
Intestinal Neurofibromatosis Is a Subtype of Familial GIST and Results From a Dominant Activating...
6
CASE REPORT Intestinal Neurofibromatosis Is a Subtype of Familial GIST and Results From a Dominant Activating Mutation in PDGFRA THOMAS DE RAEDT,* JAN COOLS,* ,‡ MARIA DEBIEC–RYCHTER,* HILDE BREMS,* NICOLE MENTENS, ‡ RAF SCIOT, § JACQUES HIMPENS, IVO DE WEVER, ¶ PATRICK SCHÖFFSKI, # PETER MARYNEN,* ,‡ and ERIC LEGIUS* *Department of Human Genetics and ‡ Flanders Interuniversity Institute for Biotechnology (VIB), Catholic University of Leuven, Leuven; § Department of Pathology, University Hospitals of Leuven, Leuven; Department of Surgery, St. Blasius Ziekenhuis, Dendermonde; ¶ Department of Oncological Surgery, University Hospitals of Leuven, Leuven; and # Department of Medical Oncology, University Hospitals of Leuven, Leuven, Belgium Background & Aims: Intestinal neurofibromatosis (Online Mendelian inheritance in Man database number 162220) is an alternate form of neurofibromatosis. Patients present with neu- rofibromas limited to the intestine in the absence of any other typical features of NF1 and NF2. At present, the molecular basis of intestinal neurofibromatosis remains elusive. The aim of the present study was to find the gene responsible for intestinal neurofibromatosis and to characterize functionally the muta- tion. Methods: Three candidate genes (NF1, KIT, and PDG- FRA) were screened for mutations in 3 sisters diagnosed with intestinal neurofibromatosis. Five tumors were available for pathologic examination. Activation (phosphorylation) of PDGFR was subsequently tested by Western blot analysis on a transfected 293T and Ba/F3 cell line. Results: We found an inherited mutation (Y555C) in the juxtamembrane domain of PDGFRA in the affected individuals. The Y555C mutation leads to autophosphorylation and thus activation of PDGFR. These observations confirm that PDGFR(Y555C) is an oncogenic kinase. The clinical phenotype in the reported family resembles the syndrome of familial gastrointestinal stromal tumors (fa- milial GIST). Somatic activating mutations in KIT and PDGFRA are frequent in sporadic GISTs, and mutations in both genes have also been described in familial GISTs. The tumors in the reported family are morphologically identical to intestinal neu- rofibromas, but, immunohistochemically, they do not express S100 or any of the known GIST markers. Conclusions: The inherited PDGFRA mutation in the reported family shows that intestinal neurofibromatosis is allelic to familial GIST caused by PDGRA mutations. We therefore propose that these tumors be classified as familial KIT-negative gastrointestinal stromal tumors. T he neurofibromatoses are a heterogeneous group of ge- netic disorders. The 2 classical types (neurofibromatosis type 1 [NF1] and neurofibromatosis type 2 [NF2]) are clinically and molecularly well-defined. Other alternate and related forms have been described, but they are often not characterized at the molecular level. One of the reported alternate forms of neuro- fibromatosis is intestinal neurofibromatosis (Online Mendelian inheritance in Man database number 162220). Intestinal neu- rofibromatosis, also called NF3b, is phenotypically described as neurofibromas strictly limited to the intestine, of adult onset with incomplete penetrance, variable expression, autosomal dominant inheritance pattern, and absence of other features of NF1 or NF2. A limited number of families with intestinal neurofibromatosis have been reported. Immunohistochemical analysis of these tumors has never been performed, and they were labeled as neurofibromas based on histopathologic ap- pearance. 1,2 Intestinal neurofibromatosis could be under diag- nosed because of the relatively mild phenotype. Intestinal neu- rofibromas are nonepithelial gastrointestinal tumors potentially resulting in intestinal obstruction. Similar nonepithelial gastro- intestinal tumors have also been observed in typical NF1 pa- tients. GISTs in NF1 patients tend to be multiple and are located predominantly within the small intestine. 3,4 The patho- genesis of GIST in NF1 is not related to KIT mutations, as is the case in 85% of the sporadic GISTs, 5,6 but to the inactivation of NF1. 7 In sporadic GIST without KIT mutations, PDGFRA mu- tations have been identified. 8,9 We tested the hypothesis that intestinal neurofibromatosis is caused by an inherited mutation in NF1, KIT, or PDGFRA and examined the (immuno)phenotype of the tumors in this condition. Materials and Methods Mutation Analysis Mutation analysis of PDGFRA and KIT was performed on genomic DNA. Each exon was polymerase chain reaction (PCR) amplified (primers available on request) and sequenced following the BigDye Terminator Sequencing protocol (Applied Biosystems, Foster City, CA). Mutation analysis of NF1 was performed on RNA from fibroblast culture. Reverse-transcrip- tion (RT) PCR was used to amplify 98.5% of the NF1 coding region (from position 48 to 8464) in 8 overlapping PCR reac- tions. Each PCR fragment was sequenced using a solid-phase sequencing protocol. 10 Exon 23a and exon 48a were sequenced starting from genomic DNA. Primer sequences and PCR con- ditions are available from the authors upon request. The pres- ence of an NF1 microdeletion was excluded after fluorescence in situ hybridization (FISH) analysis of fibroblasts (clones P1-9 and P1-12). 11 Abbreviations used in this paper: CGH, Comparative genome hybrid- ization; GIST, gastrointestinal stromal tumors; NF1, neurofibromatosis type 1; NF2, neurofibromatosis type 2. © 2006 by the AGA Institute 0016-5085/06/$32.00 doi:10.1053/j.gastro.2006.07.002 GASTROENTEROLOGY 2006;131:1907–1912
Transcript of Intestinal Neurofibromatosis Is a Subtype of Familial GIST and Results From a Dominant Activating...

