
interacções toxicológicas entre o álcool e a ecstasy. estudos ...
374
Helena de Oliveira Pontes INTERACÇÕES TOXICOLÓGICAS ENTRE O ÁLCOOL E A ECSTASY. ESTUDOS IN VIVO E IN VITRO. Orientador: Professora Doutora Maria de Lourdes Pinho de Almeida Souteiro Bastos Co-orientadores: Professor Doutor Félix Dias Carvalho Professor Doutor Fernando Manuel Gomes Remião Porto, 2009
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of interacções toxicológicas entre o álcool e a ecstasy. estudos ...

Helena de Oliveira Pontes
INTERACÇÕES TOXICOLÓGICAS ENTRE O ÁLCOOL E A ECSTASY.
ESTUDOS IN VIVO E IN VITRO. Orientador:
Professora Doutora Maria de Lourdes Pinho de Almeida Souteiro Bastos Co-orientadores:
Professor Doutor Félix Dias Carvalho
Professor Doutor Fernando Manuel Gomes Remião
Porto, 2009


Aos meus pais, Arminda e João Carlos
À minha irmã Diana


Ao Fernando


Os estudos apresentados nesta dissertação foram realizados no Laboratório
Associado REQUIMTE/Serviço de Toxicologia da Faculdade de Farmácia da
Universidade do Porto.
Estes estudos contaram com o apoio financeiro da Fundação para a Ciência e
a Tecnologia (FCT) através da atribuição de uma Bolsa de Doutoramento
(SFRH/BD/18527/2004) e do Fundo Europeu para o Desenvolvimento Regional
(FEDER) (POCI/SAU-FCF/57187/2004).


Fazem parte integrante desta dissertação os seguintes trabalhos já publicados:
Artigos em revistas internacionais com arbitragem científica
I. Pontes H, Duarte JA, Guedes de Pinho P, Soares ME, Fernandes E, Dinis-
Oliveira RJ, Sousa C, Silva R, Carmo H, Casal S, Remião F, Carvalho F,
Bastos ML (2008) Chronic exposure to ethanol exacerbates MDMA-induced
hyperthermia and exposes liver to severe MDMA-induced toxicity in CD1 mice.
Toxicology. 252: 64-71.
II. Pontes H, Santos-Marques MJ, Fernandes E, Duarte JA, Remião F,
Carvalho F, Bastos ML (2008) Effect of chronic ethanol exposure on the
hepatotoxicity of ecstasy in mice: An ex vivo study. Toxicology In Vitro. 22: 910-
20.
III. Pontes H, Guedes de Pinho P, Casal S, Carmo H, Santos A, Magalhães T,
Remião F, Carvalho F, Bastos ML (2008) Gas chromatography determination of
acetone, acetaldehyde, ethanol and methanol in biological matrices and cell
culture. Aceite para publicação no Journal of Chromatographic Science.
IV. Pontes H, Carvalho M, Guedes de Pinho P, Carmo H, Remião F, Carvalho
F, Bastos ML (2008) Ethanol, the forgotten artifact in cell culture. Archives of
Toxicology. 82: 197–8.
V. Pontes H, Sousa C, Silva R, Fernandes E, Carmo H, Remião F, Carvalho F,
Bastos ML (2008) Synergistic toxicity of ethanol and MDMA towards primary
cultured rat hepatocytes. Toxicology (doi: 10.1016/j.tox.2008.09.009)


Resumos em actas de encontros científicos publicados em revistas com
arbitragem cientifica
I. Pontes H, Marques MJ, Remião F, Carvalho F, Bastos MdL (2006) The
repeated exposure of mice to ethanol increases its susceptibility to ecstasy-
induced hepatotoxicity. Revista Portuguesa de Farmácia. 52:33
II. Pontes H, Marques MJ, Remião F, Carvalho F, Bastos ML (2006) Ethanol
and ecstasy: Allied enemies of freshly isolated mouse hepatocytes. Toxicology
Letters. 164 (Suppl 1):S205
III. Pontes H, Fernandes E, Duarte JA, Remião F, Carvalho F, Bastos ML
(2007) Ethanol and ecstasy: a dangerous cocktail to the liver? Toxicology
Letters. 172 (Suppl 1):S240
IV. Pontes H, Silva R, Fernandes E, Carmo H, Remião F, Carvalho F, Bastos
ML (2008) Ethanol increases MDMA toxicity to primary rat hepatocytes cultures.
Toxicology Letters. 180 (Suppl 1):S162


AGRADECIMENTOS
Seguir trilhos é fácil, mas quando temos que pisar terrenos onde nunca
ninguém andou, temos que ser nós a fazer o nosso caminho correndo o risco
de ter que voltar atrás algumas vezes, mas não é esse o encanto da ciência?
Um Doutoramento é, por isso, um caminho de aprendizagem que é feito pelos
nossos próprios passos, à medida que crescemos quer a nível pessoal, quer a
nível científico. Mas, a beleza da caminhada depende dos que vão connosco.
Por isso, gostaria de agradecer a todos aqueles que contribuiram para a
concretização deste trabalho, embora temendo nunca encontrar as melhores
palavras ou as palavras suficientes para exprimir a minha imensa gratidão.
Gostaria, assim, de expressar o meu mais sincero agradecimento:
À Professora Doutora Maria de Lourdes Bastos, em primeiro lugar, por
me ter aceite como aluna de Doutoramento e, sobretudo, pela orientação
rigorosa e magistral desta dissertação e de todo o trabalho científico que a ela
deu origem. Espero que, um dia, possa sentir-se muito orgulhosa desta sua
pupila.
Ao Professor Doutor Félix Dias Carvalho pela co-orientação deste
trabalho e pelo constante incentivo e apoio. Obrigada por nunca desanimar e
por aquelas palavras de alento nas horas em que remávamos contra a corrente
porque o trabalho não corria como gostaríamos.
Ao Professor Doutor Fernando Remião, pela co-orientação deste
trabalho, e pela paciência e pela disponibilidade permanente para interromper
tudo o que estivesse a fazer para ajudar a resolver algum imprevisto, para
esclarecer alguma dúvida ou para dar uma palavra de apoio.
À Engenheira Maria Elisa Soares, pelo apoio e preocupação com que
acompanha os alunos do laboratório de Toxicologia, para que consigamos
ultrapassar todas as dificuldades com que nos deparamos e encaremos cada
dia com um sorriso. É de facto como uma mãe para todos nós. Obrigada pela

amizade, pelo carinho, pela colaboração nos trabalhos experimentais, pelos
momentos de alegria e por me ter ensinado a ser uma pessoa cada vez
melhor. Nunca esquecerei os dias do famoso Coro da Toxicologia que, quando
ensaiava, não deixava ninguém indiferente...
À Professora Doutora Maria Helena Carmo pela amizade com que me
distingue, por todos os bons momentos que passámos, quer no laboratório,
quer fora dele, e pelo apoio que me deu desde o início destes trabalhos
ensinando-me que um bom trabalho surge quando somos exigentes connosco
próprios. Também quero agradecer a resposta pronta, e as palavras de apoio
que nunca vou esquecer, nos momentos menos bons. Obrigada, Boss!
Ao Professor Doutor José Alberto Duarte, pela sua valiosa colaboração
neste trabalho, por nunca dizer que não quando lhe pedi ajuda ou conselho,
mesmo que isso significasse trabalhar em dias feriados, por tudo o que me
ensinou sobre microscopia e por me ter aceite como mais um dos seus alunos.
À Doutora Paula Guedes de Pinho e à Professora Susana Casal, pelo
seu contributo na selecção, validação e aplicação das técnicas de
cromatografia gasosa essenciais à realização deste trabalho. Obrigada pelos
conhecimentos que me transmitiram!
À Renata, pela amizade que nos tornou inseparáveis, pelas gargalhadas
com ou sem motivo, pelas cantorias e pelas coreografias que nos ajudavam a
ultrapassar aqueles dias em que nada corria bem e pela ajuda quando o
trabalho ultrapassava a capacidade das minhas duas mãos. Um obrigado muito
especial por me ajudar na formatação desta dissertação. Não sei se algum dia
conseguirei retribuir tudo aquilo que recebi de ti!
À Professora Doutora Márcia Carvalho, pelo incentivo, pelos conselhos
úteis, e sobretudo, por me alertar para os pequenos detalhes que podem dar
grandes frutos.

À Professora Doutora Eduarda Fernandes, pelo incentivo, pelo apoio e
pela sua indispensável contribuição para este trabalho ao extrair a MDMA.
A todos os companheiros de laboratório (Ricardo, Miguel, Carla, Daniel,
Görkem, Maria João, João, Vera, Filipa e mais recentemente a Luciana) por
fazerem com que cada dia seja diferente do anterior, pelo companheirismo,
pelo bom ambiente de trabalho e pela amizade que irei sempre retribuir.
Às técnicas Júlia Caramez, Conceição Morais e Graziela Fernandes (já
aposentada) e à D. Celeste, pelo apoio, disponibilidade e amizade que
contribuiram para que estes trabalhos corressem sempre da melhor forma.
Aos amigos que me têm acompanhado neste percurso (Vera, Elvira,
Horácio, Dinis, Sirigaitas de Farmácia, Ema, Marisa), pela amizade, pelo
interesse demonstrado no meu trabalho e por me ajudarem a estimular o gosto
pela investigação e a curiosidade científica.
Finalmente, quero agradecer ao Fernando e à minha família, em
especial ao meus pais, Arminda e João Carlos, e à minha irmã Diana, por tudo!
Pelo apoio incondicional que me deram em todas as decisões que tomei e que,
por vezes, obrigaram a alguns sacrifícios, pela paciência, pelo amor e carinho e
pelas palavras de conforto, quando o trabalho não corria tão bem e o meu
humor não era dos melhores. Espero que vocês possam orgulhar-se de mim da
mesma forma que eu me orgulho de vocês!


ÍNDICE GERAL


_____________________________________________________ Índice Geral
I
ÍNDICE GERAL RESUMO....................................................................................................... XVII
ABSTRACT.................................................................................................. XXIII
LISTA DE ABREVIATURAS........................................................................ XXIX
1. INTRODUÇÃO ............................................................................................ 3
1.1. História da MDMA................................................................................ 4
1.2. Relação estrutura/actividade da MDMA............................................... 6
1.3. Produção de MDMA............................................................................. 7
1.4. Comercialização de MDMA.................................................................. 7
1.5. Farmacocinética da MDMA.................................................................. 8
1.5.1. Absorção....................................................................................... 8
1.5.2. Distribuição ................................................................................. 10
1.5.3. Metabolismo................................................................................ 10
1.5.4. Excreção..................................................................................... 14
1.6. Mecanismo de acção e principais efeitos da MDMA.......................... 14
1.6.1. Efeito na libertação, transporte e receptores das monoaminas
neurotransmissoras................................................................................... 15
1.6.2. Inibição da monoamina oxidase (MAO) ...................................... 19
1.6.3. Inibição da triptofano hidroxilase (TH) ........................................ 19
1.6.4. Alterações da expressão genética .............................................. 20
1.6.5. Efeito neuroendócrino................................................................. 21
1.6.6. Alterações no sistema termorregulador ...................................... 22
1.6.7. Efeito no fluxo sanguíneo e actividade cerebral.......................... 25
1.7. Efeitos tóxicos da MDMA................................................................... 25
1.7.1. Efeitos tóxicos agudos ................................................................ 27
1.7.2. Efeitos tóxicos a longo prazo ...................................................... 29
1.7.3. Mortes associadas ao consumo de MDMA................................. 31
1.8. Estudos epidemiológicos sobre a utilização da MDMA...................... 43
1.8.1. Onde? – Locais de consumo de MDMA ..................................... 53
1.8.2. Como e quando? – Padrões de consumo de MDMA.................. 53
1.9. Factores condicionantes dos efeitos da MDMA ................................. 54
1.9.1. Dose de MDMA........................................................................... 55
1.9.2. Condições ambientais no momento do consumo ....................... 59

Índice Geral _____________________________________________________
II
1.9.3. Características do consumidor de MDMA................................... 62
1.9.3.1. Idade.................................................................................... 62
1.9.3.2. Etnia .................................................................................... 64
1.9.3.3. Género................................................................................. 64
1.9.3.4. Variabilidade farmacogenética interindividual...................... 68
1.9.3.4.1. Isoenzimas do citocromo P450........................................ 68
1.9.3.4.2. Catecol-O-metiltransferase.............................................. 70
1.9.3.4.3. UDP-glucuronosiltransferase........................................... 71
1.9.3.4.4. Sulfotransferase (SULT) .................................................. 71
1.9.3.4.5. Receptores da 5-HT ........................................................ 71
1.9.3.4.6. Receptores da DA ........................................................... 72
1.9.3.4.7. Transportador da 5-HT .................................................... 72
1.9.3.5. Orientações sexuais ............................................................ 73
1.9.4. Alterações fisiológicas e fisiopatológicas .................................... 73
1.9.4.1. Gravidez .............................................................................. 74
1.9.4.2. Patologias subjacentes ao consumo de MDMA................... 76
1.9.4.2.1. Hipo/hipertiroidismo......................................................... 76
1.9.4.2.2. Insuficiência hepática ...................................................... 77
1.9.4.2.3. Insuficiência renal............................................................ 77
1.9.4.2.4. Patologias inflamatórias e infecciosas............................. 79
1.9.4.2.5. Patologia cardíaca........................................................... 79
1.9.4.2.6. Diabetes mellitus ............................................................. 80
1.9.4.2.7. Obesidade ....................................................................... 81
1.9.5. Interacções da MDMA com outros compostos............................ 81
1.9.5.1. Fármacos............................................................................. 83
1.9.5.1.1. Antidepressivos ............................................................... 93
1.9.5.1.1.1. Inibidores selectivos da recaptação da serotonina
(ISRS) .................................................................................. 93
1.9.5.1.1.2. Inibidores da monoamina oxidase (IMAO) ................ 95
1.9.5.1.2. Anti-retrovirais ................................................................. 97
1.9.5.2. Alimentos ............................................................................. 99
1.9.5.3. Contaminantes das pastilhas de ecstasy........................... 104
1.9.5.3.1. Excedentes e sub-produtos da síntese de MDMA......... 107

_____________________________________________________ Índice Geral
III
1.9.5.3.2. Outros estimulantes anfetamínicos [anfetamina, MTA,
fenfluramina, fentermina, análogos sintéticos da anfetamina (PMA, 4-
metiltioanfetamina, 2-CB) e derivados metilenodioxi (MDA,MDEA)] 109
1.9.5.3.2.1. MDA e MDEA.......................................................... 109
1.9.5.3.2.2. MTA ........................................................................ 110
1.9.5.3.2.3. Outras anfetaminas................................................. 111
1.9.5.3.3. Ketamina ....................................................................... 112
1.9.5.3.4. Cocaína ......................................................................... 114
1.9.5.3.5. Efedrina/pseudoefedrina................................................ 114
1.9.5.3.6. Cafeína .......................................................................... 115
1.9.5.3.7. Paracetamol .................................................................. 117
1.9.5.3.8. LSD ............................................................................... 118
1.9.5.3.9. Dextrometorfano (DXM)................................................. 118
1.9.5.3.10. Excipientes inertes ........................................................ 119
1.9.5.4. Drogas de abuso ilícitas co-consumidas intencionalmente
com MDMA.......................................................................................... 120
1.9.5.4.1. Derivados da Cannabis ................................................. 126
1.9.5.4.2. Estimulantes anfetamínicos........................................... 129
1.9.5.4.3. Cocaína ......................................................................... 132
1.9.5.4.4. LSD ............................................................................... 133
1.9.5.4.5. Ketamina ....................................................................... 135
1.9.5.4.6. GHB (“ecstasy líquido”) ................................................. 136
1.9.5.4.7. Outras substâncias alucinogénicas – Cogumelos
“mágicos” ...................................................................................... 137
1.9.5.5. Tabaco............................................................................... 137
1.9.5.6. Etanol ................................................................................ 139
1.9.5.6.1. Características farmacocinéticas do etanol ................... 140
1.9.5.6.1.1. Absorção................................................................. 140
1.9.5.6.1.2. Distribuição ............................................................. 141
1.9.5.6.1.3. Metabolismo............................................................ 142
1.9.5.6.1.4. Excreção ................................................................. 148
1.9.5.6.2. Mecanismos de acção e de toxicidade do etanol .......... 148
1.9.5.6.2.1. Efeitos na neurotransmissão................................... 149
1.9.5.6.2.1.1. Efeitos na transmissão glutamatérgica............. 150

Índice Geral _____________________________________________________
IV
1.9.5.6.2.1.2. Efeitos na transmissão GABAérgica................. 151
1.9.5.6.2.1.3. Efeitos nas vias serotonérgicas........................ 152
1.9.5.6.2.1.3.1. Efeitos nos níveis de 5-HT......................... 152
1.9.5.6.2.1.3.2. Efeitos nos receptores 5-HT ...................... 153
1.9.5.6.2.1.4. Aumento da libertação de DA........................... 154
1.9.5.6.2.2. Alterações no sistema termorregulador .................. 156
1.9.5.6.2.3. Alterações psicomotoras......................................... 157
1.9.5.6.2.4. Efeitos neuroendócrinos ......................................... 161
1.9.5.6.2.5. Efeitos cardiovasculares ......................................... 163
1.9.5.6.2.6. Efeitos metabólicos ................................................. 164
1.9.5.6.2.7. Efeitos no sistema imunitário .................................. 164
1.9.5.6.2.8. Efeitos no sistema hepato-biliar .............................. 166
1.9.5.6.2.9. Efeitos no tracto gastro-intestinal ............................ 169
1.9.5.6.2.10. Efeitos no sistema urinário...................................... 169
1.9.5.6.2.11. Efeitos nos músculos .............................................. 170
1.9.5.6.2.12. Efeitos teratogénicos............................................... 170
1.9.5.6.2.13. Carcinogénese........................................................ 171
1.10. Principais considerações ................................................................. 172
2. OBJECTIVOS ......................................................................................... 177
3. TRABALHO EXPERIMENTAL ............................................................... 179
Trabalho I. Optimização das doses de MDMA a administrar a ratinhos
previamente expostos repetidamente a etanol........................................ 181
Trabalho II. Chronic exposure to ethanol exacerbates MDMA-induced
hyperthermia and exposes liver to severe MDMA-induced toxicity in CD1
mice. .................................................................................................. 187
Trabalho III. Effect of chronic ethanol exposure on the hepatotoxicity of
ecstasy in mice: An ex vivo study............................................................ 183
Trabalho IV. Synergistic toxicity of ethanol and MDMA towards primary
cultured rat hepatocytes. ......................................................................... 197
Trabalho V. Interacções metabólicas entre o etanol e a MDMA em culturas
primárias de hepatócitos de rato. ............................................................ 235
Trabalho VI. Gas chromatography determination of acetone, acetaldehyde,
ethanol and methanol in biological matrices and cell culture................... 233
Trabalho VII. Ethanol, the forgotten artifact in cell culture. ..................... 251

_____________________________________________________ Índice Geral
V
4. DISCUSSÃO INTEGRADA..................................................................... 261
5. CONCLUSÕES FINAIS .......................................................................... 277
6. REFERÊNCIAS ...................................................................................... 283

Índice Geral _____________________________________________________
VI

ÍNDICE DE FIGURAS


_________________________________________________ Índice de Figuras
IX
ÍNDICE DE FIGURAS Figura 1. Estrutura química da metanfetamina, mescalina e do enantiómero
farmacologicamente mais activo da 3,4-metilenodioximetanfetamina................ 6
Figura 2. Exemplos de pastilhas e cápsulas de ecstasy. .................................. 8
Figura 3. Esquema das vias metabólicas da MDMA ....................................... 11
Figura 4. Oxidação dos metabolitos catecóis da MDMA a quinonas, semi-
quinonas e aminocromos, e formação dos conjugados com a GSH. ............... 13
Figura 5. Mecanismo de acção biológica da MDMA ao nível dos neurónios
serotonérgicos.................................................................................................. 16
Figura 6. Hipótese integrada para os efeitos termogénicos e mionecróticos da
MDMA. ............................................................................................................. 24
Figura 7. Estruturas químicas dos principais estimulantes anfetamínicos
encontrados na composição de pastilhas de ecstasy, além da MDMA.......... 109
Figura 8. Metabolismo oxidativo do etanol. ................................................... 143
Figura 9. Hipótese que sugere possíveis interacções metabólicas entre o EtOH
e a MDMA baseada na influência do primeiro nas principais isoenzimas do
CYP450 metabolizadoras deste derivado anfetamínico................................. 146
Figura 10. Mecanismos de acção biológica do etanol ao nível da
neurotransmissão glutamatérgica, GABAérgica, serotonérgica e dopaminérgica.
....................................................................................................................... 150
Figura 11. Fontes de complicações neurológicas do álcool e alcoolismo. .... 159
Figura 12. Percentagem de sobrevivência de ratinhos CD1, previamente
expostos a 12% de etanol na água de bebida durante 8 semanas, após
administração de 10 mg/kg ou 20 mg/kg de MDMA....................................... 185

Índice de Figuras _________________________________________________
X
Figura 13. Procedimento experimental utilizado para a avaliação da influência
do etanol no perfil metabólico da MDMA em culturas primárias de hepatócitos
de rato. ........................................................................................................... 226
Figura 14. Metabolização da MDMA a MDA, HMMA e HMA......................... 227
Figura 15. Preparação de amostras de meio de cultura para quantificação de
MDMA, MDA, HMMA e HMA por GC-MS....................................................... 228
Figura 16. Efeito de MDMA 1,6 mM (M) e da hipertermia na percentagem de
morte celular na presença ou na ausência de etanol, DAS e KET................. 229
Figura 17. Efeito da hipertermia e da co-incubação com etanol 300 mM, DAS
150 μM e KET 0,18 μM na formação de metabolitos da MDMA (MDA, HMMA e
HMA) em culturas primárias de hepatócitos de rato....................................... 230
Figura 18. Mecanismo de resposta ao choque térmico. ................................ 265
Figura 19. Mecanismo de activação do NF-κB.............................................. 266
Figura 20. Esquema-resumo dos resultados obtidos no estudo das interacções
hepatotóxicas entre etanol e MDMA. ............................................................. 278

ÍNDICE DE TABELAS


_________________________________________________ Índice de Tabelas
XIII
ÍNDICE DE TABELAS Tabela 1. Mortes associadas ao consumo de ecstasy de 1988 a 2007........... 34
Tabela 2. Estudos epidemiológicos acerca dos padrões de consumo da MDMA.
......................................................................................................................... 44
Tabela 3. Perfil de impurezas e conteúdo em MDMA de pastilhas vendidas
como ecstasy. .................................................................................................. 57
Tabela 4. Fármacos com potencial para estabelecer interacções
farmacocinéticas e/ou farmacodinâmicas com a MDMA.................................. 86
Tabela 5. Medicamentos, alimentos, e seus constituintes, que influenciam a
actividade e expressão das principais isoenzimas do CYP450 envolvidas no
metabolismo da MDMA. ................................................................................. 100
Tabela 6. Possíveis efeitos dos Excedentes e sub-produtos da síntese de
MDMA na toxicidade da ecstasy. ................................................................... 108
Tabela 7. Padrões de policonsumo de drogas de abuso reportados por
consumidores de ecstasy. .............................................................................. 121

Índice de Tabelas _________________________________________________
XIV

RESUMO


________________________________________________________ Resumo
XVII
RESUMO
A 3,4-metilenodioximetanfetamina (MDMA ou ecstasy) é uma droga de
abuso que tem sido amplamente consumida desde os anos 80 devido às suas
propriedades entactogénicas, empatogénicas e moderadamente
alucinogénicas, bem como ao pressuposto errado de se tratar de uma droga
com reduzida toxicidade.
Os efeitos/toxicidade da MDMA são extremamente imprevisíveis, uma
vez que são condicionados por diversos factores que vão desde a dose
consumida às características genéticas, fisiológicas e fisiopatológicas do
consumidor, às condições ambientais em que os consumidores a utilizam e
sobretudo à co-exposição a outras substâncias que podem interagir com a
MDMA. Estas interacções da MDMA podem surgir com substâncias não-
psicoactivas veiculadas em alimentos ou medicamentos ou, mais preocupante,
com substâncias psicoactivas ingeridas inadvertidamente como contaminantes
das pastilhas de ecstasy, ou intencionalmente co-consumidas para modular os
efeitos da MDMA (nesta última situação, o risco de interacção será
potencialmente maior porque as doses das substâncias psicoactivas
consumidas são, geralmente, superiores às ingeridas pelo consumo de
pastilhas de ecstasy contaminadas).
O policonsumo afirma-se, então, como o padrão mais frequente de
consumo de ecstasy, com especial prevalência do co-consumo com outras
substâncias psicoactivas, sobretudo tabaco, Cannabis e álcool que pode
ocorrer antes, durante ou pouco tempo depois da exposição à MDMA. Estes
padrões de policonsumo podem aumentar os riscos para a saúde associados
ao consumo de MDMA devido às imprevisíveis reacções químicas e
idiossincráticas que podem resultar da mistura de xenobióticos no organismo,
conduzindo potencialmente a interacções farmacocinéticas e/ou
farmacodinâmicas.
Entre os compostos co-consumidos com a MDMA, o etanol ocupa um
lugar de destaque, uma vez que devido à sua elevada acessibilidade e
aceitação social é amplamente consumido, frequentemente em grandes
quantidades, pelos indivíduos das faixas etárias mais jovens, nas quais se
regista, também, uma maior prevalência do consumo de MDMA. Por este

Resumo ________________________________________________________
XVIII
motivo, o estudo das interacções entre estes dois compostos no organismo é
de extrema importância.
Quando as experiências contempladas nesta dissertação foram
desenhadas, apenas existiam dados relativos ao co-consumo de MDMA e
etanol que se centravam nas suas mútuas alterações farmacocinéticas e nos
seus efeitos subjectivos e comportamentais em humanos. No entanto, não
existiam estudos em que se avaliasse quais os mecanismos subjacentes à
toxicidade hepática induzida pelo consumo de ambas as substâncias em
associação. Uma vez que ambas as substâncias são metabolizadas no fígado
e levam à formação de metabolitos dotados de maior toxicidade do que os seus
precursores, o estudo dos efeitos hepáticos desta mistura reveste-se de
extrema importância. Por esse motivo, o objectivo da investigação da qual
resulta a presente dissertação foi estudar os efeitos da exposição simultânea a
ecstasy e etanol a nível hepático e avaliar de que forma é que a hipertermia
(reacção potencialmente fatal frequentemente associada à exposição in vivo à
MDMA) poderia afectar os efeitos hepáticos desta mistura.
Para atingir o objectivo proposto, numa primeira fase, foram realizados
ensaios in vivo com ratinhos expostos a etanol a 12% durante 8 semanas,
seguindo-se uma administração única de MDMA numa dose de 10 mg/kg. Este
protocolo foi realizado no sentido de simular o padrão de consumo mais
habitual destas duas substâncias, no qual o início do consumo de etanol
ocorre, geralmente, antes do primeiro contacto com a MDMA. Seguidamente,
foram desenvolvidos estudos ex vivo, seguindo o mesmo protocolo de
exposição ao etanol. Nesta situação, apenas os hepatócitos isolados dos
animais expostos ao etanol foram incubados com MDMA no sentido de tentar
perceber os efeitos induzidos pela MDMA nos hepatócitos de animais expostos
repetidamente in vivo ao etanol que poderiam elucidar as alterações
observadas nos ensaios in vivo. Embora os resultados obtidos tenham
confirmado o potencial tóxico desta mistura, o modelo experimental baseado
em suspensões de hepatócitos frescos utilizado nos ensaios ex vivo tem como
limitação apenas permitir incubações máximas de 3-4 horas, o que pode
impedir a observação de determinados indicadores de dano ou reacção celular
a uma determinada agressão. Por esse motivo, foram desenvolvidos estudos in
vitro nos quais hepatócitos, em cultura primária, foram directamente expostos à

________________________________________________________ Resumo
XIX
mistura etanol+MDMA. Este último modelo foi utilizado para esclarecer os
mecanismos subjacentes à toxicidade hepática desta mistura, mas também
para avaliar uma possível interacção metabólica entre os dois compostos, já
que o etanol tem a capacidade de alterar a actividade de várias enzimas
hepáticas, incluindo aquelas envolvidas no metabolismo da MDMA.
Os resultados obtidos nos trabalhos realizados no âmbito desta
dissertação permitiram-nos constatar que: (i) a pré-exposição repetida ao
etanol tem um efeito sinérgico na hipertermia e hepatotoxicidade induzidas in
vivo pela MDMA em ratinhos Charles-River CD-1; (ii) a pré-exposição repetida
a etanol a 12% aumenta a vulnerabilidade dos hepatócitos de ratinhos Charles-
River CD-1 relativamente a agressões pró-oxidantes provavelmente por
diminuir o conteúdo celular em GSH e aumentar o conteúdo em GSSG,
aumentando a propensão para a perturbação da homeostase dos grupos tiol
intracelulares que pode resultar na perda de funcionalidades proteicas e na
iniciação de uma cascata de eventos que pode culminar na morte celular; (iii) a
co-exposição aguda a etanol+MDMA aumenta a toxicidade hepática induzida in
vitro pela MDMA, em culturas primárias de hepatócitos obtidos de ratos Wistar,
através de uma diminuição das defesas antioxidantes, diminuição das reservas
energéticas das células, aumento da formação de espécies reactivas e
aumento da morte celular por necrose, numa extensão dependente da
temperatura de incubação; (iv) a exposição aguda in vitro ao etanol, de culturas
primárias de hepatócitos obtidos de ratos Wistar, aumenta o metabolismo da
MDMA a MDA, HMA e HMMA; (v) os efeitos tóxicos da mistura etanol+MDMA
são marcadamente aumentados em condições hipertérmicas.
Em conclusão, o co-consumo de etanol e MDMA aumenta a
hepatotoxicidade da ecstasy mediante um aumento da resposta hipertérmica
associada ao consumo deste derivado anfetamínico, uma diminuição das
defesas antioxidantes e um aumento da morte celular por necrose.

Resumo ________________________________________________________
XX

ABSTRACT


________________________________________________________ Abstract
XXIII
ABSTRACT
3,4-methylenedioxymethamphetamine (MDMA or ecstasy) is a drug of
abuse that has been widely consumed since the 80s due to its entactogenic,
empathogenic and light hallucinogenic properties, as well as to the erroneous
presupposition of being considered a drug with low toxicity.
The effects/toxicity of MDMA are extremely unpredictable since they are
conditioned by several factors from the consumed dose, to the genetic,
physiologic and pathophysiologic characteristics of the consumer, the
environmental conditions where MDMA is consumed and, especially, to the co-
exposure to other substances that can interact with MDMA. These interactions
of MDMA can occur with non-psychoactive substances present in food or
pharmaceuticals or, of higher concern, with psychoactive substances
inadvertently ingested as contaminants of ecstasy pills, or intentionally
consumed to modulate MDMA effects (in this last case, the risk of interaction
will be potentially higher since the doses of psychoactive substances consumed
intentionally are, generally, higher than those taken by the consumption of
contaminated ecstasy pills).
Substances poly-consumption is the most frequent consumption pattern
of ecstasy, with high prevalence of co-consumption of other psychoactive
substances especially tobacco, Cannabis and alcohol, that can occur before,
during or in a short period after MDMA exposure. These poly-consumption
patterns can increase the health risks of MDMA consumption due to the
unpredictable chemical and idiosyncratic reactions that can result from the
mixture of xenobiotics in the body, potentially leading to pharmacokinetic and/or
pharmacodynamic interactions.
Among the compounds co-consumed with MDMA, ethanol has an
important position since, due to its high accessibility and social acceptance it is
widely consumed, often in high amounts, by the younger individuals that also
present high rates of ecstasy consumption. For this reason, the study of the
interactions between these two compounds in the body is of extreme relevance.
When the experiments included in this dissertation were designed, only
data related to the mutual effects of MDMA and ethanol co-consumption on
their pharmacokinetics and subjective and behavioural effects in humans was

Abstract ________________________________________________________
XXIV
available, but there were no studies evaluating the mechanisms underlying the
hepatic toxicity induced by the consumption of both substances in association.
Since both substances are metabolized in the liver and lead to the formation of
metabolites with higher toxicity than their parent compounds, the study of the
hepatic effects of this mixture has an extreme relevance. Thus, the aim of the
research that resulted in the present dissertation was to study the effects of the
simultaneous exposure to ecstasy and ethanol in the liver and to evaluate how
hyperthermia (a life-threatening effect frequently associated with MDMA in vivo
exposure) could affect the hepatic effects of this mixture.
To reach the proposed goal, in a first stage, in vivo studies with mice
exposed to 12% ethanol for 8 weeks followed by a single dose of 10 mg/kg
MDMA, were performed. This protocol was performed to simulate the most
common consumption pattern of these substances, characterized by an
initiation of ethanol consumption previous to the first contact with MDMA.
Subsequently, ex vivo studies, using the same protocol of ethanol exposure
were performed but, this time, only the hepatocytes isolated from the animals
exposed to ethanol were incubated with MDMA to clarify the effects induced by
MDMA to the hepatocytes obtained from animals repeatedly in vivo exposed to
ethanol that could elucidate the modifications observed in the in vivo studies.
Notwithstanding the obtained results confirmed the toxic potential of this
mixture, the experimental model based on freshly isolated mouse hepatocytes
suspensions, used in the ex vivo studies, only allow maximum incubations of 3-
4 hours which may limit the observation of certain indicators of cellular damage
or reaction towards an aggression. For this reason, in vitro studies in which
primary cultured hepatocytes were directly exposed to the ethanol+MDMA
mixture were developed. This last experimental model was used to clarify the
underlying mechanisms of the hepatic toxicity of this mixture, but also to
evaluate an eventual metabolic interaction between these two compounds,
since ethanol can alter the activity of several hepatic enzymes, including those
involved in MDMA metabolism.
The results obtained in the studies performed in the context of this
dissertation allowed us to prove that: (i) repeated pre-exposure to ethanol has a
synergistic effect on the hyperthermia and hepatotoxicity induced in vivo by
MDMA alone in male Charles-River CD-1 mice; (ii) repeated exposure to 12%

________________________________________________________ Abstract
XXV
ethanol increases the vulnerability of hepatocytes, isolated from male Charles-
River CD-1 mice, towards pro-oxidant aggression probably by decreasing the
GSH cellular content and increasing the GSSG cellular levels, increasing the
risk of impairment of intracellular thiol homeostasis that can lead to decreased
protein function and to the initiation of a cascade of events that can culminate in
cell death; (iii) acute co-exposure to ethanol+MDMA increases the hepatic
toxicity induced in vitro by MDMA, in primary cultured Wistar rat hepatocytes,
through a decrease in antioxidant defences, decrease in the cell energy stores,
increase in reactive species formation and increase in cell death by necrosis, in
an extent dependent on incubation temperature; (iv) acute in vitro exposure to
ethanol, in primary cultured Wistar rat hepatocytes, increases the metabolism of
MDMA to MDA, HMA and HMMA; (v) the toxic effects of the ethanol+MDMA
mixture are markedly increased under hyperthermic conditions.
Concluding, the co-consumption of ethanol and MDMA increases the
hepatotoxicity of ecstasy by an increase of the hyperthermic response
associated to consumption of this amphetamine derivative, a decrease of the
antioxidant defences and an increase of cell death through necrosis.

Abstract ________________________________________________________
XXVI

ABREVIATURAS


______________________________________________ Lista de Abreviaturas
XXIX
LISTA DE ABREVIATURAS ACTH hormona adrenocorticotrófica ou corticotrofina
ADH álcool desidrogenase
AG ácidos gordos
AIF factor indutor de apoptose
ALDH aldeído desidrogenase
AMPA ácido α-amino-3-hidroxi-5-metil-4-isoxazolpropiónico
AMPc adenosina monofosfato cíclica
ATP adenosina trifosfato
AUC área sob a curva
AVC acidente vascular-cerebral
BAT tecido adiposo castanho
bDNF factor neurotrófico derivado do cérebro
bFGF factor básico de crescimento dos fibroblastos
2-CB 4-bromo-2,5-dimetoxianfetamina
CD4+ células T auxiliares
CID coagulação intra-vascular disseminada
COMT catecol-O-metiltransferase
CRF factor libertador de corticotrofinas
CYP450 citocromo P450
DA dopamina
DAG diacilglicerol
DAS sulfureto dialílico
DAT transportador da dopamina
DEA Drug Enforcement Administration
DHA 3,4-diidroxianfetamina, α-metildopamina, α-MeDA
DHHA ácido 3,4-diidroxi-hipúrico
DHMA 3,4-diidroximetanfetamina, N-metil-α-metildopamina, N-Me-α-MeDA, HHMA
DHPA (3,4-diidroxifenil)acetona
DOB 4-bromo-2,5-dimetoxianfetamina
DXM dextrometorfano
ECATD Estudo sobre o Consumo de Álcool, Tabaco e Droga
ECG electrocardiograma
EMCDDA European Monitoring Center for Drugs and Drug Addiction
ESPAD European School Survey Project on Alcohol and Other Drugs
EtOH etanol, álcool etílico
EUA Estados Unidos da América
FAEE éster etílico de ácido gordo
FAS síndrome alcoólica fetal

Lista de Abreviaturas ______________________________________________
XXX
FBS soro bovino fetal
FSH hormona folículo-estimulante
GABA ácido γ-aminobutírico
GAT transportador do GABA
γ-GCS γ-glutamilcisteína sintetase
GH hormona do crescimento
GHB γ-hidroxibutirato
GI gastro-intestinal
GnRH hormona libertadora de corticotrofinas
GRF factor libertador da GH
GSH glutationa reduzida
GSSG glutationa oxidada
GST glutationa-S-transferase
HAP hidrocarbonetos aromáticos policíclicos
HAART Highly Active AntiRetroviral Therapy, terapia anti-retroviral altamente activa
HBSC/WHO Health Behaviour in School-aged Children/World Health Organization
HCV virus da hepatite C
HHMA 3,4-diidroximetanfetamina, N-metil-α-metildopamina, N-Me-α-MeDA, DHMA
5-HIAA ácido 5-hidroxi-indolacético
HIV vírus da imunodeficiência humana
HMA 4-hidroxi-3-metoxianfetamina
HMHA ácido 4-hidroxi-3-metoxi-hipúrico
HMMA 4-hidroxi-3-metoximetanfetamina
HMPA (4-hidroxi-3-metoxifenil)acetona
HSE elemento de choque térmico
HSF factor de choque térmico
HSP proteína de choque térmico
5-HT 5-hidroxitriptamina, serotonina
5-HTP 5-hidroxitriptofano
i.m. intra-muscular
i.p. intra-peritoneal
i.v. intra-venoso
IDT Instituto da Droga e da Toxicodependência
IFN-γ interferão γ
IGF-1 factor de crescimento insulin-like
IL interleuquina
IMAO inibidores da monoamina oxidase
IP3 inositol 1,4,5-trifosfato
IRC insuficiência renal crónica
ISRS inibidores selectivos da recaptação da serotonina

______________________________________________ Lista de Abreviaturas
XXXI
KET cetoconazol
LC50 concentração letal 50
LDH lactato desidrogenase
LH hormona luteinizante
LHRH hormona libertadora da hormona luteinizante
LPS lipopolissacarídeo
LSD dietilamida do ácido lisérgico
MAO monoamina oxidase
MBDB 3,4-metilenodioxi-α-etil-N-metil-fenetilamina, “Eden”
MDA 3,4-metilenodioxianfetamina
MDEA N-etil-3,4-metilenodioxianfetamina, “Eva”
MDHA ácido 3,4-metilenodioxi-hipúrico
MDMA 3,4-metilenodioximetanfetamina, ecstasy
α-MeDA α-metildopamina, 3,4-diidroxianfetamina, DHA
mEH epóxido hidrolase microssomal
MEOS sistema microssomal de oxidação do etanol
MLA metilicaconitina
MTA metanfetamina
NA noradrenalina
NAD+ dinucleótido de β-nicotinamida e adenina na forma oxidada
NADH dinucleótido de β-nicotinamida e adenina na forma reduzida
NADP+ fosfato do dinucleótido de β-nicotinamida e adenina na forma oxidada
NADPH fosfato do dinucleótido de β-nicotinamida e adenina na forma reduzida
NAPQI N-acetil-p-benzoquinonimina
NF-κB factor nuclear κB
NGF factor de crescimento neuronal
NK células exterminadoras naturais (natural killer)
NMDA N-metil-D-aspartato
N-Me-α-MeDA N-metil-α-metildopamina, 3,4-diidroximetanfetamina, HHMA, DHMA
NOS sintetase do monóxido de azoto
NT-3 neurotrofina 3
ONU Organização das Nações Unidas
p.o. per os
PAPS adenosina 3’-fosfato 5’-fosfossulfato
PCP fenciclidina
Pgp glicoproteína P
PI fosfatidilinositol
PI3K fosfatidilinositol-3-cinase
PIP2 fosfatidilinositol bifosfato
PIPAC piperonilacetona, (3,4-mitilenodioxifenil)acetona

Lista de Abreviaturas ______________________________________________
XXXII
PKA proteína cinase A
PLC fosfolipase C
PMA p-metoxianfetamina
PTH hormona da paratiróide
REDOX oxidação redução
RNS espécies reactivas de azoto
ROS espécies reactivas de oxigénio
s.c. sub-cutâneo
SERT transportador da serotonina
SIDA síndrome da imuno-deficiência adquirida
SNC sistema nervoso central
SNS sistema nervoso simpático
SOD superóxido dismutase
SULT sulfotransferase
T4 tiroxina
TG triglicerídeos
TGF-β factor de crescimento transformador β
TH triptofano hidroxilase
THC Δ9-tetraidrocanabinol
THF tetraidrofurano
TNF-α factor de necrose tumoral α
t-PA activador do plasminogénio do tipo tecidular
TR transcriptase reversa
UCP proteína desacopladora
UDP uridina difosfato
UE União Europeia
UGT uridina 5'-difosfato glucuronosiltransferase
VMAT transportador vesicular das monoaminas

PARTE I
INTRODUÇÃO e OBJECTIVOS


______________________________________________________ Introdução
3
1. INTRODUÇÃO
Desde o final da década 80, as drogas de abuso do tipo anfetamínico
têm-se tornado drogas recreativas populares 1. Dentro deste tipo de drogas, a
mais mediática é indubitavelmente a 3,4-metilenodioximetanfetamina (MDMA),
vulgarmente designada por ecstasy.
A popularidade da ecstasy pode ser explicada, em parte, pelos efeitos
que os utilizadores experimentam após o seu consumo 2 (ex.: euforia, aumento
da energia, aumento do desejo sexual, facilitação da interacção social), o que
levou a que o consumo de drogas sintéticas do tipo anfetamínico tenha vindo a
aumentar, quer entre os consumidores de droga, quer entre a população em
geral, com uma maior prevalência nas faixas etárias mais jovens.
O consumo desta droga, de fácil aquisição, constitui um risco
considerável tendo sido amplamente descritas várias reacções adversas
severas e inclusivamente fatais, que podem surgir como consequência directa
da acção tóxica deste composto, bem como devido ao facto do seu consumo
estar associado à adopção de outros comportamentos de risco (ex.: condução
sob a influência de drogas e prática de relações sexuais promíscuas e
desprotegidas). Assim, devido à sua prevalência e aos efeitos potencialmente
tóxicos associados, o consumo de MDMA é motivo de preocupação entre
educadores, meios de comunicação social, investigadores, clínicos e outros
prestadores de cuidados de saúde 2. O principal problema associado à
ocorrência de respostas tóxicas após o consumo de ecstasy reside na sua
enorme imprevisibilidade e variabilidade, o que indicia a existência de factores
de predisposição para a expressão de toxicidade. De facto, os efeitos
provocados pelo consumo de ecstasy poderão ser condicionados pela dose
ingerida, pela capacidade metabólica do indivíduo, pelas condições ambientais
em que ocorre o consumo e pelo co-consumo de outras substâncias. Estes
factores podem levar a alterações na formação de metabolitos activos e/ou
tóxicos e, potencialmente, originar interacções nefastas ou mesmo fatais. A
exposição do consumidor de ecstasy a outros compostos prende-se não só
com o padrão de policonsumo de substâncias lícitas e ilícitas que está
normalmente associado ao consumo desta droga de abuso, mas também à
ingestão de medicamentos (para intencionalmente modular os efeitos da

Introdução ______________________________________________________
4
ecstasy, ou durante regimes de tratamento) e à presença de vários compostos
(estimulantes ou não) que aparecem como contaminantes nas pastilhas de
ecstasy 3. O conhecimento da influência destes factores na toxicidade da
MDMA é fundamental para o estabelecimento de medidas de prevenção de
risco e de estratégias terapêuticas adequadas para o tratamento de
intoxicações.
Assim, na presente introdução far-se-á uma revisão da informação
disponível na literatura científica sobre as propriedades farmacocinéticas,
farmacodinâmicas e toxicológicas da MDMA, bem como da influência de vários
factores na ocorrência de reacções adversas a esta droga de abuso.
1.1. História da MDMA
A MDMA é uma das principais drogas de abuso actualmente
consumidas no cenário das festas rave, sendo conhecida por ecstasy.
Muitas publicações referem que a MDMA foi originalmente patenteada
como suppressor do apetite 4. No entanto, Cohen referiu em 1998 que a MDMA
foi patenteada na Alemanha em 1914 como um precursor de substâncias
farmacologicamente activas mas que nunca houve intenção de utilizá-la como
fármaco anoréctico 5, facto confirmado por Freudenmann e colaboradores em
2006 6.
A primeira referência acerca das propriedades psicoactivas da MDMA
em humanos foi publicada em 1978 pelo químico que a sintetizou, Alexander
Shulgin 7. Nos anos 80, a MDMA começou a ser utilizada em psicoterapia por
aumentar a auto-estima, a desinibição e a capacidade de introspecção do
paciente, facilitando a comunicação terapêutica e emocional, e criando
sentimentos de empatia entre paciente e terapeuta 5. Esta propriedade da
MDMA fez com que ela fosse incluída numa nova classe farmacológica: a dos
compostos entactogénios (do grego “contacto com o íntimo”) 8. Adicionalmente,
a MDMA tinha a vantagem de apresentar uma curta duração de acção,
adequada à duração das consultas. No entanto, ao ser administrada por via
oral em doses de 75 a 175 mg, a MDMA provocava, para além dos efeitos
entactogénicos pretendidos, efeitos simpaticomiméticos agudos como, por

______________________________________________________ Introdução
5
exemplo, aumento do ritmo cardíaco e da pressão arterial e ansiedade
transitória 9, 10.
Devido às propriedades da MDMA, a Organização das Nações Unidas
(ONU) estabeleceu, na Convenção sobre Substâncias Psicotrópicas de 1971,
que esta substância faria parte dos compostos de classe I devido ao seu
reduzido ou inexistente valor terapêutico e elevado potencial de abuso, que
constituem uma séria ameaça à saúde pública. Esta convenção estabelece,
assim a proibição da sua utilização, com a excepção de fins científicos ou de
situações médicas muito limitadas 11.
Seguindo as directivas da ONU, em 1985, a Drug Enforcement
Administration (DEA) dos Estados Unidos da América (EUA) também
classificou a MDMA como um composto de Classe I devido ao seu elevado
potencial de abuso, carência de aplicação clínica, falta de segurança para
utilização (mesmo sob supervisão médica) e também por surgirem evidências
de neurotoxicidade serotonérgica em ratos por acção da 3,4-
metilenodioxianfetamina (MDA) 12, um composto análogo e metabolito
maioritário da MDMA. Dada a controvérsia gerada em torno do seu potencial
terapêutico, esta classificação foi temporariamente alterada com a inclusão
deste composto numa classe menos restritiva (Classe III), sendo autorizado o
seu uso sob supervisão médica. No entanto, mais tarde, em 1988, a MDMA
voltou a ser incluída na Classe I, classificação que se mantém actualmente.
Desde os meados dos anos 80, a MDMA tornou-se popular como droga
de abuso, sobretudo entre as faixas etárias mais jovens, no contexto do
aumento da popularidade das festas rave ou techno onde o consumo desta
substância se generalizou, sobretudo na Europa 13. Actualmente ela pode ser
encontrada na maioria das festas rave 14. A sua grande popularidade, apenas
ultrapassada pela da Cannabis e derivados, deve-se aos seus efeitos
estimulantes, ao aumento do estado de vigília, à indução de sensação de bem-
estar e de sentimentos de confiança e empatia, à diminuição das inibições e a
uma falsa percepção de inocuidade 5.

Introdução ______________________________________________________
6
1.2. Relação estrutura/actividade da MDMA
A MDMA é um derivado anfetamínico (com substituição funcional no
anel aromático) estruturalmente semelhante à d-metanfetamina e à mescalina
(Figura 1), o que lhe confere propriedades estimulantes e alucinogénicas 15. No
entanto, estas propriedades alucinogénicas aparecem apenas para doses
muito elevadas de MDMA 16. A ausência de propriedades alucinogénicas
potentes da MDMA pode dever-se a algumas diferenças estruturais entre a
MDMA e os compostos alucinogénicos como (i) a substituição no anel
aromático nas posições 3 e 4 comparativamente às posições 3, 4 e 5 ou 2, 4 e
5, características dos compostos alucinogénicos, (ii) a presença de um grupo
metilo no grupo amina terminal (os derivados anfetamínicos mais potentes são
aminas primárias).
NHCH3
CH3
NH2H3CO
H3CO
OCH3
NHCH3
CH3
O
O
H
Figura 1. Estrutura química da metanfetamina, mescalina e do enantiómero farmacologicamente mais activo da 3,4-metilenodioximetanfetamina
Uma vez que apresenta um centro quiral, a MDMA pode existir na forma
de 2 enantiómeros distintos [S-(+)-MDMA e R-(-)-MDMA] e na forma de
racemato. O estereoisómero mais activo da MDMA é o S-(+)-MDMA
(responsável pelos efeitos psicoestimulantes e entactogénicos) enquanto que
Metanfetamina 3,4,5–trimetoxifeniletilamina (mescalina)
S-(+)-3,4–metilenodioximetanfetamina (MDMA, ecstasy)

______________________________________________________ Introdução
7
nos derivados anfetamínicos alucinogénicos, que não a MDMA, a potência do
esterioisómero R-(-) é maior 17. A presença do anel metilenodioxi aumenta a afinidade da MDMA para os
receptores serotonérgicos e diminui a afinidade para os receptores α2
adrenérgicos, relativamente à d-anfetamina 5.
A presença do grupo metilo no carbono α da cadeia alifática impede a
degradação do composto pela monoamina oxidase (MAO), o que explica a
acção mais potente das isopropilaminas (como a MDMA) relativamente aos
análogos com estrutura feniletilamina (como a mescalina) que são
preferencialmente metabolizados pela MAO-B 18.
1.3. Produção de MDMA
A produção mundial de ecstasy foi estimada em 113 toneladas em 2005 19. A Europa continua a ser o principal centro de produção de ecstasy, embora
a sua importância relativa pareça estar a decrescer, já que essa produção
alastrou a outras partes do mundo, nomeadamente à América do Norte e ao
leste e sudeste da Ásia. Na Europa, a ecstasy é sobretudo sintetizada na
Holanda (embora se observem, nesse país, alguns sinais de diminuição da
produção) e, em segundo lugar, na Bélgica 20.
1.4. Comercialização de MDMA
A ecstasy é vendida sob as designações de E, M, X, XTC, Adam, Ecky,
Bicky, rolls, beans, Clarity, lover’s speed, yaoto-wang, droga do abraço ou
droga do amor 5, na forma de pastilhas (de várias cores, formas e tamanhos),
cápsulas, pó 21 ou supositórios 22 (Figura 2). As pastilhas estão, normalmente,
identificadas com vários logótipos (ex.: Calvin Klein CK, Mitsubishi, 007, Coco
Chanel CC, Nike, Rolex, etc).

Introdução ______________________________________________________
8
Figura 2. Exemplos de pastilhas e cápsulas de ecstasy.
No período de 2000 a 2005, os preços médios de venda a retalho da
ecstasy, indexados à inflação, diminuíram na maioria dos países. Em 2005, o
custo médio na venda a retalho dos comprimidos de ecstasy na Europa variou
entre menos de 3 euros e 15 euros a unidade 20.
1.5. Farmacocinética da MDMA
A MDMA apresenta uma elevada biodisponibilidade por via oral, um
elevado volume de distribuição (cerca de 4L/kg) e uma baixa percentagem de
ligação às proteínas plasmáticas (inferior a 20%). O tempo de semi-vida da
MDMA varia entre as 6 e as 9 h, sendo a sua eliminação processada por via
hepática e renal 23-25.
Devido às suas características básicas (pKa ≅ 9.9) e ao seu peso
molecular relativamente baixo, este composto atravessa facilmente as
membranas das células e as camadas lipídicas, atingindo os tecidos ou fluidos
biológicos com pH mais acídico do que o sangue, incluindo a saliva e o suor 23.
1.5.1. Absorção
Os efeitos biológicos e as concentrações plasmáticas de MDMA
parecem seguir um perfil paralelo, com picos de concentração plasmática e de
efeito biológico a ocorrerem entre 1 e 2 horas após a ingestão e um declínio de
ambos até aos valores basais ao fim de 4 a 6 horas 24.

______________________________________________________ Introdução
9
A MDMA apresenta uma cinética linear para doses baixas, com um
aumento da concentração plasmática máxima e da área sob a curva (AUC,
concentração plasmática em função do tempo após administração) de uma
forma dependente da dose, para doses de 50, 75, 100 e 125 mg 24, 26. No
entanto, para doses de 150 mg, verificou-se que o aumento dos valores dos
parâmetros farmacocinéticos da MDMA não foi proporcional à dose
administrada, sugerindo uma cinética não linear para doses mais elevadas 26.
Esta observação pode ser explicada pela possível saturação das suas vias
metabólicas bem como pela interacção dos metabolitos com algumas das
enzimas envolvidas no metabolismo da MDMA, conduzindo à inibição das
mesmas 23. Aliás, foi já demonstrado que a MDMA em doses elevadas actua
como inibidor competitivo da CYP2D6 em microssomas de fígado humano 27,
mas que pode inclusivamente formar um intermediário metabólico por oxidação
da amina terciária ou do substituinte metilenodioxi da MDMA 28 que sofre
complexação com a enzima 29, envolvendo a formação de um carbeno e
levando a uma inibição irreversível da CYP2D6. Esta inibição pode ocorrer
durante a primeira hora após ingestão, sendo os valores basais da quantidade
de enzima apenas repostos após, pelo menos, 10 dias 30. Também a
recuperação urinária da MDMA é mais elevada para doses mais baixas, o que
se enquadra numa farmacocinética não linear 31. Esta aparente falta de
proporcionalidade entre a concentração plasmática esperada e a que é
observada após administração de doses elevadas de MDMA, aliada ao facto de
que muitos utilizadores de ecstasy a consomem em doses repetidas, faz
antever uma potenciação dos riscos de toxicidade desta substância por
aumento exagerado das suas concentrações plasmáticas 31. De facto, um
estudo clínico realizado por Farré e colaboradores em 2004 revelou que as
concentrações plasmáticas de MDMA atingidas após administração de uma
segunda dose de 100 mg de MDMA, administrada 24 h após uma dose
semelhante, não podiam ser explicadas pela simples acumulação da droga,
indiciando um fenómeno de auto-inibição metabólica com duração superior a
24 h 32. O mesmo facto foi já demonstrado em primatas não-humanos 33.
A biodisponibilidade da MDMA parece ser também condicionada por
fenómenos de estereoselectividade, uma vez que após administração oral de
uma dose única de MDMA na forma de racemato se observou que a

Introdução ______________________________________________________
10
concentração máxima plasmática do enantiómero R-(-)-MDMA era
significativamente maior do que a do enantiómero S-(+)-MDMA
(farmacologicamente mais activo), o qual era mais rapidamente eliminado 34, 35.
Estas diferenças estereoselectivas verificadas em humanos tinham sido
previamente verificadas em ratos 36.
1.5.2. Distribuição
Devido à sua baixa taxa de ligação às proteínas plasmáticas (inferior a
20%), a MDMA difunde facilmente do plasma para o compartimento extra-
vascular, apresentando um grande volume de distribuição 23. O enantiómero S-
(+)-MDMA é aquele que se distribui mais extensamente 34.
O perfil de distribuição da MDMA em humanos foi avaliado em estudos
post-mortem que mostraram que a MDMA se acumula em muitos tecidos e
órgãos onde atinge concentrações muito superiores às concentrações
plasmáticas, podendo ser até 18 vezes superiores no fígado 37, 38 e 30 vezes
superiores no cérebro 39.
1.5.3. Metabolismo
O metabolismo da MDMA (Figura 3) apresenta duas vias principais: (i)
abertura do anel metilenodioxi, seguida de metilação de um dos grupos
hidroxilo resultantes e/ou conjugação com o ácido glucurónico ou com o anião
sulfato 23, 26, 40-42 e (ii) desalquilação do grupo amina com formação da 3,4-
metilenodioxianfetamina (MDA) que retém acção biológica 43. A subsequente
desaminação e oxidação da cadeia lateral leva à formação de fenilcetonas,
posteriormente oxidadas a derivados do ácido benzóico 44, os quais, por sua
vez, são conjugados com a glicina e excretados na forma de derivados do
ácido hipúrico 31.
A principal via metabólica no rato e no homem passa pela abertura do
anel metilenodioxi da MDMA e da MDA dando origem a dois metabolitos
catecóis, a N-metil-α-metildopamina (N-Me-α-MeDA) e a α-metildopamina (α-

______________________________________________________ Introdução
11
MeDA), respectivamente 40. Ambos são posterioremente metilados pela enzima
catecol-O-metiltransferase (COMT), preferencialmente no grupo hidroxilo da
posição 4 do anel aromático. Estes metabolitos monometilados estão
maioritariamente presentes no plasma e urina na forma de conjugados com o
ácido glucurónico ou com o anião sulfato 26, 44-46.
Figura 3. Esquema das vias metabólicas da MDMA
As isoenzimas do sistema enzimático citocromo P450 (CYP450) têm um
papel preponderante na metabolização da MDMA, catalizando a
desmetilenação que leva à abertura do anel metilenodioxi e a N-desmetilação
(Figura 3). Esta desmetilenação pode ser catalizada pelas isoenzimas
CYP2D6, CYP1A2 e, em menor extensão, pelas isoenzimas CYP2B6 e
CYP3A4 40, 47, 48, mas pode também ocorrer sem catálise enzimática por um
processo de oxidação espontânea que envolve o radical hidroxilo 40, 49, 50. No
rato esta reacção é catalizada pelas isoenzimas CYP2D1 e CYP3A2 40. A N-

Introdução ______________________________________________________
12
desmetilação é catalizada, no Homem, pela isoforma CYP2B6 com
contribuição das isoenzimas CYP2D6, CYP1A2 e CYP3A4 e, no rato, pelas
isoenzimas CYP1A2 e CYP2D1 40, 47.
A presença do grupo catecol confere elevada reactividade a estes
metabolitos, que facilmente são oxidados originando as respectivas o-
quinonas. A oxidação das quinonas pode ainda levar à formação de
aminocromos 51 com subsequente formação de polímeros do tipo da melanina 52. Os ciclos de oxidação-redução associados a esta conversão metabólica 53
resultam na formação de espécies reactivas de oxigénio (ROS) e azoto (RNS)
que, tal como as quinonas, podem atacar grupos nucleófilos intracelulares
como a cisteína, a glutationa reduzida (GSH) e grupos sulfidrilo proteicos,
levando a alterações de macromoléculas importantes como proteínas, lípidos e
DNA 51, 54. Esta formação de ROS subsequente à metabolização da MDMA foi
já constatada em sinaptossomas isolados de estriado de ratinho 55, em
Escherichia coli sensiveis ao stress oxidativo por apresentarem deficiência na
proteína OxyR (importante na defesa contra agressões pró-oxidantes) 56, em
linhas celulares de neurónios granulares de cerebelo de rato 57, em células SK-
N-MC transfectadas com o transportador de serotonina humano 58, em células
Ito hepáticas 59, e também em culturas primárias de neurónios corticais obtidos
de embriões de ratos Wistar 60.
Os metabolitos quinónicos podem conjugar-se com uma molécula de
GSH formando um aducto que pode sofrer uma segunda conjugação com uma
nova molécula de GSH, num processo também acompanhado por formação de
ROS e RNS 61, 62 (Figura 4).
A metabolização da MDMA parece apresentar estereoselectividade, uma
vez que, quando se administra a mistura racémica por via subcutânea a ratos
ou por via oral a humanos, o enantiómero S-(+)- MDMA é metabolizado mais
rapidamente do que o R-(-)-MDMA 63-65. O enantiómero R-(-)-MDMA que não
for metabolizado, em conjunto com o excesso do enantiómero S-(+)-MDMA que
a enzima não conseguiu metabolizar, vão ser metabolizados por outras
isoenzimas que não a CYP2D6.
Uma vez que a MDMA é metabolizada predominantemente pela
isoforma CYP2D6 do complexo enzimático CYP450 66, está sujeita a ser alvo
de interacções farmacocinéticas/toxicocinéticas com outros compostos

______________________________________________________ Introdução
13
(fármacos, drogas de abuso, alimentos, etc) que partilhem ou afectem a
mesma via metabólica 67.
Figura 4. Oxidação dos metabolitos catecóis da MDMA a quinonas, semi-quinonas e aminocromos, e formação dos conjugados com a GSH.
A própria MDMA pode inibir a CYP2D6 mediante mecanismos que
incluem uma interacção competitiva e/ou a formação de um complexo entre os
metabolitos da MDMA e a enzima 26, 27, o que poderá explicar a sua
farmacocinética não-linear 68-70. Por este motivo, quando ingerida, a MDMA tem
a capacidade de transformar metabolizadores rápidos em metabolizadores
pobres para a dose subsequente, independentemente do genótipo.
Devido à presença do anel metilenodioxi, a MDMA sofre uma
metabolização mais extensa do que as anfetaminas-tipo, sendo menos
excretada sem biotransformação na urina 23. Cerca de 80% da MDMA é

Introdução ______________________________________________________
14
eliminada após metabolização hepática e cerca de 20% é excretada sem
biotransformação, na urina 23.
1.5.4. Excreção
A eliminação urinária da MDMA parece ser independente da dose 23,
sendo a maior parte excretada nas primeiras 24 h após o consumo 34.
O principal metabolito da MDMA excretado na urina é a 4-hidroxi-3-
metoximetanfetamina (derivado mono-metilado da N-Me-α-MeDA) seguido do
metabolito catecol N-Me-α-MeDA 71, maioritariamente na forma de conjugados
glucuronatos ou sulfatos 69. Para além da urina, a MDMA pode ser detectada
também na bile, suor, saliva, humor vítreo, cabelo e unhas 23, 68.
A excreção da MDMA é, também, estereoselectiva, sendo o isómero S-
(+)-MDMA (biologicamente mais activo) mais rapidamente excretado do que o
isómero R-(-)-MDMA 34, 65, 72.
Uma vez que a excreção renal deste composto com características de
base fraca é a principal via de eliminação, o seu tempo de semi-vida diminui
com a acidificação da urina e aumenta com a alcalinização da mesma. Por este
motivo, muitas vezes os consumidores de MDMA ingerem bicarbonato para
prolongar os efeitos da ecstasy.
1.6. Mecanismo de acção e principais efeitos da MDMA
A MDMA provoca, normalmente, um estado eufórico relaxado, incluindo
aumento da sensação de energia, abertura emocional, aumento da capacidade
de introspecção, empatia, redução de pensamentos negativos e uma
diminuição das inibições 23, 73-78. A percepção dos sons e das cores também
pode parecer mais intensa 74.
Os vários mecanismos envolvidos no aparecimento dos efeitos da MDMA
vão ser abordados de seguida.

______________________________________________________ Introdução
15
1.6.1. Efeito na libertação, transporte e receptores das monoaminas neurotransmissoras
A MDMA é um composto simpaticomimético de acção indirecta que
resulta da indução da libertação dos neurotransmissores simpáticos [serotonina
(5-HT), noradrenalina (NA), adrenalina e dopamina (DA)] a partir das vesículas
dos terminais nervosos e da medula supra-renal 79. Estes neurotransmissores
vão activar os respectivos receptores, resultando numa exacerbação da
estimulação simpática.
A MDMA induz preferencialmente a libertação de 5-
HT (Figura 5) tanto in vivo 80-82 como in vitro 83-86. A MDMA
liga-se ao transportador pré-sináptico da serotonina (SERT),
inibindo a recaptação neuronal da 5-HT presente na fenda
sináptica. Uma vez ligada ao transportador, a MDMA vai
aumentar a libertação de 5-HT por um processo de difusão por permuta 87, 88.
Após o transporte da MDMA para o interior do neurónio, o transportador fica
novamente livre para transportar a 5-HT do terminal nervoso para a fenda
sináptica 87. Este é um processo dependente do Ca2+ e é bloqueado pelos
inibidores do sistema de recaptação neuronal da 5-HT (ex.: fluoxetina,
imipramina) 87. Numa situação fisiológica, a 5-HT libertada para a fenda
sináptica deveria ser rapidamente recaptada para manter o controlo da
neurotransmissão. No entanto, quando a MDMA bloqueia o SERT, a 5-HT
acumula-se na fenda sináptica onde será degradada ou recaptada por outros
transportadores como os da dopamina (DAT) 58, 88.
Para além do efeito ao nível da recaptação neuronal da 5-HT, a MDMA
exerce também efeitos na captação vesicular da 5-HT. Em baixas
concentrações, a MDMA inibe a captação de 5-HT pelas vesículas, sendo este
efeito mais marcado para o estereoisómero S-(+)-MDMA 85, 90. Em altas
concentrações, a MDMA entra nas vesículas por difusão passiva 5 e, por ser
uma base fraca, aumenta o pH no interior das mesmas, promovendo o efluxo
de 5-HT, uma vez que esta é mantida no interior das vesículas através de um
gradiente de protões gerado por uma bomba de protões dependente de
adenosina trifosfato (ATP) 87.

Introdução ______________________________________________________
16
Figura 5. Mecanismo de acção biológica da MDMA ao nível dos neurónios serotonérgicos. A MDMA promove a libertação da 5-HT por permuta ao nível do transportador SERT (1). Para concentrações mais elevadas, a MDMA penetra nas vesículas sinápticas e promove a libertação da 5-HT nelas contida (2). O aumento da 5-HT deve-se também à inibição da degradação metabólica deste neurotransmissor pela MAO ao nível da mitocôndria (3), inibição do sistema de recaptação neuronal da 5-HT pelo SERT (4) e estimulação da libertação da 5-HT na fenda sináptica (5). Esta 5-HT libertada vai estimular receptores pós-sinápticos (6). No entanto a 5-HT tem mecanismos para evitar o aumento desmesurado dos seus níveis na fenda sináptica por auto-regulação da libertação da 5-HT pelos receptores pré-sinápticos (7), aumento da formação de metabolitos e produtos de auto-oxidação da 5-HT na fenda sináptica (8). A MDMA promove ainda a diminuição das reservas de 5-HT por provocar inibição da biossíntese da 5-HT pela triptofano hidroxilase (TH) (9). As setas descontínuas indicam inibição e as setas mais largas indicam indução. Figura adaptada de Carmo 2007 89.
A libertação de 5-HT vai activar receptores pré- e pós-sinápticos levando
à activação de múltiplos sistemas intracelulares que vão conduzir ao
aparecimento dos efeitos neuroquímicos e moleculares desta substância 88. A
activação dos receptores 5-HT1A parece ser responsável pela facilitação da
interacção social, pelo menos no rato 91. É importante referir que a estimulação
da libertação de 5-HT induzida pela MDMA provoca uma diminuição dos níveis
cerebrais de 5-HT que faz com que 2 dias depois do consumo de MDMA os
utilizadores apresentem estados de depressão 14.
Para além da acção indirecta da MDMA na estimulação da libertação de
5-HT, a própria MDMA apresenta afinidade para os receptores 5-HT podendo

______________________________________________________ Introdução
17
activá-los directamente. Esta afinidade da MDMA para os receptores da 5-HT
apresenta também estereoselectividade com maior potência do estereoisómero
R-(-)-MDMA para o receptor 5-HT2A e do estereoisómero S-(+)-MDMA para o
receptor 5-HT2C. Apesar de a activação dos receptores 5-HT2A estar
relacionada com as propriedades alucinogénicas das anfetaminas, a MDMA
tem um fraco carácter alucinogénico que pode dever-se à sua reduzida eficácia
neste receptor 5. Sendo a MDMA uma droga de abuso pró-serotonérgica, tem a
potencialidade de sofrer interacções farmacodinâmicas/toxicodinâmicas com
compostos que tenham o mesmo mecanismo de acção ou mecanismo de
acção antagónico 67, como por exemplo os antidepressivos inibidores da
recaptação da 5-HT (ex.:citalopram) ou alguns antipsicóticos (ex: clozapina),
respectivamente. Este potencial de interacção será abordado mais
detalhadamente no ponto 1.9.5.1.
O aumento de libertação da DA por acção da MDMA pode
resultar de: (i) reversão do transportador 84 e/ou (ii)
estimulação da síntese de DA através da acção agonista da
MDMA nos receptores 5-HT2A e 5-HT2C pós-sinápticos localizados nos
neurónios GABAérgicos, de cuja activação resulta uma diminuição dos níveis
do neurotransmissor ácido γ-aminobutírico (GABA) e consequente estimulação
da síntese e libertação de DA 82, 92-94. Este aumento da libertação de DA poderá
ter, também, como consequência, uma depleção dos seus níveis neuronais. De
facto, a administração de doses de 5, 10 ou 20 mg/kg de MDMA, em 4 dias
sucessivos, levou a um decréscimo nos níveis de DA e 5-HT, no núcleo
estriado de ratinho, 24 h após a última administração 95. No entanto, não se
observaram alterações quando estas doses foram administradas em dias
alternados e os níveis de monoaminas foram medidos após 48 h 95. Também
vários estudos tentaram relacionar a diminuição dos níveis estriatais de DA
com os efeitos neurotóxicos a longo termo da MDMA e verificaram uma perda
de DA estriatal dependente da dose 7 dias após o tratamento de ratinhos com
3 doses de 20 ou 30 mg/kg 96, 97.
Quanto ao sistema adrenérgico, a nível central, a
MDMA bloqueia os receptores adrenérgicos α2 pré-
sinápticos, levando ao aumento da pressão sanguínea em
humanos 5. A nível periférico, a MDMA tem uma acção agonista directa e

Introdução ______________________________________________________
18
indirecta nestes receptores, responsável por uma
vasoconstricção periférica, dilatação das pupilas, aumento
da salivação, diminuição da secreção de enzimas
pancreáticas e de insulina, retenção urinária, diminuição da motilidade gástrica
e piloerecção em humanos 98.
Devido a esta acção estimulante da MDMA na libertação de
catecolaminas, a exposição repetida a este derivado anfetamínico depleta as
reservas dos referidos neurotransmissores, diminuindo os efeitos biológicos
desta droga de abuso quando administrada repetidamente (tolerância aguda) 23.
A MDMA pode ainda activar receptores nicotínicos da acetilcolina
no músculo esquelético 99 e em várias áreas cerebrais 100, 101.
Uma vez que este tipo de receptores está implicado no
desenvolvimento de patologias psiquiátricas como a esquizofrenia
e está relacionado com a função cognitiva, a interacção das anfetaminas com
estes receptores, a nível central, poderá contribuir para as perturbações
psiquiátricas e cognitivas que aparecem após consumo crónico destas
substâncias 101, 102. A utilização de antagonistas específicos de receptores
nicotínicos contendo as subunidades β2 e α7 inibiu completamente o stress
oxidativo induzido pela MDMA em preparações de sinaptossomas de estriado
de ratinho, prevenindo a sua neurotoxicidade neste modelo animal 55, 103, 104. A
MDMA interage directamente com os receptores nicotínicos, especialmente
com os subtipos heterodiméricos, o que parece ter influência no efeito da
MDMA nos DAT, uma vez que a inibição da recaptação da DA induzida pela
MDMA é atenuada pela metilcaconitina e pela memantina (antagonistas α7), e
em menor extensão pela diidro-β-eritroidina (antagonista β2).
A hiperactividade induzida pela MDMA tem sido relacionada com a
libertação de 5-HT 5, 105, o que foi demonstrado pela utilização de vários
agonistas e/ou antagonistas da 5-HT e com receptores da 5-HT mutantes 106-
111. Existe também evidência de que os receptores da dopamina (D1, D2 e
talvez D3) contribuem para os efeitos hiperlocomotores da MDMA 112, 113, uma
vez que o aparecimento destes efeitos sofre um atraso quando se administra
um antagonista dos receptores D1 e são completamente abolidos quando se

______________________________________________________ Introdução
19
administra um antagonista dos receptores D2 114. Finalmente, uma contribuição
dos receptores α-adrenérgicos na hiperactividade induzida pela MDMA é
também possível 115 devido à activação de receptores α1-adrenérgicos ao nível
do córtex pré-frontal e das áreas ventrais-tegmentais 116.
Estas alterações comportamentais produzidas pela exposição à MDMA
são, também elas, condicionadas por variações estruturais nas moléculas dos
metabolitos da MDMA, uma vez que os conjugados da GSH com a α-MeDA (5-
glutation-S-il-α-MeDA e 6-glutation-S-il-α-Me-DA) apresentam, em ratos,
diferente actividade. Foi já verificado que apenas o regioisómero 5-GSH-α-
MeDA provoca alterações comportamentais, aparentemente de tipo
dopaminérgico (bruxismo, tremores, hiperlocomoção e clonus), sendo o
regioisómero da posição 6 comportamentalmente inactivo 117.
1.6.2. Inibição da monoamina oxidase (MAO)
Para além de aumentar a libertação e diminuir a recaptação das
catecolaminas, a MDMA, em doses elevadas, impede a sua inactivação
metabólica ao inibir competitivamente a MAO (Figura 5), prolongando, assim, a
estimulação simpática. A MDMA inibe preferencialmente a MAO-A (com mais
afinidade para a 5-HT), mas também tem capacidade para inibir a MAO-B 118.
Para doses mais baixas de MDMA (não inibitórias) a MAO está envolvida na
metabolização da DA e da 5-HT, levando à formação de espécies reactivas da
dopamina e de H2O2. Logo, concentrações mais elevadas de MDMA, capazes
de inibir a MAO, parecem diminuir a agressão oxidativa derivada da actividade
desta enzima sobre as monoaminas 55, 119.
1.6.3. Inibição da triptofano hidroxilase (TH)
A MDMA tem a capacidade de inibir a enzima limitante da síntese da 5-
HT, a triptofano hidroxilase (TH) 120 (Figura 5), provavelmente por um
mecanismo que envolve a inactivação dos grupos sulfidrilo desta enzima pelos

Introdução ______________________________________________________
20
metabolitos quinónicos da MDMA 5. Este mecanismo parece ser potenciado
pelo efeito hipertérmico induzido in vivo pela MDMA, possivelmente como
resultado do aumento da formação de espécies radicalares 5. A inibição da TH
pode perdurar durante semanas após a administração de uma única dose de
MDMA a ratos 5.
1.6.4. Alterações da expressão genética
A MDMA induz a proteína c-fos, uma proteína reguladora da expressão
de genes que codificam para neuropéptidos e proteínas essenciais a nível
neuronal e que parece estar envolvida na resposta celular ao stress e na morte
celular via apoptose 88. O aumento desta proteína ao nível das amígdalas
central e basolateral parece estar envolvido no perfil ansiogénico da MDMA em
ratinhos 121.
A MDMA parece, também, diminuir a expressão de genes como o que
codifica para a sinaptotagmina IV (proteína reguladora da secreção de
neurotransmissores, do transportador vesicular e da adesão das vesículas à
membrana plasmática) 88.
Verificou-se, em ratinhos, que a MDMA induziu o mRNA dos
transportadores do GABA (GAT1 e GAT4) 88. A expressão de outros genes
relacionados com a neurotransmissão GABAérgica, incluindo as proteínas do
citosqueleto da família das septinas e distorfinas, é diminuída pela MDMA, o
que ilustra o papel essencial deste neurotransmissor na actividade da MDMA a
nível neuronal 88.
A MDMA regula, ainda, os níveis de mRNA que codificam para a
proteína desacopladora mitocondrial UCP3 no músculo esquelético de rato 122.
O efeito da MDMA nesta proteína pode contribuir para o efeito hipertérmico da
MDMA, uma vez que a UCP3 tem um papel importante no mecanismo de
termogénese, actuando ao nível da dissipação do gradiente electroquímico de
protões na mitocôndria, favorecendo a libertação de energia sob a forma de
calor 123. Este efeito da MDMA será abordado em mais detalhe no ponto 1.6.6.

