
Kinetics of Human Immunodeficiency Virus Type 1 (HIV) DNA Integration in Acutely Infected Cells as...
9
10.1128/JVI.75.22.11253-11260.2001. 2001, 75(22):11253. DOI: J. Virol. Peng Li Nick Vandegraaff, Raman Kumar, Christopher J. Burrell and Assay for Detection of Integrated HIV DNA Infected Cells as Determined Using a Novel Type 1 (HIV) DNA Integration in Acutely Kinetics of Human Immunodeficiency Virus http://jvi.asm.org/content/75/22/11253 Updated information and services can be found at: These include: REFERENCES http://jvi.asm.org/content/75/22/11253#ref-list-1 at: This article cites 34 articles, 24 of which can be accessed free CONTENT ALERTS more» articles cite this article), Receive: RSS Feeds, eTOCs, free email alerts (when new http://journals.asm.org/site/misc/reprints.xhtml Information about commercial reprint orders: http://journals.asm.org/site/subscriptions/ To subscribe to to another ASM Journal go to: on August 22, 2014 by guest http://jvi.asm.org/ Downloaded from on August 22, 2014 by guest http://jvi.asm.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Kinetics of Human Immunodeficiency Virus Type 1 (HIV) DNA Integration in Acutely Infected Cells as...

10.1128/JVI.75.22.11253-11260.2001.
2001, 75(22):11253. DOI:J. Virol. Peng LiNick Vandegraaff, Raman Kumar, Christopher J. Burrell and Assay for Detection of Integrated HIV DNAInfected Cells as Determined Using a NovelType 1 (HIV) DNA Integration in Acutely Kinetics of Human Immunodeficiency Virus
http://jvi.asm.org/content/75/22/11253Updated information and services can be found at:
These include:
REFERENCEShttp://jvi.asm.org/content/75/22/11253#ref-list-1at:
This article cites 34 articles, 24 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://journals.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on August 22, 2014 by guest
http://jvi.asm.org/
Dow
nloaded from
on August 22, 2014 by guest
http://jvi.asm.org/
Dow
nloaded from

JOURNAL OF VIROLOGY,0022-538X/01/$04.00�0 DOI: 10.1128/JVI.75.22.11253–11260.2001
Nov. 2001, p. 11253–11260 Vol. 75, No. 22
Copyright © 2001, American Society for Microbiology. All Rights Reserved.
Kinetics of Human Immunodeficiency Virus Type 1 (HIV) DNAIntegration in Acutely Infected Cells as Determined Using a
Novel Assay for Detection of Integrated HIV DNANICK VANDEGRAAFF,1,2 RAMAN KUMAR,1 CHRISTOPHER J. BURRELL,1,2 AND PENG LI1*
National Centre for HIV Virology Research, Infectious Diseases Laboratories, Institute of Medical and Veterinary Science,1
and Department of Molecular Biosciences, University of Adelaide,2 Adelaide, Australia
Received 11 December 2000/Accepted 17 August 2001
We have developed a novel linker-primer PCR assay for the detection and quantification of integratedhuman immunodeficiency virus type 1 (HIV) DNA. This assay reproducibly allowed the detection of 10 copiesof integrated HIV DNA, in a background of 2 � 105 cell equivalents of human chromosomal DNA, withoutamplifying extrachromosomal HIV DNA. We have used this assay and a near-synchronous one-step T-cellinfection model to investigate the kinetics of viral DNA accumulation following HIV infection. We report herethat integrated HIV DNA started accumulating 1 h after the first appearance of extrachromosomal viral DNAand accounted for �10% of the total HIV DNA synthesized in the first round of viral replication. These resultshighlight the efficient nature of integrase-mediated HIV integration in infected T cells.
Integration of newly synthesized viral DNA into the host cellchromosome is common to all retroviruses and is essential fora productive human immunodeficiency virus (HIV) infection(12, 22, 28, 30). Upon reverse transcription of the viralgenomic RNA, the resulting linear DNA molecule is activelytransported to the nucleus within a complex of host and viralproteins known as the preintegration complex, which isthought to be the immediate precursor to the integration re-action (2, 3, 5, 13, 19, 24, 26). Analyses of the extrachromo-somal and total HIV DNA forms using both Southern hybrid-ization and PCR-based techniques have indicated that full-length linear DNA is first observed at approximately 3 to 4 hpostinfection (p.i.) (1, 20, 21, 23, 25). In reports on the kineticsof HIV DNA synthesis following cell-to-cell infection, the cir-cular forms of viral DNA were shown to first appear at 8 h p.i.,with the two long-terminal-repeat (2-LTR) species constitutinga minor population compared to the 1-LTR and linear formsover the course of infection (1, 25).
In contrast to investigations on both free and total viralDNA forms, little work has been performed on the accumula-tion of integrated DNA within infected cells following HIVinfection. This has been primarily due to the lack of an appro-priate assay which can selectively detect and quantify inte-grated viral DNA, as chromosomal DNA preparations isolatedfrom cells infected with HIV invariably contain significantamounts of contaminating extrachromosomal HIV DNA (1,27, 30, 34). However, two assays able to distinguish betweenthe extrachromosomal and integrated HIV DNA have recentlybeen described and used to quantify the amounts of integratedproviral HIV DNA in infected patients (6–10). Here wepresent an alternative linker-primer PCR assay (LP-PCR) de-veloped to specifically detect and quantify integrated HIV
DNA species. This assay utilizes the presence of frequentlyoccurring NlaIII restriction enzyme recognition sites in chro-mosomal DNA adjacent to the integrated provirus and atknown positions within the proviral sequence. Linkers are li-gated to the DNA termini generated by NlaIII digestion ofchromosomal DNA and serve as templates from which primingcan occur in a subsequent PCR amplifying the 5�-U3 HIVregion and upstream cellular DNA sequence. In conjunctionwith other PCR-based assays, we have used LP-PCR to studythe kinetics of total, integrated, and 2-LTR HIV DNA accu-mulation over time following a high-multiplicity infection ofHuT-78 T cells with HIVHXB2. In addition, we also presentresults comparing LP-PCR to a nested Alu PCR method forthe quantification of integrated HIV DNA.
