Issue Highlights
157
Volume 116, Issue 1; July 3, 2007,PP. 1-124 Issue Highlights Issue Highlights Circulation 2007 116: 1, doi:10.1161/CIRCULATIONAHA.107.183534 Editors' Note Gary J. Balady and Ravin Davidoff Circulation 2007 116: 2, doi:10.1161/CIRCULATIONAHA.107.184813 Editorials Cardiovascular Biomarkers: Added Value With an Integrated Approach? Wolfgang Koenig Circulation 2007 116: 3 - 5, doi:10.1161/CIRCULATIONAHA.107.707984 The ST-Segment–Elevation Myocardial Infarction Chain of Survival Joseph P. Ornato Circulation 2007 116: 6 - 9, doi:10.1161/CIRCULATIONAHA.107.710970
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of Issue Highlights

Volume 116, Issue 1; July 3, 2007,PP. 1-124
Issue Highlights Issue Highlights
Circulation 2007 116: 1, doi:10.1161/CIRCULATIONAHA.107.183534
Editors' Note Gary J. Balady and Ravin Davidoff Circulation 2007 116: 2, doi:10.1161/CIRCULATIONAHA.107.184813
Editorials Cardiovascular Biomarkers: Added Value With an Integrated Approach?
Wolfgang Koenig Circulation 2007 116: 3 - 5, doi:10.1161/CIRCULATIONAHA.107.707984
The ST-Segment–Elevation Myocardial Infarction Chain of Survival Joseph P. Ornato Circulation 2007 116: 6 - 9, doi:10.1161/CIRCULATIONAHA.107.710970

Original Articles
Arrhythmia/Electrophysiology
Common NOS1AP Variants Are Associated With a Prolonged QTc Interval in the Rotterdam Study
Albert-Jan L.H.J. Aarnoudse, Christopher Newton-Cheh, Paul I.W. de Bakker, Sabine M.J.M. Straus, Jan A. Kors, Albert Hofman, André G. Uitterlinden, Jacqueline C.M. Witteman, and Bruno H.C. Stricker Circulation 2007 116: 10 - 16; published online before print June 18 2007, doi:10.1161/CIRCULATIONAHA.106.676783
Nonsense Mutations in hERG Cause a Decrease in Mutant mRNA Transcripts by Nonsense-Mediated mRNA Decay in Human Long-QT Syndrome
Qiuming Gong, Li Zhang, G. Michael Vincent, Benjamin D. Horne, and Zhengfeng Zhou Circulation 2007 116: 17 - 24; published online before print June 18 2007, doi:10.1161/CIRCULATIONAHA.107.708818
Coronary Heart Disease
Coronary Artery Calcification Progression Is Heritable Andrea E. Cassidy-Bushrow, Lawrence F. Bielak, Patrick F. Sheedy, II, Stephen T. Turner, Iftikhar J. Kullo, Xihong Lin, and Patricia A. Peyser Circulation 2007 116: 25 - 31; published online before print June 11 2007, doi:10.1161/CIRCULATIONAHA.106.658583
Epidemiology
Association of Carotid Artery Intima-Media Thickness, Plaques, and C-Reactive Protein With Future Cardiovascular Disease and All-Cause Mortality: The Cardiovascular Health Study
Jie J. Cao, Alice M. Arnold, Teri A. Manolio, Joseph F. Polak, Bruce M. Psaty, Calvin H. Hirsch, Lewis H. Kuller, and Mary Cushman Circulation 2007 116: 32 - 38; published online before print June 18 2007, doi:10.1161/CIRCULATIONAHA.106.645606
Abdominal Visceral and Subcutaneous Adipose Tissue Compartments: Association With Metabolic Risk Factors in the Framingham Heart Study
Caroline S. Fox, Joseph M. Massaro, Udo Hoffmann, Karla M. Pou, Pal Maurovich-Horvat, Chun-Yu Liu, Ramachandran S. Vasan, Joanne M. Murabito, James B. Meigs, L. Adrienne Cupples, Ralph B. D’Agostino, Sr, and Christopher J. O’Donnell Circulation 2007 116: 39 - 48; published online before print June 18 2007, doi:10.1161/CIRCULATIONAHA.106.675355

Heart Failure
Metoprolol Reverses Left Ventricular Remodeling in Patients With Asymptomatic Systolic Dysfunction: The REversal of VEntricular Remodeling with Toprol-XL (REVERT) Trial
Wilson S. Colucci, Theodore J. Kolias, Kirkwood F. Adams, William F. Armstrong, Jalal K. Ghali, Stephen S. Gottlieb, Barry Greenberg, Michael I. Klibaner, Marrick L. Kukin, Jennifer E. Sugg on behalf of the REVERT Study Group Circulation 2007 116: 49 - 56; published online before print June 18 2007, doi:10.1161/CIRCULATIONAHA.106.666016
Negative Inotropy of the Gastric Proton Pump Inhibitor Pantoprazole in Myocardium From Humans and Rabbits: Evaluation of Mechanisms
Wolfgang Schillinger, Nils Teucher, Samuel Sossalla, Sarah Kettlewell, Carola Werner, Dirk Raddatz, Andreas Elgner, Gero Tenderich, Burkert Pieske, Giuliano Ramadori, Friedrich A. Schöndube, Harald Kögler, Jens Kockskämper, Lars S. Maier, Harald Schwörer, Godfrey L. Smith, and Gerd Hasenfuss Circulation 2007 116: 57 - 66; published online before print June 18 2007, doi:10.1161/CIRCULATIONAHA.106.666008
Interventional Cardiology
Emergency Department Physician Activation of the Catheterization Laboratory and Immediate Transfer to an Immediately Available Catheterization Laboratory Reduce Door-to-Balloon Time in ST-Elevation Myocardial Infarction
Umesh N. Khot, Michele L. Johnson, Curtis Ramsey, Monica B. Khot, Randall Todd, Saeed R. Shaikh, and William J. Berg Circulation 2007 116: 67 - 76; published online before print June 11 2007, doi:10.1161/CIRCULATIONAHA.106.677401
Contemporary Reviews in Cardiovascular Medicine The Brain–Heart Connection
Martin A. Samuels Circulation 2007 116: 77 - 84, doi:10.1161/CIRCULATIONAHA.106.678995

Cardiovascular Involvement in General Medical Conditions Chronic Kidney Disease: Effects on the Cardiovascular System
Ernesto L. Schiffrin, Mark L. Lipman, and Johannes F.E. Mann Circulation 2007 116: 85 - 97, doi:10.1161/CIRCULATIONAHA.106.678342
ACCF/AHA/SCAI Clinical Competence Statement ACCF/AHA/SCAI 2007 Update of the Clinical Competence Statement on Cardiac Interventional Procedures: A Report of the American College of Cardiology Foundation/American Heart Association/American College of Physicians Task Force on Clinical Competence and Training (Writing Committee to Update the 1998 Clinical Competence Statement on Recommendations for the Assessment and Maintenance of Proficiency in Coronary Interventional Procedures)
Circulation 2007 116: 98 - 124; published online before print June 25 2007, doi:10.1161/CIRCULATIONAHA.107.185159
Images in Cardiovascular Medicine An Unusual Site for a Common Disease
Maysaa Alzetani, Joseph J. Boyle, David Lefroy, and Petros Nihoyannopoulos Circulation 2007 116: e1, doi:10.1161/CIRCULATIONAHA.106.677120
Sine-Wave Pattern Arrhythmia and Sudden Paralysis That Result From Severe Hyperkalemia
Maurice J.H.M. Pluijmen and Ferry M.R.J. Hersbach Circulation 2007 116: e2 - e4, doi:10.1161/CIRCULATIONAHA.106.687202
Lipomatous Metaplasia in Ischemic Cardiomyopathy: A Common but Unappreciated Entity
Matthias Schmitt, Nilesh Samani, and Gerry McCann Circulation 2007 116: e5 - e6, doi:10.1161/CIRCULATIONAHA.107.690800

Correspondence Letter by Brewster and van Montfrans Regarding Article, "Risks Associated With Statin Therapy: A Systematic Overview of Randomized Clinical Trials"
Lizzy M. Brewster and Gert A. van Montfrans Circulation 2007 116: e7, doi:10.1161/CIRCULATIONAHA.107.689497
Letter by Rosenberg and Uretsky Regarding Article, "Risks Associated With Statin Therapy: A Systematic Overview of Randomized Clinical Trials"
Lauren Rosenberg and Seth Uretsky Circulation 2007 116: e8, doi:10.1161/CIRCULATIONAHA.107.690867
Response to Letters Regarding Article, "Risks Associated With Statin Therapy: A Systematic Overview of Randomized Clinical Trials"
Amir Kashani, JoAnne M. Foody, Yongfei Wang, Harlan M. Krumholz, Christopher O. Phillips, Sandeep Mangalmurti, and Dennis T. Ko Circulation 2007 116: e9, doi:10.1161/CIRCULATIONAHA.107.697227
Acknowledgment of Reviewers Acknowledgment of Reviewers
Circulation 2007 116: e10 - e21, doi:10.1161/CIRCULATIONAHA.107.184814
News From the American Heart Association News From the American Heart Association
Circulation 2007 116: 1B - 2B, doi:10.1161/CIRCULATIONAHA.107.184673
Meetings Calendar Meetings Calendar
Circulation 2007 116: 3B - 4B, doi:10.1161/CIRCULATIONAHA.107.184672

American Heart Association Newly Elected Fellows, Spring 2007 American Heart Association Newly Elected Fellows, Spring 2007
Circulation 2007 116: 5B - 6B, doi:10.1161/CIRCULATIONAHA.107.184674
European Perspectives European Perspectives
Circulation 2007 116: 1F - 6F, doi:10.1161/CIRCULATIONAHA.107.185417

ASSOCIATION OF CAROTID ARTERY INTIMA-MEDIATHICKNESS, PLAQUES, AND C-REACTIVE PROTEINWITH FUTURE CARDIOVASCULAR DISEASE ANDALL-CAUSE MORTALITY: THE CARDIOVASCULARHEALTH STUDY, by Cao et al.
There is increasing interest in methods to risk-stratify individuals’ risk forcardiovascular disease. Cao et al examined the ability of C-reactive proteinconcentrations with or without carotid intima-media thickness and carotidplaques to predict incident cardiovascular events and death in about 5000elderly participants in the Cardiovascular Health Study. The investigatorsreport that C-reactive protein was not prognostically useful withoutevidence of carotid atherosclerosis. However, they observed an interactionbetween C-reactive protein and carotid disease; increasing C-reactiveprotein concentrations were associated with a 72% and 52% increased riskof cardiovascular death and all-cause mortality, respectively, in the settingof carotid atheroslerosis. Similar to other studies, as assessed by the cstatistic, both C-reactive protein and carotid atherosclerosis added onlymodest incremental information to standard cardiovascular disease riskfactors. The study underscores the need for further statistical and clinicaltools to enhance clinical risk prediction. See p 32 (editorial p 3).
METOPROLOL REVERSES LEFT VENTRICULARREMODELING IN PATIENTS WITH ASYMPTOMATICSYSTOLIC DYSFUNCTION: THE REVERSAL OFVENTRICULAR REMODELING WITH TOPROL-XL(REVERT) TRIAL, by Colucci et al.
Until now, there has been no randomized, controlled trial data tosupport the benefit of �-blockers in patients with asymptomatic leftventricular systolic dysfunction. Colucci and colleagues investigatethis question with the REversal of VEntricular Remodeling withToprol-XL (REVERT) trial by randomly assigning patients, with aleft ventricular ejection fraction �40%, mild left ventricular dila-tion, and no symptoms of heart failure (New York Heart Associa-tion class I), to 3 treatment groups: extended-release metoprololsuccinate 200 mg or 50 mg and placebo. Echocardiographicassessment of left ventricular end-systolic volume, end-diastolicvolume, mass, and ejection fraction were performed at baseline.After 12 months, in the 200-mg group, there was a decrease inend-systolic volume index and an increase in left ventricularejection fraction. In the 50-mg group, similar effects of a lessermagnitude were observed. These results demonstrate that theantiremodeling benefits of �-blocker therapy with metoprololsuccinate extend to patients with asymptomatic left ventriculardysfunction. See p 49.
EMERGENCY DEPARTMENT PHYSICIAN ACTIVATIONOF THE CATHETERIZATION LABORATORY ANDIMMEDIATE TRANSFER TO AN IMMEDIATELYAVAILABLE CATHETERIZATION LABORATORYREDUCE DOOR-TO-BALLOON TIME IN ST-ELEVATIONMYOCARDIAL INFARCTION, by Khot et al.Guidelines recommend that hospitals strive to achieve a door-to-balloontime within 90 minutes based upon considerable observational data.Currently, most hospitals are not achieving this goal. National efforts arenow under way to improve the door-to-balloon times, but the impact ofthese efforts has not been prospectively evaluated. In this prospectiveobservational study, the impact of a protocol mandating that theemergency department physician activate the cardiac catheterizationlaboratory and transfer the patient immediately to the laboratory wasevaluated and compared with the door-to-balloon times achieved priorto the institution of the protocol. This study by Khot et al showed thatdoor-to-balloon times decreased significantly from 113.5 minutes to75.5 minutes after adoption of the protocol. Treatment within 90minutes rose from 28% to 71%. As a result, mean infarct sizedecreased, as did hospital length of stay and total hospital costs peradmission. The findings suggest that emergency department physicianactivation of the catheterization laboratory with immediate transfer tothe laboratory is highly effective in reducing door-to-balloon times andalso appears to improve outcomes and reduce cost. See p 67 (editorialp 6).
Visit http://circ.ahajournals.org:
Images in Cardiovascular MedicineAn Unusual Site for a Common Disease. See p e1.
Sine-Wave Pattern Arrhythmia and Sudden Paralysis ThatResult From Severe Hyperkalemia. See p e2.
Lipomatous Metaplasia in Ischemic Cardiomyopathy: A Com-mon but Unappreciated Entity. See p e5.
CorrespondenceSee p e7.
IIssssuuee HHiigghhlliigghhttssVol 116, No 1, July 3, 2007

Medical students often learn about the cardiovascular system as an isolated entity; in many casesa focused approach to anatomy, physiology, and pathophysiology is presented without consider-ation of other biological systems. While this may be a reasonable way to learn the fundamentalsof cardiovascular science, it becomes quite clear to these students during their clinical training thatthe cardiovascular system functions in a remarkably complex milieu in concert with other organsystems. The development of perturbations in one system often leads to responses in other systemsin an attempt to maintain functional homeostasis. Accordingly, the cardiovascular system issubject to the complex interplay among organ systems and a multitude of other factors, includingthose from the environment and the individual’s lifestyle. When problems with other organsystems develop, the initial clinical manifestations may be cardiovascular in nature (eg,abnormalities in heart rate, rhythm, and blood pressure). Understanding that today’s busypractitioner is regularly faced with patients who have many complex medical problems, theEditors of Circulation have commissioned this special series that focuses on the cardiovascularconsequences of other medical disorders.
Articles in this series, Cardiovascular Involvement in General Medical Conditions, will bepublished monthly over the next 7 months. Each article, which is written by highly respectedexperts in the field, will provide a comprehensive and insightful overview of the pathophysiology,clinical manifestations, and treatment options for a specific condition. Topics will cover thyroiddiseases, rheumatological disorders, sepsis, pulmonary diseases, cancer and chemotherapy, andalcohol use and abuse. We anticipate that this series will provide a valuable resource for theclinician, who can readily bring this information to the bedside. We also hope that the gaps in theknowledge base that are highlighted in each article of this series will inspire the researcher tomove the field forward.
Gary J. Balady, MDRavin Davidoff, MD
Series Editors, Cardiovascular Involvement in General Medical Conditions,Circulation
(Circulation. 2007;116:2.)© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.107.184813
2
Editors’ Note

Cardiovascular BiomarkersAdded Value With an Integrated Approach?
Wolfgang Koenig, MD, FRCP, FESC
In primary prevention, traditional risk factors are a usefulfirst step in the determination of who could be at risk forcardiovascular events. In the era of “global risk assessment”
scores such as the Framingham score, the Prospective Cardio-vascular Münster (PROCAM) score, or the European Society ofCardiology Systematic Coronary Risk Evaluation (SCORE),which are derived from multivariable statistical models, shouldbe used.1 However, it has been noted that a considerable numberof at-risk patients cannot be identified on the basis of traditionalrisk factors alone.2 This has prompted the search for novelmarkers of cardiovascular risk to help improve risk prediction.3
Such markers could either represent various blood biomarkersrelevant to the pathophysiology of atherothrombosis (eg, mark-ers of the inflammatory response, coagulation markers, markersof platelet aggregation, lipoproteins, or lipid-related variables),genetic markers, or markers of subclinical disease, which mayalso aid in improved risk prediction. Determination of global riskon the basis of traditional risk factors allows categorization intohigh (10-year risk, �20%), low (10-year risk, �10%), orintermediate risk (10-year risk, 10% to 20%). Subjects at highrisk should be recommended lifestyle changes or prescribed astatin. Subjects at low risk would be reevaluated 3 to 5 yearslater. Those at intermediate risk, however, who comprise up to40% of the population at risk,4 would be candidates for addi-tional testing to increase or decrease their actual risk. A largepanel of blood biomarkers are available for this purpose, butmost of them are not yet applicable in clinical practice forvarious reasons5,6
Article p 32
Emerging Blood BiomarkersAtherosclerosis is characterized by a nonspecific local inflam-matory process7 that is accompanied by a systemic response.Thus a number of prospective studies in initially healthy subjectshave convincingly demonstrated an independent associationbetween even slightly elevated concentrations of various sys-temic markers of inflammation and important cardiovascularend points. At this time, the largest database exists for C-reactiveprotein (CRP), the classic acute-phase protein.8 The measure-
ment procedure is well standardized and automated, and high-sensitive assays with sufficient precision are available. On thebasis of substantial evidence of a contribution of inflammation toatherothrombogenesis, a recent American Heart Association/Centers for Disease Control and Prevention consensus report hasrecommended the measurement of CRP in asymptomatic sub-jects at intermediate risk for future coronary events (10-year risk,10% to 20%).9 However, there are other emerging biomarkerslike lipoprotein-associated phospholipase A2 (Lp-PLA2), anenzyme that is produced by monocytes/macrophages, T-cells,and mast cells and has been found to generate proinflammatoryand proatherogenic molecules.10 Because Lp-PLA2, in contrastto CRP, does not correlate with most other risk factors, there isan additive effect of CRP and Lp-PLA2 in risk prediction.11,12
This may also apply to combinations of other biomarkers,though evidence so far is limited. In the future, we might see abiomarker profile that covers various aspects of the complexpathophysiology of the atherothrombotic process, and poten-tially, we would be able to focus on biological patterns orsystems rather than on single biomarkers. To date, however,there is no sound evidence to suggest such a procedure forclinical practice, and there is even an ongoing discussion ofwhether any of the emerging blood biomarkers alone contributesincremental information over and above the information gainedfrom available “global risk” scores.13,14
Markers of Subclinical Atherosclerotic DiseaseThere is mounting evidence that markers of subclinicaldisease (eg, intima-media thickness as assessed by high-resolution carotid ultrasound;15 coronary calcium determinedwith multislice computed tomography;16 or ankle-brachialindex, a strong marker of atherosclerotic burden17) may alsocontribute to improved risk prediction. However, the clinicalutility of multislice computed tomography needs to be furthertested, and measurement of carotid intima-media thicknessmay be burdened by considerable interobserver variabilitywhen it is used in routine clinical practice. Thus, similar toblood biomarkers, the potential incremental value of suchsurrogate markers of clinical atherosclerotic complications isnot unequivocally evident. Still, from a theoretical viewpointthe combination of blood biomarkers and markers of subclin-ical disease seems an attractive approach because this mayintegrate information on structural or functional vascular wallpathology and systemic “activity” of the disease (Figure).
However, for markers of subclinical disease as well as forblood biomarkers, controversy exists with regard to whichparameter represents the most useful one and for which timeperiod of the atherosclerotic process, and which combination ofmarkers may be most appropriate for decision making. Finally,analytical and cost considerations deserve further study.
The opinions expressed in this article are not necessarily those of theeditors or of the American Heart Association.
From the Department of Internal Medicine II, Cardiology, Universityof Ulm Medical Center, Ulm, Germany.
Correspondence to Wolfgang Koenig, MD, Department of InternalMedicine II, Cardiology, University of Ulm Medical Center, Robert-KochStr 8, D-89081 Ulm, Germany. E-mail [email protected]
(Circulation. 2007;116:3-5.)© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.orgDOI: 10.1161/CIRCULATIONAHA.107.707984
3
Editorial

Statistical Methodology: Limitationsin Assessment of Incremental
Diagnostic InformationA great deal of such uncertainty is based on the limitedavailability of adequate statistical tools to demonstrate theincremental value of an emerging biomarker in addition toglobal risk scoring. We have realized that evidence of just somemoderately strong association in epidemiological studies isinsufficient to assess the true clinical utility of a new candidatemarker. Most frequently, c statistics and area under the receiver-operating characteristic curve have been used. Risk estimatesthat would be needed here to show a clinically importantincrease in the area under the curve are usually not seen incardiovascular medicine.18 Thus, disappointingly, only a fewstudies have shown a statistically significant improvement in thearea under the curve, which, however, in most cases was toosmall to be considered clinically relevant. The aggregate expe-rience from a number of such studies demonstrates that oncethere is a single strong predictor of risk in the model, which maybe even age alone, it is extremely difficult to show a relevantcontribution of any additional variable to model prediction. This
has recently been discussed in detail by Cook,18 and alternativestatistical approaches have been suggested, such as clinical riskreclassification.19 This procedure attempts to improve risk pre-diction by development and validation of algorithms that moreprecisely allocate an individual to a risk category by use of amodel that has incorporated a new risk variable in addition toconventional risk factors, compared with a basic model thatcontains conventional risk factors alone. Such approach focusesparticularly on those subjects at intermediate risk to eitherreclassify an individual into the low- or high-risk category.
Integration of Biochemical and BioimagingMarkers: The Solution?
In the presence of such complex background, Cao andcolleagues20 present important data from the CardiovascularHealth Study in this issue of Circulation. The investigatorssimultaneously measured carotid intima-media thickness,plaque characteristics, and CRP, and related all 3 variables tothe 12-year incidence of cardiovascular disease (CVD) eventsand all-cause mortality in 5888 elderly subjects. Main resultsshowed that all parameters were correlated with one another,
Screening for subjects at risk for cardiovascular complications: blood biomarkers/risk factors and/or markers of subclinical disease. Apo indi-cates apolipoprotein; BP, blood pressure; CT, computed tomography; HDL, high-density lipoprotein; IMT, intima-media thickness; LDL, low-density lipoprotein; Lp(a), lipoprotein a; Lp-PLA2, lipoprotein-associated phospholipase A2; MRI, magnetic resonance imaging; and Syn,syndrome. Reprinted from Naghavi M et al. Am J Cardiol. 2006;98(suppl):2H–15H, with permission from Elsevier. Copyright 2006.
4 Circulation July 3, 2007

yet each parameter independently predicted risk of CVDevents and mortality in multivariable models, which includedall 3 measures and traditional risk factors. Being in the toptertile of the carotid intima-media thickness distribution wasmore predictive for various events than having CRP �3 mg/Lor than being in the high-risk group on the basis of carotidplaque characteristics. Elevated CRP was a particularly usefulpredictor in the presence of subclinical atherosclerosis with a72% increase in risk for CVD and 52% increase in totalmortality. Cumulative event rates suggested a possible addi-tive interaction for composite CVD and all-cause mortalitywith an excess risk attributable to the interaction of CRP andsubclinical atherosclerosis of 54% for CVD death and 79%for all-cause mortality. By contrast, CRP did not add predic-tive power in the absence of carotid atherosclerosis. Finally,both CRP and subclinical atherosclerosis added only modestincremental information to risk prediction when adjusted forthe effect of conventional risk factors with either c statisticsor area under the curve derived from receiver-operatingcharacteristic analysis.
ConclusionsFirst, global risk assessment, with traditional risk factors, stillrepresents the rational basis for cardiovascular risk stratification.Second, although theoretically attractive, currently availablebiomarkers, even the combination of a robust systemic marker of“disease activity” with a marker that provides information onstructural changes of the arterial vasculature, which must be seenas a surrogate/precursor of clinical disease, does not appreciablyimprove risk prediction. However, the Cardiovascular HealthStudy cohort was an elderly population and results may not begeneralizable to younger individuals with low risk, in whomCRP may work in the absence of significant atheroscleroticburden. Also, the statistical tools used, as mentioned earlier, maybe debatable. Third, in the future, despite such somewhatdisappointing information regarding single markers, the clinicalapplication of multimarker panels, for which the possibilities ofmodel improvement are greater, may still prove to be a prom-ising approach, provided that such variables show low correla-tions with conventional risk factors and with each other butprovide strong associations with clinical events. Such emergingmarkers will have to be rigorously evaluated in large cohorts fortheir clinical efficacy and effectiveness with innovative statisti-cal analytical tools. The world of proteomics and metabolomics,together with advanced imaging modalities such as functionalmolecular imaging, may offer such promising candidates.
DisclosuresNone.
References1. De Backer G, Ambrosioni E, Borch-Johnsen K, Brotons C, Cifkova R,
Dallongeville J, Ebrahim S, Faergeman O, Graham I, Mancia G, Cats VM,Orth-Gomer K, Perk J, Pyorala K, Rodicio JL, Sans S, Sansoy V, SechtemU, Silber S, Thomsen T, Wood D; European Society of Cardiology;American Heart Association; American College of Cardiology. Europeanguidelines on cardiovascular disease prevention in clinical practice. ThirdJoint Task Force of European and other Societies on Cardiovascular DiseasePrevention in Clinical Practice (constituted by representatives of eightsocieties and by invited experts). Atherosclerosis. 2004;173:381–391.
2. Khot UN, Khot MB, Bajzer CT, Sapp SK, Ohman EM, Brener SJ, EllisSG, Lincoff AM, Topol EJ. Prevalence of conventional risk factors inpatients with coronary heart disease. JAMA. 2003;290:898–904.
3. Morrow DA, ed. Cardiovascular Biomarkers. Pathophysiology andDisease Management. Totowa, New Jersey: Humana Press Inc.; 2006.
4. Greenland P, Smith SC Jr, Grundy SM. Improving coronary heart diseaserisk assessment in asymptomatic people: role of traditional risk factorsand noninvasive cardiovascular tests. Circulation. 2001;104:1863–1867.
5. Vasan RS. Biomarkers of cardiovascular disease: molecular basis andpractical considerations. Circulation. 2006;113:2335–2362.
6. Koenig W, Khuseyinova N. Biomarkers of atherosclerotic plaque insta-bility and rupture. Arterioscler Thromb Vasc Biol. 2007;27:15–26.
7. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease.N Engl J Med. 2005;352:1685–1695.
8. Ridker PM, Rifai N, eds. C-Reactive Protein and CardiovascularDisease. St-Laurent, Canada: MediEdition Inc.; 2006.
9. Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO 3rd,Criqui M, Fadl YY, Fortmann SP, Hong Y, Myers GL, Rifai N, Smith SC Jr,Taubert K, Tracy RP, Vinicor F; Centers for Disease Control and Prevention;American Heart Association. Markers of inflammation and cardiovasculardisease: application to clinical and public health practice: a statement forhealthcare professionals from the Centers for Disease Control and Preventionand the American Heart Association. Circulation. 2003;107:499–511.
10. Zalewski A, Macphee C. Role of lipoprotein-associated phospholipase A2
in atherosclerosis: biology, epidemiology, and possible therapeutic target.Arterioscler Thromb Vasc Biol. 2005;25:923–931.
11. Koenig W, Khuseyinova N, Lowel H, Trischler G, Meisinger C. Lipoprotein-associated phospholipase A2 adds to risk prediction of incident coronaryevents by C-reactive protein in apparently healthy middle-aged men from thegeneral population: results from the 14-year follow-up of a large cohort fromsouthern Germany. Circulation. 2004;110:1903–1908.
12. Ballantyne CM, Hoogeveen RC, Bang H, Coresh J, Folsom AR,Chambless LE, Myerson M, Wu KK, Sharrett AR, Boerwinkle E.Lipoprotein-associated phospholipase A2, high-sensitivity C-reactiveprotein, and risk for incident ischemic stroke in middle-aged men andwomen in the Atherosclerosis Risk in Communities (ARIC) study. ArchIntern Med. 2005;165:2479–2484.
13. Folsom AR, Chambless LE, Ballantyne CM, Coresh J, Heiss G, Wu KK,Boerwinkle E, Mosley TH Jr, Sorlie P, Diao G, Sharrett AR. Anassessment of incremental coronary risk prediction using C-reactiveprotein and other novel risk markers: the Atherosclerosis Risk in Com-munities study. Arch Intern Med. 2006;166:1368–1373.
14. Wang TJ, Gona P, Larson MG, Tofler GH, Levy D, Newton-Chen C,Jacques PF, Rifai N, Selhub J, Robins SJ, Benjamin EJ, D’Agostino RB,Vasan RS. Multiple biomarkers for the prediction of first cardiovascularevents and death. N Engl J Med. 2006;355:2631–2639.
15. Lorenz MW, Markus HS, Bots ML, Rosvall M, Sitzer M. Prediction ofclinical cardiovascular events with carotid intima-media thickness: asystematic review and meta-analysis. Circulation. 2007;115:459–467.
16. Budoff MJ, Achenbach S, Blumenthal RS, Carr JJ, Goldin JG, GreenlandP, Guerci AD, Lima JA, Rader DJ, Rubin GD, Shaw LJ, Wiegers SE;American Heart Association Committee on Cardiovascular Imaging andIntervention; American Heart Association Council on CardiovascularRadiology and Intervention; American Heart Association Committee onCardiac Imaging, Council on Clinical Cardiology. Assessment ofcoronary artery disease by cardiac computed tomography: a scientificstatement from the American Heart Association Committee on Cardio-vascular Imaging and Intervention, Council on Cardiovascular Radiologyand Intervention, and Committee on Cardiac Imaging, Council onClinical Cardiology. Circulation. 2006;114:1761–1791.
17. Heald CL, Fowkes FG, Murray GD, Price JF; Ankle Brachial Index Collab-oration. Risk of mortality and cardiovascular disease associated with theankle-brachial index: systematic review. Atherosclerosis. 2006;189:61–69.
18. Cook NR. Use and misuse of the receiver-operating characteristic curvein risk prediction. Circulation. 2007;115:928–935.
19. Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation ofimproved algorithms for the assessment of global cardiovascular risk inwomen: the Reynolds Risk Score. JAMA. 2007;297:611–619.
20. Cao JJ, Arnold AM, Manolio TA, Polak JF, Psaty BM, Hirsch CH, KullerLH, Cushman M. Association of carotid artery intima-media thickness,plaques, and C-reactive protein with future cardiovascular disease andall-cause mortality: the Cardiovascular Heath Study. Circulation. 2007;116:32–38.
KEY WORDS: Editorials � atherosclerosis � epidemiology � imaging �inflammation � risk factors
Koenig Cardiovascular Biomarkers 5

The ST-Segment–Elevation Myocardial Infarction Chainof Survival
Joseph P. Ornato, MD
The benefit of expertly performed, timely, primarypercutaneous coronary intervention (PCI) over fibri-nolysis is clear for patients with ST-segment–eleva-
tion myocardial infarction (STEMI). Primary PCI is superiorto fibrinolysis for reduction of overall short-term mortality,nonfatal reinfarction, stroke, and the combined end point ofdeath, nonfatal reinfarction, and stroke.1 These results remainvalid during long-term follow-up and are independent of boththe type of fibrinolytic used and whether the patient istransferred for primary PCI.
Article p 67Although the relationship between time delay from hospi-
tal emergency department arrival to fibrinolytic treatment andincreasing mortality has been firmly established,2 a similarrelationship for primary PCI treatment has been proven onlyrecently. De Luca et al3 assessed the relationship betweenischemic time and 1-year mortality in 1791 primary PCI-treated STEMI patients. After adjustment for age, gender,diabetes, and previous revascularization, these investigatorsshowed that every 30 minutes of primary PCI treatment delayis associated with a 7.5% (95% CI, 1.008 to 1.15; P�0.041)relative increase in 1-year mortality. With use of hierarchicalmodels adjusted for patient characteristics to evaluate theeffect of door-to-balloon time on in-hospital mortality on29 222 PCI-treated STEMI patients treated in �6 hours ofpresentation at 395 hospitals that participated in the NationalRegistry of Myocardial Infarction–3 and –4 from 1999 to2002, McNamara et al4 found that a longer door-to-balloontime interval is associated with increased in-hospital mortal-ity. Adjusted for patient characteristics, patients with adoor-to-balloon time interval �90 minutes were more likelyto die (odds ratio, 1.42; 95% CI, 1.24 to 1.62) compared withpatients who had a door-to-balloon time interval �90 min-utes. These findings provide evidence-based support for thegoal of a door-to-balloon time interval “within 90 minutes”cited in the 2004 American College of Cardiology/AmericanHeart Association (ACC/AHA) guidelines for the manage-ment of patients with STEMI5 and serve as a foundation for
the ACC’s Guidelines Applied in Practice Door-to-Balloon(GAP-D2B) campaign goal of a door-to-balloon time intervalof �90 minutes in 75% of PCI-treated STEMI patients atparticipating hospitals nationwide.6
On the ACC President’s Page, Nissen et al6 described thenew GAP-D2B campaign and stated:
In successful hospitals, the arrival of a STEMIpatient initiates a chain of well-orchestrated events,including activation of the catheterization laboratorydirectly by an emergency department physician with asingle phone call to the interventional cardiologist oncall. The catheterization laboratory team is expected toarrive within 20 to 30 minutes. Programs with the bestoutcomes employ a multidisciplinary team-based ap-proach that includes committed administrators, physi-cian champions, and nurse champions, along withmechanisms for rapid data feedback. This collabora-tion can extend to the local and regional emergencymedical systems (EMS). In some successful hospitals,the catheterization laboratory is activated based on aprehospital electrocardiography.
In this issue of Circulation, Khot et al7 report on theirexperience before and after implementation of STEMI GAP-D2B–like strategies in a 591-bed, tertiary care, Indianapolis-area, community hospital that consists of 2 separate campuses7 miles apart (a 13-minute drive). Although they began theirprogram long before the recently announced ACC initiative,Khot et al7 instituted most of the GAP-D2B recommendationson the basis of characteristics of best-performing NationalRegistry of Myocardial Infarction hospitals.8–10 Critical ele-ments of their new system included empowerment of emer-gency physicians to activate the catheterization laboratoryand team without cardiology consultation as well as imple-mentation of an in-house transfer team. Their new strategyreduced the median door-to-balloon time interval signifi-cantly during normal and off-duty work hours, even forpatients who had to be transferred physically from 1 facilityto another, which thus increased the percentage of patientstreated within the �90-minute door-to-balloon goal from28% to 71%. And, as predicted by the relationship betweentime to treatment and outcome, there were significant im-provements in mean infarct size, hospital length of stay, andtotal hospital cost per admission.
The ACC’s GAP-D2B initiative stands on even more solidground as a result of the Indiana Heart Physicians/St. FrancisHeart Center experience reported by Khot et al.7 The commonelement shared by both is choreographed multidisciplinaryteamwork with effective communication among differentdisciplines of healthcare providers, rather than the traditionallinear progression of care most patients experience as theypass through a series of hospital units that operate asindividual silos. Both initiatives are focused on the portion of
The opinions expressed in this article are not necessarily those of theeditors or of the American Heart Association.
From the Department of Emergency Medicine, Virginia Common-wealth University, Richmond.
Correspondence to Joseph P. Ornato, MD, Virginia CommonwealthUniversity, Department of Emergency Medicine, 1201 East Marshall St,AD Williams 2nd Floor, Richmond, VA 23298�0401. [email protected]
(Circulation. 2007;116:6-9.)© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.orgDOI: 10.1161/CIRCULATIONAHA.107.710970
6
Editorial

STEMI patient treatment delay that is potentially mostchangeable by hospital-based healthcare providers—thatwhich occurs after a patient presents to the hospital. This isclearly the right place to start, but it represents only part of abroader community-based opportunity to improve STEMIpatient care.
In 1991, the AHA adopted a metaphor—the “Chain ofSurvival”—to describe the sequence of events that mustoccur for the best likelihood of successful resuscitation fromhospital cardiopulmonary arrest.11 This educational construct,which consists of early access, early cardiopulmonary resus-citation, early defibrillation, and early advanced cardiac lifesupport, now serves as the structural foundation for improve-ments in the community approach to sudden cardiac deathworldwide. It may now be appropriate for the AHA toconsider adoption of a similar metaphor—the “STEMI Chainof Survival” (Figure)—that can be used to target globalimprovements in STEMI patient care. This approach is verysimilar to that which has been followed for �25 years by theAmerican College of Surgeons Committee on Trauma, as ithas led our nation to develop one of the finest and mosteffective trauma care systems in the world. The new STEMIchain begins with an emphasis on the role of the patient in therecognition of early or prodromal heart attack symptoms andimmediate request for help, preferably by calling 911 andaccessing the EMS system12,13; the chain works its waythrough the critical elements of prehospital, emergency de-partment, and reperfusion care.
There is presently no uniform national STEMI triage andtreatment system equivalent to the system that directs majortrauma victims to verified trauma centers in the United States.Because the majority of US hospitals do not have primaryPCI capability, many communities struggle to decide whetherthey should direct EMS-transported, prehospital, 12-leadECG–identified STEMI patients to only primary PCI facili-ties to bypass nonprimary PCI hospitals. Unfortunately, themajority of STEMI patients do not use the 911 ambulancesystem for transport to the hospital, the national paramedictraining curriculum considers 12-lead ECG training as anenhanced rather than core skill, and not all EMS ambulancescurrently have 12-lead ECG capability.14 Many US hospitalscontinue to use fibrinolysis as their primary reperfusionstrategy with transfer to an interventional facility for rescuewhen needed. Other hospitals transfer patients more regularlyfor primary PCI, but even the most recently publishedNational Registry of Myocardial Infarction data on 4278patients transferred to 419 hospitals for primary PCI show amedian initial hospital door-to-balloon time of 180 minutes,with only 4.2% of patients treated with reperfusion in �90minutes, the benchmark recommended by national quality
guidelines.15 Khot et al7 have shown us that this challengingtime interval also can be decreased dramatically by anorganized transfer and PCI treatment team that can beactivated by emergency physicians.
Our national challenge to provide optimal STEMI careneeds to be solved at 2 levels: the community and thehospital. We must continue to educate the public on the signsand symptoms of a myocardial infarction and reinforce theNational Heart Attack Alert Program and AHA message to“Call 911, Call Fast.”16 The community needs to be organizedinto a system of care that directs STEMI patients quickly andefficiently to primary PCI centers whenever possible, and allhospitals, whether primary PCI-capable or not, need to havea system in place to avoid unnecessary delays, just like thatwhich has been implemented by Khot et al.7
The AHA’s Acute Myocardial Infarction Advisory Work-ing Group recently released recommendations on how toincrease the number of STEMI patients who have timelyaccess to primary PCI.17 The group commissioned Pricewa-terhouseCoopers to conduct national market research, and theWorking Group interviewed a wide variety of key stakehold-ers (such as patients, physicians, nurses, EMS representa-tives, community hospitals, primary PCI facilities, payers,and evaluation/outcomes organizations such as the Agencyfor Healthcare Research and Quality, the Food and DrugAdministration, and the Joint Commission on Accreditationof Healthcare Organizations) to determine the desirability,feasibility, and potential effectiveness of establishment ofregional systems and/or centers of care for STEMI patientswith a focus on whether and how this might improve patientaccess to quality care and outcomes. The researchers foundthat key stakeholders would support a national primary PCIcertification program with the understanding that some non-primary PCI hospitals would experience a modest decline inrevenue. On the basis of these findings, the AHA hosted anational stakeholder meeting in 2006 to continue develop-ment of a more detailed plan for a national system of STEMIpatient care. As has been suggested, this is an idea whose timemay have come.18
Many communities are currently developing organizedSTEMI care plans. Three sites—a major city, a large regionof a state, and an entire state—already have model commu-nity STEMI systems in place based on the trauma care systemmodel. In 2003, Boston, Mass., implemented a comprehen-sive system of care in which STEMI patients identified byparamedics with the use of prehospital 12-lead ECGs weretransported only to designated PCI centers.19 ParticipatingPCI centers agreed to collect and submit performance mea-sures data to a Central Data Coordinating Center on all EMS-and non-EMS–transported STEMI patients. System oversight
Figure 1. The STEMI chain of survival.
Ornato STEMI Chain of Survival 7

was provided by a Steering Committee with representativesfrom 9 area hospitals and the Boston EMS, which developedtheir performance indicators and minimum standards on thebasis of nationally accepted guidelines. A central DataCoordinating Center at Tufts–New England Medical Centerreceived and tabulated data from EMS and area hospitals toprovide aggregated data reports with receiving hospitalsdesignated only A, B, C, D, etc, rather than by name. Thereports were reviewed by an independent Data and SafetyMonitoring Board composed of highly respected cardiolo-gists and a statistician. After discussion between the hospitaland Data and Safety Monitoring Board and review by theSteering Committee, any hospital that did not meet preestab-lished quality treatment, door-to-balloon, and outcome goalsfor 2 successive 6-month periods could be excluded fromreceiving EMS-transported STEMI patients for the next6-month period.
From March 2003 to May 2005, 448 STEMI patients weretransferred from 31 community hospitals by paramedic-staffed ambulance (n�149) or paramedic/critical care nurse–staffed helicopter (n�299) to the Minneapolis Heart Institutein Minneapolis, Minn., for primary PCI. A standardizedprotocol with accompanying checklists was developed on thebasis of national guidelines. Community hospitals wererequired to transfer all patients with STEMI or new leftbundle–branch block within 12 hours of symptom onset tothe regional interventional center. A level 1 myocardialinfarction protocol was developed and used to specify thesequence of events, diagnostic tests, and treatments. Patientsare preregistered by admitting personnel prior to arrival byuse of a demographic form faxed from the referring hospital.On arrival at the primary PCI center, patients are admitteddirectly to the cardiac catheterization laboratory and thusbypass the emergency department except in rare circum-stances, such as when 2 STEMI patients arrive simulta-neously. Prompt verbal and written feedback (which includes1-month and 1-year follow-up phone calls) is provided to thereferring hospital physician and nursing staff, and the timeintervals, clinical and angiographic data, and clinical out-comes are entered into a database. Time and outcomesummary reports meeting Joint Commission on Accreditationof Healthcare Organizations requirements are sent to eachcommunity hospital on a quarterly basis.
Patient treatment times and outcomes have been superbwith this regional STEMI care system. No STEMI patientswere excluded from transfer, even those with cardiogenicshock (13.7%), cardiac arrest (9.9%), and the elderly (17%,�80 years of age). No patient died during transport. Themedian total door-to-balloon time was reduced from �3hours before implementation of the regional system to 97minutes for 11 participating hospitals located �70 miles(zone 1) after implementation.18,20 The median total door-to-balloon time was 117 minutes with use of a facilitated PCIprotocol in 17 participating hospitals located �210 miles(zone 2) from the interventional center. The improvements intime to treatment were accompanied by low 30-day mortalityrates of 4.3% in zone 1 and 3.4% in zone 2.
The common denominator of these 2 models is that theyare based on a community system of care rather than centered
only on 1 hospital. A third model is the Reperfusion of AcuteMyocardial Infarction in Carolina Emergency Departments(RACE) project, which is a collaborative effort to increase therate and speed of coronary reperfusion through systematicchanges in emergency care. The project is based on thecollaborative efforts of EMS personnel, physicians, nurses,administrators, and payers from 5 regions and 68 hospitalsthroughout North Carolina. The recommendations of thisproject are based on established national guidelines, pub-lished data, and the knowledge and experience of numerousindividuals who specialize in STEMI patient care. Detailedinformation about the program, such as transfer criteria,protocols, training materials, and educational posters, areavailable on the North Carolina ACC Chapter Web site(http://www.nccacc.org/race.html).
In summary, cardiologists (and interventionalists), emer-gency physicians, nurses, and EMS providers must worktogether to establish effective regional community systems ofSTEMI patient care similar to the well-developed and highlysuccessful systems that direct major trauma victims to veri-fied trauma centers in the United States. All hospitals,whether primary PCI–capable or not, should develop aSTEMI protocol that includes procedures to expedite time toreperfusion treatment modeled after concepts inherent in theGAP-D2B program and the Indiana Heart Physicians/St.Francis Heart Center experience. A multidisciplinary com-mittee should oversee the process and provide performanceimprovement suggestions based on continuous data collectionand analysis.
DisclosuresDr Ornato has served on the science advisory board for the NationalRegistry of Myocardial Infarction, which is funded by Genentech.
References1. Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous
thrombolytic therapy for acute myocardial infarction: a quantitativereview of 23 randomised trials. Lancet. 2003;361:13–20.
2. Cannon CP, Gibson CM, Lambrew CT, Shoultz DA, Levy D, French WJ,Gore JM, Weaver WD, Rogers WJ, Tiefenbrunn AJ. Relationship ofsymptom-onset-to-balloon time and door-to-balloon time with mortalityin patients undergoing angioplasty for acute myocardial infarction.JAMA. 2000;283:2941–2947.
3. De Luca G, Suryapranata H, Ottervanger JP, Antman EM. Time delay totreatment and mortality in primary angioplasty for acute myocardialinfarction: every minute of delay counts. Circulation. 2004;109:1223–1225.
4. McNamara RL, Wang Y, Herrin J, Curtis JP, Bradley EH, Magid DJ,Peterson ED, Blaney M, Frederick PD, Krumholz HM. Effect of door-to-balloon time on mortality in patients with ST-segment elevation myo-cardial infarction. J Am Coll Cardiol. 2006;47:2180–2186.
5. Antman EM, Anbe DT, Armstrong PW, Bates ER, Green LA, Hand M,Hochman JS, Krumholz HM, Kushner FG, Lamas GA, Mullany CJ,Ornato JP, Pearle DL, Sloan MA, Smith SC Jr, Alpert JS, Anderson JL,Faxon DP, Fuster V, Gibbons RJ, Gregoratos G, Halperin JL, HiratzkaLF, Hunt SA, Jacobs AK. ACC/AHA guidelines for the management ofpatients with ST-elevation myocardial infarction: a report of theAmerican College of Cardiology/American Heart Association Task Forceon Practice Guidelines (Committee to Revise the 1999 Guidelines for theManagement of Patients with Acute Myocardial Infarction). Circulation.2004;110:e82–e292.
6. Nissen SE, Brush JE Jr, Krumholz HM. President’s page: GAP-D2B: analliance for quality. J Am Coll Cardiol. 2006;48:1911–1912.
7. Khot UN, Johnson ML, Ramsey C, Khot MB, Todd R, Shaikh SR, BergWJ. Emergency department physician activation of the catheterizationlaboratory and immediate transfer to an immediately available catheter-
8 Circulation July 3, 2007

ization laboratory reduce door-to-balloon time in ST-elevation myo-cardial infarction. Circulation. 2007;116:67–76.
8. Bradley EH, Curry LA, Webster TR, Mattera JA, Roumanis SA, RadfordMJ, McNamara RL, Barton BA, Berg DN, Krumholz HM. Achievingrapid door-to-balloon times: how top hospitals improve complex clinicalsystems. Circulation. 2006;113:1079–1085.
9. Bradley EH, Herrin J, Wang Y, McNamara RL, Radford MJ, Magid DJ,Canto JG, Blaney M, Krumholz HM. Door-to-drug and door-to-balloontimes: where can we improve? Time to reperfusion therapy in patientswith ST-segment elevation myocardial infarction (STEMI). Am Heart J.2006;151:1281–1287.
10. Bradley EH, Roumanis SA, Radford MJ, Webster TR, McNamara RL,Mattera JA, Barton BA, Berg DN, Portnay EL, Moscovitz H, Parko-sewich J, Holmboe ES, Blaney M, Krumholz HM. Achieving door-to-balloon times that meet quality guidelines: how do successful hospitals doit? J Am Coll Cardiol. 2005;46:1236–1241.
11. Cummins RO, Ornato JP, Thies WH, Pepe PE. Improving survival fromsudden cardiac arrest: the “chain of survival” concept. A statement forhealth professionals from the Advanced Cardiac Life Support Subcom-mittee and the Emergency Cardiac Care Committee, American HeartAssociation. Circulation. 1991;83:1832–1847.
12. Bahr RD. Access to early cardiac care: chest pain as a risk factor for heartattacks, and the emergence of early cardiac care centers. Md Med J.1992;41:133–137.
13. Ornato JP, Hand MM. Warning signs of a heart attack. Circulation.2001;104:1212–1213.
14. Garvey JL, MacLeod BA, Sopko G, Hand MM. Pre-hospital 12-leadelectrocardiography programs: a call for implementation by emergencymedical services systems providing advanced life support: National HeartAttack Alert Program (NHAAP) Coordinating Committee; NationalHeart, Lung, and Blood Institute (NHLBI); National Institutes of Health.J Am Coll Cardiol. 2006;47:485–491.
15. Nallamothu BK, Bates ER, Herrin J, Wang Y, Bradley EH, KrumholzHM. Times to treatment in transfer patients undergoing primary percu-taneous coronary intervention in the United States: National Registry ofMyocardial Infarction (NRMI)-3/4 analysis. Circulation. 2005;111:761–767.
16. Faxon D, Lenfant C. Timing is everything: motivating patients to call9-1-1 at onset of acute myocardial infarction. Circulation. 2001;104:1210–1211.
17. Jacobs AK, Antman EM, Ellrodt G, Faxon DP, Gregory T, Mensah GA,Moyer P, Ornato J, Peterson ED, Sadwin L, Smith SC. Recommendationto develop strategies to increase the number of ST-segment-elevationmyocardial infarction patients with timely access to primary percutaneouscoronary intervention. Circulation. 2006;113:2152–2163.
18. Henry TD, Atkins JM, Cunningham MS, Francis GS, Groh WJ, HongRA, Kern KB, Larson DM, Ohman EM, Ornato JP, Peberdy MA,Rosenberg MJ, Weaver WD. ST-segment elevation myocardialinfarction: recommendations on triage of patients to heart attack centers:is it time for a national policy for the treatment of ST-segment elevationmyocardial infarction? J Am Coll Cardiol. 2006;47:1339–1345.
19. Moyer P, Feldman J, Levine J, Beshansky J, Selker HP, Barnewolt B,Brown D, Cardoza J, Grossman S, Jacobs A, Kerman B, Kimmelstiel C,Larson R, Losordo D, Pearlmutter M, Pozner C, Ramirez A, RosenfieldK, Ryan TJ, Zane RD, Cannon CP. Implications of the mechanical (PCI)vs thrombolytic controversy for ST segment elevation myocardialinfarction on the organization of emergency medical services: the BostonEMS experience. Crit Pathways Cardiol. 2004;3:53–61.
20. Henry TD, Sharkey SW, Graham KJ, Pedersen WR, Lips DL, Wang YL,Unger BT, Henry CR, Larson DM. Transfer for direct percutaneouscoronary intervention for ST-elevation myocardial infarction: the Minne-apolis Heart Institute level 1 myocardial infarction program. Circulation.2005;112:II�620.
KEY WORDS: Editorials � infarction � myocardium
Ornato STEMI Chain of Survival 9

Common NOS1AP Variants Are Associated With aProlonged QTc Interval in the Rotterdam Study
Albert-Jan L.H.J. Aarnoudse, MD*; Christopher Newton-Cheh, MD, MPH*; Paul I.W. de Bakker, PhD;Sabine M.J.M. Straus, MD, PhD; Jan A. Kors, PhD; Albert Hofman, MD, PhD;
André G. Uitterlinden, PhD; Jacqueline C.M. Witteman, PhD; Bruno H.C. Stricker, PhD
Background—QT prolongation is an important risk factor for sudden cardiac death. About 35% of QT-interval variationis heritable. In a recent genome-wide association study, a common variant (rs10494366) in the nitric oxide synthase 1adaptor protein (NOS1AP) gene was found to be associated with QT-interval variation. We tested for association of 2NOS1AP variants with QT duration and sudden cardiac death.
Methods and Results—The Rotterdam Study is a population-based, prospective cohort study of individuals �55 years ofage. The NOS1AP variants rs10494366 T�G and rs10918594 C�G were genotyped in 6571 individuals. Heartrate–corrected QT interval (QTc) was determined with ECG analysis software on up to 3 digital ECGs per individual(total, 11 108 ECGs from 5374 individuals). The association with QTc duration was estimated with repeated-measuresanalyses, and the association with sudden cardiac death was estimated by Cox proportional-hazards analyses. Thers10494366 G allele (36% frequency) was associated with a 3.8-ms (95% confidence interval, 3.0 to 4.6; P�7.8�10�20)increase in QTc interval duration for each additional allele copy, and the rs10918594 G allele (31% frequency) wasassociated with a 3.6-ms (95% confidence interval, 2.7 to 4.4; P�6.9�10�17) increase per additional allele copy. Noneof the inferred NOS1AP haplotypes showed a stronger effect than the individual single-nucleotide polymorphisms. Therewere 233 sudden cardiac deaths over 11.9 median years of follow-up. No significant association was observed withsudden cardiac death risk.
Conclusions—Common variants in NOS1AP are strongly associated with QT-interval duration in an elderly population.Larger sample sizes are needed to confirm or exclude an effect on sudden cardiac death risk. (Circulation. 2007;116:10-16.)
Key Words: arrhythmia � death, sudden � electrocardiography � genetics � long-QT syndrome
Sudden cardiac death (SCD) claims 300 000 lives annuallyin the United States.1 Although certain high-risk groups
have been identified,2 most SCD occurs in individuals unrec-ognized to be at risk.3
Clinical Perspective p 16
Familial aggregation of SCD suggests a substantial contri-bution of genetic variation to SCD risk,4–7 but mendelianmutations identified to date individually explain little of thepopulation burden of SCD.8,9 Until recently, the search forsequence variants contributing to SCD risk has been re-stricted to candidate genes known for their role in arrhyth-mogenesis.10 The recent development of large single-nucle-otide polymorphism (SNP) databases,11 genotyping arrays of
great accuracy and genome-wide coverage of common vari-ation,12 together with analytical methods,13 has enabled un-biased surveys of most of the common variation in the humangenome. Still, the relatively small size of existing SCDcollections and etiologic heterogeneity limit the statisticalpower to detect causal variants; therefore, initial attention hasfocused on quantitative SCD risk factors in large cohorts.
The electrocardiographic QT interval is a noninvasivemeasure of ventricular repolarization. About 35% of thevariation in QT-interval duration in unselected community-based samples is heritable.14,15 Mendelian congenital long-and short-QT syndromes are both characterized by SCD fromventricular arrhythmias. Moreover, nonsyndromal long QTinterval16–19 and short QT interval20 impart increased risk of
Received November 16, 2006; accepted May 1, 2007.From the Departments of Epidemiology and Biostatistics (A.L.H.J.A., S.M.J.M.S, A.H., A.G.U., J.C.M.W., B.H.C.S.), Internal Medicine (A.G.U.,
B.H.C.S.), and Medical Informatics (J.A.K.), Erasmus Medical Center, Rotterdam, the Netherlands; Cardiology Division (C.N.-C.), Department ofMolecular Biology (P.I.W.d.B.), and Center for Human Genetics Research (P.I.W.d.B.), Massachusetts General Hospital, Boston; Program in Medicaland Population Genetics (C.N.-C., P.I.W.d.B.), Broad Institute of Harvard and MIT, Cambridge, Mass; National Heart, Lung, and Blood Institute’sFramingham Heart Study (C.N.-C.), Framingham, Mass; Department of Genetics, Harvard Medical School (P.I.W.d.B.), Boston, Mass; Inspectorate forHealth Care (A.L.H.J.A., B.H.C.S.), the Hague, the Netherlands; and Dutch Medicines Evaluation Board (S.M.J.M.S), the Hague, the Netherlands.
*The first 2 authors contributed equally to this work.Correspondence to Bruno H.C. Stricker, PhD, Department of Epidemiology and Biostatistics, Erasmus Medical Center, PO Box 2040, 3000 CA,
Rotterdam, The Netherlands. E-mail [email protected]© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.106.676783
D

SCD in unselected populations. In addition, medication-induced prolonged QT interval and ventricular arrhythmiashave led to the withdrawal of many noncardiac medications,21
making the QT interval an important phenotype to study.Previously, we identified a locus on chromosome 3 with
suggestive evidence of linkage to QT-interval duration, butthe genomic interval was large, and the finding has yet to beconfirmed.15 More recently, Arking et al22 reported thefinding from a genome-wide association study that a commonvariant (rs10494366; minor allele frequency, 38%) in theNOS1AP gene was reproducibly associated with QT-intervalvariation in several large population samples. The NOS1APgene, encoding the nitric oxide synthase 1 adaptor protein,has been found to regulate neuronal nitric oxide synthaseactivation23 and to enhance Dexras1 activation by neuronalnitric oxide synthase through a ternary complex.24 Neuronalnitric oxide synthase–knockout mice have been found to havealtered cardiac contractility, which suggests a role forNOS1AP in cardiac depolarization.25–27 Furthermore,NOS1AP is capable of interaction with ion channels throughits PDZ domain.28–30 Nevertheless, the involvement ofNOS1AP in myocardial repolarization was not known untilthe initial report of the association.
The impact of NOS1AP variants on QT-interval duration inolder populations, in whom nongenetic factors might play astronger role than heritable factors, is unknown.
The goal of the present study was to test for association ofthe NOS1AP variant with QT duration and to test for itsassociation with SCD in the Rotterdam Study.
MethodsStudy PopulationThe Rotterdam Study is a prospective population-based cohort studyof chronic diseases in the elderly. All inhabitants of Ommoord, aRotterdam suburb, �55 years of age (n�10 278), were ascertainedfrom the municipal register and invited to participate. Of them, 78%(n�7983; 58% female, 98% white) took part in the baselineexamination from March 1990 through July 1993. Second and thirdexaminations were conducted from September 1993 to August 1996and from April 1997 to December 1999, respectively. Objectives andmethods of the Rotterdam Study have been described in detail.31 Themedical ethics committee of Erasmus Medical Center (Rotterdam,the Netherlands) approved the study, and all participants providedsigned informed consent for participation, including retrieval ofmedical records, use of blood and DNA for scientific purposes, andpublication of data. DNA for genotyping is available for 6571participants (82%) from the baseline visit.
Clinical characteristics, including smoking, body mass index,hypertension, diabetes mellitus, heart failure, and myocardial infarc-tion, were ascertained as previously described.19,32–36 Active surveil-lance for incident diabetes mellitus, heart failure, and myocardial
infarction is conducted continuously between exams. In addition,exposure of study participants to medications has been gatheredcontinuously from January 1, 1991, to the present through comput-erized pharmacy records of the pharmacies in the study area.
GenotypingAll participants were genotyped for the NOS1AP SNP rs10494366T�G, previously shown to be associated with QT interval in 3independent samples.22 The correlated SNP rs10918594 C�G,which had evidence of association with QT interval in one of theoriginal samples,22 also was genotyped (see the Figure). Both weregenotyped with Taqman assays C_1777074_10 and C_1777009_10(Applied Biosystems, Foster City, Calif) in 1 ng genomic DNAextracted from leukocytes, as previously reported.37
Assessment of QTc Interval and OtherElectrocardiographic MeasurementsThe electrocardiography (ECG) phenotype studied was the heartrate–corrected QT interval (QTc) in milliseconds using Bazett’sformula (QTc�QT/�RR).38 As in previous studies of QTc in theRotterdam Study19 we used a 10-second resting 12-lead ECG(average of 8 to 10 beats), which was recorded on an ACTA ECG(ESAOTE, Florence, Italy) at a sampling frequency of 500 Hz andstored digitally. All ECGs were processed by the Modular ECGAnalysis System (MEANS) to obtain ECG measurements.39–41
MEANS determines the QT interval from the start of the QRScomplex until the end of the T wave. MEANS also determines thepresence of right or left bundle-branch block and left ventricularhypertrophy. To study the association between NOS1AP variants andQTc duration, all eligible ECGs from subjects with DNA availablewere used. ECGs with right or left bundle-branch block wereexcluded from analyses. In addition, to minimize confounding bynongenetic influences on QT duration, all ECGs taken while thesubject was on any QT-altering drugs were excluded from analyses.Drugs were considered possibly QT prolonging if they appeared onany of lists 1 through 4 at www.qtdrugs.org.42 We also excludedECGs if subjects were on flupentixol, levomepromazine, meflo-quine, olanzapine, or sertindole, which may prolong QT interval, ordigoxin, which shortens the QT interval. Up to 3 QTc measurementswere recorded across the 3 examination cycles.
Finally, in additional analyses, the mean QTc interval per individ-ual was divided into 3 gender-specific categories as previouslydescribed. For women, the cut points were �450 ms (normal), 451to 470 ms (borderline), and �470 ms (prolonged); for men, the cutpoints were �430 ms (normal), 431 to 450 ms (borderline), and�450 ms (prolonged).19,43
Adjudication of SCDFor the SCD analyses, all genotyped subjects were included. Theascertainment of SCD cases in the Rotterdam Study has beendescribed previously.19 SCDs were defined operationally as a wit-nessed natural death attributable to cardiac causes, heralded byabrupt loss of consciousness, within 1 hour of onset of acutesymptoms, or as an unwitnessed, unexpected death of someone seenin a stable medical condition �24 hours previously with no evidenceof a noncardiac cause.44,45
160,200kb 160,300kb 160,400kb 160,500kb 160,600kb
OLFML2B NOS1AP
rs10494366rs10918594
Chr. 1q23
NOS1AP and location of rs10494366 and rs10918594. The ruler indicates the physical position on chromosome 1. Thick horizontallines indicate genes in the region; thick vertical lines, NOS1AP exons; and arrows, the direction of transcription. Thick vertical lines onthe ruler indicate the positions of rs10918594 and rs10494366, which are �55 kb apart. The 2 SNPs were in linkage disequilibrium,with an r2 of 0.63 and D� of 0.89.
Aarnoudse et al NOS1AP Variants Are Associated With QTc Duration 11

Statistical AnalysisGenotype frequencies were tested for Hardy-Weinberg equilibriumwith a �2 test.
Because the QTc in subsequent ECGs of the same subject arecorrelated, we used repeated-measures analyses implemented inPROC MIXED (SAS 8.2, SAS Institute, Cary, NC). Both allelic andgeneral genotype models were tested for the 2 polymorphisms,although the allelic model was considered primary because of thepreviously reported rs10494366–QT relationship.22 Haplotypes wereestimated with the expectation-maximization algorithm implementedin PHASE 2.0 (University of Washington, Seattle),46,47 and onlyindividuals with successful genotyping for both SNPs and a posteriorprobability of P�0.95 for assigned haplotypes were included inhaplotype analyses. In total, we identified 2245 double heterozy-gotes, all of whom were phased as heterozygous haplotype TC-GGbecause these are the major haplotypes, with posterior probabilitiesin excess of 0.95. In haplotype analyses, the haplotype with majoralleles for both SNPs was considered the reference to which the other3 haplotypes were compared individually. QTc was tested forassociation with genotype as the sole predictor (crude) and withadjustment for age and gender (multivariable). To compare theoutcomes of haplotype analysis with individual SNP analysis, thelatter analyses also were performed restricting the analysis tosubjects in whom genotyping was successful for both SNPs. Finally,a sensitivity analysis was carried out, excluding ECGs with anabnormally prolonged QTc and using gender-specific cutoff pointsof �450 ms for men and �470 ms for women. Jonckheere-Terpstratests were used to test whether individuals carrying NOS1AP minoralleles had an increased frequency of borderline and abnormal meanQTc.
Hazard ratios for time to SCD from baseline were estimated withCox proportional-hazards models. Again, both allelic and generalgenotype models were tested for the 2 polymorphisms. In addition toNOS1AP genotype, known SCD risk factors—including age, gender,body mass index, smoking, hypertension, diabetes mellitus, heartfailure, and myocardial infarction at baseline and time-dependentincident diabetes mellitus, heart failure, and myocardial infarction—were included as predictors. To minimize misclassification of SCD,we additionally performed a subanalysis restricting the case defini-tion to witnessed deaths only. As we have previously shown, the riskof SCD for increasing QTc is stronger in the younger than in theolder age group,19 so we determined the hazard ratios for time toSCD separately in groups stratified by age above and below themedian age at baseline. Finally, we performed a sensitivity analysis,excluding subjects with a history of myocardial infarction at baselinefrom the analysis. All Cox proportional hazards analyses were
performed with SPSS for Windows, version 11.0 (SPSS Inc, Chi-cago, Ill).
The authors had full access to and take full responsibility for theintegrity of the data. All authors have read and agree to themanuscript as written.
ResultsStudy SubjectsBaseline characteristics for the total study population, con-sisting of all genotyped subjects from the Rotterdam Study(n�6571), are summarized in Table 1. Within the studypopulation, 12 967 ECGs were available from 6052 subjectsacross up to 3 examination cycles. After exclusion of ECGswith right or left bundle-branch block (n�640) and thoseperformed in individuals taking QT-prolonging or-shortening drugs (n�1334) or both, a total of 11 108 ECGsfrom 5374 subjects remained (on average, 2.1 ECGs perindividual). The 5374 subjects included in the QTc analyseswere 1.3 years younger at baseline, reflecting exclusionsamong older participants (Table 1). Women had an 8.9-ms-longer age-adjusted QTc interval (431.4 versus 422.5 ms;P�0.0001), as previously shown,38,48 and were 2.2 yearsolder than men (70.4 versus 68.2 years at baseline;P�0.0001). The numbers of abnormally prolonged QTc inmen and women of our study population were slightly higherthan expected on the basis of numbers from referencepopulations.48,49 However, our study population was onaverage already considerably older at baseline (69.5 versus 53and 61 years, respectively), and this mean further increasedwhen follow-up ECGs were taken.
GenotypingThe G-allele (minor) frequency of rs10494366 T�G was36.4% and of rs10918594 C�G was 31.4%. Successfulgenotype calls were made in 95.8% and 95.9% of subjects,respectively. Both SNPs were in Hardy-Weinberg equilib-rium (P�0.32 for rs10494366 and P�0.89 for rs10918594).The 2 SNPs were in linkage disequilibrium, with an r2 of 0.63and D� of 0.89. On phasing, we observed 2 common 2-SNPhaplotypes, TC (61.4%) and GG (29.1%), consisting of the 2
TABLE 1. Baseline Characteristics
Characteristic
Genotyped Sample QTc Sample SCD Cases
Men(n�2666, 40.6%)
Women(n�3905, 59.4%)
Men(n�2191, 40.8%)
Women(n�3183, 59.2%)
Men(n�116, 49.8%)
Women(n�117, 50.2%)
Age at baseline, y, meanSD 68.28.2 70.49.6 67.07.7 69.09.1 71.87.8 74.47.7
Follow-up time, y, meanSD 10.03.8 10.53.7 10.63.4 11.13.2 6.43.8 7.33.8
Current smoking, n (%) 774 (29.0) 680 (17.4) 634 (28.9) 582 (18.3%) 32 (27.6) 15 (12.8%)
Past smoking, n (%) 1635 (61.3) 1040 (26.6) 1343 (61.3) 872 (27.4) 75 (64.7) 38 (32.5%)
Body mass index, kg/m2, meanSD 25.73.0 26.74.1 25.73.0 26.74.0 25.33.0 27.33.9
Systolic blood pressure, mm Hg, meanSD 138.721.7 139.822.6 138.321.5 139.222.2 144.624.2 152.827.7
Diastolic blood pressure, mm Hg, meanSD 74.611.5 73.211.4 74.911.3 73.211.1 74.012.5 77.014.1
Hypertension, n (%) 780 (29.3) 1415 (36.2) 621 (28.3) 1102 (34.6) 53 (45.7) 65 (55.6)
Diabetes mellitus, n (%) 281 (10.5) 422 (10.8) 213 (9.7) 309 (9.7) 14 (12.1) 27 (23.1)
Myocardial infarction, n (%) 447 (16.8) 320 (8.2) 345 (15.7) 243 (7.6) 44 (37.9) 19 (16.2)
Heart failure, n (%) 81 (3.0) 131 (3.4) 34 (1.6) 75 (2.4) 17 (14.7) 7 (6.0)
Shown are characteristics of all individuals with DNA available for genotyping (genotyped sample), of the subset of genotyped subjects with ECGswithout bundle-branch block or use of a QT-prolonging drug or digoxin (QTc sample), and of the SCD cases. The SCD source sample includes allgenotyped subjects.
12 Circulation July 3, 2007
major and 2 minor alleles, respectively, and 2 remaininghaplotypes containing 1 major and 1 minor allele each, GC(7.2%) and TG (2.3%). Genotype distributions did not differbetween men and women and between quartiles of age atbaseline.
NOS1AP Polymorphisms and QTcMinor alleles of both NOS1AP SNPs were significantlyassociated with an increase in QTc duration. SNP rs10494366T�G was associated with a 3.8-ms increase in multivariable-adjusted QTc interval for each additional G allele, and SNPrs10918594 C�G was associated with a 3.6-ms increase peradditional G allele (Table 2). Additional adjustment for ECGleft ventricular hypertrophy did not alter the results (data notshown). We observed no difference in effect of the SNPsbetween men and women. A sensitivity analysis excludingECGs with an abnormally prolonged QTc (using gender-specific cut points) resulted in slightly lower estimates (2.9and 2.7 ms for the allelic models); however, the association ofNOS1AP genotypes with QTc duration remained highlysignificant (all P�10�11).
All 3 haplotypes containing 1 (GC and TG) or 2 (GG)minor alleles for the 2 SNPs were associated with increasedQTc compared with the homozygous TC reference haplotype.The GG haplotype was associated with a 4.1-ms-longer
multivariable-adjusted QTc per additional GG haplotypecopy (P�2.0�10�18) using the TC haplotype as reference.The GC and TG haplotypes were associated with a 3.2-ms-longer (P�7.0�10�4) and 4.1-ms-longer (P�0.01) multivari-able-adjusted QT interval per additional copy, respectively.None of the haplotypes had a more significant effect than theindividual SNPs.
Furthermore, rs10494366 and rs10918594 were associatedwith a larger proportion of borderline and prolonged QTcintervals using gender-specific cut points19 (test for trend,both P�0.0001; Table 3).
NOS1AP Polymorphisms and SCDWithin the study population (n�6571), we identified 233sudden cardiac deaths, 121 of which were witnessed. Base-line characteristics of all adjudicated SCD cases are shown inTable 1. After adjustment for known risk factors, theNOS1AP polymorphisms rs10494366 T�G and rs10918594C�G showed nonsignificant trends in the direction of in-creased hazard of SCD, with hazard ratios per additionalminor allele for time to SCD of 1.09 (95% confidenceinterval, 0.90 to 1.33) and 1.10 (95% confidence interval,0.90 to 1.34), respectively. In the subset of 121 adjudicatedSCD cases that were witnessed, a similar nonsignificant trendtoward increased SCD risk was found (Table 4). Stratification
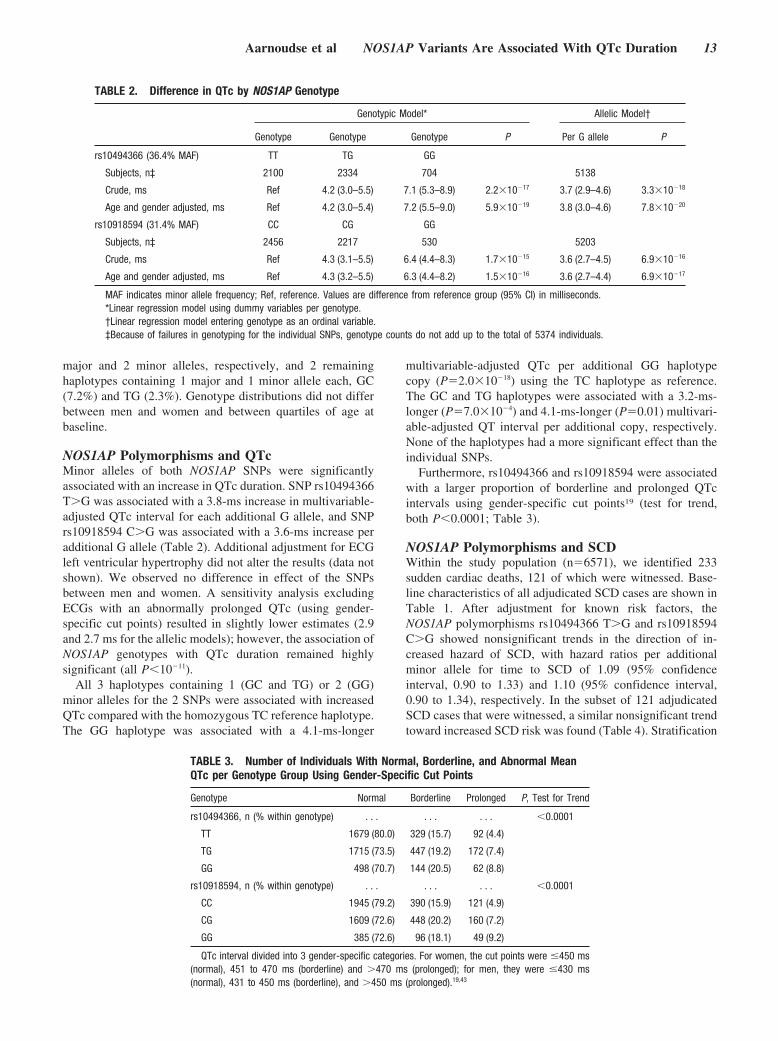
TABLE 2. Difference in QTc by NOS1AP Genotype
Genotypic Model* Allelic Model†
Genotype Genotype Genotype P Per G allele P
rs10494366 (36.4% MAF) TT TG GG
Subjects, n‡ 2100 2334 704 5138
Crude, ms Ref 4.2 (3.0–5.5) 7.1 (5.3–8.9) 2.2�10�17 3.7 (2.9–4.6) 3.3�10�18
Age and gender adjusted, ms Ref 4.2 (3.0–5.4) 7.2 (5.5–9.0) 5.9�10�19 3.8 (3.0–4.6) 7.8�10�20
rs10918594 (31.4% MAF) CC CG GG
Subjects, n‡ 2456 2217 530 5203
Crude, ms Ref 4.3 (3.1–5.5) 6.4 (4.4–8.3) 1.7�10�15 3.6 (2.7–4.5) 6.9�10�16
Age and gender adjusted, ms Ref 4.3 (3.2–5.5) 6.3 (4.4–8.2) 1.5�10�16 3.6 (2.7–4.4) 6.9�10�17
MAF indicates minor allele frequency; Ref, reference. Values are difference from reference group (95% CI) in milliseconds.*Linear regression model using dummy variables per genotype.†Linear regression model entering genotype as an ordinal variable.‡Because of failures in genotyping for the individual SNPs, genotype counts do not add up to the total of 5374 individuals.
TABLE 3. Number of Individuals With Normal, Borderline, and Abnormal MeanQTc per Genotype Group Using Gender-Specific Cut Points
Genotype Normal Borderline Prolonged P, Test for Trend
rs10494366, n (% within genotype) . . . . . . . . . �0.0001
TT 1679 (80.0) 329 (15.7) 92 (4.4)
TG 1715 (73.5) 447 (19.2) 172 (7.4)
GG 498 (70.7) 144 (20.5) 62 (8.8)
rs10918594, n (% within genotype) . . . . . . . . . �0.0001
CC 1945 (79.2) 390 (15.9) 121 (4.9)
CG 1609 (72.6) 448 (20.2) 160 (7.2)
GG 385 (72.6) 96 (18.1) 49 (9.2)
QTc interval divided into 3 gender-specific categories. For women, the cut points were �450 ms(normal), 451 to 470 ms (borderline) and �470 ms (prolonged); for men, they were �430 ms(normal), 431 to 450 ms (borderline), and �450 ms (prolonged).19,43
Aarnoudse et al NOS1AP Variants Are Associated With QTc Duration 13
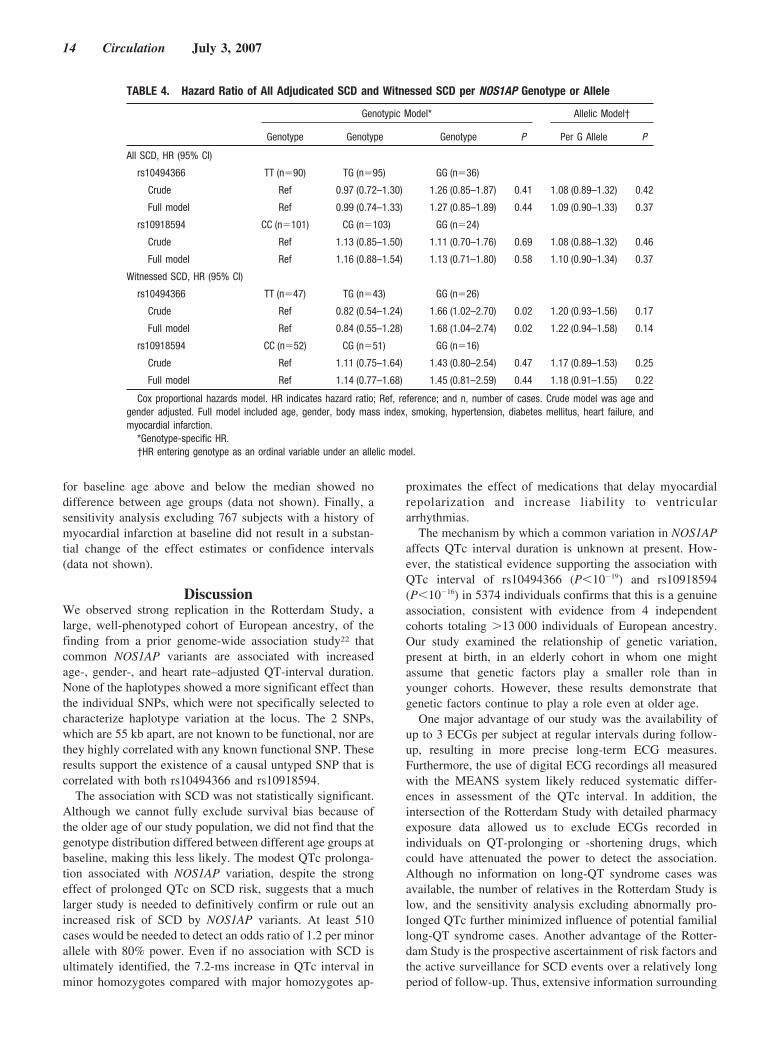
for baseline age above and below the median showed nodifference between age groups (data not shown). Finally, asensitivity analysis excluding 767 subjects with a history ofmyocardial infarction at baseline did not result in a substan-tial change of the effect estimates or confidence intervals(data not shown).
DiscussionWe observed strong replication in the Rotterdam Study, alarge, well-phenotyped cohort of European ancestry, of thefinding from a prior genome-wide association study22 thatcommon NOS1AP variants are associated with increasedage-, gender-, and heart rate–adjusted QT-interval duration.None of the haplotypes showed a more significant effect thanthe individual SNPs, which were not specifically selected tocharacterize haplotype variation at the locus. The 2 SNPs,which are 55 kb apart, are not known to be functional, nor arethey highly correlated with any known functional SNP. Theseresults support the existence of a causal untyped SNP that iscorrelated with both rs10494366 and rs10918594.
The association with SCD was not statistically significant.Although we cannot fully exclude survival bias because ofthe older age of our study population, we did not find that thegenotype distribution differed between different age groups atbaseline, making this less likely. The modest QTc prolonga-tion associated with NOS1AP variation, despite the strongeffect of prolonged QTc on SCD risk, suggests that a muchlarger study is needed to definitively confirm or rule out anincreased risk of SCD by NOS1AP variants. At least 510cases would be needed to detect an odds ratio of 1.2 per minorallele with 80% power. Even if no association with SCD isultimately identified, the 7.2-ms increase in QTc interval inminor homozygotes compared with major homozygotes ap-
proximates the effect of medications that delay myocardialrepolarization and increase liability to ventriculararrhythmias.
The mechanism by which a common variation in NOS1APaffects QTc interval duration is unknown at present. How-ever, the statistical evidence supporting the association withQTc interval of rs10494366 (P�10�19) and rs10918594(P�10�16) in 5374 individuals confirms that this is a genuineassociation, consistent with evidence from 4 independentcohorts totaling �13 000 individuals of European ancestry.Our study examined the relationship of genetic variation,present at birth, in an elderly cohort in whom one mightassume that genetic factors play a smaller role than inyounger cohorts. However, these results demonstrate thatgenetic factors continue to play a role even at older age.
One major advantage of our study was the availability ofup to 3 ECGs per subject at regular intervals during follow-up, resulting in more precise long-term ECG measures.Furthermore, the use of digital ECG recordings all measuredwith the MEANS system likely reduced systematic differ-ences in assessment of the QTc interval. In addition, theintersection of the Rotterdam Study with detailed pharmacyexposure data allowed us to exclude ECGs recorded inindividuals on QT-prolonging or -shortening drugs, whichcould have attenuated the power to detect the association.Although no information on long-QT syndrome cases wasavailable, the number of relatives in the Rotterdam Study islow, and the sensitivity analysis excluding abnormally pro-longed QTc further minimized influence of potential familiallong-QT syndrome cases. Another advantage of the Rotter-dam Study is the prospective ascertainment of risk factors andthe active surveillance for SCD events over a relatively longperiod of follow-up. Thus, extensive information surrounding
TABLE 4. Hazard Ratio of All Adjudicated SCD and Witnessed SCD per NOS1AP Genotype or Allele
Genotypic Model* Allelic Model†
Genotype Genotype Genotype P Per G Allele P
All SCD, HR (95% CI)
rs10494366 TT (n�90) TG (n�95) GG (n�36)
Crude Ref 0.97 (0.72–1.30) 1.26 (0.85–1.87) 0.41 1.08 (0.89–1.32) 0.42
Full model Ref 0.99 (0.74–1.33) 1.27 (0.85–1.89) 0.44 1.09 (0.90–1.33) 0.37
rs10918594 CC (n�101) CG (n�103) GG (n�24)
Crude Ref 1.13 (0.85–1.50) 1.11 (0.70–1.76) 0.69 1.08 (0.88–1.32) 0.46
Full model Ref 1.16 (0.88–1.54) 1.13 (0.71–1.80) 0.58 1.10 (0.90–1.34) 0.37
Witnessed SCD, HR (95% CI)
rs10494366 TT (n�47) TG (n�43) GG (n�26)
Crude Ref 0.82 (0.54–1.24) 1.66 (1.02–2.70) 0.02 1.20 (0.93–1.56) 0.17
Full model Ref 0.84 (0.55–1.28) 1.68 (1.04–2.74) 0.02 1.22 (0.94–1.58) 0.14
rs10918594 CC (n�52) CG (n�51) GG (n�16)
Crude Ref 1.11 (0.75–1.64) 1.43 (0.80–2.54) 0.47 1.17 (0.89–1.53) 0.25
Full model Ref 1.14 (0.77–1.68) 1.45 (0.81–2.59) 0.44 1.18 (0.91–1.55) 0.22
Cox proportional hazards model. HR indicates hazard ratio; Ref, reference; and n, number of cases. Crude model was age andgender adjusted. Full model included age, gender, body mass index, smoking, hypertension, diabetes mellitus, heart failure, andmyocardial infarction.
*Genotype-specific HR.†HR entering genotype as an ordinal variable under an allelic model.
14 Circulation July 3, 2007

SCD events was available, including the time between start ofsymptoms and death, which enabled rigorous adjudication ofSCD events.
One limitation of the study resides in the variety ofcompeting causes of abrupt death at increasing age, whichmay have led to misclassification, especially in cases inwhich death was unwitnessed. Because SCD coding wasblinded to NOS1AP genotype, this would likely have biasedour study to detect no effect. This might explain our findingof a slightly increased, but still nonsignificant, hazard ratiowhen the analyses were restricted to witnessed sudden car-diac deaths. Our results and those of the prior study by Arkinget al22 were restricted to population samples of Europeanancestry. Further testing in samples of African and Asianancestry is needed to establish the role of genetic variation atthe NOS1AP locus in myocardial repolarization in thesepopulation groups. Moreover, substantial frequency differ-ences are observed among European, African, and AsianHapMap samples, which raises the possibility of naturalselection in the region.50 Attempts to validate the NOS1APassociation in recently admixed populations, such as AfricanAmericans, will need to account for global and local chro-mosomal differences in ancestry because of the strong asso-ciation with continental ancestry and the risk of populationstratification.
ConclusionsWe have strongly confirmed the association of NOS1APvariants with QT-interval duration. With the limited numberof SCD cases in our cohort, it was not possible to demonstratethat this association translates into an influence on risk ofSCD, although the point estimates suggest that such a riskincrease may truly exist. Additional larger studies are re-quired to determine whether NOS1AP genotype is associatedwith SCD.
AcknowledgmentsWe thank Rowena Utberg for genotyping and Cornelis van der Hooftand Charlotte van Noord for their help with adjudication of SCDcases.
Sources of FundingDr Newton-Cheh and the genotyping were supported by a DorisDuke Charitable Foundation Clinical Scientist Development Awardand NIH K23 (HL80025).
DisclosuresNone.
References1. Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T,
Zheng ZJ, Flegal K, O’Donnell C, Kittner S, Lloyd-Jones D, Goff DC Jr,Hong Y, Adams R, Friday G, Furie K, Gorelick P, Kissela B, Marler J,Meigs J, Roger V, Sidney S, Sorlie P, Steinberger J, Wasserthiel-SmollerS, Wilson M, Wolf P. Heart disease and stroke statistics: 2006 update: areport from the American Heart Association Statistics Committee andStroke Statistics Subcommittee. Circulation. 2006;113:e85–e151.
2. Moss AJ, Zareba W, Hall WJ, Klein H, Wilber DJ, Cannom DS, DaubertJP, Higgins SL, Brown MW, Andrews ML. Prophylactic implantation ofa defibrillator in patients with myocardial infarction and reduced ejectionfraction. N Engl J Med. 2002;346:877–883.
3. Myerburg RJ, Castellanos A. Emerging paradigms of the epidemiologyand demographics of sudden cardiac arrest. Heart Rhythm. 2006;3:235–239.
4. Jouven X, Desnos M, Guerot C, Ducimetiere P. Predicting sudden deathin the population: the Paris Prospective Study I. Circulation. 1999;99:1978–1983.
5. Friedlander Y, Siscovick DS, Arbogast P, Psaty BM, Weinmann S,Lemaitre RN, Raghunathan TE, Cobb LA. Sudden death and myocardialinfarction in first degree relatives as predictors of primary cardiac arrest.Atherosclerosis. 2002;162:211–216.
6. Dekker LR, Bezzina CR, Henriques JP, Tanck MW, Koch KT, AlingsMW, Arnold AE, de Boer MJ, Gorgels AP, Michels HR, Verkerk A,Verheugt FW, Zijlstra F, Wilde AA. Familial sudden death is animportant risk factor for primary ventricular fibrillation: a case-controlstudy in acute myocardial infarction patients. Circulation. 2006;114:1140–1145.
7. Kaikkonen KS, Kortelainen ML, Linna E, Huikuri HV. Family historyand the risk of sudden cardiac death as a manifestation of an acutecoronary event. Circulation. 2006;114:1462–1467.
8. Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL,Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT. Spectrumof mutations in long-QT syndrome genes: KVLQT1, HERG, SCN5A,KCNE1, and KCNE2. Circulation. 2000;102:1178–1185.
9. Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBellWH, Song LS, Haurogne K, Kyndt F, Ali ME, Rogers TB, Lederer WJ,Escande D, Le Marec H, Bennett V. Ankyrin-B mutation causes type 4long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639.
10. Splawski I, Timothy KW, Tateyama M, Clancy CE, Malhotra A, BeggsAH, Cappuccio FP, Sagnella GA, Kass RS, Keating MT. Variant ofSCN5A sodium channel implicated in risk of cardiac arrhythmia. Science.2002;297:1333–1336.
11. International HapMap Consortium. A haplotype map of the humangenome. Nature. 2005;437:1299–1320.
12. de Bakker PI, Yelensky R, Pe’er I, Gabriel SB, Daly MJ, Altshuler D.Efficiency and power in genetic association studies. Nat Genet. 2005;37:1217–1223.
13. Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is moreefficient than replication-based analysis for two-stage genome-wide asso-ciation studies. Nat Genet. 2006;38:209–213.
14. Hong Y, Rautaharju PM, Hopkins PN, Arnett DK, Djousse L, Pankow JS,Sholinsky P, Rao DC, Province MA. Familial aggregation of QT-intervalvariability in a general population: results from the NHLBI Family HeartStudy. Clin Genet. 2001;59:171–177.
15. Newton-Cheh C, Larson MG, Corey DC, Benjamin EJ, Herbert AG, LevyD, D’Agostino RB, O’Donnell CJ. QT interval is a heritable quantitativetrait with evidence of linkage to chromosome 3 in a genome-wide linkageanalysis: the Framingham Heart Study. Heart Rhythm. 2005;2:277–284.
16. Schouten EG, Dekker JM, Meppelink P, Kok FJ, Vandenbroucke JP, PoolJ. QT interval prolongation predicts cardiovascular mortality in anapparently healthy population. Circulation. 1991;84:1516–1523.
17. Siscovick DS, Raghunathan TE, Rautaharju P, Psaty BM, Cobb LA,Wagner EH. Clinically silent electrocardiographic abnormalities and riskof primary cardiac arrest among hypertensive patients. Circulation. 1996;94:1329–1333.
18. Karjalainen J, Reunanen A, Ristola P, Viitasalo M. QT interval as acardiac risk factor in a middle aged population. Heart. 1997;77:543–548.
19. Straus SM, Kors JA, De Bruin ML, van der Hooft CS, Hofman A,Heeringa J, Deckers JW, Kingma JH, Sturkenboom MC, Stricker BH,Witteman JC. Prolonged QTc interval and risk of sudden cardiac death ina population of older adults. J Am Coll Cardiol. 2006;47:362–367.
20. Algra A, Tijssen JG, Roelandt JR, Pool J, Lubsen J. QT interval variablesfrom 24 hour electrocardiography and the two year risk of sudden death.Br Heart J. 1993;70:43–48.
21. Yap YG, Camm AJ. Drug induced QT prolongation and torsades depointes. Heart. 2003;89:1363–1372.
22. Arking DE, Pfeufer A, Post W, Kao WH, Newton-Cheh C, Ikeda M, WestK, Kashuk C, Akyol M, Perz S, Jalilzadeh S, Illig T, Gieger C, Guo CY,Larson MG, Wichmann HE, Marban E, O’Donnell CJ, Hirschhorn JN,Kaab S, Spooner PM, Meitinger T, Chakravarti A. A common geneticvariant in the NOS1 regulator NOS1AP modulates cardiac repolarization.Nat Genet. 2006;38:644–651.
23. Jaffrey SR, Snowman AM, Eliasson MJ, Cohen NA, Snyder SH.CAPON: a protein associated with neuronal nitric oxide synthase thatregulates its interactions with PSD95. Neuron. 1998;20:115–124.
Aarnoudse et al NOS1AP Variants Are Associated With QTc Duration 15

24. Fang M, Jaffrey SR, Sawa A, Ye K, Luo X, Snyder SH. Dexras1: a Gprotein specifically coupled to neuronal nitric oxide synthase viaCAPON. Neuron. 2000;28:183–193.
25. Massion PB, Pelat M, Belge C, Balligand JL. Regulation of the mam-malian heart function by nitric oxide. Comp Biochem Physiol A MolIntegr Physiol. 2005;142:144–150.
26. Ashley EA, Sears CE, Bryant SM, Watkins HC, Casadei B. Cardiac nitricoxide synthase 1 regulates basal and beta-adrenergic contractility inmurine ventricular myocytes. Circulation. 2002;105:3011–3016.
27. Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, KobeissiZA, Hobai IA, Lemmon CA, Burnett AL, O’Rourke B, Rodriguez ER,Huang PL, Lima JA, Berkowitz DE, Hare JM. Nitric oxide regulates theheart by spatial confinement of nitric oxide synthase isoforms. Nature.2002;416:337–339.
28. Murata M, Buckett PD, Zhou J, Brunner M, Folco E, Koren G. SAP97interacts with Kv1.5 in heterologous expression systems. Am J PhysiolHeart Circ Physiol. 2001;281:H2575–H2584.
29. Leonoudakis D, Mailliard W, Wingerd K, Clegg D, Vandenberg C.Inward rectifier potassium channel Kir2.2 is associated with synapse-associated protein SAP97. J Cell Sci. 2001;114:987–998.
30. Kim E, Sheng M. Differential K channel clustering activity of PSD-95and SAP97, two related membrane-associated putative guanylate kinases.Neuropharmacology. 1996;35:993–1000.
31. Hofman A, Grobbee DE, de Jong PT, van den Ouweland FA. Deter-minants of disease and disability in the elderly: the Rotterdam ElderlyStudy. Eur J Epidemiol. 1991;7:403–422.
32. Diabetes mellitus. In: Technical Reports Series 894. Geneva, Switzerland:World Health Organization; 1985.
33. Bots ML, Hoes AW, Koudstaal PJ, Hofman A, Grobbee DE. Commoncarotid intima-media thickness and risk of stroke and myocardialinfarction: the Rotterdam Study. Circulation. 1997;96:1432–1437.
34. Mosterd A, Hoes AW, de Bruyne MC, Deckers JW, Linker DT, HofmanA, Grobbee DE. Prevalence of heart failure and left ventricular dys-function in the general population: the Rotterdam Study. Eur Heart J.1999;20:447–455.
35. 1999 World Health Organization–International Society of Hypertensionguidelines for the management of hypertension: Guidelines Subcom-mittee. J Hypertens. 1999;17:151–183.
36. Bleumink GS, Knetsch AM, Sturkenboom MC, Straus SM, Hofman A,Deckers JW, Witteman JC, Stricker BH. Quantifying the heart failureepidemic: prevalence, incidence rate, lifetime risk and prognosis of heartfailure: the Rotterdam Study. Eur Heart J. 2004;25:1614–1619.
37. Fang Y, van Meurs JB, d’Alesio A, Jhamai M, Zhao H, Rivadeneira F,Hofman A, van Leeuwen JP, Jehan F, Pols HA, Uitterlinden AG.Promoter and 3�-untranslated-region haplotypes in the vitamin D receptorgene predispose to osteoporotic fracture: the Rotterdam Study. Am J HumGenet. 2005;77:807–823.
38. Bazett H. An analysis of time relations of the electrocardiogram. Heart.1920;7:353–370.
39. van Bemmel JH, Kors JA, van Herpen G. Methodology of the modularECG analysis system MEANS. Methods Inf Med. 1990;29:346–353.
40. Willems JL, Abreu-Lima C, Arnaud P, van Bemmel JH, Brohet C, DeganiR, Denis B, Gehring J, Graham I, van Herpen G, Machado H, MacfarlanePW, Michaelis J, Moulopoulos SD, Rubel P, Zywietz C. The diagnosticperformance of computer programs for the interpretation of electrocar-diograms. N Engl J Med. 1991;325:1767–1773.
41. de Bruyne MC, Kors JA, Hoes AW, Kruijssen DA, Deckers JW, GrosfeldM, van Herpen G, Grobbee DE, van Bemmel JH. Diagnostic interpre-tation of electrocardiograms in population-based research: computerprogram research physicians, or cardiologists? J Clin Epidemiol. 1997;50:947–952.
42. Drugs That Prolong the QT Interval and/or Induce Torsades de PointesVentricular Arrhythmia. Tucson, Ariz: University of Arizona Center forEducation and Research on Therapeutics, Arizona Health SciencesCenter. Available at: http://www.qtdrugs.org/medical-pros/drug-lists/drug-lists.htm. Accessed November 8, 2006.
43. The Assessment of the Potential for QT Interval Prolongation by Non-Cardiovascular Medicinal Products. London, UK: Committee for Pro-prietary Medicinal Products; 1997.
44. Myerburg RJ, Castellanos A. Cardiac arrest and sudden cardiac death. In:Zipes DP, Libby P, Bonow RO, Braunwald E, eds. Heart Disease: A TextBook of Cardiovascular Medicine. 7th ed. Philadelphia, Pa: ElsevierSaunders Co; 2004:865–908.
45. Priori SG, Aliot E, Blomstrom-Lundqvist C, Bossaert L, Breithardt G,Brugada P, Camm AJ, Cappato R, Cobbe SM, Di Mario C, Maron BJ,McKenna WJ, Pedersen AK, Ravens U, Schwartz PJ, Trusz-Gluza M,Vardas P, Wellens HJ, Zipes DP. Task Force on Sudden Cardiac Death ofthe European Society of Cardiology. Eur Heart J. 2001;22:1374–1450.
46. Stephens M, Smith NJ, Donnelly P. A new statistical method for hap-lotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989.
47. Stephens M, Donnelly P. A comparison of bayesian methods for hap-lotype reconstruction from population genotype data. Am J Hum Genet.2003;73:1162–1169.
48. Vitelli LL, Crow RS, Shahar E, Hutchinson RG, Rautaharju PM, FolsomAR. Electrocardiographic findings in a healthy biracial population: Ath-erosclerosis Risk in Communities (ARIC) Study Investigators.Am J Cardiol. 1998;81:453–459.
49. Rautaharju PM, Prineas RJ, Kadish A, Larson JC, Hsia J, Lund B. Normalstandards for QT and QT subintervals derived from a large ethnicallydiverse population of women aged 50 to 79 years (the Women’s HealthInitiative [WHI]). Am J Cardiol. 2006;97:730–737.
50. International HapMap Project. Available at: www.hapmap.org/cgi-perl/gbrowse/hapmap21_B35. Accessed November 6, 2006.
CLINICAL PERSPECTIVESudden cardiac death (SCD) claims 300 000 lives annually in the United States. The ECG QT interval is a noninvasivemeasure of ventricular repolarization, and prolongation of the QT interval is an important risk factor for SCD anddrug-induced arrhythmias. Approximately 35% of the variation in QT-interval duration is attributable to heritable factors.Until recently, the search for sequence variants contributing to QT-interval duration and SCD risk has been restricted tocandidate genes known for their role in arrhythmogenesis. However, in a recent genome-wide association study, a commonvariant in the nitric oxide synthase 1 adaptor protein (NOS1AP) gene was found to be associated with QT-interval variation.NOS1AP was not previously known to play a role in repolarization. In the present study, we have strongly confirmed theassociation of NOS1AP variants and QT-interval duration with a difference between minor homozygotes and majorhomozygotes of 7.2 ms (P�10�19). We did not find an association of NOS1AP variants with SCD cases in our cohort, butwith 228 SCD events, our study was underpowered to demonstrate such an effect. Even if no association with SCD isultimately identified, the 7.2-ms increase in QTc interval in minor allele homozygotes compared with major homozygotesapproximates the effect of medications that delay myocardial repolarization and increase liability to ventriculararrhythmias. The study underscores the power of association methods to identify novel genes and pathways involved inmyocardial repolarization and to identify genetic variants that could contribute to the risk of cardiac arrhythmias.
16 Circulation July 3, 2007

