
Synthesis and Anticancer Activity of N-Aryl-5-substituted-1,3,4-oxadiazol-2-amine Analogues
Upload
independentCategory
view
1download
0
DOI: 10.1002/chem.201202246
Immobilization of Styrene-Substituted 1,3,4-Oxadiazoles intoThermoreversible Luminescent Organogels and Their Unexpected
Photocatalyzed Rearrangement
Fr�d�ric Dumur,*[a] Emmanuel Contal,[a] Guillaume Wantz,[b] Trang N. T. Phan,[a]
Denis Bertin,[a] and Didier Gigmes[a]
Introduction
The development of low-molecular-weight linear p-conju-gated molecules that can spontaneously self-organize intowell-defined assemblies is a significant goal in relation to or-ganic electronic devices.[1] Control of the self-organization ofmolecules at the nanoscale level constitutes a powerful ap-proach for the design of new materials with tailored func-tionalities. In this regard, gel formation that solely resultsfrom van der Waals, p–p stacking, dipole–dipole, and hydro-gen-bonding interactions between molecules constitutes asimple and soft method for such self-assembly. Typically, thedriving force behind these molecular self-assemblies is thecooperative effect of noncovalent forces. The functionalproperties imparted to the generated supramolecular assem-blies are reversible as these noncovalent interactions canreadily be weakened or even destroyed.[2] Over the years,two different types of gelation have been developed, namelychemical gelation,[3] which relies on the formation of chemi-cal bonds and irreversibly constructs large polymers startingfrom a small molecule, and physical gelation,[4] which relieson the formation of intermolecular noncovalent interactions.
In this second approach, gelator molecules self-assembleinto nanoscale superstructures that further interlock intothree-dimensional networks trapping the solvent moleculesinside. The principal interest in this second type of gelsstems from the fact that their photophysical properties canbe finely modulated upon exposing them to external stimulisuch as heat, chemicals, or light.[5] Notably, when fluorescentorganogelators are employed, the supramolecular state inorganogels can significantly modify the emissive propertiesof the gelator. In this context, 1,3,4-oxadiazoles constitutean important class of heterocyclic compounds that have re-ceived considerable attention due to their potential applica-tions in medicinal chemistry[6] (antimicrobial, antifungal,anti-inflammatory, and antihypertensive activities), as fluo-rophores for metal-sensing applications,[7] and as n-typecharge carriers in organic light-emitting devices.[8] In recentyears, these fluorescent heterocycles have mostly been in-vestigated as electron transporters owing to their electrondeficiency and good thermal stability.[9] To date, 1,3,4-oxa-diazoles have only rarely been studied as organogelators,and to the best of our knowledge only five examples of gela-tors incorporating 1,3,4-oxadiazole derivatives in their scaf-folds have been reported so far (Scheme 1).
The first report mentioning the use of 1,3,4-oxadiazole de-rivatives as gelators described a C3-symmetric almost non-fluorescent molecule, OX1, that formed a highly fluorescentorganogel at 0.1 wt % in chloroform through intermolecularhydrogen bonding.[10] More recently, this approach was ex-tended to the octupolar derivatives OX2 and OX3, whichbear an additional alkyl chain on the core of the discotic ge-lator but do not contain a central hydrogen-bonding motifas in the oxadiazole OX1.[11] The gelation processes of thesedisk-shaped molecules are purely driven by p–p stacking in-
Abstract: A series of styrene-substitut-ed 1,3,4-oxadiazoles has been designedand investigated as new low-molecular-weight organogelators. The photophysi-cal properties of the resulting thermor-eversible organogels have been charac-terized by UV/Vis absorption and lu-minescence spectroscopies. Surprising-ly, the gelation ability of the oxadia-
zoles depended on the presence of thestyrene moiety as gelation of the inves-tigated oxadiazoles did not take placein its absence. Gel formation was ac-
companied by a modification of the flu-orescence of the organogelators in thesupramolecular state. UV irradiation ofthe gels caused a rearrangement of theimmobilized 1,3,4-oxadiazoles bearinga styrene moiety by a tandem [4+2]and [3+2] cascade reaction. Structuremodification and color change of thegels were also evident upon irradiation.
Keywords: aggregation · cascaderearrangement · luminescence ·organogel · oxadiazole · styrene
[a] Dr. F. Dumur, Dr. E. Contal, Dr. T. N. T. Phan, Prof. D. Bertin,Dr. D. GigmesAix-Marseille Universit�, CNRS, Institut de Chimie RadicalaireUMR 7273, 13397 Marseille Cedex (France)Fax: (+33) 2-91-28-87-58E-mail : [email protected]
[b] Dr. G. WantzUniversit� Bordeaux, IMS, UMR 5218, 33400 Talence (France)andCNRS, IMS, UMR 5218, 33400 Talence (France)
Supporting information for this article is available on the WWWunder http://dx.doi.org/10.1002/chem.201202246.