Establishment of LP-PCR for the detection and quantifica-tion of integrated HIV DNA. To specifically detect integratedHIV DNA in the presence of contaminating extrachromo-somal viral DNA forms, we modified a previously describedlinker ligation PCR protocol used for sequence analysis of thehuman T-cell leukemia virus type 1 integration junctions (32).Briefly, chromosomal DNA was digested with the restrictionenzyme NlaIII. NlaIII has a 4-bp recognition sequence andgenerates a 4-bp 3� overhang to which the specifically designedoligonucleotide linker LPNV is annealed and ligated (Fig. 1A).This linker generates a region from which priming can occur ina subsequent PCR using the same linker oligonucleotide(LPNV) in conjunction with a primer (U3NV) designed toanneal within the U3 region of the HIV LTR. Since retroviralintegration is random with respect to cellular sequences, LP-PCR generates a population of cellular-5� HIV junction DNAsequences of various lengths. A nested PCR was performed togenerate a product of a defined length, which was then quan-tified against a known set of standards (see below).
To avoid LP-PCR amplification of extrachromosomal viralDNA, the chromosomal DNA preparations were also digestedwith the restriction enzyme BglII. BglII has a recognition se-quence of 6 bp and cleaves potential LP-PCR DNA templates
* Corresponding author. Mailing address: National Centre for HIVVirology Research, Infectious Diseases Laboratories, Institute ofMedical and Veterinary Science, Frome Rd., Adelaide 5000, Australia.Phone: 61 8 82223544. Fax: 61 8 82223543. E-mail: [email protected].
11253
on August 22, 2014 by guest
http://jvi.asm.org/
Dow
nloaded from

within extrachromosomal HIV DNA forms, generating 4-bp 5�overhangs to which LPNV cannot ligate (Fig. 1B). Religationof BglII fragments was inhibited by filling in two nucleotides (Gand A) of the BglII site with Klenow (lacking 3�-5� exonucleaseactivity [3�-5� exo�]) polymerase before the ligation reactionwas done. Due to the relative sizes of the restriction enzymerecognition sites, the chances of a BglII site occurring prior toan NlaIII site in the chromosomal sequence upstream of the5�LTR is once every 16 integration events. Therefore, theoret-ically 94% of all integrated forms should be detectable by thistechnique.
The LP-PCR procedure was performed as follows: chromo-somal DNA (isolated by the method of Hirt [17, 31]) was firstdigested to completion with 20 U of BglII (New England Bio-labs) in 2� OPA Plus buffer (Pharmacia) for 3 h at 37°C in afinal volume of 20 �l. Following this digestion, buffering con-ditions were then altered to final concentrations of 1� OPAPlus, 20 mM Tris-acetate (pH 7.9), 0.1 mg of bovine serumalbumin (New England Biolabs) per ml, and 1 mM dithiothre-itol (Boehringer Mannheim) prior to the addition of 10 U ofNlaIII (New England Biolabs) and incubation at 37°C for 3 h
in a final volume of 40 �l. All digestion reactions were con-firmed to have proceeded to completion by both gel electro-phoresis and PCR-based assays (data not shown). Two nucle-otides (G and A) of the BglII overhang generated by digestionwere filled in with 5 U of Klenow (3�-5� exo�) (New EnglandBiolabs) after modification of the buffering conditions to finalconcentrations of 7.5 mM dithiothreitol, 0.25 mM dGTP (Pro-mega), and 0.25 mM dATP (Promega) and incubation at 37°Cfor 30 min in a final volume of 50 �l. Samples were thenextracted with phenol-chloroform-isoamyl alcohol (25:24:1),ethanol precipitated in the presence of glycogen (BoehringerMannheim), and washed in 70% ethanol prior to resuspensionof the pellet in water. Linker ligation reactions in 1� ligationbuffer (New England Biolabs) using 50 pmol (vast excess) ofLPNV (Table 1) were heated to 60°C for 10 min and snap-cooled to minimize inter- and intramolecular ligation of NlaIIIfragments, followed by the addition of 400 U of T4 DNA ligase(New England Biolabs) and incubation overnight at 16°C.First-round PCR was performed in 1� PCR Buffer II (Perkin-Elmer), 2 mM MgCl2, and 0.2 mM deoxynucleoside triphos-phates (dNTPs) (Promega) using 150 pmol of LPNV, 100 pmol
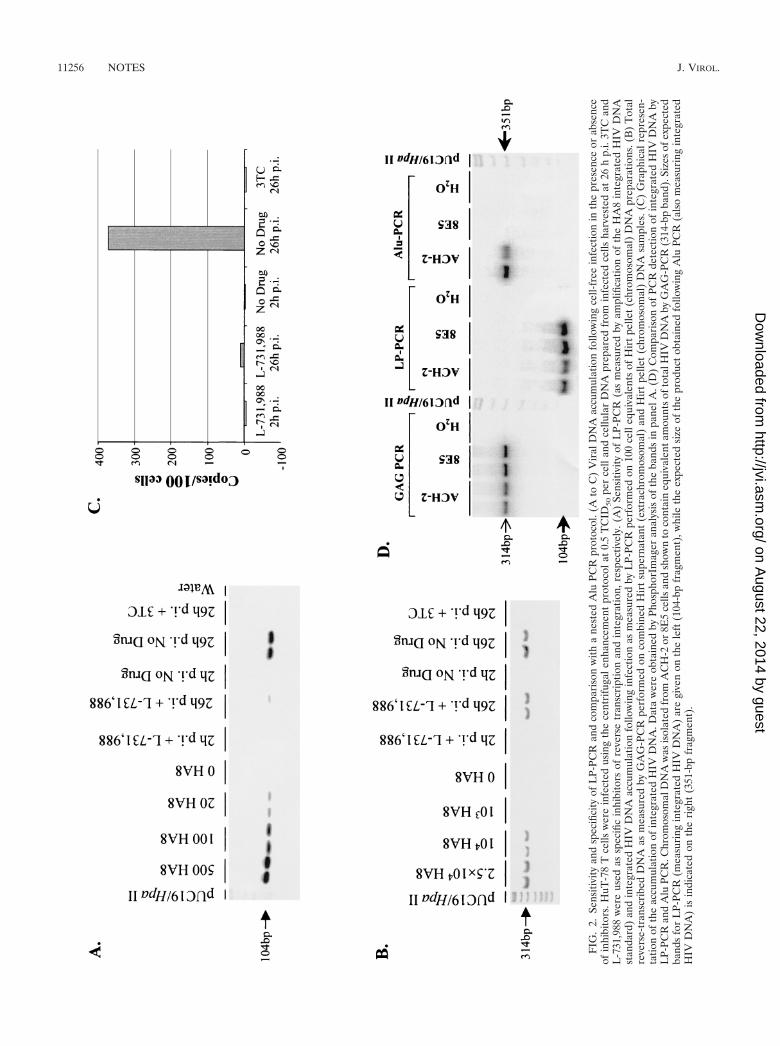
FIG. 1. LP-PCR method for detection of integrated HIV DNA. (A) LP-PCR-mediated amplification of the integrated HIV DNA forms. Thenested PCR product was detected using the U3-106 probe fragment (hatched box) (Table 1). (B) BglII-mediated selection against amplificationof the three main extrachromosomal HIV DNA forms.