Nonsense Mutations in hERG Cause a Decrease in MutantmRNA Transcripts by Nonsense-Mediated mRNA Decay in
Human Long-QT SyndromeQiuming Gong, MD, PhD; Li Zhang, MD; G. Michael Vincent, MD;
Benjamin D. Horne, PhD, MPH; Zhengfeng Zhou, MD, PhD
Background—Long-QT syndrome type 2 (LQT2) is caused by mutations in the human ether-a-go-go-related gene (hERG).More than 30% of the LQT2 mutations result in premature termination codons. Degradation of premature terminationcodon–containing mRNA transcripts by nonsense-mediated mRNA decay is increasingly recognized as a mechanismfor reducing mRNA levels in a variety of human diseases. However, the role of nonsense-mediated mRNA decay inLQT2 mutations has not been explored.
Methods and Results—We examined the expression of hERG mRNA in lymphocytes from patients carrying the R1014Xmutation using a technique of allele-specific transcript quantification. The R1014X mutation led to a reduced level ofmutant mRNA compared with that of the wild-type allele. The decrease in mutant mRNA also was observed in theLQT2 nonsense mutations W1001X and R1014X using hERG minigenes expressed in HEK293 cells or neonatal ratventricular myocytes. Treatment with the protein synthesis inhibitor cycloheximide or RNA interference–mediatedknockdown of the Upf1 protein resulted in the restoration of mutant mRNA to levels comparable to that of the wild-typeminigene, suggesting that hERG nonsense mutations are subject to nonsense-mediated mRNA decay.
Conclusions—These results indicate that LQT2 nonsense mutations cause a decrease in mutant mRNA levels bynonsense-mediated mRNA decay rather than production of truncated proteins. Our findings suggest that the degradationof hERG mutant mRNA by nonsense-mediated mRNA decay is an important mechanism in LQT2 patients withnonsense or frameshift mutations. (Circulation. 2007;116:17-24.)
Key Words: arrhythmia � ion channels � long-QT syndrome � myocytes
Long-QT syndrome is a disease associated with delayedcardiac repolarization and prolonged QT intervals on the
ECG, which can lead to ventricular arrhythmias and suddendeath.1 The inherited long-QT syndrome type 2 (LQT2) iscaused by mutations in the human ether-a-go-go-related gene(hERG), which encodes the pore-forming subunit of therapidly activating delayed rectifier K� channel (IKr) in theheart.2,3 More than 250 hERG mutations have been identifiedin patients with LQT2.4–7 The mechanisms of hERG channeldysfunction in LQT2 mutations have been studied exten-sively in the last 10 years.8–11 Most previous studies, how-ever, have focused on the analysis of mutant proteins andchannel function. More than 30% of LQT2 mutations arenonsense or frameshift mutations that introduce prematuretermination codons (PTCs).4–7 These PTC mutations gener-ally are assumed to result in truncated dysfunctional channelproteins, and several nonsense and frameshift mutations havebeen studied at the protein level.8,12–18 However, it is now
becoming clear that nonsense and frameshift mutations bear-ing PTCs can destabilize mRNA transcripts via a mechanismknown as nonsense-mediated mRNA decay (NMD) in manyhuman diseases, resulting in decreased abundance of mutantmRNA transcripts rather than in production of truncatedproteins.19,20
Clinical Perspective p 24NMD is an RNA surveillance mechanism that selectively
degrades mRNA transcripts containing PTCs resulting fromnonsense or frameshift mutations. The role of NMD as adisease-causing mechanism of PTC mutations is becomingincreasingly evident.19,20 According to the proposed rule,NMD occurs when translation terminates �50 to 55 ntupstream of the 3�-most exon-exon junction.21,22 The molec-ular mechanisms of NMD have been studied extensively.These studies have shown that pre-mRNA splicing depositsthe exon junction complex �20 to 40 nt upstream of theexon-exon junction in spliced mRNA. The exon junction
Received October 26, 2006; accepted May 8, 2007.From the Division of Cardiovascular Medicine, Department of Medicine, Oregon Health and Science University, Portland (Q.G., Z.Z.); and
Departments of Medicine and Cardiology (L.Z., G.M.V.) and Genetic Epidemiology Division (B.D.H.), LDS Hospital, Intermountain Healthcare andUniversity of Utah, Salt Lake City.
Correspondence to Dr Zhengfeng Zhou, Division of Cardiovascular Medicine, Oregon Health and Science University, 3181 SW Sam Jackson Park Rd,Portland, OR 97239. E-mail [email protected]
© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.107.708818
17

complex can recruit Upf proteins, which are required forNMD.22 Several Upf proteins (Upf1, Upf2, Upf3a, Upf3b)have been identified.23 The Upf1 protein appears to play a keyrole in the distinction between proper and improper transla-tion termination. Upf1 is a group 1 helicase that has RNA-dependent ATPase and ATP-dependent 5� to 3� helicaseactivities. Knockdown of Upf1 by RNA interference (RNAi)has been shown to inhibit NMD.24,25
The objective of this work was to determine whether NMDoccurs in hERG mutations that contain PTCs. We investi-gated 2 nonsense mutations, W1001X and R1014X, in theC-terminal region of the hERG channel. The W1001X andR1014X mutations have previously been studied at theprotein level using hERG cDNAs.14,15 It was found that bothmutations produced truncated hERG channel proteins andreduced hERG current amplitude. The R1014X mutation alsocaused a dominant-negative effect on the wild-type (WT)hERG current, which is expected to result in a severe clinicalphenotype. However, the R1014X carriers have presentedwith a mild phenotype. In the present study, we demonstratethat rather than the production of truncated proteins, theprimary defect of the W1001X and R1014X mutations is thedegradation of mutant mRNA by NMD.
MethodsSubjectsThe study was approved by the institutional review board and carriedout on receipt of informed consent. The participants were blood-related members of a large family previously identified as having theR1014X mutation.4 The pedigree included 25 blood-related familymembers in 4 generations (Figure 1). Phenotyping was performed onthe basis of the history of LQTS-related cardiac events, the assess-ment of QT intervals and T-wave morphology, and pedigree analy-sis.26 Genotyping was conducted by sequencing of DNA samplescollected from buccal swabs. Normal control subjects were unrelatedindividuals.
RNA and DNA Preparations From Blood SamplesTotal RNA was isolated from peripheral blood lymphocytes usingthe RiboPure-Blood kit (Ambion, Austin, Tex). The isolated RNA
was treated with RNase-free DNase to remove genomic DNA.Genomic DNA was isolated from lymphocytes or Epstein Barrvirus–transformed lymphoblastoid cells with the DNeasy tissue kit(Qiagen, Valencia, Calif).
Allele-Specific Quantification of RNA Transcriptsand Genomic DNAThe relative abundance of RNA transcripts from WT and R1014Xalleles was determined by a modified “hot-stop” polymerase chainreaction (PCR) method.27,28 In this assay, the regular reverse-transcription PCR was carried out using the primers in exon 13(E13-F, forward 5�-GCCTTCTCAGGAGTGTCCAA-3�) and exon14 (E14-R, reverse 5�-GAAAGCGAGTCCAAGGTGAG-3�). After35 cycles, [32P]-dCTP was added and subjected to a single cycle ofPCR.28 With hot-stop PCR, only homoduplexes incorporated 32P-labels and any heteroduplexes formed during previous cycles wereunlabeled. Thus, hot-stop PCR will prevent the detection of WT/mutant heteroduplexes, which are resistant to restriction enzymedigestion. Because hot-stop PCR analysis yields a relative measureof transcripts from 2 alleles, normalization to a reference housekeep-ing gene is unnecessary. The hERG genomic DNA was analyzed byhot-stop PCR with the same forward primer as used in reverse-transcription PCR and a reverse primer in intron 13 (I13-R, 5�-CTCCGCGCTAGAGGTGTG-3�). For analysis of allelic variation inhERG mRNA expression in normal subjects, the ratio of a commonpolymorphism 1692A/G was determined by hot-stop PCR using theprimers in exon 6 (E6-F, forward 5�-ATCAACTTCCGCACCCCTA-3�) and exon 7 (E7-R, reverse 5�-TGTGTGGCTGCTCCATGT-3�). Thelabeled PCR products were treated with TaqI or NheI restriction enzymeand analyzed by 5% PAGE and autoradiography. For quantitativeanalysis, the intensity of each band was quantified with Scion Imagesoftware (Scion Corp, Frederick, Md). The ratio of 2 alleles wascalculated, and a correction factor according to the respective GCcontent of each digested product was applied to the ratio.28
Construction of MinigenesHuman genomic DNA was used as a template for PCR amplificationof fragments spanning from hERG exons 12 to 15. The PCRproducts were cloned into pCRII vector with the TA cloning kit(Invitrogen, Carlsbad, Calif) and verified by DNA sequencing. Theminigenes were then subcloned into a mammalian expression vectorpcDNA5/FRT (Invitrogen). The N-terminus of the minigene wastagged by Myc epitope, which is in frame with the hERG translationsequence. The W1001X and R1014X mutations in the minigenes
Figure 1. Pedigree of the familywith the R1014X mutation.
18 Circulation July 3, 2007

were generated with the pAlter in vitro site-directed mutagenesissystem (Promega, Madison, Wis) and verified by DNA sequencing.
Stable Expression of Minigene Constructs inHEK293 CellsThe minigenes in pcDNA5/FRT vector were stably transfected intoHEK293 cells by using the Flp-In method (Invitrogen). In thisapproach, an FRT site sequence is integrated into the genome ofHEK293 cells and recombined by Flp recombinase with the FRT siteof the pcDNA5/FRT vector. The pcDNA5/FRT vector carries thehygromycin resistance gene, which is used for the selection of stablecell lines.
RNase Protection Assay of mRNA TranscriptsFrom Minigene Transfected CellsRNA isolation and RNase protection assay (RPA) were performed aspreviously described.29 Briefly, cytoplasmic RNA was isolated fromHEK293 cells or neonatal rat ventricular myocytes expressing hERGminigenes with the RNeasy kit (Qiagen). The antisense RNAriboprobes were transcribed in vitro in the presence of biotin-16-UTP(Roche, Indianapolis, Ind). RNA (30 �g) was analyzed with theriboprobes using the RPAIII and BrightStart BioDetect kits (Am-bion). Yeast RNA was used as control for the complete digestion ofthe probes by RNase. The expression level of the hygromycinresistance gene from the pcDNA5/FRT vector or the E2 gene fromadenovirus was used as a loading control for normalization. Theintensity of each band was quantified with Scion Image software.
RNA InterferenceTwo plasmids, pSUPERpuro-hUpf1/I and pSUPERpuro-hUpf1/II(kindly provided by Dr Oliver Mühlemann), were used to inhibitexpression of Upf1 as described by Paillusson et al.25 These plasmidscontain short hairpin RNAs targeting 2 sequences of hUpf1 (5�-GAGAATCGCCTACTTCACT-3� for pSUPERpuro-hUpf1/I and5�-GATGCAGTTCCGCTCCATT-3� for pSUPERpuro-hUpf1/II).The HEK293 cells stably expressing WT or R1014X minigenes weretransfected with a mixture of 1 �g pSUPERpuro-hUpf1/I and 1 �gpSUPERpuro-hUpf1/II or 2 �g pSUPERpuro with scrambled se-quence of hUpf1/I using LipofectAmine 2000 (Invitrogen). At 24hours after transfection, puromycin was added to the final concen-tration of 1.5 �g/mL for 48 hours to eliminate the untransfected cells.Before analysis, the cells were cultured without puromycin for atleast 24 hours to avoid potential effects of this translation inhibitoron NMD. The knockdown of the Upf1 protein was analyzed byWestern blot as described.9,25
Construction and Use of Recombinant AdenovirusThe AdEasy vector kit was used to generate WT and R1014Xminigene recombinant adenoviruses (Stratagene, La Jolla, Calif).First, the WT and R1014X minigenes were subcloned into pShuttle-CMV vector and recombined with the pAdEasy plasmid in Esche-richia coli strain BJ5183. The pAdEasy/minigene plasmids weretransfected into HEK293 cells. After 2 days, the transfected cellswere cultured in growth medium containing 1.25% Seaplaque-agarose to promote the formation of recombinant viral plaques.Approximately 2 to 3 weeks later, individual plaques were picked,amplified in HEK293 cells, and purified over a discontinuous CsClgradient.
Primary Culture of Neonatal RatVentricular MyocytesNeonatal rat ventricular myocytes were prepared as described.30
Briefly, 1- to 3-day-old Sprague-Dawley rat pups were killed underether anesthesia by decapitation, and hearts were removed through asternotomy. The ventricles were trimmed free of atria, fat, andconnective tissues. Myocytes were dissociated by several 20-minutecycles of collagenase/pancreatin treatment and serum neutralization.Myocytes were cultured in Dulbecco’s modified Eagle’s mediumwith 17% Media 199, 10% horse serum, 5% fetal bovine serum,
penicillin (100 U/mL), and streptomycin (100 �g/mL). After 1 dayin culture, myocytes were infected with the recombinantadenoviruses.
Statistical AnalysisData are presented as mean�SD for QTc intervals or mean�SEMfor PCR and RPA analyses. Statistical comparison of QTc intervalsbetween R1014X mutation carriers and noncarriers was performedwith a family-based analysis approach using the software packagePedGenie, a Monte Carlo simulation–based program.31 ANOVAwith Bonferroni correction for multiple pairwise comparisons be-tween mutation/treatment groups was used for statistical analysis ofRPA data. Values of P�0.05 were considered statisticallysignificant.
The authors had full access to and take full responsibility for theintegrity of the data. All authors have read and agree to themanuscript as written.
ResultsPatient DescriptionA total of 22 family members were tested for the presence ofthe R1014X mutation. Nine family members were identifiedas R1014X mutation carriers (Figure 1). The ECG data wereavailable for 7 of the mutation carriers, all of which showeda prolonged QTc interval and typical LQT2 ECG pattern withthe subtle bifid T waves. The mean initial QTc interval in themutation carriers was 461�7 ms (n�7) versus 420�13 ms(n�8) in noncarriers (P�0.001). Four mutation carriers hadexercise tests, with maximum QTc value of 510�10 ms. Inthis family, 89% (8 of 9) of the R1014X mutation carrierswere asymptomatic. The only person with a history of cardiacevents is the 71-year-old proband. From 32 to 42 years of age,she had multiple syncopal episodes and 1 cardiac arrest thatwere associated with the presence of hypokalemia (serum K�,2.7 mEq/L) caused by taking a dietary supplement containingpotassium-wasting diuretics or taking QT-prolonging antihis-tamines. Since then, she has remained asymptomatic bystopping the potassium-wasting diet, avoiding QT-prolongingdrugs, and taking �-blockers.
Analysis of mRNA Isolated From Blood SamplesThe R1014X mutation causes premature termination of thehERG channel protein. This mutation has previously beenstudied at the protein level.15 However, it has been knownthat nonsense and frameshift mutations that contain PTCs canlead to the degradation of mRNA transcripts by NMD inmany diseases.19,20 To determine the underlying pathogenicmechanism of the R1014X mutation, it is important to studythis mutation at the mRNA level. Because the affected hearttissue from the mutation carriers was not available for thisstudy, we analyzed hERG mRNA transcripts isolated fromthe lymphocytes of patients carrying the R1014X mutation.To distinguish between WT and R1014X alleles, we per-formed allele-specific quantification analysis using the hot-stop PCR assay. The WT allele contains a TaqI restrictionsite, which is destroyed by the R1014X mutation. Afterreverse transcription of mRNA, cDNA was amplified byhot-stop PCR. After digestion of the PCR products with TaqI,the WT allele should yield 2 fragments of 287 and 72 bp, andthe R1014X allele should give a fragment of 359 bp. Asshown in Figure 2A, cDNA from a normal subject showed a
Gong et al Nonsense-Mediated mRNA Decay in LQT2 19

single band at 287 bp, corresponding to the WT alleles,whereas in cDNA from the proband, in addition to a WT287-bp band, a weak 359-bp band from the R1014X mutantallele was observed. Quantitative analysis of the samplesfrom 3 patients carrying the R1014X mutation revealed thatthe level of the R1014X mutant was reduced to 23�1% of theWT level, suggesting that the mRNA derived from theR1014X mutant allele is decreased. As a control, we alsoanalyzed genomic DNA from these 3 patients and showedthat the ratio of R1014X to WT alleles was 1.03�0.03, veryclose to the expected ratio of 1 (Figure 2A).
The allele-specific quantification analysis depends on theassumption that there is no significant allelic variation in
hERG mRNA expression. To rule out possible allelic varia-tion in hERG expression in the general population, weexamined the allele-specific expression of hERG mRNA innormal subjects. We analyzed 3 normal subjects who areheterozygous for a common polymorphism, 1692A/G. Todistinguish between 1692A and 1692G alleles, the relativelevels of mRNA transcripts from 1692A and 1692G alleleswere measured by the hot-stop PCR assay. The 1692A allelecontains an NheI restriction site, which is absent in the 1692Gallele. Thus, digestion with NheI should allow us to determinethe relative ratio of the 2 WT alleles. After digestion of thePCR products with NheI, the 1692A allele should be cut into2 fragments of 286 and 46 bp, and the 1692G alleles shouldremain uncut (332 bp). As shown in Figure 2B, in subjects 3,4, and 5 (lanes 3 to 5), there are 2 bands of 268 and 332 bp,suggesting that they are heterozygous for the 1692A/Gpolymorphism. In these 3 normal subjects, the average ratioof 1692G to 1692A was 0.97�0.07. This result suggests thatthere is no significant allelic variation in hERG mRNAexpression in normal subjects. Subjects 1 and 2 (lanes 1 and2) are homozygous for 1692A and 1692G, respectively.
Minigene Analysis of the R1014X andW1001X MutationsTo study whether the decrease in the abundance of mRNAlevels in the R1014X mutation is due to NMD, we con-structed minigenes containing the hERG genomic sequencespanning from exon 12 to 15 and expressed the minigenes inHEK293 cells. In the minigene experiments, the 2 LQT2nonsense mutations R1014X and W1001X were analyzed byRPA. Figure 3A shows the structure of the minigene and themRNAs after splicing. The R1014X and W1001X mutationslead to a PTC in exon 13, which is expected to trigger NMD.As shown in Figure 3B, the mRNA level of the R1014Xminigene was significantly lower than that of the WTminigene. Because degradation of mRNA by NMD dependson protein synthesis, we examined whether inhibition ofprotein synthesis by cycloheximide (CHX) abrogates NMDof the mutant mRNA, as has been shown for other PTC-containing transcripts.32 The cells expressing WT andR1014X minigenes were treated with CHX for 3 hours beforeRNA isolation. Treatment with CHX had no effect on thelevel of WT mRNA but significantly increased the level ofR1014X mutant mRNA, suggesting that the mutant mRNA isdegraded by NMD. Similar results were observed in theW1001X minigene (Figure 3C), suggesting that the degrada-tion of PTC-containing mRNAs by NMD may represent acommon mechanism in LQT2 patients with nonsensemutations.
Effect of Suppression of Upf1 on NMD of theR1014X MutationRecently, the Upf1 protein has been identified as a key factorfor NMD. Reducing Upf1 expression by RNAi has been usedas a functional assay to assess the NMD sensitivity ofPTC-containing mRNA transcripts.24,25 To study the role ofUpf1 in the reduced mRNA level of the R1014X mutation,we used the RNAi method to knock down Upf1 proteinexpression. In these experiments, HEK293 cells stably ex-
Figure 2. Hot-stop PCR analysis of mRNA and genomic DNAisolated from lymphocytes. A, Analysis of WT and R1014Xmutant alleles in a normal subject (NS) and the proband. Sche-matic diagrams are shown for hot-stop PCR analysis of cDNA(left) and genomic DNA (right). In cDNA analysis, a forwardprimer in exon 13 (E13-F) and a reverse primer in exon 14(E14-R) were used, and in genomic DNA analysis, the same for-ward primer and a reverse primer in intron 13 (I13-R) were used.The position of TaqI and the size of the fragments from WT andmutant PCR products are shown. After digestion with TaqI, the32P-labeled hot-stop PCR products were analyzed by PAGE andautoradiography. In cDNA analysis (left), the bands from WT andR1014X alleles are 287 and 359 bp, respectively; in genomicDNA analysis (right), the bands from WT and R1014X alleles are275 and 347 bp, respectively (the 72-bp band ran off the gel).Similar results were obtained in 2 additional R1014X carriers,and 3 to 5 independent experiments were performed for eachpatient. B, Analysis of allelic variation of hERG expression byanalyzing 1692A/G polymorphism in 5 normal subjects. A sche-matic diagram is shown for the position of NheI and the size ofthe fragments from A and G alleles. The 32P-labeled hot-stopPCR products were digested with NheI. The 286- and 332-bpbands represent 1692A and 1692G alleles, respectively (the46-bp band ran off the gel). Results shown are representative of2 independent experiments.
20 Circulation July 3, 2007
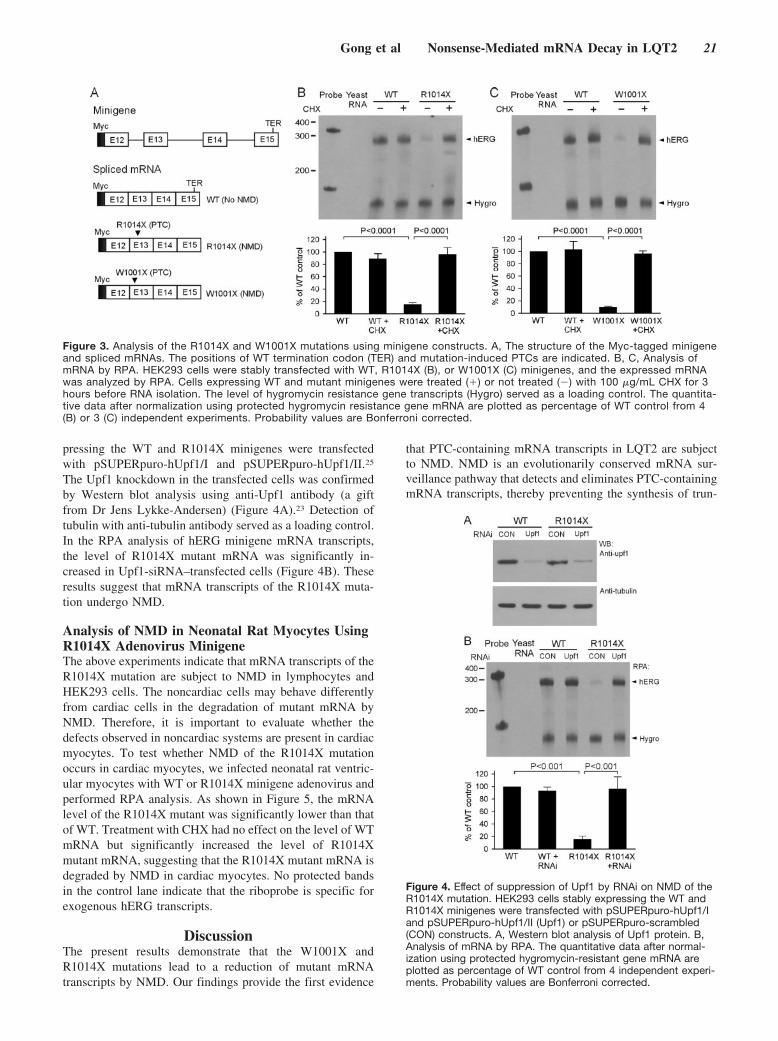
pressing the WT and R1014X minigenes were transfectedwith pSUPERpuro-hUpf1/I and pSUPERpuro-hUpf1/II.25
The Upf1 knockdown in the transfected cells was confirmedby Western blot analysis using anti-Upf1 antibody (a giftfrom Dr Jens Lykke-Andersen) (Figure 4A).23 Detection oftubulin with anti-tubulin antibody served as a loading control.In the RPA analysis of hERG minigene mRNA transcripts,the level of R1014X mutant mRNA was significantly in-creased in Upf1-siRNA–transfected cells (Figure 4B). Theseresults suggest that mRNA transcripts of the R1014X muta-tion undergo NMD.
Analysis of NMD in Neonatal Rat Myocytes UsingR1014X Adenovirus MinigeneThe above experiments indicate that mRNA transcripts of theR1014X mutation are subject to NMD in lymphocytes andHEK293 cells. The noncardiac cells may behave differentlyfrom cardiac cells in the degradation of mutant mRNA byNMD. Therefore, it is important to evaluate whether thedefects observed in noncardiac systems are present in cardiacmyocytes. To test whether NMD of the R1014X mutationoccurs in cardiac myocytes, we infected neonatal rat ventric-ular myocytes with WT or R1014X minigene adenovirus andperformed RPA analysis. As shown in Figure 5, the mRNAlevel of the R1014X mutant was significantly lower than thatof WT. Treatment with CHX had no effect on the level of WTmRNA but significantly increased the level of R1014Xmutant mRNA, suggesting that the R1014X mutant mRNA isdegraded by NMD in cardiac myocytes. No protected bandsin the control lane indicate that the riboprobe is specific forexogenous hERG transcripts.
DiscussionThe present results demonstrate that the W1001X andR1014X mutations lead to a reduction of mutant mRNAtranscripts by NMD. Our findings provide the first evidence
that PTC-containing mRNA transcripts in LQT2 are subjectto NMD. NMD is an evolutionarily conserved mRNA sur-veillance pathway that detects and eliminates PTC-containingmRNA transcripts, thereby preventing the synthesis of trun-
Figure 3. Analysis of the R1014X and W1001X mutations using minigene constructs. A, The structure of the Myc-tagged minigeneand spliced mRNAs. The positions of WT termination codon (TER) and mutation-induced PTCs are indicated. B, C, Analysis ofmRNA by RPA. HEK293 cells were stably transfected with WT, R1014X (B), or W1001X (C) minigenes, and the expressed mRNAwas analyzed by RPA. Cells expressing WT and mutant minigenes were treated (�) or not treated (�) with 100 �g/mL CHX for 3hours before RNA isolation. The level of hygromycin resistance gene transcripts (Hygro) served as a loading control. The quantita-tive data after normalization using protected hygromycin resistance gene mRNA are plotted as percentage of WT control from 4(B) or 3 (C) independent experiments. Probability values are Bonferroni corrected.
Figure 4. Effect of suppression of Upf1 by RNAi on NMD of theR1014X mutation. HEK293 cells stably expressing the WT andR1014X minigenes were transfected with pSUPERpuro-hUpf1/Iand pSUPERpuro-hUpf1/II (Upf1) or pSUPERpuro-scrambled(CON) constructs. A, Western blot analysis of Upf1 protein. B,Analysis of mRNA by RPA. The quantitative data after normal-ization using protected hygromycin-resistant gene mRNA areplotted as percentage of WT control from 4 independent experi-ments. Probability values are Bonferroni corrected.
Gong et al Nonsense-Mediated mRNA Decay in LQT2 21

cated and potentially harmful proteins.33 NMD occurs whentranslation terminates �50 to 55 nt upstream of the 3�-mostexon-exon junction.21,22 According to this rule, �90 LQT2nonsense and frameshift mutations are potential targets forNMD. Several LQT2 nonsense and frameshift mutations havebeen studied at the functional and protein levels with cDNAconstructs.8,12–18 All previous studies, however, have beencarried out under the assumption that these nonsense andframeshift mutations lead to the production of truncatedproteins. Because NMD requires introns, the absence ofintrons in cDNA constructs would preclude the degradationof PTC-containing transcripts by NMD. As a result, NMDeffects could not be observed when cDNAs were used inthese studies. In the present study, we used minigene con-structs that contain the hERG genomic DNA with both exonsand introns and showed that the W1001X and R1014Xmutations cause a marked decrease in mutant mRNA tran-scripts. Inhibition of protein synthesis by CHX or knockdownof Upf1 by RNAi results in the restoration of mutant mRNAto levels comparable to the WT minigene. These resultsstrongly suggest that the degradation of mutant mRNA byNMD is an important mechanism in LQT2 mutations carry-ing PTCs.
Previous studies have shown that different LQT2 muta-tions cause hERG channel dysfunction by different mecha-nisms. This led to a proposed classification of LQT2 muta-tions according to their underlying mechanisms.11 The
classification scheme (shown in Figure 6) illustrates themechanisms underlying LQT2 mutations. Class 1 mutationscause abnormal protein synthesis by defective transcription ortranslation. Class 2 mutations lead to defective proteintrafficking. Class 3 mutations result in abnormal gatingand/or kinetics, and class 4 mutations result in altered orabsent channel selectivity or permeability.11 In the presentstudy, we show that LQT2 nonsense mutations cause adecrease in mutant mRNAs by NMD, thereby altering theamount of mRNA available for subsequent hERG proteingeneration. We propose that the degradation of PTC-containing mRNA transcripts by NMD represents a new classof LQT2 pathogenic mechanism (class 5).
The mutations that undergo NMD will result in thedegradation of mutant mRNAs before they produce largequantities of truncated proteins. By eliminating abnormalmRNA transcripts carrying PTCs, NMD prevents the produc-tion of truncated proteins that could act in a dominant-negative manner, leading to deleterious effects on the cells.One of the physiological roles of NMD is to protect againstsevere disease phenotypes by converting the dominant-negative effect to haploinsufficiency.32 NMD as a modifier ofphenotypic severity has been reported in many human dis-eases.19,20,32,34 For example, in Marfan syndrome, anautosomal-dominant connective tissue disorder caused bymutations in the fibrillin 1 gene, nonsense mutations thatresult in reduced levels of mutant mRNA are associated witha mild phenotype. In contrast, patients with nonsense allelesthat escape NMD develop a severe phenotype as a result ofthe dominant-negative effect.19,34
Most R1014X mutation carriers in this family have pre-sented with a mild LQT2 phenotype. In contrast to patientswith pore-region mutations, who usually present with alonger QT interval and more frequent cardiac events,35 theQTc interval in the R1014X mutation carriers is only mildlyprolonged (461�7 ms), and only the proband experiencedarrhythmia-related cardiac events that were always associatedwith hypokalemia or the use of QT-prolonging drugs. Wehave previously shown that the R1014X mutation causeshERG channel dysfunction by defective trafficking of themutant protein.15 In addition, the truncated mutant proteinexhibits a dominant-negative effect on the WT hERG. Thisimplies that a severe phenotype would be expected in theR1014X mutation carriers. However, our present study re-veals that the R1014X mutant mRNA transcripts are mark-edly decreased by NMD, and as a result, the dominant-negative effect caused by the production of truncated proteinswould be minimized. Therefore, haploinsufficiency ratherthan a dominant-negative effect is probably the underlyingmechanism for the R1014X mutation, which is consistent
Figure 5. Analysis of NMD in neonatal rat ventricular myocytesusing R1014X minigene adenovirus constructs. Myocytesinfected by WT and R1014X mutant minigene adenoviruseswere treated (�) or not treated (�) with 100 �g/mL CHX for 3hours, and expressed mRNA was analyzed by RPA. The mRNAfrom uninfected myocytes was used as control (CON). The levelof E2 transcripts (E2) from adenovirus served as a loading con-trol. The quantitative data after normalization using protected E2mRNA are plotted as percentage of WT control from 4 indepen-dent experiments. Probability values are Bonferroni corrected.
Figure 6. Classification scheme for LQT2mutations.
22 Circulation July 3, 2007

with the observed clinical presentation of this family. It isinteresting to note that the W1001X mutation carriers alsopresent with a mild LQT2 phenotype.35 Moss et al35 reportedthat LQT2 patients with mutations in the pore region ofhERG have a significantly higher risk of arrhythmia-relatedcardiac events than patients with nonpore mutations. Al-though the difference may be explained by in vitro electro-physiological effects of reported hERG mutations, with poremutations having a greater negative effect on hERG currentthan nonpore mutations,35 it also is possible that NMD mayplay a role. It is noted that only 6% of LQT2 mutations in thepore region are nonsense or frameshift mutations, whereas�40% of the mutations in nonpore regions are nonsense orframeshift mutations. Clearly, further genotype-phenotypecorrelation studies are required to test whether NMD contrib-utes to the observed differences in clinical presentations ofpore and nonpore LQT2 mutations.
There are potential limitations to the present study. Ourpresent experiments analyzed endogenously expressedmRNA from patients carrying the R1014X mutation, but theRNA was isolated from lymphocytes rather than the affectedheart tissue. Although we have shown that the R1014Xmutant minigene expressed in neonatal rat ventricular myo-cytes leads to reduced mRNA levels by NMD, further studiesare required to determine whether the endogenous PTC-containing mRNA in human heart tissue is subject to NMD.Verification of our findings in human heart would strengthenthe conclusion that hERG mutations that contain PTCs canlead to degradation of the mutant mRNA by NMD.
In summary, our findings that nonsense mutations in hERGlead to a reduced level of mutant mRNA by NMD add to ourunderstanding of the disease-causing mechanisms of hERGmutations in LQT2. Thus, in studies of hERG nonsense andframeshift mutations, it is important to first analyze theabundance of mRNA to determine whether these PTC muta-tions are targeted by NMD. Obviously, this important pointhad been overlooked in previous studies that analyzed hERGPTC mutations only at the protein and functional levels.Because PTC mutations account for �30% of LQT2 muta-tions, the RNA surveillance imposed by NMD is of funda-mental importance in the pathogenesis of LQT2.
AcknowledgmentWe thank Dr Kent Thornburg for helpful comments on themanuscript.
Sources of FundingThe present study was supported in part by NIH grant HL68854 (DrZhou), Deseret Foundation grant DF400 (Dr Vincent), and NIH grant1UL1RRO24140 – 01 from the National Center for ResearchResources.
DisclosuresNone.
References1. Schwartz PJ, Periti M, Malliani A. Fundamentals of clinical cardiology:
the long QT syndrome. Am Heart J. 1975;89:378–390.2. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating
MT. A molecular basis for cardiac arrhythmia: HERG mutations causelong QT syndrome. Cell. 1995;80:795–803.
3. Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic linkbetween an inherited and an acquired cardiac arrhythmia: HERG encodesthe IKr potassium channel. Cell. 1995;81:299–307.
4. Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL,Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT. Spectrumof mutations in long-QT syndrome genes: KVLQT1, HERG, SCN5A,KCNE1, and KCNE2. Circulation. 2000;102:1178–1185.
5. Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J,Bottelli G, Cerrone M, Leonardi S. Genetic testing in the long QTsyndrome: development and validation of an efficient approach to geno-typing in clinical practice. JAMA. 2005;294:2975–2980.
6. Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium ofcardiac channel mutations in 541 consecutive unrelated patientsreferred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517.
7. Millat G, Chevalier P, Restier-Miron L, Da Costa A, Bouvagnet P,Kugener B, Fayol L, Gonzalez Armengod C, Oddou B, Chanavat V,Froidefond E, Perraudin R, Rousson R, Rodriguez-Lafrasse C. Spectrumof pathogenic mutations and associated polymorphisms in a cohort of 44unrelated patients with long QT syndrome. Clin Genet. 2006;70:214–227.
8. Sanguinetti MC, Curran ME, Spector PS, Keating MT. Spectrum ofHERG K�-channel dysfunction in an inherited cardiac arrhythmia. ProcNatl Acad Sci U S A. 1996;93:2208–2212.
9. Zhou Z, Gong Q, Epstein ML, January CT. HERG channel dysfunction inhuman long QT syndrome: Intracellular transport and functional defects.J Biol Chem. 1998;273:21061–21066.
10. Thomas D, Kiehn J, Katus HA, Karle CA. Defective protein trafficking inhERG-associated hereditary long QT syndrome (LQT2): molecularmechanisms and restoration of intracellular protein processing. Car-diovasc Res. 2003;60:235–232.
11. Delisle BP, Anson BD, Rajamani S, January CT. Biology of cardiacarrhythmias: ion channel protein trafficking. Circ Res. 2004;94:1418–1428.
12. Li X, Xu J, Li M. The human delta1261 mutation of the HERG potassiumchannel results in a truncated protein that contains a subunit interactiondomain and decreases the channel expression. J Biol Chem. 1997;272:705–708.
13. Paulussen A, Yang P, Pangalos M, Verhasselt P, Marrannes R, Ver-faille C, Vandenberk I, Crabbe R, Konings F, Luyten W, ArmstrongM. Analysis of the human KCNH2 (HERG) gene: identification andcharacterization of a novel mutation Y667X associated with long QTsyndrome and a non-pathological 9 bp insertion. Hum Mutat. 2000;15:483.
14. Kupershmidt S, Yang T, Chanthaphaychith S, Wang Z, Towbin JA,Roden DM. Defective human ether-a-go-go-related gene traffickinglinked to an endoplasmic reticulum retention signal in the C terminus.J Biol Chem. 2002;277:27442–27448.
15. Gong Q, Keeney DR, Robinson JC, Zhou Z. Defective assembly andtrafficking of mutant HERG channels with C-terminal truncations in longQT syndrome. J Mol Cell Cardiol. 2004;37:1225–1233.
16. Teng S, Ma L, Dong Y, Lin C, Ye J, Bahring R, Vardanyan V, Yang Y,Lin Z, Pongs O, Hui R. Clinical and electrophysiological characterizationof a novel mutation R863X in HERG C-terminus associated with long QTsyndrome. J Mol Med. 2004;82:189–196.
17. Paulussen AD, Raes A, Jongbloed RJ, Gilissen RA, Wilde AA, SnydersDJ, Smeets HJ, Aerssens J. HERG mutation predicts short QT based onchannel kinetics but causes long QT by heterotetrameric trafficking defi-ciency. Cardiovasc Res. 2005;67:467–475.
18. Choe CU, Schulze-Bahr E, Neu A, Xu J, Zhu ZI, Sauter K, Bahring R,Priori S, Guicheney P, Monnig G, Neapolitano C, Heidemann J, ClancyCE, Pongs O, Isbrandt D. C-terminal HERG (LQT2) mutations disrupt IKr
channel regulation through 14-3-3�. Hum Mol Genet. 2006;15:2888–2902.
19. Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in healthand disease. Hum Mol Genet. 1999;8:1893–1900.
20. Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004;36:801–808.
21. Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. TrendsBiochem Sci. 1998;23:198–199.
22. Maquat LE. Nonsense-mediated mRNA decay: splicing, translation andmRNP dynamics. Nat Rev Mol Cell Biol. 2004;5:89–99.
Gong et al Nonsense-Mediated mRNA Decay in LQT2 23

23. Lykke-Andersen J, Shu MD, Steitz JA. Human Upf proteins target anmRNA for nonsense-mediated decay when bound downstream of a ter-mination codon. Cell. 2000;103:1121–1131.
24. Mendell JT, ap Rhys CM, Dietz HC. Related separable roles forrent1/hUpf1 in altered splicing and decay of nonsense transcripts.Science. 2002;298:419–422.
25. Paillusson A, Hirschi N, Vallan C, Azzalin CM, Muhlemann O. AGFP-based reporter system to monitor nonsense-mediated mRNA decay.Nucleic Acids Res. 2005;33:e54.
26. Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC,Shen J, Splawski I, Priori S, Compton SJ, Yanowitz F, Benhorin J, MossAJ, Schwartz PJ, Robinson J, Wang Q, Zareba W, Keating M, TowbinJA, Napolitano C, Medina A. Spectrum of ST-T wave patterns andrepolarization parameters in congenital long QT syndrome: ECG findingsidentify genotype. Circulation. 2000;102:2849–2855.
27. Uejima H, Lee MP, Cui H, Feinberg AP. Hot-stop PCR: a simple andgeneral assay for linear quantitation of allele ratios. Nat Genet. 2000;25:375–376.
28. Kurreeman FA, Schonkeren JJ, Heijmans BT, Toes RE, Huizinga TW.Transcription of the IL10 gene reveals allele-specific regulation at themRNA level. Hum Mol Genet. 2004;13:1755–1762.
29. Gong Q, Keeney DR, Molinari M, Zhou Z. Degradation of trafficking-defective long QT syndrome type II mutant channels by the ubiquitin-proteasome pathway. J Biol Chem. 2005;280:19419–19425.
30. Kapiloff MS, Schillace RV, Westphal AM, Scott JD. mAKAP: anA-kinase anchoring protein targeted to the nuclear membrane of differ-entiated myocytes. J Cell Sci. 1999;112:2725–2736.
31. Allen-Brady K, Wong J, Camp NJ. PedGenie: an analysis approach forgenetic association testing in extended pedigrees and genealogies ofarbitrary size. BMC Bioinformatics. 2006;7:209–220.
32. Inoue K, Khajavi M, Ohyama T, Hirabayashi S, Wilson J, Reggin JD,Mancias P, Butler IJ, Wilkinson MF, Wegner M, Lupski JR. Molecularmechanism for distinct neurological phenotypes conveyed by allelic trun-cating mutations. Nat Genet. 2004;36:361–369.
33. Conti E, Izaurralde E. Nonsense-mediated mRNA decay: molecularinsights and mechanistic variations across species. Curr Opin Cell Biol.2005;17:316–325.
34. Dietz HC, McIntosh I, Sakai LY, Corson GM, Chalberg SC, Pyeritz RE,Francomano CA. Four novel FBN1 mutations: significance for mutanttranscript level and EGF-like domain calcium binding in the pathogenesisof Marfan syndrome. Genomics. 1993;17:468–475.
35. Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J,Towbin JA, Keating MT, Priori SG, Schwartz PJ, Vincent GM, RobinsonJL, Andrews ML, Feng C, Hall WJ, Medina A, Zhang L, Wang Z.Increased risk of arrhythmic events in long-QT syndrome with mutationsin the pore region of the human ether-a-go-go-related gene potassiumchannel. Circulation. 2002;105:794–799.
CLINICAL PERSPECTIVECongenital long-QT syndrome type 2 (LQT2) is caused by mutations in human ether-a-go-go related gene (hERG), whichencodes a voltage-gated potassium channel (IKr) in the heart. The present work demonstrates that LQT2 nonsense mutationsshow a decrease in mutant mRNA transcripts via nonsense-mediated mRNA decay (NMD), an RNA surveillancemechanism that selectively eliminates the mRNA transcripts that contain premature termination codons. These resultsindicate that, contrary to intuition, the predominant consequence of hERG nonsense mutations is not the production oftruncated proteins but rather the degradation of mutant mRNA by NMD. Given that nonsense and frameshift mutationsaccount for �30% of LQT2 mutations, the RNA surveillance imposed by NMD is of fundamental importance in thepathogenesis of LQT2. Our findings have important implications for genotype-phenotype correlation investigations inLQT2. By eliminating abnormal mRNA transcripts carrying premature termination codons, NMD prevents the productionof truncated proteins that could act in a dominant-negative manner. The clinical significance of NMD is the protectionagainst severe disease phenotypes by converting the dominant-negative effect to haploinsufficiency. Thus, NMD appearsto be an important factor in modifying phenotypic severity in LQT2.
24 Circulation July 3, 2007

Coronary Artery Calcification Progression Is HeritableAndrea E. Cassidy-Bushrow, PhD, MPH; Lawrence F. Bielak, DDS, MPH; Patrick F. Sheedy II, MD;
Stephen T. Turner, MD; Iftikhar J. Kullo, MD; Xihong Lin, PhD; Patricia A. Peyser, PhD
Background—Coronary artery calcification (CAC), a marker of coronary artery atherosclerosis, can be measuredaccurately and noninvasively with the use of electron beam computed tomography. Serial measures of CAC quantifyprogression of calcified coronary artery plaque. Little is known about the role of genetic factors in progression of CACquantity.
Methods and Results—We quantified the relative contributions of measured risk factors and unmeasured genes to CACprogression measured by 2 electron beam computed tomography examinations an average of 7.3 years apart in 877asymptomatic white adults (46% men) from 625 families in a community-based sample. After adjustment for baselinerisk factors and CAC quantity, the estimated heritability of CAC progression was 0.40 (P�0.001). Baseline risk factorsand CAC quantity explained 64% of the variation in CAC progression. Thus, genetic factors explained 14% of thevariation [(100�64)�(0.40)] in CAC progression. After adjustment for risk factors, the estimated genetic correlation(pleiotropy) between baseline CAC quantity and CAC progression was 0.80 and was significantly different than 0(P�0.001) and 1 (P�0.037). The environmental correlation between baseline CAC quantity and CAC progression was0.42 and was significantly different than 0 (P�0.006).
Conclusions—Evidence was found that many but not all genetic factors influencing baseline CAC quantity also influenceCAC progression. The identification of common and unique genetic influences on these traits will provide importantinsights into the genetic architecture of coronary artery atherosclerosis. (Circulation. 2007;116:25-31.)
Key Words: atherosclerosis � calcium � genetics � imaging � population
Coronary heart disease (CHD) is the leading cause ofdeath and disability in the United States. Despite recog-
nition of numerous factors contributing to development ofCHD, the ability to predict individuals at risk of CHD eventsremains suboptimal. More than one half of CHD deaths occurin individuals without previous symptoms.1 Traditional riskfactors (high cholesterol, high blood pressure, cigarette smok-ing, diabetes) are highly prevalent among individuals withCHD but are also prevalent in individuals without CHDevents.2
Clinical Perspective p 31Atherosclerosis is the primary cause of CHD. Coronary
artery calcification (CAC), a measure of coronary atheroscle-rosis presence and quantity, can be detected noninvasivelyand reliably with electron beam computed tomography(EBCT). CAC predicts CHD events in asymptomatic individ-uals at intermediate risk on the basis of their CHD riskfactors.3,4 EBCT can be used to serially measure the progres-sion of CAC. CAC progression is associated with CHD.5,6
Family history of premature CHD is associated with CAC.7
Unmeasured genes contribute to interindividual variation in
CAC quantity measured at a single time point across studies.Estimated heritability (�SE) was 0.42�0.13 among asymp-tomatic white individuals,8 0.40�0.08 among sibships en-hanced for hypertension,9 and 0.40�0.23 among individualsfrom families enriched for type 2 diabetes.10
No studies have focused on estimating the genetic contri-bution to CAC progression, although the complex biology ofprogression of calcium appears to be “genetically directed.”11
The purpose of the present investigation was to estimate thegenetic contribution to variation in noninvasively measuredCAC progression among an asymptomatic community-basedsample. Additionally, evidence for pleiotropy, or sharedgenetic influences, between CAC quantity at baseline andCAC progression was examined.
Methods
Study ParticipantsThe Epidemiology of Coronary Artery Calcification (ECAC) study,conducted between 1991 and 1998, examined 1240 participants aged�20 years from the Rochester Family Heart Study12,13 and 496individuals living in the vicinity of Rochester, Minn, who were not
Received August 22, 2006; accepted May 8, 2007.From the Department of Epidemiology, University of Michigan, Ann Arbor (A.E.C.-B., L.F.B., P.A.P.); Department of Biostatistics and Research
Epidemiology, Henry Ford Health System, Detroit, Mich (A.E.C.-B.); Department of Diagnostic Radiology (P.F.S.), Division of Hypertension,Department of Internal Medicine (S.T.T.), and Division of Cardiovascular Diseases (I.J.K.), Mayo Clinic and Foundation, Rochester, Minn; andDepartment of Biostatistics, Harvard University, Boston, Mass (X.L.).
Correspondence to Patricia A. Peyser, PhD, Department of Epidemiology, University of Michigan, 611 Church St, Ann Arbor, MI 48104-3028. [email protected]
© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.106.658583
25
Coronary Heart Disease

pregnant or lactating and who never had coronary or noncoronaryheart surgery.14,15 A total of 1155 ECAC study participants had afollow-up examination between December 2000 and February 2005.In general, participants were invited to return for a follow-upexamination on the basis of age (older age first) and longer timesince baseline examination. Study protocols were approved by theMayo Clinic and University of Michigan institutional review boards,and participants gave written informed consent.
One thousand fifty-five white ECAC participants had completeCAC data at baseline and follow-up and no history of myocardialinfarction, stroke, or positive angiogram at baseline or follow-up.Individuals with missing baseline or follow-up risk factor data(n�68), 79 individuals aged �45 years at follow-up, and 31individuals with outlier values (exceeding �4 SDs from samplemean) for risk factor data were excluded. Individuals were restrictedto being aged �45 years at follow-up for comparability with otherCAC heritability studies8 and because CAC prevalence in youngerindividuals, especially women, is very low.16 The final sample sizeconsisted of 877 individuals (402 men).
Risk Factor AssessmentDuring baseline and follow-up examination interviews, participantsreported current medication use, educational attainment, history ofsmoking, physician-diagnosed hypertension, myocardial infarction,angiographic evidence of a blocked coronary artery, stroke, ordiabetes. Family history of CHD was defined as self-reportedmyocardial infarction or coronary artery revascularization in a parentand/or sibling that occurred before age 60 years. Age 60 years waschosen to represent premature disease.17 Height was measured by awall stadiometer, weight was measured by electronic balance, andbody mass index (kg/m2) was calculated. Waist circumference wasmeasured at the umbilicus, hips were measured at the level ofmaximal circumference, and waist-to-hip ratio was calculated.
Standard enzymatic methods were used to measure total choles-terol, high-density lipoprotein cholesterol (HDL-C), plasma glucose,and triglycerides after overnight fasting.13 Low-density lipoproteincholesterol (LDL-C) was calculated by the Friedewald equation.18
Systolic blood pressure (SBP) and diastolic blood pressure (DBP)levels were measured in the right arm with a random-zero sphyg-momanometer (Hawksley and Sons). Three measures at least 2minutes apart were taken; the average of the second and thirdmeasurements was used. Individuals were considered hypertensive ifthey reported a prior diagnosis of hypertension and use of prescrip-tion antihypertensive medication or if the average SBP or DBP was�140 mm Hg or �90 mm Hg, respectively. Participants wereconsidered diabetic if they reported using insulin or oral hypoglyce-mic agents or if they reported a physician diagnosis of diabetes butwere not currently taking a pharmacological agent to control glucoselevels. The Framingham risk equation was used to estimate the10-year probability of CHD (10-year CHD risk) at baseline.19
Measurement of CACCAC was measured with an Imatron C-150 EBCT scanner (ImatronInc, South San Francisco, Calif). Protocols at baseline and follow-upwere identical.20 A dual-scan approach was used beginning in 1993.A scan run consisted of 40 contiguous 3-mm-thick tomographicslices from the root of the aorta to the apex of the heart. Scan timewas 100 ms per tomogram. ECG gating was used, and all imageswere triggered at end-diastole during 2 to 4 breath-holds. A radio-logical technologist scored the tomograms with an automated scoringsystem without knowledge of other EBCT examination results forthe same participant.21 CAC was defined as a hyperattenuating focuswithin 5 mm of the midline of a coronary artery, �4 contiguouspixels in size, and having CT numbers �130 Hounsfield unitsthroughout. Areas �1 mm2 for all CAC foci were summed to providea measure of CAC quantity. When 2 scan runs at a single examina-tion were available, CAC quantity was based on the average.
Statistical AnalysisBaseline CAC quantity was natural logarithm (log) transformed afteradding 1 to reduce nonnormality and is referred to as log baseline
CAC quantity. CAC progression was defined as the log annualchange in CAC area, calculated as follows: log [(difference betweenfollow-up and baseline CAC area�1)/time (in years) betweenbaseline and follow-up examinations].20 If the difference betweenfollow-up and baseline CAC area was �0, the difference was set to0 (to avoid taking the log of a negative number).
Heritability estimates (h2) were calculated for log baseline CACquantity and CAC progression with the use of a variance componentsapproach described previously8 and implemented in SOLAR.22 Fortrait y, the value of y for individual i is modeled as:
(1) yi�����jXij�gi�ei
where � is the mean of y, Xij is the j-th covariate with associatedregression coefficient �j, gi is an additive genetic effect normallydistributed with mean 0 and variance �g
2, and ei is a random residualeffect normally distributed with mean 0 and variance �e
2. It isassumed that �g
2��e2�1. Any nonadditive genetic and unmeasured
nongenetic effects (as well as measurement and random error) areincorporated into ei. Heritability is estimated by �g
2. Likelihood ratiotests are used to assess significance of a parameter of interest bycomparing the log-likelihood of the model in which the parameter isestimated with that of the model in which the parameter is fixed to0.23
Heritability estimates for CAC progression were calculated as fol-lows: (1) unadjusted; (2) adjusted for age and sex; (3) adjusted for age,sex, and the best subset of the following baseline CHD risk factors: bodymass index, waist-to-hip ratio, triglycerides, LDL-C, HDL-C, fastingglucose level, SBP, DBP, presence of diabetes, presence of hyperten-sion, college education (ie, any education beyond high school), smokinghistory, log (pack-years smoking�1), and family history of CHD; and(4) adjusted for age, sex, log baseline CAC quantity, and the best subsetof the CHD risk factors listed in step 3. Heritability estimates for logbaseline CAC quantity were calculated similarly (steps 1 to 3). Covari-ates were chosen for similarity to previous h2 studies.8 All 2-wayinteraction terms between covariates significantly associated with eitheroutcome were evaluated. The estimates of h2 and covariate varianceobtained were used to estimate the percentage of total variationexplained by genetic factors: [(1�proportion of variance explained bycovariates)�h2]�100.
The genetic correlation (�g) between log baseline CAC quantity(trait 1) and CAC progression (trait 2) was estimated to assesspleiotropic genetic effects with the use of maximum-likelihoodestimation in SOLAR.24–26 The phenotypic correlation between the2 traits is derived from the �g, the environmental correlation (�e),and the heritabilities of the 2 traits, as follows:
(2) �h12h2
2��g��1�h12��1�h2
2��e
All hypothesis tests were performed with the use of likelihood-ratio test statistics.23 The hypothesis tests of interest are whether �g
is different from 0, whether �g is different from 1, and whether �e
is different from 0. If �g is different from 0, the estimate of �g, itsSE, and test of the hypothesis �g�1 determine the magnitude of theshared genetic effects (ie, pleiotropy).27,28 If the hypothesis that�g�1 is not rejected, then all genes influencing 1 trait are assumedto also influence the other trait. Rejection of the null hypothesis that�e�0 indicates shared environmental components. Covariates sig-nificantly associated with both traits were used to adjust both traits,whereas covariates only associated with a single trait were used toadjust for that trait alone. Covariates for CAC progression werechosen from the model in which log baseline CAC quantity was notincluded as a covariate.
The authors had full access to and take responsibility for theintegrity of the data. All authors have read and agree to themanuscript as written.
ResultsMean baseline age of women was 56.4 years (range, 36.0 to82.1 years), and that of men was 54.7 years (range, 35.7 to79.0 years) (Table 1). Mean time between examinations was
26 Circulation July 3, 2007
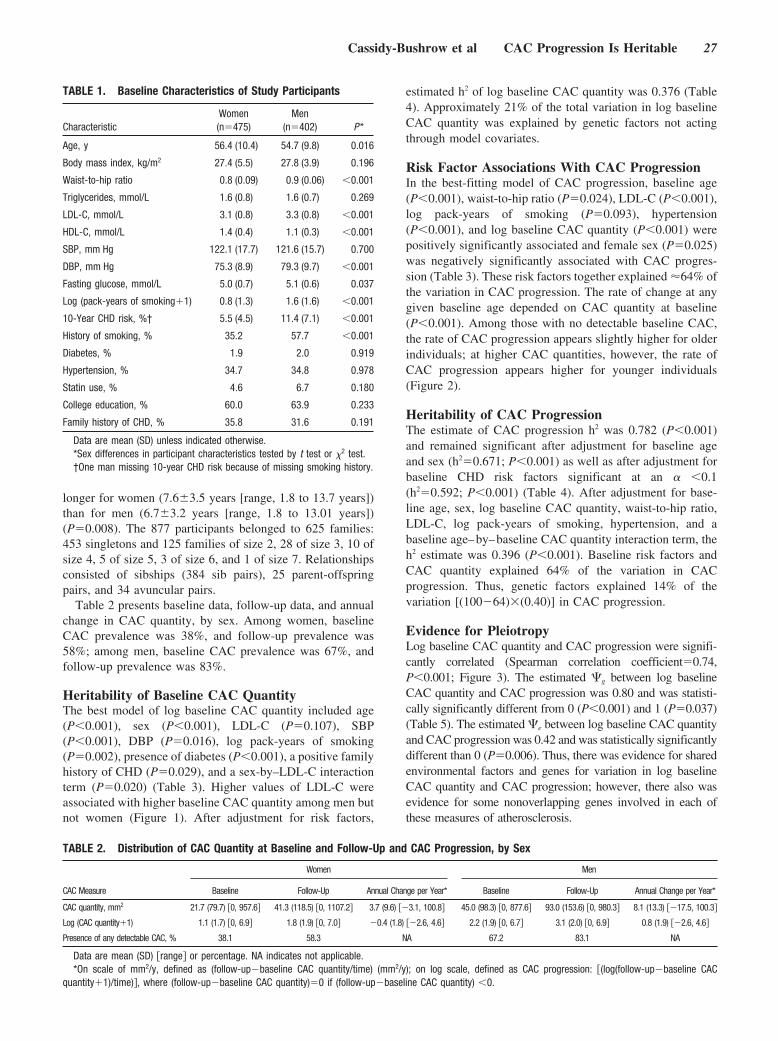
longer for women (7.6�3.5 years [range, 1.8 to 13.7 years])than for men (6.7�3.2 years [range, 1.8 to 13.01 years])(P�0.008). The 877 participants belonged to 625 families:453 singletons and 125 families of size 2, 28 of size 3, 10 ofsize 4, 5 of size 5, 3 of size 6, and 1 of size 7. Relationshipsconsisted of sibships (384 sib pairs), 25 parent-offspringpairs, and 34 avuncular pairs.
Table 2 presents baseline data, follow-up data, and annualchange in CAC quantity, by sex. Among women, baselineCAC prevalence was 38%, and follow-up prevalence was58%; among men, baseline CAC prevalence was 67%, andfollow-up prevalence was 83%.
Heritability of Baseline CAC QuantityThe best model of log baseline CAC quantity included age(P�0.001), sex (P�0.001), LDL-C (P�0.107), SBP(P�0.001), DBP (P�0.016), log pack-years of smoking(P�0.002), presence of diabetes (P�0.001), a positive familyhistory of CHD (P�0.029), and a sex-by–LDL-C interactionterm (P�0.020) (Table 3). Higher values of LDL-C wereassociated with higher baseline CAC quantity among men butnot women (Figure 1). After adjustment for risk factors,
estimated h2 of log baseline CAC quantity was 0.376 (Table4). Approximately 21% of the total variation in log baselineCAC quantity was explained by genetic factors not actingthrough model covariates.
Risk Factor Associations With CAC ProgressionIn the best-fitting model of CAC progression, baseline age(P�0.001), waist-to-hip ratio (P�0.024), LDL-C (P�0.001),log pack-years of smoking (P�0.093), hypertension(P�0.001), and log baseline CAC quantity (P�0.001) werepositively significantly associated and female sex (P�0.025)was negatively significantly associated with CAC progres-sion (Table 3). These risk factors together explained �64% ofthe variation in CAC progression. The rate of change at anygiven baseline age depended on CAC quantity at baseline(P�0.001). Among those with no detectable baseline CAC,the rate of CAC progression appears slightly higher for olderindividuals; at higher CAC quantities, however, the rate ofCAC progression appears higher for younger individuals(Figure 2).
Heritability of CAC ProgressionThe estimate of CAC progression h2 was 0.782 (P�0.001)and remained significant after adjustment for baseline ageand sex (h2�0.671; P�0.001) as well as after adjustment forbaseline CHD risk factors significant at an � �0.1(h2�0.592; P�0.001) (Table 4). After adjustment for base-line age, sex, log baseline CAC quantity, waist-to-hip ratio,LDL-C, log pack-years of smoking, hypertension, and abaseline age–by–baseline CAC quantity interaction term, theh2 estimate was 0.396 (P�0.001). Baseline risk factors andCAC quantity explained 64% of the variation in CACprogression. Thus, genetic factors explained 14% of thevariation [(100�64)�(0.40)] in CAC progression.
Evidence for PleiotropyLog baseline CAC quantity and CAC progression were signifi-cantly correlated (Spearman correlation coefficient�0.74,P�0.001; Figure 3). The estimated �g between log baselineCAC quantity and CAC progression was 0.80 and was statisti-cally significantly different from 0 (P�0.001) and 1 (P�0.037)(Table 5). The estimated �e between log baseline CAC quantityand CAC progression was 0.42 and was statistically significantlydifferent than 0 (P�0.006). Thus, there was evidence for sharedenvironmental factors and genes for variation in log baselineCAC quantity and CAC progression; however, there also wasevidence for some nonoverlapping genes involved in each ofthese measures of atherosclerosis.
TABLE 2. Distribution of CAC Quantity at Baseline and Follow-Up and CAC Progression, by Sex
Women Men
CAC Measure Baseline Follow-Up Annual Change per Year* Baseline Follow-Up Annual Change per Year*
CAC quantity, mm2 21.7 (79.7) 0, 957.6 41.3 (118.5) 0, 1107.2 3.7 (9.6) �3.1, 100.8 45.0 (98.3) 0, 877.6 93.0 (153.6) 0, 980.3 8.1 (13.3) �17.5, 100.3
Log (CAC quantity�1) 1.1 (1.7) 0, 6.9 1.8 (1.9) 0, 7.0 �0.4 (1.8) �2.6, 4.6 2.2 (1.9) 0, 6.7 3.1 (2.0) 0, 6.9 0.8 (1.9) �2.6, 4.6
Presence of any detectable CAC, % 38.1 58.3 NA 67.2 83.1 NA
Data are mean (SD) range or percentage. NA indicates not applicable.*On scale of mm2/y, defined as (follow-up�baseline CAC quantity/time) (mm2/y); on log scale, defined as CAC progression: (log(follow-up�baseline CAC
quantity�1)/time), where (follow-up�baseline CAC quantity)�0 if (follow-up�baseline CAC quantity) �0.
TABLE 1. Baseline Characteristics of Study Participants
CharacteristicWomen(n�475)
Men(n�402) P*
Age, y 56.4 (10.4) 54.7 (9.8) 0.016
Body mass index, kg/m2 27.4 (5.5) 27.8 (3.9) 0.196
Waist-to-hip ratio 0.8 (0.09) 0.9 (0.06) �0.001
Triglycerides, mmol/L 1.6 (0.8) 1.6 (0.7) 0.269
LDL-C, mmol/L 3.1 (0.8) 3.3 (0.8) �0.001
HDL-C, mmol/L 1.4 (0.4) 1.1 (0.3) �0.001
SBP, mm Hg 122.1 (17.7) 121.6 (15.7) 0.700
DBP, mm Hg 75.3 (8.9) 79.3 (9.7) �0.001
Fasting glucose, mmol/L 5.0 (0.7) 5.1 (0.6) 0.037
Log (pack-years of smoking�1) 0.8 (1.3) 1.6 (1.6) �0.001
10-Year CHD risk, %† 5.5 (4.5) 11.4 (7.1) �0.001
History of smoking, % 35.2 57.7 �0.001
Diabetes, % 1.9 2.0 0.919
Hypertension, % 34.7 34.8 0.978
Statin use, % 4.6 6.7 0.180
College education, % 60.0 63.9 0.233
Family history of CHD, % 35.8 31.6 0.191
Data are mean (SD) unless indicated otherwise.*Sex differences in participant characteristics tested by t test or �2 test.†One man missing 10-year CHD risk because of missing smoking history.
Cassidy-Bushrow et al CAC Progression Is Heritable 27
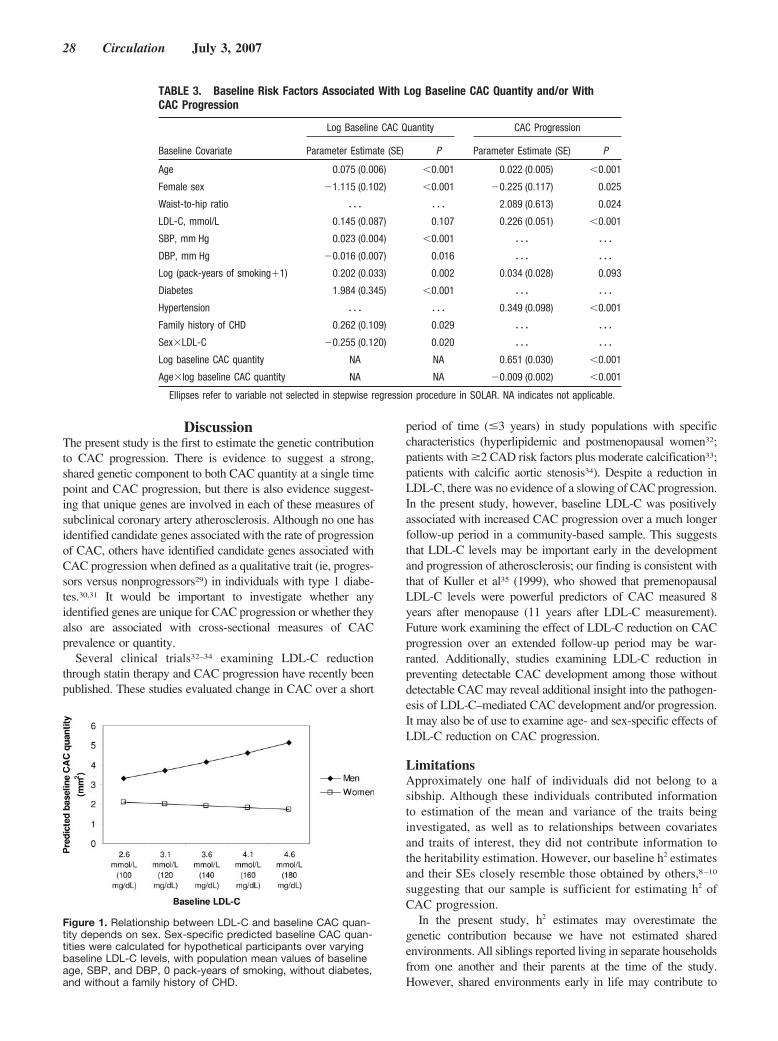
DiscussionThe present study is the first to estimate the genetic contributionto CAC progression. There is evidence to suggest a strong,shared genetic component to both CAC quantity at a single timepoint and CAC progression, but there is also evidence suggest-ing that unique genes are involved in each of these measures ofsubclinical coronary artery atherosclerosis. Although no one hasidentified candidate genes associated with the rate of progressionof CAC, others have identified candidate genes associated withCAC progression when defined as a qualitative trait (ie, progres-sors versus nonprogressors29) in individuals with type 1 diabe-tes.30,31 It would be important to investigate whether anyidentified genes are unique for CAC progression or whether theyalso are associated with cross-sectional measures of CACprevalence or quantity.
Several clinical trials32–34 examining LDL-C reductionthrough statin therapy and CAC progression have recently beenpublished. These studies evaluated change in CAC over a short
period of time (�3 years) in study populations with specificcharacteristics (hyperlipidemic and postmenopausal women32;patients with �2 CAD risk factors plus moderate calcification33;patients with calcific aortic stenosis34). Despite a reduction inLDL-C, there was no evidence of a slowing of CAC progression.In the present study, however, baseline LDL-C was positivelyassociated with increased CAC progression over a much longerfollow-up period in a community-based sample. This suggeststhat LDL-C levels may be important early in the developmentand progression of atherosclerosis; our finding is consistent withthat of Kuller et al35 (1999), who showed that premenopausalLDL-C levels were powerful predictors of CAC measured 8years after menopause (11 years after LDL-C measurement).Future work examining the effect of LDL-C reduction on CACprogression over an extended follow-up period may be war-ranted. Additionally, studies examining LDL-C reduction inpreventing detectable CAC development among those withoutdetectable CAC may reveal additional insight into the pathogen-esis of LDL-C–mediated CAC development and/or progression.It may also be of use to examine age- and sex-specific effects ofLDL-C reduction on CAC progression.
LimitationsApproximately one half of individuals did not belong to asibship. Although these individuals contributed informationto estimation of the mean and variance of the traits beinginvestigated, as well as to relationships between covariatesand traits of interest, they did not contribute information tothe heritability estimation. However, our baseline h2 estimatesand their SEs closely resemble those obtained by others,8–10
suggesting that our sample is sufficient for estimating h2 ofCAC progression.
In the present study, h2 estimates may overestimate thegenetic contribution because we have not estimated sharedenvironments. All siblings reported living in separate householdsfrom one another and their parents at the time of the study.However, shared environments early in life may contribute to
TABLE 3. Baseline Risk Factors Associated With Log Baseline CAC Quantity and/or WithCAC Progression
Log Baseline CAC Quantity CAC Progression
Baseline Covariate Parameter Estimate (SE) P Parameter Estimate (SE) P
Age 0.075 (0.006) �0.001 0.022 (0.005) �0.001
Female sex �1.115 (0.102) �0.001 �0.225 (0.117) 0.025
Waist-to-hip ratio � � � � � � 2.089 (0.613) 0.024
LDL-C, mmol/L 0.145 (0.087) 0.107 0.226 (0.051) �0.001
SBP, mm Hg 0.023 (0.004) �0.001 � � � � � �
DBP, mm Hg �0.016 (0.007) 0.016 � � � � � �
Log (pack-years of smoking�1) 0.202 (0.033) 0.002 0.034 (0.028) 0.093
Diabetes 1.984 (0.345) �0.001 � � � � � �
Hypertension � � � � � � 0.349 (0.098) �0.001
Family history of CHD 0.262 (0.109) 0.029 � � � � � �
Sex�LDL-C �0.255 (0.120) 0.020 � � � � � �
Log baseline CAC quantity NA NA 0.651 (0.030) �0.001
Age�log baseline CAC quantity NA NA �0.009 (0.002) �0.001
Ellipses refer to variable not selected in stepwise regression procedure in SOLAR. NA indicates not applicable.
Figure 1. Relationship between LDL-C and baseline CAC quan-tity depends on sex. Sex-specific predicted baseline CAC quan-tities were calculated for hypothetical participants over varyingbaseline LDL-C levels, with population mean values of baselineage, SBP, and DBP, 0 pack-years of smoking, without diabetes,and without a family history of CHD.
28 Circulation July 3, 2007
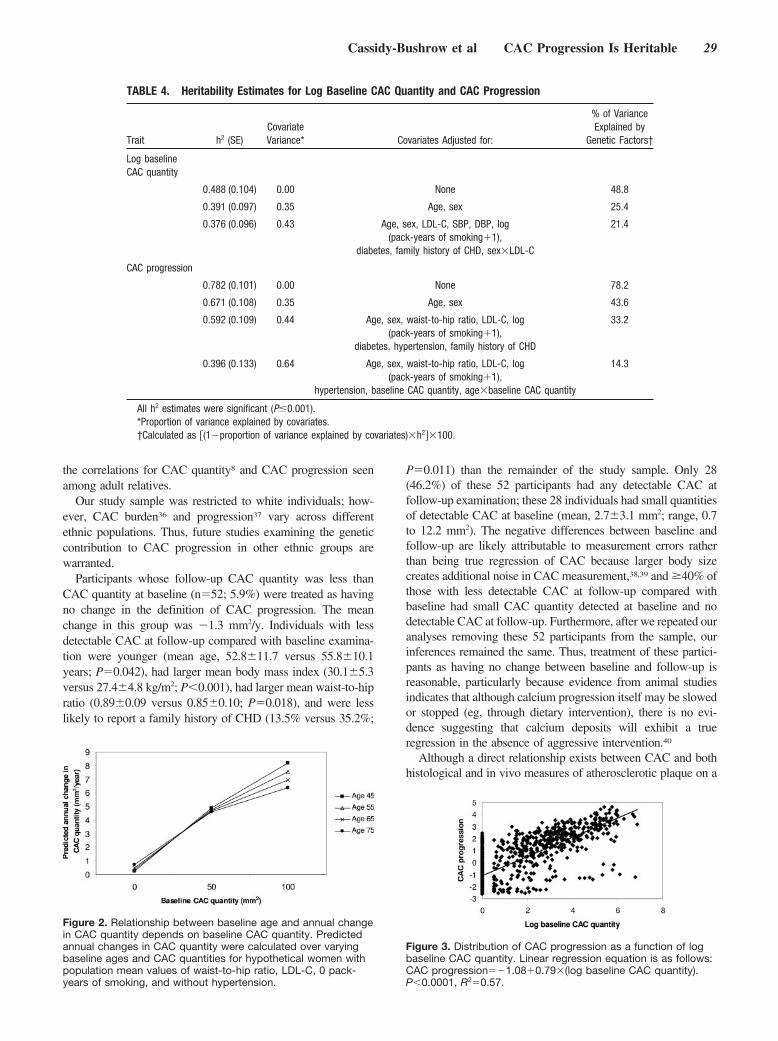
the correlations for CAC quantity8 and CAC progression seenamong adult relatives.
Our study sample was restricted to white individuals; how-ever, CAC burden36 and progression37 vary across differentethnic populations. Thus, future studies examining the geneticcontribution to CAC progression in other ethnic groups arewarranted.
Participants whose follow-up CAC quantity was less thanCAC quantity at baseline (n�52; 5.9%) were treated as havingno change in the definition of CAC progression. The meanchange in this group was �1.3 mm2/y. Individuals with lessdetectable CAC at follow-up compared with baseline examina-tion were younger (mean age, 52.8�11.7 versus 55.8�10.1years; P�0.042), had larger mean body mass index (30.1�5.3versus 27.4�4.8 kg/m2; P�0.001), had larger mean waist-to-hipratio (0.89�0.09 versus 0.85�0.10; P�0.018), and were lesslikely to report a family history of CHD (13.5% versus 35.2%;
P�0.011) than the remainder of the study sample. Only 28(46.2%) of these 52 participants had any detectable CAC atfollow-up examination; these 28 individuals had small quantitiesof detectable CAC at baseline (mean, 2.7�3.1 mm2; range, 0.7to 12.2 mm2). The negative differences between baseline andfollow-up are likely attributable to measurement errors ratherthan being true regression of CAC because larger body sizecreates additional noise in CAC measurement,38,39 and �40% ofthose with less detectable CAC at follow-up compared withbaseline had small CAC quantity detected at baseline and nodetectable CAC at follow-up. Furthermore, after we repeated ouranalyses removing these 52 participants from the sample, ourinferences remained the same. Thus, treatment of these partici-pants as having no change between baseline and follow-up isreasonable, particularly because evidence from animal studiesindicates that although calcium progression itself may be slowedor stopped (eg, through dietary intervention), there is no evi-dence suggesting that calcium deposits will exhibit a trueregression in the absence of aggressive intervention.40
Although a direct relationship exists between CAC and bothhistological and in vivo measures of atherosclerotic plaque on a
Figure 2. Relationship between baseline age and annual changein CAC quantity depends on baseline CAC quantity. Predictedannual changes in CAC quantity were calculated over varyingbaseline ages and CAC quantities for hypothetical women withpopulation mean values of waist-to-hip ratio, LDL-C, 0 pack-years of smoking, and without hypertension.
Figure 3. Distribution of CAC progression as a function of logbaseline CAC quantity. Linear regression equation is as follows:CAC progression��1.08�0.79�(log baseline CAC quantity).P�0.0001, R2�0.57.
TABLE 4. Heritability Estimates for Log Baseline CAC Quantity and CAC Progression
Trait h2 (SE)CovariateVariance* Covariates Adjusted for:
% of VarianceExplained by
Genetic Factors†
Log baselineCAC quantity
0.488 (0.104) 0.00 None 48.8
0.391 (0.097) 0.35 Age, sex 25.4
0.376 (0.096) 0.43 Age, sex, LDL-C, SBP, DBP, log(pack-years of smoking�1),
diabetes, family history of CHD, sex�LDL-C
21.4
CAC progression
0.782 (0.101) 0.00 None 78.2
0.671 (0.108) 0.35 Age, sex 43.6
0.592 (0.109) 0.44 Age, sex, waist-to-hip ratio, LDL-C, log(pack-years of smoking�1),
diabetes, hypertension, family history of CHD
33.2
0.396 (0.133) 0.64 Age, sex, waist-to-hip ratio, LDL-C, log(pack-years of smoking�1),
hypertension, baseline CAC quantity, age�baseline CAC quantity
14.3
All h2 estimates were significant (P�0.001).*Proportion of variance explained by covariates.†Calculated as (1�proportion of variance explained by covariates)�h2�100.
Cassidy-Bushrow et al CAC Progression Is Heritable 29

heart-by-heart, vessel-by-vessel, and segment-by-segment ba-sis,41–45 absence of detectable CAC with EBCT does notnecessarily indicate an absence of coronary artery atherosclero-sis. This measure likely underestimates total atherosclerosisquantity and progression in some individuals because CACquantity more closely represents calcified plaque burden ratherthan atherosclerosis.
Finally, we restricted our analyses to account for baselinemeasures of risk factors only; however, change in risk factorstatus over time may retard or accelerate CAC progression withunknown effects on estimation of the role of genetic factors.Future work should examine time-varying covariates in CACprogression.
ConclusionBoth individual and familial characteristics (eg, genes) areimportant factors in CAC progression. Importantly, there is agenetic component to CAC progression beyond that captured bybaseline risk factors (including family history of CHD) andbaseline CAC. Baseline risk factors (including family history ofCHD) and baseline CAC may provide useful tools for identify-ing individuals at otherwise low to moderate risk of a CHD eventwho may benefit from serial CAC screening for additional riskstratification and/or primary prevention of disease.
Identification of specific genes associated with increasedCAC progression may provide insights into molecular mecha-nisms of atherosclerosis, identify new targets for therapy, andlead to blood tests for early detection of susceptible individualswho would benefit from early, individualized therapeutic orlifestyle interventions for halting or slowing their CAC progression.
Sources of FundingThis research was supported by grant R01 HL46292 from theNational Institutes of Health, by a General Clinic Research Centergrant from the National Institutes of Health (MO1-RR00585)awarded to Mayo Clinic Rochester, and by National Human GenomeResearch Institute grant T32 HG00040.
DisclosuresNone.
References1. Rosamond W, Flegal K, Friday G, Furie K, Go A, Greenlund K, Haase N,
Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M,Meigs J, Moy C, Nichol G, O’Donnell CJ, Roger V, Rumsfeld J, SorlieP, Steinberger J, Thom T, Wasserthiel-Smoller S, Hong Y. Heart diseaseand stroke statistics—2007 update: a report from the American HeartAssociation Statistics Committee and Stroke Statistics Subcommittee.Circulation. 2007;115:e69–e171.
2. Greenland P, Knoll MD, Stamler J, Neaton JD, Dyer AR, Garside DB,Wilson PW. Major risk factors as antecedents of fatal and nonfatalcoronary heart disease events. JAMA. 2003;290:891–897.
3. Bielak LF, Rumberger JA, Sheedy PF II, Schwartz RS, Peyser PA.Probabilistic model for prediction of angiographically defined obstructivecoronary artery disease using electron beam computed tomographycalcium score strata. Circulation. 2000;102:380–385.
4. Budoff MJ. Atherosclerosis imaging and calcified plaque: coronary arterydisease risk assessment. Prog Cardiovasc Dis. 2003;46:135–148.
5. Raggi P, Callister TQ, Shaw LJ. Progression of coronary artery calciumand risk of first myocardial infarction in patients receiving cholesterol-lowering therapy. Arterioscler Thromb Vasc Biol. 2004;24:1272–1277.
6. Raggi P, Cooil B, Shaw LJ, Aboulhson J, Takasu J, Budoff M, CallisterTQ. Progression of coronary calcium on serial electron beam tomographicscanning is greater in patients with future myocardial infarction.Am J Cardiol. 2003;92:827–829.
7. Nasir K, Michos ED, Rumberger JA, Braunstein JB, Post WS, BudoffMJ, Blumenthal RS. Coronary artery calcification and family history ofpremature coronary heart disease: sibling history is more strongly asso-ciated than parental history. Circulation. 2004;110:2150–2156.
8. Peyser PA, Bielak LF, Chu JS, Turner ST, Ellsworth DL, Boerwinkle E,Sheedy PF II. Heritability of coronary artery calcium quantity measuredby electron beam computed tomography in asymptomatic adults. Circu-lation. 2002;106:304–308.
9. Turner ST, Peyser PA, Kardia SL, Bielak LF, Sheedy PF II, BoerwinkleE, de Andrade M. Genomic loci with pleiotropic effects on coronaryartery calcification. Atherosclerosis. 2006;185:340–346.
10. Wagenknecht LE, Bowden DW, Carr JJ, Langefeld CD, Freedman BI,Rich SS. Familial aggregation of coronary artery calcium in families withtype 2 diabetes. Diabetes. 2001;50:861–866.
11. Greenland P, Bonow RO, Brundage BH, Budoff MJ, Eisenberg MJ,Grundy SM, Lauer MS, Post WS, Raggi P, Redberg RF, Rodgers GP,Shaw LJ, Taylor AJ, Weintraub WS, Harrington RA, Abrams J, AndersonJL, Bates ER, Grines CL, Hlatky MA, Lichtenberg RC, Lindner JR,Pohost GM, Schofield RS, Shubrooks SJ Jr, Stein JH, Tracy CM, VogelRA, Wesley DJ. ACCF/AHA 2007 clinical expert consensus documenton coronary artery calcium scoring by computed tomography in globalcardiovascular risk assessment and in evaluation of patients with chestpain: a report of the American College of Cardiology Foundation ClinicalExpert Consensus Task Force (ACCF/AHA Writing Committee toUpdate the 2000 Expert Consensus Document on Electron BeamComputed Tomography). Circulation. 2007;115:402–426.
12. Turner ST, Weidman WH, Michels VV, Reed TJ, Ormson CL, Fuller T,Sing CF. Distribution of sodium-lithium countertransport and bloodpressure in Caucasians five to eighty-nine years of age. Hypertension.1989;13:378–391.
13. Kottke BA, Moll PP, Michels VV, Weidman WH. Levels of lipids,lipoproteins, and apolipoproteins in a defined population. Mayo ClinProc. 1991;66:1198–1208.
14. Maher JE, Raz JA, Bielak LF, Sheedy PF II, Schwartz RS, Peyser PA.Potential of quantity of coronary artery calcification to identify new riskfactors for asymptomatic atherosclerosis. Am J Epidemiol. 1996;144:943–953.
15. Bielak LF, Sheedy PF II, Peyser PA. Coronary artery calcificationmeasured at electron-beam CT: agreement in dual scan runs and changeover time. Radiology. 2001;218:224–229.
16. Kaufmann RB, Peyser PA, Sheedy PF II, Rumberger JA, Schwartz RS.Quantification of coronary artery calcium by electron beam computedtomography for determination of severity of angiographic coronary arterydisease in younger patients. J Am Coll Cardiol. 1995;25:626–632.
17. Shemesh J, Koren-Morag N, Apter S, Rozenman J, Kirwan BA, ItzchakY, Motro M. Accelerated progression of coronary calcification: four-yearfollow-up in patients with stable coronary artery disease. Radiology.2004;233:201–209.
18. Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concen-tration of low-density lipoprotein cholesterol in plasma, without use of thepreparative ultracentrifuge. Clin Chem. 1972;18:499–502.
19. Anderson KM, Odell PM, Wilson PW, Kannel WB. Cardiovasculardisease risk profiles. Am Heart J. 1991;121:293–298.
20. Cassidy AE, Bielak LF, Zhou Y, Sheedy PF II, Turner ST, Breen JF,Araoz PA, Kullo IJ, Lin X, Peyser PA. Progression of subclinicalcoronary atherosclerosis: does obesity make a difference? Circulation.2005;111:1877–1882.
21. Reed JE, Rumberger JA, Davitt PJ. System for quantitative analysis ofcoronary calcification via electron-beam computed tomography. In:
TABLE 5. Evidence of Pleiotropy Between Log Baseline CACQuantity and CAC Progression
Correlation (�) Estimate (SE) P for ��0 P for ��1
Genetic correlation (�g) 0.80 (0.11) �0.001 0.037
Environmental correlation (�e) 0.42 (0.11) 0.006 NA
Log baseline CAC quantity and CAC progression were both adjusted for age,sex, LDL-C, log (pack-years of smoking�1), diabetes, and family history ofCHD; log baseline CAC quantity for SBP, DBP, and sex�LDL-C; and CACprogression for waist-to-hip ratio and hypertension. NA indicates not applica-ble.
30 Circulation July 3, 2007

Hoffman EA, Acharya RS, eds. Medical Imaging 1994: Physiology andFunction From Multidimensional Images. Proc SPIE. 1994;2168:43–53.
22. Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis ingeneral pedigrees. Am J Hum Genet. 1998;62:1198–1211.
23. Self SA, Liang KY. Asymptotic properties of maximum likelihoodestimates and likelihood ratio tests under nonstandard conditions. J AmStat Assoc. 1987;82:605–610.
24. Lange K, Boehnke M. Extensions to pedigree analysis, IV: covariancecomponents models for multivariate traits. Am J Med Genet. 1983;14:513–524.
25. Boehnke M, Moll PP, Lange K, Weidman WH, Kottke BA. Univariateand bivariate analyses of cholesterol and triglyceride levels in pedigrees.Am J Med Genet. 1986;23:775–792.
26. Hokanson JE, Langefeld CD, Mitchell BD, Lange LA, Goff DC Jr,Haffner SM, Saad MF, Rotter JI. Pleiotropy and heterogeneity in theexpression of atherogenic lipoproteins: the IRAS Family Study. HumHered. 2003;55:46–50.
27. Kent JW Jr., Comuzzie AG, Mahaney MC, Almasy L, Rainwater DL,VandeBerg JL, MacCluer JW, Blangero J. Intercellular adhesionmolecule-1 concentration is genetically correlated with insulin resistance,obesity, and HDL concentration in Mexican Americans. Diabetes. 2004;53:2691–2695.
28. Comuzzie AG, Rainwater DL, Blangero J, Mahaney MC, VandeBerg JL,MacCluer JW. Shared and unique genetic effects among seven HDLphenotypes. Arterioscler Thromb Vasc Biol. 1997;17:859–864.
29. Hokanson JE, MacKenzie T, Kinney G, Snell-Bergeon JK, Dabelea D,Ehrlich J, Eckel RH, Rewers M. Evaluating changes in coronary arterycalcium: an analytic method that accounts for interscan variability. Am JRoentgenol. 2004;182:1327–1332.
30. Kretowski A, Hokanson JE, McFann K, Kinney GL, Snell-Bergeon JK,Maahs DM, Wadwa RP, Eckel RH, Ogden LG, Garg SK, Li J, Cheng S,Erlich HA, Rewers M. The apolipoprotein A-IV Gln360His poly-morphism predicts progression of coronary artery calcification in patientswith type 1 diabetes. Diabetologia. 2006;49:1946–1954.
31. Kretowski A, McFann K, Hokanson JE, Maahs D, Kinney G, Snell-Bergeon JK, Wadwa RP, Eckel RH, Ogden L, Garg S, Li J, Cheng S,Erlich HA, Rewers M. Polymorphisms of the renin-angiotensin systemgenes predict progression of subclinical coronary atherosclerosis.Diabetes. 2007;56:863–871.
32. Raggi P, Davidson M, Callister TQ, Welty FK, Bachmann GA, Hecht H,Rumberger JA. Aggressive versus moderate lipid-lowering therapy in hypercho-lesterolemic postmenopausal women: Beyond Endorsed Lipid Lowering withEBT Scanning (BELLES). Circulation. 2005;112:563–571.
33. Schmermund A, Achenbach S, Budde T, Buziashvili Y, Forster A,Friedrich G, Henein M, Kerkhoff G, Knollmann F, Kukharchuk V, LahiriA, Leischik R, Moshage W, Schartl M, Siffert W, Steinhagen-Thiessen E,Sinitsyn V, Vogt A, Wiedeking B, Erbel R. Effect of intensive versus
standard lipid-lowering treatment with atorvastatin on the progression ofcalcified coronary atherosclerosis over 12 months: a multicenter, ran-domized, double-blind trial. Circulation. 2006;113:427–437.
34. Houslay ES, Cowell SJ, Prescott RJ, Reid J, Burton J, Northridge DB,Boon NA, Newby DE. Progressive coronary calcification despiteintensive lipid-lowering treatment: a randomised controlled trial. Heart.2006;92:1207–1212.
35. Kuller LH, Matthews KA, Sutton-Tyrrell K, Edmundowicz D, BunkerCH. Coronary and aortic calcification among women 8 years aftermenopause and their premenopausal risk factors: the Healthy WomenStudy. Arterioscler Thromb Vasc Biol. 1999;19:2189–2198.
36. McClelland RL, Chung H, Detrano R, Post W, Kronmal RA. Distribution ofcoronary artery calcium by race, gender, and age: results from the Multi-EthnicStudy of Atherosclerosis (MESA). Circulation. 2006;113:30–37.
37. Kawakubo M, LaBree L, Xiang M, Doherty TM, Wong ND, Azen S,Detrano R. Race-ethnic differences in the extent, prevalence, and pro-gression of coronary calcium. Ethn Dis. 2005;15:198–204.
38. Hall EF. Use of EBCT in epidemiological studies: the effect of noiseand body size on coronary calcium scores. Int J Epidemiol. 2005;34:179 –180.
39. Wang TJ, Larson MG, Levy D, Benjamin EJ, Kupka MJ, Manning WJ, ClouseME, D’Agostino RB, Wilson PW, O’Donnell CJ. C-reactive protein is associatedwith subclinical epicardial coronary calcification in men and women: the Fra-mingham Heart Study. Circulation. 2002;106:1189–1191.
40. Stary HC. The development of calcium deposits in atherosclerotic lesionsand their persistence after lipid regression. Am J Cardiol. 2001;88:16E–19E.
41. Baumgart D, Schmermund A, Goerge G, Haude M, Ge J, Adamzik M,Sehnert C, Altmaier K, Groenemeyer D, Seibel R, Erbel R. Comparisonof electron beam computed tomography with intracoronary ultrasoundand coronary angiography for detection of coronary atherosclerosis. J AmColl Cardiol. 1997;30:57–64.
42. Kajinami K, Seki H, Takekoshi N, Mabuchi H. Coronary calcificationand coronary atherosclerosis: site by site comparative morphologic studyof electron beam computed tomography and coronary angiography. J AmColl Cardiol. 1997;29:1549–1556.
43. Mautner GC, Mautner SL, Froehlich J, Feuerstein IM, Proschan MA,Roberts WC, Doppman JL. Coronary artery calcification: assessmentwith electron beam CT and histomorphometric correlation. Radiology.1994;192:619–623.
44. Rumberger JA, Simons DB, Fitzpatrick LA, Sheedy PF II, Schwartz RS.Coronary artery calcium area by electron-beam computed tomographyand coronary atherosclerotic plaque area: a histopathologic correlativestudy. Circulation. 1995;92:2157–2162.
45. Schmermund A, Rumberger JA, Colter JF, Sheedy PF II, Schwartz RS.Angiographic correlates of “spotty” coronary artery calcium detected byelectron-beam computed tomography in patients with normal or near-normal coronary angiograms. Am J Cardiol. 1998;82:508–511.
CLINICAL PERSPECTIVENoninvasively measured progression of quantity of coronary artery calcification (CAC) provides independent information, inaddition to traditional coronary heart disease risk factors, for prediction of risk of future coronary events. Little is known aboutfactors that influence progression of CAC quantity in a community-based sample of asymptomatic adults. CAC progression over�7 years was influenced by the CAC quantity at the baseline examination as well as older age, male sex, and other traditionalcoronary heart disease risk factors (presence of hypertension, higher low-density lipoprotein cholesterol levels, higherwaist-to-hip ratio, family history of coronary heart disease, and smoking more cigarettes). Importantly, there was evidence fora genetic component unique to CAC progression beyond genes for baseline risk factors and baseline CAC quantity. Identificationof specific genes associated with increased CAC progression may provide insights into molecular mechanisms of coronaryatherosclerosis, identify new targets for therapy, and lead to blood tests for early detection of susceptible individuals who wouldbenefit from early, individualized therapeutic or lifestyle interventions for halting or slowing their CAC progression. This studyidentified measurable factors at a baseline examination that can be used immediately to identify asymptomatic adults likely tohave faster progression of subclinical coronary atherosclerosis.
Cassidy-Bushrow et al CAC Progression Is Heritable 31

Association of Carotid Artery Intima-Media Thickness,Plaques, and C-Reactive Protein With Future
Cardiovascular Disease and All-Cause MortalityThe Cardiovascular Health Study
Jie J. Cao, MD, MPH; Alice M. Arnold, PhD; Teri A. Manolio, MD, PhD;Joseph F. Polak, MD, MPH; Bruce M. Psaty, MD, PhD; Calvin H. Hirsch, MD;
Lewis H. Kuller, MD, PhD; Mary Cushman, MD, MSc
Background—Carotid atherosclerosis, measured as carotid intima-media thickness or as characteristics of plaques, hasbeen linked to cardiovascular disease (CVD) and to C-reactive protein (CRP) levels. We investigated the relationshipbetween carotid atherosclerosis and CRP and their joint roles in CVD prediction.
Methods and Results—Of 5888 participants in the Cardiovascular Health Study, an observational study of adults aged �65years, 5020 without baseline CVD were included in the analysis. They were followed up for as long as 12 years for CVDincidence and all-cause mortality after baseline ultrasound and CRP measurement. When CRP was elevated (�3 mg/L)among those with detectable atherosclerosis on ultrasound, there was a 72% (95% CI, 1.46 to 2.01) increased risk forCVD death and a 52% (95% CI, 1.37 to 1.68) increased risk for all-cause mortality. Elevated CRP in the absence ofatherosclerosis did not increase CVD or all-cause mortality risk. The proportion of excess risk attributable to theinteraction of high CRP and atherosclerosis was 54% for CVD death and 79% for all-cause mortality. Addition of CRPor carotid atherosclerosis to conventional risk factors modestly increased in the ability to predict CVD, as measured bythe c statistic.
Conclusions—In older adults, elevated CRP was associated with increased risk for CVD and all-cause mortality only inthose with detectable atherosclerosis based on carotid ultrasound. Despite the significant associations of CRP and carotidatherosclerosis with CVD, these measures modestly improve the prediction of CVD outcomes after one accounts for theconventional risk factors. (Circulation. 2007;116:32-38.)
Key Words: aging � arteriosclerosis � atherosclerosis � cardiovascular diseases � carotid arteries � inflammation
Both carotid intima-media thickness (IMT) and plaquesare measures of carotid atherosclerosis. Carotid IMT
has been linked to many cardiovascular outcomes, includ-ing cerebral and coronary events.1,2 Characteristics ofcarotid plaque have been associated with stroke risk3–5 andcoronary events6 in prospective studies. With the growinginterest in cardiovascular disease (CVD) risk stratificationby combining vascular imaging with conventional riskfactors, it is essential to understand the relationship be-tween carotid IMT and plaque and their independent andcombined contribution to the risk of coronary as wellcerebrovascular events.
In addition to ultrasonographic measures of atherosclero-sis, C-reactive protein (CRP) has been shown to be a riskfactor for CVD.7–9 Although higher CRP is associated with
Editorial p 3Clinical Perspective p 38
atherosclerosis measures such as higher carotid IMT10,11 andcomplex plaque,12,13 we have shown that the association ofCRP with stroke is more apparent in the presence of a highercarotid IMT.10 Whether the association of CRP with CVDrisk is modified by the presence of carotid atherosclerosis hasnot been explored fully.
In the present study, we evaluated the hypothesis that CRPis less predictive of CVD outcomes in the absence ofatherosclerosis by investigating the associations of carotidIMT, carotid plaque, and CRP, alone and in combination,with incident myocardial infarction, stroke, CVD death, andall-cause mortality. We also examined the roles of CRP andcarotid atherosclerosis in CVD prediction.
Received June 22, 2006; accepted April 9, 2007.From the National Heart, Lung, and Blood Institute (J.J.C.), and the National Human Genome Research Institute (T.A.M.), National Institutes of Health,
Bethesda, Md; University of Washington, Seattle (A.M.A., B.M.P.); University of California at Davis (C.H.H.); New England Medical Center, TuftsUniversity, Boston, Mass (J.F.P.); University of Pittsburgh, Pittsburgh, Pa (L.H.K.); and University of Vermont, Burlington (M.C.).
Correspondence to Jie J. Cao, MD, MPH, National Heart, Lung, and Blood Institute, National Institutes of Health, 10 Center Dr, MSC 1061, Bldg 10,Room B1D-416, Bethesda, MD 20892. E-mail [email protected]
© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.106.645606
32
Epidemiology

MethodsWe studied participants of the Cardiovascular Health Study (CHS),a population-based, prospective study of men and women aged �65years. Between 1989 and 1990, 5201 participants were enrolled fromMedicare eligibility lists in 4 counties: Forsyth County, NorthCarolina; Washington County, Maryland; Sacramento County, Cal-ifornia; and Allegheny County, Pennsylvania. To increase theirrepresentation, a second cohort of 687 black participants wasenrolled between 1992 and 1993 with the use of similar methods.Details of the study design have been published.14,15 The study wasapproved by the institutional review boards at each participatingcenter. All participants gave informed consent.
All participants underwent baseline clinical examinations, whichincluded medical history, physical examination, and carotid ultra-sound. Blood was drawn in the morning after an overnight fast.Samples were promptly centrifuged at 3000g for 10 minutes at 4°C.Aliquots of plasma were stored in a central laboratory at �70°C.CRP was measured in all stored baseline plasma samples by ahigh-sensitivity immunoassay, with an interassay coefficient ofvariation of 6.25%.16 The diagnosis of diabetes mellitus was madefollowing American Diabetes Association criteria as fasting glucose�126 mg/dL or use of insulin or oral glucose-lowering agents.Impaired fasting glucose was defined as fasting glucose �110 and�126 mg/dL.
The carotid arteries were evaluated at baseline with high-resolution B-mode ultrasonography (model SSA-270A; ToshibaAmerica Medical Systems, Tustin, Calif). One longitudinal image ofthe common carotid artery and 3 longitudinal images of the internalcarotid artery were acquired. The maximal IMT of the commoncarotid artery and of the internal carotid artery was defined as themean of the maximal IMT of the near and far walls on both the leftand right sides. Focal plaques, when present, were included in themaximum IMT measurement. Carotid IMT was defined as a com-posite measure that combined the maximum common and internalcarotid wall thickness of the left and right carotid arteries afterstandardization (subtraction of the mean and division by the standarddeviation).17 The ultrasound reading center located in Boston, Mass,was responsible for developing standardized protocols for bothscanning and interpretation of carotid sonographic images. Theultrasound protocol, including measurement and reading methods,has been published.18 The interreader variability defined by Spear-man correlation coefficients on maximum wall thickness of thecommon carotid artery was 0.91 and of the internal carotid arterywas 0.81.18 As for the detection of any carotid lesions, including thewall thickness and plaque, the � statistics for intrareader andinterreader agreement were 0.69 and 0.58 for the common carotidartery and 0.73 and 0.65 for the internal carotid artery, respectively.19
Carotid plaque, defined by the appearance of the largest focallesion, was classified by surface characteristics, echogenicity, andtexture. Surface characteristics were classified as smooth, mildlyirregular (height variations of �0.4 mm), markedly irregular (heightvariations of �0.4 mm), and ulcerated (a discrete depression of�2 mm in width extended into the media). Lesion echogenicity wascharacterized as hypoechoic, isoechoic, hyperechoic, or calcified.Lesion texture was classified as homogeneous or heterogeneous. Incase of multiple focal lesions, the largest lesion on each side wasmeasured.20 Participants were then classified as having no plaque,intermediate-risk plaque, and high-risk plaque. Those with no plaquewere defined as having a smooth intimal surface with no focalthickening. High-risk plaque was defined as presence of markedlyirregular or ulcerated surface or hypodense or heterogeneous plaquesthat occupied �50% of the total plaque volume, those featuresreportedly associated with clinical CVD.3–5,20,21 The remainingplaques, including hyperdense, calcified, or homogeneous plaques orthose with mildly irregular surface, were defined as intermediaterisk. When �1 type of plaque was detected in an individual, theplaque risk was determined by the more severe type. In someanalyses, we grouped carotid findings into binary variables: detect-able and minimal atherosclerosis. Detectable atherosclerosis wasdefined as present for participants in the upper 2 tertiles of carotidwall thickness or in the intermediate- or high-risk plaque groups.
Minimal atherosclerosis was defined as having the lowest tertile ofIMT and no plaque.
The methods of ascertainment and classification of incident strokeand myocardial infarction have been reported.22 Participants wereexamined annually at each clinical site. In addition, telephoneinterviews were alternated with clinic visits so that contacts wereevery 6 months. Follow-up was complete through June 30, 2001.Potential vascular events were validated through medical recordreview by committees. Myocardial infarction and stroke includedincident fatal and nonfatal events. Composite CVD was defined toinclude any incident myocardial infarction, stroke, or CVD death.
Of the 5888 CHS participants, 868 were excluded from analysisbecause of prebaseline myocardial infarction or stroke (n�765),missing CRP value (n�72), or missing carotid ultrasound (n�31).
The authors had full access to and take full responsibility for theintegrity of the data. All authors have read and agree to themanuscript as written.
Statistical AnalysisAnalyses were done with the use of SPSS for Windows, version11.0.1 (SPSS, Inc, Chicago, Ill) and STATA, version 9.2 (CollegeStation, Tex). Incidence rates of CVD were calculated by dividingthe total number of events by the total person-years at risk over thefollow-up time within groups defined by the carotid IMT tertile,plaque risk group, and CRP level. Pearson correlation coefficientswere computed to assess the linear relationships among carotid IMT,CRP, and plaque. CRP was log-transformed when modeled contin-uously. Hazard ratios (HRs) from multivariable Cox proportionalhazards models were used to estimate the relative risks (RRs)associated with high CRP, carotid IMT tertile, and plaque charac-teristics for CVD outcomes and all-cause mortality. The proportionalhazards assumption was assessed for the 3 measures of interest(IMT, plaque risk group, and high CRP) by testing each with aninteraction for time in a Cox model. No significant interactions withtime were found. In addition, we examined Kaplan-Meier plotsvisually to look for inconsistent effects over time. Participants whodied or were lost to follow-up before the event of interest or June 30,2001, were censored at the time of death or last follow-up. Multivari-able models were adjusted for age and sex and then further adjustedfor race, systolic and diastolic blood pressure, use of antihyperten-sive medications, body mass index, smoking (never, former, cur-rent), and amount smoked [ln(pack-years)], high-density lipoproteinand low-density lipoprotein cholesterol, and diabetes (none, im-paired fasting glucose, diabetes). All conventional risk factors weremeasured at the baseline examination and were imputed if missing,as previously described.23 The maximum percentage missing andimputed for any variable was 3.2% for pack-years of smoking. Allother variables were imputed for �0.3% of participants. Categoricalmeasures were modeled with the use of indicator variables for eachlevel compared with the lowest level, and continuous measures weremodeled linearly, per unit. We examined multiplicative and additiveinteractions of CRP with measures of atherosclerosis from ultrasound.
We tested our hypothesis that CRP confers excess risk only in thepresence of atherosclerosis by stratifying on presence of atheroscle-rosis and by computing the relative excess risk, based on an additivemodel,24 using the following 4-level variable: minimal atherosclero-sis and CRP �3 mg/L, detectable atherosclerosis and CRP �3 mg/L,minimal atherosclerosis and CRP �3 mg/L, and detectable athero-sclerosis and CRP �3 mL/L. On the basis of an additive model,Rothman24 defined no interaction if the difference in risk betweenhaving both risk factors and having neither is equal to the sum of thedifferences in risk between each risk factor alone and neither,arguing that this presents the interaction in terms of the number ofexcess cases, which is an appropriate scale for epidemiologicalstudies. Dividing by the risk when both risk factors are absentproduces an equality in terms of RR when there is no relative excessrisk due to CRP and atherosclerosis: RR(both)�1�RR(highCRP)�1�RR(atherosclerosis)�1. We used Cox proportional haz-ards models to estimate the RR due to both risk factors and each onesingly and computed the relative excess risk due to interaction(RERI), defined by RR(both)�RR(high CRP)�RR(atherosclero-
Cao et al Carotid Atherosclerosis, CRP, and CVD 33

sis)�1 for each outcome. Probability values and 95% CIs werecomputed by the delta method.25 The proportion of the diseaserelated to high CRP and atherosclerosis, either singly or in combi-nation, attributable to their interaction was calculated24 as
(1) RERI%�RERI
RR(both)�1�100.
We assessed the ability of carotid atherosclerosis and CRP topredict CVD and all-cause mortality by receiver-operating charac-teristic (ROC) curves and by the c statistic,26 a measure equivalent tothe area under the ROC curve, but allowing for time to eventanalysis. The Hosmer-Lemeshow goodness-of-fit test, which com-pares observed and predicted probabilities,27 was used to assessmodel fit. Because the probabilities were derived from a logisticregression analysis, we used occurrence of events through 8 years offollow-up, which was available for both cohorts.
ResultsAmong 5020 individuals in the analysis, the mean age was72.6�5.5 years, and 60.2% were women. Other demographicdata are shown in Table 1. A total of 593 myocardialinfarctions, 613 strokes, 696 CVD deaths, and 1844 all-causedeaths occurred during a median follow-up time of 11 years(range, 5 days to 12 years).
Correlation of Carotid IMT Category, PlaqueGroups, and CRPThe frequencies of no plaque, intermediate-risk plaques, andhigh-risk plaques were 23%, 21.4%, and 55.6%, respectively.Carotid IMT category was related to plaque risk group, withno plaque being more frequent in persons in the lowest IMTtertile and high-risk plaque more frequent in the highest IMT
tertile (Figure 1). The majority (80.9%) of persons in thehighest IMT tertile had high-risk plaques, with only 1.5%having no plaque. In contrast, among those in the lowest thirdof IMT, 27.4% had high-risk plaque, and 54.3% had noplaques. The Pearson correlation coefficient between plaquerisk group and carotid IMT was 0.51 (P�0.001).
The linear correlation between (ln)CRP level and carotid IMTwas 0.12 and between (ln)CRP and plaque group was 0.08 (bothP�0.001). Within each plaque group, higher CRP was corre-lated with higher IMT (Figure 2). For example, in theintermediate-risk plaque group, the geometric mean CRP rangedfrom 1.58 to 1.85 to 2.20 mg/L across increasing tertiles of IMT.When the comparison was made across the plaque groups, thedifference in CRP level again varied by IMT tertile.
Risk of CVD Related to Carotid IMT, PlaqueGroup, and CRPHRs increased from the lowest to the highest tertile of carotidIMT for every CVD outcome and for all-cause mortality (Table2) after adjustment for the conventional risk factors, carotidplaque groups, and CRP. The highest tertile was associated withan 84% increased risk of composite CVD events, 54% increasedrisk of all-cause mortality, and doubling of risk for CVD death.
Compared with those with no plaque, participants withintermediate- or high-risk plaque were at increased risk of everyCVD outcome and of all-cause mortality after adjustment for
TABLE 1. Descriptive Statistics for the Cohort
Age, y 72.6 (5.5)
Female, % 60
Black race, % 15
Body mass index, kg/m2 26.7 (4.7)
Current smokers, % 12
Pack-years (among ever-smokers)* 27 (12 to 49)
Diabetes status, %
Normal 73
Impaired fasting glucose 12
Diabetic 15
Systolic blood pressure, mm Hg 136 (21.7)
Diastolic blood pressure, mm Hg 70.9 (11.4)
Total cholesterol, mg/dL 212 (38.8)
High-density lipoprotein cholesterol, mg/dL 55.1 (15.8)
Low-density lipoprotein cholesterol, mg/dL 130 (35.7)
CRP, mg/L* 1.86 (0.94 to 3.31)
Common carotid IMT, mm 1.06 (0.21)
Internal carotid IMT, mm 1.40 (0.55)
Plaque risk group, %
Low 23
Intermediate 21
High 56
Values expressed as mean (SD) unless otherwise indicated.*Median (interquartile range).
Figure 1. Distribution of carotid plaque groups in carotid arteryIMT category with more complex plaque characteristics in thethicker carotid wall.
Figure 2. Relation of geometric mean CRP (mg/L) with carotidIMT in tertile and plaque risk group.
34 Circulation July 3, 2007
CVD risk factors, carotid IMT, and CRP (Table 2). Comparedwith those with no plaque, the HRs (95% CI) of composite CVDwere 1.86 (1.55 to 2.23) and 2.09 (1.78 to 2.46) for intermediate-and high-risk plaques, respectively, after adjustment for age andgender only but fell to 1.46 (1.21 to 1.77) and 1.42 (1.20 to 1.70),respectively, after further adjustment for carotid IMT. Additionaladjustment for conventional risk factors and CRP only slightlyattenuated the association, with HRs of 1.41 (1.15 to 1.72) and1.38 (1.14 to 1.67), respectively. Results were similar forindividual CVD outcomes and for all-cause mortality (Table 2).
Elevated CRP (�3 mg/L) was associated with increasedrisk of every outcome compared with CRP �3 mg/L in themultivariable-adjusted model. The magnitude of associationranged from a 26% to a 50% increased risk (Table 2).
Cardiovascular Risk Assessment by DetectableCarotid Atherosclerosis and CRPIn multivariable Cox models stratified by amount of athero-sclerosis (detectable versus minimally detectable), elevatedCRP conferred no increased hazard of composite CVDevents, CVD death, or all-cause mortality in individuals withminimally detectable atherosclerosis, with HRs of 1.05 (95%CI, 0.70 to 1.56), 1.14 (0.60 to 2.14), and 0.87 (0.62 to 1.23),respectively. In contrast, the HRs for elevated CRP weresignificant in those with detectable atherosclerosis: 1.45 (1.29to 1.62) for composite CVD events, 1.72 (1.46 to 2.01) forCVD death, and 1.52 (1.37 to 1.68) for all-cause mortality. Asignificant multiplicative interaction between CRP and pres-ence of atherosclerosis was observed for all-cause mortality.
The cumulative event rates for composite CVD and all-causemortality are shown in Figure 3. The increased rates associatedwith CRP in the presence of atherosclerosis indicated thepossibility of an additive interaction. This finding was consistentin individual CVD outcome (data not shown). For example, theincidence of composite CVD in participants with minimalatherosclerosis and CRP �3 mg/L was 13.7/1000 person-years.Among participants with detectable atherosclerosis and CRP �3mg/L, it was 32.9/1000 person-years (19.2/1000 person-yearshigher than the baseline rate), and with minimal atherosclerosisand CRP �3 mg/L, it was 14.4/1000 person-years (0.7 person-years higher than the baseline rate). If an additive model held, wewould expect the incidence rate for participants with both riskfactors to be 33.6/1000 person-years (the baseline rate of 13.7
plus 19.9 for the combination of atherosclerosis and high CRP).The observed incidence rate for participants with both riskfactors was 46.5/1000 person-years, suggestive of an excessadditive risk due to the interaction of CRP and atherosclerosis.The total excess additive risk due to CRP, atherosclerosis, andtheir interaction was (46.5�13.7)�32.8/1000 person-years, and39% of that excess risk [(32.8�19.9)/32.8] was due to theinteraction of CRP and atherosclerosis. This excess risk rose to
TABLE 2. Hazard Ratios (95% CIs)* of CVD and All-Cause Mortality by CRP, Carotid IMT, and Plaque Groups
Carotid IMT in Tertiles Plaque Groups
Event No.CRP �3 mg/L†
(n�1433)1
(n�1673)2
(n�1674)3
(n�1673)No Plaque(n�1157)
Intermediate Risk(n�1074)
High Risk(n�2789)
Myocardial infarction 595 1.33 (1.11 to 1.60) 1.00 1.41 (1.08 to 1.83) 1.80 (1.37 to 2.38) 1.00 1.41 (1.02 to 1.94) 1.46 (1.08 to 1.98)
Stroke 613 1.26 (1.05 to 1.51) 1.00 1.18 (0.92 to 1.51) 1.77 (1.36 to 2.30) 1.00 1.38 (1.03 to 1.86) 1.31 (0.99 to 1.73)
CVD death 696 1.50 (1.28 to 1.77) 1.00 1.23 (0.95 to 1.59) 2.15 (1.65 to 2.80) 1.00 1.50 (1.10 to 2.05) 1.43 (1.06 to 1.91)
Composite CVD 1904 1.33 (1.18 to 1.50) 1.00 1.28 (1.08 to 1.52) 1.84 (1.54 to 2.20) 1.00 1.41 (1.15 to 1.72) 1.38 (1.14 to 1.67)
All-cause mortality 1844 1.38 (1.25 to 1.53) 1.00 1.16 (1.01 to 1.35) 1.54 (1.32 to 1.79) 1.00 1.28 (1.08 to 1.52) 1.23 (1.04 to 1.44)
*From a model that included age, sex, race, systolic and diastolic blood pressure, use of antihypertensive medications, body mass index, smoking (never, former,current), and amount smoked (in pack-years), high-density lipoprotein and low-density lipoprotein cholesterol, diabetes (none, impaired fasting glucose, diabetes),CRP, plaque risk group, and carotid wall thickness.
†Compared with CRP �3 mg/L.
Figure 3. Kaplan-Meier plots of cumulative cardiovascularevents (A) and all-cause mortality (B) over 12-year follow-upstratified by carotid atherosclerosis and CRP level (low level �3mg/L vs high level �3 mg/L). athero indicates atherosclerosis.
Cao et al Carotid Atherosclerosis, CRP, and CVD 35