______________________________________________________ Introdução
21
1.6.5. Efeito neuroendócrino
A administração de MDMA leva a um aumento dos níveis séricos de
corticosterona, prolactina, renina, aldosterona e tiroxina (T4) em ratos 5, 122 e de
cortisol, prolactina 24, 124, 125, hormona adrenocorticotrófica (corticotrofina ou
ACTH) e hormona antidiurética (ou vasopressina) em humanos 31, 125, 126.
O aumento da secreção de corticosterona, aldosterona e renina parece
ser mediado pela estimulação serotonérgica resultante da administração de
MDMA 5, uma vez que a libertação de algumas destas hormonas também é
aumentada pela administração de agonistas serotonérgicos 127-130. Esta
resposta aos agonistas serotonérgicos está significativamente diminuída nos
consumidores crónicos de MDMA 127-130 embora, no caso particular do cortisol,
a resposta tenha sido recuperada após um ano de abstinência do uso de
MDMA 127. O mesmo não se verificou com a prolactina, o que sugere que os
efeitos neurotóxicos da MDMA nos terminais serotonérgicos são prolongados
e, provavelmente, dependentes do tipo de receptor serotonérgico em causa,
uma vez que a libertação de prolactina é regulada pela activação de receptores
5-HT1A enquanto o efeito na secreção do cortisol parece ser mediado pelos
receptores 5-HT2C 127.
Estudos in vitro utilizando hipotálamo isolado de rato indicam que a
MDMA e alguns dos seus metabolitos têm também a capacidade de aumentar
a libertação de oxitocina e vasopressina num modo dependente da dose 131.
Este aumento foi já observado em consumidores de MDMA 132. A MDMA,
mediante a activação dos receptores 5-HT1A, activa os neurónios dos núcleos
supraóptico e paraventricular do hipotálamo que contêm oxitocina, levando a
um aumento da libertação desta hormona e, consequentemente, dos seus
níveis plasmáticos. A hormona oxitocina desempenha um papel preponderante
nas relações sociais, apego e confiança 133 e a interacção oxitocina-DA pode
também influenciar a aquisição e expressão de comportamentos de procura de
drogas de abuso. Daí que a oxitocina possa desempenhar um papel
importante, embora ainda pouco explorado, na adição ao consumo de droga 134. Portanto, o efeito da MDMA na libertação da oxitocina poderá desempenhar
um papel de extrema importância nos efeitos pro-sociais da MDMA e estar na
base das propriedades de reforço positivo desta droga de abuso 135.

Introdução ______________________________________________________
22
A MDMA altera também os níveis hipotalâmicos e gonadais das
hormonas do eixo hipotálamo-hipófise-gónada em ratos macho, com
diminuição dos níveis de mRNA que codifica para a hormona hipotalâmica
libertadora de gonadotrofina (GnRH) que medeia a libertação das
gonadotrofinas FSH (hormona folículo-estimulante) e LH (hormona
luteinizante), e dos níveis séricos de testosterona 136.
No entanto, os dados obtidos por Allott e colaboradores, em 2008, não
confirmam a premissa de que o consumo de ecstasy resulta numa
desregulação mensurável do controlo serotonérgico da actividade endócrina,
uma vez que não se detectaram diferenças entre os consumidores de ecstasy
e os não-consumidores nas respostas neuroendócrinas, após estimulação
serotonérgica 137.
1.6.6. Alterações no sistema termorregulador
A MDMA interfere no sistema de termorregulação, podendo provocar
uma elevação acentuada da temperatura corporal (no rato a temperatura basal
pode sofrer um aumento de até 2ºC) cujo pico ocorre entre 40 a 60 min após a
administração 80, 138-141. O aumento da temperatura corporal após
administração de MDMA foi já observado em ratinhos 142, ratos 139, cobaios 143,
macacos 144 e humanos 31. Este efeito hipertérmico da MDMA varia com a
estirpe de animal utilizada, com a dose administrada, e é altamente
dependente da temperatura ambiente, uma vez que a mesma dose de MDMA
induz um efeito hipotérmico a baixa temperatura ambiente, e um efeito
hipertérmico a temperatura ambiente normal (20-22ºC) ou elevada (30-33ºC) 138, 139, 142.
O efeito hipertérmico da MDMA é também enantioselectivo, uma vez que
a MDMA racémica e o enantiómero S-(+)-MDMA são capazes de produzir
hipertermia, o mesmo não se verificando com o enantiómero R-(-)-MDMA 145.
A hipertermia resultante do consumo de MDMA pode dever-se à sua
acção no centro termorregulador do SNC e à vasoconstricção periférica dos
vasos sanguíneos cutâneos 146. No entanto, esta resposta hipertérmica no
homem parece ser também influenciada pelo aumento da actividade muscular

______________________________________________________ Introdução
23
associado à dança intensa, pela desidratação e pela elevada temperatura
ambiente em locais frequentemente sobrelotados 31.
Os mecanismos subjacentes a esta resposta hipertérmica podem ser
múltiplos. Apesar de normalmente o aumento da temperatura corporal envolver
a activação de vias serotonérgicas, o efeito hipertérmico da MDMA parece ser
mediado pelo aumento da libertação de DA. De facto, o aumento da
temperatura corporal foi já revertido em ratos com a administração de um
antagonista dos receptores D1 da DA 80, ao contrário do que aconteceu com os
antagonistas dos receptores serotonérgicos (ex.: ritanserina, metisergilide) e
com os inibidores da recaptação da 5-HT (ex.: fluoxetina) 80, 147-149. Da mesma
forma, a depleção dos níveis cerebrais de 5-HT não alterou a resposta
hipertérmica induzida pela MDMA 150. No entanto, a 5-HT parece ter um papel
importante na modulação da libertação da DA 151 e parece também influenciar
a resposta hipertérmica devido aos seus efeitos nos mecanismos de dissipação
de calor 149.
Mecanismos noradrenérgicos podem também contribuir para este efeito
hipertérmico da MDMA, nomeadamente via receptores α-adrenérgicos 152,
existindo evidência de que a activação de receptores GABAB reverte este efeito 153.
Adicionalmente, a MDMA induz um ligeiro aumento nos níveis séricos de
T4 em roedores e primatas, a qual desempenha um papel modulador na
termogénese induzida pela MDMA através da regulação da expressão da
UCP3 e/ou a síntese de receptores adrenérgicos 154. A activação dos
receptores adrenérgicos α1 e β3 pela NA libertada por acção da MDMA, no
músculo esquelético e no tecido adiposo castanho, por um mecanismo
dependente da adenosina monofosfato cíclica (AMPc), leva a um aumento da
disponibilidade de ácidos gordos (AG) livres e, consequentemente, à produção
de calor e desnaturação proteica pela UCP3 presente nas mitocôndrias do
músculo esquelético 155 (Figura 6).

Introdução ______________________________________________________
24
Figura 6. Hipótese integrada para os efeitos termogénicos e mionecróticos da MDMA. Neste modelo, o aumento robusto nos níveis plasmáticos de NA activa os receptores adrenérgicos α1 e β3 no tecido muscular esquelético. A activação dos receptores β3 aumenta os níveis intra-celulares de AMPc que vão activar a proteína cinase A (PKA) que, por sua vez, estimula , por fosforilação, a actividade da lipase hormono-dependente, aumentando a conversão dos triglicerídeos em ácidos gordos (AG) livres. Os AG livres fornecem os protões necessários para a ocorrência do desacoplamento mitocondrial. À medida que a temperatura do músculo aumenta, as proteínas desnaturam e pode surgir rabdomiólise como resultado da perda de integridade do músculo esquelético. A activação dos receptores α1 aumenta os níveis de cálcio intra-celulares num mecanismo dependente da fosfolipase C (PLC) que activa o fosfatidil inositol bifosfato (PIP2) a diacilglicerol (DAG) e inositol 1,4,5-trifosfato (IP3). O papel do cálcio intra-celular na activação da UCP ainda não é bem conhecido. As hormonas da tiróide desempenham um papel permissivo na regulação da expressão genética e proteica da UCP3. SNS, sistema nervoso simpático. Adaptado de Mills 2004 154.
A resposta hipertérmica requer a activação e translocação para o núcleo
de proteínas transreguladoras (os factores de choque térmico ou HSFs,
nomeadamente o HSF-1) que se ligam ao elemento do choque térmico (HSE)
do DNA, mediando a transcrição de genes responsáveis pela síntese de
proteínas de choque térmico ou HSPs 156. Assim, quando se verifica uma
resposta hipertérmica associada ao consumo de MDMA ocorre um aumento da

______________________________________________________ Introdução
25
expressão da HSPs, nomeadamente da HSP70, que pretende proteger os
terminais serotonérgicos da lesão prolongada produzida pela MDMA. Este
aumento de expressão da HSP70 depende da indução da resposta
hipertérmica pela droga e não da libertação de 5-HT nem da formação de
radicais hidroxilo 157. A resposta hipertérmica induzida pela MDMA é
potenciada quando os animais são previamente expostos a doses neurotóxicas
desta substância 147, o que pode ter implicações para os utilizadores de MDMA
que tenham danificado neurónios serotonérgicos devido ao consumo anterior
desta droga.
Em humanos, a presença de alterações prolongadas na termorregulação
associadas ao consumo prolongado ou em altas doses da MDMA pode
provocar consequências clínicas severas 158 ou mesmo fatais 159.
O aparecimento de resposta hipertérmica é extremamente variável, pois
é dependente das características ambientais e do indivíduo, uma vez que
existem casos em que (i) não se desenvolve resposta hipertérmica à MDMA,
(ii) apenas uma pastilha pode desencadear uma resposta hipertérmica fatal 14,
ou (iii) o indivíduo sobrevive, mesmo depois de dar entrada no serviço de
urgências com uma temperatura corporal de 42,9ºC após ter ingerido três
pastilhas de MDMA 160.
1.6.7. Efeito no fluxo sanguíneo e actividade cerebral
Em humanos, a MDMA tem uma capacidade moderada de alterar o fluxo
sanguíneo e a actividade cerebral de determinadas regiões do cérebro 5, 161.
Esta alteração do fluxo sanguíneo cerebral parece ser mediada pela influência
da MDMA na libertação de 5-HT, a qual está envolvida na sua regulação
fisiológica.
1.7. Efeitos tóxicos da MDMA
A MDMA tornou-se uma droga de abuso popular devido aos seus efeitos
estimulantes intensos e porque se acreditava que teria efeitos tóxicos

Introdução ______________________________________________________
26
negligenciáveis. No entanto, um crescente número de intoxicações severas e
fatais associadas ao consumo de drogas sintéticas tem sido descrito 38,
havendo também estudos que apontam para que um número significativo de
acidentes de viação possa ser causado por indivíduos intoxicados com
compostos anfetamínicos 3, 13, 162, 163.
Os efeitos tóxicos da MDMA nem sempre estão relacionados com as suas
acções biológicas, têm normalmente uma origem multifactorial e parecem ser
exacerbados pelas variáveis ambientais associadas ao contexto de utilização
desta droga (ambientes fechados, sobrelotados, com elevadas temperaturas
ambientais, com música em elevados decibéis e repetitiva) 164. No entanto,
intoxicações provocadas por exposição à MDMA fora deste contexto
constituem provas da acção tóxica directa deste composto. Por exemplo, a
intoxicação de uma criança de 8 meses 165, uma de 13 meses 166 e outra de 20
meses 167 que ingeriram acidentalmente uma pastilha/cápsula de ecstasy, ou
de um indivíduo paraplégico 168 que consumiu deliberadamente um pastilha de
ecstasy.
Aliado a esta origem multifactorial que condiciona o aparecimento de efeitos
tóxicos está, ainda, a grande variabilidade inter-individual associada ao perfil
farmacocinético desta substância. Este fenómeno pode estar na origem da
existência de intoxicações fatais com a ingestão de um único comprimido 169,
intoxicações graves em que o indivíduo sobreviveu após ingerir 30
comprimidos 170, ou 18 comprimidos 171 e de um caso praticamente
assintomático após ingestão de 42 comprimidos 159.
Assim, os efeitos adversos agudos da MDMA (incluindo a morte) estão
relacionados com as características do consumidor, a dose ingerida, a
frequência do consumo, a ingestão de outras substâncias e ainda das
condições ambientais nas quais ocorre o consumo 172.
Os principais efeitos tóxicos associados ao consumo de MDMA serão
abordados de seguida.

______________________________________________________ Introdução
27
1.7.1. Efeitos tóxicos agudos
Tipicamente, os efeitos tóxicos agudos da MDMA em humanos incluem:
aumento da pressão arterial e da frequência cardíaca 173, palpitações, náuseas,
vómitos, xerostomia, midríase, arrepios, sudação, tremores, trismo (cerrar dos
maxilares), bruxismo (ranger dos dentes que conduz a um maior desgaste
dentário nos utilizadores 174), dor ou tensão muscular, hiper-reflexia, sensação
súbita de frio ou calor, nistagmo (oscilações oculares involuntárias),
inquietação, irritabilidade e insónia 5, 13, 23, 73.
Podem também surgir intoxicações moderadas caracterizadas por:
hiperactividade, ansiedade, taquicardia, hipertensão, aumento da temperatura
corporal, confusão, ataques de pânico, delírio e episódios psicóticos com
alucinações breves que desaparecem após o término da acção da MDMA 23, 31.
Um dos principais sintomas de intoxicação aguda pela MDMA é a
hipertermia, através dos mecanismos acima referidos 23. Têm sido descritos
casos em que a temperatura corporal atingiu 43ºC 5, 175, apesar de a
temperatura de 42ºC ser considerada fatal. A hipertermia pode dar origem a
complicações graves e frequentemente fatais que incluem rabdiomiólise,
mioglobinúria, coagulação intravascular disseminada (com consequente
necrose dos tecidos) e falência renal aguda 5, 176.
Para além da hipertermia e das complicações a ela associadas, a MDMA
pode ainda provocar outros efeitos tóxicos agudos como a trombocitopenia,
leucocitose tardia, acidose, hipoglicemia, congestão pulmonar, edema e
toxicidade hepática (hepatite fulminante e necrose hepática) 159, 169, 177-181.
Outros efeitos potencialmente fatais incluem acidentes vasculares cerebrais
isquémicos e hemorrágicos (hemorragia subaracnóide, hemorragia
intracraniana, enfarte cerebral e trombose cerebral), bem como atrofia cerebral,
que podem surgir como resultado da hipertensão, angeíte cerebral ou
desidratação associadas ao consumo de MDMA 179, 182-184.
A lesão hepática induzida pela MDMA pode apresentar vários padrões
que variam de formas benignas análogas a hepatites virais 185, 186 até formas
graves com falência hepática aguda resultante de necrose hepática extensa 159,
182, 187 ou mesmo falência hepática fulminante, que obriga a recorrer a
transplante hepático ou que pode ser fatal 186-190. Este efeito pode resultar da

Introdução ______________________________________________________
28
toxicidade apresentada pela MDMA em hepatócitos 191-193 e em células Ito 59 e
podem surgir como consequência da hipertermia induzida pela MDMA que
desencadeia fenómenos de stress oxidativo com consequente oxidação lipídica
e/ou proteica, conduzindo a lesões celulares 194. No entanto, pode ocorrer
aparecimento de dano hepático sem que se verifique hipertermia e a extensão
deste dano aumenta, aparentemente, com a exposição repetida, num
fenómeno que parece estar relacionado com o desenvolvimento de
mecanismos imunológicos de tipo auto-imune 194.
As espécies reactivas formadas durante o metabolismo da MDMA e das
catecolaminas libertadas como resultado da sua acção simpaticomimética
podem levar a um aumento da activação do factor nuclear kappa-B (NF-κB),
um dos factores de transcrição envolvidos na activação de genes de resposta
imediata a estímulos deletérios e inflamatórios 195. O NF-κB pode influenciar o
desenvolvimento de processos inflamatórios, bem como a regulação da
expressão de enzimas antioxidantes, condicionando o balanço entre
manutenção da homeostasia e resposta tóxica à MDMA, tal como já foi
demonstrado a nível cardíaco 196 ou hepático 59.
A MDMA pode ainda induzir uma disfunção imunológica transitória
dependente dos seus níveis plasmáticos com diminuição do número de
linfócitos T helper (CD4+) e um aumento do número de células natural killer
(NK) 197, 198. Esta disfunção é potenciada e prolongada pela administração
repetida de MDMA 199, uma vez que os utilizadores crónicos de MDMA
apresentam uma diminuição quer do número de células NK quer do número de
células CD4+, o que permite antever uma maior susceptibilidade a infecções e
a patologias imunológicas dos indivíduos que consomem cronicamente MDMA 200.
Podem surgir, também, alterações retinianas, uma vez que a MDMA
parece levar a um aumento da apoptose em células da retina devido à
agressão pró-oxidante exercida por esta substância 201.
Um quadro de falência renal aguda é também possível 190, 202 e pode
resultar dos efeitos tóxicos da MDMA sobre as células dos túbulos renais 203.
Pode ainda surgir uma complicação rara e potencialmente fatal denominada
hiponatrémia que surge normalmente associada à ingestão excessiva de água
e/ou à secreção inapropriada de vasopressina 31, 182, 204-212. A hiponatrémia

______________________________________________________ Introdução
29
pode ocorrer porque a MDMA induz um aumento na libertação da vasopressina 213 que leva a uma diminuição plasmática de sódio, a qual pode ser agravada
com a ingestão de grandes quantidades de água e culminar em edema
cerebral 13, 214, 215. Por último, para doses mais elevadas, a MDMA pode
provocar também retenção urinária, o que poderá exacerbar a hiponatrémia 216.
Este efeito hiponatrémico da MDMA parece dependente do seu metabolismo 217, sendo a HMMA (que resulta da metilação da DHMA pela COMT) o
metabolito que parece apresentar uma maior capacidade de provocar um
aumento da libertação de vasopressina 131.
Para além destes efeitos tóxicos imediatos, a administração aguda de
MDMA pode ainda originar o aparecimento de efeitos tóxicos de longa duração
que podem persistir até sete dias após o consumo e incluem fadiga,
irritabilidade, ansiedade, temperamento depressivo, insónia, tonturas e tensão
muscular 13, 31. No entanto, estudos recentes sugerem que a maioria das
pessoas que consomem MDMA não apresentam sintomatologia depressiva a
longo prazo 218. O aparecimento de perturbações psicopatológicas (depressão,
perturbações psicóticas, alterações cognitivas, episódios bulímicos, alterações
impulsivas, crises de pânico e fobia social) parece estar relacionado com a
quantidade, a duração e a frequência do consumo de MDMA 219. Esta redução
da capacidade de interacção social após o consumo de MDMA parece não ser
mediado por alterações compensatórias dos receptores 5-HT2C, uma vez que a
capacidade de resposta a um indutor 5-HT2C é semelhante em animais
expostos e não expostos à MDMA, não se verificando diminuição da densidade
dos mesmos 220.
1.7.2. Efeitos tóxicos a longo prazo
Os efeitos tóxicos a longo prazo da MDMA surgem associados à
utilização crónica desta droga de abuso e incluem o desenvolvimento de uma
síndrome que afecta o maxilar inferior, erosão dentária e dor miofacial que
surgem em resultado do bruxismo e do trismo 179.
Existe também a possibilidade de desenvolvimento de anemia aplástica,
mas aparentemente é uma complicação reversível 179.

Introdução ______________________________________________________
30
O consumo crónico de MDMA pode ainda levar ao agravamento dos
seus efeitos hepatotóxicos com o desenvolvimento de fibrose hepática
progressiva 221 que parece estar relacionada com o aumento da produção de
colagénio tipo I pelas células Ito induzido pela MDMA, provavelmente através
de um mecanismo que envolve fenómenos de stress oxidativo nestas células 222.
O efeito tóxico a longo prazo da MDMA mais preocupante é a sua
neurotoxicidade que parece afectar selectivamente o sistema serotonérgico,
envolvendo um mecanismo independente da dopamina 223, sendo os axónios
do núcleo rafe dorsal (nucleus raphe dorsalis), ricos em receptores 5-HT2,
particularmente sensíveis 4, 224-229. Esta lesão não provoca um aumento
compensatório da síntese de 5-HT nos terminais remanescentes 230. De facto,
verifica-se uma redução da densidade dos locais de recaptação de 5-HT que
parece correlacionar-se com a duração do consumo da MDMA 226, 231-242. Estas
alterações não foram verificadas ao nível dos neurónios dopaminérgicos 234, 235,
uma vez que não se detectam défices de DA, NA e dos seus metabolitos
associados ao consumo de ecstasy 120. Existem, contudo, diferenças inter-
espécies, já que no ratinho a MDMA é uma neurotoxina dopaminérgica 243.
Há autores que defendem que a diminuição da função serotonérgica não
será uma consequência, mas sim uma causa do consumo de MDMA 244, 245,
uma vez que o comportamento impulsivo e a procura de drogas de abuso
parecem estar associados a alterações da função serotonérgica 245. Esta lesão
serotonérgica parece ser responsável pelo aparecimento de perturbações
psicopatológicas 246, 247, aparentemente reversíveis após um longo período de
abstinência, embora possam persistir algumas sequelas 227. O mecanismo
envolvido nesta neurotoxicidade ainda não foi completamente esclarecido. No
entanto, parece estar relacionado com a formação de ROS e RNA durante o
metabolismo da MDMA ou durante a auto-oxidação das monoaminas
neurotransmissoras libertadas por acção desta substância 248, e com a
hipertermia induzida pela MDMA 50, 249. Por este motivo, a indução de
hipotermia recorrendo a meios farmacológicos ou não farmacológicos tem a
capacidade de reduzir ou prevenir a neurotoxicidade da MDMA 249, 250. O stress
oxidativo causado pela MDMA, que parece estar na base da sua
neurotoxicidade, pode ser também mediado pela formação de aductos entre os

______________________________________________________ Introdução
31
metabolitos activos e a glutationa 54. Quando as alterações das funções
serotonérgicas causadas pela MDMA se verificam ao nível do lobo occipital
(responsável pela visão periférica contralateral), podem surgir alterações nos
processos visuais 251, incluindo défices na capacidade visuoespacial 252.
Outras alterações relacionadas com o consumo prolongado de MDMA
incluem a diminuição do metabolismo da glucose no hipocampo esquerdo e
alterações do EEG para padrões semelhantes aos observados em processos
de envelhecimento e demência 253, 254.
O uso crónico de MDMA foi também já associado a problemas do sono 255-257 e a vários problemas psicopatológicos, incluindo disfunções alimentares 219 e cardíacas 258.
Pode surgir falência renal crónica associada ao consumo de MDMA por
provocar angeíte necrosante a nível renal 259.
O facto da MDMA ser consumida por mulheres em idade fértil tem
suscitado preocupações relativamente às consequências de um possível efeito
tóxico a nível embrionário. Um estudo prospectivo realizado no Reino Unido em
1998 revelou a ocorrência de várias malformações e de casos de
prematuridade e aborto espontâneo, acima da média da população em geral,
nos recém-nascidos de mães consumidoras de MDMA 260. Este aumento do
risco de problemas durante a gestação quando a grávida consome anfetaminas
é já bem documentado em estudos com animais 261. Por este motivo, o abuso
destes compostos durante a gravidez, dependendo do estilo de vida da mãe,
está associado a um aumento do risco de ocorrência de parto prematuro, nado-
morto e atraso no crescimento intra-uterino 261-263.
Os fenómenos de dependência associados à MDMA são pouco
prováveis devido ao rápido desenvolvimento de tolerância aos efeitos
pretendidos, acompanhado de uma intensificação dos efeitos laterais
desagradáveis como o bruxismo, ansiedade e agitação 22, 264-266.
1.7.3. Mortes associadas ao consumo de MDMA
As mortes em que a ecstasy é mencionada como agente causal são
pouco frequentes. Como exemplo, no Reino Unido, no ano de 1996, a

Introdução ______________________________________________________
32
mortalidade de jovens entre os 15 e os 24 anos apresentou um índice de 2-53
mortes por cada 100 000 consumidores de ecstasy 267.
No entanto, estas mortes suscitaram grande preocupação quando
começaram a ser noticiadas, há alguns anos, dado ocorrerem, muitas vezes,
de forma inesperada e vitimarem jovens socialmente bem integrados. A
expressão «morte relacionada com o consumo de ecstasy» poderá significar
que a substância foi mencionada na certidão de óbito ou que foi detectada
(frequentemente em conjunto com outras drogas) através de análise
toxicológica. Embora os procedimentos de comunicação de dados não estejam
harmonizados, os dados dos relatórios nacionais Reitox relativos a 2004
sugerem que as mortes relacionadas com o consumo de ecstasy são raras na
maioria dos países da União Europeia (UE), e muito em especial os casos em
que a ecstasy é a única droga envolvida. Em 2003, vários países notificaram
mortes associadas ao consumo de ecstasy: a Áustria (uma morte unicamente
relacionada com MDMA), a República Checa (uma morte provavelmente
devida a uma sobredosagem de MDMA), a França (oito casos associados ao
consumo de MDMA), a Alemanha (dois casos associados unicamente à MDMA
e oito envolvendo MDMA combinada com outras drogas — com os valores
correspondentes de 8 e 11 em 2002), Portugal (a MDMA foi detectada em 2%
das mortes relacionadas com o consumo de droga) e o Reino Unido (a ecstasy
é mencionada em 49 certidões de óbito em 2000, 76 em 2001 e 75 em 2002) 268.
Um estudo realizado no Reino Unido entre 1994 e 2003 demonstrou que
o número de fatalidades associadas ao consumo de ecstasy tem aumentado de
ano para ano, mostrando uma correlação positiva com a prevalência do uso de
MDMA no ano anterior, o número de actos criminosos e o número de
apreensões, mas correlacionado negativamente com o preço das pastilhas de
ecstasy 269.
Os poucos dados disponíveis nos relatórios nacionais Reitox de 2006
sugerem que as mortes relacionadas com o consumo de ecstasy se mantêm
em níveis semelhantes aos mencionados nos anos anteriores. Na Europa em
geral, houve referências a 78 mortes envolvendo a ecstasy 20.
A Tabela 1 sumariza algumas das referências de mortes relacionadas
com o consumo de ecstasy desde 1988 a 2007.

______________________________________________________ Introdução
33
Todas as formas severas de toxicidade induzida pela MDMA podem ser
capazes de provocar a morte. Mas, para além desta letalidade directa, ocorrem
mortes indirectas associadas ao consumo de MDMA que podem dever-se (i) à
depressão que surge subsequentemente ao consumo de MDMA e que pode
ser suficientemente intensa para provocar suicídio 270, (ii) a uma depressão pré-
existente que leve à utilização deste tipo de droga como meio de suicídio 271,
(iii) a acidentes devidos a comportamentos de risco verificados enquanto sob
influência dos efeitos agudos da ecstasy (ex.: car surfing, andar em cima de
automóveis em movimento 272, escalar postes eléctricos 178, queimaduras 273,
condução temerária 274), a acidentes de viação que envolvem condutores e
peões incapacitados pela MDMA 159, 272, 275. As drogas do tipo anfetamínico
estiveram presentes, directa ou indirectamente, em 70 casos fatais e 467 casos
de “condução sob o efeito de drogas”, entre 1999 e 2004 na Holanda 276.
Devido ao aumento induzido pela MDMA nos comportamentos de condução
temerária, o consumo desta substância aumenta a probabilidade de estar
envolvido ou de causar um acidente de viação em 100% e 150%,
respectivamente 277.
Na maioria dos casos de intoxicação grave ou morte, os níveis
sanguíneos de MDMA variaram entre 0,5 e 10 mg/L, o que corresponde a
cerca de 40 vezes a dose recreativa habitual 270. No entanto, esta questão
agrava-se se considerarmos que, frequentemente, as drogas anfetamínicas
são consumidas em combinação com álcool e/ou outras drogas não-
anfetamínicas como cocaína ou canabinóides 276, o que pode potenciar as
alterações comportamentais, aumentando o risco de acidente, e incrementar a
toxicidade directa exercida por cada uma das drogas. Como exemplo, a MDMA
foi encontrada em 6 cadáveres autopsiados no Instituto Nacional de
Toxicologia Forense da Noruega desde 1996 a 2000. Em nenhum dos casos a
MDMA foi a única droga encontrada e apenas um dos casos correspondia a um
acidente de viação 274.

Introdução ______________________________________________________
34
Tabela 1. Mortes associadas ao consumo de ecstasy de 1988 a 2007.
Autor Ano Local Casos Idade Sexo
Causa Análise Toxicológica
postmortem
Klys et al. 278 2007 Polónia N=1 22 M Paragem cardíaca
súbita
MDMA (1,42 mg/L em S)
MDA (0,17 mg/L em S)
Libiseller et al. 279 2007 Áustria N=1 24 M Suicídio MDMA (13,33 mg/L em S;
13,0 mg/L em U)
MDEA (7,3 mg/L em S; 5,7
mg/L em U)
MDA (0,43 mg/L em S;
0,38 mg/l em U)
de Letter et al. 38 2006 Bélgica N=34 < 25 Sobredosagem com
múltiplas drogas
Combinação de
anfetaminas
Politrauma
Suicídio
Acidente de tráfego
Infracções
Falência
cardiopulmonar
aguda
Asfixia mecância
Hiperpirexia
MDMA (0,27 – 13,51 mg/L
em S)
Schifano et al.269 2006 Reino
Unido
N=394 42% das mortes
envolveram apenas MDMA
20 Acidente de viação MDMA (0.36 mg/L em S)
31 Acidente de viação MDMA (2,0 mg/L em S)
MDMA e MDA em U
Cheng et al. 280 2005 Hong
Kong
N=3
22 Acidente de viação EtOH (25 mg/100 mL em
S)
MDMA (0,68 mg/L em S)
Ketamina (0,21 mg/L em S)
Efedrina (0,40 mg/L em S)
Libiseller et al. 281 2005 Áustria N=1 19 Falência
cardiopulmonar
MDMA (3,8 mg/L em S)
Vestígios de MDA e de
THC
11-hidroxi-THC (1,6 μg/L)
11-nor-9-carboxi-THC (65
μg/L) em S
EtOH (0,1g/L em U)
CID, coagulação intra-vascular disseminada. S, sangue. U, urina. M, masculino. F, feminino. EtOH, etanol. THC, tetraidrocanabinol. MDEA, N-etil-3,4-metilenodioxianfetamina. PMA, p-metoxianfetamina. DXM, dextrometorfano.

______________________________________________________ Introdução
35
Tabela 1 (cont.). Mortes associadas ao consumo de ecstasy de 1988 a 2007.
Autor Ano Local Casos IdadeSexo
Causa Análise Toxicológica
postmortem
De Letter et al. 37
2004 Bélgica N=2 18 M Congestão
pulmonar
Edema
hemorrágico
EtOH (0,56 mg/mL em S;
1,23 mg/mL em U)
Metadona (1,1 μg/mL em
S)
MDMA (0,271 mg/L em S;
3,29 mg/L em U)
MDA (0,009 mg/L em S;
0,1 mg/L em U)
31 M Edema pulmonar MDMA (26,059 mg/L em S;
71,56 mg/L em U)
MDA (0,048 mg/L em S;
3,48 mg/L em U)
Dams et al. 282 2003 Bélgica N=1 Hiperpirexia
CID
MDMA (1,13 mg/L em S)
Anfetamina, MDA e PMA
em S
Garcia-Repetto
et al. 39
2003 Espanha N=3 19 M Hiperpirexia
Rabdomiólise
Coagulopatia
Falência multi-
orgânica
MDMA (3,18 mg/L em S;
7,99 mg/L em U)
MDA (0,06 mg/L em S;
1,18 mg/L em U)
20 M Hiperpirexia
Rabdomiólise
Coagulopatia
Falência multi-
orgânica
MDMA (0,28 mg/L em S;
32 mg/L em U)
MDA (0,05 mg/L em S;
0,74 mg/L em U)
Schifano et al. 283 2003 Reino
Unido
N=167 15-29 85,6% tinha MDMA em S
13,2% tinha MDA em S
0,6% tinha MDEA em S
0,6% tinha PMA em S
Vuori et al. 284 2003 Finlândia N=4 18 F Convulsões
Paragem
respiratória
Intoxicação
acidental por
MDMA e
moclobemida
EtOH (0.65 g/dL em S;
0,69 g/dL em U)
Anfetaminas em U
MDMA (1,6 mg/L em S)
Moclobemida (2,3 mg/L em
S)
CID, coagulação intra-vascular disseminada. S, sangue. U, urina. M, masculino. F, feminino. EtOH, etanol. THC, tetraidrocanabinol. MDEA, N-etil-3,4-metilenodioxianfetamina. PMA, p-metoxianfetamina. DXM, dextrometorfano.

Introdução ______________________________________________________
36
Tabela 1 (cont.). Mortes associadas ao consumo de ecstasy de 1988 a 2007.
Autor Ano Local Casos IdadeSexo
Causa Análise Toxicológica
postmortem
Vuori et al. 284
(cont.)
23 M Intoxicação
acidental por
MDMA e
moclobemida
Anfetamins e opiáceos em
U
MDMA (2,0 mg/L em S)
Morfina (0,08 mg/L em S)
Anfetamina (0,1 mg/L em
S)
DXM (1,1 mg/L em S)
Moclobemida (2,1 mg/L em
S)
Ciclizina (0,3 mg/L em S)
Oxazepam (1,0 mg/L em S)
Diazepam (1,0 mg/L em S)
Desmetildiazepam (0,1
mg/L em S)
Temazepam (0,2 mg/L em
S)
Vestígios de alprazolam
em S
18 M Hiperpirexia
(43,2ºC)
Intoxicação
acidental por
MDMA e
moclobemida
EtOH (0,28 g/dL em U)
Anfetaminas e
canabinóides em U
MDMA (1,7 mg/L em S)
Anfetamina (0,15 mg/L em
S)
MDA (0,05 mg/L em S)
THC (1,8 μg/L em S)
Moclobemida (7,9 mg/L em
S)
19 M MDMA (2,1 mg/L em S)
MDA (0,05 mg/L em S)
THC (3,1 μg/L em S)
Moclobemida (6,5 mg/L em
S)
Lidocaína (1,6 mg/L em S)
de Letter et al. 285 2002 Bélgica N=1 Sobredosagem MDMA (3,10 mg/L em S)
MDA em S
CID, coagulação intra-vascular disseminada. S, sangue. U, urina. M, masculino. F, feminino. EtOH, etanol. THC, tetraidrocanabinol. MDEA, N-etil-3,4-metilenodioxianfetamina. PMA, p-metoxianfetamina. DXM, dextrometorfano.

______________________________________________________ Introdução
37
Tabela 1 (cont.). Mortes associadas ao consumo de ecstasy de 1988 a 2007.
Autor Ano Local Casos IdadeSexo
Causa Análise Toxicológica
postmortem
Gill et al. 286 2002 Nova
Iorque
(EUA)
N=22 17-41 Intoxicação aguda
(n=13)
Lesão mecânica
(ex.: alvejamento)
(n=7)
Combinação de
doença anterior
com a intoxicação
aguda (n=2)
MDMA (n=22)
Opiáceos/cocaína em S
(n=7)
Fineschi et al. 287 1999 Itália N=2 19 M Hiperpirexia
CID
MDMA (7,15 mg/L em S;
31 mg/L em U)
MDA (0,25 mg/L em S;
0,85 mg/L em U)
20 M Hiperpirexia MDMA (0,18 mg/L em S;
263,13 mg/L em U)
MDA (0, 12 mg/L em S;
5,25 mg/L em U)
MDEA (1,59 mg/L em S;
183,73 mg/L em U)
O’Connor
et al. 288
1999 Nova
Zelândia
N=1 27 F Edema cerebral
Walubo et al. 289 1999 EUA N=1 53 M Suicídio
Hiperpirexia
(40,4ºC),
Rabdomiólise
CID
MDMA (3,05 mg/L em S)
Anfetaminas em U
Byard et al.290 1998 Austrália
Sul
N=2 22 F Hiperpirexia
(42,5ºC)
CID
Hipercalémia
Rabdomiólise
PMA (1,32 mg/L em S)
MDMA (0,3 mg/L em S)
Níveis elevados de MTA
em S
Canabinóides em S
26 F Cianose
Hiperpirexia
(46,1ºC)
Arritmia cardíaca
Edema pulmonar
PMA (2,2 mg/L em S)
MDMA (0,82 mg/L em S)
MTA (0,09 mg/L em S)
Henry et al. 204 1998 Reino
Unido
N=1 32 M Paragem
cardiovascular
MDMA (4,56 mg/L em S)
MDA (0,36 mg/L em S)
EtOH (0,24 mg/mL em S)
CID, coagulação intra-vascular disseminada. S, sangue. U, urina. M, masculino. F, feminino. EtOH, etanol. THC, tetraidrocanabinol. MDEA, N-etil-3,4-metilenodioxianfetamina. PMA, p-metoxianfetamina. DXM, dextrometorfano.

Introdução ______________________________________________________
38
Tabela 1 (cont.). Mortes associadas ao consumo de ecstasy de 1988 a 2007.
Autor Ano Local Casos IdadeSexo
Causa Análise Toxicológica
postmortem
Mueller et al. 291 1998 EUA N=1 20 F Hiperpirexia MDMA em S
Lora-Tamayo
et al. 275
1997 Espanha N=10 23 M Agressão por arma
branca
Choque
hipovolémico
MDMA (0,23 mg/L em S)
MDEA (0,77 mg/L em S)
MDA (0,05 mg/L em S)
Anfetamina (0,62 mg/L em
S)
17 M Salto de uma janela MDMA (0,23 mg/L em S)
Anfetamina (0,10 mg/L em
S)
MDA (0,04 mg/L em S)
MTA (0,35 mg/L em S)
32 M Atropelamento MDMA (0,27 mg/L em S)
EtOH (2,47 mg/mL em S)
39 M Morte súbita na
discoteca
Doença coronária
isquémica
Oclusão da artéria
coronária direita e
artéria circunflexa
Enfarte antigo no
ventrículo esquerdo
MDMA (0,60 mg/L em S)
MDEA (0,22 mg/L em S)
MDA (0,12 mg/L em S)
Anfetamina (0,22 mg/L em
S)
EtOH (0,54 mg/mL em S)
21 M Acidente rodoviário MDMA (0,17 mg/L em S)
MDEA (1,07 mg/L em S)
MDA (0,18 mg/L em S)
Anfetamina (0,10 mg/L em
S)
EtOH (0,71 mg/mL em S)
26 M Acidente rodoviário MDMA (0,03 mg/L em S)
MDEA (2,44 mg/L em S)
MDA (0,15 mg/L em S)
Benzilecgonina (0,05 mg/L
em S)
EtOH (0,88 mg/mL em S)
29 M Reacção adversa a
drogas
Morte em espaço
aberto
MDMA (4,07 mg/L em S)
MDA (0,49 mg/L em S)
Morfina (0,38 mg/L em S)
Alprazolam (0,1 mg/L em
S)
EtOH (0,92 mg/mL em S)
CID, coagulação intra-vascular disseminada. S, sangue. U, urina. M, masculino. F, feminino. EtOH, etanol. THC, tetraidrocanabinol. MDEA, N-etil-3,4-metilenodioxianfetamina. PMA, p-metoxianfetamina. DXM, dextrometorfano.

______________________________________________________ Introdução
39
Tabela 1 (cont.). Mortes associadas ao consumo de ecstasy de 1988 a 2007.
Autor Ano Local Casos IdadeSexo
Causa Análise Toxicológica
postmortem
Lora-Tamayo
et al. 275 (cont.)
30 M Morte em espaço
aberto
Encontrado com
seringa ao lado
Edema pulmonar
agudo
MDMA (0,98 mg/L em S)
Anfetamina (0,06 mg/L em
S)
Morfina (0,15 mg/L em S)
EtOH (0,38 mg/mL em S)
27 M Edema pulmonar
agudo
Hemorragia
pulmonar
MDMA (8 mg/L em S)
Anfetamina (0,18 mg/L em
S)
MDA (1,2 mg/L em S)
Cocaína (0,04 mg/L em S)
Benzilecgonina (0,13 mg/L
em S)
EtOH (0,9 mg/mL em S)
19 M Reacção adversa a
drogas
MDMA (0,49 mg/L em S)
MDEA (4,32 mg/L em S)
MDA (0,29 mg/L em S)
Anfetamina (0,20 mg/L em
S)
Dipirona (1,36 mg/L em S)
Parr et al. 292 1997 Austrália N=1 15 F Hiponatrémia
Edema cerebral
MDMA (0,05 mg/L em S;
430 ng/mL em U)
Coore 293 1996 Irlanda N=1 18 F Hiperpirexia (43ºC)
Lesão renal
CID
Rabdomiólise
MDA (0,246 mg/L em S)
MDMA e MDA em U
Cox et al. 294 1996 Reino
Unido
N=1 22 M Hiperpirexia
CID
MDMA (0,43 mg/L em S)
MDEA (0,30 mg/L em S)
MDA (0,25 mg/L em S)
Crifasi et al. 295 1996 EUA N=1 29 M Acidente MDMA (2,14 mg/L em S;
118,8 mg/L em U)
MDA (< 0,25 mg/L em S;
3,86 mg/L em U)
Dar et al. 296 1996 EUA N=1 17 M Hiperpirexia
Rabdomiólise
CID
CID, coagulação intra-vascular disseminada. S, sangue. U, urina. M, masculino. F, feminino. EtOH, etanol. THC, tetraidrocanabinol. MDEA, N-etil-3,4-metilenodioxianfetamina. PMA, p-metoxianfetamina. DXM, dextrometorfano.

Introdução ______________________________________________________
40
Tabela 1 (cont.). Mortes associadas ao consumo de ecstasy de 1988 a 2007.
Autor Ano Local Casos IdadeSexo
Causa Análise Toxicológica
postmortem
Ellis et al. 186 1996 Reino
Unido
N=4 21 F Hiperpirexia (41ºC)
HTA
Taquicardia
Falência renal
hiperaguda
CID
Transplante
Sepsis
MDMA (0,11 mg/L em S;
0,04 mg/L em U)
18 F Icterícia progressiva
Falência hepática
devida ao consumo
regular de ecstasy
36 F Icterícia após
consumo de 1
pastilha de ecstasy
Transplante
Sepsis
22 F Consumo de
ecstasy durante 6
meses
Icterícia
Transplante parcial
Sepsis
Fineschi et al. 297 1996 Itália N = 1 20 M Hiperpirexia
CID
MDMA (0,18 mg/L em S)
MDEA em S
Milroy et al. 182 1996 Reino
Unido
N=7 21 M Colapso na rave.
Hiperpirexia (44ºC)
Paragem cardíaca
MDMA (4,2 mg/L em S)
Anfetamina (1,4 mg/L em
S)
20 M Colapso na
discoteca
↑PA
Hiponatrémia
Intoxicação por
água
MDMA (0,04 mg/L em S)
24 M Morte na discoteca
Causa
desconhecida
MDEA (0,187 mg/L em S)
Anfetamina (0,453 mg/L
em S)
CID, coagulação intra-vascular disseminada. S, sangue. U, urina. M, masculino. F, feminino. EtOH, etanol. THC, tetraidrocanabinol. MDEA, N-etil-3,4-metilenodioxianfetamina. PMA, p-metoxianfetamina. DXM, dextrometorfano.

______________________________________________________ Introdução
41
Tabela 1 (cont.). Mortes associadas ao consumo de ecstasy de 1988 a 2007.
Autor Ano Local Casos IdadeSexo
Causa Análise Toxicológica
postmortem
Milroy et al. 182
(cont.)
21 M Morte na cama
após a festa
Causa
desconhecida
MDMA (2,1 mg/L em S)
MDEA (3,5 mg/L em S)
MDA (8,5 mg/L em S)
Anfetamina (0,256 mg/L
em S)
20 M Encontrado
inconsciente na
cama
Rigidez
Hiperpirexia
(39,5ºC)
Hipóxia cerebral
Sobrevida de 4 dias
MDMA (0,09 mg/L em S)
MDA (0,13 mg/L em S)
25 M Morte súbita na rua MDMA (vestígios em U)
MDA (vestígios em U)
23 M Icterícia progressiva
e falência hepática
Consumo
exagerado de
ecstasy
Moore et al. 72 1996 EUA N=1 20 M Intoxicação aguda
por inalação de
MDMA, cocaína e
heroína
MDMA (2,8 mg/L em S)
Benzoilecgonina (0,97
mg/L em S)
Morfina (0,11 mg/L em S)
MDMA e MDA em U
Squier et al. 298 1995 Reino
Unido
N=1 30 M Convulsões
Hiperpirexia
Necrose cerebral
MDMA, heroína,
anfetamina e EtOH em S
Forrest et al. 299 1994 Reino
Unido
N=1 21 M Asfixia
Convulsões?
MDA, MDEA e MDMA em
S
Hooft et al. 272 1994 Bélgica N=1 26 M Acidente de viação
em tentativa de car
surfing
MDMA (0,63 mg/L em S)
EtOH (1,23 g/L em S)
Anfetaminas em U
Watson et al. 300
1993
Reino
Unido
N=1 16 M Hiperpirexia
Rabdomiólise
CID
CID, coagulação intra-vascular disseminada. S, sangue. U, urina. M, masculino. F, feminino. EtOH, etanol. THC, tetraidrocanabinol. MDEA, N-etil-3,4-metilenodioxianfetamina. PMA, p-metoxianfetamina. DXM, dextrometorfano.

Introdução ______________________________________________________
42
Tabela 1 (cont.). Mortes associadas ao consumo de ecstasy de 1988 a 2007.
Autor Ano Local Casos IdadeSexo
Causa Análise Toxicológica
postmortem
Campkin et al. 301 1992 Reino
Unido
N=1 18 M Hipotensão
Taquicardia
Hiperpirexia (42ºC)
Rabdomiólise
CID
MDMA (1,26 mg/L em S)
Henry et al. 247 1992 Reino
Unido
N=6 18 M Hiperpirexia
Falência
cardiovascular
17 M Hiperpirexia
CID
Convulsões
20 M Hiperpirexia
Rabdomiólise
CID
18 M Hiperpirexia
Rabdomiólise
CID
21 M Acidente de viação
23 M Acidente de viação
Rohrig et al. 302 1992 EUA N=2 35 M
Causa
desconhecida
MDMA (2,8 mg/L em S)
F Suicídio
Depressão?
MDMA (0,58 mg/L em S)
Diazepam em S
Screaton et al. 180 1992 N=1 19 M Hiperpirexia
Rabdomiólise
CID
Chadwick
et al. 169
1991 Reino
Unido
N=1 16 F Hiperpirexia
CID
MDMA (0,424 mg/L em S)
Suarez et al. 181 1988 EUA N=1 34 M Falência
cardiovascular
MDMA (2 mg/L em S)
CID, coagulação intra-vascular disseminada. S, sangue. U, urina. M, masculino. F, feminino. EtOH, etanol. THC, tetraidrocanabinol. MDEA, N-etil-3,4-metilenodioxianfetamina. PMA, p-metoxianfetamina. DXM, dextrometorfano.

______________________________________________________ Introdução
43
1.8. Estudos epidemiológicos sobre a utilização da MDMA
Vários estudos sobre os padrões de consumo da MDMA foram já
realizados. Estes estudos são normalmente levados a cabo através de
questionários ou entrevistas e os intervenientes podem ser seleccionados por
serem consumidores, seleccionados aleatoriamente na população ou por
responderem a anúncios.
Os resultados dos diferentes estudos indicam que o consumo de MDMA
pela população jovem está em expansão. Foi já estimado que só no Reino
Unido 500.000 jovens ingerem ecstasy em cada fim-de-semana 5.
Os dados referidos coincidem, também, em que a maioria dos
consumidores de MDMA misturam as pastilhas de ecstasy com outras
substâncias psicoactivas, principalmente tabaco, Cannabis e álcool, sendo
também frequente a combinação com cocaína, outras enfetaminas ou LSD.
Uma análise mais pormenorizada destes resultados está representada na
Tabela 2.
Da análise da Tabela 2 pode deduzir-se que o uso combinado de
ecstasy pode ser feito com substâncias psicoestimulantes (cocaína,
anfetaminas, tabaco) ou com sedantes (álcool, Cannabis). Poderia especular-
se que o objectivo de ambos os padrões de consumo tem uma finalidade
moduladora dos efeitos da MDMA. No entanto, não se pode escamotear a
possibilidade de que este policonsumo se deva à presença prévia de
dependência de alguma das drogas usadas em combinação (ex.: tabaco,
álcool).

Tabela 2. Estudos epidemiológicos acerca dos padrões de consumo da MDMA.
Estudo Ano Local População-Alvo N Idades (anos)
Indivíduos que consomem MDMA (%) Nº de pastilhas
por sessão
Bellis et al. 303 2008 Europa* Frequentadores
de bares e clubs
1341 16-35 28,7% (pelo menos 1 vez na vida)
Cole et al. 304 2008 Reino Unido Consumidores
de droga
80 20-22 86,3% (pelo menos 1 vez na vida) 1-6
Mendes
et al. 305
2008 Coimbra
(Portugal)
Consumidores
de MDMA
223 13-23 53,8% consumo ocasional (0-1 vez/mês, 6,7% policonsumo)
34,5% consumo habitual (+ de 1 vez/mês, 35% policonsumo)
11,7% consumo excessivo (+ de 1 vez/semana, 58,3%
policonsumo)
Pisetsky
et al. 306
2008 EUA Estudantes 9-12º
ano
13917 4,9-7,4% (no mês anterior ao estudo)
Wilkins et al. 307 2008 Nova
Zelândia
População geral 5475 (em 1998)
5504 (em 2001)
3042 (em 2003)
1902 (em 2006)
15-45 1,5% em 1998
3,9% em 2006
Chinet et al. 308 2007 Suiça Frequentadores
de festas rave
302 16-46 40,4% (pelo menos 1 vez na vida)
14,7% (de 1 a 3 vezes no mês anterior ao do estudo)
Dresen et al. 309 2007 Alemanha Condutores
inabilitados
247 18-29 14,6% só tinha consumido MDMA
27% apresentava policonsumo de drogas sintéticas
Goudie et al. 310 2007 Reino Unido Consumidores
de drogas
40 18-45 85% (pelo menos 1 vez na vida) 1-4
* Lisboa (Portugal), Palma de Mallorca (Espanha), Veneza (Itália), Atenas (Grécia), Ljubljana (Eslovénia), Brno (República Checa), Viena (Áustria), Berlim (Alemanha), Liverpool (Reino Unido)
44
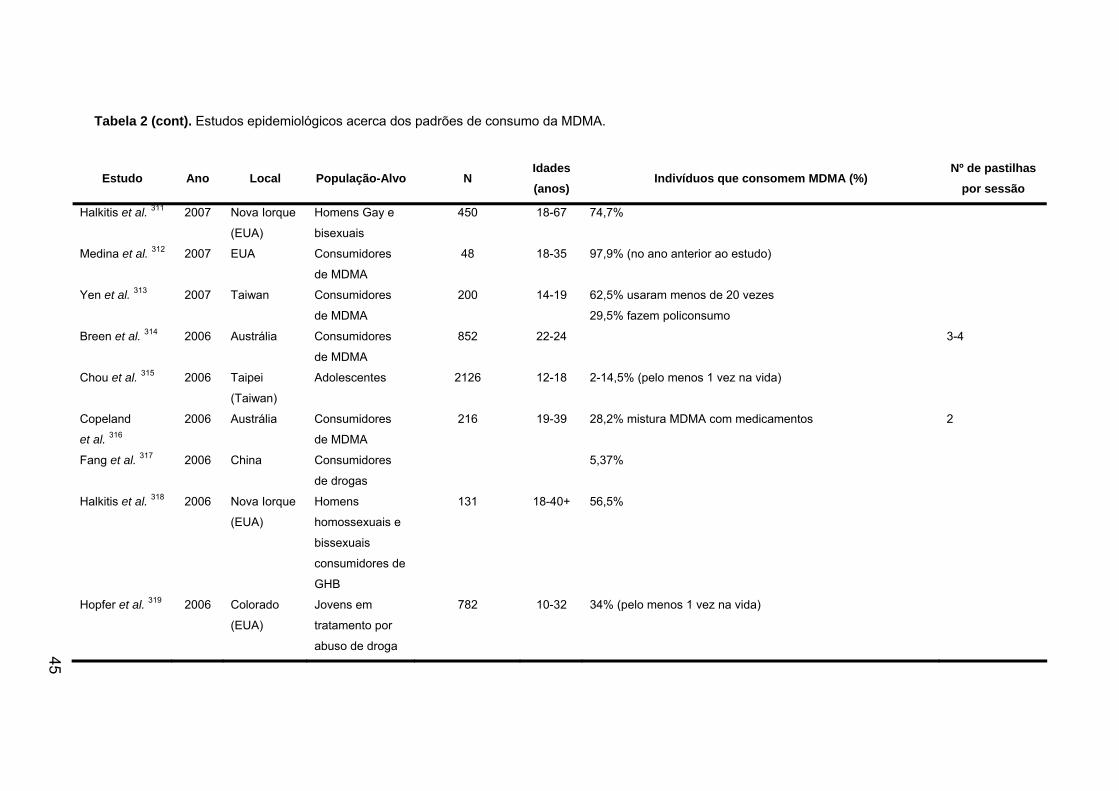
45
Tabela 2 (cont). Estudos epidemiológicos acerca dos padrões de consumo da MDMA.
Estudo Ano Local População-Alvo N Idades (anos)
Indivíduos que consomem MDMA (%) Nº de pastilhas
por sessão
Halkitis et al. 311 2007 Nova Iorque
(EUA)
Homens Gay e
bisexuais
450 18-67 74,7%
Medina et al. 312 2007 EUA Consumidores
de MDMA
48 18-35 97,9% (no ano anterior ao estudo)
Yen et al. 313 2007 Taiwan Consumidores
de MDMA
200 14-19 62,5% usaram menos de 20 vezes
29,5% fazem policonsumo
Breen et al. 314 2006 Austrália Consumidores
de MDMA
852 22-24 3-4
Chou et al. 315 2006 Taipei
(Taiwan)
Adolescentes 2126 12-18 2-14,5% (pelo menos 1 vez na vida)
Copeland
et al. 316
2006 Austrália Consumidores
de MDMA
216 19-39 28,2% mistura MDMA com medicamentos 2
Fang et al. 317 2006 China Consumidores
de drogas
5,37%
Halkitis et al. 318 2006 Nova Iorque
(EUA)
Homens
homossexuais e
bissexuais
consumidores de
GHB
131 18-40+ 56,5%
Hopfer et al. 319 2006 Colorado
(EUA)
Jovens em
tratamento por
abuso de droga
782 10-32 34% (pelo menos 1 vez na vida)

Tabela 2 (cont). Estudos epidemiológicos acerca dos padrões de consumo da MDMA.
Estudo Ano Local População-Alvo N Idades (anos)
Indivíduos que consomem MDMA (%) Nº de pastilhas
por sessão
Parsons
et al. 320
2006 Nova Iorque
(EUA)
Mulheres
frequentadoras
de discotecas
1104 18-49 44% (pelo menos 1 vez)
Pavarin 321 2006 Itália
Frequentadores
de festas rave
2015 <20-40+ 17-27,1% (pelo menos 1 vez)
9,5-16,2% (no ano anterior)
4,4%-10% (no mês anterior)
Wish et al. 322 2006 EUA Estudantes
universitários
1206 9 % (pelo menos 1 vez)
Wu et al. 323 2006 Texas (EUA) Mulheres de
baixos
rendimentos
696 18-31 15,2%(pelo menos 1 vez) (destes, 53% no ano anterior e 21%
nos 20 dias anteriores)
0,5-4
Wu et al. 324 2006 EUA Consumidores
drogas sintéticas
19084 16-23 13% (pelo menos 1 vez)
6% (no ano anterior)
Barrett et al. 325 2005 Montreal
(Canadá)
Frequentadores
de festas Rave
186 17-47 50% (pelo menos uma vez) (destes, 92,3% usa MDMA em
combinação)
1
Clatts et al. 326 2005 Nova Iorque
(EUA)
Homens jovens
homo e
bissexuais
569 17-28 47,9% (pelo menos 1 vez)
24,3% (consome cronicamente) (destes, 82.8% usou pelo
menos 1 vez e 13,3% usaram mais de 5 vezes no mês anterior)
Fernández
et al. 327
2005 EUA Homens
hispânicos homo
e bissexuais
172 14% (nos últimos 6 meses)
46

47
Tabela 2 (cont). Estudos epidemiológicos acerca dos padrões de consumo da MDMA.
Estudo Ano Local População-Alvo N Idades (anos)
Indivíduos que consomem MDMA (%) Nº de pastilhas
por sessão
Goldsamt
et al. 328
2005 Nova Iorque
(EUA)
Estudantes do
6º, 7º e 8º ano
23780 2,3% (pelo menos uma vez)
Ompad et al. 329 2005 Nova Iorque
(EUA)
Consumidores
de drogas
715 17-64 23,4% (pelo menos 1 vez)
11,9% (últimos 6 meses)
Crosby et al.330 2004 EUA Homens homo e
bissexuais
150 31-50 14% (nos últimos 3 meses)
Lora-Tamayo
et al. 331
2004 Ibiza
(Espanha)
Indivíduos
presos por actos
criminosos que
confessaram
consumo de club
drugs
70 77% (apenas MDMA na urina)
20% (MDMA + outra droga sintética)
Scholey et al.332 2004 Internautas 763 21-25 37% (pelo menos 1 vez)
“Novato” (39%, consumiu 1-9 vezes)
“Moderado” (48%, consumiu 10-99 vezes)
“Pesado”(13%, consumiu mais de 100 vezes)
“Novato” – 100%
1-2 pastilhas
“Moderado” – 3%
mais de 4
pastilhas
“Pesado” – 14%
mais de 4
pastilhas

Tabela 2 (cont). Estudos epidemiológicos acerca dos padrões de consumo da MDMA.
Estudo Ano Local População-Alvo N Idades (anos)
Indivíduos que consomem MDMA (%) Nº de pastilhas
por sessão
Van Leeuwen
et al. 333
2004 Denver,
Colorado
(EUA)
Jovens sem abrigo
e que fugiram de
casa
186 16-25 25% (nos últimos 9 meses)
Turner et al. 334 2003 Austália Mulheres 9512 22-27 15% (pelo menos 1 vez)
Fendrich
et al. 335
2003 Chicago
(EUA)
População adulta 627 18-40 49% consumiu drogas sintéticas pelo menos 1 vez
Gross et al. 336 2002 Montreal
(Canadá)
Frequentadores
festas rave
210 16-32 65,2% (pelo menos 1 vez)
National
Institute on
Drug Abuse
(NIDA) 337
2002 EUA Estudantes do 8º,
10º e 12º ano,
estudantes
universitários e
jovens adultos
44346
13-14
15-16
17-18
19-22
19-28
2,7% em 1999 para 5,2% em 2001 (pelo menos 1 vez)
6% em 1999 para 8% em 2001 (pelo menos 1 vez)
8% em 1999 para 11,7% em 2001 (pelo menos 1 vez)
2% em 1991 para 14,7% em 2001 (pelo menos 1 vez)
3,2% em 1991 para 13% em 2001 (pelo menos 1 vez)
Strote et al. 338 2002 EUA Estudantes
universitário
14000 <21-24+ 2,8% em 1997 para 4,7% em 1999
Parrott et al. 339 2001 Reino Unido
e Itália**
Jovens 768 14-28 15% (menos de 20 vezes)
15,5% (mais de 20 vezes)
Riley et al. 340 2001 Edimburgo
(Escócia)
Consumidores de
droga
122 16-47 80% (ecstasy ou anfetamina) (destes 35% toma MDMA
semanalmente)
** Roma, Pádova, Manchester e Londres
48

49
Tabela 2 (cont). Estudos epidemiológicos acerca dos padrões de consumo da MDMA.
Estudo Ano Local População-Alvo N Idades (anos)
Indivíduos que consomem MDMA (%) Nº de pastilhas
por sessão
Winstock
et al.266
2001 Reino Unido Consumidores de
droga
1151 18-29 96% (pelo menos 1 vez)
No último ano: 12% consumiu 2–3 vezes por semana e 22%
consumiu 1 vez por semana
2 ou menos
(55%)
4 ou mais (25%)
Boys et al. 341 1999 Inglaterra Sul Consumidores de
drogas e álcool
100 16-21 22% (nos últimos 90 dias)
38% (pelo menos 1 vez na vida)
Topp et al. 342 1999 Austrália Consumidores de
MDMA
329 15-46 89% (pelo menos 1 vez/mês) 1
Instituto
Nacional de
Pesquisa de
Álcool e
Drogas da
Noruega 274, 343
1998 Oslo
(Noruega)
15-20 Em Oslo:
4,8% (pelo menos 1 vez)
No total do país:
0,3% (pelo menos 1 vez, em 1994)
2,5% (pelo menos 1 vez, em 1998)
Schifano
et al.219
1998 Itália Consumidores de
MDMA
150 19-28 50% (1 ou
menos)
Lenton et al. 344
Boys et al. 345
1997 Autrália
Oeste
Frequentadores de
festas rave
83 13-48 76% (pelo menos 1 vez) (41% policonsumo)
31,3% (na última festa rave)

Tabela 2 (cont). Estudos epidemiológicos acerca dos padrões de consumo da MDMA.
Estudo Ano Local População-Alvo N Idades (anos)
Indivíduos que consomem MDMA (%) Nº de pastilhas
por sessão
Williamson
et al. 346
1997 Reino
Unido
Consumidores de
droga
158 14-59 52% usou durante o último ano
70% (no mês anterior)
2
Miller et al. 347 1996 Reino
Unido
Estudantes do
ensino secundário
7722 15-16 7,3% das raparigas (pelo menos 1 vez)
9.2% dos rapazes (pelo menos 1 vez)
Webb et al. 348 1996 Reino
Unido
Estudantes
universitários 2º
ano
3075 18-65 13% (pelo menos 1 vez)
5,2 % usou mais de 1 ou 2 vezes
2,7% consome uma vez por semana
Forsyth 349 1995 Glasgow
(Reino
Unido)
Frequentadores de
festas “rave”
135 14-44 91% (pelo menos uma vez)
Cuomo et al. 350 1994 EUA Estudantes
universitários
742 (em 1986)
1264 (em 1990)
15,5% (pelo menos 1 vez, em 1986)
24,3% (pelo menos 1 vez, em 1998)
Solowij et al. 22 1992 Sidney
(Austrália)
Consumidores de
MDMA
100 16-48 68% usou mais de 3 vezes
33% usa uma vez por mês ou por trimestre
18 % usa em ocasiões especiais
9% menos de 1
71% 1
13% 2
7% mais de 2
Peroutka 351 1987 EUA Estudantes
universitários
369 39% usou pelo menos 1 vez no ano anterior
50

______________________________________________________ Introdução
51
O mais recente relatório do Observatório Europeu da Droga e da
Toxicodependência, publicado em 2007 20, indica que em muitos países
europeus a segunda substância ilegal mais consumida é algum tipo de droga
sintética embora à escala europeia existam, presentemente, mais
consumidores de cocaína. Preocupantemente, as taxas de prevalência do
consumo destas drogas sintéticas entre os grupos etários mais jovens são
significativamente mais elevadas. A prevalência do consumo de ecstasy na UE
tem aumentado desde 1990, sendo, actualmente, a droga sintética mais
consumida em 17 países europeus dos quais se destacam a República Checa,
o Reino Unido, a Espanha e a Holanda. Na faixa etária dos 15 aos 24 anos,
encontram-se taxas mais elevadas de prevalência do consumo de ecstasy ao
longo da vida entre os homens (0,3-23,2%) do que entre as mulheres (0,3-
13,9%). Esta informação pode ser analisada de forma mais pormenorizada
acedendo à página web da European Monitoring Centre for Drugs and Drug
Addiction (EMCDDA) e seguindo o link http://www.emcdda.europa.eu/
attachements.cfm/att_44705_PT_TDAC07001PTC.pdf.
Em Portugal, o consumo de MDMA surgiu no início da década de 1990
e, à semelhança do que ocorreu nos restantes países europeus, esta droga
ganhou rapidamente popularidade, particularmente entre os jovens. Segundo o
relatório anual do Instituto da Droga e da Toxicodependência publicado em
2006, nos estudos nacionais de 2001 relativos à população portuguesa dos 15-
64 anos e à população escolar do 3.º Ciclo do Ensino Básico, a ecstasy surgiu
com prevalências de consumo muito semelhantes às de heroína e de cocaína,
chegando mesmo a ser superiores nalgumas situações, nomeadamente nos
padrões de consumo recente. No caso da população reclusa, o consumo de
ecstasy apresentou ainda bastante menos expressão que as drogas
tradicionais.
Nas populações escolares, os estudos mais recentes evidenciaram
prevalências de consumo de ecstasy relevantes, embora bastante inferiores às
de Cannabis, de acordo com rastreios do HBSC/WHO (Health Behaviour in
School-aged Children, colaboração com a Organização Mundial da Saúde), em
2002 e 2006 (respectivamente 2,2% e 1,6%), do ESPAD (European School
Survey Project on Alcohol and Other Drugs) em 2003 (4%) e do ECATD
(Estudo sobre o Consumo de Álcool, Tabaco e Droga) em 2003 (entre 1,5%

Introdução ______________________________________________________
52
dos alunos de 13 anos e 4,3% dos de 18 anos). Entre 1999 e 2003, no âmbito
do ESPAD, constatou-se um importante aumento da prevalência do consumo
de ecstasy (de 2,3% para 4%). Todavia, a nível do HBSC/WHO entre 2002 e
2006 verificou-se uma diminuição da percentagem de início do consumo de
ecstasy.
No que respeita ao estudo publicado em 2003 pelo Instituto da Droga e
da Toxicodependência (IDT) sobre consumos problemáticos de drogas em
populações que não incluem adolescentes, a ecstasy tinha pouca expressão
nas amostras das zonas down (esferas sociais marginalizadas e
estigmatizadas) e up (esferas social de elevado nível sócio-cultural), surgindo
com um consumo esporádico ou único e sendo utilizada pontualmente na zona
up como droga recreativa. Foi também assinalada a tendência do seu consumo
entre populações juvenis de bairros periféricos, o que poderia indiciar a difusão
de consumos para lá dos grupos juvenis de origem. O consumo de MDMA pela
população em geral (0,8% na população portuguesa em 2005) é habitualmente
baixo, mas estas frequências sobem se nos referirmos à população mais jovem 352.
Estas percentagens de consumo de MDMA aumentam quando os
estudos epidemiológicos se centram em populações relacionadas com a
cultura rave. Um inquérito anual realizado aos leitores da revista de música
Mixmag do Reino Unido, cujo público é constituído por pessoas que
frequentam clubes de dança regularmente, conclui que a percentagem de
pessoas definidas como grandes consumidoras de ecstasy (que consomem
normalmente mais de quatro comprimidos por sessão) aumentou para mais do
dobro entre 1999 e 2003, de 16% para 36% 353.
Num estudo realizado no Reino Unido através da internet 332 também é
mencionado um consumo de ecstasy e um policonsumo de droga cada vez
mais intenso por parte dos consumidores de ecstasy experientes.
A média das idades de início do consumo de MDMA é 19,8 anos 22, 219,
266, 303, 316, 321, 324, 329, 336, 342, 346, 354, 355. Cerca de 10% dos indivíduos com idade
inferior a 20 anos consome MDMA 5.
Geralmente, as mulheres iniciam o consumo de drogas na adolescência
e, a maioria, cessa o consumo cerca dos 25 anos, provavelmente devido ao

______________________________________________________ Introdução
53
casamento. Quando esta paragem não ocorre, normalmente o consumo é
associado a binge drinking ou a hábitos tabágicos 334.
1.8.1. Onde? – Locais de consumo de MDMA
A MDMA é, habitualmente, consumida em festas rave que,
normalmente, ocorrem em grandes espaços isolados como armazéns ou
espaços abertos, onde a supervisão está ausente ou é mínima 15.
É também consumida em festas techno e em discotecas. Estas festas
decorrem tipicamente em ambientes de elevada temperatura, sobrelotados e
ao som de música electrónica repetitiva, em elevados decibéis e acompanhada
de espectáculos de luz. Todas estas circunstâncias podem influenciar a
expressão da toxicidade da MDMA e a maior parte das intoxicações fatais
relatadas na literatura está associada ao uso da MDMA neste contexto 13.
Nestes locais, o álcool e outras drogas estão também disponíveis e são
consumidos em grandes quantidades 314. Preocupantemente, o seu consumo
tem vindo a aumentar em domicílios privados, escolas secundárias e
universidades 22, 356.
Habitualmente, este consumo ocorre preferencialmente durante o fim-
de-semana e num contexto de grupo, com os amigos. Existe uma forte relação
com o gosto pela música house, techno e trance 305.
1.8.2. Como e quando? – Padrões de consumo de MDMA
A MDMA pode ser consumida por via oral (a mais frequente), nasal
(snorting, inalação do pó obtido pelo esmagamento da pastilha 14), rectal
(supositórios, conhecida por shefting ou shelving) ou, menos frequentemente,
por via injectável. O facto de os utilizadores experimentarem diferentes vias de
administração deve-se a que 41% dos utilizadores referem diferenças nos
efeitos da MDMA, dependendo da via de administração utilizada. A via
injectável é a que provoca um início de acção mais rápido e produz uma
experiência mais intensa, embora mais curta. O snorting produz um início de

Introdução ______________________________________________________
54
efeito mais rápido que o comprimido, mas uma experiência mais curta,
enquanto que os supositórios têm um início de acção lento, produzindo, no
entanto, uma experiência mais intensa e prolongada 22. Esta diferença da
farmacocinética da MDMA em função da via de administração foi já descrita em
primatas não humanos 33.
Para aumentar a intensidade das experiências resultantes dos efeitos da
MDMA sobre as capacidades sensoriais, os utilizadores de ecstasy usam
frequentemente acessórios fluorescentes (colares, pulseiras, etc) e aplicam
cremes de menta nos lábios ou spray de menta em máscaras cirúrgicas 14. Os
consumidores de ecstasy bebem frequentemente líquidos para evitar a
hipertermia e utilizam chupa-chupas ou chupetas para aliviar o bruxismo 67,
uma vez que alguns destes utilizadores são conscientes dos efeitos deletérios
desta substância 357.
Normalmente, o consumo de MDMA é feito num padrão de policonsumo.
Num estudo australiano realizado em 2004, os utilizadores de MDMA
entrevistados referiram consumir 10 ou 11 tipos de drogas ao longo da sua vida
e 7 ou 8 nos 6 meses que antecederam a entrevista 314. Dentro das
substâncias mais frequentemente co-consumidas com a MDMA incluem-se o
tabaco, a Cannabis e o etanol 331, 332. Um outro padrão de consumo comum
envolve a utilização de ansiolíticos e sedativos (opiáceos e benzodiazepinas)
em conjunto com os estimulantes 346. Este uso concomitante de estimulantes e
sedativos está associado, de acordo com estudos epidemiológicos, ao aumento
da probabilidade do seu utilizador vir a utilizar estimulantes injectáveis, vir a
sofrer efeitos adversos e vir a ser tratado devido a problemas relacionados com
o consumo de drogas 346.
1.9. Factores condicionantes dos efeitos da MDMA
Como já referido, a MDMA provou ser tóxica em vários sistemas de órgãos
em animais e em humanos. Contudo, o tipo e a severidade desta toxicidade
são influenciados por vários factores incluindo a temperatura ambiente, o
exercício, associação de drogas e níveis de hidratação. Estas variáveis,
quando combinadas com a heterogeneidade dos compostos incluídos nas

______________________________________________________ Introdução
55
pastilhas vendidas como ecstasy, dificultam a determinação das circunstâncias
envolvidas nos efeitos tóxicos da MDMA em utilizadores humanos 358.
Os principais factores condicionantes dos efeitos agudos e tóxicos da
MDMA são abordados de seguida.
1.9.1. Dose de MDMA
Como em qualquer droga preparada e obtida ilicitamente, quer as doses
quer a pureza das pastilhas de MDMA variam substancialmente como pode ser
constatado por análise da Tabela 3.
As pastilhas de ecstasy vendidas no mercado contêm, habitualmente,
entre 80 a 150 mg de MDMA, tendo sido também encontradas dosagens
superiores (até 250 mg por pastilha) 5, podendo conter também substâncias
inertes e outras drogas. Normalmente, as pastilhas contêm a mistura racémica
dos dois enantiómeros da MDMA 359.
Como já referido, a dose ingerida de MDMA vai condicionar os seus
efeitos biológicos e a sua toxicidade, nomeadamente devido ao fenómeno de
auto-inibição metabólica induzido por esta molécula. Os efeitos pró-sociais em
ratinhos e ratos são aumentados de forma dependente da dose 360-362. Mesmo
o consumo de pequenas doses de MDMA apenas uma vez por semana pode
causar efeitos prolongados em ratos que se manifestam a nível
comportamental 363. Em humanos, o consumo de doses mais elevadas foi mais
frequentemente associado com sensação de desorientação e com mais efeitos
alucinatórios e laterais. Este padrão de consumo provoca um efeito mais
prolongado, mais intenso, com um pico de efeito mais longo e mais forte,
aparecem mais efeitos de “ressaca” e perde-se o “sentimento de estar a
controlar” 22.
No entanto, para além de influenciar a duração e intensidade dos efeitos
a dose de MDMA pode influenciar a capacidade aditiva desta substância.
Exemplo disso é o facto de, em condições de temperatura ambiente normal, a
preferência pela MDMA em macacos aumentar em função da dose, uma vez
que para uma dose de MDMA de 0,03 mg/kg, os animais preferem a ração em

Introdução ______________________________________________________
56
vez da MDMA, enquanto que para doses mais elevadas, os animais preferem a
MDMA em detrimento da ração 364.
Não apenas a quantidade de MDMA consumida em cada ocasião, mas
também a frequência do consumo, alteram o perfil de efeitos e de toxicidade
deste composto. Doses repetidas de MDMA levam ao aparecimento de
hipertermia numa forma dose-dependente em temperaturas ambientais que
não originam resposta hipertérmica quando a MDMA é administrada em doses
isoladas 147. O mesmo padrão de aumento de resposta foi verificado em ratos
para a resposta hipercinésica associada ao consumo de MDMA 375. O consumo
de várias doses de MDMA parece contribuir ainda, em primatas não-humanos,
para o aparecimento de défices serotonérgicos duradouros, mesmo com
apenas 2 doses de 4,3 mg/kg administradas per os com 3 horas de intervalo 33.
No entanto, doses sucessivas são normalmente associadas a
experiências menos intensas e de menor duração de efeitos do que a primeira
dose consumida em casa ocasião, normalmente com redução dos efeitos
agradáveis e aumento dos efeitos laterais. Este facto pode reflectir o
desenvolvimento de taquifilaxia, fenómeno de tolerância aguda normalmente
associada aos alucinogénios 22. Normalmente, aqueles que consomem um
maior número de pastilhas de MDMA durante a vida têm um maior risco de,
eventualmente, desenvolver perturbações psicopatológicas 219.
A relevância de estudos sobre o consumo da MDMA em humanos tem
vindo a aumentar devido às variações de doses consumidas 76. De facto, os
resultados de um estudo realizado em 2001 no Reino Unido indicam que
alguns utilizadores regulares de ecstasy consomem grandes doses de MDMA
que não estão muito distantes das usadas em animais para induzir
neurotoxicidade. Se considerarmos as diferenças interespécies, a maior taxa
metabólica nos pequenos roedores faz com que a dose equivalente nos
humanos seja menor, pelo que, em termos de saúde pública, um número
significativo de indivíduos estão expostos a um sério risco de danos
neurotóxicos que são ainda condicionados pela via e frequência dos consumos 266.

57
Tabela 3. Perfil de impurezas e conteúdo em MDMA de pastilhas vendidas como ecstasy.
Estudo Ano Local N Origem Dose de MDMA
Com MDMA Sem MDMA Outras substâncias presentes nas pastilhas com
MDMA
Cheng
et al. 365
2006 Hong
Kong
89 Apreensão Média 43
mg
Ketamina, cafeína, salicilatos, paracetamol,
clorfeniramina,
Metronidazol, sub-produtos de síntese
Tanner-
Smith 366
2006 EUA 1214 Submissão
anónima
39% (apenas MDMA)
15% (MDMA+X)
46% MDA, DXM, cafeína, pseudoefedrina, MTA.
Baixos teores de ketamina, DOB, Heroína, PCP, PMA
Teng
et al. 367
2006 Taiwan 181 Apreensão 16-193
mg
66-71% (apenas MDMA) 7% continha MDEA
7% continha MTA
4% continha anfetamina
18% continha cafeína
MDA, ketamina, efedrina, paracetamol, diazepam,
clorzoxazona, nicotinamida
EACD*368
2004 Nova
Zelândia
8604 Apreensão 30,2-
172,4 mg
A maioria Ketamina, cocaína,
Cheng
et al. 369
2003 Hong-
Kong
212 Apreensão 55%
MTA, MDA e
anfetamina
Ketamina (presente em 12% das pastilhas em 2000 e
42% em 2001)
Cole et al. 370
2002 Reino
Unido
80 Apreensão 20-109
mg
100% MDEA
*EACD, Expert Advisory Committee on Drugs, Nova Zelândia

Tabela 3 (cont). Perfil de impurezas e conteúdo em MDMA de pastilhas vendidas como ecstasy.
Estudo Ano Local N Origem Dose de MDMA
Com MDMA Sem MDMA Outras substâncias presentes nas pastilhas com
MDMA
Spruit 371 2001 Holanda 10000 Apreensão 48-60% (apenas MDMA)
7-13% (MDMA+X)
2-7% (MDA ou
MDEA)
MDA, MDE, anfetamina
Baggott
et al. 372
2000 EUA 107 Submissão
de
pastilhas
para
análise
63% (MDMA, MDA ou
MDEA)
29% 21% contém DXM
Cafeína, efedrina, pseudoefedrina, salicilatos
Sherlock
et al. 373
1999 Reino
Unido
25 Amnistia pombas
(19-140
mg)
outras (2-
105 mg)
64% (MDEA ou MDMA) 24% Ketamina, cafeína, MTA, anfetamina paracetamol
Schifano
et al. 219
1998 Itália 20000 Apreensão 100-150
mg
85-90% MDA, MDEA,
MBDB
Giroud
et al. 3
1997 Suiça MDA, MDEA, MBDB, 2-CB, Cafeína, efedrina, polióis,
LSD, testosterona, cloroquina, vasodilatadores
Milroy
et al. 182
1996 Reino
Unido
13 0,21-12
mg
77% (MDEA, MDMA, MDA,
ou misturas)
Renfroe 374
1986 EUA 101 58 % (apenas MDMA)
26% (MDMA+X)
16% MDA
58

______________________________________________________ Introdução
59
1.9.2. Condições ambientais no momento do consumo
O risco para a saúde associado ao consumo de substâncias de abuso
está relacionado com as condições ambientais dos locais em que elas são
usadas 376. De facto, as condições ambientais, sobretudo a temperatura,
afectam grandemente os efeitos e a toxicidade da MDMA.
Os efeitos da MDMA na temperatura corporal parecem ser dependentes
da temperatura ambiente em ratos 139, 377-379, ratinhos 142 e macacos 380, 381.
Quando administrada a ratos sob condições de temperatura ambiente elevada,
a MDMA provoca hipertermia por aumentar a produção de calor pelo tecido
adiposo castanho e diminuir a perda de calor por provocar vasoconstrição
periférica. Pelo contrário, quando administrada a ratos em ambientes frios, a
MDMA causa hipotermia inibindo a termogénese do tecido adiposo castanho e
a vasoconstrição da artéria da cauda causadas pelo frio, por um mecanismo
que parece envolver a activação de receptores 5-HT1A e D2 382. Aparentemente,
a perda de 5-HT para interagir com os receptors 5-HT1A leva a perturbações na
termorregulação em ratos colocados em ambientes com temperaturas elevadas 149. Estas alterações da temperatura corporal podem levar a alterações no
volume de distribuição, afectando a farmacocinética da MDMA 380.
Além de influenciar a resposta termogénica, a temperatura ambiente tem
também influência na intensidade reforçadora relativa da MDMA (ou seja, na
vontade de consumir novamente e em detrimento da comida) em macacos. De
facto, o aumento da temperatura ambiente (característico dos locais onde a
MDMA é consumida) aumenta a intensidade relativa de reforço de uma
injecção de MDMA na dose de 0,03 mg/kg, enquanto que a diminuição da
temperatura ambiente atenua significativamente a escolha de uma dose de
MDMA de 0,1 mg/kg 364. O mesmo aumento do reforço foi observado em ratos
que, em ambiente quente, aumentaram o número de auto-administrações de
MDMA 383. Um possível mecanismo neuroquímico para explicar este aumento
do reforço do consumo de MDMA pode ser uma potenciação do aumento dos
níveis extracelulares de DA nas áreas do cérebro envolvidas no reforço como o
nucleus accumbens 384, uma teoria confirmada em ratos 385.