11254 NOTES J. VIROL.
on August 22, 2014 by guest
http://jvi.asm.org/
Dow
nloaded from

of U3NV (Table 1), and 5 U of AmpliTaq Gold DNA poly-merase in a final volume of 100 �l. PCRs were as follows: 95°Cfor 12 min; 22 cycles of 94°C for 30 s, 58°C for 30 s, and 72°Cfor 1 min; and 72°C for 10 min. Nested PCRs were performedon 1/100 of the first-round PCR product in 1� PCR Buffer II(Perkin-Elmer), 1.5 mM MgCl2, and 0.2 mM dNTPs (Pro-mega) using 25 pmol each of primers U3.1(�) and U3-106(�)(Table 1) and 2.5 U of AmpliTaq DNA polymerase (Perkin-Elmer) in a final volume of 25 �l. PCRs were cycled as follows:94°C for 3 min; 22 cycles of 94°C for 45 s, 58°C for 30 s, and72°C for 45 s; and 72°C for 10 min. Amplified DNA wasanalyzed by subjecting 10 �l of each reaction mixture to elec-trophoresis through 8% polyacrylamide gels and then South-ern transfer (electroblot apparatus) onto Hybond N� nylonfilters (Amersham). Following denaturation and fixation using0.4 M NaOH, the filters were hybridized using the U3-106probe (Table 1) in Ultrahyb solution (Ambion). FollowingSouthern hybridization, bands were quantified using Phosphor-Imager ImageQuant analysis and a standard curve was generatedfrom the simultaneous PCR of known copy numbers of standards.
In order to assess the sensitivity of the LP-PCR procedure,an integrated proviral DNA standard (designated HA8) wasproduced by mixing 5 � 105, 1 � 106, and 1 � 106 cells of theH3B (25), ACH-2 (11), and 8E5 (14) cell lines, respectively,and preparing chromosomal DNA by the method of Hirt (17,31). These cell lines contain 2, 1, and 1 copies of the integratedHIV proviral DNA, respectively, with little or no detectableextrachromosomal forms (11, 14, 25). Rather than a single cellline, a mixture of three clonal cell lines was used as the inte-grated DNA standard to account for variations associated withdifferent integration events. DNA cell equivalents were calcu-lated based on the average signal obtained after PCR ampli-fication of the �-globin gene (31) on six chromosomal DNAextractions from cells counted independently. The HA8 stan-dards were used as copy number controls for quantifying totalHIV DNA, integrated HIV DNA, and the �-globin content ofsamples. HuT-78 (15) chromosomal DNA was used as back-ground DNA. To confirm that all four integration sites within
the HA8 cell mix could be amplified by LP-PCR, the chromo-somal sequence immediately upstream of the 5� HIV LTRregion of the integrated provirus(es) present in H3B, ACH-2,and 8E5 cells was obtained. In all cases, an NlaIII site precededthe BglII site in the flanking sequence (data not shown).
By comparison with the HA8 composite integrated HIVDNA standard, LP-PCR was shown to routinely detect 20copies of integrated HIV-1 DNA in a background of 500 cellequivalents of HuT-78 chromosomal DNA (Fig. 2A). In addi-tion, we were able to reproducibly detect 10 copies of the HA8integrated standard in the presence of 2 � 105 cell equivalentsof HuT-78 chromosomal DNA (1.2 �g) when elevated nested-PCR cycle numbers were used (data not shown). Furthermore,amplification of a construct precisely mimicking the linear viralDNA form spiked onto 1.2 �g of HuT-78 DNA routinelyresulted in a signal intensity equivalent to �7.5% of that gen-erated by an equivalent HIV DNA copy number of the HA8integrated standard. This result indicated that LP-PCR wasapproximately 15-fold more specific for the integrated thanextrachromosomal HIV DNA forms (data not shown). Sampleheating and snap-cooling in the presence of a vast excess of thelinker prior to the ligation reaction (to minimize intermolecu-lar NlaIII fragment ligation), as well as the use of a hot-startPCR (to fully dissociate NlaIII fragments and inhibit linker-mediated amplification of all NlaIII fragments), were found tobe critical to the success of this assay (data not shown). Fur-thermore, the efficiency of linker ligation to NlaIII termini wasdemonstrated to be approximately 100% (data not shown).
To further confirm that extrachromosomal DNA forms werenot detected by the LP-PCR procedure, HuT-78 cells wereinfected with HIVHXB2 (0.5 50% tissue culture infective dose[TCID50] per cell) in the presence or absence of the integra-tion inhibitor L-731,988 (16) as previously described (31). Thereverse transcriptase inhibitor lamivudine (3TC; final concen-tration, 10 �M) served as a control for inhibition of extrachro-mosomal HIV DNA synthesis prior to integration. Followinganalyses of 100 cell equivalents of cellular DNA using LP-PCRand a GAG-PCR protocol (31), strong signals correspondingto total and integrated HIV DNA were observed by 26 h p.i. indrug-free cultures, respectively (Fig. 2A and B). As expected,cultures infected in the presence of 3TC were negative for bothtotal and integrated HIV DNA. Analysis of DNA from cellsinfected in the presence of L-731,988 indicated that the accu-mulation of integrated HIV DNA had been abolished (Fig. 2Aand C), while the accumulation of extrachromosomal HIVDNA was largely unaffected (Fig. 2B). This result clearly dem-onstrates that LP-PCR specifically detects integrated HIVDNA.