50% after adjustment for CVD risk factors (Table 3). Theadjusted excess risk attributable to interaction was 54% for CVDdeath and 79% for all-cause mortality.
CVD Risk Prediction by Carotid Atherosclerosisand CRPCVD risk prediction was compared with the use of c statisticsbased on models with conventional risk factors alone andwith the sequential addition of CRP �3 mg/L, carotid IMT,and carotid plaque (Table 4). c Statistics increased onlymodestly with each additional risk factor. This observationwas consistent for every CVD outcome and for all-causemortality. The final models had excellent fit on the basis ofthe Hosmer-Lemeshow goodness-of-fit test (P�0.28) exceptfor the stroke outcome (P�0.001). As shown in Figure 4,ROC curves for composite CVD (Figure 4A) and all-causemortality (Figure 4B) overlapped for models with conven-tional risk factors alone and with the addition of CRP �3mg/L and the further addition of detectable atherosclerosis.
DiscussionIn the present large cohort study, we demonstrated that elevatedCRP, carotid IMT, and carotid plaque were all correlated withone another, yet each remained a significant risk factor for CVDoutcomes and all-cause mortality in the presence of the others.Furthermore, elevated CRP was associated with increased CVDand all-cause mortality risk only in those with detectableatherosclerosis. Addition of CRP or carotid atherosclerosis to
conventional risk factors resulted in a modest increase in theability to predict CVD, as measured by the c statistic.
Carotid IMT and plaques are both measures of atherosclerosis,perhaps having different attributes or risk associations but stillclosely related.28,29 Common and internal carotid IMT can beviewed as an estimate of atherosclerosis quantity. Sonographiccharacterization of carotid plaque can be considered a measure ofatherosclerosis quality. Both indices are associated with CVD riskfactors and outcomes. High-risk plaques as defined here were morecommon in those with thicker IMT, thus linking atherosclerosisquality with quantity. The high-risk plaque group had a higher riskof CVD outcomes in age- and gender-adjusted analyses than theintermediate-risk plaque group, but this was significantly attenuatedafter carotid IMT was taken into account. The definition of high-riskplaque in the present study was based on features previouslydemonstrated to be associated with stroke risk, and high-risk plaquewas common in this older cohort. That the RR of CVD associatedwith high-risk plaque was comparable to that of intermediate-riskplaque after accounting for wall thickness suggests that ultrasounddefinition of high-risk or vulnerable plaque can be challenging. Wesuggest that future research on plaque quality should evaluateatherosclerosis quantity when assessing risk of CVD.
CRP-related risk of CVD and all-cause mortality differed bythe severity of atherosclerosis in this cohort of older adults.Elevated CRP was not associated with increased risk of CVD orall-cause mortality in the group with minimal atherosclerosis, anobservation that was consistent with our previous report onstroke risk from the present study.10 However, there was signif-
TABLE 3. Hazard Ratios and Relative Excess Risk of Cardiovascular Outcomes With and Without Detectable Atherosclerosis andElevated CRP
Minimal Atherosclerosis Detectable Atherosclerosis Relative Excess Risk
Event No.CRP �3 mg/L
(n�686)CRP �3 mg/L
(n�223)CRP �3 mg/L
(n�2901)CRP �3 mg/L
(n�1210)
Adjusted RelativeExcess Risk(95% CI)*
% Attributableto Interaction† P
Myocardial infarction 595 1.00 1.16 2.24 3.52 0.65 (�0.14 to 1.43) 39% 0.11
Stroke 613 1.00 1.04 1.80 2.43 0.51 (�0.10 to 1.12) 49% 0.10
CVD death 696 1.00 1.16 2.22 4.06 1.14 (0.42 to 1.86) 54% 0.002
Composite CVD 1904 1.00 1.08 1.99 3.06 0.70 (0.26 to 1.14) 50% 0.002
All-cause mortality 1844 1.00 0.88 1.47 2.36 0.79 (0.47 to 1.12) 79% �0.001
*From a model that included age, sex, race, systolic and diastolic blood pressure, use of antihypertensive medications, body mass index, smoking (never, former,current), and amount smoked (in pack-years), high-density lipoprotein and low-density lipoprotein cholesterol, diabetes (none, impaired fasting glucose, diabetes),CRP, plaque risk group, and carotid wall thickness.
†Proportion of disease related to high CRP and atherosclerosis, either singly or together, that is attributable to their interaction, from the multivariable models.
TABLE 4. c Statistics for Models of Cardiovascular Outcomes and All-CauseMortality With Conventional Risk Factors and Additionally With Elevated CRP,Carotid IMT, and Carotid Plaque
Outcome Covariates* With CRP �3 mg/L With Carotid Tertile With Plaque Group
Myocardial infarction 0.6799 0.6829 0.6971 0.6981
Stroke 0.6856 0.6869 0.6984 0.6994
CVD death 0.7424 0.7485 0.7626 0.7632
Composite CVD 0.6840 0.6867 0.7009 0.7017
All-cause mortality 0.7151 0.7188 0.7247 0.7252
*Covariates included age, gender, race, body mass index, smoking status, pack-years of smoking,diabetes, systolic and diastolic blood pressure, total cholesterol, and high-density lipoprotein andlow-density lipoprotein cholesterol.
36 Circulation July 3, 2007
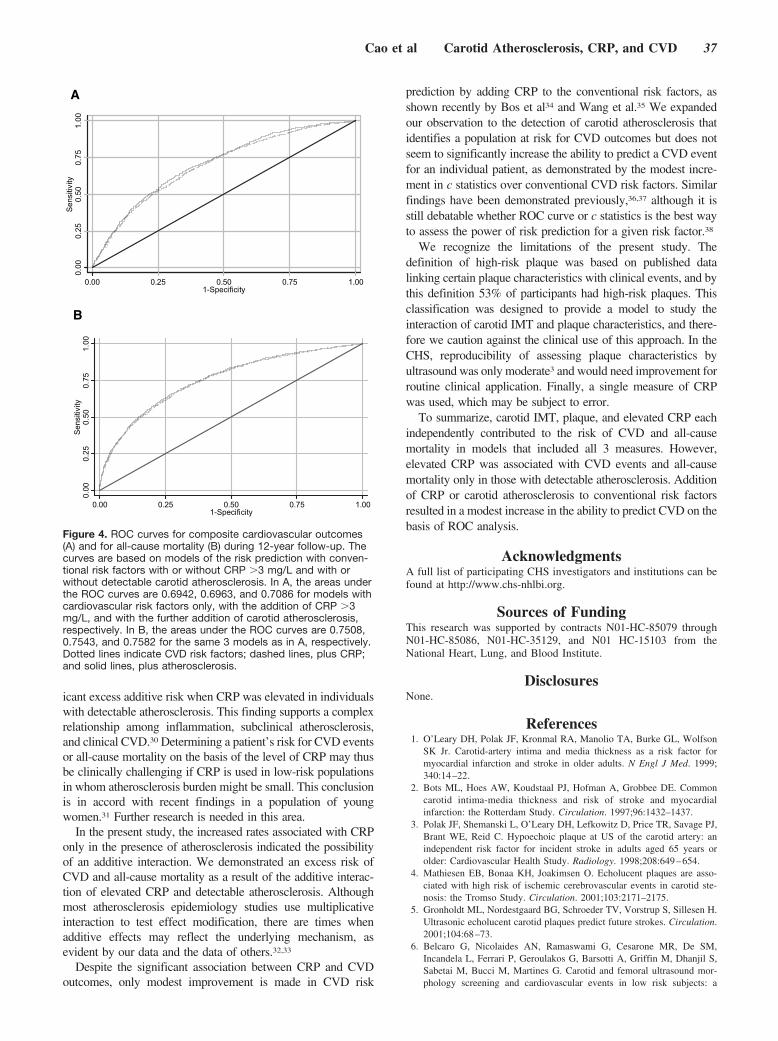
icant excess additive risk when CRP was elevated in individualswith detectable atherosclerosis. This finding supports a complexrelationship among inflammation, subclinical atherosclerosis,and clinical CVD.30 Determining a patient’s risk for CVD eventsor all-cause mortality on the basis of the level of CRP may thusbe clinically challenging if CRP is used in low-risk populationsin whom atherosclerosis burden might be small. This conclusionis in accord with recent findings in a population of youngwomen.31 Further research is needed in this area.
In the present study, the increased rates associated with CRPonly in the presence of atherosclerosis indicated the possibilityof an additive interaction. We demonstrated an excess risk ofCVD and all-cause mortality as a result of the additive interac-tion of elevated CRP and detectable atherosclerosis. Althoughmost atherosclerosis epidemiology studies use multiplicativeinteraction to test effect modification, there are times whenadditive effects may reflect the underlying mechanism, asevident by our data and the data of others.32,33
Despite the significant association between CRP and CVDoutcomes, only modest improvement is made in CVD risk
prediction by adding CRP to the conventional risk factors, asshown recently by Bos et al34 and Wang et al.35 We expandedour observation to the detection of carotid atherosclerosis thatidentifies a population at risk for CVD outcomes but does notseem to significantly increase the ability to predict a CVD eventfor an individual patient, as demonstrated by the modest incre-ment in c statistics over conventional CVD risk factors. Similarfindings have been demonstrated previously,36,37 although it isstill debatable whether ROC curve or c statistics is the best wayto assess the power of risk prediction for a given risk factor.38
We recognize the limitations of the present study. Thedefinition of high-risk plaque was based on published datalinking certain plaque characteristics with clinical events, and bythis definition 53% of participants had high-risk plaques. Thisclassification was designed to provide a model to study theinteraction of carotid IMT and plaque characteristics, and there-fore we caution against the clinical use of this approach. In theCHS, reproducibility of assessing plaque characteristics byultrasound was only moderate3 and would need improvement forroutine clinical application. Finally, a single measure of CRPwas used, which may be subject to error.
To summarize, carotid IMT, plaque, and elevated CRP eachindependently contributed to the risk of CVD and all-causemortality in models that included all 3 measures. However,elevated CRP was associated with CVD events and all-causemortality only in those with detectable atherosclerosis. Additionof CRP or carotid atherosclerosis to conventional risk factorsresulted in a modest increase in the ability to predict CVD on thebasis of ROC analysis.
AcknowledgmentsA full list of participating CHS investigators and institutions can befound at http://www.chs-nhlbi.org.
Sources of FundingThis research was supported by contracts N01-HC-85079 throughN01-HC-85086, N01-HC-35129, and N01 HC-15103 from theNational Heart, Lung, and Blood Institute.
DisclosuresNone.
References1. O’Leary DH, Polak JF, Kronmal RA, Manolio TA, Burke GL, Wolfson
SK Jr. Carotid-artery intima and media thickness as a risk factor formyocardial infarction and stroke in older adults. N Engl J Med. 1999;340:14–22.
2. Bots ML, Hoes AW, Koudstaal PJ, Hofman A, Grobbee DE. Commoncarotid intima-media thickness and risk of stroke and myocardialinfarction: the Rotterdam Study. Circulation. 1997;96:1432–1437.
3. Polak JF, Shemanski L, O’Leary DH, Lefkowitz D, Price TR, Savage PJ,Brant WE, Reid C. Hypoechoic plaque at US of the carotid artery: anindependent risk factor for incident stroke in adults aged 65 years orolder: Cardiovascular Health Study. Radiology. 1998;208:649–654.
4. Mathiesen EB, Bonaa KH, Joakimsen O. Echolucent plaques are asso-ciated with high risk of ischemic cerebrovascular events in carotid ste-nosis: the Tromso Study. Circulation. 2001;103:2171–2175.
5. Gronholdt ML, Nordestgaard BG, Schroeder TV, Vorstrup S, Sillesen H.Ultrasonic echolucent carotid plaques predict future strokes. Circulation.2001;104:68–73.
6. Belcaro G, Nicolaides AN, Ramaswami G, Cesarone MR, De SM,Incandela L, Ferrari P, Geroulakos G, Barsotti A, Griffin M, Dhanjil S,Sabetai M, Bucci M, Martines G. Carotid and femoral ultrasound mor-phology screening and cardiovascular events in low risk subjects: a
A0.
000.
250.
500.
751.
00S
ensi
tivity
0.00 0.25 0.50 0.75 1.001-Specificity
B
0.00
0.25
0.50
0.75
1.00
Sen
sitiv
ity
0.00 0.25 0.50 0.75 1.001-Specificity
Figure 4. ROC curves for composite cardiovascular outcomes(A) and for all-cause mortality (B) during 12-year follow-up. Thecurves are based on models of the risk prediction with conven-tional risk factors with or without CRP �3 mg/L and with orwithout detectable carotid atherosclerosis. In A, the areas underthe ROC curves are 0.6942, 0.6963, and 0.7086 for models withcardiovascular risk factors only, with the addition of CRP �3mg/L, and with the further addition of carotid atherosclerosis,respectively. In B, the areas under the ROC curves are 0.7508,0.7543, and 0.7582 for the same 3 models as in A, respectively.Dotted lines indicate CVD risk factors; dashed lines, plus CRP;and solid lines, plus atherosclerosis.
Cao et al Carotid Atherosclerosis, CRP, and CVD 37

10-year follow-up study (the CAFES-CAVE Study(1)). Atherosclerosis.2001;156:379–387.
7. Haverkate F, Thompson SG, Pyke SD, Gallimore JR, Pepys MB;European Concerted Action on Thrombosis and Disabilities AnginaPectoris Study Group. Production of C-reactive protein and risk ofcoronary events in stable and unstable angina. Lancet. 1997;349:462–466.
8. Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH.Inflammation, aspirin, and the risk of cardiovascular disease in apparentlyhealthy men. N Engl J Med. 1997;336:973–979.
9. Ridker PM. Role of inflammatory biomarkers in prediction of coronaryheart disease. Lancet. 2001;358:946–948.
10. Cao JJ, Thach C, Manolio TA, Psaty BM, Kuller LH, Chaves PH, PolakJF, Sutton-Tyrrell K, Herrington DM, Price TR, Cushman M. C-reactiveprotein, carotid intima-media thickness, and incidence of ischemic strokein the elderly: the Cardiovascular Health Study. Circulation. 2005;108:166–170.
11. Tracy RP, Psaty BM, Macy EM, Bovill EG, Cushman M, Cornell ES,Kuller LH. Lifetime smoking exposure affects the association ofC-reactive protein with cardiovascular disease risk factors and subclinicaldisease in healthy elderly subjects. Arterioscler Thromb Vasc Biol. 1997;17:2167–2176.
12. Lombardo A, Biasucci LM, Lanza GA, Coli S, Silvestri P, Cianflone D,Liuzzo G, Burzotta F, Crea F, Maseri A. Inflammation as a possible linkbetween coronary and carotid plaque instability. Circulation. 2004;109:3158–3163.
13. Avanzas P, Arroyo-Espliguero R, Cosin-Sales J, Quiles J, Zouridakis E,Kaski JC. Multiple complex stenoses, high neutrophil count andC-reactive protein levels in patients with chronic stable angina. Athero-sclerosis. 2004;175:151–157.
14. Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA,Kuller LH, Manolio TA, Mittelmark MB, Newman A, O’Leary D, PsatyB, Rautaharju P, Tracy R. The Cardiovascular Health Study: design andrationale. Ann Epidemiol. 1991;1:263–276.
15. Tell GS, Fried LP, Hermanson BH, Manolio TA, Newman AB, BorhaniNO. Recruitment of adults 65 years and older as participants in theCardiovascular Health Study. Ann Epidemiol. 1993;3:358–366.
16. Macy EM, Hayes TE, Tracy RP. Variability in the measurement ofC-reactive protein in healthy subjects: implication for reference intervalsand epidemiological applications. Clin Chem. 1997;43:52–58.
17. O’Leary DH, Polak JF, Kronmal RA, Savage PJ, Borhani NO, Kittner SJ,Tracy RP, Gardin JM, Price TR, Furberg CD; Cardiovascular HealthStudy Collaborative Research Group. Thickening of the carotid wall: amarker for atherosclerosis in the elderly? Stroke. 1996;27:224–231.
18. O’Leary DH, Polak JF, Wolfson SK, Bond MG, Bommer W, Sheth S,Psaty BM, Sharrett AR, Manolio TA. Use of sonography to evaluatecarotid atherosclerosis in the elderly. Stroke. 1991;22:1155–1163.
19. O’Leary DH, Bryan FA, Goodison MW, Rifkin MD, Gramiak R, Ball M,Bond MG, Dunn RA, Goldberg BB, Toole JF. Measurement variability ofcarotid atherosclerosis: real-time (B-mode) ultrasonography andangiography. Stroke. 1987;18:1011–1017.
20. Polak JF, O’Leary DH, Kronmal RA, Wolfson SK, Bond MG, Tracy RP,Gardin JM, Kittner SJ, Price TR, Savage PJ. Sonographic evaluation of
carotid artery atherosclerosis in the elderly: relationship of diseaseseverity to stroke and transient ischemic attack. Radiology. 1993;188:363–370.
21. Kitamura A, Iso H, Imano H, Ohira T, Okada T, Sato S, Kiyama M,Tanigawa T, Yamagishi K, Shimamoto T. Carotid intima-media thicknessand plaque characteristics as a risk factor for stroke in Japanese elderlymen. Stroke. 2004;35:2788–2794.
22. Psaty BM, Kuller LH, Bild D, Burke GL, Kittner SJ, Mittelmark M, PriceTR, Rautaharju PM, Robbins J. Methods of assessing prevalent cardio-vascular disease in the Cardiovascular Health Study. Ann Epidemiol.1995;5:270–277.
23. Arnold AM, Kronmal RA. Multiple imputation of baseline data in theCardiovascular Health Study. Am J Epidemiol. 2003;157:74–84.
24. Rothman KJ. Modern Epidemiology. Boston, Mass: Little, Brown; 1986.25. Hosmer D, Lemeshow S. Confidence interval estimation of interaction.
Epidemiology. 1992;3:452–456.26. Harrell FE Jr. Regression Modeling Strategies. New York, NY: Springer;
2001.27. Hosmer DW, Lemeshow S. Applied Logistic Regression. New York, NY:
Wiley & Sons Inc; 2000.28. Bonithon-Kopp C, Touboul PJ, Berr C, Leroux C, Mainard F, Courbon D,
Ducimetiere P. Relation of intima-media thickness to atheroscleroticplaques in carotid arteries: the Vascular Aging (EVA) Study. ArteriosclerThromb Vasc Biol. 1996;16:310–316.
29. Homma S, Hirose N, Ishida H, Ishii T, Araki G. Carotid plaque andintima-media thickness assessed by B-mode ultrasonography in subjectsranging from young adults to centenarians. Stroke. 2001;32:830–835.
30. Fuster V, Moreno PR, Fayad ZA, Corti R, Badimon JJ. Atherothrombosisand high-risk plaque, part I: evolving concepts. J Am Coll Cardiol.2005;46:937–954.
31. Cook NR, Buring JE, Ridker PM. The effect of including C-reactiveprotein in cardiovascular risk prediction models for women. Ann InternMed. 2006;145:21–29.
32. Li R, Chambless L. Test for additive interaction in proportional hazardsmodels. Ann Epidemiol. 2007;17:227–236.
33. Schwartz SW, Carlucci C, Chambless LE, Rosamond WD. Synergismbetween smoking and vital exhaustion in the risk of ischemic stroke:evidence from the ARIC study. Ann Epidemiol. 2004;14:416–424.
34. Bos MJ, Schipper CM, Koudstaal PJ, Witteman JC, Hofman A, BretelerMM. High serum C-reactive protein level is not an independent predictorfor stroke: the Rotterdam Study. Circulation. 2006;114:1591–1598.
35. Wang TJ, Gona P, Larson MG, Tofler GH, Levy D, Newton-Cheh C,Jacques PF, Rifai N, Selhub J, Robins SJ, Benjamin EJ, D’Agostino RB,Vasan RS. Multiple biomarkers for the prediction of first major cardio-vascular events and death. N Engl J Med. 2006;355:2631–2639.
36. Greenland P, O’Malley PG. What is a new prediction marker useful?Arch Intern Med. 2005;165:2454–2456.
37. Folsom AR, Chambless LE, Ballantyne CM, Coresh J, Heiss G, Wu KK,Boerwinkle E, Mosley TH Jr, Sorlie P, Diao G, Sharrett AR. Anassessment of incremental coronary risk prediction using C-reactiveprotein and other novel risk markers: the Atherosclerosis Risk in Com-munities Study. Arch Intern Med. 2006;166:1368–1373.
38. Cook NR. Use and misuse of the receiver operating characteristic curvein risk prediction. Circulation. 2007;115:928–935.
CLINICAL PERSPECTIVEUltrasound-determined carotid intima-media thickness and presence of plaques are measures of carotid atherosclerosis.Higher carotid intima-media thickness is a risk marker for future stroke and coronary heart disease. Characteristics ofcarotid plaque have also been associated with vascular risk. C-reactive protein (CRP) is also a risk factor for cardiovasculardisease. In the present study we examined the relationship of carotid atherosclerosis and CRP and their joint roles incardiovascular risk prediction in 5022 elderly individuals (�65 years) who were the participants of the CardiovascularHealth Study. After up to 12 years of follow-up, we found that when CRP was elevated (�3 mg/L) among those withdetectable atherosclerosis on ultrasound, there was a significantly increased risk for cardiovascular disease and all-causemortality. Elevated CRP in the absence of atherosclerosis did not increase cardiovascular or all-cause mortality risk. Weconcluded that determining a patient’s risk for cardiovascular events or all-cause mortality on the basis of the level of CRPmay be clinically challenging if CRP is used in low-risk populations in whom atherosclerosis burden might be small.
38 Circulation July 3, 2007

Abdominal Visceral and Subcutaneous AdiposeTissue Compartments
Association With Metabolic Risk Factors in the Framingham Heart Study
Caroline S. Fox, MD, MPH; Joseph M. Massaro, PhD; Udo Hoffmann, MD, MPH; Karla M. Pou, MD;Pal Maurovich-Horvat, MD; Chun-Yu Liu, PhD; Ramachandran S. Vasan, MD;
Joanne M. Murabito, MD, ScM; James B. Meigs, MD, MPH; L. Adrienne Cupples, PhD;Ralph B. D’Agostino, Sr, PhD; Christopher J. O’Donnell, MD, MPH
Background—Visceral adipose tissue (VAT) compartments may confer increased metabolic risk. The incremental utilityof measuring both visceral and subcutaneous abdominal adipose tissue (SAT) in association with metabolic risk factorsand underlying heritability has not been well described in a population-based setting.
Methods and Results—Participants (n�3001) were drawn from the Framingham Heart Study (48% women; mean age, 50years), were free of clinical cardiovascular disease, and underwent multidetector computed tomography assessment ofSAT and VAT volumes between 2002 and 2005. Metabolic risk factors were examined in relation to increments of SATand VAT after multivariable adjustment. Heritability was calculated using variance-components analysis. Among bothwomen and men, SAT and VAT were significantly associated with blood pressure, fasting plasma glucose, triglycerides,and high-density lipoprotein cholesterol and with increased odds of hypertension, impaired fasting glucose, diabetesmellitus, and metabolic syndrome (P range �0.01). In women, relations between VAT and risk factors were consistentlystronger than in men. However, VAT was more strongly correlated with most metabolic risk factors than was SAT. Forexample, among women and men, both SAT and VAT were associated with increased odds of metabolic syndrome. Inwomen, the odds ratio (OR) of metabolic syndrome per 1–standard deviation increase in VAT (OR, 4.7) was strongerthan that for SAT (OR, 3.0; P for difference between SAT and VAT �0.0001); similar differences were noted for men(OR for VAT, 4.2; OR for SAT, 2.5). Furthermore, VAT but not SAT contributed significantly to risk factor variationafter adjustment for body mass index and waist circumference (P �0.01). Among overweight and obese individuals, theprevalence of hypertension, impaired fasting glucose, and metabolic syndrome increased linearly and significantlyacross increasing VAT quartiles. Heritability values for SAT and VAT were 57% and 36%, respectively.
Conclusions—Although both SAT and VAT are correlated with metabolic risk factors, VAT remains more stronglyassociated with an adverse metabolic risk profile even after accounting for standard anthropometric indexes. Ourfindings are consistent with the hypothesized role of visceral fat as a unique, pathogenic fat depot. Measurement of VATmay provide a more complete understanding of metabolic risk associated with variation in fat distribution. (Circulation.2007;116:39-48.)
Key Words: abdominal fat � diabetes mellitus � epidemiology � hypertension � intra-abdominal fat� metabolic syndrome X � obesity
Cardiovascular disease (CVD) is the leading cause ofmorbidity and mortality in the United States, affecting
roughly 70 million people and accounting for nearly 1 milliondeaths per year.1 Improvements in CVD risk factor profileshave led to significant reductions in death from CVD over the
Clinical Perspective p 48
past 50 years,2 but recent data suggest that the increasingprevalence of obesity may have slowed this rate of decline.3
Rates of overweight and obesity continue to increase,4–6
Received November 9, 2006; accepted April 18, 2007.From the National Heart, Lung and Blood Institute’s Framingham Heart Study (C.S.F., C.J.O.), Framingham, Mass; Division of Endocrinology,
Metabolism, and Diabetes, Department of Medicine (C.S.F., K.M.P.), Brigham and Women’s Hospital and Harvard Medical School, Boston, Mass;Boston University, Department of Mathematics (J.M.M.; R.B.D.) and School of Public Health, Division of Biostatistics (C.-Y.L., L.A.C.), Boston, Mass;Radiology Department (U.H.) and Department of Medicine (J.B.M., C.J.O.), Massachusetts General Hospital, Harvard Medical School, Boston;Semmelweis University (P.M.-H.), Budapest, Hungary; and Boston University School of Medicine (R.S.V.), Boston, Mass.
Guest Editor for this article was Robert H. Eckel, MD.The online-only Data Supplement, which consists of a table, is available with this article at http://circ.ahajournals.org/cgi/
content/full/CIRCULATIONAHA.106.675355/DC1.Correspondence to Caroline S. Fox, MD, MPH, 73 Mt Wayte Ave, Ste 2, Framingham, MA 01702. E-mail [email protected]© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.106.675355

which suggests that the full impact of the obesity epidemichas yet to be realized.
Obesity, defined by a body mass index (BMI) of at least 30kg/m2, is a risk factor for multiple CVD risk factors, includinghypertension, dyslipidemia, diabetes, and the metabolic syn-drome (MetS). BMI is a useful indicator of overall adiposity,and recent National Health and Nutrition Examination Surveydata demonstrate that two thirds of the US population is eitheroverweight or obese.6 However, different fat compartmentsmay be associated with differential metabolic risk.7 In par-ticular, the visceral adipose tissue (VAT) compartment maybe a unique pathogenic fat depot.8–10 VAT has been termedan endocrine organ, in part because it secretes adipocytokinesand other vasoactive substances that can influence the risk ofdeveloping metabolic traits.10–16
Waist circumference (WC) is an easily obtainable butimprecise measure of abdominal adiposity17 because it is afunction of both the subcutaneous adipose tissue (SAT) andVAT compartments. Therefore, assessment of VAT requiresimaging with radiographic techniques such as computedtomography (CT) or magnetic resonance imaging (MRI).Available studies report relations of greater SAT and VATwith a higher prevalence of impaired fasting glucose,9,18
diabetes,9,10,19 insulin resistance,9,20,21 hypertension,22–24 lip-ids,25–29 MetS,30–32 and risk factor clustering.26 However,current imaging studies evaluating SAT and VAT are limitedto small, referral-based samples often enriched for adiposity-related traits.23,25–27,32–34 Furthermore, study samples haveoften been limited to either women or men, precluding the studyof sex differences.22,23,25–28,34–36 Some studies have focused onJapanese Americans or Southeast Asians,19,22,26,29,35,37 ethnicgroups with more visceral fat than expected for a given overallBMI.38 We recently demonstrated that multidetector CT permitshighly reproducible volumetric measurements of both SAT andVAT.39 In addition, this initial observation suggested that volu-metric fat measurements, as opposed to previous studies usingsingle-slice methodology, can accurately characterize the hetero-geneity of abdominal fat distribution between individuals andthe differences in fat distribution with age and between womenand men.
Thus, our aims in a community-based sample of womenand men free of CVD across the spectrum of BMI were toassess whether the volume of SAT and VAT are associatedwith metabolic risk factors cross-sectionally, to assesswhether VAT is more strongly associated with metabolic riskfactors than is SAT, and to determine whether sophisticatedvolumetric imaging methods of SAT and VAT provideinformation about metabolic risk other than that offered bysimpler measures such as BMI and WC.
MethodsStudy SampleParticipants for this study were drawn from the Framingham HeartStudy Multidetector Computed Tomography Study, a population-based substudy of the community-based Framingham Heart StudyOffspring and Third-Generation Study cohorts. Beginning in 1948,5209 men and women 28 to 62 years of age were enrolled in theoriginal cohort of the Framingham Heart Study. The offspring andspouses of the offspring of the original cohort were enrolled in theOffspring Study starting in 1971. Selection criteria and study design
have been described elsewhere.40,41 Beginning in 2002, 4095 ThirdGeneration Study participants, who had at least 1 parent in theoffspring cohort, were enrolled in the Framingham Heart Study andunderwent standard clinic examinations. The standard clinic exam-ination included a physician interview, a physical examination, andlaboratory tests. For the present study, the study sample consisted ofOffspring and Third Generation Study participants who were part ofthe multidetector CT substudy.
Between June 2002 and April 2005, 3529 participants (2111 thirdgeneration, 1418 offspring participants) underwent multidetector CTassessment of coronary and aortic calcium. Inclusion in this studywas weighted toward participants from larger Framingham HeartStudy families and those who resided in the greater New Englandarea. Overall, 755 families were included in our analysis. Men had tobe �35 years of age; women had to be �40 years of age and notpregnant; and all participants had to weigh �350 pounds. Of theparticipants, 433 (222 offspring and 211 third generation) wereimaged as participants in an ancillary study using an identicalimaging protocol, the National Heart, Lung, and Blood’s FamilyHeart Study.42 Of the total 3529 subjects imaged, 3394 had inter-pretable CT measures; of those, 3329 had both SAT and VATmeasured; of those, 3124 of them were free of CVD; of those, 3102attended a contemporaneous examination; and of those, 3001 had acomplete covariate profile. Thus, the overall sample size for analysisis 3001.
The study was approved by the institutional review boards of theBoston University Medical Center and Massachusetts General Hos-pital. All subjects provided written informed consent.
Volumetric Adipose Tissue ImagingSubjects underwent 8-slice multidetector CT imaging of the abdo-men in a supine position as previously described (LightSpeed Ultra,General Electric, Milwaukee, Wis).39 Briefly, 25 contiguous 5-mm-thick slices (120 kVp; 400 mA; gantry rotation time, 500 ms; tablefeed, 3:1) were acquired covering 125 mm above the level of S1.
Abdominal Adipose Tissue MeasurementsSAT and VAT volumes were assessed (Aquarius 3D Workstation,TeraRecon Inc, San Mateo, Calif). To identify pixels containing fat,an image display window width of �195 to �45 Hounsfield units(HU) and a window center of �120 HU were used. The abdominalmuscular wall separating the visceral from the subcutaneous com-partment was traced manually. Interreader reproducibility was as-sessed by 2 independent readers measuring VAT and SAT on asubset of 100 randomly selected participants.39 Interclass correla-tions for interreader comparisons were 0.992 for VAT and 0.997 forSAT. Similarly high correlations were noted for intrareadercomparisons.
Risk Factor and Covariate AssessmentRisk factors and covariates were measured at the contemporaneousexamination. BMI, defined as weight (in kilograms) divided by thesquare of height (in meters), was measured at each index examina-tion. WC was measured at the level of the umbilicus. Fasting plasmaglucose, total and high-density lipoprotein (HDL) cholesterol, andtriglycerides were measured on fasting morning samples. Diabeteswas defined as a fasting plasma glucose level �126 mg/dL at aFramingham examination or treatment with either insulin or ahypoglycemic agent. Participants were considered current smokers ifthey had smoked at least 1 cigarette per day for the previous year.Assessed through a series of physician-administered questions,alcohol use was dichotomized on the basis of consumption of �14drinks per week (in men) or 7 drinks per week (in women). Physicalactivity, assessed with a questionnaire, is a score based on theaverage daily number of hours of sleep and sedentary, slight,moderate, and heavy activity of the participant. Women wereconsidered menopausal if their periods had stopped for �1 year.Impaired fasting glucose was defined as a fasting plasma glucoselevel of 100 to 125 mg/dL among those not treated for diabetes.Hypertension was defined as systolic blood pressure �140 mm Hg,
40 Circulation July 3, 2007
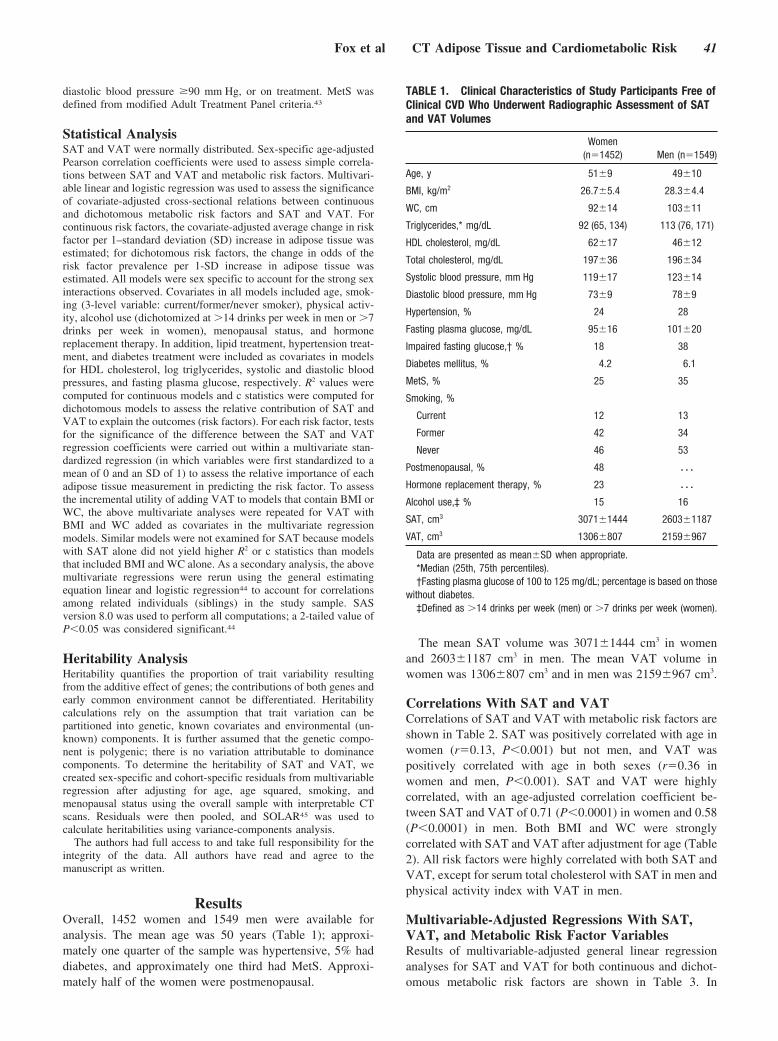
diastolic blood pressure �90 mm Hg, or on treatment. MetS wasdefined from modified Adult Treatment Panel criteria.43
Statistical AnalysisSAT and VAT were normally distributed. Sex-specific age-adjustedPearson correlation coefficients were used to assess simple correla-tions between SAT and VAT and metabolic risk factors. Multivari-able linear and logistic regression was used to assess the significanceof covariate-adjusted cross-sectional relations between continuousand dichotomous metabolic risk factors and SAT and VAT. Forcontinuous risk factors, the covariate-adjusted average change in riskfactor per 1–standard deviation (SD) increase in adipose tissue wasestimated; for dichotomous risk factors, the change in odds of therisk factor prevalence per 1-SD increase in adipose tissue wasestimated. All models were sex specific to account for the strong sexinteractions observed. Covariates in all models included age, smok-ing (3-level variable: current/former/never smoker), physical activ-ity, alcohol use (dichotomized at �14 drinks per week in men or �7drinks per week in women), menopausal status, and hormonereplacement therapy. In addition, lipid treatment, hypertension treat-ment, and diabetes treatment were included as covariates in modelsfor HDL cholesterol, log triglycerides, systolic and diastolic bloodpressures, and fasting plasma glucose, respectively. R2 values werecomputed for continuous models and c statistics were computed fordichotomous models to assess the relative contribution of SAT andVAT to explain the outcomes (risk factors). For each risk factor, testsfor the significance of the difference between the SAT and VATregression coefficients were carried out within a multivariate stan-dardized regression (in which variables were first standardized to amean of 0 and an SD of 1) to assess the relative importance of eachadipose tissue measurement in predicting the risk factor. To assessthe incremental utility of adding VAT to models that contain BMI orWC, the above multivariate analyses were repeated for VAT withBMI and WC added as covariates in the multivariate regressionmodels. Similar models were not examined for SAT because modelswith SAT alone did not yield higher R2 or c statistics than modelsthat included BMI and WC alone. As a secondary analysis, the abovemultivariate regressions were rerun using the general estimatingequation linear and logistic regression44 to account for correlationsamong related individuals (siblings) in the study sample. SASversion 8.0 was used to perform all computations; a 2-tailed value ofP�0.05 was considered significant.44
Heritability AnalysisHeritability quantifies the proportion of trait variability resultingfrom the additive effect of genes; the contributions of both genes andearly common environment cannot be differentiated. Heritabilitycalculations rely on the assumption that trait variation can bepartitioned into genetic, known covariates and environmental (un-known) components. It is further assumed that the genetic compo-nent is polygenic; there is no variation attributable to dominancecomponents. To determine the heritability of SAT and VAT, wecreated sex-specific and cohort-specific residuals from multivariableregression after adjusting for age, age squared, smoking, andmenopausal status using the overall sample with interpretable CTscans. Residuals were then pooled, and SOLAR45 was used tocalculate heritabilities using variance-components analysis.
The authors had full access to and take full responsibility for theintegrity of the data. All authors have read and agree to themanuscript as written.
ResultsOverall, 1452 women and 1549 men were available foranalysis. The mean age was 50 years (Table 1); approxi-mately one quarter of the sample was hypertensive, 5% haddiabetes, and approximately one third had MetS. Approxi-mately half of the women were postmenopausal.
The mean SAT volume was 3071�1444 cm3 in womenand 2603�1187 cm3 in men. The mean VAT volume inwomen was 1306�807 cm3 and in men was 2159�967 cm3.
Correlations With SAT and VATCorrelations of SAT and VAT with metabolic risk factors areshown in Table 2. SAT was positively correlated with age inwomen (r�0.13, P�0.001) but not men, and VAT waspositively correlated with age in both sexes (r�0.36 inwomen and men, P�0.001). SAT and VAT were highlycorrelated, with an age-adjusted correlation coefficient be-tween SAT and VAT of 0.71 (P�0.0001) in women and 0.58(P�0.0001) in men. Both BMI and WC were stronglycorrelated with SAT and VAT after adjustment for age (Table2). All risk factors were highly correlated with both SAT andVAT, except for serum total cholesterol with SAT in men andphysical activity index with VAT in men.
Multivariable-Adjusted Regressions With SAT,VAT, and Metabolic Risk Factor VariablesResults of multivariable-adjusted general linear regressionanalyses for SAT and VAT for both continuous and dichot-omous metabolic risk factors are shown in Table 3. In
TABLE 1. Clinical Characteristics of Study Participants Free ofClinical CVD Who Underwent Radiographic Assessment of SATand VAT Volumes
Women(n�1452) Men (n�1549)
Age, y 51�9 49�10
BMI, kg/m2 26.7�5.4 28.3�4.4
WC, cm 92�14 103�11
Triglycerides,* mg/dL 92 (65, 134) 113 (76, 171)
HDL cholesterol, mg/dL 62�17 46�12
Total cholesterol, mg/dL 197�36 196�34
Systolic blood pressure, mm Hg 119�17 123�14
Diastolic blood pressure, mm Hg 73�9 78�9
Hypertension, % 24 28
Fasting plasma glucose, mg/dL 95�16 101�20
Impaired fasting glucose,† % 18 38
Diabetes mellitus, % 4.2 6.1
MetS, % 25 35
Smoking, %
Current 12 13
Former 42 34
Never 46 53
Postmenopausal, % 48 � � �
Hormone replacement therapy, % 23 � � �
Alcohol use,‡ % 15 16
SAT, cm3 3071�1444 2603�1187
VAT, cm3 1306�807 2159�967
Data are presented as mean�SD when appropriate.*Median (25th, 75th percentiles).†Fasting plasma glucose of 100 to 125 mg/dL; percentage is based on those
without diabetes.‡Defined as �14 drinks per week (men) or �7 drinks per week (women).
Fox et al CT Adipose Tissue and Cardiometabolic Risk 41

women, per 1-SD increase in SAT, systolic blood pressureincreased on average 3.9�0.4 mm Hg (�1 SE), whereasVAT was 4.8�0.4 mm Hg higher. For systolic blood pressurein women, the difference between the magnitude of effect ofthe SAT versus VAT was not significant (P�0.10; Table 3).In men, the magnitude of the association of the averagesystolic blood pressure increase per 1-SD increase in VATwas larger than for SAT (3.3 versus 2.3 mm Hg, respectively;P�0.01 for difference in the regression coefficients betweenSAT and VAT). Similar results were obtained for diastolicblood pressure.
In women and men, the association of both SAT and VATwith continuous measures of metabolic risk factors washighly significant. For fasting plasma glucose, the effect ofVAT was stronger than that of SAT (P�0.0001 for differencein women, P�0.001 in men). Strong and significant resultsfor log triglycerides and HDL cholesterol followed similarpatterns (Table 3).
Highly significant associations with SAT and VAT alsowere noted for dichotomous risk factor variables. Amongwomen and men, both SAT and VAT were associated with anincreased odds of hypertension (Table 3). In women, the oddsratio of hypertension per 1-SD increase in VAT (odds ratio,2.1) was stronger than that for SAT (odds ratio, 1.7; P�0.001for difference between SAT and VAT); similar differenceswere noted for men. Similar highly significant differencesalso were noted for impaired fasting glucose, diabetes, andMetS and are presented in Table 3.
The magnitude of association between VAT and all riskfactors examined was consistently greater for women than formen (Table 3). Weaker sex differences were observed forSAT.
Residual Effect of VAT in Multivariable ModelsThat Contain BMI and WCTo address whether radiographic imaging of abdominaladipose tissue explains variation in metabolic risk factorsover and above the contribution of BMI and WC, weexamined the residual effect size of each metabolic risk factorfrom multivariable models that additionally contained BMI
and WC. Because models with BMI and WC routinelyyielded higher R2 or c statistic than models with SAT (TableI in the online-only Data Supplement), the addition of all 3variables into one model was not pursued. For example, inwomen, SAT plus covariates were associated with 21% of thevariation in log triglycerides (R2�0.21), VAT plus covariateswere associated with 30% of the variation in log triglycerides,and both BMI and WC plus the covariates were associatedwith 26% of the variation in triglycerides. Models with VAT,BMI, and WC demonstrated significant additional contribu-tion of VAT for all variables except diabetes in men.Statistically significant residual effect sizes for VAT wereobserved for all metabolic risk factors except diabetes in men(Table 3).
Risk Factor Distribution Based on Quartiles of VATBecause VAT adds to risk factor variation above and beyondBMI and WC, we assessed the impact of stratifying individ-uals by VAT quartile within clinically defined categories ofBMI (normal weight, BMI �25 kg/m2; overweight, BMI of25 to 29.9 kg/m2; and obese, BMI �30 kg/m2). Thirty-threepercent of the sample was normal weight, 41% was over-weight, and 26% was obese. Among normal-weight, over-weight, and obese individuals, there was a highly statisticallysignificant stepwise linear increase in the prevalence of theMetS across quartiles of VAT in both women and men (seethe Figure) after adjustment for age and BMI; similar rela-tions were noted for additional risk factors, including hyper-tension and impaired fasting glucose.
Secondary AnalysisWhen education status was included as an additional covari-ate, relations between SAT and VAT and the continuous anddichotomous metabolic risk factors were not materially dif-ferent (data not shown). When analyses were rerun using thegeneral estimating equation procedure, the resulting proba-bility values were not substantively changed from thosediscussed above (data not shown).
Heritability AnalysisHeritability for SAT was 57%, whereas heritability for VATwas 36%.
DiscussionIn our community-based sample, volumetric CT measures ofboth SAT and VAT were correlated with multiple metabolicrisk factors, although risk factor correlations with VAT wereconsistently significantly stronger than those for SAT. VAT,not SAT, provided information above and beyond simpleclinical anthropometrics, including BMI and WC, and con-sistently provided differences in risk factor stratificationamong individuals who were overweight and obese. VATwas more strongly associated with metabolic risk factors inwomen than in men. Finally, both SAT and VAT are heritabletraits.
VAT has traditionally been considered the more patho-genic adipose tissue compartment compared with SAT, butdata confirming these relations using high-resolution volu-metric imaging assessments in large, community-based sam-
TABLE 2. Age-Adjusted Pearson Correlation CoefficientsBetween Metabolic Risk Factors and SAT and VAT Volumes
Women Men
SAT VAT SAT VAT
Age 0.13† 0.36† 0.03 0.36†
BMI 0.88† 0.75† 0.83† 0.71†
WC 0.87† 0.78† 0.88† 0.73†
Log triglycerides 0.31† 0.46† 0.18† 0.37†
HDL cholesterol �0.25† �0.35† �0.17† �0.33†
Total cholesterol 0.11† 0.15† 0.02 0.08*
Systolic blood pressure 0.26† 0.30† 0.18† 0.24†
Diastolic blood pressure 0.26† 0.28† 0.21† 0.27†
Blood glucose 0.23† 0.34† 0.12† 0.19†
Physical activity index �0.14† �0.09* �0.08* �0.03
*P�0.01; †P�0.001.
42 Circulation July 3, 2007
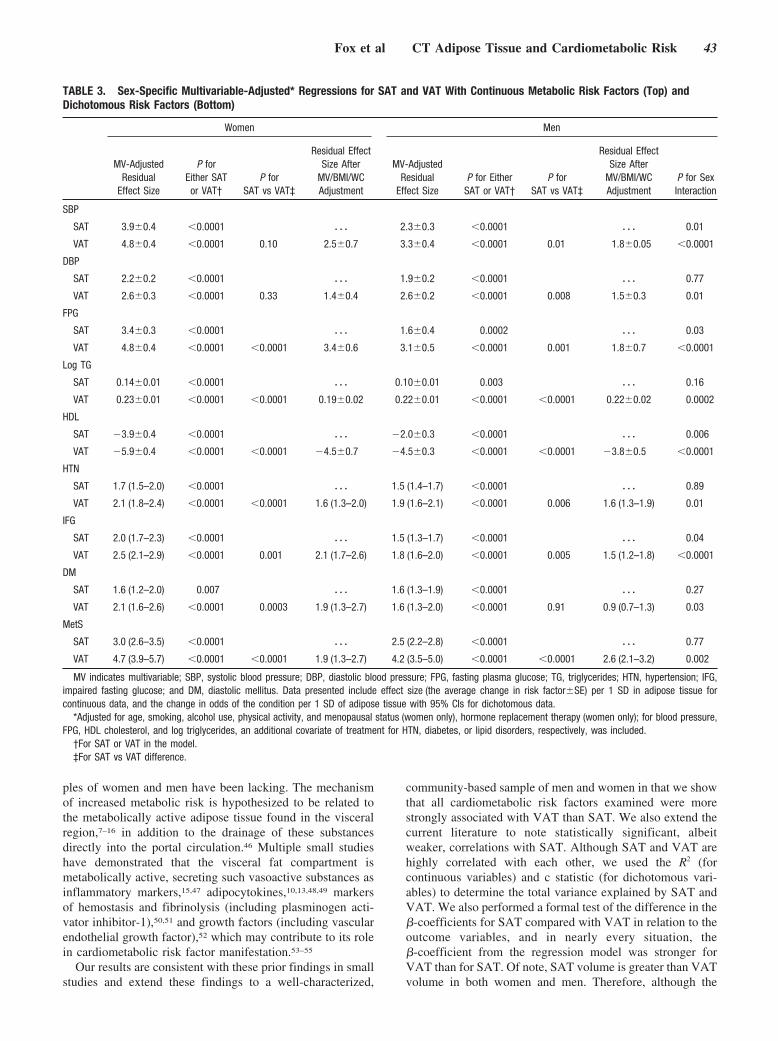
ples of women and men have been lacking. The mechanismof increased metabolic risk is hypothesized to be related tothe metabolically active adipose tissue found in the visceralregion,7–16 in addition to the drainage of these substancesdirectly into the portal circulation.46 Multiple small studieshave demonstrated that the visceral fat compartment ismetabolically active, secreting such vasoactive substances asinflammatory markers,15,47 adipocytokines,10,13,48,49 markersof hemostasis and fibrinolysis (including plasminogen acti-vator inhibitor-1),50,51 and growth factors (including vascularendothelial growth factor),52 which may contribute to its rolein cardiometabolic risk factor manifestation.53–55
Our results are consistent with these prior findings in smallstudies and extend these findings to a well-characterized,
community-based sample of men and women in that we showthat all cardiometabolic risk factors examined were morestrongly associated with VAT than SAT. We also extend thecurrent literature to note statistically significant, albeitweaker, correlations with SAT. Although SAT and VAT arehighly correlated with each other, we used the R2 (forcontinuous variables) and c statistic (for dichotomous vari-ables) to determine the total variance explained by SAT andVAT. We also performed a formal test of the difference in the�-coefficients for SAT compared with VAT in relation to theoutcome variables, and in nearly every situation, the�-coefficient from the regression model was stronger forVAT than for SAT. Of note, SAT volume is greater than VATvolume in both women and men. Therefore, although the
TABLE 3. Sex-Specific Multivariable-Adjusted* Regressions for SAT and VAT With Continuous Metabolic Risk Factors (Top) andDichotomous Risk Factors (Bottom)
Women Men
MV-AdjustedResidual
Effect Size
P forEither SATor VAT†
P forSAT vs VAT‡
Residual EffectSize After
MV/BMI/WCAdjustment
MV-AdjustedResidual
Effect SizeP for Either
SAT or VAT†P for
SAT vs VAT‡
Residual EffectSize After
MV/BMI/WCAdjustment
P for SexInteraction
SBP
SAT 3.9�0.4 �0.0001 � � � 2.3�0.3 �0.0001 � � � 0.01
VAT 4.8�0.4 �0.0001 0.10 2.5�0.7 3.3�0.4 �0.0001 0.01 1.8�0.05 �0.0001
DBP
SAT 2.2�0.2 �0.0001 � � � 1.9�0.2 �0.0001 � � � 0.77
VAT 2.6�0.3 �0.0001 0.33 1.4�0.4 2.6�0.2 �0.0001 0.008 1.5�0.3 0.01
FPG
SAT 3.4�0.3 �0.0001 � � � 1.6�0.4 0.0002 � � � 0.03
VAT 4.8�0.4 �0.0001 �0.0001 3.4�0.6 3.1�0.5 �0.0001 0.001 1.8�0.7 �0.0001
Log TG
SAT 0.14�0.01 �0.0001 � � � 0.10�0.01 0.003 � � � 0.16
VAT 0.23�0.01 �0.0001 �0.0001 0.19�0.02 0.22�0.01 �0.0001 �0.0001 0.22�0.02 0.0002
HDL
SAT �3.9�0.4 �0.0001 � � � �2.0�0.3 �0.0001 � � � 0.006
VAT �5.9�0.4 �0.0001 �0.0001 �4.5�0.7 �4.5�0.3 �0.0001 �0.0001 �3.8�0.5 �0.0001
HTN
SAT 1.7 (1.5–2.0) �0.0001 � � � 1.5 (1.4–1.7) �0.0001 � � � 0.89
VAT 2.1 (1.8–2.4) �0.0001 �0.0001 1.6 (1.3–2.0) 1.9 (1.6–2.1) �0.0001 0.006 1.6 (1.3–1.9) 0.01
IFG
SAT 2.0 (1.7–2.3) �0.0001 � � � 1.5 (1.3–1.7) �0.0001 � � � 0.04
VAT 2.5 (2.1–2.9) �0.0001 0.001 2.1 (1.7–2.6) 1.8 (1.6–2.0) �0.0001 0.005 1.5 (1.2–1.8) �0.0001
DM
SAT 1.6 (1.2–2.0) 0.007 � � � 1.6 (1.3–1.9) �0.0001 � � � 0.27
VAT 2.1 (1.6–2.6) �0.0001 0.0003 1.9 (1.3–2.7) 1.6 (1.3–2.0) �0.0001 0.91 0.9 (0.7–1.3) 0.03
MetS
SAT 3.0 (2.6–3.5) �0.0001 � � � 2.5 (2.2–2.8) �0.0001 � � � 0.77
VAT 4.7 (3.9–5.7) �0.0001 �0.0001 1.9 (1.3–2.7) 4.2 (3.5–5.0) �0.0001 �0.0001 2.6 (2.1–3.2) 0.002
MV indicates multivariable; SBP, systolic blood pressure; DBP, diastolic blood pressure; FPG, fasting plasma glucose; TG, triglycerides; HTN, hypertension; IFG,impaired fasting glucose; and DM, diastolic mellitus. Data presented include effect size (the average change in risk factor�SE) per 1 SD in adipose tissue forcontinuous data, and the change in odds of the condition per 1 SD of adipose tissue with 95% CIs for dichotomous data.
*Adjusted for age, smoking, alcohol use, physical activity, and menopausal status (women only), hormone replacement therapy (women only); for blood pressure,FPG, HDL cholesterol, and log triglycerides, an additional covariate of treatment for HTN, diabetes, or lipid disorders, respectively, was included.
†For SAT or VAT in the model.‡For SAT vs VAT difference.
Fox et al CT Adipose Tissue and Cardiometabolic Risk 43
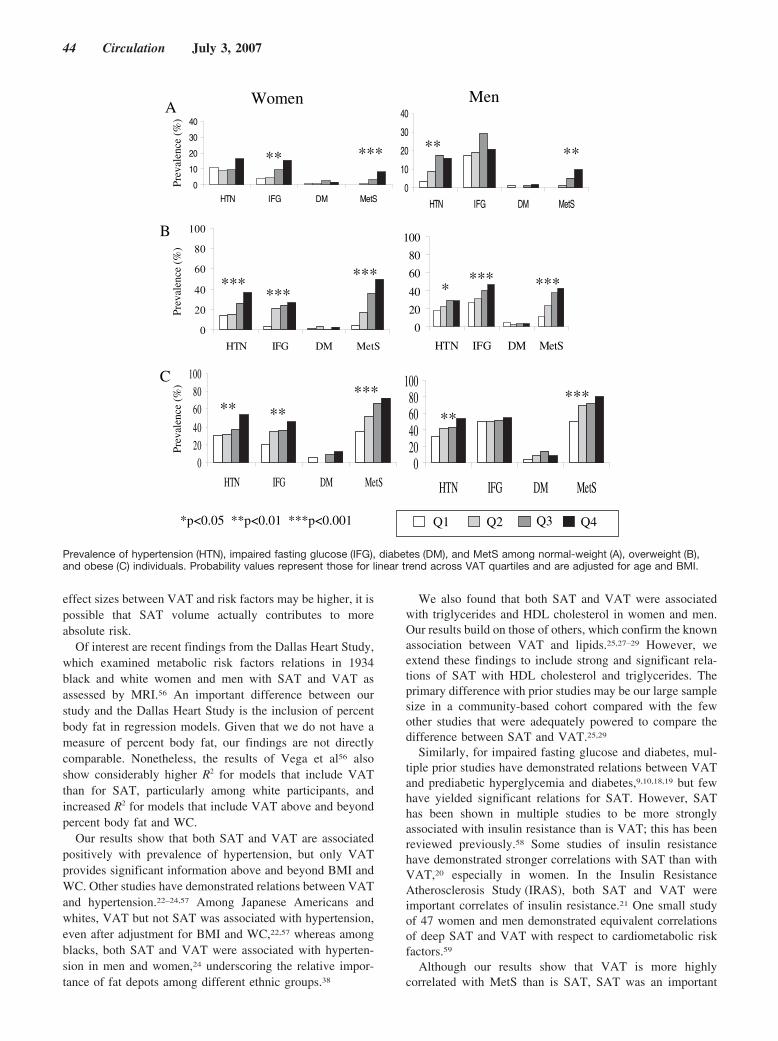
effect sizes between VAT and risk factors may be higher, it ispossible that SAT volume actually contributes to moreabsolute risk.
Of interest are recent findings from the Dallas Heart Study,which examined metabolic risk factors relations in 1934black and white women and men with SAT and VAT asassessed by MRI.56 An important difference between ourstudy and the Dallas Heart Study is the inclusion of percentbody fat in regression models. Given that we do not have ameasure of percent body fat, our findings are not directlycomparable. Nonetheless, the results of Vega et al56 alsoshow considerably higher R2 for models that include VATthan for SAT, particularly among white participants, andincreased R2 for models that include VAT above and beyondpercent body fat and WC.
Our results show that both SAT and VAT are associatedpositively with prevalence of hypertension, but only VATprovides significant information above and beyond BMI andWC. Other studies have demonstrated relations between VATand hypertension.22–24,57 Among Japanese Americans andwhites, VAT but not SAT was associated with hypertension,even after adjustment for BMI and WC,22,57 whereas amongblacks, both SAT and VAT were associated with hyperten-sion in men and women,24 underscoring the relative impor-tance of fat depots among different ethnic groups.38
We also found that both SAT and VAT were associatedwith triglycerides and HDL cholesterol in women and men.Our results build on those of others, which confirm the knownassociation between VAT and lipids.25,27–29 However, weextend these findings to include strong and significant rela-tions of SAT with HDL cholesterol and triglycerides. Theprimary difference with prior studies may be our large samplesize in a community-based cohort compared with the fewother studies that were adequately powered to compare thedifference between SAT and VAT.25,29
Similarly, for impaired fasting glucose and diabetes, mul-tiple prior studies have demonstrated relations between VATand prediabetic hyperglycemia and diabetes,9,10,18,19 but fewhave yielded significant relations for SAT. However, SAThas been shown in multiple studies to be more stronglyassociated with insulin resistance than is VAT; this has beenreviewed previously.58 Some studies of insulin resistancehave demonstrated stronger correlations with SAT than withVAT,20 especially in women. In the Insulin ResistanceAtherosclerosis Study (IRAS), both SAT and VAT wereimportant correlates of insulin resistance.21 One small studyof 47 women and men demonstrated equivalent correlationsof deep SAT and VAT with respect to cardiometabolic riskfactors.59
Although our results show that VAT is more highlycorrelated with MetS than is SAT, SAT was an important
Women Men
*p<0.05 **p<0.01 ***p<0.001 Q1 Q2 Q3 Q4
0
10
20
30
40
HTN IFG DM MetS
**
0
10
20
30
40
HTN IFG DM MetS
**
0
20
40
60
80
100
HTN IFG DM MetS
******
***
0
20
40
60
80
100
HTN IFG DM MetS
**** ***
02040
6080
100
HTN IFG DM MetS
** *****
020406080
100
HTN IFG DM MetS
*****
Prev
alen
ce (
%)
Prev
alen
ce (
%)
Prev
alen
ce (
%)
A
B
C
*****
Prevalence of hypertension (HTN), impaired fasting glucose (IFG), diabetes (DM), and MetS among normal-weight (A), overweight (B),and obese (C) individuals. Probability values represent those for linear trend across VAT quartiles and are adjusted for age and BMI.
44 Circulation July 3, 2007

correlate of the MetS. These findings are in contrast to priorstudies, which have reported that SAT is only weaklyassociated with MetS. For example, in the Health, Aging, andBody Composition (Health ABC) Study, SAT was associatedonly with MetS in normal-weight and overweight men31;however, unlike our study, the Health ABC Study focusedprimarily on older individuals.32 Therefore, SAT may be animportant adipose tissue compartment that should not beoverlooked. Only 1 very small intervention study has beenconducted to date to examine the relation of SAT reductionwith metabolic variables: In a small study of 15 women whounderwent large-volume liposuction, despite the loss ofnearly 10 kg subcutaneous fat, improvements in cardiometa-bolic risk factors were not observed.60 However, the smallsample size, associated low power, and inclusion of morbidlyobese study participants make it difficult to rule out abeneficial effect.
The strong correlation between SAT and cardiometabolicrisk factors may be driven by the results from some20,21,58 butnot all37,61 studies that have shown that insulin sensitivity isrelated to SAT and VAT. In addition to insulin resistance,relations between specific fat depots and adipocytokines maybe responsible for mediating the relations with risk factors. Inparticular, leptin has been shown to be equally, if not more,correlated to SAT compared with VAT.62 Leptin also hasbeen implicated in vascular dysfunction,63–66 which suggestsanother potential mechanism whereby SAT may be associ-ated with cardiometabolic risk factors.
Despite the statistically significant results observed withboth SAT and VAT, only VAT provided information aboveand beyond BMI and WC, suggesting that VAT may be aunique pathogenic fat depot. Similar findings have been notedamong Japanese Americans, for whom VAT but not SAT wasassociated with hypertension, even after adjustment for BMIand WC.22,57 Unfortunately, we were unable to analyze SATin the same models as BMI and WC because of the highcollinearity between the variables. In fact, the R2 of SATversus BMI/WC is much higher for SAT than for VAT (0.76versus 0.54 for men, 0.81 versus 0.64 for women).
Sex DifferencesIn our study, we found evidence for sex interactions in thatincreasing volumes of SAT and of VAT were consistentlyand more strongly associated with more adverse risk factorslevels in women than in men. To the best of our knowledge,our findings are the most comprehensive examination of sexdifferences reported to date. In the Health ABC Study, asignificant sex interaction was observed between VAT anddiabetes.10 Among women and men from the Quebec FamilyStudy and the Health, Risk factors, Exercise Training, andGenetics (HERITAGE) Family Study, only in women werehigher amounts of VAT associated with adverse CVD riskfactor profiles.67 The cause of these sex differences isuncertain but may be related to the higher amount of hepaticfree fatty acid delivery derived from lipolysis from VAT thathas been observed in women than in men.16
HeritabilityWe demonstrate moderate to high heritability for VAT andSAT, respectively, indicating that a significant portion of the
variability in these traits is familial. Two prior studies thatinvestigated the heritability of intra-abdominal fat depots68,69
have found estimates for VAT ranging from 42% to 56% andestimates for SAT of 42%. Differences between our findingsand prior published work may be due to the inclusion ofyounger participants with lower mean BMI, exclusion ofcertain metabolic conditions, and inclusion in a fitness study,which may have biased estimates. Overall, these findingssuggest that a significant proportion of variability in VAT andSAT may be due to genetic causes. Further research iswarranted to explore this further, including uncoveringgenomic regions of linkage and novel candidate genes.
ImplicationsThe relation of MetS and its components with increasingVAT quartiles, particularly in overweight and obese individ-uals, suggests that VAT in particular may confer increasingrisk within clinically defined categories of body weight. Twothirds of our study sample were either overweight or obese,statistics that mirror national data.6 Our data suggest thatfurther observational and possibly interventional studies arewarranted to test the impact of weight reduction and, inparticular, the reduction of VAT on metabolic risk factors andoverall CVD risk. Additionally, our work demonstratesstrong and significant results for SAT and VAT in relation tocardiometabolic risk factors, suggesting that SAT should notbe overlooked in regional adipose tissue research. Nonethe-less, we note that the proportion of overall variation of VATand of SAT with metabolic risk factors is moderate at best.This finding, which has been observed previously,56 suggeststhat other factors not accounted for in this study may beresponsible for the variation in metabolic risk factors. In fact,many of these traits have a substantial heritable component,with reported heritabilities for glucose being 34% to 51%70;for systolic blood pressure, 53%71; and for total cholesterol,40%.72 Therefore, the potential role of gene–adiposity inter-actions should be considered in future research.
Strengths and LimitationsStrengths of our study include the use of a community-basedsample with participants not enriched for adiposity-relatedtraits. Routine screening of metabolic risk factors was per-formed, and adjustment was made for several potentialconfounders. We used a highly reproducible volumetricmethod of SAT and VAT assessment, which accounts forheterogeneity of fat distribution throughout the abdomen. Wewere able to assess the role of SAT and VAT above andbeyond clinical anthropometry. Our study participants wereprimarily middle-aged, allowing assessment of relations be-tween fat compartments and risk factors in the absence ofsignificant comorbidity. Lastly, we have a large sample withadequate power to detect potentially smaller but significantrelations with SAT. Limitations include the cross-sectionaldesign; because the associations are not prospective, causalitycannot be inferred. Because the Framingham Offspring Studyis primarily a white sample, generalizability to other ethnicgroups is uncertain. For example, Japanese Americans andSoutheast Asians have groups with more visceral fat thanexpected for a given overall BMI,38 whereas blacks have less
Fox et al CT Adipose Tissue and Cardiometabolic Risk 45

visceral fat than do whites for a given BMI.73 Although weaccounted for sibling–sibling correlations, current analyticalmethods did not allow us to account for all familial relations.Because we did not subdivide subcutaneous fat into superfi-cial and deep compartments, we cannot comment on therelative importance of these compartments. Furthermore, wemeasured only abdominal, not truncal, SAT. Truncal SAT hasbeen shown to be more correlated to insulin resistance than isabdominal SAT in men.58,74 Finally, we had only radiograph-ic CT measures of intra-abdominal fat, not other techniquessuch as MRI or ultrasound. MRI may provide a betterresolution of fat depots, and ultrasound may be a reasonablealternative to CT75 and is less invasive. Neither MRI norultrasound places patients at risk of radiation exposure, butMRI is limited by its expense and amount of time needed toperform the actual scan.
ConclusionsBoth SAT and VAT are associated with an adverse metabolicrisk profile. However, only VAT provides information aboveand beyond easily obtainable clinical anthropometric mea-surements. Measurement of VAT may provide a more com-plete understanding of metabolic risk, and further studies arewarranted to prospectively assess the impact of the loweringof VAT and SAT on the incidence of MetS and CVD.
Sources of FundingThis work was supported by the National Heart, Lung and BloodInstitute’s Framingham Heart Study (N01-HC-25195). Dr Meigs issupported by an American Diabetes Association Career Develop-ment Award. Dr Vasan is supported in part by 2K24HL04334(National Heart, Lung, and Blood Institute, National Institutes ofHealth).
DisclosuresDr Meigs has been the recipient of research grants from GlaxoSmith-Kline, Pfizer, and Wyeth and has served on Advisory Boards forGlaxoSmithKline, Merck, Pfizer, and Lilly. The other authors reportno conflicts.
References1. Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T,
Zheng ZJ, Flegal K, O’Donnell C, Kittner S, Lloyd-Jones D, Goff DC Jr,Hong Y, Adams R, Friday G, Furie K, Gorelick P, Kissela B, Marler J,Meigs J, Roger V, Sidney S, Sorlie P, Steinberger J, Wasserthiel-SmollerS, Wilson M, Wolf P; American Heart Association Statistics Committeeand Stroke Statistics Subcommittee. Heart disease and stroke statis-tics–2006 update: a report from the American Heart Association StatisticsCommittee and Stroke Statistics Subcommittee. Circulation. 2006;113:e85–e151.
2. Fox CS, Evans JC, Larson MG, Kannel WB, Levy D. Temporal trends incoronary heart disease mortality and sudden cardiac death from 1950 to1999: the Framingham Heart Study. Circulation. 2004;110:522–527.
3. Hu FB, Stampfer MJ, Manson JE, Grodstein F, Colditz GA, Speizer FE,Willett WC. Trends in the incidence of coronary heart disease andchanges in diet and lifestyle in women. N Engl J Med. 2000;343:530–537.
4. Flegal KM, Carroll MD, Kuczmarski RJ, Johnson CL. Overweight andobesity in the United States: prevalence and trends, 1960–1994. Int JObes Relat Metab Disord. 1998;22:39–47.
5. Flegal KM, Carroll MD, Ogden CL, Johnson CL. Prevalence and trendsin obesity among US adults, 1999–2000. JAMA. 2002;288:1723–1727.
6. Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, FlegalKM. Prevalence of overweight and obesity in the United States,1999–2004. JAMA. 2006;295:1549–1555.
7. Poirier P, Despres JP. Waist circumference, visceral obesity, and cardio-vascular risk. J Cardiopulm Rehabil. 2003;23:161–169.
8. Klein S. The case of visceral fat: argument for the defense. J Clin Invest.2004;113:1530–1532.
9. Goodpaster BH, Krishnaswami S, Resnick H, Kelley DE, Haggerty C,Harris TB, Schwartz AV, Kritchevsky S, Newman AB. Associationbetween regional adipose tissue distribution and both type 2 diabetes andimpaired glucose tolerance in elderly men and women. Diabetes Care.2003;26:372–379.
10. Kanaya AM, Harris T, Goodpaster BH, Tylavsky F, Cummings SR.Adipocytokines attenuate the association between visceral adiposity anddiabetes in older adults. Diabetes Care. 2004;27:1375–1380.
11. Matsuzawa Y. Therapy insight: adipocytokines in metabolic syndromeand related cardiovascular disease. Nat Clin Pract Cardiovasc Med.2006;3:35–42.
12. Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relationto the metabolic syndrome. Endocr Rev. 2000;21:697–738.
13. Yatagai T, Nagasaka S, Taniguchi A, Fukushima M, Nakamura T, KuroeA, Nakai Y, Ishibashi S. Hypoadiponectinemia is associated with visceralfat accumulation and insulin resistance in Japanese men with type 2diabetes mellitus. Metabolism. 2003;52:1274–1278.
14. Bacha F, Saad R, Gungor N, Arslanian SA. Adiponectin in youth: rela-tionship to visceral adiposity, insulin sensitivity, and beta-cell function.Diabetes Care. 2004;27:547–552.
15. Saijo Y, Kiyota N, Kawasaki Y, Miyazaki Y, Kashimura J, Fukuda M,Kishi R. Relationship between C-reactive protein and visceral adiposetissue in healthy Japanese subjects. Diabetes Obes Metab. 2004;6:249–258.
16. Nielsen S, Guo Z, Johnson CM, Hensrud DD, Jensen MD. Splanchniclipolysis in human obesity. J Clin Invest. 2004;113:1582–1588.
17. Kuk JL, Lee S, Heymsfield SB, Ross R. Waist circumference andabdominal adipose tissue distribution: influence of age and sex. Am J ClinNutr. 2005;81:1330–1334.
18. Hayashi T, Boyko EJ, Leonetti DL, McNeely MJ, Newell-Morris L, KahnSE, Fujimoto WY. Visceral adiposity and the risk of impaired glucosetolerance: a prospective study among Japanese Americans. DiabetesCare. 2003;26:650–655.
19. Boyko EJ, Fujimoto WY, Leonetti DL, Newell-Morris L. Visceral adi-posity and risk of type 2 diabetes: a prospective study among JapaneseAmericans. Diabetes Care. 2000;23:465–471.
20. Tulloch-Reid MK, Hanson RL, Sebring NG, Reynolds JC, Premkumar A,Genovese DJ, Sumner AE. Both subcutaneous and visceral adipose tissuecorrelate highly with insulin resistance in African Americans. Obes Res.2004;12:1352–1359.
21. Wagenknecht LE, Langefeld CD, Scherzinger AL, Norris JM, HaffnerSM, Saad MF, Bergman RN. Insulin sensitivity, insulin secretion, andabdominal fat: the Insulin Resistance Atherosclerosis Study (IRAS)Family Study. Diabetes. 2003;52:2490–2496.
22. Hayashi T, Boyko EJ, Leonetti DL, McNeely MJ, Newell-Morris L, KahnSE, Fujimoto WY. Visceral adiposity is an independent predictor ofincident hypertension in Japanese Americans. Ann Intern Med. 2004;140:992–1000.
23. Sironi AM, Gastaldelli A, Mari A, Ciociaro D, Positano V, Buzzigoli E,Ghione S, Turchi S, Lombardi M, Ferrannini E. Visceral fat in hyper-tension: influence on insulin resistance and beta-cell function. Hyper-tension. 2004;44:127–133.
24. Ding J, Visser M, Kritchevsky SB, Nevitt M, Newman A, Sutton-TyrrellK, Harris TB. The association of regional fat depots with hypertension inolder persons of white and African American ethnicity. Am J Hypertens.2004;17:971–976.
25. Pascot A, Lemieux S, Lemieux I, Prud’homme D, Tremblay A, BouchardC, Nadeau A, Couillard C, Tchernof A, Bergeron J, Despres JP. Age-related increase in visceral adipose tissue and body fat and the metabolicrisk profile of premenopausal women. Diabetes Care. 1999;22:1471–1478.
26. Nagaretani H, Nakamura T, Funahashi T, Kotani K, Miyanaga M,Tokunaga K, Takahashi M, Nishizawa H, Kishida K, Kuriyama H, HottaK, Yamashita S, Matsuzawa Y. Visceral fat is a major contributor formultiple risk factor clustering in Japanese men with impaired glucosetolerance. Diabetes Care. 2001;24:2127–2133.
27. Nicklas BJ, Penninx BW, Ryan AS, Berman DM, Lynch NA, Dennis KE.Visceral adipose tissue cutoffs associated with metabolic risk factors forcoronary heart disease in women. Diabetes Care. 2003;26:1413–1420.
28. Lemieux S, Prud’homme D, Moorjani S, Tremblay A, Bouchard C,Lupien PJ, Despres JP. Do elevated levels of abdominal visceral adiposetissue contribute to age-related differences in plasma lipoprotein concen-trations in men? Atherosclerosis. 1995;118:155–164.
46 Circulation July 3, 2007

29. Kobayashi H, Nakamura T, Miyaoka K, Nishida M, Funahashi T,Yamashita S, Matsuzawa Y. Visceral fat accumulation contributes toinsulin resistance, small-sized low-density lipoprotein, and progression ofcoronary artery disease in middle-aged non-obese Japanese men. Jpn CircJ. 2001;65:193–199.
30. Mori Y, Hoshino K, Yokota K, Yokose T, Tajima N. Increased visceralfat and impaired glucose tolerance predict the increased risk of metabolicsyndrome in Japanese middle-aged men. Exp Clin Endocrinol Diabetes.2005;113:334–339.
31. Goodpaster BH, Krishnaswami S, Harris TB, Katsiaras A, KritchevskySB, Simonsick EM, Nevitt M, Holvoet P, Newman AB. Obesity, regionalbody fat distribution, and the metabolic syndrome in older men andwomen. Arch Intern Med. 2005;165:777–783.
32. Carr DB, Utzschneider KM, Hull RL, Kodama K, Retzlaff BM, BrunzellJD, Shofer JB, Fish BE, Knopp RH, Kahn SE. Intra-abdominal fat is amajor determinant of the National Cholesterol Education Program AdultTreatment Panel III criteria for the metabolic syndrome. Diabetes. 2004;53:2087–2094.
33. von Eyben FE, Mouritsen E, Holm J, Montvilas P, Dimcevski G, SuciuG, Helleberg I, Kristensen L, von Eyben R. Intra-abdominal obesity andmetabolic risk factors: a study of young adults. Int J Obes Relat MetabDisord. 2003;27:941–949.
34. Ross R, Freeman J, Hudson R, Janssen I. Abdominal obesity, musclecomposition, and insulin resistance in premenopausal women. J ClinEndocrinol Metab. 2002;87:5044–5051.
35. Fujimoto WY, Bergstrom RW, Boyko EJ, Chen KW, Leonetti DL,Newell-Morris L, Shofer JB, Wahl PW. Visceral adiposity and incidentcoronary heart disease in Japanese-American men: the 10-year follow-upresults of the Seattle Japanese-American Community Diabetes Study.Diabetes Care. 1999;22:1808–1812.
36. Hernandez-Ono A, Monter-Carreola G, Zamora-Gonzalez J, Cardoso-Saldana G, Posadas-Sanchez R, Torres-Tamayo M, Posadas-Romero C.Association of visceral fat with coronary risk factors in apopulation-based sample of postmenopausal women. Int J Obes RelatMetab Disord. 2002;26:33–39.
37. Tong J, Fujimoto WY, Kahn SE, Weigle DS, McNeely MJ, Leonetti DL,Shofer JB, Boyko EJ. Insulin, C-peptide, and leptin concentrations predictincreased visceral adiposity at 5- and 10-year follow-ups in nondiabeticJapanese Americans. Diabetes. 2005;54:985–990.
38. Park YW, Allison DB, Heymsfield SB, Gallagher D. Larger amounts ofvisceral adipose tissue in Asian Americans. Obes Res. 2001;9:381–387.
39. Maurovich-Horvat P, Massaro J, Fox CS, Moselewski F, O’Donnell CJ,Hoffmann U. Comparison of anthropometric, area- and volume-basedassessment of abdominal subcutaneous and visceral adipose tissuevolumes using multi-detector computed tomography. Int J Obes (Lond).2007;31:500–506.
40. Dawber TR, Kannel WB, Lyell LP. An approach to longitudinal studiesin a community: the Framingham Heart Study. Ann N Y Acad Sci. 1963;107:539–556.
41. Shurtleff D. Some characteristics related to the incidence of cardiovas-cular disease and death: Framingham study, 18-year follow-up. In:Kannel WB, Fordon T, eds. The Framingham Study: An EpidemiologicalInvestigation of Cardiovascular Disease. Washington, DC: Departmentof Health, Education, and Welfare; 1973. DHEW publication No. NIH74–599, section 30.
42. Hopkins PN, Ellison RC, Province MA, Pankow JS, Carr JJ, Arnett DK,Lewis CE, Heiss G, Hunt SC. Association of coronary artery calcifiedplaque with clinical coronary heart disease in the National Heart, Lung,and Blood Institute’s Family Heart Study. Am J Cardiol. 2006;97:1564–1569.
43. Executive Summary of the third report of the National Cholesterol Edu-cation Program (NCEP) Expert Panel on Detection, Evaluation, andTreatment of High Blood Cholesterol in Adults (Adult Treatment PanelIII). JAMA. 2001;285:2486–2497.
44. SAS/STAT User’s Guide, Version 8. Cary, NC: SAS Institute Inc; 2000.45. Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in
general pedigrees. Am J Hum Genet. 1998;62:1198–1211.46. Bjorntorp P. “Portal” adipose tissue as a generator of risk factors for
cardiovascular disease and diabetes. Arteriosclerosis. 1990;10:493–496.47. Lemieux I, Pascot A, Prud’homme D, Almeras N, Bogaty P, Nadeau A,
Bergeron J, Despres JP. Elevated C-reactive protein: another componentof the atherothrombotic profile of abdominal obesity. ArteriosclerThromb Vasc Biol. 2001;21:961–967.
48. Azuma K, Katsukawa F, Oguchi S, Murata M, Yamazaki H, Shimada A,Saruta T. Correlation between serum resistin level and adiposity in obeseindividuals. Obes Res. 2003;11:997–1001.
49. Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, KishimotoK, Matsuki Y, Murakami M, Ichisaka T, Murakami H, Watanabe E,Takagi T, Akiyoshi M, Ohtsubo T, Kihara S, Yamashita S, Makishima M,Funahashi T, Yamanaka S, Hiramatsu R, Matsuzawa Y, Shimomura I.Visfatin: a protein secreted by visceral fat that mimics the effects ofinsulin. Science. 2005;307:426–430.
50. Cigolini M, Targher G, Bergamo Andreis I, Tonoli M, Agostino G, DeSandre G. Visceral fat accumulation and its relation to plasma hemostaticfactors in healthy men. Arterioscler Thromb Vasc Biol. 1996;16:368–374.
51. Mertens I, Van Gaal LF. Visceral fat as a determinant of fibrinolysis andhemostasis. Semin Vasc Med. 2005;5:48–55.
52. Miyazawa-Hoshimoto S, Takahashi K, Bujo H, Hashimoto N, Saito Y.Elevated serum vascular endothelial growth factor is associated withvisceral fat accumulation in human obese subjects. Diabetologia. 2003;46:1483–1488.
53. Giusti V, Suter M, Verdumo C, Gaillard RC, Burckhardt P, Pralong FP.Molecular determinants of human adipose tissue: differences betweenvisceral and subcutaneous compartments in obese women. J Clin Endo-crinol Metab. 2004;89:1379–1384.
54. Arner P. Regional differences in protein production by human adiposetissue. Biochem Soc Trans. 2001;29:72–75.
55. Dusserre E, Moulin P, Vidal H. Differences in mRNA expression of theproteins secreted by the adipocytes in human subcutaneous and visceraladipose tissues. Biochim Biophys Acta. 2000;1500:88–96.
56. Vega GL, Adams-Huet B, Peshock R, Willett D, Shah B, Grundy SM.Influence of body fat content and distribution on variation in metabolicrisk. J Clin Endocrinol Metab. 2006;91:4459–4466.
57. Hayashi T, Boyko EJ, Leonetti DL, McNeely MJ, Newell-Morris L, KahnSE, Fujimoto WY. Visceral adiposity and the prevalence of hypertensionin Japanese Americans. Circulation. 2003;108:1718–1723.
58. Garg A. Regional adiposity and insulin resistance. J Clin EndocrinolMetab. 2004;89:4206–4210.
59. Kelley DE, Thaete FL, Troost F, Huwe T, Goodpaster BH. Subdivisionsof subcutaneous abdominal adipose tissue and insulin resistance.Am J Physiol Endocrinol Metab. 2000;278:E941–E948.
60. Klein S, Fontana L, Young VL, Coggan AR, Kilo C, Patterson BW,Mohammed BS. Absence of an effect of liposuction on insulin action andrisk factors for coronary heart disease. N Engl J Med. 2004;350:2549–2557.
61. Goodpaster BH, Kelley DE, Wing RR, Meier A, Thaete FL. Effects ofweight loss on regional fat distribution and insulin sensitivity in obesity.Diabetes. 1999;48:839–847.
62. Ruhl CE, Everhart JE, Ding J, Goodpaster BH, Kanaya AM, SimonsickEM, Tylavsky FA, Harris TB. Serum leptin concentrations and bodyadipose measures in older black and white adults. Am J Clin Nutr.2004;80:576–583.
63. Sundell J, Huupponen R, Raitakari OT, Nuutila P, Knuuti J. High serumleptin is associated with attenuated coronary vasoreactivity. Obes Res.2003;11:776–782.
64. Yamagishi S, Inagaki Y, Amano S, Okamoto T, Takeuchi M.Up-regulation of vascular endothelial growth factor and down-regulationof pigment epithelium-derived factor messenger ribonucleic acid levels inleptin-exposed cultured retinal pericytes. Int J Tissue React. 2002;24:137–142.
65. Cooke JP, Oka RK. Does leptin cause vascular disease? Circulation.2002;106:1904–1905.
66. Singhal A, Farooqi IS, Cole TJ, O’Rahilly S, Fewtrell M, Kattenhorn M,Lucas A, Deanfield J. Influence of leptin on arterial distensibility: a novellink between obesity and cardiovascular disease? Circulation. 2002;106:1919–1924.
67. Tanaka S, Togashi K, Rankinen T, Perusse L, Leon AS, Rao DC, SkinnerJS, Wilmore JH, Despres JP, Bouchard C. Sex differences in the rela-tionships of abdominal fat to cardiovascular disease risk among normal-weight white subjects. Int J Obes Relat Metab Disord. 2004;28:320–323.
68. Hong Y, Rice T, Gagnon J, Despres JP, Nadeau A, Perusse L, BouchardC, Leon AS, Skinner JS, Wilmore JH, Rao DC. Familial clustering ofinsulin and abdominal visceral fat: the HERITAGE Family Study. J ClinEndocrinol Metab. 1998;83:4239–4245.
69. Perusse L, Despres JP, Lemieux S, Rice T, Rao DC, Bouchard C. Familialaggregation of abdominal visceral fat level: results from the QuebecFamily Study. Metabolism. 1996;45:378–382.
Fox et al CT Adipose Tissue and Cardiometabolic Risk 47

70. Meigs JB, Panhuysen CI, Myers RH, Wilson PW, Cupples LA. Agenome-wide scan for loci linked to plasma levels of glucose andHbA(1c) in a community-based sample of Caucasian pedigrees: theFramingham Offspring Study. Diabetes. 2002;51:833–840.
71. Levy D, DeStefano AL, Larson MG, O’Donnell CJ, Lifton RP, Gavras H,Cupples LA, Myers RH. Evidence for a gene influencing blood pressureon chromosome 17: genome scan linkage results for longitudinal bloodpressure phenotypes in subjects from the Framingham Heart Study.Hypertension. 2000;36:477–483.
72. Mathias RA, Roy-Gagnon MH, Justice CM, Papanicolaou GJ, Fan YT,Pugh EW, Wilson AF. Comparison of year-of-exam- and age-matched
estimates of heritability in the Framingham Heart Study data. BMCGenet. 2003;4(suppl 1):S36.
73. Tittelbach TJ, Berman DM, Nicklas BJ, Ryan AS, Goldberg AP. Racialdifferences in adipocyte size and relationship to the metabolic syndromein obese women. Obes Res. 2004;12:990–998.
74. Abate N, Garg A, Peshock RM, Stray-Gundersen J, Grundy SM. Rela-tionships of generalized and regional adiposity to insulin sensitivity inmen. J Clin Invest. 1995;96:88–98.
75. Stolk RP, Wink O, Zelissen PM, Meijer R, van Gils AP, Grobbee DE. Validityand reproducibility of ultrasonography for the measurement of intra-abdominaladipose tissue. Int J Obes Relat Metab Disord. 2001;25:1346–1351.
CLINICAL PERSPECTIVEVisceral adipose tissue (VAT) compartments may confer increased metabolic risk. The incremental utility of measuringboth VAT and subcutaneous abdominal adipose tissue (SAT) in association with metabolic risk factors and underlyingheritability has not been well described in a population-based setting. Participants from the Framingham Heart Studyunderwent multidetector computed tomography assessment of SAT and VAT volumes. SAT and VAT were significantlyassociated with increased odds of hypertension, impaired fasting glucose, diabetes, and metabolic syndrome. In women,relations between VAT and risk factors were consistently stronger than in men. VAT was more strongly correlated withmost metabolic risk factors than was SAT. Furthermore, VAT but not SAT contributed significantly to risk factor variationafter adjustment for body mass index and waist circumference. Among overweight and obese individuals, the prevalenceof hypertension, impaired fasting glucose, and metabolic syndrome increased linearly and significantly across increasingVAT quartiles. Heritability values for SAT and VAT were 57% and 36%, respectively. Although both SAT and VAT arecorrelated with metabolic risk factors, VAT remains more strongly associated with an adverse metabolic risk profile evenafter accounting for standard anthropometric indices. Our findings are consistent with the hypothesized role of visceral fatas a unique, pathogenic fat depot. Measurement of VAT may provide a more complete understanding of metabolic riskassociated with variation in fat distribution.
48 Circulation July 3, 2007

