hunston DL, Williams RS. Durability of building jointsealants. In: Martin JW, Ryntz RA, Chin J, Dickie R, editors. Service lifeprediction of polymeric materials: global perspective. New York: Springer;2008. p. 115e28.
[26] Martin JW. Repeatability and reproducibility of field exposure results. In:Martin JW, Bauer DR, editors. Service life prediction: methodology andmetrologies. American Chemical Society; 2002. p. 2e22.
[27] Pospisil J, Pilar J, Billingham NC, Marek A, Horak Z, Nespurek S. Factorsaffecting accelerated testing of polymer photostability. Polym Degrad Stab2006;91(3):417e22.




















