
'Like Sheep'? Disobedience Among Soviet Tourists Travelling Abroad
Upload
independentCategory
view
1download
0
Ketoconazole increases the plasma levels of ivermectin in sheep
Michel Alvinerie, Jacques Dupuy, Solange Kiki-Mvouaka,Jean-Francois Sutra, Anne Lespine *
INRA-Toulouse, UR66 Laboratoire de Pharmacologie-Toxicologie, 180 chemin de Tournefeuille,
BP 3, F-31931 Toulouse Cedex 9, France
Received 6 March 2008; received in revised form 12 June 2008; accepted 16 June 2008
Abstract
The parasiticide ivermectin and the antifungal drug ketoconazole are drugs that interact with P-glycoprotein. We have tested the
ability of ketoconazole at a clinical dose to modify the pharmacokinetics of ivermectin in sheep. Lacaune lambs were administered
with a single oral dose of ivermectin alone at 0.2 mg/kg (n = 5) or in combination with a daily oral dose of ketoconazole (10 mg/kg)
given for 3 days before and 2 days after the ivermectin (n = 5). The plasma kinetics of ivermectin and its metabolite were followed
over 15 days by HPLC analysis. Co-administration of ketoconazole induced higher plasma concentrations of ivermectin, leading to
a substantial increase in the overall exposure of the animals to the drug. Ketoconazole did not reduce the production of the main
ivermectin metabolite but it may rather act by inhibiting P-glycoprotein, and thus increasing the absorption of ivermectin. The use of
a P-gp reversing agent such as ketoconazole could be useful tool to optimize antiparasitic therapy in the face of the worldwide
development of anthelmintic resistance.
# 2008 Elsevier B.V. All rights reserved.
Keywords: Macrocyclic lactone; Ketoconazole; P-glycoprotein; Drug–drug interaction; Resistance; Sheep
www.elsevier.com/locate/vetpar
Available online at www.sciencedirect.com
Veterinary Parasitology 157 (2008) 117–122
1. Introduction
Parasitic infections continue to play a significant role
in limiting the ability of livestock to reach full
productivity. At the present time, ivermectin (IVM), a
potent parasiticide belonging to the macrocyclic lactone
(ML) family of compounds, remains of major
importance for the treatment of both internal and
external parasites (McKellar and Benchaoui, 1996).
Their remarkable broad spectrum of activity and their
safety profile put these drugs at the cornerstone of
modern anthelmintic therapy in livestock. Several
million humans are also treated with ivermectin for
the control of onchocerciasis and lymphatic filiarisis
* Corresponding author. Tel.: +33 561285387; fax: +33 561285710.
E-mail address: [email protected] (A. Lespine).
0304-4017/$ – see front matter # 2008 Elsevier B.V. All rights reserved.
doi:10.1016/j.vetpar.2008.06.017
(Molyneux et al., 2003). The success of ivermectin and
other MLs has also been due to their broad spectrum of
activity, safety and effectiveness against benzimida-
zole-, levamisole-, and pyrantel-resistant strains of
parasites, whose emergence severely restricted the fight
against parasites in the late 1970s (Molyneux et al.,
2003). Now, however, after 25 years of intensive use of
MLs, resistance to them has also appeared, initially in
small ruminants and now in cattle (Kaplan, 2004).
The final concentration and duration of an anthelmin-
tic in the host’s systemic circulation and finally in the
parasite is a key determinant of the efficacy of that drug.
Among the factors contributing to the loss of drug
efficacy and emergence of resistance are under-dosing
and the low bioavailability of some formulations. Due to
their role of the active efflux of xenobiotics out of the
organism, P-gp and probably other ABC transporter

M. Alvinerie et al. / Veterinary Parasitology 157 (2008) 117–122118
proteins play a key role in modulating the bioavailability
of macrocyclic lactones in both host and parasite. P-gp
appears to be the ABC transporter playing the major role
in IVM transport (Pouliot et al., 1997) and body
disposition (Schinkel et al., 1994). As it is located on
the blood–brain barrier, P-gp protects mammals against
the penetration of IVM into the brain and its subsequent
neurotoxicity (Kwei et al., 1999; Mealey et al., 2001;
Roulet et al., 2003; Schinkel et al., 1994). P-gp is also
present on the surface of the intestinal epithelium and the
bile canaliculi, and thus contributes to the high faecal
elimination of MLs (Laffont et al., 2002). In addition, P-
gp homologue gene selection is one of the mechanisms
involved in avermectin resistance in trichostrongylid
nematodes (Prichard and Roulet, 2007). By expelling the
drug out of the parasite, the presence of P-gp homologues
certainly plays a key role in reducing drug activity, and
favouring the development of resistance to MLs
(Sangster et al., 1999; Xu et al., 1998). Given its central
role in controlling the efflux of ivermectin, P-gp has
emerged as a key pharmacological target and the co-
administration of P-gp inhibitors has become a challen-
ging strategy for the improvement of drug action.