Metoprolol Reverses Left Ventricular Remodeling inPatients With Asymptomatic Systolic Dysfunction
The REversal of VEntricular Remodeling with Toprol-XL(REVERT) Trial
Wilson S. Colucci, MD; Theodore J. Kolias, MD; Kirkwood F. Adams, MD;William F. Armstrong, MD; Jalal K. Ghali, MD; Stephen S. Gottlieb, MD; Barry Greenberg, MD;
Michael I. Klibaner, MD, PhD; Marrick L. Kukin, MD; Jennifer E. Sugg, MS; on behalf of theREVERT Study Group*
Background—There are no randomized, controlled trial data to support the benefit of �-blockers in patients withasymptomatic left ventricular systolic dysfunction. We investigated whether �-blocker therapy ameliorates leftventricular remodeling in asymptomatic patients with left ventricular systolic dysfunction.
Method and Results—Patients with left ventricular ejection fraction �40%, mild left ventricular dilation, and no symptomsof heart failure (New York Heart Association class I) were randomly assigned to receive extended-release metoprololsuccinate (Toprol-XL, AstraZeneca) 200 mg or 50 mg or placebo for 12 months. Echocardiographic assessments of leftventricular end-systolic volume, end-diastolic volume, mass, and ejection fraction were performed at baseline and at 6and 12 months. The 149 patients randomized to the 3 treatment groups (200 mg, n�48; 50 mg, n�48; and placebo,n�53) were similar with regard to all baseline characteristics including age (mean, 66 years), gender (74% male),plasma brain natriuretic peptide (79 pg/mL), left ventricular end-diastolic volume index (110 mL/m2), and leftventricular ejection fraction (27%). At 12 months in the 200-mg group, there was a 14�3 mL/m2 decrease (least squaremean�SE) in end-systolic volume index and a 6�1% increase in left ventricular ejection fraction (P�0.05 versusbaseline and placebo for both). The decrease in end-diastolic volume index (14�3) was different from that seen atbaseline (P�0.05) but not with placebo. In the 50-mg group, end-systolic and end-diastolic volume indexes decreasedrelative to baseline but were not different from what was seen with placebo, whereas ejection fraction increased by4�1% (P�0.05 versus baseline and placebo).
Conclusion—�-Blocker therapy can ameliorate left ventricular remodeling in asymptomatic patients with left ventricularsystolic dysfunction. (Circulation. 2007;116:49-56.)
Key Words: heart failure � receptors, adrenergic, beta � remodeling � ventricles
Asymptomatic left ventricular (LV) systolic dysfunctionis common in the general population, with a prevalence
on the order of 3%,1,2 constituting a high percentage of allpatients with LV dysfunction. For example, in the OlmsteadCounty population, less than half of all patients with an LVejection fraction (EF) �40% had been diagnosed with con-gestive heart failure (HF).2 Although asymptomatic, thesepatients are at high risk for developing clinical HF. Inasymptomatic patients with a LVEF �40% in the Framing-
ham Heart Study population, the annual incidence of symp-tomatic HF was �6%, and the annual mortality rate was�8%.1 Despite the important consequences of asymptomaticLV systolic dysfunction, very few clinical trials of therapeuticagents have been conducted in this population.
In patients with symptoms of HF due to systolic LVdysfunction, extensive clinical trial data have demonstratedthat �-blockers improve survival, decrease hospitalizationsrelated to HF, and alleviate symptoms.3–6 The improved
Continuing medical education (CME) credit is available for this article. Go to http://cme.ahajournals.org to take the quiz.Received October 5, 2006; accepted April 20, 2007.From Boston University Medical Center, Boston, Mass (W.S.C.); University of Michigan Medical Center, Ann Arbor (T.J.K., W.F.A.); University of
North Carolina School of Medicine, Chapel Hill (K.F.A.); Wayne State University, Detroit, Mich (J.K.G.); University of Maryland Hospital, Baltimore(S.S.G.); University of California at San Diego (B.G.); AstraZeneca LP, Wilmington, Del (M.I.K., J.E.S.); and St Luke’s–Roosevelt Hospitals, ColumbiaUniversity College of Physicians and Surgeons, New York, NY (M.L.K.).
*All members of the REVERT Study Group are listed in the Appendix.Clinical trial registration information—URL: http://www.clinicaltrials.gov. Unique identifier: NCT00038077.Guest Editor for this article was Martin M. LeWinter, MD.Correspondence to Wilson S. Colucci, MD, Cardiovascular Medicine, Boston University Medical Center, 88 E Newton St, Boston, MA 02118. E-mail
[email protected]© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.106.666016

outcomes are associated with amelioration of LV remodelingcharacterized by decreases in end-diastolic and end-systolicvolumes and an increase in EF.7–10 The studies that havedemonstrated these beneficial effects on outcomes and re-modeling have been performed almost exclusively in symp-tomatic patients in New York Heart Association (NYHA)functional classes II, III, or IV.
Although an exception would be the Australia/New Zea-land Carvedilol Trial, in which approximately one third ofsubjects were asymptomatic, the survival benefits of carve-dilol in that trial were not analyzed with regard to functionalclass.11 Likewise, although some patients in the CarvedilolProspective Randomized Cumulative Survival (CAPRI-CORN) trial had asymptomatic LV dysfunction,12 the impactof therapy was not analyzed with regard to functional classand would not be directly applicable to patients with chronicLV dysfunction.
There have been no randomized controlled trials of theeffects of �-blockers on clinical outcomes or remodeling inpatients who have chronic LV systolic dysfunction but arefree of symptoms. Theoretically, patients with asymptomaticLV dysfunction may be less responsive to �-blockers becausethe degree of sympathetic nervous system activation isless.13–15 Alternatively, there is reason to believe that�-blockers would be effective in such patients because somestudies have shown clinical benefit in patients with mild (eg,predominantly NYHA class II) symptoms of HF.16
Because of the lack of direct evidence from randomizedcontrolled trials, the current recommendation for the use of a�-blocker in patients with a chronic reduction in LVEF but noHF symptoms is based only on expert opinion.17 Furthermore,it is unlikely that a placebo-controlled study can be performedto test the effects of �-blockade on clinical outcomes in thispopulation.
There is evidence that the outcome benefits of �-blockersin patients with systolic LV dysfunction are related to theantiremodeling effect, which might therefore be used as areasonable surrogate for clinical benefit.18 Accordingly, wedesigned a placebo-controlled, randomized trial, REversal ofVEntricular Remodeling with Toprol-XL (REVERT), to testthe hypothesis that �-blocker therapy would ameliorate LVremodeling in asymptomatic (NYHA functional class I)patients with LV systolic dysfunction.
MethodsEntry CriteriaTo be eligible, patients must have had a LVEF �40% and mild LVdilatation (LV end-diastolic volume index [LVEDVI] �75 mL/m2)due to idiopathic, ischemic, or hypertensive cardiomyopathy andmust have had no symptoms of HF for at least 2 months. Thedetermination of LV systolic dysfunction and dilation was based ona screening echocardiogram that was performed within 14 days ofrandomization and interpreted by the core laboratory. AsymptomaticLV systolic dysfunction was defined as no limitation of ordinaryphysical activity because of fatigue or dyspnea (NYHA functionalclass I). Patients previously treated for symptomatic HF wereallowed to participate if they met the inclusion and exclusion criteriafor the study. The use of an angiotensin-converting enzyme (ACE)inhibitor or angiotensin receptor blocker for at least 3 months, astolerated, was required before enrollment. Patients must have had nochanges in the doses of their cardiovascular medications, including
ACE inhibitors, angiotensin receptor blockers, diuretics, digoxin,and/or vasodilators for at least 3 months before randomization.
Patients were excluded if they had indications for or contraindi-cations to �-blocker therapy or took �-blockers (including ophthal-mic preparations) for �1 week during the 6 months precedingrandomization. Also excluded were patients who, during the 6months before randomization, had myocardial infarction, unstableangina, coronary intervention, or hospitalization for cardiovascular-related causes, as well as patients with heart rate �60 bpm, sittingblood pressure �140/90 mm Hg, heart block greater than first degreenot treated with a permanent pacemaker, history of ventricular oratrial fibrillation, or serum creatinine �3 mg/dL. The institutionalreview boards of the participating institutions approved the protocol,and all participants provided written informed consent. The studywas conducted in accordance with the Declaration of Helsinki.
Echocardiographic MeasurementsTwo-dimensional echocardiograms with Doppler were recorded atscreening (baseline) and at 6 and 12 months, with the same scannerused for each patient. The videotapes were evaluated in a blindedfashion by the core echocardiography laboratory at the University ofMichigan.
The echocardiograms were analyzed with the use of a dedicatedoffline echocardiography analysis system (TomTec Imaging Sys-tems, Munich, Germany). LV end-systolic volume (LVESV),LVEDV, and LVEF were measured by Simpson’s method in theapical 4-chamber view, which was used for the main analyses, aswell as the apical 2-chamber view when possible. LV mass wascalculated from the 2-dimensional parasternal long-axis view withthe use of the Penn cubed formula.19 LVESV index (LVESVI),LVEDVI, and LV mass index (LVMI) were determined by dividingvolume or mass by body surface area.
Treatment RegimenPatients who met inclusion/exclusion criteria were randomized in a1:1:1 ratio to a 52-week treatment with metoprolol succinateextended-release tablets (metoprolol succinate, TOPROL-XL, Astra-Zeneca, Wilmington, Del) or placebo as follows: (1) metoprololsuccinate 200 mg/d (high-dose group); (2) metoprolol succinate 50mg/d (low-dose group); or (3) placebo. The study drug was force-titrated to the assigned dose over the first 2 months on the basis oftolerability, and the achieved dose was maintained as tolerated untilthe end of the study. Drug blinding was achieved with the use of adouble-blind, double-dummy technique. Treatment compliance wasverified by pill count of returned study medication at each visit.
Brain Natriuretic PeptideIn a substudy that enrolled 82 patients, venous blood was collectedat baseline and at 6 and 12 months for determination of plasma brainnatriuretic peptide, which was measured by radioimmunoassay(Quest Diagnostics, Van Nuys, Calif).
Statistical AnalysesThe change in LVESVI from baseline to 12 months was chosen asthe prespecified primary end point because it reflects changes in bothLV size and systolic function and has been shown to be a sensitiveindex of LV remodeling.7 The power calculation was based on a SDfor LVESVI at 12 months of 15 mL/m2 and a dropout rate of 20%.With an � of 0.050 for a 2-sided test, 150 patients would provide94% power to detect a change of 12 mL/m2 and 84% power to detecta change of 10 mL/m2. Prespecified key secondary end pointsincluded the change from baseline in LVESVI at month 6 andchanges from baseline in LVEDVI, LVMI, and LVEF at months 6and 12. Efficacy was analyzed by a modified intention-to-treatpopulation (n�149 patients) who took at least 1 dose of studymedication after randomization and had at least 1 follow-up echo-cardiogram. All available data were analyzed, and no substitutionswere made for missing values. Pairwise comparisons were madebetween the high-dose group and placebo (primary comparison) andbetween the low-dose group and placebo (secondary comparison) for
50 Circulation July 3, 2007
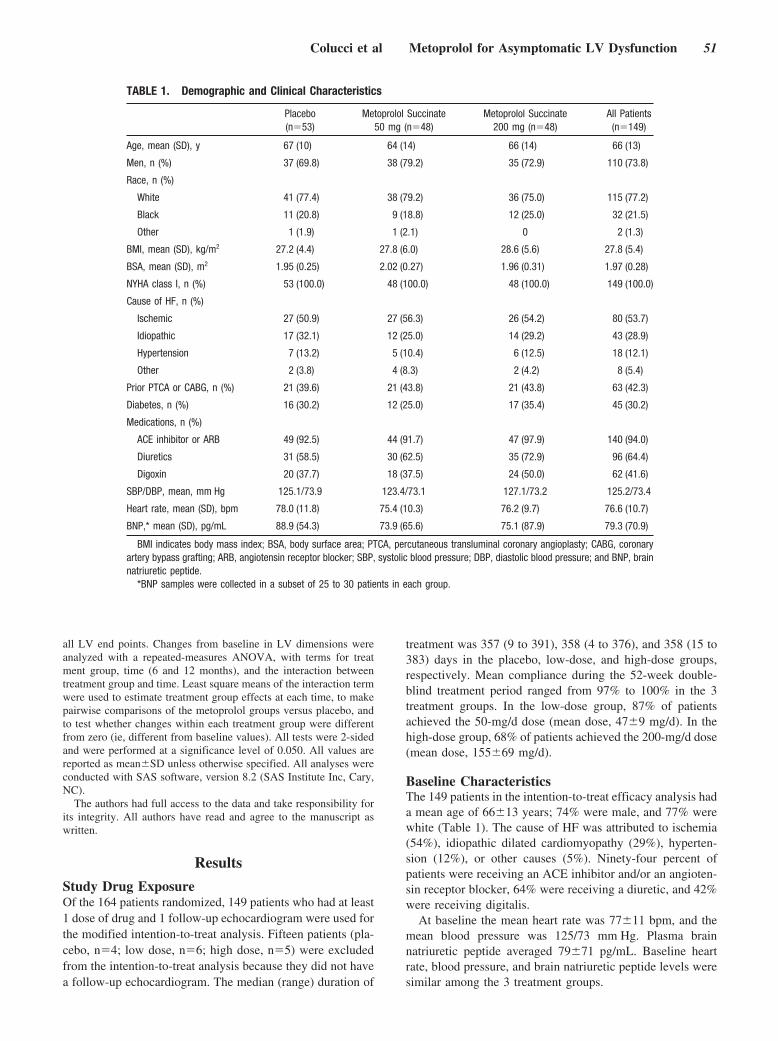
all LV end points. Changes from baseline in LV dimensions wereanalyzed with a repeated-measures ANOVA, with terms for treatment group, time (6 and 12 months), and the interaction betweentreatment group and time. Least square means of the interaction termwere used to estimate treatment group effects at each time, to makepairwise comparisons of the metoprolol groups versus placebo, andto test whether changes within each treatment group were differentfrom zero (ie, different from baseline values). All tests were 2-sidedand were performed at a significance level of 0.050. All values arereported as mean�SD unless otherwise specified. All analyses wereconducted with SAS software, version 8.2 (SAS Institute Inc, Cary,NC).
The authors had full access to the data and take responsibility forits integrity. All authors have read and agree to the manuscript aswritten.
Results
Study Drug ExposureOf the 164 patients randomized, 149 patients who had at least1 dose of drug and 1 follow-up echocardiogram were used forthe modified intention-to-treat analysis. Fifteen patients (pla-cebo, n�4; low dose, n�6; high dose, n�5) were excludedfrom the intention-to-treat analysis because they did not havea follow-up echocardiogram. The median (range) duration of
treatment was 357 (9 to 391), 358 (4 to 376), and 358 (15 to383) days in the placebo, low-dose, and high-dose groups,respectively. Mean compliance during the 52-week double-blind treatment period ranged from 97% to 100% in the 3treatment groups. In the low-dose group, 87% of patientsachieved the 50-mg/d dose (mean dose, 47�9 mg/d). In thehigh-dose group, 68% of patients achieved the 200-mg/d dose(mean dose, 155�69 mg/d).
Baseline CharacteristicsThe 149 patients in the intention-to-treat efficacy analysis hada mean age of 66�13 years; 74% were male, and 77% werewhite (Table 1). The cause of HF was attributed to ischemia(54%), idiopathic dilated cardiomyopathy (29%), hyperten-sion (12%), or other causes (5%). Ninety-four percent ofpatients were receiving an ACE inhibitor and/or an angioten-sin receptor blocker, 64% were receiving a diuretic, and 42%were receiving digitalis.
At baseline the mean heart rate was 77�11 bpm, and themean blood pressure was 125/73 mm Hg. Plasma brainnatriuretic peptide averaged 79�71 pg/mL. Baseline heartrate, blood pressure, and brain natriuretic peptide levels weresimilar among the 3 treatment groups.
TABLE 1. Demographic and Clinical Characteristics
Placebo(n�53)
Metoprolol Succinate50 mg (n�48)
Metoprolol Succinate200 mg (n�48)
All Patients(n�149)
Age, mean (SD), y 67 (10) 64 (14) 66 (14) 66 (13)
Men, n (%) 37 (69.8) 38 (79.2) 35 (72.9) 110 (73.8)
Race, n (%)
White 41 (77.4) 38 (79.2) 36 (75.0) 115 (77.2)
Black 11 (20.8) 9 (18.8) 12 (25.0) 32 (21.5)
Other 1 (1.9) 1 (2.1) 0 2 (1.3)
BMI, mean (SD), kg/m2 27.2 (4.4) 27.8 (6.0) 28.6 (5.6) 27.8 (5.4)
BSA, mean (SD), m2 1.95 (0.25) 2.02 (0.27) 1.96 (0.31) 1.97 (0.28)
NYHA class I, n (%) 53 (100.0) 48 (100.0) 48 (100.0) 149 (100.0)
Cause of HF, n (%)
Ischemic 27 (50.9) 27 (56.3) 26 (54.2) 80 (53.7)
Idiopathic 17 (32.1) 12 (25.0) 14 (29.2) 43 (28.9)
Hypertension 7 (13.2) 5 (10.4) 6 (12.5) 18 (12.1)
Other 2 (3.8) 4 (8.3) 2 (4.2) 8 (5.4)
Prior PTCA or CABG, n (%) 21 (39.6) 21 (43.8) 21 (43.8) 63 (42.3)
Diabetes, n (%) 16 (30.2) 12 (25.0) 17 (35.4) 45 (30.2)
Medications, n (%)
ACE inhibitor or ARB 49 (92.5) 44 (91.7) 47 (97.9) 140 (94.0)
Diuretics 31 (58.5) 30 (62.5) 35 (72.9) 96 (64.4)
Digoxin 20 (37.7) 18 (37.5) 24 (50.0) 62 (41.6)
SBP/DBP, mean, mm Hg 125.1/73.9 123.4/73.1 127.1/73.2 125.2/73.4
Heart rate, mean (SD), bpm 78.0 (11.8) 75.4 (10.3) 76.2 (9.7) 76.6 (10.7)
BNP,* mean (SD), pg/mL 88.9 (54.3) 73.9 (65.6) 75.1 (87.9) 79.3 (70.9)
BMI indicates body mass index; BSA, body surface area; PTCA, percutaneous transluminal coronary angioplasty; CABG, coronaryartery bypass grafting; ARB, angiotensin receptor blocker; SBP, systolic blood pressure; DBP, diastolic blood pressure; and BNP, brainnatriuretic peptide.
*BNP samples were collected in a subset of 25 to 30 patients in each group.
Colucci et al Metoprolol for Asymptomatic LV Dysfunction 51
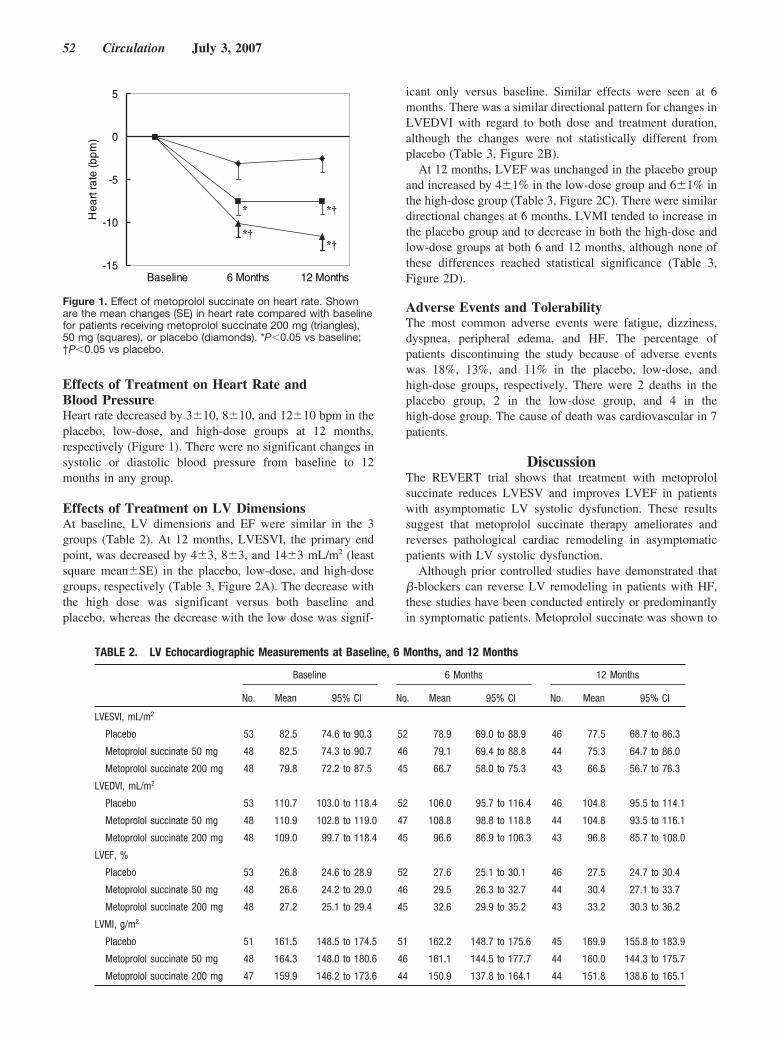
Effects of Treatment on Heart Rate andBlood PressureHeart rate decreased by 3�10, 8�10, and 12�10 bpm in theplacebo, low-dose, and high-dose groups at 12 months,respectively (Figure 1). There were no significant changes insystolic or diastolic blood pressure from baseline to 12months in any group.
Effects of Treatment on LV DimensionsAt baseline, LV dimensions and EF were similar in the 3groups (Table 2). At 12 months, LVESVI, the primary endpoint, was decreased by 4�3, 8�3, and 14�3 mL/m2 (leastsquare mean�SE) in the placebo, low-dose, and high-dosegroups, respectively (Table 3, Figure 2A). The decrease withthe high dose was significant versus both baseline andplacebo, whereas the decrease with the low dose was signif-
icant only versus baseline. Similar effects were seen at 6months. There was a similar directional pattern for changes inLVEDVI with regard to both dose and treatment duration,although the changes were not statistically different fromplacebo (Table 3, Figure 2B).
At 12 months, LVEF was unchanged in the placebo groupand increased by 4�1% in the low-dose group and 6�1% inthe high-dose group (Table 3, Figure 2C). There were similardirectional changes at 6 months. LVMI tended to increase inthe placebo group and to decrease in both the high-dose andlow-dose groups at both 6 and 12 months, although none ofthese differences reached statistical significance (Table 3,Figure 2D).
Adverse Events and TolerabilityThe most common adverse events were fatigue, dizziness,dyspnea, peripheral edema, and HF. The percentage ofpatients discontinuing the study because of adverse eventswas 18%, 13%, and 11% in the placebo, low-dose, andhigh-dose groups, respectively. There were 2 deaths in theplacebo group, 2 in the low-dose group, and 4 in thehigh-dose group. The cause of death was cardiovascular in 7patients.
DiscussionThe REVERT trial shows that treatment with metoprololsuccinate reduces LVESV and improves LVEF in patientswith asymptomatic LV systolic dysfunction. These resultssuggest that metoprolol succinate therapy ameliorates andreverses pathological cardiac remodeling in asymptomaticpatients with LV systolic dysfunction.
Although prior controlled studies have demonstrated that�-blockers can reverse LV remodeling in patients with HF,these studies have been conducted entirely or predominantlyin symptomatic patients. Metoprolol succinate was shown to
-15
-10
-5
0
5)
mpb( etar traeH
Baseline 6 Months 12 Months
*†*†
* *†
Figure 1. Effect of metoprolol succinate on heart rate. Shownare the mean changes (SE) in heart rate compared with baselinefor patients receiving metoprolol succinate 200 mg (triangles),50 mg (squares), or placebo (diamonds). *P�0.05 vs baseline;†P�0.05 vs placebo.
TABLE 2. LV Echocardiographic Measurements at Baseline, 6 Months, and 12 Months
Baseline 6 Months 12 Months
No. Mean 95% CI No. Mean 95% CI No. Mean 95% CI
LVESVI, mL/m2
Placebo 53 82.5 74.6 to 90.3 52 78.9 69.0 to 88.9 46 77.5 68.7 to 86.3
Metoprolol succinate 50 mg 48 82.5 74.3 to 90.7 46 79.1 69.4 to 88.8 44 75.3 64.7 to 86.0
Metoprolol succinate 200 mg 48 79.8 72.2 to 87.5 45 66.7 58.0 to 75.3 43 66.5 56.7 to 76.3
LVEDVI, mL/m2
Placebo 53 110.7 103.0 to 118.4 52 106.0 95.7 to 116.4 46 104.8 95.5 to 114.1
Metoprolol succinate 50 mg 48 110.9 102.8 to 119.0 47 108.8 98.8 to 118.8 44 104.8 93.5 to 116.1
Metoprolol succinate 200 mg 48 109.0 99.7 to 118.4 45 96.6 86.9 to 106.3 43 96.8 85.7 to 108.0
LVEF, %
Placebo 53 26.8 24.6 to 28.9 52 27.6 25.1 to 30.1 46 27.5 24.7 to 30.4
Metoprolol succinate 50 mg 48 26.6 24.2 to 29.0 46 29.5 26.3 to 32.7 44 30.4 27.1 to 33.7
Metoprolol succinate 200 mg 48 27.2 25.1 to 29.4 45 32.6 29.9 to 35.2 43 33.2 30.3 to 36.2
LVMI, g/m2
Placebo 51 161.5 148.5 to 174.5 51 162.2 148.7 to 175.6 45 169.9 155.8 to 183.9
Metoprolol succinate 50 mg 48 164.3 148.0 to 180.6 46 161.1 144.5 to 177.7 44 160.0 144.3 to 175.7
Metoprolol succinate 200 mg 47 159.9 146.2 to 173.6 44 150.9 137.8 to 164.1 44 151.8 138.6 to 165.1
52 Circulation July 3, 2007
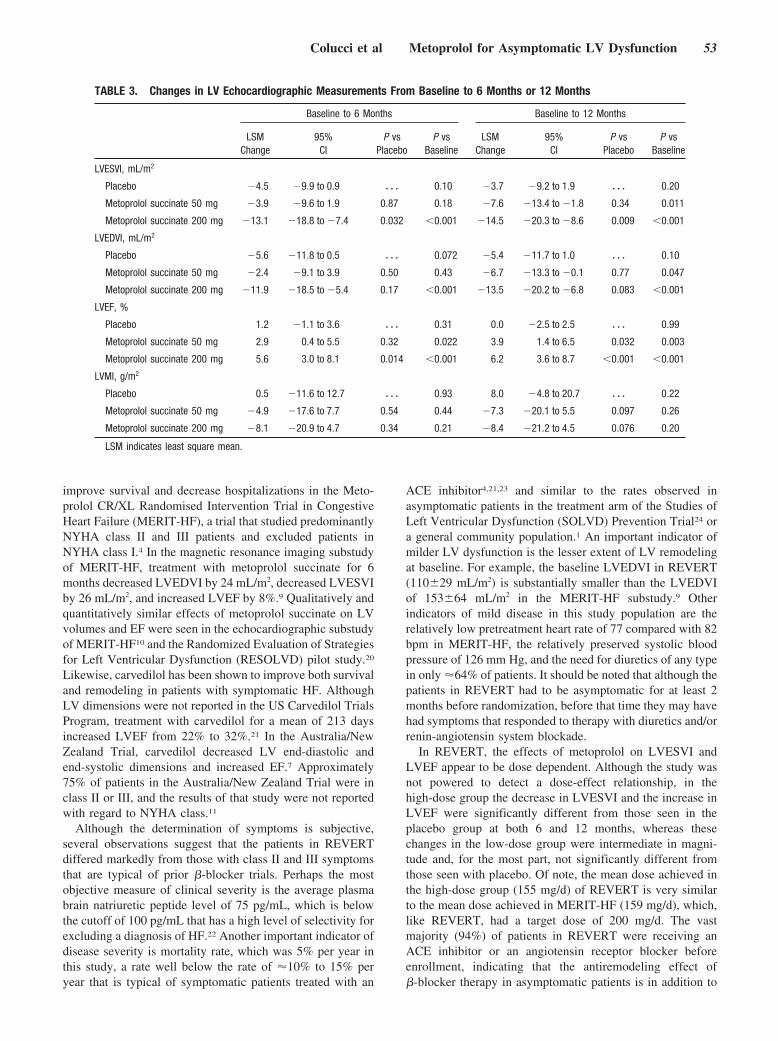
improve survival and decrease hospitalizations in the Meto-prolol CR/XL Randomised Intervention Trial in CongestiveHeart Failure (MERIT-HF), a trial that studied predominantlyNYHA class II and III patients and excluded patients inNYHA class I.4 In the magnetic resonance imaging substudyof MERIT-HF, treatment with metoprolol succinate for 6months decreased LVEDVI by 24 mL/m2, decreased LVESVIby 26 mL/m2, and increased LVEF by 8%.9 Qualitatively andquantitatively similar effects of metoprolol succinate on LVvolumes and EF were seen in the echocardiographic substudyof MERIT-HF10 and the Randomized Evaluation of Strategiesfor Left Ventricular Dysfunction (RESOLVD) pilot study.20
Likewise, carvedilol has been shown to improve both survivaland remodeling in patients with symptomatic HF. AlthoughLV dimensions were not reported in the US Carvedilol TrialsProgram, treatment with carvedilol for a mean of 213 daysincreased LVEF from 22% to 32%.21 In the Australia/NewZealand Trial, carvedilol decreased LV end-diastolic andend-systolic dimensions and increased EF.7 Approximately75% of patients in the Australia/New Zealand Trial were inclass II or III, and the results of that study were not reportedwith regard to NYHA class.11
Although the determination of symptoms is subjective,several observations suggest that the patients in REVERTdiffered markedly from those with class II and III symptomsthat are typical of prior �-blocker trials. Perhaps the mostobjective measure of clinical severity is the average plasmabrain natriuretic peptide level of 75 pg/mL, which is belowthe cutoff of 100 pg/mL that has a high level of selectivity forexcluding a diagnosis of HF.22 Another important indicator ofdisease severity is mortality rate, which was 5% per year inthis study, a rate well below the rate of �10% to 15% peryear that is typical of symptomatic patients treated with an
ACE inhibitor4,21,23 and similar to the rates observed inasymptomatic patients in the treatment arm of the Studies ofLeft Ventricular Dysfunction (SOLVD) Prevention Trial24 ora general community population.1 An important indicator ofmilder LV dysfunction is the lesser extent of LV remodelingat baseline. For example, the baseline LVEDVI in REVERT(110�29 mL/m2) is substantially smaller than the LVEDVIof 153�64 mL/m2 in the MERIT-HF substudy.9 Otherindicators of mild disease in this study population are therelatively low pretreatment heart rate of 77 compared with 82bpm in MERIT-HF, the relatively preserved systolic bloodpressure of 126 mm Hg, and the need for diuretics of any typein only �64% of patients. It should be noted that although thepatients in REVERT had to be asymptomatic for at least 2months before randomization, before that time they may havehad symptoms that responded to therapy with diuretics and/orrenin-angiotensin system blockade.
In REVERT, the effects of metoprolol on LVESVI andLVEF appear to be dose dependent. Although the study wasnot powered to detect a dose-effect relationship, in thehigh-dose group the decrease in LVESVI and the increase inLVEF were significantly different from those seen in theplacebo group at both 6 and 12 months, whereas thesechanges in the low-dose group were intermediate in magni-tude and, for the most part, not significantly different fromthose seen with placebo. Of note, the mean dose achieved inthe high-dose group (155 mg/d) of REVERT is very similarto the mean dose achieved in MERIT-HF (159 mg/d), which,like REVERT, had a target dose of 200 mg/d. The vastmajority (94%) of patients in REVERT were receiving anACE inhibitor or an angiotensin receptor blocker beforeenrollment, indicating that the antiremodeling effect of�-blocker therapy in asymptomatic patients is in addition to
TABLE 3. Changes in LV Echocardiographic Measurements From Baseline to 6 Months or 12 Months
Baseline to 6 Months Baseline to 12 Months
LSMChange
95%CI
P vsPlacebo
P vsBaseline
LSMChange
95%CI
P vsPlacebo
P vsBaseline
LVESVI, mL/m2
Placebo �4.5 �9.9 to 0.9 � � � 0.10 �3.7 �9.2 to 1.9 � � � 0.20
Metoprolol succinate 50 mg �3.9 �9.6 to 1.9 0.87 0.18 �7.6 �13.4 to �1.8 0.34 0.011
Metoprolol succinate 200 mg �13.1 �18.8 to �7.4 0.032 �0.001 �14.5 �20.3 to �8.6 0.009 �0.001
LVEDVI, mL/m2
Placebo �5.6 �11.8 to 0.5 � � � 0.072 �5.4 �11.7 to 1.0 � � � 0.10
Metoprolol succinate 50 mg �2.4 �9.1 to 3.9 0.50 0.43 �6.7 �13.3 to �0.1 0.77 0.047
Metoprolol succinate 200 mg �11.9 �18.5 to �5.4 0.17 �0.001 �13.5 �20.2 to �6.8 0.083 �0.001
LVEF, %
Placebo 1.2 �1.1 to 3.6 � � � 0.31 0.0 �2.5 to 2.5 � � � 0.99
Metoprolol succinate 50 mg 2.9 0.4 to 5.5 0.32 0.022 3.9 1.4 to 6.5 0.032 0.003
Metoprolol succinate 200 mg 5.6 3.0 to 8.1 0.014 �0.001 6.2 3.6 to 8.7 �0.001 �0.001
LVMI, g/m2
Placebo 0.5 �11.6 to 12.7 � � � 0.93 8.0 �4.8 to 20.7 � � � 0.22
Metoprolol succinate 50 mg �4.9 �17.6 to 7.7 0.54 0.44 �7.3 �20.1 to 5.5 0.097 0.26
Metoprolol succinate 200 mg �8.1 �20.9 to 4.7 0.34 0.21 �8.4 �21.2 to 4.5 0.076 0.20
LSM indicates least square mean.
Colucci et al Metoprolol for Asymptomatic LV Dysfunction 53
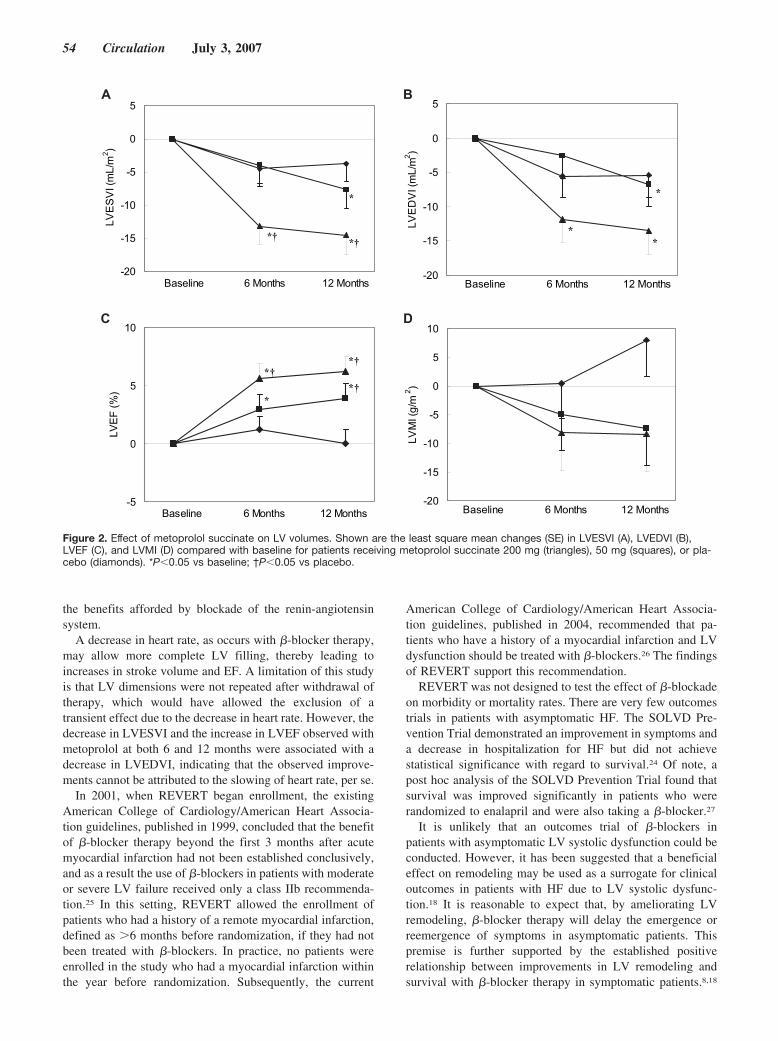
the benefits afforded by blockade of the renin-angiotensinsystem.
A decrease in heart rate, as occurs with �-blocker therapy,may allow more complete LV filling, thereby leading toincreases in stroke volume and EF. A limitation of this studyis that LV dimensions were not repeated after withdrawal oftherapy, which would have allowed the exclusion of atransient effect due to the decrease in heart rate. However, thedecrease in LVESVI and the increase in LVEF observed withmetoprolol at both 6 and 12 months were associated with adecrease in LVEDVI, indicating that the observed improve-ments cannot be attributed to the slowing of heart rate, per se.
In 2001, when REVERT began enrollment, the existingAmerican College of Cardiology/American Heart Associa-tion guidelines, published in 1999, concluded that the benefitof �-blocker therapy beyond the first 3 months after acutemyocardial infarction had not been established conclusively,and as a result the use of �-blockers in patients with moderateor severe LV failure received only a class IIb recommenda-tion.25 In this setting, REVERT allowed the enrollment ofpatients who had a history of a remote myocardial infarction,defined as �6 months before randomization, if they had notbeen treated with �-blockers. In practice, no patients wereenrolled in the study who had a myocardial infarction withinthe year before randomization. Subsequently, the current
American College of Cardiology/American Heart Associa-tion guidelines, published in 2004, recommended that pa-tients who have a history of a myocardial infarction and LVdysfunction should be treated with �-blockers.26 The findingsof REVERT support this recommendation.
REVERT was not designed to test the effect of �-blockadeon morbidity or mortality rates. There are very few outcomestrials in patients with asymptomatic HF. The SOLVD Pre-vention Trial demonstrated an improvement in symptoms anda decrease in hospitalization for HF but did not achievestatistical significance with regard to survival.24 Of note, apost hoc analysis of the SOLVD Prevention Trial found thatsurvival was improved significantly in patients who wererandomized to enalapril and were also taking a �-blocker.27
It is unlikely that an outcomes trial of �-blockers inpatients with asymptomatic LV systolic dysfunction could beconducted. However, it has been suggested that a beneficialeffect on remodeling may be used as a surrogate for clinicaloutcomes in patients with HF due to LV systolic dysfunc-tion.18 It is reasonable to expect that, by ameliorating LVremodeling, �-blocker therapy will delay the emergence orreemergence of symptoms in asymptomatic patients. Thispremise is further supported by the established positiverelationship between improvements in LV remodeling andsurvival with �-blocker therapy in symptomatic patients.8,18
02-
51-
01-
5-
0
5LV
ES
VI (
mL
/m2 )
shtnoM 21shtnoM 6enilesaB
†* †*
*
02-
51-
01-
5-
0
5
LV
ED
VI (
mL
/m2 )
shtnoM 21shtnoM 6enilesaB
*
**
5-
0
5
01
LVE
F (
%)
shtnoM 21shtnoM 6enilesaB
*
†*†*
†*
02-
51-
01-
5-
0
5
01
LVM
I (g
/m 2 )
shtnoM 21shtnoM 6enilesaB
A B
C D
Figure 2. Effect of metoprolol succinate on LV volumes. Shown are the least square mean changes (SE) in LVESVI (A), LVEDVI (B),LVEF (C), and LVMI (D) compared with baseline for patients receiving metoprolol succinate 200 mg (triangles), 50 mg (squares), or pla-cebo (diamonds). *P�0.05 vs baseline; †P�0.05 vs placebo.
54 Circulation July 3, 2007

The results of REVERT suggest that the survival benefitobserved in symptomatic patients treated with metoprololsuccinate may extend to asymptomatic patients with LVsystolic dysfunction as well.
AppendixREVERT Study GroupAlan Camp, Albert A. Carr, Barry Harold Greenberg, BruceK. Jackson, Bruce Kowalski, Chang-seng Liang, ChiayuChen, Chris Boylan, D. Marty Denny, Douglas Chapman,Freny Mody, Garo Garibian, Garrie J. Haas, David R.Richard, Inder Anand, Jalal K. Ghali, James Zebrack, JeromeLyman Anderson, John Murphy, Jose de Jesus Ortiz, JosephL. Gelormini, Larry Baruch, Lisa Mendes, Flora Sam, MarcJay Kozinn, Mark J. Geller, Marrick L. Kukin, MichaelBenjamin Honan, Michael Lesser, Nancy R. Cho, Ralph M.Vicari, Raymond Rodriguez, Robert Davidson, Robert E.Foster, Robert Michael Kipperman, Robert Weiss, RussellSilverman, Stephen Gottlieb, Steven Hutchins, Thomas D.Giles, Thomas J. Knutson, Uri Elkayam, W. David Hager,Wayne David Old, Vince Figueredo.
REVERT Steering CommitteeWilson S. Colucci (Chairman), Kirkwood F. Adams, WilliamF. Armstrong, Stephen S. Gottlieb, Barry Greenberg, MarrickL. Kukin.
Sources of FundingThe REVERT study was funded by AstraZeneca, which was respon-sible for data collection and analysis, which was conducted accord-ing to a prespecified analysis plan. Academic leadership wasprovided by the Steering Committee, which supervised the manage-ment of the study and was responsible for interpretation of the data,preparation, review, and approval of the manuscript.
DisclosuresAuthors who are employees of AstraZeneca are identified as such.All other authors have received research grants, consultant fees,and/or honoraria from AstraZeneca.
References1. Wang TJ, Evans JC, Benjamin EJ, Levy D, Leroy EC, Vasan RS. Natural
history of asymptomatic left ventricular systolic dysfunction in the com-munity. Circulation. 2003;108:977–982.
2. Redfield MM, Jacobsen SJ, Burnett JC Jr, Mahoney DW, Bailey KR,Rodeheffer RJ. Burden of systolic and diastolic ventricular dysfunction inthe community: appreciating the scope of the heart failure epidemic.JAMA. 2003;289:194–202.
3. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomisedtrial [see comments]. Lancet. 1999;353:9–13.
4. Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XLRandomised Intervention Trial in Congestive Heart Failure (MERIT-HF).Lancet. 1999;353:2001–2007.
5. Packer M, Fowler MB, Roecker EB, Coats AJ, Katus HA, Krum H,Mohacsi P, Rouleau JL, Tendera M, Staiger C, Holcslaw TL,Amann-Zalan I, DeMets DL. Effect of carvedilol on the morbidity ofpatients with severe chronic heart failure: results of the Carvedilol Pro-spective Randomized Cumulative Survival (COPERNICUS) study. Cir-culation. 2002;106:2194–2199.
6. Hjalmarson A, Goldstein S, Fagerberg B, Wedel H, Waagstein F,Kjekshus J, Wikstrand J, El Allaf D, Vitovec J, Aldershvile J, Halinen M,Dietz R, Neuhaus KL, Janosi A, Thorgeirsson G, Dunselman PH,Gullestad L, Kuch J, Herlitz J, Rickenbacher P, Ball S, Gottlieb S,Deedwania P; MERIT-HF Study Group. Effects of controlled-releasemetoprolol on total mortality, hospitalizations, and well-being in patients
with heart failure: the Metoprolol CR/XL Randomized Intervention Trialin Congestive Heart Failure (MERIT-HF). JAMA. 2000;283:1295–1302.
7. Doughty RN, Whalley GA, Gamble G, MacMahon S, Sharpe N;Australia-New Zealand Heart Failure Research Collaborative Group. Leftventricular remodeling with carvedilol in patients with congestive heartfailure due to ischemic heart disease. J Am Coll Cardiol. 1997;29:1060–1066.
8. Udelson JE. Ventricular remodeling in heart failure and the effect ofbeta-blockade. Am J Cardiol. 2004;93:43B–48B.
9. Groenning BA, Nilsson JC, Sondergaard L, Fritz-Hansen T, Larsson HB,Hildebrandt PR. Antiremodeling effects on the left ventricle during beta-blockade with metoprolol in the treatment of chronic heart failure. J AmColl Cardiol. 2000;36:2072–2080.
10. Hole T, Froland G, Gullestad L, Offstad J, Skjaerpe T. Metoprolol CR/XLimproves systolic and diastolic left ventricular function in patients withchronic heart failure. Echocardiography. 2004;21:215–223.
11. Australia-New Zealand Heart Failure Research Collaborative Group.Effects of carvedilol, a vasodilator-beta-blocker, in patients with con-gestive heart failure due to ischemic heart disease. Circulation. 1995;92:212–218.
12. Dargie HJ. Effect of carvedilol on outcome after myocardial infarction inpatients with left-ventricular dysfunction: the CAPRICORN randomisedtrial. Lancet. 2001;357:1385–1390.
13. Francis GS, Benedict C, Johnstone DE, Kirlin PC, Nicklas J, Liang CS,Kubo SH, Rudin-Toretsky E, Yusuf S. Comparison of neuroendocrineactivation in patients with left ventricular dysfunction with and withoutcongestive heart failure: a substudy of the Studies of Left VentricularDysfunction (SOLVD). Circulation. 1990;82:1724–1729.
14. Benedict CR, Johnstone DE, Weiner DH, Bourassa MG, Bittner V, KayR, Kirlin P, Greenberg B, Kohn RM, Nicklas JM; SOLVD Investigators.Relation of neurohumoral activation to clinical variables and degree ofventricular dysfunction: a report from the registry of Studies of LeftVentricular Dysfunction. J Am Coll Cardiol. 1994;23:1410–1420.
15. Benedict CR, Francis GS, Shelton B, Johnstone DE, Kubo SH, Kirlin P,Nicklas J, Liang CS, Konstam MA, Greenberg B; SOLVD Investigators.Effect of long-term enalapril therapy on neurohormones in patients withleft ventricular dysfunction. Am J Cardiol. 1995;75:1151–1157.
16. Colucci WS, Packer M, Bristow MR, Gilbert EM, Cohn JN, Fowler MB,Krueger SK, Hershberger R, Uretsky BF, Bowers JA, Sackner-BernsteinJD, Young ST, Holcslaw TL, Lukas MA; US Carvedilol Heart FailureStudy Group. Carvedilol inhibits clinical progression in patients with mildsymptoms of heart failure. Circulation. 1996;94:2800–2806.
17. Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, GaniatsTG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, RahkoPS, Silver MA, Stevenson LW, Yancy CW, Antman EM, Smith SC Jr,Adams CD, Anderson JL, Faxon DP, Fuster V, Halperin JL, Hiratzka LF,Jacobs AK, Nishimura R, Ornato JP, Page RL, Riegel B. ACC/AHA 2005guideline update for the diagnosis and management of chronic heartfailure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (WritingCommittee to Update the 2001 Guidelines for the Evaluation and Man-agement of Heart Failure): developed in collaboration with the AmericanCollege of Chest Physicians and the International Society for Heart andLung Transplantation: endorsed by the Heart Rhythm Society. Circu-lation. 2005;112:e154–e235.
18. Konstam MA, Udelson JE, Anand IS, Cohn JN. Ventricular remodelingin heart failure: a credible surrogate endpoint. J Card Fail. 2003;9:350–353.
19. Devereux RB, Reichek N. Echocardiographic determination of left ven-tricular mass in man: anatomic validation of the method. Circulation.1977;55:613–618.
20. RESOLVD Investigators. Effects of metoprolol CR in patients withischemic and dilated cardiomyopathy: the Randomized Evaluation ofStrategies for Left Ventricular Dysfunction Pilot Study. Circulation.2000;101:378–384.
21. Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM,Shusterman NH; U.S. Carvedilol Heart Failure Study Group. The effectof carvedilol on morbidity and mortality in patients with chronic heartfailure. N Engl J Med. 1996;334:1349–1355.
22. Maisel AS, McCord J, Nowak RM, Hollander JE, Wu AH, Duc P,Omland T, Storrow AB, Krishnaswamy P, Abraham WT, Clopton P, StegG, Aumont MC, Westheim A, Knudsen CW, Perez A, Kamin R,Kazanegra R, Herrmann HC, McCullough PA. Bedside B-type natriuretic
Colucci et al Metoprolol for Asymptomatic LV Dysfunction 55

peptide in the emergency diagnosis of heart failure with reduced orpreserved ejection fraction: results from the Breathing Not ProperlyMultinational Study. J Am Coll Cardiol. 2003;41:2010–2017.
23. The SOLVD Investigators. Effect of enalapril on survival in patients withreduced left ventricular ejection fractions and congestive heart failure.N Engl J Med. 1991;325:293–302.
24. The SOLVD Investigators. Effect of enalapril on mortality and the devel-opment of heart failure in asymptomatic patients with reduced left ven-tricular ejection fractions [published erratum appears in N Engl J Med.1992;327:1768]. N Engl J Med. 1992;327:685–691.
25. Ryan TJ, Antman EM, Brooks NH, Califf RM, Hillis LD, Hiratzka LF,Rapaport E, Riegel B, Russell RO, Smith EE III, Weaver WD, GibbonsRJ, Alpert JS, Eagle KA, Gardner TJ, Garson A Jr, Gregoratos G, SmithSC Jr. 1999 update: ACC/AHA guidelines for the management of patientswith acute myocardial infarction: executive summary and recommen-dations: a report of the American College of Cardiology/American HeartAssociation Task Force on Practice Guidelines (Committee on Man-
agement of Acute Myocardial Infarction). Circulation. 1999;100:1016–1030.
26. Antman EM, Anbe DT, Armstrong PW, Bates ER, Green LA, Hand M,Hochman JS, Krumholz HM, Kushner FG, Lamas GA, Mullany CJ,Ornato JP, Pearle DL, Sloan MA, Smith SC Jr, Alpert JS, Anderson JL,Faxon DP, Fuster V, Gibbons RJ, Gregoratos G, Halperin JL, HiratzkaLF, Hunt SA, Jacobs AK. ACC/AHA guidelines for the management ofpatients with ST-elevation myocardial infarction: a report of theAmerican College of Cardiology/American Heart Association Task Forceon Practice Guidelines (Committee to Revise the 1999 Guidelines for theManagement of Patients with Acute Myocardial Infarction). Circulation.2004;110:e82–e292.
27. Exner DV, Dries DL, Waclawiw MA, Shelton B, Domanski MJ. Beta-adrenergic blocking agent use and mortality in patients with asymptom-atic and symptomatic left ventricular systolic dysfunction: a post hocanalysis of the Studies of Left Ventricular Dysfunction. J Am CollCardiol. 1999;33:916–923.
Go to http://cme.ahajournals.org to take the CME quiz for this article.
56 Circulation July 3, 2007

Negative Inotropy of the Gastric Proton Pump InhibitorPantoprazole in Myocardium From Humans and Rabbits
Evaluation of Mechanisms
Wolfgang Schillinger, MD*; Nils Teucher, MD*; Samuel Sossalla, MS; Sarah Kettlewell, PhD;Carola Werner, PhD; Dirk Raddatz, MD; Andreas Elgner, MS; Gero Tenderich, MD;
Burkert Pieske, MD; Giuliano Ramadori, MD; Friedrich A. Schöndube, MD;Harald Kögler, MD; Jens Kockskämper, PhD; Lars S. Maier, MD; Harald Schwörer, MD;
Godfrey L. Smith, PhD; Gerd Hasenfuss, MD
Background—Proton pump inhibitors are used extensively for acid-related gastrointestinal diseases. Their effect oncardiac contractility has not been assessed directly.
Methods and Results—Under physiological conditions (37°C, pH 7.35, 1.25 mmol/L Ca2�), there was a dose-dependentdecrease in contractile force in ventricular trabeculae isolated from end-stage failing human hearts superfused withpantoprazole. The concentration leading to 50% maximal response was 17.3�1.3 �g/mL. Similar observations weremade in trabeculae from human atria, normal rabbit ventricles, and isolated rabbit ventricular myocytes. Real-timepolymerase chain reaction demonstrated the expression of gastric H�/K�–adenosine triphosphatase in human and rabbitmyocardium. However, measurements with BCECF-loaded rabbit trabeculae did not reveal any significantpantoprazole-dependent changes of pHi. Ca2� transients recorded from field-stimulated fluo 3–loaded myocytes (F/F0)were significantly depressed by 10.4�2.1% at 40 �g/mL. Intracellular Ca2� fluxes were assessed in fura 2–loaded,voltage-clamped rabbit ventricular myocytes. Pantoprazole (40 �g/mL) caused an increase in diastolic [Ca2�]i by33�12%, but peak systolic [Ca2�]i was unchanged, resulting in a decreased Ca2� transient amplitude by 25�8%. Theamplitude of the L-type Ca2� current (ICa,L) was reduced by 35�5%, and sarcoplasmic reticulum Ca2� content wasreduced by 18�6%. Measurements of oxalate-supported sarcoplasmic reticulum Ca2� uptake in permeabilizedcardiomyocytes indicated that pantoprazole decreased Ca2� sensitivity (Kd) of sarcoplasmic reticulum Ca2� adenosinetriphosphatase: control, Kd�358�15 nmol/L; 40 �g/mL pantoprazole, Kd�395�12 nmol/L (P�0.05). Pantoprazolealso acted on cardiac myofilaments to reduced Ca2�-activated force.
Conclusions—Pantoprazole depresses cardiac contractility in vitro by depression of Ca2� signaling and myofilamentactivity. In view of the extensive use of this agent, the effects should be evaluated in vivo. (Circulation. 2007;116:57-66.)
Key Words: calcium � contractility � heart failure � inotropic agents � pharmacology
Proton pump inhibitors (PPIs) like pantoprazole, omepra-zole, esomeprazole, lansoprazole, and rabeprazole are the
most effective pharmacological means of reducing gastricacid secretion by blocking the final step of proton secretion,ie, the gastric acid pump, H�/K�–adenosine triphosphatase(ATPase). The efficacy of this class of drugs has beendemonstrated in the treatment of a number of acid-relatedgastrointestinal diseases, in particular, gastroesophageal re-flux and ulcer disease.1 Hence, there is extensive use of these
Clinical Perspective p 66agents for patients in a variety of settings. In a US Medicaidpopulation, PPIs accounted for 5.6% of the net pharmacyexpenditures and ranked first in expenditures among all drugtherapy classes during the 12 months before the implemen-tation of the PPI prior-authorization policy.2 Drug shortagebulletins have been issued repeatedly for intravenous PPIs inthe United States.3
Received September 25, 2006; accepted May 1, 2007.From the Herzzentrum, Kardiologie und Pneumologie, Universitaet Goettingen, Goettingen, Germany (W.S., S.S., A.E., B.P., H.K., J.K., L.S.M.,
G.H.); Herzzentrum, Thorax-, Herz-, und Gefaesschirurgie, Universitaet Goettingen, Goettingen, Germany (N.T., F.A.S.); Institute of Biomedical and LifeSciences, University of Glasgow, Glasgow, UK (S.K., G.L.S.); Medizinische Statistik, Universitaet Goettingen, Goettingen, Germany (C.W.);Gastroenterologie und Endokrinologie, Universitaet Goettingen, Goettingen, Germany (D.R., G.R., H.S.); and Herz- und Diabeteszentrum Nordrhein-Westfalen, Klinik fuer Thorax- und Kardiovaskularchirurgie, Bad Oeynhausen, Germany (G.T.).
*The first 2 authors contributed equally to this work.Correspondence to Wolfgang Schillinger, MD, Herzzentrum, Kardiologie und Pneumologie, Georg-August Universitaet Goettingen, Robert-Koch
Strasse 40, 37099 Goettingen, Germany. E-mail [email protected]© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.106.666008
57

It has been shown recently that the expression of H�/K�-ATPase is not limited strictly to gastric tissue. It has also beenidentified in renal4 and colonic epithelial cells,5 vascularsmooth muscle cells,6 and other tissues. In myocardium fromrats, the expression of H�/K�-ATPase has been demonstratedat the transcriptional and protein levels.7 Biochemical evi-dence and physiological evidence for a myocardial H�/K�-ATPase have also been found.8 Furthermore, Beisvag andcoworkers7 showed a contribution from the H�/K�-ATPase of�25% of total 86Rb� uptake in rat hearts and suggested thatthe enzyme could contribute significantly to the regulation ofmyocardial K� and H� homeostasis. Inhibition of H�/K�-ATPase might therefore induce cellular acidosis, which isknown to depress myocardial contractility mainly at the levelof myofilament responsiveness to [Ca]i.9
Orally applied PPIs are considered safe1 and have beenfound advantageous in regard to cardiovascular side effectscompared with histamine type 2 (H2) receptor antagonistsbecause of lack of chronotropic and inotropic effects. Incontrast to famotidine, omeprazole did not show any changesin cardiac performance in healthy volunteers as measured byimpedance cardiography and mechanocardiography after1-week oral treatment with therapeutic doses.10 Pantoprazole,lansoprazole, and esomeprazole are currently available asintravenous formulations in the United States. The rationalefor use has come primarily with the suggested efficacy inreducing rebleeding after endoscopic hemostasis of bleedingpeptic ulcers (eg, References 11 and 12). The target goal forgastric pH in these patients has been suggested to be �6 inorder to promote hemostasis and minimize clot lysis, incontradistinction to the target pH �4 for treating patients toprevent stress ulcer or heal ulcers or reflux esophagitis.1,11,12
As such, the dosing amounts of intravenous PPI have beenhigher than that of oral PPI. Recently, omeprazole and panto-prazole have been dosed at 80 mg followed by 8 mg/h for 72hours.11,12 However, information regarding the cardiac effects ofhigh doses is lacking. Moreover, few data are available regard-ing the direct effect of such agents on the myocardium.
Hence, because of the abundant use of this drug class andbecause of the presence of a H�/K�-ATPase in myocardium,we sought to investigate the effects of PPI on contractility ofisolated human myocardium using pantoprazole, which is themost commonly used intravenous formulation. In addition,the mode of action in cardiac tissue was further analyzed inisolated cardiac tissue from rabbits.
MethodsMyocardial TissueExperiments were performed in left ventricular muscle strips from 8end-stage failing human hearts obtained from patients undergoingcardiac transplantation and right atrial trabeculae obtained from 16patients who underwent cardiac surgery. Additional right ventriculartrabeculae were obtained from adult female Chinchilla Bastardrabbits (weight, 2.0 to 2.5 kg; Charles River Deutschland, Kisslegg,Germany). Myocardial trabeculae13 and adult rabbit ventricularcardiac myocytes14 were isolated and forces of electrically stimu-lated preparations were investigated as described previously. Exper-imental procedures with human tissue were reviewed and approvedby the ethical committee of the University Clinics of Goettingen, andthe subjects gave informed consent. Procedures with rabbits were
performed in accordance with institutional guidelines for the careand use of laboratory animals.
pHi MeasurementsIntracellular pH (pHi) was measured in isolated trabeculae fromrabbit hearts as described previously.13 Briefly, trabeculae weremounted in a cylindrical glass cuvette, connected to an isometricforce transducer, and loaded with 2’,7’-bis(carboxyethyl)-5(6)-carboxyfluorescein (BCECF)-AM (15 �mol/L) in Tyrode’s solutionby 45-minute incubation at room temperature. Excitation light froma mercury lamp was passed alternately through 2 bandpass filters(450 nm/495 nm) and focused on the muscle strip. Fluorescenceemission was collected by a photomultiplier (Scientific Instruments)after passage through a bandpass filter (535�5 nm). Values of pHi
were estimated from the ratio of the BCECF fluorescence signals(F495/F450) after subtraction of background fluorescence. At the end ofeach experiment, the BCECF fluorescence ratio was calibrated in vivoby means of the high K�-nigericin method as previously reported.15
Voltage Clamp and Intracellular [Ca2�]Measurements in Rabbit CardiomyocytesThe cardiomyocytes were superfused with a solution consisting of(mmol/L) 144.0 NaCl, 5.4 KCl, 0.3 NaH2PO4, 1.0 MgCl2, 5.0HEPES, 11.1 glucose, 1.8 CaCl2, 0.1 niflumic acid, 5.0 4-AP (pH7.4) at room temperature in a chamber mounted on the stage of aninverted microscope. Voltage clamp was achieved with the use ofwhole-cell patch-clamp technique with an Axoclamp 2A amplifier(Axon Instruments, Foster City, Calif) in switch clamp (discontinu-ous) mode. Pipettes were filled with an intracellular solution of thefollowing composition (mmol/L): 20.0 KCl, 100.0 K aspartate (DL),20.0 TEA Cl, 10.0 HEPES, 4.5 MgCl2, 4.0 Na2ATP, 1.0 Na2CrP, 2.5EGTA (pH 7.25 with KOH) and were of resistance 3 to 6 mol/L�.Intracellular [Ca2�] was measured from fura 2 fluorescence signalsby a dual-wavelength spectrophotometer method as previouslydescribed.16 Cytosolic loading of fura 2 was achieved by incubatingcardiomyocytes with 5 �mol/L fura 2-AM at room temperature for12 minutes.
Electrophysiological ProtocolsIsolated rabbit cardiomyocytes were held at �80 mV, and thevoltage was stepped to �40 mV (50 ms) to inactivate the inward Na�
current before stepping to 0 mV (150 ms). Tetrodotoxin 310�5
mol/L was also used to block INa. This protocol was repeated 40times at a rate of 1 Hz to achieve steady state Ca2� transients.Sarcoplasmic reticulum (SR) Ca2� content and Na�/Ca2� exchanger(NCX) activity were then estimated by rapidly switching to10 mmol/L caffeine to cause SR Ca2� release. In the continuedpresence of caffeine (20 seconds), the SR is unable to reaccumulateCa2�, and therefore Ca2� removal is mainly via NCX. The timecourse of the decay of [Ca2�] and the NCX-mediated inward current(INCX) represent rates of extrusion of Ca2� from the cell predomi-nantly by NCX.17 These signals were fitted to exponential decaysover �80% of their amplitude. The magnitude of non-NCX Ca2�-removal mechanisms was estimated from the Ca2� decay obtained byrapidly switching to 10 mmol/L caffeine in the presence of10 mmol/L NiCl2 (which blocks NCX), in which the absence of acurrent indicated that the current obtained without NiCl2 was solelydue to NCX.
Simultaneous Measurements of MyocyteShortening and Intracellular [Ca2�]Shortening and [Ca2�]i were measured simultaneously as reportedpreviously.18 Cells were loaded with 10 �mol/L fluo 3-AM (Molec-ular Probes, Carlsbad, Calif) for 15 minutes. Fluo 3 was excited at480 nm, and fluorescence was measured at 535 nm. The field-stimulation frequency was 1 Hz (37°C, pH 7.35). Normalizedamplitude of calcium transients (F/F0) was calculated by dividing
58 Circulation July 3, 2007

fluorescence F by the baseline fluorescence F0 after subtraction ofthe background fluorescence (IonWizard, IonOptix Corp). Cellswere treated with 0/10/40 �g/mL pantoprazole followed by awashout. Fluorescence and shortening were analyzed at steady stateconditions.
Measurements of SR Ca2� Uptake CharacteristicsMeasurements of SR Ca2� uptake were performed as describedpreviously.14 Ventricular myocytes freshly dissociated from adultrabbit hearts were permeabilized with 0.1 mg/mL �-escin and kept inmock intracellular solution of the following composition (mmol/L):100 K�, 40 Na�, 25 HEPES, 100 Cl�, 0.05 EGTA, 5 ATP, 10 CrP(pH 7.0). To test the influence of pantoprazole on SR Ca2� uptake,myocytes were divided into 3 groups, and 0 (control), 10 �g/mL, and40 �g/mL pantoprazole were added into the cuvette. Oxalate(10 mmol/L) was included to maintain low and constant intra-SR[Ca2�], and ruthenium red (2.7 �mol/L) was included to block SRCa2� efflux. Myocyte suspensions were stirred and [Ca2�] wasmonitored with the use of fura 2 (10 �mol/L). The decline of Ca2�
signal was used to calculate SR Ca2� uptake characteristics.
Skinned FibersFibers dissected from rabbit right ventricles were skinned byincubation with Triton (1%) for 24 hours at 4°C. Paired measure-
ments of calcium sensitivity and force development of the myofila-ments were performed with the use of 1 relaxation and 10 activationsolutions containing pantoprazole (10/40 �g/mL) or NaCl as controlwith increasing Ca2� concentrations (from 1.6610�7 mol/L to5.1510�5 mol/L Ca2�). Active tension was measured via forcetransducer and analyzed at steady state conditions. To exclude thatchanges in the active tension were due to rundown of the preparation,each exposure to pantoprazole was preceded and followed by 1control step with NaCl 0.9%.
Transcription of H�/K�-ATPase inHuman MyocardiumRNA was isolated from myocardial tissue and gastric corpus mucosafrom humans and rabbits with the use of RNeasy Kits (Qiagen).Quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed by real-time PCR as described earlier.19 Primerpairs for H�/K�-ATPase and �-actin are summarized in the Table.Each sample was analyzed in duplicate. The investigated number ofcDNA molecules was related to �-actin cDNA molecules detected inthe same sample to normalize for the quantity of RNA extracted andthe efficiency of cDNA synthesis. The relative expression was thencalculated as described.19
Dev
elop
ed T
ensi
on (m
N/m
m²)
)Lm/gµ( noitartnecnoC
*
tuo-hsaW
muirta namuH
05525.2152.6521.352.1526.00
2
4
6
8lortnoC
)21/51 = n(
elozarpotnaP)21/61 = n(
tuo-hsaW05525.2152.6521.352.1526.000
1
2
3
4
5
6
**
elcirtnev namuH
lortnoC)5/21 = n(
elozarpotnaP)5/11 = n(
* *
tuo-hsaW
elcirtnev tibbaR04020155.252.1526.00
0
5
01
51
02
52
03
lortnoC)11/11 = n(
elozarpotnaP)11/11 = n(
tuo-hsaW
muirta namuH
05525.2152.6521.352.1526.001
2
3
4
5
6
lortnoC)21/41 = n(elozarpemosE
)5/5 = n(
BA
DC
Figure 1. Dose dependence of isometric twitch force from pantoprazole (A to C) and esomeprazole (D) in different types of myocardiumas indicated and partial reversibility after washout of the drug. The number of trabeculae and hearts used for each experiment is indi-cated in parentheses. *P�0.025 (only concentrations with potential clinical relevance have been tested).
TABLE 1. Primer Pairs Used in Real-Time PCR
Gene Sequence Gene Bank Accession No.
HumanH�/K�-ATPase
Sense: 5-CCACATCCACACAGCTGACAC-3 M63962
Antisense: 5-TCAAACGTCTGCCCTGACTG-3
Rabbit H�/K�-ATPase Sense: 5-ACTCTGCACCGACATTTTCCC-3 X64694
Antisense: 5-TCAGCCTTCTCATACGCCAAG-3
�-Actin Sense: 5-CTGGCACCCAGCACAATG-3 M10277
Antisense: 5-CCGATCCACACGGAGTACTTG-3
Schillinger et al Negative Inotropy of Pantoprazole 59
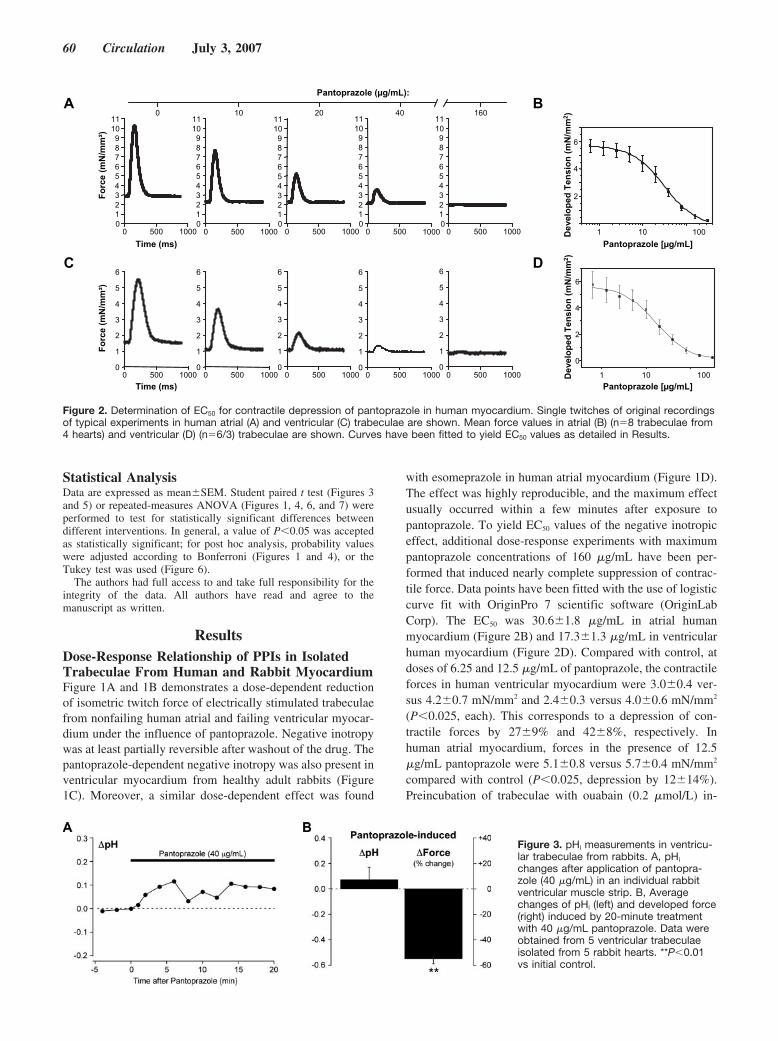
Statistical AnalysisData are expressed as mean�SEM. Student paired t test (Figures 3and 5) or repeated-measures ANOVA (Figures 1, 4, 6, and 7) wereperformed to test for statistically significant differences betweendifferent interventions. In general, a value of P�0.05 was acceptedas statistically significant; for post hoc analysis, probability valueswere adjusted according to Bonferroni (Figures 1 and 4), or theTukey test was used (Figure 6).
The authors had full access to and take full responsibility for theintegrity of the data. All authors have read and agree to themanuscript as written.
ResultsDose-Response Relationship of PPIs in IsolatedTrabeculae From Human and Rabbit MyocardiumFigure 1A and 1B demonstrates a dose-dependent reductionof isometric twitch force of electrically stimulated trabeculaefrom nonfailing human atrial and failing ventricular myocar-dium under the influence of pantoprazole. Negative inotropywas at least partially reversible after washout of the drug. Thepantoprazole-dependent negative inotropy was also present inventricular myocardium from healthy adult rabbits (Figure1C). Moreover, a similar dose-dependent effect was found
with esomeprazole in human atrial myocardium (Figure 1D).The effect was highly reproducible, and the maximum effectusually occurred within a few minutes after exposure topantoprazole. To yield EC50 values of the negative inotropiceffect, additional dose-response experiments with maximumpantoprazole concentrations of 160 �g/mL have been per-formed that induced nearly complete suppression of contrac-tile force. Data points have been fitted with the use of logisticcurve fit with OriginPro 7 scientific software (OriginLabCorp). The EC50 was 30.6�1.8 �g/mL in atrial humanmyocardium (Figure 2B) and 17.3�1.3 �g/mL in ventricularhuman myocardium (Figure 2D). Compared with control, atdoses of 6.25 and 12.5 �g/mL of pantoprazole, the contractileforces in human ventricular myocardium were 3.0�0.4 ver-sus 4.2�0.7 mN/mm2 and 2.4�0.3 versus 4.0�0.6 mN/mm2
(P�0.025, each). This corresponds to a depression of con-tractile forces by 27�9% and 42�8%, respectively. Inhuman atrial myocardium, forces in the presence of 12.5�g/mL pantoprazole were 5.1�0.8 versus 5.7�0.4 mN/mm2
compared with control (P�0.025, depression by 12�14%).Preincubation of trabeculae with ouabain (0.2 �mol/L) in-
:)Lm/gµ( elozarpotnaP
00010 005
1234567890111
0 005 0001
1234567890111
)sm( emiT
Forc
e (m
N/m
m²)
0 005 0001
1234567890111
0 005 0001
1234567890111
0 005 0001
1234567890111
0 005 00010
1
2
3
4
5
6
)sm( emiT
Forc
e (m
N/m
m²)
0 005 00010
1
2
3
4
5
6
0 005 00010
1
2
3
4
5
6
0 005 00010
1
2
3
4
5
6
0 005 00010
1
2
3
4
5
6
0610402010
2
6
4
Dev
elop
ed T
ensi
on (m
N/m
m2 )
]Lm/gµ[ elozarpotnaP001011
0
2
4
6
Dev
elop
ed T
ensi
on (m
N/m
m2 )
]Lm/gµ[ elozarpotnaP001011
BA
DC
00000
Figure 2. Determination of EC50 for contractile depression of pantoprazole in human myocardium. Single twitches of original recordingsof typical experiments in human atrial (A) and ventricular (C) trabeculae are shown. Mean force values in atrial (B) (n�8 trabeculae from4 hearts) and ventricular (D) (n�6/3) trabeculae are shown. Curves have been fitted to yield EC50 values as detailed in Results.
Figure 3. pHi measurements in ventricu-lar trabeculae from rabbits. A, pHi
changes after application of pantopra-zole (40 �g/mL) in an individual rabbitventricular muscle strip. B, Averagechanges of pHi (left) and developed force(right) induced by 20-minute treatmentwith 40 �g/mL pantoprazole. Data wereobtained from 5 ventricular trabeculaeisolated from 5 rabbit hearts. **P�0.01vs initial control.
60 Circulation July 3, 2007
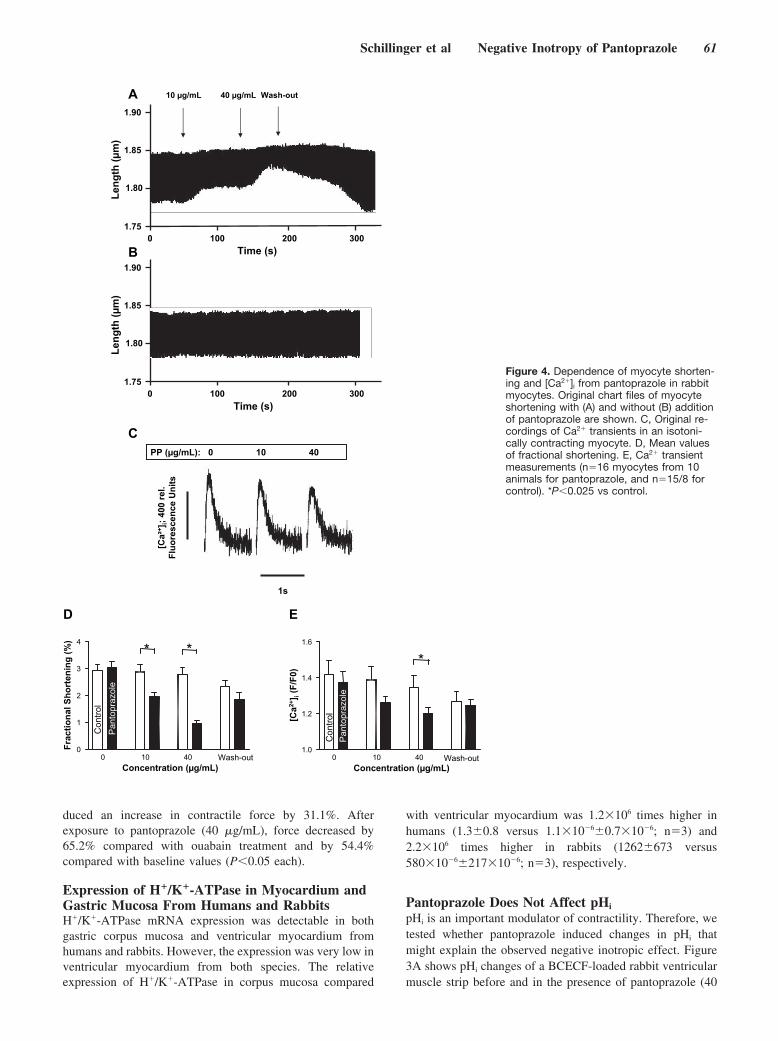
duced an increase in contractile force by 31.1%. Afterexposure to pantoprazole (40 �g/mL), force decreased by65.2% compared with ouabain treatment and by 54.4%compared with baseline values (P�0.05 each).
Expression of H�/K�-ATPase in Myocardium andGastric Mucosa From Humans and RabbitsH�/K�-ATPase mRNA expression was detectable in bothgastric corpus mucosa and ventricular myocardium fromhumans and rabbits. However, the expression was very low inventricular myocardium from both species. The relativeexpression of H�/K�-ATPase in corpus mucosa compared
with ventricular myocardium was 1.2106 times higher inhumans (1.3�0.8 versus 1.110�6�0.710�6; n�3) and2.2106 times higher in rabbits (1262�673 versus58010�6�21710�6; n�3), respectively.
Pantoprazole Does Not Affect pHi
pHi is an important modulator of contractility. Therefore, wetested whether pantoprazole induced changes in pHi thatmight explain the observed negative inotropic effect. Figure3A shows pHi changes of a BCECF-loaded rabbit ventricularmuscle strip before and in the presence of pantoprazole (40
A tuo-hsaWLm/gµ 04Lm/gµ 01
)s( emiT 001 0 003 002
57.1
08.1
58.1
09.1Le
ngth
(µm
)
001 0 003 002 57.1
08.1
58.1
09.1
Leng
th (µ
m)
)s( emiT
B
04 01 0 :)Lm/gµ( PP
s1
[Ca²
+ ]i;
400
rel.
Fluo
resc
ence
Uni
ts
C
040100
1
2
3
4* *
Pan
topr
azol
e
Con
trol
tuo-hsaW)Lm/gµ( noitartnecnoC
Frac
tiona
l Sho
rten
ing
(%)
040100.1
2.1
4.1
6.1
*
Pan
topr
azol
e
Con
trol
tuo-hsaW)Lm/gµ( noitartnecnoC
[Ca2+
] i(F
/F0)
ED
Figure 4. Dependence of myocyte shorten-ing and [Ca2�]i from pantoprazole in rabbitmyocytes. Original chart files of myocyteshortening with (A) and without (B) additionof pantoprazole are shown. C, Original re-cordings of Ca2� transients in an isotoni-cally contracting myocyte. D, Mean valuesof fractional shortening. E, Ca2� transientmeasurements (n�16 myocytes from 10animals for pantoprazole, and n�15/8 forcontrol). *P�0.025 vs control.
Schillinger et al Negative Inotropy of Pantoprazole 61
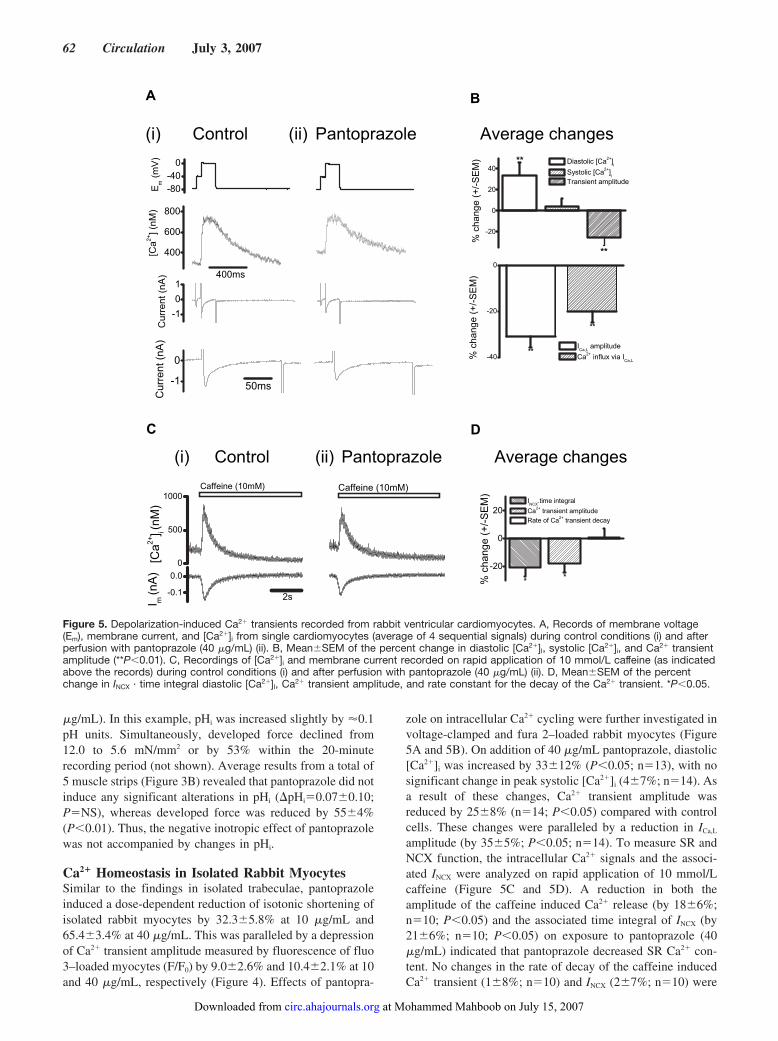
�g/mL). In this example, pHi was increased slightly by �0.1pH units. Simultaneously, developed force declined from12.0 to 5.6 mN/mm2 or by 53% within the 20-minuterecording period (not shown). Average results from a total of5 muscle strips (Figure 3B) revealed that pantoprazole did notinduce any significant alterations in pHi (�pHi�0.07�0.10;P�NS), whereas developed force was reduced by 55�4%(P�0.01). Thus, the negative inotropic effect of pantoprazolewas not accompanied by changes in pHi.
Ca2� Homeostasis in Isolated Rabbit MyocytesSimilar to the findings in isolated trabeculae, pantoprazoleinduced a dose-dependent reduction of isotonic shortening ofisolated rabbit myocytes by 32.3�5.8% at 10 �g/mL and65.4�3.4% at 40 �g/mL. This was paralleled by a depressionof Ca2� transient amplitude measured by fluorescence of fluo3–loaded myocytes (F/F0) by 9.0�2.6% and 10.4�2.1% at 10and 40 �g/mL, respectively (Figure 4). Effects of pantopra-
zole on intracellular Ca2� cycling were further investigated involtage-clamped and fura 2–loaded rabbit myocytes (Figure5A and 5B). On addition of 40 �g/mL pantoprazole, diastolic[Ca2�]i was increased by 33�12% (P�0.05; n�13), with nosignificant change in peak systolic [Ca2�]i (4�7%; n�14). Asa result of these changes, Ca2� transient amplitude wasreduced by 25�8% (n�14; P�0.05) compared with controlcells. These changes were paralleled by a reduction in ICa,L
amplitude (by 35�5%; P�0.05; n�14). To measure SR andNCX function, the intracellular Ca2� signals and the associ-ated INCX were analyzed on rapid application of 10 mmol/Lcaffeine (Figure 5C and 5D). A reduction in both theamplitude of the caffeine induced Ca2� release (by 18�6%;n�10; P�0.05) and the associated time integral of INCX (by21�6%; n�10; P�0.05) on exposure to pantoprazole (40�g/mL) indicated that pantoprazole decreased SR Ca2� con-tent. No changes in the rate of decay of the caffeine inducedCa2� transient (1�8%; n�10) and INCX (2�7%; n�10) were
004
006
008
sm004
[Ca2+
] (nM
)
1-
0
1
Cur
rent
(nA
)
1-
0
sm05
Cur
rent
(nA
)
segnahc egarevAelozarpotnaPlortnoC
02-
0
02
04
**
** aC[ cilotsaiD +2 ]i
aC[ cilotsyS +2 ]i
edutilpma tneisnarT
% c
hang
e (+
/-S
EM
)
04-
02-
0
**
**I
L,aCedutilpma
aC +2 I aiv xulfni L,aC
% c
hang
e (+
/-S
EM
)
A B
)ii()i(
08-04-0
Em (
mV
)
C D
1.0-
0.0
0
005
0001
s2
[Ca2+
] i(nM
)I m
(nA
)
)Mm01( enieffaC )Mm01( enieffaC
elozarpotnaPlortnoC
02-
0
02
**
I XCN
largetni emit.
aC +2 edutilpma tneisnart
aC fo etaR +2 yaced tneisnart
% c
hang
e (+
/-S
EM
)
segnahc egarevA)ii()i(
Figure 5. Depolarization-induced Ca2� transients recorded from rabbit ventricular cardiomyocytes. A, Records of membrane voltage(Em), membrane current, and [Ca2�]i from single cardiomyocytes (average of 4 sequential signals) during control conditions (i) and afterperfusion with pantoprazole (40 �g/mL) (ii). B, Mean�SEM of the percent change in diastolic [Ca2�]I, systolic [Ca2�]i, and Ca2� transientamplitude (**P�0.01). C, Recordings of [Ca2�]i and membrane current recorded on rapid application of 10 mmol/L caffeine (as indicatedabove the records) during control conditions (i) and after perfusion with pantoprazole (40 �g/mL) (ii). D, Mean�SEM of the percentchange in INCX · time integral diastolic [Ca2�]I, Ca2� transient amplitude, and rate constant for the decay of the Ca2� transient. *P�0.05.
62 Circulation July 3, 2007
at Mohammed Mahboob on July 15, 2007 circ.ahajournals.orgDownloaded from
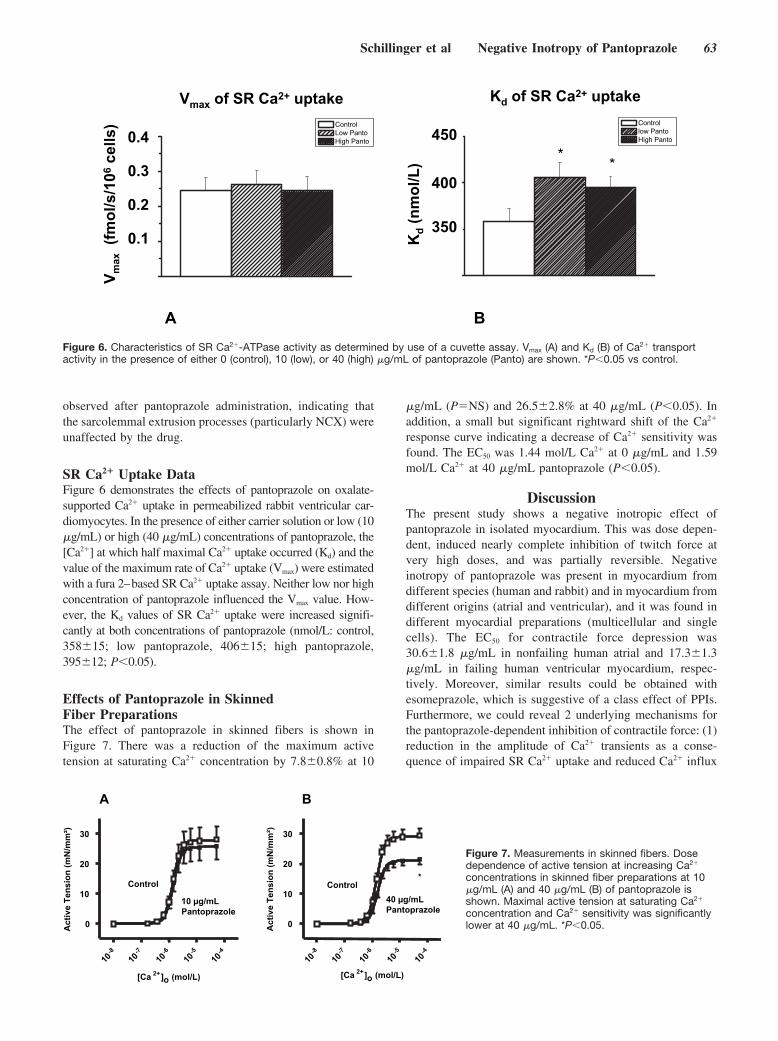
observed after pantoprazole administration, indicating thatthe sarcolemmal extrusion processes (particularly NCX) wereunaffected by the drug.
SR Ca2� Uptake DataFigure 6 demonstrates the effects of pantoprazole on oxalate-supported Ca2� uptake in permeabilized rabbit ventricular car-diomyocytes. In the presence of either carrier solution or low (10�g/mL) or high (40 �g/mL) concentrations of pantoprazole, the[Ca2�] at which half maximal Ca2� uptake occurred (Kd) and thevalue of the maximum rate of Ca2� uptake (Vmax) were estimatedwith a fura 2–based SR Ca2� uptake assay. Neither low nor highconcentration of pantoprazole influenced the Vmax value. How-ever, the Kd values of SR Ca2� uptake were increased signifi-cantly at both concentrations of pantoprazole (nmol/L: control,358�15; low pantoprazole, 406�15; high pantoprazole,395�12; P�0.05).
Effects of Pantoprazole in SkinnedFiber PreparationsThe effect of pantoprazole in skinned fibers is shown inFigure 7. There was a reduction of the maximum activetension at saturating Ca2� concentration by 7.8�0.8% at 10
�g/mL (P�NS) and 26.5�2.8% at 40 �g/mL (P�0.05). Inaddition, a small but significant rightward shift of the Ca2�
response curve indicating a decrease of Ca2� sensitivity wasfound. The EC50 was 1.44 mol/L Ca2� at 0 �g/mL and 1.59mol/L Ca2� at 40 �g/mL pantoprazole (P�0.05).
DiscussionThe present study shows a negative inotropic effect ofpantoprazole in isolated myocardium. This was dose depen-dent, induced nearly complete inhibition of twitch force atvery high doses, and was partially reversible. Negativeinotropy of pantoprazole was present in myocardium fromdifferent species (human and rabbit) and in myocardium fromdifferent origins (atrial and ventricular), and it was found indifferent myocardial preparations (multicellular and singlecells). The EC50 for contractile force depression was30.6�1.8 �g/mL in nonfailing human atrial and 17.3�1.3�g/mL in failing human ventricular myocardium, respec-tively. Moreover, similar results could be obtained withesomeprazole, which is suggestive of a class effect of PPIs.Furthermore, we could reveal 2 underlying mechanisms forthe pantoprazole-dependent inhibition of contractile force: (1)reduction in the amplitude of Ca2� transients as a conse-quence of impaired SR Ca2� uptake and reduced Ca2� influx
lortnoC otnaP woL otnaP hgiH
V xam aC RS fo +2 ekatpu
1.0
2.0
3.0
V max
(fmol
/s/1
06ce
lls)
4.0
Kd
(nm
ol/L
)
lortnoC otnaP wol otnaP hgiH
**
Kd aC RS fo +2 ekatpu
053
004
054
BAFigure 6. Characteristics of SR Ca2�-ATPase activity as determined by use of a cuvette assay. Vmax (A) and Kd (B) of Ca2� transportactivity in the presence of either 0 (control), 10 (low), or 40 (high) �g/mL of pantoprazole (Panto) are shown. *P�0.05 vs control.
Lm/gµ 01elozarpotnaP
lortnoC
01-8 01
-7 01-6 01
5- 01-4
0
01
02
03
aC[ +2 ]o )L/lom(
*
Act
ive
Tens
ion
(mN
/mm
²)
Lm/gµ 04elozarpotnaP
lortnoC
01-8 01
-7 01-6 01
-5 01-4
0
01
02
03
aC[ +2 ]o )L/lom(
BA
Act
ive
Tens
ion
(mN
/mm
²)
Figure 7. Measurements in skinned fibers. Dosedependence of active tension at increasing Ca2�
concentrations in skinned fiber preparations at 10�g/mL (A) and 40 �g/mL (B) of pantoprazole isshown. Maximal active tension at saturating Ca2�
concentration and Ca2� sensitivity was significantlylower at 40 �g/mL. *P�0.05.
Schillinger et al Negative Inotropy of Pantoprazole 63

via ICa,L and (2) reduced Ca2� responsiveness of the myofila-ments as a result of a reduced maximal active tension and aslightly lower Ca2� sensitivity. In contrast, despite the expres-sion of the H�/K�-ATPase at the transcriptional level inhuman and rabbit myocardium, no significant changes in pHi
could be detected in the presence of pantoprazole.Note that the mechanisms underlying the effects of pantopra-
zole in myocardium are completely different from the mecha-nisms of the drug in gastric parietal cells and probably do notinvolve inhibition of H�/K�-ATPase. In regard to gastric protonpump inhibition, all PPIs are prodrugs and require acid tobecome protonated and converted into the active form.1 Afterintravenous administration or intestinal absorption, when orallyadministered the lipophilic unprotonated form readily penetratescell membranes, including that of gastric parietal cells andmyocytes. In parietal cells, as it transverses the cell it is exposedto the acidic environment in the secretory canaliculus of thegastric site and becomes protonated, converting it to a hydro-philic drug that can no longer permeate cell membranes. Thedrug becomes trapped in the canaliculus of the parietal cell. Forthis reason, in parietal cells PPIs exhibit a substantial accumu-lation versus plasma at low pH, eg, 1000-fold for omeprazoleand 10000-fold for rabeprazole at a pH of 1. Moreover, proto-nation of the drug initiates a series of chemical reactions thatculminates in covalent binding of the drug with selected cysteineresidues of the H�/K�-ATPase.1
The present study is the first one reporting on the expres-sion of H�/K�-ATPase in human and rabbit ventricularmyocardium. Recently, it has been suggested that H�/K�-ATPase may contribute to pH homeostasis in rat myocardi-um.7 We therefore investigated the hypothesis that cellularacidosis subsequent to proton pump inhibition might explainthe negative inotropy of PPIs. However, our measurementswith the H�-sensitive fluorescence dye BCECF did not revealany significant influence of pantoprazole on pHi. Moreover,the absence of acidic compartments in cardiac tissue with pH�1 precludes significant accumulation of pantoprazole in themyocardium. In moderately acidic compartments like lyso-somes, slow activation of pantoprazole theoretically mayoccur with an activation half-life of 4.7 hours.20 However,negative inotropy in our experiments usually occurredquickly within several minutes. Moreover, the effect was atleast partially reversible after washout of the drug, whereasthe recovery of the gastric proton pump from pantoprazolehas a half-life of �46 hours21 and was suggested to dependmainly on the synthesis of new pump protein.1,22 In addition,the expression of H�/K�-ATPase in myocardium was verylow compared with gastric mucosa. Therefore, it is unlikelythat the negative inotropy of pantoprazole in myocardium isthe consequence of H�/K�-ATPase inhibition.
We investigated the effects of pantoprazole on intracellularCa2� homeostasis and myofilament Ca2� responsiveness,which are the 2 principal physiological mechanisms of themyocardium to alter contractility. We found a reduction in theCa2� transient amplitude in field-stimulated ventricular myo-cytes. Interpretation of this result in terms of Ca2� signaling iscomplicated by possible effects of pantoprazole on the actionpotential. With fixed depolarizing pulses in voltage-clampedmyocytes, pantoprazole caused a rise in diastolic [Ca2�]i and
minimal changes in systolic [Ca2�]i, the net effect being areduced Ca2� transient amplitude. Independent evidence forpantoprazole-dependent inhibition of SERCA as an underly-ing mechanism was obtained with a fura 2–based SR calciumuptake assay in permeabilized cardiomyocytes. Moreover,Ca2� influx via ICa,L was also depressed by pantoprazole. Thecombination of reduced SR calcium uptake and reduced Ca2�
influx would explain the reduction in SR Ca2� contentobserved and raised diastolic [Ca2�]i.
We also investigated the influence of pantoprazole inskinned fibers. These preparations allow for the investigationof drug effects directly at the myofilament level underwell-defined in vitro conditions. Hence, it can be excludedthat the observed effects are mediated by changes in [Ca2�]i orpHi. We were able to show a significant reduction in maximalactive tension of the contractile proteins at saturating Ca2�
concentrations by 26.5�2.8% at 40 �g/mL. Moreover, at 40�g/mL there was a slight but significant reduction in myo-filament Ca2� sensitivity. However, when the marked nega-tive inotropy in multicellular trabeculae is considered, theseeffects were too faint to explain entirely the mode of action ofpantoprazole in myocardium. Therefore, the negative inot-ropy of pantoprazole observed at 10 �g/mL mainly resultsfrom decreased intracellular [Ca2�]. At the higher dose (40�g/mL), depression of myofilament responsiveness appearsto contribute to the negative inotropic effect. We also suggestthat the effects of pantoprazole in myocardium depend on thenative unprotonated form of the drug and do not requireactivation by low pH because all effects occurred at pH 7.3to 7.4.
In contrast to our data, Yenisehirli and Onur23 found a positiveinotropic effect of 3 different PPIs in rat atria that was potenti-ated by pretreatment with the Na�/K�-ATPase inhibitor ouabain.Moreover, lansoprazole induced a prolongation of the actionpotential. The authors speculated that this could be mediated byinhibition of H�/K�-ATPase promoting altered intracellularCa2� handling. In our study with human and rabbit myocardium,pantoprazole decreased tension and reduced shortening and Ca2�
transient amplitude in “unclamped” trabeculae and single cells.When single rabbit myocytes were clamped with a fixed voltageclamp duration, pantoprazole decreased the Ca2� transient am-plitude to a degree similar to that seen in “unclamped” prepara-tions. This suggests that in humans and rabbits, action potentialduration changes are not instrumental in the decreased Ca2�
transient and subsequent mechanical response caused by panto-prazole application. Moreover, a significant contribution ofH�/K�-ATPase to the action potential is unlikely because of thelow degree of expression. The different responses to PPIs maytherefore be related to species. Compared with human and rabbitmyocardium, in rats the action potential is short and intracellularsodium is high. In particular, the latter condition favors Ca2�
entry by reverse-mode Na�-Ca2� exchange, which contributes toSR Ca2� load and contractility when action potential durationincreases.9 In contrast to the study by Yenisehirli and Onur, thenegative inotropic effect of pantoprazole in rabbit myocardiumobserved by us was not influenced by blockade of Na�/K�-ATPase, and the magnitude of the effect was similar to that inexperiments without ouabain.
64 Circulation July 3, 2007

Finally, although this was beyond the scope of the presentstudy, we would like to speculate whether our findings might beof clinical relevance. Because the negative inotropic effect waspartially reversible after washout of the drug and significantaccumulation in the myocardium is unlikely, the effect ofpantoprazole in vivo probably depends on plasma concentra-tions. Maximal pantoprazole plasma concentrations of 4.6 �g/mL24 and 10.4 �g/mL (Altana Pharma AG, written communi-cation, July 23, 2004) have been found after administration ofcommon oral (40 mg) and intravenous (80 mg) doses. In thepresent work, similar concentrations induced a reduction ofcontractile force of isolated trabeculae by 27�9% (6.25 �g/mL)and 42�8% (12.5 �g/mL), which might indicate a potentialclinical relevance of our findings. However, the duration ofcardiac side effects is temporally limited because all PPIs arequickly eliminated from blood with plasma elimination half-lifeperiods of �1 to 2 hours.20,24 Moreover, cardiac side effects maybe attenuated in vivo because the activity of the active freecompound may be substantially lower because of high plasmaprotein binding.20 On the other hand, particular conditions maybe associated with increased duration and intensity of sideeffects. Patients with heart failure must be investigated for PPItolerance. These patients are much more susceptible to negativeinotropic drugs because of blunted contractile reserve subse-quent to decreased sympathetic sensitivity25 or negative force-frequency relationship.26 In addition, the dependence of H�
elimination from H�/K�-ATPase may be increased in heartfailure because of the impaired function of the Na�/H� exchangesubsequent to increased [Na�]i.27 Moreover, all PPIs undergoextensive hepatic biotransformation before elimination. InCYP2C19-poor metabolizers that represent �3% to 5% ofwhites, a similar percentage of blacks, and 12% to 25% ofdifferent Asian populations, much higher plasma concentrationsand longer elimination half-life periods have been found.1 Thesame holds true for patients with severe liver impairment.28
Recent American29 and Canadian30 studies have shown thatappropriate use of intravenous PPIs was seen in less than halfof the patients. In view of our data, PPIs should be prescribedcarefully. We have recently initiated a clinical study for theinvestigation of cardiac side effects of pantoprazole inhealthy volunteers. Moreover, we encourage clinical studiesto identify individuals with increased intrinsic risk for cardiacside effects of PPIs.
AcknowledgmentsWe gratefully acknowledge the expert assistance of Hanna Schotola,Astrid Steen, Aileen Rankin, Gudrun Muller, Elke Neumann, andMichael Kothe.
Sources of FundingThis work was supported by grants from the Wellcome Trust (to DrKettlewell), the British Heart Foundation (to Dr Smith), and theDeutsche Forschungsgemeinschaft (to Dr Maier).
DisclosuresNone.
References1. Robinson M, Horn J. Clinical pharmacology of proton pump inhibitors:
what the practising physician needs to know. Drugs. 2003;63:2739–2754.2. Delate T, Mager DE, Sheth J, Motheral BR. Clinical and financial
outcomes associated with a proton pump inhibitor prior-authorizationprogram in a Medicaid population. Am J Manag Care. 2005;11:29–36.
3. American Society of Health-system Pharmacists (ASHP). Drug ProductShortages Management Resource Center. April 13, 2006. Available athttp://www.ashp.org/shortage/. Accessed May 16, 2006.
4. Wingo CS, Cain BD. The renal H-K-ATPase: physiological significanceand role in potassium homeostasis. Annu Rev Physiol. 1993;55:323–347.
5. Del CJ, Rajendran VM, Binder HJ. Apical membrane localization ofouabain-sensitive K�-activated ATPase activities in rat distal colon.Am J Physiol. 1991;261:1005–1011.
6. McCabe RD, Young DB. Evidence of a K�-H�-ATPase in vascularsmooth muscle cells. Am J Physiol. 1992;262:1955–1958.
7. Beisvag V, Falck G, Loenechen JP, Qvigstad G, Jynge P, Skomedal T, OsnesJ-B, Sandvik AK, Ellingsen Ö. Identification and regulation of the gastricH�/K�-ATPase in rat heart. Acta Physiol Scand. 2003;179:251–262.
8. Nagashima R, Tsuda Y, Maruyama T, Kanaya S, Fujino Y. Possibleevidence for transmembrane K�-H� exchange system in guinea pig myo-cardium. Jpn Heart J. 1999;40:351–364.
9. Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2nded. Dordrecht, Netherlands: Kluwer Academic Publishers; 2001.
10. Halabi A, Kirch W. Cardiovascular effects of omeprazole and famotidine.Scand J Gastroenterol. 1992;27:753–756.
11. Lau JYW, Sung JJY, Lee KKC, Yung MY, Wong SK, Wu JC, Chan FK,Ng EK, You JH, Lee CW, Chan AC, Chung SC. Effect of intravenousomeprazole on recurrent bleeding after endoscopic treatment of bleedingpeptic ulcers. N Engl J Med. 2000;343:310–316.
12. van Rensburg CJ, Hartmann M, Thorpe A, Venter L, Theron I, LühmannR, Wurst W. Intragastric pH during continuous infusion with panto-prazole in patients with bleeding peptic ulcer. Am J Gastroenterol. 2003;98:2635–2641.
13. Luers C, Fialka F, Elgner A, Zhu D, Kockskamper J, von Lewinski D,Pieske B. Stretch-dependent modulation of [Na�]i, [Ca2�]i, and pHi inrabbit myocardium: a mechanism for the slow force response. CardiovascRes. 2005;68:454–463.
14. Teucher N, Prestle J, Seidler T, Currie S, Elliott EB, Reynolds DF, SchottP, Wagner S, Kogler H, Inesi G, Bers DM, Hasenfuss G, Smith GL.Excessive sarcoplasmic/endoplasmic reticulum Ca2�-ATPase expressioncauses increased sarcoplasmic reticulum Ca2� uptake but decreasesmyocyte shortening. Circulation. 2004;110:3553–3559.
15. Hasenfuss G, Maier LS, Hermann HP, Luers C, Hunlich M, Zeitz O,Janssen PM, Pieske B. Influence of pyruvate on contractile performanceand Ca2� cycling in isolated failing human myocardium. Circulation.2002;105:194–199.
16. Eisner DA, Nichols CG, O’Neill SC, Smith GL, Valdeolmillos M. Theeffects of metabolic inhibition on intracellular calcium and pH in isolatedrat ventricular cells. J Physiol. 1989;411:393–418.
17. Diaz ME, Trafford AW, O’Neill SC, Eisner DA. Measurement of sarcoplasmicreticulum Ca2� content and sarcolemmal Ca2� fluxes in isolated rat ventricularmyocytes during spontaneous Ca2� release. J Physiol. 1997;501:3–16.
18. DeSantiago J, Maier LS, Bers DM. Frequency-dependent acceleration ofrelaxation (FDAR) in heart depends on CaMKII, but not phospholamban.J Mol Cell Cardiol. 2002;34:975–984.
19. Raddatz D, Middel P, Bockemuhl M, Benohr P, Wissmann C, SchworerH, Ramadori G. Glucocorticoid receptor expression in inflammatorybowel disease: evidence for a mucosal down-regulation in steroid-unresponsive ulcerative colitis. Aliment Pharmacol Ther. 2004;19:47–61.
20. Kromer W, Kruger U, Huber R, Hartmann M, Steinijans VW. Differencesin pH-dependent activation rates of substituted benzimidazoles and bio-logical in vitro correlates. Pharmacology. 1998;56:57–70.
21. Katashima M, Yamamoto K, Tokuma Y, Hata T, Sawada Y, Iga T.Comparative pharmacokinetic/pharmacodynamic analysis of proteinpump inhibitors omeprazole, lansoprazole, and pantoprazole, in humans.Eur J Drug Metab Pharmacokinet. 1998;23:19–26.
22. Gedda K, Scott D, Besancon M, Lorentzon P, Sachs G. Turnover of the gastricH�,K�-adenosine triphosphatase a subunit and its effect on inhibition of ratgastric acid secretion. Gastroenterology. 1995;109:1134–1141.
23. Yenisehirli A, Onur R. Positive inotropic and negative chronotropiceffects of proton pump inhibitors in isolated rat atrium. Eur J Pharmacol.2005;519:259–266.
24. Schulz M, Schmoldt. Therapeutic and toxic blood concentrations of morethan 800 drugs and other xenobiotics. Pharmazie. 2003;58:447–474.
Schillinger et al Negative Inotropy of Pantoprazole 65

25. Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS,Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catechol-amine sensitivity and beta-adrenergic receptor density in failing humanhearts. N Engl J Med. 1982;307:205–211.
26. Schillinger W, Lehnart SE, Prestle J, Preuss M, Pieske B, Maier LS,Meyer M, Just H, Hasenfuss G. Influence of SR Ca2�-ATPase andNa�-Ca2�-exchanger on the force-frequency relation. Basic Res Cardiol.1998;93(suppl 1):38–45.
27. Pieske B, Maier LS, Piacentino V III, Weisser J, Hasenfuss G, Houser S.Rate dependence of [Na�]i and contractility in nonfailing and failinghuman. Circulation. 2002;106:447–453.
28. Ferron GM, Preston RA, Noveck RJ, Pockros P, Mayer P, Getsy J,Turner M, Abell M, Paul J. Pharmacokinetics of pantoprazole inpatients with moderate and severe hepatic dysfunction. Clin Ther.2001;23:1180 –1192.
29. Guda NM, Noonan M, Kreiner MJ, Partington S, Vakil N. Use ofintravenous proton pump inhibitors in community practice: an expla-nation for the shortage? Am J Gastroenterol. 2004;99:1233–1237.
30. Kaplan GG, Bates D, McDonald D, Panaccione R, RomagnuoloJ. Inappropriate use of intravenous pantoprazole: extent of theproblem and successful solutions. Clin Gastroenterol Hepatol. 2005;3:1207–1214.
CLINICAL PERSPECTIVEThe proton pump inhibitor pantoprazole was evaluated for its effects on cardiac contractility in isolated myocardium fromhumans and rabbits. We found a dose-dependent negative inotropic effect mainly resulting from alterations in intracellularCa2� handling. At higher concentrations of the drug, depression of myofilament Ca2� responsiveness was also observed.However, despite the expression of the gastric proton pump in human and rabbit hearts, no relevant changes in pHhomeostasis could be detected. Moreover, expression of the pump was very low in myocardium compared with gastrictissue. A similar effect was observed with esomeprazole. Thus, proton pump inhibitors affect cardiac contractility byintracellular mechanisms that are distinct from their effects in gastric parietal cells. The effects in isolated myocardiumwere observed at concentrations that might be of potential clinical relevance. Thus, cardiac effects of proton pumpinhibitors should be evaluated in vivo because of their extensive use for acid-related gastrointestinal diseases.
66 Circulation July 3, 2007

Emergency Department Physician Activation of theCatheterization Laboratory and Immediate Transfer to anImmediately Available Catheterization Laboratory Reduce
Door-to-Balloon Time in ST-Elevation Myocardial InfarctionUmesh N. Khot, MD; Michele L. Johnson, RN; Curtis Ramsey, MS; Monica B. Khot, MD;
Randall Todd, MD; Saeed R. Shaikh, MD; William J. Berg, MD
Background—Consensus guidelines and hospital quality-of-care programs recommend that ST-elevation myocardialinfarction patients achieve a door-to-balloon time of �90 minutes. However, there are limited prospective data onspecific measures to significantly reduce door-to-balloon time.
Methods and Results—We prospectively determined the impact on median door-to-balloon time of a protocol mandating(1) emergency department physician activation of the catheterization laboratory and (2) immediate transfer of the patientto an immediately available catheterization laboratory by an in-house transfer team consisting of an emergencydepartment nurse, a critical care unit nurse, and a chest pain unit nurse. We collected door-to-balloon time for 60consecutive ST-elevation myocardial infarction patients undergoing emergency percutaneous intervention within 24hours of presentation from October 1, 2004, through August 31, 2005, and compared this group with 86 consecutiveST-elevation myocardial infarction patients from September 1, 2005, through June 26, 2006, after protocolimplementation. Median door-to-balloon time decreased overall (113.5 versus 75.5 minutes; P�0.0001), during regularhours (83.5 versus 64.5 minutes; P�0.005), during off-hours (123.5 versus 77.5 minutes; P�0.0001), and with transferfrom an outside affiliated emergency department (147 versus 85 minutes; P�0.0006). Treatment within 90 minutesincreased from 28% to 71% (P�0.0001). Mean infarct size decreased (peak creatinine kinase, 2623�3329 versus1517�1556 IU/L; P�0.0089), as did hospital length of stay (5�7 versus 3�2 days; P�0.0097) and total hospital costsper admission ($26 826�29 497 versus $18 280�8943; P�0.0125).
Conclusions—Emergency department physician activation of the catheterization laboratory and immediate transfer of thepatient to an immediately available catheterization laboratory reduce door-to-balloon time, leading to a reduction inmyocardial infarct size, hospital length of stay, and total hospital costs. (Circulation. 2007;116:67-76.)
Key Words: angioplasty � myocardial infarction � quality of health care � stents � quality indicators, health care
Emergency percutaneous intervention (PCI) is increas-ingly used in the management of ST-elevation myocar-
dial infarction (STEMI). The benefits of emergency PCI are timedependent, with door-to-balloon time delays associated withincreasing mortality.1 Therefore, consensus guidelines recom-mend that STEMI patients achieve a door-to-balloon time of�90 minutes.2 More recently, the American College of Cardi-ology, American Heart Association, the Centers for Medicareand Medicaid Services, and the Joint Commission on Accredi-tation of Healthcare Organizations have all included door-to-balloon time as a core hospital quality-of-care indicator.3–5
Despite the increased emphasis on achieving appropriatedoor-to-balloon times, only 32% of patients overall in theUnited States receive PCI within 90 minutes.6,7 In addition,there has been limited temporal improvement in door-to-
Editorial p 6Clinical Perspective p 76
balloon time,8 leading some to suggest that future improve-ments in door-to-balloon time are unlikely.9 Interestingly,select hospitals have achieved improvements in door-to-balloon times, and recent studies have highlighted qualitativecharacteristics unique to these institutions.10 More recently, asurvey of hospital strategies revealed that activation of thecatheterization laboratory by the emergency department phy-sician rather than cardiologist was associated with fasterdoor-to-balloon times.11 Prior studies actually implementingemergency department physician activation have shown im-provements in door-to-balloon time. One report revealed areduction in median door-to-balloon from 88 to 61 minutes.12
Received November 20, 2006; accepted April 20, 2007.From the Indiana Heart Physicians, Indianapolis (U.N.K., M.B.K., S.R.S., W.J.B.); St. Francis Hospital and Health Centers, Beech Grove (M.L.J.);
Curtis Ramsey and Associates, Indianapolis (C.S.); and Emergency Physicians of Indianapolis, Beech Grove (R.T.), Ind.Correspondence to Umesh N. Khot, MD, Indiana Heart Physicians/St. Francis Heart Center, 5330 E Stop 11 Rd, Indianapolis, IN 46237. E-mail
[email protected]© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.106.677401
67
Interventional Cardiology