C
IF
TJ*UL
BMartopntFipPtiPtoktmahrrSiibbt
Ttahmfiirnw
GASTROENTEROLOGY 2006;131:1907–1912
ASE REPORT
ntestinal Neurofibromatosis Is a Subtype of Familial GIST and Resultsrom a Dominant Activating Mutation in PDGFRA
HOMAS DE RAEDT,* JAN COOLS,*,‡ MARIA DEBIEC–RYCHTER,* HILDE BREMS,* NICOLE MENTENS,‡ RAF SCIOT,§
ACQUES HIMPENS,� IVO DE WEVER,¶ PATRICK SCHÖFFSKI,# PETER MARYNEN,*,‡ and ERIC LEGIUS*Department of Human Genetics and ‡Flanders Interuniversity Institute for Biotechnology (VIB), Catholic University of Leuven, Leuven; §Department of Pathology,
� ¶
niversity Hospitals of Leuven, Leuven; Department of Surgery, St. Blasius Ziekenhuis, Dendermonde; Department of Oncological Surgery, University Hospitals ofeuven, Leuven; and #Department of Medical Oncology, University Hospitals of Leuven, Leuven, BelgiumdNnawpnrritlgcNtiio
o(fBptrtssdesa
it
ackground & Aims: Intestinal neurofibromatosis (Onlineendelian inheritance in Man database number 162220) is an
lternate form of neurofibromatosis. Patients present with neu-ofibromas limited to the intestine in the absence of any otherypical features of NF1 and NF2. At present, the molecular basisf intestinal neurofibromatosis remains elusive. The aim of theresent study was to find the gene responsible for intestinaleurofibromatosis and to characterize functionally the muta-ion. Methods: Three candidate genes (NF1, KIT, and PDG-RA) were screened for mutations in 3 sisters diagnosed with
ntestinal neurofibromatosis. Five tumors were available forathologic examination. Activation (phosphorylation) ofDGFR� was subsequently tested by Western blot analysis on aransfected 293T and Ba/F3 cell line. Results: We found annherited mutation (Y555C) in the juxtamembrane domain ofDGFRA in the affected individuals. The Y555C mutation leadso autophosphorylation and thus activation of PDGFR�. Thesebservations confirm that PDGFR�(Y555C) is an oncogenicinase. The clinical phenotype in the reported family resembleshe syndrome of familial gastrointestinal stromal tumors (fa-
ilial GIST). Somatic activating mutations in KIT and PDGFRAre frequent in sporadic GISTs, and mutations in both genesave also been described in familial GISTs. The tumors in theeported family are morphologically identical to intestinal neu-ofibromas, but, immunohistochemically, they do not express100 or any of the known GIST markers. Conclusions: The
nherited PDGFRA mutation in the reported family shows thatntestinal neurofibromatosis is allelic to familial GIST causedy PDGRA mutations. We therefore propose that these tumorse classified as familial KIT-negative gastrointestinal stromalumors.
he neurofibromatoses are a heterogeneous group of ge-netic disorders. The 2 classical types (neurofibromatosis
ype 1 [NF1] and neurofibromatosis type 2 [NF2]) are clinicallynd molecularly well-defined. Other alternate and related formsave been described, but they are often not characterized at theolecular level. One of the reported alternate forms of neuro-
bromatosis is intestinal neurofibromatosis (Online Mendeliannheritance in Man database number 162220). Intestinal neu-ofibromatosis, also called NF3b, is phenotypically described aseurofibromas strictly limited to the intestine, of adult onset
ith incomplete penetrance, variable expression, autosomalominant inheritance pattern, and absence of other features ofF1 or NF2. A limited number of families with intestinaleurofibromatosis have been reported. Immunohistochemicalnalysis of these tumors has never been performed, and theyere labeled as neurofibromas based on histopathologic ap-earance.1,2 Intestinal neurofibromatosis could be under diag-osed because of the relatively mild phenotype. Intestinal neu-ofibromas are nonepithelial gastrointestinal tumors potentiallyesulting in intestinal obstruction. Similar nonepithelial gastro-ntestinal tumors have also been observed in typical NF1 pa-ients. GISTs in NF1 patients tend to be multiple and areocated predominantly within the small intestine.3,4 The patho-enesis of GIST in NF1 is not related to KIT mutations, as is thease in 85% of the sporadic GISTs,5,6 but to the inactivation ofF1.7 In sporadic GIST without KIT mutations, PDGFRA mu-
ations have been identified.8,9 We tested the hypothesis thatntestinal neurofibromatosis is caused by an inherited mutationn NF1, KIT, or PDGFRA and examined the (immuno)phenotypef the tumors in this condition.