Ketoconazole (KTZ), an imidazole derivative, is an
orally active, broad-spectrum systemic antifungal agent
used in veterinary medicine. Ketoconazole and its
derivatives are well known inhibitors of cytochrome
P4503A activity in human hepatic microsomes (Bourrie
et al., 1996; Maurice et al., 1992). In addition, it has
been shown that ketoconazole co-administration
increased the AUC of several P-gp substrate drugs in
rats (Kageyama et al., 2005). Itraconazole, another
member of the imidazole family, has been also shown to
increase ivermectin bioavailability in plasma and to
reduce the intestinal secretion of the drug by inhibiting
P-gp in the rat (Ballent et al., 2006), sheep (Ballent
et al., 2007) and monkey (Ward et al., 2004). In
addition, it has been shown recently that ketoconazole
modifies the disposition of ivermectin in dogs by
reducing the clearance of the drug (Hugnet et al., 2007).
The interaction of ivermectin and ketoconazole in
vivo at therapeutic doses has never been explored in
ruminants. The aim of the study was to examine the
impact of the co-administration of ketoconazole and
ivermectin on the disposition of the ivermectin in sheep.
2. Materials and methods
2.1. Materials
Ketoconazole was purchased as tablets (Nizoral1)
from Janssen Pharmaceutica (Issy les Moulineaux,
France). Ivermectin (Oramec1) was obtained from
Merial (Lyon, France). All other chemicals used as
reagents were of analytical and high-performance liquid
chromatographic (HPLC) grade. The ivermectin meta-
bolite, 3-O-desmethyl ivermectin, was kindly provided
by Merial (Iselin, USA).
2.2. Animals
Ten female Lacaune lambs (UAC, L’Isle en Dodon,
France) weighing 29.1 � 2.0 kg were bought from a
breeder. The animals were raised on a zero-grazing
system and were free of trichostrongyle infections at the
beginning of the experiment. The sheep were housed in
concrete pens and were fed hay ad libitum and
commercial concentrates once a day (DP Nutrition).
No other treatment had been given during the 90 days
prior to the study and the sheep were fasted for 12 h
before the experiment. All procedures involving the
animals were approved by the local and institutional
animal use and care committee.
2.3. Study design
The animals were allocated into two groups (IVM
and KTZ-IVM) of five. The IVM group received a
single administration of ivermectin orally (Oramec,
Merial) at a dosage of 0.2 mg/kg. Lambs from the KTZ-
IVM group received ivermectin (similar administration
than IVM group) combined with repeat doses of
ketoconazole orally at a dose of 10 mg/kg per day as
tablets dissolved in water at 40 mg/ml (10 ml per
animal). The treatment was given 1 h after each
morning meal and repeated over 5 days (3 days before
and 2 days after the ivermectin administration).
2.4. Blood sampling
Blood samples (1 ml) were collected from the
cephalic vein before the administration of ivermectin
and at 1, 2, 4, 8, 24, 32 h, and 2, 3, 4, 6, 8, 10, 13, 15 days
post-administration. Plasma was immediately separated
from whole blood by centrifugation for 10 min at
1500 � g and stored at �18 8C prior to analysis.