However, the results of that study were confounded bysimultaneous conversion of reperfusion strategy from a com-bination of thrombolytics and PCI to solely PCI. In addition,the study excluded data from a 6-month transition periodbetween strategies. In a recent retrospective study, emergencydepartment physician activation reduced door-to-balloon timefrom 118 to 89 minutes.13 There are limited prospective dataon the effect of adopting emergency department physicianactivation of the catheterization laboratory on door-to-balloontime in centers already dedicated to primary PCI. We there-fore prospectively determined the impact on door-to-balloontime of emergency department physician activation of thecatheterization laboratory, combined with a novel strategy ofimmediate physical transfer of the patient to an immediatelyavailable catheterization laboratory by in-house nursing staff.
MethodsStudy DesignThe present prospective study was conducted between October 1,2004, and June 26, 2006, at St Francis Hospital and Health Center(Beech Grove and Indianapolis, Ind), a 591-bed tertiary care com-munity hospital consisting of 2 campuses 7 miles apart (13-minutedrive). Both campuses have emergency departments staffed withemergency medicine residency–trained physicians; cardiology ser-vices are located within the Indianapolis campus. During the studyperiod, the hospital performed primary PCI for all STEMI patientspresenting at either campus. A single 20-physician group providescardiology services at both campuses (Indiana Heart Physicians,Indianapolis). Cardiologists take in-hospital call. Call consists of anoninterventional cardiologist on call with interventional cardiolo-gist on backup call at home �80% of the time; the other 20% of thetime, an interventional cardiologist takes primary call. Call patternwas unchanged during the study period. Four catheterization staffmembers take home call during off-hours and are expected to arriveto the hospital within 30 minutes of laboratory activation.
We prospectively enrolled consecutive patients who presented toeither the Beech Grove or Indianapolis emergency department withSTEMI who received PCI within 24 hours of presentation.4,5 Weexcluded STEMI patients who were hospital inpatients at the time ofdiagnosis.
Protocol During Cardiology Activation/RoutineTransfer Period (October 1, 2004, Through August31, 2005)Emergency department physicians requested immediate cardiologyevaluation for all STEMI patients. After patient evaluation, thecardiologist activated the catheterization laboratory by contacting thecatheterization laboratory coordinator during regular hours (7 AM to5 PM weekdays) or the hospital operator during off-hours (weekendsand 5 PM to 7 AM weekdays). During regular hours, the catheteriza-tion laboratory coordinator notified the emergency department totransfer the patient when a catheterization room became available.During off-hours, transfer to the catheterization laboratory occurredon arrival of 2 catheterization staff members.
Protocol During Emergency DepartmentActivation/Immediate Transfer Period (September1, 2005, Through June 26, 2006)On September 1, 2005, at 7 AM, we implemented a protocol mandating(1) emergency department physician activation of the catheterizationlaboratory and (2) immediate transfer of the patient to an immedi-ately available catheterization laboratory by an in-house emergencyheart attack response team (EHART) consisting of an emergencydepartment nurse, a critical care unit nurse, and a chest pain unitnurse. The only exceptions to immediate transfer by the nursing staffwere hemodynamic compromise (requiring pressors, temporary pac-
ing, or balloon pump) and ongoing cardiopulmonary resuscitation;these patients were prepared for immediate transfer but transferredby the nursing staff with the cardiologist. The emergency departmentphysician contacted the hospital operator to activate the catheteriza-tion laboratory. The operator subsequently paged the cardiologyphysician assistant, catheterization laboratory coordinator, and crit-ical care unit nurse during regular hours or the on-call cardiologist,interventional cardiologist, critical care unit nurse, chest pain unitnurse, and on-call catheterization team during off-hours. On cathe-terization laboratory activation, the critical care unit nurse proceededto the emergency department and subsequently transferred thepatient to the catheterization laboratory with the emergency depart-ment nurse. During transfer, in case of sustained hypotension orarrest, the critical care unit nurse could administer dopamine ornorepinephrine intravenous drips, perform defibrillation, and requestintubation by respiratory therapy, all without prior physician ap-proval. The critical care unit modified the work requirements for theEHART nurse by assigning the nurse 1 patient instead of 2 patients.
To make the catheterization laboratory immediately availableduring regular hours, the catheterization laboratory coordinatoridentified a catheterization room and staff for the patient. Thecatheterization laboratory coordinator could remove an electivepatient from the catheterization laboratory if the case had not started(defined as cardiologist fully scrubbed at bedside obtaining access).If all rooms were occupied with cases in progress, then the STEMIpatient went to the first available room. On patient placement on thecatheterization laboratory table, the EHART team members trans-ferred the patient’s nursing care to the catheterization team.
To make the catheterization laboratory immediately availableduring off-hours, the chest pain unit nurse proceeded to the cathe-terization laboratory, activated the catheterization laboratory imag-ing equipment, and confirmed that the temporary pacemaker, balloonpump, defibrillator, and activated clotting time machine were inworking order. This individual subsequently assisted the critical careunit nurse and emergency department nurse in the initial setup of thepatient, including placement on the catheterization table, monitoringequipment setup, prepping of groin, and assistance with the sterilecatheterization laboratory table. The emergency department nurseand critical care nurse monitored the patient until the third and fourthcatheterization staff members arrived and subsequently transferrednursing care to the catheterization team. If the patient was unstable,all staff attended to the patient until safe transfer of care waspossible. Table 1 compares the processes in the 2 time periods.
All activities in the emergency department, during the transfer tothe catheterization laboratory, and during initial setup in the cathe-terization laboratory did not require cardiologist presence or input(see the order set available at www.stfrancishospitals.org/heart). Thecardiologist evaluated the patient and determined the appropriatenessfor emergency catheterization in the emergency department, en routeto the catheterization laboratory, or in the catheterization laboratory.
Study End Points and Statistical AnalysisThe primary end point was median door-to-balloon time.3 Door timerepresented the arrival time at the initial emergency department.Secondary end points included the individual components of door-to-balloon time (ie, door-to-ECG time), infarct size measured bypeak creatinine kinase within the first 24 hours,14 hospital costs(total, direct, and indirect), hospital length of stay, and all-causein-hospital mortality. Hospital cost data reflect the actual costsinvolved in the delivery of care to each patient and were determinedby the cost-accounting software of the hospital (Alliance for Deci-sion Support, Avega, El Segundo, Calif). Cost data for all patients(including outliers) were analyzed. All-cause in-hospital mortalitywas presented in unadjusted fashion. We determined the prevalenceof “false-positive” activation, defined as a patient sent to thecatheterization laboratory by the emergency department physicianbut who subsequently was determined to be an inappropriateactivation by the cardiologist. All patients provided informed con-sent. Our institutional review board approved the study.
Time values are presented as medians with interquartile rangesand were analyzed using 2-sample Wilcoxon rank sum tests. Other
68 Circulation July 3, 2007
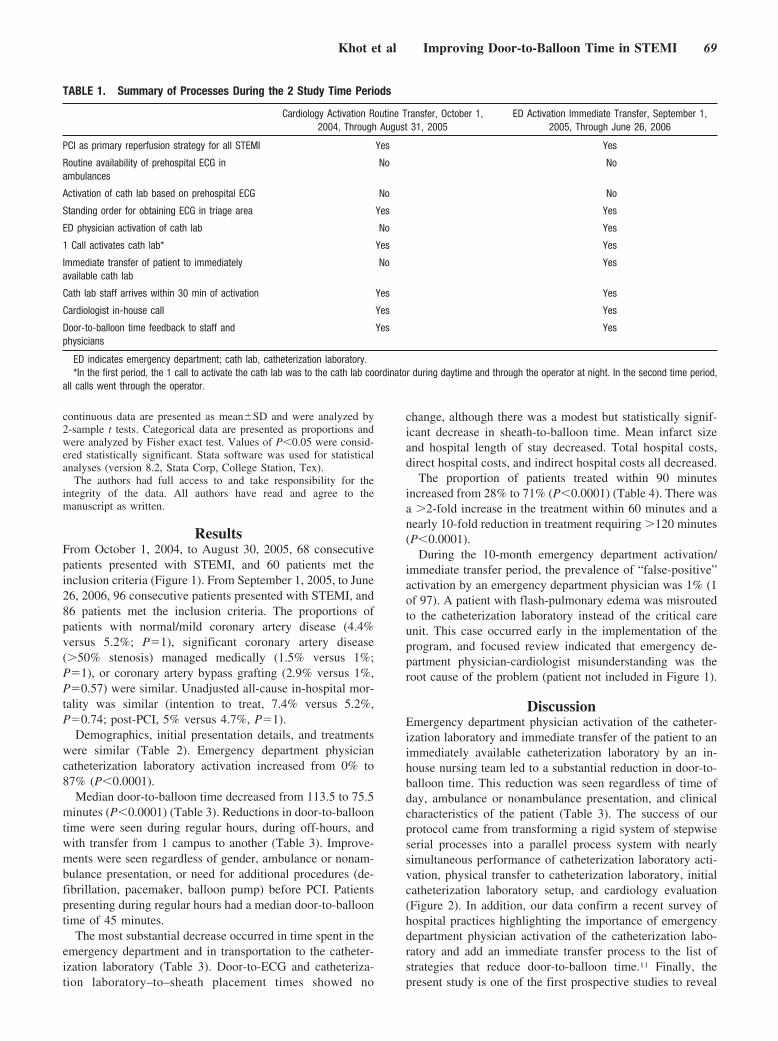
continuous data are presented as mean�SD and were analyzed by2-sample t tests. Categorical data are presented as proportions andwere analyzed by Fisher exact test. Values of P�0.05 were consid-ered statistically significant. Stata software was used for statisticalanalyses (version 8.2, Stata Corp, College Station, Tex).
The authors had full access to and take responsibility for theintegrity of the data. All authors have read and agree to themanuscript as written.
ResultsFrom October 1, 2004, to August 30, 2005, 68 consecutivepatients presented with STEMI, and 60 patients met theinclusion criteria (Figure 1). From September 1, 2005, to June26, 2006, 96 consecutive patients presented with STEMI, and86 patients met the inclusion criteria. The proportions ofpatients with normal/mild coronary artery disease (4.4%versus 5.2%; P�1), significant coronary artery disease(�50% stenosis) managed medically (1.5% versus 1%;P�1), or coronary artery bypass grafting (2.9% versus 1%,P�0.57) were similar. Unadjusted all-cause in-hospital mor-tality was similar (intention to treat, 7.4% versus 5.2%,P�0.74; post-PCI, 5% versus 4.7%, P�1).
Demographics, initial presentation details, and treatmentswere similar (Table 2). Emergency department physiciancatheterization laboratory activation increased from 0% to87% (P�0.0001).
Median door-to-balloon time decreased from 113.5 to 75.5minutes (P�0.0001) (Table 3). Reductions in door-to-balloontime were seen during regular hours, during off-hours, andwith transfer from 1 campus to another (Table 3). Improve-ments were seen regardless of gender, ambulance or nonam-bulance presentation, or need for additional procedures (de-fibrillation, pacemaker, balloon pump) before PCI. Patientspresenting during regular hours had a median door-to-balloontime of 45 minutes.
The most substantial decrease occurred in time spent in theemergency department and in transportation to the catheter-ization laboratory (Table 3). Door-to-ECG and catheteriza-tion laboratory–to–sheath placement times showed no
change, although there was a modest but statistically signif-icant decrease in sheath-to-balloon time. Mean infarct sizeand hospital length of stay decreased. Total hospital costs,direct hospital costs, and indirect hospital costs all decreased.
The proportion of patients treated within 90 minutesincreased from 28% to 71% (P�0.0001) (Table 4). There wasa �2-fold increase in the treatment within 60 minutes and anearly 10-fold reduction in treatment requiring �120 minutes(P�0.0001).
During the 10-month emergency department activation/immediate transfer period, the prevalence of “false-positive”activation by an emergency department physician was 1% (1of 97). A patient with flash-pulmonary edema was misroutedto the catheterization laboratory instead of the critical careunit. This case occurred early in the implementation of theprogram, and focused review indicated that emergency de-partment physician-cardiologist misunderstanding was theroot cause of the problem (patient not included in Figure 1).
DiscussionEmergency department physician activation of the catheter-ization laboratory and immediate transfer of the patient to animmediately available catheterization laboratory by an in-house nursing team led to a substantial reduction in door-to-balloon time. This reduction was seen regardless of time ofday, ambulance or nonambulance presentation, and clinicalcharacteristics of the patient (Table 3). The success of ourprotocol came from transforming a rigid system of stepwiseserial processes into a parallel process system with nearlysimultaneous performance of catheterization laboratory acti-vation, physical transfer to catheterization laboratory, initialcatheterization laboratory setup, and cardiology evaluation(Figure 2). In addition, our data confirm a recent survey ofhospital practices highlighting the importance of emergencydepartment physician activation of the catheterization labo-ratory and add an immediate transfer process to the list ofstrategies that reduce door-to-balloon time.11 Finally, thepresent study is one of the first prospective studies to reveal
TABLE 1. Summary of Processes During the 2 Study Time Periods
Cardiology Activation Routine Transfer, October 1,2004, Through August 31, 2005
ED Activation Immediate Transfer, September 1,2005, Through June 26, 2006
PCI as primary reperfusion strategy for all STEMI Yes Yes
Routine availability of prehospital ECG inambulances
No No
Activation of cath lab based on prehospital ECG No No
Standing order for obtaining ECG in triage area Yes Yes
ED physician activation of cath lab No Yes
1 Call activates cath lab* Yes Yes
Immediate transfer of patient to immediatelyavailable cath lab
No Yes
Cath lab staff arrives within 30 min of activation Yes Yes
Cardiologist in-house call Yes Yes
Door-to-balloon time feedback to staff andphysicians
Yes Yes
ED indicates emergency department; cath lab, catheterization laboratory.*In the first period, the 1 call to activate the cath lab was to the cath lab coordinator during daytime and through the operator at night. In the second time period,
all calls went through the operator.
Khot et al Improving Door-to-Balloon Time in STEMI 69

that decreasing door-to-balloon time leads to decreased in-farct size, length of stay, and hospital costs.
Providing timely emergency PCI is a complex undertakingdemanding rapid coordination of care by multiple physicians,nurses, and hospital staff. In prior reports, an audit process,in-depth continuous quality control improvement analysis,and multidisciplinary quality initiatives have all improveddoor-to-balloon time.15–19 However, the specific steps recom-mended have varied between these studies, making it chal-lenging for other institutions to adopt specific protocols toimprove their door-to-balloon time. In addition, the recom-
mended measures have been multiple and often complex,typically requiring months to years for full implementa-tion.18,19 In contrast, the present study showed that 2 simple,focused modifications rapidly reduced door-to-balloon timeand could be implemented rapidly within a day.
Our protocol was resisted initially because of concerns thatemergency department physician activation of the catheter-ization laboratory would not reduce door-to-balloon timesince our cardiologists took in-house call. However, thissimple change allowed catheterization laboratory staff toarrive 20 to 40 minutes earlier (Table 3), indicating that there
Figure 1. Summary of enrollment and outcomes in study during the 2 time periods. PCI denotes percutaneous intervention. Significantcoronary artery disease indicates a stenosis �50%. ED indicates emergency department; CCU, coronary care unit; CAD, coronaryartery disease; and CABG, coronary artery bypass grafting.
70 Circulation July 3, 2007

TABLE 2. Demographics, Initial Presentation Characteristics, and Treatment Outcomes
Cardiology Activation Routine Transfer, October 1,2004, Through August 31, 2005 (n�60)
ED Activation Immediate Transfer, September 1,2005, Through June 26, 2006 (n�86) P
Demographics
Age, y 58�13 60�13 0.31
Female gender 17 (28.3) 25 (29.1) 1
Health insurance
Private 28 (46.7) 48 (55.8) 0.51
Medicare 21 (35) 29 (33.7) � � �
Medicaid 3 (5) 3 (3.5) � � �
Self-pay 8 (13.3) 6 (7) � � �
Medical history
Current smoker 31 (51.7) 48 (55.8) 0.74
Diabetes 10 (16.7) 17 (19.8) 0.67
Hypertension 34 (56.7) 46 (53.5) 0.74
Hypercholesterolemia 19 (31.7) 31 (36.1) 0.60
Family history of CHD 22 (36.7) 28 (32.6) 0.72
Congestive heart failure 0 (0) 0 (0) � � �
COPD 4 (6.7) 9 (10.5) 0.56
Prior PCI 10 (16.7) 23 (26.7) 0.17
Prior CABG 5 (8.3) 5 (5.8) 0.74
PVD 3 (5) 8 (9.3) 0.53
Stroke 0 (0) 5 (5.8) 0.08
Initial presentation
Regular hours 26 (43.3) 30 (34.9) 0.39
Transferred for PCI 12 (20) 22 (25.6) 0.55
Symptom onset to arrival
�1 h 20 (33.3) 39 (45.4) 0.40
�1–2 h 14 (23.3) 22 (25.6) � � �
�2–6 h 12 (20) 8 (9.3) � � �
�6–12 h 4 (6.7) 4 (4.7) � � �
�12 h 6 (10) 6 (7) � � �
Unknown 4 (6.7) 7 (8.1) � � �
Chest pain at presentation 54 (90) 71 (82.6) 0.24
Prehospital ECG 2 (3.3) 7 (8.1) 0.31
Ambulance arrival 27 (45) 31 (36.1) 0.31
Heart rate, bpm 85�21 79�23 0.10
Systolic blood pressure, mm Hg 137�26 137�34 0.99
Diastolic blood pressure, mm Hg 83�19 83�22 0.89
Location of infarct
Anterior 24 (40) 26 (30.2) 0.29
Inferior 34 (56.7) 56 (65.1) 0.39
Lateral (isolated) 2 (3.3) 4 (4.7) 1
LBBB 2 (3.3) 0 (0) 0.17
ECG leads with ST-elevation
2 16 (27.1) 19 (22.1) 0.80
3–4 29 (49.2) 45 (52.3) � � �
�5 14 (23.7) 22 (25.6) � � �
Cardiogenic shock 4 (6.7) 5 (5.8) 1
Cath lab activation
ED physician 0 (0) 75 (87.2) �0.0001
Cardiologist 60 (100) 11 (12.8) � � �
Khot et al Improving Door-to-Balloon Time in STEMI 71
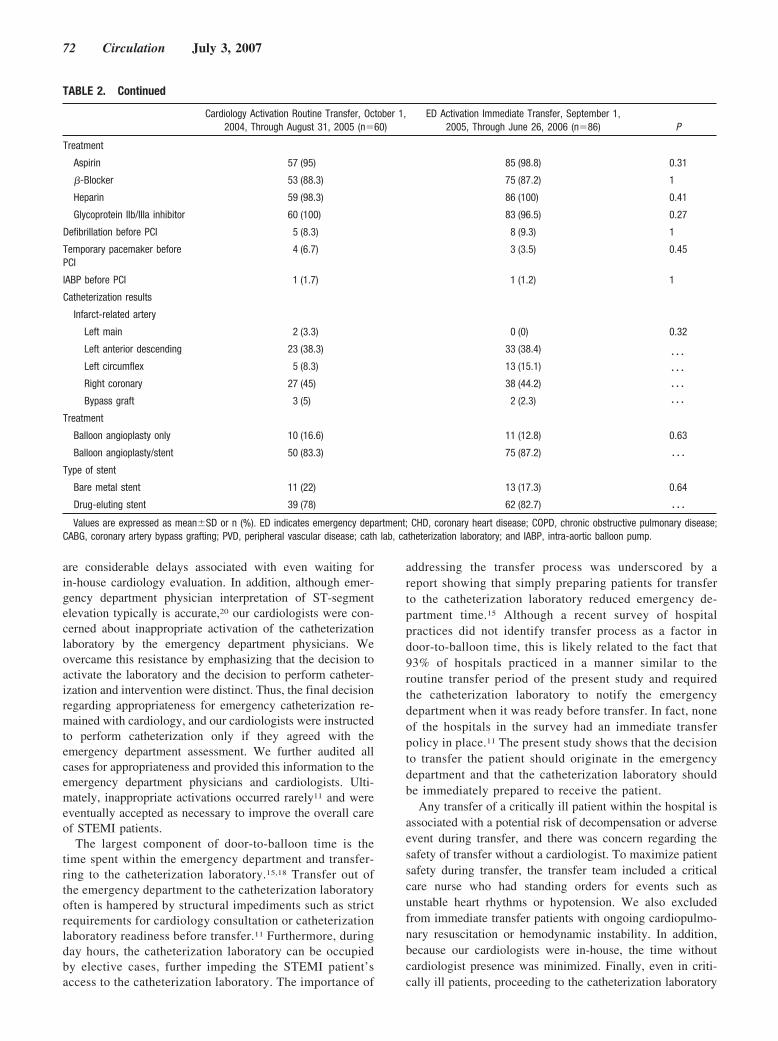
are considerable delays associated with even waiting forin-house cardiology evaluation. In addition, although emer-gency department physician interpretation of ST-segmentelevation typically is accurate,20 our cardiologists were con-cerned about inappropriate activation of the catheterizationlaboratory by the emergency department physicians. Weovercame this resistance by emphasizing that the decision toactivate the laboratory and the decision to perform catheter-ization and intervention were distinct. Thus, the final decisionregarding appropriateness for emergency catheterization re-mained with cardiology, and our cardiologists were instructedto perform catheterization only if they agreed with theemergency department assessment. We further audited allcases for appropriateness and provided this information to theemergency department physicians and cardiologists. Ulti-mately, inappropriate activations occurred rarely11 and wereeventually accepted as necessary to improve the overall careof STEMI patients.
The largest component of door-to-balloon time is thetime spent within the emergency department and transfer-ring to the catheterization laboratory.15,18 Transfer out ofthe emergency department to the catheterization laboratoryoften is hampered by structural impediments such as strictrequirements for cardiology consultation or catheterizationlaboratory readiness before transfer.11 Furthermore, duringday hours, the catheterization laboratory can be occupiedby elective cases, further impeding the STEMI patient’saccess to the catheterization laboratory. The importance of
addressing the transfer process was underscored by areport showing that simply preparing patients for transferto the catheterization laboratory reduced emergency de-partment time.15 Although a recent survey of hospitalpractices did not identify transfer process as a factor indoor-to-balloon time, this is likely related to the fact that93% of hospitals practiced in a manner similar to theroutine transfer period of the present study and requiredthe catheterization laboratory to notify the emergencydepartment when it was ready before transfer. In fact, noneof the hospitals in the survey had an immediate transferpolicy in place.11 The present study shows that the decisionto transfer the patient should originate in the emergencydepartment and that the catheterization laboratory shouldbe immediately prepared to receive the patient.
Any transfer of a critically ill patient within the hospital isassociated with a potential risk of decompensation or adverseevent during transfer, and there was concern regarding thesafety of transfer without a cardiologist. To maximize patientsafety during transfer, the transfer team included a criticalcare nurse who had standing orders for events such asunstable heart rhythms or hypotension. We also excludedfrom immediate transfer patients with ongoing cardiopulmo-nary resuscitation or hemodynamic instability. In addition,because our cardiologists were in-house, the time withoutcardiologist presence was minimized. Finally, even in criti-cally ill patients, proceeding to the catheterization laboratory
TABLE 2. Continued
Cardiology Activation Routine Transfer, October 1,2004, Through August 31, 2005 (n�60)
ED Activation Immediate Transfer, September 1,2005, Through June 26, 2006 (n�86) P
Treatment
Aspirin 57 (95) 85 (98.8) 0.31
�-Blocker 53 (88.3) 75 (87.2) 1
Heparin 59 (98.3) 86 (100) 0.41
Glycoprotein IIb/IIIa inhibitor 60 (100) 83 (96.5) 0.27
Defibrillation before PCI 5 (8.3) 8 (9.3) 1
Temporary pacemaker beforePCI
4 (6.7) 3 (3.5) 0.45
IABP before PCI 1 (1.7) 1 (1.2) 1
Catheterization results
Infarct-related artery
Left main 2 (3.3) 0 (0) 0.32
Left anterior descending 23 (38.3) 33 (38.4)� � �
� � �
� � �
� � �
Left circumflex 5 (8.3) 13 (15.1)
Right coronary 27 (45) 38 (44.2)
Bypass graft 3 (5) 2 (2.3)
Treatment
Balloon angioplasty only 10 (16.6) 11 (12.8) 0.63
Balloon angioplasty/stent 50 (83.3) 75 (87.2) � � �
Type of stent
Bare metal stent 11 (22) 13 (17.3) 0.64
Drug-eluting stent 39 (78) 62 (82.7) � � �
Values are expressed as mean�SD or n (%). ED indicates emergency department; CHD, coronary heart disease; COPD, chronic obstructive pulmonary disease;CABG, coronary artery bypass grafting; PVD, peripheral vascular disease; cath lab, catheterization laboratory; and IABP, intra-aortic balloon pump.
72 Circulation July 3, 2007
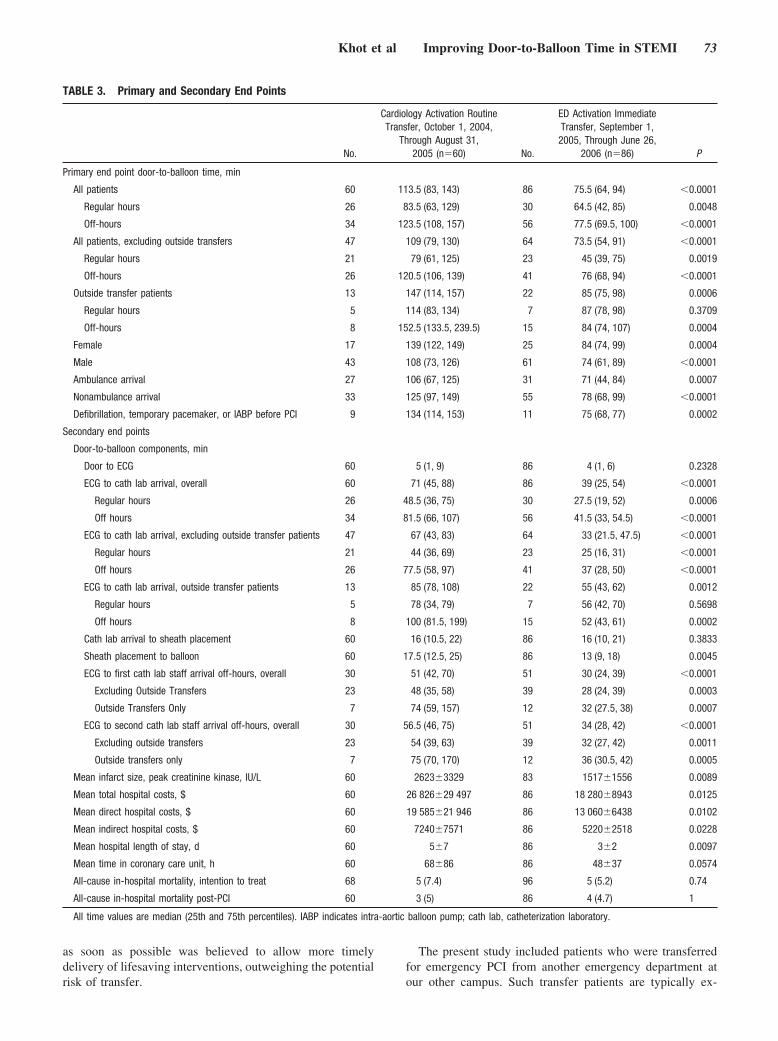
as soon as possible was believed to allow more timelydelivery of lifesaving interventions, outweighing the potentialrisk of transfer.
The present study included patients who were transferredfor emergency PCI from another emergency department atour other campus. Such transfer patients are typically ex-
TABLE 3. Primary and Secondary End Points
No.
Cardiology Activation RoutineTransfer, October 1, 2004,
Through August 31,2005 (n�60) No.
ED Activation ImmediateTransfer, September 1,2005, Through June 26,
2006 (n�86) P
Primary end point door-to-balloon time, min
All patients 60 113.5 (83, 143) 86 75.5 (64, 94) �0.0001
Regular hours 26 83.5 (63, 129) 30 64.5 (42, 85) 0.0048
Off-hours 34 123.5 (108, 157) 56 77.5 (69.5, 100) �0.0001
All patients, excluding outside transfers 47 109 (79, 130) 64 73.5 (54, 91) �0.0001
Regular hours 21 79 (61, 125) 23 45 (39, 75) 0.0019
Off-hours 26 120.5 (106, 139) 41 76 (68, 94) �0.0001
Outside transfer patients 13 147 (114, 157) 22 85 (75, 98) 0.0006
Regular hours 5 114 (83, 134) 7 87 (78, 98) 0.3709
Off-hours 8 152.5 (133.5, 239.5) 15 84 (74, 107) 0.0004
Female 17 139 (122, 149) 25 84 (74, 99) 0.0004
Male 43 108 (73, 126) 61 74 (61, 89) �0.0001
Ambulance arrival 27 106 (67, 125) 31 71 (44, 84) 0.0007
Nonambulance arrival 33 125 (97, 149) 55 78 (68, 99) �0.0001
Defibrillation, temporary pacemaker, or IABP before PCI 9 134 (114, 153) 11 75 (68, 77) 0.0002
Secondary end points
Door-to-balloon components, min
Door to ECG 60 5 (1, 9) 86 4 (1, 6) 0.2328
ECG to cath lab arrival, overall 60 71 (45, 88) 86 39 (25, 54) �0.0001
Regular hours 26 48.5 (36, 75) 30 27.5 (19, 52) 0.0006
Off hours 34 81.5 (66, 107) 56 41.5 (33, 54.5) �0.0001
ECG to cath lab arrival, excluding outside transfer patients 47 67 (43, 83) 64 33 (21.5, 47.5) �0.0001
Regular hours 21 44 (36, 69) 23 25 (16, 31) �0.0001
Off hours 26 77.5 (58, 97) 41 37 (28, 50) �0.0001
ECG to cath lab arrival, outside transfer patients 13 85 (78, 108) 22 55 (43, 62) 0.0012
Regular hours 5 78 (34, 79) 7 56 (42, 70) 0.5698
Off hours 8 100 (81.5, 199) 15 52 (43, 61) 0.0002
Cath lab arrival to sheath placement 60 16 (10.5, 22) 86 16 (10, 21) 0.3833
Sheath placement to balloon 60 17.5 (12.5, 25) 86 13 (9, 18) 0.0045
ECG to first cath lab staff arrival off-hours, overall 30 51 (42, 70) 51 30 (24, 39) �0.0001
Excluding Outside Transfers 23 48 (35, 58) 39 28 (24, 39) 0.0003
Outside Transfers Only 7 74 (59, 157) 12 32 (27.5, 38) 0.0007
ECG to second cath lab staff arrival off-hours, overall 30 56.5 (46, 75) 51 34 (28, 42) �0.0001
Excluding outside transfers 23 54 (39, 63) 39 32 (27, 42) 0.0011
Outside transfers only 7 75 (70, 170) 12 36 (30.5, 42) 0.0005
Mean infarct size, peak creatinine kinase, IU/L 60 2623�3329 83 1517�1556 0.0089
Mean total hospital costs, $ 60 26 826�29 497 86 18 280�8943 0.0125
Mean direct hospital costs, $ 60 19 585�21 946 86 13 060�6438 0.0102
Mean indirect hospital costs, $ 60 7240�7571 86 5220�2518 0.0228
Mean hospital length of stay, d 60 5�7 86 3�2 0.0097
Mean time in coronary care unit, h 60 68�86 86 48�37 0.0574
All-cause in-hospital mortality, intention to treat 68 5 (7.4) 96 5 (5.2) 0.74
All-cause in-hospital mortality post-PCI 60 3 (5) 86 4 (4.7) 1
All time values are median (25th and 75th percentiles). IABP indicates intra-aortic balloon pump; cath lab, catheterization laboratory.
Khot et al Improving Door-to-Balloon Time in STEMI 73

cluded from most analyses15,18 and are specifically excludedfrom public reporting of quality indicators despite having thelongest door-to-balloon times.4,5 In the National Registry ofMyocardial Infarction, transfer patients had a median door-to-balloon time of 180 minutes, with only 4.2% achievingreperfusion within 90 minutes.7 With our new protocol, themedian door-to-balloon time decreased to 85 minutes, and62% of these patients were treated within 90 minutes. Thus,our protocol leads to improvement in the care of STEMIpatients transferred directly for PCI.
Most patients undergoing emergency PCI present duringoff-hours, and only 26% undergo reperfusion in �90 min-utes.6 It has been suggested that hospitals that perform PCIhave catheterization staff on site 24 hours to ensure timelyrevascularization or to cross-train in-house staff to performcatheterization staff duties.6 However, even in high-volumecenters, 24-hour coverage would be prohibitively expensive,and maintaining proficiency of cross-trained staff exposed tonighttime myocardial infarction cases only would be chal-lenging. Our use of an in-house transfer team allowed us tosubstantially improve the care of off-hours patients while
maintaining the delivery of care by a highly trained catheter-ization staff.
Study LimitationsAlthough the 2 cohorts were similar, baseline differencescannot be completely accounted for between the 2 timeperiods because of the nonrandomized nature of the presentstudy. Nevertheless, the present study design reflects the“real-life” manner in which physicians and hospitals imple-ment process improvements and is similar to other processimprovement studies.21 The present study was performed in acommunity hospital setting with cardiologists taking in-housecall. However, the protocol could be adapted for cardiologiststaking home call only or for the inclusion of residents andfellows in academic settings. Our results could be explainedby increased attention to door-to-balloon time; however, ourresults during the baseline time period were widely presentedto physician and hospital staff with no door-to-balloon timeimprovement. Our transfer patients traveled a 7-mile dis-tance, and our results may not be applicable to longer transferdistances. Emergency medicine residency–trained physicians
TABLE 4. Proportion of Patients Treated Within Various Time Points
Door-to-Balloon (min)Cardiology Activation/ Routine Transfer, October
1, 2004, Through August 31, 2005 (n�60)ED Activation/Immediate Transfer, September 1,
2005, Through June 26, 2006 (n�86) P
�60 5 (8.3) 17 (19.8) �0.0001
60–90 12 (20) 44 (51.2)
91–120 16 (26.7) 20 (23.3)
�120 27 (45) 5 (5.8)
Values are expressed as n (%).
Figure 2. Serial vs parallel processing in achievingdoor-to-balloon time. Simultaneous performance ofcatheterization laboratory activation, physicaltransfer to catheterization laboratory, initial cathe-terization laboratory setup, and cardiology evalua-tion leads to a reduction in door-to-balloon time.
74 Circulation July 3, 2007

staffed our emergency departments, and staffing by a differ-ent mix of physicians may not be able to duplicate our results.Our results occurred with limited availability of prehospitalECG, but we believe the use of ECGs is complementary andcould further reduce door-to-balloon time. However, 50% ofSTEMI patients nationwide do not present by ambulance,underscoring the importance of protocols that improve thecare of all patients.22
Emergency department activation of the catheterizationlaboratory and immediate transfer of the patient to an imme-diately available catheterization laboratory by an in-housetransfer team are 2 specific measures that allow the deliveryof PCI in a timely fashion to a broad population of patients.Additional benefits include reductions in myocardial infarctsize, hospital length of stay, and total hospital costs. Wide-spread implementation of this simple strategy can substan-tially improve the quality of care of STEMI patients under-going emergency PCI. An electronic copy of the order setused in the EHART protocol is available at www.stfrancishospitals.org/heart.
AppendixThe following individuals participated in the present study.Indiana Heart Physicians, Indianapolis: A. Akinwande, M.Barron, J. Christie, H. Genovely, J. Graham, D. Hadian, M.Jones, S. Karanam, D. Kovacich, I. Labin, S. Lall, J. Mossler,G. Revytak, and R. Shea. Emergency Physicians of Indianap-olis, Beech Grove, Ind: S. Antoine, D. Blank, S. Boha, M.Brown, W. Corbett, D. Debikey, B. Dillman, K. Ernsting, M.Fitzpatrick, G. Godfrey, C. Hartman, B. Johnston, S. Kistler,H. Levitin, R. Mara, W. McDaniel, E. Olson, M. Overfelt, M.Russell, A. Stern, M. Stone, and E. Weinstein.
AcknowledgmentsWe are indebted to Mechelle L. Peck, RN; Diana L. Brown, RN;Mark Manning, CCT; Patricia L. Wray, RN; Stephen H. Kliman,MD; Juan E. Weksler, MD; Carl L. Rouch, MD; and Horace O.Hickman, MD, who were intimately involved in the implementationof the present study.
Sources of FundingIndiana Heart Physicians and St Francis Hospital and Health Centersprovided funding for the present study. The funding organizationshad no role in the design and conduct of the study; collection,management, analysis, and interpretation of the data; and prepara-tion, review, or approval of the manuscript.
DisclosuresNone.
References1. Berger PB, Ellis SG, Holmes DR, Jr., Granger CB, Criger DA, Betriu A,
Topol EJ, Califf RM. Relationship between delay in performing directcoronary angioplasty and early clinical outcome in patients with acutemyocardial infarction: results from the Global Use of Strategies to OpenOccluded Arteries in Acute Coronary Syndromes (GUSTO-IIb) trial.Circulation. 1999;100:14–20.
2. Antman EM, Anbe DT, Armstrong PW, Bates ER, Green LA, Hand M,Hochman JS, Krumholz HM, Kushner FG, Lamas GA, Mullany CJ,Ornato JP, Pearle DL, Sloan MA, Smith SC Jr, Alpert JS, Anderson JL,Faxon DP, Fuster V, Gibbons RJ, Gregoratos G, Halperin JL, HiratzkaLF, Hunt SA, Jacobs AK, Ornato JP. ACC/AHA guidelines for themanagement of patients with ST-elevation myocardial infarction: a reportof the American College of Cardiology/American Heart Association Task
Force on Practice Guidelines (Committee to Revise the 1999 Guidelinesfor the Management of Patients With Acute Myocardial Infarction). J AmColl Cardiol. 2004;44:E1–E211.
3. Krumholz HM, Anderson JL, Brooks NH, Fesmire FM, Lambrew CT,Landrum MB, Weaver WD, Whyte J, Bonow RO, Bennett SJ, Burke G,Eagle KA, Linderbaum J, Masoudi FA, Normand SL, Pina IL, RadfordMJ, Rumsfeld JS, Ritchie JL, Spertus JA. ACC/AHA clinical per-formance measures for adults with ST-elevation and non-ST-elevationmyocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Performance Measures(Writing Committee to Develop Performance Measures on ST-Elevationand Non-ST-Elevation Myocardial Infarction). J Am Coll Cardiol. 2006;47:236–265.
4. Centers for Medicare & Medicaid Services and the Joint Commission onAccreditation of Healthcare Organizations. Specifications Manual forNational Hospital Quality Measures. Baltimore, Md: Centers forMedicare and Medicaid Services and the Joint Commission on Accredi-tation of Healthcare Organizations; 2006. Available at: http://qnetexchange.org/public/hdc.do?hdcPage�hosp_quality_manual.Accessed April 7, 2006.
5. Williams SC, Schmaltz SP, Morton DJ, Koss RG, Loeb JM. Quality ofcare in U.S. hospitals as reflected by standardized measures, 2002–2004.N Engl J Med. 2005;353:255–264.
6. Magid DJ, Wang Y, Herrin J, McNamara RL, Bradley EH, Curtis JP,Pollack CV Jr, French WJ, Blaney ME, Krumholz HM. Relationshipbetween time of day, day of week, timeliness of reperfusion, andin-hospital mortality for patients with acute ST-segment elevation myo-cardial infarction. JAMA. 2005;294:803–812.
7. Nallamothu BK, Bates ER, Herrin J, Wang Y, Bradley EH, KrumholzHM. Times to treatment in transfer patients undergoing primary percu-taneous coronary intervention in the United States: National Registry ofMyocardial Infarction (NRMI)-3/4 analysis. Circulation. 2005;111:761–767.
8. McNamara RL, Herrin J, Bradley EH, Portnay EL, Curtis JP, Wang Y,Magid DJ, Blaney M, Krumholz HM. Hospital improvement in time toreperfusion in patients with acute myocardial infarction, 1999 to 2002.J Am Coll Cardiol. 2006;47:45–51.
9. National Heart Attack Alert Program Coordinating Committee. NationalHeart Attack Alert Program Coordinating Committee and SubcommitteesMeeting Summary Reports. In: Abstracts of the National Heart AttackAlert Program 10-Year Anniversary Meeting; June 25–26, 2001; Alex-andria, Va: 10.
10. Bradley EH, Curry LA, Webster TR, Mattera JA, Roumanis SA, RadfordMJ, McNamara RL, Barton BA, Berg DN, Krumholz HM. Achievingrapid door-to-balloon times: how top hospitals improve complex clinicalsystems. Circulation. 2006;113:1079–1085.
11. Bradley EH, Herrin J, Wang Y, Barton BA, Webster TR, Mattera JA,Roumanis SA, Curtis JP, Nallamothu BK, Magid DJ, McNamara RL,Parkosewich J, Loeb JM, Krumholz HM. Strategies for reducing thedoor-to-balloon time in acute myocardial infarction. N Engl J Med.2006;355:2308–2320.
12. Thatcher JL, Gilseth TA, Adlis S. Improved efficiency in acute myo-cardial infarction care through commitment to emergency department-initiated primary PCI. J Invasive Cardiol. 2003;15:693–698.
13. Jacoby J, Axelband J, Patterson J, Belletti D, Heller M. Cardiac cathe-terization lab activation by the emergency physician without prior con-sultation decreases door-to-balloon time. J Invasive Cardiol. 2005;17:154–155.
14. Haase J, Bayar R, Hackenbroch M, Storger H, Hofmann M, Schwarz CE,Reinemer H, Schwarz F, Ruef J, Sommer T. Relationship between size ofmyocardial infarctions assessed by delayed contrast-enhanced MRI afterprimary PCI, biochemical markers, and time to intervention. J IntervCardiol. 2004;17:367–373.
15. Ward MR, Lo ST, Herity NA, Lee DP, Yeung AC. Effect of audit ondoor-to-inflation times in primary angioplasty/stenting for acute myo-cardial infarction. Am J Cardiol. 2001;87:336–338, A339.
16. Shry EA, Eckart RE, Winslow JB, Rollefson WA, Simpson DE. Effect ofmonitoring of physician performance on door-to-balloon time for primaryangioplasty in acute myocardial infarction. Am J Cardiol. 2003;91:867–869.
17. Caputo RP, Ho KK, Stoler RC, Sukin CA, Lopez JJ, Cohen DJ, KuntzRE, Berman A, Carrozza JP, Baim DS. Effect of continuous qualityimprovement analysis on the delivery of primary percutaneous trans-luminal coronary angioplasty for acute myocardial infarction.Am J Cardiol. 1997;79:1159–1164.
Khot et al Improving Door-to-Balloon Time in STEMI 75

18. Caputo RP, Kosinski R, Walford G, Giambartolomei A, Grant W, RegerMJ, Simons A, Esente P. Effect of continuous quality improvementanalysis on the delivery of primary percutaneous revascularization foracute myocardial infarction: a community hospital experience. CatheterCardiovasc Interv. 64:428–433, 2005.
19. Zarich SW, Sachdeva R, Fishman R, Werdmann MJ, Parniawski M,Bernstein L, Dilella M. Effectiveness of a multidisciplinary qualityimprovement initiative in reducing door-to-balloon times in primaryangioplasty. J Interv Cardiol. 2004;17:191–195.
20. Brady WJ, Perron A, Ullman E. Errors in emergency physician interpre-tation of ST-segment elevation in emergency department chest painpatients. Acad Emerg Med. 2000;7:1256–1260.
21. Eagle KA, Montoye CK, Riba AL, DeFranco AC, Parrish R, Skorcz S,Baker PL, Faul J, Jani SM, Chen B, Roychoudhury C, Elma MA, MitchellKR, Mehta RH. Guideline-based standardized care is associated withsubstantially lower mortality in Medicare patients with acute myocardialinfarction: the American College of Cardiology’s Guidelines Applied inPractice (GAP) projects in Michigan. J Am Coll Cardiol. 2005;46:1242–1248.
22. Canto JG, Zalenski RJ, Ornato JP, Rogers WJ, Kiefe CI, Magid D,Shlipak MG, Frederick PD, Lambrew CG, Littrell KA, Barron HV. Useof emergency medical services in acute myocardial infarction and sub-sequent quality of care: observations from the National Registry ofMyocardial Infarction 2. Circulation. 2002;106:3018–3023.
CLINICAL PERSPECTIVEThe relationship between delays in door-to-balloon time and increased mortality is well established and has led toconsensus guidelines and hospital quality-of-care programs recommending that ST-elevation myocardial infarction patientsachieve a door-to-balloon time of �90 minutes. However, most patients do not receive treatment within the recommended90 minutes, and there are limited prospective data on specific measures to significantly reduce door-to-balloon time. In thepresent study, we prospectively determined the impact on median door-to-balloon time of a protocol mandating (1)emergency department physician activation of the catheterization laboratory and (2) immediate transfer of the patient to animmediately available catheterization laboratory by an in-house transfer team consisting of an emergency departmentnurse, a critical care unit nurse, and a chest pain unit nurse. With this protocol, median door-to-balloon time fellsubstantially, and this reduction was seen regardless of time of day, ambulance or nonambulance presentation, and clinicalcharacteristics of the patients. In addition, patients transferred from an outside affiliated emergency department to our mainhospital also saw substantial reductions in door-to-balloon time. Treatment within 90 minutes increased from 28% to 71%.Other benefits included reductions in mean infarct size, hospital length of stay, and total hospital costs per admission. Thepresent study identifies a simple and widely applicable strategy that leads to improvements in both door-to-balloon timeand the quality of care of ST-elevation myocardial infarction patients undergoing emergency percutaneous intervention.
76 Circulation July 3, 2007

The Brain–Heart ConnectionMartin A. Samuels, MD
Neurocardiology has many dimensions, but it may beconceptualized as divided into 3 major categories: the
heart’s effects on the brain (eg, cardiac source embolicstroke), the brain’s effects on the heart (eg, neurogenic heartdisease), and neurocardiac syndromes (eg, Friedreich dis-ease). The present review deals with the nervous system’scapacity to injure the heart. This subject is inherently impor-tant but also represents an example of a much more wide-spread and conceptually fascinating area of neurovisceraldamage in general.
History of Learning the Nature of theBrain–Heart Connection
In 1942, at the culmination of his distinguished career asProfessor of Physiology at Harvard Medical School, WalterB. Cannon published a remarkable paper entitled “‘Voodoo’Death,”1 in which he recounted anecdotal experiences,largely from the anthropology literature, of death from fright.These often remote events, drawn from widely disparate partsof the world, had several features in common. They were allinduced by an absolute belief that an external force, such asa wizard or medicine man, could, at will, cause demise andthat the victim himself had no power to alter this course. Thisperceived lack of control over a powerful external force is thesine qua non for all the cases recounted by Cannon, whopostulated that death was caused “by a lasting and intenseaction of the sympathico-adrenal system.” Cannon believedthat this phenomenon was limited to societies in which thepeople were “so superstitious, so ignorant, that they feelthemselves bewildered strangers in a hostile world. Instead ofknowledge, they have fertile and unrestricted imaginationswhich fill their environment with all manner of evil spiritscapable of affecting their lives disastrously.” Over the yearssince Cannon’s observations, evidence has accumulated tosupport his concept that “voodoo” death is, in fact, a realphenomenon but, far from being limited to ancient peoples,may be a basic biological principle that provides an importantclue to understanding the phenomenon of sudden death inmodern society as well as providing a window into the worldof neurovisceral disease (also known as psychosomaticillness).
George Engel collected 160 accounts from the lay press ofsudden death that were attributed to disruptive life events.2
He found that such events could be divided into 8 categories:
(1) the impact of the collapse or death of a close person; (2)during acute grief; (3) on threat of loss of a close person; (4)during mourning or on an anniversary; (5) on loss of status orself-esteem; (6) personal danger or threat of injury; (7) afterdanger is over; (8) reunion, triumph, or happy ending.Common to all is that they involve events impossible for thevictim to ignore and to which the response is overwhelmingexcitation, giving up, or both.
In 1957, Curt Richter reported on a series of experimentsaimed to elucidate the mechanism of Cannon’s “voodoo”death.3 Richter, a former student of Cannon, pursued anincidental discovery of an epidemic of sudden death in acolony of rodents, which was induced when a colleague,Gordon Kennedy, had clipped the whiskers of the animals toprevent contamination of the urine collection. Richter studiedthe length of time that domesticated rats could swim atvarious water temperatures and found that at a water temper-ature of 93°C these rats could swim for 60 to 80 minutes.However, if the animal’s whiskers were trimmed, it wouldinvariably drown within a few minutes. When carrying outsimilar experiments with fierce wild rats, he noted that anumber of factors contributed to the tendency for suddendeath, the most important of which was restraint, whichinvolved holding the animals and confinement in a glassswimming jar with no chance of escape. By trimming therats’ whiskers, a procedure that destroys possibly their mostimportant proprioceptive mechanism, the tendency for earlydemise was exacerbated. In the case of the calm domesticatedanimals in which restraint and confinement were apparentlynot significant stressors, removal of whiskers rendered theseanimals as fearful as wild rats with a corresponding tendencyfor sudden death. ECGs taken during the process showeddevelopment of a bradycardia prior to death, and adrenalec-tomy did not protect the animals. Furthermore, atropineprotected some of the animals, and cholinergic drugs led to aneven more rapid demise. All this was taken as evidence thatoveractivity of the sympathetic nervous system was not thecause of the death but rather it was caused by increased vagaltone.
We now believe that the apparently opposite conclusionsof Cannon and Richter are not mutually exclusive, but ratherthat a generalized autonomic storm, which occurs as a resultof a life-threatening stressor, will have both sympathetic andparasympathetic effects. The apparent predominance of one
From the Department of Neurology, Brigham and Women’s Hospital, Harvard Medical School, Boston, Mass.Correspondence to Dr Martin A. Samuels, Department of Neurology, Brigham and Women’s Hospital, Harvard Medical School, 75 Francis St, Boston,
MA 02115. E-mail [email protected](Circulation. 2007;116:77-84.)© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI:10.1161/CIRCULATIONAHA.106.678995 77
a

over the other depends on the parameter measured (eg, heartrate, blood pressure) and the timing of the observations inrelation to the stressor (eg, early events tend to be dominatedby sympathetic effects, whereas late events tend to bedominated by parasympathetic effects). Cerebral hemispheraldominance with regard to autonomic control (right predom-inantly sympathetic and left predominantly parasympathetic)probably also contributes to the dominant mechanism ofsudden death (ie, sympathetic versus vagal) in a givenperson.4
In human beings, one of the easily accessible windows intoautonomic activity is the ECG. Edwin Byer and colleaguesreported 6 patients whose ECGs showed large upright Twaves and long QT intervals.5 Two of these patients hadhypertensive encephalopathy, 1 patient had a brain stemstroke with neurogenic pulmonary edema, 1 patient had anintracerebral hemorrhage, 1 patient had a postpartum ische-mic stroke possibly related to toxemia of pregnancy, and 1patient had no medical history except a blood pressure of210/110 mm Hg. On the basis of experimental results ofcooling or warming the endocardial surface of a dog’s leftventricle, Byer and colleagues concluded that these ECGchanges were caused by subendocardial ischemia. HaroldLevine reported on several disorders other than ischemicheart disease that could produce ECG changes reminiscent ofcoronary disease.6 Among these was a 69-year-old womanwho was admitted and remained in coma. Her admissionECG showed deeply inverted T waves in the anterior andlateral precordial leads. Two days later, it showed ST seg-ment elevation with less deeply inverted T waves, a patternsuggestive of myocardial infarction. However, at autopsy aruptured berry aneurysm was found and no evidence ofmyocardial infarction or pericarditis was noted. Levine didnot propose a specific mechanism but referred to experimen-tal work on the production of cardiac arrhythmias by basalganglia stimulation and ST and T-wave changes induced byinjection of caffeine into the cerebral ventricle.
George Burch and colleagues7 reported on 17 patients whowere said to have “cerebrovascular accidents” (ie, strokes). In14 of the 17, hemorrhage was demonstrated by lumbarpuncture. It is not possible to determine which of thesepatients had hemorrhagic infarction, intracerebral hemor-rhage, and subarachnoid hemorrhage, and no data about theterritory of the strokes are available. The essential features ofthe ECG abnormalities were: (1) long QT intervals in allpatients; (2) large, usually inverted, T waves in all patients;and (3) U waves in 11 of the 17 patients.7
Cropp and Manning8 reported on the details of ECGabnormalities in 29 patients with subarachnoid hemorrhage.Twenty-two of these patients survived. Two of those whodied had no postmortem examination, which left 5 patients inwhom autopsies confirmed the presence of a ruptured cere-bral aneurysm. In 3 of these 5 patients, the heart and coronaryarteries were said to be normal, but the details of thepathological examination were not revealed. The point ismade that ECG changes seen in the context of neurologicaldisease do not represent ischemic heart disease but are merelya manifestation of autonomic dysregulation, possibly causedby a lesion that affected the cortical representation of the
autonomic nervous system. The authors argued that Brod-mann area 13 on the orbital surface of the frontal lobe andarea 24 on the anterior cingulate gyrus were the corticalcenters for cardiovascular control.
There is clear evidence that cardiac lesions can be pro-duced as the result of nervous system disease. The concept ofvisceral organ dysfunction that occurs as a result of neuro-logical stimuli can be traced to Ivan Pavlov. Hans Selye, astudent of Pavlov, described electrolyte–steroid–cardiopathywith necroses (ESCN).9 Selye’s view was that this cardiaclesion was common and often described by different names inthe literature. He argued that this lesion was distinct from thecoagulation necrosis that occurred as a result of ischemicdisease, but that it could exist in the same heart. Selyedemonstrated that certain steroids and other hormones createda predisposition for the development of ESCN, but that otherfactors were required for ESCN to develop. The mosteffective conditioning steroid was 2-�-methyl-9-�-fluorocortisol. Among the factors that led to ESCN insteroid-sensitized animals were certain electrolytes (eg,NaH2PO4), various hormones (eg, vasopressin, adrenaline,insulin, thyroxine), certain vitamins (eg, dihydrotachysterol),cardiac glycosides, surgical interventions (eg, cardiac reper-fusion after ischemia), and psychic or nervous stimuli (eg,restraint, fright). The cardiac lesions could not be preventedby adrenalectomy, which suggests that the process, if relatedto sympathetic hyperactivity, must exert its influence bydirect neural connection to the heart rather than by a blood-borne route.
Cardiac lesions may be produced in rats by pretreatmentwith either 2-�-methyl-9-�-fluorohydrocortisone (fluorocor-tisol), dihydrotachysterol (calciferol), or thyroxine and thenrestraint of the animals on a board for 15 hours or with coldstress.10 Agents that act by inhibition of the catecholamine-mobilizing reflex arc at the hypothalamic level (eg, chlor-promazine) or by blockade of only the circulating but not theneurogenic intramyocardial catecholamines (eg, dibenamine)were the least effective for protection of cardiac muscle,whereas those drugs that act by ganglionic blockade (eg,mecamylamine) or by direct intramyocardial catecholamine-depletion (eg, reserpine) were the most effective. Further-more, it is clear that blood catecholamine levels are oftennormal but that identical ECG findings are seen with highsystemic catecholamines. These clinical and pharmacologicaldata support the concept that the cardiac necrosis is caused bycatecholamine toxicity and that catecholamines released di-rectly into the heart via neural connections are much moretoxic than those that reach the heart via the bloodstream,though clearly the 2 routes could be additive in the intact,nonadrenalectomized animal. Intracoronary infusions of epi-nephrine reproduce the characteristic ECG pattern of neuro-cardiac disease, which is reminiscent of subendocardialischemia, though no ischemic lesion can be found in thehearts of dogs euthanized after several months of infusions.11
In the years that followed, numerous reports emanated fromaround the world that documented the production of cardiacrepolarization abnormalities in the context of various neuro-logical catastrophes and that proposed that this was caused byan autonomic storm. It seemed likely that the connection
78 Circulation July 3, 2007

between neuropsychiatric illness and the visceral organswould be provided by the autonomic nervous system.
Melville et al12 produced ECG changes and myocardialnecrosis by stimulation of the hypothalamus of cats. Withanterior hypothalamic stimulation, parasympathetic responsesoccurred, predominantly in the form of bradycardia. Lateralhypothalamic stimulation produced tachycardia and ST seg-ment depressions. With intense bilateral and repeated lateralstimulation, persistent irreversible ECG changes occurred andpostmortem examination revealed a stereotyped cardiac le-sion characterized by intense cytoplasmic eosinophilia withloss of cross-striations and some hemorrhage. The coronaryarteries were normal without occlusion. Although Melvillereferred to this lesion as “infarction,” it is probably best toreserve that term for coagulation necrosis caused by ischemia.This lesion is probably identical to Selye’s ESCN and wouldnow be called coagulative myocytolysis, myofibrillar degen-eration, or contraction band necrosis. More recently, Oppen-heimer and Cechetto have mapped the chronotropic organi-zational structure in the rat insular cortex, whichdemonstrates that sympathetic innervation arises from a morerostral part of the posterior insula then causes parasympa-thetic innervation.13 The insula and thalamus had alreadybeen shown to have a sensory viscerotropic representationthat included the termination of cardiopulmonary afferents.14
The central role of the insula in the control of cardiovascularfunction has been supported by a robust experimental andclinical literature.15,16
Despite the fact that myocardial damage could definitelybe produced in animals, until the mid-1960s there was littlerecognition that this actually occurred in human beings withacute neurological or psychiatric illness until Koskelo andcolleagues17 reported on 3 patients with ECG changes causedby subarachnoid hemorrhage who were noted on postmortemexamination to have several small subendocardial petechialhemorrhages. Connor18 reported focal myocytolysis in 8% of231 autopsies, with the highest incidence seen in patients whosuffered fatal intracranial hemorrhages. The lesion reportedby Connor conforms to the descriptions of Selye’s ESCN orwhat might now be called myofibrillar degeneration, coagu-lative myocytolysis, or contraction band necrosis. Connorpointed out that previous pathology reports probably over-looked the lesion because of the fact that it was multifocal,with each individual focus being quite small, which wouldrequire extensive tissue sampling. It is clear now that evenConnor underestimated the prevalence of the lesion and thatserial sections are required to rigorously exclude its presence.
Greenshoot and Reichenbach19 reported on 3 new patientswith subarachnoid hemorrhage and a review of 6 previouspatients from the same medical center. All 9 of these patientshad cardiac lesions of varying degrees of severity that rangedfrom eosinophilia with preservation of cross-striations totransformation of the myocardial cell cytoplasm into denseeosinophilic transverse bands with intervening granularity,sometimes with endocardial hemorrhages. Both the ECGabnormalities and the cardiac pathology could be reproducedin cats given mesencephalic reticular formation stimulation.Adrenalectomy did not protect the hearts, which supports the
contention that the ECG changes and cardiac lesions are dueto direct intracardiac release of catecholamines.
Hawkins and Clower20 injected blood intracranially intomice, which thereby produced the characteristic myocardiallesions. The number of lesions could be reduced but notobliterated by pretreatment with adrenalectomy and the use ofeither atropine or reserpine, which suggested that the cause ofthe lesions was in part caused by sympathetic overactivity(humoral arrival at the myocardium from the adrenal and bydirect release into the muscle by intracardiac nerves) and inpart caused by parasympathetic overactivity. This supportsthe concept that the cause is an autonomic storm with acontribution from both divisions to the pathogenesis.
Jacob et al21 produced subarachnoid hemorrhage experi-mentally in dogs and carefully studied the sequential hemo-dynamic and ultrastructural changes that occurred. The he-modynamic changes occurred in 4 stages and directlyparalleled the effects seen with intravenous norepinephrineinjections. These stages were: (1) dramatic rise in systemicblood pressure; (2) extreme sinus tachycardia with variousarrhythmias (eg, nodal or ventricular tachycardia, bradycar-dia, atrioventricular block, ventricular premature beats, ven-tricular tachycardia, ventricular fibrillation with suddendeath), all of which could be suppressed by bilateral vagot-omy or orbital frontal resection; (3) rise in left ventricularpressure parallel to rise in systemic pressure; and (4) up to2-fold increase in coronary blood flow.
Ultrastructurally, a series of 3 stereotyped events occurredthat could be imitated exactly with norepinephrine injections.These were: (1) migration of intramitochondrial granules thatcontained Ca2� to the periphery of the mitochondria; (2)disappearance of these granules; and (3) myofilament disin-tegration at the I bands while the density of the I band wasincreased in the intact sarcomeres.21
Partially successful efforts to modify the developments ofneurocardiac lesions were made with reserpine pretreatmentin mice subjected to simulated intracranial hemorrhage22 andby Hunt and Gore,23 who pretreated a group of rats withpropranolol and then attempted to produce cardiac lesionswith intracranial blood injections. No lesions were found inthe control animals, but they were found in 21 of the 46untreated rats and in only 4 of the 22 treated rats. Thissuggested that neurological influences via catecholaminesmay be at least partly responsible for cardiac cell death. Moremodern studies have confirmed the fact that myocardialinjury occurs in the context of subarachnoid hemorrhage andthat the likelihood of myocardial necrosis was correlated withthe severity of the clinical neurological state as judged by thestandard Hunt-Hess grading system for subarachnoidhemorrhage.24
The phenomenology of the various types of myocardialcell death was articulated most clearly by Baroldi,25 whopointed out that there were 3 main patterns of myocardialnecrosis: (1) coagulation necrosis, the fundamental lesion ofinfarction, in which the cell loses its capacity to contract anddies in an atonic state with no myofibrillar damage; (2)colliquative myocytolysis, in which edematous vacuolizationwith dissolution of myofibrils without hypercontraction oc-curs in the low-output syndromes; and (3) coagulative myo-
Samuels The Brain–Heart Connection 79

cytolysis, in which the cell dies in a hypercontracted statewith early myofibrillar damage and anomalous irregularcross-band formations.
Coagulative myocytolysis is seen in reperfused areasaround regions of coagulation necrosis in transplanted hearts,in “stone hearts,” in sudden unexpected and accidental death,and in hearts exposed to toxic levels of catecholamines, suchas in patients with pheochromocytoma. This is probably themajor lesion described by Selye as ESCN and is likely to bethe major lesion seen in animals and people who suffer acuteneurological or psychiatric catastrophes. Although coagula-tive myocytolysis is probably the preferred term, the termsmyofibrillar degeneration and contraction band necrosis arecommonly used in the literature. This lesion tends to calcifyearly and to have a multifocal subendocardial predisposition(Figure 1, 2, and 3).
Intense rapid calcification makes it likely that the subcel-lular mechanisms that underlie the development of coagula-tive myocytolysis involve calcium entry. Zimmerman andHulsmann26 reported that the perfusion of rat hearts withcalcium-free media for short periods of time creates asituation such that on readmission of calcium there is a
massive contracture followed by necrosis and enzyme re-lease. This phenomenon, known as the calcium paradox, canbe imitated almost exactly with reoxygenation after hypox-emia. The latter, called the oxygen paradox, has been linkedto the calcium paradox by pathological calcium entry.27 Thismajor ionic shift is probably the cause of the dramatic ECGchanges seen in the context of neurological catastrophe, a factthat could explain the phenomenon of sudden unexpecteddeath (SUD) in many contexts.
Although SUD is now recognized as a medical problem ofmajor epidemiological importance, it has generally beenassumed that neurological disease rarely results in SUD. Infact, it has been traditionally held that neurological illnessesalmost never cause sudden demise, with the occasionalpatient who dies during an epileptic convulsion or rapidly inthe context of a subarachnoid hemorrhage as the exception.Further, it has been assumed that the various SUD syndromes(eg, sudden death in middle-aged men, sudden infant deathsyndrome, sudden unexpected nocturnal death syndrome,frightened to death (“voodoo” death), sudden death during anepileptic seizure, sudden death during natural catastrophe,sudden death associated with recreational drug abuse, suddendeath in wild and domestic animals, sudden death duringasthma attacks, sudden death during the alcohol withdrawalsyndrome, sudden death during grief after a major loss,sudden death during panic attacks, sudden death from mentalstress, and sudden death during war) are entirely separate andhave no unifying mechanism. For example, it is generallyaccepted that sudden death in middle-aged men is usuallycaused by a cardiac arrhythmia (ie, ventricular fibrillation),which results in functional cardiac arrest, whereas most workon sudden infant death syndrome focuses on immaturity ofthe respiratory control systems in the brain stem.
However, the connection between the nervous system andthe cardiopulmonary system provides the unifying link thatallows a coherent explanation for most, if not all, of the formsof neurocardiac damage. Powerful evidence from multipledisparate disciplines allows for a neurological explanation formany forms of SUD.28
Figure 1. The neurocardiac lesion: Gross specimen of a patientwho died during an acute psychological stress shows freshendocardial hemorrhages (1 of many is shown by the arrow).
Figure 2. Cardiac contraction band necrosis (also known ascoagulative myocytolysis, myofibrillar degeneration). The arrowshows 2 of the contraction bands.
Figure 3. Intense mineralization within minutes of the onset ofcontraction band necrosis.
80 Circulation July 3, 2007

Neurogenic Heart DiseaseDefinition of NeurogenicElectrocardiographic ChangesA wide variety of changes in the ECG is seen in the contextof neurological disease. Two major categories of change areregularly noted: arrhythmias and repolarization changes. It islikely that the increased tendency for life-threatening arrhyth-mias found in patients with acute neurological disease is aresult of the repolarization change, which increases thevulnerable period during which an extrasystole would belikely to result in ventricular tachycardia and/or ventricularfibrillation. Thus, the essential and potentially most lethalfeatures of the ECG, which are known to change in thecontext of neurological disease, are the ST segment and Twave, which reflect abnormalities in repolarization. Mostoften, the changes are seen best in the anterolateral orinferolateral leads. If the ECG is read by pattern recognitionby someone who is not aware of the clinical history, it willoften be said to represent subendocardial infarction or antero-lateral ischemia. The electrocardiographic abnormalities usu-ally improve, often dramatically, with death by brain criteria.In fact, any circumstance that disconnects the brain from theheart (eg, cardiac transplantation, severe autonomic neurop-athies caused by amyloidosis or diabetes, stellate ganglionec-tomy for treatment of the long QT syndrome) blunts neuro-cardiac damage of any cause.
The phenomenon is not rare. In a series of 100 consecutivestroke patients, 90% showed abnormalities on the ECGcompared with 50% of a control population of 100 patientsadmitted for carcinoma of the colon.29 This of course does notmean that 90% of stroke patients have neurogenic ECGchanges. Obviously, stroke and coronary artery disease havecommon risk factors, so that many ECG abnormalities instroke patients represent concomitant atherosclerotic coro-nary disease. Nonetheless, a significant number of strokepatients have authentic neurogenic ECG changes.
Mechanism of the Production of NeurogenicHeart Disease
Catecholamine InfusionJosué30 first demonstrated that epinephrine infusions couldcause cardiac hypertrophy. This observation has been repro-duced on many occasions, which documents the fact thatsystemically administered catecholamines are not only asso-ciated with ECG changes reminiscent of widespread ischemiabut with a characteristic pathological picture in the cardiacmuscle that is distinct from myocardial infarction. An iden-tical picture may be found in human beings with chronicallyelevated catecholamines, as seen with pheochromocytoma.Patients with stroke often have elevated systemic catechol-amine levels, a fact that may in part account for the highincidence of cardiac arrhythmias and ECG changes seen inthese patients. On light microscopy, these changes range fromincreased eosinophilic staining with preservation of cross-striations to total transformation of the myocardial cellcytoplasm into dense eosinophilic transverse bands withintervening granularity. In severely injured areas, infiltration
of the necrotic debris by mononuclear cells is often noted,sometimes with hemorrhage.
Ultrastructurally, the changes in cardiac muscle are evenmore widespread than they appear to be in light microscopy.Nearly every muscle cell shows some pathological alteration,which range from a granular appearance of the myofibrils toprofound disruption of the cell architecture with relativepreservation of ribosomes and mitochondria. Intracardiacnerves can be seen and identified by their external lamina,microtubules, neurofibrils, and the presence of intracytoplas-mic vesicles. These nerves can sometimes be seen immedi-ately adjacent to an area of myocardial cell damage. Thepathological changes in the cardiac muscle are usually less ata distance from the nerve, often with a complete return tonormalcy by a distance of 2 to 4 �m away from the nerveending.21
Myofibrillar degeneration (also known as coagulativemyocytolysis and contraction band necrosis) is an easilyrecognizable form of cardiac injury, distinct in several majorrespects from coagulation necrosis, which is the major lesionof myocardial infarction.25,31 In coagulation necrosis, the cellsdie in a relaxed state without prominent contraction bands.This is not visible by any method for many hours or evendays. Calcification only occurs late, and the lesion elicits apolymorphonuclear cell response. In stark contrast, in myo-fibrillar degeneration the cells die in a hypercontracted statewith prominent contraction bands (Figures 2 and 3). Thelesion is visible early, perhaps within minutes of its onset. Itelicits a mononuclear cell response and may calcify almostimmediately.31,32
Stress Plus or Minus SteroidsA similar, if not identical, cardiac lesion can be producedwith various models of stress. This concept was applied to theheart when Selye published his monograph The ChemicalPrevention of Cardiac Necrosis in 1958.9 He found thatcardiac lesions probably identical to those described abovecould be produced regularly in animals that were pretreatedwith certain steroids, particularly 2-�-methyl-9-�-fluorohydrocortisone (fluorocortisol) and then subjected tovarious types of stress. Other hormones, such as dihydrotach-ysterol (calciferol) and thyroxine, could also sensitize animalsfor stress-induced myocardial lesions, though less potentlythan fluorocortisol. This so-called stress could be of multipletypes such as restraint, surgery, bacteremia, vagotomy, andtoxins. He believed that the first mediator in the translation ofthese widely disparate stimuli into a stereotyped cardiaclesion was the hypothalamus and that it, by its control overthe autonomic nervous system, caused the release of certainagents that were toxic to the myocardial cell. Since Selye’soriginal work, similar experiments have been repeated inmany different types of laboratory animals with comparableresults. Although the administration of exogenous steroidsfacilitates the production of cardiac lesions, it is clear thatstress alone can result in the production of morphologicallyidentical lesions.
Whether a similar pathophysiology could ever be repeatedin human beings is, of course, of great interest. Manyinvestigators have speculated on the role of stress in the
Samuels The Brain–Heart Connection 81

pathogenesis of human cardiovascular disease and, in partic-ular, on its relationship to the phenomenon of SUD. A fewautopsies on patients who experienced sudden death haveshown myofibrillar degeneration. Cebelin and Hirsch33 re-ported on a careful retrospective analysis of the hearts of 15victims of physical assault who died as a direct result of theassault, but without sustaining internal injuries. Eleven of the15 individuals showed myofibrillar degeneration. Age- andcardiac disease–matched controls showed little or no evi-dence of this change. This appears to represent human stresscardiomyopathy. Whether such assaults can be consideredmurder has become an interesting legal correlate of theproblem.
Because the myofibrillar degeneration is predominantlysubendocardial, it may involve the cardiac conducting sys-tem, which thus predisposes to cardiac arrhythmias. Thislesion, combined with the propensity of catecholamines toproduce arrhythmias even in a normal heart, may well raisethe risk of a serious arrhythmia. This may be the majorimmediate mechanism of sudden death in many neurologicalcircumstances, such as subarachnoid hemorrhage, stroke,epilepsy, head trauma, psychological stress, and increasedintracranial pressure. Even the arrhythmogenic nature ofdigitalis may be largely mediated by the central nervoussystem. Further evidence for this is the antiarrhythmic effectof sympathetic denervation of the heart for cardiac arrhyth-mias of many types.
Furthermore, it is known that stress-induced myocardiallesions may be prevented by sympathetic blockade with manydifferent classes of antiadrenergic agents, most notably,ganglionic blockers such as mecamylamine and catechol-amine-depleting agents such as reserpine.10 This suggests thatcatecholamines, which are either released directly into theheart by sympathetic nerve terminals or reach the heartthrough the bloodstream after release from the adrenal me-dulla, may be excitotoxic to myocardial cells.
Some people who are subjected to an extreme stress maydevelop an acute cardiomyopathy that presents with chestpain and/or symptoms of heart failure. This process is mostcommonly seen in older women, whose echocardiograms andventriculograms show a typical pattern of left ventricularapical ballooning, which was named takotsubo-like cardio-myopathy34 because of the similarity in the appearance theleft ventricle to the Japanese octopus trapping pot, thetakotsubo. If a lethal arrhythmia does not intervene, theprocess is potentially completely reversible. Some debateexists regarding whether this syndrome (variously describedas myocardial stunning or myocardial hibernation) could beexplained by ischemia, but it is striking that this pattern ofdysfunction is most consistent with a neural rather than avascular distribution.35,36 Wittstein and colleagues37 reporteda series of such patients and referred to the problem asmyocardial stunning. In patients in whom endocardial biop-sies were performed, contraction band lesions were found.The finding of contraction bands suggests either catechol-amine effect and/or reperfusion. The 2 mechanisms are notmutually exclusive in that a neural stimulus could produceboth catecholamine release and coronary vasospasm followedby vasodilation. There is no direct evidence that the nervous
system can cause coronary vasospasm, but the possibilityremains. Regardless of the precise mechanism, the factremains that takotsubo-like cardiomyopathy occurs after anacute psychological stress and thereby represents an exampleof a neurocardiac lesion. It seems likely that this dramaticcondition may be the tip of an iceberg under which lurks amuch larger, albeit less easily demonstrable, problem; namelyneurocardiac lesions that are not sufficiently severe andwidespread to produce gross heart failure but may predisposeto serious cardiac arrhythmias.
Nervous System StimulationNervous system stimulation produces cardiac lesions that arehistologically indistinguishable from those described forstress and catecholamine-induced cardiac damage. It has beenknown for a long time that stimulation of the hypothalamuscan lead to autonomic cardiovascular disturbances,38 andmany years ago lesions in the heart and gastrointestinal tracthave been produced with hypothalamic stimulation.39,40 It hasbeen clearly demonstrated that stimulation of the lateralhypothalamus produces hypertension and/or electrocardio-graphic changes reminiscent of those seen in patients withcentral nervous system damage of various types. Further-more, this effect on the blood pressure and ECG can becompletely prevented by C2 spinal section and stellateganglionectomy, but not by vagotomy, which suggests thatthe mechanism of the electrocardiographic changes is sym-pathetic rather than parasympathetic or humoral. Stimulationof the anterior hypothalamus produces bradycardia, an effectthat can be blocked by vagotomy. Unilateral hypothalamicstimulation does not result in histological evidence of myo-cardial damage by light microscopy, but bilateral prolongedstimulation regularly produces myofibrillar degeneration in-distinguishable from that produced by catecholamine injec-tions and stress, as previously described.41
Other methods to produce cardiac lesions of this typeinclude stimulation of the limbic cortex, the mesencephalicreticular formation, the stellate ganglion, and regions knownto elicit cardiac reflexes such as the aortic arch. Experimentalintracerebral and subarachnoid hemorrhages can also result incardiac contraction band lesions. These neurogenic cardiaclesions will occur even in an adrenalectomized animal,although they will be somewhat less pronounced.20 Thisevidence argues strongly against an exclusively humoralmechanism in the intact organism. High levels of circulatingcatecholamines exaggerate the electrocardiographic findingsand myocardial lesions, but high circulating catecholaminelevels are not required for the production of pathologicalchanges. These electrocardiographic abnormalities and car-diac lesions are stereotyped and identical to those found in thestress and catecholamine models already outlined. They arenot affected by vagotomy and are blocked by maneuvers thatinterfere with the action of the sympathetic limb of theautonomic nervous system, such as C2 spinal section, stellateganglion blockade, and administration of antiadrenergicdrugs such as propranolol.
The histological changes in the myocardium range fromnormal muscle on light microscopy to severely necrotic (butnot ischemic) lesions with secondary mononuclear cell infil-
82 Circulation July 3, 2007

tration. The findings on ultrastructural examination are in-variably more widespread, often involving nearly everymuscle cell, even when the light microscopic appearance isunimpressive. The electrocardiographic findings undoubtedlyreflect the total amount of muscle membrane affected by thepathophysiological process. Thus, the ECG may be normalwhen the lesion is early and demonstrable only by electronmicroscopy. Conversely, the ECG may be grossly abnormalwhen only minimal findings are present by light microscopy,since the cardiac membrane abnormality responsible for theelectrocardiographic changes may be reversible. Cardiacarrhythmias of many types may also be elicited by nervoussystem stimulation along the outflow of the sympatheticnervous system.
ReperfusionThe fourth and last model for the production of myofibrillardegeneration is reperfusion, as is commonly seen in patientswho die after a period of time on a left ventricular assist pumpor after they undergo extracorporeal circulation. Similarlesions are seen in hearts that were reperfused with angio-plasty or fibrinolytic therapy. The mechanism by whichreperfusion of ischemic cardiac muscle produces coagulativemyocytolysis (also known as myofibrillar degeneration andcontraction band necrosis) involves entry of calcium after aperiod of relative deprivation.41
Sudden calcium influx by one of several possible mecha-nisms (eg, a period of calcium deficiency with loss ofintracellular calcium, a period of anoxia followed by reoxy-genation of the electron transport system, a period of ische-mia followed by reperfusion, or opening of the receptor-operated calcium channels by excessive amounts of locallyreleased norepinephrine) may be the final common pathwayby which the irreversible contractures occur, which leads tomyofibrillar degeneration. Thus reperfusion-induced myocar-dial cell death may be a form of apoptosis (programmed celldeath) analogous to that seen in the central nervous system, inwhich excitotoxicity with glutamate results in a similar, if notidentical, series of events.42
The precise cellular mechanism for the electrocardio-graphic change and the histological lesion may well reflectthe effects of large volumes of norepinephrine released intothe myocardium from sympathetic nerve terminals.43 The factthat the cardiac necrosis is greatest near the nerve terminals inthe endocardium and is progressively less severe as onesamples muscle cells near the epicardium provides furtherevidence that catecholamine toxicity produces the lesion.19
This locally released norepinephrine is known to stimulatesynthesis of adenosine 3�,5�-cyclic phosphate, which in turnresults in the opening of the calcium channel with influx ofcalcium and efflux of potassium. This efflux of potassiumcould explain the peaked T waves (a hyperkalemic pattern)often seen early in the evolution of neurogenic electrocardio-graphic changes.21 The actin and myosin filaments interactunder the influence of calcium but do not relax unless thecalcium channel closes. Continuously high levels of norepi-nephrine in the region may result in failure of the calciumchannel to close, which leads to cell death, and finally toleakage of enzymes out of the myocardial cell. Free radicals
released as a result of reperfusion after ischemia or by themetabolism of catecholamines to the known toxic metabolite,adrenochrome, may contribute to cell membrane destruction,which leads to leakage of cardiac enzymes into the blood.44,45
Thus, the cardiac toxicity of locally released norepinephrinefalls on a continuum that ranges from a brief reversible burstof electrocardiographic abnormalities to a pattern that resem-bles hyperkalemia and then finally to an irreversible failure ofthe muscle cell with permanent repolarization abnormalities,or even the occurrence of transmural cardiac necrosis withenzyme (eg, troponin, creatine kinase) release and Q wavesseen on the ECG.
Histological changes would also represent a continuumthat ranges from complete reversibility in a normal heartthrough mild changes seen only by electron microscopy tosevere myocardial cell necrosis with mononuclear cell infil-tration and even hemorrhages. The amount of cardiac en-zymes released and the electrocardiographic changes wouldroughly correlate with the severity and extent of the patho-logical process. This explanation, summarized in Figure 4,rationalizes all the observations in the catecholamine infu-sion, stress plus or minus steroid, nervous system stimulation,and reperfusion models.
Concluding Remarks and Potential TreatmentsIn conclusion, there is powerful evidence to suggest thatoveractivity of the sympathetic limb of the autonomic ner-vous system is the common phenomenon that links the majorcardiac pathologies seen in neurological catastrophes. Theseprofound effects on the heart may contribute in a major wayto the mortality rates of many primarily neurological condi-tions such as subarachnoid hemorrhage, cerebral infarction,status epilepticus, and head trauma. These phenomena mayalso be important in the pathogenesis of SUD in adults,sudden infant death, sudden death during asthma attacks,cocaine- and amphetamine-related deaths, and sudden deathduring the alcohol withdrawal syndrome, all of which may belinked by stress and catecholamine toxicity.
Investigations aimed at alteration of the natural history ofthese events with catecholamine receptor blockade, calcium-channel blockers, free-radical scavengers, and antioxidants
Figure 4. Cascade of events that lead to neurocardiac damage.
Samuels The Brain–Heart Connection 83

are in progress in many centers around the world and aresummarized in Figure 5.
DisclosuresNone.
References1. Cannon WB. “Voodoo” death. Am Anthropologist. 1942;44(new series):
169–118.2. Engel G. Sudden and rapid death during psychological stress. Ann Intern
Med. 1971;74:771–782.3. Richter CP. On the phenomenon of sudden death in animal and man.
Psychosomatic Med. 1957;19:191–198.4. Oppenheimer SM, Gelb A, Girvin JP, Hachinski VC. Cardiovascular
effects of human insular cortex stimulation. Neurology. 1992;42:1727–1732.
5. Byer E, Ashman R, Toth LA. Electrocardiogram with large upright Twave and long Q-T intervals. Am Heart J. 1947;33:796–801.
6. Levine HD. Non-specificity of the electrocardiogram associated withcoronary heart disease. Am J Med. 1953;15:344–350.
7. Burch GE, Myers R, Abildskov JA. A new electrocardiographic patternobserved in cerebrovascular accidents. Circulation. 1954;9:719–726.
8. Cropp CF, Manning GW. Electrocardiographic change simulating myo-cardial ischaemia and infarction associated with spontaneous intracranialhaemorrhage. Circulation. 1960;22:25–38.
9. Selye H. The Chemical Prevention of Cardiac Necrosis. New York, NY:Ronald Press; 1958.
10. Raab W, Stark E, MacMillan WH, Gigee WR. Sympathogenic origin andanti-adrenergic prevention of stress-induced myocardial lesions.Am J Cardiol. 1961;8:203–211.
11. Barger AC, Herd JA, Liebowitz MR. Chronic catheterization of coronaryartery induction of ECG pattern of myocardial ischemia by intracoronaryepinephrine. Proc Soc Exp Biol Med. 1961;107:474–477.
12. Melville KI, Blum B, Shister HE, Silver MD. Cardiac ischemic changesand arrhythmias induced by hypothalamic stimulation. Am J Cardiol.1963;12:781–791.
13. Oppenheimer SM, Cechetto DF. Cardiac chronotropic organization of therat insular cortex. Brain Res. 1990;533:66–72.
14. Cechetto DF, Saper CB. Evidence for a viscerotopic sensory repre-sentation in the cortex and thalamus in the rat. J Comp Neurology.1987;262:27–45.
15. Cheung RTF, Hachinski V. The insula and cerebrogenic sudden death.Arch Neurol. 2000;57:1685–1688.
16. Cheshire WP, Saper CB. The insular cortex and cardiac response tostroke. Neurology. 2006;66:1296–1297.
17. Koskelo P, Punsar SO, Sipila W. Subendocardial haemorrhage and ECGchanges in intracranial bleeding. BMJ. 1964;1:1479–1483.
18. Connor RCR. Myocardial damage secondary to brain lesions. Am Heart J.1969;78:145–148.
19. Greenshoot JH, Reichenbach DD. Cardiac injury and subarachnoid haem-orrhage. J Neurosurg. 1969;30:521–531.
20. Hawkins WE, Clower BR. Myocardial damage after head trauma andsimulated intracranial haemorrhage in mice: the role of the autonomicnervous system. Cardiovasc Res. 1971;5:524–529.
21. Jacob WA, Van Bogaert A, DeGroot-Lasseel MHA. Myocardial ultra-structural and haemodynamic reactions during experimental subarachnoidhaemorrhage. J Moll Cell Cardiol. 1972;4:287–298.
22. McNair JL, Clower BR, Sanford RA. The effect of reserpine pretreatmenton myocardial damage associated with simulated intracranial haemor-rhage in mice. Eur J Pharmacol. 1970;9:1–6.
23. Hunt D, Gore I. Myocardial lesions following experimental intracranialhemorrhage: prevention with propranolol. Am Heart J. 1972;83:232–236.
24. Tung P, Kopelnik A, Banki N, Ong K, Ko N, Lawton MT, Gress D, DrewB, Foster E, Parmley W, Zaroff J. Predictors of neurocardiogenic injuryafter subarachnoid hemorrhage. Stroke. 2004;35:548–553.
25. Baroldi F. Different morphological types of myocardial cell death in man. In:Fleckstein A, Rona G, eds. Recent Advances in Studies in Cardiac Structureand Metabolism. Pathophysiology and Morphology of Myocardial CellAlteration. Vol 6. Baltimore, Md: University Park Press, 1975.
26. Zimmerman ANA, Hulsmann WC. Paradoxical influence of calcium ionson their permeability of the cell membranes of the isolated rat heart.Nature. 1966;211:616–647.
27. Hearse DJ, Humphrey SM, Bullock GR. The oxygen paradox and thecalcium paradox: two facets of the same problem? J Moll Cell Cardiol.1978;10:641–668.
28. Samuels MA. Neurally induced cardiac damage. Neurol Clin. 1993;11:273–292.
29. Dimant J, Grob D. Electrocardiographic changes and myocardial damagein patients with acute cerebrovascular accidents. Stroke. 1977;8:448–455.
30. Josué O. Hypertrophie cardiaque cause par l’adrenaline and la toxinetyphique. C R Soc Biol (Paris). 1907;63:285–287.
31. Karch SB, Billingham ME. Myocardial contraction bands revisited. HumPathol. 1986;17:9–13.
32. Rona G. Catecholamine cardiotoxicity. J Moll Cell Cardiol. 1985;17:291–306.
33. Cebelin M, Hirsch CS. Human stress cardiomyopathy. Hum Pathol.1980;11:123–132.
34. Sato H, Tateishi H, Uchida T. Takotsubo-type left ventricular dysfunctiondue to multivessel coronary spasm. In: Kodama K, Haze K, Hori M, eds.Clinical Aspects of Myocardial Injury: From Ischemia to Heart Failure.Tokyo, Japan: Kagakuhyoronsha Publishing Co; 1990:56–64.
35. Angelakos ET. Regional distribution of catecholamines in the dog heart.Circ Res. 1965;16:39–44.
36. Murphree SS, Saffitz JE. Quantitative autoradiographic delineation of thedistribution of beta-adrenergic receptors in canine and feline left ventric-ular myocardium. Circ Res. 1987;60:568–579.
37. Wittstein IS, Thiemann DR, Lima JAC, Baughman KL, Schulman SP,Gerstenblith G, Wu KC, Rade JJ, Bivalaqua TJ, Champion HC. Neuro-humoral features of myocardial stunning due to sudden emotional stress.N Engl J Med. 2005;352:539–548.
38. Dikshit BB. The production of cardiac irregularities by excitation of thehypothalamic centres. J Physiol. 1934;81:382–394.
39. Karplus JP, Kreidl A. Gehirn und Sympathicus. Sympathicusleitung imGehirn und Halsmark [German]. Pflugers Arch. 1912;143:109–127.
40. Karplus JP, Kreidl A. Gehirn und Sympathicus. Uber Beziehungen derHypothalamaszentren zu Blutdruck und innerer Sekretion [German].Pflugers Arch. 1927;215:667–674.
41. Braunwald E, Kloner RA. Myocardial reperfusion: a double-edgedsword? J Clin Invest. 1985;76:13–19.
42. Gottlieb R, Burleson KO, Kloner RA Babior BM, Engler RL. Reper-fusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest.1994;94:1621–1628.
43. Eliot RS, Todd GL, Pieper GM, Clayton FC. Pathophysiology of cate-cholamine-mediated myocardial damage. J S C Med Assoc. 1979;75:513–518.
44. Singal PK, Kapur N, Dhillon KS, Beamish RE, Dhalla NS. Role of freeradicals in catecholamine-induced cardiomyopathy. Can J PhysiolPharmacol. 1982;60:1390–1397.
45. Meerson FZ. Pathogenesis and prophylaxis of cardiac lesions in stress.Adv Myocardiol. 1983;4:3–21.
KEY WORDS: antioxidants � apoptosis � cardiomyopathy � cerebralinfarction � death, sudden � nervous system, autonomic � nervous system,sympathetic
Figure 5. Possible therapeutic approaches aimed to prevent neu-rocardiac damage. GABA indicates gamma aminobutyric acid.
84 Circulation July 3, 2007