To further characterize the LP-PCR procedure, a directcomparison of LP-PCR and a previously published method forthe detection of integrated HIV DNA (a nested Alu PCRprotocol [31]) was performed. Chromosomal preparations ofthe clonal cell lines ACH-2 and 8E5 (each containing one copyof integrated provirus) were shown to contain equivalentamounts of viral DNA by GAG-PCR (31) and then subjectedto the LP-PCR and the nested Alu PCR procedures to detectintegrated HIV DNA. While integrated DNA within theACH-2 cell line was efficiently amplified, the nested Alu PCRmethod was unable to facilitate amplification of integrated
TABLE 1. Primer sequences and probes used in this studya
Primer orprobe Sequence Coordinates
(nt)
PrimersPBS-659(�) 5�-TTTCAGGTCCCTGTTCGGGCGCCAC-3� 659–635b
R7 5�-GGGTCTCTCTGGTTAGACC-3� 454–472b
U3NV 5�-GGCTTCTTCTAACTTCTCTGGCTC-3� 179–156b
LPNV 5�-TCATGATCAATGGGACGATCACATG-3� Same asB101c
U3PNV 5�-GGTACTAGCTTGTAGCACCATCC-3� 151–129b
U3-106(�) 5�-CCTGGCCCTGGTGTGTAGTTC-3� 106–86b
U3.1(�) 5�-GGAAGGGCTAATTCACTCC-3� 2–20b
Alu164 5�-TCCCAGCTACTGGGGAGGCTGAGG-3� 164–187d
Probee
U3-106 2–106b
a For all other primer sequences and probes used in this study, see reference31.
b HIVHXB2 GenBank accession number K03455.c Reference 32.d Reference 18.e Probes were labeled with [�-32P]dATP and a commercially available kit
(Amersham Megaprime).
VOL. 75, 2001 NOTES 11255
on August 22, 2014 by guest
http://jvi.asm.org/
Dow
nloaded from
FIG
.2.
Sens
itivi
tyan
dsp
ecifi
city
ofL
P-PC
Ran
dco
mpa
riso
nw
itha
nest
edA
luPC
Rpr
otoc
ol.(
Ato
C)
Vir
alD
NA
accu
mul
atio
nfo
llow
ing
cell-
free
infe
ctio
nin
the
pres
ence
orab
senc
eof
inhi
bito
rs.H
uT-7
8T
cells
wer
ein
fect
edus
ing
the
cent
rifu
gale
nhan
cem
ent
prot
ocol
at0.
5T
CID
50pe
rce
llan
dce
llula
rD
NA
prep
ared
from
infe
cted
cells
harv
este
dat
26h
p.i.
3TC
and
L-7
31,9
88w
ere
used
assp
ecifi
cin
hibi
tors
ofre
vers
etr
ansc
ript
ion
and
inte
grat
ion,
resp
ectiv
ely.
(A)
Sens
itivi
tyof
LP-
PCR
(as
mea
sure
dby
ampl
ifica
tion
ofth
eH
A8
inte
grat
edH
IVD
NA
stan
dard
)an
din
tegr
ated
HIV
DN
Aac
cum
ulat
ion
follo
win
gin
fect
ion
asm
easu
red
byL
P-PC
Rpe
rfor
med
on10
0ce
lleq
uiva
lent
sof
Hir
tpe
llet
(chr
omos
omal
)D
NA
prep
arat
ions
.(B
)T
otal
reve
rse-
tran
scri
bed
DN
Aas
mea
sure
dby
GA
G-P
CR
perf
orm
edon
com
bine
dH
irt
supe
rnat
ant
(ext
rach
rom
osom
al)
and
Hir
tpe
llet
(chr
omos
omal
)D
NA
sam
ples
.(C
)G
raph
ical
repr
esen
-ta
tion
ofth
eac
cum
ulat
ion
ofin
tegr
ated
HIV
DN
A.D
ata
wer
eob
tain
edby
Phos
phor
Imag
eran
alys
isof
the
band
sin
pane
lA.(
D)
Com
pari
son
ofPC
Rde
tect
ion
ofin
tegr
ated
HIV
DN
Aby
LP-
PCR
and
Alu
PCR
.Chr
omos
omal
DN
Aw
asis
olat
edfr
omA
CH
-2or
8E5
cells
and
show
nto
cont
ain
equi
vale
ntam
ount
sof
tota
lHIV
DN
Aby
GA
G-P
CR
(314
-bp
band
).Si
zes
ofex
pect
edba
nds
for
LP-
PCR
(mea
suri
ngin
tegr
ated
HIV
DN
A)
are
give
non
the
left
(104
-bp
frag
men
t),w
hile
the
expe
cted
size
ofth
epr
oduc
tob
tain
edfo
llow
ing
Alu
PCR
(als
om
easu
ring
inte
grat
edH
IVD
NA
)is
indi
cate
don
the
righ
t(3
51-b
pfr
agm
ent)
.
11256 NOTES J. VIROL.
on August 22, 2014 by guest
http://jvi.asm.org/
Dow
nloaded from

DNA in the preparation of 8E5 chromosomal DNA (Fig. 2D).In contrast, the LP-PCR procedure allowed the efficient am-plification of integrated DNA present in both cell lines (Fig.2D). BLAST analyses of chromosomal sequence upstream ofHIV integration sites in the 8E5 and ACH-2 cell lines revealedthat in 8E5 cellular DNA, the Alu repeat element immediatelyupstream of the integrated DNA existed in the same orienta-tion as the PBS-659(�) primer. Consequently, amplificationbetween Alu elements upstream of the integration site in 8E5cells, instead of amplification between the Alu164 and thePBS-659(�) primers, would have occurred. In contrast, theanalogous Alu element in ACH-2 chromosomal DNA waspresent in the correct orientation for successful amplificationwith the PBS-659(�) primer (data not shown). We thereforepropose that the nested Alu PCR technique allows amplifica-tion of only those integrants inserted at chromosomal sitesimmediately adjacent to an Alu element present in an orien-tation opposite to that of the PBS-659(�) primer. Statistically,then, the nested Alu PCR approach would be expected tosuccessfully amplify at best half of all integrated proviral forms.Consequently, we believe that the LP-PCR assay is a poten-tially more appropriate protocol for detecting integrated HIVDNA. A comprehensive comparison between LP-PCR, nestedAlu PCR, and an alternative assay currently being developed inour laboratory to detect integrated proviral forms based on theuse of degenerate primers will be published elsewhere.
