
Intramolecular -Arylation of Lithiated Carbamates
385
Intramolecular -Arylation of Lithiated Carbamates A thesis submitted to the University of Manchester for the degree of Doctor of Philosophy in the Faculty of Engineering and Physical Sciences 2012 Anne FOURNIER School of Chemistry
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Intramolecular -Arylation of Lithiated Carbamates

Intramolecular -Arylation of Lithiated Carbamates
A thesis submitted to the University of Manchester
for the degree of Doctor of Philosophy
in the Faculty of Engineering and Physical Sciences
2012
Anne FOURNIER
School of Chemistry

2

3
CONTENTS
Abstract 9
Declaration 10
Copyright Statement 10
Acknowledgements 11
Abbreviations 12
Preface 17
CChhaapptteerr II:: IINNTTRROODDUUCCTTIIOONN 18
I.1 Strategies for the Asymmetric Synthesis of Tertiary Alcohols 18
I.1.1 Asymmetric addition of carbon-based organometallic nucleophiles to
ketones 18
I.1.1.1 Catalytic enantioselective arylation, alkylation and alkenylation reactions 21
I.1.1.2 Catalytic enantioselective allylation reaction 33
I.1.1.3 Catalytic enantioselective alkynylation reactions 39
I.1.2 Catalytic asymmetric aldol reactions 43
I.1.3 Kinetic resolution of tertiary alcohols 51
I.1.4 1,2-Metallate rearrangement of boronate complexes 52
I.2 Intramolecular Electrophilic Arylation of Lithiated Ureas and
Thiocarbamates 55
I.2.1 Discovery of N to C aryl migration in lithiated N-benzyl ureas 55
I.2.2 Aryl migration in lithiated N-benzyl ureas 56
I.2.3 Aryl migration in other lithiated urea analogues 60
I.2.3.1 Aryl migration of N-benzyl-N-pyridyl ureas 60
I.2.3.2 Aryl migration of N-aryl urea derivatives of hetero- or carbocyclic
amines 61
I.2.3.3 Aryl migration of N-allyl-N-aryl ureas 63
I.2.4 Enantioselective synthesis of tertiary thiols by intramolecular arylation
of lithiated thiocarbamates 63

4
I.3 Properties and Reactivity of Lithiated Carbamates 65
I.3.1 Dipole-stabilized carbanions adjacent to oxygen 65
I.3.2 Versatile reactivity and configurational stability of lithiated
O-benzylcarbamates 66
I.3.2.1 Electrophilic substitutions of lithiated O-benzylcarbamates 67
I.3.2.2 N to C arylation migration in lithiated O-benzylcarbamates 70
I.3.3 Configurational stability of lithiated O-allylcarbamates 73
I.3.4 Configurational stability of lithiated O-propargylcarbamates 78
I.4 Carbolithiation Reactions Involving Organolithiums 80
I.4.1 Overview of carbolithiation reactions 80
I.4.1.1 General aspects of carbolithiations 80
I.4.1.2 Intermolecular carbolithiation reactions 82
I.4.1.3 Intramolecular carbolithiation reactions 85
I.4.2 Intermolecular carbolithiation reactions of vinyl carbamates 87
I.4.3 Tandem β-alkylation--arylation of amines by carbolithiation and
rearrangement of vinyl ureas 89
I.5 Aims of the project 91
CChhaapptteerr IIII:: RREESSUULLTTSS AANNDD DDIISSCCUUSSSSIIOONN 94
II.1 Synthesis of Clemastine, an Antihistaminic Agent 94
II.1.1 Total synthesis and absolute configuration of clemastine via resolution 94
II.1.1.1 Ebnöther and Weber’s pioneer work on the synthesis of
(R,R)-clemastine 94
II.1.1.2 Synthesis of optically active (R)-2-(2-chloroethyl)-1-methyl-
pyrrolidine, an intermediate in the synthesis of clemastine via resolution 95
II.1.2 Stereoselective synthesis of ()-hydroxyclemastine 97
II.1.3 Novel asymmetric synthesis of ()-(S,S)-clemastine 98
II.1.3.1 Retrosynthesis of (S,S)-clemastine 98
II.1.3.2 Synthesis of pyrrolidine fragments 99
II.1.3.3 Synthesis of tertiary alcohol by aryl migration of a lithiated
carbamate 103

5
II.1.3.4 Further evidences for stereochemically invertive
rearrangement for carbamates 104
II.1.3.5 Formation of the ether linkage and isolation of clemastine 109
II.2 Spectroscopic and Computational Studies of the Mechanism 116
II.2.1 In situ IR spectroscopy 116
II.2.2 Computational studies 124
II.3 Arylation of Lithiated Carbamates by Intramolecular N to C
Aryl Migration: Scope, Stereoselectivity and Mechanism 130
II.3.1 N to C aryl migration in lithiated N-aryl-O-allylcarbamates 130
II.3.1.1 Synthesis and aryl migration of a cinnamyl carbamate 130
II.3.1.2 Synthesis and aryl migration of -methyl cinnamyl carbamates 132
II.3.1.3 Stereospecificity in the aryl migration 135
II.3.1.4 Aryl migration of simple O-allylcarbamates 142
II.3.1.5 In situ IR spectroscopy studies 143
II.3.2 N to C aryl migration in lithiated N-aryl-O-propargylcarbamates 145
II.3.2.1 Synthesis and attempted aryl migration of primary
O-propargylcarbamates 145
II.3.2.2 Synthesis and aryl migration of -methylated propargylcarbamate 146
II.3.2.3 Stereospecificity in the aryl migration 150
II.3.3 γ-deprotonation-mediated N to C aryl migration of N-aryl-Z-enol
carbamates 153
II.3.3.1 Synthesis of Z-enol carbamates 153
II.3.3.2 -Arylation of Z-enol carbamates by γ-deprotonation and
N to C aryl migration 154
II.3.3.3 In situ IR spectroscopy studies 157
II.3.4 Limits of the N to C aryl migration 159
II.3.4.1 Attempted aryl migration in lithiated carbamates containing
the electron rich heterocycle furan 159
II.3.4.2 Attempted aryl migration in lithiated O-alkylcarbamates 160
II.4 Tertiary Alcohols by Tandem β-Carbolithiation and N to C
Aryl Migration in Enol Carbamates 162
II.4.1 Carbolithiation and aryl migration of N-methyl--aryl-O-vinylcarbamate 162
II.4.2 Carbolithiation and aryl migration of N-isopropyl--aryl-O-vinylcarbamates 163

6
II.4.2.1 Optimisation of the reaction 163
II.4.2.2 Synthesis of diarylalkylalcohols 165
II.4.2.3 Enantioselectivity in the β-carbolithiation-aryl migration 167
II.4.3 Carbolithiation and aryl migration of β-substituted
N-isopropyl--aryl-O-vinylcarbamates 169
II.4.4 Carbolithiation and aryl migration of -proparyl-O-vinylcarbamates 171
II.4.4.1 Synthesis of N-isopropyl-α-propargyl-O-vinylcarbamates 171
II.4.4.2 One-pot synthesis of multiply branched arylalkynylalcohols 172
II.4.4.3 Synthesis of enantioenriched allenes 175
II.4.5 Extension of the substrate scope of the reaction 178
II.4.5.1 Carbolithiation-aryl migration of -alkenyl-O-vinylcarbamates 178
II.4.5.2 Carbolithiation-aryl migration of -silyl-O-vinylcarbamate 180
II.4.5.3 Attempted carbolithiation-aryl migration of -heteroaryl-
O-vinylcarbamates 180
II.5 Further Extension of the Methodology 182
II.5.1 N to C vinyl migration in lithiated O-benzylcarbamates 182
II.5.2 Attempted N to C alkynyl migration in lithiated O-benzylcarbamates 187
II.5.2.1 Synthesis of ynecarbamates 188
II.5.2.2 Reactivity of lithiated ynecarbamates 190
II.5.3 Attempted N to C cyano migration in lithiated O-benzylcarbamates 193
II.5.4 Attempted N to C heteroaryl migration in lithiated O-benzylcarbamates 194
II.6 Conclusions and Future Work 197
CChhaapptteerr IIIIII:: EEXXPPEERRIIMMEENNTTAALL SSEECCTTIIOONN 203
III.1 General Information 203
III.2 General Procedures 205
III.3 Experimental Procedures and Data 217
III.4 X-Ray Crystal Data 360

7
RREEFFEERREENNCCEESS && NNOOTTEESS 364
AAPPPPEENNDDIIXX 380
Appendix 1: List of publications 380
Appendix 2: Experimental procedures for in situ React IR studies 381
Appendix 3: In situ IR spectroscopy monitoring the rearrangement
of a N-allylurea 384

8
Total Word Count : 77 989

9
ABSTRACT
Intramolecular -Arylation of Lithiated Carbamates
A submission for the degree of Doctor of Philosophy at The University of Manchester
Anne Fournier
2012
Keywords: carbamates, organolithiums, arylation/vinylation, stereospecific, carbolithiation
This thesis describes research carried out on the synthesis of tertiary alcohols or
derivatives by N to C aryl/vinyl migration in lithiated carbamates.
Section II.1 describes the first enantioselective synthesis of the antihistamine agent
clemastine, as its (S,S)-stereoisomer as an illustration of the methodology. It has been
achieved by ether formation between a proline-derived chloroethylpyrrolidine and an
enantiomerically enriched tertiary alcohol. The tertiary alcohol was formed from the
carbamate derivative of -methyl-p-chlorobenzyl alcohol by invertive aryl migration on
lithiation. The (S,S)-stereochemistry of the product confirms the invertive nature of the
rearrangement in contrast with related ureas.
Modelling work to establish the origin of this stereodivergent behaviour is reported in
Section II.2. This also reports in-situ IR experiments providing evidence of the mechanistic
pathway of the rearrangement of an O-benzyl-N-aryl carbamate.
The scope of the N to C aryl migration in other stabilised organolithiums is shown in
section II.3. The rearrangement is now addressed in more systematic manner, thus
allowing the -arylation of O-allyl and O-propargylcarbamates (by -deprotonation) and
O-vinylcarbamates (by -deprotonation) to be achieved in good yields but with poor
stereoselectivity.
Section II.4 goes on to show that enol carbamates derived from aromatic or ,-
unsaturated compounds and bearing an N-aryl substituent undergo carbolithiation by
nucleophilic attack at the position of the enol double bond. The resulting carbamate-
stabilised allylic, propargylic or benzylic organolithium rearranges with N to C migration
of the N-aryl substitutent, creating a quaternary carbon to O. The products may be easily
hydrolysed to generate multiply branched tertiary alcohols in good to moderate yields in a
one-pot tandem reaction.
Finally, Section II.5 proves that the rearrangement in lithiated carbamates can be
extended to N to C vinyl transfer.

10
DECLARATION
No portion of the work referred to in this thesis has been submitted in support of an
application for another degree or qualification of this or any other university or other
institute of learning
COPYRIGHT STATEMENT
i. The author of this thesis (including any appendices and/or schedules to this
thesis) owns any copyright in it (the “Copyright”) and she has given The
University of Manchester the right to use such Copyright for any administrative,
promotional, educational and/or teaching purposes.
ii. Copies of this thesis, either in full or in extracts, may be made only in
accordance with the regulations of the John Rylands University Library of
Manchester. Details of these regulations may be obtained from the Librarian.
This page must form part of any such copies made.
iii. The ownership of any patents, designs, trade marks and any and all other
intellectual property rights except for the Copyright (the “Intellectual Property
Rights”) and any reproductions of copyright works, for example graphs and
tables (“Reproductions”), which may be described in this thesis, may not be
owned by the author and may be owned by third parties. Such Intellectual
Property Rights and Reproductions cannot and must not be made available for
use without the prior written permission of the owner(s) of the relevant
Intellectual Property Rights and/or Reproductions.
iv. Further information on the conditions under which disclosure, publication and
exploitation of this thesis, the Copyright and any Intellectual Property Rights
and/or Reproductions described in it may take place is available from the Head
of School of Chemistry (or the Vice-President).

11
ACKNOWLEDGEMENTS
I would firstly like to thank Jonathan Clayden for the opportunity to work in his group and
for his support and help throughout the course of the research and during the search of my
postdoctoral. I would like to mention that it has been really pleasant to work with. I am
also grateful to the EPSRC and GlaxoSmithKline for the provision of funds.
I thank my industrial supervisor, Chris Nichols, for his continuous support in the Ph.D.
program and his patience during my three-month placement at GSK. Thanks also to Nick
Wooster (at GSK) for his assistance for React IR studies.
I am indebted to the staff at the School of Chemistry for the provision of services. I would
especially like to thank Rohana and Gareth for mass spectrometry/HPLC.
Thanks must go to all members of the Clayden group past and present – PhD, post-doc and
MChem/summer students. “Merci” to all my compatriots, Morgan, Gilles, Julien, Gaëlle,
Vincent, Thomas, Hatice, Julie, Juliette, Alexis, Alexandra and Anne-Sophie. In no
particular order thanks also to Paul, Beckii, James, Alex, Jordi, Alberto, Steve, Jemma,
Mike, Nadia, Rob, Liam, Daniele, Matteo, Tommaso, Edmund, Simon H., Rachel, Sam,
Francis and Bryden.
A special thank also to Abby, my first lab-hood neighbour, for tolerating my “horrible
English”, Simon “Sadsack” for his attention, Dan and Nicole for teaching me climbing.
Special mention goes to Sarah who is now a very good friend, with whom I laughed a lot
and spent some good times.
I am additionally grateful to Sarah, Chris and Vittorio for proof-reading.
I really enjoyed walking in the Peak District, Lake District, Snowdonia, Scotland,…with
good and bad weather, but contrary to what we think in France the weather is not that bad,
I have never been so tanned as in UK!!.
Many thanks to my family, especially my sister and my friends even though most of you
have no idea what it is that I do.

12
ABBREVIATIONS
Å angstrom(s)
[]D specific rotation [expressed without units ; the units (deg.mL)/(g.dm) are
understood]
Ac acetyl
aq. aqueous
Ar aryl
Aro aromatic
BINOL bi-2-naphthol
Bn benzyl
Boc tert-butoxycarbonyl
BOX bisoxazoline
Br broad (spectral)
Bu, n-Bu normal (primary) butyl
s-Bu sec-butyl
t-Bu tert-butyl
Bz benzoyl
c concentration
°C degrees Celsius
CAN ceric ammonium nitrate
cat. catalyst
Cb N-N-diisopropylcarbamoyl
Cbz benzyloxycarbonyl
CI chemical ionisation
CIP contact ion pair
CIPE complex induced proximity effect
Cm centimeter(s)
cm-1
wavenumber(s)
coe cyclooctene
Conds. conditions
Conv. conversion

13
COSY correlation spectroscopy
Cp cyclopentadienyl
cPr cyclopropyl
C.T. chirality transfer
Cy cyclopentyl
chemical shift in parts per million downfield from tetramethylsilane
d doublet (spectral); day(s)
DABCO 1,4-diazabicyclo[2.2.2]octane 5-(dimethylamino)-1-naphthalenesulfonyl
DBB 4,4’-di-tert-butylbiphenyl
DCE 1,2-dichloroethane
d.e. diastereoisomeric excess
DEPT distortionless enhancement by polarization transfer
DFT density functional theory
DIBAL-H diisobutylaluminium hydride
DiPEA diisopropylethylamine
DIPT ()-diisopropyltartrate
DMAP 4-(N,N-dimethylamino)pyridine
DME 1,2-dimethoxyethane
DMF dimethylformamide
DMPU 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone
DMSO dimethyl sulfoxide
dppa diphenylphosphoryl azide
d.r. diastereomeric ratio
E or EX electrophile
e.e. enantiomeric excess
EI electron impact
equiv. or eq. equivalent(s)
e.r. enantiomeric ratio
ESI or ES electrospray ionization
Et ethyl
EWG electron-withdrawing group
fum-
fumarate
g gram(s)

14
GC gas chromatography
H hour(s)
Hex hexyl
HMBC heteronuclear multiple bond correlation
HMDS hexamethyldisilazane
HMQC heteronuclear multiple quantum correlation
HPLC high-performance liquid chromatography
HRMS high-resolution mass spectrometry
HSQC heteronuclear single quantum correlation
Hz hertz
inv inversion
IPA propan-2-ol
IR infrared
J coupling constant (in NMR spectrometry)
L ligand; liter(s)
L*
chiral ligand
LDA lithium diisopropylamide
lit. literature value
LUMO lowest unoccupied molecular orbital
LiTMP lithium 2,2,6,6-tetramethylpiperidide
micro
m multiplet (spectral) or milli or meta
M molar (moles per liter) or unspecified metal or molecular mass (mass
spectrometry)
M+
parent molecular ion
Me methyl
min minute(s)
mmol millimole(s)
mol mole(s)
m.p. melting point
MS mass spectrometry; molecular sieves
MW molecular weight
m/z mass-to-charge ratio

15
N normal (equivalents per liter)
NBS N-bromosuccinimide
nm nanometer(s)
NMP N-methylpyrrolidine
NMR nuclear magnetic resonance
nOe nuclear Overhauser effect
Np naphthyl
Nu- or Nu nucleophile
n-Pent pentyl
n.d. non determined
oct octuplet
o ortho
p para
Ph phenyl
PMP p-methoxyphenyl
ppm part(s) per million
Pr propyl
i-Pr isopropyl
Py pyridine; pyridyl
q quartet (spectral)
qn quintet (spectral)
R unspecified substituent
Ret retention
Rf retention factor
RT room temperature
s singlet (spectral); second(s)
s. selectivity
SCX strong cation exchange (cartridge)
SE’ electrophilic substitution with allylic rearrangement
SE2 bimolecular electrophilic substitution
sep septet (spectral)
S.M. starting material
SN2 bimolecular nucleophilic substitution
SNAr nucleophilic aromatic substitution

16
()-sp. ()-sparteine
SSIP solvent separated ion pair
t time
t triplet (spectral)
T or Temp. temperature
TBAF tetrabutylammonium fluoride
TBDPS tert-butyldiphenylsilyl
TBHP tert-butyl hydroperoxide
TBME tert-butylmethylether
Tf trifluoromethanesulfonyl
THF tetrahydrofuran
TIPS triisopropylsilyl
TIPT tetra(isopropoxy)titanium
TLC thin layer chromatography
TMEDA N,N,N,N-tetramethyl-1,2-ethylenediamine
TMS trimethylsilyl; tetramethylsilane
Tol tolyl or toluene
tR retention time (in chromatography)
Ts p-toluenesulfonyl
UV ultraviolet
max absorption maximum
vis visible
v/v volume per unit volume
w weak
w/w weight per unit weight

17
PREFACE
The author worked for sanofi-aventis in France for two years as a Data Quality Manger
before graduating from the University of Paris XI in Orsay (France) in 2008 with a Master
of Science degree in Organic Chemistry. Her final year research project was undertaken at
the Institut de Chimie Moléculaire et des Matériaux (ICMMO) under the supervision of Dr.
Gérard Rousseau. The project investigated the reactivity of 5-phenyl-2H-pyrrol-2-one
synthesised by a 5-endo halo cyclisation of , -ethylenic hydroxamate.
In 2008, she moved to England to join the group of Prof. Jonathan Clayden working on the
lithiation of carbamates. This research is embodied in this thesis. In May 2012, the author
will take up a post-doctoral research position with Prof. James Gleason at McGill
University in Montreal, Canada.

18
CChhaapptteerr II :: IINNTTRROODDUUCCTTIIOONN
I.1 Strategies for the Asymmetric Synthesis of Tertiary Alcohols
Tertiary alcohols are common motifs in natural products and medicinally active agents
(Figure 1). As such, roughly 20 % of the top 50 pharmaceutical contain tertiary alcohols or
their derivatives.[1]
Figure 1. Examples of Tertiary Alcohols Containing Natural Products and
Pharmaceuticals.
HN
ON
N
O OH
O
O
O
O
OH
O
OH
O
O
O
OMe
N
OH
O
O
H OO
HO2C OHCO2H
O
(CH2)11CH3
OH
O
O
HO
OH OHO
NHMe
K252a (kinase inhibitor)
Integerrimine (pheromone)
Cinatrin C3
(anti-inflammatory)
Fostriecin (anti-tumor)
Erythromycin (antibiotic)
The numerous methods for asymmetric synthesis of chiral secondary alcohols (e.g.
kinetic resolution, Noyori asymmetric hydrogenation, enantioselective addition to
aldehydes and many are commercially available) are not matched by a comparable choice
of methods for the synthesis of chiral tertiary alcohols (no oxidation of C-O possible for
kinetic resolution, no hydrogenation of carbonyl possible, ketones are less reactive and
smaller steric and electron differences between prochiral carbons).
1,2-Nucleophilic addition to carbonyl compounds is probably the most straightforward
and useful manner of achieving this goal.

19
I.1.1 Asymmetric Addition of Carbon-Based Organometallic
Nucleophiles to Ketones
Tertiary alcohols are commonly obtained by addition of an organometallic reagent to
a ketone (Scheme 1, paths a and b). Although such a process can be rendered asymmetric
through the use of chiral ligands, the process requires discrimination of the enantiotopic
faces of the ketone, which can be a significant challenge when the ketone is dialkyl or
diaryl substituted.[2]
Scheme 1. Schematic Representation of the Addition of a Nucleophile on the Prochiral
Faces of a Ketone.
Nu
OHRL
RS
ORL
RS
Path a
Nu
OHRS
RL
Path b
ML*
Nu�
Nu�
a
b
ORL
RS
Nu�
Nu�
a
b
ML*
Organometallic alkylation or arylation can be classified into three processes: (a)
substrate activation using a Lewis acid catalyst, (b) reagent activation using a Lewis base
catalyst, and (c) dual activation of the substrate and reagent using a bifunctional Lewis
acid-Lewis base catalyst (Scheme 2). In recent years, a few novel methods have been
introduced, most of them based on the double activation concept.[3]

20
Scheme 2. Catalytic Synthesis of Tertiary Alcohols from Ketones and Organometallic
Reagents.
R1 R2
O
MX
Lewisacid* R1 R2
O
Lewisbase*
(a) substrate activation with a Lewis acid catalyst
(b) reagent activation witha Lewis base catalyst
R3
MXR3
R1 R2
O
Lewisacid*
Lewisbase*
(c) dual activation of the substrate and reagentwith a bifunctional Lewis acid-Lewis base catalyst
MXR3
Although several examples involving organolithium and Grignard reagents have been
reported,[4]
these usually require greater than stoichiometric amounts of chiral ligands. In
addition, the high reactivity of these reagents precludes the presence of many functional
groups in the substrates.
In contrast, organozinc reagents are mild and exhibit excellent functional group
compatibility, making them the most popular species for carbon-carbon bond-forming
reactions on carbonyl compounds.
Organoaluminium complexes are also tolerant of a wide range of functional groups
and have been applied to the asymmetric addition to ketones.
The Lewis acidic character and nucleophile activation ability (transmetallation and
deprotonation) of copper catalysts have also been utilised in asymmetric C-C bond-
formation to ketones. Lewis acidic cationic copper-catalyzed tetrasubstituted carbon-
forming reactions generally produce excellent enantioselectivity, but substrates are
restricted to activated ketones, such as pyruvate esters. The excellent enantioselectivity is
attributed to chelate coordination of the substrates to the copper centre containing a well-

21
defined geometry. On the other hand, catalytic asymmetric C-C bond formation to simple
ketones is promoted through activation of the nucleophile via transmetallation.[5]
I.1.1.1 Catalytic Enantioselective Arylation, Alkylation and Alkenylation Reactions
Dosa and Fu reported the first catalytic enantioselective addition of organozinc
reagents to ketones in 1998.[6]
Noyori’s 3-exo-dimethylaminoisoborneol ((+)-DAIB)[7]
(15
mol %) ligand 1 allowed the addition of diphenyl zinc to aromatic and aliphatic ketones in
good to high enantiomeric excesses of up to 91 % (Scheme 3).
Scheme 3. Dosa and Fu’s Breakthrough in the Asymmetric Addition of Organozinc
Reagents on Ketones.
R1 R2
O
ZnPh2
(3.5 equiv.)
(15 mol %)
PhMe, RTMeOH (1.5 equiv.)
R1 = aryl, sec-alkyl
R2 = n-alkyl
R1 R2
OHPh
60-91% e.e.
Me2N
HO
Me
N
OZn
ZnO
Ph
PhPh
Ph
R
1
An advancement in this area came in the same year when the group of Yus reported
the first addition of dialkylzinc reagents to ketones promoted by a hydroxysulfonamide-
titanium catalyst.[8]
Although an equimolar amount of titanium(IV) isopropoxide was
required, some substrates exhibited good enantioselectivities (up to 89 % e.e.) in the
presence of 20 mol % of chiral camphorsulfonamide ligand 2 (Scheme 4).

22
Scheme 4. Asymmetric Alkylation of Ketones with Diethylzinc Developed by the Groups of
Yus and Walsh.
Me
O
Et2ZnTi(O-iPr)4, L*
PhMe
* Me
HO Et
OH
O2S NH
2
OH
O2S NH HN SO2
HO
3
O2S NH HN SO2
HO
4MeO
Yus’ first generation catalyst: 20 mol % 2, CaH2, PhMe, 4 °C, 4 d, 71 % yield, 86 % e.e.
Walsh’s catalytic system: 2 mol % 3, PhMe, RT, 29 h, 71 % yield, 96 % e.e.
Yus’ second generation catalyst: 5 mol % 4, PhMe, 25 °C, 120 h, 65 % yield, 99 % e.e.
This first generation of catalytic system inspired other groups for the design of
improved ligands and the major improvement was reported in 2002 by Walsh[9]
and in
2005 by Yus[10]
. The most successful C2-symmetric disulfonamide ligand 3 was designed
in order to induce an increased constrained geometry. In most cases, it showed improved
reactivity. The ligand loading could be reduced to 2 mol % while retaining high
enantioselectivity (up to 96 % e.e.) for the addition of diorganozinc reagents to a range of
aromatic and aliphatic ketones (Scheme 4). The results of Walsh’s study are shown in
Table 1. Yus et al. published similar data.
Table 1. Conditions, Yields and e.e. for the Asymmetric Addition of Ethyl Group to
Ketones with Ligand 3.
Entry Substrate 3 (mol %) t (h) Yield (%) e.e. (%)
O
X
1 X = H 2 29 71 96 (S)
2 X = 3-Me 10
2
12
24
82
78
99
99
3 X = 4-OMe 10 111 85 94

23
Table 1. Continued.
Entry Substrate 3 (mol %) t (h) Yield (%) e.e. (%)
4 X = 3-CF3 2 14 56 98
5 X = 2-Me 10 48 24 96
6
O
10 22 35 99
7 Bu
O
10
2
47
102
83
79
87
88
8
O
Cl
10 44 82 89
9
O
2 46 56 96
10
O
2 26 80 90
11
O
10 68 68 70
This bissulfonamide structure was also used by Ramón, Yus et al. to study various
chiral ligands,[11]
which finally gave an optimised second generation catalyst 4 with perfect
enantioselectivity ( 99 % e.e.) but only in moderate yield (65 %) for the alkylation
reaction (Scheme 4). However, the scope of this catalyst remains undefined, because only
one example has been reported. This ligand has also shown to be an excellent promoter for
the catalytic enantioselective arylation of p-bromoacetophenone (96 %, 99 % e.e.) during
which the phenyl zinc intermediate was generated from triphenylboron by transmetallation
with dimethylzinc.
In the same year, the groups of Walsh and Yus reported the asymmetric arylation of
ketones catalyzed by titanium tetraisopropoxide and the chiral ligand 3 (Scheme 5).
Significant improvements, such as a low catalyst loading (10 mol %) and the use of only
1.6 equiv. of diphenylzinc, were made.[12]
The addition of commercially available
diphenylzinc to ketones gave the expected tertiary alcohols with practically quantitative
chemical yields and enantiomeric excesses as high as previously reported results for
similar aryl transfer processes (Table 2).[13]

24
Scheme 5. Asymmetric Phenylation of Ketones with Diphenylzinc.
R1 R2
O
ZnPh2
(1.6 equiv.)
Ti(O-iPr)4
(0.6 equiv.)
3 (10 mol %)
R1 R2
HO Ph
PhMe/Hexane, 6-24 h
Table 2. Yields and e.e. for the Asymmetric Phenylation of Ketones with Diphenylzinc.
Entry R1 R
2 Yield (%) e.e. (%)
1 Et 3-ClC6H4 99 92
2 Et 4-ClC6H4 99 88
3 Me 3-CF3C6H4 93 95
4 Me 2-BrC6H4 76 95
5 Me 2-naphthyl 99 96
6 Me 1-cyclohexenyl 94 93
In the study by Yus et al., a titanium catalyst of 5 mol % 3 was employed on four
examples of ZnPh2 additions to aromatic ketones, furnishing the desired products in
enantioselectivities of 80 to 96 % e.e..[14]
Extensive applications of ligand 3 were also reported by Walsh et al. for the
asymmetric addition of a variety of carbon-based nucleophiles on ketones.[15]
Some
representative examples are displayed in Scheme 6. The catalytic system using 3 is
especially reactive toward ,-unsaturated ketones and aryl alkyl ketones. The range of
nucleophiles was also widened from simple ethyl and methyl groups to functionalised alkyl,
aryl, vinyl and dienyl groups with high enantioselectivities.
Scheme 6. Walsh’s Catalytic Method for the Asymmetric Addition of Functionalised
Organometallics to Ketones.
R1 R2
OZn(R3)2
Ti(O-iPr)4
3 (0.5-10 mol %)R1 R2
HO R3
aryl alkyl ketones and -unsaturated ketones
R3 = functionalised alkyl, aryl, vinyl and dienyl groups

25
Scheme 6. Continued.
Cl
HO Ph
92 % e.e.
HO (CH2)5-Br
90 % e.e.
HO
Bu
97 % e.e.
HO
> 99 % e.e.
OTBS
Some representative examples:
So far, enantioselective organozinc addition to ketones by double activation with
bifunctional unconjugate Lewis acid-Lewis base catalysts, in which the Lewis acid and the
Lewis base are attached to each other, has been reported. Recently, Ishihara et al.
developed a highly enantioselective organozinc addition using an active and simple chiral
phosphoramide-zinc(II) complex (S)-5 as conjugate Lewis acid-Lewis base catalyst
(Scheme 7).[16]
This type of catalysis involves an electron charge transfer at the ligand
interior, thus avoiding the direct linkage between the acid and the base, and hence
enhancing the catalytic activity. The electrophile and nucleophile are doubly activated.
From aromatic and aliphatic ketones, optically active tertiary alcohols were obtained in
excellent yields and with high enantioselectivities (91-98 % e.e.) by using 10 mol % of (S)-
5. This is the first example of highly efficient ethylation of inactive ketones under mild
conditions without titanium(IV) compounds achieved by simply mixing together the
ketone substrate, diethylzinc, and the chiral ligand in a solvent.

26
Scheme 7. Catalytic Enantioselective Organozinc Addition to Ketones using Chiral
Phosphoramide 5-Zn(II) Complex.
R1 R2
O
(R1>R2)
R3Zn heptane, RT R1 R2
HO R3
HNN P
O
(10 mol %)
N P
O
N
Zn
R3
R2
OR1Zn
R3 R3
Zn: Lewis acid, O: Lewis base
(S)-5
MeO
PhHO
91 %, 96 % e.e.
Br
PhHO
93 %, 95 % e.e.
PhHO
97 %, 97 % e.e.
PhHO
88 %, 97 % e.e.
Ph
PhHO
81 %, 98 % e.e.
HO Ph
84 %, 82 % e.e.
EtHO
80 %, 93 % e.e.
EtHO
85 %, 96 % e.e.
Cl
EtHO
85 %, 96 % e.e.
S
For phenylation, 1 equiv. of Ph2Zn and 2 equiv. of Et2Zn were used.For ethylation, 3 equiv. of Et2Zn was used.
Some representative examples:

27
Walsh and Li developed a highly enantioselective addition of vinyl groups to
aromatic, alkyl and ,-unsaturated ketones in the presence titanium(IV) compounds and 3
(Scheme 8) to furnish tertiary allylic alcohols and dienols in excellent yields and with high
enantioselectivities.[17]
Following the method developed by Wipf et al.,[18]
the in situ
preparation of alkenylzinc consisted of hydrozirconation of terminal alkynes with
Schwartz’s reagent followed by transmetallation to zinc. A variety of alkynes have been
used, indicating that the reaction is compatible with functionalised substrates (Table 3).
Scheme 8. Asymmetric Vinylation of Ketones from Alkenylzirconium Reagents.
[Cp2ZrHCl]n
(1.2 equiv.)
R3 ZrClCp2
R3
Cp2ZrMeCl ZnMe2(1.2 equiv.)
MeZnR3
1) R1R2CO, RT
2) NaHCO3, H2O
R1 R3
OHR2
Ti(O-iPr)4 ZnMe2 3 *
Ti : Zn : 3 : ketone =1.2 : 0.4 : 0.1 : 1
Table 3. e.e. for the Asymmetric Vinylation of Ketones from Alkenylzirconium Reagents.
Entry Substrate Product e.e. (yield, %)
1 Ph
O
Ph Ph
OH
87 (92)
2
O
F3C
F3C
OH
OTBDPS
90 (94)
3
O
OH
Cl
90 (98)
4 Cl
O
Cl
OH
Bu
93 (93)
5 O
Bu
OH
79 (85)
6 Ph
O
Ph Bu
OH
92 (87)

28
The reaction of carbonyl compounds 6 with Grignard reagents often gives undesired
by-products, such as reduction product 7 via -H transfer and/or self-aldol product 8 via
enolisation (Scheme 9). When Grignard reagents are strongly basic, ketones are often
enolised and recovered after an acidic workup.
Scheme 9. Addition of Grignard Reagents to Ketones.
R1 R2
O
RMgCl
6
R1 R2HO R
9
6 R1 R2HO H
7
R1 R2
O
R2
HO R1
8
To overcome these problems, stoichiometric or excess amounts of inorganic additives
is usually employed.[19]
Until the method reported in 2012 by Minnaard et al., the synthesis of
enantioenriched tertiary alcohols with readily available Grignard reagents has only been
achieved with stoichiometric amounts of a chiral ligand.[4]
They developed an efficient
asymmetric catalytic alkylation reaction to aryl alkyl ketones with branched alkyl Grignard
reagents using a copper catalyst based on a chiral Josiphos-type diphosphine ligand 10
(Scheme 10).[20]
Scheme 10. Asymmetric Copper-Catalyzed Addition of Grignard Reagents to Aryl Alkyl
Ketones.
54- 98 % e.e.71-96 % yield
R1
O
X R2MgBr
(S, RFe)-10 (6 mol %)CuBrSMe2 (5 mol %)
tBuOMe, -78 °C
R1
X
HO R2
Fe
Ph2P
Cy2P
(S, RFe)-10
(1.25 equiv.)
Proposed transition state for the reaction

29
Scheme 10. Continued.
O
X
X = H, Me, CF3, F,Br, Cl, OMe, di-Cl, di-F, di-CF3
O
CF3
OO
O
S
substrate scope
MgBr
Ph
MgBr
MgBr
Et
Et
MgBr
Bu
Et
Cy
MgBr
Me3Si
MgBr
Grignard reagent scope
Good to excellent enantioselectivities and high yields of isolated products were
obtained with no significant electronic and steric effects of the substituents on the aryl ring
to the enantioselectivity. The reaction was extended to methyl-substituted ketones, phenyl-
substituted ketones and heteroaromatic ketones. However, the reaction is restricted to aryl
alkyl ketones and bulky Grignard reagents with branched alkyl groups are required for
high enantioselectivities. They proposed the transition state depicted in Scheme 10 for the
key alkyl group transfer step.
Copper-catalyzed arylation and alkenylation reactions to produce tertiary alcohols are
currently limited to using activated ketones (such as trifluoromethyl ketones or -keto
esters) as substrates.[21]
Organoaluminium compounds are excellent nucleophiles for organic reactions, due to
their high reactivity and a greater Lewis acidic nature of the aluminium centre. As such,
Gau et al. reported the first example of asymmetric aryl additions of organoaluminium
reagents to ketones.[22]
The novel asymmetric aryl transfers from [AlAr3(THF)] to a wide
variety of ketones were catalyzed by an in situ generated titanium species with (S)-BINOL
11 as the chiral ligand (Scheme 11). The reaction afforded tertiary alcohols, in general, in
high enantioselectivities of 90 % e.e. and greater for aromatic ketones bearing either an
electron-donating or an electron-withdrawing substituent at the 2-, 3-, or 4-position on

30
the aromatic ring (Table 4, entries 1 to 7), except for 2-methoxyacetophenone substrate
(entry 8). The catalytic system also tolerated ,-unsaturated ketones and 2-acetylfuran
(entries 9 and 10). Additions of different aryl compounds, such as 2-naphthyl, 4-tolyl, or 4-
(trimethylsilyl)-phenyl, to aromatic ketones also gave the desired alcohols in high yields
with excellent enantioselectivities (90-93 % e.e.).
Scheme 11. Asymmetric [AlAr3(THF)] Addition to Ketones Catalyzed by (S)-BINOL
11/[Ti(O-iPr)4].
R1 R2
O
[AlAr3(THF)]
(2.5 equiv.)
(S)-BINOL 11 (10 mol %) [Ti(O-iPr)4] (5.0 equiv.)
PhMe, 0 °C
R1 R2
HO Ar
O
OTi
O-iPr
O-iPr
n
(S)-11/[Ti(O-iPr)4]
Table 4. Conditions, Yields and e.e. for the Asymmetric [AlAr3(THF)] Addition to Ketones
Catalyzed by (S)-BINOL 11/[Ti(O-iPr)4].
Entry Substrate Ar t (h) Yield (%) e.e. (%)
1
O
Ph 12 85 93
2
OMe
Ph 12 35 90
3
O
Cl
Ph 12 92 93
4
OBr
Ph 36 50 97
5
O
O2N
Ph 12 97 93
6
O
MeO
Ph 36 97 93
3

31
Table 4. Continued.
Entry Substrate Ar t (h) Yield (%) e.e. (%)
7
O
F3C
Ph 12 98 92
8
OOMe
Ph 12 98 30
9 Ph
O
Ph 12 96 87
10 O
O
Ph 16 95 84
11
O
4-MeOC6H4 12 87 78
Later, in order to compare AlAr3(THF) and arylzinc reagents in asymmetric aryl
additions to ketones, Gau et al. probed asymmetric AlAr3(THF) additions to ketones
catalyzed by titanium catalysts of chiral ligand 3,[23]
which have been used in titanium-
catalyzed arylzinc additions to ketones (Scheme 12).[12,13]
It was demonstrated that
inorganic salts such as MgBr2 were essential in promoting the addition reactions, thus
affording similar results to (S)-BINOL 11/[Ti(O-iPr)4] system in term of enantioselectivity.
While ZnPh2 additions to ketones require lower catalyst loadings of 3 (10 mol %) and
Ti(OiPr)4 (0.6 equiv.) and use no additive, the AlAr3(THF) system gives better yields (80-
99 %), employs shorter reaction times and the reaction can be extended to additions of
different aryl groups, such as 4-tolyl or 4-(trimethylsilyl)-phenyl.
Scheme 12. Asymmetric Aryl Addition of AlAr3(THF) to Ketones Catalyzed by the
Titanium(IV) Catalytic System of 3 Promoted by MgBr2.
R1 Me
O
AlAr3(THF)
(6.0 equiv.)
3 (20 mol %)/Ti(O-iPr)4 (10 equiv.) MgBr2 (48 mol %)
PhMe, 0 °C, 12-36 h
R1 Me
Ar OH
75-98 % e.e.
To further improve the atomic-efficiency of arylaluminium reagents, they reported
the catalytic asymmetric AlArEt2(THF) addition to ketones using a titanium catalyst of (S)-
BINOL 11 (Scheme 13).[24]
The AlArEt2(THF) compounds still proved to be excellent

32
reagents for asymmetric addition to ketones in comparison to the AlAr3(THF) reagents
affording aryl tertiary alcohols in similar yields and enantioselectivities. In addition, the
amount of Ti(O-iPr)4 was decreased from 10 equiv. to 3.5 equiv..
Scheme 13. Asymmetric Addition of AlArEt2(THF) to Ketones Catalyzed by (S)-BINOL
11/Ti(O-iPr)4 Catalyst.
R1 Me
O
AlArEt2(THF)
(2.5 equiv.)
(S)-BINOL 11 (10 mol %) Ti(O-iPr)4 (3.5 equiv.)
PhMe, 0 °C, 18-36 h
R1 Me
OH
29-94 % e.e.
Ar
Interestingly, the use of stable, commercially available aryl boronic acids in place of
common organometallic reagents has been recently considered by Xu et al. (Scheme 14).[25]
The method employed a simple, chiral N-(sulfinyl)cinnamylamine ligand 12 in a rhodium-
catalyzed asymmetric 1,2 addition of aryl boronic acids to -ketoesters 13 and -diketones
14. A variety of highly enantioenriched, tertiary -hydroxy carbonyl derivatives 15 were
easily accessed at room temperature under mild conditions in moderate to excellent yields
and with e.e. values of up to 99 %.
Scheme 14. Asymmetric 1,2-Addition of Arylboric Acids to -Ketoesters and -Diketones
Catalyzed by [{Rh(coe)2Cl}2]/12.
RX
O
O
ArB(OH)2 R Ar
HO X
O
KOH (0.1 M)/THF, RT
[{Rh(coe)2Cl}]2 (3 mol %)
Ph NH
S
O13, X = O-2-Np14, X = Ph, 4-BrC6H4,4-FC6H4, 4-MeOC6H4, Me
12
15 (up to 99 % e.e.)
HOO
O-2-Nap
F
86 %, 94 % e.e.
Some representative examples:
HOO
O-2-Nap
87 %, 91 % e.e.
HOO
O-2-Nap
72 %, 93 % e.e.
OMe
(1.5 equiv.)
12 (3.3 mol %)

33
Scheme 14. Continued.
HOO
Ph
Cl
97 %, 99 % e.e.
HOO
Ph
54 %, 95 % e.e.
Me
HOO
4-Br-C6H4
Me
93 %, 98 % e.e.
Br
Me
HOO
Me
Me
45 %, 63 % e.e.
Me
HOO
Ph
Me
85 %, 80 % e.e.
Reactions involving aryl boronic acids with different electronic and steric demands
afforded -hydroxy carbonyl products 15 with similarly high levels of enantioselectivity.
13 and 14 underwent 1,2-addition with high enantioselectivities regardless of the electronic
nature of the aryl group R. However, when aliphatic -diketone (R = X = Me) was
employed, only a moderate enantioselectivity was obtained. In the case of unsymmetrical
-diketone (R = Me, X = Ph), both regio- and enantioselectivity was observed (Scheme
14).
I.1.1.2 Catalytic Enantioselective Allylation Reactions
Another classical class of carbon-based nucleophiles for addition to carbonyl
compounds is allyl metal reagents. These have been extensively employed in asymmetric
synthesis because of the ease of their preparation and the high value in synthesis of the
tertiary homoallylic alcohols resulting from such processes.[26]
In recently reported
examples, a range of nucleophilic allyl sources have been used including stannanes, silanes,
and boranes.
The first enantioselective catalyst for the asymmetric allylation reaction was
introduced by Maruoka et al..[27]
They developed a new chiral bis-BINOL-Titanium(IV)
catalyst with dibenzofuran spacer 13 which catalyzed the allylation of acetophenone and 2-
acetonaphthone using tetraallyltin as the terminal allyl source with 90 % and 92 % e.e.,

34
respectively (Scheme 15). However, the scope of this catalyst is currently limited to these
two examples.
Scheme 15. Catalytic Enantioselective Allylation of Ketones using a bis-BINOL-Ti(IV)
Complex 13.
Ar Me
OSn
4
13 (30 mol %)
CH2Cl2, 0 °CAr
(1.5 equiv.)
HO Me
ONH HN CPh3Ph3C
O
OTi
(i-PrO)2
O
OTi
(Oi-Pr)2 Ar = Ph 95 %, 90 % e.e.Ar = 2-naphthyl 98 %, 92 % e.e.
13
Later, Woodward et al. demonstrated that (S)-MTBH2 (monothiobinaphthol) 14
promoted the allylation of acetophenone derivatives with a mixture of tri- and
tetraallylstannane, employing water as a promoter and without the use of titanium (Scheme
16).[28]
. Enantiomeric excess values as high as 92 % with acetophenone were obtained.
Two procedures were introduced: one (‘dry’ method using R = Et mix) gave higher
enantioselectivities and low yields while the other (‘wet’ method using R = Bu mix) gave
lower enantioselectivities but excellent yields.
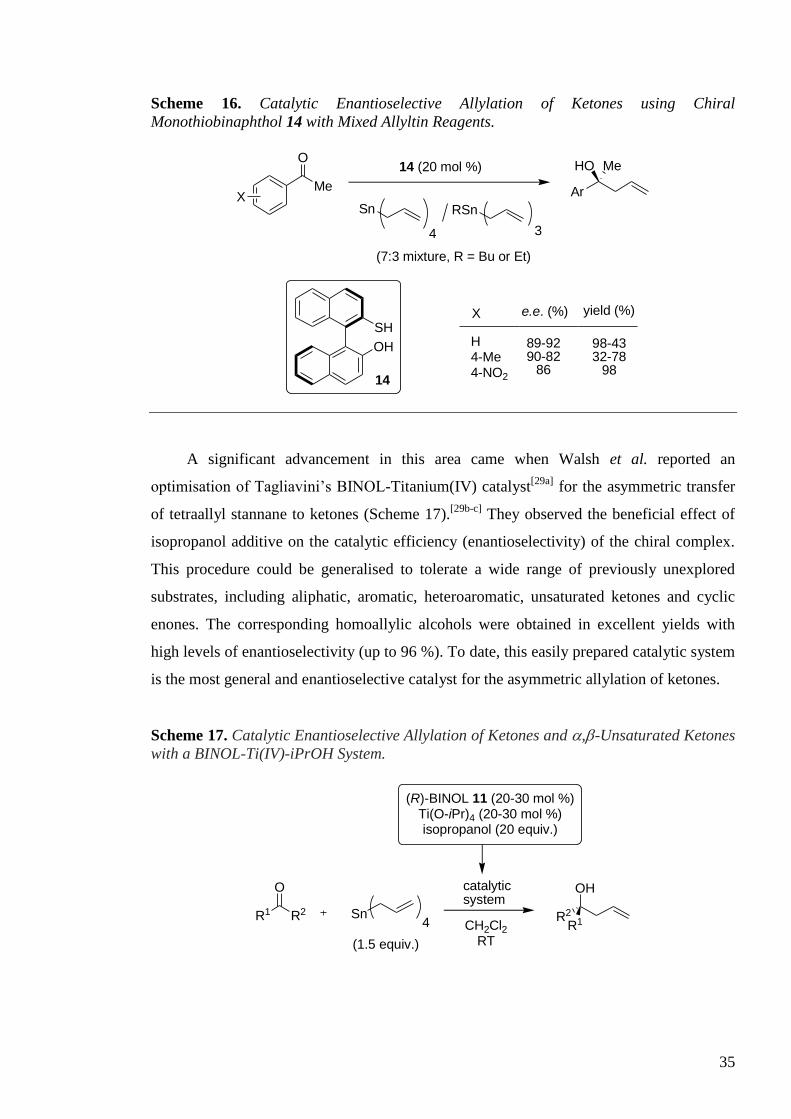
35
Scheme 16. Catalytic Enantioselective Allylation of Ketones using Chiral
Monothiobinaphthol 14 with Mixed Allyltin Reagents.
Me
O
X
14 (20 mol %)
Sn RSn
4 3
(7:3 mixture, R = Bu or Et)
Ar
SH
OH H4-Me4-NO2
89-9290-82
86
98-4332-78
98
X e.e. (%) yield (%)
HO Me
14
A significant advancement in this area came when Walsh et al. reported an
optimisation of Tagliavini’s BINOL-Titanium(IV) catalyst[29a]
for the asymmetric transfer
of tetraallyl stannane to ketones (Scheme 17).[29b-c]
They observed the beneficial effect of
isopropanol additive on the catalytic efficiency (enantioselectivity) of the chiral complex.
This procedure could be generalised to tolerate a wide range of previously unexplored
substrates, including aliphatic, aromatic, heteroaromatic, unsaturated ketones and cyclic
enones. The corresponding homoallylic alcohols were obtained in excellent yields with
high levels of enantioselectivity (up to 96 %). To date, this easily prepared catalytic system
is the most general and enantioselective catalyst for the asymmetric allylation of ketones.
Scheme 17. Catalytic Enantioselective Allylation of Ketones and ,-Unsaturated Ketones
with a BINOL-Ti(IV)-iPrOH System.
R1 R2
O
Sn4
(R)-BINOL 11 (20-30 mol %)Ti(O-iPr)4 (20-30 mol %)isopropanol (20 equiv.)
(1.5 equiv.)
catalytic system
CH2Cl2RT
R2
OH
R1

36
Scheme17. Continued.
82 %, 96 % e.e.
Me
HO HO
96 %, 95 % e.e.
Ph
HO
96 %, 80 % e.e.
Ph
HO
99 %, 96 % e.e.
O Me
HO
67 %, 84 % e.e.
n-Pen
HO
92 %, 94 % e.e.
Some representative examples:
More recently, Loh et al. reported a highly enantioselective allylation catalyzed by a
chiral indium(III) complex made from (R)-BINOL 11 and InBr3 using allyltributylstannane,
unlike other previously described asymmetric catalytic systems which demand stronger
allylation reagents such as tetraallylstannanes (Scheme 18).[30]
The allylation of aromatic,
,-unsaturated and aliphatic ketones resulted in good yields (41-82 %) and high
enantioselectivities (80-92 % e.e.).
Scheme 18. Enantioselective Allylation of Ketones Catalyzed by Chiral (R)-BINOL 11-
In(III) Complex.
R1 R2
O
SnBu3
(R)-BINOL 11-In(III) complex (20 mol %)
4Å MS / CH2Cl2
R1
OHR2
(3.0 equiv.) 80-92 % e.e.
Instead of toxic allyltin reagents with high catalyst loadings (10-30 % in most cases),
the groups of Shibasaki and Yamamoto studied the asymmetric allylation of ketones using
soft transition metal-chiral diphosphine complexes as catalysts (Scheme 19).
Shibasaki et al. used allyl boronates as nucleophiles for the allyl transfer on aliphatic
and aromatic ketones with fair to good enantioselectivities and in high yields when a chiral
copper complex was employed as a catalyst. Extensive optimisation on the catalytic system
led to the authors to select i-Pr-DuPHOS 15 (6 mol%) as a chiral ligand and La(Oi-Pr)3
(4.5 mol%) as a cocatalyst (Scheme 19).[31]

37
Yamamoto and Wadamoto later developed the silver(I)-catalyzed enantioselective
Sakurai-Hosomi allylation of ketones,[32a]
which has the advantage of using inexpensive,
non-toxic and stable allyltrialkylsilane and allyltrialkoxysilane reagents.[32b]
In the presence
of silver(I) fluoride and (R)-DIFLUOROPHOS 16 (5 mol % each) and by the addition of
methanol (1.0 equiv.), an increase in the catalyst turnover gave the highly enantioselective
allylation of a variety of ketones.(Scheme 19).
Scheme 19. Shibasaki’s and Yamamoto’s Strategies for Asymmetric Allyl Transfer on
Ketones.
R1 R2
O
Si(OMe)3
O
B O
Ag or Cuor
catalyticsystem
R1
OH
R2
P
PiPr
iPr
iPr
iPr CuF2.2H2O (3 mol %)
La(O-iPr)3 (4.5 mol %)
DMF, -40 °C
O
B O
83-99 %, 67-91% e.e.
Shibasaki et al. (2004) Yamamoto et al. (2005)
O
O
O
O
F
F
F
F
PPh2
PPh2
AgF (5 mol %)
Si(OMe)3
MeOH (1.0 equiv.)
THF, -78 °C
63-98 %, 78-96 % e.e.
(1.2 equiv.)
(2.0 equiv.)1516
(6 mol %)(5 mol %)
In the aim of improving the enantioselectivity, Shibasaki et al. designed a different
diphosphine-based ligand 17 comprising of four modules (linker, wing, chiral head, and
phosphine) (Scheme 20).[33]
In general, the enantioselectivity and catalytic activity were
significantly higher than in their previous studies using i-Pr-DuPHOS (Scheme 19).[31]

38
Scheme 20. Improved Shibasaki’s Strategy of Catalytic Asymmetric Allylation of Ketones.
R1 R2
O O
B O
CuOAc (2 mol %)17 (2.4 mol %)LiOi-Pr (0.5 equiv.)
i-PrOH (1.0 equiv.),CH2Cl2, -75 °C
R1
HO R2
O
N
O
OO
PAr2MeO
R
PAr2MeO
17, R = (S)-Ph, Ar = p-F-C6H4
phosphine
chiralhead
wing
linker
Some representative examples:
R
R = p-Me, 98 %, 92 % e.e.R = p-OMe, 90 %, 92 % e.e.
X
HO
X = CH2, 85 %, 95 % e.e. (0.1 mol% cat.)X = O, 86 %, 94 % e.e.
S
93 %, 92 % e.e. 91 %, 90 % e.e. 88 %, 83 % e.e.
HO Me
HO Me MeHO
HO Me
Another class of chiral ligand of interest for the catalytic enantioselective
allylboration of ketones and widely studied are chiral biphenols. In general, the use of
relatively high catalyst loadings is necessary. However, Schaus et al. have recently
reported an improved reaction employing 2 mol % of 18 with allyldioxaborinane and t-
BuOH (increasing enantioselectivity and the overall rate) under solvent-free reaction
conditions at room temperature to afford the products in greater than or equal to 96 % e.e.
(Scheme 21).[34]
In comparison to the reaction reported by Shibasaki,[31,33]
the
enantioselectivity was slightly superior.

39
Scheme 21. Improved Asymmetric Allylboration of Ketones Catalyzed by Chiral Biphenol
18.
R1 R2
O
BO
O
18 (2 mol %)
t-BuOH (2.0 equiv.)RT, 24 h
(1.5 equiv.)
R1
R2 OH OH
Br
OH
Br
18
> 90 % yield 96 % e.e.
I.1.1.3 Catalytic Enantioselective Alkynylation Reactions
The first general method that allowed the enantioselective addition of acetylenes to
ketones was introduced by Cozzi et al. and was based on the salen framework.[35]
An
excess of dimethylzinc was added to a mixture of the ketone substrate, a terminal alkyne
and a catalytic amount of commercially available chiral salen ligand to form the zinc
acetylide nucleophile and a zinc salen catalyst (Scheme 22). To overcome the low
reactivity of the substrates, this approach involved the concept of double activation (Lewis
acid-Lewis base) as shown in the proposed transition state (Scheme 22). For a series of
aromatic and aliphatic ketones, 20 mol % of (R,R)-N,N’-bis(3,5-di-tert-butylsalicylidene)-
1,2-cyclohexanediamine 19 effectively afforded the desired products with moderate
enantioselectivities.
Scheme 22. Cozzi’s Pioneering Work on Alkynylation of Ketones.
R1 Me
O
R2 H
Me2Zn (3.0 equiv.)
19 (20 mol %)
PhMe, RTR1 Me
OH R2
R1 = aryl, alkyl
R2 = Ph, TMS, CH2Cl
53-81 % e.e.
OH
N N
HO
(3.0 equiv.)
19

40
This first example was followed by many other examples which employed similar
concepts (chiral ligands such as BINOL, amino alcohols and amino alcohol derivatives in
combination with a metal source based on zinc, aluminium, and titanium). Only a few
methods are described in this thesis.
The method reported by Chan et al. was based on the concept of increasing the
reactivity of ketones by using a strong Lewis acid. Since copper(II) triflate in combination
with bis-oxazolidines is well established for the addition of nucleophiles to ketones,[36]
Chan studied Cu(OTf)2 with different ligands for the addition of phenylacetylene to
aromatic ketones. He found that ligand 2 (Scheme 4), which was first introduced by Yus[8]
was highly efficient for the addition of phenylacetylene to a variety of aromatic ketones
(Scheme 23). Substituents at the ortho-position of the substrate had a favourable effect on
the enantioselectivity, since the steric hindrance of the ortho substituents restricts the
orientation of the substrates. This method afforded higher enantioselectivity than Cozzi’s
reaction. The best enantioselectivity (97 %) was observed in the alkynylation of 2’-
chloroacetophenone.
Scheme 23. Catalytic Enantioselective Alkynylation of Aromatic Ketones using a Chiral
Camphorsulfonamide-copper(II) Complex.
Ar Me
O
Ph H
(2.6 equiv.)
Me2ZnCu(OTf)2 (10 mol %)
2 (10 mol %)
CH2Cl2, 0 °C, 48 h
Ar
Ph
HO Me
Ar = Ph 90 %, 82 % e.e.Ar = 2-ClC6H4 94 %, 97 % e.e.
(3.0 equiv.)
In both of these methods, the alkynyl zinc species are generated in situ by combining
an excess of Me2Zn with phenylacetylene. Unfortunately, Me2Zn is highly pyrophoric and
expensive so the addition of alkynides based on the use of Me2Zn has limited applicability.
Therefore, inspired by Seebach’s pioneering studies on alkynyl titanium reagents,[37a]
Cozzi developed a catalytic enantioselective addition of titanium(IV) phenylacetylides
prepared from lithium phenylacetylide and chlorotitanium(IV) isopropoxide using (R)-
BINOL 11 as the chiral ligand (Scheme 24).[37b]
Aromatic ketones provided good results
with moderate yields and good enantioselectivies.

41
Scheme 24. BINOL-Ti(IV)-Catalyzed Enantioselective Addition of Titanium(IV)
phenylacetylide to Aromatic Ketones.
Me
O
Ph Li
(1.5 equiv.)
ClTi(Oi-Pr)3
(R)-BINOL 11 (25 mol %)Ar
HO Me
PhX
X
4-Cl4-F4-Br4-Me
e.e. (%)
90868485
yield (%)
48842845
PhMe
Although advances have been achieved, high loadings of ligands (usually 20 mol %)
had to be used to obtain good to excellent enantioselectivities. Yet, Wang et al. reported an
example of a highly efficient addition of alkynylzinc to simple ketones with high e.e.
values (90 %) and good yields in low loading (1 mol %) of the ligand 20, an easily
prepared chiral Schiff base amino alcohol (Scheme 25, Table 5).[38]
Scheme 25. Catalytic Enantioselective Addition of Phenylacetylene to Aromatic Ketones
using Chiral Schiff Base Amino Alcohol 20.
Ar Me
O
Ph H
20 (1 mol %)
Et2Zn, hexaneAr
Ph
MeHO
Ph
OH
PhPh
N
20
ketone : Et2Zn : acetylene =1 : 2 : 2

42
Table 5. Yields and e.e. for the Asymmetric Addition of Phenylacetylene to Various
Ketones Promoted by 20.
Entry Substrate Yield (%) e.e. (%)
1 acetophenone 70 90
2 2-fluoroacetophenone 83 94
3 2-naphthacetophenone 77 95
4 1-naphthacetophenone 62 94
5 2-methoxyacetophenone 70 94
6 4-methylacetophenone 76 92
7 4-fluoroacetophenone 70 90
8 4-chloroacetophenone 63 90
9 3-methylacetophenone 76 90
To conclude, in comparison to aldehydes, catalytic asymmetric addition of
organometallic nucleophiles to ketones have proved to be more challenging owing to their
attenuated reactivity and lower binding affinity to metals. Furthermore, organometallic
reagents often cause aldol addition adducts/starting ketones due to competing enolisation
problems or competitive reduction via -H transfer to give undesired secondary alcohols,
this is especially the case of sterically hindered substrates. Moreover, the catalyst must
differentiate between the lone pairs of the carbonyl oxygen to achieve high
enantioselectivity. This task becomes difficult when the groups R’ and R of the ketone
R’RCO are similar in size. Nonetheless, successful catalysts for additions to ketones are
beginning to emerge, thus providing access to tertiary alcohols with high
enantioselectivities.
Ready et al. have reported in 2011 an alternative method, which relies on the use of
the toluene sulfinyl group as a chiral auxiliary controlling the asymmetric addition of
simple alkynyl, aryl and vinyl organometallic reagents to aryl ketones (Scheme 26).[39]

43
Scheme 26. Asymmetric Synthesis of Tertiary Benzylic Alcohols.
Me
O
SO
(p-Tol)
R M (2.0 equiv.)
-78 °C, THF, 3 h
R
SO
(p-Tol)
Me OH
M = MgBr or Li/CeCl3
Some representative examples:
S(O)Tol
Me OH
Si(i-Pr)3
87 %, >50:1 d.r. 83 %, >50:1 d.r.
Ph
S(O)Tol
Me OH
74 %, >50:1 d.r.
S(O)Tol
Me OH
79 %, >50:1 d.r.
4-MeO-Ph
S(O)Tol
Et OH
F
OHMe
95 %, >99 % e.e. OMe
OH
CF3
F3C
99 %, 92 % e.e.
Tertiary benzylic alcohols were generated in high yields and in diastereomerically
and enantiomerically pure form. In contrast to most previous studies, the methodology
utilised readily available Grignard reagents and lithium acetylides. Moreover, the sulfoxide
chiral auxiliary can be reductively removed in high yield or converted into useful
functional groups.
I.1.2 Catalytic Asymmetric Aldol Reactions
Catalytic asymmetric aldol addition to ketone acceptors has received growing
attention since the resulting tertiary chiral β-hydroxy carbonyl compounds are versatile
synthetic motifs for pharmaceutically attractive intermediates and biologically active
natural products.[40]
The first example of catalytic asymmetric aldol reactions of preactivated nucleophiles,
tert-butyl thioketene acetals 21 to activated ketones such as alkyl-substituted pyruvate

44
esters 22 and -diketones was reported by Evans et al. using chiral C2-symmetric cationic
bis(oxazolinyl)Cu(II) complex 23 (Scheme 27).[41]
The CuII
complex activates the
nucleophile through bidentate coordination to form the five-membered catalyst-substrate
chelate 24, which is a strict requirement for stereoselectivity.
Scheme 27. Chiral Lewis Acid Cu(II)-Catalyzed Aldol Reactions of Enolsilanes to
Pyruvate Esters.
R1OR2
O
O
St-Bu
R3
OTMS 1. 23 (1-10 mol %)THF or CH2Cl2
2. 1 N HCl, THF
R1OSt-Bu
O
O
R3
OHR2
(S)
2221 25
N
O
Me Me
O
N
Me3C CMe3
2 TfO-
Cu
2+
Cu(II)-box 23
catalyst complex:
substrate-catalyst complex :
xx IIIIXIIIIIIII24
A variety of alkyl groups were tolerated at the ester moiety of pyruvates (R1) (Table 6,
entries 1-4), high yields and enantioselectivities were obtained when the -position of
pyruvate ketone carbonyl (R2) was a methyl group (entries 1-3), whereas an -branched
substrate (R2 = i-Pr) produced less satisfactory results (entry 4). It was also demonstrated
that various -substituted silyl enolates could be used (R3), producing syn isomers as the
major product irrespective of the geometry of silyl enolates (entries 5-8).

45
Table 6. Scope of the Enantioselective Aldol Addition of Enolsilanes to Pyruvate Esters.
Entry R1 R
2 R
3 Yield (%) (syn/anti ratio) e.e. (%)
1 Me Me H 96 99
2 Bn Me H 95 99
3 tBu Me H 91 99
4 Et iPr H 36 36
5 Me Me (Z)-Me 96 (94:6) 96
6 Me Me (E)-Me 90 (95:5) 98
7 Me Me (Z)-i-Bu 88 (90:10) 93
8 Me Me (Z)-i-Pr 80 (90:10) 99
Since then, Pagenkopf et al. has used modified bis(oxazoline) ligands in copper(II)-
catalyzed reactions of dienolsilanes to aryl- and alkyl-substituted -ketoesters.[42]
A new
ligand class consisting of C1-symmetric aryl-bridged amino- and oxazolinyl sulfoximines
developed by Bolm et al. have also proved to be powerful ligands in copper(II)-catalyzed
enantioselective Mukaiyama and Mukaiyama vinylogous-type aldol reactions of
trimethylsilylenol ethers and n-alkyl-substituted -ketoesters.[43]
However, these catalytic
protocols are restricted to -ketoesters bearing sterically undemanding substituents. To
address this, Hoveyda et al. developed a method complementary in terms of substrate
range by employing a combination of AgF2 and chiral amino acid-based ligand 28
(Scheme 28).[44]
The catalytic process was highly effective (high yields and e.e. up to 96 %)
tolerating a variety of -ketoesters 26 containing sterically hindered alkyl, alkenyl and
aromatic substituents.
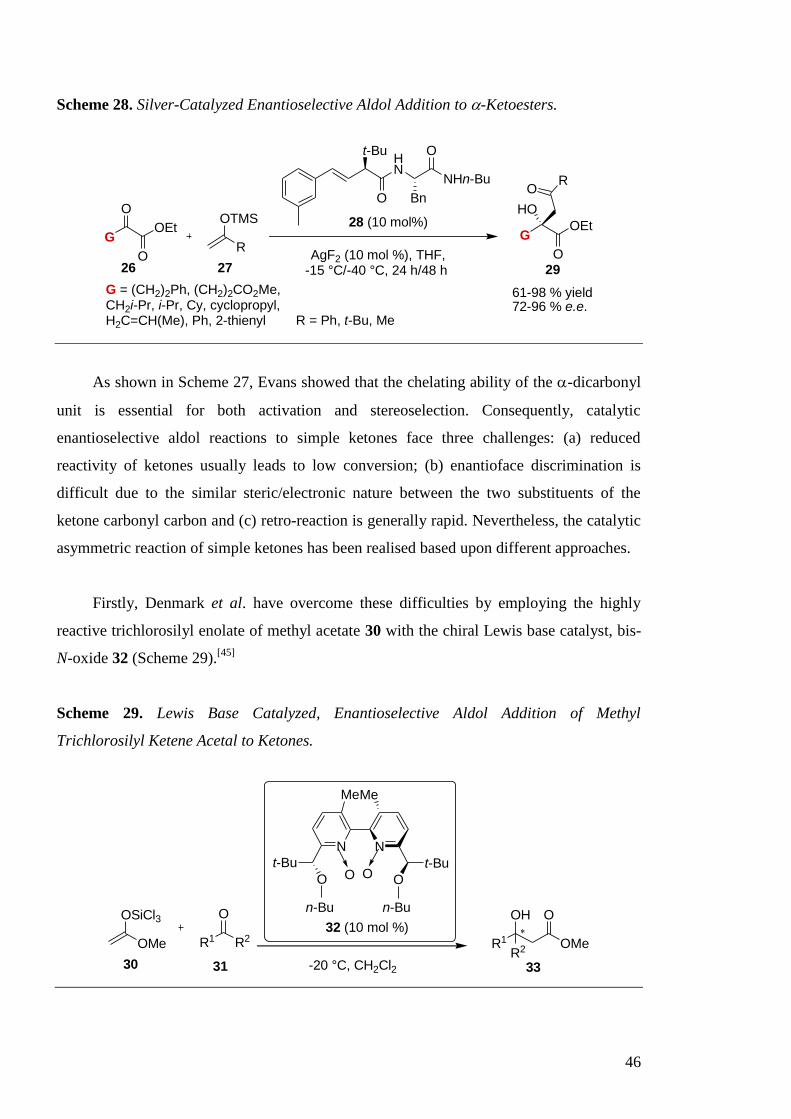
46
Scheme 28. Silver-Catalyzed Enantioselective Aldol Addition to -Ketoesters.
GOEt
O
OR
OTMS
HN
O
t-Bu
NHn-Bu
O
Bn
28 (10 mol%)
AgF2 (10 mol %), THF,-15 °C/-40 °C, 24 h/48 h
G = (CH2)2Ph, (CH2)2CO2Me, CH2i-Pr, i-Pr, Cy, cyclopropyl,H2C=CH(Me), Ph, 2-thienyl
GOEt
O
RO
HO
61-98 % yield72-96 % e.e.
R = Ph, t-Bu, Me
26 27 29
As shown in Scheme 27, Evans showed that the chelating ability of the -dicarbonyl
unit is essential for both activation and stereoselection. Consequently, catalytic
enantioselective aldol reactions to simple ketones face three challenges: (a) reduced
reactivity of ketones usually leads to low conversion; (b) enantioface discrimination is
difficult due to the similar steric/electronic nature between the two substituents of the
ketone carbonyl carbon and (c) retro-reaction is generally rapid. Nevertheless, the catalytic
asymmetric reaction of simple ketones has been realised based upon different approaches.
Firstly, Denmark et al. have overcome these difficulties by employing the highly
reactive trichlorosilyl enolate of methyl acetate 30 with the chiral Lewis base catalyst, bis-
N-oxide 32 (Scheme 29).[45]
Scheme 29. Lewis Base Catalyzed, Enantioselective Aldol Addition of Methyl
Trichlorosilyl Ketene Acetal to Ketones.
OMe
OSiCl3
R1 R2
O
-20 °C, CH2Cl2
R1
OMe
OOH
R2
N N
OO
t-Bu t-BuO O
32 (10 mol %)
n-Bun-Bu
30 31 33
MeMe

47
All reactions proceeded cleanly to give high yields of the aldol products 33 for a
range of ketone substrates. The enantioselectivity, however, was found to be highly
dependent on the structure of the ketones (Figure 2).
Figure 2. Substrate Scope in the Bis-N-Oxide Aldol Reactions.
Ph Me
O96 % yield83 % e.e.
Ph Et
O90 % yield81% e.e.
O
90 % yield80 % e.e.
O
89 % yield56 % e.e.
O
O
87 % yield49 % e.e.
Ph
O
Me
94 % yield35 % e.e.
Me
O
F3C
91 % yield76 % e.e.
Me
O
MeO
94 % yield68 % e.e.
O 86 % yield8 % e.e.
Et Me
O 84 % yield32 % e.e.
Ph Me
O97 % yield35 % e.e.
Me
O
84 % yield20 % e.e.
Me
O
91 % yield32 % e.e.
t-Bu Me
O91 % yield43 % e.e.
Later, Shibasaki et al. developed the first diastereo- and enantioselective catalytic
aldol reaction involving a chiral copper enolate intermediate, by using trimethylsilyl ketene
acetals 34 with chiral copper(I) fluoride-di-n-butylamine-type Taniaphos 35 (Scheme
30).[46]
Further acceleration of the rate-determining catalyst regeneration step was achieved
by using a combination of (EtO)3SiF and PhBF3K as a strong trapping reagent. As such,
this method almost completely overcame the reactivity problem of ketones in the aldol
reaction, producing high yields and good enantioselectivities from both aromatic and
aliphatic ketones. Moreover, the anti-isomer was the major product irrespective of the
geometry of the silyl enolate.

48
Scheme 30. Chiral Cu(I)-Catalyzed Aldol Reaction of Enolsilanes to Ketones.
R1 R2
O
OMe
OTMS
R3
1. CuF(PPh3)32EtOH (2.5 mol %)Taniaphos 35 (4 mol %)(EtO)3SiF (200 mol %)PhBF3K (10 mol %), DME
2. Et3N3HFR1
OMe
OOH
R2
R3
PCy2Fe
NnBu2PCy2
35
OH
MeOMe
O
93 %, 92 % e.e.
OH
OMe
O
MeO
88 %, 83 % e.e.
OMe
OOH
92 %, 90 % e.e.
OH
MeOMe
O
73 %, 84 % e.e.
OMe
OOH
MeMe
96 %, 91 % e.e.80/20 d.r., 75 % e.e.
OH
MeOMe
O
85 %, 87 % e.e.
Cl
3134 36
The above report demonstrated that copper enolates are sufficiently nucleophilic to
promote the addition to ketones once they are generated by transmetalation (Scheme 31
(1)). Conjugate addition of alkylcopper and copper hydride to ,β-unsaturated and allenic
carbonyl compounds is an alternative method for copper enolate formation (Scheme 31
(2)), thus overcoming the preactivation of the nucleophile.

49
Scheme 31. Two Strategies for Copper Enolate Generation.
(1) Si enolate formation via transmetalation
OR'
O
R
OR'
R
OTMS
preactivation
CuX transmetalation
OR'
R
OCu
OR'
O
•
OR'
O
orR-Cu
(2) conjugate addition
This strategy was realised in a catalytic asymmetric reductive aldol reaction reported
by Riant et al.[47]
and Shibasaki et al..[48]
Riant’s group utilised the
CuF•3PPh3•2MeOHTaniaphos 38 complex as a catalyst, PhSiH3 as a triggering
nucleophile, and methyl acrylate 37 as an acceptor for conjugate addition (Scheme 32 (1)).
Excellent enantioselectivity as well as diastereoselectivity were obtained using aromatic
ketone substrates. Shibasaki’s group employed allenic ester 40 as an acceptor. -Aldol
products 42 were produced as the major product with high diastereoselectivity when using
CuF•3PPh3•2EtOHTaniaphos 41 complex as the catalyst and pinacolborane as the
triggering reducing agent (Scheme 32 (2)).

50
Scheme 32. Catalytic Asymmetric Reductive Aldol Reactions of Ketones and Acrylate or
Allenic Esters.
R Me
O
OMe
O
•
OMe
O
Cu 3PPh3 2MeOH (1 mol %)38 (1 mol %)PhSiH3 (1.4 equiv.)
toluene, -50 °C, 1/2 h
PCy2Fe
NMe2PCy2
38
CuF3PPh3 2EtOH (2.5 mol %)41 (5 mol %)pinacolborane (1.6 equiv.)
THF, 20 °C, 16 h
PAr2Fe
PAr2
41
N
O
Ar = 3,5-xylyl
R OMe
O
Me
MeHO
anti/syn ratio up to 96/4anti adduct up to 95 % e.e.
CO2Me
OH
MeR
anti/syn ratio up to 10/1anti adduct up to 84 % e.e.
37
40
31
39
42
(1) Riant's strategy
(2) Shibasaki's strategy
To conclude, despite excellent progress in the field of metal-catalyzed
enantioselective nucleophilic addition to ketones, only limited success has been achieved
in catalytic enantioselective aldol reaction of ketones. The reported systems suffer from
either poor substrate scope and/or moderate enantioselectivity. Moreover, in most cases,
they involve silyl enolate derivatives as nucleophilic partners and thus require an additional
step for their preparation.

51
I.1.3 Kinetic Resolution of Tertiary Alcohols
Compared to secondary alcohols,[49]
nonenzymatic kinetic resolution of tertiary
alcohols has been much less studied, and there are only a few effective nonenzymatic
catalyst systems available to date.[50]
In this thesis, only a recent method reported by Fagnou et al. is discussed.[51]
They
employed commercially available (1S,2R)-N-methylephedrine 44 as the resolving agent for
the kinetic resolution of tertiary alcohols 43 arising from aldol reactions (Scheme 33).
Scheme 33. Kinetic Resolution with (1S,2R)-N-methylephedrine 44.
O
O OH
R1
R2
Me
OH
NMe2
PhMeO
O OH
R1
R2
43 racemic 44 43 enantioenriched
Temp.
Table 7. Conditions and Results for the Kinetic Resolution with (1S,2R)-N-
methylephedrine 44.
Entry Substrate Equiv. of 44 T (°C) Conv. (%) s e.e. (%)
1 O
O OH
MeCO2Et
2 60 51 38 88
2 O
O OH
PhCO2Et
2 80 58 21 96
3 O
O OH
CF3CO2Et
0.8 60 46 35 99
4 O
O OH
CF3Ph
1 RT 28 21 94
5 O
O OH
MeCOMe
1 60 51 10 70
A number of functionalities at the quaternary center may be present, including alkyl,
aryl, ester, trifluoromethyl and ketone substituents (Table 7). The resulting tertiary alcohols
43 were effectively resolved (up to 99 % e.e.). They proposed that hydrogen bonding of the

52
substrate alcohol 43 to the carbonyl group and the amine of 44 may be important for
reactivity and selectivity.
Although good selectivities for selected substrates are generally obtained, some of
the preparative applications of simple kinetic resolution are eventually replaced by
advances in enantioselective synthesis methodology with achiral substrates and chiral
catalysts, procedures that are not subject to the 50 % yield limit.
One of the most important and challenging features within the field of asymmetric
synthesis via chiral carbanions is the configurational stability of the metalated carbanionic
species. Several research groups have considered the possibility to use them as valuable
precursors for the synthesis of highly enantioenriched compounds. Aggarwal et al.
employed this strategy to make chiral tertiary alcohols as described below.
I.1.4 1,2-Metallate Rearrangement of Boronate Complexes
A breakthrough in the synthesis of optically active tertiary alcohols came when
Aggarwal et al. reported in 2008 a different and simple method to convert a chiral
secondary alcohol to either enantiomer of a tertiary alcohol.[52]
The method occurs via
lithiation of a benzylic carbamate 45Li (Scheme 34). Trapping with a borane or a boronic
ester forms a ate complex and a stereospecific 1,2-shift entailing departure of the
carbamoyloxy moiety afforded chiral boronate or borane intermediates 46 or 47. Oxidation
then gave the tertiary alcohols 48 or ent-48.

53
Scheme 34. Lithiation-Borylation of Chiral Secondary Carbamates Leading to Tertiary
Alcohols.
R1 R2
OCbs-BuLiEt2O
-78 °C20 min
Li
OCbR1
R2
RB(OR3)2 B(OR3)2
OCbR1
R2
RR
B(OR3)2R1
R2
R1 R2
R OH
Retention
Inversion
H2O2,NaOH
B(R3)2
OCbR1R2
RR
B(R3)2R1R2
RB(R3)2
R1 R2
R OH
H2O2,NaOH
Cb = N,N-diisopropylcarbamoyl
45 45Li
46
47
48
ent-48
This method allowed access to a broad range of tertiary alcohols, including alkyl,
cyclopropyl, vinyl, allyl, aryl and heterocyclic alcohols, in good yield (60-98 %) and very
high enantioselectivity (92-98 % e.e.) (Table 8).
Table 8. Examples of Tertiary Alcohols Obtained by Lithiation-Borylation of Chiral
Secondary Carbamates.
Entry Carbamate Migrating
group R
Borane/Boronic
ester Product
e.r., S:R
(Yield %)
1 Ph
OCb
Et BEt2 Ph
Et OH
99:1 (91)
2 Ph
OCb
cPr
O
OB
Ph
OH
3:97 (85)
3 Ph
OCb
vinyl
O
OB
Ph
OH
2:98 (75)
4 Ph
OCb
allyl
O
OB
Ph
OH
1:99 (95)

54
Table 8. Continued.
Entry Carbamate Migrating
group R
Borane/Boronic
ester Product
e.r., S:R
(Yield %)
5 OCb
pClC6H4 Ph BEt2
Ph OH
pClC6H4 4:96 (89)
6 OCb
pMeOC6H4 Et
O
OB
Et OH
pMeOC6H4 96:4 (87)
7 Ph
OCb
mCF3-C6H5
O
OB
Ph
OHmCF3C6H4
99:1 (92)
8 Ph
OCb
2-furyl
O
OB
Ph
OHO
98:2 (94)
9
OCb
Ph O
OB
OHPh
6:94 (73)
10
OCb
Et O
OB
OHEt
91:9 (98)
Interestingly, the use of the borane causes complete inversion of stereochemistry,
whereas the boronic ester results in almost complete retention of stereochemistry. Thus,
either enantiomer of a tertiary alcohol can be produced from just one enantiomer of the
starting material simply by the choice of an achiral reagent.
Later, with the aim of improving the enantioselectivity in the tertiary boronic esters
(or tertiary alcohols) resulting from carbamates and boronic esters with sterically
demanding groups and carbamates bearing electron-withdrawing aromatic groups, they
found that the use of either MgBr2/MeOH or neopentyl boronic esters in place of pinacol
esters provided essentially complete chirality transfer in the lithiation-borylation
reaction.[53]
They also showed that indanyl and tetralyl carbamates could be employed in this one-
pot lithiation/borylation/oxidation to give the corresponding tertiary alcohols in excellent
yields but the enatiomeric ratios were lower than their acyclic counterparts.[54]
Clayden et al. have extended the utility of the lithiated carbamates of secondary
benzylic alcohols by showing that they may be transformed into tertiary alcohols through a
N to C aryl transfer.[55]
Initially, this remarkable rearrangement was disclosed in lithiated
N-benzylic ureas[56]
and then later extended to thiocarbamates.[57]

55
I.2 Intramolecular Electrophilic Arylation of Lithiated Ureas
and Thiocarbamates
I.2.1 Discovery of N to C Aryl Migration in Lithiated N-Benzyl Ureas
While studying the regioselective lithiation of N-aryl ureas within the group,[58]
N-
benzyl urea 49 was treated with sec-BuLi and iodomethane with the aim of determining the
site of deprotonation (lateral lithiation (position 1), ortholithiation (position 2) or N-
benzylic lithiation (position 3)) (Scheme 35).[56]
However, instead, a rearrangement of 49
was observed, reminiscent of the Truce-Smiles rearrangement.[59]
Related rearrangements
of lithiated benzylamines and their derivatives are known.[60]
Lithiation at the benzylic
position was followed by transfer of the aryl group from the nitrogen to the -carbon of the
urea and subsequent quenching with iodomethane gave the unstable alkylated diarylamine
50 in a low yield. By replacing the methylation with an aqueous quench, the rearranged
product 51 was obtained in an improved yield of 89 %.
Scheme 35. Rearrangement of a Lithiated Urea.
N N
O
Me
Me2N N
O
Ph
MeHN N
O
Ph
1. s-BuLi, THF,-78 °C, 30 min2. MeI
1. s-BuLi, THF,-78 °C, 30 min2. NH4Cl
13 %
89 %
49
50
51
1
3
2

56
I.2.2 Aryl Migration in Lithiated N-Benzyl Ureas
Using the previous described conditions, a series of N-benzyl-N-aryl ureas 52
rearranged in a similar manner to provide ureas derivatives of diarylmethylamines 53
(Scheme 36) in good yields, regardless of the electronic or steric nature of the migrating
ring as shown in Table 9.[56]
Scheme 36. Synthesis of Secondary Diarylmethylamines.
N N
O
Me R1
1. s-BuLi (2.5 equiv.), THF, -78 °C, 30 min
2. H2O
MeHN N
O
R1 R2
52 53
R2
R1HNR2
A: 1. t-BuONO (6 equiv.), CH2Cl2, 24 h2. LiOH, H2O, THF, , 48 h
B: DIBAL-H, PhMe,
54
The amines 54 were returned either by hydrolysis of their N-nitroso derivatives (A)
or by reductive cleavage with DIBAL-H (B). Quenching the reaction with CD3OD
permitted the observation of the product which was deuterated at the doubly benzylic
position to nitrogen in 53. This suggests that 53 is deprotonated in the reaction
conditions meaning that the synthesis of secondary enantiopure diarylamines by this
method is not feasible.

57
Table 9. Aryl Migration in Lithiated N-Benzyl Ureas 52.
Interestingly, on treatment with sec-BuLi in THF and DMPU (dimethylhexahydro-2-
pyrimidinone) to increase the reactivity of the resulting organolithium,[61]
chiral N--
methylbenzylureas 55 formed a configurationally stable organolithium which underwent
rearrangement to give the ureas 56 with a new, fully substituted stereogenic centre in
generally good yields and with excellent enantioselectivities (Scheme 37). The reaction
tolerated both electron withdrawing and electron donating groups on the migrating ring
(Table 10). The reductive cleavage of ureas was carried out by hydrolysis of a nitroso
intermediate using LiOH to give ,-diarylethylamines 57 with an enantiomeric ratio up to
99:1, thus providing a route to amines bearing chiral tertiary substituents (Table 10).
Entry R1 R
2
Yield 53 (%)
(remaining 52)
Yield 54 (%)
(method)
1 Ph 2,6-diMe 89 -
2 Ph 4-Me 85 -
3 p-Tol H 85 -
4 Me H 78 (8) 84 (B)
5 Me 2-Me 76 (15) 72 (A)
6 Me 2-OMe 75 (21) 78 (A)
7 Me 2,4-di-Me 82 (14) 84 (A)
8 Me 4-Cl 69 (4) -
9 Me 4-OMe 76 (4) -

58
Scheme 37. Aryl Migration of Ureas Derived from -Methylbenzylamines.
N N Me
O
MeMe
R2
1. s-BuLi (2.5 equiv.), THF, DMPU (6 equiv.), -78 °C, 6 h
2. H2O
MeHNMe
R2
R1
1. t-BuONO (6 equiv.),CH2Cl2, 24 h
2. LiOH, H2O, THF, , 72 h
55
57
N N
O
DMPU
R1
MeHN N
O
MeMe
R2
R1
new quaternary centre
56
Retention ofconfiguration
Table 10. Stereospecific Aryl Migration.
Entry 55 R1 R
2 Yield 56 (%) e.r. Yield 57 (%)
1 (R)-55a 4-MeO H 56a, 82 97:3 72
3 (S)-55b 3,4-OMe H 56b, 76 98:2 -
4 (S)-55c 4-F H 56c, 34 99:1 -
5 (S)-55d 4-Cl H 56d, 51 98:2 -
6 (S)-55e 3-Cl,4-F H 56e, 69 >99:1 75
7 (S)-55f 2,3-naphtyl H 56f, n.d. 95:5 68
8 (S)-55g 3,4-naphtyl H 56g, 88 >97:3 -
9 (S)-55h H 4-MeO 56h, 73 >98:2 -
10 (S)-55i 2-Me 4-MeO 56i, 64 99:1 -
11 (R)-55j 4-Me 4-MeO 56j, 79 >97:3 -
The rearrangement presumably proceeds by the mechanism displayed in Scheme
38.[62]
Deprotonation at the benzylic position of 55 generates an organolithium
configurationally stable 55aLi on the time scale of the reaction. Ipso attack of the
organolithium centre 55bLi on the aryl ring gives a dearomatised intermediate 58, which
collapses with regeneration of aromaticity to provide lithiourea 56Li and then the urea 56

59
after protonation. The structure of the intermediate 58 has been deduced by isolation of the
crystalline enone 59 by trapping with oxygen.
X-ray crystallography of 59 confirmed the absolute stereochemistry of 56 and
provides evidence for stereochemically retentive rearrangement rather than invertive
(Figure 3).[63]
Scheme 38. Proposed Mechanism and Evidence of a Dearomatised Intermediate.
N N Me
Me
O
Me
R1 s-BuLi
R2
N N Me
O
Me
Me
Li
ArN N
O
Me Me
MeLi R1
R2
MeHN N
O
MeMe
R1
R2
MeN N
OLi
MeMe
R1
R2
N N
O
OCl
aryltransfer
H+
R1 = 2,3-benzo
R2 = p-ClO2
55 55aLi 58
59
56
Retention ofconfiguration
R1
N N
O
Ar
Li
55bLi
56Li

60
Figure 3. X-Ray Crystal Structure of 59.
I.2.3 Aryl Migration of Other Lithiated Urea Analogues
I.2.3.1 Aryl Migration of N-Benzyl-N-Pyridyl Ureas
Clayden et al. reported that this novel method allowed the -pyridylation of chiral
amines 60 via urea coupling, rearrangement and deprotection to provide functionalised
aminopyridines 63 containing a fully substituted quaternary stereogenic centre adjacent to
the pyridine ring (Scheme 39).[64]
It was found that the rearrangement was optimal when
LDA was used as the lithiating reagent instead of sec-BuLi. Moreover, the addition of
DMPU to the reaction increased both the rate and stereospecificity of the reaction,
retaining the stereochemistry of the enantiomerically pure starting material. Deprotection
of 62 by the original method reported[56]
was problematic due to the electrophilicity of the
pyridine ring and the nucleophilicity of its nitrogen. However, an alternative method was
found by refluxing the compound overnight in n-butanol to give rise to the amines 63 in
good yields.

61
Scheme 39. Synthesis of Ureas by Palladium-Catalysis Coupling and Rearrangement.
NH Me
Ar1H
Me
N N
O
Me
Ar1H
Me
N Me
N
O
Me
Ar1
Me
NH
N
HN
Me
Ar1
MeN
n-BuOH,
1. LDA (2 equiv.), THF,DMPU (10:1), -78 °C1-4 h
2. NH4Cl
1. (Cl3CO)2O2. MeNH2
6063
61 62R2 R2
R2
3. Py-Br,Pd2(dba)3 (2.5 mol %),xantphos (10 mol %)PhMe, NaOt-Bu,110 °C, 14 h
Me
Using these optimised conditions the rearrangement has been shown to work on a
range of the N-pyridyl ureas 61 as outlined in Table 11.
Table 11. Ureas 62 and Aminopyridines 63 by Stereospecific Rearrangement and
Hydrolysis.
Entry 61 Ar1 Ar
2
Yield 62
(%) e.r.
Yield 63
(%)
1 (S)-61a C6H5 2-Py 65 98:2 62a 76
2 (R)-61b 4-Cl-C6H4 2-Py 73 98:2 62b 81
3 (S)-61c C6H5 4-Me-2-Py 66 >99:1 62c -
4 (R)-61d 4-Cl-C6H4 3-OMe-2-Py 80 n.d. 62d -
5 (S)-61e C6H5 3-Py 74 99:1 62e 78
6 (S)-61f C6H5 3-Quinolyl 40 n.d. 62f -
7 (S)-61g C6H5 4-Py 83 96:4 62g 65
I.2.3.2 Aryl Migration of N-Aryl Urea Derivatives of Hetero- or Carbocyclic Amines
Since then, Clayden et al. have demonstrated that some classes of cyclic amine may
be arylated by this method.[65]

62
Unlike urea derivatives of pyrrolidine, those derivatives of 2-phenylpyrrolidine 64
rearranged successfully on treatment with LDA in THF-DMPU mixture, to offer the
arylated products 65 in good yields (Scheme 40).
When treated with either sec-BuLi or LDA in a THF-DMPU mixture all ureas
derived from aminotetralin and aminoindane 66 underwent clean aryl transfer from N to C
to give the arylated products 67 in good to excellent yields. Ureas were converted into
amines by warming to 118 °C in n-butanol for 48 h. (Scheme 40).
Treatment of tetrahydroisoquinoline 68 with sec-BuLi promoted rearrangement in the
absence of DMPU and by warming to room temperature a 78 % yield was obtained,
whereas methylated tetrahydroisoquinoline gave the corresponding rearranged product 70
in the presence of DMPU in 66 % yield whilst isoindoline failed to rearrange (Scheme 40).
It was proved that the rearrangement, unlike those of acyclic N--methylbenzylureas,
is not stereospecific.
Scheme 40. -Arylation of Cyclic Amines by Aryl Transfer in Lithiated Ureas.
NH
N
RAr
3 steps
-arylation
(Ar = 4-MeOC6H4, R = -CONHMe) 89 % (Ar = 2-pyridyl, R = Me) 62 %
N Li
ON
Ar
Me
via
Arylation of the pyrrolidine ring by rearrangement
NH2
n
3 steps
n
Ar NRMe
(n = 2, Ar = Ph, R = H) 71 %(n = 1, Ar = 4-ClC6H4, R = H) 61 %(n = 1, Ar = H, R = -CONHMe) 66 %(n = 1, Ar = 2,3-benzo, R = -CONHMe) 70 %(n = 1, Ar = 2,6-dimethyl, R = -CONHMe) 78 %
Arylation of aminoindane and aminotetralin
NH
3 steps
3 steps
N
Ph O
NHMe
69, 78 %
N
O
NHMe
70, 66 %
Ph Me
Rearrangement of tetrahydroisoquinolines
64 65
66 6768
R
(R = H or Me)

63
I.2.3.3 Aryl Migration of N-Allyl-N-Aryl Ureas
This rearrangement also turned out to be applicable to N-allyl-N-aryl ureas 71
allowing the construction of enantiomerically enriched diarylallylamine derivatives 75 by
sequential double aryl migration (Scheme 41).[66]
Upon lithiation with 2 equiv. of LDA, 71
underwent rearrangement with transfer of the aryl ring from N to the allylic carbon to
form the -arylated products 72 in good yields. A further enantioselective rearrangement
of vinyl ureas 72 using chiral lithium amides 74 afforded chiral allyl urea products 75 with
selectivities lying between 84:16 and 92:8 e.r..
Scheme 41. Sequential Double Arylation of N-Allyl-N-Aryl Ureas.
N N
Me
O
PMP
1. LDA (2 eq.),THF, -78 °C, 10 min
MeHN N
O
PMP
71
(Z)-72, 53 to 95 %
R1
R1
2. DMPU,-78 °C, 3 h3. MeOH
R1 = H, p-Me, m-Tol, p-MeO,
p-Cl, m-F, p-NC, m-CF3, Ph
Ar2Br, Pd2(dba)3
xantphos, NaOt-Butoluene,
N N
O
PMP
R1
Me
R2
R1 NR2
Me
Li
R1 N R1
R2 R2
Li
or
LiCl, THF,-78 °C, 10 min
NCONHMePMP
R1
R2
R2 = p-NC, 6-MeOPy,
p-Cl, m-F, m-MeO
73
74a
74b up to 91% yieldup to 92:8 e.r.
75
I.2.4 Enantioselective Synthesis of Tertiary Thiols by Intramolecular
Arylation of Lithiated Thiocarbamates
Asymmetric tertiary thiols are a synthetically challenging class of compounds to
prepare. The few reported methods for synthesising these species require restrictive

64
functionality to be incorporated into the products or are limited to using simple carbon
electrophiles.[67]
As a result, enantiomerically pure tertiary thiols are, despite the simplicity
of their structure, a remarkably difficult class of compounds to make.
Recently, Clayden et al. reported a solution to this problem: N-aryl S--alkylbenzyl
thiocarbamates 76 proved to be suitable substrates for the N to C aryl transfer.[57]
Lithiation
of such thiocarbamates with LiTMP in THF at -78 °C led to rearrangement with invertive
migration of the N-aryl ring and returned chiral benzylic tertiary thiol derivatives 77 in
excellent yields and high enantiomeric ratios (Scheme 42).
Scheme 42. Enantioselective Synthesis of Tertiary Thiols.
Ar1 S NMe
R O
Ar2
1. LiTMP (2.5 eq.),THF, -78 °C, 15 h
2. EtCO2HAr1 S NHMe
OR Ar2
76 77
NaOEt,EtOH
20 °C,15 min
Ar1 SH
R Ar2
78
SH
Me
63 %, 96:4 e.r.
SH
Me
97 %, 91:9 e.r.
MeO
SH
Me
Cl
97 %, 96:4 e.r.
SH
Me
41 %, 96:4 e.r.
Cl
SH
Me
CN
98 %, 97:3 e.r.
SH
Me
51 %, 96:4 e.r.
SH
97 %, 98:2 e.r.
SH
Me
76 %, 67:33 e.r.
F3C
SH
Me
OMe
94 %, 50:50 e.r.
SH
Me
75 %,50:50 e.r.
MeO
SH
89 %, 74:26 e.r.
Retention of configuration

65
I.3 Properties and Reactivity of Lithiated Carbamates
I.3.1 Dipole-Stabilized Carbanions Adjacent to Oxygen
The direct formation of stable carbanions to oxygen is not a facile process.[63a,68]
In
fact, the effect of an ether oxygen on a prospective carbanion appears to be deactivating
with respect to proton removal, due to electron pair repulsion overcoming the favourable
inductive effect. Such destabilization may provide some driving force for the Wittig
rearrangement[69]
and - and -eliminations[70]
observed for reactions in which -oxo
carbanions are possible intermediates (Scheme 43).
Scheme 43. Lithiation to Oxygen and Wittig Rearrangement.
R2O R1
H H RLi
R2O R1
H Li
LiO R1
H R2Wittig
rearrangement
Following the ideas of chelate stabilization and of proximity effects[71]
as powerful
devices for directed deprotonation, Hoppe discovered the strongly activating influence of
N,N-dialkylcarbamoyloxy groups (R2) in the removal of adjacent protons of weak CH-
acids by alkyllithiums[72]
and also prevent the Wittig rearrangement (Scheme 44). 2-
alkenyl, 2-benzyl and 2-alkynyl N,N-diisopropylcarbamates are deprotonated with great
ease by n-butyllithium at low temperatures leading to five-membered chelate complexes A
(Scheme 44).
Scheme 44. Induced Dipole Stabilization.
A
..
..
..O
Li
O
NR2
R1
L
L
B
..
..
..OO
NR2
R1
L
LLi..
C
..
..
..OO
NR2
R1
L
LLi..