Materials and MethodsMutation AnalysisMutation analysis of PDGFRA and KIT was performed
n genomic DNA. Each exon was polymerase chain reactionPCR) amplified (primers available on request) and sequencedollowing the BigDye Terminator Sequencing protocol (Appliediosystems, Foster City, CA). Mutation analysis of NF1 waserformed on RNA from fibroblast culture. Reverse-transcrip-ion (RT) PCR was used to amplify 98.5% of the NF1 codingegion (from position 48 to 8464) in 8 overlapping PCR reac-ions. Each PCR fragment was sequenced using a solid-phaseequencing protocol.10 Exon 23a and exon 48a were sequencedtarting from genomic DNA. Primer sequences and PCR con-itions are available from the authors upon request. The pres-nce of an NF1 microdeletion was excluded after fluorescence initu hybridization (FISH) analysis of fibroblasts (clones P1-9nd P1-12).11
Abbreviations used in this paper: CGH, Comparative genome hybrid-zation; GIST, gastrointestinal stromal tumors; NF1, neurofibromatosisype 1; NF2, neurofibromatosis type 2.
© 2006 by the AGA Institute0016-5085/06/$32.00
doi:10.1053/j.gastro.2006.07.002

iepitwf(
diT1mthmsrmmar
w(wMieGSptErai
m(gsPdw(asmaw
lw(m1DD1aC(pf
odc
etavoiIntodt(qpu4MiTrIioaMpgiiFi35
1908 DE RAEDT ET AL GASTROENTEROLOGY Vol. 131, No. 6
Array-Comparative Genome HybridizationAnalysisArray-Comparative genome hybridization (CGH) exper-
ments were performed as described by Menten et al12 on DNAxtracted from 3 of the intestinal tumors. The experiments wereerformed on DNA extracted from tumor samples with the
nherent risk of contamination by nontumoral cells. Therefore,he analysis of the array-CGH data were statistically analyzedith a CGH-smooth software,13 a program especially designed
or the analysis of heterogeneous samples such as tumorshttp://www.few.vu.nl/�vumarray/).
Western Blot AnalysisThe tumors were snap frozen in liquid nitrogen imme-
iately after surgery and stored at �80°C. Western blot exper-ments were performed 1 week after removal of the tumors.umor specimens were lysed in ice-cold lysis buffer containing% NP40 in TNE (50 mmol/L Tris-HCl, 150 mmol/L NaCl, 1mol/L EDTA, pH 7.6) and supplemented with complete pro-
ease inhibitor cocktail (Boehringer Mannheim GmbH, Mann-eim, Germany), 10 �g/mL leupeptin, 10 g/mL aprotinin, 5mol/L sodium fluoride, 5 mmol/L sodium PP1, 5 mmol/L
odium vanadate, and 1 mmol/L phenylmethylsulphonyl fluo-ide (Sigma-Aldrich, St Louis, MO). Lysates were rocked for 30
inutes at �4°C and then centrifuged at 21,000g for 21,00030inutes at �4°C. Supernatants were removed to a fresh tube,
nd protein content was determined using BCA protein assayeagent (Pierce, Rockford, IL).
For Western blot analysis, aliquots containing 30 �g proteinere electrophoresed and electroblotted to PDVF membranes
Amersham Pharmacia Biotechnology, UK). Ponceau S solutionas used to confirm adequate protein transfer (Sigma-Aldrich).embranes were blocked in Tris-buffered saline (TBS) contain-
ng 3% blocking reagent (NFM) and incubated sequentially withither rabbit anti-human KIT (anti-CD117, A4502; DAKO,lostrup, Denmark), rabbit anti-human PDGFRA (sc-338;anta Cruz Biotechnology, Santa Cruz, CA), rabbit antiphos-ho-PDGFRA (Santa Cruz Biotechnology), goat polyclonal an-i-PKC (Santa Cruz Biotechnology) antibodies, or antiphospho-RK1/2 (Cell Signaling, Beverly, MA) diluted in 5% blockingeagent. Membranes were then washed in TBST, probed withnti-rabbit or anti-goat immunoglobulin-HRP conjugate, andncubated with ECL substrate (Pierce).
Cell Cultures and TransfectionsBa/F3 cells were grown in RPMI1640 medium, supple-
ented with 10% fetal bovine serum (FBS), and interleukinIL)-3 (1 ng/mL) (Peprotech, Rocky Hill, NJ). 293T cells wererown in DMEM medium, supplemented with 10% FBS. Inome experiments, human PDGF-AA (Peprotech), the ligand ofDGFR�, was added. Ba/F3 cells were retrovirally transduced asescribed previously.14 For transfections of 293T cells, the cellsere seeded in 6-well plates and transfected using Fugene-6
Roche). Total DNA was constant for all transfections, but, tochieve different expression levels, the concentration of thepecific constructs was varied between 0.5 and 2 �g supple-
ented with empty vector DNA. Cells were harvested 24 hoursfter transfection. Cells were treated 90 minutes with imatinib
hen indicated. pStainingsHistopathologic examination was performed on forma-
in-fixed, paraffin-embedded tissue. Five-micrometer sectionsere used for routine H&E and immunohistochemical staining
avidin-biotin-peroxidase complex method) using the followingonoclonal (mc) and polyclonal (pc) antibodies: CD117 (pc,
/250; DAKO, Glostrup, Denmark), CD34 (mc, 1/10; Bectonickinson, San Jose, CA), SMA (mc, 1/100; DAKO, Glostrup,enmark), desmin (mc, 1/20; ICN, Aurora, OH), S-100 (pc,
/300; DAKO), epithelial membrane antigen (mc, 1/50; DAKO),nd neurofilament (mc, 1/20, Monosan, Gent, Belgium). TheD117 stain was performed with and without antigen retrieval
20 minute microwave treatment in citrate buffer, pH 6.0). Theresence of mast cells in all cases served as an internal controlor CD117 staining.