Chem. Eur. J. 2013, 19, 1373 – 1384 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 1373
FULL PAPER
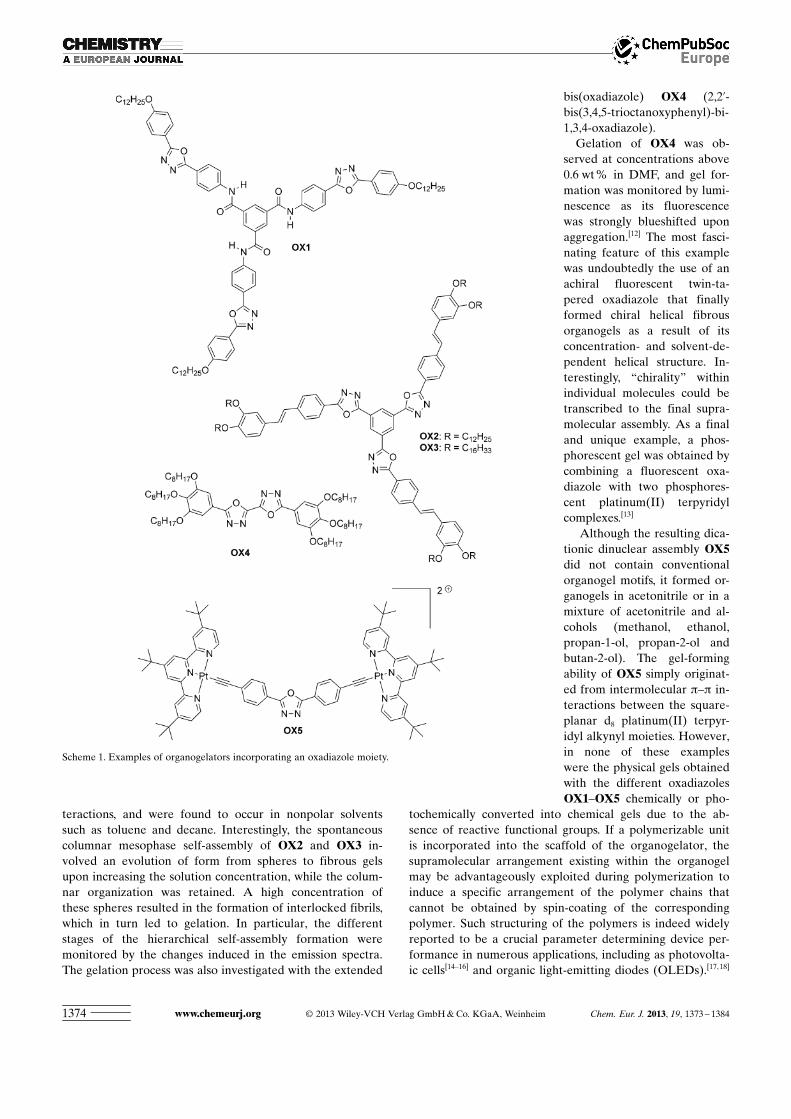
teractions, and were found to occur in nonpolar solventssuch as toluene and decane. Interestingly, the spontaneouscolumnar mesophase self-assembly of OX2 and OX3 in-volved an evolution of form from spheres to fibrous gelsupon increasing the solution concentration, while the colum-nar organization was retained. A high concentration ofthese spheres resulted in the formation of interlocked fibrils,which in turn led to gelation. In particular, the differentstages of the hierarchical self-assembly formation weremonitored by the changes induced in the emission spectra.The gelation process was also investigated with the extended
bis(oxadiazole) OX4 (2,2’-bis(3,4,5-trioctanoxyphenyl)-bi-1,3,4-oxadiazole).
Gelation of OX4 was ob-served at concentrations above0.6 wt % in DMF, and gel for-mation was monitored by lumi-nescence as its fluorescencewas strongly blueshifted uponaggregation.[12] The most fasci-nating feature of this examplewas undoubtedly the use of anachiral fluorescent twin-ta-pered oxadiazole that finallyformed chiral helical fibrousorganogels as a result of itsconcentration- and solvent-de-pendent helical structure. In-terestingly, “chirality” withinindividual molecules could betranscribed to the final supra-molecular assembly. As a finaland unique example, a phos-phorescent gel was obtained bycombining a fluorescent oxa-diazole with two phosphores-cent platinum(II) terpyridylcomplexes.[13]
Although the resulting dica-tionic dinuclear assembly OX5did not contain conventionalorganogel motifs, it formed or-ganogels in acetonitrile or in amixture of acetonitrile and al-cohols (methanol, ethanol,propan-1-ol, propan-2-ol andbutan-2-ol). The gel-formingability of OX5 simply originat-ed from intermolecular p–p in-teractions between the square-planar d8 platinum(II) terpyr-idyl alkynyl moieties. However,in none of these exampleswere the physical gels obtainedwith the different oxadiazolesOX1–OX5 chemically or pho-
tochemically converted into chemical gels due to the ab-sence of reactive functional groups. If a polymerizable unitis incorporated into the scaffold of the organogelator, thesupramolecular arrangement existing within the organogelmay be advantageously exploited during polymerization toinduce a specific arrangement of the polymer chains thatcannot be obtained by spin-coating of the correspondingpolymer. Such structuring of the polymers is indeed widelyreported to be a crucial parameter determining device per-formance in numerous applications, including as photovolta-ic cells[14–16] and organic light-emitting diodes (OLEDs).[17,18]
Scheme 1. Examples of organogelators incorporating an oxadiazole moiety.
www.chemeurj.org � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2013, 19, 1373 – 13841374

Notably, poly(oxadiazole)s are widely used as electron trans-porters in OLEDs.
In the present work, an unprecedented formation of fluo-rescent organogels from styrene-substituted oxadiazole gela-tors in polar and apolar organic solvents is reported. Highlyluminescent organogels were obtained through p–p stackinginteractions of oxadiazoles only when these precursors borea styrene substituent. The gelation ability of the oxadiazoleswas found to depend on the presence of the styrene group,which is not recognized as a usual gelation motif. Interest-ingly, photoirradiation of the gels produced a remarkablestructural modification with a concomitant color changefrom blue to yellow, whereas irradiation of a solution of thecorresponding monomer resulted in the formation of a poly-mer that retained the blue emission of the oxadiazole.
Results and Discussion
Three 1,3,4-oxadiazoles were investigated as novel lumines-cent organogelators and their structures are presented inScheme 2.
The synthetic strategy for preparing oxadiazoles 1–3 isoutlined in Scheme 3. As the structures of the three mono-mers vary only in the substitution pattern on one benzenering, an iterative parallel-synthesis approach was employed.For the series of parallel syntheses, the ester-substituted aro-matic moiety of the oxadiazole was kept constant and differ-ent substituents on the second aromatic ring of the oxadia-zole were surveyed. The presence of peripheral long-chainalkyl groups is known to keep the “nanodomains” of the ge-lator separated from one another and thus to favor self-or-ganization.[19] For comparison with monomers 1 and 2, anoxadiazole bearing a tert-butyl substituent was also synthe-sized. The synthesis of monomer 3 has previously been re-ported in the literature.[20] Acids 7 and 8 were prepared in
two and three steps, respectively, starting from 2-(4-methyl-phenyl)-4,4-dimethyl-2-oxazoline 4. Lithiation of 4 with lith-ium diisopropylamide (LDA) followed by nucleophilic sub-stitution with 1-bromododecane furnished the key substrate5 in 94 % yield. Subsequent lithiation with butyllithium se-lectively deprotonated the aromatic ring at the ortho-posi-tion with respect to the oxazoline ring, and reaction with 1-bromododecane afforded 6 in 71 % yield. The acid functionwas then regenerated in two steps, first by ring-opening ofthe oxazoline under acidic conditions and secondly by hy-drolysis of the remaining ester under basic conditions.