66
Quantitative thermodynamic stability scales of organolithium compounds derived
from tin-lithium exchange equilibria has shown that an -oxycarbanion is far better
stabilized by a carbonyl group as the O-substituent than by an alkyl or alkoxyalkyl group
(Geq is approximately 3 kcal mol-1
).[73]
The thermodynamic data and NMR studies point
to the fact that the stabilization imparted by a carbonyloxy group in “dipole-stabilized”
carbanions is at least partly derived from an electron-withdrawing effect, and not only from
the traditionally accepted effect of the chelation of the Li atom bound to the carbanionic
centre with the carbonyl oxygen as shown in Scheme 44.[74]
The favourable chelation
should contribute to the stabilization of these organolithiums since the interaction between
the cation and the oxo group increases the acceptor properties of the carbamoyloxy group
according to the resonance forms B and C.
I.3.2 Versatile Reactivity and Configurational Stability of Lithiated O-
Benzylcarbamates
Lithiation to oxygen of carbamates such as 79a generates a series of d1 reagents
79Li that may be alkylated or acylated to give 80, providing an -lithio alcohol synthetic
equivalent or homoenolate equivalent (alkenyl counterpart) (Scheme 45). Less hindered
carbamates 79b, however, are too unstable on standing at -78 °C: rearrangement by 1,2-
acyl transfer from O to C provides -hydroxyamides 81.[75]
Scheme 45. Reactivity of Lithiated O-Benzylcarbamates.
N O
R3O
R1
R2
79a, R1 = R2 = i-Pr
79b, R1 = R2 = Et
RLiN O
LiO
R1
R2 R3 R2HN O
R1O
82
R3R1 = Ar
N O
EO
R1
R2
80
R3
NR1R2O
HOR3
EX
R1 = R2 = Et
81
1,2-acyl shift
1,4-aryl transfer
79Li
R1 = R2 = i-Pr

67
I.3.2.1 Electrophilic Substitutions of Lithiated O-Benzylcarbamates
The examples of -oxy-substituted benzylic precursors leading to lithium carbanions
of considerable configurational stability are secondary carbamates,[76]
such as 83,
introduced by Hoppe et al. (Scheme 46). Deprotonation with sec-BuLi/TMEDA leads to
the chelate-stabilized lithium carbanion pair 83Li•TMEDA,[77]
configurationally stable
below -70 °C in ether or hydrocarbon solution occurring with retention of stereochemistry.
The substitution of 83Li proceeds either with retention or inversion to form the products 84
or ent-84, respectively, depending on the nature of the electrophile.[76a,b,g]
Scheme 46. Generation of a Configurationally Stable, Enantioenriched Lithiated O-
Benzylcarbamate: Stereodivergence of its Electrophilic Substitution.
i-Pr2N O
O H Me
(R)-83, 98:2 e.r.
i-Pr2N O
O Li Me
(R)-83LiTMEDA
N N
ent-84
Inversion(SE2inv)
Retention(SE2ret)
EX = MeOH (90:10 e.r.), C3H7Br (>92:8 e.r.), (MeO)2CO (97:3 e.r.), iPrCH=O (>98:2 e.r.), PhC(=O)OMe (>98:2 e.r.)
EX = HOAc (90:10 e.r.), ClCO2Me (93:7 e.r.),MeCOCl (>98:2 e.r.), MeCOCN (96:4 e.r.),PhCH2Cl (>98:2 e.r.), CO2 (92:8 e.r.),iPrNCO (>92:8 e.r.), Me3SnCl (>98:2 e.r.)
i-Pr2N O
O E Me
84
i-Pr2N O
O Me E
s-BuLi/TMEDA
hexane, -78 °C
To rationalise the stereodivergence of the electrophilic substitution, the authors
hypothesised that electrophiles, which have a high tendency to interact with the lithium
cation, such as esters, alcohols, aliphatic aldehydes and ketones, prefer to attack at the
same side as the lithium atom (retention). Electrophiles, which have an energetically low
LUMO, such as acid chlorides, heterocumulenes and trialkyltin chlorides prefer to attack
from the opposite side (inversion), which has increased electron density because of the
partially flattened nature of the carbanion 83Li.[78]
Strohmann et al. also reported that the coordination sphere of the lithium atom must
be considered to rationalise the stereochemical course of the trapping reaction.[79]
Recently,

68
Aggarwal et al. reported that the stereochemical outcome of the reaction of lithiated
alkylcarbamates with boranes depended on the steric bulk of diamine additive such as
TMEDA and sparteine, which were complexed to lithiated carbamates.[80]
On the basis of
these studies, Takeda et al. showed that the coordinating ability of electrophiles and
solvents to a lithiocarbanion generated from 1-phenylbut-2-en-1-yl diisopropylcarbamate
played a significant role in determining the stereochemical course by involving equilibrium
between a solvent-separated ion pair and contact ion pairs.[81]
The tertiary carbamoyloxy-substituted benzyllithium 83Li was also found to be
configurationally stable based on the “Hoffmann test”.[82]
Racemic benzyllithium 83Li was
generated from 83 with sec-BuLi in Et2O/TMEDA at -78 °C. Reaction with the racemic
aldehyde 85 at -78 °C furnished two diastereomeric adducts 86 and ent-86 in 30:70 ratio
(Scheme 47). Repeating the reaction with the enantiomerically pure aldehyde 85 gave the
same two products with a different ratio close to 50:50. They therefore concluded that 83Li
was configurationally stable on the time scale set by the addition to the aldehyde 85.[83]
They also proved that 83Li, trapped by tributyltin chloride and regenerated by reaction
with n-BuLi in THF, was configurationnally stable at -78 °C even in THF.
Scheme 47. Hoffmann Test on Tertiary Carbamoyloxy-substituted Benzyllithium
Compound 83Li.
i-Pr2N O
O
i-Pr2N O
O
Et2O/TMEDA,-78° C
Me
83
MeLiBu3SnCl
n-BuLi,THF, -78 °C
i-Pr2N O
O SnBu3Me
Ph
NBn2
O
H
85
Ph
O Me
OH
NBn2N
O
i-Pr2
Ph
O Me
OH
NBn2N
O
i-Pr2
86 ent-86
Et2O/TMEDA
()-85
(S)-85
(30:70)
(48:52)
93 %
86 %
()-85
(S)-85
(30:70)
(50:50)
95 %
92 %
THF
83LiTMEDA 87
s-BuLi

69
Whereas carbenoids derived from chiral secondary benzyl carbamates are
configurationally stable at -78 °C, those from primary benzyl carbamates such as 88
(Scheme 48) are not, although the same “Hoffmann test” as above demonstrated that the
enantiomerization of the complex 88Li•TMEDA is not faster than its trapping by the
aldehyde 85.[83]
Enantioselective lithiation in the presence of chiral ligands to form configurationally
labile lithiated O-benzylcarbamates and electrophiles incorporation has also been
dominated by the studies of Hoppe. The first -oxybenzyllithium compound was the
lithiated benzyl carbamate 88, but it could not be tuned to useful selectivities in the
presence of ()-sparteine 89[84]
(Scheme 48).[85]
Scheme 48. Configurationally Labile Epimeric Diamine Complexes of -Lithiated
Benzylcarbamate 88Li.
N
OO
N
i-Pr2N O
O SnBu3
i-Pr2N O
O HS HR
88
i-Pr2N O
O
(RC)-88Li90
Li90
i-Pr2N O
O
(SC)-88Li89
Li89
enantiotopic differentiation
dynamic thermodynamicresolution
Diamine, -78 °C,toluene, 2.5 h
Bu3SnCl, -78 °C, 2 h
66:34 e.r. 1:99 e.r.
8990
91 % 88 %
SEinv SEinv
91a
By changing the ligand to sterically more demanding bis(oxazoline) 90, they found
that the resulting epimeric complexes equilibrated and the RC-configured complex (RC)-
88Li•90 was strongly favored in the equilibrium (Scheme 49). After dynamic
thermodynamic resolution, the complexes could be trapped with different classes of

70
electrophiles with a similar stereodivergence in the stereochemical course to yield highly
enantioenriched secondary benzyl carbamates 91 in toluene at -78 °C.[86]
Scheme 49. Deprotonation-Substitution Sequence with O-Benzylcarbamate 88 in the
Presence of Chiral Bis(oxazoline) 90.
i-Pr2N O
O HS HR
88
i-Pr2N O
O
(RC)-8890
Li90
90, s-BuLii-Pr2N O
O
(SC)-8890
Li90
PhMe, -78 °C,2.5 h RR R
R = H, Me, OMe,2,3-benzo EX
i-Pr2N O
O
(R)-91b, 28 %, 77:23 e.r.
i-Pr2N O
OHO O
(S)-91g, 64 %, 96:4 e.r.
Et
i-Pr2N O
O
(S)-91d, 95 %, >95:5 e.r.
MeO O
i-Pr2N O
O
(S)-91e, 64 %, 96:4 e.r.
SnBu3
OMe
OH
i-Pr2N O
O
(S)-91f, 77 %, >96:4 e.r.
Me
i-Pr2N O
O
(1S,2R)-91c, 70 %, 66:34 e.r.
OHtBu
Clayden et al. reported a third mode of reactivity displayed by O-benzylcarbamates
carrying N-aryl substituents to furnish 82 with no competing 1,2-acyl shift (Scheme 45).[55]
Computational studies showed that when R1 = Ar, the calculated free energy barrier for the
attack on the aromatic ring (3.6 kJ mol-1
) is considerably lower (by 14.4 kJ mol-1
) than that
for attack on the carbonyl group (18.0 kJ mol-1
).
I.3.2.2 N to C Arylation Migration in Lithiated O-Benzylcarbamates
The novel rearrangement reaction of carbamates such as 92
(Scheme 50) allows an aromatic ring to migrate to a new
fully substituted stereogenic centre via an organolithium
intermediate in a similar way to ureas. The resulting reactions constitute (stereospecific) -
R1 H
OH
R2R1 Ar
OH
R2

71
arylations of secondary alcohols, providing a connective, “umpolung” route to (chiral)
tertiary alcohols bearing heavily substituted substituents.
Upon lithiation with LDA in a Et2O-DMPU mixture, primary O-benzylcarbamates 92
underwent the rearrangement cleanly, then by simple treatment of the rearranged
carbamates 93 with sodium ethoxide in ethanol, the secondary alcohols 94 were returned in
good yields (Scheme 50).[55]
Scheme 50. Synthesis of Diarylmethanols by Aryl Transfer.
N O
Me
OR
1. LDA (2.5 equiv.)Et2O/DMPU (4:1)-78 °C, 4 h
2. NH4ClMeHN O
O
R
92
NaOEt, EtOH
, 2 h
OH
R
94
93a R = p-Me, 62 %93b R = o-Me, 55 %93C R = p-F, 40 %
94a R = p-Me, 68 %94b R = o-Me, 83 %94c R = p-F, 67 %
A series of secondary carbamates 95 was subjected to lithiation either with sec-BuLi
in THF/DMPU (condition A) or with LDA in Et2O/DMPU (condition B) (Scheme 51).
Under either set of conditions, 1,4-aryl transfers from N to C took place, and the arylated
carbamates 96 were isolated in good to excellent yields (Table 12).[55]
Scheme 51. Aryl Transfer in Secondary Lithiated Carbamates.
N O
Me
O
MeHN O
O
R1
95 96
Me
R2
Me
R2
Conditions As-BuLi (2.5 equiv.)THF, DMPU (4:1),-78 °C, 4 h
or Conditions BLDA (2.5 equiv.),Et2O, DMPU (4:1)-78 °C, 4 h
R1

72
Table 12. Conditions and Yields for N to C Aryl Transfer in Carbamates 95.
Entry 95 R1 R
2 Conds. Yield 96 (%)
1 95a H H A 96a, 75
2 95b p-Me H B 96b, 84
3 95c o-Me H B 96c, 72
4 95d o-i-Pr H B 96d, 67
5 95e p-OMe H A 96e, 68
6 95f p-Cl H B 96f, 56
7 95g 2,3-benzo H A 96g, 85
8 95h 3,4-benzo H A 96h, 57
9 95i H p-Cl B 96i, 90
10 95j p-Me p-Cl B 96j, 82
11 95k p-OMe p-Cl A 96k, 68
12 95l 2,3-benzo p-Cl A 96l, 67
Attempts to make 96i enantioselectively by stereospecific rearrangement of (S)-95i
by using the conditions above led to a racemate. A brief look at the literature seemed to
suggest that in general organolithium species are not configurationally stable in strongly
lithium-coordinating solvents when they contain α-oxygen atoms at the benzylic
position.[76b]
This is in contrast to their nitrogen counterparts and the urea analogues have
already been shown to rearrange with minimal loss of enantiopurity. The configurational
stability of benzylic α-oxygenated organolithiums in other solvents such as hexane and
diethyl ether is greatly increased, as seen in Section I.3.2.1, a number of examples have
been shown to be stable at −78 °C in these solvents. Indeed, when (S)-95i was lithiated in
Et2O with LDA, substantial enantiomeric enrichment was preserved: (S)-96i was returned
in 85:15 e.r. (Scheme 52). Enantiomerically enriched (S)-96i was converted into alcohol
(+)-(S)-97i and allowed to conclude that unlike ureas the rearrangement proceeds with
inversion at the lithium-bearing centre by comparison with published data for optical and
chromatographic properties.[52]

73
Scheme 52. Enantioselectivity in the Rearrangement of the Carbamate 95i.
N O
O
Me
Me
Cl
(S)-95i (94:6 e.r.)
1. LDA (2.5 equiv.)Et2O, -78 °C -35 °C,24 h
2. MeOH
MeHN O
O
Me
Cl
(S)-96i (85:15 e.r.)
NaOEt,EtOH, , 2 h
(+)-(S)-97i, 77%
Cl
Me
O
NMe
(R,R)-Clemastine 98
OH
Me
Cl
Inversion ofconfiguration
(S)
(S)
(S)
,-Diaryl ethanols are valuable compounds in enantiomerically pure form.[86]
For
example, (R)-97i is an intermediate in the synthesis of the antihistamine agent (R,R)-
clemastine 98 (Scheme 52).[88]
Therefore, clemastine forms potential synthetic target of
this methodology.
Prior to this work, it was well established that not only benzylic but also allylic and
propargylic carbamates may be deprotonated to form synthetically versatile substituted
allylithiums and propargyllithiums.
I.3.3 Configurational Stability of Lithiated O-Allylcarbamates
Hoppe et al. used -lithiated 2-alkenyl N,N-diisopropylcarbamates 99Li as
homoenolate equivalents in regio and stereospecific reactions with aldehydes and ketones
after a lithium-titanium exchange (Scheme 53).[89]

74
Scheme 53. 1-Alkenyl Carbamates in Asymmetric Homoaldol Reactions.
R2
O
HR1
2. R3CH=O
3. H3O+99a R1 = R2 = Me
99b R1 = H, R2 = Me
99c R1 = H, R2 = Ph
R3
R1
OH
R2 OO
Ni-Pr2
R2
O
R1Li
O
Ni-Pr2
Ln
1. TiX4O
Ni-Pr2
10099Li
Enantioenriched (-lithio-2-alkenyl)carbamates 99Li derived from secondary
carbamates such as (E) and (Z)-2-pentenyl N,N-diisopropylcarbamates 99a, are
configurationally stable below -70 °C in nonpolar solvent. They could be generated either
by stereoretentive deprotonation of the optically active precursors by n-BuLi/TMEDA in
ether/hexane (Scheme 54) or by kinetic resolution of the racemic precursors, deprotonating
with sec-BuLi/()-sparteine in pentane (Scheme 55).[90]
The allyllithium was formed with
greater than 90:10 e.r. as estimated from trapping experiments as shown by the two
examples of stannylation depicted in Scheme 54 and 55.
Scheme 54. Configurational Stability of Lithiated (E),(Z)-2-pentenyl N,N-
diisopropylcarbamates 99aLi Generated by Deprotonation of the Optically Active
Precursors.
Me
CbO H
s-BuLi/TMEDA
hexane, -78 °C
(R,E)-99a95:5 e.r.
Me
CbO LiTMEDA
Me3SnCl Me
OCbMe3Sn
(S,Z)-101, 52 % 95:5 e.r.
Me
H OCb
(S,Z)-99a99:1 e.r.
s-BuLi/TMEDA
hexane, -78 °C
Me
CbO Li�TMEDA
Me3SnCl Me3Sn Me
OCb
(S,Z)-101, 55 % 95:5 e.r.
anti-SE'
anti-SE'
(R,E)-99aLiTMEDA
(S,Z)-99aLiTMEDA

75
Scheme 55. Configurational Stability of Lithiated (E)-2-Pentenyl N,N-
diisopropylcarbamate 99aLi Generated by Kinetic Resolution of the Racemic Precursor.
Me
OCbH
()-99a
n-BuLi/()-sparteine 89
pentane, -78 °C, 10 h
Me
OCbLi()-sparteine
OCb
MeLi()-sparteine
torsionH
OCbMe
(R)-99a41 %, 90:10 e.r.
(S)-99aLi.()-89
Me
OCbSnBu3
()-101 (R,Z)42 %, 90:10 e.r.
OCb
MeSnBu3
()-101 (S,E)6 %, 90:10 e.r.
Bu3SnCl
anti-SE'
Additional trapping experiments, in order to elucidate their stereochemical course, showed
that the lithium-titanium exchange of (S)-99aLi•TMEDA by tetra(isopropoxy)titanium
(TIPT) was accompanied by stereoretention (SEret),[91]
whereas the (S)-99aLi•()-sparteine
complex proceeded with stereoinversion (SEinv) under the same conditions.[92]
Carboxylation and methoxycarbonylation of the lithium sparteine complex (S)-99aLi•()-
89 also proceeded with stereoinversion.[93]
In contrast, (-lithio-2-alkenyl) carbamates derived from carbamate derivatives of
primary alcohols, such as (E)-2-butenyl N,N-diisopropylcarbamate 99b[94]
or (E)-cinnamyl
N,N-diisopropylcarbamate 99c,[95]
turned out to be configurationally unstable in solution
with n-BuLi/()-sparteine even at -78 °C.
In the case of (E)-2-butenyl N,N-diisopropylcarbamate 99b, however, the (S)-
configurated sparteine complex (S)-99bLi•()-89[96]
crystallised from the
pentane/cyclohexane solution with concomitant dynamic kinetic resolution,[97]
resulting in
up to 95 % d.e. in the solid (Scheme 56). Reaction of the solid with Ti(Oi-Pr)4 proceeded

76
with configurational inversion to give the allyltitanium intermediate (R)-102, which was
stable in solution up to -20 °C.[94a]
Reaction with tetrabutyltin chloride yielded
allylstannane (R)-103 with high regioselectivity and enantiomeric purity (>97:3 e.r.) via a
delithiotitanation (inversion) and detitanostannylation (anti-SE’) sequence.[94b]
Scheme 56. Configurational Stability of Lithiated (E)-2-Butenyl N,N-
diisopropylcarbamate 99bLi.
HR
OCbHS
99b
n-BuLi/pentanecyclohexane
()-sparteine,-78 °C
H
O
N(i-Pr)2
O
Li�()-sp.
(S)-99bLi89, 95 % d.e.
H
O
N(i-Pr)2
O
Li�()-sp.
(R)-99bLi89
1. epimerization2. crystallisation
HS
OCbTi(O-iPr)3
(R)-102
HR
OCbTi(O-iPr)3
(S)-102
Ti(O-iPr)4
Ti(O-iPr)4
OCb
(R)-103, 80 %, >97:3 e.r.
SEinv
SEinv
anti-SE' Bu3SnCl
Bu3Sn
Deprotonation of (E)-cinnamyl N,N-diisopropylcarbamate 99c with n-BuLi/()-
sparteine in toluene showed that the cinnamyllithium (S)-99bLi•()-89 formed upon
deprotonation epimerizes within thirty minutes at -78 °C to the thermodynamically more
stable allylithium-()-sparteine complexe (R)-99cLi•()-89 (Scheme 57). Reactions with
several electrophiles proceeded regio- and stereospecifically at the - or γ position, but at
different rates for the two diastereoisomers, leading to a decrease in enantioselectivity
compared to the diastereomeric ratio of the complexes.[95]
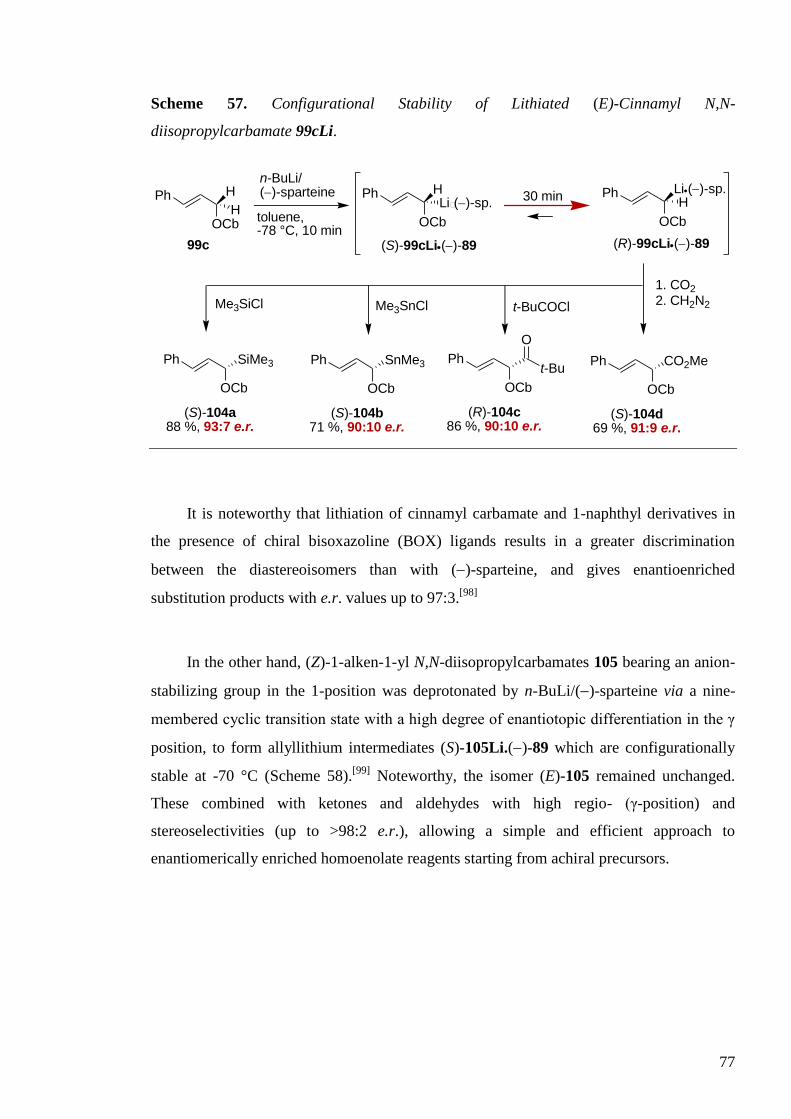
77
Scheme 57. Configurational Stability of Lithiated (E)-Cinnamyl N,N-
diisopropylcarbamate 99cLi.
Ph H
OCbH
99c
Ph H
OCb
Li�()-sp.toluene,-78 °C, 10 min
n-BuLi/()-sparteine Ph Li()-sp.
OCb
H30 min
Ph
OCb
SiMe3
(S)-104a88 %, 93:7 e.r.
Ph
OCb
SnMe3
(S)-104b71 %, 90:10 e.r.
Ph
OCb
(R)-104c86 %, 90:10 e.r.
t-Bu
O
Ph
OCb
CO2Me
(S)-104d69 %, 91:9 e.r.
1. CO2
2. CH2N2t-BuCOClMe3SnClMe3SiCl
(S)-99cLi()-89 (R)-99cLi()-89
It is noteworthy that lithiation of cinnamyl carbamate and 1-naphthyl derivatives in
the presence of chiral bisoxazoline (BOX) ligands results in a greater discrimination
between the diastereoisomers than with ()-sparteine, and gives enantioenriched
substitution products with e.r. values up to 97:3.[98]
In the other hand, (Z)-1-alken-1-yl N,N-diisopropylcarbamates 105 bearing an anion-
stabilizing group in the 1-position was deprotonated by n-BuLi/()-sparteine via a nine-
membered cyclic transition state with a high degree of enantiotopic differentiation in the γ
position, to form allyllithium intermediates (S)-105Li.()-89 which are configurationally
stable at -70 °C (Scheme 58).[99]
Noteworthy, the isomer (E)-105 remained unchanged.
These combined with ketones and aldehydes with high regio- (γ-position) and
stereoselectivities (up to >98:2 e.r.), allowing a simple and efficient approach to
enantiomerically enriched homoenolate reagents starting from achiral precursors.

78
Scheme 58. Enantiotopos-Differentiating γ-Deprotonation of 1-Alkenylcarbamates.
R1 R2
O HS HR
R1 = aryl, 1-alkynyl, 1-alkenyl, SiMe3
R2 = Me, Et
n-BuLi/(�)-sparteine
R1 R2
O H H
O
N
Li BuiPr2
105
R1 R2
OLi
NiPr2O
(S)-105Li()-89
()-sp.
toluene, -70 °C
O
i-Pr2N ()-sp.
I.3.4 Configurational Stability of Lithiated O-Alkynylcarbamates
Enantiomerically enriched, configurationally stable alkynylllithiums 106aLi.TMEDA
can be produced by deprotonation of enantiomerically pure secondary 2-alkynyl N,N-
diisopropylcarbamates 106a by n-BuLi or sec-BuLi/TMEDA in hexane at -78 °C (Scheme
59).[100]
It has been revealed by a high degree of chirality transfer (C.T.) in the formation of
allenyl carbinols 107.
Scheme 59. Configurational Stability of Lithiated Secondary Alkynyllithiums.
R1
OCb
Me
106a
Base/TMEDA
R1
Me LiTMEDA
OCb
• OCbMe
OH
R1• OCb
Me
OH
R1
syn-107 anti-107
106aLiTMEDA
R1
t-Bu
c-C6H11
CH3
PhMe2Si
t-BuCHO
C.T. (%) (Method)
0 (A), 84 (B)
0 (A), 88 (B)
Method A: Deprotonation with n-BuLi at -78 °CMethod B: In situ deprotonation with s-BuLi at -100 °C or -78 °C in the presence of aldehyde
Et2O, 20 min
100 (A)
73 (A)
In contrast, deprotonation of primary 2-alkynyl N,N-diisopropylcarbamates 106b by
n-BuLi/()-sparteine in toluene at -78 °C and trapping the intermediate ion pairs by
carboxylation or silylation led to slightly enantioenriched products. However, when the

79
deprotonation was performed in pentane, dynamic resolution of the lithium-()-sparteine
complexes (S)-106bLi.()-89 by selective crystallisation gave much higher levels of
enantioselectivity in the allenyl carbamates (M)-109a,b (92:8 e.r. and 94:6 e.r. respectively)
and the allenyl carbinols 110 (>96:4 e.r.) after transmetallation by rapid addition of
chlorotitanium tri(isopropoxide) from the solid state (Scheme 60).[101]
Scheme 60. Synthesis of Chiral Allenyl Carbamates and Carbinols.
(S)-106bLi()-89 (R)-106bLi()-89
R1
OCb
HR HS s-BuLi/()-sparteine
pentane, -78 °CR1
OCb
H Li�()-sp.
R1
OCb
Li H(-sp.
crystallisation
106b
R1
OCb
H Ti(O-iPr)3
R1
OCb
(Oi-Pr)3Ti H
(R)-108 (S)-108
ClTi(Oi-Pr)3, -78 °C
AcOH
i.R2CHO, -78 °Cii. AcOH
• HOCb
R2
R1
OH
110, >96:4 e.r.
R1 = t-Bu, Me3Si
R2 = i-Pr, Me, Ph
• HOCb
H
R1
(M)-109
a R1 = t-Bu, 80 %, 92:8 e.r.
b R1 = Me3Si, 86 %, 94:6 e.r.

80
I.4 Carbolithiation Reactions
Organolithium compounds are important intermediates in organic synthesis.[63a,102]
Various methods for generation of organolithium intermediates have been developed to
date. Among them, carbolithiation of unsaturated compounds such as alkenes,[103]
alkynes[104]
and enynes[105]
serves as an attractive method since carbon-carbon bond
formation leads to the formation of a second organolithium intermediate such as 111
(Scheme 61), which can be employed for subsequent reactions with different electrophiles,
thus allowing two new C-C bonds to be formed in a single pot. Commercially available n-
BuLi, sec-BuLi, and tert-BuLi are often utilised as starting organolithium compounds.[106]
Activation of the starting material by TMEDA, DABCO or ()-sparteine is also often
advantageous.
I.4.1 Overview of Carbolithiation Reactions Involving Organolithiums
1.4.1.1 General Aspects of Carbolithiation Reactions
For synthetically useful carbolithiation reactions,[107]
the formation of intermediate
111 must proceed much more rapidly than the next addition step in order to avoid the
polymerization (formation of 112) (Scheme 61). Therefore, 111 must markedly differ from
the starting one[108]
or special stabilization of 111 is required by using for example
cinnamyl derivatives 113 bearing a Lewis-basic substituent X (X = OR, NR2,…) as
demonstrated by Normant et al. (Scheme 61).[109]
The complex-induced proximity effect
(CIPE),[71]
resulting from the formation of the complex 114, causes a rapid addition
reaction to form the stabilized adduct 115, which can be trapped by various electrophiles.
Scheme 61. Conditions Required for Intermolecular Carbolithiation Reactions.
R1 R2 Li R1
Li
R2
R1
R2
R1
Li
111
112
Anionic Polymerization
E+
R1
E
R2
R1
n

81
Scheme 61. Continued.
Ph X
R1 LiPh X
R1 Li113
114
Ph
Li
R1
X
115
E+
Ph X
E
R1
X = OR, NR2, ...
Li = Li�(ligands)n
116
This drawback is diverted if the addition occurs on a triple bond, where a less
reactive sp2-C-Li bond is formed, or on an intramolecular double bond, where entropy
factors are favourable (Scheme 62). In the latter case, a new sp3 carbon is created.
Scheme 62. Carbolithiation on a Triple Bond and an Intramolecular Double Bond.
R3 LiR1 R2
R1
LiR3
R2
Li
Li
If the carbolithiation is now carried out on an ,β-disubstituted double bond, two
regioisomers can be formed and after reaction with an electrophilic reagent, two sp3
stereogenic centers are created (Scheme 63). So, in order to have a powerful reaction, it is
necessary to control the regio- and the diastereoselectivity of the carbolithiation reaction.
Regioselectivity is highly dependent on the substrate, but generally selectivity of the
addition is syn. Diastereoselectivity means that the configurational stability of sp3
organolithium derivatives towards electrophiles must be controlled. And, the sense of
diastereoselectivity, by which 116 is formed, depends on the configuration of the
intermediate 115 and the stereochemistry of the substitution step. All substrates should be
liable to an enantioselective version of carbolithiation, under the influence of either a chiral

82
ligand for lithium, which would promote a face selection on the double bond, or of a chiral
group already present on the substrate (diastereoselection). Carbolithiation is regiospecific,
for instance with the alkyl and aryl group adding exclusively to the double bond such that
the more mesomerically stabilized benzylic lithiated compound is formed.[110]
Scheme 63. Regio-, Diastereo- and Enantioselectivity of the Carbolithiation Reaction.
R3 Li
E+
R2R1
R3
R2R1
Li
HH
Regioselectivity
R3
R2R1
E
HH
Diastereoselectivity
Enantioselectivity
In this thesis, selected examples of substrates allowing such carbolithiations are
shown.
1.4.1.2 Intermolecular Carbolithiation Reactions
The first enantioselective carbolithiation of (E)-β-methylstyrene 117a and (E)-
stilbene 117b with alkyllithiums has been reported by Marek, Normant et al. utilising ()-
sparteine to induce asymmetry in the products (Scheme 64).[110e,f]
In these examples, the
chiral centre formed as a result of the C-C bond formation is configurationally stable but
the benzylic C-Li centre has low configurational stability. Protonation of the lithiated
intermediates 118aLi and 118bLi respectively afforded 2-methylhexylbenzene 118a and
1,2-biphenylhexane 118b in good yields and e.e.. Attempts to achieve diastereoselectivity
by treating 118aLi with methanol-d4 gave the deuterated product with only 60:40 d.r..
Scheme 64. Enantioselective Carbolithiation of (E)-β-Methylstyrene and (E)-Stilbene.
Ph
R
117a, R = Me117b, R = Ph
n-BuLi, ()-sparteine
hexane, -15 °C
Ph
R
118aLi, R = Me118bLi, R = Ph
Bu
Li
H3O+ Ph
R
118a, R = Me, 83 %, >92:8 e.r.118b, R = Ph, 81 %, 85:15 e.r.
Bu

83
The carbolithiation of (E)-cinnamyl alcohol 119 illustrates how stereocontrol over
both centres can be achieved (Scheme 65). Indeed, the enantioselective carbolithiation of
119 with n-BuLi/()-sparteine and subsequent trapping of the lithiated intermediate
120Li.()-89 with different electrophiles allowed the formation of two chiral centres in a
one-pot diastereospecific and enantioselective manner to provide the acyclic products 120,
121a, 121b and 121c.[110c]
Scheme 65. Enantioselective Carbolithiation with Diastereoselective Electrophile
Substitution.
Ph
HO
n-BuLi, (�)-sparteine
cumene, 0 °C
119
Ph
120Li()-89
Bu
LiO
(-)-spart
H3O+ BuPh
CH2OH
120, 85 %, 83 % e.e.
BuPh
CH2OH
BuPh
CH2OH
BuPh
CH2OHMe D PhS
121a 121b 121c
MeI DCI PhSSPh
d.r.> 98:263 % yield, 91:9 e.r.
d.r.> 98:261 % yield, > 91:9 e.r.
d.r.> 95:587 % yield, 92:8 e.r.
The presence of a free alcohol is not a necessity as similar e.e. was obtained with
dialkylamines[110e]
and improved e.r. (up to > 97:3 e.r.) with a tert-butyl ether.[111]
The
stereochemistry of the olefin is crucial for the enantioselectivity of the carbolithiation.
Employing the alkene of opposing stereochemistry ((Z)-cinnamyl alcohol) allowed access
to the opposite enantiomer of 120 from the same ()-sparteine chiral source.[110g]
Recently, O’Shea et al. have extended this methodology to the enantioselective
carbolithiation of various ortho-amino β-methylstyrenes and (E)-stilbenes initiating
cascade route to chiral heterocycles.[112]
Enynes are also reactive towards carbolithiation.[113]
Although the yields are variable,
the products are valuable allenyllithiums (Scheme 66).

84
Scheme 66 Intramolecular Carbolithiation of Enynes.
Li
•Li
60%
Carbolithiation of alkynes is recognised to be useful in the preparation of
stereochemically defined tri- and tetrasubstituted alkenes.
For example, O’Shea et al. employed an intermolecular carbolithiation of
diphenylacetylene 122 as the key synthetic step in a concice and highly stereoselective
synthesis of (Z)-tamoxifen 126, the most commonly utilised therapeutic agent for the
treatment of estrogen-dependent breast cancer (Scheme 67).[114]
Starting from
diphenylacetylene 122, syn addition of EtLi yielded the vinylic lithium species (Z)-123
which rapidly isomerises[115]
to the more thermodynamically favoured (E)-123 under the
strongly coordinating solvent conditions. Subsequent reaction of this intermediate with
triisopropylborate gave the (E)-vinyl boronic acid 124. (Z)-Tamoxifen 126 was generated
by Suzuki-Miyaura cross-coupling of 124 with the aryl iodide 125 in 38 % overall yield.
Scheme 67. Stereoselective Synthesis of (Z)-Tamoxifen.
PhPh
122
EtLi, THF
-10 °C, 2 h Ph
Li
Ph
(Z)-123
Ph
Ph
Li
(E)-123
1. B(Oi-Pr)3
2. HCl (aq.)52 %
Ph
Ph
B(OH)2
I
N
Pd(PPh3)4, Na2CO3
DME/H2O73 %
Ph
Ph
124N
126
125
Although simple alkyl groups on alkynes are prone to deprotonation, it has been
reported that carbolithiation of such alkynes is possible when the addition is accelerated
through intramolecular reaction[116]
or by a heteroatom directing group such as an alkoxy

85
or amino group on alkynes.[117]
Interestingly, as depicted in the Scheme 68, alkyl- and
aryllithium compounds underwent sterero- and regioselective carbolithiation reactions with
alkynes 127 having no heteroatoms under iron or iron-copper catalysis.[118]
Scheme 68. Iron-Catalyzed Alkyllithium of Alkynes followed by Methanolysis.
R3R2
R1 LiFeCl3 (5 mol %)TMEDA (20 mol %)PPh3 (10 mol %)
(Zn (20 mol %))Et2O, -20 °C
R2
LiR1
R3 R2
R3R1
Li R2
HR1
R3 R2
R3R1
H
MeOH
127 (E)-128 (Z)-128
R1 = Bu, R2 = Bu, R3 = 3-CF3C6H4, 82 %
R1 = Bu, R2 = Bu, R3 = 2-MeOC6H4, 82 %
R1 = Bu, R2 = Hex, R3 = Ph, 79 %, 93:7
R1 = Bu, R2 = i-Bu, R3 = Ph, 82 %, 94:6
R1 = i-Bu, R2 = Me, R3 = Ph, 72 %, > 99:1
1.4.1.3 Intramolecular Carbolithiation Reactions
The intramolecular carbolithiation of lithium carbanions onto multiple bonds is an
efficient method for the construction of carbocycles. These reactions usually proceed
rapidly only for the formation of five-membered rings.
In the field of diastereoselective intramolecular carbolithiations, a stereogenic center,
already present in the vicinity of the double bond, can induce a facial choice. For instance,
the epimerizable tertiary benzyllithium 130 of Scheme 69, underwent a 5-exo-trig
cyclisation under the influence of the neighboring chiral lithium alkoxide to give four
isomeric cyclopentanes 131 in a ratio of 82:8:7.5:2.5 in 90 % yield.[119]
Scheme 69. Synthesis of Optically Active Arylcyclopentanes by Carbocyclisation of
Alkenylbenzyllithiums Involving Diastereofacial Differentiation.
MeSePh OH
1. MeLi, LiBr
2. t-BuLi, Et2O
PhLi OLi
-20 °C, 1 h
H3O+
PhOH
131 (major isomer)129 130

86
It is also possible to start with an enantiomerically enriched secondary lithium
derivative, if cyclisation is fast enough, as compared to epimerization. An interesting
enantioselective cyclocarbolithiation has been disclosed by Hoppe et al., starting from the
carbamate 132 derived from indene (Scheme 70).[120]
Enantioselective deprotonation led to
two lithiodiastereoisomers (R,S)-132Li.()-89 and (S,S)-132Li.()-89. A kinetic resolution
takes place since the syn carbolithiation from (R,S)-132Li.()-89 maintains the carbamate
group in a pseudoequatorial position, whereas for (S,S)-132Li.()-89, this group would be
forced in an endo position. As a result the tricyclic adduct ()-133 was formed in high
purity after hydrolysis, whereas (S,S)-132Li.()-89 returned the optically active starting
material (S)-(+)-132.
Scheme 70. Enantioselective Cyclocarbolithiation via ()-Sparteine-Mediated Lithiation
of a Racemic Carbamate and Kinetic Resolution.
OCb
()-132
s-BuLi, ()-sparteineEt2O, -78 °C, 20 h
OCb
(R,S)-132(-89
HLiOCb
(S,S)-132(-89
HLi
LiH
OCbH2O
H
OCb
()-133one diastereoisomeryield 24 %>98:2 e.r.
H2O(S)-(+)-132yield 48 %
()-133Li
The same concept has been applied to the corresponding alkyne allowing the
synthesis of substituted enantiopure cyclopentanoid building blocks.[121]
Hoppe et al.
extended this application to the synthesis of chiral protected 2-alkylidene-cyclohexane 135
(as a single diastereoisomer) and -cyclobutane-1,3-diols 137 by the lithiodestannylation

87
and unsual intramolecular anti-selective 4- or 6-exo-dig carbolithiations of -lithiated ω-
carbamoyloxy-1-alkynylcarbamates (Scheme 71).[122]
Scheme 71. Synthesis of 2-Alkylidene-cycloalkane-1,3-diols via Enantioselective
Intramolecular Carbolithiation.
CbO
OCb
SnBu3O
(1S, 5RS)-134d.r. = 50:50 97:3 e.r.
TBS
O
OO
Li
Ni-Pr2
O
Ni-Pr2O
TBS
Li
n-BuLi, LiClTHF, -100 °C20 min
OCbH
OCbTBSO
MeOH,-100 °Cto RT
trans-135, 37 %98:2 e.r.
CbO
O
(1S, 3RS)-136d.r. = 50:50 97:3 e.r.
TBS
OCb
SnBu31.n-BuLi, LiClTHF, -40 °C3 h
2. HOAc, -40 °C to RT
OCbTBSO
OCbH
OCbTBSO
OCbH
trans-137, 25 % cis-137, 25 %
d.r. = 50:50, each >97:3 e.r.
I.4.2 Intermolecular Carbolithiation Reactions of Vinyl Carbamates
Hoppe[123]
and Snieckus[75b]
have also shown that -aryl-O-vinylcarbamates 138 are
highly receptive substrates for -carbolithiation, presumably owing to a CIPE of the
carbamate donor (as seen in Section I.3.3) and stability of a benzyl anion against
polymerization (Scheme 72).
Scheme 72. Carbolithiation of -Aryl-O-Vinyl Carbamates.
ArR
H
O O
NR1
R2Li
diamine (L2) ArR
H
O O
NR1
LiL2
R2
Ar
O
O
R1N
LiL2
ArR1
CbO E
RH
EX
138, R1 = i-Pr, Et
R2
RH
138R2LiL2
139LiL2 140

88
O-Vinyl carbamates 138 underwent facile syn-carbolithiation by
alkyllithium/TMEDA and delivered configurationally stable lithiated benzyl carbamates
139LiL2, which have been trapped with different electrophiles to give the products 140.
Selected examples are displayed in Table 13 (entries 1-6 for ref 123 and entries 7-11 for ref
75a).
However, when the reaction was performed in the presence of chiral diamines, such
as ()-sparteine or ()--isosparteine, moderate enantiofacial differentiation was observed.
Table 13. Yields for the Carbolithiation of -Aryl-O-Vinyl Carbamates 138.
Entry Ar R R2Li EX Yield (%)
1 C6H5 H t-BuLi CO2 75
2 C6H5 H n-BuLi Me3SnCl 62
3 -naphthyl H i-PrLi MeOH 91
4 2-(MeO)C6H5 H t-BuLi MeOH 91
5 C6H5 C6H5 n-BuLi Me3SiCl 51
6 C6H5 C(CH3)3 n-BuLi MeOH 84
7 C6H5 H n-BuLi Br 64
8 C6H5 H n-BuLi PhCH2Br 66
9 C6H5 H s-BuLi MeOD 96
10 C6H5 H t-BuLi EtI 71
11 (3-Et2NOC)C6H4 H t-BuLi MeI 50
In an other hand, Coudert et al. reported a carbolithiation approach on seven-
membered ring N-vinyl carbamate 141 to give the compounds 142 in high yields (Scheme
73).[124]
Scheme 73. Carbolithiation of Cyclic N-Vinyl Carbamate.
N Ph
Boc
RLi, THF
-78 °C to 0 °C NPh
Boc
R
LiH2O
N Ph
Boc
R
141 142
R = Me, 87 %R = n-Bu, 83 %R = s-Bu, 88 %
R = t-Bu, 84 %R= Ph, 95 %
142Li

89
Later, they explored the scope of this method to develop an efficient synthesis of
precursors of -amino acids 144 involving a carbolithiation reaction of acyclic N-vinyl
carbamates 143 followed by a spontaneous internal N to C alkyloxycarbonyl migration
(Scheme 74).[125]
Scheme 74. Carbolithiation of Acyclic N-Vinyl Carbamates.
Ar1
N Ar2
1. RLi, -78 °C to 0 °C
2. H2O, NH4Cl
143
Ar1
NH
CO2t-Bu
Ar2
R
Ot-BuO
144
PhNH
CO2t-Bu
N i-Pri-Pr
89 %
NH
CO2t-Bu
n-Bu
O
3-MeOC6H4
14 %
PhNH
CO2t-Bu
Ph
78 %
NH
CO2t-Bu
Me
2-MeC6H4
80 %
NH
CO2t-Bu
Me
3-MeOC6H4
56 %
Some representative examples:
Very recently, Clayden et al. applied this attractive method to vinyl ureas in order to
initiate N to C aryl transfer.[126]
I.4.3 Tandem β-Alkylation--Arylation of Amines by Carbolithiation and
Rearrangement of Vinyl Ureas
The Clayden group found that N-alkenyl ureas 145 exhibit umpolung reactivity,
undergoing addition of organolithiums to their nucleophilic β-carbons (Scheme 75). The
addition could be then coupled with N to C aryl transfer within the lithiated urea
intermediate to the rearranged products 146 in excellent yields. The products 146 were
converted to the amines 147, thus providing a valuable method for the construction of
multiply branched alkylamines.

90
Scheme 75. Umpolung Carbolithiation of Vinyl Ureas.
Ar2N N
O
Me Me
Ar1
-50 °C, 90 min
2. MeOH
R Lin-BuOH, 2.5 h
145
1. , THF
MeHN NR
O
Me
Ar1 Ar2
146
MeHNR
Ar1 Ar2
147
MeHN N
O
Me
Ph Ph
78 %
MeHN N
O
Me
Ph Ph
74 %
MeHN N
O
Me
Ph p-ClC6H4
72 %
MeHN NPh
O
Me
Ph p-FC6H5
78 %
MeHN N
O
Me
Tol Ph
75 %
MeHN N
O
Me
Tol Ph
96 %
O
Some representative examples:
Interestingly, β-carbolithiation and N to C aryl migration of (E)- and (Z)-alkenyl
ureas 148 turned out to be stereospecific[127]
at -40 °C either in THF or in toluene followed
by addition of DMPU to enforce the rearrangement after the carbolithiation was complete
(Scheme 76) to give 149 and epi-149 formed by syn-addition of R and Ar2. By simply
switching from the E to the Z form of vinyl ureas 148, both enantiomers of the tertiary
amines 150 could be prepared.
Scheme 76. Stereospecificity Reactions of (E)- and (Z)-Alkenyl Ureas.
Ar2N N
O
Me Me
Ar1 n-BuOH, 2.5 h
E-148
E
Ar2N N
O
Me Me
Ar1
Z-148
Z
1. RLi, THF,-40 °C (3-6 h)2. MeOH or
1. RLi, Tol,-40 °C, 1 h2. DMPU, 16 h3. MeOH
MeHN NMe
O
Me R
Ar2 Ar1
149, 54-85 %
MeHN NR
O
Me
Ar2 Ar1
epi-149, 44-75 %
Me
Ar1 R
Me
Ar2MeHN
150, 66-75 %
Ar1R
Me
Ar2MeHN
epi-150, 67-70 %
Ar1 = C6H5, p-ClC6H4, p-FC6H4, pMeC6H4, p-MeOC6H4
Ar2 = C6H5, p-MeOC6H4, m-MeOC6H4
R = i-Pr, n-Bu, t-Bu

91
I.5 Aims of the project
As discussed in Section I.2 and I.3.3, we have uncovered a unnoticed facet of the
reactivity of organolithiums stabilized by N-arylcarbamoyl groups, namely their tendency
to undergo migration of the N-aryl ring to the carbanion centre in what amounts to an
intramolecular nucleophilic substitution on an electron-rich aromatic ring.[56,64,65]
In the
case of N-aryl-O-benzylic carbamates, lithiation at the benzylic carbon induces a
stereochemically invertive intramolecular N to C aryl transfer, and provides a route to -
arylated tertiary alcohols in enantiomerically enriched form.[55]
Tertiary diarylated alcohol 97i is an intermediate in the synthesis of clemastine 98
(Scheme 77). Clemastine is a selective H1 antagonist with anticholinergic and sedative
effects. The synthesis of all four possible stereoisomers of clemastine was reported by
Ebnöther and Weber in 1976 (see Section II.1.1.1).[128]
The most active isomer was
identified as the (+)-(R,R)-isomer, with the stereochemistry of the quaternary center to
oxygen determining activity to a greater extent than the center in the pyrrolidine ring.
Published routes to clemastine all employ resolution,[129]
and no asymmetric synthesis of
clemastine has been described, although a synthesis of ()-hydroxyclemastine was reported
in 2007 (See Section II.1.3).[130]
Therefore, it was the first aim of the project to accomplish
the first enantioselective synthesis of clemastine as its (S,S)-enantiomer as an application
of this rearrangement. The strategy consists of the coupling of enantiomerically pure
chloroethylpyrrolidine (S)-151 derived from commercially available (S)-proline and the
enantiomerically enriched tertiary alcohol (S)-97i arising from invertive N to C aryl
migration in a lithiated carbamate.
Scheme 77. Retrosynthesis of (S,S)-clemastine.
N O
MeMe
Cl
s s
N
Me
s
Cl HOMe
Cl
s
(S,S)-clemastine 98 (S)-151 (S)-97i

92
As described in Section I.3.4 and I.3.5, the lithiated allylic and propargylic
carbamates 99 and 106 have proved their worth in stereoselective synthesis where the
organolithium is trapped by an electrophile such as a carbonyl compound or alkylating
agent (Figure 4).
Figure 4. Lithiated Allyl- and alkynylcarbamates.
Me LiR
O
NiPr2
O
N
NLi
Me
O
NiPr2
O
N
N
99LiTMEDA 106LiTMEDA
However, there are currently no methods allowing the stereoselective arylation of
lithiated allyl or propargyl carbamates. These particular nucleophiles have the potential to
react in the same manner as a benzylic organolithium species ( to the oxygen). The
second aim of the project was therefore to explore the potential of aryl migration within
these lithiated carbamates as a means to achieve this aim and thereby showing that these
arylation reactions are not confined in scope to benzyllithiums. Moreover, these substrates
are of value because the resulting alcohol products may be oxidized through to hydroxyl
acid derivatives.
One of the challenges to the extension of this mechanistically remarkable method
into a more general synthesis of hindered and/or electron rich alcohols is the fact that the
deprotonation step needed to generate the intermediate benzyllithium is relatively slow.
However, Hoppe[123]
and Snieckus[75b]
have shown that -aryl-O-alkenylcarbamates are
receptive substrates for -carbolithiation (see Section I.4.2), presumably owing to a
complex induced proximity effect arising from coordination of the organolithium to the
carbamate donor (see Section I.3.1), and the stability of benzylic anion preventing
polymerization. Judging that the carbolithiation would require some degree of stabilization
of the product anion, -aryl, alkynyl, alkenyl, and silyl-O-vinylcarbamates were prepared

93
as substrates (Scheme 78). Such a reaction would provide a remarkable connective route to
tertiary alcohols involving two new C-C bond-forming reactions in a single pot.
Scheme 78. Umpolung Carbolithiation of Vinyl Carbamates.
XR
H
O O
NR Ar1
O Li
ON
R1
R
Ar1
R
X
O Ar1
OHN
R1
R
carbolithiation
aryl migrationR1 Li R
X
X = aryl, alkynyl, alkenyl, silyl
Vinylic electrophiles cannot normally be coupled directly with organolithiums and
transmetallation is usually required to form new vinylic C-C bonds using
organolithiums.[131]
This methodology can therefore be used to provide a new approach to
the vinylation of organolithium nucleophiles that would permit the construction of
functionalised quaternary centres by vinylation of tertiary carbanions (Scheme 79).
Scheme 79. Intramolecular Vinylation of Lithiated Carbamates.
N O
Me
O Me
MeHN O
O Me
RR
vinylmigration

94
CChhaapptteerr IIII :: RREESSUULLTTSS AANNDD DDIISSCCUUSSSSIIOONN
II.1 Synthesis of Clemastine, an Antihistaminic Agent
II.1.1 Total Synthesis and Absolute Configuration of Clemastine via
Resolution
II.1.1.1 Ebnöther and Weber’s Pioneer Work on the Synthesis of (R,R)-Clemastine
The total synthesis of clemastine 98 by an optical resolution method was pioneered in
1976 by Ebnöther et al. and was followed by the determination of its absolute
configuration.[128]
Clemastine was prepared by coupling of 2-(2-chloroethyl)-1-
methylpyrrolidine 151 with -methyl-p-chlorobenzyl alcohol 97i to give an isomeric
mixture of 98 and 152. The minor structural isomer 152 is likely to be a result of an initial
intramolecular cyclisation within the pyrrolidine moiety followed by the nucleophilic ring
opening by the corresponding alcohol 97i. The corresponding diastereoisomers of 98 were
separated as hydrogenfumarate salts, and the corresponding enantiomers were resolved as
hydrogenmaleinate salts by crystallisation (Scheme 81).
Scheme 81. Ebnöther and Weber’s Synthesis of Clemastine.
NaNH2
xylene, 100 °C,16 h
Major (86 %)
Minor (14 %)
97i151
98
152
N O
MeMe
Cl
N
Me
Cl HOMe
Cl
OMe
Cl
NMe

95
The absolute configuration of clemastine has been determined by X-ray-
crystallography and also by acid catalysed hydrolysis of products 98 and 152 as illustrated
below (Scheme 82).
Scheme 82. Acid Catalysed Hydrolysis of 98 and 152.
97i 15498
152
N O
MeMe
Cl
N
Me
OHHOMe
Cl
OMe
Cl
NMe
153Cl
H+
H2O
H+
H2O
97i
HOMe
Cl153
Cl
NMe
OH
155
The ether-linkage hydrolysis of each product afforded the tertiary alcohol 97i, alkene
fragment 153, primary and secondary alcohols (154 and 155), from which the absolute
configuration of clemastine was confirmed.
II.1.1.2 Synthesis of Optically Active (R)-2-(2-Chloroethyl)-1-Methylpyrrolidine, an
Intermediate in the Synthesis of Clemastine via Resolution
Nikiforov et al. developed a new approach to synthesise the chiral pyrrolidine
fragment 151 via resolution in 1990 (Scheme 83).[129b]

96
Scheme 83. Synthesis of (R)-2-(2-Chloroethyl)-1-methylpyrrolidine 151 via Resolution.
N S
Me
N S
Me
COOCH2Ph
Br
N
Me
COOCH2Ph
N
Me
COOCH2Ph
N
Me
COOCH2PhH
N
Me
H
OHN
Me
H
Cl
BrCH2COOCH2C6H5
CH3CN
Ph3P, NEt3
CH2Cl2
96 %
NaCNBH3
MeOH
di-p-toluoyl-D-tartaric acid
EtOH
67 %
LiAlH4SOCl2
(R)-151
156 157
158 159
(R)-160 (R)-154
The synthesis began with the coupling of 1-methyl-2-thiopyrrolidinone 156 and
benzyl bromo acetate to yield the corresponding salt 157. This was followed by a sulfide
contraction reaction in the presence of triphenylphosphine and triethylamine to give the
enaminoester 158 in 96 %. The resulting product 158 was then reduced using sodium
cyanoborohydride in acidic conditions to yield the compound 159. The resolution was then
performed using di-p-toluoyl-D-tartaric acid. Several chiral acids such as tartaric, mandelic
and di-benzoyl-tartaric acids have also been investigated as alternatives but resolution
could not be achieved. The resolution involves the formation of the corresponding
diastereoisomeric salts. The diastereoisomeric salt of compound (R)-160 crystallised
selectively from 70 % ethanol and was then basified using conc. ammonia solution. The
reduction to a primary alcohol (R)-154 was carried out using lithium aluminium hydride
followed by the conversion to the chloride derivative (R)-151 to complete the synthesis.
Although this synthetic route is chemically appealing in a sense that the resolution
reagents are reusable, it suffers from the relative large number of steps. In addition, the
use of a resolution method has limited the maximum yield of the corresponding step to
50 % and results in poor overall yield.

97
II.1.2 Stereoselective Synthesis of ()-Hydroxyclemastine
Hydroxyclemastine was targeted as a versatile analogue of clemastine with H1
receptor antagonist activity.
Recently, the synthesis of ()-hydroxyclemastine 168 outlined in Scheme 84 was
achieved from the chiral auxiliary 162 via the chelation-controlled diastereoselective 1,2-
addition reaction to build the challenging quaternary centre to the oxygen, following by
O-alkylation with (R)-2-(2-chloroethyl)-1-methylpyrrolidine 151.
Scheme 84. Synthetic Route to ()-Hydroxyclemastine 168.
H
O
OO
MgBrMeO
CuI, THF/Me2S80 %
syn 99 % OMe
OH
OO
Br
NaH, DME83 %
162163
OMe
O
OO
164
O3, MeOH
CH2Cl284 %
OMe
O
OO
165
O
Ph2Zn, CH2Cl2
4-Cl-PhMgCl
87 %, 90 % d.e.
OMe
O
OO
166
PhOH
Cl
NCl2.
1. NaH, xylene
OMe
O
OO
167
O
Ph
Cl
N
Me
H CAN
aq. HCl, CH3CN45 %
HO
168
O
Ph
Cl
N
Me
H
(R)-151 Me

98
The organocopper compound, prepared from 4-methoxyphenylmagnesium bromide
and CuI in THF/Me2S, reacted with the aldehyde 162 highly stereoselectively ( 99 %)
affording syn-addition product 163 in an excellent yield of 80 %.[132]
Then, the auxiliary
163 underwent an O-alkylation with 3-bromo-2-phenylpropene in 83 % yield, followed by
ozonolysis of the alkene obtained to give the ketone 165 in 84 % yield. The key step
involves the coordination of 165 with Ph2Zn in CH2Cl2, followed by addition of 4-
chlorophenyl magnesium bromide to produce the alcohol 166 with 90 % d.e. in 87 %
yield.[133]
As previously described, the skeleton was obtained from O-alkylation of 166
with (R)-2-(2-chloroethyl)-1-methylpyrrolidine (R)-151 in 78 % yield. Finally,
deprotection of chiral auxiliary in 167 with ceric ammonium nitrate gave the desired ()-
hydroxyclemastine 168 in 45 % yield. Thus, the chiral auxiliary plays a dual role as a
chiral inducer as well as a protecting group.
Since no asymmetric synthesis of clemastine has been reported, we envisaged to
explore the potential of the rearrangement to achieve this goal.
II.1.3 Novel Asymmetric Synthesis of ()-(S,S)-Clemastine
II.1.3.1 Retrosynthesis of (S,S)-Clemastine
As revealed from the retrosynthesis (Scheme 85), clemastine 98 could be generated
by the coupling of enantiomerically pure chloropyrrolidine (S)-151 and enantiomerically
enriched tertiary alcohol (S)-97i, the key intermediate arising from the invertive N to C
aryl migration in the lithiated carbamate 95i.
The commercially available ()-(S)-N-Cbz-proline 169, as a source for one of two
chiral centres would give access to the compound (S)-174 by an Arndt-Eistert
homologation, followed by reduction of the ester (S)-174 to give alcohol (S)-175, which in
turn would be converted into chloro derivative (S)-151.

99
Scheme 85. Retrosynthesis of (S,S)-Clemastine.
N
Me
OMe
Cl
s s
N Cl
Me
s
N OMe
Cbz
sO
N CO2H
Cbz
s
HOMe
Cl
ss
MeN O
Me
Cl
O
s
Me
Cl
HO
Invertive arylmigration
Arndt-Eistert
(S,S)-clemastine 98
(S)-151
(S)-97i
(S)-169(S)-174
(S)-95i
(S)-183a
II.1.3.2 Synthesis of Pyrrolidine Fragments
II.1.3.2.1 Synthesis of the Chloroethylpyrrolidine Fragment (S)-151HCl
Construction of the fragment (S)-151 began with ()-(S)-Cbz-proline (S)-169, which
was commercially available in 99 % e.e. (Scheme 86). One-carbon homologation of (S)-
169 was achieved via the Arndt-Eistert protocol.[134]
The first step involved the formation
of acyl chloride (S)-170 with oxalyl chloride plus catalytic DMF in CH2Cl2 at 0 °C for 2 h,
which subsequently reacted with trimethylsilyldiazomethane (TMSCHN2) in the presence
of triethylamine in THF-CH3CN at 0 °C for 5 h following by an aqueous work-up to give
-diazoketone (S)-171 in 80 % yield. A small amount of (S)-172 was isolated which is the
result of the nucleophilic attack by HCl just produced on -diazoketone as observed by
Shioiri et al.[135]
. In place of the hazardous diazomethane, stable and safe TMSCHN2 was
employed.

100
Scheme 86. Synthesis of -Diazoketone (S)-171 by the Arndt-Eistert Reaction.
N COOH
Cbz
(COCl)2, DMF, CH2Cl2
0 °C, 2 hN
Cbz O
ClN
Cbz O
N2
TMSCHN2, NEt3THF/CH3CN
0 °C, 5 hN
Cbz O
(S)-169 (S)-172(S)-171, 80 %(S)-170
Cl
The key step of the Arndt-Eistert homologation is the Wolff rearrangement of
diazoketone (S)-171 to give ketene (S)-173, which was accomplished by silver(I) catalysis
(Scheme 87).[136]
The reaction was conducted in the presence of methanol as a nucleophile
to capture the ketene intermediate (S)-173 and returned the chain-extented methyl ester (S)-
174[137]
in 78 % yield. The Wolff rearrangement of the diazo-ketone was proved to proceed
with retention of configuration by [α]D values.[135]
Scheme 87. Synthesis of Ester (S)-174 by the Wolff Rearrangement.
N
Cbz O
N2
(S)-171
N
Cbz
CO
HN
Cbz
O
OMeAgObz MeOH
NEt3
(S)-174, 78 %(S)-173
RT, 3 h
Lithium aluminium hydride in THF at 45 °C reduced both the ester and the
carbamate protecting group,[138]
giving the hydroxyethylpyrrolidine derivative (S)-175[139]
in 60 % yield (Scheme 88). The alcohol was converted to the chloroethylpyrrolidine
coupling partner with thionyl chloride in chloroform at 60 °C for 2 h,[140]
which returned
the amine hydrochloride (S)-151•HCl[141]
in 92 % yield.
Scheme 88. Completion of the Synthesis of Chloroethylpyrrolidine Fragment (S)-151•HCl.
N OMe
Cbz
O
N OH
Me
N Cl
Me
·HCl
LiAlH4, THF
45 °C, 45 min
(S)-175, 60 %
SOCl2, CHCl3
60 °C, 2h
(S)-151HCl, 92 %(S)-174

101
II.1.3.2.2 Toward the Synthesis of Chloroethylpyrrolidine Fragment (R)-151•HCl
An analogous strategy could be employed for making the (R)-isomer from (+)-D-
proline. However, although the synthetic route is relatively short and attractive, (+)-D-
proline, a non-natural product is an expensive starting material and therefore not suitable
for large-scale synthesis.
Therefore, (R)-151•HCl was made previously within the group by an alternative
method. Starting with inexpensive starting material N-Boc pyrrolidine 176, which was
lithiated with sec-BuLi in the presence of ()-sparteine. The resulting complex was
quenched with dry CO2[142]
to yield N-Boc-(R)-proline (R)-177 in 55 % yield and 99:1 e.r.
after crystallisation (Scheme 89). Due to the incompatibility of the Boc group with the
conditions used for diazoketone formation, a protecting group swap was necessary, which
was achieved using 2 M HCl followed by addition of benzyl chloroformate.[143]
The
product (R)-169 was taken through the same series of transformations to yield (R)-151•HCl.
Scheme 89. Preparation of Chloroethylpyrrolidine Fragment (R)-151•HCl.
N
Boc
N
Boc
CO2H N
Cbz
CO2H
1. s-BuLi/89, Et2O
2. CO2
1. HCl (2 M),Et2O, , 2 h
2. ClCO2Bn,NaOH
(R)-177, 55 % (R)-169, 77 %
(R)-151HCl
176
A small amount of (S)-151•HCl was converted to its free base 151 by elution through
an SCX cartridge.[144]
However, we found that prolonged storage of 151 either neat or in
solution led to the formation of significant quantities of the bicyclic ammonium salt
178+
•Cl- (Scheme 90). The same cyclisation was observed in good yield on attempted
purification of 151 by distillation.[140a]
Cyclisation to 178+
•TsO- was likewise observed on attempted formation of the
tosylate derivative of 175. In general therefore we chose to store and use 151 as its stable
hydrochloride salt.

102
Scheme 90. Intramolecular Cyclisation of 151 and 175.
N
Me
Cldistillation
N
Me
X N
Me
OH
TsCl, py, DMAP
151178+
175
X = Cl- X = TsO-
In view of this instability, an alternative coupling partner lacking a basic nitrogen
atom, the Cbz-protected iodide 180 was made. This compound would avoid the unwanted
azepane ring formation and consequently the formation of the side product 152 observed in
the final O-alkylation reaction (Scheme 81). Moreover, the iodo group would favour the
SN2 substitution.
II.1.3.2.3 Synthesis of Cbz-Protected Iodoethylpyrrolidine Fragment (S)-180.
Selective reduction of (S)-174 with lithium borohydride in THF at room temperature
for 96 h gave the alcohol (S)-179 in 80 % yield (Scheme 91).[145]
Scheme 91. Synthesis of Alcohol (S)-179 by Reduction of the Ester Group.
N
Cbz
O
OMe
(S)-174
N
Cbz
OHLiBH4, THF
96 h, RT
(S)-179, 80 %
Finally, the combination of triphenylphosphine, imidazole and iodine was employed
to convert the alcohol (S)-179 into iodide derivative (S)-180 (Scheme 92). Subsequent
optimisation led to (S)-180 in 74 % yield (Table 14).
Scheme 92. Conversion of Alcohol (S)-179 into its Corresponding Iodide (S)-180.
N
Cbz
OH
(S)-179
N
Cbz
I
(S)-180
(Table 14)
PPh3, Imidazole,I2, solvent, T(°C)

103
Firstly, the conditions used commonly within the group (Table 14, entry 1) gave (S)-
180 in low yield (27 %). Treatment with the portion-wise addition of iodine at 0 °C until a
yellow colour persisted and only 2 equiv. of imidazole (entry 2) gave (S)-180 in an
increased yield of 48 %. Finally, the yield was improved greatly to 74 % when the reaction
time and the amount of PPh3 and imidazole were decreased and the reaction was carried
out in a mixture of Et2O and CH3CN (entry 3).
Table 14. Optimal Reaction Conditions.
Entry PPh3
(equiv.)
Imidazole
(equiv.) I2 Solvent T (°C) t (h)
Yield
(%)
1 2 4 4 equiv. THF/CH3CN RT 21 27
2 2 2 elemental THF/CH3CN 0 5 48
3 1.75 1.85 elemental Et2O/CH3CN 0 1 74
II.1.3.3 Synthesis of Tertiary Alcohol by Aryl Migration of a Lithiated carbamate
The tertiary alcohol (S)-97i was made from p-chloroacetophenone 181a, which was
reduced by the method of Noyori[146]
using formic acid in the presence of the ruthenium
complex (S,S)-182 to provide the alcohol (S)-183a[52]
in 91% yield and 99:1 e.r. (Scheme
93). This alcohol was converted to its carbamate derivative (S)-95i by reaction with phenyl
isocyanate in CH2Cl2 at room temperature for 15 h and methylation with sodium hydride
and methyl iodide in DMF at room temperature for 15 h in 74 % yield over 2 steps.
A previous optimisation within the group[55]
has shown that the stereospecificity of
the aryl migration in the lithiated carbamate (S)-95i is maximal if the reaction is carried out
with LDA at -78 °C, followed by slowly warming to -35 °C and quenching with MeOH
after 24 h. Accordingly, the rearranged product (S)-96i was formed in moderate yield with
84:16 e.r.. We have previously noted that the lithium-coordinating cosolvent DMPU
markedly increases the reactivity of hindered organolithiums towards nucleophilic attack
on arenes,[61]
probably by favouring the formation of solvent-separated ions pairs.[147]
Indeed, addition of DMPU to the reaction gave considerably higher yield (90 %), but
returned racemic product.
Alcoholysis of the carbamate product 96i was achieved by heating to reflux with
sodium ethoxide in ethanol for 2 h, which gave alcohol (S)-97i in 87 % yield. The
stereochemistry of the product was confirmed as (S) by comparison with literature data.[52]

104
Scheme 93. Synthesis of Tertiary Alcohol (S)-97i.
(S,S)-182 cat. HCO2H-NEt3
91%, 99:1 e.r.
1. PhNCO, NEt3, CH2Cl2,RT, 15 h
2. NaH, DMF0 °C, 30 minMeI, RT, 15 h
74 % over 2 steps, >99:1 e.r.
RT, 48 h
NH2
Ru
NPh
Ph
Ts
Cl
(S,S)-182 cat.
(S)-95i(S)-183a
1. LDA (2.5 eq.), Et2O,-78 °C-35 °C, 24 h
2. MeOH
(S)-96i, 51 %, 84:16 e.r.
NaOEt, EtOH,, 2 h
HOMe
Cl
(S)-97i, 87 %, 84:16 e.r.
Inversion of configuration
181a
N O
Me
O
Cl
Me
MeHN O
O
Me
Cl
[]D29 = +5.3 (c 0.9, CHCl3)
[lit.52 +14.8 (c 6.5, CHCl3)]
HO
Cl
Me
O
Cl
Me
II.1.3.4 Further Evidence for Stereochemically Invertive Rearrangement for
Carbamates
For the purpose of stereochemical confirmation, further rearrangement substrates (R)-
95e, (S)-95m, (S)-95n and (R)-95o (Scheme 94) were made in a similar way as (S)-95i
(Scheme 94). The chiral secondary alcohols (S)-183b and (S)-183c were prepared by
Noyori’s asymmetric reduction[146]
of the corresponding ketones 181b and 181c in 78 %
and 94 % yield with 99:1 e.r. and 97:3 e.r., respectively and subsequently converted into
the required carbamates (S)-95m and (S)-95n (Scheme 94).
Scheme 94. Preparation of enantiopure carbamates (R)-95e, (S)-95m, (S)-95n and (R)-
95o.
(S,S)-182 cat. HCO2H-NEt3
1. PhNCO, NEt3, CH2Cl2,RT, 15 h
2. NaH, DMF0 °C, 30 minMeI, RT, 15 h
RT, 48 h
(S)-183b, R1 = p-OMe,
78%, > 99:1 e.r.
(S)-183c, R1 = m-CF3,
94%, 97:3 e.r.
181b, R1 = p-OMe
181c, R1 = m-CF3
N O
Me
O Me
(R)-95e, R1 = H, R2 = p-OMe, 76 %
(S)-95m, R1 = p-OMe, R2 = H, 92 %
(S)-95n, R1 = m-CF3, R2 = H, 86 %
(R)-95o, R1 = H, R2 = m-CF3, 85 %
R1
R2
HO
Me
R1O
Me
R1

105
Under the standard lithiation conditions, LDA (2.5 equiv.) in a mixture of THF and
DMPU at -78 °C, (S)-95m failed to rearrange (Scheme 95; Table 15, entry 1). However,
the rearrangement was promoted by warming the reaction to -40 °C to give the rearranged
product 96e in low yield as a racemate (entry 2). An attempt to make the arylated
carbamate product enantioselectively, using the same successful conditions employed for
the carbamate bearing a choro group 95i, was unsuccessful (entry 3). In addition to the
starting material, only isolated products 184 and 183b resulting from nucleophilic attack
on the carbamate C=O group were identified by 1H NMR.
Scheme 95. Attempted Stereospecific Rearrangement of (S)-95m and (R)-95e.
N O
Me
O Me
R1
R2
MeHN O
O
Me
(S)-95m, R1 = p-OMe, R2 = H > 99:1 e.r.
(R)-95e, R1 = H, R2 = p-OMe, 99:1 e.r.
(Table 15)
96e
OMe
Table 15. Conditions and Yields for the Attempted Stereospecific Rearrangement of (S)-
95m and (R)-95e.
Entry S.M. Base Solvent Additive T(°C) t (h) Results
1 (S)-95m LDA THF DMPU -78 19 95m
2 (S)-95m LDA THF DMPU -78 to -40 24 96e, 19 %
3 (S)-95m LDA Et2O - -78 to -35 21 184 and 183b
4 (R)-95e s-BuLi THF DMPU -78 4 96e, 65
5 (R)-95e LDA Et2O - -78 to -35 17 -
6 (R)-95e s-BuLi Et2O - -78 to -35 24 -
NHHO
Me
OMe184 183bMe
Introduction of a p-methoxy group on the aryl group presumably leads to a decrease
in kinetic acidity of the proton adjacent to oxygen. The next attempt was therefore to
interchange the two aryl rings. The carbamate (R)-95e now rearranged successfully on

106
treatment with sec-BuLi (2.5 equiv.) in a mixture of THF and DMPU at -78 °C (Table 15,
entry 4). However, when this carbamate was subjected to lithiation, under the conditions
developed to avoid racemization, it failed to rearrange or only traces of rearranged product
96e were observed (entries 5 and 6). Indeed, the arylation involves nucleophilic attack of
the organolithium centre on the N-aryl ring, and given the nucleophilicity of the p-methoxy
aryl group it is not surprising that the rearrangement of (R)-95e was sluggish without
DMPU and THF.
We therefore turned our attention towards investigating the stereospecific aryl
migration in lithiated carbamate (S)-95n (Scheme 96).
Scheme 96. Attempted Stereospecific Rearrangement of (S)-95n.
(S)-95n, 97:3 e.r.
N O
Me
O Me
MeHN O
O
Me
CF3CF3
(Table 16)
96n
Table 16. Conditions and Yields for the Attempted Stereospecific Rearrangement of (S)-
95n.
Entry Base Solvent Additive T (°C) t (h) 96n, Yield (%) e.r.
1 LDA THF DMPU -78 4 81 50:50
2 LDA Et2O - -78 22 60 43:57
3a LDA Et2O - -78→-35 2 - -
4 LDA Et2O - -78→-35 18 64 45:55
Firstly, in order to ascertain that the rearrangement of (S)-95n occurs, the standard
lithiation conditions were used to yield the desired rearranged carbamate 96n in excellent
yield (Table 16, entry 1). Then, treatment of (S)-95n with LDA (2.5 equiv.) in Et2O at -
78 °C for 22 h allowed the rearranged product 96n to be isolated in a decreased yield of
60 %, due to the nucleophilic attack on the carbamate C=O group and with loss of
enantiomeric purity (entry 2). In order to make the rearrangement faster than the
a unintentional quench of the reaction : 95n and 96n (very low yield) = 76:24 e.r.

107
racemization of the organolithium intermediate, the temperature was raised to -35 °C
(entry 4). However, the stereochemistry was not retained.
Therefore, the rearrangement of (S)-95n competes poorly against racemization of the
organolithium intermediate, probably due to the strongly electron-withdrawing
trifluoromethyl group promoting delocalisation of the charge in the organolithium
intermediate.
As a note regarding the entry 3: a quench of the reaction occurred during the
monitoring. Nevertheless, the enantiomeric ratios of the starting material 95n and the
rearranged product 96n were determined and proved to be similar, which means that the
possibility of having a mixture of inversion and retention of configuration might be ruled
out.
The way to overcome the problem of racemization of the organolithium intermediate
would be to interchange the two aryl rings (Scheme 97).
Scheme 97. Stereospecific Rearrangement of (R)-95o.
85% over 2 steps, 98:2 e.r.
(R)-95o
N O
Me
O Me
MeHN O
O
Me
CF3(Table 17)F3C (R)
(S)-96n
Table 17. Conditions and Yields for the Stereospecific Rearrangement of (R)-95o.
Entry Base Solvent Additive T (°C) t (h) 96n, Yield (%) e.r.
1 LDA THF DMPU -78 6 46 52:48
2 LDA Et2O - -78→-35 22 - -
3 LiTMP Et2O - -78→-35 24 38 90:10
N
Li
LiTMP
N
Li
LDA
Empirical structures are shown without aggregation

108
When enantiomerically enriched carbamate (R)-95o was treated with LDA (2.5
equiv.) in THF/DMPU (Table 17, entry 1), a moderate yield of the rearranged carbamate
96n was obtained, and in essentially racemic form. Under the conditions for avoidance of
racemization, LDA in Et2O at -78 °C to -35 °C (entry 2), no sign of rearranged product 96n
was observed, only the starting material 95o and products arising from nucleophilic attack
on the carbamate C=O group were recovered. A change of the base to LiTMP (lithium
tetramethylpiperidine) led to the formation of the rearranged product 96n with a
considerable increased in enantioselectivity: 90:10 e.r and in moderate yield (entry 3).
The higher e.r. obtained with LiTMP[148]
is presumably a consequence of slower
lithiation coupled with higher configurational stability resulting from increased steric
bulk,[149]
and the electron deficient aryl ring migrates faster than the benzyllithium
intermediate racemises.
Deprotection of the rearranged carbamate 96n was achieved by refluxing with
sodium ethoxide in ethanol to yield the tertiary alcohol 97n in 75 % yield. Following
deprotection, the []D of the alcohol 97n was measured and gave a positive []D,
confirming after comparison with the literature,[52]
that the (S)-enantiomer of alcohol 97n
had been synthesised. This in turn confirmed that the (S)-rearranged carbamate 96n has
been formed and that the rearrangement proceeded with inversion of stereochemistry
(Scheme 98).
Scheme 98. Deprotection of (S)-96n.
(S)-96n, 38 %, 90:10 er
MeHN O
O
Me
CF3HO
Me
CF3(S) (S)NaOEt, EtOH,
, 3 h
(S)-97n, 75 %, 90:10 er
[]D31 = +19.2 (c 3.5, CHCl3)
[lit.52 +29.3 (c 5.5, CHCl3)]

109
II.1.3.5 Formation of the Ether Linkage and Isolation of Clemastine
II.1.3.5.1 Attempts at Formation of the Ether Linkage from Cbz-protected
Iodoethylpyrrolidine (S)-180
In an initial study, the coupling reaction via a Williamson ether reaction using
commercially available 1,1-diphenylethanol 185 was examined (Scheme 99).
Scheme 99. Coupling Reaction between (S)-180 and 1,1-Diphenylethanol 185.
N
Cbz
I
(S)-180
HO N
Cbz
OBase, Solvent
T(°C)
(S)-98185
Me Me
First of all, the conditions used by Ebnöther and Weber[128]
(Table 18, entry 1) turned
out to be unsuitable: the iodide derivative (S)-180 was prone to elimination leading to 186.
For nucleophilic substitution reactions, a wide range of solvents can be used but it should
be noted that apolar solvents tend to slow the reaction rate strongly. Therefore, the next
attempt was accomplished in a polar solvent, N-methyl-2-pyrrolidone (NMP) (entry 2), but
also resulted in the elimination product 186. Then, the use of a less basic base, KOH, was
tested in DMF and DMSO, affording the formate 187 and the elimination product 186
respectively (entries 3 and 4).
Table 18. Attempted Coupling Reaction with (S)-180.
Entry Base Solvent Temp (°C) t (h) Result
1 NaH Toluene 110 17
2 NaH NMP 100 13 186
3 KOH DMF 80 17 NCbz
O H
O
187
4 KOH DMSO 80 15 186
NCbz
186

110
Another strategy was attempted involving O-H insertion reaction of -diazo ketone
171 with tertiary alcohol 185 in the presence of indium triflate as a catalyst (Scheme
100).[140]
However, In(OTf)3 acted as an Lewis acid on the alcohol to give 189. The use of
milder Lewis acids such as ZnBr2, CuCl2 and FeCl3 led to similar results.
Scheme 100. Attempted O-H Insertion of Diazoketone 171 to Alcohol 185 Using Various
Lewis Acids.
N N2
OHO
Cbz
cat. Lewis acid
PhMe, RT
N
OCbz
O
(S)-171 185
189
188
Me Me
The last attempt was the formation of the hindered ether from the pyrrolidine
derivative 172 by using Ebnöther’s conditions (Scheme 101). Indeed, as shown by the
relative rates,[151]
the stabilization of the SN2 transition state with a carbonyl group is much
more effective than with a simple alkane or a benzene ring. However, after 16 h at reflux in
toluene in the presence of sodium hydride, 172 was returned.
Scheme 101. Attempted Coupling Reaction from 172.
Cl
O
ClMe Cl
Relative rate of SN2 reactions of alkyl
chlorides with the iodide ion
200200 100 000
N Cl
O
HO
Cbz
NaH,PhMe
110 °C, 16 hN
OCbz
O
(S)-172 185 188
Me Me

111
Since the coupling of the unstable chloroethylpyrrolidine 151 with a tertiary alcohol
is reported to lead to good yields,[128,130]
we decided to optimise this route.
II.1.3.5.2 Formation of the Ether Linkage from Chloroethylpyrrolidine (S)-151 and
Isolation of Clemastine
Enantiopure chloroethylpyrrolidine hydrochloride (S)-151HCl and enantiomerically
enriched tertiary alcohol (S)-97i (84:16 e.r.) were prepared for the challenging formation of
the hindered ether by deprotonating by free-basing (S)-151 with KOH[152]
(used
immediately) and deprotecting (S)-97i with sodium hydride (Scheme 102). Heating (S)-151
and the resulting sodium alkoxide of (S)-97i together at reflux in toluene for a period of 16
h returned a mixture of isomers: 15 % of the azepanes 190 and 48 % of the
diastereoisomers of 98.
Scheme 102. Formation of the Ether Linkage Resulting in the Synthesis of Clemastine as a
Diastereoisomeric Mixture.
PhMe, 110 °C,16 h
NCl
. HClMe
Converted to its free base form
N
Me
,NaH
Ph
Cl
ONa
98
190
N
Me
O
Cl
NO
Me
Cl
190, 15 %
Cl
HOMe
Cl
Me
s s
N
Me
O
Cl
Me
s R
48 % (ca 84:16)
(S,S)-98 (S,R)-98
(S)-151�HCl
(S)-97i
Me
[]D31 = -37.4 (c 2.04, EtOH)
[lit.128 []D20 = -33.7 for (S,S)-isomer and
-58.8 for (S,R)-isomer (c 2.00, EtOH)]
178+

112
The formation of 190 presumably arises through participation of the amine N in the
substitution to form 178+, followed by expansion of the resulting bicyclic cation. However,
as shown previously when N participation was discouraged by use of the Cbz-protected
iodopyrrolidine (S)-180 we observed no coupling of any sort, suggesting that participation
of the amine N is involved in the pathways to both products and that anchimeric assistance
to departure of Cl- is essential to the method.
The presumed ca. 84:16 mixture of diastereoisomers resulting from coupling
enantiopure (S)-151 with (S)-97i of 84:16 e.r. gave an []D of 37.4, confirming after
comparison with literature data ([α]D20
33.7 (c 2.00, EtOH) for (S,S)-isomer) [128]
that
(S,S)-clemastine has been synthesised as major isomer. The (S,S)-stereochemistry of the
product furthermore confirms the invertive nature of the rearrangement.
II.1.3.5.3 Isolation of Clemastine Fumarate
The mixture of diastereoisomers of 98 displayed a single set of peaks by 1H and
13C
NMR in both CDCl3 and CD3OH (Figure 5), and no separation into diastereoisomers was
observed by HPLC on a variety of chiral and achiral stationary phases. However, on
addition of 1 equiv. fumaric acid (Scheme 103), an 87:13 mixture of diastereoisomeric
fumarate salts 98H+
•fum- was evident by
1H NMR in CDCl3 for the set of peaks relating to
the -CH2 to the oxygen (Figure 6). This mixture was recrystallised to constant melting
point and []D, resulting in crystals which contained, by NMR, 10 % of the minor
diastereoisomer. The filtrate, by contrast, was a ca. 1:1.5 mixture of the two
diastereoisomers by 1H NMR (Figure 7).
Scheme 103. Isolation of Clemastine Fumarate 98H+
•fum-.
N
Me
O
Cl
Me
s s
N
Me
O
Cl
Me
s R
(S,S)-98
(S,R)-98
1. Fumaric acid, EtOH
2. Recrystallisation
N O
Cl
Me
s s
H Me
O2CCO2H
(S,S)-98H+fum-

113
Figure 5. 1H NMR and
13C NMR in CDCl3 of the Mixture of Diastereoisomers of
Clemastine 98.
N
Me
O
Cl
H H
MeN
Me
O
Cl
H H
Me
(S,S)-98 (S,R)-98

114
Figure 6. Sets of Peaks Assigned to –CH2 to the Oxygen of the Mixture of
Diastereoisomeric Fumarate Salts 98H+
•fum-.
Figure 7. Sets of Peaks Assigned to –CH2 to the Oxygen of Clemastine Fumarate
98H+
•fum- and the Filtrate after Recrystallisation.
After recrystallisation
crystalline powder of (S,S)-
clemastine fumarate
98H+
•fum-
powder remaining
in solution

115
The polarimetric and melting point data for the fumarate salts, and corresponding
literature values,[128]
are shown in Table 19. These are fully consistent with the hypothesis
that the initial coupling produces principally (S,S)-clemastine contamined with (S,R)-
clemastine, but that recrystallisation removes the minor diastereoisomer and returns a pure
sample of (S,S)-clemastine.
Table 19. Physical Data for Clemastine Fumarate 98H+
•fum-.
Compound m.p./°C m.p. (lit)/°C []D28c
[]D20
(lit.)c
(S,S)+(S,R)-98H+fum
- a 170-172
177-178 (S,S)
159-160 (S,R) 17.3
16.9 (S,S)
32.8 (S,R)
(S,S)-98H+fum
- b 176-177 177-178 17.1 16.9
Therefore, the first enantioselective synthesis of the antihistamine agent clemastine
98, as its (S,S)-stereoisomer, has been achieved by ether formation between a proline-
derived chloroethylpyrrolidine (S)-151 and an enantiomerically enriched tertiary alcohol
(S)-97i resulting from the carbamate (S)-95i by invertive aryl migration on lithiation. The
(S,S)-stereochemistry of the product confirms the invertive nature of the rearrangement.
This is in contrast to the retentive rearrangement of ureas.
In situ IR spectroscopy and modelling work have been performed to establish the
origin of this stereodivergent behaviour.
aFumarate salt of this mixture, which
1H NMR indicates contains an 87:13
diastereoisomeric ratio; bRecrystallised fumarate salt with 10% minor diastereoisomer
by 1H NMR.
c c = 2.0 (MeOH).

116
II.2 Spectroscopic and Computational Studies of the Mechanism
II.2.1 In Situ IR Spectroscopy
The presence of the carbonyl group makes carbamates particulary suitable for
mechanistic studies by in situ IR spectroscopy.[62,153]
Recently, for example, O’Brien
showed that the course of the lithiation of N-Boc piperidine (νC=O = 1695 cm-1
) with sec-
BuLi/TMEDA, ()-sparteine or (+)-sparteine could be followed by in situ infrared
spectroscopy. They observed directly the “pre-lithiated complex” (νC=O = 1675 cm-1
)
between the substrate and lithiating agent in TBME at -78 °C and the lithiated complex
(νC=O = 1644 cm-1
).[153d]
The existence of a prelithiation complex had previously been
deduced by kinetic studies on a related compound.[154]
Beak also monitored the lithiation of
an N-Boc allylamine through in situ IR with n-BuLi/()-sparteine in toluene at -73 °C.[153b]
They observed a similar reaction pathway involving a “prelithiated complex” at 1675 cm-1
and a lithiated complex at 1640 cm-1
. They also showed that addition of n-BuLi to N-Boc
allylamine generated a stable carbamate-n-BuLi complex at 1675 cm-1
and upon addition
of ()-sparteine deprotonation occured as evidenced by the appearance of the peak at 1640
cm-1
.
Previous studies in this area, of the rearrangements of ureas, had failed to find
evidence of either a pre-lithiation complex or of the presence of the postulated
dearomatised intermediate 58 with a benzenoid ring migrating (see Section I.2.2).[62]
We
therefore undertook detailed studies of the rearrangement of 95i by in situ IR spectroscopy.
A mixture of 95i and THF in a three-necked flask equipped with a ReactIR probe was
cooled to -60 °C. Once the temperature has stabilized, spectroscopic analysis was
commenced at a rate of one scan every 30 s: 95i showed a C=O absorbance at 1698 cm-1
(Scheme 104; Figure 8). Treatment with sec-BuLi (1.1 equiv.) transformed this signal
directly into one at 1642 cm-1
and one at 1333 cm-1
arising from C-O and C-N stretches
within the lithio carbamate function of the rearranged product 96iLi and addition of MeOH
returned the rearranged carbamate 96i with a C=O stretch of 1735 cm-1
. A similar peak at
1646 cm-1
for the intermediate 96iLi was observed with LDA (1.5 equiv.).