RNA In Situ Hybridization of DOG1The in situ hybridization experiments were performed
n the �80°C frozen tumor samples according to the protocolescribed previously.15 A sporadic GIST was used as positiveontrol.
ResultsPatient InformationThe patient family was reported before as a typical
xample of intestinal neurofibromatosis.1 The pedigree ofhe family is shown in Figure 1A. Individual I.2 died at thege of 35 years because of intestinal obstruction, and indi-idual II.3 underwent surgery for intestinal neurofibromasn 5 occasions (last surgery at the age of 72 years). We
nvestigated 3 affected sisters of this family (III.1, III.2, andII.3). All 3 complained of constipation. The 3 sisters hadormal intelligence, large hands, and broad wrists and werereated for glaucoma. It is not clear whether glaucoma is partf the phenotype because it is not reported in other familiesescribed with intestinal neurofibromatosis. Distinctive fea-ures present in this family are indicated in the family treeFigure 1A). A reciprocal balanced translocation t(12;14)(q13;13) is present in the family, and it is not associated with thehenotype or the severity of the phenotype.16 Individual III.1nderwent surgery on 4 occasions (first surgery at the age of1 years). She was in good health at the age of 71 years.ultiple small nodules of the small intestine were found in
ndividual III.2 during pelvic surgery at the age of 39 years.he tumors had a diameter ranging from 5 to 15 mm; 1 was
emoved and morphologically diagnosed as a neurofibroma.1
ndividual III.2 was last examined at the age of 66 years. Inndividual III.3, the first of 4 interventions for intestinalbstruction took place at the age of 38 years, the last at thege of 59 years. This was also the age of her last examination.ultiple tumors smaller than 10 mm in diameter were
resent in the small intestine and showed an intraluminalrowth. Only 1 tumor had a serosal location and was local-zed at the suture of a previous resection of a segment of theleum. There were no solid tumors palpable in the stomach.ive tumors were present in the 23 cm of resected terminal
leum (tumor 1: 4 mm; tumor 2: 24 � 18 � 13 mm; tumor: 45 � 25 � 25 mm; tumor 4: 18 � 14 � 17 mm; and tumor: 11 � 8 � 7 mm). These 5 tumors were available for
athologic examination and further studies. Tumor 2 had a
sh(sbf
leccMwntnw(
�a(Iod3gfa
fdim(wicotg
Ip(BesuPn
trwtt(
stdPtcib
vpft
Fittoopm
December 2006 INTESTINAL NEUROFIBROMATOSIS 1909
erosal location, but all other tumors were intraluminal andad a white, solid, and polypoid appearance. Individual II.4
father of the affected sisters) reportedly did not show anyymptoms of bowel obstruction but had large hands. He diedecause of an unrelated condition and was not available forurther study.
Pathologic Examination of IntestinalNeurofibromasFive tumors of individual III.3 were available for patho-
ogic examination. The polypoid nodules consisted of a mod-rately cellular spindle cell proliferation in a loosely textured toollagenous background, with a typical wavy aspect of theollagen fibers. There was no mitotic activity or nuclear atypia.
ast cells were numerous (Figure 2A). The histologic pictureas suggestive of neurofibromas. Immunohistochemistry didot show S100 protein expression (Figure 2B and D), indicatinghat the wavy cells were not Schwann cells as is seen in typicaleurofibromas. The myenteric plexus of the outer muscularall was used as an internal positive control for S100 positivity
igure 1. (A) Family tree: Features indicated are bowel obstruction,ntestinal tumors, large hands, and coarse face and the presence of aranslocation. Individual II.3 died from bowel obstruction and had intes-inal tumors; no data were available on the presence of the translocationr the large hands and coarse face. Individual I.2 died from bowelbstruction; all other features remain unknown. Individual III.1 is theroband investigated in Heimann et al.1 (B) The sequence shows theutation and amino acid change in individual III.1.
Figure 2B). CD117 (KIT), CD34, epithelial membrane antigen, I
-smooth muscle antigen, desmin, and neurofilament were neg-tive as well. CD117 showed a strong signal in the mast cellsFigure 2C). All stains were performed on all tumor specimens.n situ hybridization experiments did not show any expressionf DOG1, a marker for GIST (Figure 2E and F). Western blottingid not reveal any expression of S100, CD117, or PKC-� (Figure). The karyotype of short-term cultures of 3 tumors investi-ated was normal. In contrast to sporadic GIST, DNA extractedrom these tumors did not show any aberrations on array-CGHfter analysis with a CGH-smooth software (data not shown).
Mutation AnalysisMutation analysis of NF1, KIT, and PDGFRA was per-
ormed in individual III.1, and no sequence abnormalities wereetected in NF1 and KIT. A constitutional mutation was found
n exon 12 of PDGFRA (c.1664A�G; Y555C) (Figure 1B). Thisutation is also present in the 2 other affected family members
III.2 and III.3). To exclude the possibility that this mutationas a frequent polymorphism, we typed 76 alleles in random
ndividuals of the same ethnic background, and none of thesearried a G at position 1664 of PDGFRA. The 5 available tumorsf individual III.3 were investigated for additional somatic mu-ations in PDGFRA; none were found nor was loss of heterozy-osity (LOH) of PDGFRA present.