Acids 7 and 8 were obtained in yields of 82 and 67 %, re-spectively. The well-known standard procedure for the syn-thesis of 1,3,4-oxadiazoles involving the cyclodehydration ofdiacylhydrazines was then performed.[21] Acids 7–9 were firstconverted into their corresponding esters 10–12. Reactionsof 10 and 12 with an excess of hydrazine monohydrate inethanol furnished the corresponding hydrazones 13 and 15,respectively, but this standard route proved to be unsuitablefor converting 11 into the target hydrazone 14. As an alter-native synthetic pathway, acids 7–9 were converted intotheir corresponding acyl chlorides 16–18 by using neat thion-yl chloride. Subsequent reaction, without purification of theacid chlorides, with an excess of hydrazine monohydrate fur-nished the respective hydrazones 13–15 in yields rangingfrom 67 to 94 %. Further treatment of hydrazones 13–15with an equimolecular amount of methyl 4-(chlorocarbo-nyl)benzoate (19) afforded hydrazides 20–22 in yields of 84–96 %. The dehydrative cyclization reaction forming the oxa-diazole ring was then induced by heating the respectivediacyl hydrazides in anhydrous phosphorus oxychloride,[22]
whereupon the 1,3,4-oxadiazole products 23–25 were ob-tained in high yields. Finally, hydrolysis of the esters 23–25and esterification with 4-chloromethylstyrene furnished themonomers 1–3 in good yields after purification by columnchromatography. The structures and purities of all of themonomers and intermediates were thoroughly verified by1H and 13C NMR and high-resolution mass spectrometry(HRMS).
In particular, the substitution pattern of 2 could be veri-fied by NMR spectroscopy. In the 1H NMR spectrum of 2(Figure 1 b), two triplets located at d= 2.65 and 3.10 ppmconfirmed the presence of two alkyl chains on the oxadia-zole, whereas only one two-proton signal at d= 2.69 ppmcould be observed in the spectrum of the mono-substitutedoxadiazole 1 (Figure 1 a). The upfield triplet at d= 3.10 ppmcould be confidently assigned to the CH2 group in the orthoposition with respect to the oxadiazole ring. This ortho-sub-stitution on the benzene ring also gave three signals corre-sponding to the aromatic protons: one doublet at d=
7.15 ppm overlapped with a singlet at d=7.17 ppm and asecond doublet at d= 7.93 ppm (Figure 1 d). In contrast, theprotons on the corresponding aromatic ring of 1 gave rise totwo doublets of 2 H at d=7.35 and 8.05 ppm with a couplingconstant of 8.3 Hz (Figure 1 c).
Oxadiazoles 1–3 and 23–25 were tested for their gelationabilities in various polar, nonpolar, aromatic, and nonaro-
Scheme 2. Structure of the oxadiazoles investigated in this work.
Chem. Eur. J. 2013, 19, 1373 – 1384 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 1375
FULL PAPERLuminescent Organogels Based on Styrene-Substituted 1,3,4-Oxadiazoles

matic solvents by the “stable-to-inversion-of-test-tube”method. The gelation properties of all six compounds aresummarized in Table 1. Interestingly, whereas oxadiazoles23–25 esterified with a methyl group did not form gels inany of the investigated solvents, two of the oxadiazoles es-terified with 4-chloromethylstyrene were found to formwhite, almost transparent gels. Notably, styrene-appended
oxadiazole 1 was found to form stable gels in alkanes (pen-tane, heptane, dodecane, cyclohexane), whereas oxadiazole3 formed organogels in alkanes, alcohols (methanol, etha-nol, butanol), and diethyl ether at gelation concentrationsbetween 0.83 and 4 mg mL�1 (Table 1). In contrast, oxadia-zole 2, which is a structural analogue of 1 and 3, was unableto form a gel in any of the tested solvents. Indeed, this oxa-
Scheme 3. Synthesis of monomers 1–3.
www.chemeurj.org � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2013, 19, 1373 – 13841376
F. Dumur et al.

diazole proved to be highly soluble in most of the commonsolvents, except for alcohols, acetone, and DMF, from whichit precipitated. It may be noted that organogelators 1 and 3are end-capped with a styrene unit. However, despite thepresence of this polymerizable unit, gels could be formedwithout polymerization of the monomer in high-boiling al-kanes (nonane, decane, dodecane) by heating until the solidgelator had completely dissolved and subsequently coolingthe solution. The absence of polymerization was establishedby gel permeation chromatography (GPC).
As revealed by these results, the styrene moiety played animportant role in the formation of the organogels as no ge-lation occurred without its presence on the oxadiazolemoiety. To the best of our knowledge, the only previous ex-ample of organogels based on a styrene-substituted mole-cule was reported in 2005, in which divinylbenzene servedas the solvent and bis(2-ethylhexyl) sodium sulfosuccinateand 4-chlorophenol were used as mixed organogelators.[23]
Consequently, in our case, the gel-forming ability of oxadia-zoles 1 and 3 could be confidently ascribed to p–p stackinginteractions arising from the presence of the additional aro-matic ring of the styrene moiety as no gel forms without thisgroup. However, the presence of the styrene moiety on theoxadiazole is not the only requirement. Planarity of the finalmolecules is another factor of crucial importance as no gelformed with oxadiazole 2. As a result of the severe sterichindrance generated by the second alkyl chain and the tor-sion induced by its presence in the ortho-position with re-spect to the oxadiazole ring, the lack of planarity combinedwith the steric hindrance prevented any p–p stacking inter-actions.
Compared to other typical low-molecular-weight organicorganogelators, the oxadiazoles 1 and 3 investigated here re-quired similar concentrations to form gels. For instance, alloxadiazoles previously evaluated as organogelators gelifiedat concentrations between 0.05[10] and 3.5 wt %,[13] a rangethat encompasses the concentrations required with 1 and 3,supporting their satisfactory gelating properties. The criticalgelator concentration (CGC) of 1 in n-dodecane was deter-mined to be 0.62 mgmL�1, which implies that 1 can entrapapproximately 1150 solvent molecules per molecule of gela-tor. Although our gelators exhibited higher CGCs thansome of the oxadiazole-based examples reported in the liter-ature, gel 3 proved to be relatively thermally stable, whichwas advantageous since heating at temperatures higher than96 8C was necessary to completely melt the gels in n-dodec-ane and to obtain a clear and nonviscous solution. Thermor-eversibility of the gel formation was also verified by repeat-ed heating and cooling, thus providing evidence for thephysical character of the gel formation as the physical bondscould be disrupted upon heating and reformed during cool-ing.[4b] All of the organogels remained stable for months atroom temperature.