117
Scheme 104. Intermediate Detected in the Rearrangement of 95i.
N O
Me
O Me
Cl
1. s-BuLi (1.1 equiv.),
95i
MeHN O
O
Cl
MeMeN O
OLi
Cl
Me
2. MeOH
THF,-60 °C 96i96iLi
1698 cm-1 1735 cm-1
1642 cm-1
+ 1333 cm-1
Figure 8. In situ React IR Monitoring the Rearrangement of Benzylcarbamate 95i in THF
with sec-BuLi at -60 °C. (a) Three-dimensional plot of absorbance versus wavenumber
versus time. (b) Several sequential two-dimensional infrared spectra at various stages of
the reaction.
Arylcarbamate
95i in THF
sec-BuLi added
Quenched 1698cm
-1: 95i
1735cm-1
: 96i 1642cm-1
: 96iLi
(a)
(b)
1333cm-1
: 96iLi
1642cm-1
: 96iLi
Arylcarbamate in THF
sec-BuLi added
Quenched

118
Evidence that the absorption at 1642 cm-1
assigned to the product anion 96iLi was
obtained by treating 96i with sec-BuLi in THF: the spectrum assigned to 96iLi reappeared
(Scheme 105; Figure 9).
Scheme 105. Intermediate Detected in the Retreatment of 96i.
s-BuLi (1.1 equiv.),
MeHN O
O
Cl
Me
96i
MeN O
OLi
Cl
Me
96iLi
THF,-60 °C
Figure 9. In situ React IR Monitoring the Retreatment of Rearranged Carbamate 96i in
THF with sec-BuLi at -60°C.
Rearranged
carbamate 96i
in THF
sec-BuLi
added
Quenched 1735cm
-1: 96i
1642cm-1
: 96iLi

119
In the absence of THF, the rearrangement was much slower. Treatment of 95i in
TBME at -60 °C with sec-BuLi (2.0 equiv.) gave transiently an absorbance at 1686 cm-1
,
which then gave way over a period of minutes to a band at 1649 cm-1
(Scheme 106; Figure
10). Quenching the reaction with MeOH regenerated starting material 95i, we therefore
assigned the absorbance at 1649 cm-1
to 95iLi, and proposed that the transient absorption
at 1686 cm-1
is the “prelithiated complex” 95i•RLi.
Scheme 106. Intermediates Detected in the Lithiation of 95i.
N O
Me
O Me
Cl
1. s-BuLi (2.0 equiv.),
95i
N O
Me
O Me
Cl
LiR
N O
Me
O Li
Cl95iRLi 95iLi
TBME, -60 °C
R1698 cm-1 1649 cm-11686 cm-1
Figure 10. In situ React IR Monitoring the Lithiation of Benzylcarbamate 95i with sec-
BuLi in TBME at -60 °C.
Arylcarbamate
95i in TBME
Addition of
sec-BuLi
Quenched
1686cm-1
: 95i•RLi 1649cm-1
: 95iLi
1698cm-1
: 95i

120
When 95i was treated with sec-BuLi (2.0 equiv.) in toluene at -60 °C, only the absorbance
at 1686 cm-1
was formed, indicating that under these conditions, complexation may take
place, but not deprotonation (Scheme 107; Figure 11). After a reaction time of 30 min,
addition of TMEDA (1.0 equiv.) returned the starting material 95i, probably by
decomplexation of the species 95i•RLi.
Scheme 107. Intermediates Detected in the Lithiation of 95i.
N O
Me
O Me
Cl
1. s-BuLi (2.0 equiv.),
PhMe,-60 °C
95i
N O
Me
O Me
Cl
LiR
95iRLi2. TMEDA
1698 cm-11686 cm-1
Figure 11. In situ React IR Monitoring the Lithiation of Benzylcarbamate 95i with sec-
BuLi in Toluene at -60 °C.
Arylcarbamate
95i in toluene
Addition of
sec-BuLi
TMEDA
added
1698cm-1
: 95i
1686cm-1
: 95i•RLi 1649cm
-1

121
A similar outcome was obtained using LDA (3.5 equiv.) in TBME at -60 °C
displaying a band at 1690 cm-1
. After stirring for 1 h, addition of CD3OD to the reaction
mixture resulted in the recovery of starting material 95i with no deuterium incorporation
(Scheme 108; Figure 12).
Scheme 108. Intermediates Detected in the Lithiation of 95i.
1698 cm-1
1690 cm-1
N O
Me
O Me
Cl95i
LDA (3.5 eq.),TBME, -60 °C
N O
Me
O Me
Cl
LiNN
Li iPr
iPrPri
Pri
S
95iRLi
Figure 12. In situ React IR Monitoring the Lithiation of Benzylcarbamate 95i with LDA in
TBME at -60 °C.
Arylcarbamate
95i in TBME LDA
added
Quenched
with CD3OD
1690cm-1
: 95i•RLi
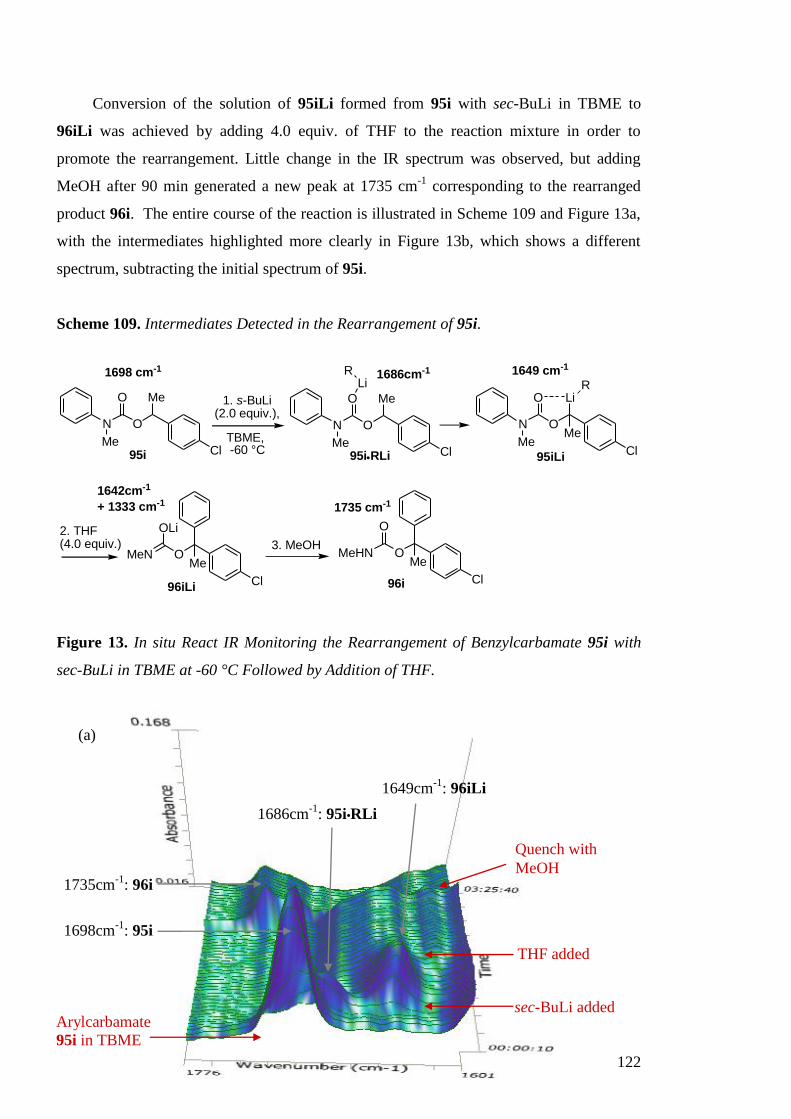
122
Conversion of the solution of 95iLi formed from 95i with sec-BuLi in TBME to
96iLi was achieved by adding 4.0 equiv. of THF to the reaction mixture in order to
promote the rearrangement. Little change in the IR spectrum was observed, but adding
MeOH after 90 min generated a new peak at 1735 cm-1
corresponding to the rearranged
product 96i. The entire course of the reaction is illustrated in Scheme 109 and Figure 13a,
with the intermediates highlighted more clearly in Figure 13b, which shows a different
spectrum, subtracting the initial spectrum of 95i.
Scheme 109. Intermediates Detected in the Rearrangement of 95i.
N O
Me
O Me
Cl
1. s-BuLi (2.0 equiv.),
95i
N O
Me
O Me
Cl
LiR
N O
Me
O Li
Cl
Me
95iRLi 95iLi
2. THF(4.0 equiv.)
MeHN O
O
Cl
Me
96i
MeN O
OLi
Cl
Me
96iLi
3. MeOH
TBME, -60 °C
R1698 cm-1 1649 cm-1
1686cm-1
1642cm-1
+ 1333 cm-1 1735 cm-1
Figure 13. In situ React IR Monitoring the Rearrangement of Benzylcarbamate 95i with
sec-BuLi in TBME at -60 °C Followed by Addition of THF.
Arylcarbamate
95i in TBME
sec-BuLi added
THF added
Quench with
MeOH
1698cm-1
: 95i
1735cm-1
: 96i
1649cm-1
: 96iLi
1686cm-1
: 95i•RLi
(a)

123
It is notable that νC=O for lithiated carbamate 95iLi and the product carbamate 96iLi
are very similar. However, the spectrum of 96iLi also showed a significant peak at 1333
cm-1
(Figure 8b) which is absent from the spectrum of 95iLi. As shown in Figure 13c,
indeed this peak grew progressively as time progressed allowing us to monitor the progress
of the rearrangement pathway. No other intermediates were observed along the reaction
pathway.
Arylcarbamate in TBME
2eq sBuLi added
2eq THF added
4eq THF added
1h30 after THF added
Quenched
(c) 1333 cm
-1 : 96iLi
1649cm-1: 96iLi 1686cm-1: 95i•RLi
1735cm-1: 96i
1698cm-1: 95i
(b)

124
II.2.2 Computational Studies
In order to understand the experimental observation of the 1,4-aryl transfer, Mark Vincent
and Ian Hillier at the University of Manchester have achieved some computational studies.
Appropriate models were constructed involving the benzylic carbamate 95a in its
metallated form 95aLi and the base LDA, with THF as solvent. The potential energy
surface for the reaction was obtained using electronic structure calculations employing
DFT. Properly characterised minima and transition structures (TS) were located at the
B3LYP/6-31G level, giving both vibrational frequencies and starting structures for
optimisation at the B3LYP/6-31++G** level. The model (Scheme 110) involves one
explicit LDA dimer coordinated to the carbonyl oxygen of the carbamate 95a•RLi (A).
They assume that deprotonation to 95aLi gives the reactant structure (B) in which the
carbanion has a solvated lithium ion bound to it.
Their strategy was to search for the transition structures, and to follow these back to the
reactants, and forward to the products.
They found three different reactant configurations, R2 leading to retention of configuration
and R3 and R4 leading to inversion, which were higher in energy than the lowest energy
reactant R1 (Scheme 110). In this latter structure (Fig. 14 (a)), the Li+ is clearly bound to
the carbanion centre (at a distance of 2.20 Å). In the three reactant configurations this
distance is increased considerably in order to free up the carbanion lone pair for
nucleophilic attack.

125
Scheme 110. Schematic Representation of the Structures Along the Computed Reaction
Pathways.
MeN O Me
O H
95aRLi (A) 95aLi (B)
Base
THF
LiR2N
NR2
Li
THF
MeN O Me
O Li
Base (THF)3
MeN O
O
Base
R1
MeN
Li
Me
O
O
(THF)3
Base
MeN O
Base
Li (THF)3
O
MeN O
BaseO
Li (THF)3
R2 R3 R4
MeN O
O
Me
Base
Li
(THF)3 P2 P4
TS2TS3 TS4
Rotation
Movement of
Li+(THF)x off C-
inversionretention inversion
MeN O
O
Me
Base
Li
Me
(THF)n
Me Me
(THF)n-Li
Both transition structures TS2 and TS3, leading to retention and inversion respectively
involve the interaction of the solvated Li+ species with the benzyl ring. Structure TS3
(Figure 14 (c)) differs from TS2 (Figure 14 (b)) in that the lithium ion is on the other side
of the phenyl group, and thus is unavailable for stabilising the developing charge on the
attacked ring. In structure TS3 the average distance of the Li to the benzyl ring is 3.32 Å,
while the length of the forming CC bond is 1.93 Å. This latter distance is shorter than in

126
transition structure (TS2) which leads to retention of stereochemistry. The transition
structure TS3 leads to inversion of stereochemistry (Figure 14 (c)) as the forming CC
bond is in the opposite direction to the original Li-carbanion bond. This is in contrast to
transition structure TS2 where the Li-phenyl interaction is in the same direction as the
original Li-carbanion bond and leads to retention of configuration.
In the third transition structure TS4 and corresponding reactant R4 (Figure 14 (d)) the
Li(THF)3+ is bonded to and in the plane of the carbamate group which requires a 1,2 shift
of the Li+. This Li
+ shift from the carbon to the in-plane oxygen lone pair requires
conformational changes in the minimum energy reactant structure so that the Li+ is now in
the plane of the carbamate and adjacent to the oxygen lone pair. We note that the other lone
pair on oxygen is not available, being conjugated with the carbonyl. This conformation has
the carbanion and the remote phenyl group neatly arranged for reaction. Indeed, the
carbanion is slightly pyramidal as is the phenyl carbon that is attacked and thus the bond
has started to form. This mechanism gives rise to inversion as moving the lithium into the
plane of the carbamate, prior to its transfer to the oxygen atom results in the Li-C bond
pointing away from the remote phenyl group.
Figure 14. Optimal structures along the reaction pathways. For clarity LDA has been
omitted.
(a) Lowest energy reactant (R1)

127
(b) Reactant (R2) and transition structure (TS2) leading to retention of stereochemistry
(c) Reactant (R3) and transition structure (TS3) leading to inversion of stereochemistry

128
(d) Reactant (R4) and transition structure (TS4) leading to inversion of stereochemistry
If we now consider the energetics of the three reactions, the transition structures for
retention (TS2), inversion via O (TS4) and inversion via phenyl (TS3), lie respectively 81,
62 and 71 kJ mol-1
, above the lowest energy reactant structure (R1) (Table 20). However,
the corresponding three reactant structures (R2, R3 and R4) are of considerable higher
energy than this lowest energy structure (R1) so that the barriers for reaction from these
structures are considerably lower, all being below 35 kJ mol-1
. Thus, the calculations
clearly predict that inversion via carbamate oxygen is the lowest energy route. However,
we note that the transition structure for inversion via benzyl ring is only 8 kJ mol-1
higher
in energy, so that inversion by both mechanisms is clearly feasible.

129
Table 20. Relative Energies (kJ mol-1
) of Stationary structures.
Stereochemistry Structure Free Energy
Lowest energy minimum R1 0.0
Retention (via Ph)
R2 81.5
TS2 81.3
P2 -66.0
Inversion (via O)
R4 45.7
TS4 62.4
P4 -67.7
Inversion (via Ph)
R3 36.0
TS3 70.7
P3 -48.3
They also determined the harmonic frequencies for the various minimum energy species
which the computations have identified along the reaction pathway of the benzyl
carbamate. In the neutral reactant, the C=O stretch is computed to occur at 1693 cm-1
, close
to the experimental band at 1698 cm-1
. The addition of base leads to a peak at 1686 cm-1
,
which correlates with the peak computed at 1672 cm-1
for structure A (Scheme 14),
corresponding to a “pre-lithiated complex” having the LDA dimer coordinated to the
carbonyl oxygen. The structure R1 is predicted to have strong peaks at 1638, 1642 and
1650 cm-1
, which are mixtures of C=O and C-H ring modes. Experimentally, a peak at
1646 cm-1
is seen to grow following the addition of LDA to the benzyl carbamate 95i
which we thus assume corresponds to the computed structure R1. For the product anion in
which the solvated THF is coordinated to the nitrogen atom of the carbamate, we predict
intense adsorptions at 1335 and 1652 cm-1
, values which are in excellent agreement with
the peaks observed experimentally at 1333 and 1642 cm-1
. After protonation of the anion,
the computed carbonyl stretch is 1745 cm-1
, close to the experimental value of 1735 cm-1
.

130
II.3 Arylation of Lithiated Carbamates by Intramolecular Aryl
N to C Aryl Migration: Scope, Stereoselectivity and Mechanism
II.3.1 N to C Aryl Migration in Lithiated N-aryl-O-Allylcarbamates
As mentioned in Section I.2.3, recent studies within the group have shown that N-allyl-
N’-aryl ureas may be lithiated to nitrogen and undergo a related rearrangement with
transfer of the aryl ring from N to the allylic carbon.[66]
We therefore considered the
possibility of extending the scope of the rearangement to lithiated N-aryl-O-allyl
carbamates.
II.3.1.1 Synthesis and Aryl Migration of a Cinnamyl Carbamate
In a preliminary experiment to assess the reactivity of lithiated N-aryl-O-
allylcarbamates towards rearrangement, the cinnamyl derivative 193 was made by standard
methods used within the group.[55]
The cinnamyl alcohol 191 was combined with phenyl
isocyanate, followed by methylation to give the desired carbamate 193 in 89 % yield over
two steps (Scheme 111).
Scheme 111. Synthesis of Cinnamyl Carbamate 193.
HO Ph
191
PhNCO,NEt3,CH2Cl2
RT, 15 hO Ph
192
NH
O1. NaH,DMF, 0 °C,30 min
2. MeI, RT,15 h
O Ph
193
N
O
Me
The rearrangement of 193 was then attempted under different conditions. Firstly, in
the presence of sec-BuLi in a mixture of THF and DMPU at -78 °C, a significant amount
of starting material remained and the cinnamyl alcohol 191 was recovered resulting from
nucleophilic attack on C=O after quenching the reaction mixture with MeOH. Under these
conditions, it appeared that sec-BuLi was acting as a nucleophile rather than lithiating at
the required centre. Addition of DMPU has been shown previously to accelerate related
rearrangements and cyclisations.[61]

131
Deprotonation of carbamate 193 by a less nucleophilic base, LDA (pKa ≈ 36),[155]
was therefore attempted. However, the TLC and 1H NMR showed a complex mixture of
products with negligible desired product.
Finally, the cinnamyl carbamate 193 was treated with sec-BuLi (2.5 equiv.) in THF
at -78 °C for 1 h. The principal product of the reaction (41 % yield) was the aryl ketone
197, a compound which can arise only by migration of the N-phenyl ring from N to C.
Evidence that N to C migration of an aryl ring is involved in the formation of 197 is
supported by the identification of a by-product 196 (9 %) from which 197 can be derived
by hydrolysis (Scheme 112).
Scheme 112. Nto C Aryl Migration of Cinnamyl Carbamate 193.
N O
O
Ph
Me
Ph
O1. s-BuLi (2.5 eq.), THF, -78 °C,1 h
2. MeOH
193 197, 41 %(Z)-196, 9 %
Ph
O O
NHMe
A plausible route to 197 is shown in Scheme 113: deprotonation to give the
allyllithium 193Li, followed by aryl migration to form 195, which undergoes a second
deprotonation to give a cinnamyllithium species 196Li, and -reprotonation on work-up to
yield the (Z)-vinyl carbamate 196. The fact that 196 was formed with Z-selectivity suggests
that 196Li exists as an endo, exo-3-allyl complex.
[66,156]
Scheme 113. Proposed Mechanism for the Formation of 197.
N O
O
Me
Phbase
193
H Ph
O
O
N
LiLn
Me
rearranges
NO
OMe
HPh
LiLn
194
H Ph
O OLi
NMe
195
Ph
O
O
N
LiLn
Me Li
MeOHPh
O O
NHMe
(Z)-196
Ph
O
197
baseMeOH
193Li
196Li
endo
exo

132
The second deprotonation could be avoided by increasing the substitution at the
position, thus creating a quaternary centre devoid of any labile protons after migration.
Therefore, we replaced the cinnamyl derivative 193 with an -methylated compound 201.
II.3.1.2 Synthesis and Aryl Migration of -Methyl Cinnamyl Carbamates
The -methyl cinnamyl carbamate 201 was prepared in 72 % over three steps using
the same method previously described, from -methyl cinnamyl alcohol 199 prepared by
addition of methyllithium to cinnamyl aldehyde 198 (Scheme 114).
Scheme 114. Synthesis of -Methyl Cinnamyl Carbamate 201.
HO Ph
NH
O
O
Ph N O
O
Ph
Me
PhNCO, NEt3, CH2Cl2
RT, 15 h
1. NaH, DMF,0 °C, 30 min
2. MeI, RT, 15 h
199
200
201, 72 % over 3 steps
Me
Me
Ph
198
H
O
1. MeLi, Et2O,-78° C, 2 h
2. NH4Cl
Me
Under the conditions previously used for deprotonation of cinnamyl carbamate 193,
sec-BuLi (2.5 equiv.) in THF at -78 °C, the resulting rearranged product underwent a
carbolithiationof the conjugated alkene[106d]
by sec-BuLi to give 202 in 23 % yield as a 1:1
diastereomeric mixture. The tertiary alcohol 203 was also isolated in poor yield (6 %)
resulting from N to C aryl migration followed by an in-situ carbamate function
deprotection (Scheme 115).
Scheme 115. Nto C Aryl Migration of -Methyl Cinnamyl Carbamate 201 upon Lithiation
with sec-BuLi.
N O
O
Ph
Me201
1. s-BuLi (2.5 eq.), THF,-78 °C, 1 h
2. MeOH
MeHN O
O
Ph
202, 23 %
Me Me
HO Ph
Me
203, 6 %

133
Instead, deprotonation with LDA (2.0 equiv.) in THF at -78 °C gave after 1 h a
compound identified as 204 in the NMR spectrum of the crude reaction mixture (Scheme
116). However, this material proved extremely unstable to purification on silica and
alumina, presumably due to the ready formation of a highly stabilized cation.
Scheme 116. N to C Aryl Migration of -Methyl Cinnamyl Carbamate 201 upon Lithiation
with LDA.
N O
O
Ph
Me 201
1. LDA (2.0 eq.), THF,-78 °C, 1 h
2. MeOH
MeHN O
O
Ph
204
Me Me
It was anticipated that deprotection of the crude carbamate 204 would result in a
product with greater stability.
It was reported that using DIBAL-H a carbamate could be reduced to give the desired
alcohol product.[157]
However, using this method, a side product 206 was isolated shown to
be an elimination product. It is likely that formation of the product was promoted by the
Lewis acidic aluminium in the DIBAL-H (Scheme 117).
Scheme 117. Attempted Deprotection of 204 by DIBAL-H.
MeHN O
O
Ph
MeDIBAL-H, THF
reflux or RT, 2 h
AlO Ph
Me
PhPh
204206205
-H2O
i-Bu2
An alternative method for the cleavage of the carbamate function would be to reflux
the product 204 in an alcohol in the presence of sodium carbonate.[64]
However, attempt to
deprotect the carbamate employing n-butanol or methanol at reflux or at room temperature
gave, in addition to the elimination product 206 in very low yield, the two isomers 207/208
and 209/210 respectively, resulting from the incorporation of the alcohol and expulsion of
the carbamate moiety (Scheme 118).

134
Scheme 118. Attempted Deprotection of 204 by Refluxing in Alcohol.
MeHN O Ph
O Me
204
Ph Ph
BuO Me
Ph Ph
Me OBu
Ph Ph
Me OMe
Ph Ph
MeO Me
207 208
209 210
40 % over 2 steps (1/1.3)
71 % over 2 steps (1.5/1)
n-BuOH,Na2CO3,reflux, 2 h
MeOH,Na2CO3,50 °Cor RT, 2 h
As an alternative, hydrolysis of the carbamate’s N-nitroso derivative 211 was
envisaged using the same method previously described in the group for ureas.[56]
However,
treatment of the rearranged product 204 with tert-butyl nitrite to make the N-nitroso
derivative 211 was unsuccessful: only the starting material 204 was recovered (Scheme
119).
Scheme 119. Attempted Deprotection of 204 by Hydrolysis of Carbamate’s N-nitroso
derivative.
MeHN O
O
Ph
Me
204
N O
O
Ph
Me
NO
HO Ph
Me1. t-BuONO
2. LiOH, H2O,THF
211 203
Me
, 48 hCH2Cl2, 24 h
As outlined in the mechanism in the Scheme 113, the rearrangement would generate
a lithiocarbamate species. Therefore, the nitrogen being more nucleophilic, N-nitrosation
could be carried out simply by terminating the rearrangement with an excess of tert-butyl
nitrite. Pleasingly, adding 6.0 equiv. of tert-butyl nitrite at the end of the reaction and
stirring the resulting basic reaction mixture at room temperature for 24 h afforded the
desired alcohol 203 in 68 % yield in a one pot reaction (Scheme 120).

135
Scheme 120. Synthesis of Tertiary Alcohol 203 by Adding t-BuONO at the End of the
Rearrangement Reaction.
N O Ph
O Me
Me
1. LDA (2.0 eq.), THF, -78 °C, 1 h
2. t-BuONO (6.0 eq.), 24 h, RT
HO Ph
Me
201 203, 68 %
II.3.1.3 Stereospecificity in the Aryl Migration
By starting with enantiomerically pure 201, we hoped to be able to form the
allyllithium intermediate as a single enantiomer which would rearrange faster than it could
racemise. Enantiomerically enriched benzylcarbamates may be lithiated and rearranged
stereospecifically, provided the conditions are carefully chosen, and carbamates closely
related to 201 have some degree of configurational stability (see Section I.3.4).
The first task was the preparation of (R)--methyl cinnamyl alcohol 199. Sharpless
Kinetic Resolution (SKR)[158]
allowed to access to the enantiopure alcohol (R)-199 in 40 %
yield and with 99:1 e.r..1 Subsequent conversion into the corresponding carbamate (R)-201,
using the same method previously described, was achieved in 89 % yield over 2 steps
(Scheme 121).
Scheme 121. Preparation of Enantiopure -Methyl Cinnamyl Carbamate (R)-201.
Ti(O-i-Pr)4 (1.0 eq.),(L)-(+)-DIPT (1.2 eq.),TBHP (0.6 eq.)
CH2Cl2, 4Å MS-20 °C, 3.5 h
HO Ph
Me
199
HO Ph
Me
(R)-199, 40 %, 99:1 e.r.
O Ph
Me
(R)-201, 89 %, 99:1 e.r.
N
O
Me
The successful reaction conditions used on the racemic material were tried on the
enantiopure starting material (R)-201 (Scheme 122; Table 21, entry 1). However, not
surprisingly, these conditions caused racemization. Interestingly, the deep red colour of the
reaction mixture indicates the high degree of ion separation (SSIP), thus decreasing the
1 Chiralcel OD-H was used as stationary chiral phase to determine the []D. The absolute stereochemistry
was confirmed by comparison with literature data[159]

136
configurational stability of the allyllithium. Rearrangements in less coordinating solvents
tend to return higher enantiomeric excesses.[160]
Therefore, the reaction was repeated in
Et2O (THF with a dipole moment of = 1.75 [D] has a stronger donor capacity compared
to that of Et2O ( = 1.15 [D])[161]
) at -78 °C which led to a yellow solution presumably
arising from the formation of a closer contact ion pair (CIP) (entries 2). However, it did not
reach completion in 2 h and gave a product with a low enantiomeric excess. Tellingly, the
recovered starting material was partially racemised too, suggesting that the lithiated
intermediate is not configurationally stable under the conditions of the reaction. Slowly
raising the temperature to -40 °C allowed the reaction to reach completion but still gave
essentially racemic product (entry 3). The challenge was therefore to find conditions under
which rearrangement is accelerated while racemization is decelerated. Diamines such as
()-sparteine, (+)-sparteine surrogate,[162]
or TMEDA are known to slow down the rate at
which organolithiums racemise.[163]
Indeed adding either ()-sparteine or TMEDA (1.0
equiv.) to the rearrangement reaction in Et2O resulted in a product retaining some degree
of enantiomeric enrichment (entries 4 to 6) but as a racemate with 2.0 equiv. of ()-
sparteine (entry 7) or in the presence of (+)-sparteine surrogate (entry 8). However, the
maximum achievable was little more than 60:40, suggesting that 80 % of the material is
racemic.
Scheme 122. Stereospecificity in the Aryl Migration of (R)-201.
O Ph
Me
(R)-201, 89 %, 99:1 e.r.
N
O
Me
1. LDA (2.0 eq.) (Table 21)
2. MeOHO Ph
(S)-204
MeHN
O Me

137
Table 21. Conditions for the Stereospecificity in the Aryl Migration of (R)-201.
Entry Solvent/Additive Temp. (°C) Time (h) e.r.a
1 THF -78 1 50:50
2 Et2O -78 2 46:54b
3 Et2O -78 to -40 3 46:54
4c Et2O + ()-sparteine (1 eq.) -45 2 37:63
5 Et2O + ()-sparteine (1 eq.) -60 3 39:61
6 Et2O + TMEDA (1 eq.) -45 2 38:62
7 Et2O + ()-sparteine (2 eq.) -60 3 48:52d
8 Et2O + (+)-sparteine
surrogate (1 eq.) -45 2 48:52
d
()-sparteine
N
HN
H
N
HN
Me
(+)-sparteine surrogate
These results point to the importance of the incorporation of the lithium cation into a
dense chelate complex which hampers the cation from changing the enantiotopic faces of
the anion.
An alternative approach for the enantioselective rearrangement could make use of a
chiral base or an alkyllithium/()-sparteine complex, the aim being to induce kinetic
resolution of the chiral starting material or dynamic kinetic or thermodynamic resolution of
the intermediate organolithium.[175]
However, racemic products were also formed when
racemic starting materials were deprotonated in the presence of chiral lithium amide 212Li
in THF or Et2O (Table 22, entries 1 and 2),[66]
or in the presence of sec-BuLi/()-sparteine
complex in Et2O at -78 °C (entry 3) or at -60 °C (entry 4) or in the presence of n-BuLi/()-
sparteine in toluene at -78 °C followed by the addition of THF (4.0 equiv.) to promote the
rearrangement (entry 5).
a e.r. determined from crude reaction.
b Remaining starting material: 77:23 e.r.
c LDA added
to a solution of carbamate and ()-sparteine. d Remaining starting material: 48:52 e.r.

138
Table 22. Enantioselectivity in the Aryl Migration of 201.
Entry Base or Additive Solvent T (°C) t (h) e.r.
1 Ph NH
Ph .n-BuLi
212
THF -78 1 50:50
2 Ph NH
Ph .n-BuLi
212
Et2O -78 1 50:50
3 ()-sparteine/s-BuLi (2.0 eq.) Et2O -78 15 49:51
4 ()-sparteine/s-BuLi (2.0 eq.) Et2O -60 15 49:51
5 ()-sparteine/n-BuLi (1.5 eq.)
then THF (4.0 eq.) PhMe -78 3 45:55
Recently, Takeda et al. examined the chirality transfer in a [2,3]-Wittig
rearrangement of 213 as a new tool for evaluation of the effect of conjugative electron-
withdrawing groups and -anion-stabilising heteroatom substitutents X on the
configurational stability of a chiral carbanion through a double bond (Scheme 123).[160]
Scheme 123. [2,3]-Wittig Rearrangement of 213 as a Tool for Estimation of the Effect of
Group X on the Configurational Stability of Chiral Carbanions.
X Ph
O
n-C5H11 X Ph
HOn-C5H11
base
solventRT
X = Me, Ph, SiMe3, CN,SO2Ph, P(O)(OEt)2, P(O)Ph2
213 214
He suggested that in the case of conjugating groups, such as a phenyl group,
racemization occurs depending on the solvent (Table 23, entries 4 to 9). In contrast, when
X is an anion-unstabilizing group such as a methyl group, little racemization was observed
regardless of the solvent used (entries 1 to 3).

139
Table 23. [2,3]-Wittig Rearrangement of 213.
Entry X Solvent Base 214, Yield
(%)
e.e. (%)
214 ent-214
1 Me 1,4-dioxane n-BuLi 77 94 98
2 Me Et2O n-BuLi 79 95 100
3 Me THF n-BuLi 87 93 96
4 Ph 1,4-dioxane n-BuLi 70 84 92
5 Ph Et2O n-BuLi 64 60 71
6 Ph THF LDA 61 3 4
7 CN 1,4-dioxane LDA 31 8 9
8 CN Et2O LDA 31 8 6
9 CN THF LDA 40 2 3
Consequently, it was worth examining the stereospecific rearrangement of -methyl
cinnamyl carbamates 217 bearing an electron-donating group (-OMe) on the phenyl group
which would increase the configurational stability of the chiral carbanion intermediate by
minimizing its delocalisation through the double bond.
(R)--Methyl cinnamyl alcohol 216 was made by Sharpless Kinetic Resolution
(SKR)[158]
in 37 % yield and with 97:3 e.r., which was converted to its carbamate
derivative (R)-217 by reaction with phenyl isocyanate and methylation with sodium
hydride and methyl iodide in 63 % yield over two steps (Scheme 124).
Scheme 124. Preparation of Enantiomerically Enriched -Methyl Cinnamyl Carbamate
(R)-217.
HO
N O
O1. PhNCO, NEt3, CH2Cl2, RT, 15 h
2. (i) NaH, DMF,0 °C, 30 min(ii) MeI, RT, 15 h
216, 87 %
(R)-217, 63 % over 2 steps, 97:3 e.r.
Me
Me
215
H
O
1. MeLi, Et2O,-78 °C, 2 h
2. NH4ClOMe OMe
OMe
HO
(R)-216, 37 %, 97:3 e.r.
Me
OMe
Ti(O-i-Pr)4 (1.0 eq.),(L)-(+)-DIPT (1.2 eq.),TBHP (0.6 eq.)
CH2Cl2, 4Å MS-20 °C, 3 days
Me

140
Scheme 125. Stereospecificity in the Aryl Migration of (R)-217.
N O
O
(R)-217, 97:3 e.r.
Me
OMeMe
O
O
MeHN
Me
OMe218
Table 24
Table 24. Conditions for the Stereospecificity in the Aryl Migration of (R)-217.
Entry Base Solvent Temp. (°C) Time (h) e.r.a
1 LDA THF -78 1 50:50
2b LDA Et2O -78 18 -
3b LDA Et2O -78 to -35 17 -
4 LDA Et2O/THF (2 eq.) -78 to -35 15 55:45
5 LDA Et2O -45 19 53:47
Firstly, in order to ascertain that the rearrangement of the carbamate 217 occurs, the
standard lithiation conditions were used yielding the desired rearranged carbamate 218
cleanly (Scheme 125; Table 24, entry 1). However, in Et2O at -78 °C, the carbamate failed
to rearrange (entry 2) as well as at higher temperature (entry 3) contrary to carbamate 201.
Two equivalents of THF (as lithium-coordinating additive) were required to promote the
aryl transfer (entry 4) but the stereochemistry is not retained under these conditions. In
order to make the rearrangement faster than the racemization of the organolithium
intermediate, the reaction was repeated at -45 °C, the rearranged product 218 was, however,
returned with a poor enantiomeric ratio (entry 5). Therefore, N to C aryl migration of the p-
methoxy analogue of 201 made no change to the previous results.
We conclude that the rearrangement of allylic carbamates cannot therefore be used to
make them in enantiomerically pure form. However, conversion of the material (S)-204
with low enantiomeric enrichment (Table 21, entry 4) to the allylic alcohol 203 by
nitrosation and base-promoted cleavage returned material whose optical rotation has been
compared with a reported value[164]
(Scheme 126). Both sign and magnitude is consistent
with the alcohol product of the reaction being of about 20 % optical purity and having (S)
a Determine from crude reaction.
b Starting material recovered.

141
absolute configuration. It appears therefore that the rearrangement of allylic carbamates,
like that of benzylic carbamates, proceeds with inversion of configuration.
Scheme 126. Confirmation of the Invertive Nature of the Aryl Migration in Allylic
Carbamates.
MeHN O
O
(S)-204, 37:63 e.r.
HO
(S)-203, 65 % over 2 steps, 37:63 e.r.
1. n-BuLi (2.0 eq.),THF, -78 °C, 30 min
2. t-BuONO (6.0 eq.),RT, 24 h
[]D25 - 1.5 (c 1.0, CHCl3)
[lit [164] []D20 - 5.33 (c 0.6, Et2O)]
Me Me
The use of ()-sparteine as a means of reducing the rate of racemization was explored
as a way of improving the results obtainable from the rearrangements of benzylic
carbamates described previously. Addition of ()-sparteine to the rearrangement of 95o
improved the e.r. of the product from 90:10 to 97:3, but at the expense of yield, while
addition of ()-sparteine to the less reactive 95i induced a remarkable change in the
mechanism of the reaction, which produced the 2-hydroxyamide 219, by a 1,2-acyl shift, in
80:20 e.r. (Scheme 127). The absolute configuration of 219 has not been determined but
Nakai et al. reported that alkyl N,N-diisopropylcarbamates underwent the 1,2-carbamoyl
migration with complete retention of configuration at the Li-bearing carbanion
terminus.[75c]
We can therefore assume that the configuration of 219 is (S).

142
Scheme 127. Effect of ()-Sparteine on the Rearrangement of Lithiated Benzylcarbamates.
N O
O Me
Me
MeHN O
O
1. LiTMP (2.5 eq.), Et2O, -78 °C to -35 °C,24 h
(R)-95o, 98:2 e.r.
F3CMe
CF3
without ()-sparteine (S)-96o, 38 %, 90:10 e.r.with ()-sparteine: (S)-96o, 18 %, 97:03 e.r.
2. MeOH
1. LDA (2.5 eq.),Et2O, -78 °C to-35 °C, 24 h2. MeOH
O
O
Cl
MeHN
(S)-96i, 51 %, 84:16 e.r.
N
Me
O
OH
Cl
(S)-219, 85 %, 80:20 e.r.
1,2-acyl shift
1,4-aryl transfer
(S)-95i, 99:1 e.r.
N O
O Me
MeCl
Me
1. LDA (2.5 eq.),Et2O, ()-sparteine,-78 °C to -35 °C, 24 h2. MeOH
Me
II.3.1.4 Aryl Migration of Simple O-Allylcarbamates
The rearrangement of simple O-allylcarbamate (N-Alloc aniline) 220 was also
attempted, with the aim of following the reaction pathway depicted in Scheme 128.
Assuming that the rearrangement of carbamate 220 occurs in the same manner as the
cinnamyl carbamate 201 previously studied, the free NH-group of the rearranged
carbamate 221 offers the possibility to couple an aryl group on the carbamate. A second
deprotonation-rearrangement sequence would lead to carbamate 223 with an interesting
diarylvinyl substituent, a possible precursor of diarylhydroxy acids.

143
Scheme 128. Proposed Route Towards Diarylvinylcarbamates 223 from a Simple Allyl
carbamate 220.
Ar1
N O
O
Me
MeHN O
O Ar1aryl migration
N O
O Ar1
Ar2
Me
MeHN O
O Ar1Ar2
N-arylation
aryl migration
221220
222 223
Upon treatment with LDA in THF at -78 °C, the less hindered N-methylated
compound 220a gave only N-methylaniline as a product (Scheme 129). While the N-
isopropyl analogue 220b was less prone to direct cleavage to the aniline, it failed to
undergo aryl migration, generating instead the 2-hydroxyamide product 226 of a 1,2-acyl
shift in 28 % yield.
Scheme 129. Attempted Aryl Migration of Simple Allylcarbamates.
N O
O
R
1. LDA (2.0 eq.), THF, -78 °C, 1 h
1,2-acyl shift
R = i-Pr
OH
226
N
O
220a, R = Me220b, R = i-Pr
2. MeOH
O
LiLnO
N
O
O
N
LiLn
28 %
224 225

144
II.3.1.5 In Situ IR Spectroscopy Studies
We also undertook detailed studies of the rearrangement of 201 (Scheme 130). A
mixture of 201 and THF in a three-necked flask equipped with a ReactIR probe was cooled
to -60 °C. Once the temperature has stabilized, spectroscopic analysis was commenced at a
rate of one scan every 1 min. The initial spectrum was recorded, before LDA (1.5 equiv.)
was added. Immediately on addition of LDA, the C=O stretching frequency of 1709 cm-1
corresponding to the starting carbamate was replaced by a peak at 1329 cm-1
and a double
peak centred approximately at 1656 cm-1
which persisted until after 45 min the reaction
was quenched with MeOH (Figure 15). At this point a peak at 1768 cm-1
appeared
corresponding to the product carbamate 204. In this reaction, under these conditions, we
saw no evidence for any other intermediates. We assume that the strong signals in the
region of 1329 and 1656 cm-1
arise from C-O and C-N stretches within the lithio carbamate
function of rearranged compound 204 which is formed by instantaneous rearrangement of
deprotonated 201.
Scheme 130. Intermediates Detected in the Rearrangement of 201.
N O
O
Me
PhLDA (1.5 eq.)
201 Me Ph
O
O
N
LiLn
Me
rearranges
Me
THF, -60 °C
MeHN O
O
Ph
204
Me
MeN O
O
Ph
MeLi MeOH
not observed
201Li
204Li
1709 cm-1
1768 cm-11656 cm-1
+ 1329 cm-1

145
Figure 15. In Situ React IR Monitoring of the Rearrangement of 201 Using LDA in THF at
-60 °C (Three-dimensional plot of absorbance versus wavenumber versus time in the 1800-
1600 cm-1
region of the IR spectrum).
Allylcarbamate 201
in THF at -60 °C
LDA added
Quench with
MeOH 1709cm
-1 : 201
1768cm-1
: 204 1664 & 1649cm
-1 : 204Li

146
II.3.2 N to C Aryl Migration in Lithiated N-Aryl-O-Propargylcarbamates
In order to broaden the scope of substrates, other stabilized organolithiums with
versatile propargylic structures was used for the rearrangement, something not studied
previously within the group.
II.3.2.1 Synthesis and Attempted Aryl Migration of Primary O-Propargylcarbamates
Primary propargyl carbamate 230a was synthesised, by the method developed within
the group, from the corresponding alcohol 227a which was converted to its carbamate
derivative by reaction with phenyl isocyanate and methylation with sodium hydride and
methyl iodide in 52 % yield over 2 steps (Scheme 131).
In contrast, the preparation of 230b by the usual method led to the formation of (E)-
4-alkylidene-2-oxazolidinone 231 as single isomer[165]
in 31 % yield and 4-
benzyloxazolone 232 in 24 % yield, arising from the deprotonation of –NH and subsequent
5-exo-dig ring closure of the intermediate sodium carbamate[166]
(Scheme 131). The base
promoted cyclisation of O-propargyl carbamates to give 4-alkylidene-2-oxazolidinones has
long been established[167]
and developed these recent years.[168]
Accordingly, 230b was
alternatively synthesised from the propargyl chloroformate 229 and N-methylaniline in
64 % yield over 2 steps.[168f]
Scheme 131. Preparation of O-Propargylcarbamates 230a and 230b.
HO
R
O
R
NH
O
PhPhNCO, NEt3
CH2Cl2, RT, 15 h227a, R = Me
227b, R = Ph
1. NaH, DMF,0 °C, 30 min2. MeI, RT, 15 h
R = Me
R = Ph
R = Ph
PhNHMe
NEt3, CH2Cl2
230a, R = Me, 52 % over 2 steps230b, R = Ph, 64 % over 2 steps
228a, R = Me228b, R = Ph
231, 31 %(E-isomer)
232, 24 %
Et2O, RT
(Cl3CO)2CO
O
Ph
Cl
O
O
R
N
O
Ph
Me229
ON
O
Ph
Ph
ON
O
Ph
Ph

147
However, as with the simple O-allylcarbamate 220a, O-propargylcarbamates 230a
and 230b, on treatment with LDA in THF at -78 °C, produced products resulting from
attack directly on the carbamate C=O group. Therefore, we turned our attention towards
investigating the rearrangement of -methylated O-propargylcarbamates.
II.3.2.2 Synthesis and Aryl Migration of -Methylated O-Propargylcarbamates
As expected, the synthesis of 237a using the initial method, returned oxazolone 235
in 52 % yield as single product (Scheme 132). Therefore, 237a and 237b were made via
chloroformate 236 in 57 % and 55 % yield over 2 steps, respectively.
Scheme 132. Preparation of -Methylated O-Propargylcarbamates 237a and 237b.
(Cl3CO)2CO,Et2O, RT
HO
Me
O
Me
NH
O
O
Me
Cl
O
O
Me
N
O
R
PhNCO, NEt3
CH2Cl2, RT233 234
236
CH2Cl2, RT
237a, R = Me 57 % over 2 steps237b, R = i-Pr 55 % over 2 steps
1. NaH, DMF,0 °C, 30 min
2. MeI, RT, 15 h
235, 52 % over 2 stepsPhNHMe, NEt3
or PhNHiPr, Na2CO3
ON
O
Me
Treatment with base led to migration of the aryl ring from N to C but was followed
by cyclisation of the resulting carbamate anion onto the triple bond to yield the
benzylidene oxazolidinones (E)-238a and (Z)-238a in a ratio highly dependent upon the
base used in the cyclisation (Scheme 133). N to C aryl migration in the presence of LDA
(2.0 equiv.) in THF at -78 °C afforded (E)-substituted 4-alkylidene-2-oxazolidinone 238a
in a 13:1 diastereoisomeric ratio in a moderate yield of 43 % due to the carbamate cleavage.
X-ray crystallography confirmed the structure of the cyclised product (E)-238a (Figure 16).
In contrast, the use of LiHMDS gave the (Z)-isomer 238a as major product (0.5:1 ratio by
1H NMR). Surprisingly, when propargyl carbamate 237a was treated with KHMDS in THF

148
at 0 °C a mixture of (E)- and (Z)-substituted 4-alkylidene-2-oxazolidinone 238a, easily
separable by column chromatography, was obtained in both 16 % yield (1.3:1 ratio by 1H
NMR). The (Z)-stereochemistry was confirmed by a nOe experiment which showed
coupling through space between the C-methyl group and the vinylic proton. The use of
NaHMDS led to the formation of (E)-substituted 4-alkylidene-2-oxazolidinone 238a in
44 % yield as major isomer (1:5 ratio by 1H NMR).Therefore, these compounds indicate
that the rearrangement can be initiated by organometallic intermediates of potassium and
sodium, which has not been observed previously.
Scheme 133. Aryl Migration of -Methylated O-Propargylcarbamate 237a.
N O
O
Me
Me
ON
O
Me
Me
ON
O
Me
Me
1. Base (2.0 equiv.)
Base: LDA, THF, -78 °C, 15 min
45 % 0 % (13:1 by 1H NMR)
KHMDS, THF, 0 °C, 15 min
NaHMDS, THF, 0 °C, 15 min
16 % 16 % (1.3:1 by 1HNMR)
7 % 44 % (1:5 by 1HNMR)
LiHMDS, THF, 0 °C, 15 min
14 % 31 % (0.5:1 by 1H NMR)
237a
(E)-238a (Z)-238a
2. MeOH
Me
O NHMe
OLi

149
Figure 16. X-Ray Crystal Structure of (E)-238a.
A number of procedures to synthesise 4-methylene-1,3-oxazolidinones have been
reported,[167,168]
while few processes to produce 5-tetrasubstituted ones have been
published.[167g,168c,b,e,g]
In addition to intramolecular nucleophilic cyclisations of propargyl
carbamates,[168f]
Schmalz et al. reported that 4-alkylidene-1,3-oxazolidin-2-ones could be
prepared from O-propargyl carbamates using catalytic AuCl in CH3CN.[168d]
In addition,
various approaches to oxazolidinones via cycloaddition reactions of carbon dioxide with
propargylic alcohols and amines have also been reported,[167f,168b,c]
which suffer from
drawbacks such as the need for toxic and noble-metal catalysts, the involvement of volatile
organic chemicals (VOCs), and harsh reaction conditions.
The cyclisation, while interesting, detracts from the possible utility of the
rearrangement. We reasoned that a triisopropylsilyl group as an acetylenic substituent
might compromise the reactivity of the acetylenic bond by decreasing its electrophilicity.
O-propargylcarbamate 241 was therefore made from 4-(trimethylsilyl)but-3-yn-2-ol 239 by
reaction with phenyl isocyanate and methylation with sodium hydride and methyl iodide to
give terminal O-propargylcarbamate 240 in 51 % yield over 2 steps. This in turn was
deprotonated with n-BuLi in THF at -78 °C and treated with i-Pr3SiCl to afford 241 in
65 % yield (Scheme 134).

150
Scheme 134. Preparation of O-Propargylcarbamate 241.
N O
O Me
H
N O
O Me
TIPS
HO
Me
TMS
1. PhNCO, Et3N,DCM, 15 h
2. NaH, DMF,0 °C, 1 h Me
1. n-BuLi, THF,-78 °C, 1 h
2. i-Pr3SiCl, RT, 5 h
Me
239 240, 51 % over 2 steps
241, 65 %
Treatment of 241 with 2.0 equiv. of LDA in THF/DMPU for 1 h gave the desired
rearranged product 242 in a promising 40 % yield accompanied with 30 % of the starting
material (Scheme 135; Table 25, entry 1). The use of 2.5 equiv. of LDA led to a further
increase in yield (66 %) with 20 % of recovered starting material (entry 2). The reaction
was completed over a longer reaction time (3 h) but in a lower yield of 54 % which could
be accounted for the formation of by-products (entry 3).
Scheme 135. Aryl Migration of O-Propargylcarbamate 241.
1. LDA, THF/DMPU,-78 °C
2. MeOHN O
O Me
TIPSMe
O
O
TIPS
MeHN
Me
241 242
Table 25. Conditions and Yields for the Aryl Migration of O-Propargylcarbamate 241.
Entry Equiv. of LDA Time (h) 242, Yield (%)
1 2.0 1 40a
2 2.5 1 66a
3 2.0 3 54
a Remaining starting material

151
II.3.2.3 Stereospecificity in the Aryl Migration
Enantiomerically enriched O-propargyl carbamate (E)-237a (Scheme 136) was made
from 4-phenylbut-3-yn-2-one, which was reduced by the method of Noyori[169]
using a
solution of KOH in 2-propanol in the presence of the ruthenium complex (S,S)-182. As
expected, in THF at -78 °C, the rearrangement/cyclisation reaction with LDA (2.0 equiv.)
gave only racemic product (Table 26, entry 1) as observed with benzylcarbamate 95i.
Replacing THF with Et2O diverted the course of the reaction towards attack directly on the
carbamate C=O group, with N-methylaniline being formed (entry 2). Attempts to reduce
the susceptibility of the carbonyl group to nucleophilic attack by replacing the N-methyl
with an N-isopropyl group ameliorated the yield in THF (70 %) (entry 3), but shut down
completely any reaction in Et2O. At various temperatures, with or without addition of ()-
sparteine, slow decay of the e.r. of the starting material, by racemization of its lithium
derivative, was also observed (entries 4 to 6).
Scheme 136. Stereospecificity in the Aryl Migration of 237a and 237b.
N O
O
R
Me ON
O
RPh
Me
(S)-237a, R = Me(S)-237b, R = i-Pr
(E)-238a, R = Me(E)-238b, R = i-Pr
(Table 26)
Table 26. Conditions and Yields for the Attempted Stereospecific Aryl Migration of 137.
Entry R Base Solvent Temp. (°C) Time Yield (%) e.r.
1 Me LDA THF -78 15 min 238a, 43 50:50
2 Me LDA Et2O -78 30 min - -
3 i-Pr LDA THF -78 15 min 238b, 70 50:50
4 i-Pr LDA Et2O -78 1 h 237b (15:85 e.r.)
5 i-Pr LDA Et2O -60 1 h 237b (27:73 e.r.)
6 i-Pr LDA + ()-
sparteine
Et2O then
THF (2 eq.) -78 1h + 4 h 237b (34:66 e.r.)

152
Again it seems likely that racemization competes with rearrangement, and no further
enantioselective reactions were pursued with these compounds.
N- or O-substituted allyllithiums may be formed either by -deprotonation of either
an allylamine/allyl alcohol derivative or alternatively by γ-deprotonation of an
enamine/enol derivative as mentioned in Section I.3.3. The advantage of the latter is the
lack of chirality in the starting material, which opens yet another mechanistic possibility
for carrying out an enantioselective rearrangement via asymmetric deprotonation.
Previously, it has been shown within the group that asymmetric deprotonation works well
in the vinyl urea series.[66]
We therefore embarked on -deprotonation-mediated N to C
aryl migration of N-aryl-Z-enol carbamates.

153
II.3.3 γ-Deprotonation-Mediated N to C Aryl Migration of N-Aryl-Z-enol
carbamates
II.3.3.1 Synthesis of Z-Enol Carbamates
The efficient method depicted below was employed to make N-aryl-Z-enol
carbamates (Scheme 137).[170]
Scheme 137. General Synthesis of (Z)-Enol Carbamates.
N O
O
R4 R3
(Z)-248
O O
DMSO, 15 h, RT
R3 R3
247
70 °C, 45 min
246
Na+N
O
R4
245
Cl
R1 R1
R1
R2R2
NH2
Me Me
O"NaBH(OAc)3"
AcOH, DCERT, 4 h
R2
HN
243
244c, R2 = p-Cl, 72 %
244d, R2 = p-Me, 86 %
244e, R2 = m-CF3, 87 %
244f, R2 = p-OMe, 90 %
Cl3CO OCCl3
OPy, CH2Cl2
RT, 2 h
248a, R1 = R2 = H, R3 = R4 = Me, 57 %
248b, R1 = R2 = H, R3 = Et, R4 = Me, 65 %
248c, R2 = H, R1 = R3 = R4 = Me, 48 %
248d, R1 = H, R2 = Cl, R3 = Et, R4 = Me, 52 %
248e, R1 = R2 = R3 = H, R4 = Me, 46 %
DMSO,NaH
248f, R1 = R2 = R3 = H, R4 = i-Pr, 62 %
248g, R2 = R3 = H, R1 = Cl, R4 = i-Pr, 60 %
248h, R1 = R3 = H, R2 = Cl, R4 = i-Pr, 50 %
248i, R1 = OMe, R3 = R2 = H, R4 = i-Pr, 37 %
248j, R1 = R2 = H, R3 = Ph, R4 = i-Pr, 55 %
248k, R1 = H, R2 = Cl, R3 = Ph, R4 = i-Pr, 46 %
R2
245a, R2 = p-Cl, R4 = Me, 87 %
245b, R2 = H, R4 = i-Pr, 60 %
245c, R2 = p-Cl, R4 = i-Pr, 74 %
245d, R2 = p-Me, R4 = i-Pr, 79 %
245e, R2 = m-CF3, R4 = i-Pr, 97 %
245f, R2 = p-OMe, R4 = i-Pr, 65 %
244
DMSO was used both as reagent and as solvent. The methyl sulfinyl carbanion
(“dimsyl anion”) was formed by reaction of the solvent with NaH at 70 °C. Corey et al.
first reported the versatility and reactivity of this base in organic synthesis.[171]
It should be
noted that the temperature should not exceed 70 °C, otherwise decomposition occurs. This
is also induced by prolonged reaction times. Upon addition of the appropriate ketone 246,
the conjugated base of DMSO afforded the corresponding sodium enolate derivative 247,
which was trapped with the desired carbamoyl chloride 245 affording the (Z)-O-

154
vinylcarbamates 248 in moderate yields. The moderate yield could be attributed to the
competing C-acylation, but also to the formation of a β-hydroxy sulfoxide adduct and a
symmetrical urea resulting from the decomposition of carbamoyl chloride. Carbamoyl
chlorides 245 were prepared by treating the amines 244 with triphosgene (safe equivalent
of phosgene) in the presence of pyridine as an HCl scavenger in good yields.[172]
The
amines 244 were obtained by direct reductive amination in excellent yields with sodium
triacetoxyborohydride, generated in situ in DCE and in the presence of acetic acid for
accelerating the reaction. This is very effective with weakly and non basic amines.[173]
The (Z)-configuration of the double bond in 248b, 248c and 248d was confirmed by
nOe experiments which did not show coupling through space between the vinylic proton
and the N-methyl protons and for 248j through a single-crystal X-ray analysis (Figure 17).
Figure 17. X-ray Crystal Structure of 248j.
II.3.3.2 -Arylation of Z-Enol Carbamates by γ-Deprotonation and N to C Aryl
Migration
(Z)-Enol carbamate 248a was firstly synthesised by the method depicted in Scheme
137 in 57 % yield. Hoppe et al. have noticed that the prerequisites for ()-sparteine-
mediated intramolecular γ-deprotonation are met only in the isomer (Z), since isomer (E)
remained unchanged.[99a]
Treatment of 248a with LDA (2.0 equiv.) in THF at -78 °C for 1
h gave the rearranged carbamate derivative of tertiary doubly benzylic alcohol 249a in
excellent yield (Scheme 138). Under the same conditions, enol carbamate (Z)-248b
underwent the rearrangement cleanly. However, when subjected to flash column
chromatography on basified silica, the resulting allylic carbamate 249b was prone to a
[3,3]-sigmatropic rearrangement to give 250 as reported by Overman.[174]
Consequently,

155
the rearranged product 249b was isolated by trituration in a mixture of hexane and Et2O in
80 % yield (Scheme 138).
Scheme 138. γ-Deprotonation-Mediated N to C Aryl Migration in Lithioallyl Carbamates.
1. LDA (2.5 eq.), THF, -78 °C, 1 hN O
O
MeO
NHMe
O
(Z)-248a, R = H
(Z)-248b, R = Me
2. MeOH R
249a, 86 %, R = H249b, 83 %, R = Me
R = Me
silica MeHN O
O Me
250
R
Z-enol carbamate 248c was then made (in 48 % yield) to examine the aryl transfer
under the influence of chiral lithium amides. Stereochemically, two extreme mechanistic
possibilities could present themselves:[175]
either the stereochemistry of the product is
determined by stereospecific rearrangement of a configurationally stable planar chiral
allyllithium or it is the result of a stereoselective reaction of a configurationally unstable
allyllithium under the kinetic or thermodynamic control of the associated chiral amine.
Vinyl carbamate (Z)-248c rearranged successfully on treatment with LDA (2.5 equiv.)
in THF at -78 °C to provide allyl carbamate 249c in 80 % yield (Scheme 139; Table 27,
entry 1). Replacing LDA with the monodentate homochiral lithium amide 252Li as its
hydrochloride salt[176]
(2.5 equiv.) led to a slower reaction, and a longer time was needed to
reach completion but the level of enantiomeric enrichment was poor (entry 2). It is
reasonable to assume that chiral lithium amides having two chiral carbons on the amide
nitrogen would be more effective as chiral bases. Based on this consideration, the
deprotonation was carried out with the bis-phenylethylamide 212 which offers the
advantage of C2-symmetry. In THF at -78 °C, the rearranged product 249c was indeed
returned with a slightly increased enantiomeric ratio of 68:32 (entry 3). The reaction was
then carried out at -60 °C (entry 4) but this led to 249c in very low enantioselectivity.
Likewise, when (Z)-248d was treated with LDA in THF at -78 °C, the rearranged product
249d was isolated in excellent yield (entry 5). However, in the presence of chiral lithium
amide 212Li, partially racemic 249d in lower yields was obtained using the conditions
attempted in entries 6 and 7.

156
Scheme 139. Enantioselectivity in the γ-Deprotonation and Aryl Migration of 248c and
248d.
N O
O
MeO
NHMe
O
R = H, R1 = Me, R2 = H, (Z)-248c
R = Me, R1 = H, R2 = Cl, (Z)-248d
R1 R1
1. chiral base, n-BuLi
2. MeOHR
R2R2
R= H, R1 = Me, R2 = H, 249c
R = Me, R1 = H, R2 = Cl, 249d
R
Table 27. Conditions, Yields and e.r. for the Asymmetric γ-Deprotonation and Aryl
Migration of 248c and 248d.
Entry S.M. Base Solvent T (°C) t (h) Yield (%) e.r.
1 (Z)-248c LDA THF -78 1 249c, 80 -
2 (Z)-248c
HN Ph
.HCl
252
THF -78 5 249c, 72 56:44
3 (Z)-248c Ph NH
Ph
212
THF -78 3 249c, 28a 68:32
4 (Z)-248c Ph NH
Ph
212
THF -60 3 249c, 50b 54:46
5 (Z)-248d LDA THF -78 1 249d, 80 -
6 (Z)-248d Ph NH
Ph
212
THF -78 5 249d, 18a 52:48
7 (Z)-248d Ph NH
Ph
212
THF -60 3 249d, 62 52:48
a Remaining starting material;
b Formation of a side-product

157
II.3.3.3 In Situ IR Spectroscopy Studies
Rearrangement of 248d gave a little more detail about the mechanistic pathway,
presumably because the allyl anion generated by deprotonation is a little more stable than
that formed from 201. Immediately on addition of LDA, a new band at 1578 cm-1
appeared
which lasted for only a few minutes before giving way to a band at 1649 cm-1
(Scheme 140;
Figure 18). This band, which corresponds to the band at 1656 cm-1
in Figure 15, persisted
until the quench, which generated the product peak at 1735 cm-1
. The band at 1649 cm-1
is
again presumably that corresponding to the product anion 251dLi, with the band at 1578
cm-1
being a precursor, probably the lithiated starting material 248dLi. A cinnamyllithium
species such as 248dLi was identified in the region of 1552 cm-1
during in situ IR studies
on the rearrangement in an N-allylurea (see Appendice 3), which suggests that the peak at
1578 cm-1
is more likely the intermediate 248dLi. Moreover, a prelithiated complex has
not been observed in THF previously.
Scheme 140. Intermediates Detected in the Rearrangement of 248d.
LDA (1.1 eq.)
THF, -60 °C
O
NMe
LiO
Cl
O
NHMe
O
Cl
O
N
OMe LiLn
Aryl migration
248dLi
Cl248d
251dLi 251d
MeOH
1731 cm-1
1735 cm-11649 cm-1
1578 cm-1
N O
O
Me
Cl

158
Figure 18. Three-Dimensional Infrared Profile for the Aryl Migration of 248d Using LDA
in THF at -60 °C.
Enolcarbamate 248d
in THF at -60°C
at -60°C
LDA added
Quench with
MeOH
1649 cm-1
: 251Li 1578 cm-1
: 248Li
1731 cm-1
: 248d
1735 cm-1
: 251d

159
II.3.4 Limits of the N to C Aryl Migration
II.3.4.1 Attempted Aryl Migration in Lithiated Carbamates Containing the Electron
Rich Heterocycle Furan
Furanyl carbamate 255a was synthesised from (furan-3-yl)methanol 254a by
standard methods in 69 % yield over 2 steps (Scheme 141). 255a failed to rearrange upon
treatment with LDA or sec-BuLi in the presence of DMPU, only the starting material was
recovered. Indeed, the deprotonation could occur on the furanyl group in the 2 or 5
position. In order to test this hypothesis, the reaction was quenched with iodomethane and
the NMR analysis showed that the methylation took place on the furanyl group.
Scheme 141. Preparation of Furanyl Carbamates.
N O
Me
O
OR
R
R = H, 255a, 69 % over 2 stepsR = Me, 255b, 71 % over 3 steps
O
HO
OR
RHO
O
253 R = H, 254aR = Me, 254b
1. LiAlH4, Et2O0 °C, 16 h, RT
2. EtOH,Rochelle's salt
1. PhNCO,NEt3, CH2Cl215 h, RT
2. NaH, DMF,MeI, 15 h, RT
Consequently, 255b with additional methyl groups on the furan ring was prepared
from dimethyl furoic acid 253 using the standard methods in 71 % yield over 3 steps. Next,
with the aim of promoting the rearrangement, a number of experiments were performed by
changing the base, the temperature and the reaction time (Scheme 142; Table 28).
However, none of these experiments resulted in the formation of the rearranged product
256b. In all cases, the starting material was recovered and in some cases, N-methylaniline
arising from nucleophilic attack on the carbamate C=O group, was also observed in the
crude 1H NMR. After quenching the reaction with MeI, no evidence of methylation was
present, suggesting that lithiation to oxygen had not occurred.

160
Scheme 142. Attempted Aryl Migration of Furanyl Carbamate 255b.
N O
Me
O
O255b
O
O
O256b
MeHN
(Table 28)
Table 28. Conditions and Results for the Attempted Aryl Migration of Furanyl Carbamate
255b.
Entry Base Equiv. Temp. (°C) Time (h) Results
1 s-BuLi 2 -78 4 255b + 184b
2a LDA 2 -78 4 255b
3 LDA 2.5 -40 6 255b
II.3.4.2 Attempted Aryl Migration in Lithiated O-Alkylcarbamates
We have shown that benzyl, alkenyl and alkynyl carbamates are deprotonated with
great ease by LDA at low temperature leading to stable five-membered chelate complexes,
which smoothly undergo N to C aryl transfer. Hoppe et al. reported that even primary alkyl
carbamates, which lack further carbanion-stabilising groups, are deprotonated by sec-
BuLi/TMEDA.[177]
Therefore, rearrangement of O-alkylcarbamate 257 was attempted,
firstly under the usual conditions for deprotonation (Scheme 143; Table 29, entry 1: LDA
in THF/DMPU at -78 °C). However, only the starting material was recovered and the same
was observed at -45 °C (entry 2). A quench with MeI indicated that lithiation to oxygen
did not occur. Repeating the reaction with a stronger base, sec-BuLi, returned 257 (entry 3).
Under the conditions used by Hoppe for lithiation, sec-BuLi in Et2O/TMEDA at -78 °C
followed by warming the reaction slowly to -15 °C to promote the rearrangement, 257 was
also isolated after a quench with MeOH (entry 4). Quenching the reaction with acetic acid
d4 after 2 h at -78 °C showed no evidence of deuterium incorporation.
a Further experiment : quenching with MeI
b Arising from nucleophilic attack on the carbamate C=O group

161
Scheme 143. Attempted Aryl Migration of Alkyl Carbamate 257.
N O
Me
O
MeHN O
O(Table 29)
257 258
Table 29. Conditions and Results for the Attempted Aryl Migration of Alkyl Carbamate
257.
Entry Base Solvent/Additive Temp. (°C) Time (h) Results
1 LDA THF/DMPU -78 2 257
2a LDA THF/DMPU -45 2 257
3 s-BuLi THF/DMPU -78 2 257
4b s-BuLi Et2O/TMEDA -78 to -15 2 + 17 257 + 258
In summary, it is clear that while enantiomeric enrichment in the product cannot be
controlled due to racemization of the intermediate organolithiums, nonetheless the
reactivity of lithiated O-benzyl, O-allyl, O-propargyl and O-vinyl carbamates, bearing N-
aryl groups, towards migration of the aryl ring from N to C provides a valuable method for
arylating an organolithium centre adjacent to oxygen in very easily made substrates.
Furthemore, enol carbamates could be attractive substrates for generation of
organolithium intermediates by β-carbolithiation. We therefore made -aryl, alkynyl,
alkenyl and silyl-O-vinylcarbamates as substrates in order to examine if the resulting
stabilized organolithiums could undergo N-aryl migration.
a Quenching with MeI: no methylation
b Quenching with acetic acid d4 at -78 °C: no deuterium incorporation

162
II.4 Tertiary Alcohols by Tandem β-Carbolithiation and N to C
Aryl Migration in Enol Carbamates
II.4.1 Carbolithiation and Aryl Migration of N-Methyl--aryl-O-
vinylcarbamate
In an initial study, N-methyl--aryl-O-vinylcarbamate 248e was treated with i-PrLi
(2.5 equiv.) in THF at -78 °C for 1 h (Scheme 144). β-Carbolithiation occurred, followed
by N to C migration of the N-phenyl ring, to return the product 259a in moderate yield
(36 %) (Table 30, entry 1). Similar yields were obtained with n-BuLi (entry 2) and sec-
BuLi (entry 3). The rearranged product 259c was obtained as a 1:1 mixture of rotamers.
This can be explained by a slow rotation due to the steric hindrance of the sec-butyl group.
The principal by-product was enamine 260 presumably resulting from a pathway initiated
by carbamate cleavage. This was highlighted by the attempted carbolithiation with MeLi
which formed only 260 (entry 4). Evidently, regioselective carbolithiation of 248e occurs
readily under these conditions, presumably yielding initially a benzylic organolithium.
Scheme 144. Carbolithiation-Aryl Migration of N-Methyl--aryl-O-vinylcarbamate 248e.
N O
Me
O
1. R-Li, THF,-78 °C, 1 h
2. MeOH
248e
O
O
259
R
Ph Ph
HN
Me
Ph
NPhMe
260
Table 30. Organolithiums and Yields for the Carbolithiation-Aryl Migration of N-Methyl-
-aryl-O-vinylcarbamate 248e.
Entry R 259, Yield (%)
1 i-Pr 259a, 36
2 n-Bu 259b, 33
3 sec-Bu 259c, 35
4 Me 259d, 80

163
The investigation was pursued by the replacement of the N-methyl group with a
bulky N-isopropyl substituent for shielding the carbonyl moiety which would lead to better
yields.
II.4.2 Carbolithiation and Aryl Migration of N-Isopropyl--aryl-O-
vinylcarbamates
II.4.2.1 Optimisation of the Reaction
The optimisation of the alkylation/arylation reactions of 248f and 248g (Scheme 145)
is displayed in Table 31.
Scheme 145. Carbolithiation-Aryl Migration of 248f and 248g.
1. R-Li
2. MeOH
248f, R1 = H
248g, R1 = Cl
262
N O
O
i-Pr
R1
PhN O
O
i-Pr
Ar
R
HN O
O
R
ArPh
i-Pr
261(Table 31)
Table 31. Optimisation of the Carbolithiation-Aryl Migration of 248f and 248g.
Entry S.M. R Solvent/Additive T (°C) t Yield (%)
1 248f i-Pr THF -78 1 h. 261a, 28 + 262a, 10
2 248f i-Pr THF -78 3 h 262a, 21
3 248f i-Pr THF -45 2 h -
4 248f i-Pr THF then DMPU -78 1 h + 2 h 262a, 62
5 248f i-Pr THF/DMPU -78 15 min 262a, 80
6 248f n-Bu THF/DMPU -78 15 min 262b, 73
7 248f sec-Bu THF/DMPU -78 5 min 262c, 80
8 248f tert-Bu THF/DMPU -78 15 min 262d, 83
9 248f Ph THF/DMPU -78 3 h 248f 10 248f Ph THF/DMPU -45 1 h 248f 11 248f Ph PhMe/TMEDA -78 5 h 248f + 261b
12 248f Me THF/DMPU -45 2 h 248f
13 248f Me THF/DMPU -20 3 h 248f
14 248f Me PhMe/TMEDA -78 3 h 248f
15 248f OEt
Li
THF/DMPU -45 3 h 248f
16 248f Li THF/DMPU -45 3 h 248f

164
Table 31. Continued.
17 248g i-Pr THF/DMPU -78 15 min 262e, 81
18 248g n-Bu THF/DMPU -78 15 min 262f, 74
19 248g sec-Bu THF/DMPU -78 15 min 262g, 75
20 248g tert-Bu THF/DMPU -78 15 min 262h, 81
Addition of i-PrLi to 248f in THF at -78 °C for 1 h and quenching with methanol led
to a mixture of a simple addition product 261a and the desired product 262a (Table 31,
entry 1). The isolation of the addition product implied a slow rearrangement, and 3 h was
required for complete migration (entry 2). However 262a was returned in low yield (21 %).
When the reaction was carried out at a higher temperature (-45 °C), the TLC and 1H NMR
showed a complex mixture of products with negligible desired product 262a (entry 3).
Adding DMPU to enforce rearrangement after the carbolithiation was complete increased
the yield to 62 % (entry 4). The best yields (73 % to 83 %) were obtained by carrying out
the reaction in a mixture of THF and DMPU (25 % v/v) at -78 °C. By following the
reaction carefully by TLC and 1H NMR, it was shown that the compounds 262 were
formed by instantaneous carbolithiation-rearrangement of 248f (entries 5 to 8). Therefore,
it is clear that replacement of the N-methyl group with an N-isopropyl substituent
improved the yield significantly.
Whatever the experimental conditions used, phenyllithium, methyllithium,
ethylvinyllithium ether[178]
and vinyllithium[179]
failed to add to 248f (entries 9 to 16). By
using TMEDA as a lithium-coordinating additive, which enhances the reactivity of
organolithium intermediate by reducing its aggregation[180]
in toluene, a single addition
product 261b was obtained with PhLi as reagent in poor yield with a significant amount of
recovered starting material (entry 11).
Likewise, the addition of iso-propyllithium, n, sec- and tert-butyllithiums coupled
with N to C aryl migration could be successfully added to O-vinylcarbamate 248g in
THF/DMPU at -78 °C for 15 min (entries 17 to 20).
In an attempt to extend the scope of the reaction by adding less reactive
organolithiums to O-vinylcarbamates, Meyers’ strategy was considered. In 1991, Meyers
et al. showed that organolithium reagents derived from lithium 4,4-di-tert-butylbiphenyl
263 (LiDBB) (Freeman’s Reagent) are uniformly superior to all other methods used for
additions to naphthalene oxazolines such as 264 (Scheme 146).[181]
Not only are the

165
reaction conditions milder and of shorter duration, but the yields are comparable in some
cases as shown by the examples in Scheme 146.
Scheme 146. Addition of LiDBB 263 to Naphthalene Oxazoline 264.
t-But-Bu2 Li+
LiDBB 263
RXRLi t-Bu
2
LiCl
NO
Ph OMe
a) RLi
b) MeI
ROXZMe
264
H2C CHSnBu3 MeLi
t-BuLi
LiDBB
Br LiDBB
RLi Preparation Temp (°C) Time (h) 265,Yield (%)
-78-0
-78- -30
-10
-30
12
10
24
12
0
78
82
70
265
Br
Br
However, attempted β-carbolithiations of 248f with 1-bromo-2-methylpropen-1-ene
and 1-bromobenzene treated with LiDBB, in THF at -20 °C for 15 h returned only the
starting material.
II.4.2.2 Synthesis of Diarylalkylalcohols
The next step was the challenging deprotection of the N-isopropyl carbamate group.
Cleavage of the carbamate group of 248f under standard conditions[55,56]
(EtONa/EtOH at
reflux, DIBAL-H/THF at room temperature and at reflux and N-nitrosation) was
accompanied by dehydration, but by quenching the carbolithiation-rearrangement reactions
with tert-butylnitrite (to form an N-nitroso carbamate in situ) and stirring the resulting
basic reaction mixture for 24 h at room temperature, diarylalkylalcohol 266a could be
obtained from 248f in a one-pot reaction (Scheme 147).

166
Scheme 147. Synthesis of Diarylalkylalcohols.
1. RLi (2.0 eq.), THF/DMPU,-78 °C, 15 min
2. t-BuONO (6.0 eq.),RT, 24 h
N O
O
i-Pr
R2
R1248f-i 266
HOR
R2
R1
The next challenge was therefore to extend the scope of the reaction by varying the
substituents on both the migrating ring and the aryl ring to oxygen.
Table 32. Yields for the Carbolithiation-Aryl Migration of 248f-i to Give 266.
Entry S.M. R1 R
2 R Product 266, Yield (%)
1 248f H H
n-Bu
HO
R2
R1
266a, 57
2 248g Cl H 266b, 55
3
4
248h
248i
H
OMe
Cl
H
266b, 77
266c, 68
5 248f H H Benzyla
HO
266d, 53b
6 248f H H
i-Pr
HO
R2
R1
266e, 73
7 248g Cl H 266f, 51
8
9
248h
248i
H
OMe
Cl
H
266f, 65
266g, 81
10 248f H H
sec-Bu
HO
R2
R1
266h, 64
11 248g Cl H 266i, 51c
12
13
248h
248i
H
OMe
Cl
H
266i, 67c
266j, 64c
14 248f H H
tert-Bu
HO
R2
R1
266k, 71
15 248g Cl H 266l, 36
16
17
248h
248i
H
OMe
Cl
H
266l, 27
266m, 67
a Obtained by metalation of toluene at RT in the presence of sparteine.
b After deprotection of the
crude rearranged product by treatment with n-BuLi in THF followed by tert-butyl nitrite quench. c 1:1 mixture of diastereoisomers.

167
Addition reactions worked best with primary and secondary alkyllithiums and gave
266 in good to excellent yields (Table 32, entries 1-13) while tert-butyllithium (entries 14-
17) gave lower yields in some cases. N to C aryl migration proceeded smoothly regardless
of the electronic nature of aryl rings Ar1 and Ar
2. Accordingly, a product could be made in
two ways by exchanging Ar1 and Ar
2.
If an efficient method would be available to render such a process asymmetric, it
would acquire utility as a method for the creation of enantioenriched tertiary multiply
branched alcohols.
II.4.2.3 Enantioselectivity in the β-Carbolithiation-Aryl Migration
The complexed conformers A/B-267, formed by the coordination of organolithium
RLi and a chiral ligand L2*, will react with the C=C bond in an intramolecular syn-addition
to form the benzyllithium derivatives (R)-268•L2* and (S)-268•L2*, which are
configurational stable and can be trapped (Scheme 148). Overall, the sequence provides
enantiofacial attack at the styrene double bond. We can note that the carbanionic centre in
the reagent is not stereogenic. Any differentiation is due to the chiral ligand L2*. A few
efficient examples for such a strategy are known.[110e,f,111,182]
Scheme 148. Enantioinduction in the Carbolithiation.
ArH
O O
NR1R2
H
ArH
O
NR1R2
H
O LiL2*
R
RLiL2*
ArH
O
NR1R2
H
O LiL2*
R
O LiL2*
O
2R1RN
Ar R
A-267 B-267
O LiL2*
O
2R1RN
Ar R
Si Re
(R)-268 L2* (S)-268 L2*

168
Normant and Marek reported that the readily available lupine alkaloid ()-sparteine
89 could serve as promoter for the enantioselective carbolithiation of cinnamyl derivatives
by addition of organolithium compounds.[107,110f,112d]
These observations led us to consider
the enantioinduction in the ()-sparteine-mediated carbolithiation reaction onto O-
vinylcarbamate 248g (Scheme 149). The data are collected in Table 33.
Scheme 149. Enantioinduction in the ()-Sparteine-Mediated Carbolithiation Reaction.
1. R-Li-sparteine
2. MeOH
262
N O
O
i-Pr
Cl
PhN O
O
i-Pr
Ar
R
HN O
O
R
PhAr
i-Pr
261(Table 33)
248g
Table 33. Conditions, Yields and e.r. for the Enantioselective Carbolithiation of 248g.
Entry RLi Solvent T (°C) t (h) Yield (%), e.r.
1 n-BuLi Toluene -78 3 248g
2 n-BuLi Toluene -50 3 261c 55, 68:38 e.r.
3 n-BuLi Cumene -50 2 261c 42, 65:35 e.r.
4 i-PrLi Cumene -50 2 248g + 261d 18, 72:28 e.r.
5 n-BuLi Toluene then
DMPU (4.0 eq.) -50 3 + 1 262f 61, 57:43 e.r.
Treating vinylcarbamate 248g with 2.0 equiv. of n-BuLi and ()-sparteine in toluene
at -78 °C for 3 h returned the starting material (Table 33, entry 1). When the reaction was
performed at higher temperature (-50 °C), a single addition product 261c was isolated in
55 % yield and a poor enantiofacial discrimination (68:38 e.r.) was revealed in the product
(entry 2). It was shown that cumene as a solvent gave high e.r. values for the
carbolithiation reaction.[110f]
However, the use of cumene in the reaction provided similar
results in terms of yield and e.r. (entry 3). The best result was obtained with i-PrLi (72:28
e.r.) returning the addition product 261d in poor yield (entry 4). However, the migration
did not take place under these conditions, and the isolation of the rearranged product 262f
was only possible by adding 4.0 equiv. of DMPU to enforce rearrangement after the
carbolithiation was complete (entry 5) but this tampered with the enantioselectivity.

169
The enantiomeric ratio obtained in entry 6 is of a similar order of magnitude as
observed by Hoppe[123]
and Snieckus.[75b]
Hoppe et al. even examined different chiral
ligands but none of them provided high enantioselectivities. They suggested that the
induction is established in the intermediate complexes A-267 and B-267 (Scheme 148).
Both complexes are in slow equilibrium and react with similar activation energies.
II.4.3 Carbolithiation and Aryl Migration of β-Substituted N-Isopropyl-
-aryl-O-vinylcarbamates
Evidently, the tandem β-carbolithiation-N to C aryl transfer of β-prostereogenic O-
vinylcarbamates can lead to two diastereoisomeric products. Therefore, it will be
interesting to examine the diastereoselectivity of the reaction.
Attempted β-carbolithiation of the β-substituted O-vinylcarbamate 248a with i-PrLi
(2.5 equiv.) in THF at -78 °C gave the alkene 269 in 57 % yield, presumably resulting
from N to C aryl migration initiated by a γ-deprotonation followed by γ-carbolithiation of
i-PrLi and subsequent β-elimination of the lithium carbamate (Scheme 150). To test this
hypothesis, the reaction was repeated with only 2.0 equiv. of i-PrLi, and indeed, in addition
to the alkene 269, the rearranged product 249a was isolated in 7 % yield.
Scheme 150. Attempted β-Carbolithiation of β-Substituted O-Vinylcarbamate 248a.
1. i-PrLi, THF, -78 °C, 3 h
2. MeOH
N O
O
Me
O
NHMe
O
2.0 eq. of i-PrLi
2.5 eq. of i-PrLi 0 % 57 %
7 % 29 %
248a
249a 269
As a note, O-vinylcarbamate 248d also failed to react upon β-carbolithiation with i-
PrLi in THF at -78 °C and -45 °C or with n-BuLi in toluene/TMEDA at -45 °C.

170
β-Carbolithiation was successful however when γ-deprotonation was blocked by a
phenyl group (Scheme 151).
Scheme 151. Stereospecificity in the Carbolithiation of 248j and 248k.
N O
O
Ar
i-Pr
Ph
Ph
(Z)-248j, Ar = Ph(Z)-248k, Ar = p-ClC6H4
HPh
PhO
BuLiO
N
i-Pr
HAr
PhO
BuPh
N
OLi
i-Pr
270
273b, Ar = p-ClC6H4
Ar
syncarbolithiation
invertivemigration
ArN O
O
i-Pr
Ph
Bu
Ph
i-PrHN O
O Ph
Bu
Ph
Ar
HO
Ph
Bu
Ph
Ar
1. n-BuLi, PhMe/TMEDA,-78 to -30 °C, 2 h
2. MeOH
2. DMPU
2. -15 °C, 15 h
3. MeOH
271a, 70 %, Ar = Ph271b, 80 %, Ar = p-ClC6H4
272a, 60 %272b, 54 %
274b, 22 % + 271b, 40 %
3. MeOH
migration +in situ carbamate
deprotection
Under the conditions used for the carbolithiation and rearrangement of β-non-
prostereogenic vinylcarbamates or at higher temperature, the starting material 248j was
returned.
A single diastereoisomer of the addition products 271 was isolated in good yields
when 248j or 248k was treated with n-BuLi/TMEDA in toluene, quenching with methanol
at -30 °C. The relative configuration of 271a was proved by an X-ray crystal structure
(Figure 19). The stereochemistry with syn-addition of the organolithium followed by
retentive protonation is in agreement with Hoppe’s related observations.[123]
This result
suggests that the substituted benzyllithium intermediate 270 is configurationally stable[183]
in toluene/TMEDA on the time scale of the reaction.

171
Figure 19. X-ray Crystal Structure of 271a.
Coordinating solvents accelerate both the aryl migration step and inversion at the
lithium center,[55]
and accordingly adding DMPU to enforce rearrangement after the
carbolithiation was complete led to the formation of the alcohol 272b but as a mixture of
diastereoisomers.[184]
Avoiding DMPU and instead warming the reaction slowly to -15 °C
and stirring for 15 h gave a 35:65 mixture of the single addition product 271b and the
rearranged product 274b in a 95:5 diastereoisomeric ratio. The relative stereochemistry of
274b was not determined because 271b and 274b were not separable by flash column
chromatography, but previous aryl migrations in lithiated carbamates have been shown to
be invertive.[55]
Having showed that β-carbolithiation-aryl migration of O-vinylcarbamates is a
straightforward method to access tertiary alcohols, we decided to investigate the scope of
the reaction by varying the migrating ring and the group at the α-position to oxygen.
II.4.4 Carbolithiation and Aryl Migration of -Alkynyl-O-
vinylcarbamates
II.4.4.1 Synthesis of N-Isopropyl-α-alkynyl-O-vinylcarbamates
The versatile method depicted in the Scheme 152 was used to prepare -alkynyl,
alkenyl, and silyl-O-vinylcarbamates.

172
Scheme 152. Synthesis of -alkynyl-O-vinylcarbamates.
O
n-BuLi
RT, 15 h
OLiDMPU, 5 h,RT
N O
i-Pr
O
R1
R2
N
i-Pr
OR2
Cl
N O
i-Pr
OR2
N O
i-Pr
O LiR2
275 276
245
277, 57-75 %
278280, 70-87 %
t-BuLi,THF
-78 °C, 1 h
I2, THF, -78 °C
C CHR1
NEt3, CuI Pd(PPh3)4,RT, 15 h
N O
i-Pr
O IR2
279
R2 = H, p-Cl, p-Me, m-CF3, p-OMe
R1 = t-Bu, Si(i-Pr)3, SiMe3, Ph
The lithium enolate 276 of acetaldehyde was generated by cleavage of THF 275
using n-BuLi,[185]
and O-carbamoylation[186]
gave simple O-vinylcarbamates 277 in good
yields (57-75 %). Under standard metalation conditions (t-BuLi, THF, -78 °C),[187]
277
underwent -lithiation and the resulting -lithio species 278 were trapped with I2 to form
vinyl iodides 279 with perfect control of regiochemistry. The alkynyl groups of 280 were
introduced by a Sonogashira reaction in excellent yields (70-87 %).[188]
II.4.4.2 One-pot Synthesis of Multiply Branched Arylalkynylalcohols
The series of -alkynyl-O-vinylcarbamates 280 were subjected to the conditions
shown to induce β-carbolithiation and N to C aryl migration in -aryl-O-vinylcarbamates
248f-i (Scheme 153).
Scheme 153. Carbolithiation-Aryl Migration of 280.
N O
i-Pr
O
R1
R2
280
Conditions A:1. RLi (2.0 eq.), THF, DMPU,-78 °C, 15 min2. t-BuONO, RT, 24 h
Conditions B:1. RLi (2.0 eq.), THF, DMPU,-45 °C, 15 min2. MeOH
R1
HO R
R2
281

173
The alkyllithium was added either in THF in the presence of DMPU at -78 °C for 15
min, followed by a tert-butyl nitrite quench, or at -45 °C, with an in situ carbamate
deprotection. The resulting alcohols 281 were isolated from this one-pot reaction in
moderate to good yields (Table 34).
Table 34. Yields for the Carbolithiation-Aryl Migration of 280.
Entry S.M. Product 281, R, Yield (%),
(conditions)
1
N O
i-Pr
O
t-Bu280a
t-Bu
HO R
281a, n-Bu, 71 (A)
281b, i-Pr, 74 (A)
281c, t-Bu, 67 (A)
2
N O
i-Pr
O
t-Bu280b
Cl
t-Bu
HO R
Cl
281d, n-Bu, 40 (A)
281e, i-Pr, 43 (A)
281f, t-Bu, 47 (A)
3
N O
i-Pr
O
t-Bu280c
Me
t-Bu
HO R
Me
281g, n-Bu, 34 (A)
281h, i-Pr, 44 (A)
4
N O
i-Pr
O
t-Bu280d
F3C
t-Bu
HO R
F3C
281i, n-Bu, 37 (A)
281j, i-Pr, 55 (A)
281k, t-Bu, 26 (A)
5
N O
i-Pr
O
t-Bu280e
MeO
t-Bu
HO R
MeO
281l, n-Bu, 40 (B)
281m, i-Pr, 47 (B)
6
N O
i-Pr
O
Si(i-Pr)3280f
Si(i-Pr)3
HO R
281n, n-Bu, 76 (A)
281o, s-Bu, 58 (A)
281p, t-Bu, 63 (A)

174
Table 34. Continued.
Entry S.M. Product 282, R, Yield (%)
7a
N O
i-Pr
O
SiMe3280g
H
CbO R
282a, n-Bu, 72
282b, i-Pr, 51
8a
SiMe3
CbO t-Bu
283, 41
H
CbO t-Bu
282c, 13
9a
N O
i-Pr
O
Ph280h
Ph
CbO i-Pr
284, 30
NO
Oi-Pr
Ph Ph
i-Pr
285, 28
As highlighted in Table 34, the migration of both electron-rich and -poor migrating
rings took place cleanly (entries 1 to 5). The reaction tolerated tert-butyl and
triisopropylsilyl groups as acetylenic substituents, but trimethylsilyl and phenyl groups
were prone to subsequent reactions. Treating O-vinylcarbamate 280g with either n-BuLi or
i-PrLi in THF at -78 °C gave the products 282a and 282b of carbolithation, N to C aryl
migration, and loss of the TMS group (entry 7). Tert-BuLi gave a mixture of 283 and
desilylated product 282c (entry 8). Treating O-vinylcarbamate 280h with i-PrLi in THF at -
78 °C led to carbolithiation, N to C aryl migration, and cyclisation of the resulting
carbamate anion of 284 onto the triple bond to yield the benzylidene oxazolidinone 285
(entry 9). These results are compatible with previous observations that a terminal phenyl
substituent accelerates the insertion of a carbon-carbon triple bond into a nitrogen-lithium
bond. In contrast, the activity of alkynes bearing an alkyl or silyl group in the terminal
position toward nucleophilic attack is significantly lowered.
It should be noted that the result of the entry 6 fulfilled our expectation: the
carbolithiation of O-enolcarbamates is a more efficient method of initiating aryl migration
than deprotonation of -methylated carbamates as seen in Section II.2.2.2.
a Cb = C(=O)NHi-Pr. Conditions : (1) RLi, THF/DMPU, -78°C, 15min. (2) MeOH.

175
The in situ carbamate deprotection at -45 °C could occur either in the presence of
excess alkyllithium or by the extrusion of the lithium alkoxide via the lithiocarbamate as
observed by Hoppe with lithiothiocarbamate (Scheme 154).[190]
Scheme 154. Rearrangement of lithiothiocarbamate on Warming.
N S
Li
O
Li
SLi
Li O
C
N
SLi
LiN
O
1. 10 min, -78 °C2. 15 min, 0 °C
3. 15 min, -78 °C
II.4.4.3 Formation of Enantioenriched Allenes
As observed before with -aryl-O-vinylcarbamate 248f, avoiding DMPU resulted in
products of carbolithiation only. Indeed, treatment of the alkynyl-O-vinylcarbamates 280a
and 280g with n-BuLi in THF at -78 °C gave allenes 286a and 286b (Scheme 155) on
protonation.[190]
Scheme 155. Formation of Enantioenriched Allenes.a
PhN O
i-Pr
O
R1
1. n-BuLi, ()-sparteine,PhMe, 4 h
2. MeOH R1•
O
H
n-Bu
N OPh
i-Pr
280a, R1 = t-Bu
280g, R1 = SiMe3
286a, R1 = t-Bu
286b, R1 = SiMe3 a Absolute stereochemistry not determined
When 280g was treated with n-BuLi in the presence of ()-sparteine (2.0 equiv.) in
toluene at -78 °C or -50 °C for 4 h, 286b was returned as a racemate (Table 35, entries 1
and 2), presumably because the silyl group stabilizes a negative charge which facilitates
racemization.[191]
In contrast, under the same conditions, 280a afforded 286a in 89:11 e.r. but in a low
yield (8 %) due to an incomplete conversion (entry 3). Raising the temperature to -50 °C
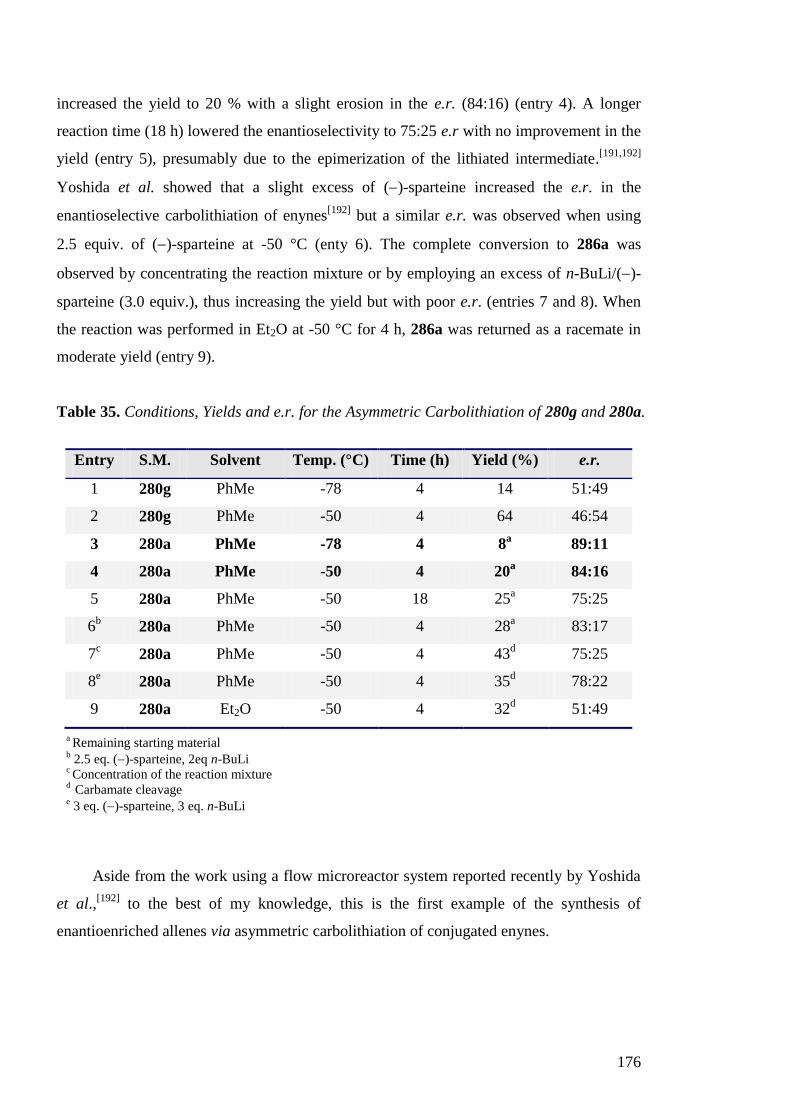
176
increased the yield to 20 % with a slight erosion in the e.r. (84:16) (entry 4). A longer
reaction time (18 h) lowered the enantioselectivity to 75:25 e.r with no improvement in the
yield (entry 5), presumably due to the epimerization of the lithiated intermediate.[191,192]
Yoshida et al. showed that a slight excess of ()-sparteine increased the e.r. in the
enantioselective carbolithiation of enynes[192]
but a similar e.r. was observed when using
2.5 equiv. of ()-sparteine at -50 °C (enty 6). The complete conversion to 286a was
observed by concentrating the reaction mixture or by employing an excess of n-BuLi/()-
sparteine (3.0 equiv.), thus increasing the yield but with poor e.r. (entries 7 and 8). When
the reaction was performed in Et2O at -50 °C for 4 h, 286a was returned as a racemate in
moderate yield (entry 9).
Table 35. Conditions, Yields and e.r. for the Asymmetric Carbolithiation of 280g and 280a.
Entry S.M. Solvent Temp. (°C) Time (h) Yield (%) e.r.
1 280g PhMe -78 4 14 51:49
2 280g PhMe -50 4 64 46:54
3 280a PhMe -78 4 8a 89:11
4 280a PhMe -50 4 20a 84:16
5 280a PhMe -50 18 25a 75:25
6b 280a PhMe -50 4 28
a 83:17
7c 280a PhMe -50 4 43
d 75:25
8e 280a PhMe -50 4 35
d 78:22
9 280a Et2O -50 4 32d 51:49
Aside from the work using a flow microreactor system reported recently by Yoshida
et al.,[192]
to the best of my knowledge, this is the first example of the synthesis of
enantioenriched allenes via asymmetric carbolithiation of conjugated enynes.
a Remaining starting material
b 2.5 eq. ()-sparteine, 2eq n-BuLi
c Concentration of the reaction mixture
d Carbamate cleavage
e 3 eq. ()-sparteine, 3 eq. n-BuLi

177
Axial chirality transfer in enantioenriched allene 286a could offer a valuable
opportunity to form the rearranged product 287 enantiomerically enriched (Scheme 156).
However, as expected, 286a lost completely its chiral information upon treatment with sec-
BuLi in THF/DMPU at -78 °C since the rearranged product 287 was returned as a
racemate.
Scheme 156. Attempted Axial Chirality Transfer in the Allene 286a.
t-Bu•
O
H
n-Bu
N OPh
i-Pr
t-Bu
OPh
n-Bu
i-PrHN
O1. sec-BuLi, THF,DMPU, -78 °C, 1 h
2. MeOH
286a, 84:16 e.r.
287, 89 %, 50:50 e.r.
As mentioned above, quenching the carbolithiation reaction with MeOH formed the
allenic adduct over the propargylic one. It therefore seemed to be interesting to examine
the regioselectivity of the electrophilic substitution of lithiocarbamate 288 with different
electrophiles (Scheme 157). The carbolithiation of 280a proceeded smoothly within 15 min
by adding a slight excess of n-BuLi to the THF solution at -78 °C. Next, addition of MeI
and CO2 gave the γ-adducts 289 and 290 while the -adduct 291 was isolated on addition
of Me3SiCl.
Lithiated propargylic derivatives exists in two tautomeric forms in equilibrium, as a
propargyl or allenyllithium reagent.[193]
Yet, we can here suspect that propargyllithium
288b should be stabilized at the cost of 288a by a chelating ligand at the 1-oxygen
atom.[193d]
Thereby, the γ-adducts was probably formed via a SE2’ process.