Kinetics of HIV-1 DNA integration following a one-stepviral infection of HuT-78 cells. To investigate the kinetics ofviral DNA accumulation following infection, a highly synchro-nous one-step infection of HuT-78 cells with cell-free HIV at amultiplicity of infection of 1 TCID50 per cell was performed aspreviously described (31). Extensive washing of cells to removeresidual noninternalized virus prior to plating minimized thechance of any additional infection events occurring after theinitial infection period. Viral release into the culture superna-tant following infection (as measured by P24 release using acommercial kit [NEN]) was evident by 26 h p.i., indicating thatone round of replication was complete by this stage (see Fig.4A). Consequently, the proportions of each viral DNA formassessed following infection were calculated at 26 h p.i. toensure that the results obtained were not skewed by secondarycell-free (or cell-to-cell) infection events.
DNA was extracted by the method of Hirt (17, 31) frominfected cells harvested at various time points following infec-tion to ensure that the bulk (80%) of extrachromosomalforms were separated from the chromosomal DNA forms.Chromosomal DNA preparations were subjected to PCR am-plification of the �-globin gene to determine the cell equivalentDNA content of each sample. Samples were volume adjustedbased on the results of initial �-globin PCR quantification, andupon reanalyses, little variation between samples was observed(Fig. 3D). Extrachromosomal DNA preparations were equal-ized between samples based on a semiquantitative PCR assay(31) measuring the mitochondrial complement of this fraction.The mitochondrial DNA PCR results showed only minor vari-ation between all samples, and therefore adjustment was notnecessary (Fig. 3E).
The total viral DNA complement was measured by mixingHirt supernatant and Hirt pellet DNA fractions from the sametime points and analyzing the pooled samples by PCR for the
presence of GAG DNA sequences that are synthesized in themid-late stages of the HIV reverse transcription process (20).PCRs were performed on 500 cell equivalents of DNA asdescribed previously (31). The results (Fig. 3A) indicated thatnear full-length viral DNA was detected approximately 3 hafter infection, which is in close agreement with previous stud-ies (1, 21, 25). PhosphorImager analysis of bands showed thattotal HIV DNA had peaked at a level of approximately 30copies/cell at 14 h p.i. and declined to levels of approximately20 copies per cell by 26 h p.i. (Fig. 4B). The reduction in thetotal viral DNA complement after 14 h p.i. (�40%) can beattributed to the degradation of extrachromosomal HIV DNAwithin the cellular environment. This result is consistent with aprevious report showing that significant proportions of reverse-transcribed viral DNA degrade within the intracellular envi-ronment following cell-to-cell infection (1). Most viral DNAhad been reverse transcribed by 5 h p.i. (Fig. 4B), providingfurther evidence that this one-step HIV infection was nearlysynchronous. Consistent with the results of P24 release follow-ing infection (Fig. 4A), the GAG signal at 50 h p.i. was higherthan at 26 h p.i., implying that some degree of either second-round cell-free or cell-to-cell infection (superinfection) mayhave occurred during this time.
Integrated HIV DNA levels were measured using both theLP-PCR procedure (Fig. 3, panel B.i) and a modified versionof the previously published nested Alu PCR protocol (31) inchromosomal DNA preparations of infected cells harvested ateach time-point (Fig. 3, panel B.ii). Integrated DNA was firstdetected by LP-PCR (performed on 100 cell equivalents ofchromosomal DNA) at 4 h p.i. (that is, 1 h after the firstappearance of newly synthesized viral DNA) (Fig. 3, comparepanels A and B.i) and the level reached approximately threecopies/cell by 26 h p.i. In contrast, when the nested Alu PCRmethod was performed (on 1,000 cell equivalents of DNA),integrated provirus was first detected considerably later andrepeatedly displayed lower levels (Fig. 3, panel B.ii) across alltime points tested. Control experiments involving first-roundamplification of 26-h-p.i. samples performed in the absence ofligation (LP-PCR) or the Alu164 primer (nested Alu PCR)(Table 1) resulted in bands of very low intensity (Fig. 3, panelsB.i and B.ii). This indicated that the signals obtained whenamplification was performed in the presence of both ligation(LP-PCR) and the Alu164 primer (nested Alu PCR) indeedresulted from the specific amplification of integrated HIVDNA and not the nested amplification of input target se-quences. The discrepancy between integration levels observedwhen either the LP-PCR or the nested Alu PCR protocol wasused was expected and was presumed to result from the abilityof the nested Alu PCR approach to detect only a small pro-portion of integration events (Fig. 2D). Therefore, integratedHIV DNA, at the end of the first round of HIV replication(i.e., 26 h p.i.), was found to account for approximately 10% ofthe total HIV DNA synthesized at the peak of viral DNAaccumulation (i.e., 14 h p.i.) (Fig. 4B).