Introdução ______________________________________________________
60
No entanto, em ratos, apesar de afectar a magnitude da resposta
hipertérmica induzida pela MDMA, a temperatura ambiente não afecta a
hipercinésia 375.
Foi também já referido que o aumento da temperatura ambiente tem
também a capacidade de aumentar a toxicidade da MDMA quer in vivo 139, 142,
quer in vitro 60, 386, 387. Por exemplo, Malberg e Seiden (1998) encontraram
marcadores de neurotoxicidade no cortex frontal, hipocampo e estriado,
quando a MDMA foi administrada a ratos a uma temperatura ambiente de 26-
30ºC, o que não se verificou quando a administração foi feita a 20-24ºC 139.
Também aqui, a DA parece ter um papel importante 93. A temperatura ambiente
elevada aumenta ainda a capacidade da MDMA inibir a triptofano hidroxilase 230, o que potencialmente irá atrasar a reposição dos níveis neuronais de 5-HT.
Um dos efeitos também aumentado em ambientes de temperatura mais
elevada é o efeito pró-social da MDMA em ratos 383. Por outro lado, a
interacção social com fêmeas, ou restrições na dissipação de calor, leva a uma
potenciação do aumento da temperatura cerebral induzido pela MDMA. Este
facto foi já confirmado em ratos 388 e pode estar envolvido na potenciação da
neurotoxicidade da MDMA.
O papel da temperatura ambiente é de tal forma importante na
intensidade dos efeitos que, em relatos subjectivos, os utilizadores de MDMA
referiram que “o calor foi o factor causal do início e manutenção dos efeitos” 364.
Também o ruído em alto volume foi já associado a um aumento da
neurotoxicidade da MDMA em ratinhos, que, nestes animais, ocorre por lesão
dos terminais dopaminérgicos 389. No entanto, o mecanismo pelo qual a
exposição a ambientes ruidosos aumenta a neurotoxicidade da MDMA não
está ainda esclarecido. O ruído pode potencialmente agravar também a
rabdomiólise ao nível do miocárdio (miocitólise), muitas vezes considerada
responsável de morte súbita subsequente ao consumo de MDMA, uma vez que
o ruído, por si só, pode lesar o miocárdio e, quando associado a um consumo
múltiplo (binge) de MDMA, origina alterações ultra-estruturais ao nível das
mitocôndrias do músculo cardíaco 390, 391.
Situações de stress (ex.: imobilização, interacção social com congéneres
agressivos, choques eléctricos nas patas, contusão da cauda, restrição de
comida, stress pré-natal) provocam um aumento das concentrações cerebrais

______________________________________________________ Introdução
61
de MDMA em ratinhos 392 e aumentam a auto-administração de
psicoestimulantes 393.
Estudos em animais demonstraram ainda que a agregação dos animais
potencia os efeitos agudos de ambos os enantiómeros da MDMA 145, 394, o que
levanta a problemática do consumo habitual desta substância em ambientes
sobrelotados. Esta agregação influencia também comportamentos aditivos
relativamente à MDMA (quer racemato quer qualquer um dos enantiómeros)
que são mais evidentes em animais isolados do que em animais agregados 395.
Também a actividade física contribui para o agravamento dos efeitos
tóxicos agudos da MDMA 394, provavelmente por aumentar a produção de calor
e contribuir, assim, para um aumento da hipertermia. De facto, utilizadores que
reportaram dançar ou fazer exercício físico quando sob os efeitos da MDMA
parecem apresentar, posteriormente, mais efeitos adversos. A imobilização de
ratinhos aos quais foi administrada uma dose de 20 mg/kg de MDMA resultou
num bloqueio da resposta hipertérmica, bem como da neurotoxicidade
observada em animais não imobilizados 243. Por outro lado, o exercício
extenuante provou já ser indutor da CYP1A2 396, o que pode conduzir a um
aumento do metabolismo da MDMA com maior taxa de formação de
metabolitos tóxicos.
A ingestão de água influencia, também, a toxicidade da MDMA em ratos
porque, se por um lado a ausência do consumo de água durante o período de
exposição à MDMA aumenta significativamente a hipertermia quando a
temperatura ambiente está elevada 375, o consumo exagerado de água pode
conduzir a uma situação de hiponatrémia que resulta em edema cerebral 13, 214,
215.
Os dados apresentados permitem, assim, inferir que ambientes quentes,
barulhentos e sobrelotados nas festas rave podem criar as condições ideais
para um aumento da resposta aguda à MDMA e, potencialmente, aumentar a
propensão para o aparecimento de sequelas neuropsicológicas adversas em
utilizadores de ecstasy 394.

Introdução ______________________________________________________
62
1.9.3. Características do consumidor de MDMA
Existem vários factores inerentes ao consumidor de ecstasy que podem
condicionar o risco a que ele está sujeito quando consome esta substância,
uma vez que condicionam a sua capacidade metabólica e a sua
susceptibilidade ao desenvolvimento dos efeitos agudos da MDMA e que,
inclusivamente, influenciam os padrões de consumo adquiridos. As principais
características do consumidor que podem influenciar os efeitos da MDMA são:
a idade, a etnia, o género, a variabilidade farmacogenética interindividual e a
orientação sexual.
1.9.3.1. Idade
O consumo de MDMA está predominantemente associado a faixas
etárias mais jovens. De facto, uma elevada proporção dos jovens com menos
de 16 anos está exposta a esta droga de abuso 13. Daí que seja importante
considerar o efeito da idade na expressão e actividade das enzimas envolvidas
no metabolismo desta substância 397. Em ratos imaturos, os níveis de CYP3A2
são elevados e, na puberdade, diferenciam-se consoante o género, sofrendo
um decréscimo selectivo nas fêmeas 398. A CYP2C11 tem níveis baixos nos
ratos imaturos e aumenta, na puberdade, nos machos 398. Em humanos,
existem poucas diferenças entre as actividades das CYP microssomais nas
diferentes faixas etárias. No entanto, a actividade da CYP1A2, CYP2B6,
CYP2C19, CYP2D6 e CYP2E1 em microssomas hepáticos humanos tende a
diminuir com a idade, enquanto que a actividade da CYP2A6 e CYP4A11 tende
a aumentar com a idade 398. Por outro lado, de acordo com o observado em
hepatócitos humanos criopreservados, a CYP3A4 tende a aumentar com o
aumento da idade, fenómeno não observado nos microssomas 398.
Globalmente, com o aumento da idade assiste-se a uma diminuição da
capacidade metabólica dos indivíduos que chega a ser de 30% após os 70
anos de idade 397. Para além disto, a clearance e o volume de distribuição de
numerosos compostos diminui com o envelhecimento, provavelmente por uma

______________________________________________________ Introdução
63
diminuição do volume do fígado e do fluxo hepático sanguíneo 399, 400, mais do
que por uma diminuição da actividade das enzimas microssomais 401.
Pensa-se que o desenvolvimento cerebral que ocorre durante a
adolescência pode resultar em respostas farmacológicas que diferem
crucialmente das dos adultos. Por exemplo, a potência da cocaína, ketamina e
MDMA para produzir estimulação motora em ratos macho é menor nos
adolescentes do que nos adultos 402. Diferenças no efeito analgésico induzido
pela MDMA (mais intenso nos animais de meia idade do que nos adolescentes)
podem ser encontradas em 3 grupos de ratos de idades diferentes e podem
relacionar-se com diferentes graus de maturação do sistema serotonérgico em
função do estadio de desenvolvimento 360. Esta diferença ao nível da
farmacodinamia da MDMA com a idade pode dever-se a (i) diferentes perfis de
diminuição dos níveis cerebrais de 5-HT e de bloqueios da recaptação da 5-HT,
que são mais intensos nos animais mais velhos; (ii) diferente tendência para
que estes efeitos sejam, ou não, persistentes 138, 403, sendo mais persistentes
as alterações verificadas nos animais mais velhos; e (iii) a uma alteração da
densidade dos SERT que manifesta uma diminuição ao longo dos anos 404.
Devido aos vários factores referidos, os adolescentes humanos parecem exibir
uma sensibilidade reduzida aos efeitos de drogas de abuso como a cocaína, a
ketamina e a MDMA, o que pode levar a uma utilização de quantidades
superiores destas substâncias por cada consumo, quando comparada com
indivíduos mais velhos 402, 405.
Por outro lado, devido ao fenómeno de tolerância, os consumidores mais
velhos estão habitualmente expostos a maiores quantidades de ecstasy em
cada ocasião e a maiores quantidade de outros tipos de drogas para
intensificar os efeitos obtidos com a MDMA 332, aumentando a propensão ao
desenvolvimento de interacções.
A adolescência é um período durante o qual os rearranjos maturativos
podem exercer uma forte influência na diferente vulnerabilidade aos
psicoestimulantes 406, 407. De facto, os efeitos da exposição repetida de ratinhos
CD-1 adolescentes à MDMA são persistentes até à idade adulta e existe uma
resposta diferente à MDMA em função da idade em que ocorre a primeira
exposição à substância 360. Este facto indica que os efeitos prolongados da
MDMA estão associados a uma utilização precoce desta substância 403, 408.

Introdução ______________________________________________________
64
1.9.3.2. Etnia
As características da resposta farmacológica e toxicológica a
xenobióticos, dependente da metabolização via CYP450, são normalmente
muito específicas para cada grupo étnico, uma vez que a frequência das
variantes alélicas que codificam para estas enzimas varia acentuadamente
entre diferentes grupos étnicos 66. Um exemplo é a CYP1A2 que apresenta
uma actividade inferior em indivíduos de raça negra relativamente aos
caucasianos 409. Os Hispânicos representam outro bom exemplo pois, quando
comparados com os Caucasianos e os Afro-Americanos, têm cerca do dobro
ou triplo de actividade de CYP2A6, CYP2B6, e CYP2C8 e metade de
actividade da CYP1A2 398.
Para além das condicionantes farmacogenéticas, a etnia parece
influenciar o padrão de consumo. Este efeito da etnia no padrão de consumo
foi evidenciada no estudo de Clatts e colaboradores os quais registaram o
consumo de club drugs entre homens homossexuais e bisexuais, tendo
apresentado diferenças entre indivíduos de raça branca (37,5% consumiu pelo
menos uma vez; 41,4% consome cronicamente), negra (13,7% consumiu pelo
menos uma vez; 11,7% consome cronicamente), hispânica (38,9% consumiu
pelo menos uma vez; 35,2 consome cronicamente) e outras (9.9% consumiu
pelo menos uma vez; 11,7% consome cronicamente) 326.
Os indivíduos de raça branca têm mais probabilidade de consumir
ecstasy pelo menos uma vez na vida do que os hispânicos e os negros (44,6%
versus 26,7% e 15,9%, respectivamente) 329.
Os negros têm demonstrado menor probabilidade de utilizar drogas
sintéticas, enquanto que os brancos, nativos americanos e jovens mestiços são
normalmente mais propensos do que os outros grupos raciais/étnicos a
consumir uma ou mais club drugs 324.
1.9.3.3. Género
Tal como já foi descrito para outras drogas de abuso 410, 411, o
mecanismo de acção e as consequências do consumo de MDMA não são

______________________________________________________ Introdução
65
idênticos entre homens e mulheres 233. A(s) causa(s) destas diferenças entre
géneros não é conhecida, mas podem dever-se a diferenças nos perfis
hormonais 412, volume e morfologia de determinadas estruturas cerebrais 413,
na transmissão monoaminérgica ou na capacidade metabólica.
A 5-HT está envolvida na modulação de uma variedade de funções,
nomeadamente na regulação do humor e está implicada na patofisiologia da
depressão e de problemas de ansiedade, patologias com maior prevalência em
indivíduos do sexo feminino 414. Curiosamente, existem também diferenças de
género relativamente a marcadores de integridade serotonérgica em
utilizadores de MDMA, indicando uma função serotonérgica diminuída nas
mulheres, quando comparada com os homens 415. Especificamente, em
indivíduos que consumiram MDMA, foram encontradas reduções nas
concentrações de ácido 5-hidroxiindolacético (metabolito da 5-HT) no fluído
cerebrospinal, mais intensas nas mulheres do que nos homens (46% vs. 20%),
o que sugere uma maior susceptibilidade das mulheres do que dos homens
aos efeitos neurotóxicos da MDMA 415. Esta hipótese foi confirmada num
estudo sobre a influência de algumas variáveis na neurotoxicidade
serotonérgica da MDMA em várias regiões cerebrais em que apenas se
registaram alterações nos neurónios serotonérgicos no grupo feminino de
consumidores de doses elevadas de MDMA, o que parece indicar que as
mulheres são aparentemente mais susceptíveis a desenvolver neurotoxicidade
devido ao consumo de MDMA do que os homens 233. De facto, o uso de MDMA
está associado a uma diminuição da densidade dos transportadores da
serotonina cerebrais nas mulheres, dependente da dose e potencialmente
reversível. Esta redução na densidade dos SERT não é significativa nos
homens 233. No entanto, em animais de laboratório a influência do género na
susceptibilidade à MDMA é bastante diferente. Em ratinhos, uma
vulnerabilidade ao aumento da actividade locomotora espontânea e à
exploração ambiental foi observada nos machos e não nas fêmeas 360.
Adicionalmente, nestes animais, a administração de MDMA na dose de 20
mg/kg está associada a uma maior letalidade nos machos, o que sugere que
eles possam ser mais sensíveis à toxicidade geral da MDMA 243. As fêmeas
parecem ser mais resistentes do que os machos à toxidade da MDMA e MDA,
provavelmente por um mecanismo de captação dos radicais livres que parecem

Introdução ______________________________________________________
66
ser responsáveis pelos efeitos letais agudos destas substâncias (ex.: radical
superóxido) pelas hormonas sexuais 416.
Também os efeitos psicoactivos da MDMA são, geralmente, mais
intensos nas mulheres do que nos homens. As mulheres têm alterações
perceptivas, problemas de raciocínio, medo de perder o controlo do corpo,
ansiedade, efeitos adversos e sequelas mais intensos, enquanto que os
homens sofrem maiores aumentos da pressão arterial 414. Aparentemente, a
dose de MDMA correlaciona-se positivamente com a intensidade das
alterações perceptivas na mulher. O facto de doses iguais de MDMA (por
quilograma de peso corporal) produzirem respostas mais intensas nas
mulheres é consistente com a ideia de uma maior susceptibilidade das
mulheres aos efeitos libertadores de 5-HT provocados pela MDMA 414. Não se
observaram, no entanto, diferenças de género nos comportamentos agressivos
manifestados pelos consumidores de ecstasy durante os dias subsequentes ao
consumo 417.
Outras diferenças de género que podem afectar as respostas biológica e
toxicológica à MDMA incluem as diferentes capacidades metabólicas. De facto,
existem factores hormonais que condicionam a expressão das isoenzimas do
citocromo P450 398. Nos humanos, coelhos, cães e macacos as diferenças de
género não influenciam a expressão microssomal do CYP450 ou de outras
enzimas metabolizadoras na mesma extensão que se verifica no rato e, menos
expressivamente, no ratinho 398. Apesar disso, as mulheres possuem menor
actividade da CYP1A2 (7-etoxiresirufina O-desalquilase) do que os homens 409,
cuja actividade se encontra também influenciada pela fase do ciclo menstrual 396. Nos ratos adultos, não se verificam diferenças de género na actividade da
CYP2D6 e CYP2E1. A CYP3A2 é uma das isoenzimas específicas dos machos
(níveis nos machos são 10 vezes maiores que nas fêmeas) devido à secreção
pulsátil de hormona do crescimento nos machos. Os níveis mais baixos de
CYP3A nas fêmeas adultas são causados por um declínio nos níveis de
CYP3A2 nas fêmeas durante a puberdade 398. A indução das isoenzimas do
CYP450 tende a ser maior nas fêmeas, o que se verifica nas isoenzimas
microssomais CYP2B e CYP3A que apresentam uma maior indutibilidade nas
fêmeas de rato do que nos ratos macho 398. Em microssomas hepáticos
humanos as actividades da CYP2B6, CYP2C19 e CYP3A4 (testosterona 6β-

______________________________________________________ Introdução
67
hidroxilase) tendem a ser mais elevadas nos microssomas de fígado de
mulheres, enquanto que a CYP1A2 e a CYP2D6 tendem a ser mais elevadas
nos microssomas hepáticos de homens 398. Estas diferenças a nível metabólico
fazem com que a metabolização da MDMA seja feita mais rapidamente nos
homens do que nas mulheres 373.
Aparentemente, as mulheres estão também mais predispostas a
desenvolverem hiponatrémia e edema cerebral após a utilização de MDMA,
uma vez que a maioria dos casos clínicos de hiponatrémia descritos na
literatura ocorreram em jovens do sexo feminino 205, 209, 210, 292, 418. Uma vez que
este efeito hiponatrémico da MDMA parece dependente do seu metabolismo 217, o seu aparecimento será provavelmente condicionado pela capacidade
metabólica individual que, como já foi descrito anteriormente, difere entre
indivíduos do sexo feminino e masculino.
Curiosamente, os indivíduos do sexo feminino são menos sensiveis aos
efeitos hipertérmicos da MDMA do que os indivíduos do sexo masculino. Este
facto foi confirmado num estudo realizado em ratos que constatou que a
incapacidade da MDMA induzir uma resposta termogénica nas fêmeas pode
dever-se ao facto de as fêmeas apresentarem (i) menor activação simpática
após administração de MDMA, (ii) vasculatura menos sensível à estimulação
adrenérgica α1, e mais sensível ao monóxido de azoto e (iii) menor expressão
de UCP3 no músculo esquelético, quando comparadas com os machos 419.
Existem ainda diferenças de género no que concerne aos padrões de
consumo da MDMA. Segundo um estudo realizado em 1999 342, as mulheres
iniciam o consumo de MDMA mais precocemente que os homens. No entanto,
os homens apresentam taxas mais elevadas de prevalência do consumo de
ecstasy ao longo da vida do que as mulheres 20.
A análise conjunta da informação reunida permite inferir que,
provalmente, enquanto as mulheres são mais susceptíveis a desenvolver os
efeitos psicoactivos e neurotóxicos da MDMA por apresentarem menor
capacidade de metabolizar esta substância, os homens, devido à sua maior
taxa metabólica e maior prevalência de consumo, estarão mais sujeitos a
desenvolver quadros de resposta tóxica disseminada subsequente ao seu
consumo, apresentando maiores taxas de letalidade.

Introdução ______________________________________________________
68
1.9.3.4. Variabilidade farmacogenética interindividual
A susceptibilidade de um ou vários indivíduos aos efeitos
biológicos/tóxicos de um determinado xenobiótico pode ser determinada por
alterações genéticas estáveis e hereditárias. Por convenção, quando uma
determinada alteração genética (mutação) atinge mais de 1% de uma
população, designa-se polimorfismo. Os polimorfismos genéticos podem
traduzir-se em alterações de proteínas que funcionam como receptores,
originando variações farmacodinâmicas 420, ou que estão associadas ao
metabolismo e transporte de xenobióticos 89, exercendo um efeito
preponderante na farmacocinética dos compostos 421.
Uma vez que a isoforma 2D6 do sistema citocromo P450 é a principal
via metabólica da MDMA, indivíduos com variações genéticas nesta enzima
(ex.: metabolizadores lentos ou metabolizadores ultra-rápidos), e noutras
isoformas que contribuem minoritariamente para o metabolismo deste
composto, vão apresentar concentrações plasmáticas variáveis do composto-
pai e de metabolitos 48, 422, o que pode condicionar a toxicidade da MDMA.
Estes polimorfismos ao nível das proteínas envolvidas na
farmacocinética e na farmacodinamia da MDMA podem explicar o facto de
alguns indivíduos poderem consumir MDMA regularmente sem dano aparente,
enquanto outros morrem ou têm reacções tóxicas severas com consumos
esporádicos ou de doses reduzidas 423.
Foram já descritos polimorfismos genéticos que afectam o perfil de
efeito/toxicidade da MDMA por alterar a expressão e/ou actividade de
isoenzimas do citocromo P450 (CYP1A2, CYP2B6, CYP2D6, CYP3A4) 398, da
COMT, da UDP-glucuronosiltransferase, da sulfotransferase, do receptor da
serotonina, do receptor da dopamina, do SERT, etc 89.
1.9.3.4.1. Isoenzimas do citocromo P450
As mutações nos genes que codificam para as enzimas do CYP450
podem traduzir-se na expressão de proteínas com actividade nula, reduzida,
aumentada ou alterada (por exemplo, perda de capacidade de ligação ao

______________________________________________________ Introdução
69
substrato 89), o que dá origem a quatro fenótipos distintos: (i) metabolizador
pobre, que não apresenta a enzima funcional; (ii) metabolizador intermédio que
apresenta uma redução da actividade enzimática; (iii) metabolizador extensivo,
que apresenta actividade enzimática normal; e (iv) metabolizador ultra-rápido
que apresenta níveis enzimáticos aumentados 424. A estes diferentes fenótipos
correspondem, geralmente, grandes variações na extensão da metabolização
de xenobióticos.
Mesmo os metabolizadores rápidos sofrem aumentos da concentração
de MDMA quando expostos a um inibidor da CYP2D6, uma vez que grandes
aumentos nas concentrações plasmáticas podem surgir devido à cinética não
linear do metabolismo da MDMA.
Nas populações caucasianas 12 a 21% dos indivíduos apresentam
fenótipo de metabolizadores pobres por possuírem o alelo CYP2D6*4. Este
alelo está praticamente ausente nas populações orientais, o que explica a
baixa incidência de metabolizadores pobres neste grupo étnico (menos de 1%) 66. No entanto, a actividade da CYP2D6 é menor nos metabolizadores
extensivos asiáticos do que nos metabolizadores extensivos caucasianos
devido à elevada frequência, entre os primeiros, do alelo CYP2D6*10 que
provoca deficiências na estrutura terciária e quaternária da proteína, diminuindo
a expressão da enzima funcional 425. A CYP2D6*10 é uma enzima com menor
afinidade e eficácia para os substratos do que a CYP2D6*1 (wildtype) 426. A
mesma capacidade metabólica diminuída foi identificada em indivíduos de raça
africana, associada à presença do alelo CYP2D6*17 que está relacionado com
uma diminuição da afinidade da enzima para os substratos 427, 428. O fenótipo
de metabolizador ultra-rápido, associado aos alelos CYP2D6*1XN,
CYP2D6*2XN e CYP2D6*35X2, surge num terço da população da Etiópia e
Arábia Saudita, em 1 a 5% dos caucasianos, 10% nas populações italiana e
turca e 5,5% da população da Europa Ocidental 424, sendo muito raro nas
populações do norte da Europa e estando praticamente ausente nos asiáticos 66.
Dada a importância da CYP2D6 no metabolismo da MDMA, foi colocada
a hipótese de que os indivíduos portadores de alelos codificantes para
isoenzimas com menor actividade poderiam apresentar um risco aumentado de
sofrer efeitos tóxicos agudos resultantes da acção farmacológica da MDMA

Introdução ______________________________________________________
70
(ex.: hipertermia, efeitos cardiovasculares) 267, 429. Em contraponto, os
indivíduos com fenótipo de metabolizador ultra-rápido estariam mais
predispostos a ser afectados por fenómenos de citotoxicidade 430 que podem
incluir neurotoxicidade, cardiotoxicidade, nefrotoxicidade e hepatotoxicidade 431
devido à previsível maior formação de metabolitos tóxicos 203, 386, 429, 432.
Assim, o estudo da influência do genótipo dos indivíduos e/ou
populações no metabolismo da MDMA permite estabelecer uma possível
relação entre o fenótipo de metabolizador pobre e a ocorrência de intoxicações
agudas devido a um decréscimo mais lento dos níveis plasmáticos de MDMA.
Também a isoforma CYP3A4 do CYP450 apresenta acentuada
variabilidade interindividual podendo o seu conteúdo hepático variar entre 40 a
50 vezes 433. Esta isoforma é a mais abundante do CYP450 humano
(representa 30% do CYP450 hepático e 70% do CYP450 intestinal) 433, 434 e é a
principal envolvida na oxidação dos xenobióticos 435.
1.9.3.4.2. Catecol-O-metiltransferase
Uma outra enzima que apresenta um grau de actividade sujeito a grande
variabilidade interindividual é a catecol-O-metiltransferase (COMT; enzima
envolvida na metilação das monoaminas neurotransmissoras, hormonas
catecóis, fármacos como a levodopa, metildopa e isoprenalina e drogas de
abuso como a MDMA 436) que é claramente evidente entre diferentes etnias.
Entre os caucasianos, 25% dos indivíduos apresentam actividade elevada,
50% apresentam actividade intermédia, e 25% apresentam actividade reduzida 436. A COMT eritrocitária apresenta uma actividade superior em indivíduos
africanos e afro-americanos (apenas 7% apresenta actividade reduzida)
relativamente aos caucasianos (27% apresenta fenótipo com actividade
reduzida) 437. Estes fenótipos de actividade enzimática reduzida parecem
apresentar risco superior para o desenvolvimento de reacções adversas às
anfetaminas 438.

______________________________________________________ Introdução
71
1.9.3.4.3. UDP-glucuronosiltransferase
A glucuronidação é a reacção de conjugação mais comum no
metabolismo humano e está envolvida na destoxificação dos metabolitos
catecóis e catecóis o-metilados da MDMA. Assim, variações farmacogenéticas
da enzima UDP-glucuronosiltransferase que condicionem a sua cinética
enzimática para determinados substratos podem afectar a toxicidade dos
mesmos se os seus metabolitos tóxicos forem destoxificados por esta via
metabólica e ela estiver comprometida 439.
Os polimorfismos mais importantes desta enzima são as variações no
gene UGT1A1 que resulta numa significativa redução da actividade da
UGT1A1. Esta variação ocorre em 7 a10% da população em geral 440.
1.9.3.4.4. Sulfotransferase (SULT)
A sulfotransferase (SULT) está também envolvida no metabolismo de
fase II dos compostos endógenos e xenobióticos, catalizando a conjugação do
composto com um grupo sulfato cedido pela adenosina 3’-fosfato 5’-
fosfossulfato (PAPS) 441. Esta enzima está envolvida na destoxificação dos
metabolitos catecóis e catecóis o-metilados da MDMA.
Também as variabilidades na actividade da SULT 441 podem estar
potencialmente associadas a alterações na toxicidade dos compostos
destoxificados por esta via.
1.9.3.4.5. Receptores da 5-HT
Foram já descritos fenómenos de polimorfismo genético associados aos
receptores 5-HT2A 442 que resultam em alterações funcionais da proteína. Uma
vez que as anfetaminas têm como alvo os receptores da 5-HT, alterações
funcionais a este nível podem condicionar a resposta biológica a este tipo de
drogas de abuso.

Introdução ______________________________________________________
72
1.9.3.4.6. Receptores da DA
Os receptores da dopamina apresentam também uma elevada
prevalência de polimorfismos que afectam a variabilidade da resposta
farmacológica a fármacos antipsicóticos e neurolépticos 443, 444, podendo
afectar também a sua afinidade para o seu substrato endógeno, a DA 445.
De facto, alguns estudos puderam já associar polimorfismos ao nível dos
receptores D2 e D4 com um risco acrescido de vulnerabilidade para o abuso e
risco de dependência de drogas psicoestimulantes 446, 447. Estes polimorfismos
podem ter um efeito aditivo aos polimorfismos da COMT no que se refere ao
risco de dependência destas drogas de abuso 447.
1.9.3.4.7. Transportador da 5-HT
O SERT (ou 5-HTT) é responsável pela manutenção das reservas
intraneuronais de serotonina mediante o transporte da mesma para os
terminais pré-sinápticos de forma a ficar disponível para subsequentes
libertações implicadas na neurotransmissão. Os polimorfismos verificados
neste transportador podem afectar o seu nível de expressão e, mais raramente,
podem alterar a própria estrutura do transportador 448. Indivíduos portadores do
polimorfismo que diminui a expressão dos SERT são mais susceptíveis aos
efeitos induzidos pela MDMA no sistema serotonérgico, havendo maior risco de
disfunção ao nível emocional 449 e cognitivo 450.
Em indivíduos portadores do genótipo 5-HTTLPR (confere
neurotransmissão serotonérica reduzida e parece aumentar a vulnerabilidade
para os efeitos da MDMA na função cognitiva), com consumo moderado de
MDMA (história de consumo de menos de 55 pastilhas ao longo da vida) não
surgem défices de memória. Para indivíduos com transportador deficiente que
consomem grandes quantidades de MDMA ou que já foram consumidores,
observa-se um desempenho inferior nas tarefas de memória relativamente ao
grupo controlo 451.

______________________________________________________ Introdução
73
1.9.3.5. Orientações sexuais
Aparentemente, a orientação sexual também condiciona o padrão de
consumo de drogas, uma vez que mulheres lésbicas/bissexuais referem níveis
de consumo de drogas (farmacêuticas ou de abuso) mais elevados do que
mulheres heterossexuais, nomeadamente de ecstasy, cocaína, MTA e LSD 320.
No entanto, não parece que a frequência do consumo difira entre os dois
grupos 452.
No caso dos homens, a frequência do consumo de MDMA é superior
nos homossexuais/bissexuais do que nos heterossexuais.
Assim, a identidade sexual foi associada com maiores taxas de consumo
de algumas drogas, embora, no geral, os padrões e a frequência do consumo
de drogas seja similar entre heterossexuais e homossexuais/bissexuais.
Contudo, a orientação sexual é um marcador para vários comportamentos de
risco que incluem utilização de drogas injectáveis, maior actividade sexual e
maior dependência de drogas anfetamínicas (ex.: MTA) 452.
Poderá ser, então, especulado que indivíduos com determinada
orientação sexual poderão estar mais predipostos a iniciar determinados
padrões de consumo de drogas e comportamentos de risco que poderão
potenciar o aparecimento de efeitos tóxicos associados às mesmas.
1.9.4. Alterações fisiológicas e fisiopatológicas
Os efeitos agudos e tóxicos da MDMA podem, também, ser
influenciados por alterações fisiológicas e fisiopatológicas que afectem o
consumidor e que podem condicionar a farmacodinamia e a farmacocinética
desta droga de abuso. São de destacar a gravidez, o hipo/hipertiroidismo, as
insuficiências hepática e renal, as patologias inflamatórias e infecciosas, as
patologias cardíacas, a diabetes mellitus e a obesidade.

Introdução ______________________________________________________
74
1.9.4.1. Gravidez
Devido ao facto da maioria dos consumidores de MDMA serem jovens
adultos, as possibilidades de surgir uma gravidez e expor os fetos aos efeitos
da MDMA é motivo de preocupação. Esta preocupação agrava-se se
considerarmos que o aparecimento de gravidezes indesejadas surge muito
associado ao policonsumo de drogas 334. Nestes casos o feto não estará
unicamente exposto à MDMA, mas sim aos efeitos deletérios de todas as
drogas consumidas, bem como às potenciais interacções que possam surgir
entre elas.
Existem evidências de que a exposição in utero dos fetos à MDMA
durante o desenvolvimento embrionário resulta numa redução dos níveis de
actividade e em deficiências de aprendizagem e memória, indicativos de dano
ao nível do hipocampo, embora não afecte o grau de ansiedade dos animais
quando estes atingem a idade adulta. A administração de MDMA às fêmeas de
rato grávidas diminui o peso das crias, e aumenta os seus níveis de
corticosterona e os níveis cerebrais do factor neurotrófico derivado do cérebro
e diminui os níveis de 5-HT. Este decréscimo de 5-HT dissipa-se inicialmente,
mas reaparece na idade adulta. Assim, parece evidente que a exposição à
MDMA tem efeitos adversos no desenvolvimento cerebral e no comportamento 453. O consumo de MDMA durante os primeiros meses de gravidez foi também
já associado ao aparecimento de situações de gastrosquise (anomalia
congénita da parede abdominal anterior através da qual ocorre herniação das
vísceras abdominais como estômago, intestino delgado, intestino grosso e
bexiga, que se desenvolvem fora da cavidade abdominal do feto) 454. Em
humanos existem ainda poucos estudos acerca do potencial teratogénico da
MDMA, no entanto, um estudo prospectivo de 136 recém-nascidos expostos à
MDMA in utero indicou que esta substância poderá estar associada com um
aumento significativo do risco de deficiências congénitas (15,4% dos nados-
vivos). Destas, as anomalias cardiovasculares (2,6% dos nados-vivos) e
musculoesqueléticas (3,8% dos nados-vivos) foram predominantes 260.
Felizmente, a prevalência do consumo de ecstasy durante a gravidez
parece ser reduzida 455. As mulheres grávidas que consomem MDMA são
normalmente jovens, solteiras, apresentam problemas psicológicos e reunem

______________________________________________________ Introdução
75
um conjunto de factores de risco que pode comprometer a gravidez e o feto
(tabagismo, consumo de álcool incluindo binge drinking, consumo de drogas
ilícitas como cocaína, Cannabis, MTA, ketamina, GHB e psilocibina) 262.
Durante a gravidez decorrem muitas alterações fisiológicas que podem
afectar a farmacocinética e a susceptibilidade da mãe aos efeitos das
substâncias consumidas durante esse período. O stress pré-natal pode
aumentar os níveis plasmáticos de MDMA, parecendo envolver uma inibição do
seu metabolismo, e aumentar as alterações motoras induzidas pelo consumo
desta substância. Estas diferenças ao nível do metabolismo da MDMA podem
afectar a propensão para sofrer reacções adversas. Uma consequência directa
seria o desenvolvimento de toxicidade com doses moderadas de MDMA, uma
vez que a droga acumular-se-ia no organismo em vez de ser metabolizada e
eliminada. Além do referido, o stress pré-natal tem a capacidade de induzir um
bloqueio prolongado da inibição reflexa (feedback) do eixo corticotrópico que
regula a secreção de corticosterona e que desempenha um papel importante
na neurotoxicidade da MDMA 456. Esta inibição associada ao facto de que, quer
o stress prolongado, quer a MDMA estimulam a libertação de corticosterona 140,
457, 458, vai aumentar os efeitos da MDMA nos animais em estado pré-natal 405,
podendo aumentar a sua neurotoxicidade.
A presença de depressões pré-natais na mulher pode também agravar
os efeitos da MDMA, uma vez que, por si só, a depressão pré-natal caracteriza-
se por aumento de cortisol e níveis baixos de dopamina e serotonina 459.
O consumo de MDMA pode ainda agravar os problemas de hipertensão
que, por vezes, surgem durante a gravidez e que são sempre extremamente
perigosos para a mãe e o feto 460, provavelmente por um mecanismo que
envolve um aumento da libertação de aldosterona induzido pela MDMA 461.
Em termos metabólicos, assiste-se durante este período a alterações da
absorção dos compostos, diminuição dos níveis de albumina e glicoproteína α1-
ácida que pode originar diminuição da ligação às proteínas plasmáticas,
aumento da actividade das isoenzimas CYP2D6 462, CYP3A4 e CYP2C9,
diminuição da actividade da CYP1A2, CYP2C19 e CYP2E1 463, aumento da
actividade da UDP-glucuronosiltransferase, aumento da clearance renal 464 e
alterações no metabolismo da vitamina D 465. O aumento da actividade da
CYP2D6 pode levar a tolerância metabólica a variados fármacos (ex.:

Introdução ______________________________________________________
76
fluoxetina) e consequente falência terapêutica 466 e pode aumentar a
metabolização da MDMA aos seus metabolitos tóxicos, aumentando a
probabilidade de fenómenos de toxicidade.
1.9.4.2. Patologias subjacentes ao consumo de MDMA
1.9.4.2.1. Hipo/hipertiroidismo
Como já foi referido, a MDMA induz um ligeiro aumento nos níveis
séricos de tiroxina em roedores e humanos 154. Esta hormona desempenha um
papel modulador na termogénese induzida pela MDMA através da regulação
da expressão da UCP3 e/ou a síntese de receptores adrenérgicos 154. Deste
modo, a existência de hipertiroidismo em roedores mimetizou os efeitos de uma
temperatura ambiente elevada na termogénese induzida pela MDMA 155. Ratos
com hipertiroidismo mostraram, em comparação com ratos eutiróidicos, um
aumento mais marcado na temperatura corporal, na expressão de UCP3 no
músculo esquelético e na letalidade consequente à exposição aguda à MDMA
(40 mg/kg s.c.), de um modo linear de acordo com os níveis plasmáticos de
tiroxina 155. O papel regulador das hormonas da tiróide na termogénese
induzida pela MDMA foi confirmado em macacos, uma vez que o tratamento
crónico de macacos com levotiroxina (3 ou 4,5 μg/kg/dia, i.m.) provocou um
aumento da resposta termogénica induzida pela MDMA quando administrada
em concentrações que habitualmente não afectavam a termorregulação. No
entanto, o tratamento com levotiroxina não teve efeito na capacidade de reforço
da MDMA (a qual já se verificou estar aumentada em ambientes quentes) 467.
Em contraponto, a remoção cirúrgica das glândulas tiróide e paratiróide
aboliu a capacidade da MDMA causar hipertermia e libertação de DA ao nível
do estriado 468. O tratamento com tiroxina repôs parcialmente a resposta
hipertérmica em animais tiróide-paratiroidectomizados 122.
Complementarmente, o hipotiroidismo crónico resulta numa resposta
hipotérmica à MDMA que é directamente proporcional à diminuição da
expressão da UCP3. Neste caso, o suplemento agudo com T4 não alterou a
expressão de UCP3 nem a resposta térmica à MDMA. Estes dados sugerem

______________________________________________________ Introdução
77
que, apesar da hipertermia induzida pela MDMA parecer resultar de níveis
aumentados de NA e AG livres, a susceptibilidade à mesma é ultimamente
determinada pela regulação da termogénese dependente da UCP3 pela T4 155.
Uma vez que a tiroxina é um importante regulador da termogénse 469, e
a intolerância ao calor pode ser associada com o hipertiroidismo em humanos 470, aparentemente, o hipertiroidismo pode predispor os indivíduos para a
hipertermia precipitada pela MDMA como parece ter acontecido numa
intoxicação fatal de um indivíduo do sexo feminino de 24 anos de idade e que
apresentava hiperplasia difusa da tiróide (Doença de Grave) 471.
1.9.4.2.2. Insuficiência hepática
A presença de patologias hepáticas crónicas prévias ao consumo de
MDMA que comprometam o funcionamento hepático pode predispor os
indivíduos para o desenvolvimento de falências hepáticas agudas. Este facto
foi já verificado num paciente HIV-positivo com esteatose não alcoólica que
sofreu falência hepática aguda após consumo de metanfetamina 472.
O fígado tem um papel preponderante no metabolismo de compostos
endógenos e exógenos. Consequentemente, a presença de determinada
patologia hepática pode diminuir selectivamente a actividade de algumas
isoformas do CYP450 (como é o caso da CYP2D6), numa extensão
dependente da severidade da patologia em causa 473. Uma vez que nos
doentes hepáticos a actividade desta enzima poderá estar diminuída, eles
serão potencialmente mais susceptíveis a desenvolver sinais de toxicidade
aguda associada à exacerbação dos efeitos agudos provocados pela MDMA
(ex.: hipertermia, taquicardia) do que indivíduos não portadores de patologias
hepáticas.
1.9.4.2.3. Insuficiência renal
A insuficiência renal crónica (IRC) corresponde a uma perda progressiva
da função renal durante um período de meses ou anos e por uma sucessão de

Introdução ______________________________________________________
78
5 estadios durante os quais ocorre um decréscimo da taxa de filtração
glomerular e da secreção tubular 474.
As principais causas de IRC são o envelhecimento, a nefropatia
diabética, a hipertensão, a glomerulonefrite e a doença vascular renal 474.
O rim tem capacidade para metabolizar determinados compostos,
apresentando uma actividade do CYP450 que corresponde a 20% da
actividade do CYP450 no fígado por grama de tecido e participando, também,
em reacções de conjugação 475. Esta capacidade metabólica do rim deteriora-
se com a progressão da IRC, levando também a uma redução da clearance
não renal (hepática, intestinal) e a uma alteração do volume de distribuição dos
compostos mediante alterações da clearance hepática, diminuição da ligação
às proteínas plasmáticas por alterações conformacionais da albumina e da
glicoproteína ácida α1 (aumentada em processos inflamatórios agudos e
crónicos) e diminuição da ligação aos tecidos, modificando a biodisponibilidade
sistémica e aumentando o risco de reacções adversas 475.
Num modelo de IRC em rato, verificou-se o aumento da actividade da
álcool desidrogenase (ADH) e uma diminuição da actividade, dos níveis de
mRNA e de expressão proteica do CYP450 a nível hepático 474, afectando
algumas isoformas envolvidas no metabolismo da MDMA como a CYP3A1 e
CYP3A2 (que correspondem à CYP3A4 no homem), mas não a CYP1A2 nem
as CYP2D 474. Esta diminuição da actividade do CYP450 correlaciona-se com a
severidade da IRC em ratos 474. Noutro estudo em ratos, a IRC não teve um
efeito nas enzimas CYP1A2 e CYP2D hepáticas, mas reduziu a actividade da
CYP3A, provavelmente por down-regulation da CYP3A2 476. No entanto há
também casos em que se detectou uma diminuição na actividade da CYP2D6
em doentes com IRC 477. Quer a severidade quer a duração da insuficiência
renal parecem influenciar o metabolismo hepático 474, 478, 479.
A IRC altera ainda a eliminação hepática e intestinal através da
glicoproteína-P (embora este transportador não tenha um papel preponderante
na absorção e distribuição da MDMA) 480.
A IRC parece também diminuir o metabolismo intestinal dos compostos,
em ratos e em humanos, reduzindo as actividades de algumas enzimas
(sacarase, maltase e xantina oxidase), bem como os níveis de mRNA e de
expressão proteica de isoenzimas do CYP450 a nível intestinal no rato, como a

______________________________________________________ Introdução
79
CYP3A2, que participa no metabolismo da MDMA e corresponde à CYP3A4 no
humano 474.
Assim, a diminuição na clearance não-renal induzida pela IRC reduzirá o
metabolismo de primeira-passagem, levando a um acentuado aumento da
biodisponibilidade de fármacos com elevado efeito de primeira passagem. O
aumento da biodisponibilidade resultará num aumento da concentração
plasmática steady-state clinicamente relevante, agravando o risco de sofrer
efeitos tóxicos de fármacos/drogas/xenobióticos metabolizados extensamente
por enzimas hepáticas. Este aumento da biodisponibilidade poderá ter ainda
maior relevância quando os compostos são metabolizados por enzimas
polimórficas, como é o caso da MDMA, uma vez que o fenótipo de
metabolizador pobre parece ser mais susceptível aos efeitos inibitórios da IRC
no metabolismo do que o metabolizador rápido 475.
1.9.4.2.4. Patologias inflamatórias e infecciosas
Algumas patologias inflamatórias e infecciosas (incluindo vacinação)
podem alterar a actividade do CYP450 481, nomeadamente da isoforma
CYP1A2 398, o que pode levar a alteração do perfil toxicocinético de compostos
metabolizados por esta isoenzima, como é o caso da MDMA.
1.9.4.2.5. Patologia cardíaca
Indivíduos que padeçam da síndrome Wolff-Parkinson-White (síndrome
de pré-excitação dos ventrículos devido a uma comunicação eléctrica anormal
proveniente da aurícula para o ventrículo) parecem estar particularmente em
risco de sofrer morte súbita após o consumo de ecstasy 301. De facto,
desfechos fatais após o consumo de MDMA parecem ser mais frequentes em
indivíduos com patologias cardíacas subjacentes 178.

Introdução ______________________________________________________
80
1.9.4.2.6. Diabetes mellitus
A diabetes, bem como a obesidade e o jejum, podem originar alterações
na secreção, sensibilidade e níveis hormonais (insulina, glucagina, hormona do
crescimento), podendo alterar a expressão de enzimas metabolizadoras 482. De
facto, na diabetes mellitus tipo I foram descritos aumentos pronunciados e
sustentados nos níveis hepáticos e extra-hepáticos de várias proteínas do
CYP450, num mecanismo aparentemente mediado pelas alterações hormonais
e pela hipercetonémia, e não pela hiperglicémia, que acompanham esta
patologia 483.
Concretamente, a diabetes pode levar à expressão aumentada de
isoenzimas do CYP450, incluindo aquelas envolvidas no metabolismo da
MDMA como a 1A2, 2B, 3A, e a 2E1, envolvida na metabolização do etanol 484
e da bilirrubina UDP-glucuronosiltransferase (UGT1A1), enquanto decresce a
expressão de outras enzimas microssomais e das sulfotransferases (SULTs),
importantes para as reacções metabólicas de fase II de que a MDMA é alvo.
Em contraste, a actividade da glutationa S-transferase (GST) in vivo,
importante para o balanço dos níveis celulares de glutationa, a qual já provou
ter um papel importante na expressão da toxicidade da MDMA, pode aumentar
ou diminuir dependendo do balanço entre factores endógenos e o grau de
stress oxidativo que pode surgir na diabetes. A síntese da glutationa (GSH)
também pode estar alterada em resposta a condições patofisiológicas como a
diabetes devido à regulação da expressão da γ-glutamilcisteína sintetase (γ-
GCS), a enzima que cataliza o primeiro passo da síntese da GSH, e que
depende da disponibilidade de cisteína para a sua síntese. No entanto, a
administração de insulina e de hormona do crescimento a ratos diabéticos
normaliza a actividade metabólica hepática 482.
Devido a esta disfunção no sistema de defesa antioxidante que se
verifica na diabetes, esta patologia pode ser um factor predisponente para um
aumento do stress oxidativo e da toxicidade induzida pela MDMA.

______________________________________________________ Introdução
81
1.9.4.2.7. Obesidade
Foi já demonstrado que a obesidade altera a disponibilidade de
determinadas substâncias em humanos, provavelmente devido a alterações na
clearance hepática de compostos que sofram metabolismo de fase I e de fase
II 485 como é o caso da MDMA. Para além disso, muitos modelos nutricionais
de roedores obesos mostraram alterações no CYP450 hepático e nos
mecanismos de conjugação, modificando a toxicidade de várias substâncias
para os seus órgãos-alvo 485, 486. A actividade e a expressão proteica da
CYP2E1, envolvida no metabolismo do etanol, estão aumentadas em fêmeas
de ratinho obesas e tinha já sido relatada em humanos 486, 487. A actividade e
expressão proteica da CYP1A2, envolvida no metabolismo da MDMA, estão
diminuídas em fêmeas e machos obesos 488.
A acrescer a estas alterações na expressão genética e actividade das
isoformas do CYP450 está o facto de que o fígado dos indivíduos obesos
apresenta frequentemente infiltrações lipídicas num quadro denominado
esteatohepatite não alcoólica. Estas lesões podem influenciar
significativamente a capacidade metabólica do fígado 489.
Assim, dependendo do grau de obesidade do indivíduo, serão de
esperar alterações ao nível do metabolismo da MDMA resultando,
provavelmente, numa exacerbação dos seus efeitos agudos.
1.9.5. Interacções da MDMA com outros compostos
Sabe-se pouco acerca das drogas rave e do seu potencial para interagir
com substâncias recreacionais ou prescritas. Infelizmente, existem ainda
poucos estudos sobre este assunto e, por isso, temos que nos basear em
casos clínicos, estudos in vitro do metabolismo destas drogas, e no
conhecimento do metabolismo de outras substâncias e do seu potencial para
induzir ou inibir o CYP450 para podermos inferir sobre possíveis interacções
farmacodinâmicas e farmacocinéticas da MDMA.
Podem surgir interacções farmacocinéticas quando um determinado
composto interfira com a absorção, distribuição, metabolismo e/ou excreção da

Introdução ______________________________________________________
82
MDMA. Por exemplo, quando o composto que é consumido simultaneamente
com a MDMA tem a capacidade de inibir a CYP2D6, pode ocorrer um aumento
das concentrações plasmáticas da MDMA, aumentando a possibilidade de se
verificar o aparecimento de sinais de toxicidade aguda associados ao seu
consumo 67. Contudo, compostos que inibam outras isoenzimas do CYP450
(ex.: 1A2, 2B6, 3A4), para além da CYP2D6, têm mais probablidade de causar
um efeito mais marcado nas concentrações de MDMA. É de especial
importância a co-administração de MDMA com fármacos de estreita margem
terapêutica que sejam substratos da CYP2D6 (ex.: alguns antidepressivos
tricíclicos, antiarrítmicos, β-bloqueadores, antipsicóticos e tramadol), uma vez
que a auto-inibição metabólica produzida pela MDMA nesta isoenzima pode
aumentar desproporcionalmente os níveis do fármaco co-administrado, levando
ao aparecimento de fenómenos de toxicidade. Outra via responsável por várias
interacções farmacocinéticas corresponde à interferência com a glicoproteína P
(Pgp), uma bomba de efluxo dependente do ATP capaz de exportar uma
grande gama de substratos e presente numa grande variedade de células (ex.:
enterócitos, hepatócitos, células dos túbulos proximais renais e células
endoteliais dos capilares presentes também na barreira hemato-encefálica). No
entanto, a MDMA e compostos análogos são apenas inibidores fracos da Pgp,
pelo que, provavelmente, não irão influenciar o acesso de outros compostos ao
cérebro. No entanto, não se pode excluir a possibilidade de que a co-
administração destes compostos com inibidores da Pgp (ex.: ritonavir ou
paroxetina) possa aumentar a biodisponibilidade da MDMA e análogos (ex.:
MDE, PMA) por diminuir a sua eliminação a nível renal e também aumentar os
seus níveis no cérebro (por diminuição da eliminação através da barreira
hemato-encefálica) levando a efeitos laterais severos 490.
As interacções farmacodinâmicas surgem quando a substância co-
consumida apresenta um mecanismo de acção semelhante ou oposto ao da
MDMA. Quando o composto que é consumido simultaneamente à MDMA
também for pró-serotonérgico, o seu efeito pode combinar-se, levando a uma
síndrome serotonérgica a nível central que inclui sintomas autónomos,
cognitivos, e neuromusculares 67. Os sintomas moderados da síndrome
serotonérgica manifestam-se com sudação, tremores, hiper-reflexia, e agitação;
os sintomas severos incluem myoclonus (contracções involuntárias de um

______________________________________________________ Introdução
83
músculo ou de um grupo de músculos), diarreia, e febre 491. Os casos mais
graves de síndrome serotonérgica podem surgir com fármacos que bloqueiem
ambas as vias que levam à diminuição dos níveis de 5-HT na fenda sináptica –
a recaptação e a monoamina oxidase.
Alguns compostos podem combinar estas duas características e ser
simultaneamente pró-serotonérgicos e inibidores da CYP2D6, tendo assim a
potencialidade de provocar interacções farmacocinéticas e farmacodinâmicas,
com consequências imprevisíveis 67. Estes compostos podem ser ingeridos
inadvertidamente na dieta, incluídos em regimes terapêuticos ou como
contaminantes das pastilhas de MDMA (ex.: anfetaminas, MDA) 492 ou
consumidos intencionalmente para aumentar os efeitos da MDMA (ex.:
cocaína, dextrometorfano) ou para diminuir os seus efeitos indesejáveis (ex.:
etanol, benzodiazepinas 316, sildenafil 493, anti-histamínicos 266, 5-
hidroxitriptofano 316). Também os fármacos pró-serotonérgicos prescritos para
tratamentos de determinadas patologias (ex.: fluoxetina, erva de S. João,
tramadol, venlafaxina, lítio, clomipramina, entre outros) podem aumentar a
propensão e a severidade dos sintomas serotonérgicos resultantes do
consumo de MDMA.
1.9.5.1. Fármacos
Evidências recentes sugerem que está a tornar-se popular entre os
utilizadores de ecstasy tentar contrariar alguns dos efeitos indesejáveis da
MDMA através do uso simultâneo de fármacos ou suplementos alimentares.
Normalmente o consumo de medicamentos é feito após o consumo da MDMA
e é mais prevalente entre os indivíduos do sexo masculino 316, havendo, no
entanto, excepções. Este fenómeno é motivo de procupação, uma vez que
algumas destas combinações ecstasy+fármacos podem ter consequências
potencialmente sérias para a saúde dos consumidores 316 por potenciarem os
efeitos agudos e os efeitos tóxicos da MDMA.
Dos medicamentos consumidos intencionalmente com MDMA, as
benzodiazepinas e o sildenafil são os mais comuns, seguidos dos
antidepressivos inibidores da monoamina oxidase (IMAO) e inibidores

Introdução ______________________________________________________
84
selectivos da recaptação da 5-HT (ISRS). O 5-hidroxitriptofano (5-HTP) é o
suplemento nutricional mais consumido (5% dos utilizadores de MDMA
consomem este suplemento).316.
No entanto, não apenas os fármacos consumidos intencionalmente para
modular os efeitos da MDMA, mas também todos os outros que os indivíduos
consumam como parte de qualquer tratamento farmacológico, podem
influenciar a toxicidade desta droga de abuso estabelecendo, com ela,
interacções farmacodinâmicas e farmacocinéticas 398.
Existem vários fármacos, pertencentes a várias classes terapêuticas que
são capazes de inibir as isoenzimas envolvidas no metabolismo da MDMA 398.
Por exemplo, o antipsicótico haloperidol e os seus metabolitos foram já
descritos como sendo potentes inibidores da CYP2D6 494. O pré-tratamento
com haloperidol (1,4 mg, i.v.), ao contrário do que se verificou com o
antidepressivo citalopram, não foi capaz de impedir o aumento da pressão
arterial e da frequência cardíaca induzido pela MDMA (1,5 mg/kg p.o.) no
homem, o que parece indicar um envolvimento da transmissão serotonérgica
na ocorrência destes efeitos tóxicos agudos induzidos pela MDMA. No entanto,
esta dose de haloperidol foi capaz de reduzir o humor positivo e a euforia
induzidos pela MDMA 77. Segundo alguns autores, os barbitúricos têm a
capacidade de reduzir ou prevenir a neurotoxicidade induzida pelos derivados
metilenodioxi da anfetamina 249, 250 tal como parece acontecer com o ácido
acetilsalicílico 249, 250 encontrados na composição de pastilhas vendidas como
ecstasy 372.
Existem, também, fármacos capazes de interagir com os mesmos
receptores, transportadores e neurotransmissores que a MDMA. Por exemplo,
os antipsicóticos atípicos clozapina e olanzapina, quando administrados
isoladamente, reduzem a temperatura corporal em humanos e animais 495, 496 e
têm a capacidade de reverter, em coelhos e ratos, a hipertermia
potencialmente fatal e a vasoconstricção cutânea quando administrados 15
minutos após uma dose de 6 mg/kg i.v. de MDMA 497. Por outro lado, a
clozapina demonstrou ainda poder reduzir, em ratos, o aumento da activação
da termogénese ao nível do tecido adiposo castanho interescapular (iBAT)
induzido pela MDMA. Este facto advoga em favor da potencial eficácia
terapêutica da clozapina e compostos análogos no tratamento da hipertermia

______________________________________________________ Introdução
85
induzida pela MDMA em humanos 498. Alguns relaxantes musculares como a
succinilcolina e alguns anestésicos (ex.: anestésicos voláteis como o halotano)
levam ao desenvolvimento de hipertermia maligna em pacientes predispostos,
o que pode potenciar a resposta hipertérmica associada à MDMA, como já foi
descrito para o halotano, uma vez que a MDMA aumentou as concentrações
mioplasmáticas de cálcio e as contracções do músculo esquelético induzidas
pelo halotano, mecanismos relacionados com a indução da hipertermia maligna 15. A pré-exposição a esteróides androgénicos anabolizantes (ex.: nandrolona)
pode modular os efeitos neuroquímicos de compensação e comportamentais
da MDMA de maneira dependente da dose 499. De facto, a nandrolona atenua o
aumento de DA a 5-HT extracelular induzido pela MDMA, quando administrada
em doses baixas a ratos, e passa a potenciar este aumento quando
administrada em doses elevadas, podendo condicionar o potencial aditivo da
MDMA 499. Por outro lado, a exposição crónica de ratinhos fêmea C57BL/6J a
níveis supra-fisiológicos de corticosterona provocou um aumento da
vulnerabilidade do núcleo estriado (onde se verificou um aumento da
destruição das células da glia), mas não do hipocampo, à neurotoxicidade
provocada pela MDMA 500. O pré-tratamento com o anti-hipertensor ketanserina
(50 mg, p.o.), reduziu as alterações perceptivas, excitabilidade emocional e
estado de vigília provocados pelo consumo de MDMA (1,5 mg/kg p.o.) em
humanos 501. Tal como já foi referido, o 5-HTP é o suplemento nutricional mais
consumido intencionalmente 316. A administração de 5-HTP-B (50 mg/kg i.p.) a
ratos repõe os níveis cerebrais de 5-HT depletados pela MDMA (3 × 7,5 mg/kg,
espaçadas 2 horas), sugerindo que esta abordagem terapêutica pode ser
clinicamente útil em utilizadores de MDMA abstinentes 502.
Os fármacos que devido às suas propriedades farmacocinéticas e/ou
farmacodinâmicas apresentem potencial para interagir com a MDMA estão
compilados na Tabela 4.
De todas estas possibilidades de interacções apresentadas, aquelas que
apresentam maior prevalência surgem em casos de associação de MDMA com
antidepressivos e com anti-retrovirais. Por esse motivo, as interacções
verificadas nestas associações serão abordadas, de seguida, mais
detalhadamente.

Tabela 4. Fármacos com potencial para estabelecer interacções farmacocinéticas e/ou farmacodinâmicas com a MDMA.
Potenciais mecanismos para o estabelecimento de interacções com a MDMA
Grupo terapêutico / Fármaco Nome comercial Farmacocinética Farmacodinamia
Antidepressivos
Tricíclicos
Nortriptilina Norterol® substrato da CYP2D6 503
Amitriptilina Adt-Zimaia®, Tryptizol® potente inibidor reversível da CYP3A4 podendo
também inibir a CYP2D6 503
Inibição não selectiva da recaptação da NA e da
5-HT
Interferência com receptores para vários outros
neurotransmissores
Inibidores selectivos da recaptação da serotonina (ISRS)
Fluoxetina Prozac®, Digassim®, Psipax®,
Selectus®
Inibidor potente da CYP2D6 podendo também
inibir, em alguma extensão, as isoformas 2B6,
2C9, 2C19 e 3ª4 do CYP450 67
Inibição da recaptação pré-sináptica da 5-HT
Paroxetina Seroxat®, Paxetil®, Denerval® Inibidor potente da CYP2D6 Inibição potente da recaptação pré-sináptica da 5-
HT
Citalopram Citalopram Genedec®
Sertralina Zoloft®
Zimelidina
Inibidores da monoamina oxidase Inibição da degradação dos neurotransmissores
na fenda sináptica
Fenelzina Nardil® (Austrália) Inibidor irreversível e não específico da MAO
Moclobemida Zorix®, Aurorix® Inibidor reversível da MAO, com maior
selectividade para a MAO-A
86

87
Tabela 4 (cont). Fármacos com potencial para estabelecer interacções farmacocinéticas e/ou farmacodinâmicas com a MDMA. Potenciais mecanismos para o estabelecimento de interacções com a MDMA
Grupo terapêutico / Fármaco Nome comercial Farmacocinética Farmacodinamia
Antidepressivos (cont.)
Atípicos
Bupropiom Zyban® Potente inibidor da CYP2D6 Inibidor da recaptação de NA e DA
Antagonista nicotínico
Hypericum perforatum Erva de S. João (contém
hiperforina, hipericina,
flavonóides, taninos, etc) 504, 505
Indução da CYP3A4 para doses baixas (devido à
hiperforina)
Inibição da CYP3A4 506) e da CYP2C9 para doses
altas
Alguma inibição da CYP2D6 506
Inibidor da recaptação da 5-HT, DA e NA 507
Fraco inibidor das MAO A e B
Afinidade para os receptores da adenosina,
GABAA, GABAB e glutamato down-regulation dos
β-adrenoreceptores
up-regulation dos receptores 5-HT2 no �urícu pré-
frontal de rato
Alteração nas concentrações de
neurotransmissores nas áreas cerebrais
implicadas na fisiopatologia da depressão 504, 505
Antipsicóticos
Haloperidol Haldol® Potente inibidor da CYP2D6 Antagonista dos receptores dopaminérgicos D2
Pimozida Orap® Potente inibidor da CYP2D6
Clozapina Leponex®
Olanzapina Zyprexa®
Elevada afinidade para determinados subtipos
dos receptores 5-HT; antagonistas 5-HT2A e 5-
HT2C 508

Tabela 4 (cont). Fármacos com potencial para estabelecer interacções farmacocinéticas e/ou farmacodinâmicas com a MDMA. Potenciais mecanismos para o estabelecimento de interacções com a MDMA
Grupo terapêutico / Fármaco Nome comercial Farmacocinética Farmacodinamia
Ansiolíticos/hipnóticos/sedativos
Fenobarbital Luminaletas®, Luminal®,
Bialminal®
Indução da CYP2B6, CYP2C9, CYP2C19 e
CYP3A4 398
Diazepam Metamidol®, Unisedil®,
Bialzepam®, Valium®, Stesolid®
Inibidor da fosfodiesterase nucleotídica cíclica do
músculo cardíaco
Potencia os efeitos inotrópicos positivos da NA e
adrenalina
Midazolam Dormicum®
Triazolam Halcion®
Alprazolam Xanax®
Substratos da CYP3A4 509
Zolpidem Stilnox®, Cymerion® Inibidor da CYP3A4 503 Ligação de grande afinidade aos receptores
GABAA com subunidades α1, aumentando a
afinidade do neurotransmissor inibitório GABA
para o seu receptor
Analgésicos opiáceos
Metadona Dolophine®, Amidone®,
Methadose®, Physeptone®,
Heptadon®
Substrato da CYP3A4 e também CYP2D6,
CYP2C9, CYP2E1 509, CYP2B6, CYP2C19 510
Potente inibidor da CYP2D6
Agonista dos receptores opióides tipo μ e
antagonista dos receptores NMDA do glutamato,
o que permite inibir o desenvolvimento e
aquisição de dependência e tolerância aos
opióides 511
88

89
Tabela 4 (cont). Fármacos com potencial para estabelecer interacções farmacocinéticas e/ou farmacodinâmicas com a MDMA. Potenciais mecanismos para o estabelecimento de interacções com a MDMA
Grupo terapêutico / Fármaco Nome comercial Farmacocinética Farmacodinamia
Analgésicos opiáceos (cont.) Codeína/Diidrocodeína Euphon®, Codipront®, Codol®,
Dafalgan Codeína®, Dol-U-
Ron®, Migraleve®, Dolviran®
Substrato da CYP2D6, CYP3A4 e UGT 512
Hidrocodona Vicodin® (EUA) Substrato da CYP2D6, CYP3A4 e UGT 512
Buprenorfina Subutex®, Buprex®, Transtec® Substrato da CYP2D6, CYP3A4 e UGT 512
Potente inibidor reversível da CYP3A4 503
Anestésicos
Succinilcolina Anectine®, Quelicin® (Canada)
Anestésicos voláteis
(ex.: halotano) Fluotane®
Desenvolvimento de hipertermia maligna em
indivíduos predispostos
Antiarrítmicos
Amiodarona Cordarone®, Miodrone® Potente inibidor das CYP 1ª2, 2C9, 2D6 e 3ª4 513 β-bloqueador e bloqueador dos canais de
potássio dos nodos sino-auricular (ou sinusal) e
aurículo-ventricular, aumentando o período
refractário via canais de sódio e potássio, e
diminuindo a condução intra-cardíaca do potencial
de acção cardíaco via canais de sódio. O
resultado é um prolongamento da fase III do
potencial de acção cardíaco.

Tabela 4 (cont). Fármacos com potencial para estabelecer interacções farmacocinéticas e/ou farmacodinâmicas com a MDMA. Potenciais mecanismos para o estabelecimento de interacções com a MDMA
Grupo terapêutico / Fármaco Nome comercial Farmacocinética Farmacodinamia
Antiarrítmicos (cont.) Quinidina Dicorantil® Potente inibidor da CYP2D6 Interfere no funcionamento dos canais de sódio,
cálcio e potássio cardíacos, prolongando o
potencial de acção cardíaco e consequentemente,
aumentando do intervalo QT do ECG
Anti-retrovirais
Inibidores das proteases
Ritonavir Norvir® Inibidor da CYP2D6, 2B6, 2C9, 2C19, 3ª4
Nelfinavir Viracept® Substrato da CYP3A4 e 2C19
Inibidores da transcriptase reversa não-nucleosídicos
Delaviridina Rescriptor® Parcialmente metabolizada pela CYP2D6 514
Efavirenz Sustiva®, Stocrin®
Inibidor ou indutor da CYP3A4 e inibidor da
CYP2B6 515
Inibidor da transcriptase reversa
Antibióticos
Rifampicina Rifadin®
Indutor da CYP1A2 516
Controla a expressão da CYP2B6, CYP2C9,
CYP2C19 e CYP3A4 398
Linezolida Zyvox®, Zyvoxid® Inibidor fraco, reversível, da MAO 517
Clotrimazol Canesten®
Itraconazol Sporanox®
Inibidores das isoenzimas do CYP450, sobretudo
a CYP2B6 518
90

91
Tabela 4 (cont). Fármacos com potencial para estabelecer interacções farmacocinéticas e/ou farmacodinâmicas com a MDMA. Potenciais mecanismos para o estabelecimento de interacções com a MDMA
Grupo terapêutico / Fármaco Nome comercial Farmacocinética Farmacodinamia
Inibidores da fosfodiesterase 5
Sildenafil Viagra® Substrato da CYP3A4 519
Anti-histamínicos de 1ª geração
Difenidramina Drenoflux®, Benaderma®
Clorfeniramina Chlor-trimeton® (EUA)
Clemastina Tavégyl®
Perfenazina Mutabon®
Hidroxizina Atarax®
Tripelenamina Benzoxale®, Cizaron® (EUA)
Inibidores da CYP2D6 520 e da CYP3A4 521
Antitússicos
Dextrometorfano Rhinathiol®, Vicks®, Drill®,
Ipésandrine®
Substrato da CYP2D6 Bloqueador do receptor N-metil-D-aspartato 372, 522
Agonista dos receptores σ1 e σ2 523
Antagonista dos receptores nicotínicos α3β4 524
Inibidor da recaptação da 5-HT 525 e,
possivelmente, da DA
Anti-hipertensores
Ketanserina Sufrexal® (Itália) antagonista dos receptors da serotonina com
maior afinidade para os receptores 5-HT2A,
podendo também ligar-se aos receptores 5-HT2C,
5-HT2B, 5-HT1D, α-adrenérgicos, e dopaminérgicos

Tabela 4 (cont). Fármacos com potencial para estabelecer interacções farmacocinéticas e/ou farmacodinâmicas com a MDMA. Potenciais mecanismos para o estabelecimento de interacções com a MDMA
Grupo terapêutico / Fármaco Nome comercial Farmacocinética Farmacodinamia
Suplementos nutricionais
5-HTP Repõe os níveis cerebrais de 5-HT
Valeriana comum Valdispert® Indutor da CYP3A4 e da CYP2D6 de maneira
linear com a dose 506
Ginkgo biloba Biloban®, Gincoben® Indutor da CYP1A2 e inibidor da CYP2D6, para
baixas concentrações
Inibidor da CYP1A2 e indutor da CYP2D6, para
altas concentrações, provavelmente por um
mecanismo de activação alostérica 506
Echinacea purpurea
Salva comum
Inibidores do CYP450
Outros
Fenitoína Hidantina® (antiepiléptico) Indutores da CYP1A2 516
Omeprazol
Proton®, Losec®, Omezolan®,
Prazolene®, Proclor®
(antiulceroso inibidor da bomba de
protões)
Contraceptivos orais Inibidores da CYP1A2 398
Clopidogrel e
ticlopidina
(antiagregantes plaquetários)
Raloxifeno (MSRE)
Inibidores da CYP2B6 518.
92

______________________________________________________ Introdução
93
1.9.5.1.1. Antidepressivos
Os antidepressivos são usados pelos consumidores de ecstasy para
prevenir potenciais efeitos neurotóxicos da MDMA, para evitar os efeitos
negativos associados à abstinência desta anfetamina, para aumentar a
intensidade dos seus efeitos e para prolongar a duração do efeito ou para
facilitar o sono 316.
Vários estudos referem que uma percentagem considerável dos
consumidores de ecstasy também consome antidepressivos. Copeland e
colaboradores referiram, em 2006, taxas de 9% 316, Degenhardt, em 2005, fala
em 5,8-22,4% 452 e Winstock e colaboradores reportam, em 2001, que 6% dos
consumidores de ecstasy tomam antidepressivos para reduzir os efeitos
desagradáveis que surgem quando os efeitos pretendidos da ecstasy
desaparecem 266.
Os antidepressivos mais usados em conjunto com a ecstasy são os
inibidores selectivos da recaptação da serotonina (ISRS) e os inibidores da
monoamina oxidase (IMAO).
1.9.5.1.1.1. Inibidores selectivos da recaptação da serotonina (ISRS)
Alguns utilizadores de MDMA fazem uma “pré-carga” com inibidores da
recaptação de 5-HT (ex.: citalopram, sertralina) porque acreditam que a co-
administração irá protegê-los de possíveis efeitos tóxicos a nível do SNC. No
entanto, apesar de já ter sido demonstrado que a administração de MDMA (1,5
mg/kg por via oral) a indivíduos pré-expostos a inibidores da recaptação da 5-
HT (citalopram, 40 mg i.v.) bloqueia os efeitos subjectivos, os efeitos
vegetativos agudos e o aumento da pressão sanguínea e do ritmo cardíaco
induzidos pela MDMA, esta associação não afecta a resposta hipertérmica 526 e
aumenta a duração média dos efeitos 527.

Introdução ______________________________________________________
94
Também o pré-tratamento de humanos com fluoxetina (20 mg durante 5
dias por via oral) atenuou a maioria dos efeitos subjectivos positivos da MDMA
(1,5 mg/kg por via oral), reduziu o aumento do ritmo cardíaco provocado pela
MDMA, mas não impediu o aumento da pressão sanguínea 528. Estes
resultados sugerem que o bloqueio da recaptação da 5-HT pela fluoxetina pode
reduzir os efeitos da MDMA, confirmando a importância do SERT nos efeitos
desta droga de abuso 528. Também o citalopram tem aparentemente a
capacidade de reduzir ou prevenir a neurotoxicidade induzida pelos derivados
metilenodioxi da anfetamina 249, 250. Já em 1998 foi descrita uma situação de
uma presumível interacção entre MDMA e exposição prévia a citalopram que
resultou em alterações comportamentais 529. Podem surgir interacções
farmacocinéticas e farmacodinâmicas simultaneamente quando o ISRS
consumido for também inibidor potente da CYP2D6 (ex.: fluoxetina,
paroxetina), aumentando quer as concentrações plasmáticas de MDMA
(farmacocinética), quer a actividade serotonérgica (farmacodinâmica). A
fluoxetina e paroxetina são, comprovadamente, inibidores do metabolismo da
MDMA in vitro 422. Concretamente, a fluoxetina é um inibidor potente da
CYP2D6 e, por esse motivo, tem a capacidade de reduzir ou prevenir a
neurotoxicidade induzida pelos metabolitos dos derivados metilenodioxi da
anfetamina 530. No entanto, este composto atrasa a eliminação da MDMA e da
MDA, aumentando o risco de desenvolvimento de sinais de toxicidade aguda,
daí que deve ter-se cuidado ao recomendar a utilização de fluoxetina para
prevenir a neurotoxicidade a longo prazo induzida pela MDMA 480. O tratamento
prolongado com fluoxetina (6 mg/kg/dia na água de bebida durante 5 semanas)
9-12 semanas após uma exposição breve de ratos Wistar à MDMA (4 doses de
5 mg/kg administradas num período de 4 horas) reverteu a ansiedade e a
depressão subsequentes ao consumo desta droga de abuso; no entanto, não
teve qualquer efeito no decréscimo da capacidade de interacção social que
surge após abstinência deste entactogénio 531. O mesmo tratamento com
fluoxetina não afectou grandemente os níveis de 5-HT, mas diminuiu
significativamente os níveis de ácido 5-hidroxindolacético (5-HIAA, principal
metabolito da 5-HT) 531. Estes resultados indicam que a fluoxetina pode ser
uma opção terapêutica válida para alguns dos efeitos deletérios a longo prazo
causados pela exposição à MDMA 531 que deverá ser confirmada em humanos.