178
Scheme 157. Divergent Regioselectivity in the Electrophilic Substitution of
Lithiocarbamate 288.
2. Me3SiCl3. MeOH
291, 67 %
1. n-BuLi, THF,
-78°C, 1h
280a
2. CO2
3. MeOH
t-Bu•
CO2H
290, 34 %
O
n-Bu
NPh
i-Pr
O
t-Bu•
Li288b
O
n-Bu
N
O
Ph
i-Pr
Li
ON
O
Ph
i-Prt-Bu
n-Bu
SiMe3
ON
O
Ph
i-Prt-Bu
n-Bu
288a
t-Bu•
Me
289, 66 %
O
n-Bu
NPh
i-Pr
O
2. MeI
PhN O
i-Pr
O
t-Bu
To further evaluate the substrate scope, the carbolithiation-aryl migration of -
alkenyl-O-vinylcarbamates, -silyl-O-vinylcarbamate and -heteroaryl-O-vinylcarbamates
was investigated.
II.4.5 Extension of the Substrate Scope of the Reaction
II.4.5.1 Carbolithiation-Aryl Migration of -Alkenyl-O-vinylcarbamates
The carbamates 292 and 293 were prepared by the strategy used for -alkynyl-O-
vinylcarbamates 280 (Scheme 158). The alkenyl groups of 292 and 293 were introduced by
a Stille reaction in 35 % yield.[194]
and by the in situ conversion of 278 into the
corresponding zinc species (ZnCl2) and subsequent Pd-catalyzed cross-coupling with
bromostyrene in 70 % yield.

179
Scheme 158. Synthesis of -Alkenyl-O-vinylcarbamates 292 and 293.
O
n-BuLi
RT, 15 h
OLiDMPU, 5 h,RT
N
i-Pr
O
Cl
N O
i-Pr
O
N O
i-Pr
O Li
275 276
245b
277a, 75 %
278
t-BuLi,THF
-78 °C, 1 h
1. ZnCl2, 0 °C
2. PhCH=CHBr,Pd(PPh3)4, RT
N O
i-Pr
O
293, 70 %
N O
i-Pr
O
292, 35 %
1. I2, THF,-78 °C
2. Pd(PPh3)2Cl2, 100 °C,20 h,
SnBu3
Under the conditions used previously for carbolithiation-N to C aryl migration of -
aryl and -alkynyl-O-vinylcarbamates (THF/DMPU, -78 °C), -alkenyl-O-
vinylcarbamates 292 and 293 failed to react, returning only degradation products and
starting material, respectively. However, the corresponding alcohol 294 was obtained in
54 % yield by carrying out the carbolithiation in toluene at -78 °C in the presence of
TMEDA, thus increasing the reactivity of the organolithium by deaggregation,[180]
then
adding DMPU and increasing the temperature to -15 °C to enforce N to C aryl migration
after the carbolithiation was complete (Scheme 159). Notably, carbolithiation was
regioselective for the enol double bond rather than the styrene double bond.[106d]
Scheme 159. Carbolithiation-Aryl Migration of -Alkenyl-O-vinylcarbamate 293.
N O
i-Pr
O
293
1. n-BuLi,PhMe, TMEDA,-78 °C, 2 h
2. DMPU, -15 °C,15 h
HO n-Bu
294, 54 %

180
II.4.5.2 Carbolithiation-Aryl Migration of -Silyl-O-vinylcarbamate
The carbamate 295 was prepared in 52 % yield from O-vinylcarbamate 277a, which
underwent an -lithiation followed by trapping with PhMe2SiCl (Scheme 160).
Scheme 160. Synthesis of -Silyl-O-vinylcarbamate 295.
N O
i-Pr
O
277a
N O
i-Pr
O
SiMe2Ph
295, 52 %
1. t-BuLi, THF,1 h
2. PhMe2SiCl,RT, 2 h
Carbolithiation of -silyl-O-vinylcarbamate 295 gave only the addition product 296
in 70 % yield upon treatment with n-BuLi in THF/DMPU at -45 °C (Scheme 161). We
were unable to induce N to C aryl migration of the intermediate, perhaps because of its
inability to form a solvent separated ion pair under the conditions of the reaction since
carbanionic centers are stabilized by -silyl groups.[195]
Scheme 161. Addition of n-BuLi to -Silyl-O-vinylcarbamate 295.
N O
i-Pr
O
SiMe2Ph
295
N O
i-Pr
O
SiMe2Ph
296, 70%
n-Bu1. n-BuLi, THF, DMPU,-45 °C, 2 h
2. MeOH
II.4.5.3 Attempted Carbolithiation-Aryl Migration of -Heteroaryl-O-
vinylcarbamates
The carbamates 299 and 300 were prepared by the method used for enolcarbamates
248 in 48 and 19 % yield, respectively (Scheme 162).

181
Scheme 162. Synthesis of -Furanyl-O-vinylcarbamates 299 and 300.
N O
i-Pr
O
O
N O
i-Pr
O
O
299, 48 %
300, 19 %
Me
O
O
O
OMe
N
i-Pr
Cl
O
DMSO, NaH
245b
297
298
Attempted carbolithiation-aryl migration of 299 with n-BuLi (2.0 equiv.) in
THF/DMPU at -78 °C for 1 h failed, only degradation products were observed by 1H-NMR.
The reaction with 300 either in THF/DMPU or in PhMe/TMEDA at -78 °C for 1 h
was unsuccessful. Only the starting material was recovered, perhaps due to the electron-
rich enol double bond or the steric hindrance of the dimethylfuranyl group.
In summary, the β-carbolithiation of a range of N-aryl-N-isopropyl-O-
vinylcarbamates, coupled with tandem N to C aryl migration, nitrosation and deprotection,
provides a method for the construction of branched tertiary benzylic, propargylic, and
allylic alcohols in a single pot from simple precursors.

182
II.5 Further Extension of the Methodology
We have showed that when carbamates bearing N’-aryl groups are lithiated, a
remarkably fast nucleophilic substitution at the ispo position of the aryl group leads to
intramolecular transfer of the aryl substituent to the carbanion centre. Given that this
intramolecular nucleophilic aromatic substitution is successful even with electron rich
rings, it seems plausible that the migration of electron-rich alkenyl groups might also be
feasible. We therefore investigated the migration of styrenyl and vinyl groups and
compared with the above methods.
II.5.1 N to C Vinyl Migration in Lithiated O-Benzylcarbamates
In a preliminary experiment, the most obvious activated styrenyl migrating group
was studied. O-benzyl-N-styrenyl carbamate 305 was made in 49 % over three steps from
-methylbenzylalcohol 183d by reaction with the alkenyl isocyanate 302 available via a
Curtius rearrangement of cinnamic acid 301 in the presence of diphenylphosphoryl azide
(dppa)[196]
followed by methylation with sodium hydride and methyl iodide (Scheme 163).
Scheme 163. Preparation of O-Benzyl-N-styrenyl carbamate 305.
OH
O
NH
O
O MeNCO
HO
Me
NEt3, CH2Cl2RT, 1 h
1. NaH, DMF, 20 min, 0 °C2. MeI, RT, 1 h
1. DPPA, NEt3,benzene2. PhMe,
301
302
183d
305, 49 % over 3 steps
N O
Me
O Me
304
When 305 was treated with LDA (2.5 equiv.) in THF/DMPU (4/1 v/v) at -78 °C for 1
h, only the starting material was recovered. On repeating the rearrangement of 305 at -

183
60 °C for 1 h, a compound identified as 307 in the 1H NMR spectrum of the crude reaction
mixture was observed (Scheme 164). We presume that the reaction occurs via the
organolithium intermediates 305Li and 306 by analogy with N to C aryl migration.
Therefore, it is an alternative way to make -aryl, -alkenyl tertiary alcohols.
Scheme 164. Rearrangement of a Styrenyl Group in Lithiated Carbamate 305.
N O
Me
O Me
MeHN O
O
N O
O
Me
LiLnMeN O
OLiN O
Me
O LiLn
Me
1. LDA (2.5 equiv.),THF/DMPU,-60 °C, 1 h
2. MeOH307305
306
deprotonation
migration
protonation
Me
MeMe
305Li
307Li
More remarkably, even the simple unactivated vinyl carbamates 308 underwent
rearrangement, allowing vinylation of the organolithium (Scheme 165). Vinyl carbamates
308 were made in two steps from commercially available starting materials, employing the
usual methods. As observed for the styrenyl group, vinyl migration seems slower than aryl
migration since at -78 °C after 2 h in THF/DMPU, the rearranged product 309 was
obtained along with a significant amount of starting material. In most cases, complete vinyl
migration was observed by performing the reaction at -45 °C in THF/DMPU for 1-4 h
affording the rearranged derivatives 309 in good to moderate yields or the in-situ
deprotected rearranged product 310a in 22 % yield (Table 36).

184
Scheme 165. Vinylation of O-Benzylcarbamates 308 from -methylbenzyl alcohols 183.
N O
Me
O Me
MeHN O
O
309308
Me
NCO
HO
Me1.
NEt3, CH2Cl2RT, 1 h
2. (i) NaH, DMF,0°C, 20 min(ii) MeI, RT, 1h
183
R
R = H, Cl, CF3,Me, OMe
R
1. LDA (2.5 eq.),THF/DMPU,-45 °C
R2. MeOH
Table 36. Yields for the Vinyl Migration of O-Benzylcarbamates 308.
Entry S.M. Yield
a
(%) Product
Time
(h)
Yield
(%)
1 N O
Me
O Me
308a
69 MeHN O
O Me
309a
1 70
2 N O
Me
O Me
308b Cl
52 MeHN O
O Me
309bCl
1 54
3 N O
Me
O Me
308c
CF3
32 MeHN O
O Me
309c
CF3
1 32
4 N O
Me
O Me
308d
Me
65 MeHN O
O Me
309d
Me
4 48
5 N O
Me
O Me
308e OMe
62 HO
Me
310a OMe
4 22b
Conversion of the crude product 309a to the -tertiary allylic alcohol 310b was
achieved simply by solvolysis in mild base. Treatment of the carbamate with EtONa in
refluxing EtOH[55]
gave 310b in 48 % over two steps in a single reaction vessel (Scheme
166).
a yields over two steps
b starting material remaining

185
Scheme 166. Conversion to -Tertiary Allylic Alcohol 310b.
N O
Me
O Me
308a
HO
310b, 48 % over 2 steps
1. (i) LDA, THF, DMPU,-45 °C, 1 h(ii) MeOH
2. EtONa, EtOH,, 3 h
The low configurational stability of benzylic organolithiums bearing -oxygen
substituents under the conditions to promote rearrangement as observed previously meant
that the product 309a was not obtained enantiomerically pure from enantiomerically pure
starting material 308a. Essentially racemic 309a or returned racemic or partially racemic
starting material 308a were obtained under all conditions attempted.
Further details of the migration of the vinyl group in carbamate 308a were provided
by in-situ IR spectroscopy (Scheme 167; Figure 20). In THF at -45 °C, the carbamate 308a
displayed two absorptions at 1716 and 1625 cm-1
which disappeared over a period of 25
min on treatment with LDA being replaced by two new intermediates with absorptions at
1660 and 1616 cm-1
. Quenching the reaction at this stage returned the starting material and
the rearranged product, so we assume that the transient absorptions arise from the lithiated
carbamate 308aLi and the lithiocarbamate 309aLi. Slowly warming the reaction to room
temperature promoted the rearrangement. At 5 °C the absorption at 1660 cm-1
began to
decrease in intensity to give, about 10 min later, only the absorption at 1616 cm-1
.
Quenching the reaction with MeOH generated a peak at 1735 cm-1
assigned to the
rearranged product 309. The lag between the disappearance of 308Li and the complete
formation of 309Li hints at the intermediacy of a cyclic structure 311, but we were unable
to identify clearly absorptions corresponding to this proposed structure.

186
Scheme 167. Intermediates Detected in the Rearrangement of 308a.
N O
Me
O Me
MeHN O
O
N O
O
Me
MeN O
OLi
N O
Me
O [Li]
Me
1. LDA (1.5 eq.),THF, -45 °C
2. MeOH
309a
308a 308aLi
311
309aLi
MeMe
Me
[Li]
-45 °C to RT
H
1716 cm-1 1656 cm-1
1735 cm-11621 cm-1
Figure 20. In Situ React IR Monitoring of the Rearrangement of 308a Using LDA at -
45 °C. (a) Three-dimensional plot of absorbance versus wavenumber versus time. (b)
Several sequential two-dimensional infrared spectra at various stages of the reaction.
Carbamate 308a in
THF at -45 °C
LDA added
at -45 °C
Warmed to RT
Quenched
1716cm-1
: 308a
1735cm-1
: 309a
1664 &
1649cm-1
: 308aLi
1627 &
1616cm-1
: 309aLi
(a)

187
II.5.2 Attempted N to C Alkynyl Migration in Lithiated O-
Benzylcarbamates
Alkynyl migration seems plausible given the reactivity of ynamides as shown in
Figure 21. The electron-donating ability of the nitrogen atom (tempered by the electron-
withdrawing group) polarizes the triple bond, which allows for a high level of reactivity
with a strong differentiation of the two sp-hybridized carbon atoms.
Figure 21. General Reactivity of Ynamides.
R N
R'
EWG
R
N•R'
EWG
NucleophilesElectrophiles
(b)
Carbamate in THF at -45 °C
LDA added at -45 °C
Warmed up to RT
At RT
Quenched

188
II.5.1 Synthesis of Ynecarbamates[197]
The use of hypervalent alkynyliodonium salts as alkynylating agents[198]
and the
copper-catalyzed coupling of amides with alkynyl halides,[199]
alkynyltrifluoroborotes,[200]
terminal alkynes,[201]
1,1-dihalo-1-alkenes,[202]
or propiolic acids[203]
have emerged as
efficient strategies for the preparation of ynamides.
In most cases, oxazolidinones are the most tolerated amide substrates for the
transformation. Other classes of amides such as acyclic carbamates were all poor coupling
partners, probably because of their increased steric hindrance compared to their cyclic
homologues and their lower acidity. Gratefully, Hsung et al. communicated an efficient
and general method for the synthesis of acyclic ynecarbamates featuring a copper sulphate-
pentahydrate-1,10-phenanthroline driven catalytic system.[199c]
The synthesis of N-methyl ynecarbamate 316a was therefore achieved in 43 % yield
by coupling N-methyl-O-benzylcarbamate 314a and phenyl alkynyl bromide 315
employing CuSO4.5H2O and 1,10-phenanthroline with K3PO4 as the base in toluene at
100 °C (Scheme 168, Strategy 1). However, when N-isopropyl-O-benzylcarbamate 316b
was engaged in the reaction under the same conditions, only a poor yield was obtained,
presumably due to the lower acidity of 314b.
Tetraisopropylsilyl alkynyl bromides were showed to be a better partner,[199d]
which
would be more versatile given the readily removable TIPS group. Pleasingly, repeating the
reaction with alkynyl bromide 318 and N-isopropyl-O-benzylcarbamate 314b or -
methylated N-isopropyl-O-benzylcarbamate 314c allowed the isolation of 319a and 319b
in good yields (Scheme 168, Strategy 2). The TIPS group was then removed with TBAF in
THF at 0 °C to give ynecarbamates 320a and 320b in excellent yields.[199b]
These products
can be further transformed to aryl-substituted ynecarbamates 316b and 316c in excellent
yields by a Sonogashira cross-coupling reaction with iodobenzene using 10 mol % of
Pd(PPh3)4 and 7 mol % of CuI in DiPEA/toluene at room temperature.[204]
O-
Benzylcarbamate 314 were made in reasonable yields from either commercially available
O-benzyl chloroformate 312a or 312b formed in-situ with triphosgene, which subsequently
reacted with isopropylamine in the presence of pyridine in CH2Cl2. Bromoalkynes 315 and
318 were readily formed in good yields by bromination of the corresponding terminal
alkynes 313 and 317 upon treatment with NBS and AgNO3 in acetone.[205]
A more direct
way of making 316c would have been the deprotonation of 316b with sec-BuLi/TMEDA
in toluene at -78 °C followed by methylation with MeI, but the reaction failed.

189
Scheme 168. Synthesis of Ynecarbamates.
N O
Ph
R1
O
ONH
O
R1 Ph
Br
Ph
H
O
O
Cl
312a
R1 NH2, Py,
CH2Cl2, RT
314a, R1 = Me, 65 %
314b, R1 = i-Pr, 78 %
313
NBS, AgNO3,acetone, RT
315, 82 %
K3PO4, CuSO4.5H2O,1,10 phenanthroline
PhMe, 100 °C, 45 h
316a, R1 = Me, 43 %
316b, R1 = i-Pr, 10 %
(a) Strategy 1: Copper-catalyzed amidation of phenyl alkynyl bromide
ONH
O
i-Pr
R2
HO
Me
1. (Cl3CO)2CO, THF,NEt3, RT2. i-PrNH2, Py,CH2Cl2, RT
183d
314b, R2 = H
314c, R2 = Me, 45 %
TIPS
Br
318, 83 %
TIPS
H317
NBS, AgNO3,acetone, RT
N O
TIPS
i-Pr
OK3PO4, CuSO4.5H2O,1,10 phenanthroline
PhMe, 100 °C, 45 h
R2
319a, R2 = H 61 %
319b, R2 = Me, 65 %
TBAF, THF,0 °C, 1 h
N O
H
i-Pr
O R2
320a, R2 = H, 88 %
320b, R2 = Me, 82 %
PhI, Pd(PPh3)4,CuI, DiPEA
PhMe, RT, 15 hN O
Ph
i-Pr
O R2
316b, R2 = H, 84 %
316c, R2 = Me, 68 %
(b) Strategy 2: TIPS group as a key in the synthesis of N-isopropyl ynecarbamates

190
II.5.2 Reactivity of Lithiated Ynecarbamates
First of all, TIPS-substituted ynecarbamates 319a and 319b turned out not to be
suitable substrates for alkynyl migration (Scheme 169). The lithiated ynecarbamate 319a
generated with 2.5 equiv. of LDA was unreactive in THF/DMPU either at -78 °C or -45 °C
(Table 37, entries 1 and 2). By concentrating the reaction and using LiCl[206]
to enhance
the reactivity of the intermediate lithium species (entries 3 and 4), the lithiated product of
319a was prone to 1,2-acyl shift giving 321a. Likewise, the product 321b was formed from
319b under the same conditions (entry 5). From these results, it can be concluded that the
excess of lithium cation might complex the carbonyl oxygen atom of the carbamate group
and facilitated the attack on the carbonyl group.
Scheme 169. Attempted Alkynyl Migration in Lithiated 319a and 319b.
N O
TIPS
i-Pr
O OH
N
O
i-Pr
TIPS
LDA (2.5 eq.),THF/DMPU
319a, R2 = H
319b, R2 = Me
(Table 37)
R2
R2
321a, R2 = H
321b, R2 = Me
Table 37. Conditions for 1,2-Acyl Shift in 319a and 319b.
Entry S.M. Temp. (°C) Time (h) Additive Result
1 319a -78 2 - 319a
2 319a -45 2 - 319a
3a 319a -45 2 LiCl 319a
4b 319a -45 2 LiCl 321a, 24 %
5b 319b -45 2 LiCl 321b, 20 % + 319b
The isolation of 321a and 321b implied that the 1,2-acyl shift competed against the
1,4-alkynyl migration. We reasoned that the replacement of the TIPS group by a phenyl
group might compromise the reactivity of the acetylenic bond and thereby its increased
electrophilicity could favour 1,4-alkynyl migration.
a c = 0.13 M;
b c = 0.22 M

191
Attempted migration of the alkynyl group in 316a upon treatment with LDA in the
presence of LiCl was unsuccessful: either starting material was recovered at -78 °C in
THF/DMPU or nucleophilic attack on the carbamate C=O group was observed at -45 °C.
Attempts to reduce the susceptibility of the carbonyl group to nucleophilic attack by
replacing the N-methyl with an N-isopropyl group enabled three oxazolidinones 322, (Z)-
323 and 324 to be isolated after quenching with MeOH (Scheme 170).
Scheme 170. Cyclisation of Ynecarbamate 316b.
N O
Ph
i-Pr
O
N O
Ph
i-Pr
O LiLnN
O
Oi-Pr
Ph
LnLi
cyclisation
NO
Oi-Pr
Ph
NO
Oi-Pr
Ph
NO
Oi-Pr
Ph
OH
1. LDA (2.5 eq.),THF/DMPU, LiCl
2. MeOH
316b
324322 (Z)-323
316aLi
oxidation-protonationdeprotonation -protonation
NO
Oi-Pr
Ph
LiLn
protonshift
322Li
Oxazolidinone 322 was produced as a major product accompanied by its isomer 323
with Z-configuration (confirmed by a nOe experiment) and an unexpected cyclised product
324, regardless of the temperature (-45 °C or -20 °C) and the time used (Table 38). X-ray
crystallography confirmed the structure of 324 (Figure 21). We presumed that the reaction
proceeds through deprotonation of the carbamate to O to give benzyllithium 316aLi,
followed by attack on the acetylenic bond to give the intermediate exo,exo-3-lithiumallyl
complex 322Li, which is protonated at its γ or position giving 322 or (Z)-323, or
oxidized at its γ position forming 324. Migration of the alkynylic substituent is therefore
interrupted by the stability of the vinylic intermediate anion 322Li. The triple bond acts as
an internal electrophile toward the organolithium intermediate and it is therefore
reasonable to say that this reaction corresponds to an intramolecular carbolithiation.

192
Table 38. Conditions and Yields for Cyclisation of Ynecarbamate 316b.
Entry Temp. (°C) Time (h) Yield (%)
1 -45 5 322, 20 + 323, 8 + 324, 18
2 -45 15 322, 30 + 323, 10 + 324, 12
3 -20 2 322, 36 + 323, 6 + 324, 20
Figure 21. X-ray Crystal Structure of 324.
Despite the failure of the alkynyl group to migrate in 316b, we examined the
behaviour of -methylated ynecarbamate 316c, which in fact cyclised in the predicted
manner as illustrated in Scheme 171. Upon treatment with LDA in THF/DMPU in the
presence of LiCl at -20 °C, three oxazolidinones were isolated in poor yields: (Z)-325
might be the result of 1,2-acyl shift followed by an anti-stereoselective 5-exo-dig
cyclisation of the oxygen nucleophile onto the triple bond (Scheme 171).[207]
However, a
syn-addition and isomerization of the double bond in the more stable vinyllithium 325aLi
cannot be excluded completely. (Z)-238, isolated in poor yield, perhaps, arises from the
vinyllithium 238bLi, which was expected to be primarily formed in the intramolecular syn-
addition of the corresponding lithiated ynecarbamate 316bLi to the triple bond.[208]
In
contrast, the product (E)-238, formed in 18 % yield, probably emerged from the
thermodynamically more stable vinyllithium species 238aLi, being in equilibrium with
238bLi.

193
Scheme 171. Proposed Pathways for the Cyclisation of -Methylated Ynecarbamate 316c.
N O
Ph
i-Pr
O
316c
Me NO
Oi-Pr
Ph
NO
Oi-Pr
Ph
NO
i-Pr
O
Ph
MePh
Me PhMe Ph
1. LDA (2 eq.)THF/DMPU,LiCl, -20 °C, 2 h
2. MeOH
(E)-238, 18 %(Z)-238(Z)-325
16 % (1:0.7) (unseparable)
316cLi
N O
O
i-Pr
Ph
LiPh
NO
Oi-Pr
Ph MePh
NO
Oi-Pr
Ph
Me Ph
238bLi 238aLi
Li
NO
i-Pr
O
Ph
Me Ph325aLi
1,2-acyl shift
Li
Li
Li
NO
i-Pr
OMe Ph
Ph
Li NO
i-Pr
OMe Ph
325bLi
LiPh
II.5.3 Attempted N to C Cyano Migration in Lithiated O-
Benzylcarbamates
N-Cyanocarbamate 326 was easily made in 50 % yield in two steps in a single
reaction vessel from O-benzyl chloroformate 312a (Scheme 172).[209]
This latter compound
was treated with an excess of monosodium cyanamide, freshly prepared by reaction with
cyanamide and sodium hydroxide in 2-propanol,[210]
to give the intermediate N-
cyanocarbamate. Given its instability at room temperature[211]
it was immediately
methylated with NaH and MeI to yield N-cyanocarbamate 326.

194
Scheme 172. Preparation of N-Cyanocarbamate 326 and Attempted Migration or
Cyclisation.
N O
O
NC
Me
OCl
O1. NaNHCN,THF, RT, 15 h
2. NaH, DMF,MeI, RT, 15 h
MeHN O
O CNN
O
OMe
HNor/and
LDA (2 eq.),THF/DMPU,LiCl, -78 °C, 3 h
HO
312a
326, 50 % over 2 steps
327 328
329
In the hope of promoting the rearrangement of the cyano group or the cyclisation
affording 327 or 328, N-cyanocarbamate 326 was treated with LDA (2.0 equiv.) in
THF/DMPU at -78 °C. However, 1H NMR showed only benzylic alcohol 329 resulting
from nucleophilic attack on the carbamate C=O group. No further studies were pursued
with this compound suspecting that the strong electron-withdrawing cyano group renders
the carbonyl group extremely sensitive to nucleophilic attack under any conditions.
II.5.4 Attempted N to C Heteroaryl Migration in Lithiated O-
Benzylcarbamates
As demonstrated previously, the formation of tertiary alcohols bearing an aryl and a
heteroaryl rings failed either by -deprotonation-aryl migration of O-heteroarylcarbamates
(see Section II.2.4.1) or by β-carbolithiation-aryl migration of -heteroaryl-O-
vinylcarbamates (see Section II.4.5.3). Another alternative to access to these alcohols
would be the migration of heteroaryl groups. We therefore embarked on the migration of
the benzothiazole group.
As reported by the group, Pd-catalyzed amidation of bromopyridines proved to be an
efficient method for the synthesis of N-pyridyl-ureas.[64]
Pleasingly, this procedure could
be successfully applied to the preparation of N-benzothiazol-O-benzylcarbamate 328 in
moderate yield (34 %) with Pd2(dba)3, Xantphos, NaOt-Bu in toluene at 100 °C (Scheme
173). O-Benzylcarbamates 314 were made by the usual method as described previously

195
and bromobenzothiazole 327 was synthesised by treatment of aminobenzothiazole 326
with t-BuONO and CuBr2 in acetonitrile in 87 % yield.[212]
Scheme 173. Preparation of N-Benzothiazol-O-benzylcarbamate 328.
ON
O
Me
N
S
S
NBr
S
NNH2
t-BuONO, CuBr2,CH3CN, 60 °C
327, 87%
ONH
O
R
O
O
Cl
312a
R1 NH2, Py,
CH2Cl2, RT
314a, R = Me, 65 %314b, R = i-Pr, 78 %
326
Pd2(dba)3, Xantphos,NaOt-Bu
PhMe,100 °C, 16 h
328, 34 %
R = Me
R = i-Pr
NH
N
Si-Pr
329
N
S
N
330
i-Pr
N
S
However, upon treatment with LDA (2.0 equiv.) in THF/DMPU at -78 °C for 1 h, N-
benzothiazol-O-benzylcarbamate 328 was cleaved. Pd-catalyzed amidation with N-
isopropylcarbamate 314b was not successful: only two side products 329 and 330 were
isolated, which nonetheless indicates that the amidation had occurred.
In summary, deprotonation of benzylic carbamates bearing N’-alkenyl substitutents
generated -O-stabilized tertiary carbanions which underwent intramolecular migration of
a styrenyl group and simple vinyl groups to the carbanionic centre. This provides an
alternative route to the synthesis of -arylated, -alkenylated tertiary alcohols, which was
unsuccessful by N to C aryl migration of simple O-allylcarbamates (see Section II.3.1.4).
However, migration of alkynyl substituents failed. Terminal substituent such as a TIPS
group is too electron-rich for being nucleophilically attacked, thereby promoting the
competing 1,2-acyl shift. In contrast, a phenyl-substituted alkyne facilitated the insertion of

196
the carbon-carbon triple bond into the carbon-lithium bond but the stability of the resulting
vinylic intermediate anion interrupted the alkynyl migration.[213]

197
II.6 Conclusions and Future Work
Many classes of electrophiles, including alkylating and acylating agents, carbonyl
compounds and halogenating agents, are compatible with organolithium chemistry.
However, competing reactions such as elimination or halogen metal exchange limit the use
of some substrates, and the direct arylation or vinylation of organolithiums is a particular
challenge typically overcome by transmetallation with copper or zinc. And, the use of
organolithiums to form C-C bonds is generally limited to quenches with alkylating agents
and carbonyl compounds, with coupling to sp2 carbon atoms to conjugate additions and
substitutions with electrophiles carrying anion stabilizing groups. The intramolecular aryl
and vinyl transfers we have described overcome these limitations.
We have successfully completed the first enantioselective synthesis of the
antihistaminic agent (S,S)-clemastine 98.[214]
The synthesis featured an application of our N
to C aryl migration in lithiated carbamate 95i, which provided the enantiomerically
enriched tertiary alcohol (S)-97i, setting one of the two chiral centres. Commercially
available (S)-N-Cbz-proline 169 was used to construct the second fragment,
enantiomerically pure chloroethylpyrrolidine (S)-151. (S,S)-Clemastine was finally made
by the challenging ether formation between these two fragments, followed by
recrystallisation from fumaric acid. Moreover, this synthesis has permitted us to bring
further evidence for the invertive nature of the rearrangement of carbamates.

198
Scheme 174. Synthesis of ()-(S,S)-Clemastine.
(S)-151
(S)-97i
(S)-169 (S)-174
(S)-95i(S)-183a
N
Me
OMe
Cl
s s
N Cl
Me
s
N OMe
Cbz
sO
N CO2H
Cbz
s
HOMe
Cl
ss
MeN O
Me
Cl
O
s
Me
Cl
HO
Arndt-Eistert
(S,S)-clemastine
invertive arylmigration
The scope of N to C aryl migration has been extended to a wide range of stabilized
lithiated carbamates. One the one hand, -deprotonation of -methylated O-allyl and O-
propargylcarbamates led to arylated allylic and propargylic alcohol derivatives 204, 218
and 242 or benzylidene oxazolidinones 238 (Scheme 175 (a)). In the other hand, -
deprotonation of O-vinylcarbamates 248 allowed the formation of diarylated allylic alcohol
derivatives 251 in excellent yield (Scheme 175 (b)). However, competition between
racemization and rearrangement of these lithiated carbamates hampered to make the
rearranged products enantioselectively by stereospecific N to C aryl migration.
Nonetheless, the reactivity of lithiated O-allyl, O-propargyl and O-vinyl carbamates
bearing N-aryl groups towards migration of the aryl ring from N to C provides a valuable
method for arylating an organolithium centre adjacent to O from very easily made
substrates.

199
Scheme 175. -Arylation of Lithiated Carbamates.
N O
O
Me
Me
R1
MeHN O
O
R1
Me
R1 = C6H5, p-OMeC6H4, TIPS
MeHN O
O Me
TIPS
ON
O
Me
Me
N O
O
Me
R1
Li
Me
MeN O
O
R1
MeLi
base
204, R1 = C6H5
218, R1 = p-OMeC6H4238, 45 to 51 %
242, 66 %
aryl migration
(a) -Deprotonation of O-allyl and O-propargylcarbamates
R1 = C6H5
R1 = TIPS
N O
O
Me O
NHMe
O
R3 R3
R1
R2R2
R1
base
(b) -Deprotonation of O-vinylcarbamates
248
251, 80 to 86 %
-Carbolithiation with simple alkyllithiums followed by N to C aryl migration of -
aryl, alkenyl, and alkynyl-O-vinylcarabamates has been accomplished to obtain multiply
branched alkylalcohols in a one pot reaction in moderate to good yields (Scheme 176).[215]

200
Scheme 176. Tertiary Alcohols by Tandem -Carbolithiation and N to C Aryl Migration in
Enol Carbamates.
N O
i-Pr
OR1
X
HO
R3
R2
R1
HO
Bu
R1
R2HO
R3
R1 R2
294, 54 %281, 40 to 76 %
R3Li
R3Li
X =
X =
X =
R2
266, 27 to 81 %
R2R2
n-BuLi
So far, we have shown that the organolithium intermediate in carbamates, generated
either by deprotonation or -carbolithiation could be stabilized by an aryl, alkenyl,
propargyl and silyl group. It would be interesting to investigate the possibility of stabilising
the intermediate by a cyano, ester or acid carboxylic group, which would provide a direct
route to hydroxyl acids 335 (Scheme 177).
Scheme 177. Aryl Reaction of Hydroxyacid Derivatives.
N O CN
Me
O
MeHN O CN
O
Me
Me
N O
Me
O Me
OR2
O
MeHN O CO2R
O
Me
332, R2 = H or t-Bu
331333
334
HO CO2HMe
335R1
R1
R1
R1
R1
Tandem -carbolithiation and N to C aryl migration could be also applied in the
synthesis of (Z)-Tamoxifen 126, a therapeutic agent for the treatment of estrogen-

201
dependent breast cancer and other emerging clinical applications,[114]
as revealed from the
proposed retrosynthesis in Scheme 179. The key would be to exploite the -elimination of
the carbamate moiety as observed in Section II.3.1.2 when attempted to deprotect
carbamates 204, 96i and 96m by DIBAL-H (Scheme 178).
Scheme 178. -Elimination of Carbamate Moiety.
MeHN O
O
Ph
Me
DIBAL-H, THF, RT
Ph
204 206
MeHN O
O
Me
R96i, R = Cl96m, R = OMe
R
336, R = Cl337, R = OMe
The intermediate 338 in the proposed synthesis of (Z)-Tamoxifen 126 could therefore
be generated by -elimination of the carbamate moiety in 339, which could be obtained by
-carbolithiation of O-vinylcarbamate 340 with EtLi followed by N to C aryl migration
(Scheme 179).
Scheme 179. Proposed Retrosynthesis of (Z)-Tamoxifen.
(Z)-Tamoxifen 126
N O
O
i-Pr
MeO
O
O
i-PrHN
MeOON
-elimination
-carbolithiation
NC aryl migration
338
339340
OMe

202
Interestingly, N to C aryl migration has been extended to the vinyl migration in
carbamates 308, easily made from commercially available secondary alcohols 303 and
vinyl isocyanate (Scheme 180), leading to the -vinylation (and -alkenylation) of
alcohols. The synthesis of quaternary centres bearing heteroatoms is in many cases
challenging, and the ability to deliver alkenyl substituents to such centres is useful.
Scheme 180. Intramolecular vinylation in lithiated tertiary carbamates.
HO Ar
Me
NCO1.
2. NaH, MeI
N O
Me
O
Ar
Me
2. MeOH MeHN O Ar
O Me
HO Ar
Me
NaOEt,EtOH,
303
308 309, 32 to 70 %
310
1.LDA, THF,DMPU,-45 °C, 1 h
vinylation
Several difficulties have been encountered: (a) deprotection of rearranged carbamate
products by usual methods returned only unwanted products. However, conversion to -
tertiary alcohols was found to be feasible in a one-pot reaction with t-BuONO; (b)
instability of some N-methyl-N-aryl carbamates under basic conditions led to carbamate
cleavage, which was solved by the introduction of an N-isopropyl group instead of the N-
methyl group; (c) the competing 1,2-acyl shift of some N-aryl carbamates hampered the
rearrangement to take place; (d) in the asymmetric version, competition between
racemization and rearrangement has not allowed the synthesis of enantiomerically enriched
tertiary alcohols. Nevertheless, this was overcome to some degree by the use of ()-
sparteine or TMEDA.

203
CChhaapptteerr IIIIII :: EEXXPPEERRIIMMEENNTTAALL SSEECCTTIIOONN
III.1 General Information
All non-aqueous reactions were conducted under an atmosphere of dry nitrogen in
oven dried glassware. The temperatures quoted are those of an external bath. Cooling baths
used are acetone/dry ice for -78 °C and acetonitrile/dry ice for -42 °C.
Reagents and solvents were used as received except where stated below. All solvents
and reagents requiring purification were done so using standard laboratory techniques.[216]
Tetrahydrofuran was distilled from sodium wire and using benzophenone as a radical
indicator under nitrogen atmosphere. The Solvent Purification System (SPS) was used for
diethyl ether and toluene. Dichloromethane and diisopropylamine were obtained by
distillation from calcium hydride under nitrogen. Petrol refers to the fraction of light
petroleum ether boiling between 40 and 65 °C. Acetonitrile and DMPU were distilled,
under reduced pressure, from calcium hydride, and stored over molecular sieves. TMEDA
was distilled over KOH. ()-Sparteine was purified by Küglerhor distillation.
Triethylamine and pyridine were stored over KOH.
n-Butyllithium was obtained from Acros as a solution in hexanes (2.5 M), sec-
butyllithium in cyclohexane/hexane (92/8) (1.3 M), isopropyllithium in pentane (0.7 M),
tert-butyllithium in pentane (1.9 M), phenyllithium in n-butylether (2.0 M), methyllithium
in diethylether (1.6 M). All the above organolithium solutions were titrated prior to use
against a solution of N-benzylbenzamide.
Analytical thin layer chromatography (TLC) was carried out on pre-coated UV254
plates (Macherey-Nagel alugram. Sil G/UV254 or Machery-Nagel polygram. Alox N/UV254),
with visualisation by UV light at 254 nm, phosphomolybdic acid dip or potassium
permanganate dip. Flash column chromatography was carried out using Fluorochem
Davisil 40 – 63 µm 60 Å silica, under a positive pressure by means of compressed air,
followed by removal of the solvent under reduced pressure after purification.
Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Varian XL
300 (300 MHz), Bruker Ultrashield 400 (400 MHz) or 500 (500 MHz) spectrometer with

204
residual non-deuterated solvent as the internal standard. Carbon nuclear magnetic
resonance (13
C NMR) spectra were recorded on a Varian XL 300 (75 MHz), Bruker
Ultrashield 400 (100 MHz) or 500 (125 MHz) spectrometer. NMR data are presented as
follows: chemical shift δ (in parts per million (ppm) downfield of trimethylsilane),
multiplicity, coupling constant J (in Hz), integration, and assignment (based on chemical
shift, integration, coupling pattern and COSY, DEPT, HMQC and HMBC NMR
experiments when necessary). Splitting patterns are abbreviated as follows: singlet (s),
doublet (d), triplet (t), quartet (q), quintet (qn), septet (sep), octet (oct), nonet (non),
multiplet (m), broad (br), or a combination of these. The solvent used was deuterated
chloroform (δH: CDCl3 7.26 ppm; δC: CDCl3 77.0 ppm; δH: CD3OD 3.31 ppm; δC: CD3OD
49.0 ppm).
Low and high resolution mass spectra were recorded by staff at the University of
Manchester. EI and CI spectra were recorded on a Fisons VG Trio 2000 and high
resolution mass spectra (HRMS) were recorded on a Kratos Concept-IS mass spectrometer,
and are accurate to 0.001 Da. For the mass spectra of chlorine and bromine containing
compounds, only 35
Cl and 80
Br isotopes are reported.
Infrared spectra were recorded on an ATi Matson Genesis Series FTIR spectrometer.
Absorptions reported are sharp and strong unless otherwise stated as broad (br) or weak
(w), only absorption maxima (max) of interest are reported and quoted as wavenumbers
(cm–1
).
Melting points (m.p.) were determined on a ‘GallenKamp Melting Point’ apparatus
or a Kofler microscope melting point machine and are uncorrected.
Optical rotations [α]DT were measured on an Optical Activity AA-100 using a cell
with a pathlength of 0.25 dm. Concentrations (c) are given in grams per 100 cm3.
Chiral HPLC measurements were carried out on a Hewlett Packard Series 1050
instrument with a Diode Array Detector, using Daicel Chiralcel OD-H, Daicel Chiralpack
AD-H or (R,R)-Whelk-O1 chiral stationary phases using a mixture of hexane and
isopropanol (IPA) as eluent. Absorption was measured at 254 or 214 nm.

205
III.2 General Procedures
General Procedure A – Preparation of racemic secondary alcohols by addition of MeLi
to aldehydes.
To a solution of aldehyde (1.0 mmol) in anhydrous Et2O (c 0.20 M) was added dropwise a
solution of methyllithium (1.5 mmol) at -78 °C under nitrogen atmosphere. The resulting
reaction mixture was stirred for 2 h after which a saturated aqueous solution of NH4Cl was
added. The organic layer was washed with brine, dried over MgSO4, filtered and
concentrated under reduced pressure. The residue was purified by flash column
chromatography.
General Procedure B – Preparation of racemic secondary alcohols by reduction with
LiAlH4.
To a suspension of lithium aluminium hydride (1.5 mmol) in anhydrous Et2O (c 0.20 M) at
0 °C under nitrogen atmosphere was slowly added a solution of ketone (1.0 mmol) in
anhydrous Et2O (c 0.50 M). The mixture reaction was stirred at room temperature for 3 h
and then slowly quenched with EtOH and a saturated aqueous solution of Rochelle’s salt at
0 °C. The organic layer was washed with brine three times, dried over MgSO4, filtered and
concentrated under reduced pressure.
General Procedure C – Preparation of enantiomerically enriched secondary alcohols via
Sharpless Kinetic Resolution.
By the method reported by Sharpless et al.:[158]
To a stirred suspension of 4 Å molecular sieves (pellets) in anhydrous CH2Cl2 (c 0.20 M),
titanium isopropoxide (1.0 mmol) was added under nitrogen atmosphere. The reaction
mixture was cooled to -20 °C and ()-diisopropyltartrate (1.2 mmol) was added and stirred
for 10 min, after which alcohol (1.0 mmol) dissolved in anhydrous CH2Cl2 (c 0.20 M) was
added and stirred at -20 °C for 30 min To the above solution tert-butyl hydroperoxide (5.5
M in decane, 0.6 mmol) was added and stirred at -20 °C for 3.5 h (for 199) or 3 days (for

206
216). After completion of half of the reaction, the reaction mixture was quenched with
10 % aqueous solution of DL-tartaric acid, after which was continued for 30 min at -20 °C
and 15 h at room temperature. The organic layer was separated, washed with water, dried
over MgSO4, filtered and concentrated under reduced pressure. The residue was diluted
with Et2O (c 0.07 M) and stirred with 1.0 M NaOH (5.0 mmol) for 1 h at 0 °C. The organic
layer was then separated, washed with brine, dried over MgSO4, filtered and concentrated
under reduced pressure. The crude compound was purified by flash column
chromatography.
General Procedure D – Preparation of enantiomerically enriched secondary alcohols via
Noyori’s asymmetric transfer hydrogenation.
By the method reported by Noyori et al.:[146]
Catalyst preparation :[146]
A mixture of [RuCl2(η6-mesitylene)]2 (80 mg, 0.13 mmol), (1S,2S)-N-p-toluenesulfonyl-
1,2-diphenylethylenediamine (TsDPEN) (100 mg, 0.26 mmol), and triethylamine (0.08
cm3, 0.52 mmol) (Ru atom:TsDPEN:NEt3 molar ratio = 1:1:2) in 2-propanol (2 cm
3) was
heated at 80 °C under nitrogen atmosphere for 1 h. The orange solution was concentrated
under reduced pressure and the solid Ru complex was collected by filtration. The crude
material was washed with a small amount of water and dried under reduced pressure for 10
h to afford (R)-RuCl[(1S,2S)-p-TsNCH(C6H5)CH(C6H5)NH2](η6-p-cymene) 182 as an
orange solid.
Reaction :[52,146]
To triethylamine (2.4 mmol) stirred at 0 °C was added dropwise formic acid (6.0 mmol)
(5:2 formic acid-NEt3 azeotropic mixture). Upon warming to room temperature, ketone
(1.0 mmol) and (R)-RuCl[(1S,2S)-p-TsNCH(C6H5)CH(C6H5)NH2](η6-p-cymene) 182 (0.5
mol %) were successively added, and the reaction mixture was stirred at room temperature
under nitrogen atmosphere for 48 h. Then it was diluted with water and extracted with
EtOAc three times. The combined organic layers were washed with a saturated aqueous
solution of NaHCO3 and brine, dried over MgSO4, filtered and concentrated under reduced
pressure.

207
General Procedure E – Preparation of carbamates from isocyanates.
By the method reported by Clayden et al.:[55]
To a solution of alcohol (1.0 mmol) in anhydrous CH2Cl2 (c 0.20 M) were added aryl
isocyanate (1.2 mmol) and triethylamine (2.0 mmol). The solution was stirred at room
temperature for 15 h (or as stated) under nitrogen atmosphere. The mixture was
concentrated under reduced pressure and the product was reacted without purification.
General Procedure F – Preparation of methylated or isopropylated carbamates from
carbamates.
By the method reported by Clayden et al.:[55]
To a solution of carbamate (1.0 mmol) in anhydrous DMF (c 0.25 M) was slowly added
sodium hydride (60 % in mineral oil, 2.0 mmol) at 0 °C under nitrogen atmosphere.
Hydrogen gas was evolved. The mixture was stirred for 30 min at 0 °C and methyl iodide
or isopropyl iodide (2.5 mmol) was added and the solution stirred for 15 h (or as stated) at
room temperature. Water was added carefully and the reaction mixture was extracted with
Et2O. The organic layer was washed with water twice, dried over MgSO4, filtered and
concentrated under reduced pressure. The crude product was purified by flash column
chromatography on silica.
General Procedure G – Preparation of propargyl chloroformates.
O
O R1
ClHO
R1
Cl3CO OCCl3
O
Et2O, C, RT
By a modified method reported by Chandrasekaran et al.:[168f]
To a stirred solution of triphosgene (0.50 mmol) in anhydrous Et2O (c 0.50 M), activated
charcoal was added and stirred for 1 h at room temperature. The solution was cooled to

208
0 °C and propargyl alcohol (1.0 mmol) in anhydrous Et2O (c 1.5 M) was added dropwise.
The resultant solution was stirred for 15 h and filtered through a Celite path. The ether
layer was concentrated under reduced pressure and the resulting oil was used for the
reactions without further purification.
General Procedure H – Preparation of propargyl carbamates from propargyl
chloroformates.
N O
R2
O R1
O
O R1
ClNH
, Na2CO3
CH2Cl2, 0 °C
or
NH
, NEt3
then RT, 15 h
To a solution of propargyl chloroformate (1.0 mmol) and amine (1.0 mmol) in anhydrous
CH2Cl2 (c 0.55 M) was added triethylamine (with N-methylaniline) or sodium carbonate
(with N-isopropylaniline) (1.5 mmol) at 0 °C. The mixture was stirred at room temperature
for 15 h, concentrated under reduced pressure and purified by flash column
chromatography.
General Procedure I – Preparation of N-isopropyl-N-aryl amines by reductive amination.
NH2 O"NaBH(OAc)3"
AcOH, DCERT, 4 h
HN
RR
By the method reported by Abdel-Magid et al.:[173]
A two-necked round bottomed flame-dried flask was equipped with a magnetic stirring bar,
a nitrogen inlet, and a digital thermometer probe and was purged with nitrogen. The flask
was then charged with sodium borohydride (1.5 mmol) and 1,2-dichloroethane (c 0.30 M),

209
then was cooled to 0 °C. Glacial acetic acid (4.5 mmol) was added dropwise maintaining
the internal temperature of the flask below 5 °C and upon completion of acid addition was
allowed to further react at 0 °C for 45 min to ensure completion of formation of sodium
triacetoxyborohydride. The flask was then brought to room temperature over the course of
30 min. Amine (1.0 mmol) and glacial acetic acid (1.5 mmol) were added to the flask and
allowed to stir for 5 min, then distilled acetone (1.0 mmol) was added to the flask dropwise.
The reaction mixture was further agitated at room temperature for a period of 4 h. The
reaction was quenched by the slow addition of a saturated aqueous solution of NaHCO3
and extracted with EtOAc three times. The resulting organic layers were combined, dried
over MgSO4, filtered and concentrated under reduced pressure and the resulting crude was
purified by flash column chromatography.
General Procedure J – Preparation of carbamoyl chlorides.
HN
R4Cl3CO OCCl3
O
Py, CH2Cl2RT., 2 h
N Cl
O
R4
R R
By the method reported by Rouden et al.:[172]
To triphosgene (1.0 mmol) in anhydrous CH2Cl2 (c 0.30 M) at -78 °C were added pyridine
(2.8 mmol) then amine (2.8 mmol) slowly under nitrogen atmosphere. The reaction
mixture was warmed to room temperature, stirred for 2 h then hydrolyzed with 1 N HCl.
The product was extracted with CH2Cl2 and the organic extract was washed with a
saturated aqueous solution of NaHCO3, dried over MgSO4, filtered and concentrated under
reduced pressure. The resulted product was reacted on without purification.

210
General Procedure K – Preparation of -aryl-O-vinylcarbamates.
N Cl
O
R4
DMSO, NaH,RT, 15 h
O N O
R4
OR1
R3
R3
R2
R2
R1
By the method reported by Feringa et al.:[170]
A two-necked round bottomed flame-dried flask was equipped with a magnetic stirring bar,
a nitrogen inlet, and a digital thermometer probe and was purged with nitrogen. The flask
was then charged with sodium hydride (60 % in mineral oil, 1.5 mmol) and anhydrous
DMSO (c 2.5 M) was slowly added under nitrogen atmosphere and under vigorous stirring.
The mixture was heated at 75 °C for 45 min and cooled to room temperature. The
completion of the reaction is shown by the disappearance of sodium hydride and the
cessation of hydrogen evolution. The final solution was milky and grey-olive in colour.
Ketone (1.0 mmol) in anhydrous DMSO (c 4.0 M) was added slowly to dimsyl anion
solution, the addition being slightly exothermic and changing the color of the solution. The
solution was stirred for 15 min at room temperature and then cooled to 10 °C. The addition
of the corresponding carbamoyl chloride (1.1 mmol) in anhydrous DMSO (c 0.10 M) was
carried out while maintaining the temperature at 10 °C. After stirring 15 h at room
temperature, the solution was carefully quenched with water, extracted with n-hexane five
times and dried over MgSO4. The solvent was removed under reduced pressure and the
residue was purified by flash column chromatography or crystallisation.
General Procedure L – Preparation of simple O-vinylcarbamates.
N Cl
O
O
n-BuLi
RT, 15 h
OLi
R2
DMPU, RT, 4 h
N O
OR2

211
By a modified method reported by Snieckus et al.:[186]
A solution of n-BuLi (1.5 mmol) in anhydrous THF (c 1.3 M) was stirred at room
temperature for 15 h. The solution was then cooled to 0 °C and the corresponding N-
isopropyl-N-aryl carbamoyl chloride (1.0 mmol) in anhydrous DMPU (c 1.7 M) was added.
The solution was allowed to stir at room temperature for 4 h, which caused the color to
change to red. The reaction was quenched with a saturated aqueous solution of NH4Cl and
after extraction with Et2O, the organic layer was washed with water twice and dried over
MgSO4. After removal of the solvent under reduced pressure, the residue was purified by
flash column chromatography.
General Procedure M – -iodonisation of simple O-vinylcarbamates.
N O
OR2
1. t-BuLi, THF, -78 °C, 1 h
2. I2, THF,RT, 30 min
N O
OR2
I
To a solution of O-vinylcarbamate (1.0 mmol) in anhydrous THF (c 0.25 M) was added
tert-BuLi (1.1 mmol) at -78 °C under nitrogen atmosphere. The yellow solution was stirred
for 1 h. A solution of iodide (3.0 mmol) in anhydrous THF (c 0.15 M) was added dropwise
at -78 °C and the resulting solution was allowed to warm up to room temperature for 30
min, then diluted with EtOAc and quenched with a 25 % aqueous solution of sodium
metabisulfite. The organic layer was washed with brine, dried over MgSO4, filtered and
concentrated under reduced pressure. The residue was purified by flash column
chromatography.
General Procedure N – Preparation of -propargyl-O-vinylcarbamates.
N O
OR2
I
H
R1 , NEt3
CuI, Pd(PPh3)4, RT
N O
OR2
R1

212
By a method reported by Hoppe et al.:[188]
To iodovinylcarbamate (1.0 mmol) in triethylamine (c 0.25 M) were added the
corresponding acetylene (1.2 mmol), copper(I) iodide (2 mol %) and tetrakis
(triphenylphosphine) palladium(0) (1 mol %) under nitrogen atmosphere. The reaction
mixture was stirred for 15 h at room temperature after which a saturated aqueous solution
of NH4Cl was added. The aqueous layer was extracted with Et2O. The organic layer was
washed with water twice, dried over MgSO4, filtered and concentrated under reduced
pressure. The residue was purified by flash column chromatography.
General Procedure O – Organolithiums mediated rearrangement or alkylation/arylation
of carbamates.
A solution of freshly prepared LDA solution in anhydrous THF (c 0.35 M) or
organolithium (equiv. as stated) was added dropwise to a precooled (-78 °C or as stated)
solution of carbamate (1.0 mmol) in anhydrous THF or Et2O (c 0.07 M) with or without
anhydrous DMPU (4:1 v/v). The resulting reaction mixture was stirred under nitrogen
atmosphere after which MeOH (10 mmol) and a saturated aqueous solution of NH4Cl were
added and the reaction mixture stirred for a further 30 min while warming to room
temperature. The reaction mixture was diluted with Et2O and the organic layer was washed
with water twice. The combined organic layers were dried over MgSO4, filtered and
concentrated under reduced pressure to give a residue which was purified by flash column
chromatography.
LDA: n-BuLi (1.0 mmol) was added dropwise to a stirred solution of distilled
diisopropylamine (1.0 mmol) in distilled THF or Et2O (c 0.35 M) at 0 °C under nitrogen
atmosphere and allowed to stir for 20 min.
General Procedure P – Organolithiums mediated rearrangement or alkylation/arylation
of carbamates and quenching with t-BuONO.
Freshly prepared LDA solution in anhydrous THF (c 0.35 M) or organolithium (equiv. as
stated) was added dropwise to a precooled (-78 °C) solution of carbamate (1.0 mmol) in
anhydrous THF (c 0.15 M). The resulting reaction mixture was stirred under nitrogen

213
atmosphere after which tert-butyl nitrite (6.0 mmol) was added and the reaction mixture
was stirred at room temperature for 24 h. The mixture was diluted with Et2O and the
organic layer was washed with saturated aqueous K2CO3 three times, dried over MgSO4,
filtered and concentrated under reduced pressure to give a residue which was purified by
flash column chromatography.
General Procedure Q – The LiTMP or LDA mediated rearrangement of carbamates in
Et2O (for avoidance of racemisation).
A solution of LiTMP or LDA (2.5 mmol) in anhydrous Et2O (c 0.25 M) was added to a
solution of carbamate (1.0 mmol) in anhydrous Et2O (c 0.10 M) with or without freshly
distilled ()-sparteine (1.0 mmol) at -78 ºC under nitrogen atmosphere. The reaction was
allowed to stir at -78 ºC for 45 min and was then warmed slowly to -35 ºC. The reaction
was maintained at this temperature for 24 h. The reaction was quenched with MeOH (10
mmol) and a saturated aqueous solution of NH4Cl, diluted with Et2O and washed with
water twice. The combined organic layers were dried over MgSO4, filtered and
concentrated under reduced pressure. The residue was purified by flash column
chromatography.
General Procedure R – Deprotection of rearranged carbamate products with NaOEt
By the method reported by Clayden et al.:[55]
Rearranged product (1.0 mmol) was dissolved in EtOH (c 0.10 M) and NaOEt (21 % w/w
in EtOH, 10 mmol) was added. The solution was heated at 78 °C for 2 h. The solution was
cooled to room temperature and neutralised with glacial acetic acid. The residue was
washed with water three times. The crude product was purified by flash chromatography
on silica.
General Procedure S – Carbolithiation reactions in toluene/TMEDA.
A solution of n-BuLi (2.5 mmol) was added dropwise to a precooled solution (-78 °C) of
carbamate (1.0 mmol) in a mixture of anhydrous toluene (c 0.14 M) and distilled TMEDA

214
(2.5 mmol) under nitrogen atmosphere. The resulting reaction mixture was warmed up to -
30 °C for 2 h after which MeOH (10 mmol) and a saturated aqueous solution of NH4Cl
were added and the reaction mixture stirred for a further 30 min while warming to room
temperature. The mixture was diluted with Et2O. The organic layer was washed with water
twice and the combined organic layers were dried over MgSO4, filtered and concentrated
under reduced pressure. The residue was purified by flash column chromatography.
General Procedure T – Alkylation/arylation reactions in toluene/TMEDA followed by
addition of DMPU.
A solution of n-BuLi (2.5 mmol) was added dropwise to a precooled solution (-78 °C) of
carbamate (1.0 mmol) in a mixture of anhydrous toluene (c 0.14 M) and distilled TMEDA
(2.5 mmol) under nitrogen atmosphere. The resulting reaction mixture was warmed up to -
30 °C for 2 h after which anhydrous DMPU (4.0 mmol) was added and stirred for 15 h.
The reaction was quenched with MeOH (10 mmol) and a saturated aqueous solution of
NH4Cl and stirred for a further 30 min while warming to room temperature. The mixture
was diluted with Et2O. The organic layer was washed with water twice and the combined
organic layers were dried over MgSO4, filtered and concentrated under reduced pressure.
The residue was purified by flash column chromatography.
General Procedure U – Alkylation reactions followed by quenching with an electrophile.
To a solution of 5,5-dimethylhex-1-en-3-yn-2-yl isopropylphenylcarbamate 280a (1.0
mmol) in anhydrous THF (0.17 M) was added n-BuLi (2.0 mmol) at -78 °C under nitrogen
atmosphere. The pale yellow solution was stirred for 15 min and the electrophile (4.0
mmol) was added at -78 °C. The reaction mixture was warmed to room temperature and
stirred for 30 min. The reaction was quenched with a saturated aqueous solution of NH4Cl
and extracted with EtOAc. The organic layer was dried over MgSO4, filtered and
concentrated under reduced pressure. The residue was purified by flash column
chromatography.

215
General Procedure V – Preparation of O-benzylcarbamates.
ONH
O
R1
O
O
ClMeNH2 or i-PrNH2
CH2Cl2, Py., 0 °C then RT, 15 h
To a solution of commercially available O-benzyl chloroformate (1.0 mmol) in anhydrous
CH2Cl2 (c 0.20 M) were added methylamine (2 M in THF, 2.0 mmol) or isopropylamine
(2.0 mmol) and pyridine (2.0 mmol) at 0 °C under nitrogen atmosphere. The reaction
mixture was stirred at room temperarure for 15 h and concentrated under reduced pressure.
The residue was purified by flash column chromatography.
General Procedure W – Preparation of bromoalkynes.
R
Br
R
H
NBS, AgNO3
acetone, RT, 2 h
By the method reported by Hofmeister et al.:[205]
To a solution of acetylene (1.0 mmol) in acetone (c 0.15 M) were added N-
bromosuccinimide (1.1 mmol) and silver(I) nitrate (5 mol %). The reaction mixture was
stirred at room temperature for 2 h (cloudy solution), concentrated under reduced pressure
and the residue was purified by flash column chromatography.
General Procedure X – Preparation of ynecarbamates.
N O
R
R1
O
ONH
O
R1R
Br
K3PO4, CuSO4.5H2O,1,10 phenanthroline
PhMe, 100 °C, 45 h
By the method reported by Hsung et al.:[199c]
To a mixture of benzylcarbamate (1.0 mmol), potassium phosphate (2.0 mmol), copper(II)
sulfate pentahydrate (0.1 mmol), and 1,10-phenanthroline (0.2 mmol) in a sealed tube was

216
added a solution of bromoalkyne (1.1 mmol) in toluene (c 0.10 M). The tube was sealed
and the mixture was stirred vigourously at 100 °C for 45 h. The mixture was cooled to
room temperature, diluted with EtOAc, filtered through Celite and concentrated under
reduced pressure. The residue was purified by flash column chromatography.
General Procedure Y – Deprotection of TIPS group in ynecarbamates.
N O
TIPS O R2TBAF, THF,0 °C, 1 h
N O
H O R2
By the method reported by Hsung et al.:[199b]
To a solution of ynecarbamate (1.0 mmol) in anhydrous THF (c 0.10 M) was added
tetrabutylammonium fluoride (1.0 M in THF, 2.0 mmol) at 0 °C. The reaction mixture was
stirred at 0 °C for 1 h after quenching with a saturated aqueous solution of NH4Cl. The
mixture was diluted with Et2O and washed with brine. The organic layer was dried over
MgSO4, filtered and concentrated under reduced pressure. The residue was purified by
flash column chromatography.
General Procedure Z – Sonogashira coupling reaction with ynecarbamates.
N O
H O R2PhI, Pd(PPh3)4,CuI, DiPEA
PhMe, RT, 15 h
N O
Ph O R2
By the method reported by Hsung et al.:[204]
To ynecarbamate (1.0 mmol), phenyl iodide (1.3 mmol), copper(I) iodide (7 mol %),
tetrakis(triphenylphosphine) palladium(0) (10 mol %) were added toluene (c 0.13 M) and
diisopropylamine (c 0.07 M). The reaction mixture was stirred at room temperature under
nitrogen atmosphere in the dark for 15 h. The mixture was filtered through Celite and
concentrated under reduced pressure. The residue was purified by flash column
chromatography.

217
III.3 Experimental Procedures and Data
1-Phenylethyl 4-methoxyphenylmethylcarbamate 95e.
N O
OO
1
23
45
6
7
8
9
10
1112
13
General procedures E and F were followed. 1-Phenylethanol 183d (1.0 g, 8.2 mmol) gave
after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound ()-95e
(1.7 g, 74 % over 2 steps) as a colourless oil.
Rf 0.38 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1703 (C=O), 1512 (C=C), 1246 (C-O-C);
1H NMR (400MHz, CDCl3) 7.31-7.14 (m, 7H, aryl), 6.88 (d, J = 8.8 Hz, 2H, H-11), 5.84
(q, J = 6.2 Hz, 1H, H-5), 3.82 (s, 3H, H-13), 3.27 (s, 3H, H-8), 1.48 (br d, 3H, H-6); 13
C
NMR (100MHz, CDCl3) 157.6 (C-12), 155.1 (C-7), 142.3 (C-4), 136.2 (C-9), 128.3,
127.4, 127.2, 125.7 (CH aryl), 113.9 (C-11), 73.5 (C-5), 55.4 (C-13), 37.9 (C-8), 22.9 (C-
6); MS m/z (ES+) 308 (100 %, M+Na+); HMRS (ES+) calcd for C17H19O3N1Na1 (M+Na
+):
308.1257, found: 308.1258.
The same procedure was used to produce the equivalent enantiopure (99:1 e.r.) carbamate,
()-(R)-1-phenylethyl 4-methoxyphenylmethylcarbamate 95e (1.8 g, 76 % over 2 steps)
from (+)-(R)-1-phenylethanol 183d (1.0 g, 8.2 mmol).
[]D31
-47.2 (c 1.00, CH2Cl2); HPLC separation conditions: Chiralcel OD-H column,
hexane:2-propanol (90:10), flow rate: 1.0 mL/min; tR 10.2 min for (R)-enantiomer (major)
and 12.0 min for (S)-enantiomer (minor).

218
1-(4-Chlorophenyl)ethyl methylphenylcarbamate 95i.
N O
O
Cl
1
2
34
5
6
7
8
9
1011
12
General procedures E and F were followed. 1-(4-Chlorophenyl)ethanol 183a (300 mg, 1.95
mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title
compound ()-95i (310 mg, 55 % over 2 steps) as a colourless oil.
Rf 0.52 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1707 (C=O); 1H-NMR (500MHz, CDCl3)
δ 7.37-7.18 (m, 9H, aryl), 5.81 (q, J = 6.2 Hz, 1H, H-5), 3.31 (s, 3H, H-8), 1.47 (d, J = 6.2
Hz, 3H, H-6); 13
C-NMR (125MHz, CDCl3) δ 154.8 (C-7), 143.1 (C-9), 140.8 (C-4), 133.3
(C-1), 128.8, 128.5, 127.2, 126.1, 125.8 (CH aryl), 73.0 (C-5), 37.6 (C-8), 22.7 (C-6); MS
m/z (ES+) 312 (100 %, M+Na+); HRMS (ES+) calcd for C16H16N1O2
35Cl1Na1 (M+Na
+):
312.0783, found: 312.0773.
The same procedure was used to produce the equivalent enantiopure (99:1 e.r.) carbamate,
(S)-1-(4-chlorophenyl)ethyl methylphenylcarbamate 95i (682 mg, 74 % over 2 steps) from
()-(S)-1-(4-chlorophenyl)ethanol 183a (1.0 g).
HPLC separation conditions: Chiralcel OD-H column, hexane:2-propanol (98:2), flow rate:
1.0 mL/min; tR 14.9 min for (S)-enantiomer (major) and 15.6 min for (R)-enantiomer
(minor).

219
1-(4-Methoxyphenyl)ethyl methylphenylcarbamate 95m.
N O
O
O12
3
45
6
7
8
9
10
11
12
13
General procedures E and F were followed. 1-(4-Methoxyphenyl)ethanol 183b (2.0 g, 13
mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title
compound ()-95m (3.9 g, 96 % over 2 steps) as a colourless oil.
Rf 0.35 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1698 (C=O), 1597, 1515 (C=C), 1247 (C-
O-C); 1H NMR (400MHz, CDCl3) 7.39-7.22 (m, 7H, aryl), 6.88 (d, J = 8.8 Hz, 2H, H-3),
5.84 (q, J = 6.2 Hz, 1H, H-6), 3.82 (s, 3H, H-1), 3.33 (s, 3H, H-9), 1.51 (d, J = 6.2 Hz, 3H,
H-7); 13
C NMR (100MHz, CDCl3) 159.0 (C-2), 155.0 (C-8), 143.4 (C-10), 134.4 (C-5),
128.9, 128.7, 127.3, 125.8 (CH aryl), 113.7 (C-3), 73.5 (C-6), 55.3 (C-1), 37.6 (C-9), 22.7
(C-7); MS m/z (ES+) 308 (100 %, M+Na+); HMRS (ES+) calcd for C17H19O3N1Na1
(M+Na+): 308.1257, found: 308.1266.
The same procedure was used to produce the equivalent enantiopure (99:1 e.r.)
carbamate, (+)-(S)-1-(4-methoxyphenyl)ethyl methylphenylcarbamate 95m (661 mg, 98 %
over 2 steps) from ()-(S)-1-(4-methoxyphenyl)ethanol 183b (385 mg, 2.53 mmol).
[]D37
+30.4 (c 1.09, CH2Cl2); HPLC separation conditions: Chiralcel OD-H column,
hexane:2-propanol (90:10), flow rate: 1.0 mL/min; tR 6.7 min for (S)-enantiomer (major)
and 7.1 min for (R)-enantiomer (minor).

220
1-(3-Trifluoromethyl)phenyl)ethyl methylphenylcarbamate 95n.
N O
O
CF3
1
2
3
4
56
78
9
10
11
12
13
14
15
General procedures E and F were followed. 1-(3-Trifluoromethyl)phenyl)ethanol 183c
(439 mg, 2.31 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 5 %)
the title compound ()-95n (557 mg, 74 % over 2 steps) as a colourless oil.
Rf 0.57 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1706 (C=O), 1336 (C-F); 1H NMR
(400MHz, CDCl3) 7.52-7.23 (m, 9H, aryl), 5.89 (q, J = 6.4 Hz, 1H, H-8), 3.32 (s, 3H, H-
11), 1.49 (br d, J = 6.4 Hz, 3H, H-9); 13
C NMR (100MHz, CDCl3) 154.6 (C-10), 143.4
(C-7), 143.0 (C-12), 130.7 (q, 2JC-F = 32 Hz, C-4), 129.1, 128.9, 128.8, 126.3, 125.9 (CH
aryl), 124.3 (q, 3JC-F = 3.6 Hz, C-3), 124.0 (q,
1JC-F = 270 Hz, C-5), 122.4 (q,
3JC-F = 3.6 Hz,
C-6), 72.9 (C-8), 37.7 (C-11), 22.9 (C-9); MS m/z (ES+) 346 (100 %, M+Na+); HMRS
(ES+) calcd for C17H16O2N1F3Na1 (M+Na+): 346.1025, found: 346.1029.
The same procedure was used to produce the equivalent enantiopure (98:2 e.r.) carbamate,
(+)-(S)-1-(3-trifluoromethyl)phenyl)ethyl methylphenylcarbamate 95n (657 mg, 86 % over
2 steps) from ()-(S)-1-(3-(trifluoromethyl)phenyl)ethanol 183c (470 mg, 2.47 mmol).
[]D29
+36.2 (c 0.95, CH2Cl2); HPLC separation conditions: Chiralpack AD-H column,
hexane:2-propanol (98:2), flow rate: 1.0 mL/min; tR 12.4 min for (R)-enantiomer (minor)
and 16.6 min for (S)-enantiomer (major).

221
1-Phenylethyl 3-(trifluoromethyl)phenylmethylcarbamate 95o.
N O
O
F3C
1
2
34
5
6
7
8
9
10
1112
13
1415
General procedures E and F were followed. 1-Phenylethanol 183d (1.0 g, 8.2 mmol) gave
after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound ()-95o
(2.1 g, 82 % over 2 steps) as a colourless oil.
Rf 0.52 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1710 (C=O), 1328 (C-F); 1H NMR
(400MHz, CDCl3) 7.55 (s, 1H, H-15), 7.47 (br, 3H, aryl), 7.36-7.26 (m, 5H, aryl), 5.88 (q,
J = 6.8 Hz, 1H, H-5), 3.36 (s, 3H, H-8), 1.53 (d, J = 6.8 Hz, 3H, H-6); 13
C NMR (100MHz,
CDCl3) 154.5 (C-7), 143.7 (C-9), 141.8 (C-4), 131.1 (q, 2JC-F = 32 Hz, C-13), 129.2,
128.5, 128.4, 127.8, 125.8 (CH aryl), 124.0 (q, 1JC-F = 270 Hz, C-14), 122.3 (q,
3JC-F = 3.6
Hz, C-12 and C-15), 74.3 (C-5), 37.3 (C-8), 22.7 (C-6); MS m/z (ES+) 346 (100 %,
M+Na+); HMRS (ES+) calcd for C17H16O2N1F3Na1 (M+Na
+): 346.1025, found: 346.1029.
The same procedure was used to produce the equivalent enantiopure (98:2 e.r.) carbamate,
()-(R)-1-phenylethyl 3-(trifluoromethyl)phenylmethylcarbamate 95o (2.2 g, 85 % over 2
steps) from (+)-(R)-1-phenylethanol 183d (1.0 g, 8.2 mmol).
[]D31
-22.6 (c 1.22, CH2Cl2); HPLC separation conditions: Chiralcel OD-H column,
hexane:2-propanol (95:5), flow rate: 1.0 mL/min; tR 5.8 min for (R)-enantiomer (major)
and 6.3 min for (S)-enantiomer (minor).

222
1-(4-Methoxyphenyl)-1-phenylethyl methylcarbamate 96e.
O
O
O
NH 12
34
56
7
89
10
11
1213
General procedure O was followed with LDA (2.5 equiv.) or sec-BuLi (2.5 equiv.) and
DMPU. 1-Phenylethyl 4-methoxyphenylmethylcarbamate 95e and 1-(4-
methoxyphenyl)ethyl methylphenylcarbamate 95m (100 mg, 0.35 mmol) gave after flash
column chromatography (SiO2, Petrol:EtOAc 20 %) the title compound 96e (19 mg, 19 %
and 65 mg, 65 %) as a colourless oil.
Rf 0.16 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3364 (br, N-H), 1700 (C=O), 1520 (Amide
II)2, 1511 (C=C), 1266 (Amide III), 1251 (C-O-C);
1H NMR (400MHz, CDCl3) 7.30-
7.22 (m, 7H, aryl), 6.83 (d, J = 8.8 Hz, 2H, H-3), 4.76 (br, 1H, -NH), 3.78 (s, 3H, H-1), 2.72
(d, J = 4.8 Hz, 3H, H-13), 2.21 (s, 3H, H-7); 13
C NMR (100MHz, CDCl3) 158.4 (C-2),
155.4 (C-12), 146.5, 138.1, 128.0, 127.3, 126.9, 125.7 (aryl), 113.3 (C-3), 83.6 (C-6), 55.1
(C-1), 27.6, 27.2 (C-7 and C-13); MS m/z (ES+) 308 (100 %, M+Na+); HMRS (ES+)
calcd for C17H19O3N1Na1 (M+Na+): 308.1257, found: 308.1263.
1-(4-Chlorophenyl)-1-phenylethyl methylcarbamate 96i.
O
O
Cl
NH 1
2
34
5
6
78
9
10
1112
General procedure O was followed with LDA (2.5 equiv.) and DMPU. 1-(4-
Chlorophenyl)ethyl methylphenylcarbamate 95i (100 mg, 0.35 mmol) gave after flash
2 For carbamate (like for amide), vibration assignement is as follows: (NH) = Amide A, (C=O) = Amide I,
(C-N) contraction + (NH) opening = Amide II, (C-N) elongation+ (NH) opening = Amide III.

223
column chromatography (SiO2, Petrol:EtOAc 10 %) the title compound ()-96i (90 mg,
90 %) as a colourless oil.
Rf 0.21 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3353 (br, N-H), 1704 (C=O), 1526 (Amide
II), 1266 (Amide III); 1H-NMR (400MHz, CDCl3) δ 7.34-7.23 (m, 9H, aryl), 4.89 (br, 1H,
-NH), 2.71 (d, J = 4.8 Hz, 3H, H-12), 2.22 (s, 3H, H-6); 13
C-NMR (100MHz, CDCl3) δ
155.2 (C-11), 145.6, 144.6, 132.8, 128.2, 128.1, 127.4, 127.1, 125.7 (aryl), 83.1 (C-5), 27.3,
27.2 (C-6 and C-12); MS m/z (ES+) 312 (M+Na+) (100 %); HRMS (ES+) calcd for
C16H16N1O235
Cl1Na1 (M+Na+): 312.0762, found: 312.0762.
General procedure Q with LDA (2.5 equiv.) was used to produce the equivalent
enantiomerically enriched (84:16 e.r.) carbamate, (S)-1-(4-chlorophenyl)-1-phenylethyl
methylcarbamate 96i (51 mg, 51 %) from (S)-1-(4-chlorophenyl)ethyl
methylphenylcarbamate 95i (100 mg).
HPLC separation conditions: Chiralcel OD-H column, hexane:2-propanol (95:5), flow rate:
1.0 mL/min; tR 12.5 min for (S)-enantiomer (major) and 13.4 min for (R)-enantiomer
(minor).
1-(3-(Trifluoromethyl)phenyl)-1-phenylethyl methylcarbamate 96n.
NH
O
O
CF3
1
2
345
6
78
9
1011
12
13
14
15
General procedure O was followed with LDA (2.5 equiv.) and DMPU. 1-(3-
Trifluoromethyl)phenyl)ethyl methylphenylcarbamate 95n (100 mg, 0.31 mmol) and 1-
phenylethyl 3-(trifluoromethyl)phenylmethylcarbamate 95o gave after flash column
chromatography (SiO2, Petrol:EtOAc 20 %) the title compound ()-96n (81 mg, 81 % and
46 mg, 46 %) as a colourless oil.
Rf 0.26 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3352 (br, N-H), 1703 (C=O), 1520 (Amide
II), 1330 (C-F), 1267 (Amide III); 1H NMR (400MHz, CDCl3) 7.65 (s, 1H, H-8), 7.51-
7.49 (m, 1H, H-11), 7.41-7.39 (m, 2H, aryl), 7.32-7.25 (m, 5H, aryl), 4.85 (br, 1H, -NH),

224
2.73 (d, J = 4.8 Hz, 3H, H-15), 2.24 (s, 3H, H-6); 13
C NMR (100MHz, CDCl3) 155.0 (C-
14), 147.2 (C-7), 145.2 (C-4), 130.3 (q, 2JC-F = 32 Hz, C-9), 129.4, 128.5, 127.3, 125.8,
125.5 (CH aryl), 123.9 (q, 3JC-F = 3.6 Hz, C-11), 122.8 (q,
1JC-F = 270 Hz, C-10), 122.4 (q,
3JC-F = 3.6 Hz, C-8), 83.1 (C-5), 27.3, 27.2 (C-6, C-15); MS m/z (ES+) 346 (100 %,
M+Na+); HMRS (ES+) calcd for C17H16O2N1F3Na1 (M+Na
+): 346.1025, found: 346.1036.
General procedure Q with LiTMP (2.5 equiv.) was used to produce the equivalent
enantiomerically enriched (90:10 e.r. or 97:3 e.r. in the presence of ()-sparteine)
carbamate, (S)-1-(3-(trifluoromethyl)phenyl)-1-phenylethyl methylcarbamate 96n (76 mg,
38 % or 36 mg, 18 %) from (R)-1-phenylethyl 3-(trifluoromethyl)phenylmethylcarbamate
95o (200 mg, 0.62 mmol).
HPLC separation conditions: Chiralcel OD-H column, hexane:2-propanol (90:10), flow
rate: 1.0 mL/min; tR 7.3 min for (S)-enantiomer (major) and 8.4 min for (R)-enantiomer
(minor).
1-(4-Chlorophenyl)-1-phenylethanol 97i.[52]
HO
Cl
1
2
34
5
6
78
9
10
General procedure R was followed. 1-(4-Chlorophenyl)-1-phenylethyl methylcarbamate
96i (50 mg, 0.17 mmol) gave after flash chromatography (SiO2, Petrol:EtOAc 2 %) the
title compound ()-97i (36 mg, 90 %) as a colourless oil.
Rf 0.66 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3411 (br, O-H); 1H-NMR (400MHz,
CDCl3) δ 7.38-7.21 (m, 9H, aryl), 2.11 (br, 1H, -OH), 1.91 (s, 3H, H-6); 13
C-NMR
(100MHz, CDCl3) δ 147.4, 146.5, 132.7, 128.3, 128.2, 127.3, 127.2, 125.7 (aryl), 75.8 (C-
5), 30.8 (C-6); MS m/z (ES+) 215 (100 %, M-H2O+H+); HRMS (ES+) calcd for
C14H13O135
Cl1 (M+): 232.0646, found: 232.0649. Spectral data matched the published
values.[52]

225
The same procedure was used to produce the equivalent enantiomerically enriched (84:16
e.r.) alcohol, (+)-(S)-1-(4-chlorophenyl)-1-phenylethanol 97i (70 mg, 87 %) from (S)-1-(4-
chlorophenyl)-1-phenylethyl methylcarbamate 96i (100 mg).
[]D29
+5.3 (c 0.9, CHCl3) [lit.[52]
[]D22
+14.8 (c 6.5, CHCl3, 98:2 e.r. (S):(R))]; HPLC
separation conditions: Chiralpack AD-H column, hexane:2-propanol (97:3), flow rate: 1.0
mL/min; tR 12.9 min for (R)-enantiomer (minor) and 14.1 min for (S)-enantiomer (major).
1-Phenyl-1-(3-trifluoromethylphenyl) ethanol 97n.[52]
HO
CF3
1
2
34
5
6
78
9
1011
12
13
General procedure R was followed. 1-(3-(Trifluoromethyl)phenyl)-1-phenylethyl
methylcarbamate 96n (35 mg, 0.11 mmol) gave after flash chromatography (SiO2,
Petrol:EtOAc 2 %) gave the title compound ()-97n (22 mg, 78 %) as a colourless oil.
Rf 0.56 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3400 (br, O-H), 1329 (C-F); 1H NMR
(400MHz, CDCl3) 7.69 (br, 1H, H-8), 7.47-7.41 (m, 2H, aryl), 7.35-7.18 (m, 6H, aryl),
2.16 (s, 1H, -OH), 1.90 (s, 3H, H-6); 13
C NMR (100MHz, CDCl3) 149.0, 147.1 (C aryl),
130.6 (q, 2JC-F = 32 Hz, C-9), 129.4, 128.6, 128.4, 127.4, 125.8 (CH aryl), 123.7 (q,
3JC-F =
3.6 Hz, C-11), 122.8 (q, 1JC-F = 270 Hz, C-10), 122.3 (q,
3JC-F = 3.6 Hz, C-8), 76.0 (C-5),
30.8 (C-6); MS m/z (ES-) 265 (100 %, M-H+); HRMS (ES-) calcd for C15H13O1F3 (M
+):
266.0907, found: 266.0911. Spectral data matched the published values.[52]
The same procedure was used to produce the equivalent enantiomerically enriched (90:10
e.r.) alcohol, (+)-(S)-1-phenyl-1-(3-trifluoromethylphenyl) ethanol 97n (21 mg, 75 %)
from (S)-1-(3-(trifluoromethyl)phenyl)-1-phenylethyl methylcarbamate 96n (34 mg, 0.10
mmol).
[]D31
+19.2 (c 3.50, CHCl3) [lit.[52]
+29.3 (c 5.50, CHCl3, 99:1 e.r. (S):(R))]; HPLC
separation conditions: Chiralcel OD-H column, hexane:2-propanol (90:10), flow rate: 1.0
mL/min; tR 5.8 min for (S)-enantiomer (major) and 6.5 min for (R)-enantiomer (minor).