The thermal properties of gels 1 and 3 in n-dodecanewere investigated by differential scanning calorimetry(DSC). Their thermograms are shown in Figure 2. The ther-mal behavior of gel 1 was very different from that of gel 3.
Figure 1. 1H NMR spectra (400 MHz) in CDCl3 at 25 8C of a) 1, b) 2, andexpanded aromatic regions of c) 1 and d) 2.
Table 1. Gelation properties of oxadiazoles 1–3 and 23–25.[a]
Solvent 1 23 2 24 3 25
chloroform S S S S S Sn-pentane G (1.67) P S P G (1.25) Ccyclohexane G (1.25) P S P G (0.83) Cn-heptane G (1) P S P G (0.83) Cn-dodecane G (0.62) P S P G (1.67) Cbenzene T S S S T Cpara-xylene T S S S T Cethyl acetate P P S P T Cdiethyl ether P P S P G (4) CTHF S S S S T Cmethanol P P P P G (2.50) Cethanol P P P P G (1.50) Cn-butanol G (0.71) P P P G (2.85) Cacetone P P P P T CDMF P P P P T C
[a] S= solution, P= precipitation, G =gelation, C=crystallization, T=
turbid. The values in parentheses denote the CGC values [mg mL�1] ofthe gelators at 25 8C.
Chem. Eur. J. 2013, 19, 1373 – 1384 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 1377
FULL PAPERLuminescent Organogels Based on Styrene-Substituted 1,3,4-Oxadiazoles

The DSC thermograms of gel 1 show an endothermic transi-tion with a maximum at 31 8C in the heating process and asharp exothermic transition with a maximum at 17 8C in thecooling process. In contrast, the DSC curves of gel 3 exhibitvery broad transitions in both the heating and cooling proc-esses, with a maximum of the melting peak at 107 8C and amaximum of the crystallization peak at 70 8C. Repeated cy-cling showed similar transition behavior. These results sug-gest that the sol–gel transitions of gels 1 and 3 in n-dodec-ane are thermodynamically reversible. We noted that thethermal transitions of gel 1 occurred at temperatures lowerthan those of gel 3. This difference can be attributed to thepresence of the C13H27 group in the structure of 1, which ismore flexible and mobile than the tert-butyl group in thestructure of 3.
To gain direct insight intothe nature of the self-assem-bled microstructures, scanningelectron microscopy (SEM)was used to observe the twogels (see Figure 3 and furtherimages in the Supporting Infor-mation). Dry gels were pre-pared by freeze-drying the or-ganogels from heptane. SEMimages of the dried gels re-vealed that the two monomershad self-assembled into twowell-defined structures of dis-tinct morphology. As shown inFigure 3 a, the organogel madeof monomer 1 consisted of anentanglement of fibers withlengths of several micrometers,generating an extended andthree-dimensional fibrillar net-work that stabilized the gelstate. Figure 3 a also clearlyshows the fibers to have veryuniform structures with rela-tively smooth surfaces. On theother hand, Figure 3 b, whichshows an electron micrographof the gel prepared with mono-mer 3, indicates that thelengths of the fibers were sig-nificantly reduced compared tothose obtained with 1 as onlyfibers of length 2 mm were visi-ble in a weakly interconnectednetwork. More surprising wasthe roughness and the presenceof microstructures on the sur-face of the fibers. These resultsclearly show the profoundeffect of the substitution pat-
tern of the monomers on the self-assembled microstructures.The presence of the flexible tail on 1 led to the formation ofa strongly interconnected and extended network, in agree-ment with the higher gelation ability seen with this mono-mer.
The macroscopic organization of the gels was also investi-gated by variable-temperature 1H NMR measurements asNMR could provide a fundamental understanding of thecharacteristics of molecular self-assembly. To this end, a gelwas prepared from 3 in deuterated cyclohexane and NMRspectra were recorded upon varying the temperature from25 to 65 8C. Surprisingly, a relatively well-defined NMRspectrum could be obtained even at room temperature (seeFigure 4 a), whereas a spectrum with relatively broad peakswas expected as a result of the anisotropy of the gel. Uponraising the temperature to 65 8C, the intensity of the peaks
Figure 2. DSC thermograms of gels formed from a) 1 and b) 3.
www.chemeurj.org � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2013, 19, 1373 – 13841378
F. Dumur et al.

increased, as is clearly shown for the singlet of the tert-butylgroup at d=1.29 ppm. Similarly, a complete change in thesignal arising from the of the oxadiazole moiety was ob-served and the initially broad multiplet at d= 7.17–7.25 ppmwas simplified and shifted to d=7.20–7.35 ppm. Higher res-olution of the spectrum upon heating can be confidently as-signed to an improvement in the thermal motion in the or-ganogel.
Based on previous reports, the observation of well-re-solved 1H NMR signals in the gel state suggests the forma-tion of a “wet gel” with gelator 3, in which solvent mole-cules are incorporated into the gel fiber.[24] With organogela-tor molecules being separated by the solvent molecules,there is still sufficient thermal motion to provide a well-re-solved 1H NMR spectrum. On the other hand, the NMR sig-nals of a “dry gel” are broadened as a result of insufficientthermal motion of the molecules and the isotropy of the gel.Similar to those observed for gelator 3, temperature-de-pendent NMR measurements indicated that gelator 1 alsoformed a wet gel (see Figure in the Supporting Informa-tion).
Gel polymerization of 1 and 3 was also considered, sincethe two oxadiazoles bear polymerizable styrene end groups.Only photopolymerization was investigated as classicalchemical polymerization techniques may require heating ofthe gels beyond their sol–gel transition temperature. Photo-
chemical polymerizations were conducted in heptane in aRayonet photochemical chamber by using light of wave-length 350 nm (2.1 J cm�2), which corresponds to the maxi-mum absorption wavelength of the two monomers. Surpris-ingly, irradiation of the initially blue-emitting gels for 16 hprovided gels that were green-yellow in appearance (seeFigure 5). Interestingly, whereas the structure of the gel pre-pared with 3 was retained, complete loss of the gel state wasobserved with 1 and only a viscous solution was recoveredafter irradiation. Modification of the emission color upon ir-radiation cannot be attributed to a polymerization of themonomer because poly(1,3,4-oxadiazole)s are known to beblue-emitting polymers.[20a,25] The change in the emissioncolor can only be attributed to a modification of the conju-gation of the emitting part of the compounds.