Chronic Kidney DiseaseEffects on the Cardiovascular System
Ernesto L. Schiffrin, MD, PhD, FRSC, FRCPC; Mark L. Lipman, MD, FRCPC; Johannes F.E. Mann, MD
Abstract—Accelerated cardiovascular disease is a frequent complication of renal disease. Chronic kidney disease promoteshypertension and dyslipidemia, which in turn can contribute to the progression of renal failure. Furthermore, diabeticnephropathy is the leading cause of renal failure in developed countries. Together, hypertension, dyslipidemia, anddiabetes are major risk factors for the development of endothelial dysfunction and progression of atherosclerosis.Inflammatory mediators are often elevated and the renin-angiotensin system is frequently activated in chronic kidneydisease, which likely contributes through enhanced production of reactive oxygen species to the acceleratedatherosclerosis observed in chronic kidney disease. Promoters of calcification are increased and inhibitors ofcalcification are reduced, which favors metastatic vascular calcification, an important participant in vascular injuryassociated with end-stage renal disease. Accelerated atherosclerosis will then lead to increased prevalence of coronaryartery disease, heart failure, stroke, and peripheral arterial disease. Consequently, subjects with chronic renal failure areexposed to increased morbidity and mortality as a result of cardiovascular events. Prevention and treatment ofcardiovascular disease are major considerations in the management of individuals with chronic kidney disease.(Circulation. 2007;116:85-97.)
Key Words: atherosclerosis � hypertension � kidney � vasculature
It is increasingly apparent that individuals with chronickidney disease (CKD) are more likely to die of cardiovas-
cular (CV) disease (CVD) than to develop kidney failure.1,2 Alarge cohort study comprising �130 000 elderly subjectsshowed that increased incidence of CV events could be inpart related to the fact that persons with renal insufficiencyare less likely to receive appropriate cardioprotective treat-ments.3 However, beyond the effects of lack of appropriatetherapy, it is clear that accelerated CVD is prevalent insubjects with CKD. The first part of the present review willtherefore focus on the epidemiological links between impair-ment of renal function and adverse CV events, betweenalbuminuria and CV events, and between serum cystatin Cand CVD. The second part of the present review will addressthe mechanisms that lead to the association of renal and CVD,which include hypertension, dyslipidemia, activation of therenin-angiotensin system, endothelial dysfunction and therole of asymmetric dimethyl arginine (ADMA), oxidativestress, and inflammation. Finally, mechanisms that are in-volved in vascular calcification often found in CKD andend-stage renal disease (ESRD) will be described. Addition-ally, ESRD is associated with several specific complicationscaused by the uremic state per se, which can contribute to thedevelopment and progression of CVD through volume over-load with consequent hypertension, anemia, uremic pericar-ditis, and cardiomyopathy. However, these issues will not be
addressed because the emphasis will be on CKD beforeESRD is reached. In addition, the CV complications associ-ated with dialysis will not be discussed. The different stagesof CKD according to the level of glomerular filtration rate(GFR) are shown in Table 1. ESRD corresponds to the stagewhere patients need renal replacement therapy (ie, dialysis orrenal transplantation), whereas stage 1 is mostly recognizedby either albuminuria or structural renal abnormality (eg,hyperechoic renal parenchyma on ultrasound). Table 2 pro-vides the approximate odds ratios (univariate) of CVDaccording to stages of CKD on the basis of the literature citedbelow. The increase in risk in comparison to people withoutCKD depends on the age of the population studied: theyounger the person, the higher the relative risk. Microalbu-minuria increases the CV risk 2- to 4-fold.
Epidemiological Links Between ImpairedGFR and Adverse Cardiovascular Events
Evidence for the relationship between renal dysfunction andadverse CV events was perhaps first recognized in thedialysis population in whom the incidence of CV death isstrikingly high. Approximately 50% of individuals withESRD die from a CV cause,2,4,5 a CV mortality that is 15 to30 times higher than the age-adjusted CV mortality in thegeneral population.4,6 This disparity is present across all ages,but it is most marked in the younger age group (25 to 34 years
From the Department of Medicine (E.L.S., M.L.L.), Sir Mortimer B. Davis Jewish General Hospital, McGill University, Montreal, Quebec, Canada;and the Department of Nephrology and Hypertension (J.F.E.M.), Schwabing General Hospital, Ludwig Maximilians University, Munich, Germany.
Correspondence to Ernesto L. Schiffrin, MD, PhD, FRSC, FRCPC, Sir Mortimer B. Davis Jewish General Hospital, B-127, 3755 Côte Ste-CatherineRd, Montreal, Quebec, Canada H3T 1E2. E-mail [email protected]
© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.106.678342
85
Cardiovascular Involvement in General Medical Conditions

old), where the CV mortality is 500-fold greater in ESRDpatients compared with age-matched controls with normalrenal function.1 It is therefore unsurprising that establishedCVD is easily demonstrable in CKD. For example, 40% ofpatients who have started dialysis treatments have evidenceof coronary artery disease, and 85% of these patients haveabnormal left ventricular structure and function.7
The relationship between renal disease and CV mortalityhas also been shown to extend to subjects with moremoderate degrees of renal functional impairment. In fact, themajority of patients with stage 3 to 4 CKD (ie, a GFR�60mL/min per 1.73 m2) die of CV causes rather than progress toESRD. Here too, objective evidence of structural and func-tional cardiac abnormalities has been demonstrated by echo-cardiography. Levin et al determined left ventricular massindex in a population of 115 men and 60 women with anaverage creatinine clearance (CrCl) of 25.5�17 mL/min with2-dimensional targeted M-mode echocardiography. The pop-ulation was stratified into 3 groups according to renalfunction. The prevalence of left ventricular hypertrophy(LVH) was 26.7% in subjects with CrCl�50 mL/min, 30.8%in those with CrCl between 25 and 49 mL/min, and 45.2% inindividuals with CrCl�25 mL/min.8
Tucker et al9 reported a similar finding in a population of85 persons with renal insufficiency. With the same echocar-diographic techniques, as well as comparable criteria for thediagnosis of LVH, these investigators found an LVH preva-lence of 16% in subjects with a CrCl�30 mL/min and 38% inthose with a CrCl�30 mL/min. These studies demonstratethat LVH is common in patients with renal insufficiency evenbefore they progress to dialysis, and that the prevalence ofLVH correlates with the degree of renal functionalimpairment.
A growing number of studies have demonstrated that therelationship between renal dysfunction and increased CVmorbidity and mortality extends across the spectrum of renaldysfunction to encompass the mildest degrees of renal im-pairment. Moreover, this relationship appears to hold acrosspopulations with widely varying degrees of baseline CVhealth.
CVD Associated With Renal Disease in theGeneral PopulationThe Framingham Heart Study was among the first to assessmild renal insufficiency and its association with death andadverse CV events in the general population.10 Of the 6233participants in the study, mild renal insufficiency was presentin 246 men and 270 women (serum creatinine, 1.4 to 3.0mg/dL). Of these individuals, 81% had no prevalent CVD atentry. Over the 15-year follow-up period, there was nosignificant association between mild renal insufficiency andeither death or adverse CV events in women. However, inmen there was a trend toward more CV events with mild renalinsufficiency, and a significant association was demonstratedwith age-adjusted all-cause mortality (hazard ratio, 1.42).Given the relatively small number of subjects followed in thisstudy and the low number of outcome events, these findingswere suggestive but not definitive of a correlation betweenmild renal dysfunction and increased CV morbidity andmortality.
More recently, Go et al11 examined the relationship ofGFR and adverse CV events in a low-risk population. Theyanalyzed the database of a large healthcare provider innorthern California and used the Modification of Diet inRenal Disease (MDRD) formula12 to estimate the baselineGFR from measurements of serum creatinine in �1.1million adults, with only those who were on dialysis orwho had undergone a kidney transplant excluded. Theprimary outcomes examined included death from anycause, CV events, and hospitalizations. The end-pointinformation was obtained from the health-plan databaseand the California death registry with a mean follow-upperiod of 2.84 years. After adjustment for age, sex, race,coexisting illnesses, and socioeconomic status, a stepwiseincrease in the rate of each of the 3 primary outcomes wasseen for every sequential decrease in GFR. With the bestcohort (GFR�60 mL/min per 1.73 m2) as the point ofreference, the adjusted hazard ratio for death from anycause and any CV event increased to 1.2 and 1.4, respec-
TABLE 1. Stages of CKD
Stage Description GFR, mL/min per 1.73 m2
1* Kidney damage with normal or increased GFR �90
2 Kidney damage with mildly decreased GFR 60 to 89
3 Moderately decreased GFR 30 to 59
4 Severely decreased GFR 15 to 29
5 Kidney failure �15 or dialysis
ESRD is defined as the need for renal replacement therapy (ie, need for dialysis or renaltransplantation).
*Stage 1 CKD is mostly recognized by either albuminuria or structural renal abnormality (eg,hyperechoic renal parenchyma on ultrasound).
TABLE 2. CV Risk According to Stages of CKD
Stage CV Risk (Odds Ratio, Univariate)
1 Depending on degree of proteinuria
2 1.5
3 2 to 4
4 4 to 10
5 10 to 50
ESRD 20 to 1000
The increase in risk in comparison with people free of CKD depends on theage of the population studied: The younger the person, the higher the relativerisk. Microalbuminuria increases the CV risk 2- to 4-fold.
86 Circulation July 3, 2007

tively, for a GFR between 45 to 59 mL/min per 1.73 m2;1.8 and 2.0 for a GFR between 30 to 44 mL/min per 1.73m2; 3.2 and 2.8 for GFR between 15 to 29 mL/min per 1.73m2; and 5.9 and 3.4 for a GFR�15 mL/min per 1.73 m2.The adjusted risk of hospitalization with a reduced GFRfollowed a similar pattern. This large study, which incor-porated a diverse population of adults, clearly demon-strated an independent and graded (inverse) correlationbetween decreasing levels of renal function and increasingevent rates of CV morbidity and death.
CVD Associated With Renal Disease inHypertensive SubjectsThe association between renal function and mortality in thehypertensive population was evaluated by the HypertensionDetection and Follow-up Program Cooperative Group, whichfollowed and treated 10 940 hypertensive subjects to comparestepped care to referred care.13 The primary end point of thestudy was all-cause mortality. Persons with a baseline serumcreatinine �1.7 mg/dL experienced an 8-year mortality ratethat was �3 times higher than that of all other participants.
Data from the Hypertension Optimal Treatment (HOT)study support this finding. In the HOT study, 18 790 hyper-tensive subjects, only 10% of whom had evidence of athero-sclerotic disease, were assigned to 3 diastolic blood pressuretarget groups and followed for a mean of 3.8 years. Personswith a serum creatinine �3 mg/dL were excluded and theCockroft-Gault14 equation was used to calculate baselineGFR. The adjusted relative risks for total mortality and formajor CV events (nonfatal myocardial infarction [MI], non-fatal stroke, CV death) were 1.65 and 1.58, respectively, insubjects with GFR�60 mL/min compared with those with aGFR�60 mL/ min.15
Effect of Renal Disease on Individuals WithPreexisting Stable CVD or Risk Factors for CVDA post hoc analysis of the Heart Outcomes and PreventionEvaluation (HOPE) study examined the impact of baselineserum creatinine on the incidence of the composite primaryoutcome (CV death, MI, or stroke).16 The HOPE populationincluded individuals with objective evidence of vasculardisease or diabetes combined with another CV risk factor andwas designed to test the benefit of add-on ramipril versusplacebo in this population. Patients with heart failure or aserum creatinine concentration �2.3 mg/dL were excluded.The follow-up period was �5 years. There were 980 subjectswith mild renal insufficiency (serum creatinine�1.4 mg/dL)and 8307 subjects with normal renal function (serum creati-nine�1.4 mg/dL). The cumulative incidence of the primaryoutcome was 22.2% in individuals with mild renal insuffi-ciency versus 15% in those with normal renal function(P�0.001). The impact of renal insufficiency was indepen-dent of both the baseline CV risk factors as well as thetreatment group.
A similar relationship between renal function and CVevents was demonstrated in the Prevention of Events withAngiotensin-Converting Enzyme Inhibition (PEACE) trial.17
In PEACE, add-on trandolapril was compared with placebo ina population with chronic stable coronary artery disease and
LVEF�40%. The primary end point was a composite ofdeath from CV causes, MI, and coronary revascularization.Patients with a serum creatinine �2.0 mg/dL were excludedand the median duration of follow-up was 4.8 years. A posthoc analysis of 8280 subjects, in whom baseline renalfunction was separated into quartiles with the MDRD for-mula, demonstrated significant stepwise increases in eventrates as the baseline GFR declined. Interestingly, unlike inHOPE, there was a significant interaction between GFR andtreatment group with respect to CV and all-cause mortality inthat the angiotensin-converting enzyme inhibitor benefitedonly those individuals with a GFR�60 mL/min per 1.73 m2.
Effect of Renal Disease in Patients With EstablishedHeart Failure or Postmyocardial InfarctionHillege et al examined whether renal dysfunction was apredictor of mortality in stable patients with advanced heartfailure.18 They studied 1906 subjects with New York HeartAssociation class III and IV heart failure and evidence of leftventricular dysfunction (LVEF�35%) who were enrolled inthe Second Prospective Randomized study of Ibopamine onMortality and Efficacy (PRIME II).19 Hillege et al correlatedbaseline GFR, as calculated with the Cockroft-Gault equa-tion, with overall mortality after a median follow-up of 277days. The authors found that patients in the lowest quartile ofGFR (�44 mL/min) had relative risk of mortality of 2.85compared with subjects in the highest quartile (�76 mL/min).Somewhat surprisingly, baseline GFR was independent ofimpaired LVEF and was a stronger predictor of mortality thaneither LVEF or New York Heart Association class. In fact,GFR was the strongest predictor of mortality of all factorsanalyzed, which included parameters of neurohormonalactivation.
Hillege et al also explored the prognostic ability of baselinerenal function to predict the development of heart failure afteran anterior-wall MI.20 Patients with a serum creatinine �180�mol/L (2.0 mg/dL) were excluded. Baseline GFR wascalculated with the Cockroft-Gault formula, and the 298patients were divided into tertiles of renal function. At 1 yearof follow-up the incidence of congestive heart failure bytertile of decreasing GFR was 24.0%, 28.9%, and 41.2%.Risk of de novo congestive heart failure was 1.86-fold higherin the lowest tertile (�81 mL/min) than in the highest tertile(�103 mL/min). As the mean GFR in the lowest tertile was67.0 mL/min, the study by Hillege et al highlights the impactof even mild GFR reductions on cardiac outcomes.
In a post hoc analysis of the Valsartan in Acute MyocardialInfarction Trial (VALIANT), Anavekar et al examined therelationship between baseline renal function and adverseoutcomes in 14 527 subjects with acute MI complicated byclinical or radiologic signs of heart failure and/or left ven-tricular dysfunction.21 Subjects were randomly assigned toreceive captopril, valsartan, or both, and they were followedfor a mean of 24.7 months. Individuals with a serumcreatinine �2.5 mg/dL were excluded from the study. Theprimary end point was death from any cause, and secondaryend points included death from CV causes, heart failure,recurrent MI, resuscitation after cardiac arrest, stroke, and acomposite of these.22 Anavekar et al21 stratified these subjects
Schiffrin et al Kidney Disease and the Cardiovascular System 87

into 4 groups; the investigators used the MDRD formula toestimate baseline GFR (mL/min per 1.73 m2) and found that,irrespective of treatment group, there was a progressiveincrease in both the primary end point as well as each of thesecondary end points as GFR declined across the 4 groups.These findings remained significant even when an extensive,70-candidate, variable model was used to adjust for highercomorbidities in patients with the poorest renal function. Ifthe group with a GFR �75 mL/min per 1.73 m2 is consideredthe reference point, the adjusted hazard ratio for adverse CVevents was 1.10 in the GFR group between 60.0 to 74.9mL/min per 1.73 m2 and 1.49 in the GFR group �45.0mL/min per 1.73 m2. When GFR was analyzed as a contin-uous variable, each decrease in GFR of 10 mL/min per 1.73m2 below 81.0 was associated with a 1.1-fold increase in riskof death and nonfatal CV complications.21
Epidemiological Links Between Albuminuriaand Adverse Cardiovascular Events
Renal disease may not only be identified by low GFR but alsoby the presence of abnormal quantities of albumin in theurine. In fact, the appearance of pathological albuminuriaoften precedes the functional deterioration that is evidencedby a decline in GFR. Importantly, albuminuria has also beenshown to be a potent independent marker of CV risk in bothdiabetic and nondiabetic persons. Similar to GFR, the linkbetween albuminuria and adverse CV events was first recog-nized in the more overt situations of macroalbuminuria (urinealbumin:creatinine ratio [ACR] �300 mg/g),23,24 and thenthis link was extended to more modest elevations such asmicroalbuminuria (ACR, 30 to 300 mg/g).25 More recently, ithas become increasingly recognized that CV risk begins torise within currently defined normal levels of albuminuria(ACR�30 mg/g). Thus, urinary albumin is a continuous CVrisk factor, whereas microalbuminuria is a designated thresh-old for renal functional deterioration in individuals with andwithout diabetes.
CVD in Patients With MacroalbuminuriaThe Irbesartan Diabetic Nephropathy Trial (IDNT) enrolledsubjects with type 2 diabetes, hypertension, and macroalbu-minuria.26 A total of 1715 subjects with mean urine ACR of1416.2 mg/g were randomized into 3 treatment groups thatreceived irbesartan, amlodipine, or placebo and were fol-lowed for a mean period of 2.6 years. The primary outcomeof the main trial was a renal-centric composite of serumcreatinine doubling, ESRD, or death. Although irbesartanproved to be the superior treatment with respect to theprimary outcome, no difference was detected between treat-ment groups on the secondary outcome of CV events. Withthis data, a post hoc analysis was performed by Anavekar etal27 to assess the relationship between baseline albuminexcretion and the CV composite (CV death, nonfatal MI,hospitalization for heart failure, stroke, amputation, andcoronary and peripheral revascularization). A univariate anal-ysis revealed that the proportion of patients who experiencedthe CV end point progressively increased with increasingquartiles of baseline urine ACR. A multivariate analysisconfirmed albuminuria as an independent risk factor for CV
events with a 1.3-fold increased relative risk for each naturallog increase of 1 U in urine ACR.
A similar population was studied in the Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losar-tan (RENAAL). Here, 1513 persons with type 2 diabetes,hypertension, and macroalbuminuria (mean baseline ACR,1810 mg/g) were randomized to either losartan or placeboand followed for a mean of 3.4 years. The primary end pointwas the same as in IDNT, namely a composite of mainlyadverse nephrological events (serum creatinine doubling,ESRD, or death), and, consistent with IDNT, the angiotensinantagonist provided superior nephroprotection but conferredno statistically significant benefit on the secondary CVoutcomes,28 although de novo heart failure was less fre-quently noted in the losartan group. Nevertheless, in a posthoc analysis of RENAAL, baseline albuminuria was againshown to be a predictor of both the prespecified compositeCV end point (composite of MI, stroke, first hospitalizationfor heart failure or unstable angina, coronary or peripheralrevascularization, or CV death) as well as of heart failurealone. With subjects stratified into 3 groups on the basis ofbaseline ACR (�1500, 1500 to 3000, �3000 mg/g), compar-ison of the highest tertile with the lowest revealed an adjustedhazard ratio of 1.92 for the composite CV end point and 2.70for heart failure. In multivariate analysis, baseline albumin-uria was the strongest independent predictor of both theseoutcomes. Perhaps more significant was the finding that thechange in urine albumin excretion from baseline to 6 monthswas the only dynamic correlate of adverse CV outcomes. A50% reduction in baseline albuminuria translated into an 18%reduction in the composite CV end point and a 27% reductionin the risk of heart failure. Thus, albuminuria is not only a riskfactor for adverse CV outcomes but may also be a therapeutictarget or an indicator of therapeutic response.29
CVD in Patients With MicroalbuminuriaMicroalbuminuria also correlates with adverse CV events. Ina multivariate analysis of CHD mortality in a type-2 diabeticpopulation, Mattock et al reported that microalbuminuria wasthe strongest predictor of adverse CV outcomes with an oddsratio of 10.02, which outranked smoking (odds ratio, 6.52),diastolic blood pressure (odds ratio, 3.20), and serum choles-terol (odds ratio, 2.32).30
The HOPE study investigators reported on the risk of CVevents associated with baseline ACR �2.0 mg/mmol (equiv-alent to 17.7 mg/g). This amount of albuminuria was presentat baseline in 1140 (32.6%) subjects of the diabetic cohortand in 823 (14.8%) subjects of the nondiabetic cohort. In theoverall population a baseline ACR �2.0 mg/mmol increasedthe adjusted relative risk of CV events (1.83), all-cause death(2.09), and hospitalization for congestive heart failure (3.23).The impact of microalbuminuria on the primary compositeoutcome (CV death, MI, or stroke) was significant in bothdiabetics (relative risk, 1.97) and nondiabetics (relative risk,1.61).31
The ability of microalbuminuria to predict adverse CVevents is not restricted to a high-risk population like that ofthe HOPE trial. In fact, Hillege et al demonstrated the abilityof microalbuminuria to predict CV and non-CV mortality in
88 Circulation July 3, 2007

the general population.32 The investigators mailed medicalquestionnaires and a vial to collect early morning urinesamples to all inhabitants of the city of Groningen between1997 and 1998. More than 40 000 subjects responded andwere followed for a mean period of 961 days. Vital statisticsand the causes of death were available from governmentregistries. The percentage of subjects who manifested base-line microalbuminuria was 22.5% in those who succumbed toCV death, 16.0% in patients who died as a result of non-CVdeath, and 7.0% in patients who remained alive at the end ofthe study period. After adjustment for other known CV riskfactors, a doubling of the urine albumin excretion rate wasassociated with a relative risk of 1.29 for CV mortality and1.12 for non-CV mortality. Here again, microalbuminuriaoutranked the predictive power of other classic CV riskfactors.
CVD in Patients With Albuminuria in theNormal RangeThe relationship between CV events and albuminuria hasbeen extended further by several studies that suggest CV riskassociated with increased levels of urinary albumin excretionbegins to emerge at levels previously defined as normal(ACR�30 mg/g). Here too, the association appears to applyto a wide spectrum of patient populations. Analysis of theHOPE study population supports albuminuria as a continuousrisk factor for adverse CV events from an ACR as low as 0.5mg/mmol (equivalent to 4.4 mg/g).33 For every subsequent0.4 mg/mmol increase in the ratio, the adjusted hazard ofmajor CV events increased by 5.9%.
Similarly, Klausen et al34 reported that the risk of CVevents in the general population began to increase at urinaryalbumin excretion levels below the defined threshold formicroalbuminuria. Klausen et al followed subjects in theThird Copenhagen City Heart Study, which included�10 200 randomly selected participants who underwent adetailed CV investigation program and provided a timedovernight urine sample. Subjects were classified into quartileson the basis of urinary albumin with a follow-up period thatranged from 5 to 7 years. A urinary albumin excretion abovethe upper quartile of 4.8 �g/min (equivalent to ACR �9mg/g) was associated with an increased adjusted relative riskof 2.0 for CHD and 1.9 for death.
A post hoc analysis of the Losartan Intervention forEndpoint Reduction in Hypertension (LIFE) study related notjust baseline albuminuria to CV risk but also the impact ofreduction of urinary albumin excretion on CV events.35 TheLIFE study followed 8206 hypertensive individuals withLVH for a mean period of 4.8 years. The principal finding ofLIFE was that losartan proved superior to atenolol in thereduction of the composite primary end point (CV death,nonfatal stroke, nonfatal MI) for the same degree of bloodpressure reduction.36 In the post hoc study by Ibsen et al,35 theLIFE population was stratified into 4 groups according tomean ACR at baseline (1.21 mg/mmol, equivalent to 10.6mg/g) and at year 1 (0.67 mg/mmol, equivalent to 5.9 mg/g).The percentage of subjects who experienced an adverse CVevent was reported on the basis of whether their ACR wasabove or below the mean values. This analysis demonstrated
a statistically significant stepwise increase in the primarycomposite end point that started with the group with the lowbaseline/low year-1 group ratios (5.5%). Intermediate riskwas found in the groups with low baseline/high year-1 (8.6%)ratios and high baseline/low year-1 ratios (9.4%). The highestrisk group had high baseline/high year-1 values (13.5%).These results were independent of in-treatment blood pres-sure and indicated that reductions in urine ACR over timetranslated into diminished CV risk.
Epidemiological Links Between Serum Cystatin Cand Adverse Cardiovascular Events
Recently, serum cystatin C has gained recognition as anexcellent endogenous marker of kidney function. Cystatin Cis a cysteine proteinase with a molecular weight of 13 kDathat is produced by almost all human cells and released intothe blood. Cystatin C is freely filtered by the glomerulus andmetabolized by proximal tubular cells, but it is not secretedinto the tubules. Cystatin C does not appear to be affected byage, gender, or muscle mass, and there is evidence to suggestthat it may be a more sensitive detector of incipient renaldysfunction than creatinine-based estimates of GFR such asthe Cockroft-Gault or MDRD formulas.37 Several recentreports have indicated that cystatin C may be a betterpredictor of adverse CV events and all-cause mortality thaneither serum creatinine or creatinine-based estimating equa-tions.38–41 Ix et al categorized a population of 990 ambulatorypersons with stable coronary heart disease into quartiles onthe basis of baseline serum cystatin C levels and followedthese subjects for a median of 37 months.42 Subjects in thehighest cystatin C quartile (�1.30 mg/dL), when comparedwith the lowest quartile (�0.91 mg/dL), had a hazard ratio of3.6 for all-cause mortality, 2.0 for CV events (composite ofCHD death, MI, and stoke), and 2.6 for incident heart failure.These statistically significant results were adjusted for tradi-tional CV risk factors. Potentially the most important findingin this study was that higher cystatin C levels were predictiveof these adverse outcomes even among people withoutmicroalbuminuria or a diminished GFR as estimated by theMDRD formula (�60 mL/min per 1.73 m2). Currentlycystatin C is not routinely measured in clinical practice.
In summary, the presence of renal dysfunction, whetherdetected by GFR, urine albumin excretion, or serum cystatinC, predicts adverse CV outcomes. These relationships appearto extend to individuals with and without diabetes, those withand without preexisting CVD, and subjects with minimal tomarked perturbations in their renal parameters.
Mechanisms of Cardiovascular Complicationsin Renal Disease
As described in the preceding paragraphs, there is growingevidence that relatively minor renal abnormalities such as aslightly reduced GFR or microalbuminuria even within thenormal range may be associated with increased risk of CVevents. One of the principal pathophysiological mechanismsinvolved in this association has been proposed to be endo-thelial dysfunction. Whether micro- or macroalbuminuria isan expression of generalized endothelial cell dysfunctionremains to be demonstrated. However, many studies have
Schiffrin et al Kidney Disease and the Cardiovascular System 89

demonstrated the correlation of albuminuria with endothelialdysfunction as measured in peripheral blood vessels. Many ofthe traditional and nontraditional CV risk factors that couldaffect endothelial function can be found in association withCKD. Related conditions such as diabetes, obesity, andhypertension, as well as the presence of renal dysfunction perse lead to activation of the renin-angiotensin system, oxida-tive stress, elevated ADMA, low-grade inflammation withincreased circulating cytokines, and dyslipidemia, which areall common pathophysiological mechanisms that play a rolein the association of renal failure and CVD.43
HypertensionHypertension in and of itself represents a powerful risk factorfor CVD in CKD and is almost invariably present in patientswith renal failure. Sodium retention and activation of therenin-angiotensin system have been considered the mostimportant mechanisms involved in the elevation of bloodpressure in subjects with kidney disease.44 Sympathetic ner-vous system activation also plays a role. Plasma catechol-amine concentrations are elevated, and increased nerve sym-pathetic traffic has been demonstrated in renal failure.45,46
The participation of the sympathetic system has become morecomplex with the recent discovery of renalase, a new regu-lator of cardiac function and blood pressure produced by thekidney. Xu et al47 screened libraries of the Mammalian GeneCollection Project and identified a 37.8-kDa oxidase, whichcontained flavin-adenine-dinucleotide, expressed mainly inglomeruli and proximal tubules of the kidney but also incardiomyocytes and other tissues; the investigators called thisoxidase renalase. Renalase metabolizes catecholamines in thefollowing order: dopamine3 epinephrine3 norepinephrine.In contrast to other oxidases, renalase is secreted into plasmaand urine of healthy persons. However, it is not detectable inuremic individuals. Recombinant renalase exerts a powerfuland rapid hypotensive effect on rats. To what extent theimpairment of renalase production contributes to sympathetichyperactivity and blood pressure elevation in CKD remains tobe established. Also, endothelial dysfunction48–52 (see below)and remodeling of blood vessels53 may participate not only invascular complications in patients with kidney disease butalso in the maintenance of elevated blood pressure.
Hypertension also plays a major role in cardiac damage inCKD via LVH induction.54,55 In addition, a reduction incoronary reserve and capillary density that occurs in CKDpatients exposes them to coronary ischemia,56 which in turnleads to worsening of ventricular dysfunction.
Endothelial Dysfunction, Nitric OxideBioavailability, and ADMA in Renal DiseaseImpairment of endothelial function is recognized as one ofthe initial mechanisms that lead to atherosclerosis. Endothe-lial dysfunction, which occurs in both large and smallarteries, is present in renal disease.51 Microalbuminuria, amarker of glomerular hyperfiltration, has been correlated withand may be a manifestation of impaired endothelial func-tion.57 Experimental evidence suggests that microvascularendothelial dysfunction participates in the mechanisms thatlead to progression of renal disease,58 which in turn may
exacerbate endothelial dysfunction and contribute to acceler-ation of atherogenesis. It has been postulated that glomerularendothelial dysfunction is an early feature of essential hyper-tension that may precede blood pressure elevation. Microal-buminuria may itself contribute to renal dysfunction, whichprogresses with uncontrolled blood pressure elevation. Endo-thelial dysfunction in turn may contribute to CV mortalityalready in mild renal insufficiency as suggested by the HoornStudy.59 Reduced bioavailability of nitric oxide (NO) appearsto be one of the main factors involved in chronic renalfailure–associated endothelial dysfunction,48,52,60 in largemeasure because of increased oxidative stress in the vascularwall (see Dyslipidemia, Inflammation, and Oxidative Stressin Renal Disease).48,49 Prevalence of impaired endothelialfunction, low-grade inflammation, and dyslipidemia associ-ated with incipient and progressive renal disease may explainthe acceleration of atherosclerosis and, together with hyper-tension, may explain the high prevalence of coronary ische-mia and CV events in CKD. The presence of hypertension,sometimes difficult to control, in subjects with the previouslymentioned risk factors may underlie the prevalence of cere-brovascular disease and stroke in patients with renal disease.Paradoxically, a recent report showed that lowest systolicblood pressure was associated with stroke in stage 3 to 4CKD.61
ADMA is a competitive inhibitor of NO synthase.62
ADMA is synthesized potentially in many tissues, but in theCV system it is produced in the heart, endothelium, andsmooth muscle cells. It is derived from the catabolism ofproteins that contain methylated arginine residues, and it isreleased as the proteins are hydrolyzed. The synthesis ofADMA requires the enzyme protein arginine methyltrans-ferase type I, which methylates arginine residues, and theprotein arginine methyltransferase type II forms symmetricdimethylarginine, which is a stereoisomer of ADMA and isnot an inhibitor of NO synthase. ADMA and symmetricdimethylarginine enter endothelial cells through the cationicamino acid y� transporter. The activity of this transportercolocalizes with caveolin-bound NO synthase, which sug-gests that y� transporter activity may be a determinant of thelocal concentrations of ADMA. The ADMA and symmetricdimethylarginine compete with each other and L-arginine fortransport into the cell. Thus, ADMA may block entry ofL-arginine, with the resulting decrease in synthesis of NO.ADMA is metabolized mainly by dimethylarginine dimeth-ylaminohydrolase and cleared by the kidney. ExogenousADMA inhibits NO generation in vitro, and in humans itreduces forearm blood flow and cardiac output and increasessystemic vascular resistance and blood pressure.63 SubpressorADMA infusion increases renovascular resistance, inducesintimal hyperplasia, and affects small and large vessels.64–66
Plasma concentrations of ADMA are increased in associationwith endothelial dysfunction and/or reduced NO production,particularly in renal failure.67,68 Increased ADMA in renalfailure may result from both increased activity of proteinarginine methyltransferase and decreased metabolism bydimethylarginine dimethylaminohydrolase.69 It is unclearwhether endogenous ADMA concentrations increase suffi-ciently to inhibit NO production in vivo. Interestingly, plasma
90 Circulation July 3, 2007

norepinephrine and ADMA concentrations are closely corre-lated in patients with ESRD and are likely to act throughcommon mechanisms that contribute to CV events.70 ADMAis now considered one of the strongest markers of atheroscle-rosis.71 Elevated plasma concentrations of ADMA are asso-ciated not only with endothelial dysfunction and atheroscle-rosis72 but predict mortality and CV complications in CKDand ESRD.68 In subjects with mild to advanced CKD, plasmaADMA was inversely related to GFR73 and was an indepen-dent risk marker for progression to ESRD and mortality.74 Inthe Mild to Moderate Kidney Disease Study, ADMA wassignificantly associated with progression of nondiabetic kid-ney disease.75 Elevated plasma ADMA has been shown to bea marker of CV morbidity in early nephropathy associatedwith type 1 diabetes.76 In the Ludwigshafen Risk and Car-diovascular Health Study, ADMA independently predictedtotal and CV mortality in individuals with angiographiccoronary artery disease.77 Although reduced bioavailability ofNO and accumulation of ADMA cause endothelial dysfunc-tion, there is little evidence for coronary artery endothelialdysfunction in renal failure. Recently, Tatematsu et al78
induced renal failure in dogs and evaluated coronary vasodi-lator response to acetylcholine, which demonstrated bluntedresponses in the CKD dogs. mRNA expression of dimethyl-arginine dimethylaminohydrolase-II and endothelial NO syn-thase in coronary arteries were downregulated, which dem-onstrated a possible mechanism for coronary endothelialdysfunction in early stages of CKD.
Dyslipidemia, Inflammation, and Oxidative Stressin Renal DiseaseIndividuals with CKD become progressively malnourished,as evidenced by low levels of albumin, prealbumin, andtransferrin, which has been suggested to be a mechanism foractivation of inflammation.79 Diseases in which low-gradeinflammation is found, such as diabetes and hypertension, areoften associated with CKD. Thus it is difficult to concludewhether there is a direct effect of renal failure on inflamma-tion in early CKD. Renal failure causes changes in plasmacomponents and endothelial structure and function that favorvascular injury, which may play a role as a trigger forinflammatory response. Dyslipidemia associated withCKD80,81 contributes to the inflammatory response in renalfailure. The changes in blood-lipid composition and theirrelation to renal dysfunction and inflammation are summa-rized in Table 3. Hepatic apolipoprotein A-I synthesis de-creases and high-density lipoprotein levels fall. High-densitylipoprotein is an important antioxidant and also protects theendothelium from the effects of proinflammatory cytokines.Apolipoprotein C-III, a competitive inhibitor of lipoproteinlipase, is increased in CKD. Serum triglyceride levels in-crease as a result of accumulation of intermediate-densitylipoprotein, which comprise very low-density lipoprotein andchylomicron remnants. These impair endothelial function andare associated with CVD.
Because dyslipidemia associated with CKD appears to playa role in the enhanced CV risk of these patients, treatment ofdyslipidemia conversely should reduce proteinuria and ame-liorate the progression of CKD. Indeed, statin therapy appears
to reduce proteinuria modestly, and results in a small reduc-tion in the rate of loss of kidney function, especially inpopulations with CVD.82
The changes in lipoprotein composition and structure aswell as angiotensin II–mediated alterations in endothelialfunction stimulate and amplify the effect of inflammatorymechanisms.83 Between 30 and 50% of CKD patients haveelevated serum levels of inflammatory markers such asC-reactive protein, fibrinogen, interleukin-6, tumor necrosisfactor-�, factor VIIc, factor VIIIc, plasmin-antiplasmin com-plex, D-dimer, and the adhesion molecules E-selectin,VCAM-1 and ICAM-1.84,85 Mechanisms are unclear butincreased inflammatory mediators have been attributed toincreased oxidative stress, accumulation of postsyntheticallymodified proteins, advanced glycation end products, andother agents normally cleared by the kidney. Thus, causes ofinflammation may include comorbidities, oxidative stress,infections, and hemodialysis-related factors that depend onmembrane biocompatibility and the dialysate.86 Progressivedeterioration of renal function in CKD may lead to dyslipid-emia or accumulation of uremic toxins, which can stimulateoxidative stress and inflammation, which in turn may con-tribute to endothelial dysfunction and progression ofatherosclerosis.
A major contributor to the increase in circulating inflam-matory biomarkers in CKD may be enhanced oxidativestress.85–87 Mechanisms of oxidative stress in uremia mayinvolve activation of reduced nicotinamide adenine dinucle-otide (NAD(P)H) oxidase, xanthine oxidase, uncoupled en-dothelial NO synthase, myeloperoxidase (MPO), and mito-
TABLE 3. Effects of Renal Failure and Inflammation onLipoprotein and Endothelial Structure and Function
HDL
Effects of renal failure: decreased synthesis of apoA-I; decreased LCATactivity; increased apoC-III; increased triglycerides; decreased levels ofmature HDL
Effects of inflammation: replacement of apoA-I with serum amyloid A;decreased levels of mature HDL; decreased paroxynase and AHH activity;decreased ability to protect against cytokine action of endothelium;decreased ability to reduce oxidized LDL
Remnants
Effects of renal failure: increased levels linked to increase apoC-III;decreased clearance; interaction with blood vessels to inducevasoconstriction
LDL
Effects of renal failure: accumulation of small dense atherogenic LDL;results in increased AngII and also upregulation of the AT1 receptor
Activation of the renin-angiotensin system
Stimulates NAD(P)H oxidase, xanthine oxidase, etc., which leads toproduction of superoxide; induces IL-6 and other cytokines as well asPAI-1 gene expression; increased superoxide leads to decreasedbioavailability of nitric oxide, endothelial dysfunction, vascular remodeling,and hypertension
HDL indicates high-density lipoprotein; apo, apolipoprotein; LACT, decreasedlecithin cholesterol ester transferase; AHH, aryl hydrocarbon hydrolase; LDL,low-density lipoprotein; AT, angiotensinogen; and PAI-1, plasminogen activatorinhibitor 1. Adapted from Kaysen and Eiserich80 with permission from theAmerican Society of Nephrology. Copyright 2004.
Schiffrin et al Kidney Disease and the Cardiovascular System 91

chondrial oxidases. NAD(P)H oxidase is probably the mostimportant source in the vasculature, and it is stimulated byangiotensin II and other agents (see Renin-Angiotensin Sys-tem).88 Increased production of reactive oxygen species(ROS) by uncoupled endothelial NO synthase49 as well asreduced inactivation of ROS by antioxidant systems such assuperoxide dismutase87 also play an important role. MPO ispresent in neutrophils and monocytes/macrophages, and hasbeen shown to be expressed to a significant degree in humanatheroma.89 It may thus play a role in the acceleratedatherosclerosis of renal failure. It has recently been reportedthat a single nucleotide polymorphism in the promoter regionof the MPO gene associated with reduced expression of MPOis accompanied by a lower prevalence of CVD in ESRDpatients.90 Active MPO is released from white blood cellsduring hemodialysis, and this could be a mechanism wherebyMPO plays a role in vascular injury in subjects with ESRD.
Renin-Angiotensin SystemActivation of the renin-angiotensin system occurs in manyforms of renal disease. Angiotensin II stimulates NAD(P)Hoxidase, which leads to generation of superoxide anion andcontributes to endothelial dysfunction and vascular remodel-ing and growth.91 Mechanisms whereby the renin-angiotensinsystem may be activated by kidney disease are multiple andbeyond the scope of the present review, but such mechanismsmay in part depend on the adaptation to loss of renal mass thatresults in changes in renal hemodynamics. When angiotensinII acts through the AT1 receptor, it stimulates generation ofROS by NAD(P)H oxidase and other enzymes systems,which leads to upregulation of inflammatory mediators,which include cytokines, chemokines, adhesion molecules,and plasminogen activator inhibitor 1, and superoxide scav-enging of NO. These events, together with the mechanismsalready mentioned, promote endothelial dysfunction, vascularremodeling, and the progression of atherosclerosis.92
Vascular Calcification, Inducers and Inhibitorsof Calcification, and the Role of Phosphate inRenal FailureAccelerated calcifying atherosclerosis and valvular heartdisease occur with high frequency in CKD.93–95 A recentstudy showed that 40% of patients with CKD and a meanGFR 33 mL/min exhibited coronary artery calcificationcompared with 13% in matched control subjects with no renalimpairment.96 Calcification can be found in atheroscleroticplaques and in the vascular media, smooth muscle cells, andelastic laminae of large elastic and medium muscular arteriesas well as in cardiac valves.93–95 Subjects with renal failurewho exhibit medial calcification are typically middle-agedand have been dialyzed for some time, although someindividuals may already have calcified vessels before dialy-sis.97 There is a specific dialysis-related type of vascularcalcification called calciphylaxis, or calcific uremic arteriopa-thy, that is characterized by diffuse calcification of the mediaof small to medium arteries and arterioles with intimalproliferation and thrombosis that results in skin ulcers98 andcan lead to life-threatening skin necrosis or acral gangrene.Calciphylaxis is the result of an elevated calcium (Ca) �
phosphate (P) product without presence of an active osteo-genic process, and it must be differentiated from other formsof calcification of the skin that do not affect blood vessels andfrom medial calcific sclerosis, which affects larger vessels. Itis a rare complication of renal failure present in up to 4% ofhemodialysis patients, typically in obese diabetic females,associated often with secondary hyperparathyroidism, hyper-calcemia, hyperphosphatemia, malnutrition, and sometimeswith warfarin therapy or hypercoagulability. However, al-though warfarin and hypercoagulability have both been im-plicated, the latter on the basis of an association of protein Cdeficiency and calciphylaxis, some studies suggest that nei-ther hypercoagulability nor warfarin play a role in this rarecondition.99 Similarly, parathyroidectomy has been reportedto lead to the resolution of the skin ulcers of calciphylaxis insome series100 but not all.101
Mechanisms involved in vascular calcification in CKDinclude passive precipitation of Ca and P in the presence ofexcessively high extracellular concentrations, effects of in-ducers of osteogenic transformation and hydroxyapatite for-mation, and deficiency of calcification inhibitors.94,102 Table4 summarizes some of the inducers and inhibitors of vascularcalcification that induce an osteoblast phenotype in vascularsmooth muscle cells in CKD. Patients with ESRD often havesevere changes in their Ca�P product, which induces a trendtoward ectopic calcification. Aortic stiffening associated withcalcification103 will cause LVH, which results in increasedCV risk. Increased phosphate levels are also a source ofincreased CV risk, probably as a result of worsening vascularcalcification.104 Precipitation associated with a raised Ca�Pproduct may contribute to soft-tissue calcification, but calci-fication of the media of blood vessels appears to involveactive transport through the Na-P cotransporter PiT-1 whichoccurs in part as a result of a phenotypic switch of vascularsmooth muscle cells into osteoblast-like cells as a conse-quence of high intracellular Ca and P, which induce osteo-genic differentiation of smooth muscle cells.94,95 In an in vitromodel, elevated Ca or P induced human vascular smooth
TABLE 4. Promoters and Inhibitors of Vascular Calcification
Promoters of calcification
Traditional factors
Older age, male gender, hypertension, diabetes, smoking high LDLcholesterol, low HDL cholesterol, genetics
Uremia-related factors
Uremic serum, hyperphosphatemia, increased Ca�P product,exogenous vitamin D therapy, elevated parathyroid hormone levels,dialysis vintage, calcium load and hypercalcemia, chronic inflammation,warfarin, elevated leptin levels
Inhibitors of calcification
Circulating inhibitors
Fetuin-A, bone morphogenetic protein-7, parathyroid hormone–relatedpeptide, HDL, magnesium
Locally acting inhibitors
Matrix Gla protein, osteopontin, pyrophosphate, osteoprotegerin,genetics
Adapted from Qunibi93 with permission from the American Society ofNephrology. Copyright 2005.
92 Circulation July 3, 2007

muscle cell calcification, which was initiated by release ofmembrane-bound matrix vesicles and apoptotic bodies.105
Vesicles released by cells exposed to Ca and P calcified to animportant degree, but those released in the presence of serumwere minimally calcified and were found to contain thecalcification inhibitors fetuin-A and matrix Gla protein(MGP) (see next paragraph). Thus, vascular calcification is acell-mediated process regulated by calcification inhibitors,functional impairment of which leads to accelerated vascularcalcification.
Among the inhibitors of calcification, fetuin-A (�2-SchmidHeremans glycoprotein; molecular weight, 60 kDa), which isproduced by the liver and circulates in blood, appears to be ofprime importance. Fetuin-A has a transforming growthfactor-� receptor II–like domain and may function as asoluble transforming growth factor-� antagonist that inter-feres with insulin receptor autophosphorylation and tyrosinekinase activity.106 It forms stable colloidal spheres with Caand P (calciprotein particles) and is the main component of ahigh molecular mass complex that contains Ca, P, andMGP.107 Low serum fetuin-A levels in subjects with CKDhave been associated with enhanced vascular calcification102
and increased CV mortality.108,109 MGP belongs to a familyof N-terminal �-carboxylated (Gla) proteins that require avitamin K–dependent �-carboxylation for their biologicalactivation and prevent bone morphogenetic protein (BMP)-2/BMP receptor-2 (BMPR2) interactions.110 The�-carboxylated MGP, but not the non–�-carboxylated MGP,is carried in plasma by fetuin-A. Mice that lack MGP developspontaneous calcification of arteries and cartilage.111 Ele-vated concentrations of MGP may be found in the vicinity ofatherosclerotic plaques112 and have been shown to be associ-ated with calcification of vascular smooth muscle cells invitro.113 MGP levels in blood have been reported to correlatenegatively with coronary artery calcification.114
Osteoprotegerin regulates osteoclast activation. It acts as asoluble decoy receptor that prevents the binding of theosteoclast stimulator receptor activator of nuclear factor-�Bligand to its receptor. Osteoprotegerin deficiency in miceleads to vascular calcification, but its mechanism of actionhas not been elucidated.115 Osteoprotegerin levels are ele-vated in ESRD,116 correlate with vascular calcification, andpredict mortality in hemodialysis patients, in particular inindividuals with high C-reactive protein levels.117 Interest-ingly, lower soluble receptor activator of nuclear factor-�Bligand concentrations were associated with betteroutcomes.117
Elevated pyrophosphate (PPi) concentrations prevent hy-droxyapatite crystal formation and calcification. PPi is syn-thesized by the rate-limiting enzyme nucleotide pyrophos-phatase phospho-diesterase-1. Mice that lack nucleotidepyrophosphatase phospho-diesterase-1 develop PPi defi-ciency, which results in an altered vascular smooth musclecell phenotype and vascular calcification.118 The cellular PPiexporter ankyrin, which is encoded by the transmembranetransporter progressive ankylosis locus, mediates PPi exitfrom cells.119 Vascular calcification may also result fromenhanced activity of the membrane-bound tissue-nonspecificalkaline phosphatase, which degrades PPi to P. PPi deficiency
may occur in ESRD as a consequence of removal of PPiduring hemodialysis,120 which may be one of the mechanismsthat contribute to accelerated vascular calcification in hemo-dialysis patients.
BMPs are important regulators of bone formation. They aremembers of the largest subclass of the transforming growthfactor-� superfamily and have been localized in areas ofvascular calcification.121 BMP-2 is generated from a 60-kDaprecursor, which is processed to an 18-kDa monomer thatassociates with another monomer to form the active ho-modimer, which then binds to its receptor. The BMPR is aheterodimer that consists of types 1 and 2 serine/threoninekinases. BMPR2 phosphorylates BMPR1, which in turnphosphorylates the Smad 1/5/8 complex, which, with Smad 4,then modulates target gene expression.122 Of the differentBMPs, BMP-2 or BMP-4 may induce osteogenic differenti-ation of vascular smooth muscle cells through induction oftranscription factors Cbfa1, osterix, and the msh homeoboxhomolog MSX-2. Other effects of BMP-2/BMP-4 that con-tribute to calcification of the vasculature are the triggering ofapoptosis and inhibitory effects on MGP. In addition, BMP-4has been shown to exert vascular effects that lead to increasedoxidative stress and impaired endothelial function, and towhat extent these effects are related to media calcificationremains to be established. BMP-7 on the other hand inhibitsvascular calcification by upregulation of �-smooth muscleactin expression via induction of p21 and upregulation ofSmad 6 and 7. BMP-7 is expressed mainly in the kidney, andits expression decreases with progression of renal failure,which results in reduction of its ability to inhibit calcification.Lowering of BMP-7 affects bone metabolism with conse-quent increase in serum phosphate levels, which adverselyaffects the Ca�P product and induces phenotypic changes invascular smooth muscle cells, which leads to metastaticcalcification.
Increased leptin levels may participate in the process ofvascular calcification in CKD because serum leptin concen-trations are increased in renal failure as a result of reducedleptin excretion. Leptin induces heterotopic calcification viaits receptors in the hypothalamus that induce an increasedsympathetic activity, which stimulates osteoblast�-adrenergic receptors.123 Leptin increases bone marrowstem cell differentiation into an osteoprogenitor phenotypeand may act on vascular smooth muscle cells to inducecalcification,124 in part by an increase in ROS generation andinduction of BMP-2.95
In summary, BMP-2/BMP-4 binds the BMPR1/BMPR2receptor complex and phosphorylates the regulatory Smads,which then signal downstream to upregulate the expression oftranscription factors Cbfa1, osterix, and MSX-2. BMP-4 alsostimulates generation of ROS. Cbfa1 expression is alsoenhanced by ROS, leptin, vitamin D, high phosphate levels,and PiT-1.87 The result is a phenotypic change in vascularsmooth muscle cells to an osteogenic phenotype. These cellsexpress alkaline phosphatase and produce hydroxyapatitecrystals. Calcification inhibitors such as fetuin-A, MGP,osteoprotegerin, osteopontin, BMP-7, and Smad 6 antagonizeBMP-2/BMP-4 signaling and metastatic calcification(Figure).
Schiffrin et al Kidney Disease and the Cardiovascular System 93

ConclusionThe present review underlines the CV risk to which patientswith CKD are exposed and summarizes some of the mecha-nisms that lead to the increased risk of adverse CV events. Itis also clear that some of this risk is modifiable and can beimproved with currently available therapy by reduction ofblood pressure according to guidelines, aggressive treatmentof dyslipidemia, control of protein intake, minimization ofbone resorption, optimization of Ca and P metabolism, andcombat of hypercoagulability, with the caveat that warfarinmay be implicated in calciphylaxis of the latter. Therapeuticaspects that may require new approaches include manage-ment of the increased oxidative stress and low-grade inflam-mation, as well as development of novel strategies to increasethe concentrations of inhibitors of calcification and to mod-erate the agents that promote calcification.
Sources of FundingThe work by Dr Schiffrin was supported by a Canada Research Chairon Hypertension and Vascular Research, and by grants 37917 and82790 from the Canadian Institutes of Health Research.
DisclosuresNone.
References1. Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm
LL, McCullough PA, Kasiske BL, Kelepouris E, Klag MJ, Parfrey P,Pfeffer M, Raij L, Spinosa DJ, Wilson PW. Kidney disease as a riskfactor for development of cardiovascular disease: a statement from theAmerican Heart Association Councils on Kidney in CardiovascularDisease, High Blood Pressure Research, Clinical Cardiology, and Epi-demiology and Prevention. Hypertension. 2003;42:1050–1065.
2. Tonelli M, Wiebe N, Culleton B, House A, Rabbat C, Fok M, McAlisterF, Garg AX. Chronic kidney disease and mortality risk: a systematicreview. J Am Soc Nephrol. 2006;17:2034–2047.
3. Shlipak MG, Heidenreich PA, Noguchi H, Chertow GM, Browner WS,McClellan MB. Association of renal insufficiency with treatment andoutcomes after myocardial infarction in elderly patients. Ann InternMed. 2002;137:555–562.
4. Foley RN, Parfrey PS, Sarnak M. Clincial epidemiology of cardiovas-cular disease in chronic renal disease. Am J Kidney Dis. 1998;32:112–119.
5. Herzog CA, Ma JZ, Collins AJ. Poor long-term survival after acutemyocardial infarction among patients on long-term dialysis. N EnglJ Med. 1998;339:799–805.
6. Parfrey PS, Foley RN. The clinical epidemiology of cardiac disease inchronic uremia. J Am Soc Nephrol. 1999;10:1606–1615.
7. Foley RN, Parfrey PS, Harnett JD. Clinical and echocardiographicdisease in patients starting end-stage renal disease therapy. Kidney Int.1995;47:186–193.
8. Levin A, Singer J, Thompson CR, Ross H, Lewis M. Prevalent LVH inthe predialysis population: identifying opportunities for intervention.Am J Kidney Dis. 1996;27:347–354.
9. Tucker B, Fabbian F, Giles M, Thuraisingham RC, Raine AE, Baker LR.Left ventricular hypertrophy and ambulatory blood pressure monitoringin chronic renal failure. Nephrol Dial Transplant. 1997;12:724–728.
10. Culleton BF, Larson MG, Wilson PWF, Evans JC, Parfrey PS, Levy D.Cardiovascular disease and mortality in a community-based cohort withmild renal insufficiency. Kidney Int. 1999;56:2214–2219.
11. Go AS, Chertow GM, Fan D, McCullock CE, Hsu CY. Chronic kidneydisease and the risks of death, cardiovascular events, and hospitalization.N Engl J Med. 2004;351:1296–1305.
12. Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A moreaccurate method to estimate glomerular filtration rate from serum cre-atinine: a new prediction equation. Modification of Diet in RenalDisease Study Group. Ann Intern Med. 1999;130:461–470.
13. Shulman NB, Ford CE, Hall WD, Blaufox MD, Simon D, Langford HG,Schneider KA. Prognostic value of serum creatinine and the effect oftreatment of hypertension on renal function. Results from the hyper-tension detection and follow-up program. The Hypertension Detectionand Follow-up Program Cooperative Group. Hypertension. 1989;13(suppl):I80–I93.
14. Cockroft DW, Gault MH. Prediction of creatinine clearance from serumcreatinine. Nephron. 1976;16:31–41.
15. Ruilope LM, Salvetti A, Jamerson K, Hanson L, Warnold I, Wedel H,Zanchetti A. Renal function and intensive lowering of blood pressure in
CMSV
muilehtodnE1-XOL
SOR
4/2-PMBP x aC
noitacificlaCsretomorp
A-niuteFGPOPGMNPO
7-PMBiPP
A-niuteF
noitacificlaCsrotibihni
TA 1R
cinegoetsOCMSV PLA
/HDANHPDANesadixo
II nisnetoignAnoitammalfnI
sisobmorhT
etitapayxordyH
1-tiP nitpeLD nimatiV
OP 4
Mechanisms depicted here are some of those involved in vascular calcification in chronic kidney disease. Activation of the renin-angiotensin system results in stimulation of AT1R, which stimulates reduced NAD(P)H oxidase, the main source of vascular ROS. BMP-2/4 binds the BMP receptor BMPR1/BMPR2 receptor complex and phosphorylates the Smad 1/5/8 complex, which, with Smad 4, sig-nals downstream to upregulate expression of transcription factors Cbfa1, osterix, and MSX-2. Cbfa1 expression is also enhanced byROS, leptin, vitamin D, increased Ca�P product, or high PO4 levels induced by Pit-1, the sodium-phosphate cotransporter, activated inpart as a result of the phenotypic switch of VSMCs into osteoblast-like cells. VSMCs that have acquired an osteogenic phenotypeexpress ALP and produce hydroxyapatite crystals. Calcification inhibitors such as PPi inhibit hydroxyapatite precipitation, whereasfetuin-A, MGP, OPG, OPN, BMP-7, and Smad 6 antagonize BMP2/4 signaling and calcification. AT1R indicates angiotensin AT1 recep-tor; NAD(P)H, nicotinamide adenine dinucleotide; ROS, reactive oxygen species; BMP, bone morphogenic protein; PO4, phosphate;VSMC, vascular smooth muscle cells; ALP, alkaline phosphatase; PPi, pyrophosphate; MGP, matrix Gla protein; OPG, osteoprotegerin;and OPN, osteopontin.
94 Circulation July 3, 2007

hypertensive participants of the Hypertension Optimal Treatment (HOT)Study. J Am Soc Nephrol. 2001;12:218–225.
16. Mann JFE, Gerstein HC, Pogue J, Bosch J, Yusuf S. Renal insufficiencyas a predictor of cardiovascular outcomes and the impact of ramipril:The HOPE randomized trial. Ann Intern Med. 2001;134:629–636.
17. Solomon SD, Rice MM, Jablonski KA, Jose P, Domanski M, SabatineM, Gersh BJ, Rouleau J, Pfeffer MA. Renal function and effectivenessof angiotensin-converting enzyme inhibitor therapy in patients withchronic stable coronary disease in the prevention of events with ACEinhibition (PEACE) trial. Circulation. 2006;114:26–31.
18. Hillege HL, Girbes AR, De Kam PJ, Boomsma F, De Zeeuw D, Char-lesworth A. Renal function, neurohormonal activation and survival inpatients with chronic heart failure. Circulation. 2000;102:203–210.
19. Hampton JR, Van Veldhuisen DJ, Kleber FX, Cowley AJ, Ardia A,Block P, Cortina A, Cserhalmi L, Follath F, Jensen GK, Lie KL, ManciaG, Skene AM. Randomised study of effect of ibopamine on survival inpatients with advanced severe heart failure: second prospective ran-domised study of ibopamine on mortality and efficacy (PRIME II)investigators. The Lancet. 1997;349:971–977.
20. Hillege HL, Van Gilst WH, Van Veldhuisen DJ, Navis G, Grobbee DE,De Graeff PA. Accelerated decline and prognostic impact of renalfunction after myocardial infarction and the benefits of ACE inhibition:the CATS randomized trial. Eur Heart J. 2003;24:412–420.
21. Anavekar NS, McMurray JJV, Velazquez EJ, Solomon SD, Kover L,Rouleau JL, White HD, Nordlander R, Maggioni A, Dickstein K, Zelen-kofske S, Leimberger JD, Califf RM, Pfeffer MA. Relation betweenrenal dysfunction and cardiovascular outcomes after myocardialinfarction. N Engl J Med. 2004;351:1285–1295.
22. Pfeffer MA, McMurray JJ, Velazquez EJ, Rouleau JL, Kober L,Maggioni AP, Solomon SD, Swedberg K, Van de Werf F, White H,Leimberger JD, Henis M, Edwards S, Zelenkofske S, Sellers MA, CaliffRM. Valsartan, captopril, or both in myocardial infarction complicatedby heart failure, left ventricular dysfunction, or both. N Engl J Med.2003;349:1893–1906.
23. Kannel WB, Stampfer MJ, Castelli WP, Verter J. The prognostic sig-nificance of proteinuria: the Framingham study. Am Heart J. 1985;108:1347–1352.
24. Grimm RH, Svendsen KH, Kasiske B, Keane WF, Wahi MM. Pro-teinuria is a risk factor for mortality over 10 years of follow-up. MRFITResearch Group. Kidney Int. 1997;63:S10–S14.
25. Keane WF, Eknoyan G, NKF PC. Proteinuria, albuminuria, risk,assessment, detection, elimination (PARADE): a position paper of theNational Kidney Foundation. Am J Kidney Dis. 1999;33:1004–1010.
26. Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, RitzE, Atkins RC, Rohde R, Raz I, Collaborative Study Group. Renopro-tective effect of the angiotensin-receptor antagonist irbesartan in patientswith nephropathy due to type 2 diabetes. N Engl J Med. 2001;345:851–860.
27. Anavekar NS, Gans DJ, Berl T, Rohde RD, Cooper W, Bhaumik A,Hunsicker LG, Rouleau JL, Lewis JB, Rosendorff C, Porush JG, DruryPL, Esmatjes E, Raz I, Vanhille P, Locatelli F, Goldhaber S, Lewis EJ,Pfeffer MA. Predictors of cardiovascular events in patients with type 2diabetic nephropathy and hypertension: A case for albuminuria. KidneyInt. 2004;66(suppl 92):S50–S55.
28. Brenner BM, Cooper ME, De Zeeuw D, Mitch WE, Parving HH,Remuzzi G, Snapinn SM, Zhang ZX, Shahinfar S, RENAAL Study.Effects of losartan on renal and cardiovascular outcomes in patients withtype 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869.
29. De Zeeuw D, Remuzzi G, Parving HH, Keane WF, Zhang ZX, ShahinfarS, Snapinn S, Cooper ME, Mitch WE, Brenner BM. Albuminuria, atherapeutic target for cardiovascular protection in type 2 diabeticpatients with nephropathy. Circulation. 2004;110:921–927.
30. Mattock MB, Barnes DJ, Viberti G, Keen H; Burt D, Hughes JM;Fitzgerald AP, Sandhu B, Jackson PG. Microalbuminuria and coronaryheart disease in NIDDM: an incidence study. Diabetes. 1998;47:1786–1792.
31. Mann JFE, Yi QL, Gerstein HC. Albuminuria as a predictor of cardio-vascular and renal outcomes in people with known atherosclerotic car-diovascular disease. Kidney Int. 2004;66:S59–S62.
32. Hillege HL, Fidler V, Diercks GFH, Van Gilst WH, De Zeeuw D, VanVeldhuisen DJ, Gans ROB, Janssen WMT, Grobbee DE, De Jong PE.Urinary albumin excretion predicts cardiovascular and noncardiovascu-lar mortality in general population. Circulation. 2002;106:1777–1782.
33. Gerstein HC, Mann JFE, Qilong Y, Zinman B, Dinneen SF, HoogwerfB, Halle JP, Young J, Rashkow A, Yoyce C, Nawaz S, Yusuf S.
Albuminuria and cardiovascular events, death and heart failure indiabetic and non-diabetic individuals. JAMA. 2001;286:421–46.
34. Klausen K, Borch-Johnsen K, Feldt-Rasmussen B, Jensen G, Clausen P,Scharling H, Appleyard M, Jensen JS. Very low levels of microalbu-minuria are associated with increased risk of coronary heart disease anddeath independently of renal function, hypertension, and diabetes. Cir-culation. 2004;110:32–35.
35. Ibsen H, Olsen MH, Wachtell K, Borch-Johnsen K, Lindholm LH,Mogensen CE, Dahlöf B, Devereux RB, De Faire U, Fyhrquist F, JuliusS, Kjeldsen SE, Lederballe-Pedersen O, Nieminen MS, Omvik P, OparilS, Wan Y. Reduction in albuminuria translates to reduction in cardio-vascular events in hypertensive patients: losartan intervention for endpoint reduction in hypertension study. Hypertension. 2005;45:198–202.
36. Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, De Faire U,Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, LindholmLH, Nieminen MS, Omvik P, Oparil S, Wedel H, LIFE Study Group.Cardiovascular morbidity and mortality in the Losartan Intervention ForEnd point reduction in hypertension study (LIFE): a randomised trialagainst atenolol. The Lancet. 2002;359:995–1003.
37. Perkins BA, Nelson RG, Ostrander BE, Blouch KL, Krolewski AS,Myers BD, Warram JH. Detection of renal function decline in patientswith diabetes and normal or elevated GFR by serial measurements ofserum cystatin C concentration: results of a 4-year follow-up study. J AmSoc Nephrol. 2005;16:1404–1412.
38. Shlipak MG, Sarnak MJ, Katz R, Seliger SL, Newman AB, SiscovickDS, Stehman-Breen C. Cystatin C and the risk of death and cardiovas-cular events among elderly persons. N Engl J Med. 2005;352:2049–2060.
39. Sarnak MJ, Katz R, Stehman-Breen CO, Fried LF, Jenny NS, Psaty BM,Newman AB, Siscovick D, Shlipak MG. Cystatin C concentration as arisk factor for heart failure in older adults. Ann Intern Med. 2005;142:497–505.
40. Fried LF, Katz R, Sarnak MJ, Shlipak MG, Chaves PH, Jenny NS,Stehman-Breen C, Gillen D, Bleyer AJ, Hirsch C, Siscovick D, NewmanAB. Kidney function as a predictor of noncardiovascular mortality. J AmSoc Nephrol. 2005;16:3728–3735.
41. Shlipak MG, Wassel Fyr CL, Chertow GM, Harris TB, Kritchevsky SB,Tylavsky FA, Satterfield S, Cummings SR, Newman AB, Fried LF.Cystatin C and mortality risk in the elderly: the health, aging, and bodycomposition study. J Am Soc Nephrol. 2006;17:254–261.
42. Ix JH, Shlipak MG, Chertow GM, Whooley MA. Association of cystatinC with mortality, cardiovascular events, and incident heart failureamong persons with coronary heart disease. Circulation. 2006;115:173–179.
43. Amann K, Wanner C, Ritz E. Cross-talk between the kidney and thecardiovascular system. J Am Soc Nephrol. 2006;17:2112–2119.
44. Guyton AC, Coleman TG, Wilcox CS. Quantitative analysis of thepathophysiology of hypertension. J Am Soc Nephrol. 1999;10:2248–2249.
45. Converse RL Jr, Jacobsen TN, Toto RD, Jost CM, Cosentino F, Fouad-Tarazi F, Victor RG. Sympathetic overactivity in patients with chronicrenal failure. N Engl J Med. 1992;327:1912–1918.
46. Neumann J, Ligtenberg G, Klein II, Koomans HA, Blankestijn PJ.Sympathetic hyperactivity in chronic kidney disease: pathogenesis,clinical relevance, and treatment. Kidney Int. 2004;65:1568–1576.
47. Xu J, Li G, Wang P, Velazquez H, Yao X, Li Y, Wu Y, Peixoto A,Crowley S, Desir GV. Renalase is a novel, soluble monoamine oxidasethat regulates cardiac function and blood pressure. J Clin Invest. 2005;115:1275–1280.
48. Wever R, Boer P, Hijmering M, Stroes E, Verhaar M, Kastelein J,Versluis K, Lagerwerf F, Van Rijn H, Koomans H, Rabelink T. Nitricoxide production is reduced in patients with chronic renal failure. Arte-rioscler Thromb Vasc Biol. 1999;19:1168–1172.
49. Vaziri ND, Ni Z, Oveisi F, Liang K, Pandian R. Enhanced nitric oxideinactivation and protein nitration by reactive oxygen species in renalinsufficiency. Hypertension. 2002;39:135–141.
50. Stehouwer CDA, Henry RMA, Dekker JM, Nijpels G, Heine RJ, BouterLM. Microalbuminuria is associated with impaired brachial artery, flow-mediated vasodilation in elderly individuals without and with diabetes:Further evidence for a link between microalbuminuria and endothelialdysfunction: the Hoorn study. Kidney Int. 2004;66:S42–S44.
51. Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am SocNephrol. 2004;15:1983–1992.
52. Passauer J, Pistrosch F, Bussemaker E, Lassig G, Herbrig K, Gross P.Reduced agonist-induced endothelium-dependent vasodilation in uremia
Schiffrin et al Kidney Disease and the Cardiovascular System 95

is attributable to an impairment of vascular nitric oxide. J Am SocNephrol. 2005;16:959–965.
53. Foley RN, Parfrey PS, Harnett JD, Kent GM, Murray DC, Barre PE.Impact of hypertension on cardiomyopathy, morbidity and mortality inend-stage renal disease. Kidney Int. 1996;49:1379–1385.
54. Locatelli F, Bommer J, London GM, Martin-Malo A, Wanner C,Yaqoob M, Zoccali C. Cardiovascular disease determinants in chronicrenal failure: clinical approach and treatment. Nephrol Dial Transplant.2001;16:459–468.
55. Amann K, Breitbach M, Ritz E, Mall G. Myocyte/capillary mismatch inthe heart of uremic patients. J Am Soc Nephrol. 1998;9:1018–1022.
56. Amann K, Miltenberger-Miltenyi G, Simonoviciene A, Koch A, Orth S,Ritz E. Remodeling of resistance arteries in renal failure: effect ofendothelin receptor blockade. J Am Soc Nephrol. 2001;12:2040–2050.
57. Stehouwer CDA, Smulders YM. Microalbuminuria and risk for cardio-vascular disease: analysis of potential mechanisms. J Am Soc Nephrol.2006;17:2106–2111.
58. Fujihara CK, De Nucci G, Zatz R. Chronic nitric oxide synthase inhi-bition aggravates glomerular injury in rats with subtotal nephrectomy.J Am Soc Nephrol. 1995;5:1498–1507.
59. Stam F, van Guldener C, Becker A, Dekker JM, Heine RJ, Bouter LM,Stehouwer CDA. Endothelial dysfunction contributes to renal function-associated cardiovascular mortality in a population with mild renalinsufficiency: the Hoorn study. J Am Soc Nephrol. 2006;17:537–545.
60. Hasdan G, Benchetrit S, Rashid G, Green J, Bernheim J, Rathaus M.Endothelial dysfunction and hypertension in 5/6 nephrectomized rats aremediated by vascular superoxide. Kidney Int. 2002;61:586–590.
61. Weiner DE, Tighiouart H, Levey AS, Elsayed E, Griffith JL, Salem DN,Sarnak MJ. Lowest systolic blood pressure was associated with stroke instage 3–4 chronic kidney disease. J Am Soc Nephrol. 2007;18:960-966.
62. Vallance P, Leiper J. Cardiovascular biology of the asymmetric dim-ethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arte-rioscler Thromb Vasc Biol. 2004;24:1023–1030.
63. Kielstein JT, Impraim B, Simmel S, Bode-Boger SM, Tsikas D, FrolichJC, Hoeper MM, Haller H, Fliser D. Cardiovascular effects of systemicnitric oxide synthase inhibition with asymmetrical dimethylarginine inhumans. Circulation. 2004;109:172–177.
64. Zoccali C, Benedetto FA, Maas R, Mallamaci F, Tripepi G, MalatinoLS, Boger R, CREED Investigators. Asymmetric dimethylarginine,C-reactive protein, and carotid intima-media thickness in end-stage renaldisease. J Am Soc Nephrol 2002;13:490–496.
65. Kielstein JT, Zoccali C. Asymmetric dimethylarginine: a cardiovascularrisk factor and a uremic toxin coming of age? Am J Kidney Dis.2005;46:186–202.
66. Yilmaz MI, Saglam M, Caglar K, Cakir E, Sonmez A, Ozgurtas T,Aydin A, Eyileten T, Ozcan O, Acikel C, Tasar M, Genctoy G, Erbil K,Vural A, Zoccali C. Pathogenesis and treatment of kidney disease andhypertension: the determinants of endothelial dysfunction in CKD: oxi-dative stress and asymmetric dimethylarginine. Am J Kidney Dis. 2006;47:42–50.
67. Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation ofan endogenous inhibitor of nitric oxide synthesis in chronic renal failure.The Lancet. 1992;339:572–575.
68. Zoccali C, Bode-Boger SM, Mallamaci F, Benedetto F, Tripepi G,Malatino L, Cataliotti A, Bellanuova I, Fermo I, Frolich J, Boger R.Plasma concentration of asymmetrical dimethylarginine and mortality inpatients with end-stage renal disease: a prospective study. The Lancet.2001;358:2113–2117.
69. Matsuguma K, Ueda S, Yamagishi SI, Matsumoto Y, Kaneyuki U,Shibata R, Fujimura T, Matsuoka H, Kimoto M, Kato S, Imaizumi T,Okuda S. Molecular mechanism for elevation of asymmetric dimethyl-arginine and its role for hypertension in chronic kidney disease. J AmSoc Nephrol. 2006;17:2176–2183.
70. Mallamaci F, Tripepi G, Maas R, Malatino L, Boger R, Zoccali C.Analysis of the relationship between norepinephrine and asymmetricdimethyl arginine levels among patients with end-stage renal disease.J Am Soc Nephrol. 2004;15:435–441.
71. Cooke JP. Asymmetrical dimethylarginine: the uber marker? Circu-lation. 2004;109:1813–1818.
72. Kielstein JT, Frolich JC, Haller H, Fliser D. ADMA (asymmetric dim-ethylarginine): an atherosclerotic disease mediating agent in patientswith renal disease? Nephrol Dial Transplant. 2001;16:1742–1745.
73. Kielstein JT, Boger RH, Bode-Boger SM, Frolich JC, Haller H, Ritz E,Fliser D. Marked increase of asymmetric dimethylarginine in patients
with incipient primary chronic renal disease. J Am Soc Nephrol. 2002;13:170–176.
74. Ravani P, Tripepi G, Malberti F, Testa S, Mallamaci F, Zoccali C.Asymmetrical dimethylarginine predicts progression to dialysis anddeath in patients with chronic kidney disease: a competing risksmodeling approach. J Am Soc Nephrol. 2005;16:2449–2455.
75. Fliser D, Kronenberg F, Kielstein JT, Morath C, Bode-Boger SM, HallerH, Ritz E. Asymmetric dimethylarginine and progression of chronickidney disease: the Mild to Moderate Kidney Disease study. J Am SocNephrol. 2005;16:2456–2461.
76. Tarnow L, Hovind P, Teerlink T, Stehouwer CDA, Parving HH.Elevated plasma asymmetric dimethylarginine as a marker of cardiovas-cular morbidity in early diabetic nephropathy in type 1 diabetes.Diabetes Care. 2004;27:765–769.
77. Meinitzer A, Seelhorst U, Wellnitz B, Halwachs-Baumann G, BoehmBO, Winkelmann BR, Marz W. Asymmetrical dimethylarginine inde-pendently predicts total and cardiovascular mortality in individuals withangiographic coronary artery disease (the Ludwigshafen Risk and Car-diovascular Health study). Clin Chem. 2007;53:273–283.
78. Tatematsu S, Wakino S, Kanda T, Homma K, Yoshioka K, Hasegawa K,Sugano N, Kimoto M, Saruta T, Hayashi K. Role of nitric oxide-producing and -degrading pathways in coronary endothelial dysfunctionin chronic kidney disease. J Am Soc Nephrol. 2007;18:741–749.
79. Stenvinkel P, Heimburger O, Lindholm B, Kaysen GA, Bergstrom J.Are there two types of malnutrition in chronic renal failure? Evidencefor relationships between malnutrition, inflammation and atherosclerosis(MIA syndrome). Nephrol Dial Transplant. 2000;15:953–960.
80. Kaysen GA, Eiserich JP. The role of oxidative stress-altered lipoproteinstructure and function and microinflammation on cardiovascular risk inpatients with minor renal dysfunction. J Am Soc Nephrol. 2004;15:538–548.
81. Trevisan R, Dodesini AR, Lepore G. Lipids and renal disease. J Am SocNephrol. 2006;17(suppl 2):S145–S147.
82. Sandhu S, Wiebe N, Fried LF, Tonelli M. Statins for improving renaloutcomes: a meta-analysis. J Am Soc Nephrol. 2006;17:2006–2016.
83. Pecoits-Filho R, Heimburger O, Barany P, Suliman M, Fehrman-Ekholm I, Lindholm B, Stenvinkel P. Associations between circulatinginflammatory markers and residual renal function in CRF patients. Am JKidney Dis. 2003;41:1212–1218.
84. Jofre R, Rodriguez-Benitez P, Lopez-Gomez JM, Perez-Garcia R.Inflammatory syndrome in patients on hemodialysis. J Am Soc Nephrol.2006;17(suppl 3):S274–S280.
85. Vaziri ND, Oveisi F, Ding YX. Role of increased oxygen free radicalactivity in the pathogenesis of uremic hypertension. Kidney Int. 1998;53:1748–1754.
86. Himmelfarb J, Stenvinkel P, Ikizler TA, Hakim RM. The elephant inuremia: oxidant stress as a unifying concept of cardiovascular disease inuremia. Kidney Int. 2002;62:1524–1538.
87. Vaziri ND, Dicus M, Ho ND, Boroujerdi-Rad L, Sindhu RK. Oxidativestress and dysregulation of superoxide dismutase and NADPH oxidasein renal insufficiency. Kidney Int. 2003;63:179–185.
88. Touyz RM, Yao G, Quinn MT, Pagano PJ, Schiffrin EL. p47phoxassociates with the cytoskeleton through cortactin in human vascularsmooth muscle cells: role in NAD(P)H oxidase regulation by angioten-sin II. Arterioscler Thromb Vasc Biol. 2005;25:512–518.
89. Daugherty A, Dunn JL, Rateri DL, Heinecke JW. Myeloperoxidase, acatalyst for lipoprotein oxidation, is expressed in human atheroscleroticlesions. J Clin Invest. 1994;94:437–444.
90. Pecoits-Filho R, Stenvinkel P, Marchlewska A, Heimburger O, BaranyP, Hoff CM, Holmes CJ, Suliman M, Lindholm B, Schalling M,Nordfors L. A functional variant of the myeloperoxidase gene is asso-ciated with cardiovascular disease in end-stage renal disease patients.Kidney Int. 2003;63:S172–S176.
91. Touyz RM, Schiffrin EL. Reactive oxygen species in vascular biology:implications in hypertension. Histochem Cell Biol. 2004;122:339–352.
92. Schiffrin EL, Touyz RM. From bedside to bench to bedside: role ofrenin-angiotensin-aldosterone system in remodeling of resistancearteries in hypertension. Am J Physiol Heart Circ Physiol. 2004;287:H435–H446.
93. Qunibi WY. Reducing the burden of cardiovascular calcification inpatients with chronic kidney disease. J Am Soc Nephrol. 2005;16(suppl2):S95–S102.
94. Ketteler M, Schlieper G, Floege J. Calcification and cardiovascularhealth: new insights into an old phenomenon. Hypertension. 2006;47:1027–1034.
96 Circulation July 3, 2007

95. Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobio-logical mechanisms and clinical implications. Circ Res. 2006;99:1044–1059.
96. Russo D, Palmiero G, De Blasio AP, Balletta MM, Andreucci VE.Coronary artery calcification in patients with CRF not undergoingdialysis. Am J Kidney Dis. 2004;44:1024–1030.
97. Blacher J, Guerin AP, Pannier B, Marchais SJ, London GM. Arterialcalcifications, arterial stiffness, and cardiovascular risk in end-stagerenal disease. Hypertension. 2001;38:938–942.
98. Budisavljevic MN, Cheek D, Ploth DW. Calciphylaxis in chronic renalfailure. J Am Soc Nephrol. 1996;7:978–982.
99. Rudwaleit M, Schwarz A, Trautmann C, Offermann G, Distler A. Severecalciphylaxis in a renal patient on long-term oral anticoagulant therapy.Am J Nephrol. 1996;16:344–348.
100. Hafner J, Keusch G, Wahl C, Sauter B, Hurlimann A, von WeizsackerF, Krayenbuhl M, Biedermann K, Brunner U, Helfenstein U. Uremicsmall-artery disease with medial calcification and intimal hyperplasia(so-called calciphylaxis): a complication of chronic renal failure andbenefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954–962.
101. Budisavljevic MN, Cheek D, Ploth DW. Calciphylaxis in chronic renalfailure. J Am Soc Nephrol. 1996;7:978–982.
102. Moe SM, Reslerova M, Ketteler M, O’Neill K, Duan D, Koczman J,Westenfeld R, Jahnen-Dechent W, Chen NX. Role of calcificationinhibitors in the pathogenesis of vascular calcification in chronic kidneydisease (CKD). Kidney Int. 2005;67:2295–2304.
103. Pannier B, Guèrin AP, Marchais SJ, Safar ME, London GM. Stiffness ofcapacitive and conduit arteries prognostic significance for end-stagerenal disease patients. Hypertension. 2005;45:592–596.
104. Tonelli M, Sacks F, Pfeffer M, Gao Z, Curhan G, for the CholesterolAnd Recurrent Events (CARE) Trial Investigators. Relation betweenserum phosphate level and cardiovascular event rate in people withcoronary disease. Circulation. 2005;112:2627–2633.
105. Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ,Proudfoot D, Jahnen-Dechent W, Weissberg PL, Shanahan CM. Humanvascular smooth muscle cells undergo vesicle-mediated calcification inresponse to changes in extracellular calcium and phosphate concen-trations: a potential mechanism for accelerated vascular calcification inESRD. J Am Soc Nephrol. 2004;15:2857–2867.
106. Demetriou M, Binkert C, Sukhu B, Tenenbaum HC, Dennis JW.Fetuin/alpha 2-HS glycoprotein is a transforming growth factor-betatype ii receptor mimic and cytokine antagonist. J Biol Chem. 1996;271:12755–12761.
107. Price PA, Thomas GR, Pardini AW, Figueira WF, Caputo JM, Wil-liamson MK. Discovery of a high molecular weight complex of calcium,phosphate, fetuin, and matrix gamma-carboxyglutamic acid protein inthe serum of etidronate-treated rats. J Biol Chem. 2002;277:3926–3934.
108. Ketteler M, Bongartz P, Westenfeld R, Wildberger JE, Mahnken AH,Bohm R, Metzger T, Wanner C, Jahnen-Dechent W, Floege J. Asso-ciation of low fetuin-A (AHSG) concentrations in serum with cardio-vascular mortality in patients on dialysis: a cross-sectional study. TheLancet. 2003;361:827–833.
109. Stenvinkel P, Wang K, Qureshi AR, Axelsson J, Pecoits-Filho R, Gao P,Barany P, Lindholm B, Jogestrand T, Heimburger O, Holmes C,Schalling M, Nordfors L. Low fetuin-A levels are associated withcardiovascular death: impact of variations in the gene encoding fetuin.Kidney Int. 2005;67:2383–2392.
110. Zebboudj AF, Imura M, Bostrom K. Matrix GLA protein, a regulatoryprotein for bone morphogenetic protein-2. J Biol Chem. 2002;277:4388–4394.
111. Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR,Karsenty G. Spontaneous calcification of arteries and cartilage in micelacking matrix GLA protein. Nature. 1997;386:78–81.
112. Shanahan CM, Cary NR, Metcalfe JC, Weissberg PL. High expressionof genes for calcification-regulating proteins in human atheroscleroticplaques. J Clin Invest. 1994;93:2393–2402.
113. Proudfoot D, Skepper JN, Shanahan CM, Weissberg PL. Calcification ofhuman vascular cells in vitro is correlated with high levels of matrix Glaprotein and low levels of osteopontin expression. Arterioscler ThrombVasc Biol. 1998;18:379–388.
114. Jono S, Ikari Y, Vermeer C, Dissel P, Hasegawa K, Shioi A, TaniwakiH, Kizu A, Nishizawa Y, Saito S. Matrix Gla protein is associated withcoronary artery calcification as assessed by electron-beam computedtomography. Thromb Haemost. 2004;91:790–794.
115. Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C,Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS.Osteoprotegerin-deficient mice develop early onset osteoporosis andarterial calcification. Genes Dev. 1998;12:1260–1268.
116. Nitta K, Akiba T, Uchida K, Kawashima A, Yumura W, Kabaya T,Nihei H. The progression of vascular calcification and serum osteopro-tegerin levels in patients on long-term hemodialysis. Am J Kidney Dis.2003;42:303–309.
117. Morena M, Terrier N, Jaussent I, Leray-Moragues H, Chalabi L, RivoryJP, Maurice F, Delcourt C, Cristol JP, Canaud B, Dupuy AM. Plasmaosteoprotegerin is associated with mortality in hemodialysis patients.J Am Soc Nephrol. 2006;17:262–270.
118. Johnson K, Polewski M, van Etten D, Terkeltaub R. Chondrogenesismediated by PPi depletion promotes spontaneous aortic calcification inNPP1-/- mice. Arterioscler Thromb Vasc Biol. 2005;25:686–691.
119. Harmey D, Hessle L, Narisawa S, Johnson KA, Terkeltaub R, Millan JL.Concerted regulation of inorganic pyrophosphate and osteopontin byakp2, enpp1, and ank: an integrated model of the pathogenesis ofmineralization disorders. Am J Pathol. 2004;164:1199–1209.
120. Lomashvili KA, Khawandi W, O’Neill WC. Reduced plasma pyro-phosphate levels in hemodialysis patients. J Am Soc Nephrol. 2005;16:2495–2500.
121. Hruska KA, Mathew S, Saab G. Bone morphogenetic proteins invascular calcification. Circ Res. 2005;97:105–114.
122. Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. GrowthFactors. 2004;22:233–241.
123. Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL,Armstrong D, Ducy P, Karsenty G. Leptin regulates bone formation viathe sympathetic nervous system. Cell. 2002;111:305–317.
124. Parhami F, Tintut Y, Ballard A, Fogelman AM, Demer LL. Leptinenhances the calcification of vascular cells: artery wall as a target ofleptin. Circ Res. 2001;88:954–960.
Schiffrin et al Kidney Disease and the Cardiovascular System 97

ACCF/AHA/SCAI 2007 Update of the Clinical CompetenceStatement on Cardiac Interventional Procedures
A Report of the American College of Cardiology Foundation/AmericanHeart Association/American College of Physicians Task Force on
Clinical Competence and Training (Writing Committee to Update the1998 Clinical Competence Statement on Recommendations for
the Assessment and Maintenance of Proficiency inCoronary Interventional Procedures)
WRITING COMMITTEE MEMBERS
Spencer B. King III, MD, MACC, FAHA, FSCAI, Chair;Thomas Aversano, MD, FACC; William L. Ballard, MD, FACC, FSCAI;
Robert H. Beekman III, MD, FACC, FAHA;Michael J. Cowley, MD, FACC, FSCAI*; Stephen G. Ellis, MD, FACC;
David P. Faxon, MD, FACC, FAHA, FSCAI*; Edward L. Hannan, PhD, FACC;John W. Hirshfeld, Jr, MD, FACC, FAHA; Alice K. Jacobs, MD, FACC, FAHA, FSCAI;
Mirle A. Kellett, Jr, MD, FACC, FSCAI; Stephen E. Kimmel, MD, FACC, FAHA;Joel S. Landzberg, MD, FACC; Louis S. McKeever, MD, FACC, FSCAI;
Mauro Moscucci, MD, FACC; Richard M. Pomerantz, MD, FACC, FSCAI;Karen M. Smith, MD, FACC, FSCAI; George W. Vetrovec, MD, FACC, FSCAI*
TASK FORCE MEMBERS
Mark A. Creager, MD, FACC, FAHA, Chair; John W. Hirshfeld, Jr, MD, FACC, FAHA†; David R.Holmes, Jr, MD, FACC; L. Kristin Newby, MD, FACC, FAHA; Howard H. Weitz, MD, FACC,
FACP; Geno Merli, MD, FACP; Ileana Piña, MD, FACC, FAHA;George P. Rodgers, MD, FACC, FAHA; Cynthia M. Tracy, MD, FACC†
*Society for Cardiovascular Angiography and Interventions Representative.†Former Task Force member during the writing effort.This document was approved by the American College of Cardiology Board of Trustees in May 2007, the American Heart Association Science
Advisory and Coordinating Committee in May 2007, and the Society for Cardiovascular Angiography and Interventions in May 2007.When this document is cited, the American College of Cardiology, American Heart Association, and Society for Cardiovascular Angiography and
Interventions would appreciate the following citation format: King SB III, Aversano T, Ballard WL, Beekman RH III, Cowley MJ, Ellis SG, Faxon DP,Hannan EL, Hirshfeld JW Jr., Jacobs AK, Kellett MA Jr., Kimmel SE, Landzberg JS, McKeever LS, Moscucci M, Pomerantz RM, Smith KM, VetrovecGW. ACCF/AHA/SCAI 2007 update of the clinical competence statement on cardiac interventional procedures: a report of the American College ofCardiology Foundation/American Heart Association/American College of Physicians Task Force on Clinical Competence and Training (WritingCommittee to Update the 1998 Clinical Competence Statement on Recommendations for the Assessment and Maintenance of Proficiency in CoronaryInterventional Procedures). Circulation. 2007;116:98–124.
This article has been copublished in the July 3, 2007, issue of the Journal of the American College of Cardiology.Copies: This document is available on the World Wide Web sites of the American College of Cardiology (www.acc.org), the American Heart
Association (www.americanheart.org), and the Society for Cardiovascular Angiography and Interventions (www.scai.org). For copies of this document,please contact Elsevier Inc. Reprint Department, fax (212) 633-3820, e-mail [email protected]. To purchase Circulation reprints, call 843-216-2533or e-mail [email protected].
Permissions: Multiple copies, modification, alteration, enhancement, and/or distribution of this document are not permitted without the expresspermission of the American College of Cardiology or the American Heart Association. Instructions for obtaining permission are located at http://www.americanheart.org/presenter.jhtml?identifier�4431. A link to the “Permission Request Form” appears on the right side of the page.
(Circulation. 2007;116:98-124.)© 2007 by the American College of Cardiology Foundation and the American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.107.185159
98
ACCF/AHA/SCAI Clinical Competence Statement

TABLE OF CONTENTS
Preamble . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .99Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .100
Purpose . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .101Writing Group Composition . . . . . . . . . . . . . . . . . . . . .101Literature Review. . . . . . . . . . . . . . . . . . . . . . . . . . . . . .101
Percutaneous Coronary Intervention . . . . . . . . . . . . . . . . .101Evolution of Competence and TrainingStandards . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .101Evolution of Coronary InterventionalCapabilities . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .102Procedural Success and Complications ofCoronary Interventional Procedures . . . . . . . . . . . . . . .102Patient, Lesion, and Institutional VariablesInfluencing Success and ComplicationRates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .103
Measures/Definitions of Success . . . . . . . . . . . . . . . .103Patient and Lesion Characteristics Relatedto Procedural Success and ComplicationRates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .104Strategies for Risk Stratification andOperator Evaluation . . . . . . . . . . . . . . . . . . . . . . . . . . . .104Impact of the Facility on ProceduralSuccess . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .104Components of Operator Competence . . . . . . . . . . . . .105
Cognitive Knowledge Base . . . . . . . . . . . . . . . . . . . .105Technical Skills . . . . . . . . . . . . . . . . . . . . . . . . . . . . .106Nonballoon Devices . . . . . . . . . . . . . . . . . . . . . . . . . .106
Relationships of Operator and InstitutionalExperience and Activity to Outcomes inCoronary Interventional Procedures . . . . . . . . . . . . . . .106
Evidence Reviewed . . . . . . . . . . . . . . . . . . . . . . . . . .106Relationship of Institutional Volume toProcedural Outcome. . . . . . . . . . . . . . . . . . . . . . . . . . . .107Volume and Outcomes Relationship forPrimary PCI in Acute MI . . . . . . . . . . . . . . . . . . . . . . .108
Relationship of Individual OperatorVolume to Procedural Outcome . . . . . . . . . . . . . . . .109
Combination of Individual OperatorVolume and Institutional Volume onProcedural Outcome. . . . . . . . . . . . . . . . . . . . . . . . . . . .110Ongoing Quality Improvement andMaintenance of Competence . . . . . . . . . . . . . . . . . . . . .111
Institutional Maintenance of Quality. . . . . . . . . . . . .111Individual Maintenance of Quality . . . . . . . . . . . . . .112
Quality Assurance . . . . . . . . . . . . . . . . . . . . . . . . . . . . .112Definition of Quality in PCI . . . . . . . . . . . . . . . . . . .112Institutional Quality AssuranceRequirement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .112Role of Risk Adjustment in AssessingQuality . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .112Challenges in Determining Quality . . . . . . . . . . . . . .112Requirement for Institutional Resourcesand Support . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .113
The Quality Assessment Process. . . . . . . . . . . . . . . .113Conclusions and Recommendationsfor PCIs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .113
Success and Complication Rates . . . . . . . . . . . . . . . .113Risk Adjustment. . . . . . . . . . . . . . . . . . . . . . . . . . . . .114Volume–Activity Relationships . . . . . . . . . . . . . . . . .114Recommendations for InstitutionalMaintenance of Quality . . . . . . . . . . . . . . . . . . . . . . .114Recommendations for IndividualMaintenance of Quality . . . . . . . . . . . . . . . . . . . . . . .114
Percutaneous Noncoronary Interventions . . . . . . . . . . . . .114Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .114Disorders of the Atrial Septum . . . . . . . . . . . . . . . . . . .115
Criteria for Competency . . . . . . . . . . . . . . . . . . . . . .115Cardiologists in Training Programs . . . . . . . . . . . . .115Cardiologists in Practice . . . . . . . . . . . . . . . . . . . . . .115Maintenance of Competency forPercutaneous ASD/PFO Closure . . . . . . . . . . . . . . . .115Quality Assurance . . . . . . . . . . . . . . . . . . . . . . . . . . .115
Hypertrophic Cardiomyopathy andAlcohol Septal Ablation . . . . . . . . . . . . . . . . . . . . . . . .116
Criteria for Competency . . . . . . . . . . . . . . . . . . . . . .116Valvular Heart Disease . . . . . . . . . . . . . . . . . . . . . . . . .116
Cognitive Knowledge Base . . . . . . . . . . . . . . . . . . . .116Criteria for Competency . . . . . . . . . . . . . . . . . . . . . .116Percutaneous Ventricular AssistDevices . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .116
Laboratory and Staff Competence. . . . . . . . . . . . . . . . .116Conclusions and Recommendations . . . . . . . . . . . . . . .117
Percutaneous NoncoronaryInterventions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .117
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .117Appendix 1. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .121Appendix 2. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .122
PreambleThe granting of clinical staff privileges to physicians is aprimary mechanism used by institutions to uphold the qualityof care. The Joint Commission on Accreditation of HealthCare Organizations requires that the granting of continuingmedical staff privileges be based on the criteria specified inthe medical staff bylaws. Physicians themselves are thuscharged with identifying the criteria that constitute profes-sional competence and with evaluating their peers accord-ingly. Yet, the process of evaluating physicians’ knowledgeand competence is often constrained by the evaluator’s ownknowledge and ability to elicit the appropriate information,problems compounded by the growing number of highlyspecialized procedures for which privileges are requested.
The American College of Cardiology Foundation/Amer-ican Heart Association/American College of Physicians(ACCF/AHA/ACP) Task Force on Clinical Compe-
King et al ACCF/AHA/SCAI Clinical Competence Statement 99

tence and Training was formed in 1998 to developrecommendations for attaining and maintaining the cogni-tive and technical skills necessary for the competent perfor-mance of a specific cardiovascular service, procedure, ortechnology. These documents are evidence based and,where evidence is not available, expert opinion is utilized toformulate recommendations. Indications and contraindica-tions for specific services or procedures are not included inthe scope of these documents. Recommendations are in-tended to assist those who must judge the competence ofcardiovascular health care providers entering practice for thefirst time and/or those who are in practice and undergoperiodic review of their practice expertise or who apply forprivileges at a new institution. The assessment of compe-tence is complex and multidimensional, therefore, isolatedrecommendations contained herein may not necessarily besufficient or appropriate for judging overall competence.The current document addresses competence in cardiacinterventional procedures and is authored by representativesof the ACCF, the AHA, and the Society for CardiovascularAngiography and Interventions (SCAI). This documentapplies to specialists trained in internal medicine and/oradult cardiology and is not meant to be a clinical compe-tence statement on procedures for congenital heart diseasein the child or young adult.
The ACCF/AHA/ACP Task Force makes every effort toavoid any actual or potential conflicts of interest that mightarise as a result of an outside relationship or personal interestof a member of the ACCF/AHA/ACP Writing Commit-tee. Specifically, all members of the Writing Committeewere asked to provide disclosure statements of all suchrelationships that might be perceived as real or potentialconflicts of interest relevant to the document topic. Thesestatements were reviewed by the Writing Committee andupdated as changes occurred. The relationships with indus-try for authors and peer reviewers are published in theappendices of the document.
Mark A. Creager, MD, FACC, FAHAChair, ACCF/AHA/ACP Task Force on
Clinical Competence and Training
Introduction
Coronary intervention has evolved from an investigationalprocedure to a widely practiced, mature mainstream clinicaltherapy (1). Conventional balloon angioplasty, while still acore procedure in interventional cardiology, has been aug-mented by adjunctive stenting, which greatly improvesprocedure efficacy and modestly reduces the risk of resteno-sis (2). Bare-metal stents have been replaced by drug-elutingstents in the majority of cases, which further reduce the riskof restenosis (3). Because stents or other interventionaldevices are commonly used, the coronary angioplasty pro-cedure is more aptly termed “percutaneous coronary inter-vention” (PCI).
The AHA estimated that more than 1,000,000 PCIswere performed in the United States in 2003 (4). Physiciansperforming these procedures represent approximately 25%of board-certified cardiologists in the United States (5).
As a result of the maturation of PCI as a discipline andthe ongoing clarification of its role in the management ofcoronary heart disease, the public can and should appropri-ately expect consistent access to high-quality PCI capability.However, there is potential for substantial variation in thequality of PCI services. PCI is often a complex, demandingprocedure. To perform PCI optimally, an operator mustpossess a substantial cognitive knowledge base as well asconsiderable technical skill. In addition, the technical diffi-culty of a particular procedure can vary greatly from onepatient to another. Furthermore, serious complications ofcoronary interventional procedures may occur unpredictablyin procedures that initially appear to be straightforward.Recognition and management of complications are criticalcomponents of PCI procedures that require skill, knowl-edge, experience, and judgment. Since there can be variationamong operators in cognitive knowledge and skill andamong procedures in technical difficulty, there is a potentialfor substantial variation in procedure safety and efficacy.
Credentialing physicians to perform procedures is theresponsibility of the governance of the local health carefacility. The Joint Commission on the Accreditation ofHealth Care Organizations requires that medical staff priv-ileges be granted to applicants only after assessment basedon professional criteria. Physicians are charged with theresponsibility to establish the criteria that constitute profes-sional competence and to evaluate their peers on the basis ofsuch criteria. The U.S. health care system relies, in part, onthis process of granting and renewing clinical privileges tomaintain quality.
The issue of determining quality standards and creden-tialing criteria has presented a major challenge to themedical profession. Developing standards has been difficultbecause, until recently, there were few data available onwhich to base them and because PCI techniques, indica-tions, and capability have evolved rapidly. During the pastseveral years, documents have been published that haveoffered guidelines and standards for the training and main-tenance of competence (6–15). Because of the paucity ofclinical data, the earlier standards were developed principallythrough observation, experience, and intuition. These stan-dards relied heavily on operator activity level as a surrogatefor skill and quality.
The most recent document published by the ACC wasbased on the information available in 1998 (16). Therecommendations of this and other similar documentsrequire updating as technology and training evolve (17).
Percutaneous noncoronary cardiac interventions, such asaortic and mitral valvuloplasty, atrial septal defect (ASD)and patent foramen ovale (PFO) closure, and alcohol septalablation therapy, were not addressed in the previous docu-ment (16). These procedures, although constituting a
100 Circulation July 3, 2007

small minority of interventional activity, are performedby interventional cardiologists and are included in theAccreditation Council on Graduate Medical Education(ACGME) curriculum and the American Board of In-ternal Medicine (ABIM) certifying exam. There havebeen no statements addressing clinical competence innoncoronary interventions.
The ACC, the ACP, the SCAI, the Society for VascularMedicine and Biology (SVMB), and the Society for Vas-cular Surgery (SVS) have jointly developed a document onacquisition and maintenance of competence in vascularmedicine and catheter-based vascular interventions (18);however, PCI and other percutaneous cardiac proceduresare not addressed by the current document. This documentis divided into 2 sections: PCI and percutaneous noncoro-nary cardiac interventions.
Purpose
This document was developed to review the currentlyavailable scientific data with the following purposes:
1. To characterize the expected success and complicationrates for coronary interventional procedures when per-formed by highly skilled operators.
2. To identify comorbidities and other risk factors that maybe used for risk adjustment when assessing procedure-specific expected success and complication rates.
3. To assess the relationship between operator activity leveland success rates in PCI procedures as assessed byrisk-adjusted outcome statistics.
4. To assess the relationship between institutional activitylevel and success rates in PCI procedures as assessed byrisk-adjusted outcome statistics.
5. To develop recommendations for standards to assessoperator proficiency and institutional program quality.These include standards for data collection to permitmonitoring of appropriateness and effectiveness of PCIprocedures both at the level of the operator and theinstitution.
6. To expand the scope of this competency document,previously limited to coronary procedures, to also includenoncoronary cardiac interventions.
Writing Group Composition
The Writing Group was selected to represent a broad rangeof experience and expertise to bear on this issue. Themembers of the Writing Group were identified on the basisof 1 or more of the following attributes: PCI operators witha broad range of experience (in practice and in academicsettings); individuals who have performed clinical researchstudying the outcome of PCI procedures; individuals whodirect catheterization laboratories with a broad cross sectionof interventional operators; and individuals with broadclinical experience who have had considerable previousinvolvement with PCI.
Literature Review
A literature search was conducted with 5 goals:
1. To identify published coronary and other cardiac inter-ventional outcomes data that could be used as bench-marks for quality assessment. In addition, the processsought to identify those risk adjustment variables thataffect the likelihood of success and complications. Thisreview focused on outcomes of coronary interventions,including the latest interventional devices as of the dateof this revision.
2. To identify data that examines the relationships betweenoperator and institutional experience, and activity levels,and their impact on procedural success and complicationrates.
3. To assess the issues and problems associated with judg-ing operator and institutional proficiency based on out-come statistics—in particular, the challenge of accuratelyassessing the performance of low-volume operators andinstitutions.
4. To expand the recommendations beyond coronary inter-ventions to other cardiac interventional procedures.
5. To identify methods for monitoring appropriateness ofperformance of PCI.
Percutaneous Coronary Intervention
Evolution of Competence and Training Standards
Initially, because experience was limited, the coronary an-gioplasty technique was disseminated informally amongphysicians who were highly experienced at diagnostic car-diac catheterization. During this period, physicians acquiredangioplasty skills through “on-the-job” experience, and nostandards existed for either training requirements or fordemonstration of competence.
As the coronary angioplasty knowledge base grew andtechniques evolved, standards were developed for training(19). Formal angioplasty training programs were first orga-nized in the early 1980s. The most recent recommendationswere published by the ACC in 1999 (20). The ABIMdeveloped an Examination in Interventional Cardiologythat was first administered in 1999. As of 2005, 5,020physicians had successfully passed the examination andbecome board certified in interventional cardiology. Cur-rently, eligibility to sit for the ABIM interventional cardi-ology examination requires completion of a fourth-yearfellowship in interventional cardiology in an ACGME-accredited program. During academic year 2004 to 2005,there were 122 accredited interventional cardiology pro-grams in the United States that had 240 filled trainingpositions.
Professional organizations have addressed the issue ofstandards and criteria for proficiency in PCI proceduressince 1986, with an increasing focus on the issue of
King et al ACCF/AHA/SCAI Clinical Competence Statement 101

maintenance of proficiency and skills (6–15). These docu-ments have universally endorsed an annual caseload goal formaintenance of proficiency. The most commonly endorsedactivity level has been 75 procedures per year per operator.This standard was initially based on general consensus ofexperts. In recent years, considerable research has examinedthe volume–outcome relationship and, in general, has af-firmed it (21,22).
Since the previous guidelines were published, there hasbeen debate over the relationship between volume andquality. While a relationship between volume and outcomesexists, volume alone does not determine quality. Also, theABIM interventional cardiology board exam has beenestablished to certify a level of knowledge and experience inthe field. This competency document addresses these factorsas they relate to determinations of overall operator andinstitutional quality.
Evolution of Coronary Interventional Capabilities
The cognitive and technical knowledge base required forproficiency in PCI has expanded. The fundamental con-cepts of coronary angioplasty technique, namely the coaxialguide catheter and the dilation catheter with a minimallycompliant cylindrical balloon, were formulated by AndreasGruntzig (23). Because of the initial comparatively primitiveequipment design and capability, coronary angioplasty wasonly applicable to readily accessible discrete proximal coro-nary stenoses. Subsequent refinement in instrumentationhas greatly enhanced procedural success and extended theindications for the performance of PCI. Complex anatomicsituations now considered technically suitable for PCIprocedures include multivessel disease (24–30), distal andbifurcation stenoses, total occlusions (31), saphenous veingraft stenoses (32), and complex stenoses. Challengingclinical situations now considered appropriate for coronaryintervention include patients with unstable angina (33,34)and myocardial infarction (MI) (35,36) and those who arenot considered candidates for coronary bypass surgery.
Nonballoon devices, including coronary stents, and direc-tional, rotational, and laser atherectomy devices, have beenintroduced. These devices augment conventional balloonangioplasty and extend its capability; however, they allrequire specific training and mentoring by a previouslyexperienced operator. To become competent in the use ofany of these newer interventional devices, an operator mustacquire the additional knowledge and technical skills spe-cific to each device.
A number of adjunctive antithrombotic and antiplateletmedications have been introduced for the purpose of reduc-ing acute thrombus-related treatment site complications.Understanding the appropriate indications for and compli-cations associated with the use of these medications, whichare powerful anticoagulants, requires knowledge of hemo-static mechanisms.
Procedural Success and Complicationsof Coronary Interventional Procedures
Recent clinical studies have demonstrated that despite acontinuing increase in clinical and angiographic complexity,procedural and clinical success rates have remained high andcomplication rates have remained low (37–45) (Table 1).Angiographic success occurs in over 95% of patients.Among patients without ST-segment elevation myocardialinfarction (STEMI), PCI is associated with an averagemortality rate of less than 1%, a Q-wave MI rate of less than1%, and an emergency coronary artery bypass surgery(CABG) rate of less than 1%. Table 1 contains data from 5large contemporary registries of PCI procedures and thefirst 2 National Heart, Lung, and Blood Institute (NHLBI)registries for historical comparison. These data constitute apoint of departure for developing benchmarking standards.
Adverse events related to PCI procedures are categorizedeither by the mechanism of the complication or by theadverse event caused by the procedure. A given adverseevent, such as death, may be caused by a variety ofcomplications.
Complications can be divided into 3 mechanistic categories:
1. Coronary vascular injury. Coronary arterial injury canoccur when devices are introduced into coronary vesselsor result from embolization of thrombotic or atheroscle-rotic material from devices or vessel walls. Examplesinclude coronary dissection, thrombosis, perforation, andembolization.
2. Other vascular events. Other vascular events are causedeither by injury to a peripheral vessel by catheter inser-tion, manipulation, or removal, or by embolization ofthrombotic or atherosclerotic material. Examples includepseudoaneurysm, retroperitoneal hemorrhage, arterio-venous fistula, and stroke.
3. Systemic nonvascular events. Systemic nonvascular ad-verse events are caused by the procedure but are not dueto vascular injury. They include all the systemic hazardsof cardiovascular radiographic angiography procedures.Examples include contrast agent-induced nephropathyand acute pulmonary vascular congestion.
For the purpose of assessing clinical competence, com-plications may be divided into 8 basic outcome categories:
1. Death: related to the procedure, regardless of mechanism2. Stroke3. MI: related to the procedure, regardless of mechanism4. Ischemia requiring emergency CABG: either as a result
of procedure failure or a procedure complication5. Vascular access site complications6. Contrast agent nephropathy7. Excessive bleeding, requiring treatment8. Other (such as coronary perforation and tamponade)
The first 4 of these categories are generally consideredmajor adverse cardiac and cerebral events (MACCE). Be-
102 Circulation July 3, 2007

cause adverse events are definite end points, they are easilyrecognized and captured for statistical summary purposes.The ACC-National Cardiovascular Data Registry(NCDR)® has developed a comprehensive data dictionarywith rigorous definitions of recognized adverse events (46).It may be impossible to determine conclusively whetherdeath or a complication was caused by a procedure. None-theless, for the purposes of monitoring performance, rate ofcomplications or deaths substantially above that expected,after adjustment for patient risk factors, is a cause forconcern.
Patient, Lesion, and Institutional VariablesInfluencing Success and Complication Rates
A number of factors have improved the overall success andcomplication rates of PCI procedures. These include in-creased operator experience, modifications in conventionalinstrumentation (balloon catheters, guide catheters, guidewires), newer interventional devices (stents and emboliza-tion protection devices), and advances in adjunctive phar-macologic therapy. Concurrently, these improvements haveled to the extension of interventional treatment to higher-risk patients with more complex coronary anatomy andcomorbid disease. These factors have influenced overallacute and long-term outcome associated with PCI procedures.
Measures/Definitions of Success
Anatomic success. The definition of anatomic successfocuses exclusively on the enlargement of the lumen at thetarget site and blood flow through the epicardial coronaryartery. Although there has been disagreement, the currentdefinition of success of PCI with stenting is the achieve-ment of a minimal diameter stenosis of less than 20% asvisually assessed by angiography and maintenance ofThrombolysis In Myocardial Infarction (TIMI) flow grade3 (15). Anatomic success of PCI without stenting is definedas stenosis diameter reduction greater than 20% with resid-ual stenosis less than 50%. Notably, there is frequently adisparity between the visual estimate of lumen diameter andquantitative measurements (47,48).
Procedural success. Procedural success has been defined asthe achievement of anatomic success of all treated lesionswithout the major complications of death, MI, or emer-gency CABG (14,40). Although emergency CABG duringhospitalization and death are easily identified end points,the definition of periprocedural MI has been more prob-lematic. Some definitions require the development of Qwaves in addition to a threshold value for creatine kinase(CK) elevation. However, more recent reports have includednon-STEMIs with CK elevations greater than 3 or 5 timesthe upper limit of normal as clinically significant, since theyhave been shown to correlate with long-term mortality (49).Although major adverse cardiac events (MACE) have beenTa
ble
1.
Cha
nges
inC
oron
ary
Inte
rven
tion
alP
ract
ice
and
Out
com
eFr
omR
egis
try
Dat
a
Var
iabl
eN
HLB
I-1(4
0)
NH
LBI-2
(38,3
9)
NH
LBI
Dyn
amic
Reg
istr
y(4
1)
AC
CN
atio
nal
Car
diov
ascu
lar
Dat
aR
egis
try
(42
)
Nor
ther
nN
ewEn
glan
dC
onso
rtiu
m(4
3)
Mic
higa
nB
lue
Cro
ssC
onso
rtiu
m(4
4)
New
Yor
kSta
teR
egis
try
(45)
Non
emer
gent
Emer
gent
Clin
ical
Cha
ract
eris
tics
Year
sof
entr
y1
97
7–1
98
11
98
5–1
98
61
99
7–2
00
21
99
8–2
00
52
00
0–2
00
42
00
22
00
1–2
00
3
No.
ofpa
tient
s1
,15
51
,80
26
,18
31
,08
2,6
90
36
,83
15
,90
11
24
,09
61
4,9
46
Ste
ntus
e(%
)0
07
89
1.6
86
84
.08
7.5
92
.7
Mea
npa
tient
age
(yrs
)5
45
86
36
16
26
36
56
0
Uns
tabl
ean
gina
(%)
37
49
44
35
43
32
.02
7.8
83
.6
ST-
segm
ent
elev
atio
nM
I(%
)0
02
51
31
21
8.9
05
3
Suc
cess
and
Com
plic
atio
nIn
dica
tors
Ang
iogr
aphi
csu
cces
s(%
)6
89
19
32
00
5:s
tent
edle
sion
s:9
9;
nons
tent
edle
sion
s:8
69
4N
A9
7.5
97
.5
Emer
genc
yCA
BG
(%)
5.8
03
.40
1.0
00
.40
.40
.51
0.2
00
.54
Mor
talit
y(%
)1
.21
.01
.33
1.2
unad
just
edra
te1
.17
1.2
70
.36
3.2
5
Rep
rinte
dw
ithpe
rmis
sion
from
Sm
ithS
CJr
.,Fe
ldm
anTE
,H
irshf
eld
JWJr
.,et
al.
ACC
/AH
A/S
CAI
2005
guid
elin
eup
date
for
perc
utan
eous
coro
nary
inte
rven
tion—
sum
mar
yar
ticle
:a
repo
rtof
the
Amer
ican
Col
lege
ofC
ardi
olog
y/Am
eric
anH
eart
Asso
ciat
ion
Task
Forc
eon
Prac
tice
Gui
delin
es(A
CC
/AH
A/S
CAI
Writ
ing
Com
mitt
eeto
Upd
ate
the
2001
Gui
delin
esfo
rPe
rcut
aneo
usC
oron
ary
Inte
rven
tion)
.J
AmC
ollC
ardi
ol2006;4
7:2
16
–35
(15).
ACC
�Am
eric
anC
olle
geof
Car
diol
ogy;
CAB
G�
coro
nary
arte
ryby
pass
graf
tsu
rger
y;M
CD
�m
ultic
ente
rda
taba
se;
MI
�m
yoca
rdia
linf
arct
ion;
NA
�no
tav
aila
ble;
NH
LBI
�N
atio
nalH
eart
,Lu
ng,
and
Blo
odIn
stitu
te.
King et al ACCF/AHA/SCAI Clinical Competence Statement 103