226
Assignment of stereochemistry of (S)-97n.
The absolute stereochemistry of ()-(S)-97n was deduced by comparison with literature
data of (a) the optical rotation and (b) relative retention times of the two enantiomers on
the chiral stationary phase Chiralcel OD-H.
Thus:
• Material obtained from the rearrangement is dextrorotatory, with []D31
= +19.2 (c
3.50, CHCl3). Aggarwal[52]
reported that (S)-97n is dextrorotatory and (R)-97n is
laevorotatory.
• The major enantiomer from the rearrangement is the faster eluting on Chiralcel OD-
H. Aggarwal[52]
reported that (S)-97n is faster eluting on Chiralcel OD-H.
()-2-{2-[1-(4-Chlorophenyl)-1-phenylethoxy]ethyl}-1-methylpyrroline (Clemastine)
98.[128]
16
15 8
9
10
11
12
13
14
1
2
34
5
6
7
N O
Cl17
1-(4-Chlorophenyl)-1-phenylethanol (S)-97i (118 mg, 0.42 mmol) and NaH (60 % in
mineral oil, 28 mg, 0.56 mmol) were dissolved in anhydrous toluene (3 cm3) and refluxed
under nitrogen atmosphere for 3 h. The solution was allowed to cool to room temperature
and 2-(2-chloroethyl)-1-methylpyrrolidine (S)-151 (45 mg, 0.28 mmol) in anhydrous
toluene (1.3 cm3) was added. The reaction mixture was stirred at reflux for 20 h. The
reaction mixture was quenched with water and diluted with EtOAc. The aqueous layer was
separated and extrated with EtOAc (2 10 cm3). The combined organic layers were dried
over MgSO4, filtered, and concentrated under reduced pressure. The crude oil was purified
by flash column chromatography (SiO2, Petrol:EtOAc:NEt3 80:20:1) to provide the title
compound 98 (50 mg, 48 %) as a colourless oil.
Rf 0.14 (Petrol:EtOAc:NEt3 60:40:1); []D31
37.4 (c 2.04, EtOH) [lit.[128]
[α]D20
33.7 (c
2.00, EtOH) for (S,S)-isomer]; 1H-NMR (400MHz, CDCl3) δ 7.35-7.20 (m, 9H, aryl), 3.34-
3.20 (m, 2H, H-11), 3.08-3.03 (m, 1H, H-16), 2.32 (s, 3H, H-17), 2.20-2.09 (m, 2H, H-16,
H-13), 2.07-1.99 (m, 1H, H-12), 1.93-1.81 (m, 1H, H-14), 1.84 (s, 3H, H-6), 1.79-1.61 (m,

227
2H, H-15), 1.51-1.37 (m, 2H, H-12, H-14); 13
C-NMR (100MHz, CDCl3) δ 146.1, 145.7,
132.4, 128.1, 128.0, 126.9, 126.7, 126.6 (aryl), 80.0 (C-5), 63.8 (C-13), 60.4 (C-11), 57.1
(C-16), 40.4 (C-17), 34.4 (C-12), 30.9 (C-14), 25.5 (C-6), 21.9 (C-15); MS m/z (ES+) 344
(100 %, M+H+); HRMS (ES+) calcd for C21H27N1O1
35Cl1 (M+H
+): 344.1776, found:
344.1787.
()-(2S)-2-{2-[(1S)-1-(4-Chlorophenyl)-1-phenylethoxy]ethyl}-1-methylpyrroline
monofumarate 98H+ (Clemastine Fumarate).
[128]
N O
ClH
O2CCO2H
2-{2-[1-(4-Chlorophenyl)-1-phenylethoxy]ethyl}-1-methylpyrroline 98 (50 mg, 0.15 mmol)
and fumaric acid (20 mg) were dissolved by heating in EtOH (0.5 cm3), then the mixture
was allowed to cool to room temperature slowly to afford 98H+
•fum- (47 mg, 66 %) as a
white powder. []D28
17.3 (c 2.08, MeOH) [lit.[128]
[α]D20
16.9 (c 2.00, MeOH) for (S,S)-
isomer]; m.p. 170-172 °C, [lit.[128]
177-178 °C]. The salt was recrystallised from EtOH to
give (S,S)-98H+
•fum- as a white powder with an []D
28 17.1 and a melting point of 176-
178 °C.
()-(S)-2-(2-Chloroethyl)-1-methylpyrrolidine Hydrochloride 151.[140]
N
Me
1
2
3
45
6
7
Cl
�HCl
To a solution of 2-(1-methylpyrrolidin-2-yl)ethanol (S)-175 (100 mg, 0.93 mmol) in
chloroform (1.5 cm3), a solution of thionyl chloride (0.16 cm
3, 2.6 mmol) in chloroform
(0.5 cm3) was added dropwise at 0 °C, and the resulting mixture was refluxed for 2 h and
concentrated under reduced pressure. The brown residue was triturated with MeOH, and a

228
small amount of decolorizing charcoal was added. The mixture was refluxed for 30 min,
filtered through Celite, and the filtrate was concentrated under reduced pressure to provide
the title compound (S)-151HCl as a yellow solid. Recrystallisation from
ethanol:diethylether provided 130 mg (92 %) of (S)-151HCl as a white solid.
Rf 0.41 (CH2Cl2:MeOH:NH4OH 95:5:1); []D31
49.8 (c 1.10, CHCl3); m.p. 119-121 °C
(EtOH/Et2O); IR max (neat/cm-1
) 3420 (br); 1H-NMR (400MHz, CDCl3) δ 12.5 (br, 1H,
N+H), 3.92-3.90 (m, 2H, H-1), 3.38-3.30 (m, 1H, H-3), 2.89-2.82 (m, 1H, H-6), 2.85-2.81
(m, 4H, H-6 and H-7), 2.58-2.39 (m, 1H, H-5), 2.48-2.39 (m, 1H, H-5), 2.36-2.23 (m, 2H,
H-2 and H-4), 2.09-1.93 (m, 2H, H-2 and H-4); 13
C-NMR (100MHz, CDCl3) δ 66.1 (C-3),
55.9 (C-1), 41.2 (C-6), 39.2 (C-7), 32.3 (C-2), 29.1 (C-4), 21.3 (C-5); MS m/z (ES+) 148
(100 %, M+H+); HRMS (ES+) calcd for C7H15N1
35Cl1 (M+H
+): 148.0885, found:
148.0888.
The free base of (S)-151 was obtained as a colourless oil by treatment with aqueous KOH,
extraction with CH2Cl2, drying over MgSO4, and concentrated under reduced pressure. The
free base is used immediately without storage.
(S)-N-Carboxylbenzyldiazoketone intermediate 171.[134,137]
N
O OO
N2
123
45
6
7
8 910
11
12
To a solution of 1-(benzyloxycarbonyl)pyrrolidine-2-carboxylic acid (S)-169 (5.0 g, 20
mmol) in anhydrous DMF (35 drops) and anhydrous CH2Cl2 (65 cm3) at 0
oC, oxallyl
chloride (2.3 cm3, 27 mmol) was added dropwise under nitrogen atmosphere. The reaction
mixture was allowed to rise to room temperature and stirred for 2 h, resulting in a pale
yellow solution. The solvent was evaporated under reduced pressure to yield a pale yellow
oil. The crude product was immediately dissolved in a mixture of THF (80 cm3) and
CH3CN (80 cm3) and cooled to 0
oC under nitrogen atmosphere. Anhydrous triethylamine

229
(5.6 cm3, 40 mmol) and 2 M trimethylsilyldiazomethane solution (22 cm
3, 44 mmol) were
added, in turn, and the reaction mixture stirred for 5 h at 0 oC. The solution was quenched
with glacial acetic acid (20 cm3) and concentrated under reduced pressure to afford a dark
brown oil. The residue was purified by flash column chromatography (SiO2, Petrol:EtOAc
30 %) to provide the title compound (S)-171 (4.4 g, 80 %) as a yellow solid.
Rf 0.35 (Petrol:EtOAc 5:5); m.p. 58-60 °C (CH2Cl2); IR max (neat/cm-1
) 2105 (C=N),
1702 (C=O, carbamate), 1698 (C=O, ketone); 1H-NMR (400MHz, CDCl3) (mixture of
rotamers) δ 7.37-7.33 (m, 5H, aryl), 5.48 (br, 0.5H, H-1, rotamer 1), 5.25 (br, 0.5H, H-1,
rotamer 2), 5.20-5.06 (m, 2H, H-8), 4.35-4.33 (m, 0.5H, H-3, rotamer 1), 4.29-4.27 (m,
0.5H, H-3, rotamer 2), 3.60-3.45 (m, 2H, H-6), 2.15-2.02 (m, 2H, H-4), 1.99-1.85 (m, 2H,
H-5); 13
C-NMR (100MHz, CDCl3) (mixture of rotamers) δ 195.2, 194.4 (C-2), 155.2,
154.5 (C-7), 136.4, 136.3 (C-9), 128.5, 128.4, 128.0, 127.9, 127.8 (CH aryl), 67.2 (C-8),
64.0, 63.9 (C-1), 53.3, 52.6 (C-3), 47.3, 46.9 (C-6), 31.2, 29.6 (C-4), 24.3, 23.5 (C-5); MS
m/z (ES+) 296 (100 %, M+Na+); HRMS (ES+) calcd for C14H16N3O3 (M+H
+): 274.1113,
found: 274.1186. Spectral data matched published values.[137]
()-(S)-2-Methoxycarbonylmethylpyrrolidine-1-carboxylic acid benzyl ester
174.[137,217]
N
O O
1
2
3
4
56
7
8
910
11
12
O
O
13
To a solution of N-Cbz-diazoketone (S)-171 (4.3 g, 16 mmol) in anhydrous MeOH (50 cm3)
at room temperature under nitrogen atmosphere, a solution of silver benzoate (0.36 g, 1.6
mmol) in anhydrous triethylamine (6.6 cm3) was added dropwise. The mixture was stirred
at room temperature for 3 h, changing colour from yellow to black as an exothermic
reaction took place, generating N2 gas. A solution of sodium metabisulfite 20 % (50 cm3)
was added and the mixture filtered to remove silver residue. The solution was extracted
with EtOAc (2 30 cm3) and the combined organic layers washed with brine (30 cm
3),
dried over MgSO4, filtered and concentrated under reduced pressure. The crude oil was

230
purified by flash column chromatography (SiO2, Petrol:EtOAc 20 %) to provide the title
compound (S)-174 (3.4 g, 78 %) as a colourless oil.
Rf 0.58 (Petrol:EtOAc 5:5); []D31
42.7 (c 1.02, CHCl3) [lit.[217]
[]D25
36.6 (c 1.33,
CHCl3, >99:1 e.r. (S):(R))]; IR max (neat/cm-1
) 1734 (C=O, ester), 1700 (C=O, carbamate);
1H-NMR (400MHz, CDCl3) (mixture of rotamers) δ 7.38-7.30 (m, 5H, aryl), 5.15 (s, 1H,
H-9, rotamer 1), 5.12 (s, 1H, H-9, rotamer 2), 4.27-4.21 (m, 1H, H-4), 3.66 (s, 1.5H, H-1,
rotamer 1), 3.63 (s, 1.5H, H-1, rotamer 2), 3.35-3.40 (m, 2H, H-7), 3.00 (dd, J = 16.0 and
4.0 Hz, 0.5H, H-3, rotamer 1), 2.82-2.77 (dd, J = 16.0 and 4.0 Hz, 0.5H, H-3, rotamer 2),
2.38-2.31 (m, 1H, H-3), 2.13-2.04 (m, 1H, H-5), 1.90-1.82 (m, 2H, H-6), 1.80-1.73 (m, 1H,
H-5); 13
C-NMR (100MHz, CDCl3) (mixture of rotamers) δ 171.9, 171.7 (C-2), 154.6 (C-8),
136.9, 136.8 (C-10), 128.4, 127.9, 127.8 (CH aryl), 66.8, 66.6 (C-9), 54.5, 53.9 (C-4), 51.5
(C-1), 46.7, 46.4 (C-7), 39.07, 38.1 (C-3), 31.3, 30.6 (C-5), 23.5, 22.7 (C-6); MS m/z (ES+)
300 (100 %, M+Na+); HRMS (ES+) calcd for C15H20N1O4 (M+H
+): 278.1314, found:
278.1387. Spectral data matched published values.[137]
()-2-((S)-1-Methylpyrrolidin-2-yl)ethanol 175.[138,139]
N
Me
1
2
3
45
6
7
OH
To a solution of 2-methoxycarbonylmethylpyrrolidine-1-carboxylic acid benzyl ester (S)-
174 (1.50 g, 5.41 mmol) in anhydrous THF (30 cm3) at 0 °C under nitrogen atmosphere,
lithium aluminium hydride (1 M in THF, 16 cm3, 16 mmol) was added dropwise. The
resulting mixture was warmed at 45 °C for 45 min. The reaction mixture was diluted with
Et2O (15 cm3) at 0 °C and quenched with water (4 cm
3). An aqueous solution of 15 %
NaOH (4 cm3) was added followed by addition of further water (2 cm
3). The resulting
mixture was stirred for 15 min and anhydrous MgSO4 was added into the mixture. Further
stirred for 15 min and the solid residue was filtered by suction. The filtrate was
concentrated under reduced pressure to afford an amber oil as a crude. The crude product
was purified by Isolute® SCX (Strong Cation Exchange) cartridge (manufactured by

231
Biotage) using CH2Cl2:MeOH:NH4OH 7 N in MeOH (90:10:1) to provide the title
compound (S)-175 (0.42 g, 60 %) as a colourless oil.
Rf 0.24 (MeOH/NH4OH 1 %); []D31
–78.9 (c 1.03, EtOH) [lit.[139]
[α]D20
58.4 (c 1.00,
acetone]; IR max (neat/cm-1
) 3287 (br, O-H), 1055 (C-O); 1H-NMR (400MHz, CDCl3) δ
5.61 (br, 1H, O-H), 4.00 (td, J = 10.9 and 2.8 Hz, 1H, H-1), 3.68 (dt, J = 10.9 and 4.4 Hz,
1H, H-1), 3.08-3.03 (m, 1H, H-6), 2.62-2.56 (m, 1H, H-3), 2.36 (s, 3H, H-7), 2.18-2.12 (m,
1H, H-6), 2.05-1.96 (ddt, J = 14.7, 10.9 and 4.4 Hz, 1H, H-2), 1.93-1.73 (m, 4H, H-4 and H-
5), 1.46 (dq, J = 12.0 and 4.4 Hz, 1H, H-2); 13
C-NMR (100MHz, CDCl3) δ 65.2 (C-1),
60.3 (C-3), 57.0 (C-6), 40.9 (C-7), 31.1 (C-2), 28.1 (C-4), 23.2 (C-5); MS m/z (ES+) 130
(100 %, M+H+); HRMS (ES+) calcd for C7H15N1O1 (M
+): 129.1152, found: 129.1148.
()-(S)-2-(2-Hydroxyethyl)pyrrolidine-1-carboxylic acid benzyl ester 179.[145,218]
N
O O
1
2
3
45
6
7
8 910
11
12
OH
2-Methoxycarbonylmethylpyrrolidine-1-carboxylic acid benzyl ester (S)-174 (3.3 g, 12
mmol) was dissolved in anhydrous THF (60 cm3) and cooled to 0
oC under nitrogen
atmosphere. Lithium borohydride (0.91 g, 42 mmol) was added and the reaction mixture
stirred for 96 h at room temperature. The solution was quenched to pH 7 with glacial acetic
acid. It was diluted with H2O (30 cm3) and extracted with EtOAc (2 30 cm
3). The
combined organic layers were dried over MgSO4, filtered and concentrated under reduced
pressure to afford a colourless oil, which was purified by flash column chromatography
(SiO2, Petrol:EtOAc 30 %) to provide the title compound (S)-179 (2.1 g, 71 %) as a
colourless oil.
Rf 0.23 (Petrol:EtOAc 5:5); []D32
7.6 (c 1.10, CHCl3) [lit.[218]
[]D25
+7.2 (c 1.00,
CHCl3), >97:3 e.r. (R):(S))]; IR max (neat/cm-1
) 3435 (O-H), 1677 (C=O); 1H-NMR
(400MHz, CDCl3) δ 7.36-7.29 (m, 5H, aryl), 5.14 (s, 2H, H-8), 4.25-4.20 (m, 1H, H-3),

232
4.14-4.11 (m, 1H, -OH), 3.60-3.57 (m, 2H, H-1), 3.44-3.38 (m, 2H, H-6), 2.05-1.95 (m, 1H,
H-4), 1.93-1.88 (m, 2H, H-5), 1.76-1.63 (m, 2H, H-4 and H-2), 1.54-1.47 (m, 1H, H-2); 13
C-
NMR (100MHz, CDCl3) δ 156.7 (C-7), 136.6 (C-9), 128.5, 128.0, 127.8 (CH aryl), 67.1
(C-8), 59.0 (C-1), 54.3 (C-3), 46.3 (C-6), 38.2 (C-2), 31.1 (C-4), 23.5 (C-5); MS m/z (ES+)
272 (100 %, M+Na+); HRMS (ES+) calcd for C14H19N1O3Na1 (M+Na
+): 272.1257, found:
272.1259. Spectral data matched published values.[218]
(S)-2-(2-Iodoethyl)pyrrolidine-1-carboxylic acid benzyl ester 180.[219]
N
O O
1
2
3
45
6
7
8 910
11
12
I
2-(2-Hydroxyethyl)pyrrolidine-1-carboxylic acid benzyl ester (S)-179 (300 mg, 1.20 mmol)
was dissolved in a mixture of anhydrous Et2O and anhydrous CH3CN (3:2, 30 cm3) under
nitrogen atmosphere and cooled to 0 °C. Imidazole (120 mg, 1.68 mmol) and
triphenylphosphine (410 mg, 1.56 mmol) were added and the mixture was treated portion-
wise with iodine, until an orange colour persisted and a white solid appeared. After stirring
for 1 h at 0 °C, the reaction was completed (TLC control). The reaction mixture was
quenched with a saturated aqueous solution of sodium metabisulfite (25 %). It was
extracted with Et2O (3 20 cm3) and the combined organic layers were washed with brine
(20 cm3), dried over MgSO4, filtered and concentrated to give a pale yellow oil. The
residue was purified by flash column chromatography (SiO2, Petrol:EtOAc 5 % + NEt3
1 %) to give the title compound (S)-180 (320 mg, 74 %) as a yellow oil.
Rf 0.65 (Petrol:EtOAc 2:1); IR max (neat/cm-1
) 1686 (C=O); 1H-NMR (400MHz, CDCl3)
(mixture of rotamers) δ 7.36-7.30 (m, 5H, aryl), 5.13-5.10 (m, 2H, H-8), 3.96-3.90 (m, 1H,
H-3), 3.50-3.35 (m, 2H, H-1), 3.19-3.01 (m, 2H, H-6), 2.38-2.22 (m, 1H, H-4), 1.99-1.85 (m,
4H, H-2 and H-5), 1.68-1.62 (m, 1H, H-4); 13
C-NMR (100MHz, CDCl3) (mixture of
rotamers) δ 155.1, 154.9 (C-7), 136.8 (C-9), 128.4, 128.1, 127.9, 127.8 (CH aryl), 66.9,
66.6 (C-8), 58.6, 57.8 (C-3), 46.5, 46.3 (C-6), 39.0, 38.7 (C-2), 30.4, 29.9 (C-4), 23.8, 23.0

233
(C-5), 1.47 (C-1); MS m/z (ES+) 382 (100 %, M+Na+); HRMS (ES+) calcd for
C14H18N1O2Na1 (M+Na+): 382.0384, found: 382.0274.
()-(S)-1-(4-Chlorophenyl)ethanol 183a.[52]
1
2
34
5
6
HO
Cl
General procedure D was followed. 1-(4-chlorophenyl)ethanone 181a (500 mg, 3.25 mmol)
gave after flash column chromatography (SiO2, Petrol:EtOAc 15 %) the title compound
(S)-183a (450 mg, 91 %, 99:1 e.r.) as a colourless oil.
Rf 0.15 (Petrol:EtOAc 8:2); []D28
36.6 (c 1.08, CH2Cl2) [lit.[52]
[]D24
45.3 (c 2.00,
CH2Cl2, 98:2 e.r. (S):(R))]; IR max (neat/cm-1
) 3364 (br, O-H), 1088 (C-O); 1H-NMR
(400MHz, CDCl3) δ 7.31 (br, 4H, aryl), 4.88 (q, J = 6.4 Hz, 1H, H-5), 1.83 (br, 1H, -OH),
1.47 (d, J = 6.4 Hz, 3H, H-6); 13
C-NMR (100MHz, CDCl3) δ 144.2 (C-4), 133.0 (C-1),
128.6, 126.8 (CH aryl), 69.7 (C-5), 25.3 (C-6). Spectral data matched the published
values.[52]
HPLC separation conditions: Chiralcel OD-H column, hexane:2-propanol (98:2), flow rate:
1.0 mL/min; tR 12.9 min for (S)-enantiomer (major) and 13.8 min for (R)-enantiomer
(minor).
()-(S)-1-(4-Methoxyphenyl)ethanol 183b.[52]
12
3
456HO
O
7
General procedure D was followed. 1-(4-Methoxyphenyl)ethanone 181b (500 mg, 3.33
mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 20 %) the title
compound (S)-183b (385 mg, 76 %, 99:1 e.r.) as a colourless oil.

234
Rf 0.43 (Petrol:EtOAc 6:4); []D37
-45.4 (c 1.03, CH2Cl2). [lit.[52]
[α]D24
-50.5 (c 1.02,
CH2Cl2, 98:2 e.r. (S):(R))]; IR max (neat/cm-1
) 3364 (br, O-H), 1615, 1514 (C=C), 1246
(C-O-C), 1035 (C-O); 1H NMR (400MHz, CDCl3) 7.31 (d, J = 8.4 Hz, 2H, H-4), 6.90 (d,
J = 8.4 Hz, 2H, H-3), 4.85 (q, J = 6.4 Hz, 1H, H-6), 3.82 (s, 3H, H-1), 2.09 (br, 1H, -OH),
1.49 (d, J = 6.4 Hz, 3H, H-7); 13
C NMR (100MHz, CDCl3) 158.9 (C-2), 138.0 (C-5),
126.7 (C-4), 113.8 (C-3), 69.9 (C-6), 55.3 (C-1), 25.0 (C-7); MS m/z (ES+) 175 (100 %,
M+Na+); HMRS (ES+) calcd for C9H12O2Na1 (M+Na
+): 175.1797, found: 175.0730.
Spectral data matched the published values.[52]
HPLC separation conditions: Chiralcel OD-H column, hexane:2-propanol (98:2), flow rate:
1.0 mL/min; tR 32.0 min for (R)-enantiomer (minor) and 37.4 min for (S)-enantiomer
(major).
1-(3-(Trifluoromethyl)phenyl)ethanol 183c.[52,220]
HOCF3
1
2
3
4
56
78
9
General procedure B was followed. 1-[3-(Trifluoromethyl)phenyl]ethanone 181c (500 mg,
2.65 mmol) gave the title compound ()-183c (491 mg) as a colourless oil which was
reacted without purification.
Rf 0.61 (Petrol:EtOAc 6:4); IR max (neat/cm-1
) 3338 (br, O-H), 1328 (C-F); 1H NMR
(400MHz, CDCl3) 7.65 (s, 1H, H-6), 7.57-7.45 (m, 3H, aryl), 4.97 (qd, J = 6.4 and 3.6 Hz,
1H, H-8), 1.91 (d, J = 3.6 Hz, 1H, -OH), 1.51 (d, J = 6.4 Hz, 3H, H-9); 13
C NMR (100MHz,
CDCl3) 146.7 (C-7), 130.9 (q, 2JC-F = 32 Hz, C-4), 128.9, 128.7 (C-1, C-2), 124.2 (q,
3JC-
F = 3.6 Hz, C-3), 124.1 (q, 1JC-F = 270 Hz, C-5), 122.2 (q,
3JC-F = 3.6 Hz, C-6), 69.8 (C-8),
25.4 (C-9); MS m/z (ES+) 173 (100 %, M-H2O+H+); HMRS (ES+) calcd for C9H8F3 (M-
H2O+H+): 173.1547, found: 173.0573. Spectral data matched published values.
[220]
General procedure D was used to produce the equivalent enantiopure (99:1 e.r.) alcohol,
()-(S)-1-(3-trifluoromethyl)phenyl)ethanol 183c (470 mg, 94 %) as a colourless oil from

235
1- (3-(trifluoromethyl)phenyl)hanone 181c (500 mg, 2.66 mmol) after flash column
chromatography (SiO2, Petrol:EtOAc 15 %).
[]D25
-27.0 (c 1.03, CH2Cl2) [lit.[52]
[α]D24
-21.9 (c 1.40, CH3OH, 92:8 e.r. (S):(R))]. HPLC
separation conditions: Chiralcel OD-H column, hexane:2-propanol (98:2), flow rate: 1.0
mL/min; tR 17.2 min for (S)-enantiomer (major) and 18.6 min for (R)-enantiomer (minor).
2-Vinyl pyrrolidine-1-carboxylic acid benzyl ester 186.[221]
N
O O
1
2
3
45
6
7
8 910
11
12
Rf 0.53 (Petrol:EtOAc 6:3); IR max (neat/cm-1
) 1686 (C=O); 1H NMR (300MHz, CDCl3)
(mixture of rotamers) 7.46-7.24 (m, 5H, aryl), 5.86-5.75 (m, 1H, H-2), 5.14 (s, 2H, H-8),
5.11-5.02 (m, 2H, H-1), 4.42 (m, 1H, H-3), 3.52-3.48 (m, 2H, H-6), 2.07-1.74 (m, 4H, H-4,
H-5); 13
C NMR (100MHz, CDCl3) (mixture of rotamers) 155.0, 154.7 (C-7), 138.4,
137.8 (C-9), 136.9 (C-2), 128.3, 128.1, 127.7 (CH aryl), 114.2 114.0 (C-1), 66.5 (C-8),
59.3, 58.9 (C-3), 46.6, 46.3 (C-6), 31.9, 31.1 (C-4), 23.3, 22.5 (C-5); MS m/z (ES+) 254
(100 %, M+Na+). Spectral data matched the published values.
[221]
2-(2-Formyloxyethyl) pyrrolidine-1-carboxylic acid benzyl ester 187.
N
O O
12
3
4
5
67
8
9
10 1112
OH
O
13
14
2-(2-Iodoethyl)pyrrolidine-1-carboxylic acid benzyl ester (S)-xx (50 mg, 0.14 mmol) gave
after flash column chromatography (SiO2, Petrol:EtOAc 5 % + NEt3 1 %) gave the title
compound 187 (20 mg, 52 %) as a colourless oil.

236
Rf 0.31 (Petrol:EtOAc 6:3); IR max (neat/cm-1
) 1723 (C=O), 1698 (C=O); 1H NMR
(400MHz, CDCl3) (mixture of rotamers) 7.90 (s, 0.6 H, H-1, rotamer 1), 7.38 (s, 0.4 H,
H-1, rotamer 2), 7.37-7.28 (m, 5H, aryl), 5.13 (s, 2H, H-10), 4.24-4.12 (m, 2H, H-3), 3.99
(m, 1H, H-5), 3.53-3.37 (m, 2H, H-8), 2.21-1.70 (m, 6H, H-4, H-6 and H-7); 13
C NMR
(100MHz, CDCl3) (mixture of rotamers) 161.1, 160.9 (C-2), 155.0 (C-9), 114.1 (C-11),
128.4, 128.0, 127.9, 127.8 (CH aryl), 66.9, 66.6 (C-10), 61.7, 61.5 (C-3), 55.3, 54.5 (C-5),
46.5, 46.2 (C-8), 33.7, 33.1 (C-4), 30.9, 30.3 (C-6), 23.8 23.0 (C-7); MS m/z (ES+) 300
(100 %, M+Na+).
4-(1,1-Diphenylethoxy)-1-methylperhydroazepine 190.
1615
8
9
10
11
1213
14
1
2
34
5
6
7
NO
Cl17
Rf 0.11 (Petrol:EtOAc:NEt3 60:40:1); 1H-NMR (400MHz, CDCl3) δ 7.33-7.19 (m, 9H),
3.63 (qn, J = 6.0 Hz, 1H, H-11), 2.71-2.64 (m, 1H, H-16), 2.58-2.55 (m, 2H, H-14), 2.43-
2.38 (m, 1H, H-16), 2.33 (s, 3H, H-15), 1.81 (s, 3H, H-6), 1.83-1.66 (m, 4H, H-12 and H-
17), 1.56-1.43 (m, 2H, H-13); MS m/z (ES+) 344 (100 %, M+H+); HRMS (ES+) calcd for
C21H27N1O135
Cl1 (M+H+): 344.1776, found: 344.1787.
(E)-Cinnamyl methylphenylcarbamate 193.
N O
O
1
2
34
5
6
7
8
9
10
11
12
13
General procedures E and F were followed. (2E)-3-Phenyl-2-propen-1-ol 191 (1.0 g, 7.5
mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 10 %) the title
compound 193 (1.7 g, 90 % over 2 steps) as a colourless oil.

237
Rf 0.61 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1707 (C=O), 1597 (C=C); 1H NMR
(300MHz, CDCl3) 7.43-7.24 (m, 10H, aryl), 6.60 (d, J = 16.5 Hz, 1H, H-5), 6.30 (dt, J =
16.5 and 5.7 Hz, 1H, H-6), 4.81 (d, J = 5.7 Hz, 2H, H-7), 3.37 (s, 3H, H-9); 13
C NMR
(100MHz, CDCl3) 154.9 (C-8), 142.9 (C-10), 136.0 (C-4), 132.9 (C-5), 128.5, 128.2,
127.5, 126.2, 125.7, 125.4, 123.5 (C-6 and CH aryl), 65.8 (C-7), 37.3 (C-9); MS m/z (ES+)
290 (100 %, M+Na+); HMRS (ES+) calcd for C17H17O2N1Na1 (M+Na
+): 290.1152, found:
290.1153.
(Z)-1,3-Diphenylprop-1-enyl methylcarbamate 196.
O O
NH
1
2
34
5
67
8
9
11
12
13
10
General procedure O was followed with sec-BuLi (2.5 equiv.). (E)-Cinnamyl
methylphenylcarbamate 193 (100 mg, 0.37 mmol) gave after flash column chromatography
(SiO2, Petrol:EtOAc 5 %) the title compound 196 (9 mg, 9 %) as a yellow solid.
Rf 0.26 (Petrol:EtOAc 8:2); m.p. 74-76 °C (CH2Cl2); IR max (neat/cm-1
) 3351 (br, N-H),
1715 (C=O), 1537 (Amide II), 1237 (Amide III); 1H NMR (400MHz, CDCl3) 7.38 (d, J
= 7.8 Hz, 2H, aryl), 7.28-7.11 (m, 8H, aryl), 5.89 (t, J = 7.6 Hz, 1H, H-6), 4.96 (br, 1H, -
NH), 3.47 (d, J = 7.6 Hz, 2H, H-5), 2.79 (d, J = 4.8 Hz, 3H, H-9); MS m/z (ES+) 290
(100 %, M+Na+); HMRS (ES+) calcd for C17H17O2N1Na1 (M+Na
+): 290.1145, found:
290.1145.
The (Z)-stereochemistry was deduced by a nOe experiment: irradiation at 5.89 ppm (CH=C)
enhanced peak at 7.38 ppm (aryl) from 2.16 %.

238
1,3-Diphenylpropan-1-one 197.[222]
O
1
2
34
56
78
9
10
11
General procedure O was followed with sec-BuLi (2.5 equiv.). (E)-Cinnamyl
methylphenylcarbamate 193 (100 mg, 0.37 mmol) gave after flash column chromatography
(SiO2, Petrol:EtOAc 5 %) the title compound 197 (32 mg, 41 %) as a yellow solid.
Rf 0.70 (Petrol:EtOAc 8:2); m.p. 66-68 °C (MeOH); 1H NMR (300MHz, CDCl3) 7.87
(d, J = 9.0 Hz, 2H, H-3), 7.49-7.10 (m, 8H, aryl), 3.22 (t, J = 7.8 Hz, 2H, H-6), 2.98 (t, J =
7.8 Hz, 2H, H-7); 13
C NMR (100MHz, CDCl3) 199.1 (C-5), 141.3 (C-8), 136.8 (C-4),
133.0, 128.5, 128.4, 128.2, 128.0, 126.1 (CH aryl), 40.4 (C-6), 30.1 (C-7); MS m/z (ES+)
233 (100 %, M+Na+). Spectral data matched the published values.
[222]
(E)-4-Phenylbut-3-en-2-ol 199.[159,223]
HO
1
2
34
5
67
8
General procedure A was followed. trans-Cinnamaldehyde 198 (1.00 g, 7.57 mmol) gave
after flash column chromatography (SiO2, Petrol:EtOAc 10 %) the title compound ()-199
(0.76 g, 68 %) as a yellow oil.
Rf 0.50 (Petrol:EtOAc 6:4); IR max (neat/cm-1
) 3350 (br, O-H); 1H NMR (400MHz,
CDCl3) 7.24-7.07 (m, 5H, aryl), 6.41 (d, J = 16.0 Hz, 1H, H-5), 6.11 (dd, J = 16.0 Hz and
6.4 Hz, 1H, H-6), 4.34 (qn, J = 6.4 Hz, 1H, H-7), 1.45 (br, 1H, -OH), 1.22 (d, J = 6.4 Hz,
3H, H-8); 13
C NMR (100MHz, CDCl3) 136.7 (C-4), 133.5 (C-5), 129.4 (C-6), 128.6,
127.7, 126.4 (CH aryl), 69.0 (C-7), 23.4 (C-8). Spectral data matched the published
values.[223]

239
General procedure C was used to produce the equivalent enantiopure (99:1 e.r.) alcohol,
(+)-(R)-(E)-4-phenylbut-3-en-2-ol 199 (120 mg, 40 %) as a white solid from (E)-4-
phenylbut-3-ene-2-ol 199 (300 mg, 2.03 mmol).
[]D28
+26.3 (c 0.85, CHCl3) [lit.[159]
[α]D20
+33.2 (c 1.02, CHCl3, >99:1 e.r. (R):(S))];
HPLC separation conditions: Chiralcel OD-H column, hexane:2-propanol (90:10), flow
rate: 1.0 mL/min, tR 15.9 min for (R)-enantiomer (major) and 10.7 min for (S)-enantiomer
(minor).
(E)-4-Phenylbut-3-en-2-yl methylphenylcarbamate 201.
N O
O
1
2
34
5
67
8
9
10
11
12
13
14
General procedures E and F were followed. (E)-4-Phenylbut-3-en-2-ol 199 (560 mg, 3.78
mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 10 %) the title
compound ()-201 (940 mg, 89 % over 2 steps) as a colourless oil.
Rf 0.67 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1699 (C=O), 1597 (C=C); 1H NMR
(500MHz, CDCl3) 7.30-7.12 (m, 10H, aryl), 6.44 (d, J = 15.5 Hz, 1H, H-5), 6.09 (dd, J =
15.5 and 5.5 Hz, 1H, H-6), 5.42 (qn, J = 5.5 Hz, 1H, H-7), 3.25 (s, 3H, H-10), 1.32 (d, J =
5.5 Hz, 3H, H-8); 13
C NMR (125MHz, CDCl3) 155.0 (C-9), 143.3 (C-11), 136.5 (C-4),
130.9 (C-5), 129.3 (C-6), 128.7, 128.5, 127.7, 126.5, 125.9, 125.7 (CH aryl), 72.4 (C-7),
37.6 (C-10), 20.7 (C-8); MS m/z (ES+) 304 (100 %, M+Na+); HMRS (ES+) calcd for
C18H19O2N1Na1 (M+Na+): 304.1308, found: 304.1307.
The same procedure was used to produce the equivalent enantiopure (99:1 e.r.) carbamate,
()-(R)-(E)-4-phenylbut-3-en-2-yl methylphenylcarbamate 201 (202 mg, 89 % over 2 steps)
from ()-(R)-(E)-4-phenylbut-3-en-2-ol 199 (120 mg, 0.81 mmol).
[]D30
-49.9 (c 1.05, CHCl3); HPLC separation conditions: Chiralcel OD-H column,
hexane:2-propanol (97:3), flow rate: 1.0 mL/min; tR 13.7 min for (R)-enantiomer (major)
and 12.09 min for (S)-enantiomer (minor).

240
3-sec-Butyl-2,4-diphenylbutan-2-yl methylcarbamate 202.
NH
O
O
1
2
34
56
78
9
10
11
12
13
14 15
16
1718
General procedure O was followed with sec-BuLi (2.5 equiv.). (E)-4-Phenylbut-3-en-2-yl
methylphenylcarbamate 201 (100 mg, 0.35 mmol) gave after flash column chromatography
(SiO2, Petrol: EtOAc 5 % to 10 %) the title compound 202 (28 mg, 23 %) as a white solid.
Rf 0.34 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3360 (br, N-H), 1701 (C=O), 1517 (Amide
II), 1258 (Amide III); 1H NMR (400MHz, CDCl3) (mixture of diastereoisomers) 7.31-
7.11 (m, 10H, aryl), 4.06 (br, 0.5H, -NH), 3.98 (br, 0.5H, -NH), 2.90 (dt, J = 14.6 and 5.9
Hz, 1H, H-6), 2.72 (d, J = 5.9 Hz, 0.5H, H-5), 2.68 (d, J = 5.9 Hz, 0.5H, H-5), 2.53 (d, J =
5.6 Hz, 1.5H, H-18), 2.59 (d, J = 5.6 Hz, 1.5H, H-18), 2.34 (t, J = 5.9 Hz, 0.5H, H-5), 2.29
(t, J = 5.9 Hz, 0.5H, H-5), 1.98 (s, 1.5H, H-16), 1.97 (s, 1.5H, H-16), 1.50-1.41 (m, 1H, H-
8), 1.32-1.25 (m, 1H, H-7), 1.03-0.92 (m, 1H, H-8), 0.86 (d, J = 76.9 Hz, 1.5H, H-16), 0.75
(t, J = 7.3 Hz, 1.5H, H-9), 0.61 (d, J = 6.9 Hz, 1.5H, H-10), 0.50 (t, J = 7.3 Hz, 1.5H, H-9);
MS m/z (ES+) 362 (100 %, M+Na+); HMRS (ES+) calcd for C22H29O2N1Na1 (M+Na
+):
362.2079, found: 362.2091.
(E)-2,4-Diphenyl-but-3-en-2-ol 203.[164,224]
HO
1
2
34
5
6
7
8
910
11
12
General procedure P was followed. (E)-4-phenylbut-3-en-2-yl methylphenylcarbamate
201 (100 mg, 0.35 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc
5 %) the title compound 203 (27 mg, 68 % over 2 steps) as a colourless oil.

241
Rf 0.54 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3397 (br, O-H); 1H NMR (400MHz,
CDCl3) 7.45 (d, J = 8.4 Hz, 2H, aryl), 7.28-7.14 (m, 8H, aryl), 6.57 (d, J = 16.0 Hz, 1H,
H-6), 6.43 (d, J = 16.0 Hz, 1H, H-5), 1.94 (s, 1H, -OH), 1.69 (s, 3H, H-8); 13
C NMR
(100MHz, CDCl3) 146.5 (C-9), 136.6 (C-4), 136.3 (C-5), 128.8, 128.5, 128.3, 127.6,
127.1, 126.5, 125.2 (C-6 and CH aryl), 74.6 (C-7), 29.7 (C-8); MS m/z (ES+) 207 (100 %,
M-H2O+H+), HRMS (ES+) calcd for C16H15 (M-H2O+H
+): 207.1168, found: 207.1173.
Spectral data matched the published values.[224]
The enantiomerically enriched alcohol ()-(S)-203 was made by following the procedure
below:
A solution of LDA (0.72 mmol) in anhydrous Et2O (2.0 cm3) was added to a solution of
carbamate (100 mg, 0.36 mmol) in anhydrous Et2O (2.5 cm3) and freshly distilled ()-
sparteine (0.08 cm3, 0.36 mmol) at -45 ºC under nitrogen atmosphere. The reaction was
stirred for 2 h was quenched slowly with MeOH (0.1 cm3) at -45 °C. The mixture was
diluted with Et2O and washed with water twice. The combined organic layers were dried
over MgSO4, filtered and concentrated under reduced pressure. The residue was dissolved
in THF (2.0 cm3) and cooled at -78 °C under nitrogen atmosphere. n-BuLi (0.28 cm
3, 0.72
mmol) was added and the yellow solution was stirred at -78 °C for 30 min after which tert-
butyl nitrite (0.25 cm3, 2.16 mmol) was added and the reaction mixture was stirred at room
temperature for 24 h. The mixture was diluted with Et2O and the organic layer was washed
with saturated aqueous K2CO3 three times, dried over MgSO4, filtered and concentrated
under reduced pressure to give a residue which was purified by flash column
chromatography (SiO2, Petrol:EtOAc 5 %) the title compound ()-(S)-203 (26 mg, 65 %
over 2 steps, 37:63 e.r.) as a colourless oil.
[]D25
1.5 (c 1.00, CHCl3) [lit.[164]
[α]D20
5.33 (c 0.60, Et2O, 95:5 e.r. (S):(R))]; HPLC
separation conditions: Chiralcel OD-H column, hexane:2-propanol (97:3), flow rate: 1.0
mL/min, tR 17.0 min for (R)-enantiomer (minor) and 20.9 min for (S)-enantiomer (major).

242
(E)-2,4-Diphenylbut-3-en-2-yl methylcarbamate 204.
NH
O
O
1
2
34
5
6
7
8
910
1112
1314
General procedure O was followed with LDA (2.0 equiv.) from (E)-4-phenylbut-3-en-2-yl
methylphenylcarbamate 201 (100 mg, 0.35 mmol).
1H NMR (400MHz, CD3OD) (crude) 7.28-7.13 (m, 10H, aryl), 6.63 (d, J = 16.4 Hz, 1H,
H-6), 6.47 (d, J = 16.4 Hz, 1H, H-5), 4.70 (br, 1H, -NH), 2.63 (d, J = 4.8 Hz, 3H, H-14),
1.91 (s, 3H, H-8); MS m/z (ES+) 226 (100 %, M-CONHCH3+) 207 (90 %, M-
OCONHCH3+); HMRS (ES+) calcd for C22H29O2N1Na1 (M+Na
+): 304.1308, found:
304.1301.
(E)-1,3-Diphenylbuta-1,3-diene 206.[225]
1
23
45
67
8
910
11
12
To a solution of crude (E)-2,4-diphenylbut-3-en-2-yl methylcarbamate 204 (~100 mg, 0.35
mmol) in THF (5 cm3) was added DIBAL-H (1 M in toluene, 0.70 cm
3, 0.70 mmol) at 0 °C
under nitrogen atmosphere. The mixture was refluxed for 2 h The reaction was then
allowed to cool and quenched with a saturated aqueous solution of Rochelle’s salt (sodium
potassium tartrate) (5 cm3). The reaction mixture was extracted with EtOAc (20 cm
3),
washed with water (2 20 cm3), dried over MgSO4, filtered and concentrated under
reduced pressure.
Rf 0.95 (Petrol:EtOAc 8:2); 1H NMR (300MHz, CDCl3) 7.32-7.11 (m, 10H, aryl), 6.95
(d, J = 16.0 Hz, 1H, H-6), 6.40 (d, J = 16.0 Hz, 1H, H-5), 5.31 (s, 1H, H-8), 5.14 (s, 1H, H-8).
Spectral data matched the published values.[225]

243
(E)-3-Butoxy-1,3-diphenylbut-1-ene 207. (E)-1-Butoxy-1,3-diphenylbut-2-ene 208.
O O
1
2
34
5
67
8
9
10
11
12
1314
15
16 1'
2'
3'4'
5'6'
7'
8' 9'
10' 11'
12'
13'14'
15'
16'
To a solution of crude (E)-2,4-diphenylbut-3-en-2-yl methylcarbamate 204 (~50 mg, 0.18
mmol) in n-butanol (5 cm3, 1 cm
3 per 10 mg) was added sodium carbonate (20 mg, 0.19
mmol) and the mixture was heated under reflux at 117 °C for 2 h. The solution was cooled
to room temperature and the solvent was evaporated under reduced pressure. The residue
was purified by flash column chromatography (SiO2, Petrol:EtOAc 5 %) to yield the two
title compounds 207 and 208 (20 mg, 40 %).
Rf 0.65 (Petrol:EtOAc 9.5:0.5); 1H NMR (300MHz, CDCl3) 7.34-7.15 (m, 20H, aryl),
6.53 (d, J = 16.4 Hz, 0.6H, H-6), 6.31 (d, J = 16.4 Hz, 0.6H, H-5), 5.88 (d, J = 8.7 Hz, 0.4H,
H-6’), 5.11 (d, J = 8.7 Hz, 0.4H, H-5’), 3.48-3.23 (m, 4H, H-9 and H-9’), 1.11 (s, 1.8H, H-
8), 1.63 (s, 1.2H, H-8’), 1.59-1.47 (m, 4H, H-10 and H-10’), 1.39-1.26 (m, 4H, H-11, H-
11’), 0.86-0.77 (m, 6H, H-12 and H-12’); 13
C NMR (100MHz, CDCl3) 145.8, 142.8 (C-
13 and C-13’), 137.0, 136.9 (C-4 and C-4’), 135.2 (C-7’), 130.7, 129.4, 128.9, 128.3, 128.1,
127.9, 127.5, 127.4, 127.0, 126.5, 126.2, 126.1, 126.0, 125.8, 125.1 (C-6, C-5, C-6’ and
CH aryl), 78.6, 78.4 (C-7 and C-5’), 68.2, 62.5 (C-9 and C-9’), 32.6, 32.0 (C-10 and C-10’),
24.9, 22.3 (C-8 and C-8’), 19.5, 16.5 (C-11 and C-11’), 14.0, 13.9 (C-12 and C-12’); MS
m/z (ES+) 207 (100 %, M-BuOH), 303 (65 %, M+Na+).

244
(E)-3-Methoxy-1,3-diphenylbut-1-ene 209. (E)-1-methoxy-1,3-diphenylbut-2-ene 210.
O O
1
2
34
5
67
89
1011
12
13 1'
2'
3'4'
5'6'
7'
8'9'
10'11'
12'
13'
To a solution of crude (E)-2,4-diphenylbut-3-en-2-yl methylcarbamate 204 (~50 mg, 0.18
mmol) in methanol (5 cm3, 1 cm
3 per 10 mg) was added sodium carbonate (20 mg, 0.19
mmol) and the reaction mixture was heated at 50 °C for 1 h. The solution was cooled to
room temperature and the solvent was evaporated under reduced pressure. The residue was
purified by flash column chromatography (SiO2, Petrol:EtOAc 95:5) to yield the 2 title
compounds 209 and 210 (30 mg, 71 %).
Rf 0.50-0.42 (Petrol:EtOAc 9.5:0.5); 1H NMR (300MHz, CDCl3) 7.40-7.15 (m, 20H,
aryl), 6.54 (d, J = 16.4 Hz, 0.6H, H-6), 6.30 (d, J = 16.4 Hz, 0.6H, H-5), 5.87 (d, J = 8.0 Hz,
0.4H, H-6’), 5.0 (d, J = 8.0 Hz, 0.4H, H-5’), 3.29 (s, 1.2H, H-8’), 3.16 (s, 1.8H, H-8), 2.11 (s,
1.2H, H-9’), 1.63 (s, 1.8H, H-9); 13
C NMR (100MHz, CDCl3) 144.7, 142.8 (C-10 and C-
10’), 137.7, 136.9 (C-4 and C-4’), 134.5 (C-7’), 129.4, 128.9, 128.6, 128.5, 128.3, 127.9,
127.6, 127.3, 127.1, 126.7, 126.6, 126.4, 125.9 (C-5, C-6, C-6’ and CH aryl), 80.2, 79.2
(C-7 and C-5’), 50.9, 56.2 (C-8 and C-8’), 24.5, 16.6 (C-9 and C-9’); MS m/z (ES+) 173
(100 %, M-MeOH), 261 (75 %, M+Na+).
(E)-4-(4-methoxyphenyl)but-3-en-2-ol 216.[226]
HO
O12
3
45
6
78
9
General procedure A was followed. trans-p-Methoxycinnamaldehyde 215 (1.00 g, 6.17
mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 20 %) the title
compound ()-216 (0.95 g, 87 %) as a white solid.

245
Rf 0.19 (Petrol:EtOAc 8:2); m.p. 67-68 °C (MeOH); IR max (neat/cm-1
) 3312 (br, O-H),
1608, 1512 (C=C), 1251 (C-O-C); 1H NMR (400MHz, CDCl3) 7.31 (d, J = 8.8 Hz, 2H,
H-4), 6.85 (d, J = 8.8 Hz, 2H, H-3), 6.51 (d, J = 16.0 Hz, 1H, H-6), 6.12 (dd, J = 16.0 and
6.8 Hz, 1H, H-7), 4.51-4.43 (m, 1H, H-8), 3.81 (s, 3H, H-1), 1.52 (d, J = 3.6 Hz, 1H, -OH)
1.36 (d, J = 6.4 Hz, 1H, H-9); 13
C NMR (100MHz, CDCl3) 159.2 (C-2), 131.3 (C-6),
129.3 (C-5), 129.0 (C-7), 127.6 (C-4), 114.0 (C-3), 69.1 (C-8), 55.3 (C-1), 23.4 (C-9).
Spectral data matched the published values.[226]
General procedure C was used to produce the equivalent enantiopure (93:7 e.r.) alcohol,
(+)-(E)-(R)-4-(4-methoxyphenyl)but-3-en-2-ol 216 (151 mg, 39 %) from (E)-4-(4-
methoxyphenyl)but-3-en-2-ol 216 (384 mg, 2.16 mmol).
[]D28
+28.3 (c 1.03, CHCl3) [lit.[227]
[α]D25
+9.4 (c 1.10, CHCl3, >99:1 e.r. (R):(S)]. HPLC
separation conditions: Chiralcel OD-H column, hexane:2-propanol (90:10), flow rate: 1.0
mL/min, tR 13.1 min for (R)-enantiomer (major) and 14.0 min for (S)-enantiomer (minor).
(E)-4-(4-Methoxyphenyl)but-3-en-2-yl methylphenylcarbamate 217.
N O
O
O12
3
45
6
78
9
10
11
12
13
14
15
General procedures E and F were followed. 4-(4-Methoxyphenyl)but-3-en-2-ol 216 (750
mg, 4.21 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the
title compound ()-217 (837 mg, 64 % over 2 steps) as a colourless oil.
Rf 0.37 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1697 (C=O), 1607, 1512 (C=C), 1248 (C-
O-C); 1H NMR (400MHz, CDCl3) 7.37-7.19 (m, 7H, aryl), 6.83 (d, J = 8.8 Hz, 2H, H-3),
6.48 (d, J = 15.6 Hz, 1H, H-6), 6.03 (dd, J = 15.6 and 6.4 Hz, 1H, H-7), 5.47 (qn, J = 6.4 Hz,
1H, H-8), 3.80 (s, 3H, H-1), 3.31 (s, 3H, H-11), 1.38 (d, J = 6.4 Hz, 3H, H-9); 13
C NMR
(100MHz, CDCl3) 159.3 (C-2), 155.0 (C-10), 143.4 (C-12), 130.5 (C-6), 129.2 (C-7),
128.7, 127.7, 125.8, 125.7 (CH aryl), 113.9 (C-3), 72.6 (C-8), 55.2 (C-1), 37.6 (C-11), 20.8

246
(C-9); MS m/z (ES+) 334 (100 %, M+Na+); HMRS (ES+) calcd for C19H21O3N1Na1
(M+Na+): 334.1414, found: 334.1409.
The same procedure was used to produce the equivalent enantiopure (93:7 e.r.) carbamate,
(S)-(E)-4-(4-methoxyphenyl)but-3-en-2-yl methylphenylcarbamate 217 (218 mg, 63 %
over 2 steps) from (R)-4-(4-methoxyphenyl)but-3-en-2-ol 216 (197 mg, 1.11 mmol).
HPLC separation conditions: Chiralpack AD-H column, hexane:2-propanol (97:3), flow
rate: 1.0 mL/min, tR 13.2 min for (S)-enantiomer (major) and 15.6 min for (R)-enantiomer
(minor).
(E)-4-(4-Methoxyphenyl)-2-phenylbut-3-en-2-yl methylcarbamate 218.
NH
O
O
12
3
45
6
7
8
9
12
13
14
O
1110
15
General procedure O was followed with LDA (2.0 equiv.) from (E)-4-(4-
Methoxyphenyl)but-3-en-2-yl methylphenylcarbamate 217 (100 mg, 0.32 mmol).
1H NMR (300MHz, CD3OD) 7.36-7.16 (m, 7H, aryl), 6.77 (d, J = 8.7 Hz, 2H, H-3), 6.53
(d, J = 16.2 Hz, 1H, H-7), 6.42 (d, J = 16.2 Hz, 1H, H-6), 4.63 (br, 1H, -NH), 3.73 (s, 3H,
H-1), 2.67 (d, J = 4.8 Hz, 3H, H-15), 1.91 (s, 3H, H-9).
(S)-2-(4-Chlorophenyl)-2-hydroxy-N-methyl-N-phenylpropanamide 219.
N
O
OH
Cl1
2
34
5
6
7
8
9
10
11
12
General procedure Q was followed with LDA (2.5 equiv.) in the presence of ()-sparteine
(1.0 equiv.). (S)-1-(4-Chlorophenyl)ethyl methylphenylcarbamate 95i (50 mg, 0.17 mmol)

247
gave after trituration in hexane the title compound 219 (42 mg, 85 %, 80:20 e.r.) as a
white solid.
Rf 0.20 (Petrol:EtOAc 8:2); 1H-NMR (400MHz, CDCl3) δ 7.22-6.92 (m, 9H, aryl), 5.24
(br, 1H, -OH), 3.23 (br, 3H, H-8), 1.62 (br, 3H, H-6); 13
C-NMR (100MHz, CDCl3) δ 174.9
(C-7), 141.9, 133.2, 128.9, 128.4 (br), 128.2, 127.9, 127.3 (aryl), 74.9 (C-5), 40.8 (C-8),
25.6 (C-6); MS m/z (ES+) 312 (M+Na+) (100 %).
HPLC separation conditions: Chiralcel OD-H column, hexane:2-propanol (95:5), flow rate:
1.0 mL/min; tR 10.5 min for (R)-enantiomer (minor) and 11.4 min for (S)-enantiomer
(major). The absolute configuration of (S)-219 remains unassigned.
Allyl methylphenylcarbamate 220a.
N O
O1
2
3
4
5
6
7
8
9
General procedures E and F were followed. 2-Propen-1-ol (1.0 g, 1.7 mmol) gave after
flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound 220a (2.9 g,
89 % over 2 steps) as a colourless oil.
Rf 0.45 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1706 (C=O); 1H NMR (400MHz, CDCl3)
7.40-7.36 (m, 2H, aryl), 7.28-7.22 (m, 3H, aryl), 5.96-5.89 (m, 1H, H-2), 5.24 (d, J = 17.6
Hz, 1H, H-1), 5.19 (d, J = 10.4 Hz, 1H, H-1), 4.64 (d, J = 5.2 Hz, 2H, H-3), 3.34 (s, 3H, H-
5); 13
C NMR (100MHz, CDCl3) 155.2 (C-4), 143.1 (C-6), 132.7 (C-2), 128.8, 126.0,
125.7 (CH aryl), 117.1 (C-1), 66.1 (C-3), 37.7 (C-5); MS m/z (ES+) 214 (100 %, M+Na+);
HMRS (ES+) calcd for C11H13O2N1Na1 (M+Na+): 214.0838, found: 214.0847.

248
Allyl isopropylphenylcarbamate 220b.
N O
O1
2
3
4
56
7
8
9
10
6
General procedures E and F were followed. 2-Propen-1-ol (1.0 g, 1.7 mmol) gave after
flash column chromatography (SiO2, Petrol:EtOAc 3 %) the title compound 220b (2.8 g,
74 % over 2 steps) as a colourless oil.
Rf 0.56 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1703 (C=O); 1H NMR (400MHz, CDCl3)
7.37-7.29 (m, 3H, aryl), 7.11 (d, J = 8.0 Hz, 2H, aryl), 5.86-5.78 (m, 1H, H-2), 5.07 (br, J
= 10.8 Hz, 2H, H-1), 4.60 (sep, J = 6.8 Hz, 1H, H-5), 4.54 (d, J = 4.8 Hz, 2H, H-3), 1.11 (d,
J = 6.8 Hz, 6H, H-6); 13
C NMR (100MHz, CDCl3) 155.2 (C-4), 138.3 (C-7), 133.0 (C-2),
129.9, 128.7, 127.4 (CH aryl), 116.4 (C-1), 65.7 (C-3), 49.0 (C-5), 21.4 (C-6); MS m/z
(ES+) 242 (100 %, M+Na+); HMRS (ES+) calcd for C13H17O2N1Na1 (M+Na
+): 242.1152,
found: 242.1152.
2-hydroxy-N-isopropyl-N-phenylbut-3-enamide 226.
OH
N
O
1
23
4
566
78
9
10
General procedure O was followed with LDA (2.0 equiv.). Allyl
isopropylphenylcarbamate 220b (50 mg, 0.23 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 15 %) the title compound 226 (14 mg, 28 %) as an
orange solid.
Rf 0.25 (Petrol:EtOAc 8:2); m.p. 48-50 °C (MeOH/Et2O); IR max (neat/cm-1
) 3409 (br, N-
H), 1648 (C=O); 1H NMR (400MHz, CDCl3) 7.42 (m, 3H, aryl), 7.15-7.10 (m, 2H, aryl),
5.58 (ddd, J = 16.8, 10.4 and 6.4 Hz, 1H, H-2), 5.02-4.92 (m, 3H, H-1, H-3), 4.31 (sep, J =

249
6.8 Hz, 1H, H-5), 3.82 (d, J = 8.0 Hz, 1H, -OH), 1.12 (d, J = 6.8 Hz, 3H, H-6), 1.04 (d, J =
6.8 Hz, 3H, H-6) 13
C NMR (100MHz, CDCl3) 171.9 (C-4), 136.2 (C-7), 135.2 (C-2),
131.5, 129.4, 128.9 (CH aryl), 117.4 (C-1), 70.2 (C-3), 47.7 (C-5), 21.1 (C-6), 20.2 (C-6);
MS m/z (ES+) 242 (100 %, M+Na+); HMRS (ES+) calcd for C13H17O2N1Na1 (M+Na
+):
242.1152, found: 242.1153.
3-Phenylprop-2-ynyl phenylcarbamate 228b.
ONH
O
1
23
45
67
8
9
1011
12
General procedure E was followed. 3-Phenylprop-2-yn-1-ol 227b (500 mg, 3.79 mmol)
gave after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound
228b (605 mg, 64 %) as a yellow solid.
Rf 0.40 (Petrol:EtOAc 8:2); 1H NMR (400MHz, CDCl3) 7.48-7.45 (m, 2H, aryl), 7.41-
7.39 (m, 2H, aryl), 7.34-7.29 (m, 5H, aryl), 7.11-7.07 (m, 1H, aryl), 6.71 (br, 1H, -NH), 5.02
(s, 2H, H-7); 13
C NMR (100MHz, CDCl3) 171.3 (C-8), 137.4 (C-9), 131.9, 129.1, 128.8,
128.3, 123.7 (CH aryl), 122.0 (C-4), 118.6 (C-10), 86.6 (C-5), 82.9 (C-6), 53.6 (C-7).
But-2-ynyl methylphenylcarbamate 230a.
N O
O
1
23
4
5
6
7
8
9
10
General procedures E and F were followed. But-2-yn-1-ol 227a (500 mg, 7.14 mmol) gave
after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound 230a
(745 mg, 52 % over 2 steps) as a colourless oil.

250
Rf 0.34 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 2237 (w, CC), 1702 (C=O); 1H NMR
(400MHz, CDCl3) 7.38-7.19 (m, 5H, aryl), 4.68 (br, 2H, H-4), 3.32 (s, 3H, H-6), 1.84 (t, J
= 2.8 Hz, 3H, H-1); 13
C NMR (100MHz, CDCl3) 155.0 (C-5), 143.1 (C-7), 128.8, 126.1,
125.7 (CH aryl), 82.6 (C-2), 73.8 (C-3), 53.9 (C-4), 37.8 (C-6), 3.72 (C-1); MS m/z (ES+)
226 (100 %, M+Na+); HMRS (ES+) calcd for C12H13O2N1Na1 (M+Na
+): 226.0838, found:
226.0831.
3-Phenylprop-2-ynyl methylphenylcarbamate 230b.
N O
O
1
2
34
56
7
8
9
10
11
12
13
General procedures G and H were followed. 3-Phenylprop-2-yn-1-ol 227b (500 mg, 3.79
mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 2 %) the title
compound 230b (640 mg, 64 % over 2 steps) as a white solid.
Rf 0.42 (Petrol:EtOAc 8:2); m.p. 46-48 °C (Et2O/Petrol); IR max (neat/cm-1
) 1711 (C=O);
1H NMR (400MHz, CDCl3) 7.46-7.22 (m, 10H, aryl), 4.96 (s, 2H, H-7), 3.35 (s, 3H, H-
9); 13
C NMR (100MHz, CDCl3) 154.8 (C-8), 142.9 (C-10), 131.9, 128.9, 128.6, 128.2,
126.2, 125.7 (CH aryl), 122.3 (C-4), 85.9 (C-5), 83.7 (C-6), 53.9 (C-7), 38.0 (C-9); MS m/z
(ES+) 288 (100 %, M+Na+); HMRS (ES+) calcd for C17H15O2N1Na1 (M+Na
+): 288.0995,
found: 288.1004.

251
(E)-4-benzylidene-3-phenyloxazolidin-2-one 231.[168f]
ON
O
12
3
4
5
67
8 9
10 11
12
General procedure F was followed. 3-Phenylprop-2-ynyl phenylcarbamate 228b (605 mg,
2.41 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 10 %) the title
compound 231 (188 mg, 31 %) as a slightly yellow solid.
Rf 0.33 (Petrol:EtOAc 8:2); m.p. 158-160 °C (Et2O/Petrol); IR max (neat/cm-1
) 1751
(C=O); 1H NMR (400MHz, CDCl3) 7.57-7.44 (m, 5H, aryl), 7.42-7.16 (m, 3H, aryl), 6.98
(d, J = 7.2 Hz, 2H, aryl), 5.65 (t, J = 2.4 Hz, 1H, H-5), 5.40 (d, J = 2.4 Hz, 2H, H-7); 13
C
NMR (100MHz, CDCl3) 155.6 (C-8), 136.9 (C-6), 135.0, 133.5, 129.9, 128.8, 127.6,
127.0, 126.1 (aryl), 101.5 (C-5), 67.4 (C-7); MS m/z (ES+) 274 (100 %, M+Na+); HMRS
(ES+) calcd for C16H13O2N1Na1 (M+Na+): 274.0838, found: 274.0833. Spectral data
matched the published values.[168f]
4-Benzyl-3-phenyloxazol-2(3H)-one 232.
ON
O
12
3
4
5
67
8 9
10 11
12
General procedure F was followed. 3-Phenylprop-2-ynyl phenylcarbamate 228b (605 mg,
2.41 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 20 %) the title
compound 232 (143 mg, 24 %) as a yellow oil.

252
Rf 0.15 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1748 (C=O); 1H NMR (400MHz, CDCl3)
7.43-7.38 (m, 3H, aryl), 7.24-7.18 (m, 5H, aryl), 7.02-7.00 (m, 2H, aryl), 6.55 (t, J = 1.4
Hz, 1H, H-7), 3.55 (d, J = 1.4 Hz, 2H, H-5); 13
C NMR (400MHz, CDCl3) 155.2 (C-8),
134.8 (C-9), 133.2 (C-4), 129.4, 128.7, 128.6, 128.5 (CH aryl), 128.4 (C-6), 127.4, 127.1
(CH aryl), 125.0 (C-7), 30.3 (C-5); MS m/z (ES+) 274 (100 %, M+Na+); HMRS (ES+)
calcd for C16H13O2N1Na1 (M+Na+): 274.0838, found: 274.0833.
()-(S)-4-Phenylbut-3-yn-2-ol 233.[169]
HO
1
23
45
67
8
By a modified method reported by Noyori et al.:[169]
A solution of [RuCl2(6-p-cymene)]2 (15 mg, 3 mol %) and (S,S)-TsDPEN (17 mg, 6
mol %) in deaerated 2-propanol (2 cm3) was stirred at 28 °C for 15 min under nitrogen
atmosphere. A 1.5 M solution of KOH in 2-propanol (0.18 cm3) and a solution of 4-
phenylbut-3-yn-2-one (1.00 g, 6.94 mmol) in deaerated 2-propanol (8 cm3) were added
sequentially and stirred at 28 °C for 5 days. The mixture was then concentrated under
reduced pressure and the residue was purified by flash column chromatography (SiO2,
Petrol:EtOAc 5 %) to give the title compound (S)-233 (0.65 g, 65 %, 97:3 e.r.) as a
colourless oil.
Rf 0.26 (Petrol:EtOAc 8:2); []D31
28 (c 1.60, CHCl3) [lit.[169]
[]D23
35.0 (c 1.00,
CHCl3, 99:1 e.r. (S):(R)]. 1H NMR (400MHz, CDCl3) 7.58-7.27 (m, 5H, aryl), 4.76 (q, J
= 6.6 Hz, 1H, H-7), 2.71 (br, 1H, -OH), 1.55 (d, J = 6.6 Hz, 3H, H-8); 13
C NMR (100MHz,
CDCl3) 132.5, 128.6, 128.3 (CH aryl), 122.5 (C-4), 90.9 (C-6), 83.9 (C-5), 58.8 (C-7),
24.3 (C-8). Spectral data matched the published values.[228]
HPLC separation conditions: Chiralcel OD-H column, hexane:2-propanol (90:10), flow
rate: 1.0 mL/min; tR 7.21 min for (S)-enantiomer (major) and 16.3 min for (R)-enantiomer
(minor).

253
4-Phenylbut-3-yn-2-yl phenylcarbamate 234.
ONH
O
1
2
34
56
7
8
9
10
11
12
13
General procedure E was followed. 3-Phenylprop-2-yn-1-ol 233 (500 mg, 3.42 mmol) gave
after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound 234 (605
mg, 64 %) as a yellow solid.
Rf 0.40 (Petrol:EtOAc 8:2); m.p. 77-80 °C (Et2O/Petrol); IR max (neat/cm-1
) 3343 (br, N-
H), 1702 (C=O), 1599 (C=C), 1527 (Amide II), 1224 (Amide III); 1H NMR (400MHz,
CDCl3) 7.47-7.41 (m, 4H, aryl), 7.34-7.29 (m, 5H, aryl), 7.08 (t, J = 7.4 Hz, 1H, aryl),
6.78 (br, 1H, -NH), 5.77 (q, J = 6.7 Hz, 1H, H-7), 1.66 (d, J = 6.7 Hz, 3H, H-8); 13
C NMR
(100MHz, CDCl3) 152.3 (C-9), 137.6 (C-10), 131.8, 129.0, 128.6, 128.2, 123.5 (CH aryl),
122.1 (C-4), 118.7 (C-11), 87.4 (C-6), 84.8 (C-5), 61.8 (C-7), 21.8 (C-8).
4-Benzyl-5-methyl-3-phenyloxazol-2(3H)-one 235.
ON
O
12
3
4
5
678
9 10
11 12
13
General procedure F was followed. 4-Phenylbut-3-yn-2-yl phenylcarbamate 234 (100 mg,
0.38 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 20 %) the title
compound 235 (52 mg, 52 %) as a yellow oil.
Rf 0.11 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1750 (C=O); 1H NMR (400MHz, CDCl3)
7.33-7.31 (m, 3H, aryl), 7.19-7.17 (m, 3H, aryl), 7.07-7.04 (m, 2H, aryl), 6.87-6.84 (m, 2H,
aryl), 3.58 (s, 2H, H-5), 2.16 (s, 3H, H-8); 13
C NMR (100MHz, CDCl3) 154.6 (C-9),

254
136.4 (C-10), 133.7 (C-4), 133.6, 129.2, 128.5, 128.4, 128.0, 127.5 (CH aryl), 126.8 (C-6),
121.0 (C-7), 28.9 (C-5), 10.2 (C-8); MS m/z (ES+) 288 (100 %, M+Na+); HMRS (ES+)
calcd for C17H15O2N1Na1 (M+Na+): 288.0995, found: 288.1006.
4-Phenylbut-3-yn-2-yl methylphenylcarbamate 237a.
ON
O
1
23
45
67
8
9
10
11
12
13
14
General procedures G and H were followed. 4-Phenylbut-3-yn-2-ol 233 (500 mg, 3.42
mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 3 %) the title
compound ()-237a (544 mg, 57 % over 2 steps) as a yellow oil.
Rf 0.30 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1702 (C=O), 2233 (w, C≡C); 1H NMR
(500MHz, CDCl3) 7.44-7.20 (10H, aryl), 5.70 (q, J = 5.3 Hz, 1H, H-7), 3.34 (s, 3H, H-10),
1.55 (br d, J = 5.3 Hz, 3H, H-8); 13
C NMR (125MHz, CDCl3) 154.5 (C-9), 143.1 (C-11),
131.8, 128.7, 128.4, 128.1, 125.9, 125.5 (CH aryl), 122.4 (C-4), 87.9 (C-6), 84.3 (C-5),
62.2 (C-7), 37.7 (C-10), 21.7 (C-8); MS m/z (ES+) 302 (100 %, M+Na+); HMRS (ES+)
calcd for C18H18O2N1 (M+H+): 280.1332, found: 280.1325.
The same procedure was used to produce the equivalent enantiopure (97:3 e.r.) carbamate,
(S)-4-phenylbut-3-yn-2-yl methylphenylcarbamate 237a (296 mg, 54 % over 2 steps) from
()-(S)- 4-phenylbut-3-yn-2-ol 233 (300 mg, 2.05 mmol).
HPLC separation conditions: Chiralcel OD-H column, hexane:2-propanol (90:10), flow
rate: 1.0 mL/min; tR 6.06 min for (R)-enantiomer (minor) and 7.27 min for (S)-enantiomer
(major).

255
4-Phenylbut-3-yn-2-yl isopropylphenylcarbamate 237b.
N O
O
1
2
34
56
7
8
9
101111
13
14
15
16
General procedures G and H were followed. 4-Phenylbut-3-yn-2-ol 233 (140 mg, 0.96
mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 2 %) the title
compound ()-237b (163 mg, 55 % over 2 steps) as a white solid.
Rf 0.53 (Petrol:EtOAc 8:2); m.p. 70-72 °C (Petrol/Et2O); IR max (neat/cm-1
) 1685 (C=O),
2235 (w, CC); 1H NMR (400MHz, CDCl3) 7.38-7.28 (m, 8H, aryl), 7.12 (d, J = 7.2 Hz,
2H, aryl), 5.66 (q, J = 6.2 Hz, 1H, H-7), 4.60 (sep, J = 6.8 Hz, 1H, H-10), 1.46 (br d, J = 6.2
Hz, 3H, H-8), 1.14 (d, J = 6.8 Hz, 3H, H-11), 1.13 (d, J = 6.8 Hz, 3H, H-11) 13
C NMR
(100MHz, CDCl3) 154.6 (C-9), 138.2 (C-13), 131.8, 129.9, 128.6, 128.3, 128.2, 127.3 (CH
aryl), 122.6 (C-4), 88.2 (C-6), 84.0 (C-5), 61.7 (C-7), 49.2 (C-10), 21.6 (C-8), 21.4 (C-11);
MS m/z (ES+) 330 (100 %, M+Na+); HMRS (ES+) calcd for C20H21O2N1Na1 (M+Na
+):
330.1465, found: 330.1460.
The same procedure was used to produce the equivalent enantiopure (97:3 e.r.) carbamate,
(S)-isopropyl phenyl carbamic acid 1-methyl-3-phenylprop-2-ynyl ester 237b (188 mg,
54 % over 2 steps) from ()-(S)- 4-phenylbut-3-yn-2-ol 233 (350 mg, 2.40 mmol).
HPLC separation conditions: Chiralcel OD-H column, hexane:2-propanol (90:10), flow
rate: 1.0 mL/min; tR 5.19 min for (R)-enantiomer (minor) and 6.25 min for (S)-enantiomer
(major).

256
(E)-4-Benzylidene-3,5-dimethyl-5-phenyloxazolidin-2-one 238a.
ON
O
12
3
4
5
6
78
9
10
11
1213
14
General procedure O was followed with LDA (2.0 equiv.). 4-Phenylbut-3-yn-2-yl
methylphenylcarbamate 237a (58 mg, 0.21 mmol) gave after flash column chromatography
(SiO2, Petrol:EtOAc 4 %) the title compound (E)-238a (25 mg, 43 %) as a yellow solid.
Rf 0.22 (Petrol:EtOAc 8:2); m.p. 112-114 °C (MeOH/Et2O); IR max (neat/cm-1
) 1751
(C=O), 1661 (C=C); 1H NMR (400MHz, CDCl3) 7.36-7.31 (m, 5H, aryl), 7.09-7.04 (m,
3H, aryl), 6.75 (dd, J = 7.2 and 1.2 Hz, 2H, aryl), 5.75 (s, 1H, H-5), 3.22 (s, 3H, H-7), 1.87
(s, 3H, H-10); 13
C NMR (100MHz, CDCl3) 155.3 (C-8), 144.2 (C-6), 140.1 (C-11), 134.0
(C-4), 129.0, 128.9, 128.7, 127.9, 126.4, 126.2 (aryl), 101.2 (C-5), 85.1 (C-9), 28.3 (C-7),
22.9 (C-10); MS m/z (ES+) 302 (100 %, M+Na+); HMRS (ES+) calcd for C18H17O2N1Na1
(M+Na+): 302.1152, found: 302.1151.
The (Z)-stereochemistry was deduced by a nOe experiment: irradiation at 5.75 ppm (CH=C)
enhanced peak at 3.22 ppm (N-CH3) from 3.10 %. The structure of the compound was
confirmed by an X-ray crystal structure.
(Z)-4-Benzylidene-3,5-dimethyl-5-phenyloxazolidin-2-one 238a.
ON
O
1
23
45
6
78
9
10
11
1213
14
General procedure O was followed with LDA (2.0 equiv.). 4-Phenylbut-3-yn-2-yl
methylphenylcarbamate 237a (50 mg, 0.18 mmol) gave after flash column chromatography
(SiO2, Petrol:EtOAc 3 %) the title compound (Z)-238a (22 mg, 44 %) as a yellow oil.

257
Rf 0.40 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1765 (C=O), 1675 (C=C); 1H NMR
(400MHz, CDCl3) 7.47-7.07 (m, 10H, aryl), 5.56 (s, 1H, H-5), 2.73 (s, 3H, H-7), 1.90 (s,
3H, H-10); 13
C NMR (125MHz, CDCl3) 156.8 (C-8), 143.6 (C-6), 141.5 (C-11), 134.2
(C-4), 129.6, 128.6, 128.5, 127.8, 126.8, 125.0 (aryl), 100.8 (C-5), 84.8 (C-9), 32.3 (C-7),
27.6 (C-10); MS m/z (ES+) 302 (100 %, M+Na+); HMRS (ES+) calcd for C18H17O2N1Na1
(M+Na+): 302.1152, found: 302.1156.
The (Z)-stereochemistry was deduced by a nOe experiment: irradiation at 5.56 ppm (CH=C)
enhanced peak at 1.90 ppm (N-CH3) from 1.44 %.
(E)-4-Benzylidene-3-isopropyl-5-methyl-5-phenyloxazolidin-2-one 238b.
ON
O
12
3
4
5
6
7
8
910
11
12
1314
815
General procedure O was followed with LDA (2.0 equiv.). 4-Phenylbut-3-yn-2-yl
isopropylphenylcarbamate 237b (50 mg, 0.16 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 3 %) the title compound (E)-238b (35 mg, 70 %) as a
white solid.
Rf 0.44 (Petrol:EtOAc 8:2); m.p. 122-124 °C (MeOH/Et2O); IR max (neat/cm-1
) 1744
(C=O), 1661 (C=C); 1H NMR (400MHz, CDCl3) 7.34-7.25 (m, 5H, aryl), 7.10-7.02 (m,
3H, aryl), 6.71 (dd, J = 6.8 and 0.8 Hz, 2H, aryl), 5.94 (s, 1H, H-5), 4.29 (sep, J = 6.8 Hz,
1H, H-7), 1.80 (s, 3H, H-11), 1.59 (d, J = 6.8 Hz, 3H, H-8), 1.58 (d, J = 6.8 Hz, 3H, H-
8)13
C NMR (100MHz, CDCl3) 154.6 (C-9), 143.3 (C-6), 140.8 (C-12), 134.3 (C-12),
129.2, 128.7, 128.5, 127.8, 126.3, 126.0 (aryl), 101.3 (C-5), 83.6 (C-10), 45.7 (C-7), 23.1
(C-11), 19.1, 18.7 (C-8)MS m/z (ES+) 330 (100 %, M+Na+); HMRS (ES+) calcd for
C20H22O2N1 (M+H+): 308.1646, found: 308.1656.

258
But-3-yn-2-methylphenylcarbamate 240.
ON
O
23
4
5
6
7
8
9
10
1
General procedures E and F were followed. 4-(Trimethylsilyl)-3-butyn-2-ol 239 (500 mg,
3.52 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title
compound 240 (364 mg, 51 % over 2 steps) as a colourless oil.
Rf 0.44 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1692 (C=O); 1H NMR (400MHz, CDCl3)
7.20-7.03 (m, 5H, aryl), 5.27 (qd, J = 6.8 and 2.0 Hz, 1H, H-3), 3.15 (s, 3H, H-6), 2.27 (d,
J = 2.0 Hz, 1H, H-1), 1.30 (br d, J = 6.8 Hz, 3H, H-4); 13
C NMR (100MHz, CDCl3)
154.5 (C-5), 143.0 (C-7), 128.8, 126.0, 125.6 (CH aryl), 82.6 (C-2), 72.7 (C-1), 61.4 (C-3),
37.7 (C-6), 21.4 (C-4); MS m/z (ES+) 226 (40 %, M+Na+); HMRS (ES+) calcd for
C12H14O2N1 (M+H+): 204.1020, found: 204.1022.
4-(Triisopropylsilyl)but-3-yn-2-yl methylphenylcarbamate 241.
ON
O
1
2
34
5
6
7
8
9
10
11
12
2
Si
To a solution of but-3-yn-2-methylphenylcarbamate 240 (228 mg, 1.12 mmol) in
anhydrous THF (10 cm3) was added n-BuLi (1.6 M in hexanes, 0.90 cm
3, 1.45 mmol) at -
78 °C. After stirring the solution for 1 h, triisopropylsilylchloride (0.30 cm3, 1.45 mmol)
was added at -78 °C and the reaction mixture was warmed to room temperature and stirred
for 5 h. The reaction was quenched with water (10 cm3) and diluted with Et2O (15 cm
3).
The organic layer was washed with H2O (20 cm3), dried over MgSO4, filtered and
concentrated under reduced pressure. The residue was purified by flash column
chromatography (SiO2, Petrol:EtOAc 3 %) to give the title compound 241 (260 mg, 65 %)
as a pale yellow oil.

259
Rf 0.70 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1709 (C=O); 1H NMR (400MHz, CDCl3)
7.28-7.10 (m, 5H, aryl), 5.42 (q, J = 6.8 Hz, 1H, H-5), 3.24 (s, 3H, H-8), 1.39 (d, J = 6.8
Hz, 3H, H-6), 0.99 (s, 21H, H-1 and H-2); 13
C NMR (100MHz, CDCl3) 154.5 (C-7),
143.1 (C-9), 128.7, 125.8, 125.6 (CH aryl), 106.3 (C-4), 85.4 (C-3), 62.3 (C-5), 37.7 (C-8),
21.9 (C-6), 18.5 (C-2), 11.0 (C-1); MS m/z (ES+) 382 (100 %, M+Na+); HMRS (ES+)
calcd for C21H33O2N1Na1Si1 (M+Na+): 382.2173, found: 382.2179.
4-(Triisopropylsilyl)-2-phenylbut-3-yn-2-yl methylcarbamate 242.
ONH
O
1
2
345
6
78
9
10
1112 2
Si
General procedure O was followed with LDA (2.5 equiv.) and DMPU. 4-
(Triisopropylsilyl)but-3-yn-2-yl methylphenylcarbamate 241 (50 mg, 0.14 mmol) gave
after flash column chromatography (SiO2, Petrol:EtOAc 4 %) the title compound 242 (33
mg, 66 %) as a white solid.
Rf 0.35 (Petrol:EtOAc 8:2); m.p. 65-68 °C (Et2O/Petrol); IR max (neat/cm-1
) 3307 (N-H),
2177 (w, CC), 1703 (C=O), 1540 (Amide II), 1272 (Amide III); 1H NMR (400MHz,
CDCl3) 7.54 (d, J = 7.4 Hz, 2H, aryl), 7.29-7.18 (m, 3H, aryl), 4.58 (br, 1H, -NH), 2.63
(br d, J = 4.3 Hz, 3H, H-12), 1.82 (s, 3H, H-6), 1.03 (s, 21H, H-1 and H-2); 13
C NMR
(100MHz, CDCl3) 154.5 (C-11), 143.2 (C-7), 128.1, 127.6, 125.0 (CH aryl), 106.9 (C-4),
88.0 (C-3), 75.8 (C-5), 32.5 (C-12), 27.3 (C-6), 18.6 (C-2), 11.2 (C-1): MS m/z (ES+) 377
(60 %, M+NH4+), 360 (40 %, M+H
+), 382 (10 %, M+Na
+); HMRS (ES+) calcd for
C21H33O2N1Na1Si1 (M+Na+): 382.2173, found: 382.2162.

260
4-Chloro-N-isopropylbenzenamine 244c.[229]
HN
Cl
1
234
5
62
General procedure I was followed. 4-Chloroaniline 243c (1.00 g, 7.87 mmol) gave after
flash column chromatography (SiO2, Petrol:EtOAc 2 %) the title compound 244c (0.95 g,
72 %) as a colourless oil.
Rf 0.38 (Petrol:EtOAc 8:2); 1H-NMR (400MHz, CDCl3) δ 7.10 (d, J = 8.8 Hz, 2H, H-5),
6.49 (d, J = 8.8 Hz, 2H, H-4), 3.58 (sep, J = 6.3 Hz, 1H, H-1), 3.44 (br, 1H, -NH), 1.20 (d, J
= 6.3 Hz, 6H, H-2) ; 13
C-NMR (125MHz, CDCl3) δ 146.1 (C-3), 129.0 (C-6), 121.4 (C-5),
114.2 (C-4), 44.4 (C-1), 22.9 (C-2); MS m/z (ES+) 170 (100 %, M+H+); HMRS (ES+)
calcd for C9H13N1Cl1 (M+H+): 170.0731, found: 170.0727. Spectral data matched the
published values.[229]
N-Isopropyl-4-methylbenzenamine 244d.[230]
HN
1
2
34
5
6
7
2
General procedure I was followed. p-Toluidine 243d (3.0 g, 28 mmol) gave after flash
column chromatography (SiO2, Petrol:EtOAc 3 %) the title compound 244d (3.6 g, 86 %)
as an orange oil.
Rf 0.48 (Petrol:EtOAc 8:2); 1
H-NMR (400MHz, CDCl3) δ 6.98 (d, J = 8.2 Hz, 2H, H-5),
6.52 (d, J = 8.2 Hz, 2H, H-4), 3.60 (sep, J = 6.3 Hz, 1H, H-1), 3.31 (br, 1H, -NH), 2.24 (s,
3H, H-7), 1.20 (d, J = 6.3 Hz, 6H, H-2) ; 13
C-NMR (100MHz, CDCl3) δ 145.2 (C-3), 129.7
(C-5), 126.2 (C-6), 113.5 (C-4), 44.5 (C-1), 23.0 (C-7), 20.3 (C-2); MS m/z (ES+) 150
(100 %, M+H+); HMRS (ES+) calcd for C10H16N1 (M+H
+): 150.1277, found: 150.1274.
Spectral data matched the published values.[230]

261
(3-Trifluoromethyl)-N-isopropylbenzenamine 244e.[230]
HNF3C
1
2
34
5
62
78
9
General procedure I was followed. 3-(Trifluoromethyl)aniline 243e (1.0 g, 6.2 mmol) gave
after flash column chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 244e (1.1
g, 87 %) as a colourless oil.
Rf 0.68 (Petrol:EtOAc 8:2); IR max (neat/cm
-1) 3417 (br, N-H), 1115 (C-F);
1H-NMR
(400MHz, CDCl3) δ 7.28 (t, J = 7.8 Hz, 1H, H-8), 6.93 (d, J = 7.8 Hz, 1H, H-7), 6.81 (s, 1H,
H-4), 6.75 (d, J = 7.8 Hz, 1H, H-9), 3.72-3.65 (m, 2H, H-1, -NH), 1.27 (d, J = 6.3 Hz, 6H,
H-2) ; 13
C-NMR (100MHz, CDCl3) δ 147.6 (C-3), 131.5 (q, 2JC-F = 32 Hz, C-5), 129.6 (C-
8), 124.0 (q, 1JC-F = 270 Hz, C-6), 116.0 (C-9), 113.1 (q,
3JC-F = 3.6 Hz, C-4), 109.1 (q,
3JC-
F = 3.6 Hz, C-7), 44.1 (C-1), 22.7 (C-2); MS m/z (ES+) 204 (100 %, M+H+), (ES-) 202
(100 %, M-H+). Spectral data matched the published values.
[230]
N-Isopropyl-4-methoxybenzenamine 244f.[229]
HN
O
1
234
5
672
General procedure I was followed. p-Anisidine 243f (3.0 g, 24 mmol) gave after flash
column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound 244f (3.6 g, 90 %)
as a yellow oil.
Rf 0.25 (Petrol:EtOAc 8:2); IR max (neat/cm
-1) 3381 (br, N-H), 1507 (C=C), 1229 (C-O-C);
1H-NMR (400MHz, CDCl3) δ 6.78 (d, J = 8.9 Hz, 2H, H-5), 6.57 (d, J = 8.9 Hz, 2H, H-4),
3.75 (s, 3H, H-7), 3.54 (sep, J = 6.3 Hz, 1H, H-1), 3.11 (br, 1H, -NH), 1.19 (d, J = 6.3 Hz,
6H, H-2) ; 13
C-NMR (100MHz, CDCl3) δ 151.8 (C-3), 141.7 (C-6), 114.8 (CH aryl), 55.7
(C-7), 45.1 (C-1), 23.0 (C-2); MS m/z (ES+) 166 (100 %, M+H+); HMRS (ES+) calcd for

262
C10H16N1O1 (M+H+): 166.1227, found: 166.1228. Spectral data matched the published
values.[229]
N-Methyl-N-4-chlorophenylcarbamoyl chloride 245a.
N Cl
O
1
34
5
6
2
Cl
General procedure J was followed. 4-Chloroaniline 243c (1.0 g, 7.1 mmol) gave the title
compound 245a (1.2 g, 87 %) as an orange solid.
Rf 0.36 (Petrol:EtOAc 8:2); m.p. 74-76 °C (CH2Cl2); IR max (neat/cm
-1) 1728 (C=O);
1H
NMR (400MHz, CDCl3) 7.40 (d, J = 8.0 Hz, 2H, H-4), 7.19 (d, J = 8.0 Hz, 2H, H-5),
3.35 (br, 3H, H-1); 13
C NMR (400MHz, CDCl3) 148.9 (C-2), 141.7 (C-3), 134.4 (C-6),
129.8, 128.8 (CH aryl), 40.3 (C-1); MS m/z (ES+) 225 (100 %, M+Na+). Spectral data
matched the published values.
Isopropyl(phenyl) carbamic chloride 245b.
N Cl
O
12
345
6
7
2
General procedure J was followed. N-isopropylaniline 244b (1.5 g, 11 mmol) gave the title
compound 245b (1.1 g, 60 %) as a white solid.
Rf 0.53 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1736 (C=O); 1H NMR (400MHz, CDCl3)
7.44-7.40 (m, 3H, aryl), 7.17-7.14 (m, 2H, aryl), 4.69 (sep, J = 6.8 Hz, 1H, H-1), 1.16 (d,
J = 6.8 Hz, 6H, H-2); 13
C NMR (400MHz, CDCl3) 148.9 (C-3), 138.3 (C-4), 130.0,
129.1, 128.8 (CH aryl), 52.3 (C-1), 20.9 (C-2); MS m/z (ES+) 220 (100 %, M+Na+).

263
(4-Chlorophenyl)(isopropyl) carbamic chloride 245c.
N Cl
OCl
12
345
6
7
2
General procedure J was followed. 4-Chloro-N-isopropylbenzenamine 244c (920 mg, 5.44
mmol) gave the title compound 245c (929 mg, 74 %) as a white solid.
Rf 0.48 (Petrol:EtOAc 8:2); m.p. 81-83 °C (CH2Cl2); IR max (neat/cm
-1) 1734 (C=O);
1H-
NMR (400MHz, CDCl3) δ 7.41 (d, J = 8.8 Hz, 2H, H-5), 7.09 (d, J = 8.8 Hz, 2H, H-6),
4.68 (sep, J = 6.8 Hz, 1H, H-1), 1.15 (d, J = 6.8 Hz, 6H, H-2) ; 13
C-NMR (100MHz, CDCl3)
δ 148.7 (C-3), 136.7 (C-4), 134.9 (C-7), 131.4, 129.4 (CH aryl), 52.3 (C-1), 20.9 (C-2);
MS m/z (ES+) 254 (60 %, M+Na+).
Isopropyl(p-tolyl) carbamic chloride 245d.
N Cl
O
12 2
345
6
7
8
General procedure J was followed. N-Isopropyl-4-methylbenzenamine 244d (3.5 g, 23
mmol) gave the title compound 245d (3.9 mg, 79 %) as a white solid.
Rf 0.66 (Petrol:EtOAc 8:2); m.p. 72-74 °C (CH2Cl2); IR max (neat/cm
-1) 1716 (C=O);
1H-
NMR (400MHz, CDCl3) δ 7.21 (d, J = 8.1 Hz, 2H, H-5), 7.02 (d, J = 8.1 Hz, 2H, H-6),
4.66 (sep, J = 6.8 Hz, 1H, H-1), 2.39 (s, 1H, H-8), 1.15 (d, J = 6.8 Hz, 6H, H-2) ; 13
C-NMR
(100MHz, CDCl3) δ 149.1 (C-3), 138.8 (C-4), 135.5 (C-7), 129.7 (CH aryl), 52.1 (C-1),
21.1 (C-8), 20.8 (C-2); MS m/z (ES+) 234 (100 %, M+Na+).

264
(3-Trifluoromethyl)phenyl)(isopropyl) carbamic chloride 245e.
N Cl
O
F3C
12 2
345
6
7
8
9
10
General procedure J was followed. (3-Trifluoromethyl)-N-isopropylbenzenamine 244e (1.0
g, 4.9 mmol) gave the title compound 245e (1.2 g, 97 %) as a white solid.
m.p. 68-70 °C (CH2Cl2); IR max (neat/cm-1
) 1753 (C=O), 1323 (C-F); 1H-NMR (400MHz,
CDCl3) δ 7.68 (d, J = 7.8 Hz, 1H, H-8), 7.57 (t, J = 7.8 Hz, 1H, H-9), 7.42 (s, 1H, H-5), 7.36
(d, J = 7.8 Hz, 1H, H-10), 4.68 (sep, J = 6.8 Hz, 1H, H-1), 1.16 (d, J = 6.8 Hz, 6H, H-2) ;
13C-NMR (100MHz, CDCl3) δ 148.2 (C-3), 138.8 (C-4), 131.9 (C-10), 130.8 (q,
2JC-F = 32
Hz, C-6), 129.8 (C-9), 126.8 (q, 3JC-F = 3.6 Hz, C-5), 125.6 (q,
3JC-F = 3.6 Hz, C-8), 123.0
(q, 1JC-F = 270 Hz, C-7), 52.5 (C-1), 20.7 (C-2); MS m/z (ES+) 288 (30 %, M+Na
+).
Isopropyl(4-methoxyphenyl) carbamic chloride 245f.
N Cl
OO
12
345
6
78
2
General procedure J was followed. N-Isopropyl-4-methoxybenzenamine 244f (3.5 g, 21
mmol) gave the title compound 245f (3.1 g, 65 %) as a pale yellow solid.
m.p. 81-83 °C (CH2Cl2); IR max (neat/cm
-1) 1731 (C=O), 1505 (C=C), 1211 (C-O-C);
1H-
NMR (400MHz, CDCl3) δ 7.02 (d, J = 8.9 Hz, 2H, H-5), 6.91 (d, J = 8.9 Hz, 2H, H-6),
4.66 (sep, J = 6.8 Hz, 1H, H-1), 3.83 (s, 1H, H-8), 1.14 (d, J = 6.8 Hz, 6H, H-2) ; 13
C-NMR
(100MHz, CDCl3) δ 159.5 (C-7), 149.4 (C-3), 131.0 (C-4), 130.8 (C-5), 114.1 (C-6), 55.4
(C-8), 52.0 (C-1), 20.9 (C-2); MS m/z (ES+) 250 (100 %, M+Na+).

265
(Z)-1-Phenylprop-1-enyl methylphenylcarbamate 248a.
N O
O
1
2
3
45
6
7
8
9
1011
12
13
General procedure K was followed. Propiophenone 246a (500 mg, 3.73 mmol) gave after
flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound (Z)-248a (568
mg, 57 %) as a white solid.
Rf 0.20 (Petrol:EtOAc 8:2); m.p. 53-55 °C (Et2O/Petrol); IR max (neat/cm-1
) 1710 (C=O);
1H NMR (400MHz, CDCl3) 7.44-7.22 (m, 10H, aryl), 5.83 (q, J = 6.8 Hz, 1H, H-6), 3.40
(br, 3H, H-9), 1.75 (d, J = 6.8 Hz, 3H, H-7); 13
C NMR (100MHz, CDCl3) 153.2 (C-8),
147.3 (C-5), 143.0 (C-10), 135.6 (C-4), 129.1, 128.4, 127.8, 126.3, 126.0, 124.3 (CH aryl),
112.7 (C-6), 38.3 (C-9), 11.5 (C-7); MS m/z (ES+) 290 (100 %, M+Na+); HMRS (ES+)
calcd for C17H17O2N1Na1 (M+Na+): 290.1152, found: 290.1145.
(Z)-1-Phenylbut-1-enyl 4-chlorophenylmethylcarbamate 248b.
14
N O
O
1
2
3
45
6
78
9
10
1112
13
General procedure K was followed. Butyrophenone 246b (500 mg, 3.38 mmol) gave after
flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound (Z)-248b (615
mg, 65 %) as a white solid.
Rf 0.33 (Petrol:EtOAc 8:2); m.p. 50-52 °C (Et2O/Petrol); IR max (neat/cm-1
) 1711 (C=O);
1H NMR (400MHz, CDCl3) 7.43-7.22 (m, 10H, aryl), 5.75 (t, J = 7.2 Hz, 1H, H-6), 3.39
(br, 3H, H-10), 2.23-2.15 (m, 2H, H-7), 1.05 (t, J = 7.6 Hz, 3H, H-8); 13
C NMR (100MHz,

266
CDCl3) 153.1 (C-9), 145.9 (C-5), 142.9 (C-11), 135.5 (C-4), 129.0, 128.2, 127.7, 126.6,
126.1, 124.2 (CH aryl), 119.7 (C-6), 38.1 (C-10), 19.4 (C-7), 13.5 (C-8); MS m/z (ES+)
304 (100 %, M+Na+); HMRS (ES+) calcd for C18H19O2N1Na1 (M+Na
+): 304.1308, found:
304.1309.
The (Z)-stereochemistry was deduced by a nOe experiment: irradiation at 5.75 ppm (CH=C)
enhanced the aromatic peaks (CHaro) from 3.16 % and did not enhance peak at 3.39 ppm
(N-CH3).
(Z)-1-p-Tolylprop-1-enyl methylphenylcarbamate 248c.
N O
O
1
23
4
56
7
8
9
10
111213
14
General procedure K was followed. 4-Methylpropiophenone 246c (500 mg, 3.38 mmol)
gave after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound (Z)-
248c (455 mg, 48 %) as a white solid.
Rf 0.22 (Petrol:EtOAc 8:2); m.p. 57-59 °C (Et2O/Petrol); IR max (neat/cm-1
) 1712 (C=O);
1H NMR (400MHz, CDCl3) 7.43-7.25 (m, 7H, aryl), 7.10 (d, J = 8.0 Hz, 2H, aryl), 5.76
(q, J = 6.8 Hz, 1H, H-7), 3.38 (br, 3H, H-10), 2.31 (s, 3H, H-1), 1.73 (d, J = 6.8 Hz, 3H, H-
8); 13
C NMR (100MHz, CDCl3) 153.2 (C-9), 147.4 (C-6), 143.1, 137.6, 132.9, 129.1,
126.6, 126.3, 126.2, 124.3 (aryl), 111.8 (C-7), 38.2 (C-10), 21.1 (C-1), 11.5 (C-8); MS m/z
(ES+) 304 (100 %, M+Na+); HMRS (ES+) calcd for C18H19O2N1Na1 (M+Na
+): 304.1308,
found: 304.1295.
The (Z)-stereochemistry was deduced by a nOe experiment: irradiation at 5.76 ppm (CH=C)
enhanced the aromatic peaks (CHaro) from 3.65 % and did not enhance peak at 3.38 ppm
(N-CH3).

267
(Z)-1-Phenylbut-1-enyl 4-chlorophenylmethylcarbamate 248d.
14
N O
O
1
2
3
45
6
78
9
10
1112
13
Cl
General procedure K was followed. Butyrophenone 246b (500 mg, 3.38 mmol) gave after
flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound (Z)-248d (548
mg, 52 %) as a yellow oil.
Rf 0.23 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1719 (C=O); 1H NMR (400MHz, CDCl3)
7.32-7.17 (m, 9H, aryl), 5.70 (t, J = 7.6 Hz, 1H, H-6), 3.32 (br, 3H, H-10), 2.12 (qn, J =
7.6 Hz, 2H, H-7), 1.00 (t, J = 7.6 Hz, 3H, H-8); 13
C NMR (100MHz, CDCl3) 153.0 (C-9),
145.8 (C-5), 141.5, 135.4, 129.2, 128.3, 127.9 (br), 127.3, 124.3 (aryl), 119.9 (C-6), 38.0
(C-10), 19.5 (C-7), 13.5 (C-8); MS m/z (ES+) 338 (100 %, M+Na+); HMRS (ES+) calcd
for C18H18O2N135
Cl1Na1 (M+Na+): 338.0918, found: 338.0911.
The (Z)-stereochemistry was deduced by a nOe experiment: irradiation at 5.70 ppm (CH=C)
enhanced the aromatic peaks (CHaro) from 3.24 % and did not enhance peak at 3.32 ppm
(N-CH3).
1-Phenylvinyl methylphenylcarbamate 248e.[231]
7
8
910
11
12
1
2
34
5
6N O
O
General procedure K was followed. Acetophenone 246e (300 mg, 2.50 mmol) gave after
crystallisation from Et2O/n-hexane (2/4) the title compound 248e (288 mg, 46 %) as a
white solid.

268
Rf 0.36 (Petrol:EtOAc 8:2); m.p. 57-60 °C (Et2O/hexane); IR max (neat/cm-1
) 1706 (C=O),
1642 (C=C); 1H-NMR (400MHz, CDCl3) δ 7.43-7.29 (m, 10H, aryl), 5.41 (br, 1H, H-6),
5.08 (br, 1H, H-6), 3.39 (br, 3H, H-8); 13
C-NMR (100MHz, CDCl3) δ 153.5, 153.2 (C-5
and C-7), 142.8, 134.8, 129.1, 128.7, 128.4, 126.4 (br), 124.9 (aryl), 101.6 (C-6), 38.2 (C-
8); MS m/z (ES+) 276 (100 %, M+Na+); HMRS (ES+) calcd for C16H15O2N1Na1 (M+Na
+):
276.0995, found: 276.1003. Spectral data matched the published values.[231]
1-Phenylvinyl isopropylphenylcarbamate 248f.
7
8
1011
12
1
2
34
5
6N O
O
9 9
13
General procedure K was followed. Acetophenone 246e (500 mg, 4.17 mmol) gave after
flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound 248f (722 mg,
62 %) as a white solid.
Rf 0.24 (Petrol:EtOAc 8:2); m.p. 75-77 °C (Et2O/hexane); IR max (neat/cm-1
) 1698 (C=O);
1H NMR (400MHz, CDCl3) 7.45-7.23 (m, 10H, aryl), 5.35 (s, 1H, H-6), 5.03 (s, 1H, H-6),
4.63 (sep, J = 6.8 Hz, 1H, H-8), 1.16 (d, J = 6.8 Hz, 6H, H-9); 13
C NMR (100MHz, CDCl3)
153.2 (C-5 and C-7), 137.9, 135.1, 129.9, 128.9, 128.5, 128.3, 127.8, 124.8 (aryl), 101.1
(C-6), 49.3 (C-8), 21.2 (C-9); MS m/z (ES+) 304 (100 %, M+Na+); HMRS (ES+) calcd for
C18H19O2N1Na1 (M+Na+): 304.1308, found: 304.1297.