______________________________________________________ Introdução
95
No entanto, a fluoxetina (10 mg/kg, i.p.) administrada 30 min antes de cada
dose de MDMA não é capaz de prevenir os défices dopaminérgicos agudos e
de longo termo provocados pela MDMA (30 mg/kg × 3, i.p.) em ratinhos, mas
consegue abolir as alterações ao nível da temperatura corporal 96.
A paroxetina é um dos mais potentes inibidores da recaptação da 5-HT.
Esta substância tem também uma grande capacidade de inibir a CYP2D6. O
pré-tratamento com este composto (20 mg/dia durante 3 dias) provoca o
decréscimo dos efeitos subjectivos e fisiológicos da MDMA (100 mg, p.o.) em
humanos, apesar de aumentar os níveis plasmáticos de MDMA em 30% (com
redução dos níveis de 4-hidroxi-3-metoximetanfetamina (HMMA)) 532 e de
aumentar os níveis cerebrais de MDMA nos ratinhos pré-tratados com
paroxetina (10 mg/kg, i.p.) 5 minutos antes da exposição à MDMA (15 mg/kg) 533. Parece assim que a paroxetina consegue interagir com a MDMA ao nível
farmacodinâmico (no SERT) e a nível farmacocinético (na CYP2D6). Esta
redução marcada nos efeitos da MDMA pode levar os utilizadores a consumir
doses mais altas de MDMA, para conseguir obter o efeito pretendido, e a correr
o risco de produzir efeitos tóxicos potencialmente letais 532. O pré-tratamento
com paroxetina tem também a capacidade de contrariar parcialmente a
diminuição temporária no número de células T-helper CD4+ e o aumento
simultâneo do número de células NK, bem como de inibir completamente a
supressão da capacidade de resposta funcional dos linfócitos à estimulação
mitogénica e as alterações na secreção das citoquinas induzidas pela MDMA
em humanos 534.
A fluoxetina, a sertralina e a zimelidina (não comercializada em Portugal)
mostraram-se capazes de contrariar o efeito hiperlocomotor induzido pela
MDMA em ratos 105.
1.9.5.1.1.2. Inibidores da monoamina oxidase (IMAO)
Os IMAO causam interacções farmacodinâmicas com a MDMA em
humanos 535 potenciando a inibição da degradação dos neurotransmissores na
fenda sináptica. Por isso mesmo, estimulantes anfetamínicos não devem ser
utilizados em conjunto, ou num intervalo inferior a 2 semanas, com qualquer

Introdução ______________________________________________________
96
IMAO. Como consequência desta interacção podem surgir reacções
hipertensivas e estados confusacionais severos 23.
A fenelzina (não comercializada em Portugal) é um inibidor irreversível e
não específico da MAO indicado para o tratamento de problemas de
ansiedade, bulimia, fobias, etc que já esteve envolvido em interacções
medicamentosas com a MDMA 536.
A moclobemida é um inibidor reversível da MAO, com maior
selectividade para a MAO-A, utilizado na terapêutica para tratar a depressão e
a ansiedade social e utilizado pelos consumidores de MDMA para potenciar os
seus efeitos 284. A moclobemida foi já associada a quatro intoxicações fatais
quando consumida simultaneamente com a MDMA, sendo a causa provável de
morte o desenvolvimento de um quadro de síndrome serotonérgica 284. Quando
administrada a ratos 60 minutos após exposição à moclobemida (20 mg/kg
i.p.), a MDMA (10 mg/kg i.p.) produz um aumento dos níveis extracelulares de
5-HT muito superior ao verificado quando ela é administrada isoladamente,
sem alterar a temperatura corporal, um fenómeno também verificado quando a
MDMA (10 mg/kg i.p.) é administrada a ratos 24 horas após a administração de
clorgilina (10 mg/kg i.p.) 537. Este facto pode levar ao aparecimento de
fenómenos tóxicos resultantes de níveis demasiado elevados de 5-HT,
habitualmente associados ao consumo de PMA, uma vez que ela própria é
inibidora da MAO-A 538. Quando o mesmo ensaio foi repetido a uma
temperatura ambiente superior, verificou-se uma potenciação no aumento dos
níveis extracelulares de 5-HT e da resposta hipertérmica induzida pela MDMA.
Então, os efeitos deletérios resultantes da exposição simultânea a MDMA e a
inibidores da MAO são fortemente potenciados por temperaturas ambiente
elevadas 539.
De facto, as intoxicações mais graves com sintomatologia da síndrome
serotonérgica ocorrem quando se consomem simultaneamente os IMAO e
compostos inibidores da recaptação de 5-HT (como é o caso da MDMA) 540,
tendo sido já descritas intoxicações fatais associadas ao consumo simultâneo
deste tipo de substâncias 284.
No entanto, contrariamente ao que se verifica com os inibidores da
MAO-A, os inibidores selectivos da MAO-B (ex.: selegilina, L-deprenilo)
parecem conferir uma certa protecção relativamente à neurotoxicidade da

______________________________________________________ Introdução
97
MDMA por vários mecanismos: (i) prevenção da depleção dos níveis cerebrais
de 5-HT 541, (ii) diminuição da formação de espécies reactivas como resultado
da acção da MAO-B sobre a DA recaptada para o neurónio pré-sináptico
através do SERT 542, (iii) diminuição do grau de peroxidação lipídica provocada
pela MDMA 541, (iv) redução do dano oxidativo mitocondrial provocado por esta
substância a nível cerebral 119. Este facto indica que a MAO-B desempenha
um papel importante no desenvolvimento da neurotoxicidade da MDMA, apesar
de não influenciar a libertação aguda de DA nem a resposta hipertérmica
induzida por esta droga de abuso, um fenómeno verificado recorrendo a uma
redução da expressão proteica da MAO-B em ratos 543 e em ratinhos knockout
para a MAO-B 544. Em ambas as situações foi observada protecção
relativamente à depleção dos níveis cerebrais de 5-HT provocada pela MDMA.
1.9.5.1.2. Anti-retrovirais
A interacção dos anti-retrovirais com a MDMA é especialmente
preocupante, uma vez que, como já foi referido, ao consumo de MDMA está
associada uma maior aquisição de comportamentos de risco, que englobam
comportamentos facilitatórios da transmissão de doenças sexualmente
transmissíveis. Por esse motivo, existem altas taxas de infecção por HIV entre
os consumidores de MDMA e análogos.
Os fármacos anti-retrovirais têm sido os principais envolvidos em
interacções potencialmente letais com a MDMA 204, 514. Interacções entre
substâncias utilizadas em contextos recreativos (ex.: benzodiazepinas,
cocaína, LSD, Cannabis e derivados, anfetamina, MDMA e opiáceos) e
fármacos anti-HIV podem provocar toxicidade e influenciar os resultados
clínicos da terapia anti-retroviral, sobretudo devido a uma interferência no
metabolismo 509, uma vez que todos os inibidores das proteases e os inibidores
não-nucleosídicos da transcriptase reversa são substratos e potentes inibidores
ou indutores do CYP450 545. Concretamente, os inibidores das proteases que
inibem a CYP2D6 e a CYP3A4 podem estar implicados no aumento das
concentrações de MDMA quando consumidos simultaneamente 545.

Introdução ______________________________________________________
98
O ritonavir (Norvir®) é um fármaco anti-retroviral, da classe dos inibidores
da protease, utilizado no tratamento das infecções por HIV e da SIDA. Devido à
sua capacidade de inibir várias isoformas do complexo CYP450, o ritonavir é
utilizado frequentemente em associação com outros anti-retrovirais numa
abordagem terapêutica para o tratamento das infecções por HIV e da SIDA
denominada HAART (Highly Active AntiRetroviral Therapy). O ritonavir vai,
assim, inibir a(s) enzima(s) que metaboliza(m) os outros inibidores das
proteases, levando a concentrações plasmáticas mais altas destes fármacos, o
que permite ao médico assistente reduzir as doses e a frequência das tomas,
aumentando a sua eficácia clínica. No entanto, o problema do ritonavir
estende-se muito para além da inibição da CYP2D6 porque sabe-se que, em
exposiação aguda, o ritonavir tem a capacidade de inibir in vitro as isoformas
2B6, 2C9, 2C19, 2D6 e 3A4 do CYP450 em microsomas preparados a partir de
fígado humano 515, 546, 547. Alguns casos de interacções muito graves ou mesmo
fatais em indivíduos que consumiram MDMA enquanto tomavam ritonavir foram
já relatados 204, 548. Concretamente, um indivíduo do sexo masculino, HIV-
positivo, que já tinha tido contacto prévio com a MDMA sem sinais de
toxicidade, após ter iniciado a sua terapia anti-retroviral que incluía ritonavir,
ingeriu cerca de 180 mg de MDMA. Em poucas horas manifestou taquipneia,
taquicardia, hipertermia, uma crise tonico-clónica e veio a falecer. Os estudos
post-mortem revelaram uma concentração plasmática de MDMA 10 vezes
superior à esperada, provavelmente devido a uma inibição da CYP2D6 pelo
ritonavir que conduziu a uma situação de sobredosagem da MDMA 204. No
entanto, a farmacocinética não linear, resultante da inibição da CYP2D6 para
concentrações elevadas de MDMA, e a disfunção hepática pré-existente neste
indivíduo podem ter também contribuído acentuadamente para o
desenvolvimento desta intoxicação 204. A inibição da CYP2D6 pelo ritonavir
parece ser causa de um aumento da toxicidade da MDMA 509, uma vez que o
ritonavir tem maior afinidade para a CYP2D6 do que as anfetaminas, levando a
um aumento dos seus níveis plasmáticos entre 3 a 10 vezes 549.

______________________________________________________ Introdução
99
1.9.5.2. Alimentos
Os alimentos ingeridos pelos consumidores de MDMA podem afectar os
seus efeitos pela ocorrência de interacções farmacodinâmicas e/ou
farmacocinéticas.
Por exemplo, suplementos alimentares à base de Ephedra são utilizados
por cerca de 1% da população para perda de peso, aumento dos níveis de
energia e performance física acrescida. No entanto, devido à acção
simpaticomimética destes compostos, têm grande potencial para interagir com
compostos com propriedades semelhantes (ex.: MDMA) aumentando o risco de
agravamento dos efeitos tóxicos derivados da Ephedra como: enfarte de
miocárdio, convulsões, acidentes vasculares-cerebrais isquémicos e
hemorrágicos potencialmente letais 550.
Outros constituintes dos alimentos podem originar interacções
farmacodinâmicas com a MDMA por serem inibidores da MAO (ex.: cumarina,
flavona, entre outros) 551. De facto, algumas xantonas são potentes inibidores
reversíveis da MAO-A 551, o que, como já referido anteriormente, pode levar a
uma potenciação da neurotoxicidade induzida pela MDMA. No entanto, outras
xantonas podem apresentar uma acção inibitória mais marcada sobre a MAO-B 552, podendo possivelmente conferir um certo grau de neuroprotecção
relativamente à toxicidade da MDMA.
As interacções farmacocinéticas podem surgir porque a dieta, incluindo
os nutracêuticos 398, pode estar na base de alterações da actividade das
enzimas do CYP450 483. Daí que seja importante saber quais os constituintes
da dieta que podem afectar as principais isoenzimas do CYP450.
Como exemplo, um indivíduo que consuma habitualmente
hidrocarbonetos aromáticos policíclicos (HAPs) veiculados pelos alimentos
grelhados em carvão vai ter um metabolismo de fase I mais rápido que um
indivíduo que não os consuma 440, com o consequente aumento da
metabolização de compostos que sejam substratos das enzimas envolvidas
nesta fase do metabolismo dos xenobióticos. Quando os compostos ingeridos
sofrem bioactivação metabólica através dos sistemas enzimáticos induzidos,
levando à formação de compostos tóxicos, a indução das enzimas de fase I

Introdução ______________________________________________________
100
pode estar associada a um maior risco de desenvolvimento de respostas
tóxicas.
Alguns flavonóides (quercetina, luteolina, apigenina, baicaleina, crisina,
flavona) têm a capacidade ainda de induzir simultaneamente enzimas de fase I
e de fase II (as UGTs e o CYP450). Outros compostos que ocorrem
naturalmente e também induzem as UGTs são: benzo[a]pireno, cumarina, α-
angelicalactona, ácido elágico, e ácido ferúlico 440.
Medicamentos, alimentos, e seus constituintes, que influenciam a
actividade e expressão das principais isoenzimas do CYP450 envolvidas no
metabolismo da MDMA estão compilados na Tabela 5.
Tabela 5. Medicamentos, alimentos, e seus constituintes, que influenciam a actividade e expressão das principais isoenzimas do CYP450 envolvidas no metabolismo da MDMA. Adaptado e Ioannides et al. 483
CYP1A2
Substratos antidepressivos (amitriptilina, imipramina, clomipramina), antipsicóticos atípicos (clozapina,
olanzapina), cafeína, anestésicos locais (ropivacaína), teofilina, agonistas dos receptores 5-
HT (zolmitriptano), ciclobenzaprina, estradiol, fluvoxamina, haloperidol, ondansetron,
mexiletina, melatonina, tamoxifeno, naproxeno, paracetamol, fenacetina, propranolol, riluzol,
ropinirol, tacrina, tizanidina, verapamilo, varfarina, zileuton, lidocaína
Indutores vegetais da família Brassicaceae (vegetais crucíferos como os brócolos e couve-de-bruxelas,
devido aos glucosinolatos 396), insulina, metilcolantreno, modafinil, nafcilina, β-naftoflavona,
omeprazol, tabaco, sumo de toranja (devido à flavonona naringenina), erva de S. João (pela
hiperforina), anticonvulsivantes (carbamazepina, fenobarbital), rifampicina, espinafres, alho
francês, cebola, salsa, pão torrado, alimentos fritos ou grelhados com carvão (devido à
formação de hidrocarbonetos aromáticos policíclicos (HAPs, ex.: benzo[a]pireno) e aminas
heterocíclicas), carne e peixe fumados, presunto, salsichas, chá verde e chá preto (por
conterem cafeína) 553, elevada ingestão de TG (por aumentar a formação de corpos
cetónicos), compostos metilenodioxifenilo (presentes na pimenta preta, canela, cravinho, noz-
moscada) como o safrol, furanocumarinas (ou psoralenos, presentes no aipo, figo, toranja),
carotenos, ruibarbo (por conter antraquinona), alicina (composto organosulfurado presente no
alho)
Inibidores Potentes: fluoroquinolonas (ciprofloxacina), fluvoxamina, verapamilo
Ligeiros: cimetidina
Outros: amiodarona e mexiletina, furafilina, interferão, metoxaleno, mibefradil, cafeína,
Echinacea, enoxacina, contracepção hormonal, zileuton, pão, batatas, arroz, plantas da
família Apiaceae (cenouras, aipo, funcho, salsa, por terem furanocumarinas), algumas
Rutaceae (frutos cítricos, toranja, por terem furanocumarinas) e carne cozida, redução de
ingestão proteica, défices de vitamina D, ingestão excessiva de colesterol, jejum, aumento do
consumo de glucose

______________________________________________________ Introdução
101
Tabela 5 (cont.). Medicamentos, alimentos, e seus constituintes, que influenciam a actividade e expressão das principais isoenzimas do CYP450 envolvidas no metabolismo da MDMA Adaptado e Ioannides et al. 483
CYP2B6
Substratos bupropion, ciclofosfamida, ifosfamida, tiotepa, efavirenz e nevirapina, alfentanilo, propofol,
tamoxifeno, valproato, artemisinina, petidina, nicotina, sertralina
Indutores fenobarbital (a sua acção indutora é inibida se houver carência em selénio), rifampicina,
carbamazepina, artemisinina, clotrimazol, ifosfamida, glucocorticóides, fenitoína, ritonavir,
dietas ricas em TG, jejum, bróculos e couves-de-bruxelas, chá preto e verde, flavonóides,
terpenóides (cânfora, mentol, pineno, limoneno), safrol, psoraleno, alho, álcool
Inibidores Potentes: orfenadrina
Outros: tiotepa, ticlopidina, paroxetina, fluoxetina, sertralina, estradiol, fenciclidina, ritonavir
CYP2D6
Substratos β-bloqueadores (metoprolol, carvedilol, timolol, alprenolol, atenolol), antiarrítmicos de classe I
(flecainide, lidocaína, propafenona, encainide, mexiletina), todos os antidepressivos tricíclicos
(ex.: imipramina, amitriptilina), maioria dos ISRS (ex.: fluoxetina, paroxetina), opióides
(codeína, tramadol), debrisoquina, dextrometorfano, venlafaxina, antipsicóticos (ex.:
haloperidol, risperidona, perfenazina, tioridazina, zuclopentixol, remoxipride, aripiprazol),
ondansetron e tropisetron, mianserina, fenformina, anfetamina, clorfenamina,
metoclopramida, tamoxifeno, alcalóides da vinca
Indutores Potentes: piperidinas e derivados (ex.: glutatimida)
Intermédios: carbamazepina
Outros: dexametasona, rifampicina
Inibidores Potentes: bupropion, ISRS (fluoxetina, paroxetina, citalopram), quinidina
Médios: duloxetina, terbinafina
Ligeiros: amiodarona, cimetidina e ranitidina, sertralina
Outros: celecoxib, antagonistas receptores H1 (clorfeniramina, clorfenamina, difenidramina),
antipsicóticos (clorpromazina, haloperidol), cinacalcet, clemastina, clomipramina e imipramina,
cocaína, doxepina, doxorrubicina, halofantrina, hidroxizina, levomepromazina, metadona,
metoclopramida, mibefradil, midodrina, moclobemide, perfenazina, ranitidina, ritonavir,
ticlopidina, tripelenamina, cloranfenicol, ranolazine, levomepromazina e pimozide e tioridazina

Introdução ______________________________________________________
102
Tabela 5 (cont.). Medicamentos, alimentos, e seus constituintes, que influenciam a actividade e expressão das principais isoenzimas do CYP450 envolvidas no metabolismo da MDMA. Adaptado e Ioannides et al.
CYP3A4
Substratos bloqueadores dos canais de cálcio (diltiazem, nifedipina, felodipina, verapamilo),
imunosupressores (ciclosporina, tacrolimus, sirolimus), quimioterápicos (ciclofosfamida,
docetaxel, doxorrubicina, etoposido, ifosfamida, paclitaxel, tamoxifeno, teniposido,
vimblastina, vindesina, gefitinib), benzodiazepinas (flunitrazepam, midazolam, alprazolam,
triazolam, clonazepam), antifúngicos azólicos (cetoconazol, itraconazol, clotrimazol),
antidepressivos tricíclicos (amitriptilina, imipramina, clomipramina), antibióticos macrólidos
(eritromicina, claritromicina), ISRSs (citalopram, fluoxetina, norfluoxetina, sertralina), estatinas
(atorvastatina, lovastatina, sinvastatina), inibidores da fosfodiesterase-5 (PDE5) (sildenafil),
buspirona, haloperidol e pimozide, venlafaxina, amiodarona, etinilestradiol e levonorgestrel,
quininas, inibidores das proteases (indinavir, ritonavir, saquinavir, nelfinavir), mirtazapina,
nefazodona, reboxetine, zopiclone, inibidores da transcriptase reversa não nucleosídicos
(nevirapina), fentanilo e alfentanilo, budesonido, donepezil, esomeprazol e omeprazol,
finasteride, glibenclamida, cisapride, terfenadina, toremifeno, barbitúricos (fenobarbital),
carbamazepina, codeína, dextrometorfano, digoxina, alcalóides da cravagem do centeio,
estradiol, ivabradina, lidocaína, metadona, mifepristona, montelucaste, ondansetron,
paracetamol, quinidina, testosterona, teofilina, valproato, varfarina, tetraidrocanabinol,
antipsicóticos atípicos (aripiprazol, risperidona, ziprasidona)
Indutores anti-retrovirais inibidores da TR não nucleosídicos (efavirenz, nevirapina, etravirina),
barbitúricos (fenobarbital; a sua acção indutora é inibida se houver carência em selénio),
anticonvulsivantes (carbamazepina, oxcarbazepina, fenitoína, felbamato, primidona,
topiramato), anti-inflamatórios (glucocorticóides, ex: dexametasona), antinarcolépticos
(modafinil), antidiabéticos orais (pioglitazona, troglitazona), antibióticos (rifabutina,
rifampicina), antidepressivos (hiperforina da erva de S. João em baixas doses 554, 555),
antifúngicos (griseofulvina), cafestol, óleo de milho, dietas com redução calórica, indóis
(presentes nos vegetais crucíferos)
Inibidores Potentes: inibidores das proteases (indinavir, nelfinavir, ritonavir, saquinavir), macrólidos
(claritromicina, eritromicina, telitromicina), antifúngicos azólicos (itraconazol, cetoconazol),
nefazodona, quercetina
Médio: aprepitante, eritromicina, fluconazol, sumo de toranja (devido à bergamotina),
verapamilo
Médio-Ligeiro: diltiazem
Ligeiro: cimetidina
Outros: amiodarona, cloranfenicol, dietil-ditiocarbamato de delaviridina, fluvoxamina,
gestodeno, imatinib, mibefradil, mifepristona, norfloxacina, norfluoxetina, carambola,
voriconazol, ciprofloxacina, ciclosporina, Echinacea, enoxacina, ergotamina, metronidazol,
inibidores da transcriptase reversa não-nucleosídicos (efavirenz, nevirapina), ISRSs
(fluoxetine), piperina, hiperforina da erva de S. João em doses elevadas 554, 555.

______________________________________________________ Introdução
103
Não devemos ainda descurar o efeito imprevisível que os xenobióticos
(ex: dioxinas, antibióticos, hormonas promotoras do crescimento, poluentes
ambientais, pesticidas) presentes nos alimentos poderão ter ao nível da
farmacocinética da MDMA.
Assim, existem vários factores nutricionais que podem condicionar a
expressão do CYP450:
a) alterações nos níveis de macronutrientes [dietas pobres em
proteínas diminuem a actividade do CYP450; elevados consumos
de glucose, frutose e sacarose diminuem o metabolismo dos
xenobióticos; a natureza e a quantidade dos lípidos da dieta altera
a actividade do CYP450 em função da espécie em causa; a
deficiência em lipótropos (metionina e colina) induz o CYP450].
b) Alterações nos níveis de micronutrientes (normalmente, a
deficiência em micronutrientes (vitaminas A, C ou E) diminui ou
suprime o metabolismo; no entanto, deficiência em tiamina
(vitamina B1) aumenta o metabolismo; carência em ferro diminui a
actividade do CYP450 intestinal e a quantidade de hemoproteína,
mas não altera CYP450 hepático; o mesmo acontece com o
selénio; deficiência em zinco diminui CYP450 hepático; deficiência
em cobre tem uma regulação diferencial das várias proteínas do
CYP450).
c) Jejum e redução do consumo calórico (a redução do aporte calórico
com redução ponderada da ingestão de hidratos de carbono,
proteínas e lípidos, mas sem alterar o aporte de micronutrientes,
leva a uma redução da actividade do CYP450; o mesmo acontece
durante o jejum) 556, 557.
d) “Não-nutrientes” presentes na dieta (constituintes de plantas,
micotoxinas, produtos de cedência do material de embalagem,
produtos gerados durante a cocção e aditivos alimentares) também
têm a capacidade de alterar a actividade do CYP450 483.

Introdução ______________________________________________________
104
1.9.5.3. Contaminantes das pastilhas de ecstasy
Apesar de as pastilhas comercializadas nos espaços rave estarem
muitas vezes identificadas com determinados logótipos com o objectivo do
comprador identificar qual a droga que está a comprar, a verdade é que elas
podem conter apenas substâncias inertes (bicarbonato de sódio, pó de talco,
açúcares), percentagens variáveis de MDMA, uma droga completamente
diferente daquela que o consumidor pretende comprar, ou a droga em causa
pode estar misturada com outros contaminantes. Existe, assim, uma enorme
heterogeneidade e variabilidade nos ingredients activos das pastilhas vendidas
como ecstasy 13.
Esta composição variável das pastilhas de ecstasy tem sido estudada
desde 1986, quando Renfroe analisou o conteúdo de várias pastilhas vendidas
como ecstasy nos EUA. Os resultados deste e dos estudos que lhe seguiram
estão compilados na Tabela 3.
A maioria dos estudos realizados conduz às mesmas conclusões: (i)
apenas uma parte das pastilhas em circulação contém MDMA ou substâncias
aparentadas (ex.: MDA, MDEA, MBDB e 2-CB) 3, 366, 369, 372, 373; (ii) a proporção
de ingredientes activos varia largamente entre pastilhas (concretamente, a
MDMA chega a variar num factor de 7 vezes entre pastilhas) 3, 366, 373; (iii) a
maior parte das pastilhas apresenta na sua constituição substâncias inertes
que funcionam como excipientes (ex.: açúcares, poliálcoois como o manitol,
talco, bicarbonato de sódio) 3; e (iv) a maioria contém outras substâncias
estimulantes ou não para além da MDMA (ex.: MTA, ketamina, cafeína,
testosterona, efedrina, pseudoefedrina, LSD, cocaína, heroína, DXM,
cloroquina, paracetamol, vasodilatadores, atropina, salicilatos, etc) 3, 366, 368, 369,
372, 558.
Nalguns destes estudos verificou-se ainda uma diminuição do teor de
MDMA e um aumento da contaminação das pastilhas de ecstasy com outras
substâncias ao longo do tempo 366, 369. No entanto, na Europa, a maior parte
dos comprimidos de ecstasy continuaram, em 2005, a conter MDMA ou outra
substância afim (MDEA, MDA), normalmente como única substância
psicoactiva presente 20.

______________________________________________________ Introdução
105
O teor em MDMA dos comprimidos de ecstasy varia muito entre os lotes
(inclusive entre os que apresentam o mesmo logótipo), e ao longo do tempo,
num mesmo país e entre países. Este fenómeno pode levar a reacções
diferentes e imprevisíveis por parte dos consumidores 366. Em 2005, o teor
médio de MDMA por pastilha de ecstasy variava entre 2 e 130 mg, embora na
maioria dos países a média se situasse entre 30 e 80 mg de MDMA 20.
Contudo, estes estudos sobre a composição e pureza das pastilhas de
ecstasy têm algumas limitações que se prendem com as fontes das pastilhas
de ecstasy analisadas: (i) quando as pastilhas são obtidas de apreensões
policiais, provêm de lotes muito homogéneos que, normalmente, são pouco
representativos, uma vez que as pastilhas contidas em cada lote apreendido
têm, geralmente, a mesma origem, (ii) quando a recolha das pastilhas para
análise é feita numa determinada população-alvo de uma dada área
geográfica, os resultados obtidos não poderão ser generalizados, uma vez que
a composição das pastilhas varia com a área geográfica 366, (iii) quando as
pastilhas são obtidas através de campanhas que incentivam as pessoas a
submeter para análise pastilhas que considerem suspeitas, os níveis de pureza
em MDMA encontrados são normalmente muito inferiores porque as pastilhas
que são submetidas são aquelas associadas à ocorrência de efeitos não
esperados (tal como se verificou no estudo de Tanner-Smith realizado em 2006
no qual se incentivava o envio para análise de pastilhas de ecstasy suspeitas
de serem falsas ou adulteradas 366). Também a precisão das informações
prestadas pela população alvo e a duração do estudo (já que a composição e
pureza das pastilhas varia ao longo dos anos 366), entre outros factores,
poderão afectar as conclusões acerca dos resultados obtidos. Estas limitações
levam ao aparecimento de resultados contraditórios.
Em 2004, Parrott fez uma revisão de alguns estudos sobre a pureza das
pastilhas de ecstasy na Europa e concluiu que, nos anos 80, as pastilhas de
ecstasy eram de elevadas qualidade e pureza (90-98% continha MDMA).
Contudo, no início dos anos 90 a pureza sofreu um declínio e apenas 50 a 80%
das pastilhas continham MDMA. No entanto, a proporção de pastilhas contendo
MDMA voltou a aumentar no final dos anos 90 e atingiu valores próximos dos
100% no início do século XXI. O declínio no conteúdo de MDMA das pastilhas
está muitas vezes associado ao aumento da procura e a uma maior dificuldade

Introdução ______________________________________________________
106
para os produtores em aceder às matérias-primas 558. Considerando as
elevadas prevalências de contaminação das pastilhas de ecstasy, estudos
comparativos parecem indicar que é mais provável adquirir uma pastilha de
ecstasy não psicoactiva (sem compostos psicoactivos ou em doses demasiado
baixas) do que comprar pastilhas contendo doses elevadas de MDMA 373.
A utilização destes contaminantes (ex.: DXM, cafeína, pseudo-efedrina)
nas pastilhas de ecstasy deve-se, provavelmente, à maior disponibilidade (uma
vez que são compostos legais) e ao menor preço destes produtos quando
comparados com a MDMA. Para além deste facto, a produção de pastilhas de
MDMA é muito interessante economicamente devido à grande expansão do
consumo destas pastilhas e ao elevado preço das mesmas, o que incita os
produtores a fabricar comprimidos para serem vendidos como ecstasy, mesmo
que estes não contenham o composto em causa 366.
Assim, a pureza das pastilhas de ecstasy varia, dependendo da
disponibilidade de MDMA pura importada, da capacidade dos mercados
produzirem MDMA pura e dos efeitos procurados pelos consumidores da droga 559.
Devido a estas impurezas, os efeitos tóxicos da MDMA são apenas uma
parte dos efeitos potencialmente letais que podem resultar do consumo deste
tipo de pastilhas, uma vez que os utilizadores de ecstasy podem
inadvertidamente ingerir outras drogas potencialmente letais para além da
MDMA. A contaminação das pastilhas de ecstasy é particularmente
problemática para utilizadores com determinados factores predisponentes para
o desenvolvimento de dependência e/ou patologias de foro psiquiátrico 560. O
aparecimento de efeitos raros ou inesperados pode ser explicado pela
presença de substâncias não estimulantes nas pastilhas de ecstasy 423.
As possíveis interacções da MDMA com outros compostos incluídos na
composição das pastilhas serão abordadas, mais detalhadamente, nos sub-
capítulos seguintes.

______________________________________________________ Introdução
107
1.9.5.3.1. Excedentes e sub-produtos da síntese de MDMA
Dependendo do procedimento utilizado para obter MDMA, entre os cerca
de 20 disponíveis, os compostos precursores da sua síntese incluem MDA,
safrol, isosafrol, piperonal e piperonil acetona. Outros reagentes usados na
síntese de MDMA são o ácido bromídrico, metilamina, cloroformato de etilo,
alumino-hidreto de lítio, tetraidrofurano, cianoboro-hidreto de sódio, metanol, N-
metilformamida, entre outros 561. Para além dos precursores de síntese
poderem potencialmente interferir com os efeitos tóxicos da MDMA é também
importante ter em conta que os compostos intermediários da síntese [isosafrol
glicol, piperonilmetilcetona, N-formil-3,4-metilenodioximetilanfetamina, N-formil-
3,4-metilenodioxianfetamina e 1-(3,4-metilenodioxifenil)-2-bromopropano],
poderão também hipoteticamente originar interferências com os efeitos da
MDMA 562. Numa síntese bem sucedida, a MDMA resultante é um pó com
cerca de 100% de pureza com um aroma característico 563. Por este motivo,
segundo alguns autores, não existe evidência analítica que confirme a
presença de materiais de partida, intermediários e sub-produtos de síntese nas
pastilhas de ecstasy, uma vez que eles não são detectados em determinados
lotes 558, 564.
No entanto, estes materiais e sub-produtos de síntese foram
encontrados em amostras de 89 aprensões realizadas em Hong Kong e este
perfil de impurezas permite inferir qual o processo de síntese utilizado 365. Este
tipo de análise qualitativa foi já proposto em 2005 por Swist e colaboradores 565,
566 para identificar o método de síntese utilizado e assim poder prever qual a
origem das pastilhas.
A presença destas impurezas é procupante, uma vez que estes
contaminantes resultantes da preparação da MDMA podem exercer um papel
importante no aparecimento de problemas de saúde associados ao consumo
de pastilhas de ecstasy 4, como se pode observar na tabela seguinte (Tabela
6):

Introdução ______________________________________________________
108
Tabela 6. Possíveis efeitos dos Excedentes e sub-produtos da síntese de MDMA na toxicidade da ecstasy. Composto Possível efeito
Materiais de partida
MDA Sinergismo com os efeitos da MDMA 567
Aumento do risco de toxicidade por formação de metabolitos tóxicos,
ROS e RNS 60, 568
Safrol e análogos Potencial carcinogénese em humanos por formação com aductos
com o DNA 569
Tetraidrofurano (THF) Narcose e disfunção hepatocelular em doses altas 570
Inibe algumas isoenzimas do CYP450 (sobretudo a CYP2E1) 570
Estimulação da absorção de metabolitos reactivos de vários
compostos (ex.: solventes) 570
Metanol Desenvolvimento de acidose metabólica e inibição da respiração
celular devido à acumulação de formato 571 que poderá diminuir o
tempo de semi-vida da MDMA por aumentar a sua eliminação urinária
N-metilformamida Desenvolvimento de hepatotoxicidade 572
Piridina 365 Indução da CYP2E1
Desenvolvimento de hepatotoxicidade e nefrotoxicidade
Diminuição da motilidade dos espermatozóides em ratinhos
Altera do estro em ratos 573
Intermediários de síntese
1,3-bis(3,4-metilenodioxifenil)-2-
propanamina e N-formil-1,3-
bis(3,4-metilenodioxifenil)-prop-
2-il-amina
Inibição dos transportadores das monoaminas numa intensidade
semelhante à da MDMA 574
Fraca/ausente capacidade de aumentar o potencial neurotóxico da
MDMA por não induzirem a libertação destes neurotransmissores 574
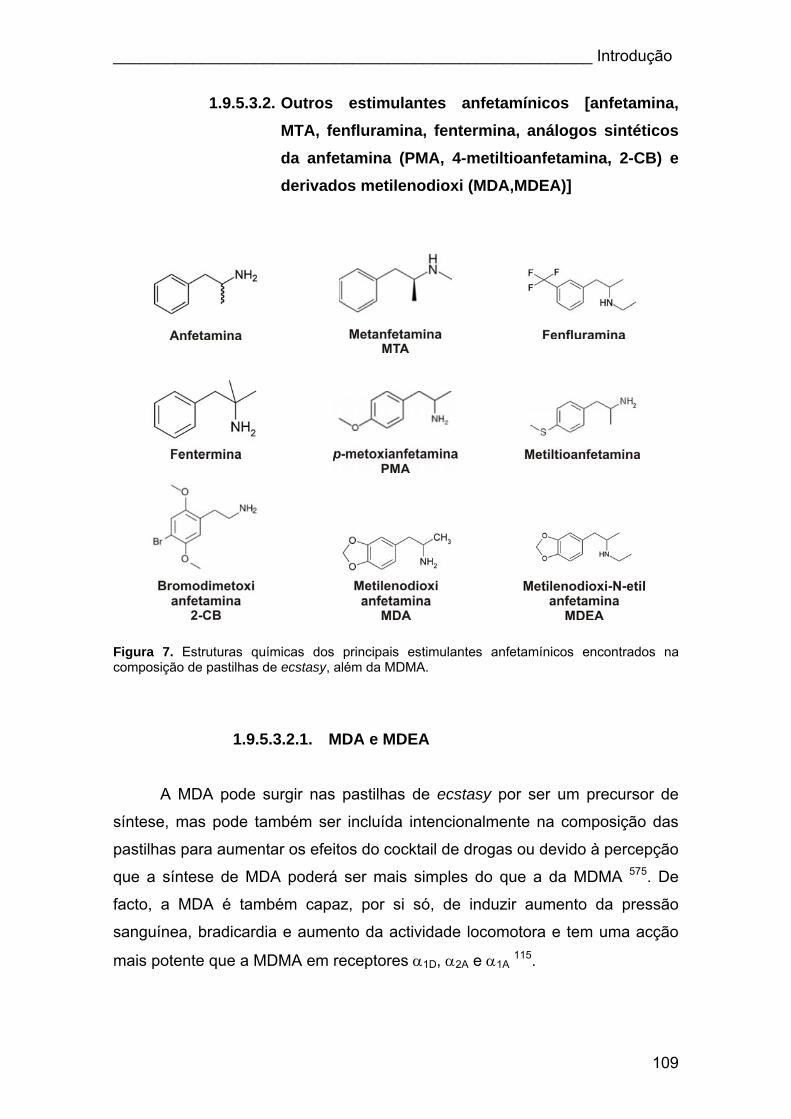
______________________________________________________ Introdução
109
1.9.5.3.2. Outros estimulantes anfetamínicos [anfetamina, MTA, fenfluramina, fentermina, análogos sintéticos da anfetamina (PMA, 4-metiltioanfetamina, 2-CB) e derivados metilenodioxi (MDA,MDEA)]
Figura 7. Estruturas químicas dos principais estimulantes anfetamínicos encontrados na composição de pastilhas de ecstasy, além da MDMA.
1.9.5.3.2.1. MDA e MDEA
A MDA pode surgir nas pastilhas de ecstasy por ser um precursor de
síntese, mas pode também ser incluída intencionalmente na composição das
pastilhas para aumentar os efeitos do cocktail de drogas ou devido à percepção
que a síntese de MDA poderá ser mais simples do que a da MDMA 575. De
facto, a MDA é também capaz, por si só, de induzir aumento da pressão
sanguínea, bradicardia e aumento da actividade locomotora e tem uma acção
mais potente que a MDMA em receptores α1D, α2A e α1A 115.

Introdução ______________________________________________________
110
No entanto, a sua presença no produto final poderá ser responsável por
um aumento da toxicidade resultante do consumo da pastilha, uma vez que a
MDA sofre um metabolismo semelhante ao da MDMA e corresponde, também,
a um dos metabolitos formados a partir da MDMA. A MDA é O-desmetilenada
ao catecol α-Me-DA que pode sofrer oxidação à correspondente o-quinona e,
posteriormente, conjugar-se com a glutationa para formar aductos. Qualquer
um destes passos metabólicos leva à formação de metabolitos com maior
toxicidade que o composto pai e é acompanhada pela libertação de ROS e
RNS 60, 568. Uma vez que a MDA também tem a capacidade de induzir uma
resposta hipertérmica semelhante à da MDMA 567, parece plausível assumir a
possibilidade de ocorrer um efeito aditivo entre os dois compostos, aumentando
os efeitos deletérios induzidos pela hipertermia.
A ingestão combinada de MDA, anfetamina, MDMA e PMA foi já
associada ao aparecimento de intoxicações fatais 282. O mesmo já se verificou
pela exposição simultânea a MDEA e MDMA 297.
1.9.5.3.2.2. MTA
Foi já demonstrado que, em ratos, a combinação da MDMA com a MTA
tem efeitos tóxicos mais marcados do que cada uma das drogas em separado 576 embora estas diferenças pareçam desaparecer num padrão de consumo
crónico 577.
A MTA e a MDMA têm afinidade para os receptores nicotínicos e são
capazes de induzir a sua up-regulation, especialmente quando se utilizam
doses mais elevadas. Este efeito pode levar a um sinergismo na
neurotoxicidade, neurotransmissão colinérgica 100.
Por outro lado, devido aos seus efeitos mais marcados no sistema
dopaminérgico, a MTA tem maior potencial de abuso do que a MDMA. Logo, a
sua inclusão nas pastilhas de ecstasy pode aumentar a prevalência de
comportamentos de adição e dependência relativamente às mesmas 100.
Enquanto a libertação aguda de 5-HT contribui para os efeitos
subjectivos da MDMA como euforia, alucinações ligeiras e sentimento de
proximidade para com os outros, o aumento da DA produzido pela MTA conduz

______________________________________________________ Introdução
111
a sentimentos de auto-confiança, energia, estimulação sexual e euforia, sendo
também responsável pelo potencial aditivo. A combinação das duas
substâncias parece provocar efeitos eufóricos e pró-sociais sinérgicos 133.
Em termos farmacocinéticos, a MTA, a MDA e a MDEA inibem in vitro a
captação e a permeação da MDMA através das células do epitélio do íleo 578,
pelo que poderão atrasar a sua absorção intestinal.
Para além disso, a izoenzima CYP2D6 tem um envolvimento primordial
na 4-hidroxilação aromática da MTA, o que faz antever uma elevada
probabilidade de interacções farmacocinéticas com outras drogas que sejam
substratos desta mesma enzima, como é o caso da MDMA 579.
1.9.5.3.2.3. Outras anfetaminas
As anfetaminas provocam neurotoxicidade serotonérgica e
dopaminérgica, a qual leva, em ratinhos Swiss Webster, a uma sensibilização
de longa duração aos efeitos locomotores de psicoestimulantes como a MDMA,
p-cloroanfetamina e cocaína 580, podendo ter efeitos aditivos com as referidas
substâncias.
Em acréscimo, todas as anfetaminas com estrutura fenilisopropilamínica
são inibidoras da CYP2D6, sendo a sua potência relativa condicionada pela
presença de outros grupos funcionais 27, o que provavelmente irá conduzir a
um aumento dos níveis plasmáticos de MDMA quando consumida
simultaneamente com estes compostos e consequentemente aumentar o risco
de desenvolvimento de toxicidade aguda.
As interacções da MDMA com outras anfetaminas também se verificam
quando estas são co-consumidas intencionalmente para modular os efeitos da
MDMA (vide secção 1.9.5.4.2).

Introdução ______________________________________________________
112
1.9.5.3.3. Ketamina
A ketamina [2-(2-clorofenil)-2-(metilamino)-
ciclohexanona] (Ketofen®, Ketalar®, Ketanest®, Ketaset®,
Ketaject®, Ketamine 500®, Imalgen®) é um anestésico derivado
da fenciclidina utilizado no contexto veterinário e clínico
(analgesia e anestesia pediátrica, anestesia de emergência, obstetrícia e nos
campos de guerra), com propriedades dissociativas, analgésicas e psicadélicas 581. Esta molécula foi já identificada em duas pastilhas de ecstasy conhecidas
como “Strawberry Milkshake” e “White Lightning” em dosagens que variaram de
185 a 197 mg 423, que correspondem a doses de cerca de 2,5 mg de ketamina
por quilo de peso corporal (para um indivíduo de 70 kg). Clinicamente, para ter
alguma acção analgésica, a ketamina é normalmente administrada via i.m.
numa dose de 0,5 mg/kg, que originaria concentrações plasmáticas
semelhantes às obtidas pela ingestão de uma destas pastilhas analisadas.
Para obter um estado de anestesia durante 10-25 minutos seria necessário
administrar uma dose de 10 mg/kg i.m., pelo que um indivíduo teria que
consumir várias pastilhas como as analisadas para ficar inconsciente. No
entanto, o consumo de algumas pastilhas ao mesmo tempo pode produzir
ataxia em alguns indivíduos. As sequelas psíquicas do consumo de ketamina
parecem ser relacionadas com a dose e aparentemente são marcadamente
reduzidas se a ketamina for utilizada em doses subanestésicas 582.
A ketamina tem um mecanismo de acção complexo que envolve
alteração da neurotransmissão glutamatérgica e monoaminérgica através de
um antagonismo, de baixa afinidade, no canal catiónico dos receptores N-metil-
D-aspartato (NMDA), um aumento da libertação de glutamato, alguma
afinidade para os receptores opióides centrais e espinais (α e μ), receptores
colinérgicos e receptores da NA e da 5-HT. Foi já descrito que a ketamina tem
a capacidade de inibir a recaptação da NA, DA e 5-HT, numa forma
dependente da dose, em células renais embrionárias humanas, tendo assim
uma acção simpaticomimética 583, que poderá estar na base de interacções
farmacodinâmicas com a MDMA. A ketamina apresenta ainda uma outra

______________________________________________________ Introdução
113
possibilidade de interacção farmacodinâmica com a MDMA, uma vez que pode
também causar aumento da temperatura corporal nalguns indivíduos 581.
Devido aos seus efeitos ao nível do sistema dopaminérgico a ketamina é
uma droga com propensão ao abuso e com a capacidade de induzir, em baixas
doses, reforços positivos que conduzam a situações de dependência (já
verificadas em modelos animais) 581. Logo, a presença de ketamina nas
pastilhas de ecstasy pode levar ao aparecimento de dependência relativamente
ao seu consumo, apesar de a MDMA propriamente dita ser uma substância
com pouca capacidade de induzir este tipo de reacção.
O consumo de ketamina veiculada nas pastilhas de ecstasy surpreende
muitas vezes os consumidores que manifestam imobilidade e alucinações
quando esperavam obter empatia e euforia 584.
Os sintomas associados à ingestão excessiva de ketamina são muito
semelhantes às da MDMA e incluem hiperactividade, ansiedade, dor no peito,
palpitações, taquicardia e, em casos mais graves, rabdomiólise. Qualquer um
destes efeitos pode ser aditivo com aqueles provocados pela MDMA,
aumentando a toxicidade das substâncias ingeridas.
Para além de poder desencadear fenómenos de interacção
farmacodinâmica com a MDMA, a ketamina tem ainda o potencial de provocar
interacções farmacocinéticas, uma vez que a maior parte do composto pai é
metabolizado via N-desmetilação catalizada pela enzyma CYP2B6 585, 586. Foi
ainda descrito que a fenciclidina (PCP, composto do qual deriva a ketamina)
tem a capacidade de reduzir, em ratos, a actividade de algumas isoenzimas do
sistema CYP450, incluindo a sub-família CYP2D 587, podendo aumentar os
níveis plasmáticos de MDMA. Embora, por outro lado, a ketamina tenha inibido
in vitro a captação e a permeação da MDMA através das células do epitélio do
íleo 578, pelo que aparentemente reduzirá a sua absorção a nível intestinal.
As interacções da MDMA que se verificam quando a ketamina é co-
consumida intencionalmente para modular os efeitos da MDMA serão
analisadas mais detalhadamente na secção 1.9.5.4.5.

Introdução ______________________________________________________
114
1.9.5.3.4. Cocaína
Contrariamente à previsão dos utilizadores 349, as
pastilhas de ecstasy têm uma reduzida probabilidade de
conter cocaína, heroína ou outros opiáceos, uma vez que a
inclusão destes ingredientes nas pastilhas seria pouco
vantajoso economicamente para os fabricantes de ecstasy 373.
No entanto, existem várias possibilidades de interacção entre estas duas
substâncias que serão abordadas adiante (vide secção 1.9.5.4.3).
1.9.5.3.5. Efedrina/pseudoefedrina
A efedrina é uma anfetamina natural encontrada na
Ephedra vulgaris (Ma-Huang ou Herbal ecstasy). O consumo
excessivo de efedrina foi já associado ao aparecimento de
sobredosagem tóxica 588.
A efedrina é um composto simpaticomimético que pode
ser usado como broncodilatador ou descongestionante nasal e
que tem propriedades semelhantes à adrenalina nos
receptores α e β adrenérgicos, e efeitos centrais semelhantes
aos de outras anfetaminas 550.
A efedrina provoca libertação de NA e, em menor
extensão, de DA e de 5-HT. No entanto, parece que esse
efeito de libertação da NA é o que está na base dos efeitos
simpaticomiméticos subjectivos desta substância 589, podendo
estar também na origem de interacções farmacodinâmicas
que potenciem os efeitos da MDMA.
O metabolismo da efedrina também envolve uma fase de N-
desmetilação a norefedrina que pode ser responsável por uma interacção
farmacocinética com a MDMA 590, uma vez que competem para as mesmas
vias metabólicas.

______________________________________________________ Introdução
115
1.9.5.3.6. Cafeína
Num estudo recente, 10% das pastilhas de ecstasy
analisadas continham esta xantina 99.
Este contaminante parece ter poucas consequências
para a saúde em doses baixas 591. No entanto, o consumo
excessivo de cafeína foi já associado ao aparecimento de
sobredosagem tóxica 588.
Além disso é importante considerar que a exposição à cafeína não é
apenas veiculada pela contaminação das pastilhas, mas também pelo consumo
de café, refrescos de cola e bebidas energéticas muito associadas à vida
nocturna e consumidas para reduzir a sonolência e o cansaço.
Este composto actua como um antagonista não-selectivo dos receptores
A1 e A2 da adenosina e como um inibidor da fosfodiesterase, aumentando a
libertação da DA dos terminais nervosos do núcleo estriado 592. O antagonismo
nos receptores 2A2 da adenosina leva a um aumento dos níveis de acetilcolina
no hipocampo, o que parece estar na base do efeito analgésico da cafeína. Por
outro lado, a MDMA também tem uma acção analgésica que parece envolver
um mecanismo serotonérgico a nível central (uma vez que o metisergide, um
antagonista não-específico dos receptores 5-HT1, 5-HT2 e 5-HT7, preveniu o
efeito analgésico da MDMA). Apesar de ambos os compostos terem uma acção
antinociceptiva, a sua co-administração a ratinhos não resultou numa
interacção sinérgica 593.
Quando ratinhos são expostos a 10 mg/kg de cafeína 20 minutos antes
de uma dose de 5 mg/kg de MDMA verifica-se uma potenciação da actividade
locomotora induzida pela MDMA, provavelmente devido ao bloqueio A2
induzido pela cafeína que vai potenciar a libertação de DA induzida pela MDMA 593. Um aumento dos níveis de DA no sistema mesolímbico pode precipitar os
movimentos voluntários, aumentar a motivação e o estado de alerta, reduzir o
apetite e induzir insónia. Esta redução de apetite poderá estar na base da
potenciação, pela cafeína, da perda de peso associada à exposição de ratos a
doses neurotóxicas de MDMA (20 mg/kg, 2 vezes por dia, durante 4 dias
consecutivos) 593. No entanto, o aumento da resposta hiper-locomotora não se

Introdução ______________________________________________________
116
verificou quando a cafeína e a MDMA foram administradas simultaneamente a
ratos 594.
A lesão neurotóxica clássica induzida pela MDMA em ratos,
caracterizada por uma redução da densidade dos SERT, é potenciada pela co-
exposição a MDMA e cafeína a qual também atrasa a recuperação dos
receptores 5-HT2 após a down-regulation induzida pela MDMA 593. Pode
especular-se que o aumento na libertação de DA induzido pela exposição a
cafeína + MDMA aumenta a disponibilidade da DA para ser transportada para o
interior dos terminais depletados em 5-HT, onde pode ser desaminada pela
MAO-B, levando à formação de espécies reactivas que destroem
selectivamente os terminais serotonérgicos 93.
Para além do já referido, em ratos, a cafeína (10 mg/kg) leva à
ocorrência de convulsões quando administrada com a MDA (7,5 mg/kg), tendo
demonstrado também aumentar profundamente a intensidade e a duração da
resposta hipertérmica associada à MDMA (15 mg/kg) e à MDA (7,5 mg/kg) 594.
No entanto, após administrações repetidas, a resposta hipertérmica à mistura
cafeína + MDMA desenvolveu tolerância, enquanto que a hipertermia
resultante da exposição a cafeína + MDA se manteve 594. Para além disso, 4
semanas após a última administração da mistura, a co-exposição a cafeína +
MDA induziu uma redução dos níveis de 5-HT e de ácido 5-hidroxindolacético
(5-HIAA) em várias áreas cerebrais, não se verificando o mesmo com a mistura
cafeína + MDMA, facto que sugere uma relação entre a manutenção dos níveis
cerebrais de 5-HT e o desenvolvimento de tolerância relativamente ao aumento
da hipertermia 594. De facto, sabe-se que a hipertermia desempenha um papel
preponderante na severidade da perda de 5-HT associada ao consumo de
anfetaminas 595. Este facto é consistente com o que se observou neste estudo
em que a hipertermia e a perda de 5-HT ocorreram de forma sequencial nos
animais expostos a cafeína + MDA. Por outro lado, o facto dos animais
expostos a cafeína + MDMA desenvolverem tolerência ao aumento da
hipertermia poderá ter contribuído para a menor depleção dos níveis de 5-HT
observada nestes animais 594.
A co-administração de uma única dose de cafeína (10 mg/kg s.c.) com
MDMA (10 mg/kg s.c.) induziu, em ratos, uma taquicardia profunda e

______________________________________________________ Introdução
117
persistente, mediada por um efeito simpaticomimético central, não observada
após administração de cada uma dos compostos em separado 596.
Se a interacção verificada no rato se generalizar ao humano, o padrão
de utilização que envolve a exposição simultânea a cafeína e anfetaminas
metilenodioxi-substituídas pode ter implicações agudas ou a longo termo,
sérias para os utilizadores de ecstasy 594.
Em termos farmacocinéticos, a cafeína (10 mM) parece não ter
capacidade para inibir in vitro a captação da MDMA (0.5 mM) pelas células do
epitélio do íleo, aumentando a permeação da MDMA do bordo apical para o
bordo basolateral de um modelo de células da parede intestinal (Caco-2), o que
leva a um aumento da AUC plasmática da MDMA quando co-administrada a
ratos com cafeína (50 mg/kg p.o.), relativamente à AUC obtida quando a
MDMA (10 mg/kg p.o.) é administrada isoladamente 578.
Provavelmente devido à confluência dos fenómenos descritos, a cafeína
(100 mg/kg i.p.) já mostrou aumentar a mortalidade após administração aguda
de anfetamina (15-42 mg/kg i.p), MDMA (15 mg/kg) e o seu principal metabolito
MDA (7,5 mg/kg) ou cocaína (35-90 mg/kg i.p.) a ratos 597. Este efeito na
letalidade da MDMA é independente da dose de cafeína. Por outro lado, a dose
de cafeína condiciona o aumento da mortalidade induzida pela MDA 594.
Assim, enquanto a cafeína é considerada como segura e livremente
acessível em bebidas como chá, café e bebidas não-alcoólicas, a sua ingestão
com MDMA pode ter o potencial para exacerbar a toxicidade aguda desta
anfetamina, constituindo um risco para a saúde dos utilizadores de ecstasy 594.
1.9.5.3.7. Paracetamol Aparentemente, o paracetamol incluído nas pastilhas
de ecstasy não se encontra em concentrações perigosas
(geralmente inferiores a 200 mg por pastilha), mesmo para
indivíduos que consumam várias pastilhas numa noite 373.
Por esse motivo, não existem ainda relatos de fatalidades associadas a
interacções entre as duas substâncias.

Introdução ______________________________________________________
118
No entanto, a oxidação do paracetamol pela CYP2E1 resulta na
formação do composto hepatotóxico N-acetil-p-benzoquinonimina (NAPQI).
Normalmente formada em pequenas quantidades, a NAPQI pode ser
destoxificada por conjugação com a GSH. No entanto, doses elevadas de
paracetamol ou uma pré-depleção da GSH (como a originada pela MDMA)
pode levar a um sinergismo de efeitos hepatotóxicos, agravando a lesão
hepática relativamente àquela que qualquer uma das substâncias causaria
isoladamente 598.
1.9.5.3.8. LSD
A dietilamida do ácido lisérgico (LSD) é uma das mais potentes
substâncias alucinogénicas conhecidas. Tendo em conta o pouco que se sabe
acerca do metabolismo e do mecanismo de acção da LSD, será de esperar que
ela desencadeie interacções farmacodinâmicas com a MDMA, potenciando o
seu efeito, sem que no entanto se observem interacções farmacocinéticas, uma
vez que o seu metabolismo parece não envolver o CYP450 (vide secção
1.9.5.4.4).
1.9.5.3.9. Dextrometorfano (DXM)
O dextrometorfano (DXM) é um agente antitússico que
foi já encontrado na constituição de pastilhas de ecstasy em
doses que variavam de 103 a 211 mg por pastilha, que
correspondem a doses consideravelmente superiores à dose
terapêutica habitual (15 a 30 mg, até 4 vezes por dia). Uma vez que o DXM é
aparentemente comum nas pastilhas de ecstasy e não é detectado por testes
rotineiros de rastreio de drogas de abuso, pode ter um papel negligenciado em
reacções adversas atribuídas à MDMA 372. De facto, doses elevadas de DXM
(superiores a 300 mg) podem causar letargia ou hiperexcitabilidade,
taquicardia, ataxia e nistagmo, bem como psicose do tipo da fenciclidina,
efeitos causados pela acção bloqueadora do receptor NMDA do seu metabolito

______________________________________________________ Introdução
119
dextorfano 372, 522. Para além do efeito ao nível dos receptores NMDA, o DXM
também actua a nível central como agonista dos receptores σ1 e σ2 523
(receptores sigma, produzem efeitos antidepressivos no ratinho e, nos
humanos, a sua activação leva a hipertonia, taquicardia, taquipneia e midríase;
a anfetamina é agonista nestes receptores), antagonista dos receptores
nicotínicos α3β4 524, inibidor da recaptação da 5-HT 525 e, possivelmente, da
DA. O DXM pode, assim, potenciar os efeitos da MDMA e a intoxicação por
DXM deve então ser considerada em pacientes que admitam consumo de
ecstasy e apresentem os referidos sintomas, particularmente se os rastreios
forem negativos para MDMA e anfetaminas.
Uma vez que quer a MDMA quer o DXM são substratos da CYP2D6 e,
uma vez que a MDMA e os seus metabolitos inibem a actividade de CYP2D6 28, a co-administração dos compostos pode levar à competição entre os dois
substratos para a enzima metabolizadora e resultar no aumento da incidência e
severidade dos efeitos adversos do DXM ou, eventualmente, no aumento dos
níveis de MDMA. No entanto, até à data, não foi descrita nenhuma intoxicação
desta natureza 23. Aliás, curiosamente, o DXM em altas doses confere
neuroprotecção, num efeito independente da metabolização do DXM a
dextrorfano 599.
1.9.5.3.10. Excipientes inertes
Como já referido, a velocidade de eliminação da MDMA está dependente
do pH dos fluidos de excreção como urina ou suor. De facto, quanto mais
ácidos forem esses fluidos, maior o grau de ionização da MDMA e maior a sua
eliminação por essas vias. Assim, a inclusão de bicarbonato de sódio nas
pastilhas de MDMA é feita com o intuito de prolongar os seus efeitos, uma vez
que, ao provocar uma alcalinização dos fluidos de excreção, conduzirá a um
atraso na eliminação deste derivado anfetamínico.

Introdução ______________________________________________________
120
1.9.5.4. Drogas de abuso ilícitas co-consumidas intencionalmente com MDMA
Os consumidores de ecstasy são frequentemente consumidores de
outras substâncias, combinando substâncias lícitas e ilícitas para aumentar os
efeitos esperados da MDMA, bem como para suavizar os sintomas de
abstinência 368. Como exemplo podem considerar-se os resultados do trabalho
desenvolvido por Schifano e colaboradores em 1998 no qual 95% dos
indivíduos consumidores de ecstasy entrevistados tinham consumido outra
droga de abuso pelo menos uma vez 219. Os estudos relativos ao co-consumo
de MDMA e outras substâncias estão compilados na Tabela 7.
Este fenómeno de co-consumo torna difícil concluir definitivamente
acerca dos efeitos tóxicos da MDMA a longo prazo e acresce o risco de
desenvolvimento de toxicidade 13. A intensidade do padrão de consumo de
MDMA altera a propensão dos indivíduos para consumir outras drogas como
cocaína, anfetamina, LSD, e cogumelos alucinogénicos (contendo psilocibina),
mas não altera o consumo de álcool, tabaco e Cannabis 332. Este facto foi já
verificado num estudo realizado em 2008 em Coimbra em que, num universo
de 223 utilizadores de MDMA, 11,7% era consumidor excessivo e, destes,
58,3% consumiam MDMA num padrão de policonsumo com outras drogas,
enquanto que consumidores de MDMA mais moderados tinham prevalências
de policonsumo inferiores 305. A ecstasy pode influenciar a utilização de outras
drogas, sobretudo cocaína e MTA. Aparentemente, o início do consumo de
ecstasy numa idade mais tardia parece diminuir o risco de consumir estas
drogas, embora não afecte o consumo de heroína 355.

121
Tabela 7. Padrões de policonsumo de drogas de abuso reportados por consumidores de ecstasy.
a Destes, 69% fazem binge drinking, ou seja, consomem mais de 5 bebidas standard por noite.
Cole et al. 370 2008
N = 80 (Reino Unido)
de Sola et al. 354 2008
N = 14 (Espanha)
Dresen et al. 309 2007
N = 247 (Alemanha)
Medina et al. 312 2007
N = 48 (EUA)
Yen et al. 313 2007
N = 200 (Taiwan)
Breen et al. 314 2006
N = 852 (Austrália)
Copeland et al. 316 2006
N = 216 (Austrália)
Estudo Associação % inquiridos % inquiridos % inquiridos % inquiridos % inquiridos % inquiridos % inquiridos
Tabaco
Cannabis 37,7 91,7 6,5 82
Etanol 93,2 93,8 57,5 65a
Cocaína 51,2 57,1 50 60 27
Anfetamina 19,8
MTA 57,1 25 4 83
Alucinogénios (ex.: LSD, PCP) 14,3 29,2 26
Ketamina 35,7 23
GHB 28,6 10
Opiáceos (ex.: heroína) 14,3 20,8 1 6
Sedativos (ex.: barbitúricos,
benzodiazepinas)
14,3 68,8 5 27 16
Cogumelos “mágicos” 50
Inalantes (“poppers”) 25
Solventes
Noz de Areca ou Betel 50
Nenhuma 14,6 70,5

Tabela 7 (cont.). Padrões de policonsumo de drogas de abuso reportados por consumidores de ecstasy.
b Percentagens de policonsumo descritas pelos utilizadores crónicos de MDMA;
Wish et al. 322 2006
N = 1206 (EUA)
Wu et al. 323 2006
N = 696 (EUA)
Wu et al. 324 2006
N = 19084 (EUA)
Barrett et al. 325 2005
N = 186 (Canadá)
Clatts et al. 326 2005
N = 569 (EUA)b
Degenhardt 452
2005 N = 852
(Austrália)
Liechti et al. 190
2005 N = 52 (Suiça)
Estudo Associação % inquiridos % inquiridos % inquiridos % inquiridos % inquiridos % inquiridos % inquiridos
Tabaco 82,1 34,6
Cannabis 98 91,4 97,4 68,5 73,8-86,6 19,2
Etanol 88,2 99,5 45,2 89,5-97,1 53,8
Cocaína 46 56,3 56,4 9,8 18,8-26,4 25
Anfetamina 31,1 36,5 48,4 14,7-20,3 19,2
MTA 1 60,5-70,6
Alucinogénios (ex.: LSD, PCP) 38 36,4 100 11,6-31,9 3,8
Ketamina 9,7 4 17,9-36,2 3,8
GHB 8,6 7,5-15,9 25
Opiáceos (ex.: heroína) 17 30,7 7,7 2,8-19,7 7,7
Sedativos (ex.: barbitúricos,
benzodiazepinas)
28,5 40,4 23,7-52,6 5,8
Cogumelos “mágicos” -
Inalantes (“poppers”) 38 45,1 13,0-31,9
Solventes
Noz de Areca ou Betel
Nenhuma 9,6
122

123
Tabela 7 (cont.). Padrões de policonsumo de drogas de abuso reportados por consumidores de ecstasy.
c Os valores indicados entre parêntesis indicam a percentagem de consumidores de MDMA que referiram a utilização da substância em causa na modulação das reacções desagradáveis que surgem durante a regressão dos efeitos da MDMA. d Destes, 70% consome etanol em doses consideradas perigosas. e Destes, 41% fazem binge drinking, ou seja, consomem mais de 5 bebidas standard por noite. f Média de 5 cidades: Coimbra, Modena, Nize, Palma de Mallorca, Utrecht.
Milani
et al. 600
2005 N = 59
(Reino Unido)
Lora-Tamayo et al. 331
2004 N = 70
(Espanha)
Scholey
et al. 332
2004 N = 763
(Internet)
Parrott
et al. 339 2001
N = 768 (Reino Unido e
Itália)
Winstock et al. 266
2001 N = 1.106
(Reino Unido)c
Topp et al. 342 1999
N = 329 (Austrália)c
Calafat et al. 601
1998 N = 801
(Europa)f
Estudo Associação % inquiridos % inquiridos % inquiridos % inquiridos % inquiridos % inquiridos %inquiridos
Tabaco 56 69,4 - 62 (54) 88.7
Cannabis 72,9 58,6 70 87,6 82 (82) 45 (64) 79.4
Etanol 79 80,4 88d (60) 40e (21) 67.8
Cocaína 55,7 56 73,1 58 (0,5) 37.6
Anfetamina 69 73,7 83 43 (7) 35.4
MTA
Alucinogénios (ex.: LSD, PCP) 60 66,3 30 13 31.7
Ketamina 14,2 14
GHB
Opiáceos (ex.: heroína) 23 25,5 (2) (5) 8.8
Sedativos (ex.: barbitúricos,
benzodiazepinas)
38 22,1 (18) (17)
Cogumelos “mágicos” 56 46,6 - 18.5
Inalantes (“poppers”) 39,1 51 12 (8)
Solventes 21 17,5
Noz de Areca ou Betel
Nenhuma - -

Tabela 7 (cont.). Padrões de policonsumo de drogas de abuso reportados por consumidores de ecstasy.
c Os valores indicados entre parêntesis indicam a percentagem de consumidores de MDMA que referiram a utilização da substância em causa na modulação das reacções desagradáveis que surgem durante a regressão dos efeitos da MDMA
Schifano et al. 219 1998
N = 150 (Itália)
Alvarez et al. 601 1997
N = 418 (Espanha)
Boys et al. 345 e Lenton et al. 344
1997 N = 83
(Austrália)
Williamson et al. 346
1997 N = 158
(Reino Unido)c
Webb et al. 348 1996
N = 3075 (Reino Unido)
Solowij et al. 22 1992
N = 100 (Austrália)
Estudo Associação % inquiridos % inquiridos % inquiridos % inquiridos % inquiridos % inquiridos
Tabaco 61.7 -
Cannabis 31 59.6 69,2 (55) 96,6 47
Etanol 39 63.6 19,2 30
Cocaína 33.7 -
Anfetamina 32.5 34,6 41
MTA
Alucinogénios (ex.: LSD, PCP) 24 31.3 26,9 19
Ketamina
GHB
Opiáceos (ex.: heroína) 6.7 -
Sedativos (ex.: barbitúricos,
benzodiazepinas)
7,7
Cogumelos “mágicos” - -
Inalantes (“poppers”) 34,6 7
Solventes
Noz de Areca ou Betel
Nenhuma 2.6 59 24
124

______________________________________________________ Introdução
125
O estudo do padrão de consumo de outras drogas de abuso pelos
consumidores habituais de MDMA 332 é consistente com a noção de limiares na
progressão do consumo de drogas desde as drogas legais, a Cannabis e seus
derivados e, posteriormente outras drogas ilícitas 602. Foi já descrito que o uso
recreativo de MDMA é normalmente precedido pelo consumo de etanol,
nicotina, Cannabis, e anfetamina, por esta ordem 602. A utilização de várias
drogas de abuso pode dever-se também a uma tolerância farmacodinâmica
apresentada por vários consumidores após o consumo regular de MDMA, que
alegam notar uma diminuição dos seus efeitos agudos após uso continuado, o
que aumenta a propensão para a utilização de outras substâncias psicoactivas
ou cocktails de drogas para obter experiências agradáveis 73, 603, 604. Algumas
drogas de abuso podem também afectar os efeitos da MDMA por alterarem a
expressão das isoenzimas do CYP450 398.
Comparado com outros policonsumidores de drogas, aqueles
policonsumidores que incluem MDMA na sua lista de consumos apresentam
dificuldade em adaptar-se a novos ambientes, em processar memórias
recentes, e em processos de julgamento social e emocional 605.
Foi também já descrito um caso de síndrome amnésica e ataxia severa
após consumo concomitante de MDMA, nitrato de amilo, LSD, Cannabis e
álcool 606.
Embora os consumidores de ecstasy demonstrem níveis elevados de
sintomas psicológicos e de disfunção executiva, estes sintomas não estão,
normalmente, estatisticamente associados com o consumo de ecstasy, mas
sim com o policonsumo de drogas que lhe está associado (álcool, Cannabis,
opióides e inalantes, etc) 312.
Drogas como a heroína parecem também potenciar a toxicidade da
MDMA, embora o mecanismo subjacente a este facto permaneça por
esclarecer 607.
O uso simultâneo de múltiplas drogas estimulantes não só aumenta o
risco de problemas relacionados com a sobre-estimulação simpaticomimética
com aumento da propensão para desidratação, hipertermia 296, 608 e
complicações cardiovasculares 182, mas pode também aumentar a
neurotoxicidade 266.

Introdução ______________________________________________________
126
1.9.5.4.1. Derivados da Cannabis
A maioria dos consumidores de ecstasy (90-98%) também consome
Cannabis 609. Tal como acontece com o etanol, o primeiro contacto com a
Cannabis é geralmente mais precoce do que o primeiro contacto com a MDMA 355. De facto, grande parte dos co-consumidores de ecstasy e Cannabis usou
derivados da Cannabis, mais ou menos regularmente, antes de iniciar o
consumo de MDMA e continua a consumir estes produtos em paralelo com o
uso de MDMA 332, 334, 610.
A Cannabis é sobretudo usada para ultrapassar os efeitos
desagradáveis que aparecem quando a euforia inicial provocada pela MDMA
se dissipa 611.
O principal componente psicoactivo da Cannabis e
seus derivados, o Δ9- tetraidrocanabinol (THC), é um agonista
directo dos receptores canabinóides CB1 endógenos,
afectando o apetite, a regulação da dor e as funções
vegetativa e motora.
O consumo regular de Cannabis e derivados é um factor de risco
conhecido para o desenvolvimento de patologias neuropsiquiátricas
(depressão, psicose e desmotivação) e défices de atenção e memória que
persistem após o desaparecimento dos efeitos agudos da droga e que podem
normalizar após semanas de abstinência 612.
A Cannabis também contribui para problemas psicológicos, deficiências
cognitivas e outros poblemas neuropsicobiológicos (perturbações do sono,
disfunção sexual, imunocompetência reduzida e aumento do stress oxidativo)
em utilizadores de MDMA 613. Em termos crónicos, ambas as drogas são
lesivas, pelo que os policonsumidores normalmente exibem défices
neurobiológicos cumulativos. 609. De facto, em humanos, o uso prolongado de
MDMA e Cannabis está associado com uma variedade de efeitos psicológicos,
incluindo aumento da impulsividade, ansiedade, queixas somáticas, padrões
obsessivo-compulsivos e comportamento psicótico. Défices neurocognitivos
(memória, aprendizagem, fluência verbal, velocidade de processamento e
destreza manual) em várias áreas cerebrais (hipocampo, lobo frontal) foram
reportados em utilizadores desta associação 614. Contudo, este assunto

______________________________________________________ Introdução
127
permanece controverso, uma vez que enquanto uns grupos defendem que o
principal responsável dos défices cognitivos associados ao consumo desta
mistura é a MDMA 615, outros grupos refutam esta hipótese e argumentam que
os défices cognitivos estão mais relacionados com o consumo de Cannabis do
que de MDMA 612. Algum do défice cognitivo atribuído à MDMA parece ser
devido ao consumo de THC 614, 616 bem como as queixas de sintomas
neurobiológicos 617. Ambas as drogas prejudicam a memória quando
administradas isoladamente, embora a deterioração devida à MDMA seja
inferior à causada pelo THC. Quando co-administradas, as drogas causam uma
deterioração sinérgica da memória de trabalho em ratos que se traduz numa
fraca capacidade de execução dos ratos no teste comportamental Y-maze 618.
Num estudo feito em ratinhos, a administração combinada de doses baixas de
MDMA (3 mg/kg) e THC (0,3 mg/kg) produziu “preferência de lugar
condicionada”, enquanto que a combinação de MDMA (10 mg/kg) com THC
(0,3 mg/kg) decresceu significantemente essa preferência. A administração de
doses baixas de THC aumentou significantemente os níveis de DA no nucleus
accumbens, enquanto que uma dose baixa de MDMA não o fez. Quando a
MDMA é administrada antes do THC, os níveis de DA aumentam relativamente
ao efeito do THC isolado. Estes resultados mostram que uma dose baixa de
THC modifica a sensibilidade dos animais aos efeitos comportamentais da
MDMA e que o THC e a MDMA convergem para um mecanismo comum que
modula o fluxo de DA no nucleus accumbens de ratinho 619.
Alguns estudos in vitro demonstraram neurotoxicidade associada a
concentrações elevadas de THC 620 contudo, a evidência de neurotoxicidade
em humanos nunca foi convincente 612. Em contraponto, vários estudos
atribuíram aos canabinóides propriedades antioxidantes, antiexcitotóxicas e
anti-inflamatórias a nível celular, bem como neuroprotecção numa variedade de
modelos de dano neuronal, quer in vitro, quer in vivo 621. Para além disso, um
estudo realizado em ratos demonstrou que quer o THC quer um agonista
sintético dos receptores canabinóides preveniram a hipertermia induzida pela
MDMA e diminuiram a depleção de 5-HT induzida pelo consumo prolongado de
MDMA em várias regiões do cérebro, bloqueando parcialmente a
neurotoxicidade induzida pela MDMA em animais de laboratório 621,
possivelmente através das propriedades antioxidantes do THC 612. Também foi

Introdução ______________________________________________________
128
já demonstrado que o THC previne completamente a agressão oxidativa da
MDMA, num efeito não inibido por um antagonista específico dos receptores
CB1 (o AM251) 55. Este resultado aponta para propriedades antioxidantes
inespecíficas do THC e não para uma interacção específica com os receptores
CB1. Devido a estas propriedades, o THC parece proteger dos efeitos
deletérios provocados pela oxidação da DA libertada em excesso devido ao
consumo de MDMA 55. O consumo moderado de Cannabis parece, assim,
melhorar ou mascarar a agressividade e os sintomas somáticos induzidos pela
MDMA. Contudo, o consumo destes compostos em grandes doses está
associado a vários problemas psicobiológicos que podem aparecer após um
período de abstinência das duas substâncias 600.
Assim, embora os efeitos agudos da Cannabis possam ser deletérios,
aparentemente os efeitos prolongados da Cannabis podem proporcionar
neuroprotecção contra os efeitos da MDMA e anfetaminas, uma vez que a
Cannabis tem propriedades relaxantes e hipotérmicas 613. A sua co-utilização
pode reflectir efeitos opostos em 3 áreas psicobiológicas: atenção, temperatura
corporal e stress oxidativo. Primeiro, a MDMA aumenta o estado de alerta
enquanto a Cannabis é sedante. Em segundo lugar, a MDMA causa
hipertermia enquanto a Cannabis causa hipotermia. Terceiro, a MDMA
aumenta o stress oxidativo, enquanto a Cannabis é antioxidante. Assim, a
Cannabis pode modular as reacções agudas e sub-agudas da MDMA, reduzir a
hipertermia e o stress oxidativo.
Os consumidores habituais de MDMA utilizam as propriedades
relaxantes da Cannabis e derivados para reduzir os sintomas típicos de
anedonia (incapacidade de sentir prazer em situações normalmente
prazenteiras) e depressão, que surgem nos dias após o consumo de ecstasy 622, 623. No entanto, a utlização de Cannabis pode provocar várias alterações
neurovasculares que podem resultar em eventos isquémicos que poderão
agravar a neurotoxicidade induzida pela MDMA 184.
As alterações endócrinas encontradas em utilizadores de MDMA
parecem também relacionadas com o co-consumo de Cannabis 611.
O consumo de MDMA com Cannabis provoca um decréscimo
significativo na IL-2 e um aumento no TGF-β1 (Transforming Growth Factor)
anti-inflamatório, juntamente com uma diminuição no número total de linfócitos

______________________________________________________ Introdução
129
(CD4+ e NK). Foi também observada uma maior frequência de infecções
ligeiras no grupo de consumidores regulares, quando comparados com os
consumidores ocasionais. Estas alterações, a longo prazo, da homeostase
imunológica pode resultar numa deterioração do estado de saúde geral e uma
maior susceptibilidade a infecções e a doenças do foro imunológico 624.
Apesar destes dados, a contribuição da utilização de Cannabis para os
efeitos prolongados da MDMA permanece inconsistente 612.
Resumindo, existem interacções importantes e complexas entre a
MDMA e a Cannabis no que diz respeito à actividade cerebral, psicopatologia e
aprendizagem 612 que se estendem também aos sistemas endócrino e
imunológico.
Neste contexto de policonsumo é preciso ter ainda em consideração a
teoria “portal de entrada” ou “oportunidade de exposição” que sugere que a
utilização de Cannabis leva ao consumo de outras drogas 625, 626 e que
complica ainda mais as possibilidades de interacções com a MDMA. De facto,
foi já demonstrado que as mulheres consumidoras de Cannabis têm mais
predisposição para utilizarem outras drogas, uma vez que a Cannabis pode ser
um ponto de partida para mulheres que estejam interessadas em experimentar
drogas de abuso, uma vez que é mais facilmente obtida e mais aceite
socialmente 334.
As interacções farmacocinéticas entre a MDMA e a Cannabis são ainda
possíveis por partilha de algumas vias metabólicas, uma vez que o
componente activo da Cannabis e seus derivados (THC) é metabolizado pela
CYP3A4 e pela CYP2C9 627.
1.9.5.4.2. Estimulantes anfetamínicos
Os estimulantes anfetamínicos também são neurotóxicos para as vias
serotonérgicas e dopaminérgicas, num mecanismo semelhante ao da MDMA
Estes estimulantes mostraram ser neurotóxicos em estudos com animais 250 e
possivelmente em humanos 612.
A MDMA e a MTA têm acções semelhantes no cérebro, aumentando os
níveis das monaminas neurotransmissoras DA, NA e 5-HT. A principal

Introdução ______________________________________________________
130
diferença é que a MDMA inibe sobretudo a recaptação e estimula a libertação
da 5-HT, enquanto a MTA inibe sobretudo a recaptação e estimula a libertação
da DA. Ambas as anfetaminas aumentam ainda os níveis destes
neurotransmissores por inibirem as enzimas responsáveis pela sua
metabolização e através da expulsão das monoaminas das vesículas
sinápticas. Esta libertação massiva de monoaminas vai levar a um défice
temporário nas suas reservas que podem necessitar de alguns dias para serem
repostas 133.
Estes estimulantes também se ligam aos transportadores pré-sinápticos
das monoaminas, induzem a libertação aguda e inibem a recaptação de DA e
5-HT. Podem, assim, exercer efeitos tóxicos sinérgicos com a MDMA, e
prolongar os seus efeitos deletérios de longa duração 612. Apesar das
alterações das funções cerebrais dos utilizadores serem de longa duração, a
literatura sobre as sequelas psicopatológicas de longa duração que resultam do
uso de estimulantes é inconclusiva 612.
O consumo repetido e/ou de grandes quantidades de anfetamina ou
MTA induzem uma degeneração generalizada dos axónios pré-sinápticos
serotonérgicos e dopaminérgicos levando a uma depleção de 5-HT e DA e a
uma diminuição da densidade dos seus transportadores (SERT e DAT) no
tecido cerebral 250, 628, 629. Consequentemente, os utilizadores crónicos de
estimulantes demonstraram défices na memória de curto prazo, controlo
executivo frontal e capacidades de planificação 612.
Assim, o co-consumo de MDMA e outros estimulantes anfetamínicos
pode aumentar a neurotoxicidade serotonérgica e os efeitos tóxicos para o
sistema dopaminérgico induzidos pela MDMA 612. Ambas as substâncias
MDMA e MTA provocam fenómenos de neurotoxicidade que, muito
provavelmente, são causados pela formação de ROS e RNS formados devido
ao metabolismo das monoaminas libertadas em excesso, ou em resposta ao
esgotamento das fontes de energia. Uma vez que ambas provocam este efeito,
combinar as duas substâncias pode produzir um padrão mais amplo de
depleção de NA e 5-HT do que o observado quando doses equivalentes são
administradas separadamente. As anfetaminas podem ainda interferir nos
efeitos da MDMA, uma vez que um grande número destes compostos são
substractos do transportador vesicular das monoaminas tipo 2 (VMAT2),

______________________________________________________ Introdução
131
depletando os neurotransmissores vesiculares por um mecanismo mais
relacionado com uma troca mediada pelo transportador do que devido às suas
propriedades de bases fracas 630. Estes factos sugerem que a ingestão
simultânea de MDMA e MTA pode ser particularmente perigosa 133.
Uma vez que, quer a MDMA, quer a MTA têm sido individualmente
associadas a efeitos adversos neuronais e comportamentais a longo prazo, é
concebível que a combinação das duas substâncias, intencional ou inadvertida,
possa ser particularmente perigosa 133. De facto, a combinação de MDMA e
MTA em ratos tem tendência também a produzir uma maior variedade e
intensidade de défices comportamentais (ansiedade social, crises de ansiedade
e sintomas depressivos) que a exposição individual 133.
A auto-administração de ambas as substâncias por animais de
laboratório é aumentada em condições de temperatura ambiente elevada, o
que pode explicar o elevado consumo destas substâncias em ambientes
quentes e sobrelotados 133. Uma vez que a temperatura ambiente influencia
largamente os efeitos tóxicos da MDMA, este maior consumo em altas
temperaturas é extremamente preocupante. A depleção de 5-HT induzida pela
MDMA pode inicialmente atrasar a aquisição de auto-administração de
anfetamina, no entanto, a pré-exposição à MDMA pode também sensibilizar os
animais para os efeitos locomotores e para o efeito da MDMA na inicialização
de comportamentos de procura de droga, aumentando a probabilidade de uma
reinstalação da auto-administração de anfetamina após um período de
abstinência 631.
Surpreendentemente, apesar das combinações de MDMA com MTA
(intencionais ou não-intencionais devido à pureza das pastilhas) serem
elevadas entre os utilizadores, uma pesquisa recente sugere que a
administração combinada de MDMA diminui o efeito de reforço positivo da MTA
em ratos testados no paradigma de auto-administração 632.
Tal como a MDMA, a anfetamina induz hipertermia dependente da
temperatura ambiente 633, embora a afinidade destas drogas para os diferentes
transportadores das monoaminas apresente algumas diferenças 634. De facto,
estudos prévios destacaram algumas diferenças nos efeitos ao nível do
sistema termorregulador da MDMA e da anfetamina ou seus derivados 635. Por
isso, outra interacção que será de esperar é ao nível da resposta hipertérmica

Introdução ______________________________________________________
132
induzida por ambas as substâncias que, quando administradas em conjunto,
deverão causar um efeito sinérgico a este nível.
Foram já descritas intoxicações fatais associadas ao consumo
simultâneo de MDMA, anfetamina, MDA e PMA que se caracterizaram por um
quadro de coagulação intra-vascular disseminada induzida por hipertermia 282.
A pré-exposição a MDA tem ainda a capacidade de inibir o metabolismo
da MDMA, aumentando as suas concentrações a nível cerebral 533.
1.9.5.4.3. Cocaína
A cocaína é frequentemente consumida com a MDMA para aumentar os
efeitos psicoestimulantes da MDMA 636.
Esta droga aumenta os níveis sinápticos de DA, 5-HT e NA por se ligar
aos transportadores destes neurotransmissores e inibir a sua recaptação para
os terminais pré-sinápticos 636.
A pré-exposição repetida de ratos adultos à MDMA mostrou aumentar a
actividade motora induzida pela cocaína e promover a subsequente aquisição
do comportamento de auto-administração de cocaína, o que demonstra os
efeitos cruzados destas duas substâncias 637.
Por outro lado, a administração simultânea de MDMA e cocaína resultou
num antagonismo da “preferência de lugar condicionada” de cada uma das
substâncias. No entanto, este efeito parece ser reversível em doses elevadas
dos compostos em causa 638.
Foram já descritas diferenças nos perfis de sensibilização
psicoestimulante relacionados com a idade, quer para a cocaína, quer para as
anfetaminas 406, 639. Apesar dos ratos adolescentes mostrarem um aumento no
reforço condicionado da cocaína após tratamento com MDMA 408, 640, um
estudo recente sugere que eles são menos vulneráveis à sensibilização
induzida pela MDMA quando administrada numa dose alta e num curto
intervalo de tempo 640.
A toxicidade dopaminérgica induzida pela MDMA em ratinhos pode ser
afectada pela co-administração de cocaína 636. Por outro lado, a administração
repetida de MDMA a ratos aumenta a libertação de DA no nucleus accumbens

______________________________________________________ Introdução
133
induzida pela cocaína. Este facto sugere que a MDMA pode alterar a
vulnerabilidade para desenvolver dependência e abuso da cocaína 641. Estudos
sugerem que a neurotoxicidade da MDMA requer um sistema serotonérgico 141
e dopaminérgico intacto e que a extensão da toxicidade pode aumentar se os
níveis de dopamina estão aumentados 228. Este aumento nos niveis
extracelulares de DA pode verificar-se após utilização de cocaína, pelo que
este estimulante pode constituir uma ameaça dupla de complicações
fisiológicas e neurotoxicológicas 266.
A cocaína é inibidora da recaptação da 5-HT e comprovadamente
inibidora do metabolismo da MDMA in vitro 422, podendo aumentar
substancialmente as concentrações de MDMA in vivo, sobretudo se consumida
em conjunto com outras substâncias que sejam potentes inibidores da
CYP2D6. A cocaína pode ter efeitos semelhantes aos induzidos pela fluoxetina
e paroxetina no que respeita às interacções com a MDMA 422.
O abuso de cocaína foi já associado ao aparecimento de acidentes
cerebrovasculares isquémicos e hemorrágicos, atrofia cerebral em caso de
abuso crónico, e a um aumento da incidência de malformações congénitas 184
daí que possa agravar as propriedades neurotóxicas e teratogénicas da
MDMA.
Tal como a MDMA, a cocaína induz hipertermia dependente da
temperatura ambiente 642, 643, embora a afinidade destas duas substâncias para
os diferentes transportadores das monoaminas não seja comparável, quer em
humanos quer em roedores 634.
1.9.5.4.4. LSD
A LSD (dietilamida do ácido d-lisérgico) foi sintetizada
por primeira vez em 1938 pela indústria farmacêutica Sandoz
(Suiça) a partir do ácido lisérgico que existe naturalmente num
fungo (Claviceps pupurea) que cresce nos grãos de trigo e
centeio, na procura de um agente com actividade oxitócica.
No entanto, como apresentava propriedades oxitócicas
inferiores a outros compostos, a sua síntese foi abandonada. As suas

Introdução ______________________________________________________
134
propriedades alucinogénicas foram descobertas pelo mesmo
cientista que a sintetizou pela primeira vez, devido a uma
ingestão acidental. A LSD consegue provocar alterações
comportamentais com dosagens de 25-50 ng 644.
A co-administração da LSD com MDMA tem vindo a
aumentar e a combinação do consumo destas duas
substâncias recebe o nome de candyflipping ou XL. Várias
páginas web referem que o uso simultâneo leva a um aumento dos efeitos
centrais destas substâncias. E, de facto, foi verificado que em ratos Fawn-
Hooded machos (uma estirpe com deficiência cerebral em 5-HT) a co-
administração de LSD (0,04 mg/kg i.p.) e MDMA (0,15 mg/kg i.p.) resultou num
sinergismo que originou uma resposta máxima do tipo da verificada para a
MDMA numa dose de 1,5 mg/kg (i.p.). Este sinergismo ocorre provavelmente
por um mecanismo que envolve os neurónios serotonérgicos centrais 645.
Aparentemente a LSD é agonista dos receptores pós-sinápticos 5-HT2 e
é antagonista parcial dos receptores pré-sinápticos 5-HT1A e 5-HT1B, tendo já
mostrado também actividade em receptores 5-HT clonados. Teoricamente, a
LSD produz libertação de glutamato nos terminais tálamo-corticais, produzindo
uma dissociação entre os centros sensoriais e a capacidade cortical devido a
uma modulação dos processos sensoriais, perceptivos, afectivos e cognitivos
mediados pelos receptores NMDA. Contudo, desconhece-se de que forma é
que o agonismo nos receptores 5-HT2C contribui para as alterações
comportamentais 644.
Assim, a combinação do aumento dos níveis de 5-HT sinápticos,
capazes de actuar em todos os receptores envolvidos nas propriedades
estimulantes da LSD e da MDMA parece ser o mecanismo subjacente à acção
sinérgica destas duas substâncias 645.
Uma vez que os efeitos tóxicos agudos deste composto também
englobam sinais cardiovasculares (taquicardia, hipertensão), rabdomiólise e
falência renal estes podem agravar a resposta tóxica da MDMA 644.
A LSD é metabolizada a compostos inactivos (N-desmetil-LSD, hidroxi-
LSD, 2-oxo-LSD, e 2-oxo-3-hidroxi-LSD) não tendo sido descrito metabolismo
via CYP450, daí que não será de esperar que ocorram interacções
farmacocinéticas com a MDMA.