2.5. Determination of plasma ivermectin
concentration
Ivermectin was analyzed in plasma by high-
performance liquid chromatography (HPLC) with
automated solid phase extraction and fluorescence
detection according to a previously described method
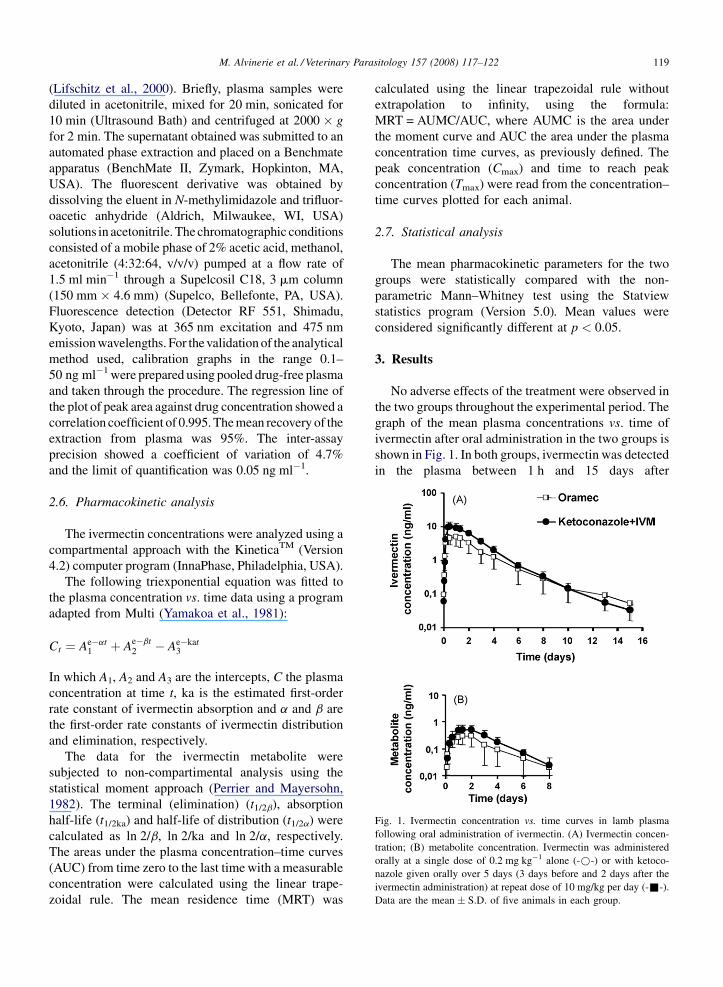
M. Alvinerie et al. / Veterinary Parasitology 157 (2008) 117–122 119
Fig. 1. Ivermectin concentration vs. time curves in lamb plasma
following oral administration of ivermectin. (A) Ivermectin concen-
tration; (B) metabolite concentration. Ivermectin was administered
orally at a single dose of 0.2 mg kg�1 alone (-*-) or with ketoco-
nazole given orally over 5 days (3 days before and 2 days after the
ivermectin administration) at repeat dose of 10 mg/kg per day (-&-).
Data are the mean � S.D. of five animals in each group.
(Lifschitz et al., 2000). Briefly, plasma samples were
diluted in acetonitrile, mixed for 20 min, sonicated for
10 min (Ultrasound Bath) and centrifuged at 2000 � g
for 2 min. The supernatant obtained was submitted to an
automated phase extraction and placed on a Benchmate
apparatus (BenchMate II, Zymark, Hopkinton, MA,
USA). The fluorescent derivative was obtained by
dissolving the eluent in N-methylimidazole and trifluor-
oacetic anhydride (Aldrich, Milwaukee, WI, USA)
solutions in acetonitrile. The chromatographic conditions
consisted of a mobile phase of 2% acetic acid, methanol,
acetonitrile (4:32:64, v/v/v) pumped at a flow rate of
1.5 ml min�1 through a Supelcosil C18, 3 mm column
(150 mm � 4.6 mm) (Supelco, Bellefonte, PA, USA).
Fluorescence detection (Detector RF 551, Shimadu,
Kyoto, Japan) was at 365 nm excitation and 475 nm
emission wavelengths. For the validation of the analytical
method used, calibration graphs in the range 0.1–
50 ng ml�1 were prepared using pooled drug-free plasma
and taken through the procedure. The regression line of
the plot of peak area against drug concentration showed a
correlation coefficient of 0.995. The mean recovery of the
extraction from plasma was 95%. The inter-assay
precision showed a coefficient of variation of 4.7%
and the limit of quantification was 0.05 ng ml�1.
2.6. Pharmacokinetic analysis
The ivermectin concentrations were analyzed using a
compartmental approach with the KineticaTM (Version
4.2) computer program (InnaPhase, Philadelphia, USA).
The following triexponential equation was fitted to
the plasma concentration vs. time data using a program
adapted from Multi (Yamakoa et al., 1981):
Ct ¼ Ae�at1 þ Ae�bt
2 � Ae�kat3
In which A1, A2 and A3 are the intercepts, C the plasma
concentration at time t, ka is the estimated first-order
rate constant of ivermectin absorption and a and b are
the first-order rate constants of ivermectin distribution
and elimination, respectively.