269
1-(4-Chlorophenyl)vinyl isopropylphenylcarbamate 248g.
7
8
1011
12
1
2
34
5
6N O
O
9 9
13
Cl
General procedure K was followed. 4-Chloroacetophenone 246g (500 mg, 3.25 mmol)
gave after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound
248g (611 mg, 60 %) as a yellow solid.
Rf 0.19 (Petrol:EtOAc 8:2); m.p. 82-84 °C (Et2O/hexane); IR max (neat/cm-1
) 1717 (C=O);
1H NMR (400MHz, CDCl3) 7.46-7.36 (m, 3H, aryl), 7.26-7.21 (m, 6H, aryl), 5.31 (s, 1H,
H-6), 5.06 (s, 1H, H-6), 4.61 (sep, J = 6.8 Hz, 1H, H-8), 1.15 (d, J = 6.8 Hz, 6H, H-9); 13
C
NMR (100MHz, CDCl3) 152.2 (C-5 and C-7), 137.8, 134.4, 133.7, 129.8, 129.0, 128.5,
127.9, 126.2 (aryl), 101.5 (C-6), 49.4 (C-8), 21.2 (C-9); MS m/z (ES+) 338 (100 %,
M+Na+); HMRS (ES+) calcd for C18H18O2N1
35Cl1Na1 (M+Na
+): 338.0918, found:
338.0908.
1-Phenylvinyl 4-chlorophenylisopropylcarbamate 248h.
7
8
1011
12
1
2
34
5
6N O
O
9 9
13
Cl
General procedure K was followed. Acetophenone 246e (400 mg, 3.33 mmol) gave after
flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound 248h (529
mg, 50 %) as a white solid.

270
Rf 0.27 (Petrol:EtOAc 8:2); m.p. 95-97 °C (Et2O/hexane); IR max (neat/cm-1
) 1698 (C=O);
1H NMR (400MHz, CDCl3) 7.41 (d, J = 8.5 Hz, 2H, aryl), 7.31-7.29 (m, 5H, aryl), 7.18 (d,
J = 8.5 Hz, 2H, aryl), 5.36 (br, 1H, H-6), 5.02 (br, 1H, H-6), 4.63 (sep, J = 6.8 Hz, 1H, H-8),
1.15 (d, J = 6.8 Hz, 6H, H-9); NMR (100MHz, CDCl3) 153.2, 153.0 (C-5 and C-7), 136.4,
134.9, 133.7, 131.1, 129.2, 128.7, 128.3, 124.8 (aryl), 101.4 (C-6), 49.3 (C-8), 21.2 (C-9);
MS m/z (ES+) 338 (100 %, M+Na+); HMRS (ES+) calcd for C18H18O2N1
35Cl1Na1
(M+Na+): 338.0919, found: 338.0905.
1-(4-Methoxyphenyl)vinyl isopropylphenylcarbamate 248i.
78
10
1112
1
23
45
6
N O
O
109
13
O
14
General procedure K was followed. p-Methoxyacetophenone 246i (500 mg, 3.33 mmol)
gave after flash column chromatography (SiO2, Petrol:EtOAc 10 %) the title compound
248i (370 mg, 37 %) as a white solid.
Rf 0.17 (Petrol:EtOAc 8:2); m.p. 81-83 °C (Et2O/hexane); IR max (neat/cm-1
) 1702 (C=O),
1245 (C-O-C); 1H NMR (400MHz, CDCl3) 7.43-7.22 (m, 7H, aryl), 6.81 (d, J = 8.4 Hz,
2H, H-3), 5.22 (s, 1H, H-7), 4.93 (s, 1H, H-7), 4.63 (sep, J = 6.8 Hz, 1H, H-9), 3.79 (s, 3H,
H-1), 1.15 (d, J = 6.8 Hz, 6H, H-10); 13
C NMR (100MHz, CDCl3) 159.8 (C-2), 153.1 (C-
6 and C-8), 142.5, 138.0, 129.9, 128.9, 127.8, 127.7, 126.2 (aryl), 113.7 (C-3), 99.3 (C-7),
55.3 (C-1), 49.2 (C-9), 21.2 (C-10)MS m/z (ES+) 334 (100 %, M+Na+); HMRS (ES+)
calcd for C18H21O3N1Na1 (M+Na+): 334.1414, found: 334.1415.

271
(Z)-1,2-Diphenylvinyl isopropylphenylcarbamate 248j.
1
2
34
56
78
910
11
1213
1415
16
17
13
N O
O
General procedure K was followed. Deoxybenzoin 246j (500 mg, 2.55 mmol) gave after
flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound (Z)-248j (485
mg, 55 %) as a white solid.
Rf 0.38 (Petrol:EtOAc 8:2); m.p. 96-98 °C (Et2O/hexane); IR max (neat/cm-1
) 1716 (C=O);
1H NMR (400MHz, CDCl3) 7.51-7.23 (m, 15H, aryl), 6.56 (s, 1H, H-6), 4.57 (sep, J = 6.8
Hz, 1H, H-12), 1.11 (d, J = 6.8 Hz, 6H, H-13); 13
C NMR (100MHz, CDCl3) 152.1 (C-11),
147.0 (C-5), 137.6, 136.5, 134.5, 129.9, 128.9, 128.8, 128.5, 128.2, 128.1, 128.0, 127.2,
124.7 (aryl), 116.6 (C-6), 49.3 (C-12), 21.1 (C-13); MS m/z (ES+) 380 (100 %, M+Na+);
HMRS (ES+) calcd for C24H23O2N1Na1 (M+Na+): 380.1621, found: 380.1625.
The Z-stereochemistry was deduced by an X-ray crystal structure.
(Z)-1,2-diphenylvinyl 4-chlorophenylisopropylcarbamate 248k.
1
2
34
56
78
910
11
1213
1415
16
17
13
N O
OCl
General procedure K was followed. Deoxybenzoin 246j (1.1 g, 5.6 mmol) gave after flash
column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound (Z)-248k (1.1 g,
46 %) as a white solid.

272
Rf 0.45 (Petrol:EtOAc 8:2); m.p. 83-85 °C (Et2O/hexane); IR max (neat/cm-1
) 1718 (C=O);
1H NMR (400MHz, CDCl3) 7.47-7.20 (m, 14H, aryl), 6.58 (s, 1H, H-6), 4.56 (sep, J = 6.8
Hz, 1H, H-12), 1.09 (d, J = 6.8 Hz, 6H, H-13); 13
C NMR (100MHz, CDCl3) 152.1 (C-11),
146.9 (C-5), 136.3, 136.2, 134.5, 131.3, 129.3, 128.8, 128.6, 128.4, 128.2, 127.3, 124.7
(aryl), 116.8 (C-6), 49.3 (C-12), 21.1 (C-13); MS m/z (ES+) 414 (100 %, M+Na+); HMRS
(ES+) calcd for C24H22O2N1Na1 (M+Na+): 414.1231, found: 414.1231.
1-1-Diphenylallyl methylcarbamate 249a.
O
NH
O123
45
6
7
8
9
General procedure O was followed with LDA (2.5 equiv.). (Z)-1-Phenylprop-1-enyl
methylphenylcarbamate 248a (50 mg, 0.19 mmol) gave after flash column chromatography
(SiO2, Petrol:EtOAc 5 % + NEt3 1 %) the title compound 249a (43 mg, 86 %) as a white
solid.
Rf 0.13 (Petrol:EtOAc 8:2); m.p. 98-100 °C (Et2O/hexane); IR max (neat/cm-1
) 3349 (br,
N-H), 1700 (C=O), 1522 (Amide II), 1262 (Amide III); 1H NMR (400MHz, CDCl3)
7.32-7.23 (m, 10H, aryl), 7.13 (dd, J = 17.4 Hz, J = 10.8 Hz, 1H, H-2), 5.42 (d, J = 10.8
Hz, 1H, H-1), 4.82 (br, 1H, -NH), 4.80 (d, J = 17.4 Hz, 1H, H-1), 2.74 (d, J = 4.8 Hz, 3H, H-
9); 13
C NMR (100MHz, CDCl3) 155.1 (C-8), 143.4 (C-4), 140.3 (C-2), 127.8, 127.3,
127.1 (CH aryl), 119.3 (C-1), 86.5 (C-3), 27.4 (C-9); MS m/z (ES+) 290 (100 %, M+Na+);
HMRS (ES+) calcd for C17H17O2N1Na1 (M+Na+): 290.1152, found: 290.1148.

273
(E)-1,1-Diphenylbut-2-enyl methylcarbamate 249b.
O
NH
O
1
23
4
9
10
56
7
8
General procedure O was followed with LDA (2.5 equiv.). (Z)-1-Phenylbut-1-enyl 4-
chlorophenylmethylcarbamate 248b (50 mg, 0.18 mmol) gave after crystallisation from
hexane:Et2O 1 % the title compound 249b (39 mg, 78 %) as a white solid.
Rf 0.18 (Petrol:EtOAc 8:2); m.p. 88-90 °C (Et2O/hexane); IR max (neat/cm-1
) 3329 (br, N-
H), 1703 (C=O), 1517 (Amide II), 1251 (Amide III); 1H NMR (400MHz, CDCl3) 7.29-
7.20 (m, 10H, aryl), 6.83 (d, J = 15.6 Hz, 1H, H-3), 5.13 (dq, J = 15.6 and 6.5 Hz, 1H, H-2),
4.80 (br, -NH), 2.70 (d, J = 4.8 Hz, 3H, H-10), 1.72 (dd, J = 6.5 and 1.6 Hz, 3H, H-1); 13
C
NMR (100MHz, CDCl3) 155.2 (C-9), 144.2 (C-5), 133.6 (C-3), 131.6 (C-2), 127.7,
127.1, 127.0 (CH aryl), 86.5 (C-4), 27.3 (C-10), 18.0 (C-1); MS m/z (ES+) 304 (100%,
M+Na+); HMRS (ES+) calcd for C18H19O2N1Na1 (M+Na
+): 304.1302, found: 304.1321.
1-Phenyl-1-p-tolylallyl methylcarbamate 249c.
O
NH
O123
45
6
7
8
910
11
12
13
14
General procedure O was followed with LDA (2.5 equiv.). (Z)-1-p-Tolylprop-1-enyl
methylphenylcarbamate 248c (50 mg, 0.18 mmol) gave after flash column chromatography
on silica (Petrol:EtOAc 10 % + NEt3 1%) the title compound 249c (40 mg, 80 %) as a
white solid.

274
Rf 0.10 (Petrol:EtOAc 8:2); m.p. 80-82 °C (Et2O/hexane); IR max (neat/cm-1
) 3353 (br, N-
H), 1700 (C=O), 1513 (Amide II), 1263 (Amide III); 1H NMR (400MHz, CDCl3) 7.30-
7.09 (m, 10H, aryl and H-2), 5.41 (d, J = 10.8 Hz, 1H, H-1), 4.84 (br, 1H, -NH), 4.81 (d, J =
17.4 Hz, 1H, H-1), 2.73 (d, J = 4.8 Hz, 3H, H-14), 2.33 (s, 3H, H-8); 13
C NMR (100MHz,
CDCl3) 155.1 (C-13), 143.5, 140.5, 140.4, 136.9, 128.6, 127.8, 127.2, 127.0, 126.9 (aryl
and C-2), 119.0 (C-1), 86.5 (C-3), 27.3 (C-14), 21.0 (C-8); MS m/z (ES+) 304 (100 %,
M+Na+); HMRS (ES+) calcd for C18H19O2N1Na1 (M+Na
+): 304.1308, found: 304.1299.
HPLC separation conditions: Chiralpack AD-H column, hexane:2-propanol (95:5), flow
rate: 1.0 mL/min, tR 13.3 min for the major enantiomer and 13.9 min for the minor
enantiomer.
(E)-1-(4-Chlorophenyl)-1-phenylbut-2-enyl methylcarbamate 249d.
O
NH
O
Cl
1
234
5 6
7
8
910
11
12
13
14
General procedure O was followed with LDA (2.5 equiv.). (Z)-1-Phenylbut-1-enyl 4-
chlorophenylmethylcarbamate 248d (50 mg, 0.16 mmol) gave after crystallisation from
hexane:Et2O 1 % the title compound 249d (40 mg, 80 %) as a white solid.
Rf 0.14 (Petrol:EtOAc 8:2); m.p. 115-117 °C (Et2O/hexane); IR max (neat/cm-1
) 3309 (br,
N-H), 1692 (C=O), 1556 (Amide II), 1264 (Amide III); 1H NMR (400MHz, CDCl3)
7.24-7.13 (m, 9H, aryl), 6.73 (d, J = 15.6 Hz, 1H, H-3), 5.11-5.02 (m, 1H, H-2), 4.73 (br,
1H, -NH), 2.66 (d, J = 4.8 Hz, 3H, H-14), 1.68 (dd, J = 5.2 and 1.6 Hz, 3H, H-1); 13
C NMR
(100MHz, CDCl3) 155.1 (C-13), 143.7, 142.9 (C-5 and C-9), 133.2 (C-3), 133.0 (C-12),
132.0 (C-2), 128.5, 128.0, 127.9, 127.3, 127.0 (CH aryl), 86.1 (C-4), 27.4 (C-14), 18.1 (C-
1); MS m/z (ES+) 338 (20 %, M+Na+), (ES-) 350 (100 %, M+Cl
-); HMRS (ES+) calcd for
C18H19O2N135
Cl1 (M+H+): 316.1099, found: 316.1112.
HPLC separation conditions: Chiralcel OD-H column, hexane:2-propanol (97:3), flow rate:
1.0 mL/min, tR 15.1 min for the minor enantiomer and 16.5 min for the major enantiomer.

275
4,4-Diphenylbut-3-en-2-yl methylcarbamate 250.
NH
O
O
5
67
8
9
10
1
23
411
12
1314
General procedure O was followed with LDA (2.5 equiv.). (Z)-1-Phenylbut-1-enyl 4-
chlorophenylmethylcarbamate 248b (50 mg, 0.18 mmol) gave after flash column
chromatography on silica (Petrol:EtOAc 5 %) the title compound 250 (37 mg, 75 %) as a
colourless oil.
Rf 0.18 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3338 (br, N-H), 1694 (C=O), 1519 (Amide
II), 1248 (Amide III); 1H NMR (400MHz, CDCl3) 7.41-7.22 (m, 10H, aryl), 6.03 (d, J =
8.8 Hz, 1H, H-10), 5.33-5.27 (m, 1H, H-11), 4.53 (br, 1H, -NH), 2.76 (d, J = 5.2 Hz, 3H, H-
14), 1.31 (d, J = 6.4 Hz, 3H, H-12); 13
C NMR (100MHz, CDCl3) 156.4 (C-13), 143.0,
141.6, 139.0 (C-4, C-5 and C-6), 129.5, 128.7, 128.3, 128.1, 127.5, 127.4, 127.3 (C-10 and
aryl), 69.8 (C-11), 27.4 (C-14), 21.4 (C-12); MS m/z (ES+) 304 (100 %, M+Na+); HMRS
(ES+) calcd for C18H19O2N1Na1 (M+Na+): 304.1308, found: 304.1302.
(2,5-Dimethylfuran-3-yl)methanol 254b.[232]
O1
2
3
45
6
1HO
Procedure B was followed. 2,5-Dimethyl-3-furoic acid 253 (500 mg, 3.57 mmol) gave the
title compound 254b (331 mg) as a yellow oil which was reacted without purification.
Rf 0.57 (Petrol:EtOAc 5:5); 1H NMR (300MHz, CDCl3) 5.94 (s, 1H, H-4), 4.40 (s, 2H,
H-6), 2.23 (s, 6H, H-1). Spectral data matched the published values.[232]

276
(Furan-3-yl)methyl methylphenylcarbamate 255a.
N O
O
O1
2
34
5
6
7
8
910
11
General procedures E and F were followed. 2-Furylmethanol 254a (1.0 g, 0.01 mmol) gave
after flash column chromatography (SiO2, Petrol:EtOAc 10 %) the title compound 255a
(1.5 g, 66 % over 2 steps) as a colourless oil.
Rf 0.52 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1703 (C=O); 1H NMR (400MHz, CDCl3)
7.39 (s, 1H, H-1), 7.34-7.30 (m, 3H, aryl, H-2), 7.23-7.16 (m, 3H, aryl), 6.39 (s, 1H, H-3),
5.02 (s, 2H, H-5), 3.31 (s, 3H, H-7); 13
C NMR (100MHz, CDCl3) 155.5 (C-6), 143.5 (C-
8), 143.2 (C-2), 141.2 (C-1), 128.9, 126.1, 125.8 (CH aryl), 121.0 (C-4), 110.5 (C-3), 59.0
(C-5), 29.7 (C-7); MS m/z (ES+) 254 (100%, M+Na+); HMRS (ES+) calcd for
C13H13O3N1Na1 (M+Na+): 254.0788, found: 254.0790.
(2,5-Dimethylfuran-3-yl)methyl methylphenylcarbamate 255b.
N O
O
O1
2
3
45
6
7
8
9
1011
12
1
General procedures E and F were followed. (2,5-Dimethylfuran-3-yl)methanol 254b (331
mg, 2.63 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 10 %) the
title compound 255b (573 mg, 62 % over 3 steps) as a yellow oil.
Rf 0.57 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1698 (C=O); 1H NMR (400MHz, CDCl3)
7.28-7.11 (m, 5H, aryl), 5.82 (br, 1H, H-4), 4.82 (s, 2H, H-6), 3.22 (s, 3H, H-8) 2.14 (s, 6H,
H-1); 13
C NMR (100MHz, CDCl3) 155.7 (C-7), 149.8 (C-3), 148.6 (C-2), 143.3 (C-9),
128.8, 125.9, 125.7 (CH aryl), 115.5 (C-5), 107.3 (C-4), 59.5 (C-6), 37.7 (C-8), 13.4, 11.6

277
(C-1); MS m/z (ES+) 282 (100 %, M+Na+); HMRS (ES+) calcd for C15H17O3N1Na1
(M+Na+): 282.1101, found: 282.1101.
Propyl methylphenylcarbamate 257.
N O
O1
2
3
4
5
6
7
8
9
General procedures E and F were followed. 1-Propanol (0.5 g, 8.3 mmol) gave after flash
column chromatography (SiO2, Petrol:EtOAc 4 %) the title compound 257 (1.2 g, 74 %
over 2 steps) as a pale yellow oil.
Rf 0.50 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1699 (C=O); 1H NMR (400MHz, CDCl3)
7.36-7.18 (m, 5H, aryl), 4.06 (t, J = 6.8 Hz, 2H, H-3), 3.30 (s, 3H, H-5), 1.64-1.57 (m, 2H,
H-2), 0.88 (t, J = 7.6 Hz, 3H, H-1); 13
C NMR (100MHz, CDCl3) 155.7 (C-4), 143.3 (C-
6), 128.7, 125.8, 125.6 (CH aryl), 67.2 (C-3), 37.5 (C-5), 22.2 (C-2), 10.3 (C-1); MS m/z
(ES+) 194 (100 %, M+H+); HMRS (ES+) calcd for C11H15O2N1Na1 (M+Na
+): 216.0995,
found: 216.0989.
3-Methyl-1,1-diphenylbutyl methylcarbamate 259a.
NH
O
O
1
2
34
5
6 8
7
8
910
2
3
1
General procedure O was followed with i-PrLi (2.5 equiv.). 1-Phenylvinyl
methylphenylcarbamate 248e (50 mg, 0.20 mmol) gave after flash column chromatography
(SiO2, Petrol:EtOAc 2 %) the title compound 259a (21 mg, 36 %) as a colourless oil.

278
Rf 0.52 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3390 (br, N-H), 1627 (C=O), 1592 (Amide
II), 1354 (Amide III); 1H NMR (400MHz, CDCl3) 7.41-7.03 (m, 10H, aryl), 5.11 (br, 1H,
-NH), 3.20 (br, 3H, H-10), 2.09-1.83 (m, 3H, H-6 and H-7), 1.04 (d, J = 5.9 Hz, 3H, H-8),
0.98 (d, J = 5.9 Hz, 3H, H-8) ; 13
C NMR (100MHz, CDCl3) 174.8 (C-9), 143.9 (C-4),
128.8, 128.1, 127.6, 127.3, 125.8 (CH aryl), 78.0 (C-5), 44.6 (C-6), 40.8 (C-10), 24.4 (C-7),
24.1 (C-8); MS m/z (ES+) 298 (100 %, M+H+); HMRS (ES+) calcd for C19H24O2N1
(M+H+): 298.1802, found: 298.1806.
1,1-Diphenylhexyl methylcarbamate 259b.
2
3
1
NH
O
O
1
2
34
5
6 7
8 9
10
1112
General procedure O was followed with n-BuLi (2.5 equiv.). 1-Phenylvinyl
methylphenylcarbamate 248e (50 mg, 0.20 mmol) gave after flash column chromatography
(SiO2, Petrol:EtOAc 2 %) the title compound 259b (22 mg, 33 %) as a colourless oil.
Rf 0.55 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3411 (br, N-H), 1634 (C=O), 1593 (amide
II), 1356 (Amide III); 1H NMR (400MHz, CDCl3) 7.30-7.00 (m, 10H, aryl), 5.28 (br, 1H,
-NH), 3.22 (br, 3H, H-12), 1.86 (br, 2H, H-6), 1.67-1.58 (m, 1H, H-7), 1.39-1.19 (m, 5H, H-
7, H-8 and H-9), 0.92 (t, J = 7.2 Hz, 3H, H-10) ; 13
C NMR (100MHz, CDCl3) 174.5 (C-
11), 143.4 (C-4), 128.8, 128.4, 128.1, 127.8, 127.4, 126.0 (CH aryl), 77.8 (C-5), 39.1 (C-6),
36.3 (C-12), 32.1, 23.2, 22.7 (C-7, C-8 and C-9), 14.1 (C-10); MS m/z (ES+) 334 (100 %,
M+Na+); HMRS (ES+) calcd for C20H25O2N1Na1 (M+Na
+): 334.1778, found: 334.1768.

279
3-Methyl-1,1-diphenylpentyl methylcarbamate 259c.
NH
O
O
1
2
34
5
67
8
910
1112
12
3
General procedure O was followed with sec-BuLi (2.5 equiv.). 1-Phenylvinyl
methylphenylcarbamate 248e (50 mg, 0.20 mmol) gave after flash column chromatography
(SiO2, Petrol:EtOAc 2 %) the title compound 259c (23 mg, 35 %) as a colourless oil.
Rf 0.45 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3400 (br, N-H), 1628 (C=O), 1594 (Amide
II), 1369 (Amide III); 1H NMR (400MHz, CDCl3) (mixture of rotamers) 7.18-6.99 (br,
10H, aryl), 5.07 (br, 0.5H, -NH, rotamer 1), 4.97 (br, 0.5H, -NH, rotamer 1), 3.18 (br, 3H,
H-12), 2.15-1.79 (2 br, 2H, H-6), 1.74-1.60 (m, 1H, H-7), 1.53-1.42 (m, 1H, H-9), 1.31-1.20
(m, 1H, H-9), 1.00 (d, J = 6.8 Hz, 1.5H, H-8, rotamer 1), 0.98 (d, J = 6.8 Hz, 1.5H, H-8,
rotamer 2), 0.93 (t, J = 7.6 Hz, 1.5H, H-10, rotamer 1), 0.86 (t, J = 7.6 Hz, 1.5H, H-10,
rotamer 2); 13
C NMR (100MHz, CDCl3) (mixture of rotamers) 174.9, 174.8 (C-11),
143.8 (C-4), 128.9, 128.1, 127.5, 127.2, 125.9, 125.8 (CH aryl), 77.8 (C-5), 43.0 (C-6),
31.0, 30.9 (C-9), 29.8 (C-12), 20.3 (C-7), 11.4 (C-8), 11.0 (C-10); MS m/z (ES+) 334
(100 %, M+Na+); HMRS (ES+) calcd for C20H25O2N1Na1 (M+Na
+): 334.1778, found:
334.1771.

280
3-Methyl-1-phenylbutyl isopropylphenylcarbamate 261a.
N O
O
1
2
34
5
67
8
9
1011
12
13
14
8
11
15
General procedure O was followed with i-PrLi (2.5 equiv.). 1-Phenylvinyl
isopropylphenylcarbamate 248f (50 mg, 0.18 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 261a as a colourless oil.
Rf 0.64 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1703 (C=O); 1H NMR (400MHz, CDCl3)
7.33-7.00 (m, 10H, aryl), 5.64 (dd, J = 8.8 and 5.6 Hz, 1H, H-5), 4.50 (sep, J = 6.8 Hz, 1H,
H-10), 1.62-1.51 (m, 1H, H-6), 1.44-1.25 (m, 2H, H-6 and H-7), 1.03 (d, J = 6.8 Hz, 3H, H-
11), 0.98 (d, J = 6.8 Hz, 3H, H-11), 0.80 (d, J = 6.0 Hz, 3H, H-8), 0.77 (d, J = 6.0 Hz, 3H,
H-8); 13
C NMR (100MHz, CDCl3) 154.9 (C-9), 142.1 (C-12), 138.3 (C-4), 130.1, 128.5,
128.2, 127.3, 127.2, 126.0 (CH aryl), 75.5 (C-5), 48.6 (C-10), 46.1 (C-6), 24.7 (C-7), 22.8,
22.4 (C-8), 21.4, 21.3 (C-11); MS m/z (ES+) 348 (100 %, M+Na+); HMRS (ES+) calcd for
C21H27O2N1Na1 (M+Na+): 348.1934, found: 348.1940.
1-(4-Chlorophenyl)hexyl isopropylphenylcarbamate 261c.
N O
O
Cl1
2
34
5
67
8 9
10
11
1213
14
1516
17
13
General procedure S was followed. 1-(4-Chlorophenyl)vinyl isopropylphenylcarbamate
248g (50 mg, 0.16 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc
1 %) the title compound 261b as a colourless oil.

281
Rf 0.55 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1699 (C=O); 1H NMR (500MHz, CDCl3)
7.40-7.03 (m, 9H, aryl), 5.59 (t, J = 6.5 Hz, 1H, H-5), 4.56 (sep, J = 6.8 Hz, 1H, H-12),
1.69-1.45 (m, 2H, H-6), 1.22-1.10 (m, 6H, H-7, H-8 and H-9), 1.07 (d, J = 6.8 Hz, 6H, H-
13), (t, J = 6.5 Hz, 3H, H-10); 13
C NMR (125MHz, CDCl3) 154.7 (C-11), 140.3 (C-14),
138.2 (C-4), 133.0 (C-1), 130.0, 128.6, 128.3, 127.5, 127.4 (CH aryl), 76.1 (C-5), 48.7 (C-
12), 36.6 (C-6), 31.3, 24.7, 22.4 (C-7, C-8 and C-9), 21.3 (C-13), 13.9 (C-10); MS m/z
(ES+) 396 (100 %, M+Na+); HMRS (ES+) calcd for C22H28O2N1
35Cl1Na1 (M+Na
+):
396.1701, found: 396.1714.
3-Methyl-1,1-diphenylbutyl isopropylcarbamate 262a.
NH
O
O
1
2
34
5
67
8
8
9
10
11
11
General procedure O was followed with i-PrLi (2.0 equiv.) and DMPU. 1-Phenylvinyl
isopropylphenylcarbamate 248f (50 mg, 0.18 mmol) gave after recrystallisation from
hexane:Et2O 1 % the title compound 262a (46 mg, 80 %) as a white solid.
Rf 0.58 (Petrol:EtOAc 8:2); m.p. 154-156 °C (Hexane/Et2O); 1H NMR (400MHz, CDCl3)
7.34 (d, J = 7.4 Hz, 4H, aryl), 7.26 (t, J = 7.4 Hz, 4H, aryl), 7.17 (t, J = 7.4 Hz, 2H, aryl),
4.68 (br d, J = 6.6 Hz, 1H, -NH), 3.70 (oct, J = 6.6 Hz, 1H, H-10), 2.74 (d, J = 5.6 Hz, 2H,
H-6), 1.56-1.48 (m, 1H, H-7), 1.11 (d, J = 6.6 Hz, 6H, H-11), 0.81 (d, J = 6.4 Hz, 6H, H-8);
13C NMR (100MHz, CDCl3) 153.5 (C-9), 146.0 (C-4), 127.9, 126.5, 125.8 (CH aryl),
85.3 (C-5), 44.7 (C-6), 42.6 (C-10), 24.0 (C-8), 23.8 (C-11), 23.0 (C-7); MS m/z (ES+) 298
(100 %, M+Na+); HMRS (ES+) calcd for C19H24O2N1Na1 (M+Na
+): 298.1802, found:
298.1806.

282
1,1-Diphenylhexyl isopropylcarbamate 262b.
NH
O
O
1
2
34
5
67
8 9
10
11
12
13
13
General procedure O was followed with n-BuLi (2.0 equiv.) and DMPU. 1-Phenylvinyl
isopropylphenylcarbamate 248f (50 mg, 0.18 mmol) gave after recrystallisation from
hexane:Et2O 1 % the title compound 262b (44 mg, 73 %) as a white solid.
Rf 0.50 (Petrol:EtOAc 8:2); m.p. 126-128 °C (Hexane/Et2O); IR max (neat/cm-1
) 3280 (br,
N-H), 1683 (C=O), 1535 (Amide II), 1258 (Amide III); 1H NMR (400MHz, CDCl3)
7.34-7.17 (m, 10H, aryl), 4.68 (br d, J = 6.6 Hz, -NH), 3.71 (oct, J = 6.6 Hz, 1H, H-12),
2.75 (t, J = 7.6 Hz, 2H, H-6), 1.24-1.21 (m, 6H, H-7, H-8 and H-9), 1.11 (d, J = 6.6 Hz, 6H,
H-13), 0.81 (t, J = 6.8 Hz, 6H, H-10); 13
C NMR (100MHz, CDCl3) 153.6 (C-11), 145.7
(C-4), 127.9, 126.6, 125.9 (CH aryl), 85.3 (C-5), 42.7 (C-12), 37.4 (C-6), 31.9, 23.0 (C-13),
22.8, 22.5 (C-7, C-8 and C-9), 13.9 (C-10); MS m/z (ES+) 362 (100 %, M+Na+); HMRS
(ES+) calcd for C22H29O2N1Na1 (M+Na+): 362.2091, found: 362.2082.
3-Methyl-1,1-diphenylpentyl isopropylcarbamate 262c.
2
3
1
4NH
O
O
1
23
45
67
8
9
10
11
12
13
13
General procedure O was followed with sec-PrLi (2.0 equiv.) and DMPU. 1-Phenylvinyl
isopropylphenylcarbamate 248f (50 mg, 0.18 mmol) gave after recrystallisation from
hexane:Et2O 1 % the title compound 262c (48 mg, 80 %) as a white solid.

283
Rf 0.42 (Petrol:EtOAc 8:2); m.p. 133-135 °C (Hexane/Et2O); 1H NMR (400MHz, CDCl3)
7.35-7.24 (m, 8H, aryl), 7.16 (t, J = 7.2 Hz, 2H, aryl), 4.67 (br d, J = 6.6 Hz, -NH), 3.68
(oct, J = 6.6 Hz, 1H, H-12), 2.77 (dd, J = 14.0 and 3.2 Hz, 1H, H-6), 2.69 (dd, J = 14.0 and
6.8 Hz, 1H, H-6), 1.27-1.16 (m, 3H, H-7 and H-9), 1.13 (d, J = 6.6 Hz, 3H, H-13), 1.11 (d, J
= 6.6 Hz, 3H, H-13), 0.74-0.70 (m, 6H, H-8 and H-10); 13
C NMR (100MHz, CDCl3)
153.5 (C-11), 146.1, 146.0 (C-4), 127.8, 126.5, 126.4, 125.9, 125.8 (CH aryl), 85.2 (C-5),
42.9 (C-6), 42.6 (C-12), 30.8 (C-9) 29.8 (C-7), 22.9 (C-13), 20.2 (C-8), 11.1 (C-10); MS
m/z (ES+) 362 (100 %, M+Na+); HMRS (ES+) calcd for C22H29O2N1Na1 (M+Na
+):
362.2091, found: 362.2103.
3,3-Dimethyl-1,1-diphenylbutyl isopropylcarbamate 262d.
NH
O
O
1
2
34
5
67
8
9
10
11
11
General procedure O was followed with tert-BuLi (2.0 equiv.) and DMPU. 1-Phenylvinyl
isopropylphenylcarbamate 248f (50 mg, 0.18 mmol) gave after recrystallisation from
hexane:Et2O 1 % the title compound 262d (40 mg, 83 %) as a white solid.
Rf 0.60 (Petrol:EtOAc 8:2); m.p. 198-200 °C (Hexane/Et2O); IR max (neat/cm-1
) 3333 (br,
N-H), 1688 (C=O), 1521 (Amide II), 1253 (Amide III); 1H NMR (400MHz, CDCl3) 7.35
(d, J = 7.6 Hz, 4H, aryl), 7.24 (t, J = 7.6 Hz, 4H, aryl), 7.15 (t, J = 7.6 Hz, 2H, aryl), 4.69 (br
d, J = 6.6 Hz, -NH), 3.70 (oct, J = 6.6 Hz, 1H, H-10), 2.90 (s, 2H, H-6), 1.11 (d, J = 6.6 Hz,
6H, H-11), 0.77 (s, 9H, H-8) ; 13
C NMR (100MHz, CDCl3) 153.5 (C-9), 147.0 (C-4),
127.9, 126.3, 125.6 (CH aryl), 84.7 (C-5), 47.2 (C-6), 42.6 (C-10), 31.5 (C-7), 31.0 (C-8),
23.0 (C-11); MS m/z (ES+) 362 (100 %, M+Na+); HMRS (ES+) calcd for C22H29O2N1Na1
(M+Na+): 362.2091, found: 362.2092.

284
1-(4-Chlorophenyl)-3-methyl-1-phenylbutyl isopropylcarbamate 262e.
NH
O
O
Cl1
2
34
5
67
8
8
910
11
12
13
14
15
15
General procedure O was followed with i-PrLi (2.0 equiv.) and DMPU. 1-(4-
Chlorophenyl)vinyl isopropylphenylcarbamate 248g (50 mg, 0.16 mmol) gave after
recrystallisation from hexane:Et2O 1 % the title compound 262e (43 mg, 81 %) as a white
solid.
Rf 0.30 (Petrol:EtOAc 8:2); m.p. 154-156 °C (Hexane/Et2O); 1H NMR (400MHz, CDCl3)
7.30-7.16 (m, 9H, aryl), 4.66 (br d, J = 6.6 Hz, -NH), 3.68 (oct, J = 6.6 Hz, 1H, H-14),
2.71 (dd, J = 14.0 and 6.1 Hz, 1H, H-6), 2.65 (dd, J = 14.0 and 5.6 Hz, 1H, H-6), 1.53-1.46
(m, 1H, H-7), 1.10 (d, J = 6.6 Hz, 6H, H-15), 0.81 (d, J = 6.7 Hz, 3H, H-8), 0.77 (d, J = 6.7
Hz, 3H, H-8); 13
C NMR (100MHz, CDCl3) 153.4 (C-13), 145.4, 144.7, 132.3, 128.1,
128.0, 127.3, 126.7, 125.7 (aryl), 84.8 (C-5), 44.7 (C-6), 42.7 (C-14), 24.0, 23.9 (C-8),
23.7 (C-15), 22.9 (C-7); MS m/z (ES+) 382 (100 %, M+Na+); HMRS (ES+) calcd for
C22H26O2N135
Cl1Na1 (M+Na+): 382.1544, found: 382.1539.
1-(4-Chlorophenyl)-1-phenylhexyl isopropylcarbamate 262f.
NH
O
O
Cl
1
2
345
67
8 9
10
1112
13
14
1516
17
17
General procedure O was followed with n-BuLi (2.0 equiv.) and DMPU. 1-(4-
Chlorophenyl)vinyl isopropylphenylcarbamate 248g (50 mg, 0.16 mmol) gave after

285
recrystallisation from hexane:Et2O 1 % the title compound 262f (44 mg, 74 %) as a white
solid.
Rf 0.70 (Petrol:EtOAc 8:2); m.p. 133-135 °C (Hexane/Et2O); IR max (neat/cm-1
) 3330 (br,
N-H), 1686 (C=O), 1538 (Amide II), 1254 (Amide III); 1H NMR (400MHz, CDCl3)
7.30-7.19 (m, 9H, aryl), 4.69 (br d, J = 6.6 Hz, -NH), 3.71 (oct, J = 6.6 Hz, 1H, H-16),
2.79-2.65 (m, 2H, H-6), 1.27-1.23 (m, 6H, H-7, H-8 and H-9), 1.12 (d, J = 6.6 Hz, 6H, H-
17), 0.81 (t, J = 6.9 Hz, 3H, H-10); 13
C NMR (100MHz, CDCl3) 153.4 (C-15), 145.1,
144.3, 132.5, 128.1, 128.0, 127.4, 126.8, 125.8 (aryl), 84.9 (C-5), 42.7 (C-16), 37.4 (C-6),
31.8, 23.0 (C-17), 22.7, 22.5 (C-7, C-8 and C-9), 13.9 (C-10); MS m/z (ES+) 396 (100 %,
M+Na+); HMRS (ES+) calcd for C22H28O2N1
35Cl1Na1 (M+Na
+): 396.1701, found:
396.1686.
1-(4-Chlorophenyl)-3-methyl-1-phenylpentyl isopropylcarbamate 262g.
NH
O
O
Cl1
2
34
5
6
7
8
9
10
1112
13
14
1516
17
17
General procedure O was followed with sec-BuLi (2.0 equiv.) and DMPU. 1-(4-
Chlorophenyl)vinyl isopropylphenylcarbamate 248g (50 mg, 0.16 mmol) gave after
recrystallisation from hexane:Et2O 1 % the title compound 262g (44 mg, 75 %) as a white
solid.
Rf 0.48 (Petrol:EtOAc 8:2); m.p. 138-140°C (Hexane/Et2O); IR max (neat/cm-1
) 3304 (br,
N-H), 1688 (C=O), 1527 (Amide II), 1241 (Amide III); 1H NMR (400MHz, CDCl3)
(mixture of diastereoisomers) 7.28-7.14 (m, 9H, aryl), 4.64 (br d, J = 6.6 Hz, -NH), 3.67
(oct, J = 6.6 Hz, 1H, H-16), 2.74 (dd, J = 14.0 and 3.6 Hz, 0.5H, H-6), 2.66 (d, J = 5.2 Hz,
1H, H-6), 2.59 (dd, J = 14.0 and 6.8 Hz, 0.5H, H-6), 1.32-1.07 (m, 3H, H-7, H-9), 1.08 (br,
6H, H-17), 0.73-0.65 (m, 6H, H-8 and H-10); 13
C NMR (100MHz, CDCl3) (mixture of
diastereoisomers) 153.3 (C-15), 145.5, 145.4, 144.8, 144.7, 132.4, 132.3, 128.0, 127.9,

286
127.5, 127.3, 126.8, 126.7, 125.8, 125.7 (aryl), 84.8 (C-5), 42.8, 42.7 (C-6), 42.6 (C-16),
30.8, 30.7 (C-9), 29.8 (C-7), 23.0 (C-17), 20.3 (C-8), 11.1 (C-10); MS m/z (ES+) 396
(100 %, M+Na+); HMRS (ES+) calcd for C22H28O2N1
35Cl1Na1 (M+Na
+): 396.1701, found:
396.1698.
1-(4-Chlorophenyl)-3,3-dimethyl-1-phenylbutyl isopropylcarbamate 262h.
NH
O
O
Cl1
2
34
5
67
8
910
11
12
13
14
15
15
General procedure O was followed with tert-BuLi (2.0 equiv.) and DMPU. 1-(4-
Chlorophenyl)vinyl isopropylphenylcarbamate 248g (50 mg, 0.16 mmol) gave after
recrystallisation from hexane:Et2O 1 % the title compound 262h (48 mg, 81 %) as a white
solid.
Rf 0.60 (Petrol:EtOAc 8:2); m.p. 182-183 °C (Hexane/Et2O); IR max (neat/cm-1
) 3323 (br,
N-H), 1689 (C=O), 1524 (Amide II), 1255 (Amide III); 1H NMR (500MHz, CDCl3)
7.32-7.14 (m, 9H, aryl), 4.68 (br d, J = 6.6 Hz, -NH), 3.70 (oct, J = 6.6 Hz, 1H, H-14),
2.91 (d, J = 14.5 Hz, 1H, H-6), 2.81 (d, J = 14.5 Hz, 1H, H-6), 1.12 (d, J = 6.6 Hz, 6H, H-
15), 0.76 (s, 9H, H-8) ; 13
C NMR (125MHz, CDCl3) 153.4 (C-13), 146.4, 145.8, 132.1,
128.1, 128.0, 127.1, 126.5, 125.5 (aryl), 84.3 (C-5), 47.2 (C-6), 42.8 (C-14), 31.5 (C-7),
31.0 (C-8), 23.0, 22.8 (C-15); MS m/z (ES+) 396 (100 %, M+Na+); HMRS (ES+) calcd for
C22H29O2N135
Cl1 (M+H+): 374.1881, found: 374.1874.

287
1,1-Diphenylhexan-1-ol 266a.
OH
1
2
34
5
6
78
910
General procedure P was followed with n-BuLi (2.0 equiv.). 1-Phenylvinyl
isopropylphenylcarbamate 248f (50 mg, 0.18 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 266a (25 mg, 57 %) as a
colourless oil.
Rf 0.61 (Petrol:EtOAc 8:2); 1H NMR (400MHz, CDCl3) 7.43-7.20 (m, 10H, aryl), 2.29-
2.25 (m, 2H, H-6), 2.09 (s, 1H, -OH), 1.30-1.27 (m, 6H, H-7, H-8 and H-9), 0.85 (t, J = 6.8
Hz, 3H, H-10); 13
C NMR (125MHz, CDCl3) 147.2 (C-4), 128.1, 126.7, 126.0 (CH aryl),
78.3 (C-5), 41.9 (C-6), 32.2, 23.4, 22.2 (C-7, C-8 and C-9), 14.0 (C-10); MS m/z (ES+)
254 (100 %, M+); HMRS (ES+) calcd for C18H22O1 (M
+): 254.1665, found: 254.1665.
1-(4-Chlorophenyl)-1-phenylhexan-1-ol 266b.
OH
1
2
34
5
6
78
910
1112
13
14Cl
General procedure P was followed with n-BuLi (2.0 equiv.). 1-(4-Chlorophenyl)vinyl
isopropylphenylcarbamate 248g and 1-phenylvinyl 4-chlorophenylisopropylcarbamate
248h (50 mg, 0.16 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc
1 %) the title compounds 266b (35 mg, 77 % and 25 mg, 55 %) as a colourless oil.
Rf 0.65 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3464 (br, O-H); 1H NMR (400MHz,
CDCl3) 7.40-7.21 (m, 9H, aryl), 2.24 (t, J = 7.6 Hz, 2H, H-6), 2.08 (s, 1H, -OH), 1.30-
1.18 (m, 6H, H-7, H-8 and H-9), 0.85 (t, J = 6.9 Hz, 3H, H-10); 13
C NMR (100MHz,

288
CDCl3) 146.8 (C-11), 145.7 (C-4), 132.5 (C-1), 128.2, 128.2, 127.5, 127.0, 125.9 (CH
aryl), 77.9 (C-5), 41.8 (C-6), 32.1, 23.3, 22.5 (C-7, C-8 and C-9), 14.0 (C-10); MS m/z
(ES-) 287 (100 %, M-H+); HMRS (ES+) calcd for C18H21O1
35Cl1 (M
+): 288.1275, found:
288.1276.
1-(4-Methoxyphenyl)-1-phenylhexan-1-ol 266c.
OH
12
3
45
6
7
89
10
11
1213
14
15O
General procedure P was followed with n-BuLi (2.0 equiv.). 1-(4-Methoxyphenyl)vinyl
isopropylphenylcarbamate 248i (50 mg, 0.16 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 266c (31 mg, 68 %) as a
colourless oil.
Rf 0.46 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3492 (br, O-H), 1610, 1510 (C=C), 1245
(C-O-C); 1H NMR (500MHz, CDCl3) 7.39 (d, J = 5.9 Hz, 2H, aryl), 7.33-7.20 (m, 5H,
aryl), 6.84 (d, J = 8.8 Hz, 2H, H-3), 3.79 (s, 3H, H-1), 2.24 (t, J = 6.1 Hz, 2H, H-7), 2.06 (s,
1H, -OH), 1.29-1.24 (m, 6H, H-7, H-9 and H-10), 0.85 (t, J = 5.4 Hz, 3H, H-11); 13
C NMR
(125MHz, CDCl3) 158.3 (C-2), 147.3 (C-12), 139.5 (C-5), 128.0, 127.3, 126.6, 125.9
(CH aryl), 113.4 (C-3), 78.0 (C-6), 55.2 (C-1), 42.0, 32.2, 23.5 (C-7, C-8 and C-9), 22.5
(C-10), 14.0 (C-11); MS m/z (ES+) 267 (100 %, M-H2O+H+), (ES-) 283 (30 %, M-H
+);
HMRS (ES+) calcd for C19H23O1 (M-H2O+H+): 267.1744, found: 267.1754.

289
1,1,3-Triphenylpropan-1-ol 266d.
OH
1
23
45
6
7
8
910
11
To a mixture of anhydrous toluene (0.10 cm3) and distilled ()-sparteine (0.10 cm
3, 0.45
mmol) was added sec-BuLi (1.20 M, 0.35 cm3, 0.43 mmol) at -45 °C under nitrogen
atmosphere, then the mixture reaction was stirred at room temperature for 1 h. To a
solution of 1-phenylvinyl isopropylphenylcarbamate 248f (50 mg, 0.18 mmol) in
anhydrous THF (1 cm3) and DMPU (0.25 cm
3) was added the above yellow solution
dropwise at -78 °C under nitrogen atmosphere. The resulting reaction mixture was stirred
at -78 °C under nitrogen atmosphere after which MeOH (0.1 cm3) and a saturated aqueous
solution of NH4Cl were added and the reaction mixture stirred for a further 30 min while
warming to room temperature. The mixture was partitioned between Et2O (10 cm3) and
water. The organic layer was washed with water (3 30 cm3), dried over MgSO4, filtered
and concentrated under reduced pressure. The residue was dissolved in anhydrous THF (1
cm3), and n-BuLi (0.14 cm
3, 0.27 mmol) was added, followed by tert-butylnitrite (0.11 cm
3,
1.44 mmol) under nitrogen atmosphere. The reaction was stirred 24 h. The mixture was
partitioned between Et2O (10 cm3) and a saturated aqueous solution of K2CO3. The organic
layer was washed with saturated aqueous K2CO3 (3 20 cm3), dried over MgSO4, filtered
and concentrated under reduced pressure to give a residue which was purified by flash
column chromatography (SiO2, Petrol:EtOAc 2 %) to give the title compound 266d (27 mg,
53 % over 2 steps) as a colourless oil.
Rf 0.26 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3444 (br, O-H); 1H NMR (400MHz,
CDCl3) 7.49 (d, J = 7.3 Hz, 4H, aryl), 7.38-7.19 (m, 11H, aryl), 2.65 (s, 4H, H-6 and H-7),
2.20 (s, 1H, -OH), 13
C NMR (100MHz, CDCl3) 146.8 (C-4), 142.3 (C-8), 128.4, 128.3,
128.2, 126.9, 126.0, 125.8 (CH aryl), 78.2 (C-5), 44.0 (C-6), 30.2 (C-7); MS m/z (ES+)
311 (100 %, M+Na+); HMRS (ES+) calcd for C21H20O1Na1 (M+Na
+): 311.1404, found:
311.1405.

290
3-Methyl-1,1-diphenylbutan-1-ol 266e.
OH
1
2
34
5
6
78 8
General procedure P was followed with i-PrLi (2.0 equiv.). 1-Phenylvinyl
isopropylphenylcarbamate 248f (45 mg, 0.16 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 266e (28 mg, 73 %) as a
colourless oil.
Rf 0.53 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3472 (br, O-H); 1H NMR (500MHz,
CDCl3) 7.43 (d, J = 7.7 Hz, 4H, aryl), 7.32-7.20 (m, 6H, aryl), 2.25 (d, J = 6.6 Hz, 2H, H-
6), 2.04 (s, 1H, -OH), 1.71 (non, J = 6.6 Hz, 1H, H-7), 0.88 (d, J = 6.6 Hz, 6H, H-8); 13
C
NMR (125MHz, CDCl3) 147.6 (C-4), 128.0, 126.7, 126.0 (CH aryl), 78.8 (C-5), 50.3 (C-
6), 24.6 (C-8), 24.3 (C-7); EI GC/MS 183 (M+-C4H9).
1-(4-Chlorophenyl)-3-methyl-1-phenylbutan-1-ol 266f.
OH
1
23
45
6
78
910
11
12
8
Cl
General procedure P was followed with i-PrLi (2.0 equiv.). 1-(4-Chlorophenyl)vinyl
isopropylphenylcarbamate 248g and 1-phenylvinyl 4-chlorophenylisopropylcarbamate
248h (50 mg, 0.16 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc
1 %) the title compound 266f (28 mg, 65 % and 22 mg, 51 %) as a colourless oil.
Rf 0.55 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3464 (br, O-H); 1H NMR (400MHz,
CDCl3) 7.42-7.22 (m, 9H, aryl), 2.22 (d, J = 6.2 Hz, 2H, H-6), 2.01 (s, 1H, -OH), 1.69
(non, J = 6.2 Hz, 1H, H-7), 0.89 (d, J = 6.2 Hz, 3H, H-8), 0.87 (d, J = 6.2 Hz, 3H, H-8); 13
C

291
NMR (100MHz, CDCl3) 147.2 (C-9), 146.0 (C-4), 132.4 (C-1), 128.2, 128.1, 127.5,
127.0, 125.9 (CH aryl), 78.4 (C-5), 50.2 (C-6), 24.6, 24.5 (C-8), 24.2 (C-7); MS m/z (ES-)
273 (100 %, M-H+); HMRS (ES+) calcd for C17H19O1
35Cl1 (M
+): 274.1107, found:
274.1119.
1-(4-Methoxyphenyl)-3-methyl-1-phenylbutan-1-ol 266g.
OH
12
3
45
6
7
89
1011
12
13
9
O
General procedure P was followed with i-PrLi (2.0 equiv.). 1-(4-Methoxyphenyl)vinyl
isopropylphenylcarbamate 248i (50 mg, 0.16 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 266g (35 mg, 81 %) as a
colourless oil.
Rf 0.39 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3473 (br, O-H), 1610, 1509 (C=C), 1245
(C-O-C); 1H NMR (400MHz, CDCl3) 7.41 (d, J = 7.3 Hz, 2H, aryl), 7.34-7.19 (m, 5H,
aryl), 6.83 (d, J = 8.8 Hz, 2H, H-3), 3.78 (s, 3H, H-1), 2.21 (d, J = 6.2 Hz, 2H, H-7), 2.00 (s,
1H, -OH), 1.69 (non, J = 6.2 Hz, 1H, H-8), 0.89 (d, J = 6.2 Hz, 3H, H-9), 0.85 (d, J = 6.2 Hz,
3H, H-9); 13
C NMR (100MHz, CDCl3) 158.2 (C-2), 147.7 (C-10), 140.0 (C-5), 128.0,
127.2, 126.5, 126.0 (CH aryl), 113.3 (C-3), 78.5 (C-6), 55.2 (C-1), 50.4 (C-7), 24.6 (C-9),
24.3 (C-8); MS m/z (ES+) 253 (100 %, M-H2O+H+), (ES-) 269 (40 %, M-H
+); HMRS
(ES+) calcd for C18H21O1 (M-H2O+H+): 253.1587, found: 253.1592.

292
3-Methyl-1,1-diphenylpentan-1-ol 266h.
OH
1
2
34
5
6
7 89
10
General procedure P was followed with sec-BuLi (2.0 equiv.). 1-Phenylvinyl
isopropylphenylcarbamate 248f (50 mg, 0.18 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 266h (29 mg, 64 %) as a
colourless oil.
Rf 0.29 (Petrol:EtOAc 9:1); 1H NMR (400MHz, CDCl3) 7.44 (d, J = 8.0 Hz, 4H, aryl),
7.33-7.20 (m, 6H, aryl), 2.36 (dd, J = 14.3 and 4.1 Hz, 1H, H-6), 2.15 (dd, J = 14.3 and 6.8
Hz, 1H, H-6), 2.03 (s, 1H, -OH), 1.54-1.45 (m, 1H, H-7), 1.38-1.28 (m, 1H, H-9), 1.22-1.12
(m, 1H, H-9), 0.83-0.77 (m, 6H, H-8 and H-10); 13
C NMR (100MHz, CDCl3) 147.8,
147.4 (C-4), 128.0, 126.7, 126.6, 126.1, 126.0 (CH aryl), 78.7 (C-5), 48.3 (C-6), 31.2 (C-9),
30.2 (C-7), 21.2 (C-8), 11.2 (C-10); MS m/z (ES-) 253 (100 %, M-H+).
1-(4-Chlorophenyl)-3-methyl-1-phenylpentan-1-ol 266i.
OH
1
2
34
5
6
789
10
1112
13
14Cl
General procedure P was followed with sec-BuLi (2.0 equiv.). 1-(4-Chlorophenyl)vinyl
isopropylphenylcarbamate 248g and 1-phenylvinyl 4-chlorophenylisopropylcarbamate
248h (50 mg, 0.16 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc
1 %) the title compound 266i (30 mg, 67 % and 23 mg, 51 %) as a yellow oil.
Rf 0.55 (Petrol:EtOAc 8:2); 1H NMR (500MHz, CDCl3) (mixture of diastereoisomers)
7.42-7.22 (9H, aryl), 2.33 (dd, J = 14.2 and 4.0 Hz, 1H, H-6), 2.12 (dd, J = 14.2 and 5.6 Hz,

293
1H, H-6), 2.00 (s, 1H, -OH), 1.56-1.44 (m, 1H, H-7), 1.39-1.27 (m, 1H, H-9), 1.23-1.12 (m,
1H, H-9), 0.85-0.78 (m, 6H, H-8 and H-10); 13
C NMR (125MHz, CDCl3) (mixture of
diastereoisomers) 147.5, 147.1 (C-11), 146.3, 145.9 (C-4), 132.5, 132.4 (C-1), 128.2,
128.1, 127.6, 127.5, 127.0, 126.9, 126.0, 125.9 (CH aryl), 78.5, 78.4 (C-5), 48.2, 48.1 (C-
6), 31.2, 31.1 (C-9), 30.3, 30.2 (C-7), 21.1, 21.0 (C-8), 11.1 (C-10); MS m/z (ES-) 287
(100 %, M-H+); HMRS (ES+) calcd for C18H21O1
35Cl1 (M
+): 288.1275, found: 288.1276.
1-(4-Methoxyphenyl)-3-methyl-1-phenylpentan-1-ol 266j.
OH
12
34
56
7
8910
11
1213
14
15O
General procedure P was followed with sec-BuLi (2.0 equiv.). 1-(4-Methoxyphenyl)vinyl
isopropylphenylcarbamate 248i (50 mg, 0.16 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 266j (29 mg, 64 %) as a
colourless oil.
Rf 0.44 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3475 (br, O-H), 1682, 1510 (C=C), 1245
(C-O-C); 1H NMR (500MHz, CDCl3) (mixture of diastereoisomers) 7.41 (d, J = 7.3 Hz,
2H, aryl), 7.28-7.19 (m, 5H, aryl), 6.83 (d, J = 8.8 Hz, 2H, H-3), 3.79 (s, 1.5H, H-1), 3.78 (s,
1.5H, H-1), 2.32 (dd, J = 14.3 and 4.0 Hz, 1H, H-7), 2.11 (dd, J = 14.3 and 6.7 Hz, 1H, H-7),
1.98 (s, 1H, -OH), 1.53-1.44 (m, 1H, H-8), 1.38-1.21 (m, 1H, H-10), 1.21-1.10 (m, 1H, H-
10), 0.84-0.75 (m, 6H, H-9 and H-11); 13
C NMR (125MHz, CDCl3) (mixture of
diastereoisomers) 158.3, 158.2 (C-2), 147.9, 147.6 (C-12), 140.2, 139.8 (C-5), 128.0,
127.3, 127.2, 126.6, 126.5, 126.0, 125.9 (CH aryl), 113.3 (C-3), 78.5, 78.4 (C-6), 55.2 (C-
1), 48.4, 48.3 (C-7), 31.2, 31.1 (C-10), 30.3, 30.2 (C-8), 21.1, 21.0 (C-9), 11.2, 11.1 (C-11);
MS m/z (ES+) 267 (100 %, M-H2O+H+), (ES-) 283 (20 %, M-H
+); HMRS (ES+) calcd for
C19H23O1 (M-H2O+H+): 267.1744, found: 267.1747.

294
3,3-Dimetyl-1,1-diphenylbutan-1-ol 266k.
OH
1
2
34
5
6
7
8
General procedure P was followed with tert-BuLi (2.0 equiv.). 1-Phenylvinyl
isopropylphenylcarbamate 248f (50 mg, 0.18 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 266k (32 mg, 71 %) as a
white solid.
Rf 0.29 (Petrol:EtOAc 9:1); m.p. 70-72 °C (MeOH/Et2O); 1H NMR (400MHz, CDCl3)
7.49 (d, J = 7.2 Hz, 4H, aryl), 7.31-7.18 (m, 6H, aryl), 2.43 (s, 2H, H-6), 2.05 (s, 1H, -OH),
0.86 (s, 9H, H-8); 13
C NMR (100MHz, CDCl3) 148.5 (C-4), 127.9, 126.4, 125.8 (CH
aryl), 78.5 (C-5), 53.0 (C-6), 31.9 (C-7), 31.7 (C-8); MS m/z (ES-) 253 (100 %, M-H+).
1-(4-Chlorophenyl)-3,3-dimethyl-1-phenylbutan-1-ol 266l.
OH
1
23
4
6
5
7
8
910
11
12Cl
General procedure P was followed with tert-BuLi (2.0 equiv.). 1-(4-Chlorophenyl)vinyl
isopropylphenylcarbamate 248g and 1-phenylvinyl 4-chlorophenylisopropylcarbamate
248h (50 mg, 0.16 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc
1 %) the title compound 266l (12 mg, 27 % and 16 mg, 36 %) as a yellow oil.
Rf 0.63 (Petrol:EtOAc 8:2); 1H NMR (500MHz, CDCl3) 7.46-7.42 (m, 4H, aryl), 7.31-
7.19 (m, 5H, aryl), 2.40 (s, 2H, H-6), 2.03 (s, 1H, -OH), 0.86 (s, 9H, H-8); 13
C NMR
(125MHz, CDCl3) 148.3 (C-9), 146.9 (C-4), 132.2 (C-1), 128.1, 128.0, 127.3, 126.7,

295
125.6 (CH aryl), 78.2 (C-5), 53.0 (C-6), 31.9 (C-7), 31.7 (C-8); MS m/z (ES-) 287 (60 %,
M-H+).
1-(4-Methoxyphenyl)-3,3-dimethyl-1-phenylbutan-1-ol 266m.
OH
12
3
45
6
7
8
1011
12
13
9
O
General procedure P was followed with tert-BuLi (2.0 equiv.). 1-(4-Methoxyphenyl)vinyl
isopropylphenylcarbamate 248i (50 mg, 0.16 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 266m (30 mg, 67 %) as a
yellow oil.
Rf 0.33 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3494 (br, O-H), 1608, 1510 (C=C), 1244
(C-O-C); 1H NMR (400MHz, CDCl3) 7.45 (d, J = 7.3 Hz, 2H, H-11), 7.37 (d, J = 8.8 Hz,
2H, H-4), 7.28 (t, J = 7.3 Hz, 2H, H-12), 7.18 (t, J = 7.3 Hz, 1H, H-13), 6.81 (d, J = 8.8 Hz,
2H, H-3), 3.77 (s, 3H, H-1), 2.38 (s, 2H, H-7), 2.00 (s, 1H, -OH), 0.85 (s, 9H, H-9); 13
C
NMR (100MHz, CDCl3) 158.1 (C-2), 148.6 (C-10), 141.0 (C-5), 127.9, 127.0, 126.3,
125.8 (CH aryl), 113.2 (C-3), 78.3 (C-9), 55.1 (C-1), 53.1 (C-7), 31.9 (C-9), 31.7 (C-8);
MS m/z (ES+) 267 (100 %, M-H2O+H+), (ES-) 283 (50 %, M-H
+); HMRS (ES+) calcd for
C19H23O1 (M-H2O+H+): 267.1744, found: 267.1745.

296
(1S*,2S*)-1,2-Diphenylhexyl isopropylphenylcarbamate 271a.
N O
O
1
2
34
5
67
8 9
10
1112
13
14
15
1617
18
1920
21
17
General procedure S was followed. (Z)-1,2-Diphenylvinyl isopropylphenylcarbamate 248j
(50 mg, 0.14 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 1 %) the
title compound 271a (40 mg, 70 %) as a white solid.
Rf 0.50 (Petrol:EtOAc 8:2); m.p. 80-82°C (Petrol/Et2O); IR max (neat/cm-1
) 1688 (C=O);
1H NMR (400MHz, CDCl3) 7.41-7.38 (m, 3H, aryl), 7.18-6.91 (m, 10H, aryl), 6.60 (m,
2H, aryl), 5.81 (d, J = 6.4 Hz, 1H, H-5), 4.48 (sep, J = 6.8 Hz, 1H, H-16), 2.70 (br, 1H, H-6),
1.53-1.41 (m, 2H, H-7), 1.24-1.07 (m, 4H, H-8, H-9), 1.02 (d, J = 6.8 Hz, 3H, H-17), 0.96
(d, J = 6.8 Hz, 3H, H-17), 0.74 (t, J = 7.2 Hz, 3H, H-10); 13
C NMR (100MHz, CDCl3)
154.4 (C-15), 140.7 (C-4), 140.3 (C-11), 138.0 (C-18), 130.3, 128.9, 128.6, 127.8, 127.5,
127.3, 127.2, 126.5, 125.9 (aryl), 79.8 (C-5), 52.0 (C-6), 48.3 (C-16), 31.7, 29.4, 22.5 (C-7,
C-8 and C-9), 21.2 (C-17), 13.8 (C-10); MS m/z (ES+) 438 (100 %, M+Na+), (ES-) 414
(100 %, M-H+); HMRS (ES+) calcd for C28H33O2N1Na1 (M+Na
+): 438.2404, found:
438.2387.
The syn-stereochemistry was proved by an X-ray crystal structure.

297
(1S*,2S*)-1,2-Diphenylhexyl 4-chlorophenylisopropylcarbamate 271b.
N O
O
1
2
34
5
67
8 9
10
1112
13
14
15
1617
18
1920
21
17
Cl
General procedure S was followed. (Z)-1,2-diphenylvinyl 4-
chlorophenylisopropylcarbamate 248k (50 mg, 0.13 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 271b (44 mg, 80 %) as a
white solid.
Rf 0.64 (Petrol:EtOAc 8:2); m.p. 93-95 °C (Petrol/Et2O); IR max (neat/cm-1
) 1687 (C=O);
1H NMR (400MHz, CDCl3) 7.35 (d, J = 8.5 Hz, 2H, aryl), 7.23-7.09 (m, 6H, aryl), 6.98
(m, 2H, aryl), 6.80 (d, J = 8.5 Hz, 2H, aryl), 6.71 (m, 2H, aryl), 5.82 (d, J = 7.2 Hz, 1H, H-5),
4.46 (sep, J = 6.8 Hz, 1H, H-16), 2.73 (br, 1H, H-6), 1.55-1.38 (m, 2H, H-7), 1.21-0.96 (m,
4H, H-8 and H-9), 0.99 (d, J = 6.8 Hz, 3H, H-17), 0.93 (d, J = 6.8 Hz, 3H, H-17), 0.74 (t, J
= 7.2 Hz, 3H, H-10); 13
C NMR (100MHz, CDCl3) 154.2 (C-15), 140.8 (C-4), 140.2 (C-
11), 136.5 (C-18), 133.2 (C-21), 131.5, 128.8, 128.7, 127.9, 127.7, 127.4, 126.6, 126.1
(aryl), 80.1 (C-5), 52.0 (C-6), 48.3 (C-16), 31.8, 29.3, 22.4 (C-7, C-8 and C-9), 21.1 (C-17),
13.8 (C-10); MS m/z (ES+) 472 (100 %, M+Na+); HMRS (ES+) calcd for
C28H33O2N135
Cl1 (M+H+): 450.2194, found: 450.2205.

298
1,1,2-Triphenylhexan-1-ol 272a.
1
2
34
5
6
7
8
9
10
11
1213
14
1516
18
19
17
OH
General procedure T was followed. (Z)-1,2-Diphenylvinyl isopropylphenylcarbamate 248j
(50 mg, 0.14 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 1 %) the
title compound 272a (28 mg, 60 %) as a white solid.
Rf 0.61 (Petrol:EtOAc 8:2); m.p. 99-101°C (MeOH/Et2O); 1H NMR (400MHz, CDCl3)
8.61-8.00 (m, 15H, aryl), 4.71 (dd, J = 11.6 and 2.8 Hz, 1H, H-6), 3.45 (s, 1H, -OH),
2.91-2.72 (m, 2H, H-7), 2.35-2.03 (m, 4H, H-8, H-9), 1.78 (t, J = 7.2 Hz, 3H, H-10); 13
C
NMR (400MHz, CDCl3) 146.4, 146.0 (C-4 and C-15), 140.0 (C-11), 130.0, 128.2, 127.7,
127.5, 126.7, 126.3, 126.2, 126.1, 125.6 (aryl), 80.9 (C-5), 54.1 (C-6), 30.2, 29.8, 22.6 (C-
7, C-8 and C-9), 13.9 (C-10); MS m/z (ES+) 353 (100 %, M+Na+); HMRS (ES+) calcd for
C24H26O1Na1 (M+Na+): found: 353.1876.
1-(4-Chlorophenyl)-1,2-diphenylhexan-1-ol 272b.
1
2
34
5
6
7
8
9
10
11
1213
14
1516
18
19
17
OH
Cl
General procedure T was followed. (Z)-1,2-diphenylvinyl 4-
chlorophenylisopropylcarbamate 248k (50 mg, 0.13 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 272b (25 mg, 54 %) as a
colourless oil.

299
Rf 0.34 (Petrol:EtOAc 8:2); 1H NMR (400MHz, CDCl3) (mixture of diastereoisomers)
7.50-6.93 (m, 14H, aryl), 3.59 (t, J = 12.0 Hz, 0.5H, H-6), 3.58 (t, J = 12.0 Hz, 0.5H, H-
6), 2.37 (s, 0.5H, -OH), 2.35 (s, 0.5H, -OH), 1.80-1.56 (m, 2H, H-7), 1.24-0.94 (m, 4H, H-8,
H-9), 0.68 (t, J = 7.0 Hz, 3H, H-10); 13
C NMR (100MHz, CDCl3) (mixture of
diastereoisomers) 145.9, 145.8, 145.0, 144.5 (C-4 and C-15), 139.8, 139.6 (C-11), 132.5,
131.8 (C-18), 130.0, 129.9, 128.4, 128.2, 127.8, 127.7, 127.6, 127.1, 126.9, 126.5, 126.4,
126.3, 126.1, 125.6 (aryl), 80.7, 80.6 (C-5), 54.0, 54.0 (C-6), 30.1, 29.8, 29.7, 22.6, 22.5
(C-7, C-8 and C-9), 13.9 (C-10); MS m/z (ES+) 363 (100 %, M+H+); HMRS (ES+) calcd
for C28H33N1O2Na135
Cl1 (M+Na+): 438.2404, found: 438.2387.
Vinyl isopropylphenylcarbamate 277a.
N O
O
1
2
3
45
67
8
9
5
General procedure L was followed. Isopropyl(phenyl) carbamic chloride 245b (1.00 g,
5.08 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 1 %) the title
compound 277a (0.80 g, 75 %) as a white solid.
Rf 0.41 (Petrol:EtOAc 8:2); m.p. 33-35°C (Petrol); IR max (neat/cm-1
) 1719 (C=O), 1645
(C=C); 1H-NMR (400MHz, CDCl3) δ 7.42-7.26 (m, 4H, aryl and H-2), 7.12 (d, J = 7.0 Hz,
2H, aryl), 5.70 (br, 1H, H-1), 5.39 (br, 1H, H-1), 4.58 (sep, J = 6.8 Hz, 1H, H-4), 1.15 (d, J =
6.8 Hz, 6H, H-5); 13
C-NMR (100MHz, CDCl3)3 δ 142.3 (C-2), 129.7, 128.7, 127.7 (CH
aryl), 95.4 (C-1), 49.4 (C-4), 21.1 (C-5); MS m/z (ES+) 228 (100 %, M+Na+); HMRS
(ES+) calcd for C12H15O2N1Na1 (M+Na+): 228.0995, found: 228.0992.
3 Quaternary carbons were not clearly detectable in the
13C NMR spectrum.

300
Vinyl 4-chlorophenylisopropylcarbamate 277b.
N O
OCl
1
2
3
45
6
5
7
8
9
General procedure L was followed. (4-Chlorophenyl)(isopropyl) carbamic chloride 245c
(1.20 g, 5.19 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 1 %) the
title compound 277b (0.85 g, 67 %) as a pale yellow oil.
Rf 0.35 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1715 (C=O), 1645 (C=C); 1H-NMR
(400MHz, CDCl3) δ 7.36 (d, J = 8.5 Hz, 2H, H-7), 7.17 (dd, J = 14.0 and 6.0 Hz, 1H, H-2),
7.05 (d, J = 8.5 Hz, 2H, H-8), 4.59 (sep, J = 6.8 Hz, 1H, H-4), 4.51 (br, 1H, H-1), 4.38 (br,
1H, H-1), 1.13 (d, J = 6.8 Hz, 6H, H-5); 13
C-NMR (100MHz, CDCl3)
§ δ 142.2 (C-2), 133.6,
131.0, 129.0 (CH aryl), 95.7 (C-1), 49.4 (C-4), 21.2 (C-5); MS m/z (ES+) 262 (100 %,
M+Na+); HMRS (ES+) calcd for C12H14O2N1Na1
35Cl1 (M+Na
+): 262.0606, found:
262.0598.
Vinyl isopropyl-p-tolylcarbamate 277c.
N O
O
1
2
3
45
67
5
8
9
10
General procedure L was followed. Isopropyl(p-tolyl) carbamic chloride 245d (250 mg,
1.18 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 2 %) the title
compound 277c (148 mg, 57 %) as a colourless oil.
Rf 0.48 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1713 (C=O), 1646 (C=C); 1H-NMR
(400MHz, CDCl3) δ 7.18-7.16 (m, 3H, H-7 and H-2), 6.99 (d, J = 7.8 Hz, 2H, H-8), 4.58
(sep, J = 6.8 Hz, 1H, H-4), 4.48 (br, 1H, H-1), 4.34 (br, 1H, H-1), 2.38 (s, 3H, H-10), 1.12 (d,
J = 6.8 Hz, 6H, H-5) ; 13
C-NMR (100MHz, CDCl3) § δ 142.4 (C-2), 137.5, 129.4 (CH aryl),

301
95.4 (C-1), 49.2 (C-4), 24.3 (C-10), 21.1 (C-5); MS m/z (ES+) 242 (100 %, M+Na+);
HMRS (ES+) calcd for C13H17O2N1Na1 (M+Na+): 242.1152, found: 242.1156.
Vinyl 3-(trifluoromethyl)phenylisopropylcarbamate 277d.
NF3C O
O
1
2
3
45
67
89
1011
12
5
General procedure L was followed. (3-Trifluoromethyl)phenyl)(isopropyl) carbamic
chloride 245e (1.00 g, 3.77 mmol) gave after flash column chromatography (SiO2,
Petrol:EtOAc 2 %) the title compound 277d (0.68 g, 63 %) as a colourless oil.
Rf 0.68 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1717 (C=O), 1648 (C=C), 1317 (C-F); 1H-
NMR (400MHz, CDCl3) δ 7.62 (d, J = 7.9 Hz, 1H, H-10), 7.52 (t, J = 7.9 Hz, 1H, H-11),
7.39 (s, 1H, H-7), 7.32 (d, J = 7.9 Hz, 1H, H-12), 7.18 (dd, J = 14.0 and 6.2 Hz, 1H, H-2),
4.61 (sep, 3J = 6.8 Hz, 1H, H-4), 5.56 (br, 1H, H-1), 4.40 (br, 1H, H-1), 1.15 (d,
3J = 6.8 Hz,
6H, H-5) ; 13
C-NMR (100MHz, CDCl3) δ 152.3 (C-3), 142.1 (C-2), 138.3 (C-6), 133.2 (C-
12), 131.4 (q, 2JC-F = 32 Hz, C-8), 129.4 (C-11), 126.5 (q,
3JC-F = 3.6 Hz, C-7), 125.5 (q,
1JC-F = 270 Hz, C-9), 124.6 (q,
3JC-F = 3.6 Hz, C-10), 95.9 (C-1), 49.7 (C-4), 21.2 (C-5);
MS m/z (ES+) 296 (100 %, M+Na+); HMRS (ES+) calcd for C13H15O2N1F3 (M+H
+):
274.1050, found: 274.1051.
Vinyl isopropyl-4-methoxyphenylcarbamate 277e.
N O
OO
1
2
3
455
678
910
General procedure L was followed. Isopropyl(4-methoxyphenyl) carbamic chloride 245f
(1.00 g, 4.40 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 2 %) the
title compound 277e (0.71 g, 69 %) as a colourless oil.

302
Rf 0.52 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1712 (C=O), 1645, 1509 (C=C), 1245 (C-
O-C); 1H-NMR (400MHz, CDCl3) δ 7.16 (br dd, J = 13.0 and 4.9 Hz, 1H, H-2), 7.00 (d, J
= 8.8 Hz, 2H, H-7), 6.87 (d, J = 8.8 Hz, 2H, H-8), 4.58 (sep, J = 6.8 Hz, 1H, H-4), 4.46 (d, J
= 13.0 Hz, 1H, H-1), 4.30 (br, 1H, H-1), 3.80 (s, 3H, H-10), 1.09 (d, J = 6.8 Hz, 6H, H-5) ;
13C-NMR (100MHz, CDCl3) δ 158.7 (C-9), 152.7 (C-3), 142.3 (C-2), 130.6 (C-7), 129.7
(C-6), 113.7 (C-8), 95.2 (C-1), 55.2 (C-10), 49.0 (C-4), 20.9 (C-5); MS m/z (ES+) 258
(100 %, M+Na+); HMRS (ES+) calcd for C13H18O3N1 (M+H
+): 236.1282, found: 236.1288.
1-Iodovinyl isopropylphenylcarbamate 279a.
N O
O I
123
45
67
8
9
5
General procedure M was followed. Vinyl isopropylphenylcarbamate 277a (500 mg, 2.44
mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title
compound 279a (715 mg, 88 %) as a white solid.
Rf 0.40 (Petrol:EtOAc 8:2); m.p. 62-64°C (Petrol); IR max (neat/cm-1
) 1719 (C=O), 1624
(C=C), 1H-NMR (400MHz, CDCl3) δ 7.42-7.26 (m, 3H, aryl), 7.12 (d, J = 7.0 Hz, 2H, aryl),
5.70 (br, 1H, H-1), 5.39 (br, 1H, H-1), 4.58 (sep, J = 6.8 Hz, 1H, H-4), 1.15 (d, J = 6.8 Hz,
6H, H-5) ; 13
C-NMR (100MHz, CDCl3) δ 150.3 (C-3), 135.0 (C-6), 129.7, 128.9, 128.0
(CH aryl), 118.0 (C-1), 102.0 (C-2), 50.1 (C-4), 21.0 (C-5); MS m/z (ES+) 354 (100 %,
M+Na+); HMRS (ES+) calcd for C12H14O2N1Na1I1 (M+Na
+): 353.9961, found: 353.9958.

303
1-Iodovinyl 4-chlorophenylisopropylcarbamate 279b.
N O
O ICl
123
45
6
5
7
8
9
General procedure M was followed. Vinyl 4-chlorophenylisopropylcarbamate 277b (606
mg, 2.53 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 2 %) the
title compound 279b (470 mg, 51 %) as a white solid.
Rf 0.40 (Petrol:EtOAc 8:2); m.p. 108-110°C (Petrol); IR max (neat/cm-1
) 1715 (C=O),
1618 (C=C); 1
H-NMR (400MHz, CDCl3) δ 7.37 (d, J = 8.5 Hz, 2H, H-7), 7.06 (d, J = 8.5
Hz, 2H, H-8), 5.71 (br, 1H, H-1), 5.41(br, 1H, H-1), 4.56 (sep, J = 6.8 Hz, 1H, H-4), 1.14 (d,
J = 6.8 Hz, 6H, H-5) ; 13
C-NMR (100MHz, CDCl3) δ 150.1 (C-3), 135.5, 133.9, 131.0,
129.2 (aryl), 118.2 (C-1), 101.6 (C-2), 50.0 (C-4), 21.1 (C-5); MS m/z (ES+) 388 (100 %,
M+Na+); HMRS (ES+) calcd for C12H13O2N1
35Cl1Na1I1 (M+Na
+): 387.9577, found:
387.9572.
1-Iodovinyl isopropyl-p-tolylcarbamate 279c.
N O
O I
123
45
67
8
9
10
5
General procedure M was followed. Vinyl isopropyl-p-tolylcarbamate 277c (332 mg, 1.52
mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 2 %) the title
compound 279c (191 mg, 37 %) as a yellow solid.
Rf 0.40 (Petrol:EtOAc 8:2); m.p. 92-94°C (Petrol); IR max (neat/cm-1
) 1729 (C=O), 1623
(C=C); 1
H-NMR (400MHz, CDCl3) δ 7.19 (d, J = 7.8 Hz, 2H, H-7), 6.99 (d, J = 7.8 Hz,
2H, H-8), 5.69 (br, 1H, H-1), 5.38 (br, 1H, H-1), 4.57 (sep, J = 6.8 Hz, 1H, H-4), 2.38 (s, 3H,

304
H-10), 1.14 (d, J = 6.8 Hz, 6H, H-5) ; MS m/z (ES+) 368 (100 %, M+Na+); HMRS (ES+)
calcd for C13H16O2N1Na1I1 (M+Na+): 368.0119, found: 368.0117.
1-Iodovinyl 3-(trifluoromethyl)phenylisopropylcarbamate 279d.
N O
O I
F3C 123
455
67
89
10
11
12
General procedure M was followed. Vinyl 3-(trifluoromethyl)phenylisopropylcarbamate
277d (668 mg, 2.45 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc
3 %) the title compound 279d (390 mg, 40 %) as a white solid.
Rf 0.50 (Petrol:EtOAc 8:2); m.p. 58-60 °C (Petrol); IR max (neat/cm-1
) 1722 (C=O), 1620
(C=C), 1309 (C-F); 1
H-NMR (400MHz, CDCl3) δ 7.63 (d, J = 7.9 Hz, 1H, H-10), 7.54 (t,
J = 7.9 Hz, 1H, H-11), 7.39 (s, 1H, H-7), 7.33 (d, J = 7.9 Hz, 1H, H-12), 5.74 (br, 1H, H-1),
5.43 (br, 1H, H-1), 4.58 (sep, J = 6.8 Hz, 1H, H-4), 1.17 (d, J = 6.8 Hz, 6H, H-5) ; 13
C-
NMR (100MHz, CDCl3) δ 150.8 (C-3), 137.9 (C-6), 132.1 (C-12), 131.2 (q, 2JC-F = 32 Hz,
C-8), 129.6 (C-11), 126.5 (q, 3JC-F = 3.6 Hz, C-7), 125.2 (q,
1JC-F = 270 Hz, C-9), 124.9 (q,
3JC-F = 3.6 Hz, C-10), 118.4 (C-1), 101.2 (C-2), 50.3 (C-4), 21.1 (C-5); MS m/z (ES+) 422
(100 %, M+Na+); HMRS (ES+) calcd for C13H13N1O2F3Na1I1 (M+Na
+): 421.9835, found:
421.9824.
1-Iodovinyl isopropyl-4-methoxyphenylcarbamate 279e.
N O
OO
I
123
45
67
8
910
5
General procedure M was followed. Vinyl isopropyl-4-methoxyphenylcarbamate 277e
(1.00 g, 4.25 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 2 %) the
title compound 279e (0.71 g, 69 %) as a colourless oil.

305
Rf 0.52 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1719 (C=O), 1618, 1512 (C=C), 1245 (C-
O-C); 1
H-NMR (400MHz, CDCl3) δ 7.02 (d, J = 8.8 Hz, 2H, H-7), 6.89 (d, J = 8.8 Hz, 2H,
H-8), 5.69 (br, 1H, H-1), 5.38 (br, 1H, H-1), 4.58 (sep, J = 6.8 Hz, 1H, H-4), 3.83 (s, 3H, H-
10), 1.12 (d, J = 6.8 Hz, 6H, H-5) ; 13
C-NMR (100MHz, CDCl3) δ 159.0 (C-9), 151.5 (C-
3), 130.7 (C-6), 129.4 (C-7), 118.0 (C-8), 114.0 (C-1), 102.3 (C-2), 55.4 (C-10), 49.8 (C-4),
21.0 (C-5); MS m/z (ES+) 384 (100 %, M+Na+); HMRS (ES+) calcd for C13H16N1O3Na1I1
(M+Na+): 384.0068, found: 384.0057.
5,5-Dimethylhex-1-en-3-yn-2-yl isopropylphenylcarbamate 280a.
N O
O
1
2
3
4
5
67
89 9
1011
12
13
General procedure N was followed. 1-Iodovinyl isopropylphenylcarbamate 279a (710 mg,
2.15 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 2 % then 4 %)
the title compound 280a (533 mg, 87 %) as a white solid.
Rf 0.56 (Petrol:EtOAc 8:2); m.p. 68-70 °C (Et2O/Petrol); IR max (neat/cm-1
) 2211 (w,
C≡C), 1712 (C=O); 1H-NMR (400MHz, CDCl3) δ 7.39-7.30 (m, 3H, aryl), 7.13 (d, J = 7.0
Hz, 2H, H-11), 5.04 (br, 1H, H-6), 4.99 (br, 1H, H-6), 4.59 (sep, J = 6.8 Hz, 1H, H-8), 1.24
(s, 9H, H-1), 1.14 (d, J = 6.8 Hz, 6H, H-9); 13
C-NMR (100MHz, CDCl3) δ 152.8 (C-7),
138.0 (C-10), 137.1 (C-5), 129.7, 128.7, 127.5 (CH aryl), 109.2 (C-6), 98.4 (C-3), 73.8 (C-
4), 49.6 (C-8), 30.5 (C-1), 27.7 (C-2), 21.2 (C-9); MS m/z (ES+) 308 (100 %, M+Na+);
HMRS (ES+) calcd for C18H23O2N1Na1 (M+Na+): 308.1621, found: 308.1623.

306
5,5-Dimethylhex-1-en-3-yn-2-yl 4-chlorophenylisopropylcarbamate 280b.
N O
O
1
2
3
4
5
67
89 9
1011
12
13
Cl
General procedure N was followed. 1-Iodovinyl 4-chlorophenylisopropylcarbamate 279b
(470 mg, 1.29 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 2 %
then 3 %) the title compound 280b (302 mg, 74 %) as a white solid.
Rf 0.40 (Petrol:EtOAc 8:2); m.p. 75-77 °C (Et2O/Petrol); IR max (neat/cm-1
) 2209 (w,
C≡C), 1702 (C=O); 1H-NMR (400MHz, CDCl3) δ 7.34 (d, J = 8.5 Hz, 2H, H-12), 7.07 (d,
J = 8.5 Hz, 2H, H-11), 5.06 (br, 1H, H-6), 4.99 (br, 1H, H-6), 4.58 (sep, J = 6.8 Hz, 1H, H-
8), 1.24 (s, 9H, H-1), 1.38 (d, J = 6.8 Hz, 6H, H-9); 13
C-NMR (100MHz, CDCl3) δ 152.5
(C-7), 137.0 (C-10), 136.5 (C-5), 133.4 (C-13), 131.0, 129.0 (CH aryl), 109.3 (C-6), 98.6
(C-3), 73.6 (C-4), 49.5 (C-8), 30.5 (C-1), 27.7 (C-2), 21.2 (C-9); MS m/z (ES+) 342
(100 %, M+Na+); HMRS (ES+) calcd for C18H22O2N1Na1
35Cl1 (M+Na
+): 342.1232, found:
342.1233.
5,5-Dimethyl-1-en-3-yn-2-yl isopropyl-p-tolylcarbamate 280c.
N O
O
1
2
3
4
5
67
89 9
1011
12
13
14
General procedure N was followed. 1-Iodovinyl isopropyl-p-tolylcarbamate 279c (184 mg,
0.53 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title
compound 280c (112 mg, 70 %) as a white solid.

307
Rf 0.33 (Petrol:EtOAc 8:2); m.p. 45-47 °C (Et2O/Petrol); IR max (neat/cm-1
) 2215 (w,
C≡C), 1720 (C=O); 1H-NMR (400MHz, CDCl3) δ 7.17 (d, J = 7.8 Hz, 2H, H-11), 7.00 (d,
J = 7.8 Hz, 2H, H-12), 5.04 (br, 1H, H-6), 4.98 (br, 1H, H-6), 4.58 (sep, J = 6.8 Hz, 1H, H-
8), 2.37 (s, 3H, H-14), 1.24 (s, 9H, H-1), 1.12 (d, J = 6.8 Hz, 6H, H-9); 13
C-NMR (100MHz,
CDCl3) δ 153.2 (C-7), 137.3, 137.2 (C-5 and C-10), 135.0 (C-13), 129.4, 129.3 (CH aryl),
109.2 (C-6), 98.3 (C-3), 73.9 (C-4), 49.4 (C-8), 30.5 (C-1), 27.7 (C-2), 21.2 (C-14),
21.1 (C-9); MS m/z (ES+) 322 (100 %, M+Na+); HMRS (ES+) calcd for C19H25O2N1Na1
(M+Na+): 322.1778, found: 322.1772.
5,5-Dimethyl-1-en-3-yn-2-yl 3-(trifluoromethyl)phenylisopropylcarbamate 280d.
N O
O
1
2
3
4
5
67
89 9
10
11
1213
14
1516
F3C
General procedure N was followed. 1-Iodovinyl 3-
(trifluoromethyl)phenylisopropylcarbamate 279d (369 mg, 0.92 mmol) gave after flash
column chromatography (SiO2, Petrol:EtOAc 2 % then 4 %) the title compound 280d (245
mg, 75 %) as a yellow oil.
Rf 0.61 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 2216 (w, C≡C), 1725 (C=O), 1322 (C-F);
1H-NMR (400MHz, CDCl3) δ 7.60 (d, J = 7.9 Hz, 1H, H-13), 7.52 (t, J = 7.9 Hz, 1H, H-
12), 7.40 (s, H, H-16), 7.33 (d, J = 7.9 Hz, 1H, H-11), 5.07 (br, 1H, H-6), 5.01 (br, 1H, H-6),
4.60 (sep, J = 6.8 Hz, 1H, H-8), 1.24 (s, 9H, H-1), 1.16 (d, J = 6.8 Hz, 6H, H-9); 13
C-NMR
(100MHz, CDCl3) δ 152.4 (C-7), 138.8 (C-10), 136.9 (C-5), 133.1 (C-11), 131.4 (q, 2JC-F =
32 Hz, C-14), 129.4 (C-12), 126.5 (q, 3JC-F = 3.6 Hz, C-16), 127.0 (q,
1JC-F = 270 Hz, C-15),
124.4 (q, 3JC-F = 3.6 Hz, C-13), 109.4 (C-6), 98.8 (C-3), 73.5 (C-4), 49.9 (C-8), 30.4 (C-1),
27.7 (C-2), 21.2 (C-9); MS m/z (ES+) 376 (100 %, M+Na+); HMRS (ES+) calcd for
C19H22O2N1F3Na1 (M+Na+): 376.1495, found: 376.1487.

308
5,5-Dimethylhex-1-en-3-yn-2-yl isopropyl-4-methoxyphenylcarbamate 280e.
N O
O
1
2
3
4
5
67
89 9
1011
12
13
O14
General procedure N was followed. 1-Iodovinyl isopropyl-4-methoxyphenylcarbamate
279e (506 mg, 1.40 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc
5 %) the title compound 280e (325 mg, 74 %) as an orange oil.
Rf 0.64 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 2215 (w, C≡C), 1719 (C=O), 1632, 1510
(C=C), 1245 (C-O-C); 1H-NMR (400MHz, CDCl3) δ 7.01 (d, J = 8.8 Hz, 2H, H-11), 6.88
(d, J = 8.8 Hz, 2H, H-12), 5.03 (br, 1H, H-6), 4.96 (br, 1H, H-6), 4.58 (sep, J = 6.8 Hz, 1H,
H-8), 3.82 (s, 3H, H-14), 1.24 (s, 9H, H-1), 1.11 (d, J = 6.8 Hz, 6H, H-9); 13
C-NMR
(100MHz, CDCl3) δ 158.7 (C-13), 152.9 (C-7), 137.2 (C-5 and C-10), 130.7 (C-11), 113.9
(C-12), 109.2 (C-6), 98.3 (C-3), 73.9 (C-4), 55.3 (C-14), 49.2 (C-8), 30.5 (C-1), 27.7 (C-2),
21.1 (C-9); MS m/z (ES+) 338 (100 %, M+Na+); HMRS (ES+) calcd for C19H25O3N1Na1
(M+Na+): 338.1727, found: 338.1729.
4-(Triisopropylsilyl)but-1-en-3-yn-2-yl isopropylphenylcarbamate 280f.
112
N O
O
3
4
5
67
89 9
1011
12
13
Si
General procedure N was followed. 1-Iodovinyl isopropylphenylcarbamate 279a (416 mg,
1.26 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 2 %) the title
compound 280f (363 mg, 75 %) as a colourless oil.

309
Rf 0.63 (Petrol:EtOAc 8:2) ; IR max (neat/cm-1
) 1725 (C=O); 1H-NMR (400MHz, CDCl3)
δ 7.37-7.30 (m, 3H, aryl), 7.12 (d, J = 7.6 Hz, 2H, H-10), 5.17 (br, 1H, H-6), 5.10 (br, 1H,
H-6), 4.58 (sep, J = 6.8 Hz, 1H, H-8), 1.13 (d, J = 6.8 Hz, 6H, H-9), 1.09 (s, 21H, H-1 and
H-2); 13
C-NMR (100MHz, CDCl3) δ 152.5 (C-7), 138.0 (C-10), 136.6 (C-5), 129.7, 128.7,
127.6 (CH aryl), 110.6 (C-6), 100.6 (C-4), 91.6 (C-3), 49.7 (C-8), 21.1 (C-9), 18.5 (C-1),
11.1 (C-2); MS m/z (ES+) 408 (100%, M+Na+); HMRS (ES+) calcd for C23H36O2N1Si1
(M+H+): 386.2510, found: 386.2506.
4-(Trimethylsilyl)but-1-en-3-yn-2-yl isopropylphenylcarbamate 280g.
N O
O
Si1
2
34
56
78 8
910
11
12
General procedure N was followed. 1-Iodovinyl isopropylphenylcarbamate 279a (596 mg,
1.80 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 2%) the title
compound 280g (402 mg, 74 %) as a white solid.
Rf 0.39 (Petrol:EtOAc 8:2); m.p. 73-75 °C (Petrol/Et2O); IR max (neat/cm-1
) 2166 (w,
C≡C), 1714 (C=O); 1H-NMR (400MHz, CDCl3) δ 7.40-7.31 (m, 3H, aryl), 7.14 (d, J = 7.0
Hz, 2H, H-10), 5.20 (br, 1H, H-5), 5.10 (br, s, 1H, H-5), 4.59 (sep, J = 6.8 Hz, 1H, H-7),
1.14 (d, J = 6.8 Hz, 6H, H-8), 0.20 (s, 9H, H-1); 13
C-NMR (100MHz, CDCl3) δ 152.6 (C-6),
137.8 (C-9), 136.6 (C-4), 129.7, 128.7, 127.6 (CH aryl), 111.5 (C-5), 98.5 (C-3), 95.0 (C-
2), 49.7 (C-7), 21.2 (C-8), -0.40 (C-1); MS m/z (ES+) 324 (80 %, M+Na+), 302 (20 %,
M+H+); HMRS (ES+) calcd for C17H24O2N1 (M+H
+): 302.1571, found: 302.1573.