We also monitored accumulation of the 2-LTR viral DNAforms using a specific PCR amplification protocol with primersflanking the dual-repeat cassette within the circular form. Toallow quantification of 2-LTR viral DNA levels, a control con-struct was generated by PCR amplification of Hirt supernatantsamples taken from a cell-free infection of HuT-78 cells at 26 h
VOL. 75, 2001 NOTES 11257
on August 22, 2014 by guest
http://jvi.asm.org/
Dow
nloaded from
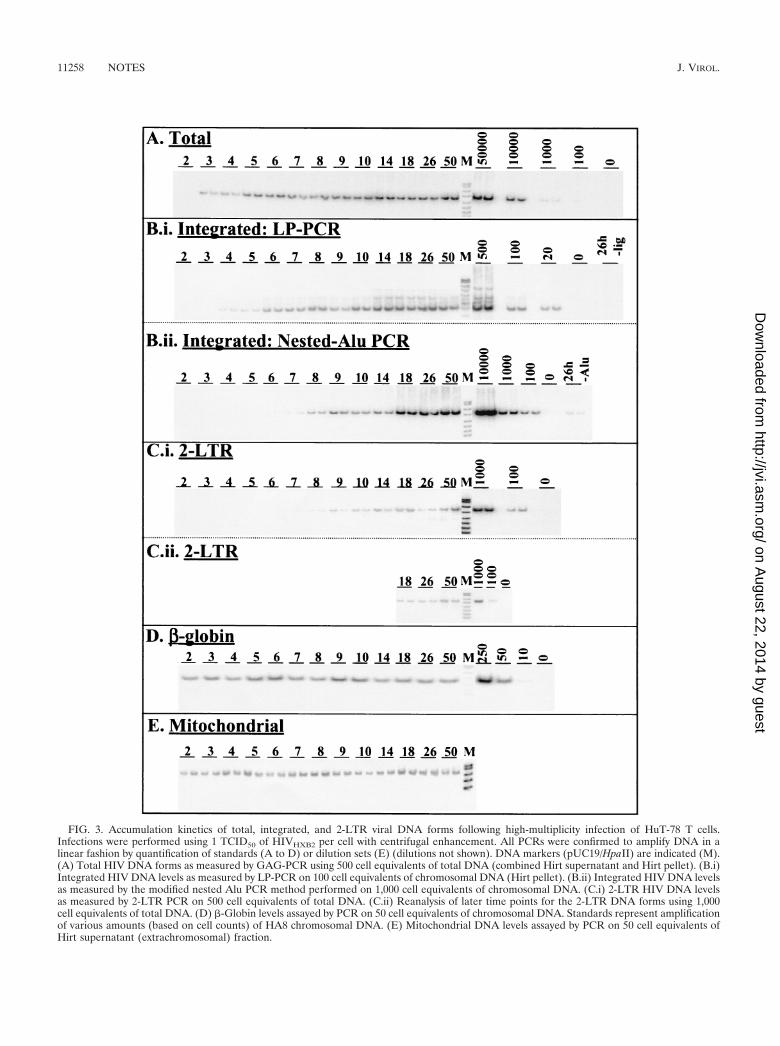
FIG. 3. Accumulation kinetics of total, integrated, and 2-LTR viral DNA forms following high-multiplicity infection of HuT-78 T cells.Infections were performed using 1 TCID50 of HIVHXB2 per cell with centrifugal enhancement. All PCRs were confirmed to amplify DNA in alinear fashion by quantification of standards (A to D) or dilution sets (E) (dilutions not shown). DNA markers (pUC19/HpaII) are indicated (M).(A) Total HIV DNA forms as measured by GAG-PCR using 500 cell equivalents of total DNA (combined Hirt supernatant and Hirt pellet). (B.i)Integrated HIV DNA levels as measured by LP-PCR on 100 cell equivalents of chromosomal DNA (Hirt pellet). (B.ii) Integrated HIV DNA levelsas measured by the modified nested Alu PCR method performed on 1,000 cell equivalents of chromosomal DNA. (C.i) 2-LTR HIV DNA levelsas measured by 2-LTR PCR on 500 cell equivalents of total DNA. (C.ii) Reanalysis of later time points for the 2-LTR DNA forms using 1,000cell equivalents of total DNA. (D) �-Globin levels assayed by PCR on 50 cell equivalents of chromosomal DNA. Standards represent amplificationof various amounts (based on cell counts) of HA8 chromosomal DNA. (E) Mitochondrial DNA levels assayed by PCR on 50 cell equivalents ofHirt supernatant (extrachromosomal) fraction.
11258 NOTES J. VIROL.
on August 22, 2014 by guest
http://jvi.asm.org/
Dow
nloaded from
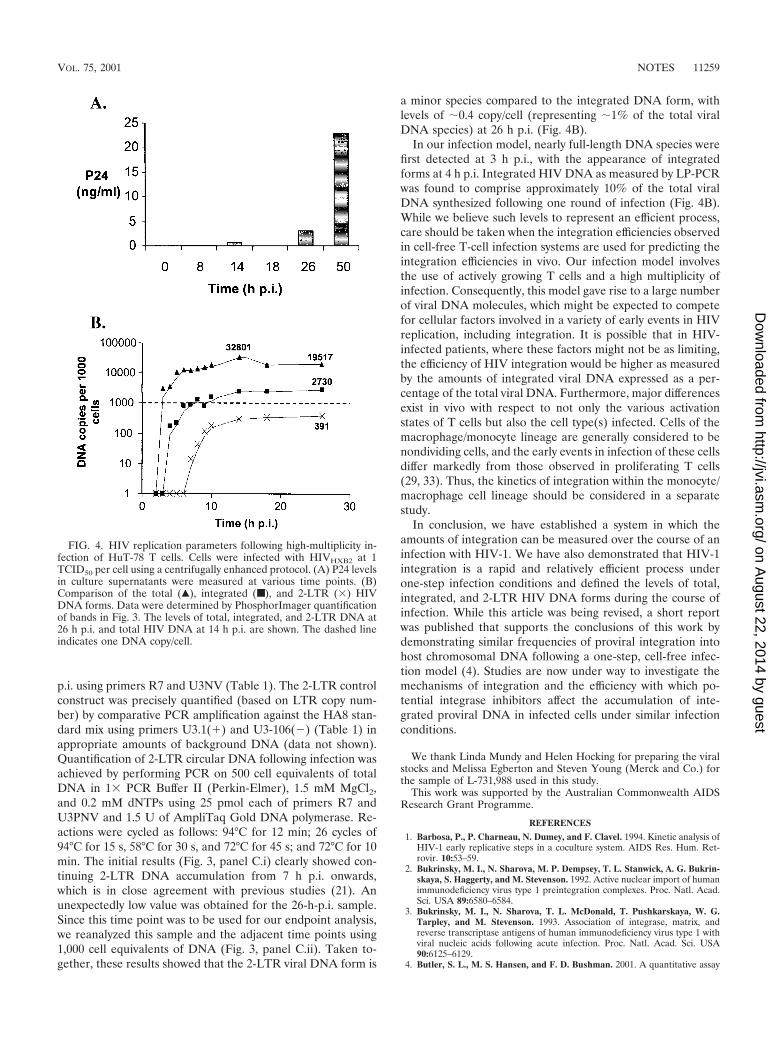
p.i. using primers R7 and U3NV (Table 1). The 2-LTR controlconstruct was precisely quantified (based on LTR copy num-ber) by comparative PCR amplification against the HA8 stan-dard mix using primers U3.1(�) and U3-106(�) (Table 1) inappropriate amounts of background DNA (data not shown).Quantification of 2-LTR circular DNA following infection wasachieved by performing PCR on 500 cell equivalents of totalDNA in 1� PCR Buffer II (Perkin-Elmer), 1.5 mM MgCl2,and 0.2 mM dNTPs using 25 pmol each of primers R7 andU3PNV and 1.5 U of AmpliTaq Gold DNA polymerase. Re-actions were cycled as follows: 94°C for 12 min; 26 cycles of94°C for 15 s, 58°C for 30 s, and 72°C for 45 s; and 72°C for 10min. The initial results (Fig. 3, panel C.i) clearly showed con-tinuing 2-LTR DNA accumulation from 7 h p.i. onwards,which is in close agreement with previous studies (21). Anunexpectedly low value was obtained for the 26-h-p.i. sample.Since this time point was to be used for our endpoint analysis,we reanalyzed this sample and the adjacent time points using1,000 cell equivalents of DNA (Fig. 3, panel C.ii). Taken to-gether, these results showed that the 2-LTR viral DNA form is
a minor species compared to the integrated DNA form, withlevels of �0.4 copy/cell (representing �1% of the total viralDNA species) at 26 h p.i. (Fig. 4B).