______________________________________________________ Introdução
135
No entanto, a LSD parece induzir tolerância cruzada à mescalina, pelo
que se pode esperar que o mesmo ocorra relativamente à MDMA, uma vez que
é estruturalmente semelhante.
1.9.5.4.5. Ketamina
O uso recreativo da ketamina foi reportado pela primeira vez em 1971 na
América do Norte e apareceu no Reino Unido nos anos 90 associado à cultura
dance gay 423. Em termos recreacionais, a ketamina é frequentemente
consumida por via nasal na forma de pó (100-400 mg por dose) (conhecida
como K, Super K, Special K, Vitamina K, Green, Mean Green, kit-kat, keets,
super acid, cat valiums e Jet), mas é também comercializada para ser fumada
ou na forma de spray nasal ou de líquido, pó ou cápsulas para ingestão (350-
500 mg por dose) 423.
De acordo com o já referido no sub-capítulo 1.9.5.3.3., o consumo de
ketamina pode ainda indirectamente afectar a toxicidade associada ao
consumo de outras substâncias, incluindo a MDMA, devido ao facto de
provocar amnésia, dificultando o controlo do número de doses consumidas
durante a noite e aumentando a probabilidade de ocorrência de uma
sobredosagem e intoxicação.
O consumo simultâneo da ketamina com outras drogas de abuso deve
ser cuidadosamente considerado, uma vez que as mortes causadas pelo
consumo de ketamina em Nova Iorque foram todas associadas ao consumo de
múltiplas substâncias (sobretudo opióides e anfetaminas) e não ao consumo de
ketamina per se 646. O risco de super-estimulação cardíaca é uma das
consequências potencialmente fatais de combinar ketamina com estimulantes
(ex.: anfetaminas, cocaína).

Introdução ______________________________________________________
136
1.9.5.4.6. GHB (“ecstasy líquido”)
O GHB (γ-hidroxibutirato, oxibato de sódio, G, ecstasy
líquido, Grievous Bodily, harm, gib, sabão, scoop, nitro) é um
potente agente hipnótico/sedativo derivado do
neurotransmissor inibitório ácido γ-aminobutírico e tem a capacidade de
atenuar os efeitos desagradáveis ou disfóricos da MDMA devido ao seu efeito
no sistema dopaminérgico a nível central 647. O GHB é um pó que se dissolve
em água e é vendido como droga recreativa 648 por 5 a 10 USD a dose. No
início dos anos 90 era também vendido em lojas de alimentação saudável
como suplemento para atletas e culturistas 648. A probabilidade de ocorrência
de sobredosagem é elevada porque a concentração da solução é muitas vezes
desconhecida (em 2000 eram já 60 as mortes provocadas por sobredosagem
deste composto 14). O sabor desagradável da solução de GHB é muitas vezes
mascarado com bebidas alcoólicas, o que aumenta a toxicidade do GHB,
devido ao facto do etanol ser também um depressor central 14.
Apesar de, em 2000, ter sido classificada como uma substância de
Classe I devido ao elevado número de mortes e ao facto de ser considerada
uma “droga de violação”, em 2002, o GHB, na forma de oxibato de sódio,
passou a fazer parte da Classe III, sendo utilizado para o tratamento da
narcolepsia e estando a ser estudada a sua utilização na cessação alcoólica 649, 650.
O GHB produz euforia, progredindo em doses mais altas para tonturas,
hiper-salivação, hipotonia, amnésia 14, sedação profunda, coma e mesmo
morte que surge, habitualmente, devido a depressão do sistema respiratório 651.
Interacções metabólicas com a MDMA parecem pouco prováveis, uma
vez que os dois compostos não partilham vias metabólicas.

______________________________________________________ Introdução
137
1.9.5.4.7. Outras substâncias alucinogénicas – Cogumelos “mágicos”
Alguns cogumelos possuem propriedades alucinogénicas devido ao seu
conteúdo em psilocibina. No entanto, alguns autores defendem que a
psilocibina não é o componente activo destes cogumelos, mas sim que ela é
convertida no seu metabolito activo psilocina por hidrólise do grupo fosfato,
quimicamente (pH ácido) ou numa reacção catalizada por fosfatases. A
psilocibina sofre, assim, um forte efeito de primeira passagem a nível hepático
e a sua principal via de eliminação é a eliminação urinária do seu conjugado
com o ácido glucurónico 652.
As reacções mais comuns após ingestão oral incluem relaxamento
muscular, arrefecimento dos braços e abdómen, dilatação das pupilas. À
medida que o efeito fica mais forte podem aparecer alterações de humor. Se a
dose for suficientemtente elevada, os efeitos podem incluir distorções visuais e
auditivas e alucinações.
A psilocibina/psilocina interage sobretudo com os receptores da 5-HT (5-
HT1A, 5-HT1D, 5-HT2A e 5-HT2C). Liga-se com grande afinidade aos receptores
5-HT2A pré-sinápticos e, em menor extensão aos 5-HT1A. A psilocibina e o seu
metabolito activo, psilocina, não apresentam afinidade para os receptores D2
da dopamina, mas levam à sua activação através de uma acção
simpaticomimética com aumento de libertação de neurotransmissores para a
fenda sináptica (ao contrário do que acontece com a LSD) 652.
Devido ao seu mecanismo de acção, a psilocibina/psilocina tem um forte
potencial de interacção com a MDMA.
1.9.5.5. Tabaco
O tabaco é preparado através do processamento de
folhas das plantas do género Nicotiniana.
O tabaco tem a capacidade de induzir significativamente,
e independentemente do sexo do indivíduo, algumas

Introdução ______________________________________________________
138
isoenzimas do sistema CYP450 como a CYP1A2 398 e a
CYP2E1 e, ainda, a uridina difosfato
glucuronosiltransferase, resultando esta última indução
numa mais rápida glucuronidação de algumas substâncias 653. Estas propriedades indutoras do tabaco eram
inicialmente atribuídas aos hidrocarbonetos aromáticos policíclicos (ex.:
benzo[a]pireno) e outros produtos de combustão. No entanto, a nicotina per se
parece induzir in vivo a CYP2E1 e inibir in vitro a CYP2A6, principal enzima
envolvida no metabolismo da nicotina a cotinina 653. Outras enzimas envolvidas
no metabolismo da nicotina são a CYP2B6 e a CYP2E1 653.
O mecanismo de acção da nicotina passa pela sua
ligação aos receptores nicotínicos que levam à libertação de
DA, NA, acetilcolina, 5-HT, GABA, glutamato e endorfinas 653,
o que pode estar na base de interacções farmacodinâmicas
que potenciem os efeitos da MDMA.
Devido ao papel importante dos receptores nicotínicos contendo
subunidades β2 na adição à nicotina 654 e devido à elevada afinidade da MDMA
para receptores contendo essa subunidade, pode esperar-se que a MDMA
module processos de adição e dependência à nicotina 55.
O rápido desenvolvimento de tolerância aos efeitos da nicotina faz com
que os utilizadores rapidamente aumentem as doses consumidas para manter
o efeito desejado e evitar os sintomas de abstinência. Quanto maior a dose de
nicotina a que o fumador se expõe, maiores serão, potencialmente, as
possibilidades de interacção com a MDMA.
O problema desta interacção é uma questão importante, uma vez que os
consumidores de ecstasy apresentam uma maior prevalência de fumadores
(quando comparados com consumidores de Cannabis, ou não consumidores
de drogas) e têm um início do consumo de tabaco mais precoce que se situa,
em termos médios, nos 14,3 anos de idade 354.

______________________________________________________ Introdução
139
1.9.5.6. Etanol
Uma das substâncias mais fequentemente
consumidas com a MDMA é o etanol (EtOH) (65% dos
consumidores habituais de ecstasy consomem
simultaneamente etanol 314), uma vez que é facilmente acessível e legal na
maioria dos países 331, 332 e o seu consumo é encorajado por fortes medidas
publicitárias empreendidas por produtores e distribuidores nos ambientes onde
habitualmente se consome ecstasy (discotecas, nightclubs, afterhours, etc).
Embora os dados disponíveis sobre o consumo combinado de drogas e
de álcool continuem a ser limitados, o consumo de álcool, muitas vezes em
quantidades consideradas perigosas para a saúde e em combinação com
drogas estimulantes, constitui um motivo de preocupação crescente 20.
Dos consumidores de ecstasy que bebem EtOH, uma grande parte fá-lo
num padrão agudo chamado binge drinking (ex.: 69% dos utilizadores de
MDMA que co-consomem EtOH referiram consumir mais de 5 bebidas por
episódio 314).
A utilização de EtOH sob a influência de drogas estimulantes leva a que
se consumam maiores quantidades de etanol sem que se sintam efeitos
imediatos. Contudo, apesar da ausência de efeitos, os riscos fisiológicos e
comportamentais associados a este consumo podem ainda verificar-se 314.
O consumo quer de MDMA quer de EtOH foi já associado a uma
elevada prevalência de comportamentos de risco como consumo de doses
excessivas de ambas as substâncias, promiscuidade sexual sem medidas de
protecção 303, condução sob a influência de drogas e viajar com condutores
embriagados 314. São bem conhecidos episódios de comportamento violento
muitas vezes relacionados com o consumo de álcool 655, bem como uma maior
prevalência de acidentes e lesões entre aqueles que consomem EtOH em
maior quantidade (binge drinkers) 314. Estes comportamentos de risco são
agravados quando ambas as substâncias são consumidas simultaneamente.
O EtOH parece contribuir numa maior proporção para comportamentos
sexuais de risco do que qualquer outra substância 656. Entre outras
consequências, o aumento de práticas sexuais perigosas quando o EtOH é

Introdução ______________________________________________________
140
consumido com ecstasy resulta no aumento da disseminação de doenças
sexualmente transmissíveis 314.
Normalmente, o primeiro contacto com o álcool ocorre antes do início do
consumo de ecstasy 657, sendo a idade média de início do consumo de EtOH
de 14,5 anos 303, 336, 355. Este facto pode levar à ocorrência de fenómenos de
tolerância ao EtOH antes do primeiro contacto com a MDMA que podem estar
na base de consumos de quantidades ainda maiores de bebidas alcoólicas,
bem como de susceptibilidades aumentadas do organismo aos efeitos
deletérios da MDMA. É importante também referir que dentro de uma mesma
população de estudo, aqueles que consomem ecstasy tiveram um primeiro
contacto com álcool mais precoce (14,8 anos) do que os não-consumidores de
MDMA (16,5 anos) 354 e que o uso precoce de EtOH está associado com um
uso precoce de cocaína, psilocibina, LSD e MDMA, sugerindo o seu potencial
para funcionar como droga “de entrada” 336.
Foi já descrito que as interacções dos estimulantes anfetamínicos com o
EtOH levam a interacções farmacodinâmicas relevantes e a ligeiras interacções
farmacocinéticas 23. Por este motivo, torna-se importante analisar as principais
características farmacocinéticas e farmacodinâmicas do etanol para se poder
inferir acerca do seu potencial para interagir com a MDMA, o que corresponde
ao objectivo da presente dissertação.
1.9.5.6.1. Características farmacocinéticas do etanol
1.9.5.6.1.1. Absorção
Em jejum, o álcool ingerido é absorvido maioritariamente no duodeno e
jejuno devido à rápida passagem pelo estômago vazio e à elevada superfície
de absorção que existe nesta zona pela presença de microvilosidades nas
células da mucosa. Nestas condições, o tempo para alcançar a concentração
plasmática máxima varia entre os 30 e os 90 minutos. Os alimentos sólidos e
as bebidas hipertónicas atrasam o esvaziamento gástrico. Nestas condições,
uma quantidade considerável de álcool pode ser absorvido no estômago (até
70% do álcool ingerido) e a concentração plasmática máxima pode ser atingida

______________________________________________________ Introdução
141
com um atraso de 4 a 5 horas 658. Esta absorção mais lenta confere maior
relevância ao efeito de primeira passagem, uma vez que o atraso no
esvaziamento gástrico provocado pela presença de alimentos faz com que
exista menos EtOH no duodeno e jejuno, a acção da álcool desidrogenase
(ADH) gástrica seja mais efectiva, menos EtOH atinja o fígado (uma vez que o
sangue drenado do tracto GI é encaminhado ao fígado através do sistema
porta, sendo depois enviado ao coração e finalmente à circulação sistémica),
resultando numa menor passagem de álcool para a corrente sanguínea. Para
doses de álcool baixas ou moderadas, o fenómeno de primeira passagem é o
principal factor responsável por uma maior ou menor quantidade e rapidez com
que o álcool atinge a circulação sistémica e o cérebro 659.
Assim, a concentração plasmática máxima e o tempo necessário para
atingi-la variam amplamente entre indivíduos e dependem de vários factores: a
quantidade de EtOH consumido, a velocidade do consumo, o tipo de bebida
(cerveja, vinho, bebidas espirituosas, cocktails) e, sobretudo a velocidade de
esvaziamento gástrico 658. Quando a ingestão de álcool é feita muito
rapidamente e em grande quantidade, os níveis cerebrais de EtOH aumentam
rapidamente, activando o reflexo fisiológico do vómito 658.
1.9.5.6.1.2. Distribuição
O EtOH distribui-se na água total, o que faz com que, em situação de
equilíbrio (que se atinge até às 2 horas após o consumo), as concentrações de
EtOH a nível extra- e intra-celular sejam aproximadamente iguais. Contudo,
este equilíbrio atinge-se mais rapidamente nos órgãos com maior
vascularização (ex.: cérebro, fígado, pulmões e rins). O volume de distribuição
do EtOH é influenciado pela percentagem de água e de gordura corporal.
Apesar da percentagem de água corporal ser relativamente constante nos
humanos (67 a 77%) existem situações que podem modificá-lo como: a fase do
ciclo menstrual, a gravidez, edemas ou ascites. A gordura corporal apresenta
uma grande variabilidade interindividual (5 a 50%), por exemplo entre os
géneros, com uma maior percentagem nas mulheres do que nos homens 659.
Por estas razões, indivíduos mais corpulentos ou com maior quantidade de

Introdução ______________________________________________________
142
água corporal, vão apresentar concentrações sanguíneas de EtOH inferiores
aos indivíduos mais magros (que consumiram a mesma dose de EtOH no
mesmo intervalo de tempo) porque nestes o EtOH ingerido se distribui num
menor volume. O volume aparente de distribuição é aproximadamente de 30 L /
70 kg 660. A taxa de ligação às proteínas plasmáticas é nula 658.
1.9.5.6.1.3. Metabolismo
Em condições fisiológicas, aproximadamente 90% do álcool ingerido é
metabolizado pela ADH (Km para o etanol de 2 a 5 mg/dL) a acetaldeído,
acompanhado da redução do NAD+ a NADH 658 (Figura 8).
A ADH é uma enzima citosólica presente em vários tecidos como o
fígado (onde existem os níveis mais elevados) o rim, o pulmão e a mucosa
gástrica. A ADH humana é uma proteína dimérica constituída por 2
subunidades que podem ser do tipo α, β, γ, π, χ, ou de um sexto tipo conhecido
por σ or µ. Conhecem-se mais de 20 isoenzimas ADH que diferem nas suas
propriedades farmacocinéticas e estão agrupadas em 5 classes diferentes 661.
Para o metabolismo do EtOH e de outros álcoois alifáticos de cadeia
curta as isoenzimas mais importantes pertencem à classe I que engloba as
isoenzimas ADH1, ADH2 e ADH3 formadas pela associação aleatória das
subunidades α, β e γ, respectivamente. Estas isoenzimas da classe I da ADH
estão expressas em grande quantidade no fígado e glândulas supra-renais, e
em baixa quantidade no rim, pulmão, vasos sanguíneos (no caso da ADH2) e
noutros tecidos, mas não no cérebro 662.
O metabolismo do EtOH dependente da ADH ocorre principalmente no
fígado e é menos importante na mucosa gástrica (onde predomina a classe IV
da ADH) 658. No entanto, a ADH3 presente no fígado e no estômago tem um
papel pouco relevante na metabolização do EtOH quando este se encontra em
concentrações inferiores a 50 mM no fígado, mas pode ter um papel importante
no efeito de primeira passagem do EtOH a nível gástrico, onde o EtOH pode
atingir concentrações molares 661.

______________________________________________________ Introdução
143
Figura 8. Metabolismo oxidativo do etanol. As enzimas álcool desidrogenase (ADH), citocromo P450 2E1 (CYP2E1), e catalase contribuem para o metabolismo oxidativo do álcool. A ADH, presente no citosol, transforma o etanol em acetaldeído. Esta reacção envolve um intermediário portador de electrões, dinucleótido nicotinamida adenina (NAD+), que é reduzida por dois electrões para formar NADH. A catalase, localizada nos peroxissomas requer peróxido de hidrogénio para oxidar o etanol. A CYP2E1, presente predominantemente nos microssomas, assume um papel importante na metabolização do etanol a acetaldeído quando o etanol está presente em concnetrações elevadas. O acetaldeído é metabolizado, sobretudo, pela aldeído desidrogenase 2 (ALDH2) na mitocôndria para formar acetato e NADH. ROS, espécies reactivas de oxigénio. RNS, espécies reactivas de azoto. Adaptado de Klaassen C.D., “Casarett & Doull's Toxicology: The Basic Science of Poisons”, McGraw-Hill, 2001 e de Zakhari S., “Overview: how is alcohol metabolized by the body?”, Alcohol Research & Health (2006) 29:245-54.
Os restantes 10% do metabolismo do álcool dependem de enzimas do
CYP450 e da catalase, podendo ocorrer no fígado (onde as enzimas do
CYP450 constituem o sistema microssomal de oxidação do etanol MEOS,
Microsomal Ethanol Oxidising System), mas também em tecidos extra-
hepáticos que não contêm ADH, como o cérebro. O sistema microssomal
hepático MEOS, converte o EtOH em acetaldeído, acompanhado da conversão
de NADPH em NADP+ e de O2 em água 663. Esta via metabólica adquire
especial importância em situações de ingestão excessiva de álcool. A
isoenzima do CYP450 maioritariamente implicada é a CYP2E1 (com Km para o
etanol de 60 a 80 mg/dL, superior ao da ADH) 398, embora outras também
possam contribuir para o metabolismo do álcool, como a CYP1A2 e CYP3A4 659 658, 664. Para além disso, a actividade da CYP2E1 aumenta após uma
exposição continuada a grandes doses de EtOH 665 devido a uma rápida

Introdução ______________________________________________________
144
síntese de novo da enzima que leva ao desenvolvimento de tolerância
metabólica provocada por um aumento de 2 ou 3 vezes da taxa de eliminação
do EtOH da corrente sanguínea 658, 666. A existência deste sistema enzimático e
a sua capacidade de ser induzido, bem como a indução cruzada de outras
enzimas microssomais, pode explicar os fenómenos de tolerância metabólica
ao álcool pelos alcoólicos e a tolerância a muitos fármacos, mas também a
particular susceptibilidade dos alcoólicos aos efeitos adversos de solventes
industriais, anestésicos, fármacos de uso corrente, analgésicos não sujeitos a
receita médica, agentes hepatotóxicos, carcinogénios e mesmo factores
nutricionais como a vitamina A 658, 667, 668, uma vez que aumenta a bioactivação
de numerosos xenobióticos com formação dos respectivos metabolitos tóxicos 669. Esta indução enzimática leva a um maior gasto energético e a um aumento
na produção de acetaldeído 667 que é altamente tóxico para o organismo,
mesmo em baixas concentrações 670.
Outra enzima, a catalase, localizada nos peroxissomas é capaz de
oxidar o EtOH em presença de peróxido de hidrogénio. Esta é considerada
uma via minoritária de oxidação do EtOH, excepto quando este é consumido
em jejum. Este facto deve-se a que em jejum a manutenção das reservas
energéticas do organismo é assegurada pela mobilização dos depósitos
lipídicos e β-oxidação dos ácidos gordos, quando na forma de derivados acil-
coenzima A. Os compostos acil-coenzima A de cadeia longa sofrem β-oxidação
nos peroxissomas através de uma acil CoA oxidase que consome O2 e produz
H202, o que aumenta a preponderância da catalase no metabolismo do EtOH,
devido à maior quantidade de H2O2 formado 671. Por outro lado, o consumo
crónico de EtOH demonstrou provocar um aumento na produção de H2O2 nas
zonas centrilobulares do fígado e na actividade da catalase, em ratos 661.
O acetaldeído, formado por uma ou outra via, desempenha um papel
importante na expressão da toxicidade do EtOH 672, sendo depois metabolizado
a ácido acético pela aldeído desidrogenase (ALDH) 659, localizada na
mitocôndria, com formação de NADH que é depois utilizado como primeiro
dador de electrões da cadeia transportadora de electrões mitocondrial 661.
Quando produzido em excesso, por exemplo quando se ingere EtOH em
grandes quantidades, o NADH leva a um aumento da produção do radical
anião superóxido ao nível dos complexos I e III da mitocôndria, originando H2O2

______________________________________________________ Introdução
145
e outras ROS e RNS, potencialmente lesivas para a mitocôndria e, em última
instância, para a célula, se não forem eficazmente destoxificadas 673. A maioria
do acetato é depois transportada pela corrente sanguínea a outras partes do
organismo, onde vai fazer parte de ciclos metabólicos fisiológicos levando à
produção de energia ou de moléculas úteis para a manutenção da homeostasia 670.
O acetaldeído é um electrófilo que causa dano celular pela formação de
aductos com proteínas, levando à produção de anticorpos contra esses
aductos imunogénicos, inactivação enzimática, diminuição da reparação do
DNA e alterações nos microtúbulos, membrana plasmática e mitocôndria com
um marcado comprometimento da utilização de oxigénio. O acetaldeído causa,
ainda, depleção de glutationa e peroxidação lipídica e estimula a síntese de
colagénio a nível hepático, promovendo o desenvolvimento de fibrose 667.
Variações genéticas nas enzimas envolvidas no metabolismo do EtOH
(ADH, CYP2E1 e catalase) influenciam o consumo, a toxicidade e a
dependência do álcool 661.
O EtOH pode ainda sofrer, em quantidade mínima, metabolismo não
oxidativo que pode ocorrer por 2 mecanismos distintos: (i) formação de etil
ésteres de ácidos gordos por reacção do EtOH com ácidos gordos e (ii)
formação de fosfatidil etanol num mecanismo que envolve a fosfolipase D 661. A
expressão das vias não-oxidativas aumenta quando as vias de metabolização
oxidativa do EtOH estão comprometidas 661.
A indução do sistema MEOS está também associada a um aumento na
produção de ROS e de peroxidação lipídica, bem como a uma diminuição da
capacidade de protecção contra radicais livres, enzimática e não-enzimática 669,
674, 675. Este facto pode agravar as alterações do estado redox provocadas
durante o metabolismo da MDMA, potenciando o risco de desenvolvimento de
toxicidade resultante da co-exposição a EtOH e MDMA, sobretudo a nível
hepático onde ocorre a maior parte do metabolismo destes dois compostos
(Figura 9).

Introdução ______________________________________________________
146
Figura 9. Hipótese que sugere possíveis interacções metabólicas entre o EtOH e a MDMA baseada na influência do primeiro nas principais isoenzimas do CYP450 metabolizadoras deste derivado anfetamínico. As setas a cheio indicam as vias metabólicas principais e, destas, as mais largas indicam as vias metabólicas mais expressivas. As setas a tracejado indicam vias metabólicas menos expressivas. As setas rosa indicam as possibilidades de interacção metabólica entre o etanol e a MDMA. O sinal (-) traduz diminuição da actividade e o sinal (+) traduz aumento de actividade.
Além de ter a capacidade de induzir a isoforma CYP2E1, o EtOH
também mostrou induzir a CYP3A em vários modelos experimentais 676 como
culturas primárias de hepatócitos de rato 677, linhas celulares de hepatoma de
rato 678 e ensaios in vivo em ratos 679. O EtOH apresenta, ainda, num sistema in
vitro de CYP2D6 supersomes®, um efeito bifásico na actividade da CYP2D6,
aumentando inicialmente a sua actividade (aumento em 175% relativamente ao
controlo para uma concentração de EtOH de 1,1%) para depois inibir
linearmente a actividade desta isoenzima 506. De facto, foi já descrito o
aumento da actividade da CYP2D6, avaliada pela capacidade de O-
desmetilação do DXM a dextrorfano através da determinação destes
compostos no sangue, em consumidores crónicos de EtOH 680. Para além
destas isoenzimas, em concentrações de 0,1% o EtOH inibe a CYP1A1,
CYP2B6, e CYP2C19 em 20 a 30%, enquanto em concentrações de 1% não
inibe CYP1A2, CYP3A4, CYP2C8, e CYP2C9 681. Em ratinhos, o EtOH leva a

______________________________________________________ Introdução
147
um aumento dos níveis de MDMA no núcleo estriado e tem um efeito
neuroprotector nesta área cerebral, evitando a depleção dopaminérgica e a
diminuição da actividade da tirosina hidroxilase (enzima limitante da síntese da
DA) induzidas pela MDMA 392. Aparentemente, em ensaios in vivo em ratos,
este aumento da concentração plasmática da MDMA provocado pela exposição
ao EtOH leva a uma diminuição de aproximadamente 16% na produção de
MDA quando comparada com animais expostos unicamente à MDMA 682.
Estudos clínicos em voluntários humanos demonstraram que as
concentrações plasmáticas de MDMA aumentaram 13% quando se combinou
uma dose de 100 mg de MDMA com 0,8 g/kg de EtOH 683 e um aumento de 7 e
8,7% quando doses de 75 e 100 mg de MDMA foram combinadas com álcool
(concentração de álcool em sangue = 0,06 g/dL) 684, enquanto que as
concentrações plasmáticas de EtOH diminuiram, indicando uma possível
interacção farmacocinética 683.
Devido à elevada taxa metabólica do EtOH, o seu efeito de primeira
passagem assume um papel muito importante na expressão da sua toxicidade,
uma vez que leva a uma diminuição dos níveis de EtOH capazes de atingir a
circulação sistémica, diminuindo, assim, os seus efeitos a nível central. No
entanto, o efeito de primeira passagem do álcool é extremamente variável:
quando o EtOH é ingerido em pequenas quantidades ou depois de uma
refeição, este fenómeno pode chegar a ser superior a 75%; para grandes
doses de EtOH, este efeito pode descer até 10% ou menos. Outros factores
que influenciam este efeito são o género, a etnia ou padrão de consumo de
álcool. Por exemplo, as mulheres apresentam níveis mais baixos de ADH
gástrica do que os homens, e o mesmo acontece nos indivíduos de raça
oriental comparativamente aos caucasianos, ou nos alcoólicos relativamente
aos não alcoólicos. Convém também recordar que a administração aguda de
álcool pode resultar em inibição metabólica, enquanto que o seu consumo
crónico pode induzir o metabolismo microssomal, afectando o efeito de primeira
passagem. A maior parte deste efeito de primeira passagem ocorre nos
hepatócitos, e apenas uma pequena parte ocorre na mucosa gástrica.

Introdução ______________________________________________________
148
1.9.5.6.1.4. Excreção
A taxa de eliminação do álcool apresenta uma grande variabilidade inter-
individual (70-150 mg/kg/h), mas é independente da dose (uma vez que
obedece a uma cinética de ordem zero) e apresenta pouca variabilidade intra-
individual 659. As variações inter-individuais na taxa de eliminação do álcool são
influenciadas por factores como: consumo crónico de álcool, dieta, jejum ou
não, idade, tabagismo, e momento do dia em que ocorre o consumo 661.
O álcool não metabolizado pode ser eliminado pela urina, ar expirado ou
suor numa percentagem que varia entre 2 e 10%, dependendo da quantidade
ingerida, e uma fracção muito reduzida é conjugada com o ácido glucurónico 685. Praticamente a metade deste álcool é eliminado através do ar expirado
devido à sua elevada volatilidade 660. A importância desta via metabólica radica
na sua utilidade toxicológica, uma vez que permite determinar indirectamente o
grau de alcoolémia recorrendo a aparelhos denominados alcoolímetros 601.
1.9.5.6.2. Mecanismos de acção e de toxicidade do etanol
O EtOH é sobretudo um depressor do SNC e, por isso, caracteriza-se
pela produção de efeitos sedativos e deterioração das funções cognitivas e do
rendimento psicomotor que envolvem uma interacção com receptores de
membrana associados com a transmissão glutamatérgica e GABAérgica a nível
cerebral 658, 686. Contudo, o álcool tem efeitos não só nos componentes
principais do SNC (cérebro e medula espinal), mas também ao nível do sistema
nervoso periférico 687.
As consequências neurológicas do alcoolismo apresentam uma grande
variedade inter-individual, uma vez que são condicionadas por vários factores
como doenças co-existentes, nomeadamente malnutrição e doença hepática, a
idade de início do consumo abusivo, o género do alcoólico, e a história familiar
de alcoolismo 687.
Para além dos seus efeitos ao nível da neurotransmissão, o EtOH pode
afectar a fluidez transmembranar das membranas celulares por alterar o seu
conteúdo em colesterol, por afectar o grau de acilação dos fosfolípidos

______________________________________________________ Introdução
149
membranares 674 e por levar à inclusão de ésteres etílicos de ácidos gordos
(FAEEs) nas membranas 688. Estas alterações membranares aumentam a
susceptibilidade da membrana a agressões oxidativas e podem ter graves
implicações que condicionam a comunicação e a sobrevivência celular como,
por exemplo, a transmissão sináptica do impulso nervoso, a actividade dos
transportadores membranares com acção Ca2+, Na+ e K+-ATPásica 674 ou o
transporte de GSH para a mitocôndria, essencial à sobrevivência da célula 670.
Os mecanismos envolvidos nos efeitos e na toxicidade do EtOH podem
estar na origem de interacções farmacodinâmicas entre o álcool e outras
substâncias co-consumidas.
1.9.5.6.2.1. Efeitos na neurotransmissão
O etanol é capaz de produzir uma grande variedade de efeitos a nível
cerebral por interacção com múltiplos alvos farmacológicos ilustrados na Figura
10.
Alguns estudos atribuem ao EtOH, tanto propriedades sedativas, como
estimulantes, dependendo da dose consumida (quanto maiores os níveis de
alcoolémia, maior será o grau de depressão central), das expectativas do
indivíduo e dos factores ambientais 691, 692. De facto, os efeitos centrais do
EtOH correlacionam-se com os níveis de alcoolémia, apesar de existir uma
grande variabilidade inter-individual.

Introdução ______________________________________________________
150
Figura 10. Mecanismos de acção biológica do etanol ao nível da neurotransmissão glutamatérgica, GABAérgica, serotonérgica e dopaminérgica. O etanol bloqueia não-competitivamente os efeitos do glutamato ao nível dos seus receptores NMDA e reduz a sua libertação causando lapsos de memória associados à intoxicação, hiperactividade durante a abstinência após consumo prolongado, e dano cerebral causado pelo consumo crónico 689. No sistema GABAérgico, o etanol tem um efeito potenciador nos receptores GABAA, favorecendo a ligação do neurotransmissor inibitório GABA ao receptor e aumentando o influxo de iões pelo canal de cloreto activado pelo GABA para a célula pós-sináptica, o que poderá estar na base dos efeitos ansiolíticos e depressores desta substância 658, 659. O etanol pode aumentar os níveis cerebrais de dopamina directamente ou por mecanismos indirectos como a inibição do transportador da DA ou como o aumento da libertação de 5-HT a nível central que influencia o sistema GABAérgico e aumenta a síntese da DA e pode ainda activar os receptores 5-HT3 levando a uma rápida abertura do canal transmembranar causando um aumento na condutância Na+/K+ e um subsequente rápido influxo do Ca2+ extracelular 690. As setas descontínuas indicam inibição e as setas mais largas indicam indução. DAT, transportador da DA; DA, dopamina; 5-HT, 5-hidroxitriptamina.
1.9.5.6.2.1.1. Efeitos na transmissão glutamatérgica
O glutamato é o principal neurotransmissor excitatório a nível cerebral 687.
Em termos farmacodinâmicos, o EtOH actua, tal como a ketamina, por
bloquear os efeitos do glutamato ao nível dos seus receptores NMDA de um
modo não competitivo e dependente da concentração e reduz a sua libertação
ao nível do hipocampo 689 (Figura 10). Este bloqueio ocorre desde
concentrações sanguíneas de álcool associadas com consumo social 689 até
concentrações associadas a intoxicações em humanos (0,2 – 4,6 g/L) 581. Os
efeitos do EtOH no sistema neurotransmissor do glutamato são responsáveis

______________________________________________________ Introdução
151
pelos lapsos de memória associados à intoxicação, hiperactividade durante a
abstinência após consumo prolongado, e dano cerebral causado pelo consumo
crónico 689.
Devido ao seu efeito inibitório na transmissão glutamatérgica, o consumo
crónico de EtOH leva a uma up-regulation dos receptores do glutamato no
hipocampo, uma área cerebral crucial para a memória e envolvida nas
convulsões epilépticas. Por isso, durante a abstinência alcoólica estes
receptores do glutamato adaptados à presença de elevados níveis de EtOH
podem tornar-se hiper-reactivos podendo levar a ataques e a convulsões que
parecem também ser influenciados pela deficiência em tiamina e magnésio,
comuns em alcoólicos como resultado da sua frequente malnutrição 687.
1.9.5.6.2.1.2. Efeitos na transmissão GABAérgica
O neurotransmissor ácido γ-aminobutírico (GABA) é o principal
neurotransmissor inibitório do SNC 687.
O EtOH tem um efeito potenciador nos receptores GABAA, favorecendo
a ligação do neurotransmissor inibitório GABA ao receptor e aumentando o
influxo de iões pelo canal de cloreto activado pelo GABA para a célula pós-
sináptica, o que poderá estar na base dos efeitos ansiolíticos e depressores
desta substância 658, 659 (Figura 10).
Este efeito ao nível do GABA explica a tolerância cruzada observada
com outras substâncias como as benzodiazepinas e os barbitúricos, que
também se ligam ao complexo receptor do GABA para produzir os seus efeitos 658.
A MDMA contraria este efeito do EtOH devido à acção agonista da
MDMA nos receptores 5-HT2A e 5-HT2C pós-sinápticos localizados nos
neurónios GABAérgicos, de cuja activação resulta uma diminuição dos níveis
do GABA e consequente estimulação da síntese e libertação de DA 82, 92-94.
Este antagonismo pode ser responsável pela diminuição da sensação de
sedação produzida pelo álcool descrita pelos indivíduos que consomem
simultaneamente as duas substâncias 683.

Introdução ______________________________________________________
152
O consumo crónico de EtOH leva a uma diminuição da sensibilidade
deste receptor ao EtOH e a uma down-regulation do mesmo, o que,
indirectamente, vai aumentar a actividade do glutamato. Este fenómeno
também pode contribuir para o efeito proconvulsivante observado durante a
abstinência de EtOH em alcoólicos 601.
Uma vez que a transmissão GABAérgica parece ter um papel importante
no mecanismo de acção da MDMA 88, as interferências do EtOH a este nível
poderão influenciar os efeitos resultantes do consumo de ecstasy,
inclusivamente o seu efeito hipertérmico, o qual pode ser revertido pela
activação de receptores GABAB 153.
1.9.5.6.2.1.3. Efeitos nas vias serotonérgicas
A interferência com as vias serotonérgicas é um ponto comum dos
mecanismos de acção da MDMA e do EtOH, podendo estar na base de
interacções farmacodinâmicas entre os dois compostos.
1.9.5.6.2.1.3.1. Efeitos nos níveis de 5-HT
O EtOH conduz a uma libertação de 5-HT a nível central (Figura 10).
Esta 5-HT pode influenciar o sistema GABAérgico e aumentar a síntese da DA 689, tal como acontece com a MDMA.
Por outro lado, o uso continuado de EtOH parece provocar um défice da
função serotonérgica, observado em indivíduos alcoólicos, e que se traduz
numa diminuição dos níveis de triptofano no plasma e de 5-HIAA (metabolito da
5-HT) no líquido cefalo-raquidiano 601.
Existem teorias que implicam o défice cerebral de 5-HT no
desenvolvimento do alcoolismo e nos transtornos comportamentais associados
ao consumo de álcool e ao alcoolismo, reforçadas pelo facto da administração
de inibidores da recaptação ou estimulantes da libertação de 5-HT diminuírem
o consumo de álcool 693. No entanto, curiosamente, a diminuição farmacológica
da função serotonérgica não leva a um aumento do consumo de álcool 601.

______________________________________________________ Introdução
153
Neste sentido, poder-se-ia especular que a MDMA, devido à sua acção pró-
serotonérgica, poderia inibir o desejo de consumir EtOH 601, facto confirmado
em ratos 694, 695. Contudo, 2 dias depois do consumo de MDMA surge uma
diminuição dos níveis cerebrais de 5-HT devido à estimulação da libertação da
5-HT induzida pela MDMA, que desencadeia estados de depressão 14, que
podem ser potenciados pelo efeito depressor do EtOH.
A disfunção na função serotonérgica que surge como consequência da
diminuição dos níveis de triptofano pode levar ao desenvolvimento de vertigens 601.
1.9.5.6.2.1.3.2. Efeitos nos receptores 5-HT
O receptor 5-HT1B parece estar envolvido no consumo de álcool e nos
mecanismos de recompensa associados a este consumo, uma vez que os
agonistas 5-HT1B produzem, em ratos, efeitos semelhantes aos do álcool 696 e
que ratinhos knockout para este tipo de receptores apresentam menor
sensibilidade aos efeitos de recompensa do álcool, sem alterar os seus efeitos
aversivos 697. No entanto, o sistema serotonérgico parece ter também um papel
importante na sensibilidade e/ou tolerância à ataxia induzida pelo EtOH como
se pode confirmar pelo facto de ratinhos knockout para os receptores 5-HT1B
serem menos sensíveis a este efeito induzido pelo EtOH e desenvolverem
tolerância mais lentamente 698.
Por outro lado, a administração prolongada (60 dias) de EtOH a ratos
não altera o número de recptores 5-HT2, mas diminui a sua funcionalidade,
uma vez que diminui a capacidade de hidrólise do fosfatidilinositol (PI) pela
fosfolipase C (PLC) que é activada pela proteína G acoplada a estes
receptores, diminuindo assim a produção de inositol trifosfato (IP3) e de
diacilglicerol (DAG), mensageiros intracelulares secundários envolvidos no
controlo de múltiplos processos celulares 699. A exposição crónica ao EtOH
parece também causar um aumento de receptores 5-HT2C no plexus
choroideus associada a um aumento da hidrólise do PI estimulada pela 5-HT,
não observada no córtex nem no hipocampo de cérebro de rato 699.
Contrariamente a este facto, concentrações elevadas de EtOH que se podem

Introdução ______________________________________________________
154
atingir em situações de intoxicação podem inibir a actividade dos receptores 5-
HT2C 699. Foi, também, observado que a abstinência de EtOH após consumo
crónico resulta numa diminuição do número de receptores 5-HT2A, não tendo
qualquer efeito na afinidade dos mesmos. A diminuição dos receptores 5-HT2A
está também associada a uma diminuição da hidrólise do PI estimulada pela
serotonina no cortex de rato 700. Estas alterações ao nível dos receptores 5-
HT2A e também nos 5-HT2C sugerem que estes receptores poderão estar
envolvidos na patogénese da dependência e da síndrome de privação do álcool 699. Estes efeitos poderão ainda provocar interacções farmacodinâmicas entre o
EtOH e a MDMA, uma vez que ambos actuam nos mesmos alvos.
O EtOH pode ainda activar os receptores 5-HT3 (canais iónicos
membranares que estão ligados ao sistema de sinalização celular do PI no
cérebro) levando a uma rápida abertura do canal transmembranar causando
um aumento na condutância Na+/K+ e um subsequente rápido influxo do Ca2+
extracelular (Figura 10). A activação dos receptores 5-HT3 pelo EtOH pode
afectar a libertação de DA em áreas ricas neste neurotransmissor como o
nucleus accubens e o tuberculum olfactorium. A interacção entre o sistema
dopaminérgico e os receptores 5-HT3 poderá estar envolvida na dependência
alcoólica 699.
1.9.5.6.2.1.4. Aumento da libertação de DA
O EtOH tem a capacidade de elevar os níveis cerebrais de DA por
mecanismos directos e indirectos (Figura 10). Este aumento é, aparentemente,
responsável pelos efeitos de recompensa ou reforço positivo do álcool, estando
na base do desenvolvimento de adição e/ou dependência relativamente a esta
substância e pode condicionar uma potenciação dos efeitos da MDMA, uma
vez que este derivado anfetamínico também aumenta a libertação de DA.
A administração de EtOH a ratos aumenta também a transmissão
dopaminérgica nas vias mesolímbicas e aumenta a frequência de disparo dos
neurónios dopaminérgicos, o que aumenta a quantidade de dopamina libertada
no nucleus accumbens 701.

______________________________________________________ Introdução
155
O aumento da libertação directa de DA no nucleus accumbens e na área
ventral tegmentada parece ocorrer por um mecanismo independente do cálcio.
Este facto foi confirmado por um estudo em ratos em que uma elevada
concentração de EtOH administrada por microdiálise aumentava os níveis de
DA na área ventral tegmentada (área de origem das fibras dopaminérgicas),
aumento que não foi atenuado por administração de uma solução alcoólica
isenta de cálcio 690. Noutro estudo em ratos, apenas concentrações elevadas
de EtOH (mais de 170 mM administrados por microdiálise) conseguiam
aumentar os níveis extracelulares de DA no nucleus striatum (área com
elevado conteúdo em terminais dopaminérgicos) 702. Os dados destes estudos
sugerem que as concentrações que provocam intoxicações moderadas, não
afectariam directamente os níveis extracelulares de DA.
Como referido anteriormente, a libertação indirecta de DA pode ocorrer
por activação dos receptores 5-HT3 pelo EtOH. Neste sentido, um estudo de
microdiálise em ratos in vivo mostrou que o aumento na libertação da DA
induzido pelo EtOH foi prevenido pela perfusão de um antagonista dos
receptores 5-HT3 703.
Este aumento da libertação de DA pode, ainda, ser mediado pelo
aumento da libertação de opióides endógenos (endorfinas e encefalinas) por
acção do álcool, que vão também modular a libertação de DA.
Tal como a MDMA, o EtOH pode ainda atrasar a eliminação da DA do
espaço extracelular por interferência com o DAT, cuja função é depurar o
espaço sináptico da DA libertada 704, 705.
Este sinergismo do EtOH e da MDMA no sistema dopaminérgico pode
resultar numa potenciação dos efeitos hiperlocomotores da MDMA, uma vez
que existe evidência de que os receptores da dopamina (D1, D2 e talvez D3)
contribuem para estes efeitos 112, 113.
O acetaldeído produzido pela metabolização do EtOH tem a capacidade
de formar aductos com a DA para formar salsolinol, o qual pode contribuir para
a dependência do álcool 706.

Introdução ______________________________________________________
156
1.9.5.6.2.2. Alterações no sistema termorregulador
Normalmente, a ingestão de pequenas quantidades de EtOH está
associada a um decréscimo da temperatura corporal por indução de
vasodilatação periférica com aumento das perdas de calor 687, 707, por um
mecanismo que envolve o aumento da libertação e monóxido de azoto com
acção a nível endotelial 708, inibição do ritmo normal de actividade mecânica
espontânea do tecido muscular liso vascular (vasomoção) e diminuição das
respostas contrácteis a mediadores neuro-humorais endógenos que
desempenham um papel importante na manutenção do tónus vascular e na
regulação do fluxo sanguíneo 709. No entanto, estudos experimentais em
animais demonstraram que doses elevadas de EtOH em condições extremas
de temperatura provocam um efeito poiquilotérmico, ou seja, um transtorno do
mecanismo de adaptação tanto ao frio como ao calor 601. Este efeito do ácool
depende basicamente de 3 factores: da temperatura ambiente, da quantidade
de álcool ingerida ou administrada e do exercício ou actividade física. Por
exemplo, em condições de temperatura ambiente baixa, exercício físico intenso
e ingestão abundante de álcool, será de esperar uma descida marcada da
temperatura corporal devido a uma inibição dos tremores (shivering) e
consequente diminuição da termogénese, ao que se acrescenta um aumento
das perdas de calor por aumento da vasodilatação cutânea podendo levar a
uma hipotermia potencialmente fatal 687. Contudo este efeito hipotérmico do
EtOH é apenas evidente para administrações agudas de EtOH, sendo afectado
por fenómenos de tolerância que podem surgir rapidamente, mesmo entre duas
exposições consecutivas espaçadas 24 horas 710.
Quando se avalia a acção conjunta do EtOH e da MDMA em ratos
mantidos a uma temperatura ambiente de 21-23ºC, o EtOH (1,5 g/kg/dia i.p.
durante 4 dias) potenciou dramaticamente a hiperlocomoção induzida pela
MDMA (6,6 mg/kg em quatro dias não consecutivos ou 10 mg/kg em 4 dias
consecutivos) (um efeito não observado em primatas 144), mas
surpreendentemente, atenuou o seu efeito hiperpirético 633, 711, 712. Para além
disso, a magnitude deste último efeito aumentou com injecções repetidas (ao
4º, 6º, 11º e 13º dia da experiência) de EtOH 1,5 g/kg + MDMA 6,6 mg /kg,
sugerindo sensibilização, o que leva a que à quarta injecção se observe uma

______________________________________________________ Introdução
157
completa prevenção da hipertermia 633. No entanto, para doses mais baixas de
MDMA (3,3 mg/kg), a administração de EtOH chega mesmo a provocar
hipotermia à 2ª injecção. Esta observação indica que os efeitos piréticos
induzidos pela combinação MDMA+EtOH estão largamente dependentes da
dose de MDMA. No entanto, num estudo recente, realizado a uma temperatura
ambiente de 32ºC, uma dose de 6,6 mg/kg de MDMA na presença ou na
ausência de EtOH matou todos os animais em menos de 2 h sem sinais de
atenuação da hipertermia por parte do EtOH 713.
1.9.5.6.2.3. Alterações psicomotoras
Os efeitos comportamentais do EtOH são dependentes da dose e o
consumo de pequenas quantidades produz, inicialmente, sentimentos de
euforia seguidos de depressão e estupor após ingestão de doses elevadas.
Devido ao rápido desenvolvimento de tolerância, os consumidores de
EtOH inexperientes desenvolvem menos efeitos agradáveis e mais disforia e
embriaguez do que os indivíduos experientes 659.
Indivíduos não alcoólicos com antecedentes familiares de alcoolismo
manifestam efeitos estimulantes mais pronunciados e menos efeitos sedantes 659.
Os primeiros efeitos negativos sobre o rendimento psicomotor
(sobretudo nos comportamentos complexos que requerem estado de alerta e a
tomada de decisões rápidas) surgem em torno dos 0,4 g/kg, que correponde a
uma alcoolémia de 40-50 mg/dL 659. Doses de EtOH iguais ou superiores a
0,75 g/kg (alcoolémia superior a 60 mg/dL), provocam uma deterioração
semelhante à produzida por outras substâncias como os opióides ou as
benzodiazepinas hipnóticas 601.
As áreas cerebrais mais vulneráveis ao dano produzido pelo alcoolismo
crónico incluem o cerebelo, o sistema límbico (incluindo o hipocampo e a
amígdala que estão envolvidos nos processos de memória, funcionamento
emocional e olfacto), o diencéfalo (incluindo o tálamo e o hipotálamo que
também contribuem para os processos de memória) e o córtex cerebral
(envolvido no raciocínio, aprendizagem e memória espacial, visual e táctil).

Introdução ______________________________________________________
158
Algumas destas áreas são também as mais afectadas pela neurotoxicidade da
MDMA, podendo ocorrer um agravamento mútuo dos efeitos psicomotores
destas duas subtâncias.
Os sintomas mais comuns que aparecem pouco tempo após o consumo
de álcool são: linguagem confusa, descoordenação motora (devido à
ocorrência de danos no cerebelo), marcha instável, nistagmo, diminuição da
atenção ou da memória, diminuição das capacidades visuoespaciais (também
provocadas pela MDMA) e, em última instância, estupor e coma.
Outros efeitos do EtOH sobre o SNC incluem (i) encefalopatia alcoólica
(ou de Wernicke) devida à deficiência em tiamina (síndrome aguda
caracterizada por nistagmo, parálise ocular, ataxia, confusão, letargia,
indiferença, delírio ligeiro, ansiedade e insónia), (ii) transtornos amnésicos
alcoólicos [demência tipo Korsakoff; síndrome aguda devida à deficiência em
tiamina que leva ao dano de determinadas áreas cerebrais conduzindo ao
aparecimento de dificuldade em processar informação nova (amnésia
anterógrada) e, mais raramente, de amnésia retrógrada], e (iii) alucinações
agudas. A longo prazo, podem surgir transtornos afectivos (ex.: zelotipia),
perturbações do sono (podem surgir para doses relativamente baixas de EtOH
e incluem diminuição da latência, diminuição da fase REM, despertar
prematuro), ansiedade, depressão, fadiga, excitabilidade, fraqueza muscular e
controlo deficiente da temperatura corporal que podem durar 2-3 meses ou
tornar-se crónicos 601, podendo ser agravados pela co-exposição à MDMA,
uma vez que este derivado anfetamínico também está associado ao
aparecimento deste tipo de efeitos. Estes fenómenos podem surgir como
consequência de anomalias cerebrais como atrofia neuronal e diminuição do
tamanho do cérebro observadas em indivíduos alcoólicos recorrendo a técnicas
neuropatológicas e de imagiologia 687.
O consumo crónico de EtOH pode ainda levar a uma redução do fluxo
sanguíneo e da capacidade metabólica de determinadas áreas cerebrais, tal
como acontece com a MDMA 5, 161. Se estas alterações provocadas pelo EtOH
se localizarem ao nível do hipocampo, podem potenciar as lesões retinianas
induzidas pela MDMA. Menores fluxos sanguíneos cerebrais foram associados
a uma maior gravidade de alcoolismo e a capacidade cognitiva mais
comprometida 687.

______________________________________________________ Introdução
159
Os indivíduos mais velhos são mais susceptíveis aos efeitos
neurotóxicos do EtOH devido ao processo fisiológico de envelhecimento
subjacente e também devido à maior probabilidade de desenvolvimento de
interacções medicamentosas 687.
Tal como o da MDMA, o potencial neurotóxico do EtOH parece dever-se
ao seu metabolismo que leva à formação de radicais livres, mas também à
possível implicação de alguns factores de cresciento como o NGF (Nerve
Growth Factor), o bDNF (Brain-Derived Neurotrophic Factor), a neurotrofina 3
(NT-3) e o factor de crescimento dos fibroblastos (bFGF), mas pode também
dever-se indirectamente ao dano noutros órgãos (ex.: fígado) que
subsequentemente interferem com o funcionamento cerebral 687 (Figura 11).
Figura 11. Fontes de complicações neurológicas do álcool e alcoolismo. Adaptado de Oscar-Berman M., “Impairments of brain and behavior. The neurological effects of alcohol”, Alcohol research and Health (1997), 21: 65-75.
Os efeitos do EtOH ao nível do sistema nervoso periférico incluem
neuropatia periférica (com torpor e fraqueza nas mãos e pés) como
consequência da malnutrição frequente em alcoólicos e a “paralisia de sábado
à noite” quando o indivíduo intoxicado exerce pressão em nervos
vulnerabilizados ficando incapaz de distender o braço durante dias ou semanas 687.
Tal como acontece com a MDMA, o EtOH aumenta o risco de lesões,
acidentes de viação e ainda de comportamentos violentos 714.
O EtOH mostrou ter um efeito potenciador na hiperactividade induzida
pela MDMA 682 que, tal como a potenciação do efeito hipertérmico, parece estar
sujeito a sensibilização, para ambas as doses de MDMA testadas (3,3 e 6,6
mg/kg), um efeito que não ocorre quando a MDMA é administrada

Introdução ______________________________________________________
160
isoladamente. Uma vez que o aumento do efeito locomotor da MDMA pelo
EtOH pode dever-se a uma potenciação da libertação de 5-HT e de DA ao nível
do estriado, que foi demonstrada em cérebro de rato 715, será de esperar uma
redução da potenciação da hiperlocomoção pelo EtOH em animais depletados
em 5-HT 633.
Isto significa que anteriores intoxicações alcoólicas graves podem
incrementar o aumento na libertação de 5-HT induzido pela MDMA 716, o qual,
dependendo da dose e/ou do perfil de exposição, pode induzir toxicidade
serotonérgica de longa duração, detectável 6-7 dias após a administração da
droga 717, 718.
A combinação de EtOH e MDMA leva também a uma dissociação entre
a sedação objectiva e subjectiva provocada pelo EtOH, o que faz com que os
utilizadores tenham uma sensação de euforia mais prolongada e uma falsa
sensação de aumento da aptidão para realizar determinadas tarefas, quando
esta na realidade está comprometida 683. Quanto à alteração da percepção
objectiva e subjectiva da capacidade de condução, a MDMA moderou
significativamente a deterioração da capacidade de seguir a estrada induzida
por doses baixas de EtOH, mas não afectou a deterioração da capacidade de
seguimento de outro veículo e dos testes comportamentais realizados em
laboratório com simuladores 719. Este facto é de extrema importância, uma vez
que o consumo de ecstasy é frequentemente combinado com o consumo de
álcool e uma proporção substancial dos utilizadores de MDMA revela conduzir
sob os efeitos da MDMA e/ou outras drogas. 719. De facto, foram já reportados
vários acidentes rodoviários fatais, nos quais a MDMA foi encontrada no
plasma dos condutores, isoladamente ou com outras drogas ou álcool 720.
Apesar do consumo de MDMA ser normalmente associado a um aumento de
comportamentos impulsivos e de risco durante os períodos de abstinência,
durante a exposição aguda a MDMA não induz comportamentos de
impulsividade e pode inclusivamente reduzi-los. O mecanismo farmacológico
que está na base desta melhoria do controlo dos impulsos pode estar
relacionado com um suprimento serotonérgico e dopaminérgico agudo após
uma dose única de MDMA. No entanto, quando se avalia o efeito agudo
conjunto de doses moderadas de EtOH com MDMA, a MDMA não consegue
mitigar o aumento da impulsividade e da aquisição de comportamentos de risco

______________________________________________________ Introdução
161
provocado pelo EtOH. Este resultado parece indicar que o efeito estimulador
central da MDMA não é suficiente para ultrapassar os efeitos induzidos pelo
EtOH na aquisição de comportamentos impulsivos 684. Alguns utilizadores de
MDMA referem que o EtOH elimina ou contraria os efeitos da MDMA e
aumenta os efeitos laterais e os vómitos 22.
O EtOH parece também contribuir para um aumento da propensão para
o desenvolvimento de perturbações psicopatológicas associadas ao consumo
de MDMA, uma vez que os consumidores de MDMA que consomem álcool
simultaneamente têm um risco 2,5 vezes superior de desenvolver este tipo de
perturbações do que os consumidores abstémios 219.
1.9.5.6.2.4. Efeitos neuroendócrinos
O EtOH aumenta a libertação do factor libertador de corticotrofinas
(CRF) pelo hipotálamo, facto confirmado pela inibição da estimulação do eixo
hipotálamo-hipófise-adrenal pelo EtOH quando se destrói o núcleo
paraventricular (área hipotalâmica onde se produz o CRF), quando se
neutraliza o CRF circulante com anticorpos específicos, ou quando se
bloqueiam os receptores do CRF.
O consumo de EtOH, tal como de MDMA, está associado a um aumento
dos níveis plasmáticos de cortisol. Este efeito é mais evidente quando se
verificam perturbações gastro-intestinais induzidas pelo EtOH. Neste sentido,
um estudo em voluntários saudáveis revelou que a ingestão de 1 g/kg de álcool
aumentava significativamente os níveis de cortisol, ACTH e vasopressina,
apenas nos indivíduos que sofreram naúseas e vómitos após o consumo de
EtOH 721. Este efeito ao nível da libertação de cortisol e ACTH, em ratos, é
geralmente mais expressivo em fêmeas do que em machos 722 e pode ocorrer
por activação directa das glândulas que os produzem ou através de activação
do eixo hipotálamo-hipófise que regula a sua libertação 722.
A diurese associada à intoxicação alcoólica tem sido relacionada
classicamente com a inibição da vasopressina, produzida pela hipófise. Esta
teoria é confirmada por estudos em humanos nos quais se verificou uma
diminuição significativa dos níveis desta hormona após a administração de uma

Introdução ______________________________________________________
162
dose moderada de álcool (0,5 g/kg) a voluntários saudáveis 723. Este efeito do
EtOH poderá, eventualmente, contrariar o efeito libertador da vasopressina da
MDMA quando as duas substâncias forem consumidas em simultâneo.
O consumo agudo de EtOH diminui a libertação, pelo hipotálamo, da
hormona libertadora da hormona luteinizante (LHRH), o que vai levar a uma
menor libertação de hormona luteinizante (LH) pela hipófise e a um decréscimo
nos níveis de testosterona, uma vez que as gónadas não são estimuladas para
a sua produção devido a uma acção directa do EtOH sobre as células de
Leydig (células testiculares produtoras de testosterona). Esta inibição do eixo
hipotálamo-hipófise-gónada mantém-se com o consumo continuado de EtOH 722 e pode ser potenciado pela co-exposição à MDMA. Nas mulheres, o
consumo agudo de EtOH aumenta os níveis de estrogénios e suprime a
produção de progesterona pelo corpo lúteo (o que aumenta o risco de aborto
espontâneo). O consumo regular de 3 ou mais bebidas alcoólicas por dia está
associado a atrasos ou ausência de ovulação (o que pode provocar
infertilidade), existindo uma relação dose-resposta entre o consumo de álcool e
o aparecimento de problemas menstruais.
O EtOH, en doses orais entre 0,75 e 1,3 g/kg leva a um aumento dos
níveis plasmáticos de prolactina (hormona produzida pela hipófise anterior,
responsável pela lactação e amamentação) 724 cuja intensidade é maior se os
consumidores forem descendentes de indivíduos alcoólicos 725. Este efeito do
EtOH pode potenciar os aumentos dos níveis de prolactina induzidos pela
MDMA.
A exposição aguda e crónica ao EtOH foi já associada a uma diminuição
dos níveis séricos de hormona do crescimento (GH) e de IGF-1 (insulin-like
growth factor 1, uma substância que medeia muitas das acções da GH, como
formação proteica e crescimento celular, ao nível dos tecidos) e da capacidade
de resposta da GH a factores envolvidos na regulação da sua libertação.
Verifica-se também um consequente aumento dos níveis séricos do factor
libertador da GH (GRF).

______________________________________________________ Introdução
163
1.9.5.6.2.5. Efeitos cardiovasculares
O álcool pode ser benéfico ou prejudicial para o sistema cardiovascular,
dependendo da quantidade consumida e das características do consumidor. Os
efeitos benéficos do EtOH podem dever-se a numerosos mecanismos que
incluem: (i) aumento das lipoproteínas de elevada densidade, (ii) prevenção da
aterosclerose por interferir com os processos inflamatórios ao nível endotelial,
(iii) inibição da produção de tromboxano A2, reduzindo a eficácia plaquetária na
coagulação sanguínea, (iv) estimulação da dissolução de coágulos sanguíneos
por aumento dos níveis sanguíneos de t-PA (tissue-type plasminogen activator)
que estimula a conversão do plasminogénio na sua forma activa, plasmina, a
qual aumenta a dissolução dos coágulos sanguíneos 726.
Alguns destes efeitos do EtOH na coagulação sanguínea parecem ter
potencial para diminuir o risco de coagulação intravascular disseminada que
pode surgir após consumo de MDMA.
Apesar do consumo de EtOH reduzido ou moderado poder conferir
protecção contra doenças coronárias, o consumo de grandes quantidades de
EtOH pode danificar o sistema cardiovascular, resultando em cardiomiopatias,
arritmias, hipertensão arterial e enfarte que podem potenciar a cardiotoxicidade
induzida pela MDMA.
Vários mecanismos têm sido propostos para explicar a hipertensão
provocada pelo consumo crónico de EtOH, incluindo: (i) aumento da actividade
do sistema nervoso simpático que vai aumentar a vasoconstricção e a força
contráctil do coração, (ii) aumento dos níveis plasmáticos de catecolaminas, (iii)
diminuição da sensibilidade dos baroreceptores das paredes arteriais e (iv)
diminuição dos níveis plasmáticos de magnésio ionizado, o qual relaxa a
parede dos vasos sanguíneos e estabelece um equilíbrio com os iões cálcio de
forma a manter o tónus vascular 726.
Tal como a MDMA, o EtOH pode exercer efeitos agudos ou crónicos a
nível cardíaco. Os efeitos agudos do EtOH incluem a vasodilatação periférica, a
hipotensão arterial e a taquicardia 727. O consumo crónico de EtOH pode
chegar a produzir miocardite alcoólica que se traduz por lesões do miocárdio
detectadas por análise bioquímica e anatomopatologica 601.

Introdução ______________________________________________________
164
A hipertensão arterial associada ao consumo crónico de EtOH pode
aumentar o risco de desenvolvimento de AVC isquémico ou hemorrágico,
sobretudo nas mulheres 714 e se consumido com substâncias também
associadas a um aumento do risco de AVC como é o caso da MDMA. Contudo,
em indivíduos com tensão arterial normal, o risco de AVC isquémico pode estar
diminuído pela aparente capacidade do álcool de diminuir o dano dos vasos
sanguíneos provocado pelos depósitos lipídicos e reduzir a coagulação
sanguínea (o que pode aumentar o risco de AVC hemorrágico) 714.
1.9.5.6.2.6. Efeitos metabólicos
O consumo de EtOH em jejum (quando as reservas de glicogénio estão
diminuídas) leva ao aparecimento de hipoglicemia por inibição da
gluconeogénese hepática, acidose láctica, hiperuricémia, cetose alcoólica e
hiperlipidémia. Quando se consome EtOH com alimentos, verifica-se um
aumento da glicémia. Assim, a ocorrência destes efeitos depende muito da
dose de álcool ingerida e das condições de consumo (ex.: em jejum, consumo
crónico, etc) 601.
Em diabéticos, o consumo de EtOH leva a um aumento da hemoglobina
glicosilada, a uma diminuição da produção endógena de insulina e a um
aumento da resistência à insulina 722.
Devido às alterações no balanço NAD+/NADH produzidas durante o
metabolismo do EtOH pela ADH, o consumo de grandes quantidades de EtOH
pode levar a transtornos metabólicos como a hiperlactiacidémia e a
hiperuricémia.
Assim, o EtOH, sobretudo se consumido em jejum, pode agravar a acidose e a
hipoglicemia provocadas pelo consumo de MDMA.
1.9.5.6.2.7. Efeitos no sistema imunitário
Apesar do sistema imunitário constituir uma protecção eficaz contra
substâncias estranhas, microrganismos e processos carcinogénicos, em

______________________________________________________ Introdução
165
determinadas circunstâncias, como o consumo exagerado de álcool, o
processo inflamatório pode ameaçar os tecidos do próprio organismo. Por
exemplo, as prostaglandinas e as prostaciclinas podem proteger as células de
determinados tipos de agressão, enquanto que os tromboxanos, por
produzirem vasoconstricção, podem promover fenómenos de hipoxia ou causar
directamente inflamação ou hipóxia. Por sua vez, os leucotrienos (ex.:
leucotrieno B4) podem causar lesão hepática por atrair e activar neutrófilos. O
consumo crónico de EtOH, diminui a produção de prostaglandinas e
prostaciclinas protectoras e aumenta a produção de tromboxanos e
leucotrienos, aumentando o risco de dano celular 670.
O consumo agudo de álcool produz ma diminuição significativa do
número absoluto de linfócitos T-helper (ou CD4+) e de linfócitos B 198, 728. Este
consumo está também associado a uma diminuição dos níveis das citoquinas
pró-inflamatórias TNFα e IL-1β, bem como da IL-12, responsável pela
activação dos linfócitos T. Verifica-se, ainda, um incremento nos níveis de
TNFα, de citoquinas imunoreguladoras TGFβ e IL-10 e um efeito bifásico no
IFNγ, que inicialmente aumenta para depois diminuir 728.
No entanto, o consumo crónico de EtOH leva a uma activação crónica
do sistema imunitário, a qual está relacionada com o desenvolvimento dos
problemas hepáticos provocados pelo álcool 729. Por seu lado, a administração
aguda de MDMA produz uma disfunção imune transitória, dependente da dose,
com diminuição da razão CD4/CD8 dos linfócitos T devido a uma diminuição do
número de células CD4 T-helper e a um aumento das células natural killer
(NK), apesar da contagem total leucocitária se manter inalterada 728. A
combinação aguda de MDMA e EtOH causa um maior efeito supressor nas
células T CD4+ e um maior aumento nas células NK em humanos. A exposição
a estas substâncias leva, também, a um aumento da produção das citoquinas
imunossupressoras (TGF-β e IL-10) e uma troca de citoquinas tipo Th1 (IL-1 e
IFN-γ) por citoquinas tipo Th-2 (IL-4 e IL-10). Observa-se assim uma
desregulação da produção de citoquinas pro- e anti-inflamatórias com um
desiquilíbrio no sentido da resposta anti-inflamatória. Em humanos, a função
imunitária tende a voltar aos valores basais 24 horas após a exposição à
MDMA 199. Este efeito transiente na homeostase imunológica, se repetida

Introdução ______________________________________________________
166
temporariamente, pode alterar a resposta immune, com risco para o estado
geral de saúde do consumidor 198, 728.
1.9.5.6.2.8. Efeitos no sistema hepato-biliar
O fígado é particularmente sensível ao dano provocado pelo EtOH, uma
vez que é o principal local onde ocorre o metabolismo alcoólico 670. Sendo
também a MDMA uma substância capaz de induzir hepatotoxicidade, o co-
consumo de ambas poderá aumentar o risco de aparecimento de efeitos
tóxicos ao nível do sistema hepato-biliar.
Os mecanismos envolvidos na toxicidade hepática exercida pelo EtOH
incluem: (i) formação de espécies reactivas de oxigénio produzidas durante o
metabolismo do EtOH (ex.: anião superóxido, peróxido de hidrogénio) que
levam a fenómenos de peroxidação lipídica com consequente lesão da
membrana celular e da mitocôndria; (ii) formação do radical hidroxietilo que, por
ser ainda muito reactivo e ter maior semi-vida do que o radical hidroxilo, pode
lesar uma maior quantidade de estruturas celulares 730, (iii) aumento da razão
NADH/NAD+, indicativa do estado redox das células, que leva a uma indução
da xantina oxidase, aumentando a produção de espécies reactivas 730; (iv)
aumento da produção de espécies reactivas de azoto devido à indução da
sintetase do monóxido de azoto indutível (iNOS), (v) diminuição dos níveis de
antioxidantes intracelulares (ex.: GSH, vitaminas A e E), e das actividades das
enzimas antioxidantes (ex.: GPx, SOD), aumentando a susceptibidade celular à
agressão de ROS e RNS; (vi) diminuição dos níveis de ATP devida à lesão
mitocondrial provocada pelas espécies reactivas, (vii) aumento do consumo de
oxigénio pelas células da zona periportal e secreção de endotelina pelas
células endoteliais dos sinusóides (provoca vasoconstricção), que vão reduzir a
disponibilidade de oxigénio para as células centrilobulares, desencadeando
uma situação de hipóxia que faz com que esta última zona do lóbulo hepático
seja mais atingida pela toxicidade do EtOH; (viii) aumento da produção de
tromboxanos e leucotrienos que vão aumentar a hipoxia e o dano resultante da
activação leucocitária; (ix) aumento da libertação de lipopolissacarídeo (LPS)
pelas bactérias intestinais resultando num aumento da activação do factor

______________________________________________________ Introdução
167
nuclear kappa-B (NF-κB) nas células Kupffer, aumentando a produção de
TNFα que vai lesar as células directamente ou indirectamente através da
estimulação da produção de leucotrienos730; (x) aumento da permeabilidade
intestinal à passagem dos LPS libertados pelas bactérias da flora intestinal que,
ao atingir o fígado, vão activar as células de Kupffer e aumentar a libertação de
substâncias promotoras de hipoxia e inflamação; (xi) formação de aductos
entre o acetaldeído ou as espécies reactivas formadas durante o metabolismo
do EtOH com proteínas endógenas, levando à formação de moléculas
imunogénicas que vão activar uma resposta imunológica que vai lesar as
células onde estes aductos se formam; (xii) estimulação da produção de
colagénio pelas células Ito (ou hepatic stellate cells ou células armazenadoras
de gordura) pelo acetaldeído e seus aductos, pelo TGFβ (transforming growth
factor beta) produzido pelas células Kupffer ou pelas espécies reactivas
formadas durante o metabolismo do EtOH. Qualquer um destes mecanismos
pode interagir com os mecanismos de hepatotoxicidade da MDMA, causando
um agravamento da lesão hepática, podendo acelerar a progressão para
necrose hepática ou hepatite fulminante.
O EtOH interfere com a transferência da GSH sintetizada no citoplasma
para a mitocôndria, o que pode conduzir a dano mitocondrial e morte celular 670.
O consumo crónico de EtOH pode levar ao desenvolvimento de (i)
esteatose (acumulação reversível de gordura ao nível da zona periportal do
lóbulo hepático), (ii) hepatite alcoólica (reacção inflamatória aguda reversível,
caracterizada por febre, icterícia e dor abdominal, que leva a necrose das
células hepáticas as quais, sendo substituídas por tecido fibroso, originam um
quadro de fibrose hepática), (iii) hepatite crónica e (iv) cirrose (fibrose severa
irreversível em que ocorre destruição da estrutura hepática e comprometimento
da funcionalidade do fígado) 670.
O EtOH é hepatotóxico devido à produção de acetaldeído (potencial
carcinogénico em humanos) e a alterações redox produzidas pelo NADH
gerado durante a sua oxidação via ADH, o que, por sua vez, afecta o
metabolismo dos lípidos (inibindo o metabolismo dos ácidos gordos ao nível da

Introdução ______________________________________________________
168
mitocôndria, o que faz com que eles se acumulem no citoplasma), hidratos de
carbono, proteínas e purinas 667.
Uma dieta rica em gordura pode, também, incrementar o potencial
hepatotóxico do EtOH, uma vez que o excesso de consumo de gorduras
aumenta a actividade de CYP2E1 e, simultaneamente, altera a composição das
membranas celulares, tornando-as mais susceptíveis à peroxidação provocada
pelas espécies reactivas formadas aquando da actuação da referida enzima
sobre o EtOH 670.
Este dano a nível hepático pode também indirectamente ser responsável
pela neurotoxicidade 687 e nefrotoxicidade 670 do EtOH.
O risco de desenvolvimento de dano hepático em alcoólicos depende de
factores como: (i) o estado nutricional do consumidor (a deficiência em
determinados antioxidantes e vitaminas favorece o dano hepático 663), (ii) o
género (as mulheres são, aparentemente, mais susceptíveis do que os homens
às lesões hepáticas provocadas pelo EtOH, possivelmente por terem menores
níveis de ADH gástrica e menor activação de mecanismos alternativos para
contrariar a inibição do metabolismo dos ácidos gordos induzida pelo EtOH),
(iii) a quantidade e padrão de consumo (quanto maior a quantidade e a
frequência de consumo de EtOH, maior o risco de desenvolver
hepatotoxicidade 714), (iv) os polimorfismos genéticos na actividade das
enzimas envolvidas no metabolismo do EtOH (indivíduos com fenótipo de
metabolizador pobre associado à ALDH e de metabolizador rápido para a
CYP2E1 e ADH acumulam maiores quantidades de acetaldeído e são mais
propensos a desenvolver hepatotoxicidade), (v) a infecção pelo vírus da
hepatite C (HCV) (a infecção por HCV aumenta o risco de dano hepático nos
alcoólicos), (vi) o tabagismo (os alcoólicos que fumam mais de 1 maço de
cigarros por dia têm um risco de desenvolver cirrose 3 vezes superior ao dos
não fumadores) e (vii) o consumo de café (os alcoólicos que consomem 4 ou
mais cafés por dia têm 5 vezes menos incidência de cirrose do que aqueles
que não consomem café, embora o mecanismo subjacente a este facto
continue por esclarecer) 670.
Uma condição associada com doença hepática avançada, incluindo a
doença hepática alcoólica, é a encefalopatia hepática (também denominada
encefalopatia portal-sistémica), uma perturbação metabólica hepática

______________________________________________________ Introdução
169
progressiva que afecta o funcionamento intelectual. Os alcoólicos com
encefalopatia hepática têm o fígado de tal forma afectado por fenómenos
cirróticos que a entrada de sangue venoso no fígado está obstruída, permitindo
que substâncias tóxicas (ex.: amónia, manganês) e produtos intermediários do
metabolismo entrem na corrente sanguínea e atinjam o cérebro onde vão
interferir com as acções dos neurotransmissores 687.
1.9.5.6.2.9. Efeitos no tracto gastro-intestinal
Em indivíduos não alcoólicos, o consumo agudo de EtOH pode levar ao
aparecimento de sintomas sugestivos de irritação da mucosa gástrica que pode
levar a gastrite crónica.
O aparecimento de lesões mais importantes, como a pancreatite
alcoólica, requer um historial de alcoolismo de mais de 10 anos de evolução.
O consumo crónico de EtOH promove a absorção intestinal de ferro e
favorece o armazenamento de ferro, importante catalizador da formação de
ROS e RNS, no fígado 670.
O álcool compromete, também, a absorção intestinal de cálcio no jejuno
e íleo, resultando numa diminuição dos níveis séricos de cálcio que exerce um
feedback negativo nas glândulas paratiróides, aumentando a libertação da
hormona da paratiróide (PTH) que, por sua vez, vai provocar reabsorção de
cálcio ao nível dos ossos, levando a um quadro de desmineralização óssea que
pode culminar em osteoporose, a qual pode ser agravada por uma inibição
directa dos osteoblastos (células formadoras de osso) e pela diminuição dos
níveis de testosterona induzidos pelo EtOH 722.
1.9.5.6.2.10. Efeitos no sistema urinário
O consumo agudo e crónico de EtOH podem comprometer a função
renal, particularmente se houver doença hepática estabelecida. Foram
observadas alterações na estrutura e função dos rins e diminuição da sua
capacidade para regular o volume e a composição do fluído e electrólitos no

Introdução ______________________________________________________
170
organismo. Alcoólicos podem apresentar níveis sanguíneos baixos de
electrólitos e alterações do balanço ácido-base. Para além disso, o EtOH pode
alterar os mecanismos de controlo hormonal que controlam a função renal. Ao
promover a doença hepática, o consumo crónico de EtOH tem consequentes
efeitos deletérios nos rins, incluindo desregulação do teor em sódio e fluidos e
mesmo falência renal aguda 731.
Estes efeitos deletérios exercidos pelo EtOH no rim podem aumentar o
risco de desenvolvimento de retenção urinária, hiponatrémia ou falência renal
aguda ou crónica como consequência do consumo de MDMA.
1.9.5.6.2.11. Efeitos nos músculos
O efeito do EtOH no aumento da PTH pode estar na base de efeitos
deletérios a nível muscular, uma vez que resulta num aumento da excreção de
fósforo, levando a um quadro de hipofosfatémia que pode resultar num
enfraquecimento dos músculos das articulações do ombro e da pélvis,
dificultando movimentos tão simples como levantar-se de uma cadeira e subir
escadas.
Este efeito a nível muscular pode ter especial importância quando o
EtOH é consumido com a MDMA, já que esta última é também capaz de
provocar danos a nível muscular que incluem rabdiomiólise e consequente
mioglobinúria.
1.9.5.6.2.12. Efeitos teratogénicos
Tal como a MDMA, o EtOH também está relacionado com o
aparecimento de malformações fetais 714, pelo que a sua utilização
concomitante pode agravar o risco de desenvolvimento das referidas
malformações.
O stress oxidativo e o metabolismo não oxidativo do EtOH
desempenham um papel importante no dano induzido pelo EtOH ao feto em
desenvolvimento, estando relacionado com o desenvolvimento de problemas

______________________________________________________ Introdução
171
de nascença provocados pelo EtOH, incluindo a síndrome alcoólica fetal (FAS) 661.
1.9.5.6.2.13. Carcinogénese
O consumo de álcool tem sido relacionado com o desenvolvimento de
carcinomas na cabeça e pescoço (boca, faringe, laringe e esófago), tracto
digestivo (estômago, cólon e recto) e mama 732.
O acetaldeído formado durante o metabolismo do EtOH tem a
capacidade de formar aductos com o DNA, formando compostos
carcinogénicos como a 1,N2-propanodesoxiguanosina 661.
Para além disso, a CYP2E1, induzida pelo consumo de EtOH, activa
numerosos pró-carcinogénios presentes no fumo do tabaco 661.
Assim, na análise das interacções da MDMA com o EtOH deve entrar-se
em linha de conta com: (1) variações na formação de metabolitos com diferente
actividade/toxicidade quando co-administrada com EtOH, o que, em termos
gerais, poderá intensificar, prolongar, contrabalançar ou não alterar os efeitos
do composto original; (2) alterações na biodisponibilidade da MDMA na
presença de EtOH; (3) sinergismo ou antagonismo, mais ou menos
pronunciado, entre os efeito da MDMA e do EtOH na libertação de
monoaminas e, particularmente, DA e 5-HT; (4) interacções entre os efeitos
produzidos pela libertação de monoaminas induzida pela MDMA e os efeitos do
EtOH em variados receptores envolvidos na modulação desta libertação (ex.:
receptores da adenosina, receptores NMDA, e mesmo receptores nicotínicos);
e, (5) vasodilatação, mais ou menos eficiente, induzida pelo EtOH que pode
levar a um aumento da dissipação de calor que pode contrabalançar
parcialmente os efeitos vasoconstrictores da MDMA 633.
Por este motivo, o consumo de EtOH, particularmente quando se
consome ecstasy ou outras drogas ilícitas, deve ser desencorajado, tendo em
conta os efeitos do uso combinado de ecstasy e álcool, incluindo desidratação 314. Aliás, foram já descritas várias mortes após consumo de MDMA em que o
consumo de EtOH foi também verificado (vide Tabela 1).