Notably, electron-deficient 1,3,4-oxadiazoles have beenwidely reported to take part in intermolecular inverse-elec-tron-demand Diels–Alder reactions with electron-rich dieno-
Figure 3. SEM images obtained from gels made of a) 1 and b) 3 afterdrying. Scale bar is 2 mm.
Figure 4. Variable-temperature 1H NMR spectra of the organogel madeof 3 (10 mg mL�1) in [D12]cyclohexane.
Chem. Eur. J. 2013, 19, 1373 – 1384 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 1379
FULL PAPERLuminescent Organogels Based on Styrene-Substituted 1,3,4-Oxadiazoles

philes.[26] Briefly, in a cascade mechanism, initial reaction ofthe oxadiazole with an olefinic dienophile proceeds througha [4+2] cycloadduct that loses N2 and provides a cyclic car-bonyl ylide that reacts in a second step with the olefin in a1,3-dipolar cycloaddition (see Scheme 4). Interestingly, these
transformations did not occur when the molecules were dis-solved in organic solvents at the same concentration leveland then irradiated. The results indicate that due to thepresence of J-aggregates in the solid state, intermolecular[4+2] cycloadditions and/or intermolecular cascade cycload-dition reactions between the oxadiazole rings and styrenemoieties of different molecules could be photochemically in-duced, these reactions being favored by the close proximityof the reactive functionalities in the solid state.
A careful survey of the literature revealed that irreversi-ble color changes of organogels and photopolymerizationsof gels have scarcely been reported to date. In most cases,
gelators were designed with adiacetylenic group covalentlylinked to an additional groupsuch as urea,[27] acrylic acid,[28]
oligoethylene oxide,[29] or anaryl ring.[30] Depending on thespacers introduced betweenthe second group and the buta-diyne moiety, gel states wereeither retained or lost uponphotopolymerization. In allcases, photopolymerization ofcolorless translucent organo-gels produced polydiacetylenechromophores that absorbedstrongly in the UV/Vis region.Light-induced rearrangement
of gels has also been accomplished by the metathesis of al-kenes[31] and by the dimerization of anthracene.[32]
The effects of irradiation on the microstructures of thetwo gels were examined by SEM (see Figure 6). Significantmodifications of the two structures were observed. In thecase of 1, the microstructure evolved towards a denser fi-brous network with thinner fibers, which is consistent withthe lowered stability of the gel after irradiation. Duringstanding at room temperature, solvent was progressively
Figure 5. Gels prepared with organogelators 1 and 3 : a) before irradiation in a Rayonet photochemical cham-ber at 350 nm for 16 h (left) and the same gel after irradiation (right); b) color change around the beam ob-tained in a spectrophotometer cell on leaving the cuvette in the spectrophotometer.
Scheme 4. Mechanism of the cascade reactions reported with electron-de-ficient 1,3,4-oxadiazoles and electron-rich dienophiles.
Figure 6. SEM images of the gels made of a) 1 and b) 3 after overnight ir-radiation at 350 nm. Scale bar is 2 mm.
www.chemeurj.org � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2013, 19, 1373 – 13841380
F. Dumur et al.

exuded and the unstable gel liquefied upon ageing. Con-versely, in the case of monomer 3, the lengths of the fibersincreased and the resulting network consisted of well-sepa-rated fibers tens of micrometers in length. The roughness ofthe fibers also decreased as a result of their collapse upon ir-radiation.
In order to characterize and identify the different prod-ucts formed during irradiation, the yellow-emitting residuewas subjected to gel permeation chromatography. The gelswere dissolved in THF and the molecular-weight distributionwas analyzed. The obtained chromatogram clearly showedthat reaction of monomer 3 formed mainly dimers and trim-ers (see Figure 7). According to the respective intensities, asmall proportion of the molecules also formed oligomerswith chains comprising between 4 and 14 monomers. Theseresults clearly indicated that a chemical reaction occurredupon irradiation, producing new species that were responsi-ble for the new emission color of the gel. Similar behavior
was seen with 1, irradiation of the gel mainly producingdimers and trimers. The stabilities of the irradiated gels canbe ascribed to their contents of unreacted monomer, whichmaintains the supramolecular architecture. Surprisingly,upon irradiating solutions of 1 and 3 in chloroform at thesame concentrations as those used for the gel formation andfor the same time, the blue emissions of the two solutionswere maintained. On examining the products formed duringirradiation, dimers and trimers were again detected as themain species. However, oligomers are formed by a com-pletely different mechanism to that previously observed inthe gels. Since the blue emissions of the solutions are pre-served, the formation of dimers and trimers can be confi-dently assigned to photopolymerization of the monomersand not to a photochemical rearrangement of the oxadia-zoles (see Scheme 4), the oxadiazole core not being affectedduring oligomer formation.
As a result of the simultaneous generation of numerousradicals, the presence of oxygen that can also initiate theformation of radicals, and the lack of stirring, there is littleor no propagation of the polymerization and a rapid recom-bination of radicals is observed, resulting in the formationof oligomers of low molecular weight. Final proof of poly-merization was provided by NMR analyses (see the NMRspectra in the Supporting Information). On examining theNMR spectra of the two irradiated solutions, a significantdecrease in the intensity of the vinylic signals compared tothat of the parent monomers was apparent, confirming thepolymerization. The concomitant appearance of new signalsin the aliphatic region corresponding to the formation ofCH2-CH(R)- bonds between the styrene units was also ob-served. No modifications of the oxadiazole signals were ob-served, either by 1H or 13C NMR (see the spectra in the Sup-porting Information).
The optical properties of 1–3 were investigated by UV/Visabsorption and photoluminescence spectroscopies. The ab-sorption and emission spectra of 1–3 in chloroform areshown in Figure 8. Compounds 1–3 show similar absorptionspectra with a strong band centered at 300 nm attributableto p–p* transitions resulting from the conjugation. Whenmonomers 1–3 were excited at 300 nm, their emission peakswere observed at 370, 368, and 376 nm, respectively. The sol-ution-to-gel transition was accompanied by a redshift of theemission maxima (370 and 387 nm, respectively). After over-night irradiation in a Rayonet photochemical chamber(lexc = 350 nm), light-induced cycloaddition resulted in a sig-nificant modification of the gel emission with the appear-ance of distinct emission peaks at higher wavelength. Thepeak of oxadiazole 1 appeared slightly redshifted (397 nminstead of 387 nm). However, this apparent shift was due tothe overlap of numerous bands at wavelengths above387 nm. Furthermore, a broad and intense peak with maxi-mum emission centered at 496 nm was observed. This newpeak was indicative of photochemical conversion of mole-cules into oligomers, these being responsible for the newemission color of the material (see Figure 9 a).