The data for the ivermectin metabolite were
subjected to non-compartimental analysis using the
statistical moment approach (Perrier and Mayersohn,
1982). The terminal (elimination) (t1/2b), absorption
half-life (t1/2ka) and half-life of distribution (t1/2a) were
calculated as ln 2/b, ln 2/ka and ln 2/a, respectively.
The areas under the plasma concentration–time curves
(AUC) from time zero to the last time with a measurable
concentration were calculated using the linear trape-
zoidal rule. The mean residence time (MRT) was
calculated using the linear trapezoidal rule without
extrapolation to infinity, using the formula:
MRT = AUMC/AUC, where AUMC is the area under
the moment curve and AUC the area under the plasma
concentration time curves, as previously defined. The
peak concentration (Cmax) and time to reach peak
concentration (Tmax) were read from the concentration–
time curves plotted for each animal.
2.7. Statistical analysis
The mean pharmacokinetic parameters for the two
groups were statistically compared with the non-
parametric Mann–Whitney test using the Statview
statistics program (Version 5.0). Mean values were
considered significantly different at p < 0.05.
3. Results
No adverse effects of the treatment were observed in
the two groups throughout the experimental period. The
graph of the mean plasma concentrations vs. time of
ivermectin after oral administration in the two groups is
shown in Fig. 1. In both groups, ivermectin was detected
in the plasma between 1 h and 15 days after
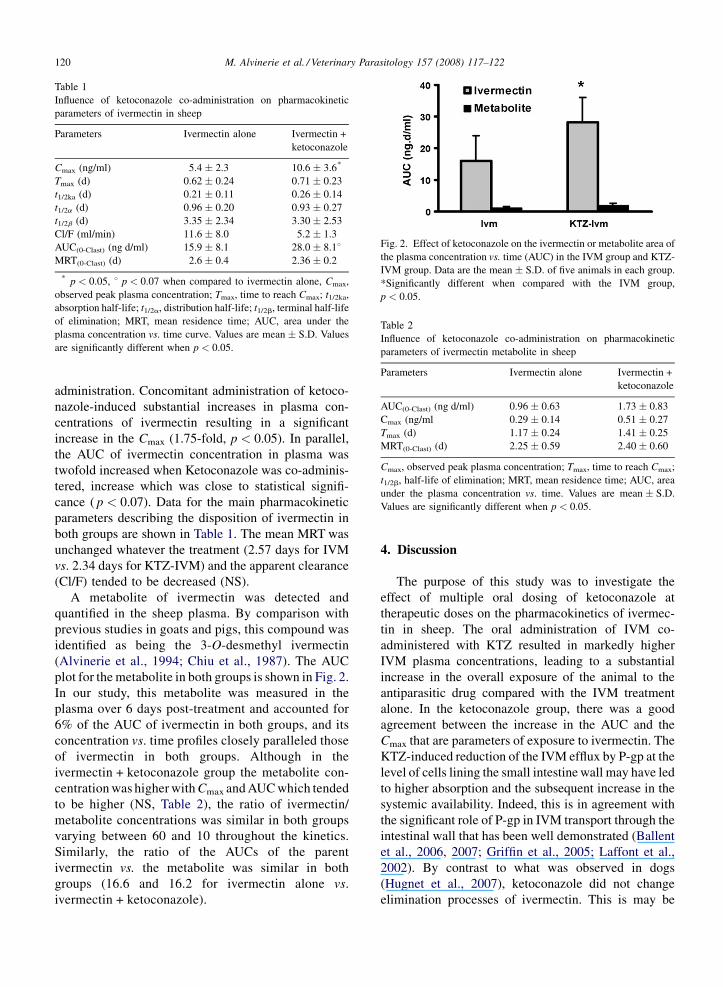
M. Alvinerie et al. / Veterinary Parasitology 157 (2008) 117–122120
Table 1
Influence of ketoconazole co-administration on pharmacokinetic
parameters of ivermectin in sheep
Parameters Ivermectin alone Ivermectin +
ketoconazole
Cmax (ng/ml) 5.4 � 2.3 10.6 � 3.6*
Tmax (d) 0.62 � 0.24 0.71 � 0.23
t1/2ka (d) 0.21 � 0.11 0.26 � 0.14
t1/2a (d) 0.96 � 0.20 0.93 � 0.27
t1/2b (d) 3.35 � 2.34 3.30 � 2.53
Cl/F (ml/min) 11.6 � 8.0 5.2 � 1.3
AUC(0-Clast) (ng d/ml) 15.9 � 8.1 28.0 � 8.18MRT(0-Clast) (d) 2.6 � 0.4 2.36 � 0.2
* p < 0.05, 8 p < 0.07 when compared to ivermectin alone, Cmax,
observed peak plasma concentration; Tmax, time to reach Cmax; t1/2ka,
absorption half-life; t1/2a, distribution half-life; t1/2b, terminal half-life
of elimination; MRT, mean residence time; AUC, area under the
plasma concentration vs. time curve. Values are mean � S.D. Values
are significantly different when p < 0.05.