Introdução ______________________________________________________
172
É, pois, de extrema importância, e por isso constitui o objectivo desta
dissertação, o esclarecimento dos mecanismos envolvidos nas interacções
entre estes dois compostos, sobretudo a nível hepático onde ambos são
metabolizados, dando origem a metabolitos dotados de maior toxicidade do
que os compostos que lhes deram origem.
1.10. Principais considerações
De toda a informação reunida ao longo desta introdução é facilmente
inferido que os efeitos causados pela ingestão de uma pastilha de ecstasy
poderão ser condicionados por uma plêiade de factores que convergem num
determinado indivíduo e num determinado momento, o que dificulta a atribuição
dos efeitos observados nos consumidores apenas à MDMA. Particularmente
preocupantes são as interacções que podem surgir entre a MDMA e outras
drogas co-consumidas, propositada ou inadvertidamente, e que podem ser
potencialmente fatais.
A eventual presença de substâncias estranhas nas pastilhas de ecstasy
é preocupante porque, se considerarmos que algumas dessas substâncias em
pequenas doses podem ser menos perigosas que a MDMA (ex.: cafeína),
outras podem ser mais tóxicas e produzir efeitos devastadores prolongados.
É, então, simples perceber que os utilizadores de MDMA podem estar a
sujeitar-se a um risco acrescido de danos físicos e/ou psicológicos induzidos
pelas drogas como ansiedade, alterações psicóticas e do humor, e risco
aumentado de psicose em utilizadores com alterações de personalidade
esquizóide/esquizotípica pré-mórbida 560, ao ingerirem outras substâncias para
além da MDMA (ex.: uso repetido de MTA, pseuso-efedrina, cafeína, cocaína,
opióides) 366.
Outro problema relacionado com o decréscimo da pureza dos
comprimidos de ecstasy é que, à medida que o conteúdo em MDMA decresce,
os utilizadores não obtêm o efeito desejado e tendem a aumentar o número de
pastilhas consumidas 733 e, como consequência da contaminação das
pastilhas, o risco de efeitos prejudiciais associados aos contaminantes.

______________________________________________________ Introdução
173
Infelizmente, este padrão de consumo de múltiplas pastilhas para atingir a
reacção física esperada é muito comum entre os consumidores de MDMA 219.
Conhecer quais os contaminantes presentes nas pastilhas de ecstasy é
essencial também para os terapeutas e prestadores de cuidados de saúde
para, mais facilmente poderem diagnosticar problemas relacionados com
outras substâncias para além da MDMA e melhorar o tratamento e os cuidados
a prestar ao paciente.
Esta contaminação das pastilhas de ecstasy também dificulta o estudo
dos efeitos da MDMA nos utilizadores frequentes, uma vez que os efeitos
observados após a ingestão de uma pastilha podem dever-se a outra(s)
substância(s) e não à MDMA.
As pastilhas de ecstasy nem sempre são aquilo que se propõem ser. No
mercado de drogas elícitas não existe controlo de qualidade e os consumidores
devem estar alertados para esta ausência 423. Apesar de, aparentemente,
serem conscientes dos riscos para a saúde associados ao consumo de ecstasy 22, 357, os consumidores de ecstasy não são provavelmente conscientes dos
riscos inerentes às substâncias que inadvertidamente ingerem misturadas nas
pastilhas de ecstasy 366.
Quanto aos riscos da adopção de padrões de policonsumo de drogas,
parece legítimo assumir que serão mais preocupantes as associações com
drogas de fácil aquisição e elevada acessibilidade (ex.: álcool e tabaco) devido
à maior propensão do seu consumo atingir taxas de prevalência mais
generalizadas. No entanto, obviamente não se pode descurar o co-consumo
com drogas ilegais já que habitualmente lhe está associado um padrão de
agravamento de toxicidade mais marcado e potencialmente mais letal.
É então essencial desenvolver programas de prevenção para aumentar
o esclarecimento acerca dos riscos associados ao consumo de ecstasy em
conjunto com outras drogas de abuso e enfatizar a impureza das pastilhas de
ecstasy, focando o impacto destas substâncias contaminantes na saúde,
especialmente para a população em risco elevado de vir a sofrer de problemas
derivados do consumo de drogas 366.
Devem também ser desenhados estudos longitudinais e prospectivos
utilizando grupos controlo adequados para reproduzir os padrões de consumo
de múltiplas drogas manifestados pelos consumidores de ecstasy 612.

Introdução ______________________________________________________
174
O co-consumo de EtOH com MDMA é dos mais preocupantes, uma vez
que se trata de uma substância amplamente disponível e acessível, cujo
potencial tóxico é muitas vezes negligenciado pelos consumidores. Para
agravar esta situação, o EtOH tem ainda potencial para interagir com a MDMA,
afectando os efeitos e a toxicidade deste derivado anfetamínico. Por este
motivo, o estudo das possíveis interacções entre estas duas substâncias, e dos
mecanismos que lhe estão subjacentes, são o objectivo primordial do trabalho
experimental desenvolvido no âmbito da presente dissertação.

PARTE II
TRABALHO EXPERIMENTAL


______________________________________________________ Objectivos
177
2. OBJECTIVOS
A presente dissertação pretende contribuir para o esclarecimento das
possíveis interacções toxicológicas entre o álcool e a ecstasy, e dos
mecanismos que lhes estão subjacentes.
Deste modo, os estudos realizados no âmbito da presente dissertação
tiveram como principais objectivos:
• Avaliação do efeito da co-exposição ou da prévia exposição repetida ao
etanol na toxicidade aguda induzida pela MDMA utilizando modelos in
vivo, in vitro e ex vivo, nomeadamente através da análise dos seguintes
parâmetros:
o Alterações histológicas a nível hepático
o Estado redox das células hepáticas (quantificação dos níveis de
GSH e GSSG, de ROS e RNS, e de enzimas envolvidas na
homeostasia da GSH como a γ-GCS )
o Capacidade energética dos hepatócitos
o Danificação mitocondrial a nível hepático
o Morte celular (necrose e apoptose)
o Perfil metabólico da MDMA na presença de etanol
• Avaliação, in vitro, da influência da hipertermia induzida pela MDMA na
toxicidade da mistura álcool + ecstasy
Para atingir estes objectivos foram, paralelamente desenvolvidos os
seguintes estudos:
• Determinação da concentração de etanol em meio de cultura de células
recorrendo a um método de GC-FID previamente validado para essa
matriz
• Avaliação da contribuição do etanol utilizado na esterilização de
superfícies na concentração deste composto nos meios de incubação de
células

Objectivos ______________________________________________________
178

_____________________________________________ Trabalho Experimental
179
3. TRABALHO EXPERIMENTAL
Apresentam-se neste capítulo a descrição detalhada dos protocolos
experimentais, os resultados obtidos e o respectivo tratamento estatístico, bem
como a discussão dos resultados no âmbito de cada trabalho, sob a forma das
publicações que originaram.
São também descritos alguns resultados que aguardam publicação.
Trabalho I. Optimização das doses de MDMA a administrar a ratinhos previamente expostos repetidamente a etanol.
Trabalho II. Chronic exposure to ethanol exacerbates MDMA-induced hyperthermia and exposes liver to severe MDMA-induced toxicity in CD1 mice. Reprinted from Toxicology (doi:10.1016/j.tox.2008.07.064). Copyright
(2008) (with kind permission of Elsevier)
Trabalho III. Effect of chronic ethanol exposure on the hepatotoxicity of ecstasy in mice: An ex vivo study. Reprinted from Toxicology In Vitro, 22:
910-20. Copyright (2008) (with kind permission of Elsevier)
Trabalho IV. Synergistic toxicity of ethanol and MDMA towards primary cultured rat hepatocytes. Reprinted from Toxicology (DOI:
10.1016/j.tox.2008.09.009). Copyright (2008) (with kind permission of Elsevier)
Trabalho V. Interacções metabólicas entre o etanol e a MDMA em culturas primárias de hepatócitos de rato.
Trabalho VI. Gas chromatography determination of acetone, acetaldehyde, ethanol and methanol in biological matrices and cell culture. Aceite para
publicação no Journal of Chromatoraphic Science.
Trabalho VII. Ethanol, the forgotten artifact in cell culture. Reprinted from
Archives of Toxicology. 82: 197–8. Copyright (2008) (with kind permission of
Springer)

Trabalho Experimental _____________________________________________
180

Trabalho I. Optimização das doses de MDMA a administrar a ratinhos previamente
expostos repetidamente a etanol.


_______________________________________________________ Trabalho I
183
Trabalho I. Optimização das doses de MDMA a administrar a ratinhos previamente expostos repetidamente a etanol. a) Introdução Os efeitos induzidos in vivo pela MDMA foram já alvo de vários estudos
realizados inclusivamente pelo nosso grupo de trabalho 142 que utilizaram
doses de MDMA de 5, 10 e 20 mg/kg e que serviram de base para a escolha
das doses de MDMA a utilizar no presente trabalho. No entanto, a avaliação da
influência in vivo de uma prévia exposição repetida ao etanol nos referidos
efeitos ainda não foi descrita.
b) Objectivo Comparar, in vivo, os efeitos de duas doses de MDMA (10 mg/kg e 20
mg/kg) administradas a ratinhos Charles River CD-1 previamente expostos a
12% de etanol na água de bebida, durante 8 semanas.
c) Metodologia Foram utilizados 8 grupos de 6 ratinhos cada (30-40 g). Destes grupos, 4
foram expostos a 5% de etanol ad libitum, na água de bebida, durante 1
semana. Após este período de adaptação, a solução de bebida foi substituída
por 12% de etanol, durante 8 semanas. Os restantes 4 grupos tiveram acesso
a água ad libitum durante o mesmo período. Findo este período, foram
definidos os seguintes grupos:

Trabalho I _______________________________________________________
184
As administrações de MDMA realizadas após as 8 semanas de pré-
exposição foram feitas por via intra-peritoneal e num volume de 0,1 mL de
solução por 30 g de peso corporal. As soluções de MDMA foram preparadas
por dissolução da mesma em soro fisiológico (NaCl 0,9%).
A temperatura corporal e o comportamento dos animais foram
observados durante as 24 horas seguintes à administração de MDMA.
d) Resultados e discussão Após a administração de qualquer uma das doses consideradas, os
grupos expostos unicamente à MDMA (M) apresentaram uma reacção
anfetamínica típica, caracterizada por hiperactividade e hipertermia (atingiu
40,5ºC nos animais expostos a 20 mg/kg de MDMA e 39,5ºC nos animais
expostos a 10 mg/kg de MDMA). Nos animais expostos a etanol e MDMA
(E+M), verificou-se, também, um aumento marcado da temperatura corporal
(atingiu 40,5ºC nos animais expostos a 20 mg/kg de MDMA e 40,3ºC nos
animais expostos a 10 mg/kg de MDMA), observando-se, no entanto, uma
prostração marcada que poderá dever-se unicamente ao aumento da
temperatura corporal, ou eventualmente a outros mecanismos como: (i) uma
diminuição das reservas de serotonina ao nível do SNC induzida pela
exposição crónica ao etanol 734, diminuindo a disponibilidade de serotonina
para ser libertada por acção da MDMA e, consequentemente, reduzindo a
activação de receptores 5-HT1B que parecem medear, pelo menos em parte, as
respostas locomotoras induzidas pelas MDMA 111; e/ou (ii) uma interacção
toxicodinâmica entre o etanol e a MDMA devido ao facto do etanol contrariar a
hiperlocomoção induzida pela MDMA através da activação de receptores
GABAA que diminuem a actividade neuronal 735.
A administração de uma dose de 20 mg/kg de MDMA a ratinhos
previamente expostos a 12% de etanol provocou um marcado aumento da
mortalidade induzida pela MDMA, que não se verificou no caso da
administração da dose mais baixa, como se pode observar na Figura 12:

_______________________________________________________ Trabalho I
185
% de sobrevivência 24 horas após administraçãode 10 mg/kg de MDMA
C E M E+M0
50
100
Grupo
% S
obre
vivê
ncia
% de sobrevivência 24 horas após administraçãode 20 mg/kg de MDMA
C E M E+M0
50
100
Grupo
% S
obre
vivê
ncia
Figura 12. Percentagem de sobrevivência de ratinhos CD1, previamente expostos a 12% de etanol na água de bebida durante 8 semanas, após administração de 10 mg/kg ou 20 mg/kg de MDMA.
Apesar da dose de MDMA inicialmente testada (20 mg/kg) ter sido
escolhida com base em trabalhos anteriores realizados pelo nosso grupo 142, o
aumento da mortalidade verificado quando os animais expostos a esta dose
tinham sido previamente expostos a 12% de etanol, impossibilitou a
continuidade do ensaio devido à ausência de um grupo estatisticamente
representativo. Por esse motivo, a dose de 10 mg/kg de MDMA foi utilizada
para a continuação dos estudos in vivo sobre as interacções toxicológicas entre
o etanol e a MDMA a nível hepático e que deram origem ao Trabalho II
apresentado de seguida. As alterações da temperatura corporal resultantes da
exposição à MDMA serão também alvo de discussão no Trabalho II.

Trabalho I _______________________________________________________
186

Trabalho II. Chronic exposure to ethanol exacerbates MDMA-induced hyperthermia
and exposes liver to severe MDMA-induced toxicity in CD1 mice.


Author's personal copy
Toxicology 252 (2008) 64–71
Contents lists available at ScienceDirect
Toxicology
journa l homepage: www.e lsev ier .com/ locate / tox ico l
Chronic exposure to ethanol exacerbates MDMA-induced hyperthermia andexposes liver to severe MDMA-induced toxicity in CD1 mice
Helena Pontesa,∗, José Alberto Duarteb, Paula Guedes de Pinhoa, Maria Elisa Soaresa,Eduarda Fernandesc, Ricardo Jorge Dinis-Oliveiraa,d,e, Carla Sousaa, Renata Silvaa,Helena Carmoa, Susana Casal f, Fernando Remiãoa, Félix Carvalhoa, Maria Lourdes Bastosa,∗
a REQUIMTE, Toxicology Department, Faculty of Pharmacy, University of Porto, Rua Aníbal Cunha 164, 4099-030 Porto, Portugalb CIAFEL, Faculty of Sport, University of Porto, Rua Dr. Plácido Costa 91, 4200-450 Porto, Portugalc REQUIMTE, Physical-Chemistry Department, Faculty of Pharmacy, University of Porto, Rua Aníbal Cunha 164, 4099-030 Porto, Portugald Clinical Analysis and Public Health Department, Cooperativa de Ensino Superior, Politécnico e Universitário, CRL Rua José António Vidal,81, 4760-409 Vila Nova de Famalicão, Portugale Institute of Legal Medicine, Faculty of Medicine, University of Porto, Jardim Carrilho Videira, 4050-167 Porto, Portugalf REQUIMTE, Bromatology Department, Faculty of Pharmacy, University of Porto, Rua Aníbal Cunha 164, 4099-030 Porto, Portugal
a r t i c l e i n f o
Article history:Received 17 June 2008Received in revised form 24 July 2008Accepted 26 July 2008Available online 7 August 2008
Keywords:Ethanol3,4-Methylenedioxymethamphetamine(MDMA)HyperthermiaOxidative stressHepatotoxicity
a b s t r a c t
3,4-Methylenedioxymethamphetamine (MDMA; ecstasy) is an amphetamine derivative drug with entac-togenic, empathogenic and hallucinogenic properties, commonly consumed at rave parties in a polydrugabuse pattern, especially with cannabis, tobacco and ethanol. Since both MDMA and ethanol may causedeleterious effects to the liver, the evaluation of their putative hepatotoxic interaction is of great interest,especially considering that most of the MDMA users are regular ethanol consumers.
Thus, the aim of the present study was to evaluate, in vivo, the acute hepatotoxic effects of MDMA(10 mg/kg i.p.) in CD-1 mice previously exposed to 12% ethanol as drinking fluid (for 8 weeks). Bodytemperature was continuously measured for 12 h after MDMA administration and, after 24 h, hepaticdamage was evaluated.
The administration of MDMA to non pre-treated mice resulted in sustained hyperthermia, which wassignificantly increased in ethanol pre-exposed mice. A correspondent higher increase of hepatic heatshock transcription factor (HSF-1) activation was also observed in the latter group. Furthermore, MDMAadministration resulted in liver damage as confirmed by histological analysis, slight decrease in liverweight and increased plasma transaminases levels. These hepatotoxic effects were also exacerbated whenmice were pre-treated with ethanol. The activities of some antioxidant enzymes (such as SOD, GPx andCatalase) were modified by ethanol, MDMA and their joint action. The hepatotoxicity resulting from thesimultaneous exposure to MDMA and ethanol was associated with a higher activation of NF-�B, indicatinga pro-inflammatory effect in this organ.
In conclusion, the obtained results strongly suggest that the consumption of ethanol increases thehyperthermic and hepatotoxic effects associated with MDMA abuse.
© 2008 Elsevier Ireland Ltd. All rights reserved.
Abbreviations: MDMA, 3,4-methylenedioxymethamphetamine, ecstasy; MDA,methylenedioxyamphetamine; ROW, relative organ weight; %DLWC, percent drylipid weight content; HSF-1, heat shock factor 1; NF-�B, nuclear factor kappa-B; GOT,glutamic oxalic transaminase; GPT, glutamic pyruvic transaminase; SOD, superoxidedismutase; GPx, glutathione peroxidase; GST, glutathione-S-transferase; GSH, glu-tathione (reduced form); GSSG, glutathione (oxidised form); TBARS, thiobarbituricacid reactive substances; IHC, immunohistochemistry.
∗ Corresponding authors. Tel.: +351 222078979; fax: +351 222003977.E-mail addresses: [email protected] (H. Pontes), [email protected] (M.L. Bastos).
1. Introduction
3,4-Methylenedioxymethamphetamine (MDMA; ecstasy) is anamphetamine derivative drug with entactogenic, empathogenicand hallucinogenic properties, commonly consumed at rave par-ties in a polydrug abuse pattern, especially with cannabis,amphetamine, cocaine, tobacco and ethanol (Green et al., 2003;Schifano, 2004; Breen et al., 2006; Barrett et al., 2006). The com-mon concomitant use of ethanol and MDMA has a great interactionpotential since both compounds are metabolized in the liver to hep-atotoxic metabolites (Gemma et al., 2006; Green et al., 2003; Carmoet al., 2006) with higher toxicity than their parent compounds
0300-483X/$ – see front matter © 2008 Elsevier Ireland Ltd. All rights reserved.doi:10.1016/j.tox.2008.07.064
Helena
Rectangle
Helena
Text Box
189

Author's personal copy
H. Pontes et al. / Toxicology 252 (2008) 64–71 65
(Carvalho et al., 2004; Meier and Seitz, 2008). This metabolic acti-vation observed with MDMA seems to be a common feature ofamphetamine derivatives, since it was already described for com-pounds such as 4-MTA (Carmo et al., 2004b, 2007), 2-CB (Carmo etal., 2004a, 2005) which showed a variable toxicity profile accord-ingly to the individual metabolic capacity. In addition, ethanol canalter the expression and/or activity of some drug-metabolizingenzymes (Jang and Harris, 2007), including those involved inMDMA metabolism. Furthermore, MDMA users are regular ethanolconsumers before their first contact with MDMA (Ben Hamida etal., 2006), which may represent a putative risk factor for the toxicinteraction between these two drugs. In fact, our group has recentlydemonstrated that the repeated exposure of CD1 mice to ethanolseems to increase the vulnerability of freshly isolated hepatocytestowards pro-oxidant conditions (Pontes et al., 2008). Additionally,results reported by Pacifici and Hernández-Lopes in human sub-jects (Hernández-López et al., 2002; Pacifici et al., 2001) showedthat also the physiologic and psychopathologic effects of MDMAcould be increased in presence of ethanol. Thus, the objective of thepresent study was to evaluate, in vivo, the acute hepatotoxic effectsof MDMA (10 mg/kg i.p.) in CD-1 mice previously exposed to 12%ethanol as drinking fluid (for 8 weeks). This experimental approachwas designed to simulate the most common pattern of ethanol andMDMA abuse, in which the individuals are exposed to ethanol priorto the first contact with MDMA. To our knowledge, experimentalwork addressing the in vivo effects of chronic ethanol exposure onMDMA-induced hepatotoxicity has not been previously reported.
2. Materials and methods
2.1. Chemicals
All the reagents used in this study were of analytical grade. Bovine serumalbumin (fraction V), �-nicotinamide adenine dinucleotide phosphate reducedform (�-NADPH), pyruvic acid, 2-vinylpyridine, glutathione reduced form (GSH),glutathione oxidised form (GSSG), glutathione reductase (EC 1.6.4.2), 5,5-dithiobis-2-nitrobenzoic acid (DTNB), guanidine hydrochloride, 2,4-dinitrophenylhydrazine,1-chloro-2,4-dinitrobenzene (CDNB), xanthine, nitrotetrazolium blue chloride, xan-thine oxidase from bovine milk and hydrogen peroxide solution were obtained fromSigma–Aldrich Co. (St. Louis, MO, USA). The injectable solution of sodic heparinewas obtained from B. Braun (Lisbon, Portugal). Perchloric acid, trichloroacetic acid,2-thiobarbituric acid, Folin Ciocalteau reagent and all other chemicals were pur-chased from Merck (Darmstadt, Germany). 3,4-Methylenedioxymethamphetamine(HCl salt) was extracted and purified from high purity MDMA tablets that wereprovided by the Portuguese Criminal Police Department. The obtained salt waspure and fully characterized by NMR and mass spectrometry methodologies(purity > 99%).
2.2. Animals
Adult male CD1 mice (Charles-River Laboratories, Barcelona, Spain), weighing30–40 g, were used in all experiments. For at least 1 week prior to the treatment,animals were acclimatized in polyethylene cages with wire-mesh tops, lined withwood shavings, at an ambient temperature of 20 ± 2 ◦C, humidity between 40 and60% and 12 h/12 h light/dark cycle (light on from 8.00 to 20.00 h), having standardchow and tap drinking water ad libitum. After this period, animals were dividedinto two main groups: ethanol (n = 12) and non-ethanol (n = 12). In the ethanol maingroup the tap drinking water was replaced by a 5% ethanol solution for 1 weekand, afterwards, by a 12% ethanol solution for 8 weeks. The replacement of waterfor 5% ethanol before 12% ethanol corresponds to a required adaptation period, toavoid aversion of mice to a high ethanol concentration in the drinking fluid, sincethese animals do not have natural preference for 12% ethanol. The non-ethanol maingroup had drinking tap water ad libitum for the same period. There were no differ-ences in body weight between the main groups after the 8 weeks of the experiment(data not shown). During the 24-h period after MDMA administration, animals weredeprived of food but the drinking solution was allowed ad libitum. Surgical proce-dures for blood collection and liver excision were performed under anaesthesia andcarried out between 9 and 11 a.m. Animal experiments were licensed by PortugueseGeneral Directorate of Veterinary Medicine. Housing and experimental treatmentof animals were in accordance with the Guide for the Care and Use of LaboratoryAnimals from the Institute for Laboratory Animal Research (Institute for LaboratoryAnimal Research, 1996). The experiments complied with the current Portugueselaws.
2.3. MDMA challenge
Mice were housed in four groups of six animals (control, ethanol, MDMA andethanol + MDMA groups) and were injected intraperitoneally with saline (0.9% NaCl)or 10 mg/kg MDMA (dissolved in saline) in a volume of 0.1 mL/30 g body weightbetween 9:00 and 10:00 a.m. The experiment was performed under controlledambient temperature fixed at 20 ◦C (±2 ◦C). Subcutaneous temperature of animalswas measured repeatedly after MDMA administration (see measurement of bodytemperature). After 24 h, animals were anaesthetized and blood was removed fromthe inferior vena cava and collected into heparinized tubes. The liver was excisedand weighed, after body perfusion with 0.9% NaCl solution, and used for biochemi-cal analysis. Samples were kept frozen (−80 ◦C) until assay. Liver sections were alsocollected for histological examination. The relative organ weight (ROW), an indica-tor of tissue harm, was also calculated for each animal as a percentage of the totalbody weight at the sacrifice day.
2.4. Measurement of body temperature
Subcutaneous temperatures of mice were measured using BioMedic pro-grammable temperature transponders (microchips BMDS IPTT-100, Plexx BV, 6660AE ELST, Netherlands). The microchips were implanted subcutaneously between theshoulder blades, using the BioMedic implant device, 2 days before the experiment.Readings of subcutaneous temperature were taken immediately prior to MDMA orsaline injection and for 12 h thereafter using a hand held Biomedic Pocket Scanner(DAS-5007). Temperature was recorded every 15 min during the first hour, every30 min during the second and third hours and every hour thereafter.
2.5. Plasma biochemical analysis
2.5.1. Plasma transaminasesPlasma activities of glutamic oxalic transaminase (GOT) and glutamic pyru-
vic transaminase (GPT) were evaluated as biomarkers hepatic damage. Thesedeterminations were performed in a Reflotron Analyser (Boehringer Mannheim,Indianapolis, IN) using 32 �L of plasma for each analysis (GOT, Cat no. 10745120,Roche Diagnostics; GPT, Cat no. 10745138, Roche Diagnostics).
2.5.2. Oxidative stress parameters2.5.2.1. Sample preparation. For the quantification of protein carbonyl groups, lipidperoxidation extent and GSH and GSSG concentrations, tissue samples were homog-enized (1:4 m/v) in ice-cold 5% perchloric acid with an Ultra-Turrax® homogenizer.After the homogenization, aliquots were taken to determine protein carbonylgroups content and protein levels. The remaining homogenates were centrifugedat 3200 × g for 10 min at 4 ◦C and the supernatants were used for the quantificationof lipid peroxidation and GSH/GSSG.
For measurement of antioxidant enzymes activities, liver samples were homoge-nized (1:4 m/v) in ice cold 0.1% Triton X-100 in 50 mM phosphate buffer (pH 7.4). Thehomogenates were centrifuged at 3200 × g for 10 min at 4 ◦C, and the supernatantscollected for the enzymatic and protein assays.
2.5.2.2. Protein carbonyl groups. Protein carbonyl groups (ketones and aldehydes)were quantified according to Levine et al. (1994). Results were expressed as nmol ofDNPH incorporated per mg of protein, calculated by using an extinction coefficientof 2.2 × 104 M−1 cm−1.
2.5.2.3. Non-specific lipid peroxidation. The extent of lipid peroxidation was mea-sured by the thiobarbituric acid reactive substances (TBARS) assay at 535 nm (Buegeand Aust, 1978; Carvalho et al., 1997). TBARS were expressed as malondialdehydeequivalents per milligram of protein, calculated by using an extinction coefficient of1.56 × 105 M−1 cm−1.
2.5.2.4. GSH and GSSG. Tissue GSH and GSSG contents were determined by theDTNB-GSSG reductase recycling assay as described before (Pontes et al., 2008).
2.5.2.5. Catalase. Catalase activity was measured according to the method of Aebi(1984), by monitoring the decomposition of H2O2 at 240 nm, calculated by usingan extinction coefficient of 39.4 M−1 cm−1). The enzyme activity was expressed as�mol of H2O2 consumed per min and per mg of protein.
2.5.2.6. Glutathione-S-transferase (GST). Glutathione-S-transferase (GST) activitywas assayed according to the method of Habig et al. (1974). The formation of GSHconjugate with 1-chloro-2,4-dinitrobenzene was monitored for 5 min at 340 nm.The GST activity was calculated by using an extinction coefficient of 9.6 mM−1 cm−1
and expressed as �mol per min per mg of protein.
2.5.2.7. Selenium-dependent glutathione peroxidase (GPx). Selenium-dependentglutathione peroxidase (GPx) activity was assayed by NADPH oxidation at 340 nm
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
190

Author's personal copy
66 H. Pontes et al. / Toxicology 252 (2008) 64–71
(Flohe and Gunzler, 1984). The enzyme activity was calculated by using an extinc-tion coefficient of 6.22 mM−1 cm−1 and expressed as nmol of NADPH oxidized permin per mg of protein.
2.5.2.8. Superoxide dismutase (SOD). Copper/zinc superoxide dismutase (CuZnSOD)and manganese superoxide dismutase (MnSOD) were assayed using the methodof Flohé and Ötting (1984), with some modifications previously reported before(Dinis-Oliveira et al., 2006).
2.5.3. Protein quantificationProtein was determined by the Lowry method (Lowry et al., 1951) using bovine
serum albumin as the standard.
2.5.4. Dry lipid weight contentThe total liver lipid content was determined according to Folch et al. (1957) and
expressed on a dry weight percentage (%DLWC).
2.6. Nuclear extracts and fEMSA
Transcriptional activations of NF-�B and HSF-1 were determined by fluorescentelectrophoretic mobility shift assay (fEMSA) using nuclear protein extracts accord-ing to a previously reported method (Dinis-Oliveira et al., 2007). The nuclear proteinextracts were obtained from tissue homogenates (1:2.5 m/v) in cell lysis buffer (ACBuffer) containing 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM EDTA, 0.1 mMEGTA, 1 mM ditiothreitol (DTT), 0.25 mM phenyl methylsulfonyl fluoride (PMSF) and20 �L/mL igepal (pH 7.9).
To obtain the nuclear extracts, the homogenates were exposed to a freeze/thawcycle and centrifuged for 10 min at 850 × g, 4 ◦C. The pellets were washed againwith AC Buffer, incubated for 15 min on ice, and centrifuged at 16,000 × g, 4 ◦C, for30 s. The obtained nuclear pellets were resuspended in complete lysis Buffer (BCBuffer, containing 20 mM HEPES, 20% (m/v) Glycerol, 420 mM NaCl, 1.5 mM MgCl2,0.5 mM EDTA, 1 mM DTT, 0.25 mM PMSF, 20 �L/mL igepal and 5 �g/mL of a proteaseinhibitors cocktail containing 1 mg/mL aprotinin, 1 mg/mL leupeptin and 1 mg/mLpepsatin), incubated for 30 min on ice on a rocking platform at 150 rpm and cen-trifuged for 10 min at 16,000 × g at 4 ◦C. Aliquots of the supernatant were stored at−80 ◦C until assay. Protein quantification was determined by the Lowry assay andeach extract aliquot was diluted to 2 mg/mL of protein.
Thereafter, to perform the fEMSA electrophoresis, 20 �g of protein from nuclearextracts were incubated with Cy5-labeled NF-�B or HSF-1 consensus probe in assaybuffer (200 mM HEPES, 500 mM KCl, 10 mM EDTA and 10 mM DTT) supplementedwith glycerol, poly(dI-dC), DTT and igepal, at 4 ◦C over-night. Specificity of theDNA–protein complex was confirmed by the addition of a 50-fold excess of an unla-beled specific competitor (SC) (equal to the specific probe but without the Cy5 label).Nine microliters of each mixture were loaded on a 5% non-denaturating polyacry-lamide gel at 10 ◦C, 800 V, 50 mA and 30 W, in TBE buffer 1× (90 mM Tris base, 90 mMboric acid, 2 mM EDTA, pH 8.3) using an ALFexpress II DNA Analyser (AmershamPharmacia Biotech, Sweden). The temperature was regulated by an external ALF-express II Cooler system (Amersham Pharmacia Biotech, Sweden). The followingCy5-labeled specific double stranded probes were used for NF-�B and HSF-1 gelassays: NF-�B, 5′-GCC TGG GAA AGT CCC CTC AAC T-3′; HSF-1, 5′-GAT CCT CGA ATGTTC GCG AAA AG-3′ .
2.7. Tissue preparation for light and transmission electron microscopy
For light microscopy, after excision, liver was sliced in 2–4 mm3 pieces, approxi-mately, and fixed in 4% formaldehyde. The fixed pieces were, then, dehydrated withgraded ethanol and included in paraffin blocks. The compound used in the transitionbetween dehydration and impregnation was benzene. Semi-thin sections (4 �m)were cut in a microtome, applied on silane-coated slides and deparaffinated. Thedeparaffinated sections were treated for different staining protocols (Haematoxylin-Eosin, Van Gieson) and for immunohistochemistry assays for the detection of NF-�B.
The haematoxylin–eosin staining was performed by immersion in Mayer’shaematoxylin solution for 3–4 min followed by immersion in 1% eosin solution for7 min, dehydration with graded alcohols through xylene, and mounting with DPX.
To perform the NF-�B immunohistochemical detection, NF-�B p50 (NLS) rabbitpolyclonal antibody (SC-114, 1:20, Santa Cruz Biotechnology Inc., California, USA)was applied on deparaffinated liver sections and these samples were incubatedat 37 ◦C for 2 h in a humidified chamber. The same samples were then incubatedwith a secondary anti-immunoglobulin goat anti-rabbit IgG, F(ab′)2 conjugatedwith alkaline phosphatase (SC-3838, 1:50, Santa Cruz Biotechnology Inc., California,USA), under the same conditions, for 1 h. SIGMAFAST® Fast Red TR/Naphthol AS-MXTablets were used as substrate according to manufacturer’s instructions. The sec-tions were counterstained with Mayer’s haematoxylin. The primary antibody wasreplaced by PBS for negative control sections. Wistar rat 14th day placenta slideswere used as positive control. All stained sections were mounted on glass slidesusing Aquatex® . For the light microscopy study, an optic photomicroscope Nikon,Model Eclipse E400 was used.
For electron microscopy, 1 mm3 tissue pieces were fixed in 2% gluteraldehyde (in0.2 M sodium cacodylate) for 2 h at 4 ◦C. After three 15 min washes with 0.2 M sodiumcacodylate buffer at 4 ◦C, the pieces were post-fixed with 2% osmium tetroxide (in
0.2 M sodium cacodylate), for 2 h at 4 ◦C. After a new wash with sodium cacody-late (5 min), the samples were submitted to a dehydration process using increasingconcentrations of ethanol (from 50% to 100%) for 90 min. In the impregnation stage,the pieces were placed in mixtures containing absolute ethanol and LR White, withincreasing concentrations of resin (50% and 75% (v/v) of absolute ethanol) for 30 mineach. In the next stage, samples were kept for two periods of 30 min each, in 100% LRWhite, and afterwards were mounted on covered gelatine capsules. The inclusionwas processed in a drying oven at 60 ◦C for 22–26 h. Ultra-thin (thickness equal to500–600 Å) sections were cut and contrasted with a saturated aqueous solution ofuranyl acetate, for 30 min, and with a solution of lead citrate, for 15 min, with washesat the beginning and at the end of each one of these procedures. For the electronmicroscopy study a transmission electron microscope Zeiss EM 10 A, at 60 Kvoltswas used.
2.8. Statistical analysis
Results are presented as mean ± S.E.M. (from six animals) and were tested fornormality with the Kolmogorov–Smirnov test. For temperature analysis, compar-isons were made by analysis of variance (ANOVA) with repeated measures, followedby the Bonferroni’s post-hoc test. The other statistical comparisons were performedby one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisontest. Significance was accepted at P < 0.05.
3. Results
3.1. Body temperature
After administration of 10 mg/kg MDMA, animals from theMDMA group showed a typical amphetaminic reaction char-acterized by hyperthermia and hyperactivity. However, in theethanol + MDMA animals, hyperactivity was not present and amarked prostration was observed.
The ethanol group presented a similar body temperature pro-file to that of the control group. However, the ethanol + MDMAgroup showed a sustained increase in body temperature that wassignificantly higher than that observed for MDMA alone (Fig. 1A).Statistical differences were found during the first 7 h after MDMAadministration.
3.2. Heat shock factor-1 (HSF-1) activation
The ethanol + MDMA group presented a significantly higherincrease of heat shock transcription factor (HSF-1) activation(Fig. 1B, lane 4), when compared to that induced by MDMA alone(Fig. 1B, lane 3).
3.3. Nuclear factor kappa-B (NF-�B) activation
As shown in Fig. 2, the ethanol + MDMA group presented asignificant sharp activation of NF-�B in mouse liver (lane 4) com-paratively to control (lane 1), ethanol (lane 2) and MDMA (lane3) groups. This increase in NF-�B activation was also visible inthe immunohistochemistry for NF-�B in liver sections since in theethanol + MDMA group a high number of red stained nucleus, cor-responding to activated NF-�B, was observed when compared tothe other groups (Fig. 2).
3.4. Structural and ultra-structural analysis
The histological analysis of hepatic sections by light and electronmicroscopy (Fig. 3) revealed, in all groups, a preserved lobular struc-ture and ultrastructure. The control group presented no relevantchanges in the various cellular organelles while the ethanol, MDMAand ethanol + MDMA groups showed diffuse cytoplasmic vacuoliza-tion, lysosomal activation, mitochondrial swelling and a dilatationof the cytoplasmic reticulum and the perinuclear cisterna, alter-ations particularly evident in the ethanol + MDMA group. A severedilatation of the hepatic centrilobular sinusoids was evident inethanol and ethanol + MDMA groups. All the groups presentedabundant cytoplasmic lipid droplets although in the control and
Helena
Note
Marked set by Helena
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
191

Author's personal copy
H. Pontes et al. / Toxicology 252 (2008) 64–71 67
Fig. 1. Effect of ethanol pre-treatment on the hyperthermic effect induced by MDMA (A) and in the consequent HSF-1 transcriptional activation (B) in the liver, detected bysemi-quantitative fluorescent electrophoretic mobility shift assay (fEMSA). The illustrated gel is representative of the specific HSF-1 bands (highlighted by the dotted line)obtained for the control (C), ethanol (E), MDMA (M) and ethanol + MDMA (E + M) groups by using fluorescence labeled specific probes (SP). Lane 5 represents a competitiveexperiment with a 50-fold molar excess of a specific competitor (SC, unlabeled specific probe). The optical density (OD) of the bands ± S.E.M. of six animals is indicatedbelow. Body temperature results are presented as mean ± S.E.M. of six animals. *P < 0.05, **P < 0.01, ***P < 0.001, compared to control group; #P < 0.05, ##P < 0.01, ###P < 0.001,compared to ethanol group; @P < 0.05, @@P < 0.01, compared to MDMA group.
MDMA groups the distribution of these droplets was preferentiallycentrilobular, while in the ethanol and ethanol + MDMA groups thisdistribution had a diffuse pattern along the lobule. In control andMDMA groups the interstitial space and the hepatocytes microvilli
of the bile canaliculi and of the sinusoids presented a normal struc-ture while the ethanol and the ethanol + MDMA groups presented apronounced deposition of collagen fibres between the hepatocytesand the endothelial cells of the sinusoids, resulting in an apparent
Fig. 2. Effect of ethanol pre-treatment on NF-�B transcriptional activation in the liver detected by semi-quantitative fluorescent electrophoretic mobility shift assay (fEMSA).The illustrated gel is representative of the specific NF-�B bands (highlighted by the dotted line) obtained for the control (C), ethanol (E), MDMA (M) and ethanol + MDMA(E + M) groups by using fluorescence labeled specific probes (SP). Lane 5 represents a competitive experiment with a 50-fold molar excess of a specific competitor (SC,unlabeled specific probe). The optical density (OD) of the bands ± S.E.M. of six animals is indicated below. ***P < 0.001, compared to control group; ###P < 0.001, comparedto ethanol group; @@@P < 0.001, compared to MDMA group. Representative light micrographs of hepatic immunohistochemistry for NF-�B detection are also shown (themagnification bar represents 20 �m). A cytoplasmic red staining is observed in all images, being more pronounced in the E, M and E + M pictures where some red stainednuclei are also depicted (arrow).
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
192

Author's personal copy
68 H. Pontes et al. / Toxicology 252 (2008) 64–71
Fig. 3. Transmission electron micrographs and light microscopy images (haematoxylin–eosin stain and Van Gieson’s stain) of liver sections from control, ethanol, MDMA andethanol + MDMA groups. In the light microscopy images, a preserved lobular structure is observed. The ethanol illustrations present dilatation of the centrilobular sinusoids.Necrotic cells are punctually observed in MDMA images (arrow). More confluent necrotic areas are evident in the ethanol + MDMA slide (arrow). In the slides stained with VanGieson, collagen content is poorly pronounced in control and MDMA images and more marked in ethanol exposed groups (arrow). The transmission electron micrographsreveal a mainly lipidic cytoplasmic vacuolization and mitochondrial swelling in all pictures, more pronounced in ethanol, MDMA and ethanol + MDMA images. Large collagencontents between endothelial and parenchymal cells are depicted in the ethanol picture (arrow). In the MDMA micrograph numerous nucleoli are also observed. An activatedKupffer cell is also shown in the ethanol + MDMA picture (arrow). The magnification bar on the light microscopy images represents 20 �m. The magnification bar on theelectron microscopy images represents 2 �m.
reduction of the size and density of the hepatocytes microvilli in thesinusoids and in the bile canaliculi. This high collagen depositionwas also evidenced by the Van Gieson stain, which revealed a higherdeposition of collagen fibres (coloured in red) in both ethanol andethanol + MDMA groups. In the ethanol group some nuclei pre-sented chromatin condensation on their peripheral regions. In theMDMA group, nuclei with numerous and voluminous nucleoli wereobserved and in both ethanol and MDMA groups it was also noticedthe existence of osmiophobic confluent areas, suggesting intracel-lular oedema. In the MDMA group, some isolated necrotic cellswere observed, while in the ethanol + MDMA group some appar-ently necrotic regions were found, as well as cell fragments in the
interstitial space. Kupffer cells with indicative signs of activation,suggested by high amounts of lysosomes, were also observed in theethanol + MDMA group.
3.5. Plasma transaminases
Concerning to GOT levels (Fig. 4A), they were slightly increasedin the ethanol group compared to the control group. The adminis-tration of MDMA produced a significant increase of plasma levelsof this transaminase in both MDMA and ethanol + MDMA groups,the increase in this last group being significantly higher (increaseof approximately 35%) than in the MDMA group (increase of
Fig. 4. Effect of 3,4-methylenedioxymethamphetamine (MDMA) on plasma levels of glutamic oxalic transaminase (GOT (A)), glutamic pyruvic transaminase (GPT (B)) ofcontrol and ethanol pre-treated animals. C: control, E: ethanol, M: MDMA and E + M: ethanol + MDMA groups. Results are presented as mean ± S.E.M. from six animals.*P < 0.05, ***P < 0.001.
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
193

Author's personal copy
H. Pontes et al. / Toxicology 252 (2008) 64–71 69
Table 1Hepatic percent dry lipid weight content (%DLWC) and liver relative organ weight(ROW) of mice from the control, ethanol, MDMA and ethanol + MDMA groups
Group Liver
ROW %DLWC
Control 4.359 ± 0.198 22.38 ± 1.89Ethanol 5.582 ± 0.293** 19.91 ± 1.57MDMA 3.686 ± 0.165### 30.93 ± 2.546*,##
Ethanol + MDMA 4.222 ± 0.261## 21.84 ± 1.53@
Results are presented as mean ± S.E.M. from six animals. All groups were compared,however, only the statistically differences were indicated. *P < 0.05, **P < 0.01, com-pared to control group; ##P < 0.01, ###P < 0.001, compared to ethanol group; @P < 0.05,compared to MDMA group.
approximately 30%) when compared to ethanol and control groups,respectively.
The levels of GPT (Fig. 4B) were significantly increased in MDMAand ethanol + MDMA groups, especially in the ethanol + MDMAwhich presented a twofold increase in GPT levels when compared tothe ethanol group. The increase of GPT present in the MDMA groupcorresponds to a 50% increase when compared to control group.
3.6. Relative organ weight (ROW) and hepatic percent dry lipidweight content (%DLWC)
As it can be observed in Table 1, the ethanol group presenteda significant increase in liver ROW, and the ethanol + MDMA groupshowed a marked decrease of this parameter when compared tothe ethanol group. The MDMA group presented a slight decreasein liver ROW without statistical significance. The MDMA group alsopresented a significant increase in the liver %DLWC when comparedto both control and ethanol groups.
3.7. Oxidative stress biomarkers (TBARS, carbonyl groups,GSH/GSSG)
No significant changes in hepatic lipid peroxidation wereobserved between control and MDMA groups, but a significantincrease in TBARS levels was observed in liver of animals fromthe ethanol group (compared to control group) that was not vis-ible in the ethanol + MDMA group (Table 2). The same occurs withthe hepatic GSH/GSSG ratio that suffers a significant reduction inthe ethanol group (comparing to control group), not observed inthe ethanol + MDMA group.
Additionally, there were found no important changes in hepaticprotein carbonyl groups.
3.8. Hepatic anti-oxidant enzymes
The MDMA group presented a significant increase in GST activ-ity while the ethanol + MDMA group had a significantly reducedGST activity compared to control group (Table 3). Hepatic GPx andcatalase activities did not present significant differences betweenthe control, ethanol and MDMA groups, but evidenced a significant
Table 2Oxidative stress parameters in the control, ethanol, MDMA and ethanol + MDMAgroups
Groups GSHred/GSSGratio
TBARS (nmolMDA/mg protein)
Carbonyl groups(nmol/mg protein)
Control 9.35 (0.62) 0.080 (0.004) 5.36 (0.33)Ethanol 6.73 (0.47)** 0.129 (0.015)** 5.76 (0.28)MDMA 7.75 (0.88) 0.097 (0.004) 4.63 (0.18)Ethanol + MDMA 7.43 (0.26) 0.073 (0.004)@ 5.04 (0.37)
Results are presented as mean ± (S.E.M.) from six animals. All groups were com-pared, however, only the statistically differences were indicated. *P < 0.05, **P < 0.01,***P < 0.001, compared to control group; #P < 0.05, ##P < 0.01, ###P < 0.001, comparedto ethanol group; @P < 0.05, @@P < 0.01, compared to MDMA group. MDA: malondi-aldehyde.
decrease in the ethanol + MDMA group. Ethanol group presentedreduced activities of both MnSOD and Cu/ZnSOD, when comparedto control group, and the ethanol + MDMA group showed a signifi-cant enhancement in MnSOD and Cu/ZnSOD activities.
4. Discussion
The results obtained in the present study clearly demonstratethat the chronic exposure to 12% ethanol prior to MDMA admin-istration potentiates the hyperthermic and hepatotoxic effectsinduced by MDMA, which was evidenced by a significant increasein several biochemical and histopathological biomarkers of toxicity.
The administration of MDMA was followed by a sharp increasein body temperature. This is an expected effect (Carvalho et al.,2002), which can be attributed to several central and peripheralmechanisms (Walubo and Seger, 1999; Dafters and Lynch, 1998).However, this is the first report of a potentiation of MDMA-relatedhyperthermia by ethanol pre-treatment resulting not only in signif-icantly higher body temperature scores but also in more sustainedhyperthermia than the one induced by MDMA alone. A corre-sponding higher increase of hepatic heat shock transcription factor(HSF-1) activation was observed (Fig. 1). This reflects a natural reac-tion of the liver to high temperatures and functions as a defencemechanism towards the hyperthermic aggression by increasingthe synthesis of heat shock proteins (HSPs). HSPs are highly con-served proteins with an important role in protein folding (actinglike molecular chaperones), signal transduction, cell growth anddifferentiation, and in the regulation of the actin cytoskeleton,contributing for cellular homeostasis (Pirkkala et al., 2001), coun-teracting heat shock and developing adaptation to oxidative stressto avoid cell death (Santos-Marques et al., 2006). This defence sys-tem may be disrupted if the hyperthermic effect surpasses thenormal physiological variations, or in the presence of other riskfactors. Indeed, animals exposed simultaneously to ethanol andMDMA and affected by sustained hyperthermia, may be more proneto deleterious phenomena.
The enhancement of the MDMA-induced hyperthermicresponse by ethanol may seem unexpected since ethanol is knownto induce vasodilatation and decrease body temperature (Huttunen
Table 3Hepatic antioxidant enzymes activities in the control, ethanol, MDMA and ethanol + MDMA groups
Groups GST (U/mg protein) GPx (U/mg protein) Catalase (U/mg protein) SOD (U/mg protein)
Mn SOD Cu/Zn SOD
Control 1112 ± 64 252.5 ± 21.4 182.9 ± 5.9 0.233 ± 0.024 3.071 ± 0.180Ethanol 1080 ± 123 191.2 ± 11.5 187.7 ± 9.5 0.150 ± 0.011** 2.252 ± 0.128**MDMA 1626 ± 121*,# 231.5 ± 19.3 183.1 ± 7.1 0.239 ± 0.020# 3.463 ± 0.185###
Ethanol + MDMA 885 ± 109@@ 173.9 ± 13.0* 149.0 ± 13.6# 0.259 ± 0.010## 3.123 ± 0.214#
Results are presented as mean ± S.E.M. from six animals. All groups were compared, however, only the statistically differences were indicated. *P < 0.05, **P < 0.01, ***P < 0.001,compared to control group; #P < 0.05, ##P < 0.01, ###P < 0.001, compared to ethanol group; @P < 0.05, @@P < 0.01, compared to MDMA group.
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
194

Author's personal copy
70 H. Pontes et al. / Toxicology 252 (2008) 64–71
et al., 1998). However, this hypothermic effect is only evident foracute ethanol administration and it is affected by tolerance phe-nomena that can appear as early as between two consecutiveexposures with a delay of 24 h after the first one (Khanna et al.,1996). The result obtained in the present study is in agreement witha previous report of Cassel et al., who showed that ethanol inhibitsthe MDMA-induced hyperthermia by the first day of treatment,but not on subsequent treatment days, suggesting a toleranceeffect on ethanol-induced hypothermia (Cassel et al., 2004).
Ethanol pre-treatment was also able to exacerbate MDMA-induced hepatotoxicity, which could be ascertained by thesignificant increase of plasma transaminases activities, biomark-ers of hepatic lesion, when animals were exposed to ethanoland MDMA. The increase in GOT and GPT activities were alreadydescribed for both compounds in humans (Ellis et al., 1996; Yue etal., 2006) and rats (Beitia et al., 2000; Montet et al., 2002). This hep-atotoxic effect was also confirmed by the decrease in liver weightwhen MDMA was administered to ethanol pre-exposed mice andby histological analysis of liver sections by light and electronmicroscopy, which gives further evidences that the concomitantexposure to MDMA and ethanol results in a marked aggravation ofthe hepatotoxic effects exerted by each one of the compounds atisolated exposures.
The marked increase in the activation of NF-�B, one of thetranscription factors involved in the activation of immediate earlyresponse genes in response to injurious and inflammatory stim-uli (Chen and Shi, 2002), indicates a pro-inflammatory effect asascertained by the observation of activated Kupffer cells and cor-responds to another cellular defence mechanism by increasing theactivity of antioxidant enzymes such as SOD (Lenart et al., 2007).However, the activation of both NF-�B and HSF-1 was not suffi-cient to protect cells from the toxicity of MDMA and ethanol despitebeing efficient in avoiding sharp modifications in oxidative stressparameters such as GSH/GSSG ratio, protein carbonyl groups con-tent and TBARS. Unchanged glutathione levels were also detectedin rats after a single dose of MDMA by Beitia et al. (2000). Theincrease in SOD activity due to MDMA administration verified inthis study was previously reported in the kidney by Ninkovic etal. (2008). The decrease of SOD activity following chronic ethanolconsumption is also in accordance with previous reports by Chenet al. (2002). The increase in SOD activity is a specific adaptivemechanism to a stress condition due to an increased superoxidegeneration associated with MDMA exposure. Increased expressionof SOD leads to removal of reactive superoxide radical anions, min-imizing the generation of cytotoxic peroxynitrite and terminatinglipid peroxidation-induced chain reactions.
Catalase and GPx are other antioxidant enzymes whose activi-ties protect cells from the toxicity of H2O2. The observed decreaseof their activities in the ethanol + MDMA group evidenced thatthe hepatic oxidative injury was significantly more intense whenMDMA was administered to ethanol pre-exposed mice when com-pared to the other studied groups. The effect of MDMA itself on theactivities of antioxidant enzymes confirmed the results previouslyobtained by our group showing no significant changes in the activ-ities of hepatic Mn SOD, Cu/Zn SOD, GPx and Catalase by 10 mg/kgMDMA (Carvalho et al., 2002). The only exception was GST, whichpresented an increase in its activity never reported before. The pre-treatment with 12% ethanol seems to avoid the observed increaseon GST activity provoked by the administration of MDMA, whichcan be explained by the GST-lowering effect already described forethanol (Pari and Suresh, 2008).
Our results also confirm the results of previous reports thataddressed an increase in hepatic lipid content to MDMA consump-tion (Beitia et al., 2000) probably due to an increase of �-adrenergicreceptors-dependent lipolysis in the skeletal muscle tissue (Unger,
1995), and the subsequent increase of circulating levels of free fattyacids (Sprague et al., 2007) that are thereafter taken up by the liver.However, in the present study, ethanol was not able to modify thiseffect despite being widely described that the chronic exposure toethanol leads to an increase in hepatic lipid content (de la Monteet al., 2008; Pritchard and Nagy, 2005).
In conclusion, the obtained results strongly suggest that the con-sumption of ethanol increases the toxic effects induced by MDMAexposure, with special relevance to its hyperthermic and hepato-toxic effects. If fact, signs of hepatic necrosis and inflammatoryresponse were evident and consistent with drug related hepatitis.
These results will certainly contribute for the understandingof the health risks undertaken by polydrug abusers who combineecstasy and ethanol and who were already associated with MDMA-related fatalities (Schifano et al., 2003).
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Acknowledgements
This work was supported by a project from FCT and POCI-2010, Portugal, by FEDER European Community co-funding(POCI/SAU-FCF/57187/2004). Helena Pontes was the recipientof a PhD grant from “Fundacão para a Ciência e Tecnologia”(SFRH/BD/18527/2004).
The authors would like to thank Doctor Georgina Correia-da-Silva from the Biochemistry Department of the Faculty of Pharmacyof the University of Porto for kindly provide the Wistar rat 14th dayplacenta slides.
References
Aebi, H., 1984. Catalase in vitro. Methods Enzymol. 105, 121–126.Barrett, S.P., Darredeau, C., Pihl, R.O., 2006. Patterns of simultaneaous polysubstance
use in drug using university students. Hum. Psychopharmacol. 21, 255–263.Beitia, G., Cobreros, A., Sainz, L., Cenarruzabeitia, E., 2000. Ecstasy-induced toxicity
in rat liver. Liver 20, 8–15.Ben Hamida, S., Bach, S., Plute, E., Jones, B.C., Kelche, C., Cassel, J.C., 2006. Ethanol-
ecstasy (MDMA) interactions in rats: preserved attenuation of hyperthermiaand potentiation of hyperactivity by ethanol despite prior ethanol treatment.Pharmacol. Biochem. Behav. 84, 162–168.
Breen, C., Degenhardt, L., Kinner, S., Bruno, R., Jenkinson, R., Matthews, A., Newman,J., 2006. Alcohol use and risk taking among regular ecstasy users. Subst. UseMisuse 41, 1095–1109.
Buege, J.A., Aust, S.D., 1978. Microsomal lipid peroxidation. Methods Enzymol. 52,302–310.
Carmo, H., Brulport, M., Hermes, M., Oesch, F., de Boer, D., Remião, F., Carvalho, F.,Schön, M.R., Krebsfaenger, N., Doehmer, J., Bastos, M.L., Hengstler, J.G., 2007.CYP2D6 increases toxicity of the designer drug 4-methylthioamphetamine (4-MTA). Toxicology 229, 236–244.
Carmo, H., Brulport, M., Hermes, M., Oesch, F., Silva, R., Ferreira, L.M., Branco,P.S., Boer, D., Remião, F., Carvalho, F., Schön, M.R., Krebsfaenger, N., Doehmer,J., Bastos, M.L., Hengstler, J.G., 2006. Influence of CYP2D6 polymorphism on3,4-methylenedioxymethamphetamine (‘Ecstasy’) cytotoxicity. Pharmacogenet.Genomics 16, 789–799.
Carmo, H., de Boer, D., Remião, F., Carvalho, F., dos Reys, L.A., de Lour-des Bastos, M., 2004a. Metabolism of the designer drug 4-bromo-2,5-dimethoxyphenethylamine (2C-B) in mice, after acute administration. J.Chromatogr. B Anal. Technol. Biomed. Life Sci. 811, 143–152.
Carmo, H., Hengstler, J.G., de Boer, D., Ringel, M., Carvalho, F., Fernandes, E., Remião, F.,dos Reys, L.A., Oesch, F., de Lourdes Bastos, M., 2004b. Comparative metabolismof the designer drug 4-methylthioamphetamine by hepatocytes from man, mon-key, dog, rabbit, rat and mouse. Naunyn Schmiedebergs Arch Pharmacol. 369,198–205.
Carmo, H., Hengstler, J.G., de Boer, D., Ringel, M., Remião, F., Carvalho, F., Fernandes,E., dos Reys, L.A., Oesch, F., de Lourdes Bastos, M., 2005. Metabolic pathways of4-bromo-2,5-dimethoxyphenethylamine (2C-B): analysis of phase I metabolismwith hepatocytes of six species including human. Toxicology 206, 75–89.
Carvalho, F., Remião, F., Soares, M.E., Catarino, R., Queiroz, G., Bastos, M.L., 1997. d-Amphetamine-induced hepatotoxicity: possible contribution of catecholaminesand hyperthermia to the effect studied in isolated rat hepatocytes. Arch. Toxicol.71, 429–436.
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
195

Author's personal copy
H. Pontes et al. / Toxicology 252 (2008) 64–71 71
Carvalho, M., Carvalho, F., Remiao, F., Pereira, M.L., Pires-das-Neves, R., Bastos, M.L.,2002. Effect of 3,4-methylenedioxymethamphetamine (“ecstasy”) on body tem-perature and liver antioxidant status in mice: influence of ambient temperature.Arch. Toxicol. 76, 166–172.
Carvalho, M., Remião, F., Milhazes, N., Borges, F., Fernandes, E., Carvalho, F., Bastos,M.L., 2004. The toxicity of N-methyl-alpha-methyldopamine to freshly isolatedrat hepatocytes is prevented by ascorbic acid and N-acetylcysteine. Toxicology200, 193–203.
Cassel, J.C., Jeltsch, H., Koenig, J., Jones, B.C., 2004. Locomotor and pyretic effects ofMDMA—ethanol associations in rats. Alcohol 34, 285–289.
Chen, F., Shi, X., 2002. Signaling from toxic metals to NF-kappaB and beyond: notjust a matter of reactive oxygen species. Environ. Health Perspect. 110, 807–811.
Chen, L.H., Thielen, V., Ciccia, R., Langlais, P.J., 2002. Effects of chronic ethanol feedingand thiamin deficiency on antioxidant defenses in kidney and lung of rats. Nutr.Res. 22, 835–845.
Dafters, R.I., Lynch, E., 1998. Persistent loss of thermoregulation in the rat inducedby 3,4-methylenedioxymethamphetamine (MDMA or “Ecstasy”) but not by fen-fluramine. Psychopharmacology (Berlin) 138, 207–212.
de la Monte, S.M., Yeon, J.E., Tong, M., Longato, L., Chaudhry, R., Pang, M.Y., Duan,K., Wands, J.R., 2008. Insulin resistance in experimental alcohol-induced liverdisease. J. Gastroenterol. Hepatol., doi:10.1111/j.1440-1746.2008.05339.x.
Dinis-Oliveira, R.J., Remião, F., Duarte, J.A., Ferreira, R., Sánchez Navarro, A., Bas-tos, M.L., Carvalho, F., 2006. P-glycoprotein induction: an antidotal pathway forparaquat-induced lung toxicity. Free Radic. Biol. Med. 41, 1213–1224.
Dinis-Oliveira, R.J., Sousa, C., Remião, F., Duarte, J.A., Navarro, A.S., Bastos, M.L.,Carvalho, F., 2007. Full survival of paraquat-exposed rats after treatment withsodium salicylate. Free Radic. Biol. Med. 42, 1017–1028.
Ellis, A.J., Wendon, J.A., Portmann, B., Williams, R., 1996. Acute liver damage andecstasy ingestion. Gut 38, 454–458.
Flohe, L., Gunzler, W.A., 1984. Assays of glutathione peroxidase. Methods Enzymol.105, 114–121.
Flohé, L., Ötting, F., 1984. Superoxide dismutase assays. Methods Enzymol. 105,93–104.
Folch, J., Lees, M., Sloane-Stanley, G.H., 1957. A simple method for the isolation andpurification of total lipids from animal tissues. J. Biol. Chem. 226, 497–509.
Gemma, S., Vichi, S., Testai, E., 2006. Individual susceptibility and alcoholeffects:biochemical and genetic aspects. Ann. Ist. Super Sanita 42, 8–16.
Green, A.R., Mechan, A.O., Elliott, J.M., O’Shea, E., Colado, M.I., 2003. The phar-macology and clinical pharmacology of 3,4-methylenedioxymethamphetamine(MDMA, “ecstasy”). Pharmacol. Rev. 55, 508–563.
Habig, W.H., Pabst, M.J., Jakoby, W.B., 1974. Glutathione S-transferases. The firstenzymatic step in mercapturic acid formation. J. Biol. Chem. 249, 7130–7139.
Hernández-López, C., Farré, M., Roset, P.N., Menoyo, E., Pizerro, N., Ortuno, J., Tor-rens, M., Cami, J., de la Torre, R., 2002. 3,4-Methylenedioxymethamphetamine(ecstasy) and alcohol interactions in humans: psychomotor performance, sub-jective effects, and pharmacokinetics. J. Pharmacol. Exp. Ther. 300, 236–244.
Huttunen, P., Sämpi, M., Myllylä, R., 1998. Ethanol-induced hypothermia and ther-mogenesis of brown adipose tissue in the rat. Alcohol 15, 315–318.
Institute for Laboratory Animal Research, 1996. Guide for the Care and Use of Labo-ratory Animals. National Academy Press, Washington, D.C.
Jang, G.R., Harris, R., 2007. Drug interactions involving ethanol and alcoholic bever-ages. Expert Opin. Drug Metab. Toxicol. 3, 719–731.
Khanna, J.M., Chau, A., Shah, G., 1996. Characterization of the phenomenon of rapidtolerance to ethanol. Alcohol 13, 621–628.
Lenart, J., Dombrowski, F., Görlach, A., Kietzmann, T., 2007. Deficiency of man-ganese superoxide dismutase in hepatocytes disrupts zonated gene expressionin mouse liver. Arch. Biochem. Biophys. 462, 238–244.
Levine, R.L., Williams, J.A., Stadtman, E.R., Shacter, E., 1994. Carbonyl assays for deter-mination of oxidatively modified proteins. Methods Enzymol. 233, 346–357.
Lowry, O.H., Rosebrough, N.J., Farr, A.L., Randall, R.J., 1951. Protein measurement withthe Folin phenol reagent. J. Biol. Chem. 193, 265–275.
Meier, P., Seitz, H.K., 2008. Age, alcohol metabolism and liver disease. Curr. Opin.Clin. Nutr. Metab. Care 11, 21–26.
Montet, A.M., Oliva, L., Beaugé, F., Montet, J.C., 2002. Bile salts modulate chronicethanol-induced hepatotoxicity. Alcohol Alcoholism 37, 25–29.
Ninkovic, M., Selakovic, V., Ethukic, M., Milosavljevic, P., Vasiljevic, I., Jovanovic,M., Malicevic, Z., 2008. Oxidative stress in rat kidneys due to 3,4-methylenedioxymetamphetamine (ecstasy) toxicity. Nephrology (Carlton) 13,33–37.
Pacifici, R., Zuccaro, R., Hernandez-Lopez, C., Pichini, S., Di Carlo, S., Farre, M.,Roset, P.N., Ortuno, J., Segura, J., Torre, R.L., 2001. Acute effects of 3,4-methylenedioxymethamphetamine alone and in combination with ethanol onthe immune system in humans. J. Pharmacol. Exp. Ther. 296, 207–215.
Pari, L., Suresh, A., 2008. Effect of grape (Vitis vinifera L.) leaf extract on alcoholinduced oxidative stress in rats. Food Chem. Toxicol. 46, 1627–1634.
Pirkkala, L., Nykänen, P., Sistonen, L., 2001. Roles of the heat shock transcrip-tion factors in regulation of the heat shock response and beyond. FASEB J. 15,1118–1131.
Pontes, H., Santos-Marques, M.J., Fernandes, E., Duarte, J.A., Remião, F., Carvalho, F.,Bastos, M.L., 2008. Effect of chronic ethanol exposure on the hepatotoxicity ofecstasy in mice. An ex-vivo study. Toxicol. In Vitro 22, 910–920.
Pritchard, M.T., Nagy, L.E., 2005. Ethanol-induced liver injury: potential roles foregr-1. Alcohol. Clin. Exp. Res. 29, 146S–150S.
Santos-Marques, M.J., Carvalho, F., Sousa, C., Remião, F., Vitorino, R., Amado, F., Fer-reira, R., Duarte, J.A., Bastos, M.L., 2006. Cytotoxicity and cell signalling inducedby continuous mild hyperthermia in freshly isolated mouse hepatocytes. Toxi-cology 224, 210–218.
Schifano, F., 2004. A bitter pill. Overview of ecstasy (MDMA, MDA) related fatalities.Psychopharmacology (Berlin) 173, 242–248.
Schifano, F., Oyefeso, A., Corkery, J., Cobain, K., Jambert-Gray, R., Martinotti, G.,Ghodse, A.H., 2003. Death rates from ecstasy (MDMA, MDA) and polydrug usein England and Wales 1996–2002. Hum. Psychopharmacol. 18, 519–524.
Sprague, J.E., Yang, X., Sommers, J., Gilman, T.L., Mills, E.M., 2007. Roles of nore-pinephrine, free fatty acids, thyroid status, and skeletal muscle uncouplingprotein 3 expression in sympathomimetic-induced thermogenesis. J. Pharmacol.Exp. Ther. 320, 274–280.
Unger, R.H., 1995. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM.Genetic and clinical implications. Diabetes 44, 863–870.
Walubo, A., Seger, D., 1999. Fatal multi-organ failure after suicidal overdose withMDMA, ‘ecstasy’: case report and review of the literature. Hum. Exp. Toxicol. 18,119–125.
Yue, M., Ni, Q., Yu, C.H., Ren, K.M., Chen, W.X., Li, Y.M., 2006. Transient elevation ofhepatic enzymes in volunteers after intake of alcohol. Hepatobiliary Pancreat.Dis. Int. 5, 52–55.
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
196

Trabalho III. Effect of chronic ethanol exposure on the
hepatotoxicity of ecstasy in mice: An ex vivo study.