In our infection model, nearly full-length DNA species werefirst detected at 3 h p.i., with the appearance of integratedforms at 4 h p.i. Integrated HIV DNA as measured by LP-PCRwas found to comprise approximately 10% of the total viralDNA synthesized following one round of infection (Fig. 4B).While we believe such levels to represent an efficient process,care should be taken when the integration efficiencies observedin cell-free T-cell infection systems are used for predicting theintegration efficiencies in vivo. Our infection model involvesthe use of actively growing T cells and a high multiplicity ofinfection. Consequently, this model gave rise to a large numberof viral DNA molecules, which might be expected to competefor cellular factors involved in a variety of early events in HIVreplication, including integration. It is possible that in HIV-infected patients, where these factors might not be as limiting,the efficiency of HIV integration would be higher as measuredby the amounts of integrated viral DNA expressed as a per-centage of the total viral DNA. Furthermore, major differencesexist in vivo with respect to not only the various activationstates of T cells but also the cell type(s) infected. Cells of themacrophage/monocyte lineage are generally considered to benondividing cells, and the early events in infection of these cellsdiffer markedly from those observed in proliferating T cells(29, 33). Thus, the kinetics of integration within the monocyte/macrophage cell lineage should be considered in a separatestudy.
In conclusion, we have established a system in which theamounts of integration can be measured over the course of aninfection with HIV-1. We have also demonstrated that HIV-1integration is a rapid and relatively efficient process underone-step infection conditions and defined the levels of total,integrated, and 2-LTR HIV DNA forms during the course ofinfection. While this article was being revised, a short reportwas published that supports the conclusions of this work bydemonstrating similar frequencies of proviral integration intohost chromosomal DNA following a one-step, cell-free infec-tion model (4). Studies are now under way to investigate themechanisms of integration and the efficiency with which po-tential integrase inhibitors affect the accumulation of inte-grated proviral DNA in infected cells under similar infectionconditions.
We thank Linda Mundy and Helen Hocking for preparing the viralstocks and Melissa Egberton and Steven Young (Merck and Co.) forthe sample of L-731,988 used in this study.
This work was supported by the Australian Commonwealth AIDSResearch Grant Programme.
REFERENCES
1. Barbosa, P., P. Charneau, N. Dumey, and F. Clavel. 1994. Kinetic analysis ofHIV-1 early replicative steps in a coculture system. AIDS Res. Hum. Ret-rovir. 10:53–59.
2. Bukrinsky, M. I., N. Sharova, M. P. Dempsey, T. L. Stanwick, A. G. Bukrin-skaya, S. Haggerty, and M. Stevenson. 1992. Active nuclear import of humanimmunodeficiency virus type 1 preintegration complexes. Proc. Natl. Acad.Sci. USA 89:6580–6584.
3. Bukrinsky, M. I., N. Sharova, T. L. McDonald, T. Pushkarskaya, W. G.Tarpley, and M. Stevenson. 1993. Association of integrase, matrix, andreverse transcriptase antigens of human immunodeficiency virus type 1 withviral nucleic acids following acute infection. Proc. Natl. Acad. Sci. USA90:6125–6129.
4. Butler, S. L., M. S. Hansen, and F. D. Bushman. 2001. A quantitative assay
FIG. 4. HIV replication parameters following high-multiplicity in-fection of HuT-78 T cells. Cells were infected with HIVHXB2 at 1TCID50 per cell using a centrifugally enhanced protocol. (A) P24 levelsin culture supernatants were measured at various time points. (B)Comparison of the total (Œ), integrated (■), and 2-LTR (�) HIVDNA forms. Data were determined by PhosphorImager quantificationof bands in Fig. 3. The levels of total, integrated, and 2-LTR DNA at26 h p.i. and total HIV DNA at 14 h p.i. are shown. The dashed lineindicates one DNA copy/cell.
VOL. 75, 2001 NOTES 11259
on August 22, 2014 by guest
http://jvi.asm.org/
Dow
nloaded from

for HIV DNA integration in vivo. Nat. Med. 7:631–634.5. Chen, H., and A. Engelman. 1998. The barrier-to-autointegration protein is
a host factor for HIV type 1 integration. Proc. Natl. Acad. Sci. USA 95:15270–15274.
6. Chun, T. W., L. Carruth, D. Finzi, X. Shen, J. A. DiGiuseppe, H. Taylor, M.Hermankova, K. Chadwick, J. Margolick, T. C. Quinn, Y. H. Kuo, R. Brook-meyer, M. A. Zeiger, P. Barditch-Crovo, and R. F. Siliciano. 1997. Quanti-fication of latent tissue reservoirs and total body viral load in HIV-1 infec-tion. Nature 387:183–188.
7. Chun, T. W., D. Engel, M. M. Berrey, T. Shea, L. Corey, and A. S. Fauci.1998. Early establishment of a pool of latently infected, resting CD4(�) Tcells during primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 95:8869–8873.
8. Chun, T. W., and A. S. Fauci. 1999. Latent reservoirs of HIV: obstacles to theeradication of virus. Proc. Natl. Acad. Sci. USA 96:10958–10961.
9. Chun, T. W., D. Finzi, J. Margolick, K. Chadwick, D. Schwartz, and R. F.Siliciano. 1995. In vivo fate of HIV-1-infected T cells: quantitative analysis ofthe transition to stable latency. Nat. Med. 1:1284–1290.
10. Chun, T. W., L. Stuyver, S. B. Mizell, L. A. Ehler, J. A. Mican, M. Baseler,A. L. Lloyd, M. A. Nowak, and A. S. Fauci. 1997. Presence of an inducibleHIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl.Acad. Sci. USA 94:13193–13197.