Fig. 2. Effect of ketoconazole on the ivermectin or metabolite area of
the plasma concentration vs. time (AUC) in the IVM group and KTZ-
IVM group. Data are the mean � S.D. of five animals in each group.
*Significantly different when compared with the IVM group,
p < 0.05.
Table 2
Influence of ketoconazole co-administration on pharmacokinetic
parameters of ivermectin metabolite in sheep
Parameters Ivermectin alone Ivermectin +
ketoconazole
AUC(0-Clast) (ng d/ml) 0.96 � 0.63 1.73 � 0.83
Cmax (ng/ml 0.29 � 0.14 0.51 � 0.27
Tmax (d) 1.17 � 0.24 1.41 � 0.25
MRT(0-Clast) (d) 2.25 � 0.59 2.40 � 0.60
Cmax, observed peak plasma concentration; Tmax, time to reach Cmax;
t1/2b, half-life of elimination; MRT, mean residence time; AUC, area
under the plasma concentration vs. time. Values are mean � S.D.
Values are significantly different when p < 0.05.
administration. Concomitant administration of ketoco-
nazole-induced substantial increases in plasma con-
centrations of ivermectin resulting in a significant
increase in the Cmax (1.75-fold, p < 0.05). In parallel,
the AUC of ivermectin concentration in plasma was
twofold increased when Ketoconazole was co-adminis-
tered, increase which was close to statistical signifi-
cance ( p < 0.07). Data for the main pharmacokinetic
parameters describing the disposition of ivermectin in
both groups are shown in Table 1. The mean MRT was
unchanged whatever the treatment (2.57 days for IVM
vs. 2.34 days for KTZ-IVM) and the apparent clearance
(Cl/F) tended to be decreased (NS).
A metabolite of ivermectin was detected and
quantified in the sheep plasma. By comparison with
previous studies in goats and pigs, this compound was
identified as being the 3-O-desmethyl ivermectin
(Alvinerie et al., 1994; Chiu et al., 1987). The AUC
plot for the metabolite in both groups is shown in Fig. 2.
In our study, this metabolite was measured in the
plasma over 6 days post-treatment and accounted for
6% of the AUC of ivermectin in both groups, and its
concentration vs. time profiles closely paralleled those
of ivermectin in both groups. Although in the
ivermectin + ketoconazole group the metabolite con-
centration was higher with Cmax and AUC which tended
to be higher (NS, Table 2), the ratio of ivermectin/
metabolite concentrations was similar in both groups
varying between 60 and 10 throughout the kinetics.
Similarly, the ratio of the AUCs of the parent
ivermectin vs. the metabolite was similar in both
groups (16.6 and 16.2 for ivermectin alone vs.
ivermectin + ketoconazole).
4. Discussion
The purpose of this study was to investigate the
effect of multiple oral dosing of ketoconazole at
therapeutic doses on the pharmacokinetics of ivermec-
tin in sheep. The oral administration of IVM co-
administered with KTZ resulted in markedly higher
IVM plasma concentrations, leading to a substantial
increase in the overall exposure of the animal to the
antiparasitic drug compared with the IVM treatment
alone. In the ketoconazole group, there was a good
agreement between the increase in the AUC and the
Cmax that are parameters of exposure to ivermectin. The
KTZ-induced reduction of the IVM efflux by P-gp at the
level of cells lining the small intestine wall may have led
to higher absorption and the subsequent increase in the
systemic availability. Indeed, this is in agreement with
the significant role of P-gp in IVM transport through the
intestinal wall that has been well demonstrated (Ballent
et al., 2006, 2007; Griffin et al., 2005; Laffont et al.,
2002). By contrast to what was observed in dogs
(Hugnet et al., 2007), ketoconazole did not change
elimination processes of ivermectin. This is may be

M. Alvinerie et al. / Veterinary Parasitology 157 (2008) 117–122 121
related to a lower persistence of ketoconazole in
polygastric. The significant increase in the plasma
levels of ivermectin shows that ketoconazole increased
the exposure of the animals to the drug mainly by
increasing drug absorption.