Figure 7. GPC traces of 1 (top) and 3 (bottom) in CHCl3 before irradia-tion, after overnight irradiation of the solution at 350 nm, and after over-night irradiation of the gel at 350 nm.
Chem. Eur. J. 2013, 19, 1373 – 1384 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 1381
FULL PAPERLuminescent Organogels Based on Styrene-Substituted 1,3,4-Oxadiazoles

The broadness of this new emission band may be attribut-ed to the formation of the numerous oligomers during irra-diation, as shown by GPC analysis. Interestingly, the solu-tion-to-gel transition of 3 was not accompanied by a redshiftof the emission as the maximum emission wavelength re-mained unchanged at 368 nm. Similarly to what was ob-served with 1, irradiation of the gel prepared from 3 resultedin the formation of new species, as is evident from Fig-ure 9 b. The emission spectrum of 3 was dominated by amain band at 368 nm with a second peak centered at496 nm. Interestingly, completely different behavior was ob-served after irradiation of solutions of the monomers.Whereas irradiation of the gels resulted in the appearanceof a new emission band in the yellow region, no new emis-sion peaks could be detected after overnight irradiation ofsolutions of the same concentration as those used for gelformation. However, shifts of the emission peaks were seen,with the emission maxima shifting from 368 to 394 and385 nm for monomers 1 and 3, respectively. Broadening of
the two signals could be confi-dently assigned to the forma-tion of a wide range of oligom-ers as a result of the radicalpolymerization of 1 and 3, andnot to a rearrangement of thetwo monomers.
Conclusion
Six oxadiazoles have beenevaluated as new luminescentorganogelators. Interestingly,the presence of the styrenemoiety on the oxadiazoles wasrequired for gelation, provid-ing evidence that the styreneunit serves as a gelation auxili-ary. Complete planarity of themolecules was also necessaryfor gelation, clearly demon-strating that the driving forcefor supramolecular aggregationrelies on p–p overlap. Notably,the steric constraints imposedby the additional alkyl chainon 2 prevented any supramo-lecular self-assembly. Finally,investigation of the effects ofirradiation on solutions of themonomers and on the two gelsgenerated contrasting results.First, irradiation of the gels ob-tained with 1 and 3 in alkaneswith light of wavelength350 nm induced a photochemi-cal reaction that transformed
Figure 8. UV/Vis absorption and photoluminescence spectra of 1–3 inchloroform (lexc =300 nm).
Figure 9. Photoluminescence spectra in chloroform, in the gel state, before and after irradiation (lexc =300 nm)of a) 1 and b) 3.
www.chemeurj.org � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2013, 19, 1373 – 13841382
F. Dumur et al.

the blue photoluminescent organogels into yellow ones. Onthe other hand, irradiation of the two solutions under thesame conditions (concentration, reaction time) resulted inphotopolymerization of the monomers with the formation oflow-molecular-weight oligomers.
Experimental Section
General : Mass spectrometry was performed at the Spectropole of Aix-Marseille University (Marseille). ESI mass spectral analyses were per-formed on a 3200 QTRAP (Applied Biosystems SCIEX) mass spectrom-eter. HRMS analyses were performed with a QStar Elite (Applied Bio-systems SCIEX) mass spectrometer. Solid-state absorption and emissionspectra were recorded with a UV MC2 spectrophotometer from SAFASMonaco and a Photon Technology International spectrofluorimeter. 1Hand 13C NMR spectra were determined at room temperature from sam-ples in 5 mm o.d. tubes on a Bruker Avance 400 spectrometer at theSpectropole: 1H (400 MHz) and 13C (100 MHz). 1H chemical shifts werereferenced to the solvent peak of residual CHCl3 in CDCl3 (d=
7.26 ppm), and 13C chemical shifts were likewise referenced to CDCl3
(d=77.0 ppm). For differential scanning calorimetry measurements, aDSC 2920 from TA Instruments was used. Gels of 1 and 3 formed in do-decane at 5% (w/v) were sealed in a hermetic aluminium pan. The heat-ing and cooling traces were recorded at a temperature ramp of5 8C min�1. A Philips XL 30 ESEM scanning electron microscope (SEM)was used to acquire morphological images. The accelerating voltage ofthe SEM was 20 kV (WD�10 nm). All starting materials and solventswere purchased from Aldrich and Alfa Aesar and were used as received.2-(4-Methylphenyl)-4,4-dimethyl-2-oxazoline (4)[33] and methyl 4-(chloro-carbonyl)benzoate (16)[34] were both prepared in three steps according toliterature procedures and were obtained in yields similar to those report-ed. 4-Vinylbenzyl 4-{5-[4-(tert-butyl)phenyl]-1,3,4-oxadiazol-2-yl}benzoate(3) was prepared in six steps according to a procedure previously report-ed in the literature.[20a] The syntheses of the various molecules are descri-bed in detail in the Supporting Information.
Gelation test of organic fluids : The gelator and the selected solvent wereplaced in a capped test tube and the mixture was degassed with argon for10 min. The test tube was then heated under argon until the solid hadcompletely dissolved. The capped test tube was then cooled and left over-night under ambient conditions. The state of the material was evaluatedby the “stable-to-inversion-of-a-test-tube” method. The critical gelatorconcentration (CGC) was determined from the minimum amount of gela-tor required for the formation of a gel at room temperature.
Acknowledgements
The authors thank the Centre National de la Recherche Scientifique(CNRS), Aix-Marseille University, and the Institut Carnot STAR for fi-nancial support.
[1] a) S. Allard, M. Forster, B. Souharce, H. Thiem, U. Scherf, Angew.Chem. 2008, 120, 4138 –4167; Angew. Chem. Int. Ed. 2008, 47, 4070 –4098; b) J. Roncali, P. Leriche, A. Cravino, Adv. Mater. 2007, 19,2045 – 2060.