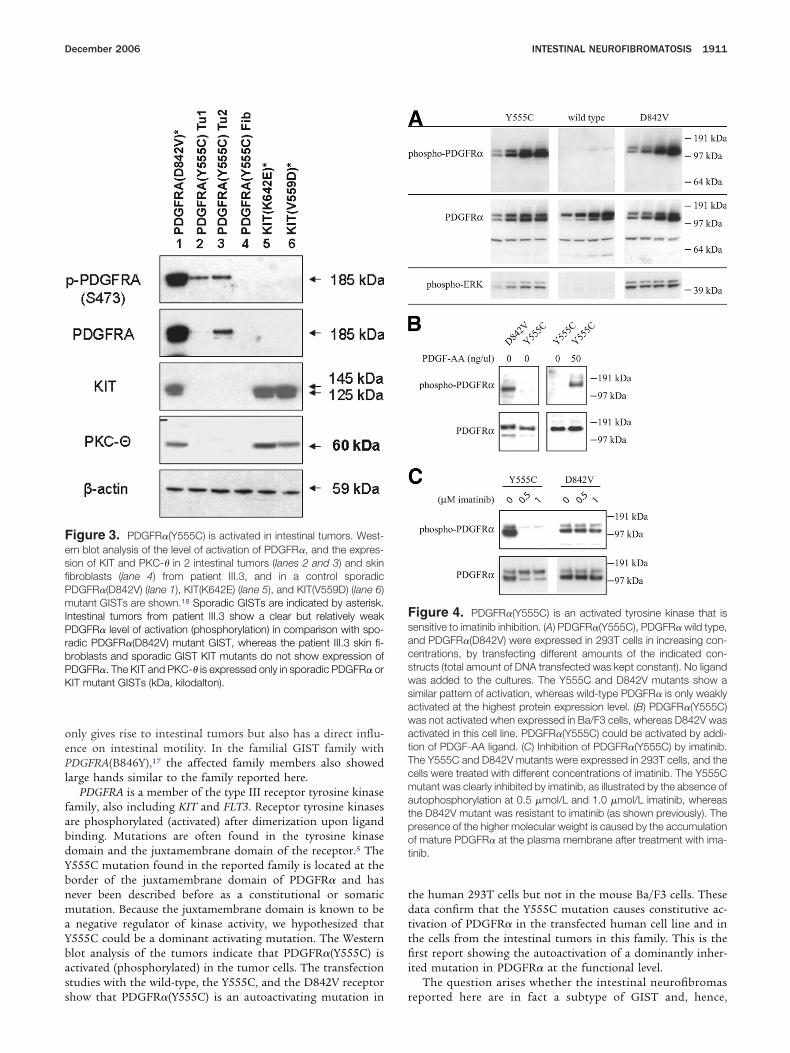
Functional Analysis of PDGFR� Y555CProtein extracts of 2 of the 5 tumors from individual
II.3 were further investigated and showed the presence ofhosphorylated (activated) PDGFR� on Western blot analysis
Figure 3). The PDGFR�(Y555C) mutant was expressed ina/F3 (a mouse pro-B-cell cell line) and 293T cells (a humanmbryonic kidney cell line). A PDGFR� with a known activatingomatic mutation (D842V) common in sporadic GIST16 wassed as a positive control for activated (phosphorylated)DGFR�. The wild-type PDGFR� receptor was expressed as aegative control.
In 293T cells, autophosphorylation was observed for bothhe D842V and the Y555C mutant but not for the wild-typeeceptor (Figure 4A). Treatment of the transfected 293T cellsith imatinib, a known PDGFR kinase inhibitor, confirmed
hat the Y555C mutant was sensitive to this drug, whereashe D842V mutant was resistant, as previously described16
Figure 4C).In Ba/F3 cells, strong autophosphorylation was only ob-
erved for the PDGFR�(D842V) mutant and not in the wildype or Y555C receptor (Figure 4B). Ba/F3 cells are IL-3 depen-ent for their growth, but expression of an activated form ofDGFR�, such as the PDGFR�(D842V) mutant, results inransformation of the cells to IL-3-independent growth.16 Inontrast to the PDGFR�(D842V) mutant, Ba/F3 cells express-ng the wild-type PDGFR� or PDGFR�(Y555C) mutant did notecome IL-3 independent (data not shown).