310
4-Phenylbut-1-en-3-yn-2-yl isopropylphenylcarbamate 280h.
N O
O
1
2
34
5
6
7
89
101111
1213
14
15
General procedure N was followed. 1-Iodovinyl isopropylphenylcarbamate 279a (394 mg,
1.19 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 1 %) the title
compound 280h (293 mg, 81 %) as a white solid.
Rf 0.36 (Petrol:EtOAc 8:2); m.p. 75-77 °C (Petrol/Et2O); IR max (neat/cm-1
) 2207 (w,
C≡C), 1713 (C=O); 1H-NMR (400MHz, CDCl3) δ 7.48-7.30 (m, 8H, aryl), 7.18 (d, J = 7.0
Hz, 2H, aryl), 5.27 (br, 1H, H-8), 5.17 (br, 1H, H-8), 4.63 (sep, J = 6.8 Hz, 1H, H-10), 1.16
(d, J = 6.8 Hz, 6H, H-11); 13
C-NMR (100MHz, CDCl3) δ 152.7 (C-9), 137.8 (C-12), 136.8
(C-7), 131.8, 129.7, 128.9, 128.8, 128.2, 127.7 (CH aryl), 122.0 (C-4), 111.2 (C-8), 89.0
(C-5), 83.6 (C-6), 49.6 (C-10), 21.2 (C-11); MS m/z (ES+) 328 (100 %, M+Na+); HMRS
(ES+) calcd for C20H19O2N1Na1 (M+Na+): 328.1308, found: 328.1307.
2,2-Dimethyl-5-phenyldec-3-yn-5-ol 281a.
23
45
6
78
910
1112
13
14
HO
1
General procedure P was followed with n-BuLi (2.0 equiv.). 5,5-Dimethylhex-1-en-3-yn-
2-yl isopropylphenylcarbamate 280a (50 mg, 0.17 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281a (32 mg, 71 %) as a
colourless oil.

311
Rf 0.55 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3424 (br, O-H), 2220 (w, CC); 1H-NMR
(400MHz, CDCl3) δ 7.62 (d, J = 7.1 Hz, 2H, aryl), 7.37-7.25 (m, 3H, aryl), 2.29 (s, 1H, -
OH), 1.94-1.86 (m, 1H, H-6), 1.83-1.76 (m, 1H, H-6), 1.52-1.40 (m, 1H, H-7), 1.34-1.22 (m,
5H, H-7, H-8 and H-9), 1.29 (s, 9H, H-1), 0.86 (t, J = 6.8 Hz, 3H, H-10); 13
C-NMR
(100MHz, CDCl3) δ 145.5 (C-11), 127.9, 127.3, 125.5 (CH aryl), 95.1 (C-3), 81.0 (C-4),
73.3 (C-5), 45.6 (C-6), 31.6 (C-7), 31.0 (C-1), 27.5 (C-2), 24.4 (C-8), 22.4 (C-9), 13.9 (C-
10); MS m/z (ES+) 281 (100 %, M+Na+); HMRS (ES+) calcd for C18H26O1Na1 (M+Na
+):
281.1876, found: 281.1866.
2,7,7-Trimethyl-4-phenyloct-5-yn-4-ol 281b.
23
45
6
788
910
11
12
HO
1
General procedure P was followed with i-PrLi (2.0 equiv.). 5,5-Dimethylhex-1-en-3-yn-2-
yl isopropylphenylcarbamate 280a (50 mg, 0.17 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281b (31 mg, 74 %) as a
colourless oil.
Rf 0.55 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3436 (br, O-H), 2200 (w, CC); 1H-NMR
(400MHz, CDCl3) δ 7.64 (d, J = 7.1 Hz, 2H, aryl), 7.37-7.26 (m, 3H, aryl), 2.26 (s, 1H, -
OH), 1.88-1.75 (m, 3H, H-6 and H-7), 1.30 (s, 9H, H-1), 0.98 (d, J = 6.4 Hz, 3H, H-8), 0.84
(d, J = 6.4 Hz, 3H, H-8) ; 13
C-NMR (100MHz, CDCl3) δ 145.9 (C-9), 128.0, 127.3, 125.5
(CH aryl), 95.1 (C-3), 81.4 (C-4), 73.0 (C-5), 53.9 (C-6), 30.9 (C-1), 27.5 (C-2), 25.1 (C-7),
24.1 (C-8), 23.9 (C-8); MS m/z (ES+) 267 (100 %, M+Na+); HMRS (ES+) calcd for
C17H24O1Na1 (M+Na+): 267.1719, found: 267.1727.

312
2,2,7,7-Tetramethyl-4-phenyloct-5-yn-4-ol 281c.
23
45
6
7
8
910
11
12
HO
1
General procedure P was followed with tert-BuLi (2.0 equiv.). 5,5-Dimethylhex-1-en-3-
yn-2-yl isopropylphenylcarbamate 280a (50 mg, 0.17 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281c (30 mg, 67 %) as a
colourless oil.
Rf 0.67 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3443 (br, O-H), 2220 (w, CC); 1H-NMR
(400MHz, CDCl3) δ 7.66 (d, J = 7.1 Hz, 2H, aryl), 7.36-7.25 (m, 3H, aryl), 2.21 (s, 1H, -
OH), 1.94 (d, J = 14.4 Hz, 1H, H-6), 1.83 (d, J = 14.4 Hz, 1H, H-6), 1.28 (s, 9H, H-1), 0.98
(s, 9H, H-8) ; 13
C-NMR (100MHz, CDCl3) δ 147.2 (C-9), 127.9, 127.2, 125.6 (CH aryl),
95.6 (C-3), 82.1 (C-4), 72.5 (C-5), 57.2 (C-6), 31.4 (C-2), 30.9 (C-1), 30.7 (C-8), 27.5 (C-
7); MS m/z (ES+) 281 (100 %, M+Na+); HMRS (ES+) calcd for C18H26O1Na1 (M+Na
+):
281.1876, found: 281.1877.
5-(4-Chlorophenyl)-2,2-dimethyldec-3-yn-5-ol 281d.
23
45
6
78
910
1112
13
14
HO
1
Cl
General procedure P was followed with n-BuLi (2.0 equiv.). 5,5-Dimethylhex-1-en-3-yn-
2-yl 4-chlorophenylisopropylcarbamate 280b (50 mg, 0.16 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281d (20 mg, 40 %) as a
colourless oil.

313
Rf 0.57 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3393 (br, O-H), 2240 (w, C≡C); 1H-NMR
(400MHz, CDCl3) δ 7.53 (d, J = 8.8 Hz, 2H, H-12), 7.30 (d, J = 8.8 Hz, 2H, H-13), 2.27 (s,
1H, -OH), 1.90-1.82 (m, 1H, H-6), 1.79-1.72 (m, 1H, H-6), 1.48-1.21 (m, 6H, H-7, H-8 and
H-9), 1.27 (s, 9H, H-1), 0.85 (t, J = 6.8 Hz, 3H, H-10); 13
C-NMR (100MHz, CDCl3)
δ 144.1, 133.1, 128.0, 127.0 (aryl), 95.4 (C-3), 80.7 (C-4), 72.8 (C-5), 45.6 (C-6), 31.6,
30.9 (C-1), 27.4 (C-2), 24.3, 22.4 (C-7, C-8 and C-9), 13.9 (C-10); MS m/z (ES+) 275
(100 %, M-H2O+H+); HMRS (ES+) calcd for C18H24
35Cl1 (M-H2O+H
+): 275.1561, found:
275.1563.
4-(4-Chlorophenyl)-2,7,7-trimethyloct-5-yn-4-ol 281e.
23
45
6
788
910
11
12
HO
1
Cl
General procedure P was followed with i-PrLi (2.0 equiv.). 5,5-Dimethylhex-1-en-3-yn-2-
yl 4-chlorophenylisopropylcarbamate 280b (50 mg, 0.16 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281e (12 mg, 43 %) as a
colourless oil.
Rf 0.64 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3409 (br, O-H), 2240 (w, C≡C); 1H-NMR
(400MHz, CDCl3) δ 7.55 (d, J = 8.8 Hz, 2H, H-10), 7.30 (d, J = 8.8 Hz, 2H, H-11), 2.20 (s,
1H, -OH), 1.82-1.70 (m, 3H, H-6 and H-7), 1.27 (s, 9H, H-1), 0.96 (d, J = 6.4 Hz, 3H, H-8),
0.83 (d, J = 6.4 Hz, 3H, H-8) ; 13
C-NMR (100MHz, CDCl3) δ 144.5, 133.1, 128.1, 127.0
(aryl), 95.5 (C-3), 81.0 (C-4), 72.6 (C-5), 53.9 (C-6), 30.8 (C-1), 27.5 (C-2), 25.1 (C-7),
24.1 (C-8), 23.9 (C-8); MS m/z (ES+) 261 (100 %, M-H2O+H+); HMRS (ES+) calcd for
C17H2235
Cl1 (M-H2O+H+): 261.1405, found: 261.1401.

314
4-(4-Chlorophenyl)-2,2,7,7-tetramethyloct-5-yn-4-ol 281f.
23
45
6
7
8
910
11
12
HO
1
Cl
General procedure P was followed with tert-BuLi (2.0 equiv.). 5,5-Dimethylhex-1-en-3-
yn-2-yl 4-chlorophenylisopropylcarbamate 280b (50 mg, 0.16 mmol) gave after flash
column chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281f (21 mg, 47 %)
as a colourless oil.
Rf 0.55 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3434 (br, O-H), 2240 (w, CC); 1H-NMR
(400MHz, CDCl3) δ 7.56 (d, J = 8.8 Hz, 2H, H-10), 7.29 (d, J = 8.8 Hz, 2H, H-11), 2.21 (s,
1H, -OH), 1.89 (d, J = 14.4 Hz, 1H, H-6), 1.78 (d, J = 14.4 Hz, 1H, H-6), 1.26 (s, 9H, H-1),
0.97 (s, 9H, H-8) ; 13
C-NMR (100MHz, CDCl3) δ 145.8, 133.0, 128.0, 127.1 (aryl), 95.9
(C-3), 81.7 (C-4), 72.1 (C-5), 57.2 (C-6), 31.4 (C-2), 31.0 (C-1), 30.6 (C-8), 27.5 (C-7);
MS m/z (ES+) 275 (100 %, M-H2O+H+); HMRS (ES+) calcd for C18H24
35Cl1 (M-H2O+H
+):
275.1561, found: 275.1550.
2,2-Dimethyl-5-p-tolyldec-3-yn-5-ol 281g.
15
23
45
6
78
910
1112
13
14
HO
1
General procedure P was followed with n-BuLi (2.0 equiv.). 5,5-Dimethyl-1-en-3-yn-2-yl
isopropyl-p-tolylcarbamate 280c (50 mg, 0.17 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281g (17 mg, 34 %) as a
colourless oil.

315
Rf 0.65 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3426 (br, O-H), 2242 (w, C≡C); 1H-NMR
(400MHz, CDCl3) δ 7.50 (d, J = 8.1 Hz, 2H, H-12), 7.15 (d, J = 8.1 Hz, 2H, H-13), 2.35 (s,
3H, H-15), 2.22 (s, 1H, -OH), 1.92-1.85 (m, 1H, H-6), 1.82-1.74 (m, 1H, H-6), 1.51-1.26 (m,
6H, H-7, H-8 and H-9), 1.28 (s, 9H, H-1), 0.85 (t, J = 6.8 Hz, 3H, H-10); 13
C-NMR
(100MHz, CDCl3) δ 142.7, 137.0, 128.6, 125.4 (aryl), 94.9 (C-3), 81.1 (C-4), 73.2 (C-5),
45.5 (C-6), 31.6, 31.0 (C-1), 27.4 (C-2), 24.4, 22.4 (C-7, C-8 and C-9), 21.0 (C-15),
13.9 (C-10); MS m/z (ES+) 255 (90 %, M-H2O+H+), 295 (60 %, M+Na
+); HMRS (ES+)
calcd for C19H27 (M-H2O+H+): 255.2108, found: 255.2151.
2,7,7-Trimethyl-4-p-tolyloct-5-yn-4-ol 281h.
13
23
45
6
788
910
11
12
HO
1
General procedure P was followed with i-PrLi (2.0 equiv.). 5,5-Dimethyl-1-en-3-yn-2-yl
isopropyl-p-tolylcarbamate 280c (50 mg, 0.17 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281h (18 mg, 44 %) as a
colourless oil.
Rf 0.68 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3429 (br, O-H), 2239 (w, C≡C); 1H-NMR
(400MHz, CDCl3) δ 7.51 (δ, Ξ = 8.1 Ηζ, 2Η, Η-10), 7.15 (δ, Ξ = 8.1 Ηζ, 2Η, Η-11), 2.34 (σ,
3Η, Η-13), 2.17 (σ, 1Η, -ΟΗ), 1.85-1.73 (μ, 3Η, Η-6 ανδ Η-7), 1.27 (σ, 9Η, Η-1), 0.97 (δ, Ξ
= 6.4 Ηζ, 3Η, Η-8), 0.83 (δ, Ξ = 6.4 Ηζ, 3Η, Η-8); 13
C-NMR (100MHz, CDCl3) δ 143.1,
137.0, 128.6, 125.4 (aryl), 95.0 (C-3), 81.5 (C-4), 72.9 (C-5), 53.8 (C-6), 30.9 (C-1), 27.4
(C-2), 25.2 (C-7), 24.1, 24.0 (C-8), 21.0 (C-13); MS m/z (ES+) 281 (100 %, M+Na+), 241
(70 %, M-H2O+H+); HMRS (ES+) calcd for C18H25 (M-H2O+H
+): 241.1951, found:
241.1962.

316
5-(3-(Trifluoromethyl)phenyl)-2,2-dimethyldec-3-yn-5-ol 281i.
1314
15
23
45
6
78
9
16
10
1112
HO
1
F3C
17
General procedure P was followed with n-BuLi (2.0 equiv.). 5,5-Dimethyl-1-en-3-yn-2-yl
3-(trifluoromethyl)phenylisopropylcarbamate 280d (50 mg, 0.14 mmol) gave after flash
column chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281i (17 mg, 37 %)
as a colourless oil.
Rf 0.70 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3426 (br, O-H), 2242 (w, CC), 1327 (C-
F); 1H-NMR (400MHz, CDCl3) δ 7.91 (s, 1H, H-12), 7.80 (d, J = 7.8 Hz, 1H, H-17), 7.52
(d, J = 7.8 Hz, 1H, H-15), 7.45 (t, J = 7.8 Hz, 1H, H-16), 2.31 (s, 1H, -OH), 1.92-1.84 (m,
1H, H-6), 1.81-1.74 (m, 1H, H-6), 1.49-1.21 (m, 6H, H-7, H-8, H-9), 1.28 (s, 9H, H-1),
0.85 (t, J = 6.8 Hz, 3H, H-10); 13
C-NMR (100MHz, CDCl3) δ 146.7 (C-11), 130.2 (q, 2JC-F
= 32 Hz, C-13), 129.0 (C-17), 128.4 (C-16), 124.2 (q, 1JC-F = 270 Hz, C-14), 124.2 (q,
3JC-F
= 3.6 Hz, C-15), 122.5 (q, 3JC-F = 3.6 Hz, C-12), 95.8 (C-3), 80.4 (C-5), 73.0 (C-4), 45.6
(C-6), 31.5, 30.9 (C-1), 27.5 (C-2), 24.3, 22.4 (C-7, C-8 and C-9), 13.9 (C-10); MS m/z
(ES+) 309 (100 %, M-H2O+H+).
4-(3-(Trifluoromethyl)phenyl)-2,7,7-trimethyloct-5-yn-4-ol 281j.
1314
152
34
5
6
788
910
11
12 HO
1
F3C
General procedure P was followed with i-PrLi (2.0 equiv.). 5,5-Dimethyl-1-en-3-yn-2-yl 3-
(trifluoromethyl)phenylisopropylcarbamate 280d (50 mg, 0.14 mmol) gave after flash

317
column chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281j (24 mg, 55 %)
as a orange oil.
Rf 0.66 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3307 (br, O-H), 2240 (w, C≡C), 1325 (C-
F); 1H-NMR (400MHz, CDCl3) δ 7.92 (s, 1H, H-10), 7.80 (d, J = 7.8 Hz, 1H, H-15), 7.53
(d, J = 7.8 Hz, 1H, H-13), 7.45 (t, J = 7.8 Hz, 1H, H-14), 2.28 (s, 1H, -OH), 1.84-1.71 (m,
3H, H-6 and H-7), 1.28 (s, 9H, H-1), 0.98 (d, J = 6.4 Hz, 3H, H-8), 0.86 (d, J = 6.4 Hz, 3H,
H-8) ; 13
C-NMR (100MHz, CDCl3) δ 147.2 (C-9), 130.3 (q, 2JC-F = 32 Hz, C-11), 129.0
(C-15), 128.5 (C-14), 124.2 (q, 1JC-F = 270 Hz, C-12), 124.2 (q,
3JC-F = 3.6 Hz, C-13),
122.5 (q, 3JC-F = 3.6 Hz, C-10), 95.9 (C-3), 80.8 (C-4), 72.8 (C-5), 53.9 (C-6), 30.8 (C-1),
27.5 (C-2), 25.2 (C-7), 24.1, 23.9 (C-8); MS m/z (ES+) 295 (100 %, M-H2O+H+); HMRS
(ES+) calcd for C18H22F3 (M-H2O+H+): 295.1668, found: 295.1659.
4-(3-(Trifluoroethyl)phenyl)-2,2,7,7-tetramethyloct-5-yn-4-ol 281k.
1314
152
34
5
6
7
8
910
1112 HO
1
F3C
General procedure P was followed with tert-BuLi (2.0 equiv.). 5,5-Dimethyl-1-en-3-yn-2-
yl 3-(trifluoromethyl)phenylisopropylcarbamate 280d (50 mg, 0.14 mmol) gave after flash
column chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281k (12 mg, 26 %)
as a yellow oil.
Rf 0.82 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3446 (br, O-H), 2239 (w, C≡C), 1326 (C-
F); 1H-NMR (400MHz, CDCl3) δ 7.94 (s, 1H, H-10), 7.81 (d, J = 7.8 Hz, 1H, H-15), 7.52
(d, J = 7.8 Hz, 1H, H-13), 7.45 (t, J = 7.8 Hz, 1H, H-14), 2.24 (s, 1H, -OH), 1.90 (d, J =
14.5 Hz, 1H, H-6), 1.78 (d, J = 14.5 Hz, 1H, H-6), 1.27 (s, 9H, H-1), 1.00 (s, 9H, H-8); 13
C-
NMR (100MHz, CDCl3) δ 148.5 (C-9), 130.4 (q, 2JC-F = 32 Hz, C-11), 129.0 (C-15),
128.4 (C-14), 124.2 (q, 1JC-F = 270 Hz, C-12), 124.1 (q,
3JC-F = 3.6 Hz, C-13), 122.6 (q,
3JC-F = 3.6 Hz, C-10), 96.4 (C-3), 81.5 (C-4), 72.2 (C-5), 57.2 (C-6), 31.6 (C-2), 31.0 (C-1),
30.6 (C-8), 27.5 (C-7); MS m/z (ES+) 309 (60 %, M-H2O+H+).

318
5-(4-Methoxyphenyl)-2,2-dimethyldec-3-yn-5-ol 281l.
152
34
5
6
78
910
1112
13
14
HO
1
O
General procedure O was followed with n-BuLi (2.0 equiv.) at -45°C. 5,5-Dimethylhex-1-
en-3-yn-2-yl isopropyl-4-methoxyphenylcarbamate 280e (50 mg, 0.16 mmol) gave after
flash column chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281l (12 mg,
40 %) as a colourless oil.
Rf 0.57 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3334 (br, O-H), 2239 (w, C≡C), 1611,
1511 (C=C), 1251 (C-O-C); 1H-NMR (400MHz, CDCl3) δ 7.46 (d, J = 8.8 Hz, 2H, H-12),
6.80 (d, J = 8.8 Hz, 2H, H-13), 3.74 (s, 3H, H-15), 2.13 (s, 1H, -OH), 1.92-1.85 (m, 1H, H-
6), 1.81-1.74 (m, 1H, H-6), 1.42-1.31 (m, 1H, H-7), 1.21-1.17 (m, 5H, H-7, H-8 and H-9),
1.19 (s, 9H, H-1), 0.78 (t, J = 6.8 Hz, 3H, H-10); 13
C-NMR (100MHz, CDCl3) δ 158.9 (C-
14), 137.7 (C-11), 126.8 (C-12), 113.2 (C-13), 95.0 (C-3), 81.1 (C-4), 73.0 (C-5), 55.2 (C-
15), 45.5 (C-6), 31.6, 31.0 (C-1), 27.5 (C-2), 24.5, 22.4 (C-7, C-8 and C-9), 13.9 (C-10);
MS m/z (ES+) 271 (100 %, M-H2O+H+); HMRS (ES+) calcd for C19H27O1 (M-H2O+H
+):
271.2057, found: 271.2065.
4-(4-Methoxyphenyl)-2,7,7-trimethyloct-5-yn-4-ol 281m.
23
45
6
788
910
11
1213
HO
1
O
General procedure O was followed with i-PrLi (2.0 equiv.) at -45°C. 5,5-Dimethylhex-1-
en-3-yn-2-yl isopropyl-4-methoxyphenylcarbamate 280e (50 mg, 0.16 mmol) gave after

319
flash column chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281m (20 mg,
47 %) as a colourless oil.
Rf 0.53 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3331 (br, O-H), 2232 (w, C≡C), 1611,
1511 (C=C), 1251 (C-O-C); 1H-NMR (400MHz, CDCl3) δ 7.54 (d, J = 8.8 Hz, 2H, H-10),
6.87 (d, J = 8.8 Hz, 2H, H-11), 3.81 (s, 3H, H-13), 2.17 (s, 1H, -OH), 1.86-1.69 (m, 3H, H-6
and H-7), 1.28 (s, 9H, H-1), 0.97 (d, J = 6.4 Hz, 3H, H-8), 0.81 (d, J = 6.4 Hz, 3H, H-8) ;
13C-NMR (100MHz, CDCl3) δ 158.8 (C-12), 138.1 (C-9), 126.8 (C-10), 113.2 (C-11),
95.1 (C-3), 81.5 (C-4), 72.7 (C-5), 55.2 (C-13), 53.9 (C-6), 30.9 (C-1), 27.5 (C-2), 25.2 (C-
7), 24.1, 23.9 (C-8); MS m/z (ES+) 257 (100 %, M-H2O+H+); HMRS (ES+) calcd for
C18H25O1 (M-H2O+H+): 257.1900, found: 257.1906.
1-(Triisopropylsilyl)-3-phenyloct-1-yn-3-ol 281n.
2
34
2
5
6
78
9
1112
13
14
10
1Si
HO
General procedure P was followed with n-BuLi (2.0 equiv.). 4-(Triisopropylsilyl)but-1-en-
3-yn-2-yl isopropylphenylcarbamate 280f (50 mg, 0.13 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281n (36 mg, 76 %) as a
colourless oil.
Rf 0.82 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3425 (br, O-H), 2161 (w, C≡C); 1H-NMR
(400MHz, CDCl3) δ 7.66 (d, J = 7.1 Hz, 2H, H-12), 7.38-7.26 (m, 3H, aryl), 2.36 (s, 1H, -
OH), 1.99-1.91 (m, 1H, H-6), 1.88-1.80 (m, 1H, H-6), 1.48-1.42 (m, 1H, H-7), 1.42-1.33 (m,
1H, H-7), 1.34-1.23 (m, 4H, H-8, H-9), 1.12 (s, 21H, H-1, H-2), 0.85 (t, J = 6.8 Hz, 3H, H-
10) ; 13
C-NMR (100MHz, CDCl3) δ 144.8 (C-11), 128.0, 127.5, 125.5 (CH aryl), 109.8
(C-4), 86.8 (C-3), 73.9 (C-5), 45.6 (C-6), 31.6, 24.5, 22.5 (C-7, C-8 and C-9), 18.6 (C-2),
13.9 (C-10), 11.2 (C-1); MS m/z (ES+) 381 (100 %, M+Na+); HMRS (ES+) calcd for
C23H42O1N1Si1 (M+NH4+): 376.3030, found: 376.3034.

320
1-(Triisopropylsilyl)-5-methyl-3-phenylhept-1-yn-3-ol 281o.
2
34
2
5
6
7
8
9
1112
13
14
10
1Si
HO
General procedure P was followed with sec-BuLi (2.0 equiv.). 4-(Triisopropylsilyl)but-1-
en-3-yn-2-yl isopropylphenylcarbamate 280f (50 mg, 0.13 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281o (27 mg, 58 %) as a
colourless oil.
Rf 0.46 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3442 (br, O-H), 2163 (w, C≡C); 1H-NMR
(400MHz, CDCl3) (mixture of diastereoisomers) δ 7.61 (d, J = 7.4 Hz, 2H, H-12), 7.29-
7.18 (m, 3H, aryl), 2.23 (s, 0.5H, -OH), 2.22 (s, 0.5H, -OH), 1.92-1.68 (m, 2H, H-6), 1.64-
156 (m, 1H, H-7), 1.47-0.98 (m, 2H, H-8), 1.04 (s, 21H, H-1, H-2), 0.93 (d, J = 6.6 Hz,
1.5H, H-10), 0.76 (t, J = 7.4 Hz, 1.5H, H-9), 0.69 (t, J = 7.4 Hz, 1.5H, H-9), 0.65 (d, J
= 6.6 Hz, 1.5H, H-10); 13
C-NMR (100MHz, CDCl3) (mixture of diastereoisomers)
δ 145.5, 145.3 (C-11), 128.0, 127.6, 127.5, 125.6, 125.5 (CH aryl), 110.1, 110.0 (C-4),
87.2, 87.1 (C-3), 74.1, 73.8 (C-5), 51.9, 51.8 (C-6), 31.7, 31.3 (C-7), 30.7, 30.6 (C-8), 20.6,
20.4 (C-9, C-10), 18.6 (C-2), 11.2 (C-1); MS m/z (ES+) 381 (100 %, M+Na+); HMRS
(ES+) calcd for C23H42O1N1Si1 (M+NH4+): 376.3030, found: 376.3028.

321
1-(Triisopropylsilyl)-5,5-dimethyl-3-phenylhex-1-yn-3-ol 281p.
2
34
88
2
5
6
7
8
111213
141
Si
HO
General procedure P was followed with tert-BuLi (2.0 equiv.). 4-(Triisopropylsilyl)but-1-
en-3-yn-2-yl isopropylphenylcarbamate 280f (50 mg, 0.13 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 281p (29 mg, 63 %) as a
colourless oil.
Rf 0.58 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3440 (br, O-H), 2160 (w, C≡C); 1H-NMR
(400MHz, CDCl3) δ 7.62 (d, J = 7.1 Hz, 2H, H-12), 7.28-7.17 (m, 3H, aryl), 2.20 (s, 1H, -
OH), 1.89 (d, J = 14.5 Hz, 1H, H-6), 1.79 (d, J = 14.5 Hz, 1H, H-6), 1.03 (s, 21H, H-1, H-2),
0.93 (s, 9H, H-8) ; 13
C-NMR (100MHz, CDCl3) δ 146.6 (C-11), 128.0, 127.4, 125.6 (CH
aryl), 110.7 (C-4), 88.3 (C-3), 73.1 (C-5), 56.9 (C-6), 31.5 (C-7), 31.0 (C-8), 18.6 (C-2),
11.2 (C-1); MS m/z (ES+) 381 (100 %, M+Na+); HMRS (ES+) calcd for C23H42O1N1Si1
(M+NH4+): 376.3030, found: 376.3027.
3-Phenyloct-1-yn-3-yl isopropylcarbamate 282a.
23
4
13
14
15
12
5
67
8
11
11
9
10
1NH
O
O
H
General procedure O was followed with n-BuLi (2.0 equiv.) and DMPU. 4
(Trimethylsilyl)but-1-en-3-yn-2-yl isopropylphenylcarbamate 280g (50 mg, 0.17 mmol)

322
gave after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound
282a (34 mg, 72 %) as a white solid.
Rf 0.17 (Petrol:EtOAc 8:2); m.p. 76-78 °C (Petrol/Et2O); IR max (neat/cm-1
) 3308 (C-H),
3285 (br, N-H), 1698 (C=O), 1532 (Amide II), 1256 (Amide III); 1H-NMR (400MHz,
CDCl3) δ 7.52 (d, J = 7.6 Hz, 2H, H-13), 7.35-7.24 (m, 3H, aryl), 4.59 (br, 1H, -NH), 3.73
(br, 1H, H-10), 2.81 (s, 1H, H-1), 2.08 (t, J = 11.8 Hz, 1H, H-4), 1.89 (t, J = 11.8 Hz, 1H, H-
4), 1.56-1.43 (m, 1H, H-5), 1.25-1.22 (m, 5H, H-5, H-6 and H-7), 1.13 (d, J = 6.0 Hz, 3H,
H-11), 1.08 (d, J = 6.0 Hz, 3H, H-11), 0.83 (t, J = 6.8 Hz, 3H, H-8) ; 13
C-NMR (100MHz,
CDCl3) δ 153.3 (C-9), 141.9 (C-12), 128.1, 127.5, 125.0 (CH aryl), 82.5 (C-2), 78.3 (C-3),
75.9 (C-1), 44.6 (C-4), 42.8 (C-10), 31.5, 23.8, 23.0 (C-11), 22.4 (C-5, C-6 and C-7),
13.9 (C-8); MS m/z (ES+) 310 (10 0%, M+Na+), (ES-) 286 (100 %, M-H
+); HMRS (ES+)
calcd for C18H25O2N1Na1 (M+Na+): 310.1778, found: 310.1781.
5-Methyl-3-phenylhex-1-yn-3-yl isopropylcarbamate 282b.
23
4
13
7
12
5
6
6
8
9
11
9
10 1NH
O
O
H
General procedure O was followed with i-PrLi (2.0 equiv.) and DMPU. 4-
(Trimethylsilyl)but-1-en-3-yn-2-yl isopropylphenylcarbamate 280g (50 mg, 0.17 mmol)
gave after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound
282b (23 mg, 51 %) as a colourless oil.
Rf 0.21 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3308 (C-H and br N-H), 1691 (C=O),
1531 (Amide II), 1248 (Amide III); 1H-NMR (400MHz, CDCl3) δ 7.54 (d, J = 7.6 Hz, 2H,
H-11), 7.35-7.24 (m, 3H, aryl), 4.55 (br, 1H, -NH), 3.74 (br, 1H, H-8), 2.83 (s, 1H, H-1),
2.05-1.99 (m, 1H, H-4), 1.88-1.85 (m, 2H, H-4 and H-5), 1.13 (d, J = 6.0 Hz, 3H, H-9), 1.08
(d, J = 6.0 Hz, 3H, H-9), 0.97 (d, J = 6.4 Hz, 3H, H-6), 0.76 (d, J = 6.4 Hz, 3H, H-6) ; 13
C-
NMR (100MHz, CDCl3) δ 153.2 (C-7), 142.3 (C-10), 128.1, 127.5, 125.0 (CH aryl), 82.7

323
(C-2), 78.3 (C-3), 76.2 (C-1), 52.7 (C-4), 42.8 (C-8), 24.9 (C-5), 24.0, 23.9 (C-6), 23.0 (C-
9); MS m/z (ES+) 296 (100 %, M+Na+), (ES-) 272 (100 %, M-H
+); HMRS (ES+) calcd for
C17H23O2N1Na1 (M+Na+): 296.1621, found: 296.1610.
5,5-Dimethyl-3-phenylhex-1-yn-3-yl isopropylcarbamate 282c.
23
4
13
7
12
5
6
8
9
11
9
10 1NH
O
O
H
General procedure O was followed with tert-BuLi (2.0 equiv.) and DMPU. 4-
(Trimethylsilyl)but-1-en-3-yn-2-yl isopropylphenylcarbamate 280g (50 mg, 0.17 mmol)
gave after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound
282c (6 mg, 13 %) as a white solid.
Rf 0.19 (Petrol:EtOAc 8:2); m.p. 127-129 °C (Petrol/Et2O); IR max (neat/cm-1
) 3309 (C-
H), 3286 (br, N-H), 1698 (C=O), 1534 (Amide II), 1258 (Amide III); 1H-NMR (400MHz,
CDCl3) δ 7.57 (d, J = 7.6 Hz, 2H, H-11), 7.35-7.24 (m, 3H, aryl), 4.52 (br, 1H, -NH), 3.75
(br, 1H, H-8), 2.87 (s, 1H, H-1), 2.13 (d, J = 14.5 Hz, 1H, H-4), 1.94 (d, J = 14.5 Hz, 1H, H-
4), 1.15 (d, J = 6.0 Hz, 3H, H-9), 1.09 (d, J = 6.0 Hz, 3H, H-9), 0.96 (s, 9H, H-6) ; 13
C-
NMR (100MHz, CDCl3) δ 153.0 (C-7), 143.3 (C-10), 128.1, 127.5, 125.0 (CH aryl), 83.3
(C-2), 78.9 (C-3), 76.8 (C-1), 56.3 (C-4), 42.8 (C-8), 31.6 (C-5), 31.0 (C-6), 23.0 (C-9);
MS m/z (ES+) 310 (100 %, M+Na+), (ES-) 286 (100 %, M-H
+); HMRS (ES+) calcd for
C18H25O2N1Na1 (M+Na+): 310.1778, found: 310.1771.

324
5,5-Dimethyl-1-(trimethylsilyl)-3-phenylhex-1-yn-3-yl isopropylcarbamate 283.
234
13
12
5
6
811
9
10
1
14
NH
O
O
Si
10
7
General procedure O was followed with tert-BuLi (2.0 equiv.) and DMPU. 4-
(Trimethylsilyl)but-1-en-3-yn-2-yl isopropylphenylcarbamate 280g (50 mg, 0.17 mmol)
gave after flash column chromatography (SiO2, Petrol:EtOAc 4 %) the title compound 283
(24 mg, 41 %) as a white solid.
Rf 0.34 (Petrol:EtOAc 8:2); m.p. 88-90 °C (Petrol/Et2O); IR max (neat/cm-1
) 3303 (br, N-
H), 1696 (C=O), 1531 (Amide II), 1248 (Amide III); 1H-NMR (400MHz, CDCl3) δ 7.57
(d, J = 8.8 Hz, 2H, H-12), 7.36-7.24 (m, 3H, aryl), 4.53 (br, 1H, -NH), 3.72 (br, 1H, H-9),
2.16 (d, J = 14.5 Hz, 1H, H-5), 1.90 (d, J = 14.5 Hz, 1H, H-5), 1.11 (br d, 6H, H-10), 0.98
(s, 9H, H-7), 0.24 (s, 9H, H-1) ; 13
C-NMR (100MHz, CDCl3) δ 152.9 (C-8), 143.6 (C-11),
128.0, 127.3, 125.2 (CH aryl), 104.6 (C-3), 94.1 (C-2), 78.2 (C-4), 56.4 (C-5), 42.9 (C-9),
31.5 (C-6), 31.0 (C-7), 22.9 (C-10), -0.34 (C-1); MS m/z (ES+) 382 (100 %, M+Na+);
HMRS (ES+) calcd for C21H33O2N1Na1Si1 (M+Na+): 382.2173, found: 382.2177.
5-Methyl-1,3-diphenylhex-1-yn-3-yl isopropylcarbamate 284.
17
2
34
13
712
5
8
11
9
10
6
1
NH
O
O
10
13 1415
16
General procedure O was followed with i-PrLi (2.0 equiv.) and DMPU. 4-Phenylbut-1-en-
3-yn-2-yl isopropylphenylcarbamate 280h (50 mg, 0.16 mmol) gave after flash column

325
chromatography (SiO2, Petrol:EtOAc 2 %) the title compound 284 (15 mg, 30 %) as a
colourless oil.
Rf 0.40 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3279 (br, N-H), 1712 (C=O); 1H-NMR
(400MHz, CDCl3) δ 7.62-7.27 (m, 10H, aryl), 4.55 (br, 1H, -NH), 3.74 (br, 1H, H-12),
2.16-2.10 (m, 1H, H-8), 1.96-1.93 (m, 2H, H-8 and H-9), 1.13 (d, J = 5.5 Hz, 3H, H-13),
1.10 (d, J = 5.5 Hz, 3H, H-13), 1.02 (d, J = 6.4 Hz, 3H, H-10), 0.83 (d, J = 6.4 Hz, 3H, H-
10) ; 13
C-NMR (100MHz, CDCl3) δ 153.2 (C-11), 142.9, 131.9, 128.4, 128.2, 128.1, 127.5,
125.2, 122.8 (aryl), 87.9, 79.0 (C-5 and C-6), 77.7 (C-7), 52.9 (C-8), 42.8 (C-12), 25.0 (C-
9), 24.1, 23.9 (C-10), 23.0 (C-13); MS m/z (ES+) 372 (50 %, M+Na+).
(E)-4-Benzylidene-5-isobutyl-3-isopropyl-5-phenyloxazolidin-2-one 285.
2
34
13
7
1712
58
11
9
102
6
1
7
14 15
16
NO
O
General procedure O was followed with i-PrLi (2.0 equiv.) and DMPU. 4-Phenylbut-1-en-
3-yn-2-yl isopropylphenylcarbamate 280h (50 mg, 0.16 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 4 %) the title compound 285 (14 mg, 28 %) as a
white solid.
Rf 0.26 (Petrol:EtOAc 8:2); 1H-NMR (400MHz, CDCl3) δ 7.32-7.25 (m, 5H, aryl), 7.12-
7.03 (m, 3H, aryl), 6.70 (d, J = 7.0 Hz, 2H, aryl), 5.98 (s, 1H, H-9), 4.32 (sep, J = 6.8 Hz,
1H, H-6), 2.08 (dd, J = 14.7 and 8.0 Hz, 1H, H-3), 1.97 (dd, J = 14.7 and 3.8 Hz, 1H, H-3),
1.87-1.77 (m, 1H, H-1), 1.56 (d, J = 6.8 Hz, 3H, H-7), 1.55 (d, J = 6.8 Hz, 3H, H-7), 0.96
(d, J = 6.6 Hz, 3H, H-2), 0.78 (d, J = 6.6 Hz, 3H, H-2) ; 13
C-NMR (100MHz, CDCl3) δ
154.9 (C-5), 141.9 (C-8), 141.4 (C-14), 134.5 (C-10), 129.1, 128.6, 128.5, 127.8, 126.4,
126.1 (CH aryl), 101.5 (C-9), 86.6 (C-4), 45.7 (C-6), 42.2 (C-3), 24.5 (C-1), 23.6 (C-7),
19.1, 18.7 (C-2); MS m/z (ES+) 330 (100 %, M+Na+); HMRS (ES+) calcd for C20H22O2N1
(M+H+): 308.1646, found: 308.1656.

326
2,2-Dimethyldeca-3,4-dien-5-yl isopropylphenylcarbamate 286a.
23
4
13
7
13
5
8
11
9
10
6
14
15
16
1
12
O •
H
N
O17
General procedure O was followed with n-BuLi (2.0 equiv.). 5,5-Dimethylhex-1-en-3-yn-
2-yl isopropylphenylcarbamate 280a (50 mg, 0.17 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 286a (44 mg, 73 %) as a
colourless oil.
Rf 0.64 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1977 (w, C=C=C), 1715 (C=O); 1H-NMR
(400MHz, CDCl3) δ 7.37-7.28 (m, 3H, aryl), 7.10 (d, J = 7.0 Hz, 2H, H-15), 5.61 (t, J =
3.1 Hz, 1H, H-3), 4.58 (sep, J = 6.8 Hz, 1H, H-12), 2.14 (br, 2H, H-6), 1.33-1.12 (m, 6H,
H-7, H-8, H-9), 1.12 (d, J = 6.8 Hz, 3H, H-13), 1.11 (d, J = 6.8 Hz, 3H, H-13), 1.05 (s, 9H,
H-1), 0.84 (t, J = 6.8 Hz, 3H, H-10) ; 13
C-NMR (100MHz, CDCl3) δ 192.1 (C-4), 153.6
(C-11), 138.3 (C-14), 129.8, 128.6, 127.3 (CH aryl), 126.3 (C-5), 113.9 (C-3), 49.1 (C-12),
33.4 (C-2), 31.8 (C-6), 31.1, 29.7 (C-1), 26.0, 22.4 (C-7, C-8 and C-9), 21.3, 21.2 (C-13),
13.9 (C-10); MS m/z (ES+) 366 (100 %, M+Na+); HMRS (ES+) calcd for C22H25O2N1Na1
(M+Na+): 366.2404, found: 366.2418.
The equivalent enantioenriched (84:16 e.r.) allene 286a (12 mg, 20 %) was made by
following the procedure below:
To a solution of distilled ()-sparteine (0.08 cm3, 0.34 mmol) in anhydrous toluene (2.0
cm3) was added n-BuLi (2.5 M, 0.14 cm
3, 0.34 mmol) at -50 °C. The resulting yellow
solution was stirred for 15 min after which a solution of 5,5-dimethylhex-1-en-3-yn-2-yl
isopropylphenylcarbamate 280a (50 mg, 0.17 mmol) in anhydrous toluene (0.50 cm3) was
added and stirred at -50 °C for 4 h. The reaction was quenched with MeOH (0.10 cm3) and
a saturated aqueous solution of NH4Cl (1 cm3) and the reaction mixture stirred for a further
30 min while warming to room temperature. The mixture was diluted with Et2O (5 cm3)
and the organic layer was washed with an aqueous solution of CuSO4 (10 cm3) and water

327
(2 x 10 cm3), dried over MgSO4, filtered and concentrated under reduced pressure. The
residue was purified by flash column chromatography (SiO2, Petrol:EtOAc 1 %) to give
the enantioenriched allene 286a as a colourless oil.
HPLC separation conditions: (R,R)-Whelk-O1 column, hexane:2-propanol (98:2), flow rate:
1.0 mL/min; tR 6.3 min for major enantiomer and 8.0 min for minor enantiomer.
1-(Trimethylsilyl)octa-1,2-dien-3-yl isopropylphenylcarbamate 286b.
23413
7
12
5
8
11
9
10
614
15
16
O •
H
SiN
O
1
12
General procedure O was followed with n-BuLi (2.0 equiv.). 4-(Trimethylsilyl)but-1-en-3-
yn-2-yl isopropylphenylcarbamate 280g (50 mg, 0.17 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 286b (38 mg, 64 %) as a
colourless oil.
Rf 0.46 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1694 (C=O); 1H-NMR (400MHz, CDCl3)
δ 7.37-7.26 (m, 3H, aryl), 7.10 (d, J = 7.0 Hz, 2H, H-14), 5.68 (t, J = 4.0 Hz, 1H, H-2),
4.58 (sep, J = 6.8 Hz, 1H, H-11), 2.15 (br, 2H, H-5), 1.25-1.21 (m, 6H, H-6, H-7 and H-8),
1.11 (d, J = 6.8 Hz, 3H, H-12), 1.10 (d, J = 6.8 Hz, 3H, H-12), 0.84 (t, J = 6.8 Hz, 3H, H-
9), 0.11 (s, 9H, H-1); 13
C-NMR (125MHz, CDCl3) δ 205.6 (C-3), 153.7 (C-10), 138.4 (C-
13), 129.8, 128.5, 127.3 (CH aryl), 123.4 (C-4), 98.0 (C-2), 49.2 (C-11), 31.2 (C-5), 31.0,
26.0, 22.4 (C-6, C-7 and C-8), 21.3, 21.2 (C-12), 14.0 (C-9), -1.07 (C-1); MS m/z (ES+)
382 (100 %, M+Na+); HMRS (ES+) calcd for C21H33O2N1Si1Na1 (M+Na
+): 382.2173,
found: 382.2178.

328
2,2-Dimethyl-5-phenyldec-3-yn-5-yl isopropylcarbamate 287.
ONH
O
23
45
67
89
10
11
12
13
14
15
16
171
13
General procedure O was followed with sec-BuLi (2.0 equiv.) and DMPU. 2,2-
Dimethyldeca-3,4-dien-5-yl isopropylphenylcarbamate 286a (45 mg, 0.13 mmol) gave
after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound 287 (40
mg, 89 %) as a white solid.
Rf 0.44 (Petrol:EtOAc 8:2); m.p. 110-113 °C (Petrol/Et2O); IR max (neat/cm-1
) 3307 (br,
N-H), 2241 (w, CC), 1698 (C=O), 1528 (Amide II), 1254 (Amide III); 1H-NMR
(400MHz, CDCl3) δ 7.52 (d, J = 7.2 Hz, 2H, H-15), 7.34-7.22 (m, 3H, aryl), 4.57 (br, 1H, -
NH), 3.71 (sep, J = 6.6 Hz, 1H, H-12), 2.10-2.03 (m, 1H, H-6), 1.86-1.79 (m, 1H, H-6),
1.49-1.43 (m, 1H, H-7), 1.30 (s, 9H, H-1), 1.13-1.10 (m, 5H, H-7, H-8 and H-9), 1.06 (d, J
= 6.6 Hz, 6H, H-13), 0.84 (t, J = 6.8 Hz, 3H, H-10); 13
C-NMR (100MHz, CDCl3) δ 153.3
(C-11), 143.0 (C-14), 127.9, 127.2, 125.2 (CH aryl), 96.9 (C-3), 79.1 (C-4), 77.5 (C-5),
44.9 (C-6), 42.8 (C-12), 31.5, 30.9 (C-1), 27.6 (C-2), 23.9, 22.9 (C-13), 22.3 (C-7, C-8 and
C-9), 13.9 (C-10); MS m/z (ES+) 366 (100 %, M+Na+); HMRS (ES+) calcd for
C22H25O2N1Na1 (M+Na+): 366.2404, found: 366.2418.

329
2,2,3-Trimethyldeca-3,4-dien-5-yl isopropylphenylcarbamate 289.
234
18 7
13
5
8
11
910
6
14
15
16
1
12 O •N
O
17
14
General procedure U was followed with iodomethane as electrophile. 5,5-Dimethylhex-1-
en-3-yn-2-yl isopropylphenylcarbamate 280a (50 mg, 0.17 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 4 %) the title compound 289 (45 mg, 66 %) as a
colourless oil.
Rf 0.68 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1972 (w, C=C=C), 1711 (C=O); 1H-NMR
(400MHz, CDCl3) δ 7.37-7.27 (m, 3H, aryl), 7.10 (d, J = 7.0 Hz, 2H, H-16), 4.59 (sep, J =
6.8 Hz, 1H, H-13), 2.19-2.07 (m, 2H, H-7), 1.76 (s, 3H, H-1), 1.31-1.12 (m, 6H, H-8, H-9
and H-10), 1.11 (d, J = 6.8 Hz, 3H, H-14), 1.10 (d, J = 6.8 Hz, 3H, H-14), 1.06 (s, 9H, H-
2), 0.85 (t, J = 6.8 Hz, 3H, H-11); 13
C-NMR (100MHz, CDCl3) δ 190.5 (C-5), 153.9 (C-
12), 138.5 (C-15), 129.8, 128.5, 127.2 (CH aryl), 123.9 (C-4), 120.5 (C-6), 49.0 (C-13),
35.0 (C-3), 31.9 (C-7), 31.2, 28.9 (C-2), 26.0, 22.5 (C-8, C-9 and C-10), 21.4, 21.3 (C-14),
16.0 (C-1), 14.0 (C-11); MS m/z (ES+) 358 (100 %, M+H+); HMRS (ES+) calcd for
C23H36O2N1 (M+H+): 358.2741, found: 358.2741.
2-tert-Butyl-4-(isopropyl(phenyl)carbamoyl)nona-2,3-dienoic acid 290.
234
18 7
13
5
8
11
910
6
14
15
16
1
12 O •
CO2H
N
O
17
14
General procedure U was followed with dry ice as electrophile. 5,5-Dimethylhex-1-en-3-
yn-2-yl isopropylphenylcarbamate 280a (50 mg, 0.17 mmol) gave after flash column

330
chromatography (SiO2, Petrol:EtOAc 4 %) the title compound 290 (23 mg, 34 %) as a
white solid.
Rf 0.60 (Petrol:EtOAc 8:2); m.p. 84-88 °C (MeOH/Et2O); IR max (neat/cm-1
) 3087 (br, O-
H), 1712 (C=O, acid), 1687 (C=O, carbamate); 1H-NMR (400MHz, CDCl3) δ 11.4 (br, 1H,
-OH), 7.44-7.10 (m, 5H, aryl), 4.61 (sep, J = 6.8 Hz, 1H, H-13), 2.06-2.04 (m, 2H, H-7),
1.25-1.07 (m, 21H, H-1, H-8, H-9, H-10 and H-14), 0.83 (t, J = 6.8 Hz, 3H, H-11); 13
C-
NMR (100MHz, CDCl3) δ 199.4 (C-5), 165.0 (C-1), 153.9 (C-12), 141.9 (C-15), 136.9 (C-
4), 129.4, 129.0, 128.2 (CH aryl), 121.2 (C-6), 50.1 (C-13), 35.0 (C-3), 31.6 (C-7), 29.1
(C-2), 25.2, 24.0, 22.2 (C-8, C-9 and C-10), 21.0, 20.9 (C-14), 13.8 (C-11); MS m/z (ES+)
410 (30 %, M+Na+), (ES-) 386 (100 %, M-H
+); HMRS (ES+) calcd for C23H34O4N1
(M+H+): 388.2483, found: 388.2486.
2,2-Dimethyl-5-(trimethylsilyl)dec-3-yn-5-yl isopropylphenylcarbamate 291.
6
23
4
18 7
13
5
8
11
910
14
15
16
12 ON
O
17
141
Si
General procedure U was followed with trimethylsilylchoride as electrophile. 5,5-
Dimethylhex-1-en-3-yn-2-yl isopropylphenylcarbamate 280a (50 mg, 0.17 mmol) gave
after flash column chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 291 (48
mg, 67 %) as a colourless oil.
Rf 0.68 (Petrol:EtOAc 9:1); IR max (neat/cm-1
) 1702 (C=O); 1H-NMR (400MHz, CDCl3)
δ 7.39-7.28 (m, 3H, aryl), 7.11 (d, J = 7.0 Hz, 2H, H-16), 4.60 (sep, J = 6.8 Hz, 1H, H-13),
1.92-1.74 (m, 2H, H-7), 1.35-1.19 (m, 6H, H-8, H-9 and H-10), 1.25 (s, 9H, H-1), 1.15 (d,
J = 6.8 Hz, 3H, H-14), 1.13 (d, J = 6.8 Hz, 3H, H-14), 0.89 (t, J = 6.8 Hz, 3H, H-11), 0.00
(s, 9H, H-6); 13
C-NMR (100MHz, CDCl3) δ 154.4 (C-12), 138.8 (C-15), 130.2, 128.4,
126.9 (CH aryl), 96.8 (C-3), 78.6 (C-4), 72.9 (C-5), 48.2 (C-13), 36.5 (C-7), 32.0, 31.1 (C-
1), 27.6 (C-2), 24.5, 22.4 (C-8, C-9 and C-10), 21.5, 21.3 (C-14), 14.0 (C-11), -2.53 (C-6);

331
MS m/z (ES+) 438 (100 %, M+Na+) 416 (85 %, M+H
+); HMRS (ES+) calcd for
C25H42O2Si1 (M+H+): 416.2980, found: 416.2965.
Buta-1,3-dien-2-yl isopropylphenylcarbamate 292.
N O
O1
23
4
5
67 7
8
9
10
11
By the method reported by Duchêne et al.:[194]
Dichlorobis(triphenylphosphine)palladium(II) (5 mol %, 44 mg) was added to an
anhydrous DMF (1.5 cm3) solution of 1-iodovinyl isopropylphenylcarbamate 278a (370
mg, 1.12 mmol) in a sealed tube under nitrogen atmosphere, and tributylvinyltin (0.75 cm3,
1.23 mmol) was added after stirring for 15 min at room temperature. The mixture was
stirred at 100 °C for 20 h. After cooling, the reaction mixture was filtered through a Celite
path and then treated with a 1.0 M solution of potassium fluoride and EtOAc. The aqueous
layer was extracted with Et2O. The organic layer was washed with brine, dried over
MgSO4, filtered and concentrated under reduced pressure. The residue was purified by
flash column chromatography (SiO2, Petrol:EtOAc 3 %) to give the title compound 292
(65 mg, 35 %) as a pale orange oil.
Rf 0.52 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1714 (C=O); 1H-NMR (400MHz, CDCl3)
δ 7.42-7.17 (m, 5H, aryl), 6.17 (dd, J = 17.0 and 11.2 Hz, 1H, H-2), 5.03 (br, 2H, H-1), 5.00
(br, 1H, H-4), 4.89 (br, 1H, H-4), 4.63 (sep, J = 6.8 Hz, 1H, H-6), 1.15 (d, J = 6.8 Hz, 6H,
H-7); 13
C-NMR (100MHz, CDCl3) δ 152.9 (C-5), 152.0 (C-3), 137.8 (C-8), 131.7 (C-2),
129.8, 128.8, 127.7 (CH aryl), 114.8 (C-1), 105.0 (C-4), 49.2 (C-6), 21.2 (C-7); MS m/z
(ES+) 254 (100 %, M+Na+); HMRS (ES+) calcd for C14H17O2N1Na1 (M+Na
+): 254.1152,
found: 254.1152.

332
(E)-4-Phenylbuta-1,3-dien-2-yl isopropylphenylcarbamate 293.
N O
O
1
2
34
5
67
8
9
101111
12
1314
15
To a solution of vinyl isopropylphenylcarbamate 277a (200 mg, 0.98 mmol) in anhydrous
THF (4 cm3) was added t-BuLi (1.9 M, 0.56 cm
3, 1.08 mmol) at -78 °C. After 1 h, a
solution of ZnCl2 (1.0 M in Et2O, 1.1 cm3, 1.08 mmol) was added dropwise. The mixture
was stirred for an additional 30 min at -78 °C then at 0 °C for 45 min. The resulting
solution was transferred to a flask containing bromostyrene (0.14 cm3, 1.08 mmol) and
tetrakis(triphenyl)palladium(0) (60 mg, 5 mol %) in THF (4 cm3) and it was stirred at room
temperature for 4 h. The reaction was then quenched with a saturated aqueous solution of
NH4Cl, extracted with Et2O twice and the combined organic layers were washed with
water, brine and dried over MgSO4, filtered and concentrated under reduced pressure. The
residue was purified by flash column chromatography (SiO2, Petrol:EtOAc 5 %) to afford
the title compound 293 (210 mg, 70 %) as a pale yellow solid.
Rf 0.55 (Petrol:EtOAc 8:2); m.p. 71-73 °C (Petrol/Et2O); IR max (neat/cm-1
) 1720 (C=O);
1H-NMR (400MHz, CDCl3) δ 7.48-7.22 (m, 10H, aryl), 6.51 (d, J = 15.8 Hz, 1H, H-5),
6.27 (br, 1H, H-6), 5.13 (s, 1H, H-8), 4.97 (s, 1H, H-8), 4.68 (sep, J = 6.8 Hz, 1H, H-10),
1.18 (d, J = 6.8 Hz, 6H, H-11); 13
C-NMR (100MHz, CDCl3) δ 152.6 (C-9), 151.8 (C-7),
137.8 (C-12), 136.1 (C-4), 129.8, 129.3, 128.9, 128.5, 127.9, 127.8, 126.6 (C-6 and aryl),
123.4 (C-5), 104.5 (C-8), 49.1 (C-10), 21.1 (C-11); MS m/z (ES+) 330 (100 %, M+Na+);
HMRS (ES+) calcd for C20H21O2N1Na1 (M+Na+): 330.1465, found: 330.1470.

333
(E)-1,3-Diphenyloct-1-en-3-ol 294.
2
34
713
58
11
9 10
1
6
14
15
16
12
HO
A solution of n-BuLi (1.8 M, 0.18 cm3, 0.32 mmol) was added dropwise to a precooled
solution (-78 °C) of (E)-4-phenylbuta-1,3-dien-2-yl isopropylphenylcarbamate 293 (50 mg,
0.16 mmol) in a mixture of anhydrous toluene (1.0 cm3) and distilled TMEDA (0.05 cm
3,
0.32 mmol) under nitrogen atmosphere. The resulting reaction mixture was stirred at -
78 °C for 2 h after which anhydrous DMPU (0.10 cm3, 0.64 mmol) was added, warmed up
to -15 °C and stirred for 15 h. The reaction was quenched with MeOH (0.1 cm3) and a
saturated aqueous solution of NH4Cl (1 cm3) and stirred for a further 30 min while
warming to room temperature. The mixture was diluted with Et2O (5 cm3). The organic
layer was washed with an aqueous solution of CuSO4 (10 cm3) and water (2 x 10 cm
3),
dried over MgSO4 and concentrated under reduced pressure. The residue was purified by
flash column chromatography (SiO2, Petrol:EtOAc 2 %) to give the title compound 294
(24 mg, 54 %) as a colourless oil.
Rf 0.68 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3410 (br, O-H); 1H-NMR (400MHz,
CDCl3) δ 7.47 (d, J = 7.2 Hz, 2H, aryl), 7.38-7.18 (m, 8H, aryl), 6.62 (d, J = 16.0 Hz, 1H,
H-6), 6.51 (d, J = 16.0 Hz, 1H, H-5), 2.04-1.90 (m, 2H, H-8), 1.93 (s, 1H, -OH), 1.32-1.20
(m, 6H, H-9, H-10 and H-11), 0.83 (t, J = 6.8 Hz, 3H, H-12); 13
C-NMR (100MHz, CDCl3)
δ 145.8 (C-13), 136.8 (C-4), 136.0 (C-5), 128.5, 128.2, 127.8, 127.5, 126.8, 126.5, 125.4
(C-6 and CH aryl), 76.9 (C-7), 42.5 (C-8), 32.1, 23.3, 22.5 (C-9, C-10, C-11), 14.0 (C-12);
MS m/z (ES+) 263 (80 %, M+H2O-H+); HMRS (ES+) calcd for C20H23 (M+H2O-H
+):
263.1795, found: 263.1800.

334
1-(Dimethyl(phenyl)silyl)vinyl isopropylphenylcarbamate 295.
N O Si
O
2
5
348 6
10
7
11
12
10 1
5
9
1314
To a solution of vinyl isopropylphenylcarbamate 277a (170 mg, 0.83 mmol) in dry THF (3
cm3) was added t-BuLi (1.9 M, 0.48 cm
3, 0.91 mmol) at -78 °C under nitrogen atmosphere.
The mixture was stirred for 1 h at -78 °C after which chloro(dimethyl)phenylsilane (0.17
cm3, 1.0 mmol) was added. The reaction was stirred at -78 °C for 15 min and then at room
temperature for 2 h. A saturated aqueous solution of NH4Cl was added and the mixture was
diluted with Et2O. The organic layer was washed with water and dried over MgSO4,
filtered and concentrated under reduced pressure to give a residue which was purified by
flash column chromatography (SiO2, Petrol:EtOAc 1 %) to give the title compound 295
(145 mg, 52 %) as a colourless oil.
Rf 0.67 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1716 (C=O); 1H-NMR (400MHz, CDCl3)
δ 7.50 (d, J = 7.2 Hz, 2H, aryl), 7.35-7.29 (m, 6H, aryl), 6.90 (d, J = 7.2 Hz, 2H, aryl), 5.44
(br, 1H, H-7), 5.01 (br, 1H, H-7), 4.47 (sep, J = 6.8 Hz, 1H, H-9), 1.01 (d, J = 6.8 Hz, 6H,
H-10), 0.41 (s, 6H, H-5); 13
C-NMR (100MHz, CDCl3) δ 162.0 (C-8), 138.0 (C-11), 136.9
(C-6), 134.1, 129.8, 129.1, 128.6, 127.6, 127.4 (CH aryl), 115.0 (C-7), 48.9 (C-9), 21.1 (C-
10), -2.30 (C-5); MS m/z (ES+) 362 (100 %, M+Na+); HMRS (ES+) calcd for
C20H25O2N1Na1Si1 (M+Na+): 362.1552, found: 362.1554.

335
1-(Dimethyl(phenyl)silyl)hexyl isopropylphenylcarbamate 296.
2
34
7
13
5
8
11
910
1
6
14
15
16
12
17
N O Si
O
5
14
18
General procedure O was followed with n-BuLi (2.0 equiv.) and DMPU. 1-
(Dimethyl(phenyl)silyl)vinyl isopropylphenylcarbamate 295 (50 mg, 0.15 mmol) gave
after flash column chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 296 (40
mg, 70 %) as a colourless oil.
Rf 0.77 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1694 (C=O); 1H-NMR (400MHz, CDCl3)
δ 7.17-7.05 (m, 8H, aryl), 6.81, (d, J = 8.0 Hz, 2H, aryl), 4.59 (t, J = 6.8 Hz, 1H, H-6), 4.38
(sep, J = 6.8 Hz, 1H, H-13), 1.19 (m, 2H, H-7), 1.08-0.93 (m, 6H, H-8, H-9 and H-10),
0.89 (d, J = 6.8 Hz, 3H, H-14), 0.86 (d, J = 6.8 Hz, 3H, H-14), 0.63 (t, J = 6.8 Hz, 3H, H-
11), 0.00 (s, 3H, H-5), -0.05 (s, 3H, H-5); 13
C-NMR (100MHz, CDCl3) δ 156.0 (C-12),
136.5, 134.0, 130.2, 129.0, 128.5, 127.5, 127.1 (aryl), 69.1 (C-6), 48.5 (C-13), 31.6 (C-7),
31.2, 26.4, 22.4 (C-8, C-9 and C-10), 21.4, 21.3 (C-14), 14.0 (C-11), -4.69, -5.11 (C-5);
MS m/z (ES+) 398 (100 %, M+H+), (ES-) 396 (100 %, M-H
+); HMRS (ES+) calcd for
C24H35O2N1Si1Na1 (M+Na+): 420.2330, found: 420.2322.
1-(Furan-2-yl)vinyl isopropylphenylcarbamate 299.
N O
O
O
12
3
45
6
7
89 9
10
1112
13
General procedure K was followed. 2-Acetylfuran 297 (250 mg, 2.27 mmol) gave after
flash column chromatography (SiO2, Petrol:EtOAc 3 %) the title compound 299 (298 mg,
48 %) as a white solid.

336
Rf 0.48 (Petrol:EtOAc 8:2); m.p. 62-64 °C (Petrol/Et2O); IR max (neat/cm-1
) 1706 (C=O);
1H NMR (400MHz, CDCl3) 7.44-7.34 (m, 3H, aryl), 7.32 (br, 1H, H-3), 7.21 (d,
3J = 7.2
Hz, 2H, H-11), 6.32 (br, 1H, H-2), 6.05 (br, 1H, H-1), 5.35 (br, 1H, H-6), 5.01 (br, 1H, H-
6), 4.63 (sep, J = 6.8 Hz, 1H, H-8), 1.17 (d, J = 6.8 Hz, 6H, H-9); 13
C NMR (100MHz,
CDCl3) 152.8 (C-7), 149.2 (C-5), 144.6 (C-4), 142.6 (C-3), 137.7 (C-10), 129.7, 128.9,
127.8 (CH aryl), 111.1 (C-2), 107.0 (C-1), 99.3 (C-6), 49.4 (C-8), 21.2 (C-9); MS m/z
(ES+) 294 (100 %, M+Na+); HMRS (ES+) calcd for C16H17O3N1Na1 (M+Na
+): 294.1101,
found: 294.1099.
1-(2,5-Dimethylfuran-3-yl)vinyl isopropylphenylcarbamate 300.
N O
O
O1
2
3
45
6
7
8
109
10
11
1213
314
General procedure K was followed. 3-Acetyl-2,5-dimethylfuran 298 (250 mg, 1.81 mmol)
gave after flash column chromatography (SiO2, Petrol:EtOAc 3 %) the title compound 300
(100 mg, 19 %) as a pale yellow oil.
Rf 0.53 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1714 (C=O); 1H NMR (400MHz, CDCl3)
7.41-7.32 (m, 3H, aryl), 7.17 (d, J = 7.2 Hz, 2H, H-12), 5.82 (br, 1H, H-1), 4.87 (br, 1H,
H-7), 4.83 (br, 1H, H-7), 4.62 (sep, J = 6.8 Hz, 1H, H-9), 2.18 (s, 6H, H-3), 1.14 (d, J = 6.8
Hz, 6H, H-10); 13
C NMR (100MHz, CDCl3) 149.5 (C-8), 148.5 (C-6), 147.4 (C-5),
137.8 (C-11), 129.8, 128.8, 127.7 (CH aryl), 117.0 (C-2), 113.2 (C-4), 105.3 (C-1), 100.5
(C-7), 49.3 (C-9), 21.2 (C-10), 13.2 (C-3), 13.0 (C-3); MS m/z (ES+) 322 (100 %, M+Na+);
HMRS (ES+) calcd for C18H21O3N1Na1 (M+Na+): 322.1414, found: 322.1409.

337
1-Phenylethyl methylstyrylcarbamate 305.
2
34
7 5
8
9
10
1
6
N O
O11
12
13
14
A suspension of trans-cinnamic acid 301 (500 mg, 3.38 mmol) in dry benzene (5.0 cm3)
was stirred in the presence of triethylamine (0.47 cm3, 3.38 mmol) at 0 °C under nitrogen
atmosphere. Diphenylphosphoryl azide (0.73 cm3, 3.38 mmol) was added slowly and the
reaction was stirred at room temperature for 3 h. The reaction was quenched with a
saturated aqueous solution of NH4Cl (5 cm3) and then washed with a 1 N aqueous solution
of KHSO4 (20 cm3), water (20 cm
3), a saturated aqueous solution of NaHCO3 (20 cm
3) and
brine (20 cm3). The organic phase was concentrated under reduced pressure at room
temperature.[196]
The resulting isocyanate 302 was solubilised directly in anhydrous
CH2Cl2 (12 cm3) and 1-phenylethanol 183d (300 mg, 2.70 mmol) and triethylamine (0.70
cm3, 6.08 mmol) were added. The reaction mixture was stirred at room temperature for 15
h under nitrogen atmosphere and the solvent was evaporated under reduced pressure. The
crude mixture was solubilised in anhydrous DMF (10 cm3), NaH (60 % in mineral oil, 180
mg, 6.76 mmol) was added slowly at 0 °C and the mixture was stirred for 30 min under
nitrogen atmosphere. Methyl iodide (0.35 cm3, 8.45 mmol) was added and the reaction was
stirred at room temperature for 15 h. Water (10 cm3) was added and the reaction mixture
was extracted with Et2O (20 cm3). The organic layer was then washed with water (2 x 30
cm3), dried over MgSO4, filtered and evaporated under reduced pressure. The crude
product was purified by flash column chromatography on silica (SiO2, Petrol:EtOAc 3 %)
to give the title compound 305 (465 mg, 49 % overall yield) as a colourless oil.
Rf 0.56 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1703 (C=O), 1643 (C=C); 1H NMR
(300MHz, CDCl3) 7.75 (d, J = 14.7 Hz, 1H, H-9), 7.39-7.16 (m, 10H, aryl), 5.91 (q, J =
6.6 Hz, 1H, H-5), 5.83 (d, J = 14.7 Hz, 1H, H-10), 3.20 (s, 3H, H-8), 1.62 (d, J = 6.6 Hz,
3H, H-6); 13
C NMR (100MHz, CD3OD) 141.7 (C-7), 140.8, 137.0, 129.6, 128.6, 128.5,
128.0, 126.1, 126.0, 125.4 (C-9 and aryl), 109.4 (C-10), 74.7 (C-5), 31.2 (C-8), 22.6 (C-6);
MS m/z (ES+) 304 (100 %, M+Na+); HMRS (ES+) calcd for C18H19O2N1Na1 (M+Na
+):
304.1308, found: 304.1302.

338
1-Phenylethyl methylvinylcarbamate 308a.
2
34
7 5
8
9
10
1
6
N O
O
General procedures E and F were followed (stirred at room temperature for 1 h). 1-
phenylethanol 183d (100 mg, 0.82 mmol) gave after flash column chromatography (SiO2,
Petrol:EtOAc 4%) the title compound 308a (100 mg, 69 % over 2 steps) as a colourless oil.
Rf 0.72 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1705 (C=O), 1625 (C=C); 1H NMR
(500MHz, CDCl3) (mixture of rotamers) 7.29-7.20 (m, 5H, aryl), 7.16 (dd, J = 15.8 and
9.2 Hz, 1H, H-9), 5.80 (q, J = 6.6 Hz, 1H, H-5), 4.23 (d, J = 15.8 Hz, 1H, H-10), 4.16 (d, J
= 9.2 Hz, 1H, H-10), 3.00 (s, 1.2H, H-8, rotamer 1), 2.97 (s, 1.8H, H-8, rotamer 2), 1.51 (d,
J = 6.6 Hz, 3H, H-6); 13
C NMR (125MHz, CDCl3) (mixture of rotamers) 153.7, 153.1
(C-7), 141.7 (C-4), 133.5 (C-9), 128.5, 127.8, 125.9 (CH aryl), 91.6 (C-10), 74.3, 74.1 (C-
5), 30.1 (C-8), 22.5 (C-6); MS m/z (ES+) 228 (100 %, M+Na+); HMRS (ES+) calcd for
C12H15O2N1Na1 (M+Na+): 228.0995, found: 228.0993.
1-(4-Chlorophenyl)ethyl methylvinylcarbamate 308b.
2
34
7 5
8
9
101
6
N O
O
Cl
General procedures E and F were followed (stirred at room temperature for 1 h). 1-(4-
Chlorophenyl)ethanol 183a (200 mg, 1.28 mmol) gave after flash column chromatography
(SiO2, Petrol:EtOAc 2 %) the title compound 308b (158 mg, 52 % over 2 steps) as a
colourless oil.
Rf 0.68 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1705 (C=O), 1625 (C=C); 1H NMR
(500MHz, CDCl3) (mixture of rotamers) 7.33-7.28 (m, 4H, aryl), 7.19 (dd, J = 15.8 and
8.8 Hz, 1H, H-9), 5.83 (q, J = 6.6 Hz, 1H, H-5), 4.32 (d, J = 15.8 Hz, 1H, H-10), 4.16 (d, J

339
= 8.8 Hz, 1H, H-10), 3.07 (s, 1.2H, H-8, rotamer 1), 3.04 (s, 1.8H, H-8, rotamer 2), 1.56 (d,
J = 6.6 Hz, 3H, H-6); 13
C NMR (125MHz, CDCl3) (mixture of rotamers) 153.1 (C-7),
140.3 (C-4), 133.6 (C-1), 133.4 (C-9), 128.7, 127.3 (CH aryl), 91.9 (C-10), 73.7, 73.5 (C-
5), 30.1 (C-8), 22.4 (C-6); MS m/z (ES+) 262 (100 %, M+Na+); HMRS (ES+) calcd for
C12H14O2N135
Cl1Na1 (M+Na+): 262,0606, found: 262.0612.
1-(3-(Trifluoromethyl)phenyl)ethyl methylvinylcarbamate 308c.
2
3
4
75
8
11
9
10
1
6
N O
O
CF313
12
General procedures E and F were followed (stirred at room temperature for 1 h). 1-(3-
Trifluoromethylphenyl)ethanol 183c (224 mg, 1.18 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 2 %) the title compound 308c (102 mg, 32 % over 2
steps) as a colourless oil.
Rf 0.69 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1709 (C=O), 1628 (C=C), 1317 (C-F); 1H
NMR (500MHz, CDCl3) (mixture of rotamers) 7.55 (s, 1H, H-6), 7.54-7.46 (m, 3H, aryl),
7.21 (dd, J = 15.8 and 9.2 Hz, 1H, H-12), 5.92 (q, J = 6.6 Hz, 1H, H-8), 4.34 (d, J = 15.8
Hz, 1H, H-13), 4.29 (d, J = 9.2 Hz, 1H, H-13), 3.10 (s, 1.3H, H-11, rotamer 1), 3.06 (s,
1.7H, H-11, rotamer 2), 1.60 (d, J = 6.6 Hz, 3H, H-9); 13
C NMR (125MHz, CDCl3)
(mixture of rotamers) 153.5, 153.0 (C-10), 142.8 (C-7), 134.0, 133.8 (C-12), 130.6 (q,
2JCF = 32 Hz, C-4), 129.3, 129.1 (CH aryl), 124.0 (q,
1JCF = 270 Hz, C-5), 124.7 (q,
3JCF =
3.6 Hz, C-3), 122.6 (q, 3JCF = 3.6 Hz, C-6), 92.2 (C-13), 73.6, 73.4 (C-8), 30.2, 30.1 (C-11),
22.5 (C-9); MS m/z (ES+) 296 (100 %, M+Na+); HMRS (ES+) calcd for
C13H14O2N1F3Na1 (M+Na+): 296.0869, found: 296.0862.

340
1-m-Tolylethyl methylvinylcarbamate 308d.
13
12
23
4
7 58
11
9
10
1
6
N O
O
General procedures E and F were followed (stirred at room temperature for 1 h). 1-(3-
Methylphenyl)ethanol 183e (203 mg, 1.49 mmol) gave after flash column chromatography
(SiO2, Petrol:EtOAc 2 %) the title compound 308d (209 mg, 65 % over 2 steps) as a
colourless oil.
Rf 0.69 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1707 (C=O), 1625 (C=C); 1H NMR
(400MHz, CDCl3) (mixture of rotamers) 7.32-7.15 (m, 5H, aryl and H-12), 5.89 (q, J =
6.6 Hz, 1H, H-8), 4.35 (d, J = 15.8 Hz, 1H, H-13), 4.29 (d, J = 9.0 Hz, 1H, H-13), 3.13 (s,
1.2H, H-11, rotamer 1), 3.10 (s, 1.8H, H-11, rotamer 2), 1.62 (d, J = 6.6 Hz, 3H, H-9); 13
C
NMR (100MHz, CDCl3) (mixture of rotamers) 153.2 (C-10), 141.7 (C-7), 138.1 (C-4),
134.2, 133.6 (C-12), 128.7, 128.4, 126.6, 122.9 (CH aryl), 91.5 (C-13), 74.2, 74.4 (C-8),
30.1 (C-11), 22.5 (C-9), 21.4 (C-5); MS m/z (ES+) 242 (100 %, M+Na+); HMRS (ES+)
calcd for C13H17O2N1Na1 (M+Na+): 242.1152, found: 242.1146.
1-(4-Methoxyphenyl)ethyl methylvinylcarbamate 308e.
2
34
7
58
11
9
10
1
6N O
O
O
General procedures E and F were followed (stirred at room temperature for 1 h). 1-(3-
Methoxyphenyl)ethanol 183b (229 mg, 1.51 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 5 %) the title compound 308e (220 mg, 62 % over 2
steps) as a colourless oil.
Rf 0.50 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1707 (C=O), 1625, 1513 (C=C), 1246 (C-
O-C); 1H NMR (300MHz, CDCl3) 7.31 (d, J = 8.8 Hz, 2H, H-4), 7.20 (dd, J = 15.8 and

341
8.8 Hz, 1H, H-10), 6.89 (d, J = 8.8 Hz, 2H, H-3), 5.84 (q, J = 6.6 Hz, 1H, H-6), 4.28 (d, J =
15.8 Hz, 1H, H-11), 4.22 (d, J = 8.8 Hz, 1H, H-11), 3.80 (s, 3H, H-1), 3.04 (s, 3H, H-9),
1.57 (d, J = 6.6 Hz, 3H, H-7); 13
C NMR (75MHz, CDCl3) 159.2 (C-2), 153.2 (C-8),
134.2 (C-5), 133.8 (C-10), 127.4 (C-4), 113.8 (C-3), 91.4 (C-11), 74.0 (C-6), 55.2 (C-1),
30.0 (C-9), 22.2 (C-7); MS m/z (ES+) 258 (100 %, M+Na+).
2-Phenylbut-3-en-2-yl methylcarbamate 309a.
2
34
7
5
8
910
1
6
NH
O
O
General procedure O was followed with LDA (2.0 equiv.) and DMPU at -45°C. 1-
Phenylethyl methylvinylcarbamate 308a (43 mg, 0.21 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 5 %) the title compound 309a (30 mg, 70 %) as a
colourless oil.
Rf 0.27 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3355 (br, N-H), 1698 (C=O), 1618 (C=C),
1520 (Amide II), 1262 (Amide III); 1H NMR (400MHz, CDCl3) 7.39-7.23 (m, 5H, aryl),
6.30 (dd, J = 17.4 and 10.8 Hz, 1H, H-7), 5.25 (d, J = 17.4 Hz, 1H, H-8), 5.23 (d, J = 10.8,
1H, H-8), 4.70 (br, 1H, -NH), 2.74 (d, J = 4.8 Hz, 3H, H-10), 1.89 (s, 3H, H-6); 13
C NMR
(100MHz, CDCl3) 155.5 (C-9), 144.3 (C-4), 142.1 (C-7), 128.1, 127.1, 125.1 (CH aryl),
114.0 (C-8), 82.2 (C-5), 27.2 (C-10), 25.8 (C-6); MS m/z (ES+) 228 (100 %, M+Na+);
HMRS (ES+) calcd for C12H15O2N1Na1 (M+Na+): 228.0995, found: 228.0985.
2-(4-Chlorophenyl)but-3-en-2-yl methylcarbamate 309b.
2
34
7
5
8
910
1
6
NH
O
O
Cl
General procedure O was followed with LDA (2.0 equiv.) and DMPU at -45°C. 1-(4-
Chlorophenyl)ethyl methylvinylcarbamate 308b (50 mg, 0.21 mmol) gave after flash

342
column chromatography (SiO2, Petrol:EtOAc 3 %) the title compound 309b (27 mg, 54 %)
as a colourless oil.
Rf 0.30 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3352 (br, N-H), 1697 (C=O), 1620 (C=C),
1520 (Amide II), 1263 (Amide III); 1H NMR (400MHz, CDCl3) 7.36-7.28 (m, 4H, aryl),
6.26 (dd, J = 17.4 and 10.8 Hz, 1H, H-7), 5.24 (d, J = 17.4 Hz, 1H, H-8), 5.23 (d, J = 10.8
Hz, 1H, H-8), 4.69 (br, 1H, -NH), 2.73 (d, J = 4.8 Hz, 3H, H-10), 1.89 (s, 3H, H-6); 13
C
NMR (100MHz, CDCl3) 155.3 (C-9), 142.9 (C-4), 141.7 (C-7), 133.0 (C-1), 128.3,
126.7 (CH aryl), 114.5 (C-8), 81.7 (C-5), 27.3 (C-10), 25.8 (C-6); MS m/z (ES+) 262
(100 %, M+Na+); HMRS (ES+) calcd for C12H14O2N1Na1
35Cl1 (M+Na
+): 262.0606, found:
262.0604.
2-(3-(Trifluoromethyl)phenyl)but-3-en-2-yl methylcarbamate 309c.
1213
2
3
47
5
8
910
1
6
11
NH
O
O
CF3
General procedure O was followed with LDA (2.0 equiv.) and DMPU at -45°C. 1-(3-
(Trifluoromethyl)phenyl)ethyl methylvinylcarbamate 308c (50 mg, 0.18 mmol) gave after
flash column chromatography (SiO2, Petrol:EtOAc 3 %) the title compound 309c (16 mg,
32 %) as a colourless oil.
Rf 0.23 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3353 (br, N-H), 1699 (C=O), 1625 (C=C),
1521 (Amide II), 1328 (C-F), 1263 (Amide III); 1H NMR (500MHz, CDCl3) 7.62 (s, 1H,
H-6), 7.52-7.43 (m, 3H, aryl), 6.28 (dd, J = 17.4 and 10.8 Hz, 1H, H-10), 5.28 (d, J = 17.4
Hz, 1H, H-11), 5.27 (d, J = 10.8 Hz, 1H, H-11), 4.75 (br, 1H, -NH), 2.74 (d, J = 4.8 Hz,
3H, H-13), 1.89 (s, 3H, H-9); 13
C NMR (125MHz, CDCl3) 155.2 (C-12), 145.5 (C-7),
141.3 (C-10), 130.5 (q, 2JCF = 32 Hz, C-4), 128.6, 128.7 (CH aryl), 124.1 (q,
1JCF = 270 Hz,
C-5), 124.0 (q, 3JCF = 3.6 Hz, C-3), 122.0 (q,
3JCF = 3.6 Hz, C-6), 114.9 (C-11), 81.6 (C-8),
27.2 (C-13), 25.9 (C-9); MS m/z (ES+) 296 (100 %, M+Na+), (ES-) 272 (50 %, M-H
+);
HMRS (ES+) calcd for C13H14O2N1Na1 (M+Na+): 296.0869, found: 296.0870.

343
2-m-Tolylbut-3-en-2-yl methylcarbamate 309d.
1312
2
3
4
75
8
910
1
6
11
NH
O
O
General procedure O was followed with LDA (2.0 equiv.) and DMPU at -45°C. 1-m-
Tolylethyl methylvinylcarbamate 308d (50 mg, 0.23 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 3 %) the title compound 309d (24 mg, 48 %) as a
colourless oil.
Rf 0.25 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3355 (br, N-H), 1698 (C=O), 1619 (C=C),
1520 (Amide II), 1252 (Amide III); 1H NMR (400MHz, CDCl3) 7.28-7.05 (m, 4H, aryl),
6.29 (dd, J = 17.4 and 10.8 Hz, 1H, H-10), 5.25 (d, J = 17.4 Hz, 1H, H-11), 5.22 (d, J =
10.8 Hz, 1H, H-11), 4.75 (br, 1H, -NH), 2.73 (d, J = 4.8 Hz, 3H, H-13), 2.35 (s, 3H, H-5),
1.88 (s, 3H, H-9); 13
C NMR (100MHz, CDCl3) 155.5 (C-12), 144.2 (C-7), 142.1 (C-10),
137.6 (C-4), 128.8, 128.0, 125.7, 122.1 (CH aryl), 113.8 (C-11), 82.1 (C-8), 27.2 (C-13),
25.7 (C-9), 21.6 (C-5); MS m/z (ES+) 242 (100 %, M+Na+); HMRS (ES+) calcd for
C13H17O2N1Na1 (M+Na+): 242.1152, found: 242.1143.
2-(4-Methoxyphenyl)but-3-en-2-ol 310a.
2
3
4
7
5
89
1
6HO
O
General procedure O was followed with LDA (2.0 equiv.) and DMPU at -45°C. 1-(4-
Methoxyphenyl)ethyl methylvinylcarbamate 308e (50 mg, 024 mmol) gave after flash
column chromatography (SiO2, Petrol:EtOAc 3 %) the title compound 310a (10 mg, 22 %)
as a colourless oil.

344
Rf 0.17 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3414 (br, O-H), 1610, 1509 (C=C), 1244
(C-O-C); 1H NMR (400MHz, CDCl3) 7.39 (d, J = 9.0 Hz, 2H, H-4), 6.87 (d, J = 9.0 Hz,
2H, H-3), 6.15 (dd, J = 17.2 and 10.6 Hz, 1H, H-8), 5.28 (dd, J = 17.2 and 1.1 Hz, 1H, H-
9), 5.13 (dd, J = 10.6 and 1.1 Hz, 1H, H-9), 3.80 (s, 3H, H-1), 1.82 (br, 1H, -OH), 1.64 (s,
3H, H-7); 13
C NMR (100MHz, CDCl3) 158.5 (C-2), 145.0 (C-8), 138.5 (C-5), 126.4 (C-
4), 113.5 (C-3), 112.0 (C-9), 74.4 (C-6), 55.3 (C-1), 29.3 (C-7); MS m/z (ES+) 161 (100 %,
M-H2O+H+); HMRS (ES+) calcd for C11H13O1 (M-H2O+H
+): 161.0961, found: 161.0961.
2-Phenylbut-3-en-2-ol 310b.[52]
2
34
7
5
8
1
6
HO
General procedures O and R were followed. 1-Phenylethyl methylvinylcarbamate 308a
(200 mg) gave after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title
compound 310b (96 mg, 48 % over 2 steps) as a yellow oil.
Rf 0.39 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3395 (br, O-H); 1H NMR (400MHz,
CDCl3) 7.38 (d, J = 8.0 Hz, 2H, H-3), 7.26 (t, J = 8.0 Hz, 2H, H-2), 7.17 (t, J = 8.0 Hz,
1H, H-1), 6.08 (dd, J = 17.2 and 10.8 Hz, 1H, H-7), 5.21 (dd, J = 17.2 and 1.2 Hz, 1H, H-
8), 5.06 (dd, J = 10.8 and 1.2 Hz, 1H, H-8), 1.92 (s, 1H, -OH), 1.57 (s, 3H, H-6); 13
C NMR
(100MHz, CDCl3) 146.3 (C-4), 144.8 (C-7), 128.2, 126.9, 125.1 (CH aryl), 112.3 (C-8),
74.7 (C-5), 29.3 (C-6); GC/MS 148.1 (M+). Spectral data matched the published data.
[52]

345
Benzyl methylcarbamate 314a.[233]
NH
O
O
1
2
34
5
67
General procedure V was followed. Benzyl chloroformate 312a (1.00 g, 5.88 mmol) gave
after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound 314a
(0.63 g, 65 %) as a colourless oil.
Rf 0.59 (Petrol:EtOAc 7:3); IR max (neat/cm-1
) 3339 (br, N-H), 1697 (C=O), 1529 (Amide
II), 1254 (Amide III); 1H NMR (400MHz, CDCl3) 7.38-7.28 (m, 5H, aryl), 5.10 (s, 2H,
H-5), 4.10 (br, 1H, -NH), 2.81 (d, J = 4.8 Hz, 3H, H-7); 13
C NMR (100MHz, CDCl3)
157.0 (C-6), 136.6 (C-4), 128.5, 128.1, 126.9 (CH aryl), 66.0 (C-5), 27.5 (C-7); MS m/z
(ES+) 188 (100 %, M+Na+); HMRS (ES+) calcd for C9H12O2N1 (M+H
+): 166.0863, found:
166.0864. Spectral data matched the published values.[233]
Benzyl isopropylcarbamate 314b. [234]
NH
O
O
1
2
34
5
6
7
8
8
General procedure V was followed. Benzyl chloroformate 312a (500 mg, 2.94 mmol) gave
after flash column chromatography (SiO2, Petrol:EtOAc 5 %) the title compound 314b
(439 mg, 78 %) as a white solid.
Rf 0.48 (Petrol:EtOAc 8:2); m.p 58-60 °C (Et2O/Petrol); IR max (neat/cm-1
) 3326 (br, N-
H), 1681 (C=O), 1528 (Amide II), 1255 (Amide III); 1H NMR (400MHz, CDCl3) 7.36-
7.29 (m, 5H, aryl), 5.08 (s, 2H, H-5), 4.59 (br d, J = 6.6 Hz, 1H, -NH), 3.84 (oct, J = 6.6
Hz, 1H, H-7), 1.16 (d, J = 6.6 Hz, 6H, H-8); 13
C NMR (100MHz, CDCl3) 155.5 (C-6),
136.6 (C-4), 128.4, 128.1, 128.0 (CH aryl), 66.4 (C-5), 43.1 (C-7), 23.0 (C-8); MS m/z

346
(ES+) 216 (100 %, M+Na+); HMRS (ES+) calcd for C11H16O2N1 (M+H
+): 194.1176,
found: 194.1170. Spectral data matched the published values.[234]
1-Phenylethyl isopropylcarbamate 314c.
NH
O
O
1
2
34
5
6
78
9
9
To a solution of 1-phenylethanol 183d (1.00 g, 8.20 mmol) in anhydrous THF (130 cm3)
were added triethylamine (1.1 cm3, 8.20 mmol) and a solution of triphosgene (2.40 g, 9.02
mmol) in anhydrous THF (30 cm3) dropwise at 0 °C. The reaction mixture was stirred at
room temperature for 15 h under nitrogen atmosphere. The solvent was evaporated under
reduced pressure and the residue was dissolved in anhydrous CH2Cl2 (40 cm3).
Isopropylamine (1.4 cm3, 16.4 mmol) and pyridine (1.2 cm
3, 16.4 mmol) were added
dropwise at 0 °C. The reaction mixture was stirred at room temperature and concentrated
after 15 h. The residue was purified by flash column chromatography (SiO2, Petrol:EtOAc
5 %) to give the title compound 314c (0.76 g, 45 % over 2 steps) as a white solid.
Rf 0.50 (Petrol:EtOAc 8:2); m.p 63-65 °C (Et2O/Petrol); IR max (neat/cm-1
) 3327 (br, N-
H), 1689 (C=O), 1525 (Amide II), 1242 (Amide III); 1H NMR (400MHz, CDCl3) 7.27-
7.17 (m, 5H, aryl), 5.71 (q, J = 6.8 Hz, 1H, H-5), 4.53 (br d, J = 6.6 Hz, 1H, -NH), 3.73
(oct, J = 6.6 Hz, 1H, H-8), 1.45 (d, J = 6.8, 3H, H-6), 1.07 (d, J = 6.6 Hz, 3H, H-9), 1.03 (d,
J = 6.6 Hz, 3H, H-9); 13
C NMR (100MHz, CDCl3) 155.0 (C-7), 142.2 (C-4), 128.4,
127.6, 125.9 (CH aryl), 72.2 (C-5), 42.9 (C-8), 22.9 (C-9), 22.4 (C-6); MS m/z (ES+) 230
(100 %, M+Na+); HMRS (ES+) calcd for C12H17O2N1Na1 (M+Na
+): 230.1152, found:
230.1158.

347
1-(2-Bromoethynyl)benzene 315.[235]
1
23
4
5
6
Br
General procedure W was followed. Phenylactetylene 313 (1.0 g, 9.8 mmol) gave after
flash column chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 315 (1.5 g,
82 %) as a yellow oil.
Rf 0.70 (Petrol:EtOAc 9:1); IR max (neat/cm-1
) 2196 (CC); 1H NMR (400MHz, CDCl3)
7.47-7.44 (m, 2H, aryl), 7.35-7.29 (m, 3H, aryl); 13
C NMR (100MHz, CDCl3) 132.0,
128.7, 128.3 (CH aryl), 122.6 (C-3), 80.0 (C-2), 49.7 (C-1). MS m/z (ES+) 180 (100 %,
M+). Spectral data matched the published values.
[235]
Benzyl methyl-2-phenylethynylcarbamate 316a.
1
23
45
6N O
O
7
89
10
11
12
13
General procedure X was followed. Benzyl methylcarbamate 314a (300 mg, 1.82 mmol)
gave after flash column chromatography (SiO2, Petrol:EtOAc 2 %) the title compound
316a (207 mg, 43 %) as a yellow solid.
Rf 0.54 (Petrol:EtOAc 8:2); m.p 46-48 °C (Et2O/Petrol); IR max (neat/cm-1
) 2249 (CC),
1722 (C=O); 1H NMR (400MHz, CDCl3) 7.33-7.15 (m, 10H, aryl), 5.16 (s, 2H, H-5),
3.16 (s, 3H, H-7); 13
C NMR (100MHz, CDCl3) 155.2 (C-6), 135.6 (C-4), 130.9, 128.4,
128.2, 128.2, 127.6, 127.4 (CH aryl), 123.1 (C-10), 85.1 (C-8), 83.8 (C-9), 68.4 (C-5), 37.7
(C-7); MS m/z (ES+) 283 (70 %, M+Na+), 266 (20 %, M+H
+); HMRS (ES+) calcd for
C17H16O2N1 (M+H+): 266.1176, found: 266.1172.