used to judge success, some recent studies also includeMACCE.
Short-term clinical success. Short-term clinical successrequires, in addition to procedural success, the relief of signsand symptoms of myocardial ischemia.
Longer-term clinical success. Longer-term clinical successrequires that the initial clinical success remains durable andthat the patient has persistent relief of signs and symptomsof myocardial ischemia for 6 to 9 months after the proce-dure. Restenosis remains the principal cause of a lack ofclinical success over the first year following a successfulprocedure. This directly leads to target lesion revasculariza-tion (TLR), target vessel revascularization, and target vesselfailure. Thereafter, clinical events are usually caused byprogression of disease at other sites. Clinically importantrestenosis may be judged by the frequency with whichsubsequent TLR procedures are performed after the indexprocedure. Incomplete revascularization, new lesion forma-tion, and stent thrombosis may also limit long-term clinicalsuccess, especially in subsequent years (50).
Patient and Lesion Characteristics Related toProcedural Success and Complication Rates
Angioplasty procedural success and complication rates areinfluenced by a variety of patient and target lesion charac-teristics. These characteristics must be taken into consider-ation through risk adjustment when assessing adverse eventrates. In addition, they must also be weighed in determiningprocedure appropriateness.
Patient clinical characteristics. The clinical factors associ-ated with an increased risk of an adverse outcome afterintervention include advanced age, female gender, acutecoronary syndrome (especially STEMI), chronic renal in-sufficiency, heart failure, and multivessel coronary disease(7,12,14,15). Patients with impaired renal function, partic-ularly patients with diabetes, are at increased risk forcontrast-induced nephropathy (51).
Target lesion anatomic factors. Particular lesion morpho-logic characteristics are predictive of immediate outcomewith coronary intervention (7,12,14,52). Lesion length,presence of thrombus, and degenerated saphenous veingrafts are independently associated with abrupt vessel clo-sure and major ischemic complications. Chronic total oc-clusions (greater than or equal to 3 months) are associatedwith a lower procedural success rate. On the basis of theseobservations, a previous ACC/AHA Clinical Task Force onClinical Privileges in Cardiology (13) proposed a classifica-tion scheme based on lesion morphology to estimate thelikelihood of procedural success and complications. Thisscheme was subsequently modified by others (52) and hasserved as a useful guide for assessing the risk of an adverseoutcome associated with a particular lesion. More recentexperience indicates that improved devices and techniques
have higher success rates in more complex lesions (53–56).As a result, lesion morphology may be less predictive ofcomplications currently than it has been in the past (57).
Strategies for Risk Stratificationand Operator Evaluation
Several large retrospective studies of patients undergoingPCI have identified clinical and angiographic characteristicsthat correlate with procedural success, in-hospital morbid-ity, and mortality (21,22,44) (Table 2). These observationshave been used to develop multivariate logistic regressionmodels that can stratify patients before the procedure.Model reliability is best assessed by relative predictiveaccuracy (C-statistic: moderate is greater than 0.80, excel-lent is greater than 0.90) and scaling accuracy (the Hosmer-Lemeshow statistic). Several models predict periproceduralmortality with C-statistic greater than 0.80 (Table 2).Prediction of other events is typically less accurate (58–60).Model utility also must consider the frequency and clinicalimportance of the event measured. Very infrequently occur-ring events, even if severe, may not allow adequate evalua-tion of operators with low volume. Results of several years ofexperience should be considered in order to have sufficientnumbers of events to be adequately assessed from a statis-tical standpoint. Operators and catheterization laboratoriesshould be encouraged to submit information to large data-bases that allow for evaluation of risk-adjusted outcomes.
Impact of the Facility on Procedural Success
Physical facility requirements. The physical facility inwhich interventional procedures are performed has an im-portant impact on procedural success. The facility mustprovide radiologic equipment, monitoring, and patient sup-port equipment to enable operators to perform at the best oftheir ability. The video and “cine” image quality of radio-logic imaging equipment must be optimal to facilitateaccurate catheter and device placement and enable properassessment of procedure results. Physiologic monitoringequipment must provide continuous, accurate informationabout the patient’s condition. Requisite support equipmentmust be available and in good operating order to respond toemergency situations.
Overall institutional system requirements. The interven-tional laboratory must have an extensive support system ofspecifically trained laboratory personnel. Cardiothoracicsurgical, respiratory, and anesthesia services should be avail-able to respond to emergency situations in order to mini-mize detrimental outcomes. The institution should havesystems for credentialing, governance, data gathering, andquality assessment. Prospective, unbiased collection of keydata elements on consecutive patients and consistent feed-back of results to providers brings important quality controlto the entire interventional program. The ACC/AHA/SCAI 2005 Guideline Update for PCI (15) recommendsthat each interventional program performing elective PCI
104 Circulation July 3, 2007
should have in-house surgical support. Institutions that donot have in-house surgical support and are performingprimary PCI only for STEMI, should have an established,well-organized system for emergency transfer to surgery atanother institution.
Components of Operator Competence
Cognitive Knowledge Base
The knowledge needed to perform PCI, including thatexpected to be acquired in ACGME-approved interven-tional training programs, has been addressed by expertpanels (7,8,20,67,68). The core knowledge is now tested bythe ABIM Interventional Cardiology certifying examina-tion which has been administered since 1999. Through2003, physicians trained by a nontraditional pathway wereeligible to take the examination based on either practice-based procedure activity and experience or by completion ofan interventional training program. Since 2003, only indi-viduals who have completed an ABIM-qualified training
program are eligible to take the certifying examination.Individuals who train in interventional cardiology shouldbecome ABIM certified in interventional cardiology.
Training programs and the qualifying examination(20,69) require that interventional cardiologists be knowl-edgeable in anatomy, physiology, and pathophysiology ofthe cardiovascular system. In particular, one should under-stand the biology of coronary artery disease, be knowledge-able about the pathophysiology of myocardial ischemia andMI, and understand the dynamics of cardiac dysfunction.Interventionalists should possess a fundamental knowledgeof stents and be familiar with the polymers and drugs thatare incorporated into stents, coagulation cascade, thrombo-sis, and the pharmacology, therapeutic application, and risksof antiplatelet, antithrombin, and fibrinolytic drugs that areused in association with PCI. Competent operators musthave knowledge of the indications for PCI and adjunctiveand alternative use of medical therapy and surgery forpatients with coronary artery disease based on an in-depth
Table 2. Odds Ratios* for Significant Independent Risk Factors† for Short-Term Mortality Related to PCI
SourceNew York
StateNorthern
New EnglandMichigan
BMC2 ACC-NCDR ACC-NCDR Update COAPNo. of patients 50,046 15,331 10,796 100,253 No acute MI
(142,817)Acute MI(30,926)
19,358
Incidence (%) 0.58 1.1 1.6 1.4 N/A N/A 1.6
Years 2003 1994–1996 1997–1999 1998–2000 1998–2001 1998–2001 1999–2000
Clinical
Acute MI less than 12–24 h 8.6 5.5 2.8 1.3 �
Age � � � � � � �
Cardiac arrest 3.7
CHF 3.2 8.6 1.6
COPD 1.3 1.7 1.5 1.8
Diabetes 1.4 1.25
Female 1.5 1.8 1.4
IABP pre 26.2 1.7 1.9
Peripheral vascular disease 2.6 3.3 1.6 1.6
Prior CABG 1.4
Priority (salvage, emergent urgent, elective) � � � � �
Renal insufficiency 3.1 6.4 5.5 3.0 3.5 2.0 3.5
Shock 22.1 32.2 11.5 8.5 9.8 8.8 9.8
Anatomic
ACC lesion score, C 2.9
Ejection fraction � � � � � �
LMT lesion 2.0 1.5 2.1
Number of diseased vessels � �
Prox LAD lesion 2.0 1.3 1.3 �
SCAI lesion score � � �
Thrombus �
Procedural
Lytic use 1.4 1.25
Nonstent use 1.6 1.6 1.4
C-statistic 0.905 0.88 0.90 0.89 0.85 0.87 0.87
*Values are odds ratios for binary variables unless otherwise noted; †specific definitions of risk factors may vary from series to series; �relationship exists for continuous or ordinal variables (61–66).ACC-NCDR � American College of Cardiology National Cardiovascular Data Registry; BMC2 � Blue Cross Blue Shield of Michigan Cardiovascular Consortium; CABG � coronary artery bypass graft;
CHF � congestive heart failure; COAP � clinical outcome assessment program; COPD � chronic obstructive pulmonary disease; IABP � intra-aortic balloon pump; LAD � left anterior descending; LMT� left main trunk; MI � myocardial infarction; PCI � percutaneous coronary intervention; SCAI � Society for Cardiovascular Angiography and Interventions.
King et al ACCF/AHA/SCAI Clinical Competence Statement 105

understanding of published clinical trials. Coronary inter-ventionalists must understand the role of primary angio-plasty compared with fibrinolytic therapy for STEMI andthe alternative therapeutic approaches for treating STEMIthat depend upon the time of presentation, anticipateddoor-to-balloon time, and the presence or absence ofongoing symptoms and/or electrocardiographic abnormalities.
Cognitive knowledge must be bolstered by clinical skillsand experience that support the rational selection of optimaltreatment strategies for each patient. Such decisions arebased on symptoms, anatomy, and associated risk factors.Thus, equally important to knowing the indications for PCIis an understanding of its limitations and contraindications,particularly as these relate to comorbid systemic diseases andspecial anatomical subsets. Physicians performing theseprocedures should be conversant with the applicable guide-lines (e.g., PCI, CABG, STEMI, unstable angina/NSTEMI [15,70–72]).
Coronary interventionalists must also have a thoroughknowledge of specialized equipment, techniques, and de-vices used to perform PCI competently, including:
1. The theoretical and practical aspects of X-ray imaging,radiation physics and safety, and other equipment togenerate digital images; quality control of images; imagearchiving; consequences of exposure of patients andpersonnel to ionizing radiation; and methods of reducingpatient and staff radiation exposure (73).
2. Specialized catheterization recording and safety equip-ment (physiological data recorders, pressure transducers,blood gas analyzers, defibrillators) (74).
3. Catheters, guide wires, balloon catheters, stents,atherectomy devices, ultrasound catheters, intra-aorticballoon pumps, puncture site sealing devices, contrastagents, distal protection devices, and thrombus extrac-tion devices.
Operators must be knowledgeable about the prevention,prompt recognition, and treatment of procedural complica-tions. It is extremely important to have the knowledge andskills to diagnose and manage vessel perforation, no reflow,coronary dissection, expanding hematoma, pseudoaneu-rysm, arterial venous fistulas, and retroperitoneal hemor-rhage. Interventionalists must also be cognizant of systemiccomplications, including cerebrovascular events andcontrast-related nephropathy.
Technical Skills
Many of the skills required to perform coronary interven-tional procedures are closely related to those needed toperform diagnostic cardiac catheterization and coronaryangiography. These include manual dexterity and the abilityto obtain percutaneous arterial and venous access andmaintain sterile surgical technique.
Most of the other required technical skills are unique tocoronary interventional procedures and can only be acquired
during training and by performing actual procedures underthe direction of an experienced interventionalist. Theseinclude the manipulation and operation of guide catheters,coronary angioplasty guide wires, coronary angioplasty bal-loon catheters, specialized atherectomy devices, stents, andintracoronary ultrasound catheters. Such training appro-priately occurs in standardized training programs that areACGME-approved and lead to eligibility for boardcertification.
Nonballoon Devices
A special area of competence involves use of lesion assess-ment tools. Intracoronary devices commonly used by inter-ventional cardiologists for assessment of intraluminal coro-nary anatomy and/or physiology include intravascularultrasound (IVUS) or intracoronary ultrasound (ICUS),Doppler flow wires, and pressure wires. Competency in theuse of angioscopy, optical coherence tomography, spectros-copy, intravascular thermography, and intravascular mag-netic resonance imaging is beyond the scope of this docu-ment. Expertise in device manipulation and imageinterpretation is required to use these intravascular assess-ment devices safely and effectively. The risks of these devicesis the same as those with PCI and include vessel spasm;myocardial ischemia; coronary artery dissection; plaquedisruption; thrombosis; air, plaque, or thrombotic emboli-zation; acute occlusion; coronary artery perforation; andcontrast nephropathy, stroke, and access site complications.Therefore, only an interventional cardiologist skilled intransluminal coronary techniques such as balloon angio-plasty and stenting who is able to diagnose and treatcomplications of interventional procedures should employthese devices. Recommendations regarding the use ofIVUS, Doppler flow wires, and pressure wires are publishedin Appendix C of the ACC/AHA Guidelines for CoronaryAngiography (75).
It is also important to ensure quality image acquisition,measurement, and reporting for each of the intravascularassessment devices. For ICUS, the reader is referred to theACC Clinical Expert Consensus Document on Standardsfor Acquisition, Measurement and Reporting of Intravas-cular Ultrasound Studies (76). No such documents areavailable for Doppler analysis of coronary flow reserve andpressure wire analysis of fractional flow reserve, but many ofthe general principles in the IVUS document may be ofsome benefit in guiding appropriate use of these othermodalities.
Relationships of Operator and InstitutionalExperience and Activity to Outcomesin Coronary Interventional Procedures
Evidence Reviewed
Computerized literature searches of English language pub-lications, review of recent abstract publications, and solici-tation of manuscripts under review for publication from
106 Circulation July 3, 2007
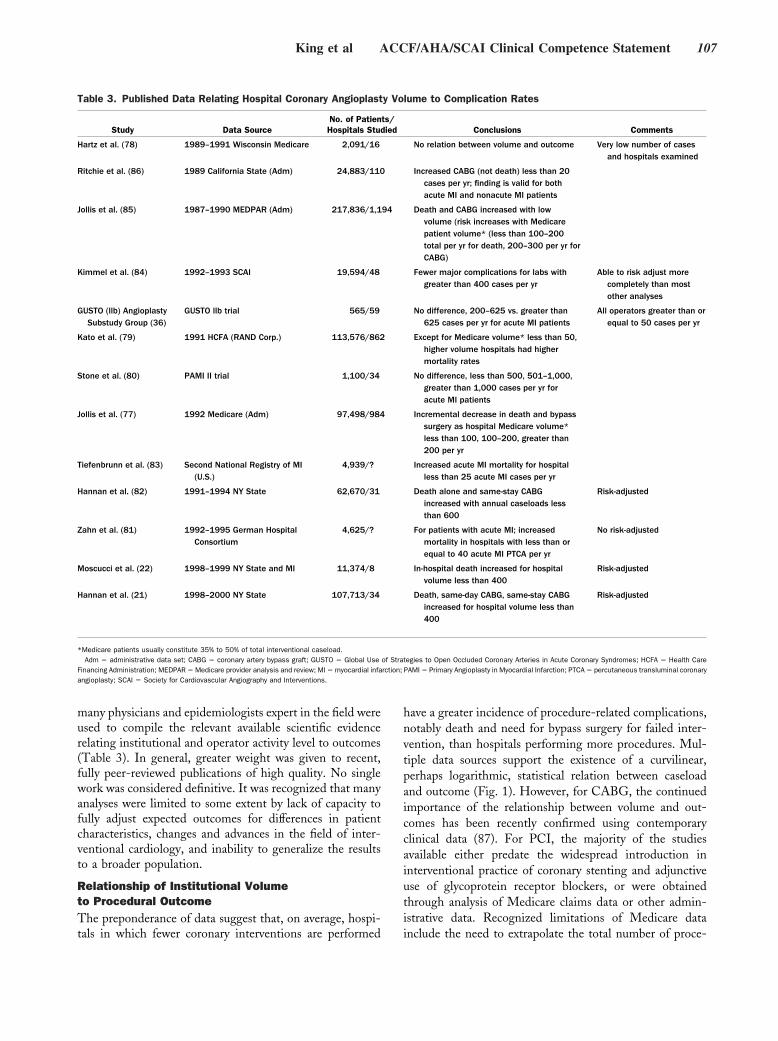
many physicians and epidemiologists expert in the field wereused to compile the relevant available scientific evidencerelating institutional and operator activity level to outcomes(Table 3). In general, greater weight was given to recent,fully peer-reviewed publications of high quality. No singlework was considered definitive. It was recognized that manyanalyses were limited to some extent by lack of capacity tofully adjust expected outcomes for differences in patientcharacteristics, changes and advances in the field of inter-ventional cardiology, and inability to generalize the resultsto a broader population.
Relationship of Institutional Volumeto Procedural OutcomeThe preponderance of data suggest that, on average, hospi-tals in which fewer coronary interventions are performed
have a greater incidence of procedure-related complications,notably death and need for bypass surgery for failed inter-vention, than hospitals performing more procedures. Mul-tiple data sources support the existence of a curvilinear,perhaps logarithmic, statistical relation between caseloadand outcome (Fig. 1). However, for CABG, the continuedimportance of the relationship between volume and out-comes has been recently confirmed using contemporaryclinical data (87). For PCI, the majority of the studiesavailable either predate the widespread introduction ininterventional practice of coronary stenting and adjunctiveuse of glycoprotein receptor blockers, or were obtainedthrough analysis of Medicare claims data or other admin-istrative data. Recognized limitations of Medicare datainclude the need to extrapolate the total number of proce-
Table 3. Published Data Relating Hospital Coronary Angioplasty Volume to Complication Rates
Study Data SourceNo. of Patients/Hospitals Studied Conclusions Comments
Hartz et al. (78) 1989–1991 Wisconsin Medicare 2,091/16 No relation between volume and outcome Very low number of casesand hospitals examined
Ritchie et al. (86) 1989 California State (Adm) 24,883/110 Increased CABG (not death) less than 20cases per yr; finding is valid for bothacute MI and nonacute MI patients
Jollis et al. (85) 1987–1990 MEDPAR (Adm) 217,836/1,194 Death and CABG increased with lowvolume (risk increases with Medicarepatient volume* (less than 100–200total per yr for death, 200–300 per yr forCABG)
Kimmel et al. (84) 1992–1993 SCAI 19,594/48 Fewer major complications for labs withgreater than 400 cases per yr
Able to risk adjust morecompletely than mostother analyses
GUSTO (llb) AngioplastySubstudy Group (36)
GUSTO llb trial 565/59 No difference, 200–625 vs. greater than625 cases per yr for acute MI patients
All operators greater than orequal to 50 cases per yr
Kato et al. (79) 1991 HCFA (RAND Corp.) 113,576/862 Except for Medicare volume* less than 50,higher volume hospitals had highermortality rates
Stone et al. (80) PAMI II trial 1,100/34 No difference, less than 500, 501–1,000,greater than 1,000 cases per yr foracute MI patients
Jollis et al. (77) 1992 Medicare (Adm) 97,498/984 Incremental decrease in death and bypasssurgery as hospital Medicare volume*less than 100, 100–200, greater than200 per yr
Tiefenbrunn et al. (83) Second National Registry of MI(U.S.)
4,939/? Increased acute MI mortality for hospitalless than 25 acute MI cases per yr
Hannan et al. (82) 1991–1994 NY State 62,670/31 Death alone and same-stay CABGincreased with annual caseloads lessthan 600
Risk-adjusted
Zahn et al. (81) 1992–1995 German HospitalConsortium
4,625/? For patients with acute MI; increasedmortality in hospitals with less than orequal to 40 acute MI PTCA per yr
No risk-adjusted
Moscucci et al. (22) 1998–1999 NY State and MI 11,374/8 In-hospital death increased for hospitalvolume less than 400
Risk-adjusted
Hannan et al. (21) 1998–2000 NY State 107,713/34 Death, same-day CABG, same-stay CABGincreased for hospital volume less than400
Risk-adjusted
*Medicare patients usually constitute 35% to 50% of total interventional caseload.Adm � administrative data set; CABG � coronary artery bypass graft; GUSTO � Global Use of Strategies to Open Occluded Coronary Arteries in Acute Coronary Syndromes; HCFA � Health Care
Financing Administration; MEDPAR � Medicare provider analysis and review; MI � myocardial infarction; PAMI � Primary Angioplasty in Myocardial Infarction; PTCA � percutaneous transluminal coronaryangioplasty; SCAI � Society for Cardiovascular Angiography and Interventions.
King et al ACCF/AHA/SCAI Clinical Competence Statement 107
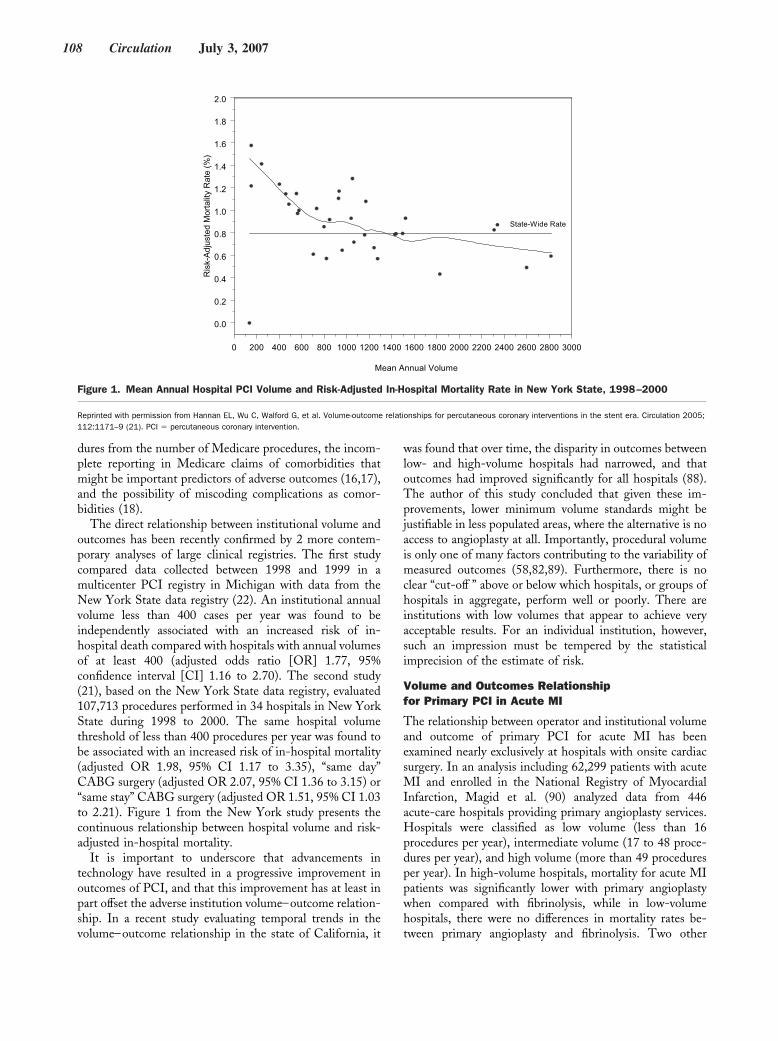
dures from the number of Medicare procedures, the incom-plete reporting in Medicare claims of comorbidities thatmight be important predictors of adverse outcomes (16,17),and the possibility of miscoding complications as comor-bidities (18).
The direct relationship between institutional volume andoutcomes has been recently confirmed by 2 more contem-porary analyses of large clinical registries. The first studycompared data collected between 1998 and 1999 in amulticenter PCI registry in Michigan with data from theNew York State data registry (22). An institutional annualvolume less than 400 cases per year was found to beindependently associated with an increased risk of in-hospital death compared with hospitals with annual volumesof at least 400 (adjusted odds ratio [OR] 1.77, 95%confidence interval [CI] 1.16 to 2.70). The second study(21), based on the New York State data registry, evaluated107,713 procedures performed in 34 hospitals in New YorkState during 1998 to 2000. The same hospital volumethreshold of less than 400 procedures per year was found tobe associated with an increased risk of in-hospital mortality(adjusted OR 1.98, 95% CI 1.17 to 3.35), “same day”CABG surgery (adjusted OR 2.07, 95% CI 1.36 to 3.15) or“same stay” CABG surgery (adjusted OR 1.51, 95% CI 1.03to 2.21). Figure 1 from the New York study presents thecontinuous relationship between hospital volume and risk-adjusted in-hospital mortality.
It is important to underscore that advancements intechnology have resulted in a progressive improvement inoutcomes of PCI, and that this improvement has at least inpart offset the adverse institution volume–outcome relation-ship. In a recent study evaluating temporal trends in thevolume–outcome relationship in the state of California, it
was found that over time, the disparity in outcomes betweenlow- and high-volume hospitals had narrowed, and thatoutcomes had improved significantly for all hospitals (88).The author of this study concluded that given these im-provements, lower minimum volume standards might bejustifiable in less populated areas, where the alternative is noaccess to angioplasty at all. Importantly, procedural volumeis only one of many factors contributing to the variability ofmeasured outcomes (58,82,89). Furthermore, there is noclear “cut-off ” above or below which hospitals, or groups ofhospitals in aggregate, perform well or poorly. There areinstitutions with low volumes that appear to achieve veryacceptable results. For an individual institution, however,such an impression must be tempered by the statisticalimprecision of the estimate of risk.
Volume and Outcomes Relationshipfor Primary PCI in Acute MI
The relationship between operator and institutional volumeand outcome of primary PCI for acute MI has beenexamined nearly exclusively at hospitals with onsite cardiacsurgery. In an analysis including 62,299 patients with acuteMI and enrolled in the National Registry of MyocardialInfarction, Magid et al. (90) analyzed data from 446acute-care hospitals providing primary angioplasty services.Hospitals were classified as low volume (less than 16procedures per year), intermediate volume (17 to 48 proce-dures per year), and high volume (more than 49 proceduresper year). In high-volume hospitals, mortality for acute MIpatients was significantly lower with primary angioplastywhen compared with fibrinolysis, while in low-volumehospitals, there were no differences in mortality rates be-tween primary angioplasty and fibrinolysis. Two other
0 200 400 600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000
Mean Annual Volume
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
Ris
k-A
djus
ted
Mor
talit
yR
ate
(%)
State-Wide Rate
Figure 1. Mean Annual Hospital PCI Volume and Risk-Adjusted In-Hospital Mortality Rate in New York State, 1998–2000
Reprinted with permission from Hannan EL, Wu C, Walford G, et al. Volume-outcome relationships for percutaneous coronary interventions in the stent era. Circulation 2005;112:1171–9 (21). PCI � percutaneous coronary intervention.
108 Circulation July 3, 2007

analyses from the same registry and 2 studies using the NewYork State data registry have shown a direct relationshipbetween hospital volume of primary angioplasty and mor-tality. In the analysis by Canto et al. (91), hospital volumewas divided in quartiles. In-hospital mortality was 28%lower in patients treated in the highest volume quartile(greater than 33 primary PCIs per year) when comparedwith patients treated in the lowest volume quartile (less than12 primary PCIs per year). Similar results were obtained byCannon et al. (92). In this analysis, a procedure volumegreater than 3 PCIs per month was found to be associatedwith a lower in-hospital mortality rate when compared witha procedure volume of less than 1 PCI per month, or witha procedure volume between 1 and 3 PCIs per month.
Recently, Hannan et al. (21) reported data from the NewYork State Coronary Angioplasty Reporting System Regis-try collected in the years 1998 to 2000, a period whenstenting was used in a large majority of the STEMIpatients. A trend toward an increased odds ratio of in-hospital mortality was observed for low-volume operatorswhen compared with high-volume operators both for avolume cut of 8 procedures per year (OR 1.40, 95% CI 0.89to 2.20) and with a volume cut of 10 procedures per year(OR 1.27, 95% CI 0.87 to 1.87). Importantly, a significantincrease in the odds of in-hospital mortality was observedwith lower institutional volume of primary PCI, regardlessof whether the institutional volume cut point was set at 36procedures per year (OR 2.01, 95% CI 1.27 to 3.17), 40procedures per year (OR 1.73, 95% CI 1.1 to 2.71), or 60procedures per year (OR 1.45, 95% CI 1.01 to 2.09).
Volume and outcomes relationship for PCI in hospitalswithout onsite cardiac surgery. There is only 1 reportindicating a relationship between institutional PCI volumeand outcome in hospitals without onsite cardiac surgery.Wennberg et al. (93) reported that among Medicare recip-ients, there was no difference in mortality after primary/rescue PCI (emergency procedure on the same day forSTEMI) performed at hospitals with or without cardiacsurgery onsite. However, they did report a higher mortalityfor PCI patients, excluding primary/rescue PCI, at hospitalswithout cardiac surgery onsite (adjusted OR 1.38, 95% CI1.14 to 1.67; p � 0.001). The relationship between insti-tutional volume and PCI affecting this outcome was con-fined mainly to hospitals without cardiac surgery onsiteperforming 50 or fewer nonprimary/rescue PCIs in Medi-care recipients per year. Among hospitals performing morethan 100 PCIs in Medicare recipients, mortality was nothigher in hospitals without surgery onsite (adjusted OR0.76, 95% CI 0.52 to 1.11; p � 0.16). These hospitals likelyperform more than 200 PCIs per year based on theassumption that 100 Medicare PCIs represent approxi-mately 200 total PCIs per year.
Taken together, these data suggest that the relationshipbetween institutional volume of PCI patients (excludingprimary/rescue PCI) and mortality in hospitals without
surgery onsite may be similar to the relationship in hospitalswith surgery onsite. For facilities without onsite surgery, itis mandatory that there be an established, well-organizedplan for transfer for surgery if needed.
Relationship of Individual Operator Volume toProcedural Outcome
Several large studies have assessed the potential relationbetween individual operator caseload and procedural com-plications (93). Recently, McGrath et al. (94) analyzedrelatively contemporary data (calendar year 1997) from theMedicare database. Based on a slightly different assumptionthan Wennberg et al. (93) that Medicare patients represent35% to 45% of total PCI procedure volume, they estimatedthat 30 PCIs per operator per year on Medicare patientscould be extrapolated to a total procedure volume of 70PCIs per operator per year (94). A significant relationshipbetween operator volume and outcomes was also reported intheir study, with better outcomes observed in patientstreated by high-volume operators when compared withpatients treated by low-volume operators. Similar resultswere obtained in the study by Hannan et al. (21) in theanalysis of data collected from the 107,713 proceduresperformed in the 34 hospitals performing PCI in New YorkState during 1998 to 2000. Operator volume thresholdswere set at 75 procedures per year based on ACC/AHArecommendations, and at slightly higher levels of 100 and125 procedures per year. There were no differences inrisk-adjusted mortality between patients undergoing PCIperformed by lower volume operators and patients under-going PCI performed by higher volume operators for any ofthe 3 volume thresholds that were examined. However, forall 3 volume thresholds, significant differences for “sameday” CABG surgery and for “same stay” CABG surgerywere observed. For example, patients undergoing PCI withoperators performing less than 75 procedures per year had a65% increased odds of undergoing same-day CABG sur-gery, and a 55% increased odds of undergoing “same-stay”CABG surgery.
Further confirmation of the adverse operator volume–outcome relationship with contemporary PCI comes froman analysis by Moscucci et al. (95) of another regional,audited, clinical PCI registry. In that analysis including18,504 procedures performed in 14 Michigan hospitals incalendar year 2002, operator volume was subdivided inquintiles (1 to 33 PCIs per year, 34 to 89 PCIs per year, 90to 139 PCIs per year, 140 to 206 PCIs per year, and 207 to582 PCIs per year). The primary end point was a compositeof MACE, including death, CABG, stroke, transient isch-emic attack, MI, and repeat PCI at the same lesion site.Stent utilization was greater than 80%, and greater than70% of patients received a glycoprotein (GPIIb/IIIa) receptorinhibitor. After adjustment for comorbidities, patientstreated by operators in the 2 lower volume quintiles (Quin-tiles 1 and 2) had a 63% increase in the odds of MACE
King et al ACCF/AHA/SCAI Clinical Competence Statement 109
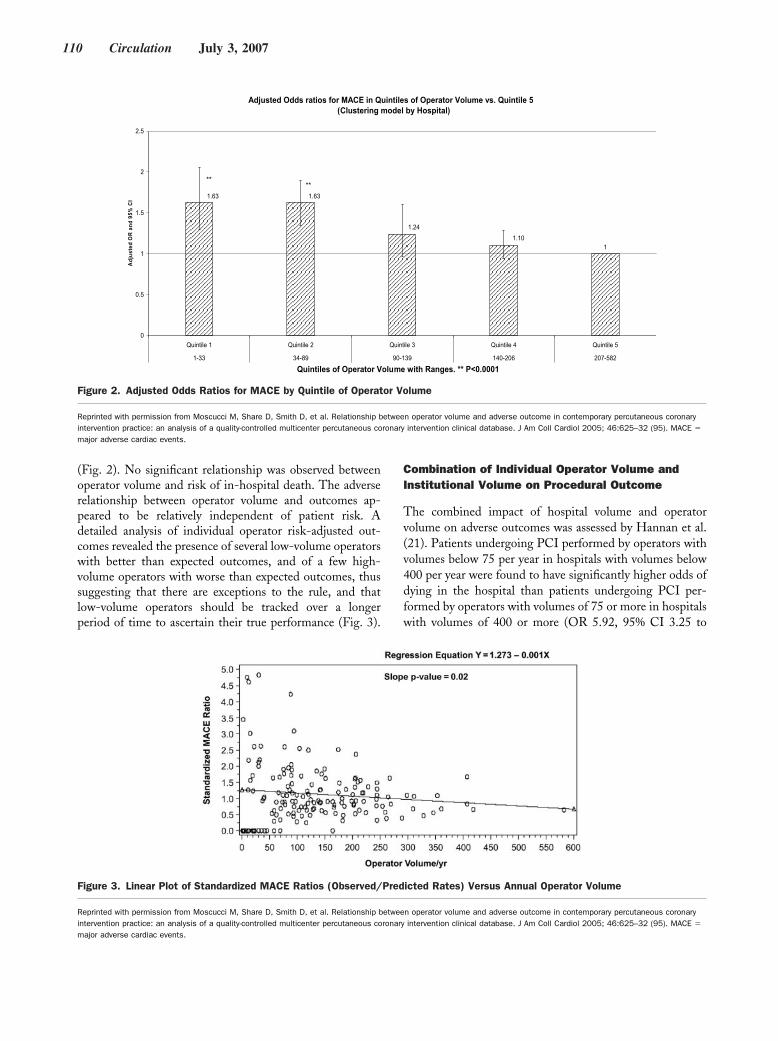
(Fig. 2). No significant relationship was observed betweenoperator volume and risk of in-hospital death. The adverserelationship between operator volume and outcomes ap-peared to be relatively independent of patient risk. Adetailed analysis of individual operator risk-adjusted out-comes revealed the presence of several low-volume operatorswith better than expected outcomes, and of a few high-volume operators with worse than expected outcomes, thussuggesting that there are exceptions to the rule, and thatlow-volume operators should be tracked over a longerperiod of time to ascertain their true performance (Fig. 3).
Combination of Individual Operator Volume andInstitutional Volume on Procedural Outcome
The combined impact of hospital volume and operatorvolume on adverse outcomes was assessed by Hannan et al.(21). Patients undergoing PCI performed by operators withvolumes below 75 per year in hospitals with volumes below400 per year were found to have significantly higher odds ofdying in the hospital than patients undergoing PCI per-formed by operators with volumes of 75 or more in hospitalswith volumes of 400 or more (OR 5.92, 95% CI 3.25 to
Adjusted Odds ratios for MACE in Quintiles of Operator Volume vs. Quintile 5 ( Clustering model by Hospital)
11.10
1.24
1.631.63
0
0.5
1
1.5
2
2.5
Quintile 1 Quintile 2 Quintile 3 Quintile 4 Quintile 5
1-33 34-89 90-139 140-206 207-582
Quintiles of Operator Volume with Ranges. ** P<0.0001
Adj
uste
d O
R a
nd 9
5%C
I
****
Figure 2. Adjusted Odds Ratios for MACE by Quintile of Operator Volume
Reprinted with permission from Moscucci M, Share D, Smith D, et al. Relationship between operator volume and adverse outcome in contemporary percutaneous coronaryintervention practice: an analysis of a quality-controlled multicenter percutaneous coronary intervention clinical database. J Am Coll Cardiol 2005; 46:625–32 (95). MACE �
major adverse cardiac events.
Figure 3. Linear Plot of Standardized MACE Ratios (Observed/Predicted Rates) Versus Annual Operator Volume
Reprinted with permission from Moscucci M, Share D, Smith D, et al. Relationship between operator volume and adverse outcome in contemporary percutaneous coronaryintervention practice: an analysis of a quality-controlled multicenter percutaneous coronary intervention clinical database. J Am Coll Cardiol 2005; 46:625–32 (95). MACE �
major adverse cardiac events.
110 Circulation July 3, 2007

10.97). Also, patients undergoing PCI performed by oper-ators with annual volumes of below 75 in hospitals withannual volumes below 400 experienced significantly highersame-day CABG rates than patients with high-volumeoperators (greater than 75 annually) in high-volume hospi-tals (greater than 400 annually), with an OR of 4.02. Forsame-stay CABG surgery the respective OR was 3.19. Itshould be noted that the magnitude of these ORs demon-strates that the increase in adverse outcomes compoundwhen patients undergo PCIs performed by low-volumeoperators (less than 75 annually) in low-volume hospitals(less than 400 annually).
In summary, analysis of more contemporary data supportsthe hypothesis that technological advancements have notcompletely offset the influence of “practice” in determiningproficiency of contemporary PCIs. However, procedurevolume is only a poor substitute for quality and outcomes;therefore, it should not be used as a replacement forappropriately risk-adjusted outcomes. Nevertheless, it iseasy to measure, and its potential implications are easilyunderstood by patients undergoing PCI. As such, it seemsappropriate to continue to include procedure volume amongthe several indirect quality indicators of contemporary PCIpractice.
However, it is also important to underscore that there aresignificant limitations to the simplistic interpretation ofprocedure volume statistics as a measure of competence andquality. First, it is uncertain whether this relationship is aresult of the “practice makes perfect” principle, or the factthat patients are more frequently referred to high-qualityoperators. Second, it remains unclear where the “cut-off”number should be set. Third, studies have shown significantvariability in the volume–outcome relationship within thesame registry, with some low-volume operators havingbetter than expected outcomes, and a few high-volumeoperators having worse-than-expected outcomes. Further-more, at present, few or no data exist linking operatorvolume to case selection, appropriateness of procedures,periprocedural MI, long-term clinical outcome, or cost-effectiveness, each of which measures a component ofquality of care, or linking clinical outcomes to operatorexperience as measured by the number of years in practice,total procedure volume over a lifetime career, or boardcertification.
The development of national, regional, and state regis-tries for outcome assessment is also promoting a shift of theparadigm surrounding quality of PCI from a mere collectionof procedure volume to objective assessment of clinicaloutcomes. In addition, the past decade has been character-ized by substantial advancement in methodology, scientificrigor, and acceptance of risk adjustment. Factors related toin-hospital mortality following PCI are now well defined,and progress is being made toward the development ofstatistical models for other outcomes. Clearly, the calcula-tion of risk-adjusted outcomes using data from clinicalregistries is a more accurate way to assess outcomes than
using volume as a surrogate, and as more registry databecome available, procedure volume will likely no longer beused as a replacement or a surrogate for quality assessment.
Yet, limitations related to the effect of random variationand to the evaluation of rare events continue to exist. Theselimitations make it difficult to assess the true performance ofvery-low-volume operators. In such situations, close scru-tiny of case selection and close monitoring of outcomes ona case-by-case basis might serve as a substitute/complementto risk adjustment.
In summary, while there are inherent limitations in usingprocedure volume as a surrogate of quality and outcomes,recent data suggest that there is still a relationship betweenexperience and outcomes. In the analysis of the New YorkState data, the relationship appeared to be at a level of 75procedures per year, with further improvement in outcomesobserved at a volume threshold of greater than 100 proce-dures per year. In the analysis of the Michigan data, therelationship was at a level of 100 procedures per year. Onthe basis of these data, it is recommended that the operatorvolume threshold continue to be 75 procedures per year.Independent of procedure volume, all operators shouldparticipate in a regional or national program for outcomeassessment and quality improvement. In addition, it isrecognized that there are limitations in the application ofthe risk-adjustment methodology in the evaluation of rareevents and of low-volume operators, and that there might besubstantial variations in the volume–outcome relationship.For operators that do not meet a threshold of 75 cases peryear measured in 2-year intervals, it is recommended that acase-by-case review, case selection, and prior experienceincluding the total number of cases in a lifetime career beincluded in their evaluation. They could also partner withhigher volume operators to perform cases together to gainfurther experience.
Ongoing Quality Improvement andMaintenance of Competence
Maintenance of competence in interventional proceduresshould be accomplished for both the individual physicianoperator and for the institutions in which cardiac interven-tional procedures are performed. The goals in settingcriteria for maintaining competence include:
1. Ensuring quality of patient care and outcomes;2. Enabling quality interventionalists and institutions to
continue to perform PCI;3. Providing standards that all institutions and operators
should strive to achieve.
Institutional Maintenance of Quality
It is recommended that all institutions have a regular (atleast monthly) catheterization laboratory conference. Theopportunity for ongoing dialogue and collaboration amongangiographers, interventional operators, and cardiothoracicsurgical colleagues is highly desirable. New developments in
King et al ACCF/AHA/SCAI Clinical Competence Statement 111

the angioplasty literature should be reviewed, and proce-dural complications should be discussed.
Maintenance of competence also requires that patientoutcomes be determined longitudinally for each procedureby the institution’s quality assessment program. Participa-tion in a state, regional, or national database is highlyencouraged. This allows institutions to measure risk-adjusted outcomes and compare them to regional andnational benchmarks for improving quality of care.
It is recommended that lower volume institutions (lessthan 400 interventions per year) consider holding confer-ences with a partnering, more highly experienced institu-tion. It is also recommended that any institution that fallsoutside the risk-adjusted national benchmarks in mortalityor emergency same-stay CABG during 2 of 3 contiguous6-month periods have an external audit looking for oppor-tunities to improve quality of care.
Individual Maintenance of Quality
To maintain a cognitive knowledge base, it is recommendedthat individual operators attend at least 30 h of interven-tional cardiology continuing medical education (CME)every 2 years. This could include catheterization conferencesand PCI meetings in addition to expanding the use ofsimulation cases for procedure use and competence.
To ensure appropriate patient selection and quality oftechnical skills, it is recommended that all operators have 5randomly selected cases and all major complications re-viewed each year by the catheterization laboratory directoror a Quality Assessment Committee at the institution. Anyoperator performing less than 75 cases per year should have10 cases reviewed per year. These performance evaluationsshould include feedback to the operator. If it is determinedthat the quality of PCI care being provided does not meetnational benchmarks, the catheterization laboratory directorshould have the discretion of making recommendations forimproving quality and reassessing over the next 6 months. Ifdisagreements concerning corrective action occur, externalreview is often helpful.
Quality Assurance
Definition of Quality in PCI
Satisfactory quality in PCI may be defined as selectingpatients appropriately for the procedure and achievingrisk-adjusted outcomes that are comparable to nationalbenchmark standards in terms of procedure success andadverse event rates. To achieve optimal quality and out-comes in PCI it is necessary that both the physician operatorand the supporting institution be appropriately skilled andexperienced.
Institutional Quality Assurance Requirement
In the United States, responsibility for quality assurance isvested in the health care institution that is responsible to the
public to ensure that patient care conducted under itsjurisdiction is of acceptable quality. Quality assessmentreview should be conducted both at the level of the entireprogram and at the level of the individual practitioner.
Each institution that performs PCI must establish anongoing mechanism for valid peer review of its quality andoutcomes. The program should provide an opportunity forinterventionalists as well as physicians who do not performangioplasty, but are knowledgeable about it, to review itsoverall results on a regular basis. The review process shouldtabulate the results achieved both by individual physicianoperators and by the overall program and compare them tonational benchmark standards with appropriate risk adjust-ment. Valid quality assessment requires that the institu-tion maintain meticulous and confidential records thatinclude the patient demographic and clinical characteris-tics necessary to assess appropriateness and to conductrisk adjustment.
Role of Risk Adjustment in Assessing Quality
A raw adverse event rate that is not appropriately riskadjusted has little meaning. Data compiled from largeregistries of procedures performed in recent years havegenerated multivariate risk adjustment models for adverseevent rates for PCI in the current era. Six multivariatemodels of the risk of mortality following PCI have beenpublished (62,64,96–99).
Although these models differ somewhat, they are consis-tent in identifying acute MI, shock, and age as importantrisk stratification variables for mortality. The ACC-NCDR® reported an univariate in-hospital mortality of0.5% for patients undergoing elective PCI, mortality of5.1% for patients undergoing primary PCI within 6 h of theonset of STEMI and mortality of 28% for patients under-going PCI for cardiogenic shock (64). Thus, it is clear that,in order to assess PCI mortality rates, patients should bestratified by whether they are undergoing elective PCI,primary PCI for acute STEMI without shock, or primaryPCI for STEMI with shock.
Challenges in Determining Quality
Given the complexity of case selection and procedureconduct, quality is difficult to measure in PCI and is notdetermined solely by adverse event rates even when properlyrisk adjusted. Accurate assessment of quality becomes moreproblematic for low-volume operators and institutions be-cause absolute event rates are expected to be small. Thus,particularly in low-volume circumstances, quality may bebetter assessed by an intensive case-review process con-ducted by recognized experts who can properly judge all ofthe facets of the conduct of a case. Case review also hasmerit in high-volume situations as it can identify subtletiesof case selection and procedure conduct that may not bereflected in pooled statistical data.
112 Circulation July 3, 2007

Requirement for Institutional Resources and Support
A high-quality PCI program requires appropriately trained,experienced, and skilled physician operators. However, theoperator does not work in a vacuum. An operator needs awell-maintained high-quality cardiac catheterization facilityto practice effectively. In addition, the operator depends ona multidisciplinary institutional infrastructure for supportand response to emergencies. Thus, to provide quality PCIservices, the institution must ensure that its catheterizationfacility is properly equipped and managed, and that all of itsnecessary support services, including data collection, are ofhigh quality and are readily available.
The Quality Assessment Process
Quality assessment is a complex process that includes morethan a mere tabulation of success and complication rates.Components of quality in coronary interventional proce-dures include appropriateness of case selection; quality ofprocedure execution; proper response to intraproceduralproblems; accurate assessment of procedure outcome bothshort- and long-term; and appropriateness of postproceduremanagement. It is important to consider each of theseparameters when conducting a quality assessment review. Aquality program performs appropriately selected procedureswhile achieving risk-adjusted outcomes, in terms of proce-dure success and complication rates, that are comparable tonational benchmark standards. It is accepted that qualityassurance monitoring is best conducted through the peer-review process despite the political challenges associatedwith colleagues evaluating each other. There has beenconsiderable controversy surrounding efforts to define stan-dards, criteria, and methodologies for conducting qualityassessment. There are many challenges to conducting thisprocess in a fair and valid manner.
The cornerstone of quality assurance monitoring is theassessment of procedure outcomes in terms of success andadverse event rates. Other components of quality assurancemonitoring include establishing criteria for assessing proce-dure appropriateness and applying proper risk adjustment tointerpret adverse event rates. As adverse events should be
rare, a valid estimate of a properly risk-adjusted adverseevent rate generally requires tabulating the results of a largenumber of procedures. This adds an additional challenge tothe valid assessment of low-volume operators and institu-tions. The responsible supervising authority should monitorthe issues outlined in Table 4.
In addition, mere tabulation of adverse event rates, evenwith appropriate risk adjustment, is inadequate to judgeoperator or program quality. Such tabulations do not ad-dress numerous other quality issues—in particular, appro-priateness. Thus, the quality assessment process should alsoconduct detailed reviews of both cases that have adverseoutcomes, to determine the cause(s) of the adverse event,and of uncomplicated cases, in order to judge case selectionappropriateness and procedure execution quality. Thesereviews should be conducted by recognized experiencedinterventionalists, drawn either from within the institutionor externally, if a requisite number of appropriately qualifiedunconflicted individuals are not available.
Conclusions and Recommendations for PCIs
In formulating conclusions and recommendations it isimportant to emphasize that the ultimate goal of settingstandards is to facilitate the attainment of optimal patientoutcomes. Optimal outcome is most likely when operatorsselect clinically appropriate patients for interventional pro-cedures and perform these procedures at a requisite level ofproficiency. Institutional and programmatic quality is ulti-mately determined by its success in achieving that goal.
Success and Complication Rates
Coronary interventional procedures may be complex andtechnically demanding to perform. Complications of theseprocedures may be life-threatening and can occur unpre-dictably. Nonetheless, recent clinical studies have demon-strated that despite increased clinical and angiographiccomplexity, procedural and clinical success has remainedhigh and complications have remained low. Angiographicsuccess (at least 1 lesion successfully dilated by greater than20%, with a residual stenosis of less than 50%), excluding
Table 4. Key Components of a Quality Assurance Program
Clinical proficiency
● General indications/contraindications
● Institutional and individual operator complication rates, mortality, and emergency coronary artery bypass grafting
● Institutional and operator procedure volumes
● Training and qualifications of support staff
Equipment maintenance and management
● Quality of laboratory facility (see ACC/SCAI Expert Consensus Document on Catheterization Laboratory Standards [100])
Quality improvement process
● Establishment of an active concurrent database to track clinical and procedural information and patient outcomes for individual operators and the institution.Participation in multicenter database is highly encouraged.
Radiation safety
● Educational program in the diagnostic use of X-ray
● Patient and operator exposure
King et al ACCF/AHA/SCAI Clinical Competence Statement 113

STEMI patients, occurs in over 95% with an averagemortality rate of less than 1%, a Q-wave MI rate of less than1%, and an emergency CABG rate of less than 1%.
Risk Adjustment
Several large retrospective studies have identified bothclinical and angiographic characteristics of PCI that corre-late with procedural success, hospital morbidity, and mor-tality. These studies have been used to develop multivariatelogistic regression models that can stratify patients into riskgroups before the procedure which have moderate predictivevalue for mortality (C-statistic 0.85 to 0.90), and slightlyless predictive value for morbidity (C-statistic 0.67 to 0.78).
Volume–Activity Relationships
Analysis of more contemporary data supports the hypothesisthat technological advancements have not offset the influ-ence of “practice” in determining proficiency of contempo-rary PCIs. There are statistical associations between activitylevels and short-term complication rates (emergency CABGand mortality) (17,58,85,89,97,101) for both institutionsand for individual operators. In particular, low-volumeoperators operating at low-volume hospitals had an in-creased mortality rate. However, procedural volume is onlyone of many factors contributing to the variability ofmeasured outcomes. Furthermore, there is no clear “cut-off”above or below which hospitals or individual operatorsperform well or poorly. Procedural volume continues to becorrelated with outcomes, but should not serve as a substi-tute for a well-controlled analysis of results and does notensure quality. The development of national, regional andstate registries for outcome assessment is promoting objec-tive assessment of clinical outcomes.
The expected low complication rate for coronary inter-ventional procedures presents a major statistical powerproblem when attempting to estimate the true complicationrate of the low-volume operator with meaningful precision.In such situations, close scrutiny of case selection and closemonitoring of outcomes on a case-by-case basis would serveas a complement to risk adjustment.
Highly complex procedures require much more skill andexperience, and should be undertaken by operators possess-ing these attributes. Complex cases appropriate for inter-ventions should be referred, not denied.
Recommendations for Institutional Maintenanceof Quality
It is recommended that all institutions have a regular (atleast monthly) catheterization laboratory conference. Pa-tient outcomes should be determined longitudinally for eachprocedure by the institution’s quality assessment program.Participation in a state, regional, or national registry ishighly encouraged to allow institutions to measure risk-adjusted outcomes and compare them to national bench-marks for improving quality of care.
For both institutional and individual volume assessments,ongoing 2-year volumes should be measured, then averagedto arrive at annual statistics. It is recommended that lowervolume institutions (less than 400 per year) consider holdingconferences with a more experienced partnering institution,with all staff expected to attend on a regular basis.
It is also recommended that any institution that falls morethan 2 standard deviations outside the risk-adjusted nationalbenchmarks in mortality or emergency same-stay CABGduring 2 of 3 contiguous 6-month periods have an externalaudit looking for opportunities to improve quality of care.
An institution offering coronary interventional proce-dures should have a physician-director who is responsiblefor the program’s overall quality. The director should becertified in interventional cardiology by the ABIM, with acareer experience of more than 500 procedures. The directorshould perform procedures at the facility that he or shedirects.
Recommendations for Individual Maintenanceof Quality
To maintain an appropriate cognitive knowledge base forPCIs, it is recommended that individual operators attend atleast 30 h of PCI CME every 2 years. The overall perfor-mance of physicians whose complication rates exceed na-tional benchmark standards for 2 of 3 contiguous 6-monthperiods should be reviewed by the program director, withcareful attention to statistical power and risk-adjustmentissues. It is recommended that the operator volume thresh-old continue to be 75 procedures per year. Monitoring ofphysicians with an annual procedural volume of less than 75should be particularly detailed because of the difficulty ofestimating their true complication rate. These performanceevaluations should include feedback to the operator.
If it is determined that the quality of PCI care beingprovided does not meet national benchmarks, the catheter-ization laboratory director should have the discretion ofmaking recommendations for improving quality and reas-sessing over the next 6 months. These recommendationscould include establishing a defined mentoring relationshipwith an experienced operator. If the operator in questiondisputes this assessment, then external review may behelpful in determining the most appropriate methods ofassuring quality performance.
Percutaneous Noncoronary Interventions
Introduction
Noncoronary interventions are a growing and importantcontribution to the field of interventional cardiology. Themajority of procedures have had their origin in the pediatricpopulation, and several have expanded to the adult patient.The purpose of this section is to discuss the training andexperience necessary for the safe and successful performance
114 Circulation July 3, 2007

of valvuloplasty, alcohol septal ablation, and percutaneousrepair of ASD/PFO.
The knowledge, skills, and training necessary for compe-tency in noncoronary interventional procedures are differentfrom that required for coronary interventions. Therefore,special study of the anatomy, physiology, and pathology ofthese conditions is a prerequisite for safe and effectivetreatment. Furthermore, an in-depth understanding of theclinical indications for treatment and the unique complica-tions of these treatments are essential.
Although the scope of this document is focused oncompetency, this section will expand the discussion some-what to describe some anatomical and procedural details.Such details are well known for PCI, and their performanceis widespread, whereas these noncoronary procedures, in theestimation of this Writing Committee, warrant some dis-cussion of background information and procedural alternatives.
Disorders of the Atrial Septum
Criteria for Competency
The knowledge base required for performing PCI is differ-ent than that required for percutaneous closure of ASD andPFO. Extensive knowledge of structural cardiac anatomy,especially that of the atrial septum and the adjacent struc-tures, is required, as is the understanding of the impact ofabnormal anatomy and function, and the relative value oftherapeutic options (85,101–105). Therefore, specific train-ing and experience is necessary to safely and successfullytreat this subgroup of patients. The Food and Drug Ad-ministration guidelines on the use of device closure of PFOsin these patients state that only patients who have failedanticoagulation or have a compelling medical reason to notbe anticoagulated are appropriate for device closure. Theseguidelines should be fully discussed with patients during theinformed consent process. In addition, complications suchas cardiac perforation, device embolization, thrombus for-mation on the device, infective endocarditis, arrhythmias,and early as well as late erosion of the device through theatrial wall or aorta should be disclosed. Currently, 2 studiesare underway comparing percutaneous closure of PFO tostandard oral anticoagulation, which should clarify theindications for interventional treatment.
Since these procedures are relatively new to intervention-alists trained in adult cardiology, no pre-existing guidelinesare available on which to base current opinion. In theabsence of such guidelines, we arrived at these recommen-dations from discussions with colleagues actively performingthese percutaneous closures.
Cardiologists in Training Programs
Acquisition of the knowledge and skills necessary to per-form percutaneous procedures to treat ASD and PFOshould be incorporated into the formal training of interven-tional cardiologists. There are no data regarding the mini-
mum number of cases required for maintenance of compe-tency and proficiency. A survey of Pediatric CardiologyInterventional Catheterization training programs concludedthat a minimum of 10 percutaneous ASD closures isnecessary for a trainee to gain clinical competence with theprocedure (102).
With this in mind, it is recommended that interventionalcardiologists who intend to perform these procedures inde-pendently, should be involved in these procedures duringtraining with at least 10 of these cases being secundum ASDclosures. Furthermore, as part of the procedure, the fellowshould be fully conversant in the use of transesophagealechocardiography and/or intracardiac echocardiography. Heor she should understand how to obtain the appropriateviews to image necessary structures in order to perform theprocedure safely and to exclude other anatomical problemssuch as a primum or sinus venosus ASD, anomalouspulmonary venous drainage, fenestrated or multiple ASD,or lipomatous hypertrophy of the septum. Obviously, not allfellows in training will be able to gain this experience and,therefore, concentrating the experience in training should belimited to a few trainees.
Cardiologists in Practice
Interventional cardiologists in practice who were not specifi-cally trained in ASD/PFO closure but would like to performthese procedures should be fully credentialed in interventionaltechniques in their institution. The first several cases should bedone with a proctor. To ensure safety and success, it seemsprudent that the first 10 cases be proctored by someone fullycredentialed in these techniques such as a pediatric cardiologistor adult cardiologist trained in congenital heart disease. Proc-tors should also be present for the first 3 to 5 cases if a differentdevice is to be used after the initial credentialing proctorship.
Maintenance of Competency for PercutaneousASD/PFO Closure
To maintain physician proficiency and competency in percu-taneous ASD/PFO closure, a minimum of 10 cases per year isrecommended. Similarly, to maintain catheterization labora-tory proficiency, a minimum of 10 cases per year should beperformed in each institution each year. To achieve thisexperience, it may be necessary to concentrate the proceduresin the hands of only a few operators. A multidisciplinaryprogram, including neurology consultation for PFO closure,prospective evaluation of case selection, and evaluation ofclinical outcomes is critical to ensure appropriateness andmaintain safety and efficacy. Laboratories and individual oper-ators that are not active enough to maintain quality outcomesshould reconsider treating these patients.
Quality Assurance
The quality improvement process used for oversight of ASD/PFO closure should include concurrent case review, and willalso benefit from regular case conferences to discuss indica-tions, procedural techniques, and case outcomes. It is particu-
King et al ACCF/AHA/SCAI Clinical Competence Statement 115

larly useful in any developing procedural area to share resultswith other institutions through informal and formal confer-ences. Because there are, as of yet, no large databases ofoutcomes for these procedures, participation in local, regional,and national registries is encouraged. Focusing the perfor-mance of these procedures in the hands of a few experiencedoperators is also recommended.
Hypertrophic Cardiomyopathy andAlcohol Septal Ablation
Hypertrophic cardiomyopathy is the most common geneticcardiovascular disease, with a prevalence in the general popu-lation estimated to be 0.2% (103). Physicians performing theseprocedures should have extensive knowledge of the outcomes,limitations and complications of medical therapy (104), dualchamber pacing and surgical myectomy (105–107), and alcoholseptal ablation (105–114). No comparative trial against surgicalmyectomy has been performed.
Criteria for Competency
Acquisition of competence. It is strongly recommended thatalcohol septal ablation be offered within a multidisciplinaryprogram that includes the contribution of experienced cardiacsurgeons, echocardiographers, general cardiologists, and elec-trophysiologists. Although there are currently no data regard-ing the minimum number of procedures required for trainingand for credentialing, a minimum number of 10 proceduresseems to be appropriate.
Maintenance of competence. It is recommended that indi-vidual operators perform a minimum of 6 cases per year tomaintain competence in performance of septal ablation forhypertrophic cardiomyopathy. Each institution should employa multidisciplinary program with prospective evaluation of caseselection and clinical outcomes. Such an approach is critical forany institution offering alcohol septal ablation as a treatmentoption for symptomatic patients with hypertrophic obstructivecardiomyopathy.
Quality assurance. Quality assurance in such low-volumeprocedures requires an approach similar to that outlined forASD and PFO closures, as previously described.
Valvular Heart Disease
Cognitive Knowledge Base
Physicians performing invasive procedures on stenotic cardiacvalves must have extensive knowledge of the pathoanatomy,the hemodynamic alterations, the clinical course, and theoutcomes of various therapeutic options. Complications ofaortic (115,116) and mitral (117–119) valvuloplasty should bewell understood.
Criteria for Competency
Acquisition of competence. Mitral valvuloplasty is one ofthe most challenging cardiac procedures. The presence of a“learning curve” has been well described (120,121). Thus,training in the performance of mitral valvuloplasty requires the
acquisition of clinical skills for the evaluation of indications forthe procedure and the assessment of suitable valve morphology.It requires the development of proficiency in the performanceof transseptal cardiac catheterization, device manipulation, andonline evaluation of hemodynamic parameters. The interven-tionalist must be able to recognize and manage complicationsspecific to mitral valvuloplasty, including acute mitral regurgi-tation, cardiac perforation, pericardial tamponade, and stroke.Although a learning curve has been well described, there arecurrently no specific data regarding the minimum numbersneeded for competency. Nonetheless, 5 to 10 cases should bedone with an experienced colleague before attempting toperform balloon valvuloplasty independently. Any programoffering mitral valvuloplasty as an alternative to mitral valvereplacement or surgical commissurotomy for the treatment ofmitral stenosis should include a thorough quality assuranceprogram and close monitoring of case selection and clinicaloutcomes. As with other infrequently performed procedures,concentration of experience among a small subset of interven-tional cardiologists within an institution is appropriate.
Maintenance of competence. With the low prevalence ofmitral stenosis in the United States, maintaining experienceis difficult. Given this limitation, concentration of thisexperience among institutional and perhaps regional centersmay be appropriate.
Quality assurance. Quality assurance in such low-volumeprocedures requires an approach similar to that outlined forASD and PFO closures, as previously described.
Percutaneous Ventricular Assist Devices
Percutaneous ventricular assist devices are becoming avail-able. They require training and proctored supervision toattain competence, as well as periodic use or refresher drillsto maintain competence. As with other seldom-used tech-niques, experience should be concentrated among a limitednumber of operators and laboratory staff who have receivedappropriate training.
Laboratory and Staff Competence
In order for laboratories to become competent in theperformance of noncoronary cardiac procedures, the super-vising or performing operator should be fully credentialed inthe procedure. Initially, this may require off-site training,simulation training, a visiting proctor, or a combination ofthese approaches. The operator responsible for the perfor-mance of the procedure in the catheterization laboratoryshould supervise the staff in acquiring the necessary skillsand equipment for the procedure. As is the case for theoperators of lower volume procedures, there should be asmall number of dedicated staff members trained to performspecific noncoronary interventions, concentrating the expe-rience. If and when a specific procedure becomes morecommon, then the training may be expanded to the remain-der of the staff and operators.
116 Circulation July 3, 2007

Conclusions and Recommendations
Percutaneous Noncoronary Interventions
Noncoronary cardiac interventions require special trainingthat is not possible for all operators to obtain because of thesmall number of these procedures. Therefore, it is necessaryto concentrate the activity both in training and practice sothat adequate experience can be obtained to allow for qualityperformance. Hospitals should develop clear credentialingcriteria, despite the small number of cases and empiric datafrom which to judge appropriateness, as well as success andcomplication rates of these procedures.
The quality improvement process used for oversight ofpercutaneous noncoronary interventions should includeconcurrent case review, and will also benefit from regularcase conferences to discuss indications, procedural tech-niques, and case outcomes. It is particularly useful in anydeveloping procedural area to share results with otherinstitutions through informal and formal conferences. Sincethere are, as of yet, no large databases of outcomes for theseprocedures, participation in local, regional, and nationalregistries is encouraged. Focusing the performance of theseprocedures in the hands of a few experienced operators isalso recommended.
Staff
American College of Cardiology FoundationJohn C. Lewin, MD, Chief Executive OfficerThomas E. Arend, Jr, Esq., Chief Operating OfficerLisa Bradfield, Associate Director, Practice GuidelinesErin A. Barrett, Senior Specialist, Clinical Policy andDocumentsAmerican Heart AssociationM. Cass Wheeler, Chief Executive OfficerRose Marie Robertson, MD, FACC, FAHA, ChiefScience OfficerKathryn A. Taubert, PhD, FAHA, Senior Scientist
REFERENCES
1. Gruntzig A. Transluminal dilatation of coronary-artery stenosis.Lancet 1978;1:263.
2. Fischman DL, Leon MB, Baim DS, et al. A randomized comparisonof coronary-stent placement and balloon angioplasty in the treatmentof coronary artery disease. Stent Restenosis Study Investigators.N Engl J Med 1994;331:496–501.
3. Moses JW, Leon MB, Popma JJ, et al. Sirolimus-eluting stents versusstandard stents in patients with stenosis in a native coronary artery.N Engl J Med 2003;349:1315–23.
4. Thom T, Haase N, Rosamond W, et al. Heart disease and strokestatistics—2006 update: a report from the American Heart Associa-tion Statistics Committee and Stroke Statistics Subcommittee. Cir-culation 2006;113:e85–151.
5. American Board of Internal Medicine Certification Examination.Available at: http://www.abim.org/resources/statcert.shtm. Last up-date 2005.
6. Conti CR, Faxon DP, Gruentzig A, Gunnar RM, Lesch M, ReevesTJ. Training in cardiac catheterization. Bethesda Conference 17:Adult Cardiology Training Task Force III. J Am Coll Cardiol1986;7:1205–6.
7. Cowley M, Faxon DP, Holmes DR Jr. Guidelines for training,credentialing, and maintenance of competence for the performance ofcoronary angioplasty: a report from the Interventional CardiologyCommittee and the Training Program Standards Committee of theSociety for Cardiac Angiography and Interventions. Cathet Cardio-vasc Diagn 1993;30:1–4.
8. Cowley MJ, King SB. ACC/AHA guidelines for credentialing andfacilities for performance of coronary angioplasty. Circulation 1988;15:136–8.
9. Douglas JSJ, Levin DC, Pepine CJ, et al. Recommendations fordevelopment and maintenance of competence in coronary interven-tional procedures. American College of Cardiology Cardiac Cathe-terization Committee. J Am Coll Cardiol 1993;22:629–31.
10. Parker DJ, Birkhead JS, Balcon R, et al. Planning for coronaryangioplasty: Guidelines for training and continuing competence.British Cardiac Society (BCS) and British Cardiovascular Interven-tion Society (BCIS) Working Group on Interventional Cardiology.Heart 1996;75:419–25.
11. Pepine CJ, Babb JD, Brinker JA, et al. Guidelines for training inadult cardiovascular medicine. Core Cardiology Training Symposium(COCATS). Task Force 3: training in cardiac catheterization andinterventional cardiology. J Am Coll Cardiol 1995;25:14–6.
12. Ryan TJ, Faxon DP, Gunnar RM, et al. Guidelines for percutaneoustransluminal coronary angioplasty: a report of the American Collegeof Cardiology/American Heart Association Task Force on Assess-ment of Diagnostic and Therapeutic Cardiovascular Procedures(Subcommittee on Percutaneous Transluminal Coronary Angio-plasty). J Am Coll Cardiol 1988;12:529–45.
13. Ryan TJ, Klocke FJ, Reynolds WA. Clinical competence in percu-taneous transluminal coronary angioplasty: a statement for physiciansfrom the ACP/ACC/AHA Task Force on Clinical Privileges inCardiology. Circulation 1990;81:2041–6.
14. Ryan TJ, Bauman WB, Kennedy JW, et al. Guidelines for percuta-neous transluminal coronary angioplasty: a report of the AmericanCollege of Cardiology/American Heart Association Task Force onAssessment of Diagnostic and Therapeutic Cardiovascular Proce-dures (Committee on Percutaneous Transluminal Coronary Angio-plasty). J Am Coll Cardiol 1993;22:2033–54.
15. Smith SC Jr., Feldman TE, Hirshfeld JW Jr., et al. ACC/AHA/SCAI 2005 guideline update for percutaneous coronary interven-tion—summary article: a report of the American College of Cardi-ology/American Heart Association Task Force on PracticeGuidelines (ACC/AHA/SCAI Writing Committee to Update the2001 Guidelines for Percutaneous Coronary Intervention). J Am CollCardiol 2006;47:216–35.
16. Hirshfeld JW Jr., Ellis SG, Faxon DP. Recommendations for theassessment and maintenance of proficiency in coronary interventionalprocedures: atatement of the American College of Cardiology. J AmColl Cardiol 1998;31:722–43.
17. Jollis JG, Peterson ED, Nelson CL, et al. Relationship betweenphysician and hospital coronary angioplasty volume and outcome inelderly patients. Circulation 1997;95:2485–91.
18. Creager MA, Goldstone J, Hirshfeld JW Jr., et al. ACC/ACP/SCAI/SVMB/SVS clinical competence statement on vascular med-icine and catheter-based peripheral vascular interventions: a report ofthe American College of Cardiology/American Heart Association/American College of Physician Task Force on Clinical Competence(ACC/ACP/SCAI/SVMB/SVS Writing Committee to Develop aClinical Competence Statement on Peripheral Vascular Disease).J Am Coll Cardiol 2004;44:941–57.
19. Hodgson JM, Tommaso CL, Watson RM, Weiner BH. Corecurriculum for the training of adult invasive cardiologists: report ofthe Society for Cardiac Angiography and Interventions Committeeon Training Standards. Cathet Cardiovasc Diagn 1996;37:392–408.
20. Hirshfeld JW Jr., Banas JS Jr., Brundage BH, et al. American Collegeof Cardiology training statement on recommendations for the struc-ture of an optimal adult interventional cardiology training program: areport of the American College of Cardiology task force on clinicalexpert consensus documents. J Am Coll Cardiol 1999;34:2141–7.
21. Hannan EL, Wu C, Walford G, et al. Volume-outcome relationshipsfor percutaneous coronary interventions in the stent era. Circulation2005;112:1171–9.
22. Moscucci M, Eagle KA, Share D, et al. Public reporting and caseselection for percutaneous coronary interventions: an analysis from
King et al ACCF/AHA/SCAI Clinical Competence Statement 117

two large multicenter percutaneous coronary intervention databases.J Am Coll Cardiol 2005;45:1759–65.
23. Gruntzig A. Percutaneous dilatation of experimental coronary arterystenosis—description of a new catheter system. Klin Wochenschr1976;54:543–45.
24. CABRI Trial Participants. First-year results of CABRI (CoronaryAngioplasty versus Bypass Revascularisation Investigation). Lancet1995;346:1179–84.
25. The Bypass Angioplasty Revascularization Investigation (BARI)Investigators. Comparison of coronary bypass surgery with angio-plasty in patients with multivessel disease. N Engl J Med 1996;335:217–25.
26. Writing Group for the Bypass Angioplasty Revascularization Inves-tigation (BARI) Investigators. Five-year clinical and functional out-come comparing bypass surgery and angioplasty in patients withmultivessel coronary disease: a multicenter randomized trial. JAMA1997;277:715–21.
27. King SB III, Lembo NJ, Weintraub WS, et al. A randomized trialcomparing coronary angioplasty with coronary bypass surgery. EmoryAngioplasty versus Surgery Trial (EAST). N Engl J Med 1994;331:1044–50.
28. Serruys PW, Unger F, Sousa JE, et al. Comparison of coronary-arterybypass surgery and stenting for the treatment of multivessel disease.N Engl J Med 2001;344:1117–24.
29. Sharma S, Forrester J, Makkar RM. The SoS trial. Lancet 2003;361:614–5.
30. Hamm CW, Reimers J, Ischinger T, Rupprecht HJ, Berger J,Bleifeld W. A randomized study of coronary angioplasty comparedwith bypass surgery in patients with symptomatic multivessel coro-nary disease. German Angioplasty Bypass Surgery Investigation(GABI). N Engl J Med 1994;331:1037–43.
31. Myler RK, Shaw RE, Stertzer SH, et al. Lesion morphology andcoronary angioplasty: current experience and analysis. J Am CollCardiol 1992;19:1641–52.
32. de Feyter PJ, van Suylen RJ, de Jaegere PP, Topol EJ, Serruys PW.Balloon angioplasty for the treatment of lesions in saphenous veinbypass grafts. J Am Coll Cardiol 1993;21:1539–49.
33. Ambrose JA, Torre SR, Sharma SK, et al. Adjunctive thrombolytictherapy for angioplasty in ischemic rest angina: results of a double-blind randomized pilot study. J Am Coll Cardiol 1992;20:1197–204.
34. Perry RA, Seth A, Hunt A, Shiu MF. Coronary angioplasty inunstable angina and stable angina: a comparison of success andcomplications. Br Heart J 1988;60:367–72.
35. Grines CL, Browne KF, Marco J, et al. A comparison of immediateangioplasty with thrombolytic therapy for acute myocardial infarc-tion. The Primary Angioplasty in Myocardial Infarction StudyGroup. N Engl J Med 1993;328:673–9.
36. GUSTO (IIb) Angioplasty Substudy Group. The Global Use ofStrategies to Open Occluded Coronary Arteries in Acute CoronarySyndromes: a clinical trial comparing primary coronary angioplastywith tissue plasminogen activator for acute myocardial infarction.N Engl J Med 1997;336:1621–8.
37. Bittl JA. Advances in coronary angioplasty. N Engl J Med 1996;24;335:1290–302.
38. Detre KM, Holubkov R, Kelsey S, et al. Percutaneous transluminalcoronary angioplasty in 1985–1986 and 1977–1981: the NationalHeart, Lung, and Blood Institute Registry. N Engl J Med 1988;318:265–70.
39. Holmes DRJ, Holubkov R, Vlietstra RE, et al. Comparison ofcomplications during percutaneous transluminal coronary angioplastyfrom 1977 to 1981 and from 1985 to 1986: the National Heart,Lung, and Blood Institute Percutaneous Transluminal CoronaryAngioplasty Registry. J Am Coll Cardiol 1988;12:1149–55.
40. Kent KM, Bentivoglio LG, Block PC, et al. Percutaneous translu-minal coronary angioplasty: report from the Registry of the NationalHeart, Lung, and Blood Institute. Am J Cardiol 1982;49:2011–20.
41. Abbott JD, Choi EJ, Selzer F, Srinivas VS, Williams DO. Impact ofcoronary collaterals on outcome following percutaneous coronaryintervention (from the National Heart, Lung, and Blood InstituteDynamic Registry). Am J Cardiol 2005;96:676–80.
42. Hirshfeld JW Jr. ACC-National Cardiovascular Data Registry®,CathPCI data repository, 1998–2005. November 28, 2005.
43. Malenka DJ, Leavitt BJ, Mearne MS, et al. Northern New EnglandCardiovascular Disease Study Group. Comparing long-term survival
of patients with multivessel coronary disease after CAGB or PCI:analysis of BARI-like patients in northern New England. Circulation2005;112:I371–6.
44. Moscucci M, Rogers EK, Montoye C, et al. Association of acontinuous quality improvement initiative with practice and outcomevariations of contemporary percutaneous coronary interventions.Circulation 2006;113:814–22.
45. Percutaneous Coronary Interventions (PCI) in New York State,2001–2003. New York State Department of Health. Available at:http://www.health.state.ny.us/nysdoh/heart/pdf/pci_2001–2003.pdf.Last update 2005.
46. Weintraub WS, McKay CR, Riner RN, et al. The American Collegeof Cardiology National Database: progress and challenges. AmericanCollege of Cardiology Database Committee. J Am Coll Cardiol1997;29:459–65.
47. Brown BG, Bolson EL, Dodge HT. Quantitative computer tech-niques for analyzing coronary arteriograms. Prog Cardiovasc Dis1986;28:403–18.
48. Detre KM, Wright E, Murphy ML, Takaro T. Observer agreementin evaluating coronary angiograms. Circulation 1975;52:979–86.
49. Califf RM, Abdelmeguid AE, Kuntz RE, et al. Myonecrosis afterrevascularization procedures. J Am Coll Cardiol 1998;31:241–51.
50. Cutlip DE, Chhabra AG, Baim DS, et al. Beyond restenosis:five-year clinical outcomes from second-generation coronary stenttrials. Circulation 2004;110:1226–30.
51. Mehran R, Aymong ED, Nikolsky E, et al. A simple risk score forprediction of contrast-induced nephropathy after percutaneous cor-onary intervention: development and initial validation. J Am CollCardiol 2004;44:1393–9.
52. Ellis SG, Vandormael MG, Cowley MJ, et al. Coronary morphologicand clinical determinants of procedural outcome with angioplasty formultivessel coronary disease. Implications for patient selection. Mul-tivessel Angioplasty Prognosis Study Group. Circulation 1990;82:1193–2.
53. Altmann DB, Racz M, Battleman DS, et al. Reduction in angioplastycomplications after the introduction of coronary stents: results from aconsecutive series of 2242 patients. Am Heart J 1996;132:503–7.
54. Williams DO, Holubkov R, Yeh W, et al. Percutaneous coronaryintervention in the current era compared with 1985–1986: theNational Heart, Lung, and Blood Institute Registries. Circulation2000;102:2945–51.
55. Malenka DJ, Wennberg DE, Quinton HA, et al. Gender-relatedchanges in the practice and outcomes of percutaneous coronaryinterventions in Northern New England from 1994 to 1999. J AmColl Cardiol 2002;40:2092–101.
56. Krone RJ, Kimmel SE, Laskey WK, et al. Evaluation of the Societyfor Coronary Angiography and Interventions lesion classificationsystem in 14,133 patients with percutaneous coronary interventionsin the current stent era. Catheter Cardiovasc Interv 2002;55:1–7.
57. Krone RJ, Shaw RE, Klein LW, et al. Evaluation of the AmericanCollege of Cardiology/American Heart Association and the Societyfor Coronary Angiography and Interventions lesion classificationsystem in the current “stent era” of coronary interventions (from theACC-National Cardiovascular Data Registry). Am J Cardiol 2003;92:389–94.
58. Ellis SG, Omoigui N, Bittl JA, et al. Analysis and comparison ofoperator-specific outcomes in interventional cardiology: from a mul-ticenter database of 4860 quality-controlled procedures. Circulation1996;93:431–9.
59. Krone RJ, Laskey WK, Johnson C, et al. A simplified lesionclassification for predicting success and complications of coronaryangioplasty. Registry Committee of the Society for Cardiac Angiog-raphy and Intervention. Am J Cardiol 2000;85:1179–84.
60. Singh M, Lennon RJ, Holmes DR Jr., Bell MR, Rihal CS.Correlates of procedural complications and a simple integer risk scorefor percutaneous coronary intervention. J Am Coll Cardiol 2002;40:387–93.
61. Maynard C, Goss JR, Malenka DJ, Reisman M. Adjusting forpatient differences in predicting hospital mortality for percutaneouscoronary interventions in the Clinical Outcomes Assessment Pro-gram. Am Heart J 2003;145:658–64.
62. Moscucci M, Kline-Rogers E, Share D, et al. Simple bedside additivetool for prediction of in-hospital mortality after percutaneous coro-nary interventions. Circulation 2001;104:263–8.
118 Circulation July 3, 2007

63. O’Connor GT, Malenka DJ, Quinton H, et al. Multivariate predic-tion of in-hospital mortality after percutaneous coronary interven-tions in 1994–1996. Northern New England Cardiovascular DiseaseStudy Group. J Am Coll Cardiol 1999;34:681–91.
64. Shaw RE, Anderson HV, Brindis RG, et al. Development of a riskadjustment mortality model using the American College ofCardiology-National Cardiovascular Data Registry (ACC-NCDR)experience: 1998–2000. J Am Coll Cardiol 2002;39:1104–12.
65. Shaw RE, Anderson HV, Brindis RG, et al. Updated risk adjustmentmortality model using the complete 1.1 dataset from the AmericanCollege of Cardiology National Cardiovascular Data Registry (ACC-NCDR). J Invasive Cardiol 2003;15:578–80.
66. Wu C, Hannan EL, Walford G, et al. A risk score to predictin-hospital mortality for percutaneous coronary interventions. J AmColl Cardiol 2006;47:654–60.
67. Pepine CJ, Babb JD, Brinker JA, et al. Guidelines for training inadult cardiovascular medicine. Core Cardiology Training Symposium(COCATS). Task Force 3: training in cardiac catheterization andinterventional cardiology. J Am Coll Cardiol 1995;25:14–6.
68. Beller GA, Bonow RO, Fuster V. ACC revised recommendations fortraining in adult cardiovascular medicine. Core Cardiology TrainingII (COCATS 2). (Revision of the 1995 COCATS training state-ment). J Am Coll Cardiol 2002;39:1242–6.
69. Smith SC Jr., Dove JT, Jacobs AK, et al. ACC/AHA guidelines ofpercutaneous coronary interventions (revision of the 1993 PTCAguidelines)—executive summary. A report of the American Collegeof Cardiology/American Heart Association Task Force on PracticeGuidelines (Committee to Revise the 1993 Guidelines for Percuta-neous Transluminal Coronary Angioplasty). J Am Coll Cardiol2001;37:2215–39.
70. Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guide-lines for the management of patients with ST-elevation myocardialinfarction: a report of the American College of Cardiology/AmericanHeart Association Task Force on Practice Guidelines (Committee toRevise the 1999 Guidelines for the Management of Patients WithAcute Myocardial Infarction). J Am Coll Cardiol 2004;44:E1–211.
71. Braunwald E, Antman EM, Beasley JW, et al. ACC/AHA 2002guideline update for the management of patients with unstableangina and non-ST-segment elevation myocardial infarction—summary article: a report of the American College of Cardiology/American Heart Association task force on practice guidelines (Com-mittee on the Management of Patients With Unstable Angina). J AmColl Cardiol 2002;40:1366–74.
72. Eagle KA, Guyton RA, Davidoff R, et al. ACC/AHA 2004 guidelineupdate for coronary artery bypass graft surgery: summary article. Areport of the American College of Cardiology/American HeartAssociation Task Force on Practice Guidelines (Committee toUpdate the 1999 Guidelines for Coronary Artery Bypass GraftSurgery). J Am Coll Cardiol 2004;44:e213–e310.
73. Hirshfeld JW Jr., Balter S, Brinker JA, et al. ACCF/AHA/HRS/SCAI clinical competence statement on physician knowledge tooptimize patient safety and image quality in fluoroscopically guidedinvasive cardiovascular procedures: a report of the American Collegeof Cardiology Foundation/American Heart Association/AmericanCollege of Physicians Task Force on Clinical Competence andTraining. J Am Coll Cardiol 2004;44:2259–82.
74. Kern MJ, Lim M. Evaluation of myocardial blood flow and metab-olism. In: Baim DS, editor. Grossman’s Cardiac Catheterization,Angiography, and Intervention. Philadelphia, PA: Lippincott Wil-liams & Wilkins, 2006:335–70.
75. Scanlon PJ, Faxon DP, Audet AM, et al. ACC/AHA guidelines forcoronary angiography. A report of the American College of Cardi-ology/American Heart Association Task Force on Practice Guide-lines (Committee on Coronary Angiography). Developed in collab-oration with the Society for Cardiac Angiography and Interventions.J Am Coll Cardiol 1999;33:1756–824.
76. Mintz GS, Nissen SE, Anderson WD, et al. American College ofCardiology clinical expert consensus document on standards foracquisition, measurement and reporting of intravascular ultrasoundstudies (IVUS). A report of the American College of CardiologyTask Force on Clinical Expert Consensus Documents. J Am CollCardiol 2001;37:1478–92.
77. Jollis JG, Peterson ED, Nelson CL, et al. Relationship betweenphysician and hospital coronary angioplasty volume and outcome inelderly patients. Circulation 1997;95:2485–91.
78. Hartz AJ, Kuhn EM, Kayser KL, et al. Assessing providers ofcoronary revascularization: a method for peer review organizations.Am J Public Health 1992;82:1631–40.
79. Kato NS, Ergun ME, Carter GM. Health policy implications ofvolume recommendations on percutaneous transluminal coronaryangioplasty in the United States (abstr). Circulation 1996;94:I532.
80. Stone GW, Marsalese D, Brodie BR, et al., on behalf of the SecondPrimary Angioplasty In Myocardial Infarction (PAMI-II) TrialInvestigators. A prospective, randomized evaluation of prophylacticintraaortic balloon counterpulsation in high risk patients with acutemyocardial infarction treated with primary angioplasty. J Am CollCardiol 1997;29:1459–67.
81. Zahn R, Vogt A, Seidl K, et al, Balloon dilatation in acute myocardialinfarct in routine clinical practice: results of the register of theWorking Society of Leading Cardiologic Hospital Physicians in4,625 patients. Z Kardiol 1997;86:712–21.
82. Hannan E, Racz M, Ryan TJ, et al. Coronary angioplasty volume-outcome relationships for hospitals and operators in New York State:1991–1994. JAMA 1997;277:892–8.
83. Tiefenbrunn AJ, Chandra NC, French WJ, Gore JM, RogersWJ. Clinical experience with primary PTCA compared with alteplase(rt-PA) in patients with acute myocardial infarction: a report fromthe Second National Registry of Myocardial Infarction (NRMI 2).J Am Coll Cardiol 1998;31:1240–5.
84. Kimmel SE, Berlin JA, Strom BL, Laskey WK. Development andvalidation of simplified predictive index for major complications incontemporary percutaneous transluminal coronary angioplasty prac-tice. The Registry Committee of the Society for Cardiac Angiogra-phy and Interventions. J Am Coll Cardiol 1995;26:931–8.
85. Jollis JG, Peterson ED, DeLong ER, et al. The relation between thevolume of coronary angioplasty procedures at hospitals treatingMedicare beneficiaries and short-term mortality. N Engl J Med1994;331:1625–29.
86. Ritchie JL, Phillips KA, Luft HS. Coronary angioplasty: statewideexperience in California. Circulation 1993;88:2735–43.
87. Peterson ED, Coombs LP, DeLong ER, Haan CK, Ferguson TB.Procedural volume as a marker of quality for CABG surgery. JAMA2004;291:195–201.
88. Ho V. Evolution of the volume-outcome relation for hospitalsperforming coronary angioplasty. Circulation 2000;101:1806–11.
89. Kimmel SE, Berlin JA, Laskey WK. The relationship betweencoronary angioplasty procedure volume and major complications.JAMA 1995;274:1137–42.
90. Magid DJ, Calonge BN, Rumsfeld JS, et al. Relation betweenhospital primary angioplasty volume and mortality for patients withacute MI treated with primary angioplasty vs thrombolytic therapy.JAMA 2000;284:3131–8.
91. Canto JG, Every NR, Magid DJ, et al. The volume of primaryangioplasty procedures and survival after acute myocardial infarction.National Registry of Myocardial Infarction 2 Investigators. N EnglJ Med 2000;342:1573–80.
92. Cannon CP, Gibson CM, Lambrew CT, et al. Relationship ofsymptom-onset-to-balloon time and door-to-balloon time with mor-tality in patients undergoing angioplasty for acute myocardial infarc-tion. JAMA 2000;283:2941–7.
93. Wennberg DE, Lucas FL, Siewers AE, Kellett MA, Malenka DJ.Outcomes of percutaneous coronary interventions performed atcenters without and with onsite coronary artery bypass graft surgery.JAMA 2004;292:1961–8.
94. McGrath PD, Wennberg DE, Dickens JD Jr., et al. Relationbetween operator and hospital volume and outcomes followingpercutaneous coronary interventions in the era of the coronary stent.JAMA 2000;284:3139–44.
95. Moscucci M, Share D, Smith D, et al. Relationship between operatorvolume and adverse outcome in contemporary percutaneous coronaryintervention practice: an analysis of a quality-controlled multicenterpercutaneous coronary intervention clinical database. J Am CollCardiol 2005;46:625–32.
96. Ellis SG, Weintraub W, Holmes D, Shaw R, Block PC, King SB III.Relation of operator volume and experience to procedural outcome of
King et al ACCF/AHA/SCAI Clinical Competence Statement 119

percutaneous coronary revascularization at hospitals with highinterventional volumes. Circulation 1997;95:2479–84.
97. Hannan EL, Racz M, Ryan TJ, et al. Coronary angioplasty volume-outcome relationships for hospitals and cardiologists. JAMA 1997;277:892–8.
98. Holmes DR, Selzer F, Johnston JM, et al. Modeling and riskprediction in the current era of interventional cardiology: a reportfrom the National Heart, Lung, and Blood Institute DynamicRegistry. Circulation 2003;107:1871–6.
99. McGrath PD, Malenka DJ, Wennberg DE, et al. Changing out-comes in percutaneous coronary interventions: a study of 34,752procedures in northern New England, 1990 to 1997. Northern NewEngland Cardiovascular Disease Study Group. J Am Coll Cardiol1999;34:674–80.
100. Bashore TM, Bates ER, Berger PB, et al. American College ofCardiology/Society for Cardiac Angiography and Interventions clin-ical expert consensus document on cardiac catheterization laboratorystandards: a report of the American College of Cardiology TaskForce on Clinical Expert Consensus Documents. J Am Coll Cardiol2001;37:2170–214.
101. Ellis SG, Weintraub W, Holmes DR Jr., et al. Relation of operatorvolume and experience to procedural outcome of percutaneouscoronary revascularization at hospitals with high interventional vol-umes. Circulation 1997;95:2479–84.
102. Beekman RH III, Hellenbrand WE, Lloyd TR, et al. ACCF/AHA/AAP recommendations for training in pediatric cardiology.Task force 3: training guidelines for pediatric cardiac catheteriza-tion and interventional cardiology endorsed by the Society forCardiovascular Angiography and Interventions. J Am Coll Cardiol2005;46:1388 –90.
103. Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, BildDE. Prevalence of hypertrophic cardiomyopathy in a general popu-lation of young adults. Echocardiographic analysis of 4111 subjects inthe CARDIA study. Coronary Artery Risk Development in (Young)Adults. Circulation 1995;92:785–9.
104. Maron BJ, McKenna WJ, Danielson GK, et al. American College ofCardiology/European Society of Cardiology clinical expert consensusdocument on hypertrophic cardiomyopathy: a report of the AmericanCollege of Cardiology Foundation Task Force on Clinical ExpertConsensus Documents and the European Society of CardiologyCommittee for Practice Guidelines. J Am Coll Cardiol 2003;42:1687–713.
105. Maron BJ. Surgery for hypertrophic obstructive cardiomyopathy:alive and quite well. Circulation 2005;111:2016–8.
106. Williams WG, Wigle ED, Rakowski H, Smallhorn J, LeBlanc J,Trusler GA. Results of surgery for hypertrophic obstructive cardio-myopathy. Circulation 1987;76:V104–8.
107. Woo A, Williams WG, Choi R, et al. Clinical and echocardiographicdeterminants of long-term survival after surgical myectomy in obstruc-tive hypertrophic cardiomyopathy. Circulation 2005;111:2033–41.
108. Bhargava B, Narang R, Aggarwal R, Bahl VK, Manchanda SC.Conduction blocks following transcatheter septal ablation for hyper-trophic cardiomyopathy. Eur Heart J 1997;18:2011–2.
109. Boltwood CM Jr., Chien W, Ports T. Ventricular tachycardiacomplicating alcohol septal ablation. N Engl J Med 2004;351:1914–5.
110. Faber L, Meissner A, Ziemssen P, Seggewiss H. Percutaneoustransluminal septal myocardial ablation for hypertrophic obstructivecardiomyopathy: long term follow up of the first series of 25 patients.Heart 2000;83:326–31.
111. Fernandes VL, Nagueh SF, Wang W, Roberts R, Spencer WH III.A prospective follow-up of alcohol septal ablation for symptomatichypertrophic obstructive cardiomyopathy—the Baylor experience(1996–2002). Clin Cardiol 2005;28:124–30.
112. Kern MJ, Holmes DG, Simpson C, Bitar SR, Rajjoub H. Delayedoccurrence of complete heart block without warning after alcoholseptal ablation for hypertrophic obstructive cardiomyopathy. Cathe-ter Cardiovasc Interv 2002;56:503–7.
113. Lakkis NM, Nagueh SF, Kleiman NS, et al. Echocardiography-guided ethanol septal reduction for hypertrophic obstructive cardio-myopathy. Circulation 1998;98:1750–5.
114. Nagueh SF, Ommen SR, Lakkis NM, et al. Comparison of ethanolseptal reduction therapy with surgical myectomy for the treatment ofhypertrophic obstructive cardiomyopathy. J Am Coll Cardiol 2001;38:1701–6.
115. Kuntz RE, Tosteson AN, Berman AD, et al. Predictors of event-freesurvival after balloon aortic valvuloplasty. N Engl J Med 1991;325:17–23.
116. Safian RD, Berman AD, Diver DJ, et al. Balloon aortic valvuloplastyin 170 consecutive patients. N Engl J Med 1988;319:125–30.
117. The National Heart, Lung, and Blood Institute Balloon Valvulo-plasty Registry Participants. Multicenter experience with balloonmitral commissurotomy. NHLBI Balloon Valvuloplasty RegistryReport on immediate and 30-day follow-up results. Circulation1992;85:448–61.
118. Complications and mortality of percutaneous balloon mitral commis-surotomy. A report from the National Heart, Lung, and BloodInstitute Balloon Valvuloplasty Registry. Circulation 1992;85:2014 –24.
119. Feldman T. Hemodynamic results, clinical outcome, and complica-tions of Inoue balloon mitral valvotomy. Cathet Cardiovasc Diagn1994;Suppl 2:2–7.
120. Rihal CS, Nishimura RA, Holmes DR Jr. Percutaneous balloonmitral valvuloplasty: the learning curve. Am Heart J 1991;122:1750–6.
121. Sanchez PL, Harrell LC, Salas RE, Palacios IF. Learning curve ofthe Inoue technique of percutaneous mitral balloon valvuloplasty.Am J Cardiol 2001;88:662–7.
120 Circulation July 3, 2007
APPENDIX 1. AUTHOR RELATIONSHIPS WITH INDUSTRY—ACCF/AHA/SCAI WRITING COMMITTEE TO UPDATETHE CLINICAL COMPETENCE STATEMENT ON CARDIAC INTERVENTIONAL PROCEDURES
Name ConsultantResearch
GrantScientific
Advisory BoardSpeakers’
BureauSteering
Committee Stock Holder Other
Dr. ThomasAversano
None None None None None None None
Dr. William L.Ballard
None None None None None None None
Dr. Robert H.Beekman, III
● AGA Medical None None None None None None
Dr. Michael J.Cowley
None None None None None None None
Dr. Stephen G.Ellis
● BostonScientific
● Celera● Cordis● Guidant● Viacon
● Celera● Centacor/Lilly● Cordis
● Boston Scientific● Cordis● Viacon
None None None None
Dr. David P.Faxon
● Bristol-MyersSquibb/Sanofi
None ● Boston Scientific None None ● Medical TechnologyInformational
None
Dr. Edward L.Hannon
None None None None None None None
Dr. John W.Hirshfeld, Jr.
None None ● Bracco Inc.● Bristol-MyersSquibb/Sanofi
None None None None
Dr. Alice K.Jacobs
None None None None None None ● Wyeth–Spouse’sEmployer
Dr. Mirle A.Kellett, Jr.
None None None None None None None
Dr. Stephen E.Kimmel
None None None None None None None
Dr. Spencer B.King, III
● Bristol-MyersSquibb
● CV Therapeutics● Sanofi/Aventis
None ● Medtronic ● Bristol-MyersSquibb
● Sanofi
None None ● Novoste–Royalties
Dr. Joel S.Landzberg
None None None None None None None
Dr. Louis S.McKeever
None None None None None None None
Dr. MauroMoscucci
None ● BlueCross/BlueShield
None ● Aventis● Pfizer
None None ● Cordis–FellowshipTraining Grant
Dr. Richard M.Pomerantz
● Medacorp.● Sanofi/Aventis
None None ● Sanofi/Aventis None ● Pfizer None
Dr. Karen M.Smith
None None None None None None None
Dr. George W.Vetrovec
Merck ● Cordis/Johnson& Johnson
● NHLBI
None ● Lilly● Pfizer
None ● Johnson & Johnson None
This table represents the relationships of committee members with industry that were reported orally at the initial writing committee meeting and updated in conjunction with all meetings and conference
calls of the writing committee during the document development process. It does not necessarily reflect relationships with industry at the time of publication.
King et al ACCF/AHA/SCAI Clinical Competence Statement 121

APPENDIX 2. PEER REVIEWER RELATIONSHIPS WITH INDUSTRY—ACCF/AHA/SCAI 2007 UPDATE OF THECLINICAL COMPETENCE STATEMENT ON CARDIAC INTERVENTIONAL PROCEDURES
Name Representation Consultant Research Grant
ScientificAdvisoryBoard
Speakers’Bureau
SteeringCommittee
StockHolder Other
Dr. John C. Giacomini ● Official–AHA None None None None None None None
Dr. Lawrence Laslett ● Official–ACCBoard ofGovernors
None None None None None ● GeneralElectric
None
Dr. Carl J. Pepine ● Official–ACCBoard ofTrustees
● Abbott● CV Therapeutics
● Abbott● AstraZeneca● BerlexLaboratories
● Pfizer
None None None None ● Educational grant–AstraZeneca,CV Therapeutics,GlaxoSmithKline,KingPharmaceuticals,MonarchPharmaceuticals,Pfizer,Sanofi-Aventis,Schering-Plough,Wyeth-AyerstLaboratories
Dr. Albert P. Rocchini ● Official–AHA None None None None None None None
Dr. Samuel J. Shubrooks ● Official–ACCBoard ofGovernors
None None None None None None None
Dr. Alan Yeung ● Official–AHA ● Medtronic ● Abbott● BostonScientific
● Abbott● BostonScientific
● Cordis
None None ● BostonScientific
None
Dr. Gregory Dehmer ● Organizational–Society forCardiovascularAngiographyandInterventions
None None None None None None None
Dr. John Hodgson ● Organizational–Society forCardiovascularAngiographyandInterventions
● Volcano Corp ● BostonScientific
● GE Medical● Lilly● RADI Medical● Volcano Corp
● Volcano Corp ● GE Medical● Pfizer
None ● TechnologySolutionsGroup
● BioInfoAcceleratorFund
● VolcanoCorp
● Management–MytogenTechnologySolutions Group
● BioInfo AcceleratorFund
Dr. Morton J. Kern ● Organizational–Society forCardiovascularAngiographyandInterventions
● Bracco Inc.● Meritt Medical● Therox Inc.
None None ● RADIMedical
None None None
Dr. Ronald Krone ● Organizational–Society forCardiovascularAngiographyandInterventions
None None None None None None None
Dr. Douglass A. Morrison ● Organizational–Society forCardiovascularAngiographyandInterventions
None None None None None None None
Dr. Mark Reisman ● Organizational–Society forCardiovascularAngiographyandInterventions
None None ● Abbott● BostonScientific
● Cordis● Medtronic
● BostonScientific
● Cordis
None None None
Dr. Barry Uretsky ● Organizational–Society forCardiovascularAngiographyandInterventions
None None None None None None None
Dr. Mazen Abu-Fadel ● Content–ACCFCardiacCatheterizationandInterventionCommittee
None None None None None None None
Dr. Peter Berger ● Content–IndividualReviewer
● BostonScientific
● Cordis/Johnson& Johnson
● Genentech● Guilford
● Cardiokinetix● Conor● Cordis/Johnson& Johnson
● Datascope● Guilford● Lilly● The MedicineCompany
● Medtronic● Sankyo● Sanofi-Aventis
● Arginox● Bristol-MyersSquibb
● Sanofi-Aventis
● Schering-Plough
None None ● Lumen, Inc None
Continued on next page
122 Circulation July 3, 2007
APPENDIX 2. Continued
Name Representation Consultant Research Grant
ScientificAdvisoryBoard
Speakers’Bureau
SteeringCommittee
StockHolder Other
Dr. Robert O. Bonow ● Content–PCIGuidelineWritingCommittee
None None None None None None None
Dr. Jose G. Diez ● Content–ACCFCardiacCatheterizationandInterventionCommittee
None None None None None None None
Dr. Ted E. Feldman ● Content–ACCFCardiacCatheterizationandInterventionCommittee
● BostonScientific
● CardiacDimensions
● Cordis● Myocor
● Abbott● Atritech● BostonScientific
● CardiacDimensions
● Cordis● Evalve
None None None None None
Dr. James Ferguson ● Content–IndividualReviewer
None None None None None None None
Dr. Tommaso Gori ● Content–AHADiagnostic &InterventionalCardiacCatherizationCommittee
None None None None None None None
Dr. Hani Jneid ● Content–AHADiagnostic &InterventionalCardiacCatheterizationCommittee
None Pfizer None None None None None
Dr. Fred M. Krainin ● Content–ACCFCardiacCatheterizationandInterventionCommittee
None None None None None ● BostonScientific
● Johnson &Johnson
● Medtronic
None
Dr. Glenn Levine ● Content–AHADiagnostic &InterventionalCardiacCatheterizationCommittee
None None None None None None None
Dr. Charanjit S. Rihal ● Content–ACCFCardiacCatheterizationandInterventionCommittee
None None None None None None None
Dr. Dan M. Roden ● Content–IndividualReviewer
● Abbott● Alza● Arpida● AstraZeneca● Bristol-MyersSquibb
● CV Therapeutics● EBR Systems● First GeneticTrust
● GlaxoSmithKline● Genzyme● Johnson &Johnson
● Lexicon● Lundbeck● Medtronic● Merck● NPSPharmaceuticals
● Novartis● Pfizer● Sanofi-SynthelaboGroupe
● Solvay● ThorntonMedical
● Wyeth● Yamanouchi
None None None None None None
Dr. Carlos Ruiz ● Content–ACCFCardiacCatheterizationandInterventionCommittee
None None None None None None None
Dr. Michael J. Silka ● Content–IndividualReviewer
None None None None None None ● General Electric
Continued on next page
King et al ACCF/AHA/SCAI Clinical Competence Statement 123
APPENDIX 2. Continued
Name Representation Consultant Research Grant
ScientificAdvisoryBoard
Speakers’Bureau
SteeringCommittee
StockHolder Other
Dr. Thoralf M. Sundt ● Content–ACCFCardiacCatheterizationandInterventionCommittee
None None None None None ● Medtronic(son hasstock)
None
Dr. Cynthia M. Tracy ● Content–IndividualReviewer
None ● Guidant Corp● Medtronic
None None None None None
Dr. E. Murat Tuzcu ● Content–AHADiagnostic &InterventionalCardiacCatheterizationCommittee
None None None None None None None
Dr. Matthew Wolff ● Content–ACCFCardiacCatheterizationandInterventionCommittee
None None None None None None None
Dr. Yerem Yeghazarans ● Content–IndividualReviewer
None None None ● Pfizer● Sanofi-Aventis
None None None
This table represents the relationships of peer reviewers with industry that they reported as relevant to this topic. It does not necessarily reflect relationships with industry at the time of publication.Participation in the peer review process does not imply endorsement of the document. Names are listed in alphabetical order within each category of review.
124 Circulation July 3, 2007

An Unusual Site for a Common DiseaseMaysaa Alzetani, MRCP, MSc; Joseph J. Boyle, MRCP, PhD;
David Lefroy, MA, MB, BChir, FRCP; Petros Nihoyannopoulos, MD, FRCP
A 75-year-old Asian woman presented with a 5-monthhistory of night sweats, lethargy, and malaise. On
admission she was found to have low-grade pyrexia andelevated inflammatory markers. Septic screen, which in-cluded repeated blood cultures and chest x-ray (Figure 1),were negative. A transthoracic and transesophageal echocar-diography revealed a doughnut-shaped mass (online DataSupplement Movie I) that surrounded the mitral valve annu-lus and extended up to the left atrial walls and atrial septum.A surgical biopsy was taken (Figure 2), which showedepithelioid and langhans giant cell granulomas with centralcaseating necrosis consistent with tuberculosis. Special stain-ing with high-sensitivity immunoperoxidase confirmed thediagnosis of tuberculosis. The patient was treated for tuber-culosis with complete resolution of her symptoms. Repeatedechocardiography 6 months later showed a dramatic reduc-tion of the mass size (Data Supplement Movies II and III).
Isolated cardiac tuberculosis is extremely rare. However itshould be included in the differential diagnosis of intracardiacmasses.
Sources of FundingDr Boyle has received research support from the British HeartFoundation, KRUK, HHRTC, and the Broad Foundation.
DisclosuresDr Boyle has received honoraria from the International Journal ofExperimental Pathology, has served as a speaker for the HistochemicalSociety and the British Atherosclerosis Society as a member of theeditorial board for the International Journal of Experimental Pathology,and as an expert witness to UK coroners and mesothelioma panels.
Figure 1. Posterior-anterior chest x-ray, taken on admission,shows clear lung fields and no signs of infection.
Figure 2. Biopsy taken from the mass that surrounded themitral valve annulus and extended up to the left atrial wallsand atrial septum. Hematoxylin and eosin staining. Magnifica-tion, �20. Ep indicates epithelioid macrophages; L, Langhansgiant cells; and N, necrosis. Inset, immunohistochemistry withmonoclonal antibody to mycobacterium (Dako, Glostrup, Den-mark) and immunoperoxidase (Menarini Diagnostic, Woking-ham, UK). B indicates bacillus; M, macrophage. Magnification,�100 (cropped for space).
From the Hammersmith Hospital NHS Trust (M.A.), and the Histopathology Department (J.J.B.) and Hammersmith Hospital (D.L., P.N.), ImperialCollege, London, UK.
The online-only Data Supplement, consisting of movies, is available with this article at http://circ.ahajournals.org/cgi/content/full/116/1/e1/DC1.Correspondence to Dr Maysaa Alzetani, 42 Tryfan Close, Ilford, London IG4 5JY, United Kingdom. E-mail [email protected](Circulation. 2007;116:e1.)© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.106.677120