11. Clouse, K. A., D. Powell, I. Washington, G. Poli, K. Strebel, W. Farrar, P.Barstad, J. Kovacs, A. S. Fauci, and T. M. Folks. 1989. Monokine regulationof human immunodeficiency virus-1 expression in a chronically infectedhuman T cell clone. J. Immunol. 142:431–438.
12. Englund, G., T. S. Theodore, E. O. Freed, A. Engleman, and M. A. Martin.1995. Integration is required for productive infection of monocyte-derivedmacrophages by human immunodeficiency virus type 1. J. Virol. 69:3216–3219.
13. Farnet, C. M., and F. D. Bushman. 1997. HIV-1 cDNA integration: require-ment of HMG I(Y) protein for function of preintegration complexes in vitro.Cell 88:483–492.
14. Folks, T. M., D. Powell, M. Lightfoote, S. Koenig, A. S. Fauci, S. Benn, A.Rabson, D. Daugherty, H. E. Gendelman, and M. D. Hoggan. 1986. Biolog-ical and biochemical characterization of a cloned Leu-3� cell survivinginfection with the acquired immune deficiency syndrome retrovirus. J. Exp.Med. 164:280–290.
15. Gazdar, A. F., D. N. Carney, P. A. Bunn, E. K. Russell, E. S. Jaffe, G. P.Schechter, and J. G. Guccion. 1980. Mitogen requirements for the in vitropropagation of cutaneous T-cell lymphomas. Blood 55:409–417.
16. Hazuda, D. J., P. Felock, M. Witmer, A. Wolfe, K. Stillmock, J. A. Grobler,A. Espeseth, L. Gabryelski, W. Schleif, C. Blau, and M. D. Miller. 2000.Inhibitors of strand transfer that prevent integration and inhibit HIV-1replication in cells. Science 287:646–650.
17. Hirt, B. 1967. Selective extraction of polyoma DNA from infected mouse cellcultures. J. Mol. Biol. 26:365–369.
18. Jurka, J., and T. Smith. 1988. A fundamental division in the Alu family ofrepeated sequences. Proc. Natl. Acad. Sci. USA 85:4775–4778.
19. Karageorgos, L., P. Li, and C. Burrell. 1993. Characterization of HIV rep-
lication complexes early after cell-to-cell infection. AIDS Res. Hum. Retro-vir. 9:817–823.
20. Karageorgos, L., P. Li, and C. J. Burrell. 1995. Stepwise analysis of reversetranscription in a cell-to-cell human immunodeficiency virus infection mod-el: kinetics and implications. J. Gen. Virol. 76:1675–1686.
21. Kim, S. Y., R. Byrn, J. Groopman, and D. Baltimore. 1989. Temporal aspectsof DNA and RNA synthesis during human immunodeficiency virus infection:evidence for differential gene expression. J. Virol. 63:3708–3713.
22. LaFemina, R. L., C. L. Schneider, H. L. Robbins, P. L. Callahan, K. LeGrow,E. Roth, W. A. Schleif, and E. A. Emini. 1992. Requirement of active humanimmunodeficiency virus type 1 integrase enzyme for productive infection ofhuman T-lymphoid cells. J. Virol. 66:7414–7419.
23. Li, G., M. Simm, M. J. Potash, and D. J. Volsky. 1993. Human immunode-ficiency virus type 1 DNA synthesis, integration, and efficient viral replicationin growth-arrested T cells. J. Virol. 67:3969–3977.
24. Li, L., K. Yoder, M. S. Hansen, J. Olvera, M. D. Miller, and F. D. Bushman.2000. Retroviral cDNA integration: stimulation by HMG I family proteins.J. Virol. 74:10965–10974.
25. Li, P., and C. J. Burrell. 1992. Synthesis of human immunodeficiency virusDNA in a cell-to-cell transmission model. AIDS Res. Hum. Retrovir. 8:253–259.
26. Miller, M. D., C. M. Farnet, and F. D. Bushman. 1997. Human immunode-ficiency virus type 1 preintegration complexes: studies of organization andcomposition. J. Virol. 71:5382–5390.
27. Pauza, C. D., and J. Galindo. 1989. Persistent human immunodeficiencyvirus type 1 infection of monoblastoid cells leads to accumulation of self-integrated viral DNA and to production of defective virions. J. Virol. 63:3700–3707.
28. Sakai, H., M. Kawamura, J. Sakuragi, S. Sakuragi, R. Shibata, A. Ishimoto,N. Ono, S. Ueda, and A. Adachi. 1993. Integration is essential for efficientgene expression of human immunodeficiency virus type 1. J. Virol. 67:1169–1174.
29. Sonza, S., A. Maerz, N. Deacon, J. Meanger, J. Mills, and S. Crowe. 1996.Human immunodeficiency virus type 1 replication is blocked prior to reversetranscription and integration in freshly isolated peripheral blood monocytes.J. Virol. 70:3863–3869.
30. Stevenson, M., T. L. Stanwick, M. P. Dempsey, and C. A. Lamonica. 1990.HIV-1 replication is controlled at the level of T cell activation and proviralintegration. EMBO J. 9:1551–1560.
31. Vandegraaff, N., R. Kumar, H. Hocking, T. Burke, J. Mills, D. Rhodes, C. J.Burrell, and P. Li. 2001. Specific inhibition of human immunodeficiencyvirus type 1 (HIV-1) integration in cell culture: putative inhibitors of HIV-1integrase. Antimicrob. Agents Chemother. 45:2510–2516.
32. Wattel, E., J. P. Vartanian, C. Pannetier, and S. Wain-Hobson. 1995. Clonalexpansion of human T-cell leukemia virus type I-infected cells in asymptom-atic and symptomatic carriers without malignancy. J. Virol. 69:2863–2868.
33. Weinberg, J. B., T. J. Matthews, B. R. Cullen, and M. H. Malim. 1991.Productive human immunodeficiency virus type 1 (HIV-1) infection of non-proliferating human monocytes. J. Exp. Med. 174:1477–1482.
34. Zanussi, S., M. T. Bortolin, M. Giacca, and P. De Paoli. 2000. Quantitativeassessment of integrated and episomal HIV DNA. AIDS 16:931–933.
11260 NOTES J. VIROL.
on August 22, 2014 by guest
http://jvi.asm.org/
Dow
nloaded from




