348
Benzyl isopropyl-2-phenylethynylcarbamate 316b.
1
23
45
6N O
O
8
910
11
1213
7
14
8
General Procedure Z was followed. Benzyl ethynylisopropylcarbamate 320a (195 mg, 0.90
mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 2 %) the title
compound 316b (222 mg, 84 %) as a colourless oil.
Rf 0.68 (Petrol:EtOAc 9:1); IR max (neat/cm-1
) 2247 (CC), 1716 (C=O); 1H NMR
(500MHz, CDCl3) 7.34-7.14 (m, 10H, aryl), 5.17 (s, 2H, H-5), 4.34 (sep, J = 6.8 Hz, 1H,
H-7), 1.21 (d, J = 6.8 Hz, 6H, H-8); 13
C NMR (125MHz, CDCl3) 154.7 (C-6), 135.8 (C-
4), 130.8, 128.5, 128.2, 128.1, 127.6, 127.2 (CH aryl), 123.5 (C-11), 79.8 (C-9), 73.0 (C-
10), 68.2 (C-5), 49.3 (C-7), 20.3 (C-8); MS m/z (ES+) 316 (100 %, M+Na+); HMRS (ES+)
calcd for C19H20O2N1 (M+H+): 294.1489, found: 294.1482.
1-Phenylethyl isopropyl-2-phenylethynylcarbamate 316c.
15
1
23
45
6
N O
O
9 9
1011
12
13
7
14
8
General Procedure Z was followed. 1-Phenylethyl ethynylisopropylcarbamate 320b (336
mg, 1.45 mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 3 %) the
title compound 316c (304 mg, 68 %) as a colourless oil.
Rf 0.68 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 2245 (CC), 1715 (C=O); 1H NMR
(400MHz, CDCl3) 7.43-7.27 (m, 10H, aryl), 5.88 (q, J = 6.4 Hz, 1H, H-5), 4.40 (sep, J =
6.8 Hz, 1H, H-8), 1.62 (d, J = 6.4 Hz, 3H, H-6), 1.32 (d, J = 6.8 Hz, 3H, H-9), 1.28 (d, J =

349
6.8 Hz, 3H, H-9); 13
C NMR (100MHz, CDCl3) 154.2 (C-7), 141.7 (C-4), 130.6, 128.4,
128.2, 127.8, 127.1, 125.7 (CH aryl), 123.7 (C-12), 80.1 (C-10), 75.1 (C-5), 73.0 (C-11),
49.0 (C-8), 22.9 (C-6), 20.3 (C-9); MS m/z (ES+) 330 (100 %, M+Na+); HMRS (ES+)
calcd for C20H21O2N1Na1 (M+Na+): 330.1465, found: 330.1466.
(2-Bromoethynyl)triisopropylsilane 318. [235]
12
3 44
Si
Br
General procedure W was followed. (Triisopropylsilyl)acetylene 317 (2.0 g, 11 mmol)
gave after flash column chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 318
(2.4 g, 83 %) as a colourless oil.
Rf 0.81 (Petrol); IR max (neat/cm-1
) 2120 (CC); 1H NMR (400MHz, CDCl3) 1.07 (s,
21H, H-3 and H-4); 13
C NMR (100MHz, CDCl3) 83.4 (C-2), 61.7 (C-1), 18.5 (C-4), 11.2
(C-3); MS m/z (ES+) 260 (100 %, M+). Spectral data matched the published values.
[235]
Benzyl isopropyl-2-(triisopropylsilyl)ethynylcarbamate 319a.
1
23
45
6N O
OSi
8
910
1112
12
78
General procedure X was followed. Benzyl methylcarbamate 314b (522 mg, 2.70 mmol)
gave after flash column chromatography (SiO2, Petrol:EtOAc 2 %) the title compound
319a (615 mg, 61 %) as a colourless oil.
Rf 0.71 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 2171 (CC), 1722 (C=O); 1H NMR
(400MHz, CDCl3) 7.41-7.31 (m, 5H, aryl), 5.20 (s, 2H, H-5), 4.34 (sep, J = 6.8 Hz, 1H,

350
H-7), 1.26 (d, J = 6.8 Hz, 6H, H-8), 1.06 (s, 21H, H-11 and H-12); 13
C NMR (100MHz,
CDCl3) 155.0 (C-6), 135.6 (C-4), 128.4, 128.1, 127.9 (CH aryl), 93.4 (C-9), 70.9 (C-10),
68.2 (C-5), 48.7 (C-7), 20.2 (C-8), 18.6 (C-12), 11.4 (C-11); MS m/z (ES+) 374 (100 %,
M+H+); HMRS (ES+) calcd for C22H36O2N1Si1 (M+H
+): 374.2510, found: 374.2495.
1-Phenylethyl isopropyl-2-(triisopropylsilyl)ethynylcarbamate 319b.
1
23
45
6
N O
OSi
9 9
1011
12
13
7
13
8
General procedure X was followed. 1-Phenylethyl isopropylcarbamate 314c (738 mg, 3.56
mmol) gave after flash column chromatography (SiO2, Petrol:EtOAc 1 %) the title
compound 319b (890 mg, 65 %) as a colourless oil.
Rf 0.81 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 2172 (CC), 1721 (C=O); 1H NMR
(400MHz, CDCl3) 7.40-7.27 (m, 5H, aryl), 5.84 (q, J = 6.4 Hz, 1H, H-5), 4.29 (sep, J =
6.8 Hz, 1H, H-8), 1.57 (d, J = 6.4 Hz, 3H, H-6), 1.26 (d, J = 6.8 Hz, 3H, H-9), 1.20 (d, J =
6.8 Hz, 3H, H-9), 1.11 (s, 21H, H-12 and H-13); 13
C NMR (100MHz, CDCl3) 154.5 (C-
7), 141.7 (C-4), 128.4, 127.7, 125.8 (CH aryl), 93.7 (C-10), 74.9 (C-5), 70.7 (C-11), 48.5
(C-8), 23.0 (C-6), 20.2 (C-9), 18.7 (C-13), 11.4 (C-12); MS m/z (ES+) 410 (100 %,
M+Na+); HMRS (ES+) calcd for C23H38O2N1Si1 (M+H
+): 388.2667, found: 388.2673.
Benzyl ethynylisopropylcarbamate 320a.
1
23
45
6N O
O9
8
10
78
H
General procedure Y was followed. Benzyl isopropyl-2-
(triisopropylsilyl)ethynylcarbamate 319a (667 mg, 1.90 mmol) gave after flash column

351
chromatography (SiO2, Petrol:EtOAc 3 %) the title compound 320a (343 mg, 88 %) as a
pale yellow oil.
Rf 0.40 (Petrol:EtOAc 9:1); IR max (neat/cm-1
) 3291 (C-H), 2135 (CC), 1719 (C=O);
1H NMR (500MHz, CDCl3) 7.40-7.31 (m, 5H, aryl), 5.23 (s, 2H, H-5), 4.32 (sep, J = 6.8
Hz, 1H, H-7), 2.85 (s, 1H, H-10), 1.24 (d, J = 6.8 Hz, 6H, H-8); 13
C NMR (125MHz,
CDCl3) 155.0 (C-6), 135.7 (C-4), 128.5, 128.2, 127.8 (CH aryl), 73.1 (C-9), 68.3 (C-5),
61.0 (C-10), 48.9 (C-7), 20.0 (C-8); MS m/z (ES+) 240 (100 %, M+Na+); HMRS (ES+)
calcd for C13H15O2N1Na1 (M+Na+): 240.0995, found: 240.0993.
1-Phenylethyl ethynylisopropylcarbamate 320b.
1
23
45
6
N O
O
9 9
1011
7
8
H
General procedure Y was followed. 1-Phenylethyl isopropyl-2-
(triisopropylsilyl)ethynylcarbamate 319b (890 mg, 2.30 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 2 %) the title compound 320b (436 mg, 82 %) as a
colourless oil.
Rf 0.64 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3297 (C-H), 2138 (CC), 1716 (C=O);
1H NMR (400MHz, CDCl3) 7.40-7.27 (m, 5H, aryl), 5.86 (q, J = 6.4 Hz, 1H, H-5), 4.29
(sep, J = 6.8 Hz, 1H, H-8), 2.87 (s, 1H, H-11), 1.59 (d, J = 6.4 Hz, 3H, H-6), 1.24 (d, J =
6.8 Hz, 3H, H-9), 1.21 (d, J = 6.8 Hz, 3H, H-9); 13
C NMR (100MHz, CDCl3) 154.5 (C-
7), 141.5 (C-4), 128.5, 127.8, 125.7 (CH aryl), 94.8 (C-10), 75.1 (C-5), 60.8 (C-11), 48.6
(C-8), 22.8 (C-6), 20.0 (C-9); MS m/z (ES+) 254 (100 %, M+Na+); HMRS (ES+) calcd for
C14H17O2N1Na1 (M+Na+): 254.1152, found: 254.1154.

352
2-Hydroxy-N-isopropyl-N-(2-(triisopropylsilyl)ethynyl)-2-phenylacetamide 321a.
1
2
34
56
8
91011
12
12
78
N
OH
OSi
General Procedure O was followed with LDA (2.5 equiv.), LiCl and DMPU. Benzyl
isopropyl-2-(triisopropylsilyl)ethynylcarbamate 319a (50 mg, 0.13 mmol) gave after flash
column chromatography (SiO2, Petrol:EtOAc 2 %) the title compound 321a (12 mg, 0.03
mmol, 24 %) as a colourless oil.
Rf 0.57 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 3469 (br, O-H), 2164 (CC), 1680 (C=O);
1H NMR (400MHz, CDCl3) 7.40-7.29 (m, 5H, aryl), 5.73 (d, J = 8.4 Hz, 1H, -OH), 4.69
(sep, J = 6.8 Hz, 1H, H-7), 3.82 (d, J = 8.4 Hz, 1H, H-5), 1.26 (d, J = 6.8 Hz, 3H, H-8),
1.14 (d, J = 6.8 Hz, 3H, H-8), 1.04, 1.00 (m, 21H, H-11, H-12); 13
C NMR (100MHz,
CDCl3) 174.4 (C-6), 138.5 (C-4), 128.6, 128.4, 127.1 (CH aryl), 92.7 (C-9), 76.0 (C-10),
72.3 (C-5), 47.5 (C-7), 19.8, 19.7 (C-8), 18.6 (C-12), 11.2 (C-11); MS m/z (ES+) 396
(100 %, M+Na+), (ES-) 372 (100 %, M-H
+).
2-Hydroxy-N-isopropyl-N-(2-(triisopropylsilyl)ethynyl)-2-phenylpropanamide 321b.
1
2
34
5
6
9 9
101112
13
7
8
N
OH
OSi
13
General Procedure O was followed with LDA (2.5 equiv.), LiCl and DMPU. 1-Phenylethyl
isopropyl-2-(triisopropylsilyl)ethynylcarbamate 319b (50 mg, 0.13 mmol) gave after flash
column chromatography (SiO2, Petrol:EtOAc 1 %) the title compound 321b (10 mg, 0.03
mmol, 20 %) as a colourless oil.

353
Rf 0.78 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 2174 (CC), 1632 (C=O); 1H NMR
(400MHz, CDCl3) 7.54 (d, J = 7.2 Hz, 2H, H-3), 7.39-7.27 (m, 3H, aryl), 4.21 (sep, J =
6.8 Hz, 1H, H-8), 3.41 (s, 1H, -OH), 1.74 (s, 3H, H-6), 1.44 (d, J = 6.8 Hz, 6H, H-9), 1.10-
1.07 (m, 21H, H-12 and H-13); 13
C NMR (100MHz, CDCl3) 172.3 (C-7), 139.4 (C-4),
128.3, 128.0, 124.7 (CH aryl), 92.8 (C-10), 82.3 (C-5), 76.4 (C-11), 45.2 (C-8), 25.0 (C-6),
18.9 (C-9), 18.8 (C-13), 11.7 (C-12); MS m/z (ES+) 388 (100 %, M+H+).
4-Benzyl-3-isopropyl-5-phenyloxazol-2(3H)-one 322.
1
2
345
6
9
10
7
8
14
13 1112
NO
O
8
General Procedure O was followed with LDA (2.5 equiv.), LiCl and DMPU. Benzyl
isopropyl-2-phenylethynylcarbamate 316b (100 mg, 0.26 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 5 %) the title compound 322 (36 mg, 0.12 mmol,
36 %) as a yellow oil.
Rf 0.44 (Petrol:EtOAc 8:2); IR max (neat/cm-1
) 1743 (C=O); 1H NMR (400MHz, CDCl3)
7.41-7.17 (m, 10H, aryl), 3.97 (s, 2H, H-10), 3.70 (sep, J = 6.8 Hz, 1H, H-7), 1.24 (d, J =
6.8 Hz, 6H, H-8); 13
C NMR (100MHz, CDCl3) 153.8 (C-6), 135.9, 135.8, 129.1, 128.8
(aryl), 128.1 (C-5), 127.9, 127.6, 127.3, 125.0 (CH aryl), 120.6 (C-9), 46.5 (C-7), 29.3 (C-
10), 19.5 (C-8); MS m/z (ES+) 316 (100 %, M+Na+).

354
(Z)-4-Benzylidene-3-isopropyl-5-phenyloxazolidin-2-one 323.
1
2
345
69
7
8
NO
O
10
8
14
1311
12
General Procedure O was followed with LDA (2.5 equiv.), LiCl and DMPU. Benzyl
isopropyl-2-phenylethynylcarbamate 316b (100 mg, 0.26 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 5 %) the title compound 323 (6 mg, 0.02 mmol, 6 %)
as a yellow oil.
Rf 0.26 (Petrol:EtOAc 8:2); 1H NMR (400MHz, CDCl3) 7.56-7.25 (m, 10H, aryl), 5.60
(s, 1H, H-10), 5.07 (s, 1H, H-5), 4.32 (sep, J = 6.8 Hz, 1H, H-7), 1.53 (d, J = 6.8 Hz, 3H,
H-8), 1.51 (d, J = 6.8 Hz, 3H, H-8); MS m/z (ES+) 316 (100 %, M+Na+).
The (Z)-stereochemistry was deduced by a nOe experiment: irradiation at 5.60 ppm (CH=C)
did not enhance peak at 4.32 ppm (N-CH).
4-(Hydroxy(phenyl)methyl)-3-isopropyl-5-phenyloxazol-2(3H)-one 324.
1
2
34
5
68
9
10
7
8
14
13
11
12
NO
O
HO
General Procedure O was followed with LDA (2.5 equiv.), LiCl and DMPU. Benzyl
isopropyl-2-phenylethynylcarbamate 316b (100 mg, 0.26 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 10 %) the title compound 324 (21 mg, 0.07 mmol,
20 %) as white solid.

355
Rf 0.15 (Petrol:EtOAc 8:2); m.p. 146-148 °C (MeOH/Et2O); IR max (neat/cm-1
) 3369 (br,
O-H), 1722 (C=O); 1H NMR (400MHz, CDCl3) 7.47-7.32 (m, 10H, aryl), 6.20 (d, J =
3.6 Hz, 1H, H-10), 3.92 (sep, J = 6.8 Hz, 1H, H-7), 2.51 (d, J = 3.6 Hz, 1H, -OH), 1.40 (d,
J = 6.8 Hz, 3H, H-8), 0.94 (d, J = 6.8 Hz, 3H, H-8); 13
C NMR (100MHz, CDCl3) 153.8
(C-6), 139.0, 136.6, 128.8, 128.7, 128.6 (aryl), 127.9 (C-5), 126.3, 125.4 (aryl), 123.8 (C-
9), 65.6 (C-10), 47.7 (C-7), 19.0 (C-8), 18.2 (C-8); MS m/z (ES+) 310 (100 %, M+H+), 332
(50 %, M+Na+); HMRS (ES+) calcd for C19H19O3N1Na1 (M+Na
+): 332.1258, found:
332.1269.
The structure was confirmed by an X-ray crystal structure.
(Z)-2-Benzylidene-3-isopropyl-4-methyl-4-phenyloxazolidin-5-one 325. and (Z)-4-
Benzylidene-3-isopropyl-5-methyl-5-phenyloxazolidin-2-one 238.
1
23
45
6
9
71413
11
12
10
8
11
15
NO
O
7'
8'
15'
ON
O
1'
2'3'
4'5'
6'8'
9'10'
11'
12'
13'14'
General Procedure O was followed with LDA (2.0 equiv.), LiCl and DMPU. 1-Phenylethyl
isopropyl-2-phenylethynylcarbamate 316c (100 mg, 0.34 mmol) gave after flash column
chromatography (SiO2, Petrol:EtOAc 2 %) the title compounds 325 and 238 (16 mg, 16 %).
Rf 0.60 (Petrol:EtOAc 8:2); 1H NMR (500MHz, CDCl3) 7.54-7.03 (m, 20H, aryl), 5.42
(s, 0.4H, H-5’), 4.97 (s, 0.6H, H-5), 4.19 (sep, J = 6.8 Hz, 0.6H, H-10), 3.60 (sep, J = 6.8
Hz, 0.4H, H-7’), 1.87 (s, 1.2H, H-11’), 1.81 (s, 1.8H, H-9), 1.40 (d, J = 6.8 Hz, 3.6H, H-
11), 1.26 (d, J = 6.8 Hz, 1.2H, H-8’), 1.12 (d, J = 6.8 Hz, 1.2H, H-8’); 13
C NMR (125MHz,
CDCl3) 171.5 (C-7), 155.7 (C-9’), 149.0 (C-6), 143.6 (C-6’), 141.8, 139.2, 135.5, 134.9,
128.7, 128.6, 128.5, 128.4, 128.3, 128.2, 127.0, 126.9, 125.1, 124.6 (aryl), 100.8 (C-5’),
84.6, 83.6 (C-8 and C-10’), 82.4 (C-5), 47.6, 45.4 (C-7’ and C-10), 27.3, 25.1 (C-9 and C-
11’), 19.2, 19.0 (C-8’), 18.7 (C-11).
The (Z)-stereochemistry of xx was deduced by a nOe experiment: irradiation at 4.95 ppm
(CH=C) enhanced peak at 4.19 ppm (N-CH) from 3.47 %.

356
Benzyl cyanomethylcarbamate 326.[209]
10
1
2
34
5
6
9
CN O
ON
To a suspension of NaNHCN[210]
(1.13 g, 17.6 mmol) in anhydrous THF (30 cm3) was
added a solution of benzyl chloroformate 312a (1.00 g, 5.88 mmol) in anhydrous THF (15
cm3) dropwise at 0 °C under nitrogen atmosphere. The reaction mixture was stirred at
room temperature for 15 h, quenched with water (20 cm3) and extracted with EtOAc (20
cm3). The aqueous phase was acidified with 1 N HCl until pH 1 and extracted with EtOAc
(2 × 10 cm3). The combined organic phases were dried over MgSO4 and concentrated
under reduced pressure. The crude residue (0.96 g, 5.47 mmol) was dissolved in anhydrous
DMF (27 cm3) and NaH (60 % in mineral oil, 0.44 g, 10.9 mmol) was added slowly at 0°C
under nitrogen atmosphere. The reaction mixture was stirred at 0 °C for 30 min and methyl
iodide (0.85 cm3, 13.7 mmol) was added. The reaction was stirred at 16 °C for 21 h. Water
was added and the reaction mixture was extracted with Et2O (30 cm3). The organic layer
was then washed with water (3 × 20 cm3), dried over MgSO4, filtered and concentrated
under reduced pressure. The crude product was purified by flash column chromatography
(SiO2, Petrol: EtOAc 5 % then 7 %) to give the title compound 326 (0.55 g, 50 % over 2
steps) as a white solid.
Rf 0.32 (Petrol:EtOAc 8:2); m.p. 38-40 °C (Et2O/Petrol); IR max (neat/cm-1
) 2245 (CN),
1748 (C=O); 1H NMR (400MHz, CDCl3) 7.40-7.36 (m, 5H, aryl), 5.28 (s, 2H, H-5),
3.22 (s, 3H, H-9); 13
C NMR (100MHz, CDCl3) 152.6 (C-6), 134.1 (C-4), 128.9, 128.7,
128.4 (CH aryl), 109.5 (C-10), 70.0 (C-5), 35.4 (C-9); MS m/z (ES+) 213 (100 %, M+Na+);
HMRS (ES+) calcd for C10H10O2N2Na1 (M+Na+): 213.0635, found: 213.0634.

357
2-Bromobenzo[d]thiazole 327. [236]
75
6
23
41
S
NBr
By the modified method reported by Dehaen et al.[212]
:
To a solution of 2-amino-benzothiazole 326 (250 mg, 1.67 mmol) and copper(II) bromide
(575 mg, 2.50 mmol) in anhydrous MeCN (15 cm3) was added tert-butyl nitrite (0.30 cm
3,
2.50 mmol). The mixture was stirred at 80 °C in a sealed tube for 18 h. After cooling to
room temperature, the mixture was taken up in EtOAc (15 cm3) and washed with a 3 N
aqueous HCl solution (15 cm3), a saturated aqueous solution of NaHCO3 (15 cm
3) and
brine (15 cm3). The organic layer was dried over MgSO4, filtered and concentrated under
reduced pressure. The residue was purified by flash column chromatography (SiO2,
Petrol:EtOAc 1 %) to give the title compound 327 (310 mg, 87 %) as a pale yellow solid.
Rf 0.66 (Petrol:EtOAc 8:2); m.p. 44-46 °C (EtOH); 1H NMR (400MHz, CDCl3) 7.99 (d,
J = 7.8 Hz, 1H, H-6), 7.80 (d, J = 7.8 Hz, 1H, H-3), 7.49-7.39 (m, 2H, H-4 and H-5); 13
C
NMR (100MHz, CDCl3) 152.3 (C-2), 138.9 (C-1), 137.3 (C-7), 126.6 (C-3), 125.7 (C-6),
122.8, 120.9 (C-4 and C-5); MS m/z (ES+) 214 (100 %, M+H+); HMRS (ES+) calcd for
C7H5N2S280
Br1 (M+H+): 213.9321, found: 213.9324. Spectra data matched the published
values.[236]
Benzyl benzo[d]thiazol-2-ylmethylcarbamate 328.
13 145
6
7
8
9
1011
12
34
2
1
ON
ON
S
By the method reported by Clayden et al.:[64]
O-Benzylcarbamate 314a (258 mg, 1.56 mmol), 2-bromobenzo[d]thiazole 327 (516 mg,
2.34 mmol), sodium tert-butoxide (300 mg, 3.12 mmol), 4,5-bis(diphenylphosphino)-9,9-

358
dimethylxanthenene (XantPhos) (93 mg, 0.156 mmol, 10 mol %) and tris-
(benzylideneacetone)dipalladium(0) (37 mg, 0.039 mmol, 2.5 mol %) were dissolved in
toluene (18 cm3) in a sealed tube. The solution was degassed for 30 min and heated under
reflux (110 °C) for 16 h under nitogen atmosphere. The mixture was cooled to room
temperature and a saturated aqueous solution of NH4Cl (10 cm3) was added. The mixture
was extracted with Et2O (2 10 cm3), the combined organic phases were washed with
brine and dried over MgSO4, filtered and concentrated under reduced pressure. The residue
was purified by flash column chromatography (SiO2, Petrol:EtOAc 5 %) to give the title
compound 328 (157 mg, 34 %) as a white solid.
Rf 0.72 (Petrol:EtOAc 7:3); m.p. 146-148 °C (Et2O/Petrol); IR max (neat/cm-1
) 1713
(C=O); 1H NMR (400MHz, CDCl3) 7.86 (d, J = 7.8 Hz, 1H, H-13), 7.78 (d, J = 7.8 Hz,
1H, H-10), 7.47-7.27 (m, 7H, aryl), 5.36 (s, 2H, H-5), 3.73 (s, 3H, H-7); 13
C NMR
(100MHz, CDCl3) 161.2 (C-8), 150.0 (C-9), 149.1 (C-6), 135.0 (C-4), 132.1 (C-14),
128.7, 128.4, 126.3, 125.8, 123.3, 121.1, 120.9 (CH aryl), 68.9 (C-5), 38.9 (C-7); MS m/z
(ES+) 321 (100 %, M+Na+); HMRS (ES+) calcd for C16H15O2N2S1 (M+H
+): 299.0849,
found: 299.0835.
N-Isopropylbenzo[d]thiazol-2-amine 329.
56
7
89
1
34 2
1
S
NNH
Rf 0.27 (Petrol:EtOAc 8:2); 1
H NMR (400MHz, CDCl3) 7.47 (dd, J = 7.8 and 0.6 Hz, 1H,
H-8), 7.42 (dd, J = 7.8 and 0.6 Hz, 1H, H-5), 7.18 (t, J = 7.8 Hz, 1H, H-6), 6.96 (t, J = 7.8
Hz, H-7), 5.31 (br, 1H, -NH), 3.81 (br, 1H, H-2), 1.21 (d, J = 6.8 Hz, 6H, H-1) ; 13
C NMR
(100MHz, CDCl3) 166.6 (C-3), 152.5 (C-4), 130.4 (C-9), 125.9, 121.4, 120.8, 118.8 (CH
aryl), 47.6 (C-2), 23.0 (C-1).

359
N-(Benzo[d]thiazol-2-yl)-N-isopropylbenzo[d]thiazol-2-amine 330.
N
S
N N
S
5
6
78
9
1
34
21
Rf 0.78 (Petrol:EtOAc 8:2); 1
H NMR (300MHz, CDCl3) 7.77 (d, J = 7.8 Hz, 2H, H-8),
7.70 (d, J = 7.8 Hz, 2H, H-5), 7.35 (t, J = 7.8 Hz, 2H, H-6), 7.20 (t, J = 7.8 Hz, 2H, H-7),
5.01 (sep, J = 6.8 Hz, 1H, H-2), 1.68 (d, J = 6.8 Hz, 6H, H-1); 13
C NMR (100MHz, CDCl3)
162 (C-3), 150.0 (C-4), 132.3 (C-9), 129.0, 126.1, 123.5, 121.1 (CH aryl), 57.2 (C-2),
20.0 (C-1) ; MS m/z (ES+) 348 (100 %, M+Na+).
III.4 X-Ray Crystal Data

360
III.4.1 X-Ray Crystal Data and Structure Refinement for 238a.
ON
O
Identification code s3385m
Empirical formula C18 H17 N O2
Formula weight 279.33
Temperature 100(2) K
Wavelength 0.71073 A
Crystal system, space group Monoclinic, P2(1) /c
Unit cell dimensions a = 6.2019(5) A alpha = 90 deg.
b = 30.774(3) A beta = 92.856(2) deg.
c = 7.4657(6) A gamma = 90 deg.
Volume 1423.1 (2) A^3
Z, Calculated density 4, 1.304 Mg/m^3
Absorption coefficient 0.085 mm^-1
F(000) 592
Crystal size 0.30 x 0.25 x 0.20 mm
Theta range for data collection 1.32 to 28.28 deg.
Limiting indices -8<=h<=7, -40<=k<=40, -9<=1<=9
Reflections collected / unique 12137 / 3372 [R(int) = 0.0396]
Completeness to theta = 25.03 100.0 %
Absorption correction None
Max. and min. transmission 0.9832 and 0.9750
Refinement method Full-matrix least-squares on F^2
Data / restraints / parameters 3372 / 0 / 192
Goodness-of-fit in F^2 1.048
Final R indices [I>2sigma(I)] R1 = 0.0502, wR2 = 0.1097
R indices (all data) R1 = 0.0674, wR2 = 0.1264
Largest diff. peak and hole 0.37 and -0.226 e.A^-3

361
III.4.2 X-Ray Crystal Data and Structure Refinement for 248j.
N O
O
Identification code s3378m
Empirical formula C24 H23 N O2
Formula weight 357.43
Temperature 180(2) K
Wavelength 0.71073 A
Crystal system, space group Triclinic, P-1
Unit cell dimensions a = 9.9252(18) A alpha = 67.709(3) deg.
b = 10.0516(18) A beta = 69.292(3) deg.
c = 11.659(2) A gamma = 66.289(3) deg.
Volume 957.3 (3) A^3
Z, Calculated density 2, 1.240 Mg/m^3
Absorption coefficient 0.078 mm^-1
F(000) 380
Crystal size 0.30 x 0.25 x 0.10 mm
Theta range for data collection 1.94 to 28.34 deg.
Limiting indices -12<=h<=13, -12<=k<=12, -15<=1<=14
Reflections collected / unique 8288 / 4346 [R(int) = 0.0497]
Completeness to theta = 25.03 99.0 %
Absorption correction None
Max. and min. transmission 0.9922 and 0.9769
Refinement method Full-matrix least-squares on F^2
Data / restraints / parameters 4346 / 0 / 246
Goodness-of-fit in F^2 0.883
Final R indices [I>2sigma(I)] R1 = 0.0530, wR2 = 0.0721
R indices (all data) R1 = 0.1111, wR2 = 0.0853
Largest diff. peak and hole 0.142 and -0.163 e.A^-3

362
III.4.3 X-Ray Crystal Data and Structure Refinement for 271j.
N O
O
Identification code s3418
Empirical formula C28 H33 N O2
Formula weight 415.55
Temperature 100(2) K
Wavelength 0.71073 A
Crystal system, space group Monoclinic, P2(1) /n
Unit cell dimensions a = 9.807(4) A alpha = 90 deg.
b = 23.295(10) A beta = 90.032(9) deg.
c = 21.312(9) A gamma = 90 deg.
Volume 4869 (4) A^3
Z, Calculated density 8, 1.134 Mg/m^3
Absorption coefficient 0.070 mm^-1
F(000) 1792
Crystal size 0.60 x 0.20 x 0.20 mm
Theta range for data collection 2.10 to 25.03 deg.
Limiting indices -10<=h<=11, -16<=k<=27, -23<=1<=25
Reflections collected / unique 23859 / 8592 [R(int) = 0.1512]
Completeness to theta = 25.03 99.9 %
Absorption correction None
Refinement method Full-matrix least-squares on F^2
Data / restraints / parameters 8592 / 0 / 565
Goodness-of-fit in F^2 0.905
Final R indices [I>2sigma(I)] R1 = 0.0806, wR2 = 0.1116
R indices (all data) R1 = 0.2003, wR2 = 0.1473
Largest diff. peak and hole 0.237 and -0.241 e.A^-3

363
III.4.4 X-Ray Crystal Data and Structure Refinement for 324.
NO
O
HO
Identification code s3628m
Empirical formula C19 H19 N O3
Formula weight 309.35
Temperature 100(2) K
Wavelength 0.71073 A
Crystal system, space group Monoclinic, P2(1) /c
Unit cell dimensions a = 13.3028(10) A alpha = 90 deg.
b = 8.5645(7) A beta = 97.5880(10) deg.
c = 14.3329(11) A gamma = 90 deg.
Volume 1618.7 (2) A^3
Z, Calculated density 4, 1.269 Mg/m^3
Absorption coefficient 0.086 mm^-1
F(000) 656
Crystal size 0.40 x 0.35 x 0.33 mm
Theta range for data collection 2.78 to 28.34 deg.
Limiting indices -17<=h<=17, -11<=k<=11, -18<=1<=18
Reflections collected / unique 13525 / 3839 [R(int) = 0.0475]
Completeness to theta = 25.03 99.9 %
Absorption correction None
Max. and min. transmission 0.9722 and 0.9665
Refinement method Full-matrix least-squares on F^2
Data / restraints / parameters 3839 / 0 / 211
Goodness-of-fit in F^2 1.123
Final R indices [I>2sigma(I)] R1 = 0.0523, wR2 = 0.1145
R indices (all data) R1 = 0.0586, wR2 = 0.1181
Largest diff. peak and hole 0.329 and -0.232 e.A^-3

364
REFERENCES & NOTES
1. See http://cbc.arizona.edu/njardarson/group/top-pharmaceuticals-poster.
2. Schmidt, F.; Stemmler, R. T.; Rudolph, J.; Bolm, C. Chem. Soc. Rev. 2006, 35, 454.
3. For reviews, see: (a) Kanai, M.; Kato, N.; Ichikawa, E.; Shibasaki, M. Synlett 2005,
1491. (b) Yamamoto, H.; Futatsugi, K. Angew. Chem. Int. Ed. 2005, 44, 1924.
4. (a) Inch, T. D.; Lewis, G. J.; Sainsbury, G. L.; Sellers, D. J. Tetrahedron Lett. 1969,
41, 3657. (b) Nozaki, H.; Aratani, T.; Toraya, T.; Noyori, R. Tetrahedron 1971, 27,
905. (c) Noyori, R.; Suga, S.; Kawai, K.; Okada, S.; Kitamura, M. Pure Appl. Chem.
1988, 60, 1597. (d) Weber, B.; Seebach, D. Angew. Chem. Int. Ed. Engl. 1992, 31,
84. (e) Weber, B.; Seebach, D. Tetrahedron 1994, 50, 6117. (f) For a review, see:
Luderer, M. R.; Bailey, W. F.; Luderer, M. R.; Fair, J. D.; Dancer, R. J.; Sommer,
M. B. Tetrahedron Asymmetric 2009, 20, 981.
5. Shibasaki, M.; Kanai, M. Chem. Rev. 2008, 108, 2853.
6. Dosa, P. I.; Fu, G. J. Am. Chem. Soc. 1998, 120, 445.
7. Noyori, R.; Kitamura, M. Angew. Chem. Int. Ed. Engl. 1991, 30, 49.
8. (a) Ramón, D. J.; Yus, M. Tetrahedron Lett. 1998, 39, 1239. (b) Ramón, D. J.; Yus,
M. Tetrahedron 1998, 54, 5651.
9. (a) Garcia, C.; Larochelle, L. K.; Walsh, P. J. J. Am. Chem. Soc. 2002, 124, 10970.
(b) For a review, see: Betancort, J. M.; Garcia, C.; Walsh, P. J. Synlett 2004, 749.
10. (a) Yus, M.; Ramón, D. J.; Prieto, O. Tetrahedron Asymmetric 2002, 13, 2291. (b)
Ramón, D. J.; Yus, M. Angew. Chem. Int. Ed. 2004, 43, 284.
11. (a) Forrat, V. J.; Ramón, D. J.; Yus, M. Tetrahedron Asymmetric 2005, 16, 3341. (b)
Forrat, J. V.; Prieto, O.; Ramón, D. J.; Yus, M. Chem. Eur. J. 2006, 12, 4431.
12. Garcia, C.; Walsh, P. J. Org. Lett. 2003, 5, 3641.
13. (a) Zhuang, W.; Gathergood, N.; Hazell, R. G.; Jørgensen, K. A. J. Org. Chem.
2001, 66, 1009. (b) Corma. A.; Garcia, H.; Moussaif, A.; Sabatier, M. J.; Zniber, R.;
Redouane, A. Chem. Commun. 2002, 1058.
14. Prieto, O.; Ramón, D. J.; Yus, M. Tetrahedron Asymmetric 2003, 14, 1955.
15. (a) Jeon, S.-J.; Walsh, P. J. J. Am. Chem. Soc. 2003, 125, 9544. (b) Garcia, C.;
Libra, E. R.; Carroll, P. J.; Walsh, P. J. J. Am. Chem. Soc. 2003, 125, 3210. (c)
Anaya de Parrodi, C.; Walsh, P. J. Synlett 2004, 2417. (d) Li, H.; Walsh, P. J. J. Am.
Chem. Soc. 2004, 126, 6538. (e) Li, H.; Walsh, P. J. J. Am. Chem. Soc. 2005, 127,

365
8356. (f) Jeon, S.-J.; Li, H.; Garcia, C.; LaRochelle, L. K.; Walsh, P. J. J. Org.
Chem. 2005, 70, 448. (g) Jeon, S.-J.; Li, H.; Garcia, L. K.; Walsh, P. J. J. Am. Chem.
Soc. 2005, 127, 16416.
16. Hatano, M.; Miyamoto, T.; Ishihara, K. Org. Lett. 2007, 9, 4535.
17. Li, H.; Walsh, P. J. J. Am. Chem. Soc. 2004, 126, 6538.
18. (a) Wipf, P.; Ribe, S. J. Org. Chem. 1998, 63, 6454. (b) Wipf, P.; Kendall, C. Chem.
Eur. J. 2002, 8, 1778. (c) Wipf. P.; Nunes, R. L. Tetrahedron 2004, 60, 1269.
19. Some selected examples: CeCl3: (a) Imamoto, Y.; Takiyama, N.; Nakamura, K.;
Hatajima, T.; Kamiya, Y. J. Am. Chem. Soc. 1989, 111, 4392. LiCl: (b) Krasovskiy,
A.; Knochel, P. Angew. Chem. Int. Ed. 2004, 43, 3333. FeCl3: (c) Fürstner, A.;
Krause, H.; Lehmann, C. W. Angew. Chem. Int. Ed. 2006, 45, 440. LaCl3: (d)
Metzger, A.; Gavryushin, A.; Knochel, P. Synlett 2009, 1433. ZnCl2: (e) Hatano,
M.; Ito, O.; Suzuki, S.; Ishihara, K. J. Org. Chem. 2010, 15, 5008.
20. Madduri, A. V. R.; Harutyunyan, S. R.; Minnaard, A. J. Angew. Chem. Int. Ed.
2012, DOI:10.1002/anie.201109040.
21. (a) Zhuang, W.; Gathergood, N.; Hazell, R. G.; Jørgensen, K. A. J. Org. Chem.
2001, 66, 1009. (b) Lyle, M. P. A.; Draper, N. D.; Wilson, P. D. Org. Lett. 2005, 7,
901. (c) Tomita, D.; Wada, R.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2005,
127, 4138. (d) Zhao, J.-L.; Liu, L.; Sui, Y.; Liu, Y.-L.; Wang, D.; Chen, Y.-J. Org.
Lett. 2006, 8, 6127. (e) Motoki, R.; Tomita, D.; Kanai, M.; Shibasaki, M.
Tetrahedron Lett. 2006, 47, 8083.
22. Chen, C.-A.; Wu, K.-H.; Gau, H.-M. Angew. Chem. Int. Ed. 2007, 46, 5373.
23. Chen, C.-A.; Wu, K.-H.; Gau, H.-M. Adv. Synth. Catal. 2008, 350, 1626.
24. Zhou, S.; Wu, K.-H.; Chen, C.-A.; Gau, H.-M. J. Org. Chem. 2009, 74, 3500.
25. Zhu, T.-S.; Jin, S.-S.; Xu, M.-H. Angew. Chem. Int. Ed. 2011, 50, 780.
26. Denmark, S. E.; Fu, J. Chem. Rev. 2003, 103, 2763.
27. Hanawa, H.; Kii, S.; Maruoka, K. Adv. Synth. Catal. 2001, 343, 57.
28. Cunningham, A.; Woodward, S. Synthesis 2002, 43.
29. (a) Casolari, S.; D’Addiaro, D.; Tagliavini, E. Org. Lett. 1999, 1, 1061. (b) Waltz,
K. M.; Gavenonis, J.; Walsh, P. J. Angew. Chem. Int. Ed., 2002, 41, 3697. (c) Kim,
J. G.; Waltz, K. M.; Garcia, I. F.; Kwiatkowski, D.; Walsh, P. J. J. Am. Chem. Soc.
2004, 126, 12580. (d) Wooten, A. J.; Kim, J. G.; Walsh, P. J. Org. Lett. 2007, 9,
381.
30. Teo, Y.-C.; Goh, J.-D.; Loh, T.-P. Org. Lett. 2005, 7, 2743.

366
31. Wada, R.; Oisaki, K.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2004, 126, 8910.
32. (a) Hosomi, A.; Shirahata, A.; Sakurai, H. Tetrahedron Lett. 1978, 33, 3043. (b)
Wadamoto, M.; Yamamoto, H. J. Am. Chem. Soc. 2005, 127, 14556.
33. Shi, S.-L.; Xu, L.-W.; Oisaki, K.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2010,
132, 6638.
34. Barnett, D. S.; Moquist, P. N.; Schaus, S. E. Angew. Chem. Int. Ed. 2009, 48, 8679.
35. Cozzi, P. G. Angew. Chem. Int. Ed. 2003, 42, 2895.
36. Lu, G.; Li, X.; Jia, X.; Chan, W. L.; Chan, A. S. C. Angew. Chem. Int. Ed. 2003, 42,
5057.
37. (a) Krause, N.; Seebach, D. Chem. Rev. 1987, 120, 1845. (b) Cozzi, P. G.; Alesi, S.
Chem. Commun. 2004, 2448.
38. Chen, C.; Hong, L.; Xu, Z.-Q.; Liu, L.; Wang, R. Org. Lett. 2006, 8, 2277.
39. Antczak, M. I.; Cai, F.; Ready, J. M. Org. Lett. 2011, 13, 184.
40. For some selected references, see: (a) Senanayake, C. H.; Fang, K.; Grover, P.;
Bakale, R. P.; Vandenbossche, C. P.; Wald, S. A. Tetrahedron Lett. 1999, 40, 819.
(b) Grover, P. T.; Bhongle, N. N.; Wald, S. A.; Senanayake, C. H. J. Org. Chem.
2000, 65, 6283. (c) Masumoto, S.; Suzuki, M.; Kanai, M.; Shibasaki, M.
Tetrahedron Lett. 2002, 43, 8647. (d) Gupta, P.; Fernandes, R. A.; Kumar, P.
Tetrahedron Lett. 2003, 44, 4231. (e) Masumoto, S.; Suzuki, M.; Kanai, M.;
Shibasaki, M. Tetrahedron 2004, 60, 10497.
41. (a) Evans, D. A.; Burgey, C. S.; Kozlowski, M. C.; Tregay, S. W. J. Am. Chem. Soc.
1999, 121, 686. (b) Evans, D. A.; Johnson, J. Accounts Chem. Res. 2000, 33, 325.
42. Le, J. C.-D.; Pagenkopf, B. L. Org. Lett. 2004, 6, 4097.
43. (a) Langner, M.; Bolm, C. Angew. Chem. Int. Ed. 2004, 43, 5984. (b) Langner, M.;
Rémy, P.; Bolm, C. Chem. Eur. J. 2005, 11, 6254. (c) Rémy, P.; Langmer, M.;
Bolm, C. Org. Lett. 2006, 6, 1209. (d) Sedelmeier, J.; Hammerer, T.; Bolm, C. Org.
Lett. 2008, 10, 917.
44. Akullian, L. C.; Snapper, M. L.; Hoveyda, A. H. J. Am. Chem. Soc. 2006, 128,
6532.
45. (a) Denmark, S. E.; Fan, Y. J. Am. Chem. Soc. 2002, 124, 4233. (b) Denmark, S. E.;
Fan, Y.; Eastgate, M. D. J. Org. Chem. 2005, 70, 5235.
46. Oisaki, K.; Zhao, D.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2006, 128, 7164.
47. Deschamp, J.; Chuzel, O.; Hannedouche, J.; Riant, O. Angew. Chem. Int. Ed. 2006,
45, 1292.

367
48. (a) Zhao, D.; Oisaki, K.; Kanai, M.; Shibasaki, M. Tetrahedron Lett. 2006, 47, 1403.
(b) Zhao, D.; Oisaki, K.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2006, 128,
14440. (c) Oisaki, K.; Zhao, D.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2007,
129, 7439.
49. (a) Vedejs, E.; Jure, M. Angew. Chem. Int. Ed. 2005, 44, 3974. (b) Robinson, D. E.
J.; Bull, S. D. Tetrahedron Asymmetric 2003, 14, 1407.
50. (a) Angione, M. C.; Miller, S. J. Tetrahedron 2006, 62, 5254. (b) Zhao, Y.; Mitra,
A. W.; Hoveyda, A. H.; Snapper, M. L. Angew. Chem. Int. Ed. 2007, 46, 8471. (c)
Karatas, B.; Rendler, S.; Fröhlich, R.; Oestreich, M. Org. Biomol. Chem. 2008, 6,
1435. (d) Shintani, R.; Takatsu, T.; Hayashi, T. Org. Lett. 2008, 10, 1191. (e) Hara,
K.; Tosaki, S.; Gnanadesikan, V.; Morimoto, S.; Harada, M.; Sugita, M.;
Yamagiwa, N.; Matsunaga, S.; Shibasaki, M. Tetrahedron 2009, 65, 5030.
51. Schipper, D. J.; Rousseaux, S.; Fagnou, K. Angew. Chem. Int. Ed. 2009, 48, 8343.
52. Stymiest, J. L.; Bagutski, V.; French, R. M.; Aggarwal, V. K. Nature (London)
2008, 456, 778.
53. Bagutski, V.; French, R. M.; Aggarwal, V. K. Angew. Chem. Int. Ed. 2010, 49,
5142.
54. Scott, H. K.; Aggarwal, V. K. Chem. Eur. J. 2011, 17, 13124.
55. Clayden, J.; Farnaby, W.; Grainger, D. M.; Hennecke, U.; Mancinelli, M.; Tetlow,
D. J.; Hillier, I. H.; Vincent, M. A. J. Am. Chem. Soc. 2009, 131, 3410.
56. Clayden, J.; Dufour, J.; Grainger, D. M.; Helliwell, M. J. Am. Chem. Soc. 2007, 129,
7488.
57. MacLellan, P.; Clayden, J. Chem. Commun. 2011, 3395.
58. Clayden, J.; Dufour, J. Tetrahedron Lett. 2006, 47, 6945.
59. Snape, T. J. Chem. Soc. Rev. 2008, 37, 2452.
60. (a) Eisch, J. J.; Kovacs, C. A. J. Organomet. Chem. 1971, 30, C97. (b) Dannecker,
W.; Fariborz, M. Z. Naturforsch. 1974, 29b, 578. (c) Eisch, J.; Dua, S. K.; Kovacs,
C. A. J. Org. Chem. 1987, 52, 4437.
61. DMPU has been shown previously to accelerate cyclisations, see: Clayden, J.;
Knowles, F. E.; Menet, C. J. Synlett 2003, 1701.
62. For a study of the mechanism of the arylation, see: Grainger, D. M.; Campbell
Smith, A.; Vincent, M. A.; Hillier, I. H.; Wheatley, A. E. H.; Clayden, J. Eur. J.
Org. Chem. 2012, 731.

368
63. In general, benzyllithiums react stereospecifically, but with the sense of retention or
inversion being strongly dependent on conditions and electrophile. For a discussion,
see: (a) Clayden, J. Organolithiums: Selectivity for Synthesis; Pergamon: Oxford,
2002, pp. 241-258. (b) ref 52 (c) Scott, H. K.; Aggarwal, V. K. Chem. Eur. J. 2011,
17, 13124. (d) Sonawane, R. P.; Jheengut, V.; Rabalakos, C.; Larouche-Gauthier,
R.; Scott, H. K.; Aggarwal, V. K.; Angew. Chem. Int. Ed. 2011, 50, 3760. (e)
Gawley, R. E. Tetrahedron Lett. 1999, 40, 4297. (f) Gawley, R. E.; Low, E.; Zhang,
Q.; Harris, R. J. Am. Chem. Soc. 2000, 122, 3344.
64. Clayden, J.; Hennecke, U. Org. Lett. 2008, 10, 3567.
65. Basch, R.; Clayden, J.; Hennecke, U. Synlett 2009, 421.
66. Tetlow, D. J.; Hennecke, U.; Raftery, J.; Waring, M. J.; Clarke, D. S.; Clayden, J.
Org. Lett. 2010, 12, 5442.
67. (a) Kellogg, M.; Strijtveen, B. Tetrahedron 1987, 43, 5039. (b) McFadden, J. M.;
Frehywot, G. L.; Townsend, C. A. Org. Lett. 2002, 4, 3859. (c) Palomono, C.;
Oiarbide, M.; Dias, F.; Lopez, R.; Linden, A. Angew. Chem. Int. Ed. 2004, 43, 3307.
(d) Mukaiyama, Y.; Ikegai, K.; Pluempanupat, W. Chem. Lett. 2005, 34, 638. (e)
Palomono, C.; Oiarbide, M.; Dias, F.; Lopez, R.; Gonzalez, P. B.; Gomez-Bengoa,
E.; Saa, J. M.; Linden, A. J. Am. Chem. Soc. 2006, 128, 15236. (f) Mukaiyama, Y.;
Ikegai, K.; Pluempanupat, W. Bull. Chem. Soc. Jpn. 2006, 79, 780.
68. Hoppe, D.; Marr, F.; Brüggermann, M. Topics Organomet. Chem. 2003, 5, 61.
69. (a) Garst, J. F.; Smith, C. D. J. Am. Chem. Soc., 1976, 98, 1526. (b) Hodgson, D.
M.; Tomooka, K.; Gras, E. Topics Organomet. Chem. 2003, 5, 217.
70. (a) Meyers, A. I.; Adickers, H. W. Tetrahedron Lett. 1969, 5151. (b) Schdllkopf, U.;
Hoppe, I. Angew. Chem. Int. Ed. Engl. 1975, 14, 765. (c) Barber, G. N.; Olofson, R.
A. Tetrahedron Lett. 1976, 3783.
71. (a) Rondan, N. G.; Houk, K. N.; Beak, P.; Zajdel, W. J.; Chandrasekhar, J.;
Scheleyer, P. R. J. Org. Chem. 1981, 46, 4108. (b) Beak, P.; Meyers, A. I. Accounts
Chem. Res. 1986, 19, 356. (c) Hay, D. R.; Song, Z.; Smith, S. G.; Beak, P. J. Am.
Chem. Soc. 1988, 110, 8145.
72. For a review, see: Hoppe, D.; Krämer, T.; Schwark, J.-R.; Zschage, O. Pure Appl.
Chem. 1990, 62, 1999.
73. Graa, P.; Paleo, M. R.; Sardina; F. J. J. Am. Chem. Soc. 2002, 124, 12511.
74. Beak, P.; Reitz, D. B. Chem. Rev. 1978, 78, 275.

369
75. (a) Zhang, P.; Gawley, R. E. J. Org. Chem. 1993, 58, 3223. (b) Superchi, S.;
Sotomayo, N.; Miao, G.; Babu, J.; Campbell, M. G.; Snieckus, V. Tetrahedron Lett.
1996, 37, 6061. (c) Tomooka, K.; Shimizu, H.; Inoue, T.; Shibata, H.; Nakai, T.
Chem. Lett. 1999, 759. (d) Slana, G. B. C.; de Azevedo, M. S.; Lopes, R. S. C.;
Lopes, C. C.; Cardoso, J. N. Beilstein J. Org. Chem. 2002, 2, 1.
76. (a) Hoppe, D.; Carsten, A.; Krämer, T. Angew. Chem. Int. Ed. Engl. 1990, 29, 1414.
(b) Carsten, A.; Hoppe, D. Tetrahedron 1994, 50, 6097. (c) Aggarwal, V. K. Angew.
Chem. Int. Ed. Engl. 1994, 33, 175. (d) Kaiser, B.; Hoppe, D. Angew. Chem. Int. Ed.
Engl. 1995, 34, 323. (e) Hammerschmicht, F.; Hanninger, A. Chem. Ber. 1995, 128,
1069. (f) Derwing, C.; Hoppe, D. Synthesis 1996, 149. (g) Hammerschmidt, F.;
Hanniger, A.; Völlenkle, H. Chem Eur. J. 1997, 3, 1728. (h) Hammerschmidt, F.;
Hanniger, A.; Simov, B. P.; Völlenkle, H.; Werner, A. Eur. J. Org. Chem. 1999,
3511. (i) Derwing, C.; Frank, H.; Hoppe, D. Eur. J. Org. Chem. 1999, 3519.
77. Contrary to alkylic carbanion, the mesomerically stabilized benzylic carbanion is
partially flattened, see: Lange, H.; Huenerbein, R.; Fröhlich, R.; Grimme, S.; Hoppe,
D. Chem. Asian J. 2008, 3, 500.
78. For X-ray structure analyses of monomeric benzyllithium-solvent adducts, see:
Zarges, W; Marsch, M.; Harms, K.; Boche, G. Chem. Ber. 1989, 122, 2303.
79. Strohmann, C.; Abele, B. C.; Lehmen, K.; Schildbach, D. Angew. Chem. Int. Ed.
2005, 44, 3136.
80. Binanzer, M.; Fang, G. Y.; Aggarwal, V. K. Angew. Chem. Int. Ed. 2010, 49, 4264.
81. Ikemoto, H.; Sasaki, M.; Kawahata, M.; Yamaguchi, K.; Takeda, K. Eur. J. Org.
Chem. 2011, 6553.
82. (a) Hoffmann, R. W.; Lanz, J.; Metternich, R.; Tarara, G.; Hoppe, D. Angew. Chem.
Int. Ed. Engl. 1987, 26, 1145. (b) Hirsch, R.; Hoffmann, R. W. Chem. Ber. 1992,
125, 975.
83. Hoffmann, R. W.; Rühl, T.; Harbach, J. Liebigs Ann. Chem. 1992, 725.
84. The alkaloid is readily available and can be isolated in significant quantities from
several species of papilionaceous plants: Couch, J. F. J. Am. Chem. Soc. 1936, 58,
1296. Although it barely misses the C2-symmetry it is ideally suited for acting as a
bidentate ligand for aminophilic cations.
85. Lange, H.; Huenerbein, R.; Fröhlich, R.; Grimme, S.; Hoppe, D. Chem. Asian J.
2008, 3, 78.

370
86. Lange, H.; Huenerbein, R.; Wibbeling, B.; Fröhlich, R.; Grimme, S.; Hoppe, D.
Synthesis 2008, 2905.
87. Schmidt, F.; Stemmler, R. T.; Rudolph, J.; Bolm, C. Chem. Soc. Rev. 2006, 35, 454.
88. Nelson, W. L., In Williams, D. A., Thomas, L. L. Foye’s Principles of Medicinal
Chemistry; Lippincott Williams & Wilkins: Philadelphia, 2002, pp. 794-818.
89. For a review, see: Ahlbrecht, H.; Beyer, U. Synthesis 1999, 365.
90. Zschage, O.; Schwark, J.-R.; Krämer, T.; Hoppe, D. Tetrahedron 1992, 48, 8377.
91. (a) Krämer, T.; Hoppe, D. Tetrahedron Lett. 1987, 28, 5149. (b) Zschage, O.;
Schwark, J.-R.; Hoppe, D. Angew. Chem. Int. Ed. Engl. 1990, 29, 296.
92. Hoppe, D.; Zschage, O. Angew. Chem. Int. Ed. Engl. 1989, 28, 69.
93. Zschage, O.; Hoppe, D. Tetrahedron 1992, 48, 8389.
94. (a) Zschage, O.; Hoppe, D. Tetrahedron 1992, 48, 5657. (b) Paulsen, H.; Graeve, C.;
Hoppe, D. Synthesis 1996, 141.
95. Behrens, K.; Fröhlich, R.; Meyer, O.; Hoppe, D. Eur. J. Org. Chem. 1998, 2397.
96. For an X-ray crystal structure analysis of the γ-silyl derivative, see: Marsch, M.;
Harms, K.; Zschage, O.; Hoppe, D.; Boche, G. Angew. Chem. Int. Ed. Engl. 1991,
30, 321.
97. This special case of a dynamic kinetic resolution through crystallisation is also
referred to as “asymmetric transformation of the second order, see: Caddick, S.;
Jenkins, K.; Chem. Soc. Rev. 1996, 25, 447.
98. Hémery, T.; Huenerbein, R.; Fröhlich, R.; Grimme, S.; Hoppe, D. J. Org. Chem.
2010, 75, 5716.
99. (a) Seppi, M.; Kalkofen, R.; Reupohl, J.; Fröhlich, R.; Hoppe, D. Angew. Chem. Int.
Ed. 2004, 43, 1423. (b) Reuber, J.; Fröhlich, R.; Hoppe, D. Org. Lett. 2004, 6, 783.
(c) Chedid, R. B.; Fröhlich, R.; Wibbeling, B.; Hoppe, D. Eur. J. Org. Chem. 2007,
3179. (d) Würthwein, E.-U.; Hoppe, D. J. Org. Chem. 2008, 73, 9055.
110. (a) Bartlett, P. D.; Friedman, S.; Stiles M. J. Am. Chem. Soc. 1953, 75, 1771. (b)
Bartlett, P. D.; Tauber, S. J.; Weber, W. P. J. Am. Chem. Soc. 1969, 91, 6362. (c)
Klein, S.; Marek, I.; Poisson, J. F. J. Am. Chem. Soc. 1995, 117, 8853. (d) Wie, X.;
Taylor, R. J. K. Chem. Commun. 1996, 187. (e) Norsikian, S.; Marek, I.; Normant,
J. F. Tetrahedron Lett. 1997, 38, 7523. (f) Norsikian, S.; Marek, I.; Klein, S.;
Poisson, J. F.; Normant, J. F. Chem. Eur. J. 1999, 5, 2055. (g) Wei, X.; Johnson, P.;
Taylor, R. J. K. Chem. Soc., Perkin Trans 1 2000, 1109.

371
111. Norsikian, S.; Marek, I.; Poisson, J. F.; Normant, J. F. J. Org. Chem. 1997, 62,
4898.
112. (a) Hogan, A.-M. L.; O’Shea, D. F. J. Am. Chem. Soc. 2006, 128, 10360. (b)
Hogan, A.-M. L.; O’Shea, D. F. Org. Lett. 2006, 8, 3769. (c) Hogan, A.-M. L.;
Tricotet, T.; Meek, A.; Kokhar, S. S.; O’Shea, D. F. J. Org. Chem. 2008, 73, 6041.
(d) Hogan, A.-M. L.; O’Shea, D. F. J. Org. Chem. 2008, 73, 2503. (e) Tricotet, T.;
Cotter, J.; Fleming, P.; Hogan, A.-M. L.; Strohmann, C.; Gessner, V. H.; O’Shea, D.
F. J. Am. Chem. Soc. 2009, 131, 3142. (f) Gessner, V. H.; Koller, S. G.; Strohmann,
C.; Hogan, A.-M. L.; O’Shea, D. F. Chem. Eur. J. 2011, 17, 2996.
113. (a) Lorthiois, E.; Marek, I.; Normant, J. F. Tetrahedron Lett. 1996, 37, 6693. (b)
Tomida, Y.; Nagaki, A.; Yoshida, J.-I. J. Am. Chem. Soc. 2011, 133, 3744.
114. McKinley, N. F.; O’Shea, D. F. J. Org. Chem. 2006, 71, 9552.
115. (a) Mulvaney, J. E.; Gardlund, Z. G.; Gardlund, S. L. J. Am. Chem. Soc. 1963, 85,
3897. (b) Mulvaney, J. E.; Newton, D. J. J. Org. Chem. 1969, 34, 1936. (c) Olsson,
L.-I.; Claesson, A.; Tetrahedron Lett. 1974, 2161. (d) Knorr, R.; Lattke, E.
Tetrahedron Lett. 1977, 45, 3969. (e) Bauer, W.; Feigel, M.; Müller, G.; Schleyer,
P. von R. J. Am. Chem. Soc. 1988, 110, 6033.
116. (a) Bailey, W. F.; Ovaska, T. V.; Leipert, T. K. Tetrahedron Lett. 1989, 30, 3901.
(b) Wu, G.; Cederbaum F. E.; Neghishi, E. Tetrahedron Lett. 1990, 31, 493. (c)
Bailey, W. F.; Ovaska, T. V. Tetrahedron Lett. 1990, 31, 627. (d) Bailey, W. F.;
Ovaska, T. V. J. Am. Chem. Soc. 1993, 115, 3080. (e) Oestreich, M.; Fröhlich, R.;
Hoppe, D. Tetrahedron Lett. 1998, 39, 1745. (f) Oestreich, M.; Fröhlich, R.; Hoppe,
D. J. Org. Chem. 1999, 64, 8616.
117. (a) Olsson, L.-I.; Claesson, A. Tetrahedron Lett. 1974, 15, 2161. (b) Hojo, M.;
Murakami, Y.; Aihara, H.; Sakuragi, R.; Baba, Y.; Hosomi, A. Angew. Chem. Int.
Ed. 2001, 40, 621. (c) Shirakawa, E.; Yamagami, T.; Kimura, T.; Yamaguchi, S.;
Havashi, T. J. Am. Chem. Soc. 2005, 127, 17164. (d) Zhang, D.; Ready, J. M. J.
Am. Chem. Soc. 2006, 128, 15050. (e) Yamagami, T.; Shintani, R.; Shirakawa, E.;
Hayashi, T. Org. Lett. 2007, 9, 1045.
118. Shirakawa, E.; Ikeda, D.; Ozawa, T.; Watanabe, S.; Hayashi, T. Chem. Commun.
2009, 1885.
119. Krief, A.; Bousbaa, J. Synlett 1996, 1007.

372
120. Laqua, H.; Fröhlich, R.; Wibbeling, B.; Hoppe, D. J. Organomet. Chem. 2001, 624,
96.
121. (a) Oestreich, M.; Fröhlich, R.; Hoppe, D. Tetrahedron Lett. 1998, 39, 1745. (b)
Oestreich, M.; Fröhlich, R.; Hoppe, D. Tetrahedron Lett. 1999, 40, 1881. (c)
Oestreich, M.; Fröhlich, R.; Hoppe, D. J. Org. Chem. 1999, 64, 8616. (d) Gralla, G.;
Wibbeling, B.; Hoppe, D. Org. Lett. 2002, 4, 2193.
122. Gralla, G.; Wibbeling, B.; Hoppe, D. Tetrahedron Lett. 2003, 44, 8979.
123. Peters, J. G.; Seppi, M.; Fröhlich, R.; Wibbeling, B.; Hoppe, D., Synthesis 2002,
381.
124. Lepifre, F.; Cottineau, B.; Mousset, D.; Bouyssou, P.; Coudert, G. Tetrahedron Lett.
2004, 45, 483.
125. (a) Cottineau, B.; Gillaizeau, I.; Farard, J.; Auclair, M.-L.; Coudert, G. Synlett 2007,
1925. Similar N-C acyl migration was found by Rouden, see: (b) Rouden, J.; Ragot,
A.; Gouault, S.; Cahard, D.; Plaquevent, J.-C.; Lasne, M.-C. Tetrahedron
Asymmetric 2002, 13, 1299.
126. Clayden, J.; Donnard, M.; Lefranc, J.; Minassi, A.; Tetlow, D. J. J. Am. Chem. Soc.
2010, 132, 6624.
127. In the sense of Zimmerman, see: Zimmerman, H. E.; Singer, L.; Thyagarajan, B. S.
J. Am. Chem. Soc. 1959, 81, 108.
128. Ebnöther, A.; Weber, H.-P. Helv. Chim. Acta 1976, 59, 2462.
129. (a) Tokaoka, M., Japan patent JP53012857, 1978. (b) Nikiforov, T.; Stanchev, S.;
Milenkov, B.; Dimitrov, V. Synthetic Commun. 1990, 20, 1977. (c) Parvez, M.;
Wendling, M. A., Acta Crystallogr. 1991, C47, 613.
130. Jung, J. W.; Kim, H.-D. Arch. Pharm. Res. 2007, 30, 1521.
131. For a review, see: O’Brien, P.; Bilke, J. L. Angew. Chem. Int. Ed. 2008, 47, 2734.
For recent representative examples, see: (a) Tomooka, K.; Shimizu, H.; Nakai, T. J.
Organomet. Chem. 2001, 624, 364. (b) Papillon, J. P. N.; Taylor, R. J. K. Org. Lett.
2002, 4, 119. (c) Dieter, R. K.; Watson, R. T.; Goswami, R. Org. Lett. 2004, 6, 253.
(d) Falck, J. R.; Patel, P. K.; Bandyopadhyay, A. J. Am. Chem. Soc. 2007, 129, 790.
132. Sato, F.; Kobayashi, Y.; Takahashi, O.; Chiba, T.; Takeda, Y.; Kusakabe, M. Chem.
Commun. 1985, 22, 1636.
133. Jung, J. E.; Ho, H.; Kim, H.-D. Tetrahedron Lett. 2000, 41, 1793.
134. (a) Aoyama, T.; Shioiri, T. Chem. Pharm. Bull. 1981, 29, 3249. (b) Podlech, J.;
Seebach, S. Liebigs Ann. 1995, 1217.

373
135. Aoyama, T.; Shioiri, T. Chem. Pharm. Bull. 1981, 29, 3249.
136. Newman, M. S.; Beal III, P. F. J. Am. Chem. Soc. 1950, 72, 5163.
137. Hanessian, S.; Sharma, R. Heterocycles 2000, 52, 1231.
138. Soai, K.; Ookawa, A.; Kaba, T.; Ogawa, K. J. Am. Chem. Soc. 1987, 109, 7111.
139. Nikiforov, T.; Stanchev, S.; Milenkov, B.; Dimitrov, V. Heterocycles 1986, 24,
1825.
140. Bourquin, J. P.; Schwarb, G.; Gamboni, G.; Fischer, R.; Ruesch, L.; Guldimann, S.;
Theus, V.; Schenker, E.; Renz, J. Helv. Chim. Acta 1958, 151, 1072.
141. Yoshihiro, N.; Hiroyuki, O. Japan patent JP53046967, 1978.
142. Beak, P.; Kerrick, S. T.; Wu, S.; Chu, J. J. Am. Chem. Soc., 1994, 116, 3231.
143. Rispens, M. T.; Gelling, O. J.; de Vries, A. H. M.; Meetsma, A.; van Bolhuis, F.;
Feringa, B. L. Tetrahedron, 1996, 52, 3521.
144. Strong cation exchange (SCX) is a form of ion chromatography which retains basic
compounds via ionic interactions. The SCX cartridges used were Isolute SCX
columns (manufactured by Biotage) which contain a benzene sulfonic acid based
SCX sorbent.
145. Campbell, J. A.; Rapoport, H. J. Org. Chem. 1996, 61, 6313.
146. Fujii, A.; Hashiguchi, S.; Uematsu, N.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc.
1996, 118, 2521.
147. (a) Mukhopadhyay, T.; Seebach, D. Helv. Chim. Acta 1982, 65, 385. (b) Sikorski,
W. H.; Reich, H. J. Am. Chem. Soc. 2001, 123, 6527.
148. Remenar, J. F.; Lucht, B. L.; Kruglyak, D.; Romesberg, F. E.; Gilchirst, J. H.;
Collum, D. B. J. Org. Chem. 1997, 62, 5748.
149. (a) Hoffmann, R. W.; Dress, R. K.; Ruhland, T. Angew. Chem. Int. Ed. Engl. 1993,
32, 1467. (b) Hoffmann, R. W.; Dress, R. K.; Ruhland, T.; Wenzel, A. Chem. Ber.
1995, 128, 861. (c) Kaiser, B.; Hoppe, D. Angew. Chem. Int. Ed. Engl. 1995, 34,
232.
150. (a) Muthusamy, S.; Babu, S. A.; Gunanathan, C. Tetrahedron Lett. 2002, 43, 3133.
(b) Sosa, J. R.; Tudjarian, A. A.; Minehan, T. G. Org. Lett. 2008, 21, 5091.
151. Clayden, J.; Greeves, N.; Warren, S.; Wothers, P. Organic Chemistry; Oxford
University Press: New York, 2001; Chapter 17, pp. 425.
152. Back, T. G.; Nakajima, K. J. Org. Chem. 2000, 65, 4543.
153. For recent studies of organolithium reactions using React-IR, see: (a) Hay, D. R.;
Song, Z.; Smith, S. G.; Beak, P. J. Am. Chem. Soc. 1988, 110, 8145. (b) Pippel, D. J.;

374
Weisenburger, G. A.; Faibish, N. C.; Beak, P. J. Am. Chem. Soc. 2001, 123, 4919. (c)
Rutherford, J. L.; Hoffmann, D.; Collum, D. B. J. Am. Chem. Soc. 2002, 124, 264.
(d) Stead, D.; Carbone, G.; O’Brien, P.; Campos, K. R.; Coldham, I.; Sanderson, A.
J. Am. Chem. Soc. 2010, 132, 7260. (e) Barker, G.; McGrath, J. L.; Klapars, A.;
Stead, D.; Zhou, G.; Campos, K. R.; O’Brien, P. J. Org. Chem. 2011, 76, 5936.
154. Gallagher, D. J.; Beak, P. J. Org. Chem. 1995, 60, 7092.
155. Reich, H. J. Lithium Amide Bases – A primer 2002.
156. For the structure of allylithiums, see: (a) Neugebauer, W.; von Rague Schleyer, P. J.
Organomet. Chem. 1980, 198, C1. (b) Balzer, H.; Berger, S. Chem. Rev. 1992, 125,
733. The preference of the allyllithium species 193Li for the Z geometry is
presumably due to intramolecular Li-O coordination, see: (a) Hoppe, D. Angew.
Chem. Int. Ed. Engl. 1984, 23, 932. (b) Schlosser, M.; Desponds, O.; Lehmann, R.;
Moret, E.; Rauchschwalbe, G. Tetrahedron 1993, 49, 10175. (c) Margot, C.;
Maccaroni, P.; Leroux, F.; Schlosser, M. Tetrahedron 1998, 54, 12853. (d) Lefranc,
J.; Tetlow, D. J.; Donnard, M.; Minassi, A.; Gálvez, E.; Clayden, J. Org. Lett. 2010,
13, 296.
157. Barner, B. A.; Mani, R. S. Tetrahedron. Lett. 1989, 30, 5413.
158. (a) Martin, V. S.; Woodward, S. S.; Katsuki, T.; Yamada, Y.; Ikeda, M.; Sharpless,
K. B. J. Am. Chem. Soc. 1981, 103, 6237. (b) George, S.; Narina, S. V.; Sudalai, A.
Tetrahedron 2006, 62, 10202.
159. Titu, D.; Chadha, A. Tetrahedron Asymmetry 2008, 19, 1698.
160. Takeda showed that the configurational stability of chiral carbanions is greatly
affected by the choice of solvent and additive, see: (a) Sasaki, M.; Ikemoto, H.;
Kawahata, M.; Yamaguchi, K.; Takeda, K. Chem. Eur. J. 2009, 15, 4663. (b)
Ikemoto, H.; Sasaki, M.; Takeda, K. Eur. J. Org. Chem. 2010, 6643.
161. Lide, D. R. CRC Handbook of Chemistry and Physics, 87th ed.; Taylor & Francis,
Ed.; Boca Raton: FL, 2006.
162. (a) Dearden, M. J.; Firkin, C. R.; Hermet, J.-P. R.; O’Brien, P. J. Am. Chem. Soc.
2002, 124, 11870. (b) O’Brien, P. Chem. Commun. 2008, 655. (+)-sparteine
surrogate behaves in an enantiocomplementary fashion to ()-sparteine.
163. Yousaf, T. I.; Williams, R. L.; Coldham, I.; Gawley, R. E. Chem. Commun. 2008,
97.
164. Li, H.; Walsh, P. J. J. Am. Chem. Soc. 2005, 127, 8355.

375
165. The (E)-stereochemistry was confirmed by comparison with published data, see ref
168f.
166. In contrast to the original Baldwin rules, see: Baldwin, J. E. Chem. Commun. 1976,
734., endo-dig cyclisation are less favourable than the competing exo-dig closures,
see: Alabugin, I. V.; Gilmore, K.; Manoharan, M. J. Am. Chem. Soc. 2011, 133,
12608.
167. (a) Shapiro, S. L.; Bandurco, V.; Freedman, L. J. Org. Chem. 1961, 26, 3710. (b)
Sisido, K.; Hukuoka, K.; Tuda, M.; Nozaki, H. J. Org. Chem. 1962, 27, 2663. (c)
Easton, N. R.; Cassady, D. R.; Dillard, R. D. J. Org. Chem. 1962, 27, 2927. (d)
Shachat, N.; Bagnell, J. J. Org. Chem. 1963, 28, 991. (e) Stoffel, P. J.; Speziale, A.
J. J. Org. Chem. 1963, 28, 2814. (f) Fournier, J.; Bruneau, C.; Dixneuf, P. H.
Tetrahedron Lett. 1990, 31, 1721. (g) Kimura, M.; Kure, S.; Yoshida, Z.; Tanaka,
S.; Fugami, K.; Tamaru, Y. Tetrahedron Lett. 1990, 31, 4887. (h) Tamaru, Y.;
Kimura, M.; Tanaka, S.; Kure, S.; Yoshida, Z. Bull. Chem. Soc. Jpn. 1994, 67, 2838.
(i) Bouyssi, D.; Cavicchioli, M.; Balme, G. Synlett 1997, 944. (j) Arcadi, A. Synlett
1997, 941.
168. (a) Shi, M.; Shen, Y. M.; Chen, Y. J. Heterocycles 2002, 57, 245. (b) Gu, Y. L.;
Zhang, Q. H.; Duan, Z. Y.; Zhang, J.; Zhang, S. G.; Deng, Y. Q. J. Org. Chem.
2005, 70, 7376. (c) Zhang, Q. H.; Shi, F.; Gu, Y. L.; Yang, J.; Deng, Y. Q.
Tetrahedron Lett. 2005, 46, 5907. (d) Ritter, S.; Horino, Y.; Lex, J.; Schmalz, H.
Synlett 2006, 3309. (e) Jiang, H. F.; Zhao, J. W.; Wang, A. Z. Synthesis 2008, 763.
(f) Ramesh, R.; Chandrasekaran, Y.; Megha, R.; Chandrasekaran, S. Tetrahedron
2007, 63, 9153. (g) Jiang, H.-F.; Zhao, J.-W. Tetrahedron Lett. 2009, 50, 60.
169. Matsumura, K.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1997, 119,
8738.
170. Jiang, X-B.; van den Berg, M.; Minnaard, A. J.; Feringa, B. L.; de Vries, J. G.
Tetrahedron Asymmetric 2004, 15, 2223.
171. Corey, E. J.; Chaykovsky, M. J. Am. Chem. Soc. 1965, 87, 1345.
172. Lemoucheux, L.; Rouden, J.; Ibazizene, M; Sobrio, F.; Lasne, M.-C. J. Org. Chem.
2003, 68, 7289.
173. Abdel-Magid, A. F.; Carson, K. G.; Harris, B. D.; Maryanoff, C. A.; Shah, R. D. J.
Org. Chem. 1996, 61, 3849.
174. Overman, L. E.; Campbell, C. B.; Knoll, F. M. J. Am. Chem. Soc. 1978, 4822.
175. Basu, A.; Thayumanavan, S. Angew. Chem. Int. Ed. 2002, 41, 716.

376
176. Formation of the lithium amides directly from the ammonium salts rather than from
the free bases has been shown to be preferable, presumably a selectivity-enhancing
effect of the resulting LiCl, see ref 66.
177. (a) Hoppe, D.; Hintze, F.; Tebben, P. Angew. Chem. Int. Ed. Engl. 1990, 29, 1422.
(b) Hintze, F.; Hoppe, D. Synthesis 1992, 1216. (c) Hoppe, D.; Hintze, F.; Tebben,
P.; Paetow, M.; Ahrens, H.; Schwerdtfeger, J.; Sommerfeld, P.; Haller, J.; Guarnieri,
W.; Kolczewski, T.; Hense, T.; Hoppe, I. Pure Appl. Chem. 1994, 66, 1479. (d)
Hoppe, D.; Ahrens, H.; Guarnieri, W.; Helmke, H.; Kolczewski, S. Pure Appl.
Chem. 1996, 68, 613.
178. The α-ethoxyvinyllithium reagent has been made following the procedure described
by Roush et al., see: Savall, B. M.; Blanchard N.; Roush, W. R. Org. Lett. 2003, 5,
377.
179. The vinyllithium reagent has been made following the procedure described by
Weiner et al., see: Seyferth, D.; Weiner, M. A. Chem. Ind. 1959, 402.
180. (a) Seebach, D. Angew. Chem. Int. Engl. 1988, 100, 1685. (b) For a review on the
role of TMEDA on organolithium reactivity, see: Collum, D. B. Accounts Chem.
Res. 1992, 25, 448. (c) Rutherford, J. L.; Hoffmann, D.; Collum, D. B. J. Am. Chem.
Soc. 2002, 124, 264.
181. Rawson, D. J.; Meyers, A. I. Tetrahedron Lett. 1991, 32, 2095.
182. (a) Bailey, W. F.; Mearly, M. J. J. Am. Chem. Soc. 2000, 122, 6787. (b) Sanz Gil,
G.; Groth, U. M. J. Am. Chem. Soc. 2000, 122, 6789.
183. Basu, A.; Thayumanavan, S. Angew. Chem. Int. Ed. 2002, 41, 716.
184. The lithium-coordinating additive DMPU decreases the configurational stability of
O-substituted benzyllithiums, see: ref 55.
185. (a) Bates, R. B.; Kroposki, L. M.; Potter, D. E. J. Org. Chem. 1972, 37, 560. (b)
Jung, M. E.; Blum, R. B. Tetrahedron Lett. 1977, 43, 3791. (c) Clayden, J.; Yasin,
S. A. New J. Chem. 2002, 26, 191.
186. Sengupta, S; Snieckus, V. J. Org. Chem. 1990, 55, 5680.
187. Snieckus, V. Pure Appl. Chem. 1990, 62, 2047.
188. Chedid, R. B.; Brummer, M.; Wibbeling, B.; Fröhlich, R.; Hoppe, D. Angew. Chem.
Int. Ed. 2007, 46, 3131.
189. Marr, F.; Fröhlich, R.; Wibbeling, B.; Diedrich, C. Hoppe, D. Eur. J. Org. Chem.
2002, 2970.

377
190. Review on allenes: Modern Allene Chemistry; Krause, N.; Hashimi, A. S. K., Eds.;
Wiley-VCH: Weinheim, 2004; Vols. 1 and 2. Reviews on the synthesis applications
of allenes: (a) Ma, S. Accounts Chem. Res. 2003, 36, 701. (b) Ma, S. Chem. Rev.
2005, 105, 2829. (c) Ma. S. Accounts Chem. Res. 2009, 42, 1679. Reviews on
allenic natural products and pharmaceuticals: Hoffmann-Röder, A.; Krause, N.
Angew. Chem. Int. Ed. 2004, 43, 1196. Reviews on the synthesis of allenes: (a)
Krause, N.; Hoffmann-Röder, A. Tetrahedron 2004, 60, 11671. (b) Brummond, K.
M.; DeForrest, J. E. Synthesis 2007, 795. (c) Yu, S.; Ma, S. Chem. Commun. 2011,
5384. For selected examples on the synthesis of enantioselective allenes, see: (a)
Dreller, S.; Dyrbusch, M.; Hoppe, D. Synlett 1991, 397. (b) Schultz-Fademrecht, C.;
Wibbeling, B.; Fröhlich, R.; Hoppe, D. Org. Lett. 2001, 3, 1221. (c) Deutsch, C.;
Lipshutz, B. H.; Krause, N. Angew. Chem. Int. Ed. 2007, 46, 1650. (d) Ogasawara,
M.; Okada, A.; Subbarayan, V.; Sörgel, S.; Takahashi, T. Org. Lett. 2010, 12, 5736.
(e) Nishimura, T.; Makino, H.; Nagaosa, M.; Hayashi, T. J. Am. Chem. Soc. 2010,
132, 12865.
191. Dreller, S.; Dyrbusch, M.; Hoppe, D. Synlett 1991, 397.
192. Tomida, Y.; Nagaki, A.; Yoshida, J.-I. J. Am. Chem. Soc. 2011, 133, 3744.
193. (a) Huynh, C.; Linstrumelle, G. Chem. Commun. 1983, 1133. (b) Lambert, C.;
Schleyer, P. von R.; Würthwein, E.-U. J. Org. Chem. 1993, 58, 6377. (c) Reich, H.
J.; Holladay, J. E.; Mason, J. D.; Sikorski, W. H. J. Am. Chem. Soc. 1995, 117,
12137. (d) Reich, J. H.; Holladay, J. E. J. Am. Chem. Soc. 1995, 117, 8470. (e)
Reich, J. H.; Holladay, J. E.; Walker, T. G.; Thompson, J. L. J. Am. Chem. Soc.
1999, 121, 9769. (f) Reich, H. J.; Thompson, J. L. Org. Lett. 2000, 2, 783.
194. Lamandé-Langle, S.; Abarbri, M.; Thibonnet, J. ; Duchêne, A. J. Organomet. Chem.
2009, 694, 2368.
195. (a) Weber, W. P. Silicon Reagents for Organic Synthesis; Springer-Verlag: Berlin,
1983. (b) Ott, H.; Daschlein, C.; Leusser, D.; Schildbach, S.; Seibel, T.; Stalke, D.
Strohmann, C. J. Am. Chem. Soc. 2008, 130, 11901.
196. Greco, M. N.; Hawkins, M. J.; Powell, E. T.; Almond, H. R.; de Garavilla, L.; Hall,
J.; Minor, L. K.; Wang, Y.; Corcoran, T. W.; Di Cera, E.; Cantwell, A. M.;
Savvides, S. N.; Damiano, B. P.; Maryanoff, B. E. J. Med. Chem. 2007, 50, 1727.
197. Reviews for ynamides, see: (a) Mulder, J. A.; Kurtz, K. C. M.; Hsung, R. P. Synlett
2003, 1379. (b) DeKorver, K. A.; Li, H.; Lohse, A. G.; Hayashi, R.; Lu, Z.; Zhang,

378
Y.; Hsung, R. P. Chem. Rev. 2010, 110, 5064. (c) Evano, G.; Coste, A.; Jouvin, K.
Angew. Chem. Int. Ed. 2010, 49, 2840.
198. Wittulski, B.; Stengel, T. Angew. Chem. Int. Ed. 1998, 37, 489.
199. (a) Frederick, M. O.; Mulder, J. A.; Tracey, M. R.; Hsung, R. P.; Huang, J.; Kurtz,
K. C. M.; Shen, L.; Douglas, C. J. J. Am. Chem. Soc. 2003, 125, 2368. (b) Dunetz, J.
R.; Danheiser, R. L.; Org. Lett. 2003, 5, 4011. (c) Zhang, Y.; Hsung, R. P.; Tracey,
M. R.; Kurtz, K. C. M.; Vera, E. L. Org. Lett. 2004, 6, 1151. (d) Zhang, X.; Zhang,
Y.; Huang, J.; Hsung, R. P.; Kurtz, K. C. M.; Oppenheimer, J.; Petersen, M. E.;
Sagamanova, I. K.; Shen, L.; Tracey, M. R. J. Org. Chem. 2006, 71, 4170. (e) Yao,
B.; Liang, Z.; Niu, T.; Zhang, Y. J. Org. Chem. 2009, 74, 4630.
200. Jouvin, K.; Couty, F.; Evano, G. Org. Lett. 2010, 12, 3272.
201. Hamada, T.; Ye, X.; Stahl, S. S.; J. Am. Chem. Soc. 2008, 130, 833.
202. Coste, A.; Karthikeyan, G.; Couty, F.; Evano, G. Angew. Chem. Int. Ed. 2009, 48,
4381.
203. Jia, W.; Jiao, N. Org. Lett. 2010, 12, 2000.
204. Tracey, M. R.; Zhang, Y.; Frederick, M. O.; Mulder, J. A.; Hsung, R. P. Org. Lett.
2004, 6, 2209.
205. Jiang, M. X.; Rawat, M.; Wulff, W. D. J. Am. Chem. Soc. 2004, 126, 5970.
206. Hevia, E.; Mulvey, R. E. Angew. Chem. Int. Ed. 2011, 50, 6448.
207. Vasilevsky, S. F.; Baranov, D. S.; Mamatyuk, V. I.; Gatilov, Y. V.; Alabugin, I. V.
J. Org. Chem. 2009, 74, 6143. They suggested that this effect is due to
hyperconjugative stabilization of the anion with antiperiplanar *(C-O)-orbital.
208. Usually the intramolecular addition reaction of lithium carbanions onto phenyl-
substituted multiple bonds occurs with complete syn-selectivity, see ref 121.
209. Know, C.-H.; Nagasawa, H. T.; DeMaster, E. G.; Shirota, F. N.; J. Med. Chem.
1986, 29, 1922.
210. Weiss, S.; Krommer, H. Angew. Chem. Int. Ed. 1974, 13, 546.
211. Kwon, C.-H.; Nagasawa, H. T. J. Org. Chem. 1990, 55, 3403.
212. Metten, B.; Smet, M.; Boens, N.; Dehaen, W. Synthesis 2005, 1838.
213. Oestreich, M.; Fröhlich, R.; Hoppe, D. J. Org. Chem. 1999, 64, 8616.
214. Fournier, A. M.; Brown, R. A.; Farnaby, W.; Miyatake-Ondozadal, H.; Clayden, J.
P. Org. Lett. 2010, 12, 2222.
215. Fournier, A.; Clayden, J. Org. Lett. 2012, 14, 142.

379
216. Perrin, D. D.; Armarego, W. L. F. Purification of Laboratory Chemicals. 3rd Ed,;
Pergamon press, 1988.
217. Hanessian, S; Sharma, R. Heterocycles 2000, 52, 1231.
218. Carlson, E. C.; Rathbone, L. K.; Yang, H.; Collett, N. D.; Carter, R. G. J. Org.
Chem. 2008, 73, 5155.
219. Enders, D.; Kirchhoff, J. H. Synthesis 2000, 2099.
220. Kantam, M. L.; Laha, S.; Yadav, J.; Likhar, P. R.; Sreedhar, B.; Jha, S.; Bhargava,
S.; Udayakiran, M.; Jagadeesh, B. Org. Lett. 2008, 10, 2979.
221. Zhang, Z.; Bender, C. F.; Widenhoefer, R. A. Org. Lett. 2007, 9, 2887.
222. Kotani, S.; Osakama, K.; Sugiura, M.; Nakajima, M. Org. Lett. 2011, 13, 3968.
223. Kamal, A.; Sandbhor, M.; Shaik, A. A.; Sravanthi, V. Tetrahedron Asymmetry 2003,
14, 2839.
224. Mahler, H.; Braun, M. Chem. Ber. 1991, 124, 1379.
225. Six, Y. Eur. J. Org. Chem. 2003, 1157.
226. Wang, D.; Chen, D.; Haberman, J. X.; Li, C.-J. Tetrahedron 1998, 54, 5129.
228. Tanaka, K.; Shoji, T. Org. Lett. 2005, 7, 3561.
229. Larrosa, M.; Guerrero, C.; Rodríhuez, R.; Cruces, J. Synlett, 2010, 2101.
230. Saidi, O.; Williams, L. M. J.; Blacker, A. J.; Farah, M. M.; Marsden, S. P. Angew.
Chem. Int. Ed. 2009, 48, 7375.
231. Enthaler, S.; Erre, G.; Junge, K.; Michalik, D.; Spannenberg, A.; Marras, F.;
Gladiali, S.; Beller, M. Tetrahedron Asymmetric 2007, 18, 1288.
232. Trahanovsky, W. S.; Chou, C.-H.; Cassady, T. J. J. Org. Chem. 1994, 59, 2613.
233. Artuso, E.; Degani, I.; Fochi, R.; Magistris, C. Synthesis 2008, 10, 1612.
234. Benalil, A.; Roby, P.; Carboni, B.; Vaultier, M. Synthesis 1991, 787.
235. Jiang, M. X.; Rawat, M.; Wulff, W. D. J. Am. Chem. Soc. 2004, 126, 5970.
236. Do, H.-Q.; Daugulis, O. Org. Lett. 2009, 11, 421.

380
AAPPPPEENNDDIIXX
APPENDIX 1: List of Publications
1. Fournier, A. M.; Brown, R. A.; Farnaby, W.; Miyatake-Ondozadal, H.; Clayden, J.
P. Synthesis of ()-(S,S)-Clemastine by Invertive Aryl Migration in a Lithiated
Carbamate, Org. Lett. 2010, 12, 2222.
2. Fournier, A.; Clayden, J. Tertiary Alcohols by Tandem β-Carbolithiation and N→C
Aryl Migration in Enol Carbamates, Org. Lett. 2012, 14, 142.
3. Lefranc, J.; Fournier, A.; Mingat, G.; Herbert, S.; Marcelli, T.; Clayden, J.
Intramolecular Vinylation of Secondary and Tertiary Organolithiums, J. Am. Chem.
Soc. Communication, accepted.
4. Fournier, A. M.; Nichols, C.; Vincent, M. A.; Hillier, I. H.; Clayden, J. P. Lithium
Choreography:Intramolecular Arylations of Carbamate-Stabilized Carbanions and
their Mechanisms Probed by in-situ IR and DFT, J. Am. Chem. Soc., Article,
submitted.

381
APPENDIX 2: Experimental Procedures for the in situ React IR Studies.
An FTIR analyzer, ReactIRTM
iC10 with a DiComp (Diamond) probe connected to an
MCT detector with a DS Series 9.5mm AgXFiberConduitTM
from Mettler Toledo was used
for all experiments. The iC IRTM
software was used to control the spectrometer and collect
spectra. Nitrogen purge on the IR system was maintained throughout any experiments and
nitrogen background was used in computing the absorbance spectra. Each spectrum
represent 30 co-added scans measured at a spectral resolution of 8 cm-1
in the 2000 to 650
cm-1
range with the Happ-Genzel apodization.
II.1 In situ React IR monitoring the rearrangement of benzylcarbamate 95i with sec-
BuLi in THF.
Anhydrous THF (4 cm3, 0.26 M) was added to benzylcarbamate 95i (300 mg) in a 25 cm
3
three-neck flask equipped with a stirrer bar, thermometer and React IR probe under N2.
The solution was cooled in an acetone/CO2 bath and aged for 15 min (for stability of
readout on React IR). Then, sec-BuLi (0.81 cm3, 1.1 equiv., 1.4 M in cyclohexane) was
added dropwise. The reaction was aged for 1 h at -60 °C before quenching with MeOH.
II.2 In situ React IR monitoring the retreatment of rearranged carbamate 96i.
Anhydrous THF (4 cm3, 0.26 M) was added to rearranged product 96i (300 mg) in a 25
cm3 three-neck flask equipped with a stirrer bar, thermometer and React IR probe under
N2. The solution was cooled in an acetone/CO2 bath and aged for 15 min (for stability of
readout on React IR). Then, sec-BuLi (0.81 cm3, 1.1 equiv., 1.4 M in cyclohexane) was
added dropwise. The reaction was aged for 15 min at -60 °C before quenching with MeOH.

382
II.3 In situ React IR monitoring the rearrangement of benzylcarbamate 95i with sec-
BuLi in TBME.
Anhydrous TBME (4 cm3, 0.26 M) was added to benzylcarbamate 95i (300 mg) in a 25
cm3 three-neck flask equipped with a stirrer bar, thermometer and React IR probe under N2.
The solution was cooled in an acetone/CO2 bath and aged for 15 min (for stability of
readout on React IR). Then, sec-BuLi (1.40 cm3, 2.0 equiv., 1.4M in cyclohexane) was
added dropwise. The reaction was aged for 1 h at -60 °C before quenching with MeOH OR
anhydrous THF (0.34 cm3, 4.0 equiv.) was added and the reaction was stirred for further 1
h 30 before quenching with MeOH.
II.4 In situ React IR monitoring the rearrangement of benzylcarbamate 95i with sec-
BuLi in toluene.
Anhydrous toluene (4 cm3, 0.26 M) was added to benzylcarbamate 95i (300 mg) in a 25
cm3 three-neck flask equipped with a stirrer bar, thermometer and React IR probe under N2.
The solution was cooled in an acetone/CO2 bath and aged for 15 min (for stability of
readout on React IR). Then, sec-BuLi (1.4 cm3, 2.0 equiv., 1.4 M in cyclohexane) was
added dropwise. The reaction was aged for 1 h at -60 °C before adding TMEDA (0.15 cm3,
1.1 equiv.). The reaction mixture was stirred for 30 min before quenching with MeOH.
II.5 In situ React IR monitoring the rearrangement of benzylcarbamate 95i with LDA
in TBME.
Anhydrous TBME (4 cm3, 0.26 M) was added to benzylcarbamate 95i (300 mg) in a 25
cm3 three-neck flask equipped with a stirrer bar, thermometer and React IR probe under N2.
The solution was cooled in an acetone/CO2 bath and aged for 15 min (for stability of
readout on React IR). Then, LDA (1.80 cm3, 3.5 equiv., 2 M in THF/heptane/ethylbenzene)
was added dropwise. The reaction was aged for 1 h at -60 °C before quenching with
MeOH.
II.6 In situ React IR monitoring the rearrangement of allylcarbamate 201 with LDA
in THF.
Anhydrous THF (4 cm3, 0.27 M) was added to allylcarbamate 201 (300 mg) in a 25 cm
3
three-neck flask equipped with a stirrer bar, thermometer and React IR probe under N2.
The solution was cooled in an acetone/CO2 bath and aged for 15 min (for stability of
readout on React IR). Then, LDA (0.80 cm3, 1.5 equiv., 2 M in THF/heptane/ethylbenzene)

383
was added dropwise. The reaction was aged for 1 h at -60 °C before quenching with
MeOH.
II.7 In situ React IR monitoring the rearrangement of benzylcarbamate 308 with
LDA in THF.
Anhydrous THF (5 cm3, 0.26 M) was added to benzylcarbamate 308 (400 mg) in a 25 cm
3
three-neck flask equipped with a stirrer bar, thermometer and React IR probe under N2.
The solution was cooled in an acetone/CO2 bath and aged for 15 min (for stability of
readout on React IR). Then, LDA (1.5 cm3, 1.5 equiv., 2 M in THF/heptane/ethylbenzene)
was added dropwise. The reaction was aged for 45 min at -60 °C and then warmed slowly
to room temperature and stirred at room temperature for 30 min before quenching with
MeOH.

384
APPENDIX 3: In Situ IR Spectroscopy Monitoring the Rearrangement
of a N-allylurea
Anhydrous THF (4 cm3, 0.25M) was added to allylurea 341 (300 mg) in a 25 mL three-
neck flask equipped with a stirrer bar, thermometer and React IR probe under N2. The
solution was cooled in an acetone/CO2 bath and aged for 15 min (for stability of readout on
React IR). Then, LDA (1.0 cm2, 2.0 equiv., 2 M in THF/heptane/ethylbenzene) was added
dropwise. The reaction was aged for 1 h at -60 °C before quenching with MeOH and
warming up to room temperature.
Scheme 181. Intermediates Detected in the Rearrangement of 341.
PhN N
Me
O
PMP LDA (1eq.),THF, -60 °C
NN
O LiLnMe
PhPMP
N
OLi
PMPPh
MeN
N
O LiLn
PMPPh
MeN
Li
MeHN N
O
PMP
Ph
LDA (1eq.),THF, -60 °C
341
343
MeOH
341Li
343Li
342Li

385
Figure 22. Characteristic changes in the 1800-1400 cm-1
region of the IR spectrum during
the rearrangement of 341. Five spectra taken at different stages in the experiment are
overlaid. Dark blue: urea prior to lithiation; green: after addition of 1 eq. of LDA at -60 °C;
red: after addition of a further equivalent of LDA; light blue: after addition of MeOH at -
60 °C; pink: after warming to 25 °C.
Figure 23. Perspective view of the changes in the IR spectrum during the rearrangement.
LDA added
Quench with
MeOH Allylurea 341
in THF at -60 °C
Warming up
to 25 °C
1657 cm-1
: 341
1683 cm-1
: 343
1552 cm-1
: 343Li
1605 cm-1
: 342Li
1595 cm-1
: 343Li




