Sine-Wave Pattern Arrhythmia and Sudden Paralysis ThatResult From Severe HyperkalemiaMaurice J.H.M. Pluijmen, MD; Ferry M.R.J. Hersbach, MD
A 54-year-old man with a history of end-stage renaldisease treated with hemodialysis presented to the emer-
gency department because of a sudden inability to move hislimbs.
Physical examination revealed a complete quadriplegia.Blood pressure was 145/90 mm Hg, with a regular pulse of 45beats per minute. Initial 12-lead electrocardiography showeda sinus bradycardia with atrial, atrioventricular, and intraven-tricular conduction delay (Figure 1). Subsequently, the pa-tient developed several episodes of a nonsustained wide-complex tachycardia with a right bundle-branch blockconfiguration that gradually evolved into and from a “sine-wave” pattern (Figures 2 and 3). Remarkably, the patientremained hemodynamically stable. Because hyperkalemiawas suspected, serum potassium was determined in venous aswell as arterial blood samples to be 9.9 mmol/L. Calciumgluconate was administered, and emergency hemodialysiswas performed. After normalization of the serum potassiumconcentration, sinus rhythm was maintained, the cardiacconduction times returned to normal (Figure 4), and thequadriplegia resolved completely.
This case illustrates some typical features of severe hyper-kalemia. Initial characteristic electrocardiographic abnormal-ities in hyperkalemia are tall and peaked T waves, followedby an increasing cardiac conduction delay. As demonstratedin this case, this results in flattened and broadened P waves,an atrioventricular block of first or higher degrees, andwidening of the QRS complex. In rare instances, ST-segmentelevation may occur, which leads to a “pseudoinfarction”
pattern.1 Progression of hyperkalemia causes further widening ofthe QRS complex, often with the configuration of a left or rightbundle-branch block. Eventually, merger of QRS complex and Twave will lead to the appearance of a typical sine-wave pattern.Contrary to our patient, a sine-wave pattern often precedesventricular fibrillation or asystole.2 Furthermore, rapidly pro-gressing flaccid motor weakness may result in a quadriplegia,which was the presenting symptom in this case and is anuncommon manifestation of severe hyperkalemia that mayultimately result in respiratory failure.2,3
Recognition of the combination of sudden paralysis andelectrocardiographic abnormalities as demonstrated in thiscase can lead to early diagnosis and treatment of severehyperkalemia.
DisclosuresNone.
References1. Sims DB, Sperling LS. Images in cardiovascular medicine. ST-segment
elevation resulting from hyperkalemia. Circulation. 2005;111:e295–e296.
2. 2005 American Heart Association Guidelines for CardiopulmonaryResuscitation and Emergency Cardiovascular Care, Part 10.1. Life-threatening electrolyte abnormalities. Circulation. 2005;112:IV121–IV125.
3. Freeman SJ, Fale AD. Muscular paralysis and ventilatory failure causedby hyperkalaemia. Br J Anaesth. 1993;70:226–227.
From the Department of Cardiology, Medisch Centrum Rijnmond-Zuid, Rotterdam, the Netherlands.Correspondence to Dr Maurice J.H.M. Pluijmen, Groene Hilledijk 315, 3075EA Rotterdam, The Netherlands. E-mail [email protected](Circulation. 2007;116:e2-e4.)© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.106.687202
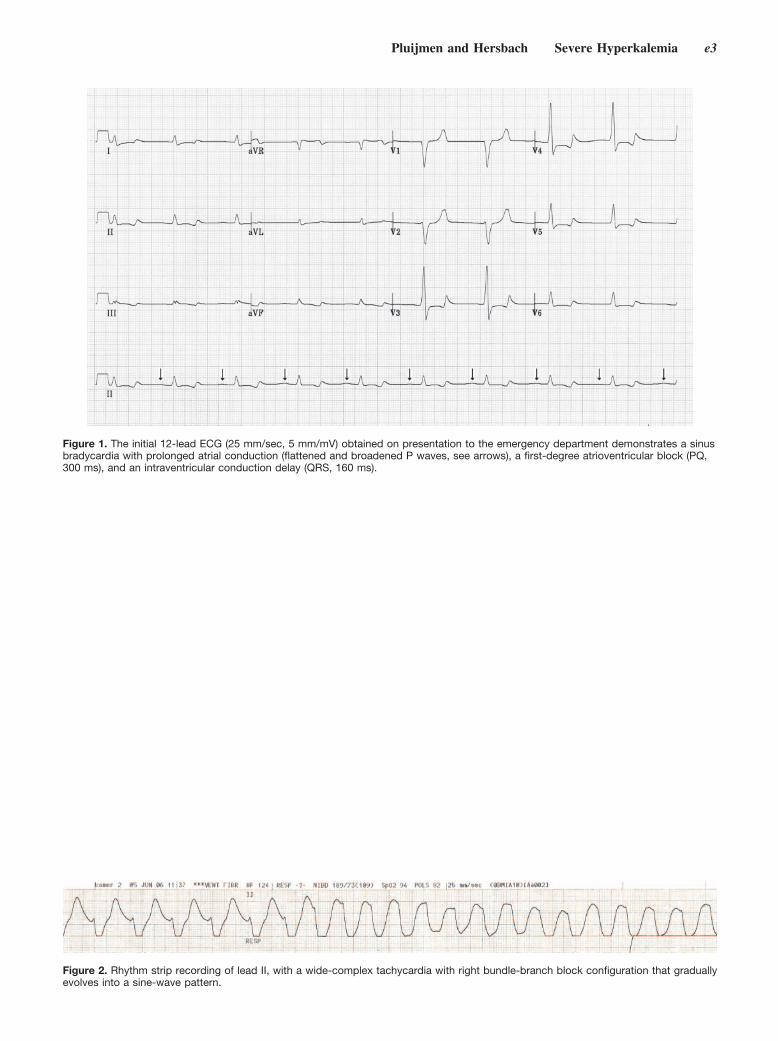
Figure 1. The initial 12-lead ECG (25 mm/sec, 5 mm/mV) obtained on presentation to the emergency department demonstrates a sinusbradycardia with prolonged atrial conduction (flattened and broadened P waves, see arrows), a first-degree atrioventricular block (PQ,300 ms), and an intraventricular conduction delay (QRS, 160 ms).
Figure 2. Rhythm strip recording of lead II, with a wide-complex tachycardia with right bundle-branch block configuration that graduallyevolves into a sine-wave pattern.
Pluijmen and Hersbach Severe Hyperkalemia e3

Figure 3. This 12-lead ECG (25 mm/sec, 10 mm/mV) of the wide-complex tachycardia (QRS, 240 ms) demonstrates the right bundle-branch block configuration that evolves into and from a sine-wave pattern.
Figure 4. The 12-lead ECG (25 mm/sec, 5 mm/mV) after normalization of the serum potassium concentration reveals the normalizationof atrial, atrioventricular (PQ, 140 ms), and intraventricular (QRS, 80 ms) conduction times, as well as a preexistent left ventricular hy-pertrophy pattern.
e4 Circulation July 3, 2007

Lipomatous Metaplasia in Ischemic CardiomyopathyA Common but Unappreciated Entity
Matthias Schmitt, MD, PhD, MRCP; Nilesh Samani, BSc, MB, ChB, MD, FRCP, FmedSci;Gerry McCann, BSc, MB, ChB, MD, MRCP
A68-year-old man with a 14 1year prior history of anteriormyocardial infarction was referred for viability assess-
ment. He had established 3-vessel coronary artery diseasewith a proximally occluded left anterior descending coronaryartery. He complained of worsening shortness of breath anddiminishing exercise tolerance. Cardiac magnetic resonanceimaging demonstrated a nondilated left ventricle with minoraneurysmal transformation that affected the mid-anterior walland extended into mid- and antero-septum as well as the apex.Gradient (Figure, A) and T1-weighted spin-echo imagesdemonstrated bright signal intensity in the mid-myocardiumof the anterior wall (see arrows in Figure, B), which disap-peared with fat saturation (see arrows in Figure, C and D).These findings are indicative of lipomatous metaplasia (LM).The term LM describes fat that is present in and seeminglyreplaces scar tissue in the myocardium. The exact etiology ofLM is unknown, but it is not seen in the absence ofsubstitutive myocardial fibrosis. Histological evidence of LMhas been found in up to 68%, 24%, and 37% of areas of leftventricular myocardial scars in explanted hearts of patientswho underwent transplantation for ischemic heart disease,
idiopathic dilated cardiomyopathy, or chronic valvulopathy,respectively.1 Myocardial perfusion imaging (lipohilic myo-cardial perfusion agents such as tetrofosmin and sestamibimay well be taken up into fat cells) and echocardiography failto diagnose LM (which accounts for the lack of recognition ofthis not uncommon entity) with the consequent implicationsfor overestimation of viable myocardium or underestimationof scar size.
Importantly, LM on magnetic resonance imaging must notbe mistaken for late enhancement after gadolinium contrast.
DisclosuresNone.
References1. Baroldi G, Silver MD, De Maria R, Parodi O, Pellegrini A. Lipo-
matous metaplasia in left ventricular scar. Can J Cardiol. 1997;13:65–71.
From the Department of Cardiology, Glenfield Regional Cardiac Centre, Leicester University Hospital Trust, Leicester, UK.The online-only Data Supplement, consisting of movies, is available with this article at http://circ.ahajournals.org/cgi/content/full/116/1/e5/DC1.Correspondence to Dr Matthias Schmitt, Department of Cardiology, Glenfield Regional Cardiac Centre, Leicester University Hospital Trust, Groby
Road, Leicester LE3 9QP, UK. E-mail [email protected](Circulation. 2007;116:e5-e6.)© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.107.690800
e5
Images in Cardiovascular Medicine

Short-axis images at mid-cavity level (Aand C) and long-axis images (B and D).White arrows mark high signal intensity(no gadolinium given) in the anterior wall(A and B) that turns black in correspond-ing fat suppression sequence (C and D).
e6 Circulation July 3, 2007

Letter by Brewster and van MontfransRegarding Article, “Risks Associated WithStatin Therapy: A Systematic Overview ofRandomized Clinical Trials”To the Editor:
Kashani et al1 report that in randomized trials, statintherapy (with the exclusion of cerivastatin) did not result insignificant absolute increases in myalgias (risk difference per1000 patients, 2.7 (95% CI, �3.2 to 8.7), or mild creatinekinase (CK) elevations 0.2 (95% CI, �0.6 to 0.9). The authorsused data from published trials to reach this conclusion.
However, Kashani et al did not take into account that manystatin trials disclose that eligible patients with muscle complaints,previous adverse responses to cholesterol-lowering therapy, oreven a mild asymptomatic elevation of CK (�1.5 to 6.0 times theupper limit of normal) are not randomized.2–5
On the basis of the complete exclusion criteria, up to 76% ofthe screened participants in statin trials are not randomized andexcluded.2–5 Thus, the incidence of hyperCKemia with the use ofstatins that emerges from these trials may mainly concernsubjects in the lower part of the CK distribution, with a lowa priori risk to develop highly elevated CK levels.
The exclusion of these patients before randomization may leadto biased reports on the frequency of occurrence of side effectswith statin use. This should be acknowledged to be a limitationwhen adverse effects associated with statins are assessed inpublished trials.
DisclosuresNone.
Lizzy M. Brewster, MDGert A. van Montfrans, MD
Departments of Internal and Vascular MedicineAcademic Medical Center F4-222
University of AmsterdamAmsterdam, The Netherlands
1. Kashani A, Phillips CO, Foody JM, Wang Y, Mangalmurti S, Ko DT,Krumholz HM. Risks associated with statin therapy: a systematic overviewof randomized clinical trials. Circulation. 2006;114:2788–2797.
2. LaRosa JC, Grundy SM, Waters DD, Shear C, Barter P, Fruchart JC, GottoAM, Greten H, Kastelein JJ, Shepherd J, Wenger NK; Treating to NewTargets (TNT) Investigators. Intensive lipid lowering with atorvastatin inpatients with stable coronary disease. N Engl J Med. 2005;352:1425–1435.
3. Heart Protection Study Collaborative Group. MRC/BHF Heart ProtectionStudy of cholesterol lowering with simvastatin in 20,536 high-risk indi-viduals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22.
4. Shepherd J, Blauw GJ, Murphy MB, Bollen EL, Buckley BM, Cobbe SM,Ford I, Gaw A, Hyland M, Jukema JW, Kamper AM, Macfarlane PW,Meinders AE, Norrie J, Packard CJ, Perry IJ, Stott DJ, Sweeney BJ,Twomey C, Westendorp RG; PROSPER Study Group. Pravastatin inelderly individuals at risk of vascular disease (PROSPER): a randomisedcontrolled trial. Lancet. 2002;360:1623–1630.
5. Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW,McKillop JH, Packard CJ. Prevention of coronary heart disease withpravastatin in men with hypercholesterolemia. West of Scotland CoronaryPrevention Study Group. N Engl J Med. 1995;333:1301–1307.
(Circulation. 2007;116:e7.)© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.107.689497
e7
Correspondence

Letter by Rosenberg and Uretsky Regarding Article,“Risks Associated With Statin Therapy: ASystematic Overview of Randomized Clinical Trials”To the Editor:
In regard to the article by Kashani et al,1 the authorsconclude that “...statin therapy is associated with small excessrisk of transminase elevations, but not of myalgias, creatinekinase elevations, rhabdomyolysis...”. We think this conclu-sion is misleading. On closer inspection of the data, atorva-statin has 3 times the occurrence of myalgias compared withplacebo (P�0.04). According to the authors’ data, theseadverse events seem to be unique to atorvastin and were notobserved with other statins. Admittedly the absolute risk issmall; however, when one considers that in the US alone �5million patients are presently treated with atorvastatin, this
drug could still account for myalgias in �250 000 patients—not quite an insignificant number!
DisclosuresNone.
Lauren Rosenberg, MDSeth Uretsky, MD
St Luke’s-Roosevelt Medical CenterColumbia University College of Physicians and Surgeons
New York, NY
1. Kashani A, Phillips CO, Foody JM, Wang Y, Mangalmurti S, Ko DT, KrumholzHM. Risks associated with statin therapy: a systematic overview of randomizedclinical trials. Circulation. 2006;114:2788–2797.
(Circulation. 2007;116:e8.)© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.107.690867
e8
Correspondence

Response to Letters Regarding Article, “RisksAssociated With Statin Therapy: A SystematicOverview of Randomized Clinical Trials”
The letters that address our analysis of risks associated with statintherapy1 raise 2 important points: the generalizability of the data andthe excess incidence of myalgias observed with atorvastatin therapy.Although inclusion/exclusion criteria required for clinical trials limitgeneralizability to patients in clinical practice, randomized con-trolled trials remain the best unbiased source of data to assessadverse effects.2,3 However, there remains a need for additional,large, safety studies in populations previously not studied.
In their letter, Drs Brewster and van Montfrans indicate thatsome statin trials exclude patients with elevated creatine kinaselevels (�1.5 to 6 times the upper limit of normal). We believethat these patients are appropriately excluded, as patients withextreme creatine kinase elevations have an underlying pathologyand may represent a population inappropriate for statin therapy.
The issue of excess incidence of myalgia observed withatorvastatin, raised in the letter from Drs Rosenberg and Uretsky,merits further investigation. This observation reached marginalstatistical significance (P�0.04) and was based on only 567patients (19 of 375 patients versus 3 of 192 patients in thetreatment and placebo groups, respectively). Accordingly, thisfinding is worth further pursuit, but should not be considereddefinitive at this time.
DisclosuresDr Foody received honoraria from and served as a consultant/advisory board member for Merck, BMS/Sanofi, and Pfizer. Theother authors have nothing to disclose.
Amir Kashani, MS, MDJoAnne M. Foody, MD
Yongfei Wang, MSHarlan M. Krumholz, MD, SM
Section of Cardiovascular MedicineDepartment of Medicine
Yale University School of MedicineNew Haven, Conn
Christopher O. Phillips, MD, MPHDepartment of General Internal Medicine
The Cleveland Clinic FoundationCleveland, Ohio
Sandeep Mangalmurti, MDAmbulatory Health Clinic
United States NavyGroton, Conn
Dennis T. Ko, MDSchlich Heart Centre
Sunnybrook Health Sciences CentreOntario, Canada
1. Kashani A, Phillips CO, Foody JM, Wang Y, Mangalmurti S, Ko DT,Krumholz HM. Risks associated with statin therapy: a systematicoverview of randomized clinical trials. Circulation. 2006;114:2788 –2797.
2. Curtin F, Altman DG, Elbourne D. Meta-analysis combining paralleland cross-over clinical trials. I: Continuous outcomes. Stat Med.2002;21:2131–2144.
3. Curtin F, Elbourne D, Altman DG. Meta-analysis combining parallel andcross-over clinical trials. III: The issue of carry-over. Stat Med.2002;21:2161–2173.
(Circulation. 2007;116:e9.)© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.107.697227
e9
Correspondence

The editors express appreciation to the following referees who served from July 1, 2006, though December 31, 2006.
Einari AavikInmaculada B. AbanNicola AbateAmr E. AbbasAntonio AbbateJinnette Dawn AbbottMohamed Abdel-WahabHazem Abdul HussienE. Dale AbelJamil A. AboulHosnM. Roselle AbrahamNader G. AbrahamTheodore P. AbrahamHugues AbrielElias AbrutynStephan AchenbachPaul E. AchouhMichael AckerMichael J. AckermanVolker AdamsPhilip A. AdesSrilakshmi M. AdhyapakGail K. AdlerVahid Afshar-KharghanSalvatore AgatiPiergiuseppe AgostoniEustachio AgricolaPietro Maria G. AgricolaDavid AguilarSeyedhossein AharinejadFerhaan AhmadAli AhmedAlsir A.M. AhmedIsmayil AhmetYoungkeun AhnSeema S. AhujaDiana AicherElena AikawaMasanori AikawaRyuichi AikawaJudith A. AireyAnthony AizerAdesuyi A. AjayiNadine AjzenbergTankut Hakki AkayShahab A. AkhterOlakunle O. AkinboboyeMasahiro AkishitaAmin Al-AhmadDagmar G. AlberAlexander AlbertChristine M. AlbertJeffrey AlbertMichelle AlbertGeorge AlbertiGabriel AldeaYvette B.J. Aldenhoff
Michael H. AldermanSalvatore AlesciAnthony AletrasBarbara AlexanderM. Yvonne AlexanderMark E. AlexanderR. Wayne AlexanderAndrei V. AlexandrovAyyaz A. AliZiad A. AliKari AlitaloLindsey D. AllanLarry Alexander AllenPaul D. AllenMaurits A. AllessieMatthew A. AllisonThomas G. AllisonCarlos Alonso-VillaverdeNicholas AlpBahaaldin AlsoufiPeter A. AltmanSalomon AmarPierre AmarencoJohn A. AmbroseGiuseppe AmbrosioPeter AmmannEnrico AmmiratiEzra AmsterdamDhakshinamurthy Vijay AnandInder S. AnandSonia S. AnandNagesh Sadanand AnavekarUlla Overgaard AndersenGarnet L. AndersonH. Vernon AndersonJeffrey L. AndersonKelley P. AndersonMark E. AndersonPeter G. AndersonRobert H. AndersonTodd J. AndersonStefan AndreasArne Kristian AndreassenMaria Grazia AndreassiFelicita AndreottiDouglas AndresDominick J. AngiolilloStefan D. AnkerBrian H. AnnexBenjamin AnsellJack AnsellAmedeo AnselmiDavid AntoniucciJovan P. AntovicCharles AntzelevitchPiero AnversaAni Anyanwu
Toshihisa AnzaiCaroline M. ApovianChristina L. AquilanteAndrew E. AraiJack L. ArbiserAbbas ArdehaliRoss ArenaMiguel A. AriasRobert A. AriensChiara ArmaniGary C. ArmitageEhrin Johnson ArmstrongPaul W. ArmstrongDonna K. ArnettDoron AronsonRishi AroraRohit AroraUmesh AroraHiroshi AsanumaRaimondo AscioneMuhammad AshrafSamuel J. AsirvathamPeter AspelinGerd AssmannBirgit AssmusNimer N. AssyCarmela AsteriaAnne Sofie AstrupBela F. AsztalosDianne L. AtkinsJohn P. AtkinsonPavan AtluriDouwe E. AtsmaHavard AttramadalAndrew M. AtzAngelo AuricchioGerard P. AurigemmaMichael V. AutieriPablo AvanzasMaurizio R. AvernaAntonio AversaNili AvidanAbraham AvivAngelo AvogaroAlberto AvolioOla A. AwadVitor M.P. Azevedo
Gerard BabatasiFritz H. BachRichard G. BachEmile BachaStephen L. BacharachRobert J. BacheJean E. BachetMarkus Michael BachschmidCarl Backer
Peter BackxLarry BaddourMichael BaderNitish BadhwarJuan Jose BadimonEmilio BadoerHala M. BadranStephen F. BadylakMan Jong BaekEmilia BagiellaSteven BaileyJean-Patrice BaillargeonDonald S. BaimAlison E. BairdAndrew H. BakerGeorge L. BakrisStephan BaldusH. Scott BaldwinChristie M. BallantyneJean-Luc BalligandScott W. BallingerAndriy BandosEddy BaraschJohn C. BarbatoSilvia Stella BarbieriJohn C. BarefootAmber E. BarnatoAdrian G. BarnettFrancesco Barone-AdesiJose A. BarrabesRobyn J. BarstPhilip BarterJohn R. BartholomewMatthias BartonPeter BartschRiyaz BashirThomas M. BashoreTheodore A. BassEric R. BatesPhilip M.W. BathD. BattlePaulo Fernando Dotto BauRobert BauernschmittJohann BauersachsKenneth Lee BaughmanJan H. BaumertDaniela BaumgartnerHelmut BaumgartnerIris BaumgartnerRalf W. BaumgartnerWilliam BaumgartnerJeroen J. BaxJeroen BaxB. Timothy BaxterGary F. BaxterAntoni Bayés de LunaW. Scott Beattie
e10
Acknowledgment of Reviewers

James BeckDiane M. BeckerEdmund R. BeckerRichard C. BeckerJoshua A. BeckmanThomas R. BehrenbeckVidar BeisvagBernard BelhassenJonathan N. BellaCarsten J. BellerGeorge A. BellerMarek BelohlavekLotfi Ben MimeDavid G. BendittUmberto BenedettoFederico Jose BenettiYanai Ben-GalFrank M. BengelIvor J. BenjaminRalf BenndorfJoel S. BennettD. Woodrow BensonRobert A. BergAlan K. BergerMartin W. BergmannZekarias BerhaneBradford C. BerkDaniel S. BermanJose M. BernalSheilah A. BernardLuciano BernardiMichael C. BerndtSusannah BernheimDaniel BernsteinStuart S. BerrDonald M. BersGiuseppe S. BertonAlain G. BertoniGabriele B. BertoniJoline W.J. BeulensSteve BevanFriedhelm BeyersdorfConnie R. BezzinaDwaipayan BharadwajAruni BhatnagarDeepak BhatnagarDeepak L. BhattJinsong BianGiuseppe BianchiGiorgio M. BiasiLuigi Marzio BiasucciKirsten Bibbins-DomingoJ. Thomas BiggerDiane E. BildGeorge E. BillmanOfer BinahChristian BinggeliAndreia BioloRainer BirckYochai BirnbaumJonathan Birns
Joyce BischoffNanette H. BishopricGianluigi BisleriJohn A. BittlVera BittnerMartin BjörckHenry W. BlackburnEugene H. BlackstoneStefan BlankenbergJames C. BlankenshipAndrew D. BlannSabine BleizifferRudiger BlindtWilhelm BlochPeter C. BlockHanna BloomfieldDavid A. BluemkeElizabeth D. BlumeRoger S. BlumenthalYuri V. BobryshevJorge B. BoczkowskiChristoph BodeWilliam E. BodenJohannes BoehmMatthijs BoekholdtCornelis BoersmaEric BoersmaDaniel J. BoffaRainer H. BogerFrank BogunBarry Anthony BoilsonWilliam BoisvertAnn BolgerRoberto BolliMarvin O. BoluytMassimo BonacchiNikolaos BonarosMark BondEnzo BonoraRobert O. BonowMunir BoodhwaniGeorge W. BoozJeffrey S. BorerMichael A. BorgerSusanna Etje BorggreveClaudio BorghiChristina A. BorosDirk BöseKritina BostromRené M. BotnarMichiel L. BotsMorten BottcherElias H. BotvinickTarek BouhaliAnne BouloumieMartial G. BourassaPascal BousquetEdward L. BoveThierry BoveFrieke BoxPenelope A. Boyden
Mark R. BoyettAndrew J. BoyleE. H. BradleyT. Douglas BradleySusan BrainRandy W. BraithStefan-Martin Brand-HerrmannRuediger C. Braun-DullaeusEugene BraunwaldAlan C. BravermanGeorge A. BrayBernhard R. BrehmOle A. BreithardtSorin J. BrenerRobert T. BrennanHermann BrennerKate M. BrettSally E. BrettMartin BreuerGregorio BrevettiCharles R. BridgesDavid BriegerFrancoise BrietJurgen BrinckmannMichael R. BristowSusanne BroCraig BrobergOtto-Erich BroddeSergey V. BrodskyUlrich BroeckelRobert D. BrookMaria Mori BrooksJames M. BrophyMeg E. BrousseauIngeborg A. BrouwerGregory L. BrowerB. Greg BrownClive M. BrownDavid A. BrownJeremiah R. BrownNancy J. BrownMartina BrueckmannMartina BruecknerRamon BrugadaEric J. BrunnerChris L. BrysonMichael R. BuchananV.M. BuckalewMatthew J. BudoffBrian BuijsseL. Maximilian BujaHarry R. BullerT. Jared BunchAllen P. BurkeGregory L. BurkeJohn C. BurnettMichel BurnierIvo BuschmannSebastian J. BussRudi BussePeter M. Buttrick
Denis BuxtonGraham B. Byrnes
Candido CaboHoward CabralEvren CaglayanMichael E. CainFrancesc CalafellAntonio Maria CalafioreA. Louise CalderJames H. CaldwellMary CaldwellDavid A. CalhounRobert M. CaliffHugh CalkinsRonald CallahanDavid J. CallansFrancois A. CambienVicky A. CameronPaolo G. CamiciA. John CammDuncan J. CampbellJulie H. CampbellUmberto CampiaAnn E. CanfieldChristopher P. CannonRichard O. CannonJohn G. CantoNoel M. CapliceMaurizo C. CapogrossiThomas P. CappolaMassimo CaputoBlase A. CarabelloChristopher M. CarlinMark David CarlsonRobert M. CarneyJohn Alfred CarrThierry P. CarrelOscar A. CarreteroAlain CarrieMichel CarrierJohn D. CarrollAndrew J. CarterAngela M. CarterRaymond CartierBarbara CasadeiJuan Pablo CasasIvan Pearse CasserlyDaniel F. CatanzaroMartha CathcartChristophe CaussinErdal CavusogluSerghei CebotariFrank CecchinDavid S. CelermajerAntonio CerielloMatteo CesariJuan C. ChachquesClaudia U. ChaeBernard R. ChaitmanSubrata ChakrabartiAravinda Chakravarti
Acknowledgment of Reviewers e11

Lorraine ChalifourJohn ChalmersHunter Clay ChampionWing Bun ChanKrishnaswamy ChandrasekaranY. ChandrashekharChih-Jen ChangJoyce Chung-Chou Ho ChangRuey-Kang R. ChangKeith M. ChannonJean-Pierre ChanoineMiguel ChaputMarietta CharakidaFadi Joseph CharcharIsrael F. CharoJohn C. ChathamKanu ChatterjeeSarwat ChaudhryIrshad H. ChaudryVijay S. ChauhanMelvin D. CheitlinAlex F. ChenChunguang ChenFrederick Y.Y. ChenHua Yun ChenIan Y. ChenJiu-Chiuan ChenJu ChenKai ChenPeng-Sheng ChenShih-Ann ChenYiu-Fai ChenBin ChengDebbie ChengTsung O. ChengYu ChengGlenn M. ChertowAdrian H. ChesterBernard M.Y. CheungMordechai ChevionShu ChienJohn S. ChildWilliam M. ChilianMichael T. ChinRandolph W. Chitwood-JrRay C.J. ChiuAram V. ChobanianYeon Hyeon ChoeMichel B. ChoncholMagdalena Chottova DvorakovaBenjamin J.W. ChowPhil ChowienczykTorsten ChristGeir ChristensenTimothy F. ChristianKarkos D. ChristosSumeet S. ChughMoo K. ChungJuan CincaFrancesco CipolloneMarco Cirillo
Leslie L. ClarkRobert ClarkeMatthias A. ClaussJohn G.F. ClelandDenis L. ClementRichard T. ClementsTon J. CleophasJack P.M. CleutjensRoger L. ClickWilliam T. ClusinMichael A. CoadyChristopher S. CoffeyDavid J. CohenHillel W. CohenMeryl S. CohenMichael V. CohenRichard A. CohenSmadar CohenStanley N. CohenJay N. CohnLawrence H. CohnWilliam E. CohnDuncan Robert ColesAndrea ColliAlan R. CollinsRobert W. ColmanAntonio ColomboCatherine CommunalGianluigi CondorelliPaul R. ConlinJohn E. ConnettAgostino ConsoliNancy R. CookStephen R. CookThomas CookJohn P. CookeJoshua M. CooperLeslie T. CooperMark E. CooperRhonda M. Cooper-DeHoffJames CoromilasDomenico CorradoEgle CorradoJames P. CorsettiRoberto CortiFrancesco CosentinoMarco A. CostaSalvatore CostaNathalie Costedoat-ChalumeauWilliam G. CottsPatrick Anthony CoughlinDavid CouperFrancis CouturaudLinda D. CowanAllen W. CowleyDermot CoxCiprian M. CrainiceanuSybil CrawfordFilippo CreaMark A. CreagerHarry Crijns
Michael H. CiquiJulia Alison CritchleyBernard Lewis CroalCarrollE. CrossLuigi X. CubedduBruce F. CulletonWilliam C. CulpAnne B. CurtisRicardo C. CuryMary CushmanDaniele M. CusiJeffrey A. CutlerDonald E. Cutlip
Michael W. DaeMat J.A.P. DaemenMichael DaffertshoferMarinos C. DalakasDarshan DalalJames E. DalenRonald L. DalmanCaroline A. DalyJan Kristian DamasDorte DamgaardMichael DandelGeorge DangasJean-Marie Daniel LamaziereStephen R. DanielsA.H. Jan DanserLara Danziger-IsakovJeanine M. D’ArmientoDipak K. DasSandeep DasKaberi DasguptaJames DaubertJean Claude DaubertSiamak DavaniAnthony P. DavenportTirone E. DavidCharles J. DavidsonKarina W. DavidsonMichael H. DavidsonPeter F. DaviesVictor G. Davila-RomanBryce H. DavisRoger B. DavisRobin L. DavissonKevin P. DavyBuddhadeb DawnJeffrey D. DawsonJonathan R.S. DayMariza de AndradeDirk De BacquerJacques de BakkerBernard De BruyneMarco De CarloRaffaele De CaterinaCarlos R. De DiegoSarah D. de FerrantiPim J. de FeyterDominique P. de KleijnPeter W. de Leeuw
James A. de LemosMoniek P.M. de MaatRamon de NooijerAnne-Cornelie J.M. de PontAlbert de RoosGiovanni de SimoneBianca L. De StavolaPieter P. de TombeRoberto De VogliLeon J. De WindtRobbert J. de WinterDick de ZeeuwBarbara J. DealJohn Eric DeanfieldArjun DebG. William DecJeanne M. DeCaraCarole J. DeckerUlrich K.M. DeckingPrakash C. DeedwaniaCarolin DeinerEtienne DelacretazBrian P. DelisleMario DelmarAnthony N. DeMariaLinda L. DemerNikolaos DemirisMartin den HeijerMario C. DengDavid DeNofrioChristophe DepreAkshay S. DesaiMilind DesaiAnne M. DeschampsChristopher A. DeSouzaJean-Pierre DespresAlexander DetenChristian DetterMarcelo F. Di CarliMichele Di MauroNuno V. DiasDavid A. DichekHans DieplingerD.B. DiercksRodney J. DilleyWolfgang H. DillmannVasken DilsizianJ. Michael DiMaioJohn P. DiMarcoPawel Petkow DimitrowStefanie DimmelerRobert A.E. DionDonald J. DiPetteDobromir DobrevDouglas W. DockeryFrances DockeryJean-Michel P.N. DognéAnuja DokrasRaul J. DomenechJ. Kevin DonahueRosario Donato
e12 Acknowledgment of Reviewers

Chunming DongG.A. DonnanMarjo DonnersVincent DorAndrea DoriaPaul DorianPedro D’orléans-JusteGerald W. DornMirko DossJohn S. DouglasPamela S. DouglasJames M. DowneyLuciano Ferreira DragerChristopher DrakeMark H. DraznerHelmut DrexlerDaniel L. DriesDayue DuanNaihua DuanRaghvendra K. DubeyAnique DucharmeStephen J. DuffyJean G. DumesnilBrian W. DuncanJ. Michael DuncanDaniel DuprezJocelyn DupuisWilliam DuranteGreg DustingLance D. DworkinJason R. DyckPeter Dyck
Kim A. EagleMark J. EarleyCara A. EastRobert T. EberhardtFranz R. EberliShah EbrahimDwain L. EckbergRobert H. EckelElazer R. EdelmanSarah EderThomas S. EdgingtonRichard EdwardsIgor R. EfimovKensuke EgashiraHannelore EhrenreichOliver EickelbergAndreas EickenBenjamin W. EidemJohn F. EidtJohn W. EikelboomMichael EikmansGraeme EisenhoferDavid A. EisnerTony EissaDaniel T. EitzmanUlf EkelundGarabed EknoyanAmir ElamiJohn A. Elefteriades
Suzette Elias-SmaleUri ElkayamRonald C. ElkinsKenneth A. EllenbogenC. Gregory ElliottStephen G. EllisR Curtis EllisonCostanza EmanueliMichael EmersonMary EmondJean-Philippe EmpanaMasao EndohMatthias EndresS. EngeliRichard M. EngelmanMarguerite M. EnglerRobert L. EnglerMark L. EntmanAndrew EpsteinStephen E. EpsteinRaimund R. ErbelSandra ErbsJohn M. EriksonUrs K. ErikssonAycan F. ErkanKorhan ErkanliDavid ErlingeSabine ErnstGeorg ErtlNilda Gladys EspinolaChristine Espinola-KleinKatherine EspositoBarry C. EsrigFaadiel EssopMohammed Rafique EssopN.A. Mark EstesAnthony L. EstreraSusan P. EtheridgeCharles J. EverettGordon A. EwyVernat ExilMichael D. Ezekowitz
Gianluca FaggioliColomba FalconePierre-Emmanuel FalcozErling FalkRodney H. FalkJames A. FallavollitaJohn T. FallonJames C. FangJames I. FannSøren FanøFrank FaraciAndrew FarbJeronimo FarreMarc FatarFarzin F. Fath-OrdoubadiDiane FatkinRossella FattoriDesiderio FavaratoWilliam P. Fay
Zahi A. FayadSergio FazioWilliam F. FearonPaul W.M. FedakMartin FeelischJeffrey A. FeinsteinFrederick FeitArthur M. FeldmanDavid S. FeldmanTed FeldmanPeter FerdinandyMaros FerencikJames J. FergusonT. Bruce FergusonFrancisco Fernandez-AvilesOlivier FeronChristiane FerranCarlos M. FerrarioVictor A. FerrarisPaolo FerrazziRobert E. FerrellJulio J. Ferrer-HitaAndreas FestaLoren J. FieldDavid S. FienoHans R. FigullaMarcin FijalkowskiJanos G. FilepKristian B. FilionJeffrey R. FinemanToren FinkelJoel FinkelsteinLouis D. FiorePaolo FiorinaChristian FirschkePeter FischbachRoman FischbachMarcus FischerUwe M. FischerRodolphe FischmeisterCeline FisetMichael C. FishbeinEdward A. FisherMarc FisherPatrick W. FisherSteven A. FisherGarret A. FitzGeraldPeter J. FitzgeraldGreg C. FlakerScott D. FlammMarcus D. FlatherJerome L. FlegLee A. FleisherIngrid FlemingDanilo FliserJohn S. FlorasAlan M. FogelmanRobert N. FoleyWarren FoltzGregg C. FonarowGuo-Hua Fong
Thomas Joseph ForbesThomas ForceIan FordDaniel FormanMyriam FornageJames S. ForresterTom ForsenElyse FosterJean-Claude FouronCaroline S. FoxKeith A.A. FoxHarry A. FozzardGabriele FragassoRenerio FraguasAlain FraisseMark W. FramptonGary S. FrancisVeronica FrancoJohn V. FrangioniNikolaos G. FrangogiannisJohn A. FrangosBarry A. FranklinCharles FraserSohrab FratzGunilla Nordin FredriksonDavid S. FreedmanS. Ben FreedmanBrent A. FrenchJohn K. FrenchMichael P. FrenneauxUlrich H. FreyGary S. FriedmanMatthias G. FriedrichSoren FriisM. Kent FrobergVictor FroelicherEdward D. FrohlichJiri J. FrohlichPeter C. FrommeltAlexandra Theresia FuchsFlavio Danni FuchsRobert C. FuhlbriggeBianca FuhrmanYoshihiro FukumotoJohn W. FunderColin D. FunkCurt D. FurbergGeorge FustValentin Fuster
William H. GaaschAlain-Pierre GadeauJames V. GainerPeter A. GainesMaurizio GalderisiCatharine R. GaleZorina S. GalisWilliam GalloApoor S. GamiRonald GangnonPeter GanzFeng Gao
Acknowledgment of Reviewers e13

Susan M. GapsturMario J. GarciaJulius M. GardinTimothy J. GardnerAlistair N. GarrattPeter GarredDaniel J. GarryJean-Michel Théodule GaspozMichael A. GatzoulisGlenn R. GaudetteKimberlee GauvreauHaralambos P. GavrasIrene GavrasJ. William GaynorDavid C. GazeThomas GazianoRaul J. GazmuriCarmine GazzarusoCarolyn L. GeczyBruce D. GelbCaroline GencoYong-Jian GengThomas L. GentlesAlfred L. GeorgeJames F. GeorgeSarah Jane GeorgeBernhard L. GerberBernard J. GershWelton M. GersonyGary GerstenblithEdward P. GerstenfeldRobert E. GersztenLeonard S. GettesGodfrey S. GetzTal GevaHenry GewirtzMichael GewitzMihai GheorghiadeGiorgio GhilardiRaymond J. GibbonsC. Michael GibsonFrank C. GibsonSamuel S. GiddingStephan GielenWayne R. GilesLinda D. GillamBrenda W. GillespieA. Marc GillinovMatthew W. GillmanRichard F. GillumRobert F. GilmourJeffrey M. GimbleFrank J. GiordanoDomenico GirelliAdriana C. Gittenberger-de
GrootRobert P. GiuglianoCarla GiustettoMichael M. GivertzDavid GjertsonMark T. Gladwin
Stanton A. GlantzRuchira GlaserChristopher GlassStephen J. GlattDavid K. GloverPeter D. GluckmanRobert J. GlynnAlan S. GoDavid C. GoffNoyan GokceDiane R. GoldJeffrey P. GoldAlexander GoldbergAndrew P. GoldbergJeffrey J. GoldbergerIlan GoldenbergJoshua I. GoldhaberSamuel Z. GoldhaberLee GoldmanMartin E. GoldmanSteve GoldmanJames A. GoldsteinLarry B.GoldsteinSidney GoldsteinPaolo GolinoJonathan GolledgeMichael H. GollobGershon GolombAna M. GomezCelso E. Gomez-SanchezPhilimon GonaIsabel GoncalvesMario D. GonzalezMark O. GoodarziJohn GorcsanTommaso GoriAgnes GorlachJoseph H. GormanShinya GotoRoberta A. GottliebK. Lance GouldAndrew A. GraceJuan F. GranadaScott GrandyChristopher B. GrangerAugustus O. GrantRichard A. GrayWilliam A. GrayPaul A. GrayburnJ. Thomas GraystonSally C. GreavesBarry GreenbergRoy K. GreenbergStephen E. GreenwaldDarren C. GreenwoodKathy K. GriendlingBrian P. GriffinJohn H. GriffinCindy L. GrinesSteven K. GrinspoonJohannes C. Grisar
William J. GrohChristian GroheGarrett J. GrossGil J. GrossPaul D. GrossfeldEugene A. GrossiBlair P. GrubbEberhard GrubeScott M. GrundyGary L. GrunkemeierWilliam H. GuilfordChristian GuilleminaultGerard Marcel GuiraudonGiosue GulliPaul A. GurbelGeoffrey C. GurtnerBjörn I. GustafssonDavid D. GuttermanRobert A. GuytonTomasz J. Guzik
Constance Kay HaanJudith HaendelerSteven M. HaffnerDominik Georg HaiderDavid E. HainesMichel HaissaguerreKatherine HajjarRoger J. HajjarJulian P.J. HalcoxCharles A. HalesAndrew HalestrapMichael E. HalkosHermann HallerMichael HallmanAlfred P. HallstromNaomi M. HamburgMohamed H. HamdanChristian W. HammWayne W. HancockAase HandbergAnthony J. HanleyEdward L. HannanTim HansonGoran K. HanssonTomonori HaraguchiViolet I. HaraszthyJoshua M. HareRobert A. HarringtonWilliam S. HarrisDavid G. HarrisonPaul HarrisonDavid HasdaiGerd HasenfussVic HasselbladRichard N.W. HauerPaul J. HauptmanDerek J. HausenloyRichard J. HavelAxel HaverichEdward P. HavranekNat Hawkins
Robert A. HaworthKenshi HayashiToshio HayashiMichael R. HaydenDavid L. HayesDaniel HayozStanley L. HazenMary Fran HazinskiGuo-Wei HeJiang HeGeoffrey HeadJohn P. HeadrickSusan R. HeckbertMarkus HeckerTimothy HeerenChristopher HeeschenRobert A. HegelePaul A. HeidenreichJay W. HeineckeChristian HeissClaes HeldJohanna HelmerssonHarry HemingwayJeroen HendrikseMarc HendrikxTimothy D. HenryMoonseong HeoDirk HermannRamon C. HermidaAdrian F. HernandezVictoria L.M. HerreraAmy HerringDavid M. HerringtonHoward C. HerrmannRay E. HershbergerCharles A. HerzogOtto M. HessGerd HeuschStephane HeymansWilliam R. HiattCharles B. HigginsRobert HigginsThomas HilbergKarl F. HilgersJohn S. HillJoseph A. HillHans L. HillegeL. David HillisGerhard HindricksAroon D. HingoraniRabea HinkelLoren F. HiratzkaYoshitaka HirookaKaren K. HirschiValeria HirschlerJohn W. HirshfeldMark A. HlatkyMichael HoJudith S. HochmanMorrison HodgesBarbara H. Hoffmann
e14 Acknowledgment of Reviewers

Martin H. HoffmannUdo HoffmannPeter HöglundThomas HohlfeldStefan H. HohnloserBrian D. HoitFernando HolguinDavid R. HolmesPaul HolvoetMichael HolzerShunichi HommaYuling HongL.N. HopkinsPaul N. HopkinsRichard HopkinsSusan HopkinsMaria T.E. HopmanMasatsugu HoriMasatsugu HoriuchiBenjamin D. HorneJohn D. HorowitzKeith A. HorvathLawrence D. HorwitzAkiko HoslerBarbara V. HowardGeorge HowardVicky Y. HoymansPatrick C.H. HsiehDaphne T. HsuFang-Chi HsuPriscilla HsueChengcheng HuGang HuHoward HuRae-Chi HuangJohnny HuardSally Ann HuberSusanna Y. HuhJames C. HuhtaHeikki V. HuikuriP.P. HujoelRoger HullinPer M. HumpertWilliam Gregory HundleyJudy HungSharon Ann HuntStephen HunyorAhsan Husain
Gianluca IacobellisIoannis IakovouFumito IchinoseRaymond E. IdekerMasaaki IiJohn S. IkonomidisErkki IlveskoskiArmin ImhofAkihiro InazuRobin IngallsJulie R. IngelfingerErik IngelssonJoanne S. Ingwall
John P. IoannidisCarlos IribarrenThomas A. IschingerShun IshibashiMasaharu IshiharaTetsuya IshikawaAmi E. IskandrianHiroyasu IsoMitsuaki IsobeHideki ItohBernard IungD. Dunbar IvyKohichiro Iwasaki
Wael A. JaberEdwin K. JacksonAlice K. JacobsDavid R. JacobsMarshall L. JacobsDonald W. JacobsenPaul JacquesSae Young JaeMichael R. JaffAllan S. JaffeFarouc A. JafferMukesh Kumar JainJose JalifeRichard W. JamesKonrad JamrozikIk-Kyung JangJoseph S. JanickiWarren R. JanowitzMaurits A. JansenStefan P. JanssensCraig T. JanuaryJames Louis JanuzziPatrick Y. JayJohn JefferiesJ. Richard JenningsJong Hyeon JeongJamie Yancey JeremyTomas JernbergMichael Jerosch-HeroldPaula Jerrard-DunneAshish K. JhaIshwarlal JialalHongyu JiangZhihua JiangZhezhen JinHanjoong JoMagnus Carl JohanssonRoger A. JohnsArnold JohnsonB. Delia JohnsonJason L. JohnsonLynne L. JohnsonPaula A. JohnsonRichard J. JohnsonRobert L. JohnsonRobert Graham JohnsonTimothy D. JohnsonJames G. Jollis
Gregory T. JonesRobert H. JonesSteven P. JonesHabo J. JongsmaJens JordanMarit Eika JørgensenJacob JosephMark E. JosephsonPekka JousilahtiMichael J. JoynerJ. Wouter JukemaPhilip JungSuh-Hang Hank Juo
Stefan KaabRamanathan KadirvelAlan H. KadishHiroyuki KageyamaRichard KahnHisashi KaiJan KajsturaAfksendiyos KalangosJonathan M. KalmanTimothy J. KampDavid E. KandzariSachiko Kanki-HorimotoPrince J. KannankerilRonald J. KanterJørgen K. KantersEmmanouil Ioannis KapetanakisTara KaramlouRichard H. KarasElissavet KardamiTom R. KarlJohan KarlbergJoel S. KarlinerAly KarsanS. Ananth Ananth KarumanchiDavid Alan KassRobert S. KassJerome P. KassirerAdnan KastratiSekar KathiresanMasahiko KatoTomohiro KatsuyaHugo A. KatusZvonimir S. KatusicStuart D. KatzMarc P. KaufmanPhilipp A. KaufmannPadma KaulSanjiv KaulRae-Ellen KaveyChuichi KawaiZiya KayaDavid M. KayeMark T. KearneyBernard KeavneyDaniel P. KellyDarren J. KellyRalph A. KellyKenneth M. Kent
Rosemary J. KeoghRichard E. KerberDean J. KereiakesKarl B. KernMorton J. KernSteven J. KeteyianPaul KhairyAmit KheraAli Reza KhoshdelStefan KiechlJan T. KielsteinShinji KiharaLois A. KillewichPhilip J. KilnerDavid KilpatrickHyo-Soo KimShokei Kim-MitsuyamaSpencer B. KingBronwyn A. KingwellScott KinlayMargaret L. KirbyPaulus F. KirchhofJames KirklinLorrie A. KirshenbaumToru KitaMasafumi KitakazeMichelle KittlesonArthur L. KlatskyNeal S. KleimanAllan L. KleinGeorge J. KleinLloyd W. KleinMichael D. KleinCharles S. KleinmanPaul D. KligfieldUwe KlimaFrancis J. KlockeRobert A. KlonerJohn L. KnightAnne A. KnowltonJuhani KnuutiJon A. KobashigawaLars KoberColleen Gorman KochWalter J. KochWerner KochPatrick M. KochanekPaul V. KochupuraTodd M. KoellingWolfgang KoenigTheo KofidisKwang Kon KohMartin KöhrmannPipin KojodjojoPappachan E. KolattukudyTheodore J. KoliasFrank D. KolodgieMichelM. K. KomajdaTatsuya KomaruMasashi KomedaIssei Komuro
Acknowledgment of Reviewers e15

Takahisa KondoMarvin A. KonstamStavros V. KonstantinidesMichael C. KontosMarianne Eline KooiWillem J. KopBruce A. KoplanGideon KorenAndreas KosterThomas Erling KottkeDarrell N. KottonAlexei KouroedovPetri T. KovanenPeter R. KoweyJun KoyamaAndrew D. KrahnDara L. KraitchmanAldi KrajaChristopher M. KramerEvangelia G. KraniasWilliam E. KrausRonald M. KraussEswar KrishnanSteen Dalby KristensenMichael H. KrollIrving L. KronMitchell W. KrucoffWarren KrugerHenry KrumIsao KubotaKaren KuehlNino KuenzliDonald M. KuhnHelena KuivaniemiThomas J. KulikLewis H. KullerIftikhar J. KulloKoichiro KumagaiCheng-Deng KuoChristian KupattSabina KupershmidtHiromi KurosawaTobias KurthIrving KushnerJohanna KuusistoJeffrey T. KuvinMasafumi KuzuyaMartijn KwaijtaalRaymond Y. KwongYoshiki KyoAlan Patrick Kypson
David E. LaaksonenCarlos Alberto LabarrereVinod LabhasetwarArthur LabovitzDaniel LacklandKarl J. LacknerPeter S. LacyStephanie LaeerHuichuan LaiShenghan Lai
Wyman W. LaiEdward G. LakattaEvanthia LallaKaren S.L. LamBenoit LamarcheJohn J. LambertiMichael J. LaMonteRachel LampertKathryn G. LampingSteve LancelR. Clive LandisUlf LandmesserMichael J. LandzbergFlorian LangIrene Marthe LangRoberto M. LangJonathan LangbergElisabetta LapennaGina LaRoccaJohn C. LaRosaMartin LarsonWarren K. LaskeyRobert D. LasleyJohan P.E. LassusRoberto LatiniWei C. LauMichael S. LauerUlrich LaufsJari A. LaukkanenLenore J. LaunerGeoffrey J. LaurentKenneth R. LauritaPeter C. LaussenMichael LaValleyCarl LavieDebbie A. LawlorHarold L. LazarMitchell A. LazarThierry H. Le JemtelBruce J. LeavittAlexander Wolfgang LeberNathan K. LeBrasseurSandrine LecourAmanda J. LeeDouglas S. LeeDuk-Hee LeeHon-Chi LeeI-Min LeeKerry L. LeeRichard T. LeeFrans H.H. LeenenDavid J. LeferMichael H. LehmannStephan E. LehnartStephanie LehouxJames M. LeiperPaul LeLorierKarl B. LemstromSteven R. LentzDavid A. LeonPhilipp M. Lepper
Amir LermanBruce B. LermanLilach O. LermanMichelle LetarteHanno H. LeuchteAdeera LevinMax LevinBenjamin D. LevineR.J. LevineRobert A. LevineSidney LevitskyBodo LevkauBruce D. LevyDaniel LevyFinn Olav LevyJerrold H. LevyRobert J. LevyWayne C. LevyMarilyne LevyElad I. LevyE. Douglas LewandowskiMartin M. LeWinterEldrin F. LewisGary F. LewisChuanfu LiFan LiGui-Rong LiLiang LiNa LiNan LiRen-Ke LiRonald A. LiChang-seng LiangJames K. LiaoRonglih LiaoPeter LibbyAndrew H. LichtmanDavid Richard LightChee Chew LimValter C. LimaMarian C. LimacherJing Ping LinJulie LinJoAnn LindenfeldMarshall D. LindheimerJes S. LindholtJonathan R. LindnerKarl H. LindnerKen A. LindstedtMark S. LinkAxel LinkeMacRae F. LintonGregory Y.H. LipSteven E. LipshultzLewis A. LipsitzRichard LiptonWilliam C. LittleLaszlo LittmannKiang LiuDonald M. Lloyd-JonesCecilia Wen-Ya Lo
Eng H. LoAmanda LochnerJames E. LockAndrew J. LodgeFrank W. LoGerfoBarry LondonGerard M. LondonEva M. LonnGary D. LopaschukMatthias W. LorenzDouglas W. LosordoStavros P. LoukogeorgakisGordon D. LoweTse Min LuRussell V. LuepkerFriedrich C. LuftKetil LundeKathryn LunettaKeith G. LurieThomas F. LuscherAldons J. LusisDeborah LynJohn W. Lynch
Xin Liang MaChristoph MaackDavid M. MaahsCharles A. MackMichael J. MackWendy J. MackTodd A. MacKenzieRachel H. MackeyNigel MackmanMichael I. MacknessKenneth N. MacLeanWilliam Robb MacLellanPaolo MadedduAldo P. MaggioniWilliam T. MahleWilliam T. MahleJonathan D. MahnkenLynn MahonyLars S. MaierWillibald MaierFrancesco MaisanoWilliam H. MaiselAmy S. MajorKoon-Hou MakJonathan C. MakielskiMarek MalikRobert T. MalletGiuseppe ManciaDonna M. ManciniG.B. John ManciniKaushik MandalRavi MandapatiJayawant N. MandrekarDennis T. ManganoArduino A. MangoniDouglas L. MannGiovanni E. MannStewart Mann
e16 Acknowledgment of Reviewers

Warren J. ManningTeri A. ManolioNicolas MansencalAlberto MantovaniMichael Stephen MarberFrancis E. MarchlinskiFrank I. MarcusAndrew O. MareeRaffaele MarfellaJames R. MargolisKenneth B. MarguliesDaniel B. MarkRoger MarkwaldJonathan D. MarmurBarry J. MaronDavid J. MaronMartin MaronLuc MaroteauxPhilip A. MarsdenSteven P. MarsoRandolph P. MartinUlrich MartinThomas H. MarwickGerald R. MarxNikolaus MarxDaniele MaselliAttilio MaseriFrederick A. MasoudiJoseph M. MassaroBarry M. MassieSerge MassonKazuko MasuoFiona MathewsKnut MatreHiroaki MatsubaraHikaru MatsudaAkira MatsumoriFrancesco U.S. Mattace-RasoChristian M. MatterMarco L.S. MatteucciNilanjana MaulikMathew S. MaurerLaura MauriSimon MaxwellCharles MaynardBongani Mawethu MayosiTodor N. MazgalevPatrickM. McCarthyMichael V. McConnellMichael Leon McCormickBrian W. McCrindlePeter A. McCulloughDavid H. McDermottMary McGrae McDermottDoff B. McElhinneyCarmelM. McEnieryEdward Mcfalls M.D.Daniel McGeeJohn C. McGiffJohn L. McGregorDarren K. McGuire
William J. McKennaVallerie V. McLaughlinC. Alex McMahanJohn J.V. McMurrayElizabeth M. McNallyRobert L. McNamaraHelene McNultyCharles F. McTiernanMandeep R. MehraRoxana MehranJawahar L. MehtaShamir R. MehtaBernhard MeierJames B. MeigsSilke MeinersCynthia J. MeiningerChrista MeisingerDaniel MeldrumRussell H. MellorPhilippe MenascheUlrike MendeJean-Jacques MercadierPatrick MercieYahye MerhiAlan F. MerryC. Noel MerzEmmanuel MessasFranz H. MesserliLuisa MestroniPeter MeyerTheo E. MeyerJ.A. MichaelsEvangelos D. MichelakisErin Donnelly MichosShigetoshi MienoRichard V. MilaniD. Douglas MillerD. Craig MillerJordan D. MillerLeslie W. MillerTodd D. MillerVirginia M. MillerSusumu MinamisawaL. LuAnn MinichKenji MinoguchiGary S. MintzIsrael MirskyManisha MishraSeema MitalBrett M. MitchellGary F. MitchellR. Scott MitchellRichard N. MitchellArnold MitnitskiSuneet MittalFriedrich MittermayerMurray A. MittlemanHiroto MiuraMihaela M. MocanuPeter MohlerEmile R. Mohler III
Ali H. MokdadErnesto MolinaGiuseppe MolinariDavid J. MoliternoJeffery D. MolkentinNico R. MolletTom E. MollnesDonald A. MolonyLaurent MonassierNicola MontanoFarouk MookadamJeffrey MooreSamia MoraMartin MoradFred MoradyPierre MoreauL.A. MorenoMarie-Claude MoriceCarlos A. MorilloRyuichi MorishitaToshisuke MoritaGregory E. MorleyNicholas W. MorrellBrian J. MorrisLaurie J. MorrisonDavid A. MorrowRichard MortensenRalph S. MoscaMauro MoscucciJeffrey W. MosesIvan P. MoskowitzArthur J. MossKaren S. MoultonJ. Paul MounseyMatthew MovsesianDariush MozaffarianThomas MuenzelAndreas MuggeAndrew MugglinDebabrata MukherjeeRupak MukherjeeDouwe J. MulderJames E. MullerJochen Muller-EhmsenMichael J. MulvanyTomoatsu MunePaul MuntnerElizabeth MurphyTimothy P. MurphyCharles E. MurryAnthony J. MuslinAviva MustRobert J. MyerburgDaniel D. MyersJonathan MyersLeann Myers
Elizabeth G. NabelKoonlawee NademaneeRyozo NagaiShoichiro NagasakaEike Nagel
Sherif F. NaguehMatthias NahrendorfSamer NajjarHiroshi NakagawaBrahmajee K. NallamothuBin NanClaudio NapoliCarlo NapolitanoSanjiv M. NarayanJagat NarulaDavid NashAndrea NatalePeter NathanielszStanley NattelMatthew T. NaughtonMohamad NavabFrank NayaMona NemerDario NeriRichard W. NestoStefan NeubauerDonna S. NeubergEllis J. NeufeldFranz-Josef NeumannJoachim NeumannPeter NewburgerDavid E. NewbyL. Kristin NewbyJohn H. NewmanMark F. NewmanChristopher H. Newton-ChehLudwig NeysesDusko G. NezicStephen James NichollsWilmer W. NicholsGeorg NickenigMartin John NicklinAlfred C. NicolosiJames T. NiemannChristoph A. NienaberPetros NihoyannopoulosSigrid NikolRick A. NishimuraSteven E. NissenDorothea NitschTimothy David NoakesYoshihiro NojiGeorg NollertFumikazu NomuraLars NorgrenSharon-Lise T. NormandKari E. NorthGavin R. NortonMichael G.A. NorwoodAnn-Trude With NotøGian M. NovaroWilliam C. NugentSatoshi NumataJurg NussbergerPirjo Nuutila
Acknowledgment of Reviewers e17

Timothy Daniel O’ConnellChristopher M. O’ConnorGerald T. O’ConnorChristopher J. O’DonnellPeter OettgenPatrick T. O’GaraJae K. OhSeil OhTakahiro OharaAnn M. O’HarePatrick OhlmannE. Magnus OhmanJohn OhrvikPeter M. OkinJohannes OldenburgDonal S. O’LearyJeffrey W. OlinMichael Hecht OlsenEric N. OlsonLyle J. OlsonAnders G. OlssonRay A. OlssonPatrick G. O’MalleyEileen O’MearaSteve R. OmmenJames O. O’NeillHenry OoiSuzanne OparilLionel H. OpieHakan OralE. John OravJose M. OrdovasJoseph P. OrnatoMichael F. O’RourkeLeiv OseClive OsmondYutaka OtsujiDavid OttHarald C. OttCatherine M. OttoNoriyuki OuchiMichel OvizeAl Ozonoff
Pal PacherSandosh PadmanabhanFrancis D. PaganiMassimo PaganiRichard L. PageJennifer PaiPaolo PalatiniWulf PalinskiJulio C. PalmazColin N.A. PalmerDemosthenes PanagiotakosNatesa G. PandianJames S. PankowNazareno PaolocciDomenico PaparellaCarlo PapponeThomas G. ParkerJuan C. Parodi
Michele PasottiGerard PasterkampAmit N. PatelAnushka Alankar PatelAyan PatelJeetesh V. PatelCarlo PatronoRichard D. PattenCam PattersonWalter J. PaulusAimee D.C. PaulussenJeffrey M. PearlJeremy D. PearsonDaniel PellaPatricia A. PellikkaMichael PencinaMarc S. PennDudley J. PennellCarl J. PepinePaul E. PeppardEmerson C. PerinJohn PernowJames C. PerryStephen D. PersellSharina D. PersonG. Rutger PerssonInga PeterKarlheinz PeterNicholas S. PetersEric D. PetersonLinda R. PetersonPamela PetersonEva PetkovaMichael E. PhelpsGerald B. PhillipsRichard P. PhippsColin K. PhoonRobert N. PianaPhilippe PibarotEugenio PicanoMichael H. PicardJ. Geoffrey PickeringThomas G. PickeringLuc A. PierardBurkert PieskeBruce PihlstromNico H.J. PijlsLouise PiloteDavid R. PimentelIleana PinaH. Michael PiperTobias PischonBertram PittJeffrey L. PlattJonathan F. PlehnMark J. PletcherJorge PlutzkyBruno K. PodesserGerald M. PohostPaul PoirierJoseph F. Polak
Don PoldermansJaimie W. PolsonGiulio PompilioPhilip A. Poole-WilsonBarry M. PopkinThomas R. PorterWendy S. PostLuciano PotenaLincoln PotterJeffrey T. PottsNeil PoulterAndrew J. PowellJanet T. PowellAshwin PrakashAbhiram PrasadSanjay K. PrasadSusan J. PresslerJack F. PriceRonald PrineasFrits W. PrinzenSilvia G. PrioriKirkwood A. Pritchard, Jr.Vincent ProbstKarin PrzyklenkWilliam T. PuJohn D. PuskasReed PyeritzKalevi Pyorala
Zhaohui Steve QinMiguel A. Quiñones
Ton J. RabelinkAlejandro A. RabinsteinVittorio RaccaFrank E. RademakersDaniel J. RaderR. RadermeckerMartha J. RadfordPaolo RaggiShahbudin H. RahimtoolaLeopoldo RaijElaine W. RainesOlli T. RaitakariSanjay RajagopalanNalini M. RajamannanVenkatesh RajapurohitamHarry RakowskiVivek RaoUrsula RauchUrsula RavensReza S. RazaviPeter RazeghiRichard RePatricia ReantRita F. RedbergMargaret M. RedfieldJosep RedonThomas C. RegisterJalees RehmanNathaniel ReichekMuredach Reilly
Azaria J.J.T. ReinOlaf ReinhartzSteven E. ReisWillem J. RemmeSerge C. RenaudFrederic S. ResnicKathyrn M. RexrodeMatthew R. ReynoldsShereif H. RezkallaEdward K. RheeJonathan RhodesJorge P. RibeiroFernando F. Ribeiro-FilhoPaul M. RibislKen RiceJean-Paul RichaletVincent RichardPaul M RidkerJohannes RiegerNader RifaiCharanjit S. RihalEric B. RimmP.A. RinglebRasmus Sejersten RipaJames M. RitterEberhard RitzAlain RivardJeffrey RobbinsRobert RobertsWilliam C. RobertsSander J. RobinsJennifer G. RobinsonSimon C. RobsonFrederic RocheDan M. RodenBrian RodriguesAlfredo E. RodriguezAlicia Rodriguez-PlaMatthew T. RoeMark RoestMarco RoffiVeronique L. RogerCampbell RogersJoseph G. RogersMary J. RomanMats RönnbackDieter RopersWayne D. RosamondEric A. RoseNoel R. RoseMichael R. RosenDavid S. RosenbaumGary A. RosenbergMichael E. RosenfeldRobert S. RosensonDavid N. RosenthalAnthony RosenzweigBernard RosnerAllan M. RossDavid L. RossGian Paolo D. Rossi
e18 Acknowledgment of Reviewers

Marco L. RossiJ.E. RossouwStephen J. RothDietrich RothenbacherRichard B. RothmanPeter M. RothwellJoris RotmansMelvyn RubenfireFrederick L. RubergLewis J. RubinIsrael RubinsteinNeil B. RudermanGoran RudezMarc RuelLuis M. RuilopeCarlos E. RuizPilar Ruiz-LozanoJohn S. RumsfeldMarschall S. RungeFrank RuschitzkaRaymond R. RussellVincenzo RussoWolfgang RutschCarolyn RutterElfriede RuttmannThomas RyanThomas J. RyanJack RychikLars Ryden
Tobias SaamSamir SabaHani N. SabbahJoseph F. SabikRalph L. SaccoMichael N. SackFrank M. SacksMichael S. SacksJ. Evan SadlerJunichi SadoshimaMichel E. SafarJeffrey E. SaffitzRobert D. SafianAlexander SagieDavid J. SahnGenichi SakaguchiSanjeev SaksenaTomas A. SalernoVeikko SalomaaFlora SamFrederick F. SamahaNilesh J. SamaniJeffrey SametJonathan M. SametJane-Lise SamuelTimothy Allen SanbornDirk SanderMikael SanderStephen P. SandersJohn E. SandersonAnthony J. SanfilippoMichael C. Sanguinetti
John Lewis SappDennis SarabiWim SarisFerdinando Carlo SassoMasataka SataNaveed SattarKurt W. SaupeGiorgio SavazziStephen G. SawadaTatsuya SawamuraDouglas B. SawyerLeslie A. SaxonTiziano ScarabelliPierre-Yves ScarabinVolker SchachingerThomas SchachnerAlvin SchadenbergHartzell V. SchaffMartin J. SchalijBernhard SchallerWolfgang SchaperDoug E. SchaubelPatrick SchauerteDebra Ann SchaumbergDierk ScheinertMelvin M. ScheinmanSebastian M. SchellongBenjamin J. ScherlagRalph Theo SchermulyBernhard SchiefferGiuseppe SchillaciMartin SchillingerAlexandru SchiopuThomas SchlosserAlvin SchmaierAxel SchmermundClaudia SchmidtkeNeil SchneidermanAlbert SchoemigFrederick J. SchoenMark Howard SchoenfeldJurgen SchraderRichard B. SchuesslerGerhard C. SchulerKevin A. SchulmanHeinz-Peter SchultheissRichard SchulzEric Schulze-BahrHeribert SchunkertArnold SchwartzGary L. SchwartzKenneth A. SchwartzPeter J. SchwartzRobert Stockton SchwartzDavid S. SchwartzmanErnst R. SchwarzP.E. SchwarzKarie ScroginPaola SebastianiChristine E. SeidmanJonathan G. Seidman
Christian SeilerAkira SekikawaDonald F. SellittiFrank W. SellkeElizabeth SelvinAndrew P. SelwynCraig H. SelzmanGregg L. SemenzaMarc J. SemigranChris SemposChandan K. SenRoxy SeniorVictor L. SerebruanyCharles N. SerhanPatrick W. SerruysMarc J. ServantSudha SeshadriHoward D. SessoMagnus SettergrenRalph ShabetaiRobert E. ShaddyAjay M. ShahMaully J. ShahPravin M. ShahPrediman K. ShahDavid M. ShahianCatherine M. ShanahanRichard P. ShannonBehrooz G. SharifiArya M. SharmaSamin K. SharmaKen SharpePalma ShawAmanda M. ShearmanMichael ShechterSoren P. SheikhPrem S. ShekarRhidian John SheltonStanton K. ShernanMark V. SherridGuo-Ping ShiWeibin ShiRei ShibataMei-Chiung ShihKoichi ShimizuTatsuya ShimizuWataru ShimizuHiroaki ShimokawaIchiro ShiojimaGirish S. ShiraliKalyanam ShivkumarMichael G. ShlipakYehuda ShoenfeldStephen R. ShorofskyMatthias SiepeHans-H. SieversUlrich SigwartDonald S. SilverbergDavid I. SilvermanJean-Sebastien SilvestreRobert D. Simari
Daniel I. SimonJoel A. SimonMichael SimonsMaarten L. SimoonsPaul C. SimpsonRoss J. SimpsonAlan R. SinaikoMervyn SingerKrishna SinghMichael N. SinghSteven N. SinghTajinder P. SinghAlbert J. SinusasDeborah A. SiwikAllan C. SkanesSusan A. SlaugenhauptKaren SliwaRichard W. SmallingOtto A. SmisethCraig SmithGeorge Davey SmithGrace L. SmithJonathan SmithNicholas L. SmithStephen Mark SmithTimothy William SmithWarren Morrison SmithRyszard T. SmolenskiDavid B. SneadAllan D. SnidermanBurton E. SobelBirgitta SöderStefan SoderbergKyoko SoejimaManoocher SoleimaniScott D. SolomonVirend K. SomersRobert J. SommerAli SonelDan SorescuVincent L. SorrellP.C. SouvereinArthur A. SpectorJ. David SpenceMarkus SperandioJohn A. SpertusMartin SpieckerBruce SpiessFrancis G. SpinalePaolo SpiritoDavid H. SpodickMartinus SpoorMatthew L. SpringerHenri M.H. SpronkFrancesco SquadritoIain B. SquireRay W. SquiresV.S. SrinivasMartin G. St. John SuttonEarl R. StadtamJan A. Staessen
Acknowledgment of Reviewers e19

Meir J. StampferKenneth StanleyWilliam C. StanleyWilliam StanleyAlice V. StantonNorbert StefanMichael W. SteffesPhilippe Gabriel StegCoen D. StehouwerHelmut O. SteinbergJulia SteinbergerRobin H. SteinhornSteve R. SteinhublKurt R. StenmarkAndrew SteptoeMichael P. SternLynne Warner StevensonWilliam G. StevensonDuncan J. StewartJulian M. StewartRalph A.H. StewartSimon StewartRoland StockerKaren StokesMonika StollGregg W. StonePeter H. StoneRoslyn A. StoneJulie St-PierreRuth H. StrasserBodo E. StrauerH. William StraussBruno H. StrickerErik S.G. StroesMatthias StuberChristian StumpfRodney SturdivantYan Ru SuKrishnankutty SudhirCathie L.M. SudlowKoichi SughimotoM.-Saadeh SuleimanJerome L. SullivanLisa M. SullivanJack C.J. SunThoralf M. Sundt IIIH. Robert SuperkoMark A. SussmanThomas M. SuterAllison J. SutherlandKim Sutton-TyrrellErik J. SuuronenAlan F. SvedLars G. SvenssonElisabet SvenungssonMichael O. SweeneyE.R. SwensonBernard Swynghedauw
Koichi TabayashiStefano TaddeiHeinrich Taegtmeyer
David P. TaggartPeter TaggartMasato (Mike) TakahashiShinji TakaiSatoshi TakeshitaJohanna J.M. TakkenbergWilliam T. TalmanMasashi TanakaToshihiro TanakaLilong TangRajendra K. TangiralaLloyd Y. TaniLaszlo B. TankoFelix C. TannerMasaya TannoYoshihisa TanoueKahraman TanriverdiVictor F. TapsonJean-Claude TardifGiovanni TargherMark B. TaubmanAhmed TawakolAllen J. TaylorAndrew M. TaylorAnne L. TaylorAlain TedguiUsha TedrowJohn R. TeerlinkPaul S. TeirsteinDavid F. TeitelGeorge TellidesMartin TepelMasahiro TerashimaRonald L. TerjungNorma TerrinDellara F. TerryPierre TherouxAravinda ThiagalingamPerumal ThiagarajanRavi R. ThiagarajanChris ThiemermannGaetano ThieneAnita C. ThomasJames D. ThomasRandal J. ThomasShane R. ThomasWilliam ThomasDouglas ThompsonMaryLou ThompsonPaul D. ThompsonRichard ThompsonJens Jakob ThuneJohan ThybergLu TianRong TianTomasz TimekBrian TimmonsLaurence TiretAsa TivestenGeoffrey H. ToflerCheng-Hock Toh
Gordon F. TomaselliNaruya TomitaMarcello TonelliAndrew M. TonkinEric J. TopolPer TornvallRobert Daniel TotoRhian M. TouyzJeffrey A. TowbinDwight A. TowlerJonathan N. TownendKazunori ToyodaRussell P. TracyJohn K. TriedmanHan-Mou TsaiThomas T. TsaiTeresa S.M. TsangSotirios TsimikasTim K. TsoYukiomi TsujiIgor TudorachePaul A. TunickJosé TuñónTanya N. TuranFiona TurnbullAlexander G.G. TurpieE. Murat TuzcuVolkan TuzcuJames S. TweddellChristophe Tzourio
Thomas UngerZoltan UngvariPhilip UrbanZsolt UrbanBarry F. UretskyMasuko Ushio-Fukai
Viola VaccarinoMiguel ValderrabanoMarco ValgimigliPatrick J.T. VallanceJesus G. VallejoJoannis E. VamvakopoulosEric Van BellePeter Van BurenRuud M.A. Van de WalFrans J. Van de WerfGreta Van den BergheJohanna Gerarda van der bomYvonne T. van der SchouwErnst E. van der WallBerry M. van GelderBethany Van GuelpenGeorge F. Van HareLinda Van HornJohannes J. van LieshoutJoost P. van MelleA.M. van RijJ. Peter van TintelenDirk J. van VeldhuisenDavid R. van Wagoner
Thomas E. VanheckeMani A. VannanCristina Varas-LorenzoNerea VaroMariuca Vasa-NicoteraThomas A. VassiliadesDorothy E. VatnerStephen F. VatnerMary S. Vaughan SarrazinHector O. VenturaPaolo VerdecchiaFreek W.A. VerheugtSubodh VermaCees VermeerRichard L. VerrierSara VeselyGeorge W. VetrovecVictoria VetterAristidis VevesFlordeliza S. VillanuevaFrancisco VillarrealKaren A. VincentJakob Vinten-JohansenRenu VirmaniSami ViskinEric VittinghoffGus J. VlahakesRobert A. VogelManfred VogtPierre VoisineArnold von EckardsteinFinn Edler von EybenMarc A. VosRobert VoswinckelSari VoutilainenNaren Vyavahare
Bernard WaeberAnja WagnerRon WaksmanAlbert L. WaldoLars WallentinSusanna M. WallerstedtReidar WallinEdward P. WalshThomas WaltherPaul J. WangPing H. WangQing WangThomas J. WangWenyu WangXuejun WangYun WangJames W. WarnicaManabu WatanabeMari A. WatanabeNozomi WatanabeDavid D. WatersSergio WaxmanW. Douglas WeaverCatherine WebbDavid J. Webb
e20 Acknowledgment of Reviewers

Gary D. WebbJohn G. WebbSteven A. WebberChristian WeberKarl T. WeberMichael A. WeberMark W.I. WebsterKevin WeiL. WeiWei WeiFranz WeidingerDorothee WeihrauchMax Harry WeilMyron WeinbergerNeal L. WeintraubWilliam S. WeintraubRichard D. WeiselMary C. Weiser-EvansEric S. WeissHarvey Richard WeissRobert G. WeissRobert M. WeissNeil J. WeissmanJeffrey I. WeitzCarrie WelchDavid J. WelshFrederick G. WeltPeter WenaweserNanette Kass WengerJolanda J. WentzelNikos S. WernerMalcolm WestCynthia M. WesterhoutDirk WestermannCornelia M. Weyand
Andrew S. WeyrichGillian A. WhalleyJohn WhartonChristopher J. WhiteGuy StJ. WhitleyJ. Lindsay WhittonMark H. WholeySamuel A. WicklineJulian WidderSusan E. WiegersWilliam WijnsDavid J. WilberArthur A.M. WildeIan B. WilkinsonBruce L. WilkoffAndrew R. WillanWalter C. WillettBryan WilliamsDavid O. WilliamsDavid M. WilliamsMark A. WilliamsScott R. WilloughbyAndrew M. WilsonPeter W. WilsonStephan WindeckerKarl WinklerJeffrey A. WinklesJonathan Allan WinstonJacqueline C.M. WittemanJanet WittesJoseph L. WitztumStephen D. WiviottPhilip A. WolfEugene E. WolfelMichael S. Wolin
Paul WolkowiczKai C. WollertLennie WongNathan D. WongJohn C. WoodMark A. WoodElizabeth A. WoodcockAngela WoodiwissR. Scott WrightAlan H.B. WuChuntao WuJoseph C. WuKenneth K. WuD. George Wyse
Xiao XiaoSusan Xu
Magdi H. YacoubHitoshi YakuHafize YalinizNorikazu YamadaYoshiji YamadaTakashi YamakiClyde W. YancyHomer YangQinglin YangZhihong YangKatsusuke YanoMasafumi YanoJack YanovskiDerek M. YellonMidori Anne YenariSeppo Yla-HerttualaPaul G. YockMervin C. Yoder
Mitsuhiro YokoyamaShi-Joon YooYoung-sup YoonLawrence H. YoungCheuk-Man YuChun Yuan
Ian C. ZacharyRalf ZahnPeter ZahradkaAndrew ZalewskiFaiez ZannadWojciech ZarebaBarry L. ZaretAlan M. ZaslavskyCuihua ZhangJianyi ZhangRong ZhangShetuan ZhangZefeng ZhangLi-Ru ZhaoShankuan ZhuXinsheng ZhuFelix ZijlstraMichael R. ZileThomas ZimmerM. Bridget ZimmermanJean-Marc ZinggDouglas P. ZipesEdgar ZitronCarmine ZoccaliIrving H. ZuckerJay L. Zweier
Acknowledgment of Reviewers e21

AHA Issues New ProductsThe following new products for the public and the healthcareprofessional are available through your local American HeartAssociation or by calling 1-800-AHAUSA1.
● AHA Conference Proceedings: Understanding the Com-plexity of Trans Fatty Acid Reduction in the American Diet:American Heart Association Trans Fat Conference 2006.Read about the current status and future implications ofreducing trans fatty acids without increasing saturated fatsin the food supply, while functionality and consumeracceptance of packaged, processed, and prepared foods aremaintained. Product code 71-0326.
● AHA Guideline: Evidence-Based Guidelines for Cardio-vascular Disease Prevention in Women: 2007 Update. Thisupdate provides the most current clinical recommendationsfor the prevention of cardiovascular disease in women �20years of age and is based on a systematic search of thehighest-quality science, interpreted by experts in the fieldsof cardiology, epidemiology, family medicine, gynecology,internal medicine, neurology, nursing, public health, statis-tics, and surgery. These guidelines also cover the primaryand secondary prevention of chronic atherosclerotic vascu-lar diseases. Product code 71-0401.
● AHA Guideline: Prevention of Infective Endocarditis. Thisguideline updates the recommendations for the preventionof infective endocarditis that were last published in 1997.Product code 71-0407.
● AHA Policy Statement: Nonfinancial Incentives for Qual-ity. Four principles were crafted to guide the structure andmetrics used in pay-for-quality programs, and they identi-fied at least 6 areas that required additional research toserve as criteria that should be considered when designingand evaluating pay-for-quality programs. Product code71-0387.
● AHA Scientific Statement: Acute Coronary Care in theElderly, Part I: Non–ST-Segment-Elevation Acute Coro-nary Syndromes. The first part of this 2-part statementsummarizes evidence on patient heterogeneity, clinicalpresentation, and treatment of non–ST-segment elevationacute coronary syndromes in relation to age (�65, 65 to 74,75 to 84, and �85 years). Product code 71-0404.
● AHA Scientific Statement: Acute Coronary Care in theElderly, Part II: ST-Segment-Elevation Myocardial Infarc-tion. This second part summarizes evidence on presentationand treatment of ST-segment–elevation myocardial infarc-tion in relation to age (�65, 65 to 74, 75 to 84, and �85years). Product code 71-0405.
● AHA Scientific Statement: Cardiovascular Risk Reductionin High-Risk Pediatric Patients. A panel of experts re-viewed what is known about very premature cardiovasculardisease in 8 high-risk pediatric diagnoses and, from thescience base, developed practical recommendations formanagement of cardiovascular risk. Product code 71-0378.
● AHA Scientific Statement: Drug Therapy of High-RiskLipid Abnormalities in Children and Adolescents. Thisstatement examines new evidence on the association of
lipid abnormalities with early atherosclerosis, discuss chal-lenges with previous guidelines, and highlight results ofclinical trials with statin therapy in children and adoles-cents with familial hypercholesterolemia or severe hyper-cholesterolemia. Product code 71-0406.
● AHA Scientific Statement: Essential Features of a Surveil-lance System to Support the Prevention and Managementof Heart Disease and Stroke. This statement provides abrief overview of the Healthy People 2010 goals, preven-tion and management strategies, and the role of surveil-lance in monitoring the impact of prevention and treatmentefforts. It also provides a review of the existing surveil-lance system for monitoring progress toward preventingheart disease and stroke in the United States and recom-mendations for filling important gaps in that system.Product code 71-0386.
● AHA Scientific Statement: Exercise and Acute Cardiovas-cular Events: Placing the Risks Into Perspective. Thisscientific statement discusses the potential cardiovascularcomplications of exercise, their pathological substrate, andtheir incidence and suggests strategies to reduce thesecomplications. Product code 71-0400.
● AHA Scientific Statement: Genetic Basis for CongenitalHeart Defects: Current Knowledge. This statement pro-vides the clinician with a summary of what is currentlyknown about the contribution of genetics to the origin ofcongenital heart disease. Product code 71-0376.
● AHA Scientific Statement: Indications for Heart Trans-plantation in Pediatric Heart Disease. Learn about theevaluation that led to the development and refinement ofindications for heart transplantation for patients with con-genital heart disease and pediatric cardiomyopathies inaddition to indications for pediatric heart retransplantation.Product code 71-0393.
● AHA Scientific Statement: Noninherited Risk Factors andCongenital Cardiovascular Defects: Current Knowledge.This statement summarizes the currently available litera-ture on potential fetal exposures that might alter risk forcardiovascular defects. Product code 71-0377.
● AHA Scientific Statement: Physical Activity InterventionStudies: What We Know and What We Need to Know. Anoverview is provided of existing physical activity interven-tion research, focusing on subpopulations and interventionmodalities. New ideas and recommendations are alsooffered to improve the state of the science within each areaand, where possible, to propose ideas to help bridge thegaps between these existing categories of research. Productcode 71-0369.
● AHA Scientific Statement: Recommendations and Consid-erations Related to Preparticipation Screening for Cardio-vascular Abnormalities in Competitive Athletes: 2007 Up-date. Preparticipation cardiovascular screening is thesystematic practice of medically evaluating large, generalpopulations of athletes before participation in sports for thepurpose of identifying (or raising suspicion of) abnormal-ities that could provoke disease progression or suddendeath. Identifying the relevant diseases may prevent some
B1
m

instances of sudden death after temporary or permanentwithdrawal from sports or targeted treatment interventions.Product code 71-0399.
● AHA Scientific Statement: Relevance of Genetics andGenomics for Prevention and Treatment of CardiovascularDisease. Approaches that researchers are using to advanceunderstanding of the genetic basis of cardiovascular dis-ease are discussed, as well as details of the current state ofknowledge regarding the genetics of myocardial infarction,atherosclerotic cardiovascular disease, hypercholesterol-emia, and hypertension. Product code 71-0410.
● AHA Scientific Statement: Treatment of Hypertension inthe Prevention and Management of Ischemic Heart Dis-ease. This statement summarizes the published data relat-ing to the treatment of hypertension in the context of coronaryartery disease prevention and management and attempts, onthe basis of the best available evidence, to develop recom-mendations that will be appropriate for blood pressure reduc-tion and the management of coronary artery disease in itsvarious manifestations. Product code 71-0412.
● AHA Scientific Statement: Use of Nonsteroidal Antiinflam-matory Drugs: An Update for Clinicians. Read about thecurrent evidence that indicates that selective COX-2 inhib-itors have important adverse cardiovascular effects, whichinclude increased risk for myocardial infarction, stroke,heart failure, and hypertension. Product code 71-0396.
● AHA/ASA Guideline. Guidelines for the Early Managementof Adults With Ischemic Stroke. This guideline is anoverview of the evaluation and treatment of adults withacute ischemic stroke for physicians and other emergencyhealthcare providers who treat patients within the first 48hours after stroke. Information for healthcare policy mak-ers is included. Recommendations for management fromthe first contact by emergency medical services personnelthrough initial admission to the hospital are also provided.Product code 71-0398.
● AHA/ASA Guideline: Guidelines for the Management ofSpontaneous Intracerebral Hemorrhage in Adults: 2007Update. Current and comprehensive recommendations arepresented for the diagnosis and treatment of acute sponta-neous intracerebral hemorrhage. Product code 71-0411.
● AHA/AACVPR Scientific Statement: Core Components ofCardiac Rehabilitation/Secondary Prevention Programs:2007 Update. This updated statement presents currentinformation on the evaluation, interventions, and expectedoutcomes in each of the core components of cardiacrehabilitation/secondary prevention programs, in agree-ment with the 2006 update of the American Heart Associ-
ation/American College of Cardiology Secondary Preven-tion Guidelines, including baseline patient assessment,nutritional counseling, risk factor management (lipids,blood pressure, weight, diabetes mellitus, and smoking),psychosocial interventions, and physical activity counsel-ing and exercise training. Product code 71-0394.
● AHA/ACC/HRS Scientific Statement: Recommendations forthe Standardization and Interpretation of the Electrocar-diogram: Part I: The Electrocardiogram and Its Technol-ogy. This statement examines the relation of the restingECG to its technology and to foster an understanding ofhow the modern ECG is derived and displayed so thatstandards are established that will improve the accuracyand usefulness of the ECG in practice. Product code71-0389.
● AHA/ACC/HRS Scientific Statement: Recommendations forthe Standardization and Interpretation of the Electrocar-diogram: Part II: Electrocardiography Diagnostic State-ment List. This statement provides a concise list of diag-nostic terms for ECG interpretation that can be shared bystudents, teachers, and readers of electrocardiography. Anintended outcome of this statement list is greater unifor-mity of ECG diagnosis and a resultant improvement inpatient care. Product code 71-0390.
● AHA/ACC/SCAI/ACS/ADA Science Advisory: Preventionof Premature Discontinuation of Dual Antiplatelet Therapyin Patients With Coronary Artery Stents. This advisorystresses the importance of dual antiplatelet therapy afterplacement of a drug-eluting stent and educating the patientand healthcare providers about hazards of premature dis-continuation. Product code 71-0395.
● AHA/ADA Scientific Statement: Primary Prevention ofCardiovascular Diseases in People With Diabetes Mellitus.The ADA and AHA have issued separate recommendationsfor each of the cardiovascular risk factors in patients withdiabetes. This statement attempts to harmonize the recom-mendations of both organizations where possible andrecognizes areas in which AHA and ADA recommenda-tions differ. Product code 71-0379.
● AHA/HRS Scientific Statement: Addendum to “Personaland Public Safety Issues Related to Arrhythmias That MayAffect Consciousness: Implications for Regulation andPhysician Recommendations: A Medical/Scientific State-ment From the American Heart Association and the NorthAmerican Society of Pacing and Electrophysiology”: PublicSafety Issues in Patients With Implantable Defibrillators. Thisstatement extends the original 1996 recommendations andprovides specific recommendations on driving for individualswith implantable cardioverter-defibrillators (implanted forprimary prevention). Product code 71-0392.
B2 News

AHA MeetingsMany of these meetings are sponsored by the AmericanHeart Association (AHA) and its Scientific Councils. Forinformation, contact AHA, Scientific Meetings, 7272Greenville Avenue, Dallas, TX 75231-4596; Fax 214-373-3406; E-mail [email protected]; orvisit the Web site http://my.americanheart.org/portal/professional/conferencesevents
2007July 30–Aug 2: 4th Annual Symposium of the Ameri-
can Heart Association Council on Basic Cardio-vascular Sciences: Cardiovascular Repair andRegeneration: Structural and Molecular Ap-proaches in the Cellular Era. Keystone, Colo. This3.5-day conference is a multifaceted symposiumhighlighting research under development in the car-diovascular community targeted at slowing and/orreversing the pathogenesis of disease. The confer-ence will focus on how cellular-based approachesare being manipulated to enhance the repair andregeneration capabilities of the cardiovascular sys-tem with the goal of therapeutic based interventions.See Web site: http://www.heart.org/presenter.jhtml?identifier�3044056
Sept 26–29: 61st Annual High Blood Pressure ResearchConference 2007. Tucson, Ariz. The 2.5-day scien-tific program gives physicians and research investi-gators an opportunity to enhance their knowledge,advance their skills, and learn about the latestdevelopments in research pertaining to hyperten-sion, stroke, kidney function, obesity, and genetics.The program will include state-of-the-art lecturesand more than 350 oral and poster abstract presenta-tions and discussions led by authorities. See Web site:http://www.heart.org/presenter.jhtml?identifier�3043476
Nov 4–7: Scientific Sessions 2007. Orlando, Fla. Scien-tific Sessions encompasses 4 days of invited lecturesand investigative reports. Simultaneous presenta-tions represent all fields of cardiovascular and re-lated disciplines. The program will include morethan 4,000 basic, clinical, and population scienceabstract presentations; plenary, special, and how-tosessions, morning programs and cardiovascularseminars; clinical practice sessions focusing on cur-rent standards of care for practicing clinicians;translational science sessions that bring togetherbasic scientists and clinicians; and Ask the Expertsluncheons and in-depth subspecialty updates. There willalso be 7 pre-Sessions symposia. See Web site: http://scientificsessions.americanheart.org
2008Feb 20–22: International Stroke Conference 2008. New
Orleans, La. This 2.5-day conference provides a
forum in which to present recent scientific workrelated to stroke and cerebrovascular disease. Morethan 600 abstract presentations and lectures will befeatured. This year, special symposia will focus onnumerous topics, including controversies in vascularcognitive impairment, genetics of stroke, gettingtherapies across the blood–brain barrier, metabolicdown regulation in cerebral ischemia, stroke inneonates, stroke in women, platelet resistance instroke prevention, diagnosis and management ofAVM, aneurysm and intracranial hemorrhage, therole of exercise in stroke rehabilitation, the latest instroke prevention, advancing stroke systems of care,and other informative symposia. Sessions in clinicalcategories will center on diagnosis, acute manage-ment, in-hospital treatment, rehabilitation and recov-ery, pediatric stroke, prevention and community/riskfactors, outcomes, vascular cognitive impairment,and systems of stroke care. Experimental categorieswill address neurons/glia/inflammation and vascularpathophysiology/thrombosis. See Web site:http://www.heart.org/presenter.jhtml?identifier�3045505
Mar 11–15: Joint Conference – 48th CardiovascularDisease Epidemiology and Prevention and theNutrition, Physical Activity, and MetabolismConference 2008. Colorado Springs, Colo. TheAnnual Conference offers participants the opportu-nity to learn about: population trends in cardiovas-cular diseases and their risk factors; causes andmechanisms of atherosclerosis and other vasculardiseases; results of cardiovascular disease treatmentand prevention trials; methods of population surveil-lance for cardiovascular disease and risk factors;techniques in preventive cardiology nutrition andcardiovascular disease; and outcomes research incardiovascular disease. See Web site: http://www.heart.org/presenter.jhtml?identifier�3046690
Other Meetings of Interest—Domestic
2007Sept 5–8: 8th Annual New Cardiovascular Horizons
and Management of the Diabetic Foot & WoundHealing. New Orleans, La. For more information,contact [email protected], phone337-261-0944, or fax 337-572-9778. See Web site:http://www.newcvhorizons.com
Other Meetings of Interest—International
2007Sept 17–20: 4th European Meeting on Vascular Biology
and Medicine. Bristol, United Kingdom. Keynoteand plenary speakers’ topics include atherosclerosistreatment, heart disease treatment, clinical modula-
B3
Meetings Calendar

tion of angiogenesis, vulnerable plaque, and pheno-type of vascular cells. Tracks include atherosclero-sis, endothelium and angiogenesis, and cellulardysfunction. For more information, [email protected], phone �44-1922-457-984, or fax �44-1922-455-238. See Web site:http://2007.emvmb.org
Oct 7–10: 7th International Congress on CoronaryArtery Disease–From Prevention to Intervention(ICCAD 7). Venice, Italy. The meeting will followthe format of the very successful previous ICCADCoronary Artery Disease meetings and will providea comprehensive update on coronary disease in allits aspects. Keynote lectures will be delivered by adistinguished international faculty, while a largenumber of selected free communications will reportnew data from basic research laboratories and clin-ical centers around the globe. The program willinclude sessions on molecular mechanisms, genetherapy and cell therapy, epidemiology and preven-tion, and clinical aspects. There will be a majorfocus on new frontiers in interventional cardiologyand to the surgical management of coronary disease.A new feature will be a fast track for recent and“about to break” clinical trials. For more informa-tion, contact [email protected] or phone �41-22-908-0488. See Web site: http://www.kenes.com/cad7
Oct 7–10: 20th Annual Congress of the EuropeanSociety of Intensive Care Medicine. Berlin, Ger-
many. A series of thematic lectures will run through-out the congress that describe progresses and inno-vations from 14 separate topics. Pre-conferencemeetings are also offered. Educational sessions willtake the form of lectures, round tables, pro-condebates, clinical presentations, core competencies,tutorials, and interactive education. For more infor-mation, contact [email protected], phone �32-2-559-03-55, or fax �32-2-527-00-62. See Web site:http://www.esicm.org
Oct 14–16: 5th International Meeting on IntensiveCardiac Care. Tel Aviv, Israel. Three parallelsessions, with more 100 presentations, will be fea-tured. A parallel nursing stream will also be pro-vided. For details, contact [email protected]. SeeWeb site: http://isas.co.il/cardiaccare2007
Nov 8 –11: Fifth International Congress on VascularDementia. Budapest, Hungary. Attendees shallhave an opportunity to deliberate on large andsmall vessel brain diseases and how they contrib-ute to cognitive decline. There will also be anopportunity to identify the specific psychologicalmarkers, if any, of vascular dementia, and also thegenetic factors involved. The overlap with Alz-heimer’s disease will be a central issue, as willbe the white matter changes frequently seen invascular dementia. For details, [email protected]. See Web site: http://www.kenes.com/vascular
The American Heart Association welcomes announcements of interest to physicians, scientists, researchers, andothers concerned with cardiovascular and cerebrovascular medicine. All copy is reviewed by the ScientificPublishing Department. Content may be edited for style, clarity, and length. Copy should be sent to Publications–AHA News & Meetings Calendar, American Heart Association, 7272 Greenville Ave, Dallas, TX 75231-4596; Fax214-691-6342; E-mail [email protected]
B4 Meetings Calendar

Sixty-six Premium Professional Members were elected Fellows and International Fellows of the American HeartAssociation (AHA) in the spring 2007. Fellows are elected on the basis of their outstanding credentials, achievements, andcommunity contributions to the study of cardiovascular disease and stroke. Persons elected to fellowship are entitled to useFAHA, Fellow of the American Heart Association, as a professional designation. Fellows who reside outside the UnitedStates and Canada are designated International Fellows. For more information on the AHA Fellowship program, please visitour Web site at http://www.americanheart.org/presenter.jhtml?identifier�3033104
COUNCIL ON CLINICALCARDIOLOGY
MARTIN R. BERK, MD, FAHAPartner/PhysicianCardiology & InterventionalVascular AssociatesDallas, Tex
David J. D’Agate, DO,FACC, FCCP, FAHASuffolk Heart Group, LLPSmithtown, NY
Harold L. Dauerman, MD,FAHADirector, CV CatheterizationLabsUniversity of VermontBurlington, Vt
Mark H. Drazner, MD, MSc,FAHAAssociate Professor/MedicalDirectorUniversity of Texas SouthwestMedical CenterDallas, Tex
Mark B. Effron, MD, FAHAMedical FellowLilly Corporate CenterIndianapolis, Ind
Victor A. Ferrari, MD,FAHAAssociate Director, NonInvasive ImagingUniversity of PennsylvaniaMedical CenterPhiladelphia, Pa
Robert A. Harrington, MD,FAHAProfessor of MedicineDirector, Duke ClinicalResearch InstituteDuke University MedicalCenterDurham, NC
John A. Hildreth, MD,FAHAMedical Director, FPL GroupPalm Beach Gardens, Fla
Eileen Michelle Hsich, MD,FAHAAssistant ProfessorCase Western ReserveUniversityCleveland, Ohio
Atul R. Hulyalkar, MD,FAHACardiologistNorth Ohio Heart Center, Inc.Westlake, Ohio
Birgit Kantor, MD, PhD,FAHASenior Associate Consultant IIIMayo Clinic, RochesterRochester, Minn
Kelley D. Kennedy, MD,FAHACardiologistBloomington Heart InstituteNormal, Ill
Daniel M. Kolansky, MD,FAHAAssociate Professor ofMedicineUniversity of PennsylvaniaPhiladelphia, Pa
Robert J. Lederman, MD,FAHAInvestigator, CV BranchNational Heart, Lung, andBlood InstituteNational Institutes of HealthDivision of IntramuralResearchBethesda, Md
Pedro Lozano, MD, FAHAAssistant Professor of MedicineUniversity of Oklahoma HealthSciences CenterOklahoma City, Okla
Wayne L. Miller, MD, PhD,FAHAAssociate Professor ofMedicineMayo Clinic and FoundationRochester, Minn
Ira S. Nash, MD, FAHAAssociate ProfessorCardiovascular InstituteMount Sinai Medical CenterNew York, NY
Jeffrey W. Olin, DO, FAHAProfessor and Director,Vascular MedicineMount Sinai School of MedicineNew York, NY
Armen Ovsepian, MD, FAHAStaff PhysicianSuffolk Heart Group, LLPSmithtown, NY
Robert A. Pelberg, MD,FAHAOhio Heart and VascularCenterCincinnati, Ohio
Jan J. Piek, MD, PhD, FAHAProfessor, CardiologyAcademic Medical CenterAmsterdam, the Netherlands
Manuel A. Quiles-Lugo, MD,FACC, FAHACardiologistSan Juan, Puerto Rico
Srinivas Ramaka, MBBS,MD, DM, FAHAConsultant CardiologistSrivinvas Heart CentreIndia
Dimitrius Richter, MD,FAHAHead of Cardiac DepartmentAthens GurolliwicAthens, Greece
Spanos Vassilios, MD, FAHAConsultant, InterventionalCardiologyEuroclinic HospitalAthens, Greece
Laurence S. Sperling, MD,FAHADirector, Preventive CardiologyEmory University Hospital/TheEmory ClinicAtlanta, Ga
Martin St. John Sutton,MBBS, FAHADirector, Cardiovascular ImagingDepartment ofMedicine/CardiologyHospital of the University ofPennsylvaniaPhiladelphia, Pa
Jon Walter Wahrenberger,MD, FAHAAssistant Professor of MedicineDartmouth Hitchcock MedicalCenterLebanon, NH
Pavlos Toutouzas, MD, FAHAAthens, Greece
Dimitrios N. Tziakas, MD,FAHAAssistant Professor in CardiologyAlexandropolis, Greece
Yee Guan Yap, BMedSci,MBBS, MD, FAHAAssoc. Professor/Head ofCardiologyUniversity of Putra MalaysiaPetaling Jaya, Selangor,Malaysia
COUNCIL ONCARDIOVASCULARDISEASEIN THE YOUNG
Luis E. Alday, MD, FAHAHead, Division of CardiologyHospital AcronauticoArgentina
Stuart Berger, MD, FAHAMedical Director, Herma HeartCenterProfessor of PediatricsMedical College of WisconsinMilwaukee, Wis
David Jonathan Sahn, MD,FAHAProfessor, Pediatrics(Cardiology)Oregon Health and ScienceUniversityPortland, Ore
B5
American Heart Association Newly Elected Fellows, Spring 2007

Craig Andrew Sable, MD,FAHADirector, Echocardiography,Fellowship Training, andTelemedicineChildren’s National MedicalCenterWashington, DC
L. LuAnn Minich, MD, FAHAUniversity of Utah School ofMedicineChildren’s Medical CenterPediatricsSalt Lake City, Utah
COUNCIL ONCARDIOVASCULARRADIOLOGY ANDINTERVENTION
David A. Bluemke, MD, PhD,MSB, FAHAClinical Director, MRIAssociate Professor, Radiologyand MedicineJohns Hopkins UniversitySchool of MedicineBaltimore, Md
Seung Woon Rha, MD, PhD,FAHAProfessorKorea University Guro HospitalSeoul, South Korea
COUNCIL ON CVSURGERYAND ANESTHESIA
Jerrold Henry Levy, MD,FAHAProfessor and Director, CTAnesthesiaEmory HealthcareAtlanta, Ga
Yoshitaka Hayashi, MD,PhD, FAHAOverseas SurgeonDepartment of CardiothoracicSurgeryMonash Medical CentreMonash UniversityClayton, Victoria, Australia
Hitoshi Hirose, MD, PhD,FAHAFaculty StaffCardiothoracic SurgeryDrexel University College ofMedicinePhiladelphia, Pa
Madhav Swaminathan, MD,FASE, FAHAAssistant ProfessorDuke University MedicalCenterDurham, NC
Yasuyuki Shimada, MD,PhD, FAHADirector, Cardiovascular SurgeryYuri General HospitalJapan
COUNCIL ONEPIDEMIOLOGY ANDPREVENTION
Javed Butler, MD, MPH,FAHAAssociate Professor ofMedicineDepartment ofMedicine/CardiologyEmory UniversityAtlanta, Ga
J. Jeffrey Carr, MD, MSCE,FAHAProfessor/Vice ChairDivision of RadiologicSciences/Clinical ResearchWake Forest UniversityDurham, NC
Ellen Demerath, PhD, FAHAAssociate ProfessorCommunity HealthBoonshoft School of MedicineWright State UniversityKettering, Ohio
James M. Galloway, MD,FAHADirector, Native AmericanCardiologyProgram & Senior CardiologistIndian Health ServiceFlagstaff, Ariz
Jiang He, MD, PhD, FAHAProfessor and ChairDepartment of EpidemiologyTulane University School ofPublic Healthand Tropical MedicineNew Orleans, La
Angela Dorthea Liese, PhD,MPH, FAHAAssociate DirectorEpidemiology and BiostatisticsUniversity of South CarolinaColumbia, SC
Hirotsugu Ueshima, MD,PhD, FAHAProfessor, Health SciencesShiga University of MedicalScienceTsukinowa-cho, OtsuJapan
COUNCIL FOR HIGHBLOOD PRESSURERESEARCH
Keiichiro Atarashi, MD, PhD,FAHAPhysician of the CrownPrince’s HouseholdImperial Household AgencyTokyo, Japan
Olakunle O. Akinboboye,MD, MPH, MBA, FAHAAssociate DirectorNew York Hospital/QueensRoslyn, NY
Anil K. Bidani, MD, FAHAProfessor of Medicine andDivision DirectorLoyola University MedicalCenter andHines VA HospitalMaywood, Ill
STROKE COUNCIL
Erfan A. Albakri, MD,FAHAMedical DirectorFlorida Neuro VascularInstituteTampa, Fla
Andrew Butler, PhD, PT,FAHAEmory University School ofMedicineAtlanta, Ga
Joseph Y. Chu, MD, FRCPC,FACP, FAHANeurologistUniversity of TorontoToronto, Ontario, Canada
Niloufar Hadidi, RN, MS,CNS, FAHANeuroscience Clinical NurseSpecialistUniversity of MinnesotaMedical CenterShoreview, Minn
Sherene Schlegel, AssociateRN, FAHAStroke Program ClinicalEffectiveness CoordinatorSwedish Medical CenterSeattle, Wash
Tammy L. Cress, RN, BSN,MSN, FAHAStroke Program ManagerSwedish Medical CenterSeattle, Wash
Afshin Andre Divani, MSc,PhD, FAHADirector, Zeenat QureshiStroke Research CenterUMDNJ – New Jersey MedicalSchoolNewark, NJ
Wende N. Fedder, RN, BSN,MBA, FAHADirector of Nursing, StrokeCenterAlexian Brothers HospitalNetworkChicago, Ill
Elias A. Giraldo, MD, FAHADirector, UTHSC StrokeProgramDepartment of NeurologyUniversity of TennesseeMemphis, Tenn
Keith Lowell Hull, Jr, MD,FAHAPartner, Raleigh NeurologyAssociates, PARaleigh, NC
Robert H. Rossenwasser,MD, FAHAProfessor and ChairmanThomas Jefferson UniversityPhiladelphia, Pa
Sandeep Sachdeva, MBBS,MD, FAHALead Hospitalist StrokeProgramSwedish Medical CenterSeattle, Wash
Sean Isaac Savitz, MD, FAHANeurologist, Stroke ServiceAssistant Professor, NeurologyBeth Israel Deaconess MedicalCenterHarvard Medical SchoolBoston, Mass
B6 AHA Newly Elected Fellows, Spring 2007