DiscussionAlthough only limited information is available on indi-
idual II.4 (father of the affected sisters), substantial evidence isresent that the mutation in PDGFRA(Y555C) found in thisamily, previously identified as having intestinal neurofibroma-osis, is dominantly inherited. One paternal aunt (individual
I.3) suffered from bowel obstruction and had intestinal tu-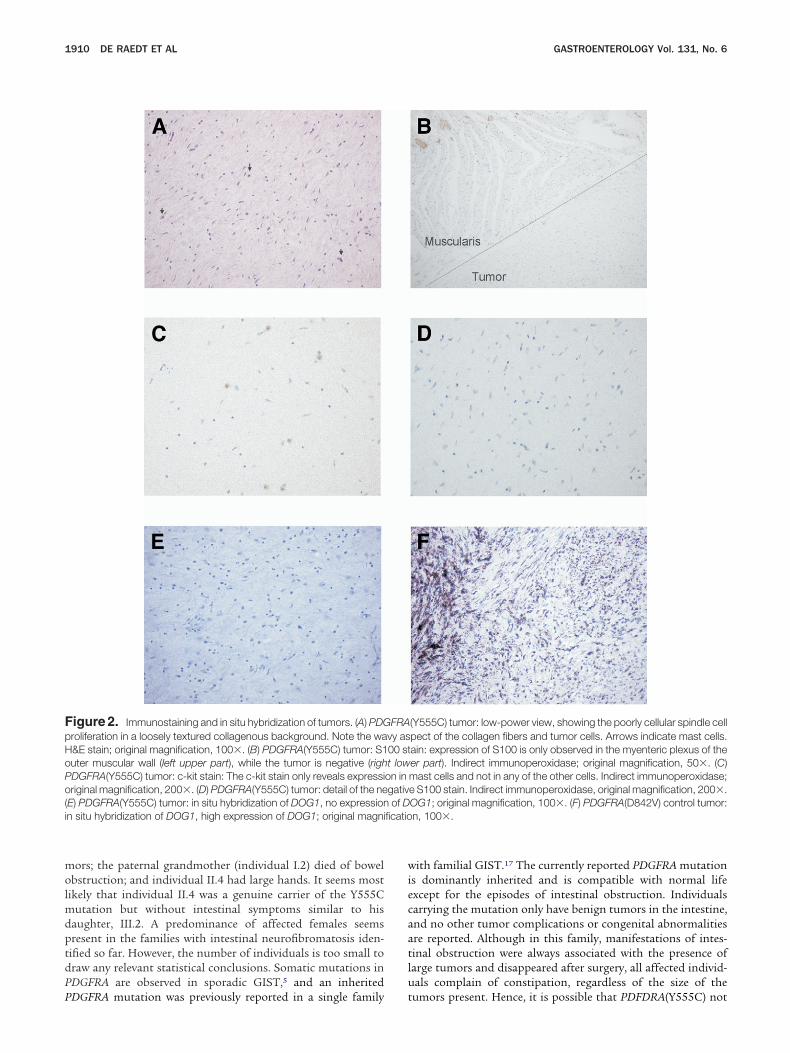
molmdptdPP
wiecaatlu
FpHoPo(i ficatio
1910 DE RAEDT ET AL GASTROENTEROLOGY Vol. 131, No. 6
ors; the paternal grandmother (individual I.2) died of bowelbstruction; and individual II.4 had large hands. It seems most
ikely that individual II.4 was a genuine carrier of the Y555Cutation but without intestinal symptoms similar to his
aughter, III.2. A predominance of affected females seemsresent in the families with intestinal neurofibromatosis iden-ified so far. However, the number of individuals is too small toraw any relevant statistical conclusions. Somatic mutations inDGFRA are observed in sporadic GIST,5 and an inherited
igure 2. Immunostaining and in situ hybridization of tumors. (A) PDGroliferation in a loosely textured collagenous background. Note the wa&E stain; original magnification, 100�. (B) PDGFRA(Y555C) tumor: S1uter muscular wall (left upper part), while the tumor is negative (righDGFRA(Y555C) tumor: c-kit stain: The c-kit stain only reveals expressiriginal magnification, 200�. (D) PDGFRA(Y555C) tumor: detail of the ne
E) PDGFRA(Y555C) tumor: in situ hybridization of DOG1, no expressionn situ hybridization of DOG1, high expression of DOG1; original magni
DGFRA mutation was previously reported in a single family t
ith familial GIST.17 The currently reported PDGFRA mutations dominantly inherited and is compatible with normal lifexcept for the episodes of intestinal obstruction. Individualsarrying the mutation only have benign tumors in the intestine,nd no other tumor complications or congenital abnormalitiesre reported. Although in this family, manifestations of intes-inal obstruction were always associated with the presence ofarge tumors and disappeared after surgery, all affected individ-als complain of constipation, regardless of the size of the
(Y555C) tumor: low-power view, showing the poorly cellular spindle cellpect of the collagen fibers and tumor cells. Arrows indicate mast cells.ain: expression of S100 is only observed in the myenteric plexus of theer part). Indirect immunoperoxidase; original magnification, 50�. (C)mast cells and not in any of the other cells. Indirect immunoperoxidase;e S100 stain. Indirect immunoperoxidase, original magnification, 200�.OG1; original magnification, 100�. (F) PDGFRA(D842V) control tumor:n, 100�.
FRAvy as00 stt lowon ingativof D
umors present. Hence, it is possible that PDFDRA(Y555C) not
oePl
fabdYbnmaYbass
tdttfii
FesfiPmIPrbPK
FsacswsawatTcmatpot
December 2006 INTESTINAL NEUROFIBROMATOSIS 1911
nly gives rise to intestinal tumors but also has a direct influ-nce on intestinal motility. In the familial GIST family withDGFRA(B846Y),17 the affected family members also showed
arge hands similar to the family reported here.PDGFRA is a member of the type III receptor tyrosine kinase
amily, also including KIT and FLT3. Receptor tyrosine kinasesre phosphorylated (activated) after dimerization upon ligandinding. Mutations are often found in the tyrosine kinaseomain and the juxtamembrane domain of the receptor.5 The555C mutation found in the reported family is located at theorder of the juxtamembrane domain of PDGFR� and hasever been described before as a constitutional or somaticutation. Because the juxtamembrane domain is known to benegative regulator of kinase activity, we hypothesized that
555C could be a dominant activating mutation. The Westernlot analysis of the tumors indicate that PDGFR�(Y555C) isctivated (phosphorylated) in the tumor cells. The transfectiontudies with the wild-type, the Y555C, and the D842V receptor
igure 3. PDGFR�(Y555C) is activated in intestinal tumors. West-rn blot analysis of the level of activation of PDGFR�, and the expres-ion of KIT and PKC-� in 2 intestinal tumors (lanes 2 and 3) and skinbroblasts (lane 4) from patient III.3, and in a control sporadicDGFR�(D842V) (lane 1), KIT(K642E) (lane 5), and KIT(V559D) (lane 6)utant GISTs are shown.18 Sporadic GISTs are indicated by asterisk.