Since ketoconazole is also known to inhibit both
CYP3A and the drug efflux transporter P-glycoprotein,
the increase in elimination pathways might also
contribute to the increase in the exposure of the sheep
to ivermectin. While the apparent clearance (Cl/F) of
ivermectin tended to be lower in the group receiving the
drug combination, this was not statistically significant.
Due to the inhibitory effect on CYP3A, the adminis-
tration of ketoconazole might be expected to decrease
the production of the ivermectin metabolite, thus
explaining the increase in ivermectin in the systemic
circulation. Indeed, the co-administration of ketoco-
nazole increases the bioavailability of highly metabo-
lized drugs such as quinidine (Kuroha et al., 2004),
mefloquine (Ridtitid et al., 2005), fexofenadine
(Ogasawara et al., 2007), quetiapine (Grimm et al.,
2006) and everolimus (Kovarik et al., 2005) in humans.
In the present experiment, a metabolite was identified
as 3-O-desmethyl ivermectin, which has been pre-
viously reported as the main ivermectin metabolite in
goats (Alvinerie et al., 1994) and pigs (Chiu et al.,
1987). This is the first report describing such metabolite
in sheep. Nevertheless, the concentration of this
metabolite was low in both experimental groups, never
exceeding 6% of the total ivermectin in plasma,
confirming that ivermectin is a poorly metabolized
drug as previously shown in several animal species
(Chiu et al., 1987). Moreover, the ratio of parent
ivermectin to metabolite concentration in plasma is
similar in the two groups. These results clearly
demonstrate that the increase in the exposure to
ivermectin observed in animals receiving both iver-
mectin and ketoconazole was not due to the inhibition
of ivermectin biotransformation.
The results suggest that the plasma and tissue
distribution of ivermectin was strongly influenced by P-
gp activity as previously shown (Kwei et al., 1999). The
strategic distribution of P-gp on the biliary canicular
membrane of hepatocytes and on the apical surface of
enterocytes means that this ABC-transporter is a key
player in both the biliary and the intestinal secretion of
ivermectin. Indeed, the disposition of ivermectin in the
intestinal fluid and tissue has been shown to be
significantly modified in the presence of P-gp mod-
ulators under both in vitro and in vivo conditions
(Ballent et al., 2006; Laffont et al., 2002). Subsequently,
enhanced ivermectin bioavailability in monogastric as
well as in polygastric animals has also been observed in
vivo with the concurrent administration of ivermectin
with other P-gp inhibitors such as verapamil, a well-
known competitive substrate for the P-gp drug-binding
site, in rats (Alvinerie et al., 1999) and sheep (Molento
et al., 2004) and with itraconazole in rats (Ballent et al.,
2006) and sheep (Ballent et al., 2007) or with
loperamide in rats (Lifschitz et al., 2004).
Our results thus provide a mechanistic explanation
for the increase in the systemic concentration of
ivermectin in sheep treated with ketoconazole: keto-
conazole does not affect the production of ivermectin
metabolite or the elimination processes in our experi-
mental conditions, but increases the absorption of the
parent drug through its potential to inhibit the activity of
P-gp that normally limits the absorption of ivermectin at
the intestinal level.
In conclusion, considering the observed increase in
the systemic bioavailability of ivermectin induced by
ketoconazole and the well-established correlation
between ivermectin plasma profiles and those achieved
in the tissues where parasites are located (Lifschitz
et al., 2000), the use of P-gp reversing agents such as
ketoconazole could be useful tools to optimize
antiparasitic therapy in the face of the worldwide
development of anthelmintic resistance. Further work to
evaluate the clinical relevance of this drug combination
is ongoing by using an optimized larval feeding
inhibition assay with the concomitant addition of
ketoconazole and ivermectin, in order to evaluate the
increase insusceptibility to ivermectin of resistant
nematodes.
Acknowledgement
This work was supported by the European Project
PARASOL CT-200X-022851.
References
Alvinerie, M., Dupuy, J., Eeckhoutte, C., Sutra, J.F., 1999. Enhanced
absorption of pour-on ivermectin formulation in rats by co-admin-
istration of the multidrug-resistant-reversing agent verapamil.
Parasitol. Res. 85, 920–922.
Alvinerie, M., Tardieu, D., Sutra, J., D, Bojensen, G., Galtier, P., 1994.
Metabolic profile of ivermectin in goats: an ‘‘in vivo’’ and ‘‘in
vitro’’ evaluation (ed.) 1994. EAVPT International Congress,
Edinburgh, Scotland. August 7–11, 1994.