Author's personal copy
Effect of chronic ethanol exposure on the hepatotoxicity of ecstasyin mice: An ex vivo study
Helena Pontes a,*, Maria Joao Santos-Marques a, Eduarda Fernandes b, Jose Alberto Duarte c,Fernando Remiao a, Felix Carvalho a, Maria Lourdes Bastos a,*
a REQUIMTE, Toxicology Department, Faculty of Pharmacy, University of Porto, Rua Anıbal Cunha 164, 4099-030 Porto, Portugalb REQUIMTE, Physical-Chemistry Department, Faculty of Pharmacy, University of Porto, Rua Anıbal Cunha 164, 4099-030 Porto, Portugal
c CIAFEL, Sport Biology Department, Faculty of Sport Science, University of Porto, Rua Dr. Placido Costa 91, 4200-450 Porto, Portugal
Received 19 October 2007; accepted 14 January 2008Available online 26 January 2008
Abstract
3,4-Methylenedioxymethamphetamine (MDMA) is frequently consumed at ‘‘rave” parties by polydrug users that usually take thisdrug in association with ethanol. In addition, many young people are repeatedly exposed to ethanol, which likely leads to tolerance phe-nomena. Both compounds are metabolized in the liver, with formation of hepatotoxic metabolites, which gives high relevance to theevaluation of their putative toxicological interaction. Therefore, the aim of this study was to evaluate the toxicity induced by 0.8 and1.6 mM MDMA to freshly isolated hepatocytes obtained from ethanol-treated mice whose tap drinking water was replaced by a 5% eth-anol solution for 1 week and, afterwards, by a 12% ethanol solution for 8 weeks (ethanol group) comparatively to non-treated animals(non-ethanol group). The hepatocytes were incubated under normothermic and hyperthermic conditions in order to simulate in vitro thehyperthermic response induced in vivo by MDMA, a condition that has been recognized as a life-threatening effect associated withMDMA exposure and implicated in its hepatotoxicity. Six mice treated under the same protocol as the ethanol group were used for his-tological analysis, and compared to non-ethanol-treated animals. The pre-treatment of mice with ethanol caused a significant decrease inthe hepatocytes yield in the isolation procedure comparatively to the non-ethanol group, which can be explained by an increase in col-lagen deposition along the hepatic parenchyma as observed in the histological analysis. The initial cell viability of hepatocytes suspen-sions was similar between ethanol and non-ethanol groups. However, the ethanol group showed a higher GSH oxidation rate, which wasenhanced under hyperthermia. Additionally, a concentration-dependent MDMA-induced loss of cell viability and ATP depletion wasobserved for both groups, at 41 �C. In conclusion, the repeated treatment with ethanol seems to increase the vulnerability of freshly iso-lated mice hepatocytes towards pro-oxidant conditions, as ascertained by the increase in collagen deposition, lower hepatocyte yield anddecreased glutathione levels. However, MDMA toxicity to the isolated hepatocytes was independent of ethanol pre-treatment, while sig-nificantly dependent on incubation temperature.� 2008 Elsevier Ltd. All rights reserved.
Keywords: Ethanol, 3,4-Methylenedioxymethamphetamine (MDMA); Hepatocytes; Oxidative stress
1. Introduction
3,4-Methylenedioxymethamphetamine (MDMA orecstasy), is a commonly used drug of abuse, whose con-
sumption appears to be spreading in many parts of theworld, with a high prevalence among young people(EMCDDA, 2005; SAMHSA, 2005). MDMA consump-tion results in a plethora of systemic and organ-specificeffects, including hepatic toxicity (Maurer et al., 2004)and also hyperthermia (Crean et al., 2006; Izco et al.,2007b) that can be, at least in part, responsible for thehepatic cellular damage (Santos-Marques et al., 2006).Various factors may contribute to MDMA-induced liver
0887-2333/$ - see front matter � 2008 Elsevier Ltd. All rights reserved.
doi:10.1016/j.tiv.2008.01.010
* Corresponding authors. Tel.: +351 222078979; fax: +351 222003977(H. Pontes).
E-mail addresses: [email protected] (H. Pontes), [email protected](M.L. Bastos).
www.elsevier.com/locate/toxinvit
Available online at www.sciencedirect.com
Toxicology in Vitro 22 (2008) 910–920
Helena
Rectangle
Helena
Text Box
199

Author's personal copy
toxicity, including the metabolism of MDMA (de la Torreet al., 2004), the increased efflux of neurotransmitters (de laTorre et al., 2004), the oxidation of biogenic amines (Carv-alho et al., 2004b), and hyperthermia (Green et al., 2004).Hyperthermia corresponds to a well known life-threateningeffect of MDMA in vivo (Hall and Henry, 2006) that ishighly dependent on ambient temperature (Carvalhoet al., 2002a; Von Huben et al., 2007). Our group hasalready shown that the in vitro toxic effects induced byMDMA in isolated mouse hepatocytes are exacerbatedunder hyperthermic conditions (Carvalho et al., 1997;Carvalho et al., 2001).
MDMA consumers are often polydrug abusers. Ethanol(EtOH) is, by far, the most popular drug among youth(Smart and Ogborne, 2000; Marques-Vidal and Dias,2005) and it is often consumed in large amounts along withMDMA use (Tossmann et al., 2001; Barrett et al., 2006). Infact, EtOH is often taken with MDMA, especially at thebeginning of the night, to get a stronger/better ‘‘high”
and, in the last part of the night, EtOH is taken in high dos-ages to decrease the long-lasting effects of MDMA such asirritability and restlessness that persist beyond its empath-ogenic and entactogenic effects (Schifano, 2004).
To study the putative interaction between these twocompounds, it is crucial to consider that many young peo-ple are exposed to EtOH early in life, sometimes evenrepeatedly, which means that many may have developedsome tolerance to EtOH when taking MDMA for the firsttime and concomitantly with EtOH (Hamida et al., 2006).Thus, the study of EtOH–MDMA interaction should notbe restricted to the acute consumption of these substances.It is of extreme importance to develop an experimentalmodel considering this common chronic abuse pattern,since the chronic exposure to EtOH prior to the first con-tact with MDMA may induce deleterious effects that caninfluence MDMA toxicity.
Both compounds, EtOH and MDMA, are metabolizedin the liver leading to the formation of hepatotoxic metab-olites. Most EtOH biotransformation occurs in the liver,mainly via oxidation catalyzed by alcohol dehydrogenase(ADH) and by a cytochrome P450 isoform (CYP2E1) intothe highly toxic metabolite, acetaldehyde (Gemma et al.,2006). The metabolism of MDMA involves N-demethyla-tion to 3,4-methylenedioxyamphetamine (MDA) and bothMDMA and MDA are O-demethylenated to catechols [N-methyl-a-methyldopamine (N-Me-a-MeDA) and a-meth-yldopamine (a-MeDA), respectively] that can undergo oxi-dation into the corresponding o-quinones (Carvalho et al.,2004b). These highly reactive molecules can alkylate crucialcellular proteins and/or DNA and further: (i) redox cycleoriginating semiquinone radicals, leading to the generationof reactive oxygen species (ROS) and reactive nitrogen spe-cies (RNS); (ii) undergo irreversible 1,4-intramolecularcyclization with subsequent formation of aminochromesand (iii) conjugate with glutathione (GSH) to form a gluta-thionyl adduct, which remains redox active and can furtherreact with GSH and protein thiols (Carvalho et al., 2004b).
Previous studies on MDMA and EtOH interactionshave focused on pharmacokinetic (Bilsky et al., 1990; Rez-vani et al., 1992; Hernandez-Lopez et al., 2002; Ramaekersand Kuypers, 2006), behavioural (Cassel et al., 2004; Casselet al., 2005; Hamida et al., 2006; Ben Hamida et al., 2007;Izco et al., 2007a) and physiologic and psychologic (Pacificiet al., 2001) consequences of these drugs combination. Nohepatic toxicological interactions between these com-pounds have been reported yet.
Thus, the aim of the present study was to evaluate,under normothermic (37 �C) and hyperthermic (41 �C)conditions, the toxic effects of MDMA in an ex vivo model,trying to simulate the most common pattern of EtOH andMDMA use and the hyperthermic response that occursin vivo as a consequence of MDMA consumption. For thispurpose, the toxicity induced by MDMA to freshly isolatedhepatocytes obtained from EtOH-treated mice was evalu-ated. The study included the quantification of the followingparameters: hepatocytes yield in the isolation procedureand initial cell viability, reduced (GSH) and oxidisedglutathione (GSSG) content, adenosine triphosphate(ATP) levels and lactate dehydrogenase (LDH) leakage.Furthermore, one separate group of ethanol-treated mice(that followed the same administration protocol as theethanol group) was used for histological analysis, andcompared to the control group.
2. Materials and methods
2.1. Chemicals
All reagents used in this study were of analyticalgrade. Collagenase (type I), bovine serum albumin (frac-tion V), 4-(2-hydroxyethyl)-piperazine-1-ethanesulfonicacid (HEPES), ethyleneglycol-bis-(b-aminoethylether)-N,N,N0,N0-tetraacetic acid (EGTA), b-nicotinamide ade-nine dinucleotide reduced form (b-NADH), b-nicotinamideadenine dinucleotide phosphate reduced form (b-NADPH), pyruvic acid, 2-vinylpyridine, reduced glutathi-one (GSH), oxidised glutathione (GSSG), glutathionereductase (EC 1.6.4.2), 5,5-dithiobis-2-nitrobenzoic acid(DTNB), trypan blue solution, trizma� hydrochloride, D-luciferin sodium salt and luciferase (EC 1.13.12.7) wereobtained from Sigma-Aldrich Co. (St. Louis, MO, USA).Perchloric acid, trichloroacetic acid, 2-thiobarbituric acid,absolute ethanol (EtOH) and all other chemicals were pur-chased from Merck (Darmstadt, Germany). 3,4-Methylen-edioxymethamphetamine (HCl salt) was extracted andpurified from high purity MDMA tablets that were pro-vided by the Portuguese Criminal Police Department.The obtained salt was pure and fully characterized byNMR and mass spectrometry methodologies.
2.2. Animals
Animal experiments were licensed by the PortugueseGeneral Directorate of Veterinary Medicine. Housing
H. Pontes et al. / Toxicology in Vitro 22 (2008) 910–920 911
Helena
Rectangle
Helena
Rectangle
Helena
Inserted Text
Dissertação de candidatura ao grau de Doutor em Toxicologia apresentada à Faculdade de Farmácia da Universidade do Porto Helena de Oliveira Pontes INTERACÇÕES TOXICOLÓGICAS ENTRE O ÁLCOOL E A ECSTASY. ESTUDOS IN VIVO E IN VITRO.
Helena
Text Box
200

Author's personal copy
and experimental treatment of animals were in accordancewith the Guide for the Care and Use of Laboratory Ani-mals from the Institute for Laboratory Animal Research(ILAR, 1996).
Adult male CD1 mice (Charles-River Laboratories, Bar-celona, Spain), weighing 35–45 g were used. For at least1 week prior to use, the animals were acclimatized in poly-ethylene cages, lined with wood shavings, with wire-meshtops, at an ambient temperature of 20 ± 2 �C, humiditybetween 40% and 60% and 12 h/12 h light/dark cycle (lighton from 8.00 to 20.00 hours), in our animal facilities, hav-ing standard chow and tap drinking water ad libitum. Afterthis adaptation period, the animals were divided into twogroups: EtOH and non-EtOH groups. In the EtOH groupthe tap drinking water was replaced by a 5% EtOH solu-tion for 1 week and, afterwards, by a 12% EtOH solutionfor 8 weeks until the hepatocytes isolation procedure. Thereplacement of water for 5% ethanol before 12% ethanolcorresponds to a required adaptation period, to avoid aver-sion of mice to a high ethanol concentration in the drinkingfluid, since these animals do not have natural preference for12% ethanol. The non-EtOH group remained drinking tapwater ad libitum, during the same period. Surgical proce-dures for the isolation of hepatocytes were performedunder anaesthesia and carried out between 10 a.m. and11 a.m.
Six mice from the EtOH group and six mice from thenon-EtOH group were used for histological analysis. After8 weeks of treatment, livers were excised under anaesthesiaand samples were processed for optic microscopy as indi-cated in 2.5.
2.3. Hepatocytes isolation and incubation
Hepatocytes isolation was performed by collagenaseperfusion as previously described (Carvalho et al.,2004b). Briefly, after anaesthesia, a cannula was introducedin the hepatic portal vein and a Hank solution with EGTAand albumin was perfused to remove the blood from thesinusoids and to chelate calcium, cleaving the hepatic des-mossomes. In a second stage, the liver was perfused with aHank solution including collagenase and its co-factor cal-cium. This enzyme hydrolyses the collagen molecules thatensure the mechanical stability of the liver. The third stepof this protocol consisted in a soft mechanical liver dissoci-ation in Krebs–Henseleit solution with albumin and thecellular suspension obtained was then purified by filtration,centrifugation and washings with Krebs–Henseleit buffersupplemented with 12.5 mM HEPES (pH 7.4) to removenon-parenchymatous cells, non-viable hepatocytes andother contaminants from the suspension. The viability ofthe isolated hepatocytes was estimated by the trypan blueexclusion test and was always higher than 80%.
Aliquots of isolated hepatocytes suspensions containing106 viable cells per mililiter in Krebs–Henseleit solutionsupplemented with 12.5 mM HEPES (pH 7.4) were pre-incubated, before the beginning of the experiments, in a
shaking water bath (90 oscillations/min) for 30 min at37 �C and continuously gassed with carbogen (95% O2/5% CO2). After this pre-incubation period, cells weredivided and the experiments were performed, in parallel,at 37 �C and 41 �C. Sample aliquots were collected at time0 and then the cells were incubated with MDMA hydro-chloride at final concentrations of 0.8 and 1.6 mM. Thetested MDMA concentrations were chosen based on previ-ous studies (Carvalho et al., 2001; Carvalho et al., 2004b)and on preliminary experiments, using this experimentalmodel. Additionally, liver concentrations of MDMA andits metabolites have been found to be substantially higher(up to 18 times) than blood concentrations (Garcia-Rep-etto et al., 2003). Taking into account that most cases ofserious toxicity or fatality have involved MDMA bloodlevels ranging from 2.5 to 50 lM, the concentrations usedin the present study are most likely attained in the in vivo
situation. On the other hand, it is important to evaluatethe interactions of MDMA and its metabolites with cellularcomponents, which is only possible using worst-caseapproach concentrations. The control cells were incubatedunder the same conditions with an equivalent volume ofKrebs–Henseleit solution. Additional sample aliquots werecollected every hour, during a period of incubation of 3 hand were used for the evaluation of cell viability, and quan-tification of GSH, GSSG, and ATP. The evaluation of cellviability was accomplished in the same day of the experi-ment, and the remaining samples were kept frozen(�80 �C) until assay.
2.4. Biochemical analysis
During the course of the experiments, cell viability wasdetermined by the lactate dehydrogenase (LDH) leakagemethod.
The GSH and GSSG contents in cell suspensions weredetermined by the DTNB-GSSG reductase recycling assayas described before (Carvalho et al., 2004a), with somemodifications. Briefly, cell suspension aliquots were precip-itated with equal volume of 10% HClO4 and centrifugedfor 2 min at 16,000g (4 �C). The supernatants were keptfrozen at –80 �C until quantification. The thawed acidicsupernatant was neutralized with equal volume of 0.76 MKHCO3 and centrifuged for 2 min at 16,000g (4 �C). Formeasurement of total glutathione (GSHt), 100 lL/well ofthe neutralized supernatants, standards or blank wereadded in triplicate to 96-well microtiter plates, followedby 65 lL/well of freshly prepared reagent containing0.69 mM NADPH and 4 mM DTNB in 72 mM phosphatebuffer. Plates were then incubated at 30 �C in a plate reader(BioTek Instruments, Vermont, US), for 15 min prior tothe addition of 40 lL/well of a 10 IU/mL glutathionereductase (GR) solution in phosphate buffer. The stoich-metric formation of 5-thio-2-nitrobenzoic acid (TNB) wasfollowed for 3 min at 415 nm and compared to a standardcurve. For the determination of GSSG, 10 lL of 2-vinyl-pyridine were added to 200 lL aliquots of acidic
912 H. Pontes et al. / Toxicology in Vitro 22 (2008) 910–920
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
201

Author's personal copy
supernatant and mixed continuously for 1 h for derivatiza-tion of GSH. GSSG was then measured as described abovefor total glutathione. The molar GSH levels were calcu-lated by subtracting the GSSG content from the total glu-tathione content (GSH = GSHt � 2 � GSSG).
ATP levels were evaluated through a bioluminescenttechnique based in an enzyme, luciferase, that catalysesthe formation of light from ATP and luciferin, as describedbefore (Capela et al., 2007). The emitted light intensity islinearly related to the ATP concentration and was mea-sured using a 96-well Microplate Luminometer (BioTekInstruments, Vermont, US). Samples in 5% HClO4 wereneutralized with 0.76 M KHCO3. After centrifugation for2 min at 16,000g (4 �C), 100 lL of the neutralized superna-tants were added to each well of a 96-well microplate con-taining 100 lL of luciferin–luciferase assay solution[0.15 mM luciferin, 300,000 light units of luciferase fromPhotinus pyralis (American firefly), 50 mM glycine,10 mM MgSO4, 1 mM Tris, 0.55 mM EDTA, 1%BSA(pH 7.6)]. ATP calibration curves were routinely performed(ATP standard stocks in HClO4 5% were kept at �80 �Cuntil the assay).
2.5. Tissue preparation for optic microscopy – histological
analysis
For optic microscopy, after excision, livers were sliced in2–4 mm3 pieces, approximately, and fixed in 4% formalde-hyde. The fixed pieces were dehydrated with graded etha-nol and included in paraffin blocks. Benzene was used inthe transition between dehydration and impregnation.Semi-thin sections (4 lm) were cut in a microtome, appliedon silane-coated slides and deparaffinized. The deparaffi-nated sections were stained for collagen detection withWeigert’s haematoxylin, acid alcohol (for differentiation)and Van Gieson’s staining solution. This stain evidencescollagen fibres with a red coloration.
2.6. Statistical analysis
Results are presented as mean ± S.E.M. (from at leastfive experiments of different preparations of hepatocytes).For hepatocyte yield analysis, comparisons were made byunpaired t-test with Welch’s correction. All other statisticalcomparisons were performed by one-way analysis of vari-ance (ANOVA) followed by the Bonferroni’s post-hoc test.Significance was accepted at P < 0.05.
3. Results
3.1. Hepatocytes yield and initial cell viability
During the perfusion step of the isolation procedure, thelivers of EtOH pre-treated animals offered high resistanceto perfusion, reducing the efficiency of this process, andresulting in significantly fewer viable cells at the end ofthe isolation procedure [42.7 ± 2.8 millions for the non-
EtOH group versus 31.1 ± 2.4 millions for the EtOH group(P < 0.01)]. Accordingly, in the histological analysis it wasobserved that liver sections of EtOH pre-treated animalspresented a higher deposition of collagen fibres than thecontrol animals along the hepatic parenchyma (Fig. 1).However, there were no statistical differences between cellviabilities of the hepatocytes suspensions obtained frommice of non-EtOH (87.8 ± 1.3%) or EtOH pre-treated(86.3 ± 1.1%) groups.
3.2. Toxicity parameters
The toxicity of MDMA to freshly isolated hepatocytesobtained from non-EtOH mice and mice repeatedlyexposed to 12% EtOH was evaluated, under normothermicand hyperthermic conditions, by measuring (i) the redoxstatus of the hepatocytes suspensions by the quantificationof total glutathione (GSHt), GSH and GSSG levels, (ii) theenergy status by determination of cellular ATP concentra-tions and (iii) the cell death-rate through the quantificationof LDH leakage to the suspension buffer.
3.2.1. Redox status of the hepatocytes suspensions3.2.1.1. Effect of EtOH pre-treatment. Both GSH andGSSG levels presented variations, which were dependenton mice EtOH pre-treatment, MDMA concentration, tem-perature and time of incubation (Fig. 2). The initial levelsof total and reduced glutathione were significantlydecreased in the hepatocytes from the EtOH-treated group(Fig. 2b and d). The non-EtOH group presented initialGSHt levels of 162.3 ± 11.4 nmol/106 cells and137.6 ± 11.2 nmol/106 cells of GSH while the EtOH grouppresented 101.1 ± 5.9 nmol/106 cells of GSHt (P < 0.01)and 71.0 ± 6.2 nmol/106 cells of GSH (P < 0.01). Theseresults correspond to a depletion of the GSHt levels andGSH levels of approximately 40% and 50%, respectively,in the EtOH-group. In accordance, the GSSG levels inthe EtOH group were significantly increased, around 25%(15.1 ± 1.0 nmol of GSSG/106 cells for the EtOH groupversus 12.4 ± 0.6 nmol of GSSG/106 cells for the non-EtOH group). However, it is interesting to notice that thevariation trends of GSH and GSSG levels during the 3 hperiod of hepatocytes incubation were similar betweenthe hepatocytes obtained from EtOH and non-EtOHgroups. In both groups, GSH decreased and GSSGincreased, in a time-, concentration- and temperature-dependent manner, although to a different extension.
3.2.1.2. Effect of hyperthermia. GSH oxidation was clearlyenhanced under hyperthermic conditions and significantlyincreased when mice were pre-treated with EtOH. In fact,after 3 h of incubation, the hepatocytes obtained fromthe non-EtOH group showed a GSH depletion more severeat 41 �C than at 37 �C (135.5 ± 48.0 nmol/106 cells at 37 �Cversus 87.6 ± 30.3 nmol/106 cells at 41 �C, for controlcells). Considering the EtOH-group, under normothermicconditions the GSH concentrations in the hepatocytes are
H. Pontes et al. / Toxicology in Vitro 22 (2008) 910–920 913
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
202

Author's personal copy
approximately half of the concentrations of the non-EtOHgroup (55.2 ± 21.2 nmol GSH/106 cells for EtOH groupversus 135.5 ± 48.0 nmol GSH/106 cells for non-EtOHgroup, considering control cells at 37 �C and t = 3 h).Under hyperthermia, these concentrations can reach values6 times lower than those in the hepatocytes obtained frommice not exposed to EtOH (19.9 ± 7.4 nmol GSH/106 cellsfor EtOH group versus 87.6 ± 30.3 nmol GSH/106 cells fornon-EtOH group, considering control cells at 41 �C andt = 3 h) (Fig. 2).
3.2.1.3. Effect of MDMA. Hepatocytes incubation withMDMA also increases their oxidative stress status. The cellsobtained from the non-EtOH group showed a GSH oxida-tion dependent on MDMA concentration, incubation timeand temperature conditions (Fig. 2). Under normothermicconditions there was a significant depletion of GSHobserved after 3 h of hepatocytes incubation with 1.6 mMMDMA [from 137.6 ± 11.2 nmol GSH/106 cells, att = 0 h, to 58.9 ± 16.7 nmol GSH/106 cells, at t = 3 h(P < 0.05)]. This GSH depletion was enhanced by hyper-thermia. In fact, at 41 �C, the GSH depletion was observed
earlier for 1.6 mM MDMA [GSH levels varied from137.6 ± 11.2 nmol GSH/106 cells at t = 0 h to 66.2 ± 20.1nmol GSH/106 cells at t = 2 h (P < 0.05) and to20.1 ± 19.0 nmol GSH/106 cells at t = 3 h (P < 0.001)] andalso observed after 3 h for hepatocytes incubation with0.8 mM MDMA [GSH levels varied from 137.6 ± 11.2nmol GSH/106 cells at t = 0 h to 61.6 ± 8.4 nmol GSH/106 cells (P < 0.05)]. In hepatocytes obtained from EtOHpre-treated mice, the GSH depletion induced by MDMAwas more pronounced, especially under hyperthermic con-ditions (44.4 ± 17.1 nmol GSH/106 cells at 37 �C versus
18.3 ± 5.8 nmol GSH/106 cells at 41 �C, for 1.6 mMMDMA at t = 3 h).
Differences in GSH depletion between MDMA concen-trations were not evident. However, hepatocytes obtainedfrom the EtOH-treated group showed, for both MDMAconcentrations, a higher depletion of GSH cellular levelscompared to the non-EtOH group. The initial levels ofGSSG were significantly higher for the EtOH-treatedgroup [15.1 ± 1.0 nmol GSSG/106 cells for EtOH groupversus 12.4 ± 0.6 nmol GSSG/106 cells for non-EtOHgroup (P < 0.01)]. A time-dependent GSSG increase was
Fig. 1. Histologycal analysis of liver sections obtained from control (a and b) and EtOH pre-treated animals (c and d) with Van Gieson staining forcollagen. The deposition of collagen fibres is evidenced by an increase of red coloured filaments.
914 H. Pontes et al. / Toxicology in Vitro 22 (2008) 910–920
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
203

Author's personal copy
observed in both groups (51.0 ± 7.8 nmol GSSG/106 cellsfor non-EtOH group and 1.6 mM MDMA at 37 �C andt = 3 h) and were significantly potentiated by hyperthermiaon the non-EtOH group (93.1 ± 8.0 nmol GSSG/106 cellsfor the non-EtOH group and 1.6 mM MDMA at 41 �Cand t = 3 h).
3.2.2. Cellular energy status
Fig. 3 illustrates the energy status of the isolated hepato-cytes under the different experimental conditions evaluatedby the quantification of ATP levels. No significant differ-ences were observed in the initial levels of ATP betweennon-EtOH and EtOH groups. In both groups, under nor-mothermic conditions, ATP levels remained unchangedduring all the experiment for control cells and bothMDMA concentrations tested. Under hyperthermic condi-tions ATP levels showed a significant concentration- andtime-dependent decrease. However, no differences wereobserved between EtOH and non-EtOH groups. In fact,for both groups, the ATP concentration, at t = 0 h, wasabout 45 lM decreasing, at t = 3 h, to levels nearly20 lM for control cells, 10 lM for cells incubated with
0.8 mM MDMA and 2 lM for cells incubated with1.6 mM MDMA.
3.2.3. Cell death-rateFig. 4 represents the death-rate of hepatocytes obtained
from non-EtOH and EtOH-treated mice, incubated underthe different conditions considered in this study, as evalu-ated by the LDH leakage. When hepatocytes were incu-bated at 37 �C (Fig. 4a and b), LDH leakage was similarto the control for EtOH and non-EtOH groups and forboth MDMA concentrations tested. The hepatocytes sus-pensions showed a time-dependent loss of cell viabilityexpressed by an increase of LDH leakage, which reachedsignificance at the 3 hour time-point (23.9 ± 0.9% ofLDH leakage at t = 0 h, compared to 43.2 ± 1.4% att = 3 h for control cells). Under hyperthermic conditions,the time-dependent loss of cell viability was aggravatedand dependent on MDMA concentration. For example,LDH leakage results were 55.1 ± 3.3%, 82.3 ± 3.9% and91.2 ± 0.4% for cells incubated with 0, 0.8 and 1.6 mMMDMA respectively, for EtOH group at t = 3 h(P < 0.001 comparing to t = 0 h and to normothermic
0 1 2 3 0 1 2 3 0 1 2 30
100
200
3001600μM MDMAControl 800μM MDMA
k k k
k k k
a abbbb
b a
bc c cc
c
Time (h)
Tota
lGSH
(nm
ol/m
illio
n ce
lls)
0 1 2 3 0 1 2 3 0 1 2 30
100
200
3001600μM MDMAControl 800μM MDMA
a
c
a
a c
cf c
fcfc
f
cf
cf
cd
Time (h)
Tota
lGSH
(nm
ol/m
illio
n ce
lls)
0 1 2 3 0 1 2 3 0 1 2 30
100
200
3001600μM MDMAControl 800μM MDMA
k k k
k k k
a
bb
c c
cc
Time (h)
Tota
l GSH
(nm
ol/m
illio
n ce
lls)
a
c
GSH GSSG
Non-EtOH group EtOH group
Nor
mot
herm
ia(3
7ºC
)H
yper
ther
mia
(41º
C)
GSH GSSG
GSH GSSG
GSH GSSG
0 1 2 3 0 1 2 3 0 1 2 30
100
200
3001600μM MDMAControl 800μM MDMA
a
c
c
c
ccc
b
Time (h)
Tota
lGSH
(nm
ol/m
illio
n ce
lls)
b
d
Fig. 2. Effect of MDMA and hyperthermia (c and d) on GSH and GSSG levels in freshly isolated mouse hepatocytes from non-EtOH (a and c) and EtOHgroups (b and d). Results are represented as mean ± SEM from at least five experiments. a P < 0.05, b P < 0.01, c P < 0.001, compared to time zero.d P < 0.05, f P < 0.001, compared to 37 �C. k P < 0.01, compared to non-EtOH group.
H. Pontes et al. / Toxicology in Vitro 22 (2008) 910–920 915
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
204

Author's personal copy
conditions). However, there were no significant differencesbetween the non-EtOH and EtOH groups.
4. Discussion
Our experimental observations clearly showed that therepeated in vivo treatment of mice with 12% EtOH for8 weeks compromised the hepatocyte yield after isolation.This handicap for the isolation of hepatocytes from etha-nol-treated animals was not reported before, although thislower hepatocyte yield can be explained by the occurrenceof collagen deposition during chronic EtOH consumption(Seitz and Stickel, 2006) as confirmed by the histologicaldata presented in Fig. 1, which can affect the hepatic perfu-sion of the collagenase solution and, therefore, the hepato-cytes isolation. In spite of the well known LDH releasingeffects of ethanol (Henzel et al., 2004), in the present study,the pre-treatment of mice with EtOH did not result in sta-tistical differences between the initial viability of hepato-cytes, obtained from EtOH and non-EtOH groups, whichis likely due to a selective isolation of EtOH tolerant hepa-
tocytes since, during the washing steps of the isolation pro-cedure, damaged cells were removed.
The toxic effects of EtOH pre-treatment were also evi-denced by the GSH content at the beginning of the exper-iment (137.6 ± 11.2 nmol GSH/106 cells for non-EtOHgroup versus 71.0 ± 6.2 nmol GSH/106 cells for EtOHgroup) and during the hepatocytes incubation period asexpressed by a severe depletion of glutathione levels, whichwere further depleted upon MDMA exposure. In the non-EtOH group, the cells incubated with MDMA underhyperthermic conditions presented a higher depletion ofGSH levels and a higher increase of GSSG levels thanthe correspondent control cells (neither exposed to ethanolnor to MDMA). GSH depletion after MDMA exposurewas previously reported in isolated rat (Carvalho et al.,2004b) and mouse (Carvalho et al., 2001) hepatocytes, asit happened in the present study. However, the GSH deple-tion induced by MDMA and/or hyperthermia was notaccompanied by a proportional increase in GSSG levels,which means that this GSH decrease was due, not onlyto glutathione oxidation, but mainly to the formation ofglutathione conjugates with MDMA reactive metabolites
0 1 2 3-5
0
5
10
15
20
25
30
35
40
45
50
55
c,f,i
c,f,i
d
c,f,g
a
c,d
a
Non-EtOH group Control Non-EtOH group 800μM MDMA Non-EtOH group 1600μM MDMA
ATP
Con
cent
ratio
n (μ
M)
Time (h)0 1 2 3
-5
0
5
10
15
20
25
30
35
40
45
50
55
a,fb,f
c,f,i
b,f,gc,f,g
c,f
EtOH group Control EtOH group 800μM MDMA EtOH group 1600μM MDMA
ATP
Con
cent
ratio
n (μ
M)
Time (h)
0 1 2 3-5
0
5
10
15
20
25
30
35
40
45
50
55
b
ATP
Con
cent
ratio
n (μ
M)
Time (h)
Non-EtOH group Control Non-EtOH group 800μM MDMA Non-EtOH group 1600μM MDMA
0 1 2 3-5
0
5
10
15
20
25
30
35
40
45
50
55
EtOH group Control EtOH group 800μM MDMA EtOH group 1600μM MDMA
ATP
Con
cent
ratio
n (μ
M)
Time (h)
a b
c d
Non-EtOH group EtOH group
Nor
mot
herm
ia(3
7ºC
)H
yper
ther
mia
(41º
C)
Fig. 3. Effect of MDMA and hyperthermia (c and d) on ATP levels in freshly isolated mouse hepatocytes from non-EtOH (a and c) and EtOH groups(b and d). Results are represented as mean ± SEM from at least five experiments. a P < 0.05, b P < 0.01, c P < 0.001, compared to time zero. d P < 0.05,f P < 0.001, compared to 37 �C. g P < 0.05, iP < 0.001 compared to control cells.
916 H. Pontes et al. / Toxicology in Vitro 22 (2008) 910–920
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
205

Author's personal copy
(Carvalho et al., 2002b; Carvalho et al., 2004a). Since GSHis a crucial endogenous antioxidant in cell protection (Hanet al., 2006), its depletion may increase the cell susceptibil-ity to the deleterious effects of reactive compounds, ROSand RNS, formed within the cells during the metabolismof MDMA and EtOH. However, the GSH depletion elic-ited by EtOH pre-treatment (Fig. 2) did not aggravatethe MDMA-induced cell death as demonstrated by thesimilar LDH leakage in both groups (Fig. 4). A possibleexplanation is the decreased formation of toxic GSH con-jugates with the catechol metabolites of MDMA, since lessGSH is available for conjugation. As already confirmed byour group, these conjugates are more toxic than their par-ent compounds for primary cultures of rat and humanrenal proximal tubular cells (Carvalho et al., 2002b) andfor rat cortical neuronal serum-free cultures (Capelaet al., 2006).
The GSH depletion that results from the EtOH pre-treatment is in accordance with the results previouslyobtained by other groups, which evidenced that long-term
incubation of isolated rat hepatocytes (Cobreros et al.,1997) and primary cultures of human and rat hepatocytes(Castilla et al., 2004; Yang et al., 2005) with high concen-trations of EtOH (P100 mM) increased oxidative stressmarkers such as lipid peroxidation and GSH depletion.Other studies demonstrated that either acute or chronicEtOH administration to experimental animals decreasedtissue levels of antioxidants, including GSH, in the liver(Nordmann et al., 1992), suggesting that the impairmentof cellular antioxidant defences along with the formationof ROS plays a role in causing oxidative damage associatedwith EtOH hepatotoxicity (Yang et al., 2005). On the otherhand, recent reports indicate that low EtOH concentra-tions (65 mM) were able to reduce the basal level of intra-cellular oxidative stress markers in primary cultures ofhuman and rat hepatocytes (Castilla et al., 2004) while,Deaciuc and colleagues described that GSH and GSSGcontents in hepatocytes were not affected when rats werefed during 7 weeks with an alcohol-containing liquid diet(Deaciuc et al., 1999).
0 1 2 3
10
20
30
40
50
60
70
80
90
100
c
a
cc
Non-EtOH group Control Non-EtOH group 800 μM MDMA Non-EtOH group 1600 μM MDMA
% L
DH
Lea
kage
Time (h)0 1 2 3
10
20
30
40
50
60
70
80
90
100
bbc
EtOH group Control EtOH group 800 μM MDMA EtOH group 1600 μM MDMA
% L
DH
Lea
kage
Time (h)
0 1 2 3
10
20
30
40
50
60
70
80
90
100
c,f,i
c,e,i
c,d,h
c,f,i
c
ca
Non-EtOH group Control Non-EtOH group 800 μM MDMA Non-EtOH group 1600 μM MDMA
% L
DH
Lea
kage
Time (h)0 1 2 3
10
20
30
40
50
60
70
80
90
100
c,f,i
c,f,h
a
c,f,i
c,f,g c,e
a
% L
DH
Lea
kage
Time (h)
EtOH group Control EtOH group 800 μM MDMA EtOH group 1600 μM MDMA
a b
c d
Non-EtOH group EtOH group
No
rmo
ther
mia
(37º
C)
Hyp
erth
erm
ia(4
1ºC
)
Fig. 4. Effect of MDMA and hyperthermia (c and d) on LDH leakage in freshly isolated mouse hepatocytes from non-EtOH (a and c) and EtOH groups(b and d). Results are represented as mean ± SEM from at least five experiments. a P < 0.05, b P < 0.01, c P < 0.001, compared to time zero. d P < 0.05,e P < 0.01, f P < 0.001, compared to 37 �C. g P < 0.05, h P < 0.01, i P < 0.001 compared to control cells.
H. Pontes et al. / Toxicology in Vitro 22 (2008) 910–920 917
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
206

Author's personal copy
Another toxic event observed in cells exposed toMDMA and EtOH was a concentration- and time-depen-dent depletion of cellular ATP levels that was significantunder hyperthermic conditions. This depletion confirmedthe results already described for MDMA alone (Carvalhoet al., 2004b) and denotes an inhibition of mitochondrialfunction under these experimental conditions (Burrowset al., 2000; Wu and Cederbaum, 2003; Quinton andYamamoto, 2006). This effect may result from altered thiolhomeostasis, and toxic metabolites formation fromMDMA and/or EtOH that can affect mitochondrial energyprocesses by damaging mitochondrial membranes and/orproteins (Seitz and Stickel, 2006). Additionally, the mito-chondrial dysfuntion underlying the observed ATP deple-tion could be directly instigated by hyperthermicconditions, as already described for tumoral cell lines andcardiomyocytes (Yuen et al., 2000; Qian et al., 2004; Zhaoet al., 2006).
Concluding, the results from the present study evi-denced, for the first time, that mice pre-treatment with12% EtOH during 8 weeks increases the vulnerability offreshly isolated mouse hepatocytes towards pro-oxidantconditions, as ascertained by the lower hepatocytes yieldand decreased GSH levels and increased GSSG levels. Thisdepletion of GSH content in the isolated hepatocytes alterstheir susceptibility to one of the early consequences ofMDMA metabolism (disruption of thiol homeostasis),which may result in loss of protein function and the initia-tion of a cascade of events leading to cellular damage.However, MDMA toxicity to the isolated hepatocytes,evaluated by LDH leakage, was independent of EtOHpre-treatment, while significantly dependent on incubationtemperature. Thus, the in vitro mimicking of the hyperther-mia induced in vivo by MDMA consumption was ofextreme importance since it significantly enhanced the del-eterious effects induced by MDMA at 37 �C for both non-EtOH and EtOH groups.
Notwithstanding the obtained results, caution should beexercised in comparing in vitro and in vivo situations,because there are some in vivo factors that could not bereproduced in this experimental model, such as uptakeand metabolism of different cell types, blood flux varia-tions, hormones and liver zonal differences in oxygen ten-sion and enzymatic distribution.
The relevance of this study is unequivocal because thesimultaneous consumption of MDMA and EtOH has beenassociated with MDMA-related fatalities (Schifano et al.,2003) and road accidents (Drummer et al., 2003; Chenget al., 2005; Kuypers et al., 2006). To our knowledge thisis the first report on the hepatic toxicological interactionof MDMA and EtOH, whose hepatic metabolism to toxicderivatives is already extensively described. Previous stud-ies on the association of these drugs have mainly focusedon physiologic or psychologic consequences to humans(Pacifici et al., 2001; Hernandez-Lopez et al., 2002; Ramae-kers and Kuypers, 2006).
Thus, the possible relationship of such findings requiresfurther in vivo testing to clarify the putative effects exertedby the combined use of MDMA and EtOH.
Acknowledgement
Helena Pontes thanks FCT for financial support(SFRH/BD/18527/2004).
References
Barrett, S.P., Darredeau, C., Pihl, R.O., 2006. Patterns of simultaneouspolysubstance use in drug using university students. Human Psycho-pharmacology 21, 255–263.
Ben Hamida, S., Plute, E., Bach, S., Lazarus, C., Tracqui, A., Kelche, C.,de Vasconcelos, A.P., Jones, B.C., Cassel, J.C., 2007. Ethanol–MDMA interactions in rats: the importance of interval betweenrepeated treatments in biobehavioral tolerance and sensitization to thecombination. Psychopharmacology (Berl) 192, 555–569.
Bilsky, E.J., Hui, Y.Z., Hubbell, C.L., Reid, L.D., 1990. Methylenedi-oxymethamphetamine’s capacity to establish place preferences andmodify intake of an alcoholic beverage. Pharmacology Biochemistryand Behavior 37, 633–638.
Burrows, K.B., Gudelsky, G., Yamamoto, B.K., 2000. Rapid andtransient inhibition of mitochondrial function following methamphet-amine or 3,4-methylenedioxymethamphetamine administration. Euro-pean Journal of Pharmacology 398, 11–18.
Capela, J.P., Meisel, A., Abreu, A.R., Branco, P.S., Ferreira, L.M., Lobo,A.M., Remiao, F., Bastos, M.L., Carvalho, F., 2006. Neurotoxicity ofecstasy metabolites in rat cortical neurons, and influence of hyperthermia.Journal of Pharmacology and Experimental Therapeutics 316, 53–61.
Capela, J.P., Macedo, C., Branco, P.S., Ferreira, L.M., Lobo, A.M.,Fernandes, E., Remiao, F., Bastos, M.L., Dirnagl, U., Meisel, A.,Carvalho, F., 2007. Neurotoxicity mechanisms of thioether ecstasymetabolites. Neuroscience 146, 1743–1757.
Carvalho, F., Remiao, F., Soares, M.E., Catarino, R., Queiroz, G.,Bastos, M.L., 1997. D-Amphetamine-induced hepatotoxicity: possiblecontribution of catecholamines and hyperthermia to the effect studiedin isolated rat hepatocytes. Archives of Toxicology 71, 429–436.
Carvalho, M., Carvalho, F., Bastos, M.L., 2001. Is hyperthermia thetriggering factor for hepatotoxicity induced by 3,4-methylenedioxy-methamphetamine (ecstasy)? An in vitro study using freshly isolatedmouse hepatocytes. Archives of Toxicology 74, 789–793.
Carvalho, M., Carvalho, F., Remiao, F., Pereira, M.L., Pires-das-Neves,R., Bastos, M.L., 2002a. Effect of 3,4-methylenedioxymethamphet-amine (‘‘ecstasy”) on body temperature and liver antioxidant status inmice: influence of ambient temperature. Archives of Toxicology 76,166–172.
Carvalho, M., Hawksworth, G., Milhazes, N., Borges, F., Monks, T.J.,Fernandes, E., Carvalho, F., Bastos, M.L., 2002b. Role of metabolitesin MDMA (ecstasy)-induced nephrotoxicity: an in vitro study using ratand human renal proximal tubular cells. Archives of Toxicology 76,581–588.
Carvalho, M., Milhazes, N., Remiao, F., Borges, F., Fernandes, E.,Amado, F., Monks, T.J., Carvalho, F., Bastos, M.L., 2004a. Hepa-totoxicity of 3,4-methylenedioxyamphetamine and alpha-methyldop-amine in isolated rat hepatocytes: formation of glutathione conjugates.Archives of Toxicology 78, 16–24.
Carvalho, M., Remiao, F., Milhazes, N., Borges, F., Fernandes, E.,Carvalho, F., Bastos, M.L., 2004b. The toxicity of N-methyl-alpha-methyldopamine to freshly isolated rat hepatocytes is prevented byascorbic acid and N-acetylcysteine. Toxicology 200, 193–203.
Cassel, J.C., Jeltsch, H., Koenig, J., Jones, B.C., 2004. Locomotor andpyretic effects of MDMA – ethanol associations in rats. Alcohol 34,285–289.
918 H. Pontes et al. / Toxicology in Vitro 22 (2008) 910–920
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
207

Author's personal copy
Cassel, J.C., Riegert, C., Rutz, S., Koenig, J., Rothmaier, K., Cosquer, B.,Lazarus, C., Birthelmer, A., Jeltsch, H., Jones, B.C., Jackisch, R.,2005. Ethanol, 3,4-methylenedioxymethamphetamine (ecstasy) andtheir combination: long-term behavioral, neurochemical and neuro-pharmacological effects in the rat. Neuropsychopharmacology 30,1870–1882.
Castilla, R., Gonzalez, R., Fouad, D., Fraga, E., Muntane, J., 2004. Dualeffect of ethanol on cell death in primary culture of human and rathepatocytes. Alcohol and Alcoholism 39, 290–296.
Cheng, J.Y., Chan, D.T., Mok, V.K., 2005. An epidemiological study onalcohol/drugs related fatal traffic crash cases of deceased drivers inHong Kong between 1996 and 2000. Forensic Science International153, 196–201.
Cobreros, A., Sainz, L., Lasheras, B., Cenarruzabeitia, E., 1997. Hepa-totoxicity of ethanol: protective effect of calcium channel blockers inisolated hepatocytes. Liver 17, 76–82.
Crean, R.D., Davis, S.A., Von Huben, S.N., Lay, C.C., Katner, S.N.,Taffe, M.A., 2006. Effects of (±)3,4-methylenedioxymethamphet-amine, (±)3,4-methylenedioxyamphetamine and methamphetamineon temperature and activity in rhesus macaques. Neuroscience 142,515–525.
de la Torre, R., Farre, M., Roset, P.N., Pizarro, N., Abanades, S., Segura,M., Segura, J., Cami, J., 2004. Human pharmacology of MDMA.Pharmacokinetics, metabolism and disposition. Therapeutic DrugMonitoring 26, 137–144.
Deaciuc, I.V., Fortunato, F., D’Souza, N.B., Hill, D.B., Schmidt, J., Lee,E.Y., McClain, C.J., 1999. Modulation of caspase-3 activity and Fasligand mRNA expression in rat liver cells in vivo by alcohol andlipopolysaccharide. Alcoholism: Clinical and Experimental Research23, 349–356.
Drummer, O.H., Gerostamoulos, J., Batziris, H., Chu, M., Caplehorn,J.R., Robertson, M.D., Swann, P., 2003. The incidence of drugs indrivers killed in Australian road traffic crashes. Forensic ScienceInternational 134, 154–162.
EMCDDA, 2005. Annual report 2005: The state of the drugs problem inEurope.
Garcia-Repetto, R., Moreno, E., Soriano, T., Jurado, C., Gimenez, M.P.,Menendez, M., 2003. Tissue concentrations of MDMA and itsmetabolite MDA in three fatal cases of overdose. Forensic ScienceInternational 135, 110–114.
Gemma, S., Vichi, S., Testai, E., 2006. Individual susceptibility andalcohol effects: biochemical and genetic aspects. Annali dell’IstitutoSuperiore di Sanita 42, 8–16.
Green, A.R., O’shea, E., Colado, M.I., 2004. A review of the mechanismsinvolved in the acute MDMA (ecstasy)-induced hyperthermicresponse. European Journal of Pharmacology 500, 3–13.
Hall, A.P., Henry, J.A., 2006. Acute toxic effects of ‘Ecstasy’ (MDMA)and related compounds: overview of pathophysiology and clinicalmanagement. British Journal of Anaesthesia 96, 678–685.
Hamida, S.B., Bach, S., Plute, E., Jones, B.C., Kelche, C., Cassel, J.C.,2006. Ethanol-ecstasy (MDMA) interactions in rats: preserved atten-uation of hyperthermia and potentiation of hyperactivity by ethanoldespite prior ethanol treatment. Pharmacology Biochemistry andBehavior 84, 162–168.
Han, D., Hanawa, N., Saberi, B., Kaplowitz, N., 2006. Mechanisms ofliver injury. III. Role of glutathione redox status in liver injury.American Journal of Physiology – Gastrointestinal and Liver Phys-iology 291, G1–G7.
Henzel, K., Thorborg, C., Hofmann, M., Zimmer, G., Leuschner, U.,2004. Toxicity of ethanol and acetaldehyde in hepatocytes treated withursodeoxycholic or tauroursodeoxycholic acid. Biochimica et Biophy-sica Acta 1644, 37–45.
Hernandez-Lopez, C., Farre, M., Roset, P.N., Menoyo, E., Pizerro, N.,Ortuno, J., Torrens, M., Cami, J., de la Torre, R., 2002. 3,4-Methylenedioxymethamphetamine (ecstasy) and alcohol interactionsin humans: psychomotor performance, subjective effects, and phar-macokinetics. Journal of Pharmacology and Experimental Therapeu-tics 300, 236–244.
ILAR, 1996. Guide for the Care and Use of Laboratory Animals.National Academy Press, Washington, DC.
Izco, M., Marchant, I., Escobedo, I., Peraile, I., Delgado, M., Higuera-Matas, A., Olias, O., Ambrosio, E., O’Shea, E., Colado, M.I., 2007a.Mice with decreased cerebral dopamine function following a neuro-toxic dose of MDMA (3,4-methylenedioxymethamphetamine,‘‘Ecstasy”) exhibit increased ethanol consumption and preference.Journal of Pharmacology and Experimental Therapeutics 322, 1003–1012.
Izco, M., Orio, L., O’shea, E., Colado, M.I., 2007b. Binge ethanoladministration enhances the MDMA-induced long-term 5-HT neuro-toxicity in rat brain. Psychopharmacology (Berl) 189, 459–470.
Kuypers, K.P.C., Samyn, N., Ramaeckers, J.G., 2006. MDMA andalcohol effects, combined and alone, on objective and subjectivemeasures of actual driving performance and psychomotor function.Psychopharmacology 187, 467–475.
Marques-Vidal, P., Dias, C.M., 2005. Trends and determinants of alcoholconsumption in Portugal: results from the national health surveys 1995to 1996 and 1998 to 1999. Alcoholism: Clinical and ExperimentalResearch 29, 89–97.
Maurer, H.H., Kraemer, T., Springer, D., Staack, R.F., 2004. Chemistry,pharmacology, toxicology, and hepatic metabolism of designer drugsof the amphetamine (ecstasy), piperazine, and pyrrolidinophenonetypes: a synopsis. Therapeutic Drug Monitoring 26, 127–131.
Nordmann, R., Ribiere, C., Rouach, H., 1992. Implication of free radicalmechanisms in ethanol-induced cellular injury. Free Radical Biologyand Medicine 12, 219–240.
Pacifici, R., Zuccaro, R., Hernandez-Lopez, C., Pichini, S., Di Carlo, S.,Farre, M., Roset, P.N., Ortuno, J., Segura, J., Torre, R.L., 2001. Acuteeffects of 3,4-methylenedioxymethamphetamine alone and in combi-nation with ethanol on the immune system in humans. Journal ofPharmacology and Experimental Therapeutics 296, 207–215.
Qian, L., Song, X., Ren, H., Gong, J., Cheng, S., 2004. Mitochondrialmechanism of heat stress-induced injury in rat cardiomyocyte. CellStress & Chaperones 9, 281–293.
Quinton, M.S., Yamamoto, B.K., 2006. Causes and consequences ofmethamphetamine and MDMA toxicity. American Association ofPharmaceutical Scientists Journal 8, E337–E347.
Ramaekers, J.G., Kuypers, K.P., 2006. Acute effects of 3,4-methylenedi-oxymethamphetamine (MDMA) on behavioral measures of impulsiv-ity: alone and in combination with alcohol. Neuropsychopharmacology31, 1048–1055.
Rezvani, A.H., Garges, P.L., Miller, D.B., Gordon, C.J., 1992. Attenu-ation of alcohol consumption by MDMA (ecstasy) in two strains ofalcohol-preferring rats. Pharmacology Biochemistry and Behavior 43,103–110.
SAMHSA, 2005. 2005 National Survey on Drug Use and Health:National Findings.
Santos-Marques, M.J., Carvalho, F., Sousa, C., Remiao, F., Vitorino, R.,Amado, F., Ferreira, R., Duarte, J.A., Bastos, M.L., 2006. Cytotox-icity and cell signalling induced by continuous mild hyperthermia infreshly isolated mouse hepatocytes. Toxicology 224, 210–218.
Schifano, F., 2004. A bitter pill. Overview of ecstasy (MDMA, MDA)related fatalities. Psychopharmacology (Berl) 173, 242–248.
Schifano, F., Oyefeso, A., Corkery, J., Cobain, K., Jambert-Gray, R.,Martinotti, G., Ghodse, A.H., 2003. Death rates from ecstasy(MDMA, MDA) and polydrug use in England and Wales 1996–2002. Human Psychopharmacology 18, 519–524.
Seitz, H.K., Stickel, F., 2006. Risk factors and mechanisms of hepatocar-cinogenesis with special emphasis on alcohol and oxidative stress.Biological Chemistry 387, 349–360.
Smart, R.G., Ogborne, A.C., 2000. Drug use and drinking among studentsin 36 countries. Addictive Behaviours 25, 455–460.
Tossmann, P., Boldt, S., Tensil, M.D., 2001. The use of drugs within thetechno party scene in European metropolitan cities. European Addic-tion Research 7, 2–23.
Von Huben, S.N., Lay, C.C., Crean, R.D., Davis, S.A., Katner, S.N.,Taffe, M.A., 2007. Impact of ambient temperature on hyperthermia
H. Pontes et al. / Toxicology in Vitro 22 (2008) 910–920 919
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
208

Author's personal copy
induced by (±)3,4-methylenedioxymethamphetamine in Rhesus Maca-ques. Neuropsychopharmacology 32, 673–681.
Wu, D., Cederbaum, A.I., 2003. Alcohol, oxidative stress, and free radicaldamage. Alcohol Research & Health 27, 277–284.
Yang, S.S., Huang, C.C., Chen, J.R., Chiu, C.L., Shieh, M.J., Lin, S.J.,Yang, S.C., 2005. Effects of ethanol on antioxidant capacity in isolatedrat hepatocytes. World Journal of Gastroenterology 11, 7272–7276.
Yuen, W.F., Fung, K.P., Lee, C.Y., Choy, Y.M., Kong, S.K., Ko, S.,Kwok, T.T., 2000. Hyperthermia and tumour necrosis factor-alphainduced apoptosis via mitochondrial damage. Life Sciences 67, 725–732.
Zhao, Q.L., Fujiwara, Y., Kondo, T., 2006. Mechanism of cell deathinduction by nitroxide and hyperthermia. Free Radical Biology andMedicine 40, 1131–1143.
920 H. Pontes et al. / Toxicology in Vitro 22 (2008) 910–920
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
209

Helena
Text Box
210

Trabalho IV. Synergistic toxicity of ethanol and MDMA towards primary
cultured rat hepatocytes.


Author's personal copy
Toxicology 254 (2008) 42–50
Contents lists available at ScienceDirect
Toxicology
journa l homepage: www.e lsev ier .com/ locate / tox ico l
Synergistic toxicity of ethanol and MDMA towards primary cultured rathepatocytes
Helena Pontesa,∗, Carla Sousaa, Renata Silvaa, Eduarda Fernandesb, Helena Carmoa,Fernando Remiãoa, Félix Carvalhoa, Maria Lourdes Bastosa,∗
a REQUIMTE, Toxicology Department, Faculty of Pharmacy, University of Porto, Rua Aníbal Cunha 164, 4099-030 Porto, Portugalb REQUIMTE, Physical-Chemistry Department, Faculty of Pharmacy, University of Porto, Rua Aníbal Cunha 164, 4099-030 Porto, Portugal
a r t i c l e i n f o
Article history:Received 14 July 2008Received in revised form 3 September 2008Accepted 4 September 2008Available online 20 September 2008
Keywords:Ethanol3,4-MethylenedioxymethamphetaminePrimary cultured rat hepatocytesHepatotoxicityOxidative stressHyperthermia
a b s t r a c t
Ethanol is frequently consumed along with 3,4-methylenedioxymethamphetamine (MDMA; ecstasy).Since both compounds are hepatotoxic and are metabolized in the liver, an increased deleterious interac-tion resulting from the concomitant use of these two drugs seems plausible. Another important featureof MDMA-induced toxicity is hyperthermia, an effect known to be potentiated after continuous exposureto ethanol. Considering the potential deleterious interaction, the aim of the present study was to evaluatethe hepatotoxic effects of ethanol and MDMA mixtures to primary cultured rat hepatocytes and to elu-cidate the mechanism(s) underlying this interaction. For this purpose, the toxicity induced by MDMA toprimary cultured rat hepatocytes in absence or in presence of ethanol was evaluated, under normothermic(36.5 ◦C) and hyperthermic (40.5 ◦C) conditions. While MDMA and ethanol, by themselves, had discreteeffects on the analysed parameters, which were slightly aggravated under hyperthermia, the simultaneousincubation of MDMA and ethanol for 24 h, resulted in high cell death ratios accompanied by a significantdisturbance of cellular redox status and decreased energy levels. Evaluation of apoptotic/necrotic fea-tures provided clear evidences that the cell death occurs preferentially through a necrotic pathway. Allthe evaluated parameters were dramatically aggravated when cells were incubated under hyperthermia.In conclusion, co-exposure of hepatocytes to ethanol and MDMA definitely results in a synergism of thehepatotoxic effects, through a disruption of the cellular redox status and enhanced cell death by a necroticpathway in a temperature-dependent extent.
© 2008 Elsevier Ireland Ltd. All rights reserved.
1. Introduction
3,4-Methylenedioxymethamphetamine (ecstasy; MDMA) con-sumption patterns have been widely described in literature(EMCDDA, 2005; Lora-Tamayo et al., 2004; SAMHSA, 2005;Schifano et al., 2006; Topp et al., 1999; Tossmann et al., 2001;Winstock et al., 2001) and are generally characterized as a polydrugconsumption scenario (Gouzoulis-Mayfrank and Daumann, 2006;Hopper et al., 2006; Schifano et al., 1998, 2003), often related withfatalities (Barrett et al., 2006; Liechti et al., 2005; Oyefeso et al.,2006; Sanjurjo et al., 2004; Schifano, 2004; Schifano et al., 2003,
Abbreviations: MDMA, 3,4-methylenedioxymethamphetamine; EtOH, ethanol;LDH, lactate dehydrogenase; AO/EB, acridine orange/ethidium bromide; ROS, reac-tive oxygen species; RNS, reactive nitrogen species; GSH, reduced glutathione; GSSG,oxidised glutathione; ATP, adenosine-5′-triphosphate; �-GCS, �-glutamylcysteinesynthetase; AIF, apoptosis-inducing factor.
∗ Corresponding authors. Tel.: +351 222078979; fax: +351 222003977.E-mail addresses: [email protected] (H. Pontes), [email protected] (M.L. Bastos).
2006). One of the most commonly consumed drugs, along withecstasy, is ethanol (EtOH) (Breen et al., 2006; Smart and Ogborne,2000; Tossmann et al., 2001), probably due to the fact that EtOHis legal, cheap and widely available. In fact, EtOH is often takenprior or along with MDMA, to increase the acute pleasant effects ofMDMA and to decrease the irritability and restlessness that persistafter the end of the empathogenic and entactogenic effects asso-ciated with ecstasy consumption (Schifano, 2004). Of note, whenEtOH is consumed to decrease the long-lasting unpleasant effectsof MDMA, it is taken in large amounts (Schifano, 2004) probablybecause ecstasy users were more likely to engage in several riskybehaviours such as binge drinking (Boyd et al., 2003).
Liver toxicity resulting from the concomitant consumption ofthese two drugs is of higher concern since both compounds aremetabolized in the liver leading to the formation of hepatotoxiccompounds (Gemma et al., 2006; Green et al., 2003). Accordingly,our group has recently demonstrated that hepatocytes isolatedfrom mice exposed to 12% EtOH for 8 weeks, as drinking fluid,showed increased vulnerability to MDMA-induced hepatotoxicity(Pontes et al., 2008b). EtOH pre-exposure has also been shown
0300-483X/$ – see front matter © 2008 Elsevier Ireland Ltd. All rights reserved.doi:10.1016/j.tox.2008.09.009
Helena
Rectangle
Helena
Text Box
213

Author's personal copy
H. Pontes et al. / Toxicology 254 (2008) 42–50 43
to modify MDMA pharmacokinetics, which results in increasedMDMA plasma concentrations in humans (Hernández-López et al.,2002). Another important feature of MDMA-induced toxicity ishyperthermia, an effect known to be potentiated after continuousexposure to ethanol (Pontes et al., 2008a).
Considering the potential synergic effects of EtOH and MDMAto the liver, the aim of the present study was to evaluate the puta-tive hepatotoxic interaction between EtOH and MDMA in primarycultured rat hepatocytes and to elucidate the mechanism(s) under-lying the observed effects. For this purpose, the toxicity inducedby MDMA to primary cultured rat hepatocytes in absence or inpresence of EtOH was evaluated, under normothermic (36.5 ◦C)and hyperthermic (40.5 ◦C) conditions, to simulate the hyperther-mic response induced in vivo by MDMA consumption. The studyincluded the evaluation of the type of cell death [acridine orangeand ethidium bromide (AO/EB) staining, lactate dehydrogenase(LDH) leakage, caspase-3 activation, Bax translocation to the mito-chondria and AIF and cytochrome c release from the mitochondria],oxidative stress parameters [reactive oxygen and nitrogen species(ROS and RNS) formation, reduced (GSH) and oxidised glutathione(GSSG) contents, �-glutamylcysteine synthetase (�-GCS) and GSTactivities] as well as cell energy status [adenosine triphosphate(ATP) levels].
2. Materials and methods
2.1. Chemicals
All chemicals used in this study were of analytical grade. Collagenase(type I), bovine serum albumin (fraction V), 4-(2-hydroxyethyl)-piperazine-1-ethanesulfonic acid (HEPES), ethyleneglycol-bis-(�-aminoethylether)-N,N,N′ ,N′-tetraacetic acid (EGTA), �-nicotinamide adenine dinucleotide reduced form(�-NADH), �-nicotinamide adenine dinucleotide phosphate reduced form (�-NADPH), pyruvic acid, 2-vinylpyridine, reduced glutathione (GSH), oxidisedglutathione (GSSG), glutathione reductase (EC 1.6.4.2), 5,5-dithiobis-2-nitrobenzoicacid (DTNB), trypan blue solution, trizma® hydrochloride, d-luciferin sodiumsalt and luciferase (EC 1.13.12.7), antibiotic–antimycotic solution, insulin solu-tion, dexamethasone, acridine orange, ethidium bromide, dihydrorhodamine123, mono(cyclohexylammonium)phosphoenolpyruvate, l-�-amino-n-butyrate, l-glutamate, pyruvate kinase from rabbit muscle and l-lactic dehydrogenasefrom rabbit muscle, adenosine-5′-triphosphate (ATP), N-Acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) were obtained from Sigma–Aldrich Co. (St. Louis, MO,USA). Perchloric acid, EtOH and all other chemicals were purchased from Merck(Darmstadt, Germany). William’s E medium, l-glutamine, foetal bovine serum(FBS) and phosphate buffer solution (PBS) were purchased from Lonza Ltd. (Basel,Switzerland). Collagen G was purchased from Biochrom AG (Berlin, Germany).3,4-Methylenedioxymethamphetamine (HCl salt) was extracted and purified fromhigh purity MDMA tablets that were provided by the Portuguese Criminal PoliceDepartment. The obtained salt was pure and fully characterized by NMR and massspectrometry methodologies.
2.2. Animals
Animal experiments were licensed by the Portuguese General Directorate ofVeterinary Medicine. Housing and experimental treatment of animals were in accor-dance with the Guide for the Care and Use of Laboratory Animals from the Institutefor Laboratory Animal Research (Institute for Laboratory Animal Research, 1996).
Adult male Wistar rats (Charles-River Laboratories, Barcelona, Spain), weighing250–300 g, were used. For at least 1 week prior to use, animals were acclimatized inpolyethylene cages, lined with wood shavings, with wire-mesh tops, at an ambienttemperature of 20 ± 2 ◦C, humidity between 40% and 60% and 12/12 h light/dark cycle(light on from 8.00 to 20.00 h), in our animal facilities, having standard chow and tapdrinking water ad libitum. Surgical procedures for the isolation of hepatocytes wereperformed under anaesthesia and carried out between 10.00 h a.m. and 11.00 h a.m.
2.3. Hepatocytes isolation and primary culture
Hepatocytes isolation was performed by collagenase perfusion as previouslydescribed (Pontes et al., 2008b) with some modifications. Briefly, after perfusionwith a chelation agent to allow the cleavage of the hepatic desmosomes, hepatic col-lagen was hydrolysed by ex situ perfusion with a collagenase solution supplementedwith its co-factor (calcium) and hepatocytes were dissociated in Krebs–Henseleitbuffer.
The obtained hepatocyte suspension was purified by low-speed centrifugationsand incubated for 30 min, at 4 ◦C, with 500 U/mL penicillin G, 0.5 mg/mL strepto-mycin sulfate and 1.25 �g/mL amphotericin B. The viability of isolated hepatocyteswas estimated by the trypan blue exclusion test. Subsequently, a suspension of0.5 × 106 viable cells/mL in complete culture medium (William’s E medium supple-mented with 2 mM l-glutamine, 0.86 �M insulin, 0.5 nM dexamethasone, 10 mMHEPES, 100 U/mL penicillin G, 0.1 mg/mL streptomycin sulfate, 0.25 �g/mL ampho-tericin B, 5% FBS) was seeded in previously collagen-coated culture flasks in a densityof 6 × 104 viable cells/cm2. Cells were incubated at 36.5 ◦C with 5% CO2, for 3–4 hfor cell adhesion, washed twice with PBS, and incubated over-night at 36.5 ◦C withculture medium without FBS. The day after, cells were added five different concen-trations of MDMA (0, 0.2, 0.4, 0.8 and 1.6 mM) in the presence or in the absence of1.4% (m/v) EtOH, and incubated with 5% CO2 at 36.5 or 40.5 ◦C. The tested MDMA con-centrations were chosen on the basis of previous studies from our group (Carvalhoet al., 2001, 2004b) and also in previous reports that found tissue concentrations ofMDMA and its metabolites substantially higher (up to 18 times) than blood concen-trations (Garcia-Repetto et al., 2003). Considering that in most of MDMA fatalities,blood levels ranged from 2.5 to 50 �M, the concentrations used in the present studyare most likely attained in the in vivo situation. On the other hand, the evaluationof the interactions of MDMA and its metabolites with cellular components is onlypossible using worst-case approach concentrations. After 24 h incubation, AO/EBstaining was performed, and samples were collected to perform Western blottinganalysis and to quantify LDH leakage, ROS and RNS formation, ATP levels, GSH andGSSG concentrations and �-GCS, GST and caspase-3 activities.
The evaluation of cell viability, the AO/EB staining, the quantification of ROS andRNS and the preparation of mitochondrial protein extracts to perform Western blotwere accomplished immediately after the 24-h incubation period. The remainingsamples were kept frozen (−80 ◦C) until assay.
2.4. Acridine orange and ethidium bromide staining
The fluorescent DNA-binding dyes ethidium bromide and acridine orange wereused to distinguish necrotic from apoptotic cells. After the 24-h incubation periodwith MDMA, EtOH or MDMA + EtOH, cells were incubated with 2 �g/mL acridineorange and 2 �g/mL ethidium bromide for 5 min before imaging, using a fluores-cence microscope with a standard fluorescein excitation filter (Eclipse E400 Nikon,Japan).
2.5. Biochemical analysis
2.5.1. LDH LeakageLDH leakage to the culture media was evaluated as previously reported
(Bergermeyer and Bernt, 1974). The LDH activity was determined by following therate of oxidation of NADH, measured at 340 nm. Results are presented as percentageof control conditions at 36.5 ◦C.
2.5.2. ROS and RNS quantificationDetection and quantification of intracellular reactive species, including RNS and
ROS were performed by the dihydrorhodamine 123 (DHR-123) assay as previouslyreported (Capela et al., 2007). Briefly, cells were pre-incubated with 100 �M DHR-123 for 60 min. At the end of the incubation period, cells were washed 2 times withPBS and added with new medium containing the tested compounds. The oxidationof DHR-123 to its fluorescent product, Rhodamine 123, was measured 24 h afterincubation with EtOH, MDMA, or EtOH + MDMA, using a fluorescence plate reader(BioTek Instruments, Winooski, VT, USA) (baseline: 485 nm excitation and 528 nmemission). Results are presented as percentage of control conditions at 36.5 ◦C.
2.5.3. GSH/GSSG quantificationThe cellular GSH and GSSG levels were determined, after cell suspension in PBS,
by the DTNB–GSSG reductase recycling assay as described before (Anderson, 1985),with some modifications (Pontes et al., 2008b). The stoichiometric formation of 5-thio-2-nitrobenzoic acid (TNB) was followed for 3 min at 415 nm and compared toa standard curve. The molar GSH levels were calculated by subtracting the GSSGcontent, determined after derivatization of GSH with 2-vinylpyridine, from the totalglutathione content (GSH = GSHt − 2 × GSSG). Results are presented as percentageof control conditions at 36.5 ◦C.
2.5.4. ATP quantificationCellular ATP levels were evaluated through a luciferase-based bioluminescent
technique, as previously reported (Pontes et al., 2008b). Sample ATP levels are pro-portional to the intensity of the light emitted by luciferine, in a reaction catalyzedby luciferase, that was measured using a 96-well Microplate Luminometer (BioTekInstruments, Vermont, US). Results are presented as percentage of control conditionsat 36.5 ◦C.
2.5.5. �-GCS activity�-Glutamylcysteine synthetase (�-GCS), also known as glutamate cysteine ligase
(GCL), is the first enzyme in the glutathione biosynthesis pathway, and its rate-limiting step. The activity of �-GCS was evaluated as previously described by Seelig
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
214

Author's personal copy
44 H. Pontes et al. / Toxicology 254 (2008) 42–50
et al. (1984) with some modifications (Piner et al., 2007). �-GCS activity was mea-sured from the rate of ADP formation (assumed to be equal to the rate of oxidation ofNADH) as calculated from the change in absorbance at 340 nm in reaction mixtures at37 ◦C. Reaction medium included 0.1 M pH 7.75 Tris buffer, 150 mM KCl, 2 mM phos-phoenolpyruvate, 10 mM l-glutamate, 20 mM MgCl2, 10 mM l-�-amino-n-butyrate,2 mM EDTA, 0.2 mM NADH, 5 mM ATP, 2 U/mL pyruvate kinase, and 2 U/mL lactatedehydrogenase. Results are presented as percentage of control conditions at 36.5 ◦C.
2.5.6. GST activityGlutathione-S-transferase (GST) activity was assayed according to the method
of Habig et al. (1974). The formation of GSH conjugate with 1-chloro-2,4-dinitrobenzene was monitored for 5 min at 340 nm. The GST activity was calculatedby using an extinction coefficient of 9.6 mM−1 cm−1 and expressed as �mol/min permg of protein. Results are presented as percentage of control conditions at 36.5 ◦C.
2.5.7. Caspase-3 activationCaspase-3 assay was based on a colorimetric assay by following the hydrolysis
of the peptide substrate Ac-DEVD-pNA by caspase 3, which results in the release ofp-nitroaniline (pNA), with high absorbance at 405 nm (Jiménez et al., 2004). Briefly,a lysate of approximately 3 × 106 cells, prepared with an appropriated lysis buffer[50 mM Hepes, 1 mM DTT, 0.1 mM EDTA, 0.1% CHAPS (pH 7.4)], was added to anassay buffer [100 mM NaCl, 50 mM Hepes, 10 mM DTT, 1 mM EDTA, 10% glycerol,0.1% CHAPS (pH 7.5)] containing 16 �M of the caspase-3 substrate. After incubationat 37 ◦C for 3 h, absorbance was measured in a microplate reader at 405 nm.
2.5.8. Protein quantificationProtein was quantified through Lowry’s assay (Lowry et al., 1951).
2.6. Western blot analysis
Equal amounts of proteins were separated using a 12.5% SDS-PAGE gel, followedby blotting on a nitrocellulose membrane (Hybond-ECL; Amersham PharmaciaBiotech). After blotting, non-specific binding was blocked with 5% non-fat dry milk inTTBS [Tris-buffered saline (TBS) with Tween 20] and the membrane was incubatedwith anti-Bax (1:1000; sc-493 rabbit polyclonal IgG; Santa Cruz Biotechnology),anti-AIF (1:2000; sc-13116 mouse monoclonal IgG2b; Santa Cruz Biotechnology) oranti-cytochrome c (1:2000; sc-99 mouse monoclonal IgG1; Santa Cruz Biotechnol-ogy) or anti-�-tubulin (1:1000; T6074 mouse monoclonal IgG1; Sigma) antibodiesduring 2 h at room temperature, washed and incubated with secondary horseradishperoxidase-conjugated anti-mouse or anti-rabbit IgG antibodies (Amersham Phar-macia Biotech) during 1 h. �-Tubulin Western blot is included as a loading proteincontrol. The membrane was then washed and developed with Western blottingchemiluminescence reagents (Amersham Pharmacia Biotech) according to manu-facturer’s instructions, followed by exposure to X-ray films (Sigma, Kodak BiomaxLight Film, St. Louis, USA). Optical density was measured and results of all groupswere presented as percentage of controls.
2.7. Statistical analysis
Results are presented as means ± SEM (from at least five experiments of differentpreparations of hepatocytes). Statistical comparisons were performed by one-wayanalysis of variance (ANOVA) followed by the Bonferroni’s post-hoc test. Significancewas accepted at P < 0.05.
3. Results
3.1. Acridine orange and ethidium bromide staining
The ethidium homodimer cannot penetrate intact cellular mem-branes and, therefore, stains the nucleus of cells in red onlywhen the membranes are disrupted, whereas acridine orange ismembrane-permeable and stains living cells in green. Therefore,under the ethidium bromide/acridine orange staining (Fig. 1), liv-ing cells appear as cells with a regular-sized green fluorescentnucleus, whereas early apoptotic cells have a green fluorescentcondensed, shrunken, or fragmented nucleus, and late apoptoticcells have a red fluorescent condensed, shrunken, or fragmentednucleus. Necrotic cells exhibit a red fluorescent regular-sized orincreased nucleus. The pictures in Fig. 1 suggest that MDMA, byitself, did not produce evident changes in cell viability, whileEtOH exposure was able to induce some degree of necrotic celldeath, especially notorious at hyperthermic conditions. This EtOH-induced necrotic cell death was aggravated by the co-exposurewith MDMA under hyperthermic conditions. It was also observed a
decrease in cell density due to high rate of cell death, leading to celldetachment.
3.2. LDH leakage
Cells exposed under normothermic conditions to several MDMAconcentrations suffered a slight loss of viability at the highest con-centration tested [down to 90.5 ± 3.8% of control (P < 0.01 at 1.6 mMMDMA)] (Fig. 2). Loss of cell viability was significantly aggravatedunder hyperthermia [down to 77.1 ± 4.2% of control (P < 0.001) at1.6 mM MDMA]. EtOH, by itself, did not have a significant effecton LDH leakage but potentiated MDMA-induced loss of cell via-bility, which was observed for MDMA concentrations as low as0.4 and 0.1 mM, at normothermia and hyperthermia, respectively.The extension of this synergic effect was, therefore, dependent onMDMA concentration and on incubation temperature [at 36.5 ◦C,EtOH + 1.6 mM MDMA decreased cell viability down to 71.3 ± 5.8%of control (P < 0.05), while at 40.5 ◦C this co-exposure resulted in acell viability down to 11.1 ± 1.8% of control (P < 0.001)].
3.3. ROS and RNS formation
When cells were incubated under normothermic conditions,none of the treatments resulted in detectable modification in theformation of reactive species (Fig. 3). However, after a 24-h incu-bation period at 40.5 ◦C, EtOH increased ROS and RNS formation[to 128.7 ± 8.2% of control (P < 0.01)] and this increase was signifi-cantly potentiated by simultaneous exposure to EtOH and MDMA[to 192.3 ± 17.2% of control (P < 0.001)].
3.4. GSH/GSSG quantification
The changes in GSH/GSSG levels are presented in Fig. 4.Under normothermic conditions, MDMA exposure caused a sig-nificant decrease of intracellular total glutathione (tGSH) andreduced glutathione (redGSH) at the highest concentration [theredGSH levels decreased from 94.2 ± 0.5% in control cells to82.3 ± 10.1% in cells exposed to 1.6 mM MDMA (P < 0.05) and tGSHdecreased to 92.8 ± 11.2%]. This decrease was also verified whencells were co-exposed to EtOH [redGSH decreased from 97.9 ± 8.4%in cells exposed only to EtOH to 51.2 ± 7.4% in cells exposed toEtOH + 1.6 mM MDMA (P < 0.05) and tGSH levels decreased from103.0 ± 8.9% in cells exposed only to EtOH to 62.7 ± 9.0% in cellsexposed to EtOH + 1.6 mM MDMA (P < 0.05)]. This decrease was onlyaggravated by hyperthermia in cells simultaneously exposed toMDMA and EtOH [redGSH decreased from 84.7 ± 10.2% when cellswere exposed only to EtOH at 40.5 ◦C to 1.78 ± 0.92% when cellswere exposed to EtOH + 1.6 mM MDMA (P < 0.001)]. This decreasein redGSH was accompanied by a tGSH decrease [from 93.4 ± 11.0%to 5.49 ± 0.95% (P < 0.01)]. As expected, the intracellular levels ofGSSG did not present changes among the considered experimen-tal conditions since, once formed, it is excreted to the extracellularmedium (Leier et al., 1996).
3.5. ATP quantification
At 36.5 ◦C, when cells were incubated with four differentMDMA concentrations, ATP levels remained unchanged compara-tively to control cells (Fig. 5). However, co-incubation with EtOHinduced a significant decrease in ATP levels whose extensionwas dependent on MDMA concentration [ATP levels decreasedfrom 87.4 ± 4.1% when cells were exposed only to EtOH, to62.3 ± 4.3% when cells were exposed to EtOH + 1.6 mM MDMA(P < 0.01)]. Under hyperthermia (40.5 ◦C), MDMA itself was ableto significantly decrease ATP levels at the highest concentration
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
215

Author's personal copy
H. Pontes et al. / Toxicology 254 (2008) 42–50 45
Fig. 1. Fluorescence microscope photographs from ethidium bromide/acridine orange staining of primary rat hepatocytes cultures exposed for 24 h to 1.4% (m/v) EtOH,1.6 mM MDMA or both compounds simultaneously. The upper row corresponds to cells incubated under normothermia (36.5 ◦C) and the lower row corresponds to cellsincubated under hyperthermia (40.5 ◦C). The columns correspond, from left to right, to control cells, and cells incubated with EtOH, MDMA and EtOH + MDMA, respectively.For each condition, the left image corresponds to a 200 times magnification, and the right image corresponds to a 400 times magnification.
Fig. 2. Effect of MDMA and hyperthermia on viability of primary cultured rat hepatocytes in presence or in absence of EtOH (W/EtOH and W/O EtOH, respectively). Resultsare represented as mean ± SEM of the percentage of control cells (exposed neither to MDMA nor to EtOH at 36.5 ◦C) from at least five experiments. Control cells presented88.1 ± 2.3% of cell viability. (*) Compared to control cells at 36.5 ◦C; (#) compared to 0 mM MDMA of the respective curve; (×) compared to normothermia; (+) compared toW/O EtOH.
Fig. 3. Effect of MDMA and hyperthermia on reactive species production (ROS and RNS) in primary cultured rat hepatocytes in presence or in absence of EtOH (W/EtOHand W/O EtOH, respectively). Results are represented as mean ± SEM of the percentage of control cells (exposed neither to MDMA nor to EtOH at 36.5 ◦C) from at least fiveexperiments. (*) Compared to control cells at 36.5 ◦C; (#) compared to 0 mM MDMA of the respective curve; (×) compared to normothermia; (+) compared to W/O EtOH.
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
216