[2] a) Comprehensive Supramolecular Chemistry (Ed.: J.-M. Lehn), Per-gamon Press, Oxford, 1996 ; b) J. W. Goodby, I. M. Saez, S. J. Cowl-ing, V. Gçrtz, M. Draper, A. W. Hall, S. Sia, C. Cosquer, S. E. Lee,E. P. Raynes, Angew. Chem. 2008, 120, 2794 –2828; Angew. Chem.Int. Ed. 2008, 47, 2754 – 2787.
[3] a) T. Tanaka, Sci. Am. 1981, 244, 124; b) D. Derossi, Y. Kajiwara, Y.Osada, A. Yamauchi, Polymer Gels: Fundamentals and BiomedicalApplications, Plenum Press, New York, 1991; c) ACS SymposiumSeries, Vol. 833, Polymer Gels: Fundamentals and Applications
(Eds.: H. B. Bohidar, P. Dubin, Y. Osada), American Chemical Soci-ety, Washington, DC, 2003.
[4] For a representative collection of review articles, see: a) E. Ostuni,P. Kamaras, R. G. Weiss, Angew. Chem. 1996, 108, 1423 – 1425;Angew. Chem. Int. Ed. Engl. 1996, 35, 1324 –1326; b) P. Terech,R. G. Weiss, Chem. Rev. 1997, 97, 3133 –3159; c) D. J. Abdallah,R. G. Weiss, Adv. Mater. 2000, 12, 1237 –1247; d) J. H. van Esch,B. L. Feringa, Angew. Chem. 2000, 112, 2351 –2354; Angew. Chem.Int. Ed. 2000, 39, 2263 –2266; e) Macromolecular Symposia, Vol. 200,Functional Networks and Gels (Ed.: E. Geissler), Wiley-VCH, Wein-heim, 2002 ; f) L. A. Estroff, A. D. Hamilton, Chem. Rev. 2004, 104,1201 – 1217; g) P. Xie, R. Zhang, J. Mater. Chem. 2005, 15, 2529 –2550; h) M. George, R. Mathew, R. G. Weiss, Molecules MolecularGels 2006, 11, 449 – 551; i) M. George, R. G. Weiss, Acc. Chem. Res.2006, 39, 489 –497; j) D. K. Smith, Chem. Commun. 2006, 34– 44;k) A. Ajayaghosh, V. K. Praveen, C. Vijayakumar, Chem. Soc. Rev.2008, 37, 109 – 122; l) L. Magginia, D. Bonifazi, Chem. Soc. Rev.2012, 41, 211 –241; m) B. Xu, Langmuir 2009, 25, 8375 – 8377.
[5] a) N. M. Sangeetha, U. Maitra, Chem. Soc. Rev. 2005, 34, 821 – 836;b) X. Yang, G. Zhang, D. Zhang, J. Mater. Chem. 2012, 22, 38– 50;c) Y. Li, K. M.-C. Wong, A. Y.-Y. Tam, L. Wu, V. W.-W. Yam, Chem.Eur. J. 2010, 16, 8690 –8698; d) M.-O. M. Piepenbrock, N. Clarke,J. W. Steed, Langmuir 2009, 25, 8451 – 8456.
[6] a) B. S. Holla, R. Gonsalves, S. Shenoy, Eur. J. Med. Chem. 2000, 35,267 – 271; b) W. R. Tully, C. R. Gardner, R. J. Gillespie, R. West-wood, J. Med. Chem. 1991, 34, 2060 –2067; c) E. Palaska, G. Sahin,P. Kelicen, N. T. Durlu, G. Altinok, Farmaco 2002, 57, 101 –107;d) A. Husain, A. Ahmad, M. M. Alam, M. Ajmal, P. Ahuja, Eur. J.Med. Chem. 2009, 44, 3798 – 3804; e) S. L. Gaonkar, K. M. L. Rai, B.Prabhuswamy, Eur. J. Med. Chem. 2006, 41, 841 –846.
[7] S. H. Mashraqui, S. Sundaram, A. C. Bhasikuttann, S. Kapoor, A. V.Sapre, Sens. Actuators B 2007, 122, 347 – 350.
[8] A. R. Brown, D. D. C. Bradley, P. L. Burns, J. H. Burroughes, R. H.Friend, N. C. Greenham, P. L. Burn, A. B. Holmes, A. Kraft, Appl.Phys. Lett. 1992, 61, 2793 –2795.
[9] a) A. P. Kulkarni, C. J. Tonzola, A. Babel, S. A. Jenekhe, Chem.Mater. 2004, 16, 4556 – 4573; b) A. Kraft, A. C. Grimsdale, A. B.Holmes, Angew. Chem. 1998, 110, 416 –443; Angew. Chem. Int. Ed.1998, 37, 402 – 428; c) K. Tsuchiya, H. Kasuga, A. Kawakami, H.Taka, H. Kita, K. Ogino, J. Polym. Sci. Part A 2010, 48, 1461 – 1468.
[10] S. Y. Ryu, S. Kim, J. Seo, Y.-W. Kim, O.-H. Kwon, D.-J. Jang, S. Y.Park, Chem. Commun. 2004, 70 –71.
[11] a) S. Varghese, N. S. Saleesh Kumar, A. Krishna, D. S. Shankar Rao,S. K. Prasad, S. Das, Adv. Funct. Mater. 2009, 19, 2064 –2073; b) S. S.Babu, K. K. Kartha, A. Ajayaghosh, J. Phys. Chem. Lett. 2010, 1,3413 – 3424.
[12] S. Qu, L. Wang, X. Liu, M. Li, Chem. Eur. J. 2011, 17, 3512 – 3518.[13] W. Lu, Y.-C. Law, J. Han, S. S.-Y. Chui, D.-L. Ma, N. Zhu, C.-M.
Che, Chem. Asian J. 2008, 3, 59– 69.[14] Y.-C. Tseng, S. B. Darling, Polymers 2010, 2, 470 – 489.[15] H. Xin, O. G. Reid, G. Ren, F. S. Kim, D. S. Ginger, S. A. Jenekhe,
ACS Nano 2010, 4, 1861 –1872.[16] M.-C. Wu, H.-C. Liao, H.-H. Lo, S. Chen, Y.-Y. Lin, W.-C. Yen, T.-
W. Zeng, C.-W. Chen, Y.-F. Chen, W.-F. Su, Solar Energy Mater.Solar Cells 2009, 93, 961 – 965.
[17] S. Jeon, J.-W. Kang, H.-D. Park, J.-J. Kim, J. R. Youn, J. Shim, J.-H.Jeong, D.-G. Choi, K.-D. Kim, A. O. Altun, S.-H. Kim, Y.-H. Lee,Appl. Phys. Lett. 2008, 92, 223307.