Ballent, M., Lifschitz, A., Virkel, G., Sallovitz, J., Lanusse, C., 2006.
Modulation of the P-glycoprotein-mediated intestinal secretion of
ivermectin: in vitro and in vivo assessments. Drug Metab. Dispos.
34, 457–463.
Ballent, M., Lifschitz, A., Virkel, G., Sallovitz, J., Lanusse, C., 2007.
Involvement of P-glycoprotein on ivermectin kinetic behaviour in

M. Alvinerie et al. / Veterinary Parasitology 157 (2008) 117–122122
sheep: itraconazole-mediated changes on gastrointestinal disposi-
tion. J. Vet. Pharmacol. Ther. 30, 242–248.
Bourrie, M., Meunier, V., Berger, Y., Fabre, G., 1996. Cytochrome
P450 isoform inhibitors as a tool for the investigation of metabolic
reactions catalyzed by human liver microsomes. J. Pharmacol.
Exp. Ther. 277, 321–332.
Chiu, S.H., Taub, R., Sestokas, E., Lu, A.Y., Jacob, T.A., 1987.
Comparative in vivo and in vitro metabolism of ivermectin in
steers, sheep, swine, and rat. Drug Metab. Rev. 18, 289–302.
Griffin, J., Fletcher, N., Clemence, R., Blanchflower, S., Brayden, D.J.,
2005. Selamectin is a potent substrate and inhibitor of human and
canine P-glycoprotein. J. Vet. Pharmacol. Ther. 28, 257–265.
Grimm, S.W., Richtand, N.M., Winter, H.R., Stams, K.R., Reele, S.B.,
2006. Effects of cytochrome P450 3A modulators ketoconazole
and carbamazepine on quetiapine pharmacokinetics. Br. J. Clin.
Pharmacol. 61, 58–69.
Hugnet, C., Lespine, A., Alvinerie, M., 2007. Multiple oral dosing of
ketoconazole increases dog exposure to ivermectin. J. Pharm.
Pharm. Sci. 10, 311–318.
Kageyama, M., Namiki, H., Fukushima, H., Ito, Y., Shibata, N.,
Takada, K., 2005. In vivo effects of cyclosporin A and ketoco-
nazole on the pharmacokinetics of representative substrates for P-
glycoprotein and cytochrome P450 (CYP) 3A in rats. Biol. Pharm.
Bull. 28, 316–322.
Kaplan, R.M., 2004. Drug resistance in nematodes of veterinary
importance: a status report. Trends Parasitol. 20, 477–481.
Kovarik, J.M., Beyer, D., Bizot,M.N., Jiang,Q., Shenouda, M., Schmou-
der, R.L., 2005. Blood concentrations of everolimus are markedly
increased by ketoconazole. J. Clin. Pharmacol. 45, 514–518.
Kuroha, M., Shirai, Y., Shimoda, M., 2004. Multiple oral dosing of
ketoconazole influences pharmacokinetics of quinidine after intra-
venous and oral administration in beagle dogs. J. Vet. Pharmacol.
Ther. 27, 355–359.
Kwei, G.Y., Alvaro, R.F., Chen, Q., Jenkins, H.J., Hop, C.E., Keohane,
C.A., Ly, V.T., Strauss, J.R., Wang, R.W., Wang, Z., Pippert, T.R.,
Umbenhauer, D.R., 1999. Disposition of ivermectin and cyclos-
porin A in CF-1 mice deficient in mdr1a P-glycoprotein. Drug
Metab. Dispos. 27, 581–587.
Laffont, C.M., Toutain, P.L., Alvinerie, M., Bousquet-Melou, A.,
2002. Intestinal secretion is a major route for parent ivermectin
elimination in the rat. Drug Metab. Dispos. 30, 626–630.
Lifschitz, A., Virkel, G., Sallovitz, J., Sutra, J.F., Galtier, P., Alvinerie,
M., Lanusse, C., 2000. Comparative distribution of ivermectin and
doramectin to parasite location tissues in cattle. Vet. Parasitol. 87,
327–338.
Lifschitz, A.L., Virkel, G.L., Sallovitz, J.M., Pis, A., Imperiale, F.A.,
Lanusse, C.E., 2004. Loperamide modifies the tissue disposition
kinetics of ivermectin in rats. J. Pharm. Pharmacol. 56, 61–67.