ntestinal tumors from patient III.3 show a clear but relatively weakDGFR� level of activation (phosphorylation) in comparison with spo-
adic PDGFR�(D842V) mutant GIST, whereas the patient III.3 skin fi-roblasts and sporadic GIST KIT mutants do not show expression ofDGFR�. The KIT and PKC-� is expressed only in sporadic PDGFR� orIT mutant GISTs (kDa, kilodalton).
how that PDGFR�(Y555C) is an autoactivating mutation in r
he human 293T cells but not in the mouse Ba/F3 cells. Theseata confirm that the Y555C mutation causes constitutive ac-ivation of PDGFR� in the transfected human cell line and inhe cells from the intestinal tumors in this family. This is therst report showing the autoactivation of a dominantly inher-
ted mutation in PDGFR� at the functional level.The question arises whether the intestinal neurofibromas
igure 4. PDGFR�(Y555C) is an activated tyrosine kinase that isensitive to imatinib inhibition. (A) PDGFR�(Y555C), PDGFR� wild type,nd PDGFR�(D842V) were expressed in 293T cells in increasing con-entrations, by transfecting different amounts of the indicated con-tructs (total amount of DNA transfected was kept constant). No ligandas added to the cultures. The Y555C and D842V mutants show aimilar pattern of activation, whereas wild-type PDGFR� is only weaklyctivated at the highest protein expression level. (B) PDGFR�(Y555C)as not activated when expressed in Ba/F3 cells, whereas D842V wasctivated in this cell line. PDGFR�(Y555C) could be activated by addi-ion of PDGF-AA ligand. (C) Inhibition of PDGFR�(Y555C) by imatinib.he Y555C and D842V mutants were expressed in 293T cells, and theells were treated with different concentrations of imatinib. The Y555Cutant was clearly inhibited by imatinib, as illustrated by the absence of
utophosphorylation at 0.5 �mol/L and 1.0 �mol/L imatinib, whereashe D842V mutant was resistant to imatinib (as shown previously). Theresence of the higher molecular weight is caused by the accumulationf mature PDGFR� at the plasma membrane after treatment with ima-inib.
eported here are in fact a subtype of GIST and, hence,

wGlTitiwaoptwiuvw
Pfcc
1
1
1
1
1
1
1
1
1
HE
rV(gACa
s
r(
1912 DE RAEDT ET AL GASTROENTEROLOGY Vol. 131, No. 6
hether intestinal neurofibromatosis is a form of familialIST resulting from a mutation in PDGFRA. At the morpho-
ogic level, the investigated tumors resemble neurofibromas.his explains the diagnosis of intestinal neurofibromatosis
n the previous report on this family.1 However, in contrasto typical neurofibromas, these tumors were S100 negative bymmunostaining. In addition, all GIST markers investigatedere also negative. It has been reported that a small percent-ge of sporadic PDGFRA mutant GISTs are negative for KITn immunostaining.5,18 The clinical phenotype in the re-orted family together with the inherited activating muta-ion in PDGFRA suggests that these tumors are GISTs, ande would label these tumors as familial KIT-negative gastro-
ntestinal stromal tumors. It is interesting to note that,nlike the family described here, the tumors from the pre-iously reported familial GIST with a mutation in PDGFRAere KIT positive.17
Based on the presence of a constitutional mutation inDGFRA, a gene known to be involved in both sporadic andamilial forms of GIST, and the clinical phenotype, we con-lude that intestinal neurofibromatosis most likely is a spe-ific subtype of familial GIST.
References
1. Heimann R, Verhest A, Verschraegen J, Grosjean W, Draps JP,Hecht F. Hereditary intestinal neurofibromatosis. I. A distinctivegenetic disease. Neurofibromatosis 1988;1:26–32.
2. Lipton S, Zuckerbrod M. Familial enteric neurofibromatosis. MedTimes 1966;94:544–548.
3. Andersson J, Sihto H, Meis-Kindblom JM, Joensuu H, NupponenN, Kindblom LG. NF1-associated gastrointestinal stromal tumorshave unique clinical, phenotypic, and genotypic characteristics.Am J Surg Pathol 2005;29:1170–1176.
4. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, LongleyBJ, Miettinen M, O’Leary TJ, Remotti H, Rubin BP, Shmookler B,Sobin LH, Weiss SW. Diagnosis of gastrointestinal stromal tu-mors: a consensus approach. Hum Pathol 2002;33:459–465.
5. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinalstromal tumors. J Clin Oncol 2004;22:3813–3825.
6. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, IshiguroS, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad TunioG, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y. Gain-of-function mutations of c-kit in human gastrointestinal stromaltumors. Science 1998;279:577–580.
7. Maertens O, Prenen H, Debiec-Rychter M, Wozniak A, Sciot R,Pauwels P, De Wever I, Vermeesch JR, de Raedt T, De Paepe A,Speleman F, van Oosterom A, Messiaen L, Legius E. Molecularpathogenesis of multiple gastrointestinal stromal tumors in NF1patients. Hum Mol Genet 2006;15:1015–1023.
8. Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ,Joseph N, Singer S, Griffith DJ, Haley A, Town A, Demetri GD,Fletcher CD, Fletcher JA. PDGFRA activating mutations in gastro-intestinal stromal tumors. Science 2003;299:708–710.
9. Hirota S, Ohashi A, Nishida T, Isozaki K, Kinoshita K, ShinomuraY, Kitamura Y. Gain-of-function mutations of platelet-derivedgrowth factor receptor � gene in gastrointestinal stromal tumors.Gastroenterology 2003;125:660–667.
0. Legius E, Wu R, Eyssen M, Marynen P, Fryns JP, Cassiman JJ.Encephalocraniocutaneous lipomatosis with a mutation in the
NF1 gene. J Med Genet 1995;32:316–319.1. Leppig KA, Viskochil D, Neil S, Rubenstein A, Johnson VP, Zhu XL,Brothman AR, Stephens K. The detection of contiguous genedeletions at the neurofibromatosis 1 locus with fluorescence insitu hybridization. Cytogenet Cell Genet 1996;72:95–98.
2. Menten B, Maas N, Thienpont B, Buysse K, Vandesompele J,Melotte C, de Ravel T, Van Vooren S, Balikova I, Backx L, Jans-sens S, De Paepe A, De Moor B, Moreau Y, Marynen P, Fryns JP,Mortier G, Devriendt K, Speleman F, Vermeesch JR. Emergingpatterns of cryptic chromosomal imbalances in patients withidiopathic mental retardation and multiple congenital anomalies:a new series of 140 patients and review of the literature. J MedGenet 2006; e-pub.
3. Jong K, Marchiori E, Meijer G, Vaart AV, Ylstra B. Breakpointidentification and smoothing of array comparative genomic hy-bridization data. Bioinformatics 2004;20:3636–3637.
4. Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J,Kutok J, Clark J, Galinsky I, Griffin JD, Cross NC, Tefferi A, MaloneJ, Alam R, Schrier SL, Schmid J, Rose M, Vandenberghe P,Verhoef G, Boogaerts M, Wlodarska I, Kantarjian H, Marynen P,Coutre SE, Stone R, Gilliland DG. A tyrosine kinase created byfusion of the PDGFRA and FIP1L1 genes as a therapeutic targetof imatinib in idiopathic hypereosinophilic syndrome. N EnglJ Med 2003;348:1201–1214.
5. West RB, Corless CL, Chen X, Rubin BP, Subramanian S, Mont-gomery K, Zhu S, Ball CA, Nielsen TO, Patel R, Goldblum JR,Brown PO, Heinrich MC, van de Rijn M. The novel marker, DOG1,is expressed ubiquitously in gastrointestinal stromal tumors irre-spective of KIT or PDGFRA mutation status. Am J Pathol 2004;165:107–113.
6. Verhest A, Heinmann R, Verschraegen J, Vamos E, Hecht F.Heredity intestinal neurofibromatosis. II. Translocation betweenchromosomes 12 and 14. Neurofibromatosis 1988;1:33–36.
7. Chompret A, Kannengiesser C, Barrois M, Terrier P, Dahan P,Tursz T, Lenoir GM, Bressac-De Paillerets B. PDGFRA germlinemutation in a family with multiple cases of gastrointestinal stro-mal tumor. Gastroenterology 2004;126:318–321.
8. Debiec-Rychter M, Wasag B, Stul M, De Wever I, Van Oosterom A,Hagemeijer A, Sciot R. Gastrointestinal stromal tumours (GISTs)negative for KIT (CD117 antigen) immunoreactivity. J Pathol2004;202:430–438.
Received March 24, 2006. Accepted June 8, 2006.Address requests for reprints to: Eric Legius, MD, Center of
uman Genetics, Herestraat 49, 3000 Leuven, Belgium. e-mail:[email protected]; fax: (32) 16 346051.Supported by the Emmanuel Vanderschueren Fonds (to T.D.); by
esearch grants from the Fonds voor Wetenschappelijk Onderzoeklaanderen (G.0096.02, to E.L.), (G.0286.05, to M.D-R.), andG.0507.04, to P.M.); by the Interuniversity Attraction Poles (IAP)ranted by the Federal Office for Scientific, Technical and Culturalffairs, Belgium (2002-2006; P5/25) (to P.M. and E.L.); and by aoncerted Action Grant from the K.U. Leuven and the Belgian Feder-tion against Cancer (SCIE2003-19, to J.C.; SCIE2003-33, to E.L.).The authors thank the family members who collaborated in this
tudy and Dr Thomy de Ravel for critically reading the manuscript.J.C. is a postdoctoral researcher, and E.L. is a part-time clinical
esearcher of the Fonds voor Wetenschappelijk Onderzoek VlaanderenFWO).
T.D.R. and J.C. contributed equally to this article.




