[18] E. Moulin, J.-J. Cid, N. Giuseppone, Adv. Mater. , DOI: 10.1002/adma.201201949.
[19] F. Fages, Angew. Chem. 2006, 118, 1710 – 1712; Angew. Chem. Int.Ed. 2006, 45, 1680 –1682.
[20] H. Sato, Y. Sakaki, K. Ogino, Y. Ito, Polym. Adv. Technol. 1997, 8,454 – 458.
[21] a) J. Hill, Comprehensive Heterocyclic Chemistry, Vol. 6 (Eds.: A. R.Katritzky, C. W. Rees), Pergamon Press, Oxford, 1984, pp. 427 – 446,and references therein; b) F. Bentiss, M. Lagren�e, J. Heterocycl.Chem. 1999, 36, 1029 –1032.
Chem. Eur. J. 2013, 19, 1373 – 1384 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 1383
FULL PAPERLuminescent Organogels Based on Styrene-Substituted 1,3,4-Oxadiazoles

[22] A. B. Theocharis, N. E. Alexandrou, J. Heterocycl. Chem. 1990, 27,1685 – 1688.
[23] G. Tan, M. Singh, J. He, V. T. John, G. L. McPherson, Langmuir2005, 21, 9322 –9326.
[24] a) Y. E. Shapiro, Prog. Polym. Sci. 2011, 36, 1184 – 1253; b) K. Sakur-ai, Y. Jeong, K. Koumoto, A. Friggeri, O. Gronwald, S. Sakurai, S.Okamoto, K. Inoue, S. Shinkai, Langmuir 2003, 19, 8211 –8217.
[25] C. A. Breen, S. Rifai, V. Bulovic, T. M. Swager, Nano Lett. 2005, 5,1597 – 1601.
[26] a) G. I. Elliott, J. Velcicky, H. Ishikawa, Y. K. Li, D. L. Boger,Angew. Chem. 2006, 118, 636 –638; Angew. Chem. Int. Ed. 2006, 45,620 – 622; b) G. D. Wilkie, G. I. Elliott, B. S. J. Blagg, S. E. Wolken-berg, D. R. Soenen, M. M. Miller, S. Pollack, D. L. Boger, J. Am.Chem. Soc. 2002, 124, 11292 – 11294; c) Y. Sasaki, D. Kato, D. L.Boger, J. Am. Chem. Soc. 2010, 132, 13533 – 13544; d) G. I. Elliott,J. R. Fuchs, B. S. J. Blagg, H. Ishikawa, H. Tao, Z.-Q. Yuan, D. L.Boger, J. Am. Chem. Soc. 2006, 128, 10589 – 10595; e) D. Kato, Y.Sasaki, D. L. Boger, J. Am. Chem. Soc. 2010, 132, 3685 –3687;f) Z. Q. Yuan, H. Ishikawa, D. L. Boger, Org. Lett. 2005, 7, 741 –744.
[27] O. J. Dautel, M. Robitzer, J.-P. L�re-Porte, F. Serein-Spirau, J. J. E.Moreau, J. Am. Chem. Soc. 2006, 128, 16213 –16223.
[28] S. H. Kang, B. M. Jung, J. Y. Chang, Adv. Mater. 2007, 19, 2780 –2784.
[29] A. Perino, M. Schmutz, S. Meunier, P. J. M�sini, A. Wagner, Lang-muir 2011, 27, 12149 – 12155.
[30] J. R. N�abo, K. I. Sika Tohoundjona, J.-F. Morin, Org. Lett. 2011, 13,1358 – 1361.
[31] J. R. Moffat, I. A. Coates, F. J. Leng, D. K. Smith, Langmuir 2009,25, 8786 –8793.
[32] C. Wang, D. Zhang, J. Xiang, D. Zhu, Langmuir 2007, 23, 9195 –9200.
[33] a) F. B. Mallory, K. E. Butler, A. C. Evans, E. J. Brondyke, C. W.Mallory, C. Yang, A. Ellenstein, J. Am. Chem. Soc. 1997, 119, 2119 –2124; b) T. D. Harris, B. Neuschwander, V. Boekelheide, J. Org.Chem. 1978, 43, 727 –730; c) A. I. Meyers, D. L. Temple, D. Haidu-kewych, E. D. Mihelich, J. Org. Chem. 1974, 39, 2787 –2793.
[34] A. Konosonoks, P. J. Wright, M.-L. Tsao, J. Pika, K. Novak, S. M.Mandel, J. A. Krause Bauer, C. Bohne, A. D. Gudmundsdottir, J.Org. Chem. 2005, 70, 2763 –2770.
Received: June 25, 2012Revised: October 7, 2012
Published online: November 30, 2012
www.chemeurj.org � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2013, 19, 1373 – 13841384
F. Dumur et al.
Copyright © 2022 FDOKUMEN







![1,3,4-oxadiazol-2-yl]sulfanyl}acetamides as suitable ther](https://static.fdokumen.com/doc/165x107/6319f8931e5d335f8d0b61c0/134-oxadiazol-2-ylsulfanylacetamides-as-suitable-ther.jpg)








![Iminophosphorane-mediated Synthesis of Fused [1,3,4] Thiadiazoles: Preparation of Imidazo[2,1-b][1,3,4]thiadiazoles and [1,3,4]Thiadiazolo[2,3-c][1,2,4]triazine Derivatives](https://static.fdokumen.com/doc/165x107/6344d54f596bdb97a908a4fa/iminophosphorane-mediated-synthesis-of-fused-134-thiadiazoles-preparation-of.jpg)



![Condensed bridgehead nitrogen heterocyclic system: Synthesis and pharmacological activities of 1,2,4-triazolo-[3,4- b]-1,3,4-thiadiazole derivatives of ibuprofen and biphenyl-4-yloxy](https://static.fdokumen.com/doc/165x107/632834412089eb31f609dd2b/condensed-bridgehead-nitrogen-heterocyclic-system-synthesis-and-pharmacological.jpg)