Maurice, M., Pichard, L., Daujat, M., Fabre, I., Joyeux, H., Domergue,
J., Maurel, P., 1992. Effects of imidazole derivatives on cyto-
chromes P450 from human hepatocytes in primary culture.
FASEB J. 6, 752–758.
McKellar, Q.A., Benchaoui, H.A., 1996. Avermectins and milbemy-
cins. J. Vet. Pharmacol. Ther. 19, 331–351.
Mealey, K.L., Bentjen, S.A., Gay, J.M., Cantor, G.H., 2001. Ivermec-
tin sensitivity in collies is associated with a deletion mutation of
the mdr1 gene. Pharmacogenetics 11, 727–733.
Molento, M.B., Lifschitz, A., Sallovitz, J., Lanusse, C., Prichard, R.,
2004. Influence of verapamil on the pharmacokinetics of the
antiparasitic drugs ivermectin and moxidectin in sheep. Parasitol.
Res. 92, 121–127.
Molyneux, D.H., Bradley, M., Hoerauf, A., Kyelem, D., Taylor, M.J.,
2003. Mass drug treatment for lymphatic filariasis and onchocer-
ciasis. Trends Parasitol. 19, 516–522.
Ogasawara, A., Kume, T., Kazama, E., 2007. Effect of oral ketoco-
nazole on intestinal first-pass effect of midazolam and fexofena-
dine in cynomolgus monkeys. Drug Metab. Dispos. 35, 410–418.
Perrier, D., Mayersohn, M., 1982. Noncompartmental determination
of the steady-state volume of distribution for any mode of admin-
istration. J. Pharm. Sci. 71, 372–373.
Pouliot, J.F., L’Heureux, F., Liu, Z., Prichard, R.K., Georges, E., 1997.
Reversal of P-glycoprotein-associated multidrug resistance by
ivermectin. Biochem. Pharmacol. 53, 17–25.
Prichard, R.K., Roulet, A., 2007. ABC transporters and beta-tubulin in
macrocyclic lactone resistance: prospects for marker develop-
ment. Parasitology 134, 1123–1132.
Ridtitid, W., Wongnawa, M., Mahatthanatrakul, W., Raungsri, N.,
Sunbhanich, M., 2005. Ketoconazole increases plasma concentra-
tions of antimalarial mefloquine in healthy human volunteers. J.
Clin. Pharm. Ther. 30, 285–290.
Roulet, A., Puel, O., Gesta, S., Lepage, J.F., Drag, M., Soll, M.,
Alvinerie, M., Pineau, T., 2003. MDR1-deficient genotype in
Collie dogs hypersensitive to the P-glycoprotein substrate iver-
mectin. Eur. J. Pharmacol. 460, 85–91.
Sangster, N.C., Bannan, S.C., Weiss, A.S., Nulf, S.C., Klein, R.D.,
Geary, T.G., 1999. Haemonchus contortus: sequence heterogene-
ity of internucleotide binding domains from P-glycoproteins. Exp.
Parasitol. 91, 250–257.
Schinkel, A.H., Smit, J.J., van Tellingen, O., Beijnen, J.H., Wagenaar,
E., van Deemter, L., Mol, C.A., van der Valk, M.A., Robanus-
Maandag, E.C., te Riele, H.P., et al., 1994. Disruption of the
mouse mdr1a P-glycoprotein gene leads to a deficiency in the
blood–brain barrier and to increased sensitivity to drugs. Cell 77,
491–502.
Ward, K.W., Stelman, G.J., Morgan, J.A., Zeigler, K.S., Azzarano,
L.M., Kehler, J.R., McSurdy-Freed, J.E., Proksch, J.W., Smith,
B.R., 2004. Development of an in vivo preclinical screen model to
estimate absorption and first-pass hepatic extraction of xenobio-
tics. II. Use of ketoconazole to identify P-glycoprotein/CYP3A-
limited bioavailability in the monkey. Drug Metab. Dispos. 32,
172–177.
Xu, M., Molento, M., Blackhall, W., Ribeiro, P., Beech, R., Prichard,
R., 1998. Ivermectin resistance in nematodes may be caused by
alteration of P-glycoprotein homolog. Mol. Biochem. Parasitol.
91, 327–335.
Yamakoa, K., Tanigavara, K., Nokaguana, K., Unot, T., 1981. A
pharmacokinetic analysis program (multi) for microcomputer. J.
Pharmacobiol. Dyn. 4, 879–885.
Copyright © 2022 FDOKUMEN




