Author's personal copy
46 H. Pontes et al. / Toxicology 254 (2008) 42–50
Fig. 4. Effect of MDMA and hyperthermia on tGSH, GSH and GSSG levels in primary cultured rat hepatocytes in presence or in absence of EtOH (W/EtOH and W/O EtOH,respectively). Results are represented as mean ± SEM of the percentage of control cells (exposed neither to MDMA nor to EtOH at 36.5 ◦C) from at least five experiments.Control cells presented 63.5 ± 5.4 nmol of tGSH/mg protein, 59.9 ± 5.3 nmol of redGSH/mg protein and 1.80 ± 0.16 nmol of GSSG/mg protein. (*) Compared to control cells at36.5 ◦C; (#) compared to 0 mM MDMA of the respective curve; (×) compared to normothermia; (+) compared to W/O EtOH.
Fig. 5. Effect of MDMA and hyperthermia on ATP levels in primary cultured rat hepatocytes in presence or in absence of EtOH (W/EtOH and W/O EtOH, respectively). Resultsare represented as mean ± SEM of the percentage of control cells (exposed neither to MDMA nor to EtOH) from at least five experiments. Control cells presented 16.5 ± 2.0 nmolof ATP/mg protein. (*) Compared to control cells at 36.5 ◦C; (#) compared to 0 mM MDMA of the respective curve; (×) compared to normothermia; (+) compared to W/OEtOH.
[from 108.7 ± 9.6% in control cells to 76.0 ± 6.3% when cells wereexposed to 1.6 mM MDMA (P < 0.05)] being this decrease drasti-cally potentiated by EtOH co-exposure [ATP levels decreased to0.25 ± 0.16% when cells were co-exposed to EtOH and 1.6 mMMDMA (P < 0.001)].
3.6. �-GCS activity
As it can be observed in Fig. 6, MDMA and EtOH, by themselves,did not change �-GCS activities, neither at 36.5 ◦C nor 40.5 ◦C, at anyof the tested concentrations. The co-incubation resulted in a slight
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
217
Author's personal copy
H. Pontes et al. / Toxicology 254 (2008) 42–50 47
Fig. 6. Effect of MDMA and hyperthermia on �-GCS activity in primary cultured rat hepatocytes in presence or in absence of EtOH (W/EtOH and W/O EtOH, respectively).Results are represented as mean ± SEM of the percentage of control cells (exposed neither to MDMA nor to EtOH at 36.5 ◦C) from at least five experiments. Control cellspresented 39.8 ± 8.5 mU of �-GCS/mg protein. (*) Compared to control cells at 36.5 ◦C; (#) compared to 0 mM MDMA of the respective curve; (×) compared to normothermia;(+) compared to W/O EtOH.
Fig. 7. Effect of MDMA and hyperthermia on GST activity in primary cultured rat hepatocytes in presence or in absence of EtOH (W/EtOH and W/O EtOH, respectively).Results are represented as mean ± SEM of the percentage of control cells (exposed neither to MDMA nor to EtOH at 36.5 ◦C) from at least five experiments. Control cellspresented 190.6 ± 62.8 U of GST/mg protein. (*) Compared to control cells at 36.5 ◦C; (#) compared to 0 mM MDMA of the respective curve; (×) compared to normothermia;(+) compared to W/O EtOH.
tendency for a decrease of �-GCS activity (down to 70.7 ± 6.3%),reaching statistical significance under hyperthermic conditions, inwhich �-GCS activities practically disappeared at the two highestMDMA concentrations tested (P < 0.001).
3.7. GST activity
The effects of ethanol, MDMA, as well as their interaction undernormothermia and hyperthermia on GST activity are presented inFig. 7. In the absence of ethanol, GST activity remained unchanged,independently on MDMA concentration and on incubation temper-ature. When cells were exposed to ethanol, a significant decreasein GST activity was observed (down to 70.7 ± 6.2%) when comparedto control (P < 0.05). Under hyperthermic conditions, co-exposureto ethanol and MDMA was able to aggravate the decrease inducedby ethanol, in an extent dependent on MDMA concentration [GSTactivity decreased to 62.7 ± 13.0% when cells were exposed toethanol + 0.4 mM MDMA (P < 0.01), to 44.4 ± 15.6% when cells wereexposed to ethanol + 0.8 mM MDMA (P < 0.001) and to 3.88 ± 1.95%when cells were exposed to ethanol + 1.6 mM MDMA (P < 0.001)].
3.8. Western blot analysis
At 36.5 ◦C, the release of mitochondrial components (AIF andcytochrome c) seems to be inexistent. However, at 40.5 ◦C, a sig-nificant release of AIF and cytochrome c from the mitochondrialextract was observed, under MDMA EtOH co-exposure (Fig. 8).
There was no translocation of Bax molecule from the cytoplasmto the mitochondria (data not shown).
3.9. Caspase-3 activity
There were no statistical differences in caspase-3 activitiesamong the different incubation conditions (data not shown).
4. Discussion
The data obtained in this study clearly demonstrated that thesimultaneous incubation of primary cultured rat hepatocytes withMDMA and EtOH for 24 h, leads to high cell death ratios, mainlythrough necrosis, accompanied by a significant disturbance of cel-lular redox status and decreased energy levels.
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
218

Author's personal copy
48 H. Pontes et al. / Toxicology 254 (2008) 42–50
Fig. 8. Western blot analysis of the effect of MDMA and hyperthermia on AIF and cytochrome c release from the mitochondria of primary cultured rat hepatocytes in presenceor in absence of EtOH (W/EtOH and W/O EtOH, respectively). Above the histogram, the panel shows a representative Western blotting of AIF and of cytochrome c, respectively,for each condition. Values are mean ± SEM and are expressed as percentage of control. �-Tubulin was used as an internal loading control. C, control; M, MDMA; E, EtOH;E + M, EtOH + MDMA.
Both EtOH and MDMA are metabolized in the liver, where EtOHcan alter the expression or activity of some drug-metabolizingenzymes (Jang and Harris, 2007) and where both drugs share somemetabolic pathways involving the cytochrome P450 isoenzymesand originating metabolites (Gemma et al., 2006; Green et al., 2003)with higher toxicity than their parent compounds, as previouslyreported for other amphetamines (Carmo et al., 2007). Notwith-standing the knowledge about the decrease of the activities ofcytochrome P450 system isoenzymes during the culture of rat hep-atocytes, the present experimental model is adequately adapted tothe study of the interactions between ethanol and MDMA since theremaining CYP enzyme activities are still sufficient to study drugmetabolism in vitro during the 24-h incubation period used in thepresent study (Gebhardt et al., 2003; Hewitt et al., 2007). Whilethis metabolic interaction may represent an explanation for mostof the synergic toxic effects found, other factors are also involvedand the hyperthermic effect contributes significantly to the aggra-vation of the observed toxicity. The MDMA-induced decrease onGSH levels is in agreement with results previously reported byour group on freshly isolated mouse hepatocytes (Pontes et al.,2008b) and can be explained by oxidation of GSH to GSSG by reac-tive oxygen and nitrogen species, and formation of GSH adductswith MDMA metabolites by GST (Hiramatsu et al., 1990), but alsoby impaired GSH synthesis, which is also a known effect associ-ated with EtOH exposure (Lauterburg et al., 1984). In the presentstudy, we demonstrated, for the first time, that the impairment of
GSH synthesis mediated by ethanol was dramatically aggravatedby MDMA co-exposure under hyperthermic conditions, with analmost absolute inhibition of �-GCS activity for the two highestMDMA concentrations. The decrease on GST activity can be relatedto the oxidation or alkylation of sulfhydryl groups present in criticalaminoacid residues within the enzyme active site, an effect alreadyreported for MDMA metabolites (Carvalho et al., 2004a) and forquinones and catecholamines (Monks and Lau, 1992; Bindoli et al.,1992; van Ommen et al., 1991), formed after exposure to MDMA.A significant decrease in GST activity was also previously reportedfor ethanol (Gyamfi and Wan, 2006). GST catalyzes the nucleophilicattack of GSH on reactive electrophilic substrates, thereby protect-ing various cell components from their potentially damaging effects(Mannervik and Danielson, 1988). Thus, its higher inactivation bysimultaneous exposure to ethanol and MDMA can increase cell sus-ceptibility to the deleterious effects of reactive electrophiles. Theseimpairments in glutathione homeostasis induced by ethanol andMDMA mixtures can contribute to their synergistic toxicity.
It was previously reported that either EtOH or MDMA is able toincrease ROS and RNS formation, in a variety of systems, cells, andspecies (Das and Vasudevan, 2007; Quinton and Yamamoto, 2006).The increased formation of reactive species after incubation withethanol may be derived from the induction of cytochrome P450 2E1activity (Morimoto et al., 1993) or by mitochondrial dysfunctionafter reduction of GSH content and ATP generation (Cunninghamet al., 1990; Colell et al., 1998). MDMA can lead to the formation of
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
219

Author's personal copy
H. Pontes et al. / Toxicology 254 (2008) 42–50 49
ROS and RNS during the redox cycles associated with its metabolicconversion (Colado et al., 1997) or during the conjugation of MDMAmetabolites with GSH molecules, a pathway also accompanied bythe formation of reactive species (Hiramatsu et al., 1990; Monkset al., 2004). However, a synergism in the ROS and RNS formation,when cells were co-exposed to both compounds, was also neverreported before.
Concerning ATP levels, MDMA was previously reported todecrease ATP in rat hepatocytes (Carvalho et al., 2004b; Beitia etal., 1999) and EtOH leads to decreased ATP levels and ATP syn-thesis in rats (Helzberg et al., 1987; Piquet et al., 2004). The ATPdepletion caused by ethanol is probably due to mitochondrial dam-age induced by an overproduction of anion superoxide (O2
•−) atmitochondrial complexes I and III, and consequently of H2O2 andother reactive oxygen species (ROS). This effect results from theexcessive NADH formation during acetaldehyde metabolism in livermitochondria that is used by mitochondria complex I (NADH dehy-drogenase) as the first electron donor of the electron transportchain (Sastre et al., 2007). The potentiation of this depletion bythe co-incubation with EtOH and MDMA is in accordance withour previous ex-vivo study (Pontes et al., 2008b). This depletionis an indicator of probable mitochondrial damage confirmed by thedecrease of cytochrome c and AIF in the mitochondrial extract.
The depletion of GSH and ATP levels was accompanied by a lossin cell viability. Evaluation of apoptotic/necrotic features providedclear evidences that the cell death resulting from the co-incubationwith EtOH and MDMA occurs through a necrotic pathway, con-firmed by a significant increase of LDH leakage, nuclei stainingby the ethidium bromide and acridine orange technique and AIFrelease that was reported to be on the basis of a necrotic pro-grammed cell death pathway, explored by the groups of Boujrad etal. (2007) and Lorenzo and Susin (2007). Hepatic necrosis was alsopreviously reported after in vitro exposure of mouse hepatocytes(Pontes et al., 2008b), and in vivo exposure of mice (Johnson et al.,2002) and humans to MDMA (Milroy et al., 1996) or after the expo-sure of human and rat hepatocytes to high EtOH concentrations(Castilla et al., 2004). In this particular case of association betweenMDMA and EtOH in hepatocytes the classic apoptotic via, alreadydescribed for MDMA in neural cells (Capela et al., 2007, Alvaro etal., 2008) and hepatic stellate cells (Montiel-Duarte et al., 2004),is discarded since there is no translocation of BAX to mitochondriaand no activation of effectors apoptotic pathways such as caspase 3,notwithstanding the cytochrome c release from the mitochondria(Fadeel et al., 2008). Therefore, the type of cell death subsequent toMDMA incubation is probably affected by EtOH co-exposure. Note-worthy, hepatocytes exposed to 1 mM MDMA concentrations werepreviously reported to suffer an apoptotic type of cell death andnot a necrotic cell death as reported in the present study (Montiel-Duarte et al., 2002). This apparent discrepancy can be explained bythe ATP requirements of apoptosis, especially in the steps mediatedby protein kinases that use ATP as a phosphoryl donor for phospho-rylation of some proteins involved in apoptosis (Skulachev, 2006).Since in the present study, a dramatic decrease in ATP levels wasobserved, these apoptotic pathways are probably impaired.
All the evaluated parameters were significantly aggravated inthe hepatocytes cultured under hyperthermia, which is known tobe an in vivo life-threatening effect associated with ecstasy con-sumption (Green et al., 2004) and exacerbates the toxicity of manytypes of drugs and environmental toxicants (Gordon, 2007), includ-ing MDMA (Carvalho et al., 2001). The increase of EtOH and MDMAtoxicity by hyperthermia is particularly notorious in the presentstudy by the dramatic decrease in cell viability, GSH content, �-GCSactivity and ATP.
In conclusion, EtOH associated with MDMA definitely enhancesthe MDMA-related toxicity towards hepatocytes through a
decrease in antioxidant defences, an increase in reactive speciesformation, and enhanced cell death through a necrotic pathway ina temperature-dependent extent.
These findings represent an important step in the elucidationof the mechanisms underlying the EtOH-induced increase on thetoxicity to hepatocytes caused by MDMA consumption.
Conflict of interest
None.
Acknowledgements
This work was supported by a project from FCT and POCI-2010, Portugal, by FEDER European Community co-funding(POCI/SAU-FCF/57187/2004). Helena Pontes was the recipientof a PhD grant from “Fundacão para a Ciência e Tecnologia”(SFRH/BD/18527/2004).
References
Alvaro, A.R., Martins, J., Costa, A.C., Fernandes, E., Carvalho, F., Ambrósio, A.F.,Cavadas, C., 2008. Neuropeptide Y protects retinal neural cells against cell deathinduced by ecstasy. Neuroscience 152, 97–105.
Anderson, M.E., 1985. Determination of glutathione and glutathione disulfide inbiological samples. Methods Enzymol. 113, 548–555.
Barrett, S.P., Darredeau, C., Pihl, R.O., 2006. Patterns of simultaneous polysubstanceuse in drug using university students. Hum. Psychopharmacol. 21, 255–263.
Beitia, G., Cobreros, A., Sainz, L., Cenarruzabeitia, E., 1999. 3,4-Methylenedioxymethamphetamine (ecstasy)-induced hepatotoxicity: effect oncytosolic calcium signals in isolated hepatocytes. Liver 19, 234–241.
Bergermeyer, H.U., Bernt, E., 1974. UV-assay for lactate dehydrogenase with pyruvateand NADH. In: Bergermeyer, H.U. (Ed.), Methods of Enzymatic Analysis. VerlagChemie GmbH, Weinheim.
Bindoli, A., Rigobello, M.P., Deeble, D.J., 1992. Biochemical and toxicological prop-erties of the oxidation products of catecholamines. Free Radic. Biol. Med. 13,391–405.
Boujrad, H., Gubkina, O., Robert, N., Krantic, S., Susin, S.A., 2007. AIF-mediated pro-grammed necrosis: a highly regulated way to die. Cell Cycle 6, 2612–2619.
Boyd, C.J., McCabe, S.E., d’Arcy, H., 2003. Ecstasy use among college undergraduates:gender, race and sexual identity. J. Subst. Abuse Treat. 24, 209–215.
Breen, C., Degenhardt, L., Kinner, S., Bruno, R., Jenkinson, R., Matthews, A., Newman,J., 2006. Alcohol use and risk taking among regular ecstasy users. Subst. UseMisuse 41, 1095–1109.
Capela, J.P., Macedo, C., Branco, P.S., Ferreira, L.M., Lobo, A.M., Fernandes, E., Remião,F., Bastos, M.L., Dirnagl, U., Meisel, A., Carvalho, F., 2007. Neurotoxicity mecha-nisms of thioether ecstasy metabolites. Neuroscience 146, 1743–1757.
Carmo, H., Brulport, M., Hermes, M., Oesch, F., de Boer, D., Remião, F., Carvalho, F.,Schön, M.R., Krebsfaenger, N., Doehmer, J., Bastos, M.L., Hengstler, J.G., 2007.CYP2D6 increases toxicity of the designer drug 4-methylthioamphetamine (4-MTA). Toxicology 229, 236–244.
Carvalho, M., Carvalho, F., Bastos, M.L., 2001. Is hyperthermia the triggering factor forhepatotoxicity induced by 3,4-methylenedioxymethamphetamine (ecstasy)?An in vitro study using freshly isolated mouse hepatocytes. Arch. Toxicol. 74,789–793.
Carvalho, M., Milhazes, N., Remião, F., Borges, F., Fernandes, E., Amado,F., Monks, T.J., Carvalho, F., Bastos, M.L., 2004a. Hepatotoxicity of 3,4-methylenedioxyamphetamine and alpha-methyldopamine in isolated rathepatocytes: formation of glutathione conjugates. Arch. Toxicol. 78, 16–24.
Carvalho, M., Remião, F., Milhazes, N., Borges, F., Fernandes, E., Carvalho, F., Bastos,M.L., 2004b. The toxicity of N-methyl-alpha-methyldopamine to freshly isolatedrat hepatocytes is prevented by ascorbic acid and N-acetylcysteine. Toxicology200, 193–203.
Castilla, R., González, R., Fouad, D., Fraga, E., Muntane, J., 2004. Dual effect of ethanolon cell death in primary culture of human and rat hepatocytes. Alcohol Alcohol.39, 290–296.
Colado, M.I., O’Shea, E., Granados, R., Murray, T.K., Green, A.R., 1997. In vivo evidencefor free radical involvement in the degeneration of rat brain 5-HT followingadministration of MDMA (’ecstasy’) and p-chloroamphetamine but not thedegeneration following fenfluramine. Br. J. Pharmacol. 121, 889–900.
Colell, A., García-Ruiz, C., Miranda, M., Ardite, E., Marí, M., Morales, A., Corrales, F.,Kaplowitz, N., Fernández-Checa, J.C., 1998. Selective glutathione depletion ofmitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gas-troenterology 115, 1541–1551.
Cunningham, C.C., Coleman, W.B., Spach, P.I., 1990. The effects of chronic ethanolconsumption on hepatic mitochondrial energy metabolism. Alcohol Alcohol. 25,127–136.
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
220

Author's personal copy
50 H. Pontes et al. / Toxicology 254 (2008) 42–50
Das, S.K., Vasudevan, D.M., 2007. Alcohol-induced oxidative stress. Life Sci. 81,177–187.
EMCDDA, 2005. Annual report 2005: The state of the drugs problem in Europe.Fadeel, B., Ottosson, A., Pervaiz, S., 2008. Big wheel keeps on turning: apoptosome
regulation and its role in chemoresistance. Cell Death Differ. 15, 443–452.Garcia-Repetto, R., Moreno, E., Soriano, T., Jurado, C., Gimenez, M.P., Menendez, M.,
2003. Tissue concentrations of MDMA and its metabolite MDA in three fatalcases of overdose. Forensic Sci. Int. 135, 110–114.
Gebhardt, R., Hengstler, J.G., Müller, D., Glöckner, R., Buenning, P., Laube, B.,Schmelzer, E., Ullrich, M., Utesch, D., Hewitt, N., Ringel, M., Hilz, B.R., Bader,A., Langsch, A., Koose, T., Burger, H.J., Maas, J., Oesch, F., 2003. New hepatocytein vitro systems for drug metabolism: metabolic capacity and recommenda-tions for application in basic research and drug development, standard operationprocedures. Drug Metab. Rev. 35, 145–213.
Gemma, S., Vichi, S., Testai, E., 2006. Individual susceptibility and alcohol effects:biochemical and genetic aspects. Ann. Ist. Super. Sanita 42, 8–16.
Gordon, C.J., 2007. Thermophysiological responses to hyperthermic drugs: extrapo-lating from rodent to human. Prog. Brain Res. 162, 63–79.
Gouzoulis-Mayfrank, E., Daumann, J., 2006. The confounding problem of polydruguse in recreational ecstasy/MDMA users: a brief overview. J. Psychopharmacol.20, 188–193.
Green, A.R., Mechan, A.O., Elliott, J.M., O’Shea, E., Colado, M.I., 2003. The phar-macology and clinical pharmacology of 3,4-methylenedioxymethamphetamine(MDMA, “ecstasy”). Pharmacol. Rev. 55, 508–563.
Green, A.R., O’shea, E., Colado, M.I., 2004. A review of the mechanisms involved inthe acute MDMA (ecstasy)-induced hyperthermic response. Eur. J. Pharmacol.500, 3–13.
Gyamfi, M.A., Wan, Y.J., 2006. The effect of ethanol, ethanol metabolizing enzymeinhibitors, and Vitamin E on regulating glutathione, glutathione S-transferase,and S-adenosylmethionine in mouse primary hepatocyte. Hepatol. Res. 35,53–61.
Habig, W.H., Pabst, M.J., Jakoby, W.B., 1974. Glutathione S-transferases. The firstenzymatic step in mercapturic acid formation. J. Biol. Chem. 249, 7130–7139.
Helzberg, J.H., Brown, M.S., Smith, D.J., Gore, J.C., Gordon, E.R., 1987. Metabolic state ofthe rat liver with ethanol: comparison of in vivo 31 phosphorus nuclear magneticresonance spectroscopy with freeze clamp assessment. Hepatology 7, 83–88.
Hernández-López, C., Farré, M., Roset, P.N., Menoyo, E., Pizerro, N., Ortuno, J., Tor-rens, M., Cami, J., de la Torre, R., 2002. 3,4-Methylenedioxymethamphetamine(ecstasy) and alcohol interactions in humans: psychomotor performance, sub-jective effects, and pharmacokinetics. J. Pharmacol. Exp. Ther. 300, 236–244.
Hewitt, N.J., Lechón, M.J., Houston, J.B., Hallifax, D., Brown, H.S., Maurel, P., Kenna,J.G., Gustavsson, L., Lohmann, C., Skonberg, C., Guillouzo, A., Tuschl, G., Li, A.P.,LeCluyse, E., Groothuis, G.M., Hengstler, J.G., 2007. Primary hepatocytes: currentunderstanding of the regulation of metabolic enzymes and transporter proteins,and pharmaceutical practice for the use of hepatocytes in metabolism, enzymeinduction, transporter, clearance, and hepatotoxicity studies. Drug Metab. Rev.39, 159–234.
Hiramatsu, M., Kumagai, Y., Unger, S.E., Cho, A.K., 1990. Metabolism of methylene-dioxymethamphetamine: formation of dihydroxymethamphetamine and aquinone identified as its glutathione adduct. J. Pharmacol. Exp. Ther. 254,521–527.
Hopper, J.W., Su, Z., Looby, A.R., Ryan, E.T., Penetar, D.M., Palmer, C.M., Lukas, S.E.,2006. Incidence and patterns of polydrug use and craving for ecstasy in reg-ular ecstasy users: An ecological momentary assessment study. Drug AlcoholDepend. 85, 221–235.
Institute for Laboratory Animal Research, 1996. Guide for the Care and Use of Labo-ratory Animals. National Academy Press, Washington, D.C.
Jang, G.R., Harris, R., 2007. Drug interactions involving ethanol and alcoholic bever-ages. Expert Opin. Drug Metab. Toxicol. 3, 719–731.
Jiménez, A., Jordà, E.G., Verdaguer, E., Pubill, D., Sureda, F.X., Canudas, A.M., Escubedo,E., Camarasa, J., Camins, A., Pallàs, M., 2004. Neurotoxicity of amphetaminederivatives is mediated by caspase pathway activation in rat cerebellar granulecells. Toxicol. Appl. Pharmacol. 196, 223–234.
Johnson, E.A., Shvedova, A.A., Kisin, E., O’Callaghan, J.P., Kommineni, C., Miller, D.B.,2002. d-MDMA during vitamin E deficiency: effects on dopaminergic neurotox-icity and hepatotoxicity. Brain Res. 933, 150–163.
Lauterburg, B.H., Davies, S., Mitchell, J.R., 1984. Ethanol suppresses hepatic glu-tathione synthesis in rats in vivo. J. Pharmacol. Exp. Ther. 230, 7–11.
Leier, I., Jedlitschky, G., Buchholz, U., Center, M., Cole, S.P., Deeley, R.G., Keppler, D.,1996. ATP-dependent glutathione disulphide transport mediated by the MRPgene-encoded conjugate export pump. Biochem. J. 14, 433–437.
Liechti, M.E., Kunz, I., Kupferschmidt, H., 2005. Acute medical problems due toEcstasy use. Case-series of emergency department visits. Swiss Med. Wkly. 135,652–657.
Lora-Tamayo, C., Tena, T., Rodriguez, A., Moreno, D., Sancho, J.R., Ensenat, P., Muela,F., 2004. The designer drug situation in Ibiza. Forensic Sci. Int. 140, 195–206.
Lorenzo, H.K., Susin, S.A., 2007. Therapeutic potential of AIF-mediated caspase-independent programmed cell death. Drug Resist. Updat. 10, 235–255.
Lowry, O.H., Rosebrough, N.J., Farr, A.L., Randall, R.J., 1951. Protein measurement withthe Folin phenol reagent. J. Biol. Chem. 193, 265–275.
Mannervik, B., Danielson, U.H., 1988. Glutathione transferases—structure and cat-alytic activity. CRC Crit. Rev. Biochem. 23, 283–337.
Milroy, C.M., Clark, J.C., Forrest, A.R., 1996. Pathology of deaths associated with“ecstasy” and “eve” misuse. J. Clin. Pathol. 49, 149–153.
Monks, T.J., Jones, D.C., Bai, F., Lau, S.S., 2004. The role of metabolism in 3,4-(+)-methylenedioxyamphetamine and 3,4-(+)-methylenedioxymethamphetamine(ecstasy) toxicity. Ther. Drug Monit. 26, 132–136.
Monks, T.J., Lau, S.S., 1992. Toxicology of quinone-thioethers. Crit. Rev. Toxicol. 22,243–270.
Montiel-Duarte, C., Ansorena, E., López-Zabalza, M.J., Cenarruzabeitia, E., Iraburu,M.J., 2004. Role of reactive oxygen species, glutathione and NF-kappaB in apop-tosis induced by 3,4-methylenedioxymethamphetamine (“Ecstasy”) on hepaticstellate cells. Biochem. Pharmacol. 67, 1025–1033.
Montiel-Duarte, C., Varela-Rey, M., Osés-Prieto, J.A., López-Zabalza, M.J., Beitia, G.,Cenarruzabeitia, E., Iraburu, M.J., 2002. 3,4-Methylenedioxymethamphetamine(“Ecstasy”) induces apoptosis of cultured rat liver cells. Biochim. Biophys. Acta1588, 26–32.
Morimoto, M., Hagbjörk, A.L., Nanji, A.A., Ingelman-Sundberg, M., Lindros, K.O., Fu,P.C., Albano, E., French, S.W., 1993. Role of cytochrome P4502E1 in alcoholic liverdisease pathogenesis. Alcohol 10, 459–464.
Oyefeso, A., Schifano, F., Ghodse, H., Cobain, K., Dryden, R., Corkery, J., 2006. Fatalinjuries while under the influence of psychoactive drugs: a cross-sectionalexploratory study in England. BMC Public Health 6, 148.
Piner, P., Sevgiler, Y., Uner, N., 2007. In vivo effects of fenthion on oxidative pro-cesses by the modulation of glutathione metabolism in the brain of Oreochromisniloticus. Environ. Toxicol. 22, 605–612.
Piquet, M.A., Roulet, M., Nogueira, V., Filippi, C., Sibille, B., Hourmand-Ollivier, I.,Pilet, M., Rouleau, V., Leverve, X.M., 2004. Polyunsaturated fatty acid deficiencyreverses effects of alcohol on mitochondrial energy metabolism. J. Hepatol. 41,721–729.
Pontes, H., Duarte, J.A., Guedes de Pinho, P., Soares, M.E., Fernandes, E., Dinis-Oliveira,R.J., Sousa, C., Silva, R., Carmo, H., Casal, S., Remião, F., Carvalho, F., Bastos, M.L.,2008a. Chronic exposure to ethanol exacerbates MDMA-induced hyperthermiaand exposes liver to severe MDMA-induced toxicity in CD1 mice. Toxicology 252,64–71.
Pontes, H., Santos-Marques, M.J., Fernandes, E., Duarte, J.A., Remião, F., Carvalho, F.,Bastos, M.L., 2008b. Effect of chronic ethanol exposure on the hepatotoxicity ofecstasy in mice: An ex vivo study. Toxicol. In Vitro 22, 910–920.
Quinton, M.S., Yamamoto, B.K., 2006. Causes and consequences of metham-phetamine and MDMA toxicity. AAPS J. 8, E337–E347.
SAMHSA, 2005. 2005 National Survey on Drug Use and Health: National Findings.Sanjurjo, E., Nogue, S., Miro, O., Munne, P., 2004. Analysis of patients attended in
an emergency department due to ecstasy consumption. Med. Clin. (Barc.) 123,90–92.
Sastre, J., Serviddio, G., Pereda, J., Minana, J.B., Arduini, A., Vendemiale, G., Poli, G.,Pallardo, F.V., Vina, J., 2007. Mitochondrial function in liver disease. Front. Biosci.12, 1200–1209.
Schifano, F., 2004. A bitter pill. Overview of ecstasy (MDMA, MDA) related fatalities.Psychopharmacology (Berl.) 173, 242–248.
Schifano, F., Corkery, J., Deluca, P., Oyefeso, A., Ghodse, A.H., 2006. Ecstasy (MDMA,MDA, MDEA, MBDB) consumption, seizures, related offences, prices, dosagelevels and deaths in the UK (1994–2003). J. Psychopharmacol. 20, 456–463.
Schifano, F., Di Furia, L., Forza, G., Minicuci, N., Bricolo, R., 1998. MDMA (’ecstasy’)consumption in the context of polydrug abuse: a report on 150 patients. DrugAlcohol Depend. 52, 85–90.
Schifano, F., Oyefeso, A., Corkery, J., Cobain, K., Jambert-Gray, R., Martinotti, G.,Ghodse, A.H., 2003. Death rates from ecstasy (MDMA, MDA) and polydrug usein England and Wales 1996–2002. Hum. Psychopharmacol. 18, 519–524.
Seelig, G.F., Simondsen, R.P., Meister, A., 1984. Reversible dissociation of gamma-glutamylcysteine synthetase into two subunits. J. Biol. Chem. 259, 9345–9347.
Skulachev, V.P., 2006. Bioenergetic aspects of apoptosis, necrosis and mitoptosis.Apoptosis 11, 473–485.
Smart, R.G., Ogborne, A.C., 2000. Drug use and drinking among students in 36 coun-tries. Addict. Behav. 25, 455–460.
Topp, L., Hando, J., Dillon, P., Roche, A., Solowij, N., 1999. Ecstasy use in Australia:patterns of use and associated harm. Drug Alcohol Depend. 55, 105–115.
Tossmann, P., Boldt, S., Tensil, M.D., 2001. The use of drugs within the techno partyscene in European metropolitan cities. Eur. Addict. Res. 7, 2–23.
van Ommen, B., Ploemen, J.H., Bogaards, J.J., Monks, T.J., Gau, S.S., van Bladeren, P.J.,1991. Irreversible inhibition of rat glutathione S-transferase 1-1 by quinones andtheir glutathione conjugates. Structure-activity relationship and mechanism.Biochem. J. 276, 661–666.
Winstock, A.R., Griffiths, P., Stewart, D., 2001. Drugs and the dance music scene: asurvey of current drug use patterns among a sample of dance music enthusiastsin the UK. Drug Alcohol Depend. 64, 9–17.
Helena
Rectangle
Helena
Rectangle
Helena
Text Box
221

Helena
Text Box
222

Trabalho V. Interacções metabólicas entre o etanol e a MDMA em
culturas primárias de hepatócitos de rato.


______________________________________________________ Trabalho V
225
Trabalho V. Interacções metabólicas entre o etanol e a MDMA em culturas primárias de hepatócitos de rato.
a) Introdução O estudo da influência do etanol no metabolismo da MDMA é de
extrema importância, uma vez que (i) estas duas substâncias são
frequentemente co-consumidas e o seu consumo simultâneo foi já verificado
em intoxicações fatais subsequentes ao consumo de ecstasy 37, 281, (ii) ambas
as substâncias são metabolizadas a nível hepático, onde inclusivamente
partilham algumas vias metabólicas, originando compostos dotados de maior
toxicidade que o composto pai 5, 736 e (iii) o etanol tem ainda a capacidade de
alterar a expressão e/ou actividade de enzimas com actividade metabólica
como as isoenzimas do sistema CYP450 668, incluindo aquelas envolvidas no
metabolismo da MDMA. Esta indução do CYP450 pelo etanol está na base de
múltiplas interacções farmacocinéticas com o etanol aumentando a
bioactivação de numerosos xenobióticos com formação dos respectivos
metabolitos tóxicos 669 e está também associada com um aumento na produção
de ROS e na peroxidação lipídica, bem como com uma diminuição da
capacidade de neutralização, enzimática e não enzimática, de radicais livres 669. Este facto pode agravar as alterações no estado redox verificadas durante
o metabolismo da MDMA, potenciando o risco do desenvolvimento de
toxicidade, especialmente a nível hepático, onde decorre a maior parte do
metabolismo destes dois compostos.
Outros estudos acerca das interacções farmacocinéticas entre o etanol e
a MDMA centraram-se no efeito do etanol nas concentrações plasmáticas de
MDMA 683, 684, no entanto a influência do etanol nas vias metabólicas da MDMA
ainda não foi descrita.
b) Objectivo
Determinar se o etanol afecta o metabolismo da MDMA, se a CYP2E1
e/ou a CYP3A1 estão envolvidas numa eventual interacção metabólica, e de
que forma é que a incubação de hepatócitos de rato em culturas primárias em

Trabalho V ______________________________________________________
226
condições hipertérmicas pode afectar esta hipotética interacção metabólica,
uma vez que a exposição à MDMA in vivo resulta, frequentemente, num
aumento da temperatura corporal 595.
b) Metodologia Neste trabalho foram utilizados hepatócitos de rato Wistar adulto obtidos
pelo mesmo procedimento referido no Trabalho IV e ilustrado na Figura 13
.
Figura 13. Procedimento experimental utilizado para a avaliação da influência do etanol no perfil metabólico da MDMA em culturas primárias de hepatócitos de rato. FBS, soro bovino fetal.
Os hepatócitos assim obtidos e colocados em cultura foram expostos a
300 mM de etanol, 1,6 mM de MDMA (M) ou à combinação de ambos, em
condições de normotermia (36,5 ± 0,5ºC) e de hipertermia (40,5 ± 0,5ºC). O
mesmo desenho experimental foi depois repetido, mas, desta vez, a incubação
com etanol, MDMA ou a mistura dos dois, foi precedida de uma incubação de
uma hora com 0,18 μM de cetoconazol (KET, inibidor da CYP3A1) ou 150 μM
de sulfureto dialílico (DAS, inibidor competitivo da CYP2E1).
Após 24 horas de incubação, foi avaliada a viabilidade celular, através
da quantificação da libertação de lactato desidrogenase (LDH) para o meio de

______________________________________________________ Trabalho V
227
cultura. Foram quantificados, por GC-MS, recorrendo a uma técnica
previamente validada pelo nosso grupo (resultados não publicados), os níveis
de três importantes metabolitos da MDMA no meio de cultura: o metabolito
resultante da N-desmetilação da MDMA (a 3,4-metilenodioxianfetamina ou
MDA) e os metabolitos catecóis o-metilados da MDMA e da MDA (4-hidroxi-3-
metoximetanfetamina ou HMMA e 4-hidroxi-3-metoxianfetamina ou HMA,
respectivamente) (Figura 14).
Figura 14. Metabolização da MDMA a MDA, HMMA e HMA.
Para podermos quantificar estes metabolitos por GC-MS foi necessário
recorrer a uma reacção de derivatização por fluoroacetilação de modo a
conferir volatilidade aos compostos em estudo. As amostras foram tratadas
como indicado na Figura 15.

Trabalho V ______________________________________________________
228
Figura 15. Preparação de amostras de meio de cultura para quantificação de MDMA, MDA, HMMA e HMA por GC-MS.

______________________________________________________ Trabalho V
229
c) Resultados e discussão
c.1) Viabilidade celular As células expostas a 1,6 mM de MDMA em condições normotérmicas
sofreram uma perda de viabilidade de cerca de 10% relativamente às células
controlo (mais 9,8 ± 2,6% de morte celular do que o controlo) (Figura 16), a
qual foi significativamente agravada em condições hipertérmicas, atingindo
valores próximos dos 25% [21,7 ± 2,8% acima do controlo a 40,5ºC (P < 0,05)].
A co-exposição ao etanol teve um efeito sinérgico na morte celular
induzida pela MDMA, provocando 33,9 ± 11,5% de morte celular acima do
controlo em normotermia (P < 0,05 quando comparado com M a 36,5ºC) ou
89,7% ± 1,6% em hipertermia (P < 0,01 quando comparado com M a 40,5ºC).
A pré-incubação com DAS ou KET não reverteu os efeitos da MDMA e
do etanol na percentagem de morte celular.
Normotermia (36,5ºC)
M DAS+M KET+M0
25
50
75
100
++
+*
**
GruposMor
te c
elul
ar (%
aci
ma
do c
ontr
olo)
Hipertermia (40,5ºC)
M DAS+M KET+M0
20
40
60
80
100Sem etanolCom etanol
*
x
x
*** *** ***xxx xx xx
++ ++ ++
GruposMor
te c
elul
ar (%
aci
ma
do c
ontr
olo)
Figura 16. Efeito de MDMA 1,6 mM (M) e da hipertermia na percentagem de morte celular na presença ou na ausência de etanol, DAS e KET. Os resultados estão apresentados como média ± erro padrão da média da percentagem acima das células controlo (não expostas a etanol nem a MDMA) de três ensaios independentes. * Comparado com M a 36,5ºC; × Comparado com normotermia; + Comparado com “Sem etanol”.
c.2) Formação de metabolitos da MDMA Em condições normotérmicas e na ausência de etanol, a pré-incubação
das células em cultura com DAS ou KET não afectou o metabolismo da MDMA
a MDA e HMA (Figura 17A e E). No entanto, a pré-incubação com DAS
provocou uma diminuição significativa da formação de HMMA [de 10,50 ± 0,56
nmol / 106 células para 7,75 ± 0,46 nmol / 106 células (P < 0,05)] (Figura 17C).

Trabalho V ______________________________________________________
230
Por outro lado, a pré-incubação com KET em condições normotérmicas
provocou um aumento significativo da formação de MDA (Figura 17A).
MDA
Normotermia (36,5ºC)
M DAS+M KET+M0.00
0.05
0.10
0.15
0.20
0.252.2
2.4
**
+ +
Groups
mm
ol d
e M
DA
por
106 c
élul
as
Hipertermia (40,5ºC)
M DAS+M KET+M0.0
0.4
0.8
1.2
1.6
2.0
2.4
+++
+
§§§
§§§
***
***×××
×
º º
Sem etanolCom etanol
Gruposm
mol
de
MD
A po
r 10
6 cél
ulas
HMMA
Normotermia (36,5ºC)
M DAS+M KET+M0
4
8
12
16
20220
240
*
+
Grupos
nmol
de
HM
MA
por
10
6 cél
ulas
Hipertermia (40,5ºC)
M DAS+M KET+M0
40
80
120
160
200
240
ººº ººº
+++
+++ +++
§§§
§§§
***
*** ***
×××
××× ×××
º ºº
Sem etanolCom etanol
Grupos
nmol
de
HM
MA
por
10
6 cél
ulas
HMA
Normotermia (36,5ºC)
M DAS+M KET+M0.0
0.5
1.0
1.512
14
++++
Grupos
nmol
de
HM
A p
or 1
06 c
élul
as
Hipertermia (40,5ºC)
M DAS+M KET+M0
2
4
6
8
10
12
14
+++
ºººººº+++
+++
§§§
***
******
×××
××××××
Sem etanolCom etanol
Grupos
nmol
de
HM
A p
or 1
06 c
élul
as
Figura 17. Efeito da hipertermia e da co-incubação com etanol 300 mM, DAS 150 μM e KET 0,18 μM na formação de metabolitos da MDMA (MDA, HMMA e HMA) em culturas primárias de hepatócitos de rato. Os resultados são apresentados como média ± erro padrão da média da quantidade de metabolitos formados por milhão de células vivas de três ensaios independentes. * Comparado com M a 36,5ºC; × Comparado com normotermia; + Comparado com “Sem etanol”; º Comparado com M a 40,5ºC; § Comparado com etanol+MDMA a 40,5ºC.
A B
DC
FE

______________________________________________________ Trabalho V
231
A co-exposição ao etanol aumentou significativamente o metabolismo da
MDMA a MDA e a HMA a 36,5ºC [a MDA aumentou de 0,124 ± 0,006 para
0,180 ± 0,023 mmol / 106 células (P < 0,05) e a HMA aumentou de 0,69 ± 0,03
para 1,23 ± 0,13 nmol / 106 células (P < 0,01)] não afectando a formação de
HMMA. A pré-incubação com DAS ou KET foi capaz de impedir o aumento da
formação de HMA induzido pelo etanol, mas não impediu o aumento na
formação de MDA.
Em condições hipertérmicas, o etanol aumentou significativamente a
formação dos três metabolitos considerados. A co-incubação de etanol e
MDMA em condições hipertérmicas resultou num aumento de 6 vezes na
formação de MDA [de 0,248 ± 0,015 para 1,467 ± 0,121 mmol / 106 células (P <
0,001)], de 16 vezes na formação de HMMA [de 8,92 ± 0,40 para 145,7 ± 11,9
nmol / 106 células (P < 0,001)] e de 10 vezes na formação de HMA [de 0,818 ±
0,090 para 8,462 ± 0,752 nmol / 106 células (P < 0,001)]. A pré-incubação com
DAS impediu parcialmente esta indução resultante da co-exposição ao etanol.
A pré-incubação com KET também foi capaz de reverter o aumento da
formação de metabolitos provocado pelo etanol, à excepção da formação de
HMA.
Esta maior actividade metabólica dos hepatócitos expostos ao etanol
poderá ser devida à indução de várias isoenzimas do sistema citocromo P450
como a CYP2E1 e a CYP3A previamente atribuída ao etanol 676, 677. De facto, o
etanol provou induzir a actividade da CYP2E em culturas de hepatócitos de
rato (após 48 horas de exposição) e em ensaios in vivo em ratos 677. Por seu
lado, a indução da CYP3A pelo etanol foi demonstrada, in vitro, em linhas
celulares HepG2 que expressam a CYP3A4 humana e, in vivo, em ratos
alimentados com a dietas contendo etanol 676. A maior formação de metabolitos
dotados de maior toxicidade do que a própria MDMA 5 pode estar na base do
aumento da mortalidade celular quando as células são co-expostas a etanol e a
MDMA. No entanto, é importante salientar que outros factores, para além do
aumento do metabolismo da MDMA, contribuem para o sinergismo na
toxicidade do etanol e da MDMA. Destes, a hipertermia, efeito potencialmente
letal associado com a exposição in vivo à MDMA, tem uma acção
preponderante, uma vez que agrava significativamente a influência do etanol

Trabalho V ______________________________________________________
232
no metabolismo da MDMA aumentando a formação de metabolitos
hepatotóxicos. No entanto, o aumento da citotoxicidade verificado em
condições hipertérmicas não se deve unicamente à maior formação de
metabolitos, uma vez que, mesmo diminuindo a quantidade de metabolitos
formados, recorrendo a uma pré-incubação com inibidores da CYP2E1 e da
CYP3A1, não se verifica uma diminuição da percentagem de morte celular a
qual se mantém elevada como nas células que não foram pré-incubadas com
os inibidores, o que ilustra a importância da resposta hipertérmica na
expressão da hepatotoxicidade da MDMA.
Assim, dos resultados deste trabalho, podemos concluir que o etanol
induz um aumento do metabolismo da MDMA , especialmente em condições
hipertérmicas e, possivelmente, devido à indução de isoenzimas do sistema
citocromo P450, incluindo a CYP2E1 e a CYP3A1.
Para confirmar esta indução estão programados estudos, já fora do
âmbito desta dissertação, que englobam a determinação da actividade,
expressão e transcrição da CYP2E1 e da CYP3A1 no modelo experimental
utilizado neste estudo.

Trabalho VI. Gas chromatography determination of acetone, acetaldehyde,
ethanol and methanol in biological matrices and cell culture


_____________________________________________________ Trabalho VI
235
Trabalho VI. Gas chromatography determination of acetone, acetaldehyde, ethanol and methanol in biological matrices and cell culture.
Autores: Helena Pontesª, Paula Guedes de Pinhoª, Susana Casalb, Helena
Carmoª, Agostinho Santosc, Teresa Magalhãesc, Fernando Remiãoª,
Félix Carvalhoª, Maria Lourdes Bastosª a REQUIMTE, Toxicology Department, Faculty of Pharmacy, University of Porto, Rua Aníbal Cunha 164, 4099-030 Porto, Portugal b REQUIMTE, Bromatology Department, Faculty of Pharmacy, University of Porto, Rua Aníbal Cunha 164, 4099-030 Porto, Portugal c National Institute of Legal Medicine I.P., Faculty of Medicine, University of Porto, Alameda Prof. Hernâni Monteiro, 4200 - 319 Porto, Portugal
a) Abstract A Gas Chromatographic with Flame Ionization Detection method (GC-
FID) with direct injection, using a capillary column, was validated to determine
ethanol, acetaldehyde, methanol and acetone in different human matrices, such
as whole blood, vitreous humour and urine, with clinical and forensic interest.
This method was also employed to quantify these compounds in cell culture
medium, thus being useful in basic research. A good peak resolution was
achieved, with linear correlation between concentration and peak areas for all
the compounds in all the matrices. The inter-day and the intra-day precision
(CV%) of the method was always under 15% and 10%, respectively. The
accuracy (A%) of the method, calculated as the percentage of target
concentration, was within the acceptable limits. The obtained limit of detection
was below 0.85 mg/L for acetaldehyde and below 0.75 mg/L for the other
considered compounds. The small injection volume and the high split ratios
applied, allied to the high performance of the GC column resulted in very good
peak resolution and high sensitivities. This method is easy to perform, making it
suitable for the routine of clinical biochemistry and forensic laboratories.

Trabalho VI _____________________________________________________
236
b) Introduction Gas chromatographic methodologies have been reported, being the
volatile fraction analysed by diversified techniques [1-5]. Chronologically, the
direct sample injection in packed columns [1, 3] became obsolete and was
gradually substituted by headspace techniques in capillary columns [6] and,
more recently, by the selective headspace injection using Solid Phase Micro-
Extraction (SPME) fibres [5, 7, 8]. However, both headspace methodologies
require an accurate time- and temperature-controlled sample heating. When the
automatic injectors are not available, reproducibility problems are difficult to
overcome. Moreover, the influence of the biological specimen on the partitioning
of the volatile compounds between liquid and headspace vapour and the
selectivity of the SPME fibres increase both the complexity of method
development and the total analysis time [9]. In addition, the headspace
techniques usually require larger sample volumes and have higher detection
limits.
Ethanol, acetaldehyde, methanol and acetone are volatile compounds
whose detection and quantification in biological matrices can be used as
biomarkers of several diseases and/or intoxications.
Ethanol consumption is under strict regulations in many circumstances,
and legal Blood Alcohol Concentration (BAC) limits for driving are well
established. Over-consumption of alcoholic beverages and drunkenness are
almost always closely related with fatal accidents, trauma deaths, drowning,
suicide and violent crimes [10]. These considerations justify the importance of
the determination of ethanol levels in ante-mortem and post-mortem specimens
with great importance in the forensic domain [4].
Humans are frequently exposed to naturally occurring acetaldehyde that
can exist in the air and that can be ingested as a contaminant of food and
alcoholic drinks since it is the main compound formed during ethanol
metabolism. Additionally, humans are also exposed to acetaldehyde coming
from automobile exhaust, cigarette smoke, fireplaces, and occupational
settings. This compound has been associated with cancer development in
experimental animals, being classified as a possible carcinogenic to humans by
the International Agency for Research on Cancer (IARC) of the World Health

_____________________________________________________ Trabalho VI
237
Organization [11]. Thus, from a clinical point of view, its quantification in
biological samples is of paramount importance.
The quantification of methanol in body fluids is very important for
confirmation of methanol intoxication-related deaths [12] and can also be a
biomarker of intentional [13], accidental (for example, by ingestion in
adulterated drinks) [14] or occupational exposure [15].
Acetone can be a biomarker of ketoacidosis, which can help to diagnose
ante-mortem diabetes mellitus, though it can also be a likely cause of death in
binge drinkers and alcoholics [16], and can be very useful in the diagnosis of
death from hypothermia [17]. Ketoacidosis is diagnosed by analysis of high
levels of ketone bodies in body fluids, namely acetone, acetoacetate and
particularly β-hydroxybutyrate [18]. Additionally, the quantification of acetone in
biological fluids can be a biomarker of occupational exposure [15].
Given the importance of the quantification of these compounds in blood
and urine from ante-mortem and post-mortem specimens we validated the
present method in these matrices. However, as post-mortem compounds levels
in blood do not necessarily reflect the concentration at the time of death, due to
drug instability and post-mortem redistribution phenomena [19], measurements
of vitreous humour (VH) concentrations could be of interest for predicting the
blood concentration at the time of death in humans [20]. VH is a very useful
matrix because the vitreous fluid is less influenced by autolytic processes, is
simple to collect and is not affected by hemolysis [10]. Additionally, it is a clean
fluid that contains less protein than urine and exhibits high stability [21]. Thus,
VH has been recently demonstrated to be a suitable alternative specimen to
post-mortem blood and urine, not only for the analysis of ethanol intoxications,
but also for other drugs as well as endogenous biochemical constituents of the
body to detect ante-mortem diseases [10]. VH is, additionally, more resistant to
putrefactive changes than other specimens such as blood [8, 22]. Due to these
advantages, formerly, VH determinations have been performed in order to
detect various drugs, in particular ethanol [10] and also morphine [23], cocaine
[24] and amitriptyline [25].
The validation of this method for its application in cell culture medium
samples is also important due to the need of controlling the levels of these

Trabalho VI _____________________________________________________
238
compounds when cell culture is used to study their effects on cell physiology
and toxicity [26].
Thus, there is an obvious need to develop a cheaper, sensitive, rapid and
reliable alternative gas chromatographic method. In this study it is proposed a
gas chromatographic direct injection in capillary columns, to quantify ethanol,
acetaldehyde, methanol and acetone, and to apply it to different matrices such
as whole blood, vitreous humour, urine and cell culture medium.
Based on the above mentioned rationale, the aim of this work was to
develop an easy direct injection gas-chromatographic methodology to
determine simultaneously some of the most important volatiles found in
biological samples with clinical, forensic and research interest and to apply it to
several matrices.
c) Materials and Methods
c.1) Reagents All the used chemicals were of analytical grade: Ethanol (> 99.9%,
Panreac, Spain), methanol (> 99.9%, Merck, Germany), 1-propanol (> 99%,
Sigma-Aldrich Co., USA), acetone (> 99.9%, Merck, Germany), acetaldehyde (>
99.9%, Fluka, USA), Triton X-100 (Sigma-Aldrich Co. USA), acetonitrile (>
99.9%, Merck, Germany). William’S E culture medium was obtained from Lonza
(Belgium) and supplemented with 2 mM L-glutamine (Lonza, Belgium), 5 μg/mL
insulin (Sigma-Aldrich Co., USA), 5 nM dexamethasone (Sigma-Aldrich Co.,
USA), 10 mM Hepes (Lonza, Belgium), 100 units/mL penicillin G, 100 μg/mL
streptomycin sulphate and 250 ng/mL amphotericin B (Sigma-Aldrich Co.,
USA).
c.2) Biological matrices characterization and preparation Human blood and urine samples were collected from healthy volunteers.
In the experiments with whole blood, EDTA-blood was used. Post-mortem
human vitreous humour was collected from autopsy samples at the North
Delegation of the National Institute of Legal Medicine I.P., after accomplishment
of all legal procedures to post-mortem samples collection. Prior to analysis,

_____________________________________________________ Trabalho VI
239
each sample was injected into the GC-FID to confirm the absence of each
studied compound. Additionally, the interference between the tested
compounds and some other volatile biomarkers of some diseases and with
great forensic interest such as formaldehyde, methyl and ethyl formate,
ethylene glycol, propylene glycol, glycerol, 1,4-butanediol and 2,3-butanediol
was excluded.
Volatile-free Blood (B), Urine (U), Vitreous Humour (VH) and Cell Culture
Medium (CCM) were used to prepare the control samples and calibration
curves.
c.3) Calibration procedures Concentrated solutions of 500 g/L ethanol, 50 g/L methanol, 50 g/L
acetaldehyde and 50 g/L acetone were prepared daily by dilution of the
commercial solutions in deionised water. Daily prepared 2.2 g/L 1-propanol in
deionised water was used as internal standard (IS).
A stock calibrator containing 10 g/L ethanol, 1 g/L methanol, 1 g/L
acetaldehyde and 1 g/L acetone was prepared daily in each tested matrix, from
the concentrated solutions. The calibration curves were prepared as shown in
Figure 1.
These procedures resulted in final concentration of 7.5, 15, 30, 60, 120
and 240 mg/L of methanol, acetone and acetaldehyde, and with 75, 150, 300,
600, 1200 and 2400 mg/L of ethanol. Theses calibration standards, in blood and
vitreous humour, undergone a 5 times dilution with a solution containing 1.2% of
Triton X-100 and 1.8 g/L of acetonitrile in water, to decrease sample viscosity
and therefore, facilitate volume measurements and sample injection, according
to the method described by Dubowski [27].
Each tube was vortex-mixed and centrifuged at 16,000 g for 5 min at 4
ºC. A fixed volume of supernatant (0.5 μL) was injected into the
chromatographic system.

Trabalho VI _____________________________________________________
240
Figure 1 – Flowchart for preparation of the calibration curves.
c.4) Samples preparation
Blood or vitreous humour samples (100 μL) were mixed with 10 μL of IS.
The samples were diluted to 500 μL with the Triton X-100 and acetonitrile
solution, centrifuged and 0.5 μL of the supernatant were directly injected into
the gas chromatograph as described below. Concerning urine and cell culture
medium, after adding the internal standard, samples were centrifuged and 0.5
μL of the supernatant were directly injected into the gas chromatograph.
c.5) Analytical Instrument settings The gas chromatograph used was a TermoFinnigan Model Focus GC
equipped with a flame ionization detector. The injection port of the
chromatograph was installed with a glass liner (5 mm ID) appropriated for split
analysis, to prevent the contamination of the gas chromatographic column with

_____________________________________________________ Trabalho VI
241
non-volatile material from the tested matrices. For whole blood and vitreous
humour, the liner was replaced after 50 injections. For cell culture medium and
for urine, the liner was replaced each 100 injections.
The analyses were performed under the following chromatographic
conditions: Column: CPWax 57 CB, (WCOT Fused Silica), 25 m × 0.25 mm id,
DF = 0.2, from Varian (USA). The temperature of the FID detector was 220ºC
and the injector temperature was 220ºC. The oven temperature was
programmed to 40ºC (for 2 min), followed by an increase of 5ºC/min until
200ºC. Carrier gas was helium with a flow of 1.5 mL/min. The injection of whole
blood and vitreous humour was performed by means of a 10 μL Hamilton
syringe (Model 701 RN) with a removable needle (needle gauge 22S), cleaned
under vacuum between each injection with the Triton X-100 and acetonitrile
solution. On the other hand, the injection of cell culture medium and urine was
performed by means of a 5 μL SGE syringe (Model 5F-GP) cleaned under
vacuum between each injection with deionised water. The volume of injection
was 0.5 μL, with a split ratio of 100 and a split flow of 120 mL/min for blood and
vitreous humour, and a split ratio of 60 and a split flow of 90 mL/min for urine
and cell culture medium.
c.6) Effect of the split ratio on the sensitivity of the method The effect of the split ratio on the sensitivity of the method was studied
for whole blood with 60 mg/L acetaldehyde, acetone and methanol and 600
mg/L ethanol. Split ratios between 1:40 and 1:500 were tested: 1:40, 1:60,
1:100, 1:150, 1:200, 1:300, 1:400, 1:500, which corresponds to 60 mL/min, 90
mL/min, 120 mL/min, 150 mL/min, 300 mL/min, 450 mL/min, 600 mL/min, 750
mL/min, respectively.
c.7) Validation experiments and acceptance criteria
c.7.1) Method linearity Method linearity was determined by evaluating the regression curve and
is indicated by the square correlation coefficient (R2). The line of best fit for the

Trabalho VI _____________________________________________________
242
relationship between the ratio of peak area and internal standard area and
concentration of analytes in the samples was determined by linear regression
performing calibration curves in the considered concentration ranges (7.5-240
mg/L for acetaldehyde, acetone and methanol and 75-2400 mg/L for ethanol).
For urine and cell culture medium, the slopes were calculated taking into
account the mean of 5 calibration curves prepared in each matrix on 5
consecutive days. For whole blood and vitreous humour, the slopes were based
on the mean of 3 curves prepared in these matrices on 3 consecutive days.
Linearity was achieved with a minimal R2 of 0.99.
c.7.2) Precision For urine and cell culture medium, the intra-day precision of the method
was determined by injecting, on the same day, 5 different replicate calibrators of
each one of the 7 points of the calibration curves; the intra-day precision of the
apparatus was determined by analysing 5 times, on the same day, the 7
calibrators of one of the calibration curves; the inter-day precision of the method
was determined by analyzing, for 5 consecutive days, daily prepared calibrators
of the 7 concentrations considered for the calibration curves.
For blood and vitreous humour, due to the complexity of these samples,
the intra-day precision of the method was determined injecting, on the same
day, 6 independent calibrators containing 1200 mg/L of ethanol and 120 mg/L of
acetaldehyde, acetone and methanol; the intra-day precision of the apparatus
was determined injecting 6 times, on the same day, one of the concentrations
contemplated in the calibration curves (1200 mg/L for ethanol and 120 mg/L for
acetaldehyde, acetone and methanol); the inter-day precision of the method
was evaluated by analyzing, for 3 consecutive days, daily prepared calibrators
of all the 7 concentrations considered for the calibration curves. Precision was
assessed by calculating the mean, standard deviation and coefficient of
variation (CV%) of the observed values.
c.7.3) Limits of detection and quantification To determine the sensitivity of the method, the calibrators with the lowest
concentrations (7.5 mg/L for methanol, acetaldehyde and acetone and 75 mg/L
for ethanol) of each matrix (urine, blood, vitreous humour and cell culture

_____________________________________________________ Trabalho VI
243
medium) were progressively diluted to determine the detection (LOD) and
quantification limits (LOQ). A signal-to-noise ratio of 3 was considered
acceptable for estimating the detection limit [28]. This concentration that
originated the peak with a signal-to-noise ratio of 3 was injected 5 times.
The quantification limit (LOQ) for each matrix was estimated based on a
signal-to-noise ratio of 10 obtained for calibration solutions containing the
compounds of interest. The LOQ corresponds to the lowest concentration
obtained by successive dilutions of standards that originate a sharp and
symmetrical chromatographic peak, required for routine analysis. Peaks that
were excessively broad, showing tailing or shoulders, or that did not resolve to
within 10% baseline were not considered [29].
c.7.4) Accuracy (A%) Accuracy was calculated in terms of bias as the percent deviation of the
mean calculated concentration at each concentration level from the
corresponding theoretical concentration. Accuracy (%) = (mean calculated
concentration – nominal concentration) / (nominal concentration) × 100.
d) Results d.1) Gas Chromatographic Separation
Interferents were ruled out by verifying the absence of peaks in the
retention times of the studies analytes (Figure 2A and 2B).
As shown in Figure 2 the retention times for acetaldehyde, acetone,
methanol and ethanol in all tested matrices (Figure 2C and 2D) were 1.66, 2.02,
2.70 and 3.16 min, respectively. The internal standard (1-propanol) retention
time was 5.26 min. The peak at 3.71 minutes in the chromatogram 2B (whole
blood and vitreous humour) corresponds to the peak of acetonitrile, which is
part of the triton X-100 and acetonitrile solution used to dilute these complex
matrices.

Trabalho VI _____________________________________________________
244
Figure 2 – Representative chromatograms obtained from blood sample without IS (2A), blood sample with IS (2B), calibrator solutions prepared in cell culture medium or urine (2C) and in blood or vitreous humour (2D) with peak identification and retention times of the quantified compounds.
d.2) Method validation
d.2.1) Linearity Regression analysis of calibration data achieved satisfactory linearity
over the considered concentration range. Square correlation coefficients (R2)
were always > than 0.99, indicating a linear relationship from 7.5 to 240 mg/L
for acetaldehyde, methanol and acetone and from 75 to 2400 mg/L for ethanol
in all the studied matrices.
The slopes and square correlation coefficients are presented in Figure
3A for cell culture medium, 3B for urine, 3C for blood and 3D for vitreous
humour.

_____________________________________________________ Trabalho VI
245

Trabalho VI _____________________________________________________
246
Figure 3 – Calibration curves of acetaldehyde, acetone, methanol and ethanol on the 4 different considered matrices: cell culture medium (3A), urine (3B), blood (3C) and vitreous humour (3D). Results are presented as mean ± SD of 5 different injections of each calibrator of cell culture medium and urine and of 3 different injections of each calibrator of whole blood and vitreous humour.

_____________________________________________________ Trabalho VI
247
d.2.2) Sensitivity The LOD and LOQ for each compound in each matrix are shown in Table
1.
Table1. Limits of Detection (LOD) and Quantification (LOQ) in mg/L.
Acetaldehyde Acetone Methanol Ethanol LOD LOQ LOD LOQ LOD LOQ LOD LOQ
Blood 0.84 2.8 0.75 2.5 0.75 2.5 0.75 2.5
Cell Culture medium 0.15 0.5 0.38 1.23 0.75 2.5 0.38 1.23
Vitreous humor 0.15 0.5 0.15 0.5 0.75 2.5 0.25 0.83
Urine 0.15 0.5 0.75 2.5 0.5 1.67 0.25 0.83
d.2.3) Effect of the split ratio on the sensitivity of the method The effect of the split ratio on the sensitivity of the method is illustrated
on Figure 4.
Methanol
Acetone
Acetaldehyde
0
50000
100000
150000
200000
250000
300000
0 50 100 150 200 250 300 350 400 450 500Split ratio
Chro
mat
ogra
phic
Pea
k A
rea
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
0 50 100 150 200 250 300 350 400 450 500
Split ratio
Chr
omat
ogra
phic
Pea
k Ar
ea
Δ
♦ Ethanol Methanol
Acetone
Acetaldehyde
0
50000
100000
150000
200000
250000
300000
0 50 100 150 200 250 300 350 400 450 500Split ratio
Chro
mat
ogra
phic
Pea
k A
rea
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
0 50 100 150 200 250 300 350 400 450 500
Split ratio
Chr
omat
ogra
phic
Pea
k Ar
ea
Δ
♦ Ethanol
Figure 4 – Variation of Chromatographic peak area in blood matrix with the injector split ratio.
For the blood matrix, higher split ratios were needed to protect liners and
the chromatographic column. However, if lower detection limits are needed, the
split ratio can be decreased until 1:50. For the other matrices, a good peak
resolution can still be obtained for ratios up to 1:30 (data not shown).

Trabalho VI _____________________________________________________
248
d.2.4) Inter-day precision (CV%) and accuracy (A%) of the method The results for the 4 matrices and for 3 representative concentrations are
presented in Table 2. For cell culture medium and urine, the simplest matrices,
the obtained coefficients of variation (CV%) were always lower than 10%. For
vitreous humour and blood, due to sample manipulations, the CV% was less
favourable. The average coefficients of variation (ACV%) of all the tested
concentrations are presented in Table III and were always lower than 15%.
The accuracy (A%) of the method for the 4 matrices and for 3
representative concentration of each compound, calculated as the percentage
of target concentration, is indicated in Table 2.
d.2.5) Intra-day precision of the method and of the apparatus The intra-day precision of the method and of the apparatus for the 4
tested compounds in the 4 considered matrices are presented in Table 3 as
average coefficients of variation (ACV%) that are always lower than 10%.
Concerning the intra-day precision of the apparatus, the average
coefficients of variation (ACV%) were always between 2% and 9% being the
lowest ACV% obtained for ethanol in urine and the highest for acetaldehyde in
blood. The ACV% values obtained for whole blood and vitreous humour were
higher than those obtained for cell culture medium and urine due to the need of
more sample manipulations (Table 3).
Concerning the intra-day precision of the method, the average
coefficients of variation (ACV%) were always between 3% and 9% being the
lowest ACV% obtained for ethanol in blood and the highest for methanol in
blood (Table 3).

249
Table 2. Inter-day precision (CV%) and accuracy (A%) of the method for the 4 matrices and for 3 representative concentrations.
ª Mean of A/A(IS) ± Standard Deviation b Reproducibility = (Standard Deviation / Mean) × 100 c Accuracy = (Mean Calculated Concentration-Nominal Concentration) / (Nominal Concentration) × 100 d Mean of 5 replicates e Mean of 3 replicates
Acetaldehyde Acetone Methanol Ethanol
Conc (mg/L)
Mean ± SDª CV%b A%c Mean ± SD CV% A% Mean ± SD CV% A% Conc (mg/L)
Mean ± SD CV% A%
15 0.258 ± 0.021 8.094 -0.3 0.326 ± 0.025 7.588 -9.4 0.249 ± 0.020 7.920 2.8 150 3.359 ± 0.213 6.327 2.0 60 0.788 ± 0.063 8.060 -10.5 1.099 ± 0.100 9.088 -1.0 0.799 ± 0.038 4.718 -10.7 600 10.951 ± 0.955 8.722 -22.3 C
Md
120 1.830 ± 0.148 8.075 7.1 2.288 ± 0.184 8.061 5.1 1.813 ± 0.034 1.878 2.8 1200 29.410 ± 1.562 5.311 3.2
15 0.248 ± 0.009 3.787 6.2 0.319 ± 0.021 6.464 -3.0 0.221 ± 0.008 3.449 -5.2 150 3.738 ± 0.276 7.385 3.3 60 0.859 ± 0.085 9.869 -1.1 1.262 ± 0.101 8.045 1.2 0.818 ± 0.061 7.413 6.5 600 12.284 ± 1.183 9.629 -1.2 U
d
120 1.579 ± 0.084 5.304 -7.9 2.444 ± 0.131 5.351 -1.1 1.582 ± 0.039 2.445 6.7 1200 23.732 ± 2.179 9.180 -1.9
15 0.077 ± 0.007 9.113 18.0 0.058 ± 0.002 3.483 -9.1 0.026 ± 0.003 10.687 -21.0 150 0.401 ± 0.055 13.720 -13.7 60 0.193 ± 0.006 3.078 18.0 0.166 ± 0.016 9.648 -11.7 0.122 ± 0.019 15.286 -2.9 600 1.685 ± 0.177 10.483 0.3 B
e
120 0.278 ± 0.041 14.592 -5.9 0.393 ± 0.030 7.511 6.9 0.257 ± 0.056 21.934 10.4 1200 3.622 ± 0.420 11.584 9.8
15 0.054 ± 0.009 10.886 -21.6 0.066 ± 0.003 5.026 19.0 0.030 ± 0.001 3.498 -16.2 150 0.644 ± 0.078 12.046 11.0 60 0.128 ± 0.029 22.461 -13.5 0.149 ± 0.012 9.412 -23.4 0.112 ± 0.005 4.210 -20.1 600 1.751 ± 0.114 6.496 -17.0 VH
e
120 0.284 ± 0.002 0.818 -2.8 0.387 ± 0.047 12.066 -1.3 0.275 ± 0.006 2.005 -1.2 1200 4.310 ± 0.750 17.413 3.9

Table 3. Compilation of the average coefficients of variation (ACV%) obtained for the evaluation of the intra-day precision of the GC-FID apparatus, the intra-day precision of the method, and the inter-day precision of the method.
Acetaldehyde Acetone Methanol Ethanol Cell Culture Medium 5.138 5.168 6.292 3.404
Urine 2.322 2.060 3.839 1.957
Blood 9.017 4.134 6.342 2.202
Intra-day precision of the
GC-FID
Vitreous Humour 4.718 2.410 6.663 4.518
Cell Culture Medium 5.978 5.042 5.697 6.207
Urine 5.699 4.485 7.003 6.235
Blood 3.797 5.262 8.829 2.973
Intra-day precision of the
method
Vitreous Humour 8.051 7.871 8.267 5.901
Cell Culture Medium 9.025 7.526 5.179 6.668
Urine 5.543 5.253 6.802 8.336
Blood 11.572 8.621 10.728 8.684
Inter-day precision of the
method
Vitreous Humour 12.254 10.759 8.777 12.854
250

_____________________________________________________ Trabalho VI
251
e) Discussion Several methods for the analysis of ethanol that are also concerned with
the simultaneous monitoring of acetaldehyde, methanol or acetone
concentrations have been reported [9, 30-32]. GC-MS methods have also been
applied to measure ethanol concentrations in different biological matrices [31,
33, 34]. However, GC-MS is a much more complex technique than GC-FID and
requires highly trained personnel. In consonance with these previous methods,
the technique described in the present study enables the GC-FID analysis of
blood, urine, vitreous humour and cell culture medium for the presence of
ethanol, acetaldehyde, acetone and methanol without any pre-treatment, in the
case of urine, and cell culture medium, and with a simple dilution with triton X-
100 and acetonitrile for vitreous humour and blood.
As shown in Figure 2, the selected chromatographic conditions resulted
in a good chromatographic resolution, with good peak separation. In addition,
the good chromatographic separation between the studied compounds and
some other important volatiles such as formaldehyde, methyl and ethyl formate,
ethylene glycol, propylene glycol, glycerol, 1,4-butanediol and 2,3-butanediol,
recognized as biomarkers of some diseases and/or having great forensic
interest, evince the interest of this method for a future adaptation to quantify
these volatiles. This fact is a great advantage of this method, indicating its
possible broad applicability.
A very good linear correlation (R2>0.99) between the concentration and
the ratio between the compound peak and the IS peak was obtained for all the
tested compounds in all the tested matrices.
The proposed acceptance limits for the accuracy were 100 ± 20% [28]
and, only in the case of VH, and for some concentrations, the obtained
accuracy results were out of these limits.
The injection of 0.5 μL of blank matrices showed no interferences with
other constituents from whole blood, vitreous humour, cell culture medium, and
urine, allowing the detection of very small amounts of the studied compounds.
The sensitivity depends on the volume injected and depends on the split
ratio. In this work, and in the case of whole blood, to prevent the shelf-life of the

Trabalho VI _____________________________________________________
252
chromatographic column, the volume injected was 0.5 μL and the split ratio was
1:100.
For more sensitive determinations, 1 μL or even larger volumes can be
injected and the split ratio can also be decreased, easily decreasing de LOD to
levels lower than 0.1 mg/L. For the suggested chromatographic conditions, the
LOD obtained with whole blood were: 0.84 mg/L for acetaldehyde, and 0.75
mg/L for acetone, methanol and ethanol. However, by changing the split ratio,
the LOD can be easily decreased 10 times, which means: 0.075 mg/L for
ethanol, acetone and methanol, and 0.084 mg/L for acetaldehyde [3].
f) Conclusion In conclusion, the direct GC-FID injection method using capillary columns
presented here is a highly sensitive, rapid and reliable procedure to determine a
plethora of volatile compounds in various biological samples. For the first time,
a method was validated for the simultaneous determination of acetaldehyde,
acetone, methanol and ethanol in four different matrices (human blood, urine
and vitreous humour and cell culture medium). The small amounts of sample
injected (0.5 μL) and the high split ratios applied (1:60 and 1:100), allied to the
high performance of the GC column, result in very good peaks resolution and
high sensitivities. This method is easy to perform and does not require highly
and specifically trained personnel, making it suitable to the routine of clinical
biochemistry and forensic laboratories.
g) Acknowledgements Helena Pontes thanks FCT for financial support (SFRH/BD/18527/2004).
h) References 1. Ferrari LA, Arado MG, Nardo CA, and Giannuzzi L. Post-mortem analysis of formic acid
disposition in acute methanol intoxication. Forensic Sci Int;133:152-158 (2003). 2. Fukunaga T, Sillanaukee P, and Eriksson CJ. Problems involved in the determination of
endogenous acetaldehyde in human blood. Alcohol Alcohol;28:535-41 (1993). 3. Tangerman A. Highly sensitive gas chromatographic analysis of ethanol in whole blood,
serum, urine, and fecal supernatants by direct injection method. Clinical Chem;43:1003-9 (1997).
4. Zilly M, Langmann P, Lenker U, Satzinger V, Schirmer D, and Klinker H. Highly sensitive gas chromatographic determination of ethanol in human urine samples. J Chromatogr B Analyt Technol Biomed Life Sci;798:179-186 (2003).

_____________________________________________________ Trabalho VI
253
5. Zuba D, Parczewski A, and Reichenbacher M. Optimization of solid phase microextraction conditions for gas chromatographic determination of ethanol and other volatile compounds in blood. J Chromatogr B Analyt Technol Biomed Life Sci;773:75-82 (2002).
6. Wasfi IA, Al-Awadhi AH, Al-Hatali ZN, Al-Rayami FJ, and Al Katheeri NA. Rapid and sensitive static headspace gas chromatography-mass spectrometry method for the analysis of ethanol and abused inhalants in blood. J Chromatogr B Analyt Technol Biomed Life Sci;799:331-6 (2004).
7. De Martinis BS and Martin CC. Automated headspace solid-phase microextraction and capillary gas chromatography analysis of ethanol in postmortem specimens. Forensic Sci Int; 128:115-9 (2002).
8. De Martinis BS, Ruzzene MAM, and Martin CCM. Determination of ethanol in human blood and urine by automated headspace solid-phase microextraction and capillary gas chromatography. Analytica Chem Acta;522:163-168 (2004).
9. Watts MT and McDonald OL. The effect of biologic specimen type on the gas chromatographic headspace analysis of ethanol and other volatile compounds. Am J Clin Pathol;87:79-85 (1987).
10. Kugelberg FC and Jones AW. Interpreting results of ethanol analysis in postmortem specimens: a review of the literature. Forensic Sci Int;165:10-29 (2007).
11. WHO-IARC. Re-Evaluation of Some Organic Chemicals, Hydrazine and Hydrogen Peroxide. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans;71:14-18 (1999).
12. Pla A, Hernandez AF, Gil F, Garcia-Alonso M, and Villanueva E. A fatal case of oral ingestion of methanol. Distribution in postmortem tissues and fluids including pericardial fluid and vitreous humor. Forensic Sci Int;49:193-6 (1991).
13. Hantson PE. Acute methanol intoxication: physiopathology, prognosis and treatment. Bull Mem Acad R Med Belg;161:425-34 (2006).
14. Paine A and Davan AD. Defining a tolerable concentration of methanol in alcoholic drinks. Hum Exp Toxicol;20:563-8 (2001).
15. Imbriani M and Ghittori S. Gases and organic solvents in urine as biomarkers of occupational exposure: a review. Int Arch Occup Environ Health;78:1-19 (2005).
16. Thomsen JL and Frohlich B. Drug abuse and intoxication in alcoholics. Alcohol Alcohol;30:379-83 (1995).
17. Teresiński G, Buszewicz G, and Madro R. The influence of ethanol on the level of ketone bodies in hypothermia. Forensic Sci Int;127:88-96 (2002).
18. Brinkmann B, Fechner G, Karger B, and DuChesne A. Ketoacidosis and lactic acidosis--frequent causes of death in chronic alcoholics? Int J Legal Med;111:115-9 (1998).
19. Pounder DJ and Jones GR. Post-mortem drug redistribution--a toxicological nightmare. Forensic Sci Int;45:253-63 (1990).
20. De Letter EA, De Paepe P, Clauwaert KM, Belpaire FM, Lambert WE, Van Bocxlaer JF, et al. Is vitreous humour useful for the interpretation of 3,4-methylenedioxymethamphetamine (MDMA) blood levels? Experimental approach with rabbits. Int J Legal Med;114:29-35 (2000).
21. Sebag J. Age-related differences in the human vitreoretinal interface. Arch Ophthalmol.;109:966-71 (1991).
22. Miekisch W, Schubert JK, Vagts DA, and Geiger K. Analysis of volatile disease markers in blood. Clinical Chem;46:1053-1060 (2001).
23. Wyman J and Bultman S. Postmortem distribution of heroin metabolites in femoral blood, liver, cerebrospinal fluid, and vitreous humor. J Anal Toxicol;28:260-3 (2004).
24. Fernández P, Aldonza M, Bouzas A, Lema M, Bermejo AM, and Tabernero MJ. GC-FID determination of cocaine and its metabolites in human bile and vitreous humor. J Appl Toxicol;26:253-7 (2006).
25. Tracqui A, Kintz P, Ritter-Lohner S, Mangin P, Lugnier A, and Chaumont A. Toxicological findings after fatal amitriptyline self-poisoning. Hum Exp Toxicol;9:257-61 (1990).
26. Schaffert CS, Toderoa SL, McVickera BL, Tumac PL, Sorrella MF, and Tumaa D. WIF-B cells as a model for alcohol-induced hepatocyte injury. Biochem Pharmacol;67:2167-2174 (2004).
27. Dubowski KM. Methods for specific substances: Ethanol. in Methodology for Analytical Toxicology, Sunshine I, Editor CRC Press Ed: Cleveland:149-154 (1975).
28. EMEA. Note for guindance on validation of analytical procedures: Text and methodology.;CPMP/ICH/381/95:1-15 (1995).

Trabalho VI _____________________________________________________
254
29. Armbruster DA, Tillman MD, and Hubbs LM. Limit of Detection (LOD)/Limit of Quantification (LOQ): Comparison of the Empirical and the Statistical Methods Exemplified with GC-MS Assays of Abused Drugs. Clin Chem;40:1233-1238 (1994).
30. Hernandez-Munoz R, Ma XL, Baraona E, and Lieber CS. Method of acetaldehyde measurement with minimal artifactual formation in red blood cells and plasma of actively drinking subjects with alcoholism. J Lab Clin Med;120:35-41 (1992).
31. Liebich HM and Wöll J. Volatile substances in blood serum: profile analysis and quantitative determination. J Chromatogr;142:505-16 (1977).
32. Varga M, Somogyi G, Posta J, and Buris L. Effect of different columns and internal standards on the quality assurance of the gas chromatographic determination of blood ethanol. Eur J Clin Chem Clin Biochem;31:773-6 (1993).
33. Liebich HM, Buelow HJ, and Kallmayer R. Quantification of endogenous aliphatic alcohols in serum and urine. J Chromatogr;239:323-9 (1982).
34. Tang BK. Detection of ethanol in urine of abstaining alcoholics. Can J Physiol Pharmacol;65:1225-7 (1987).

Trabalho VII. Ethanol, the forgotten artifact in cell culture.


Arch Toxicol (2008) 82:197–198
DOI 10.1007/s00204-008-0285-yLETTER TO THE EDITOR
Ethanol, the forgotten artifact in cell culture
Helena Pontes · Márcia Carvalho · Paula Guedes de Pinho · Helena Carmo · Fernando Remião · Félix Carvalho · Maria Lourdes Bastos
Received: 16 November 2007 / Accepted: 15 January 2008 / Published online: 6 February 2008© Springer-Verlag 2008
Those who work with cell cultures are certainly familiar-ized with the ritual of sanitizing everything that goesinto the cabinet or into the incubators with 70% ethanol,as is recommended by all the rules of aseptic techniquesand good cell culture practices. In spite of its eVective-ness in preventing microbiological contamination of thecultures, this gesture may have some undesirableconsequences.
Probably, the research groups that study the eVects ofethanol on cell cultures are more conscious about therisks of sanitizing all the material with 70% ethanolbecause they need to guarantee that they have ethanol-free control samples. Thus, these groups routinely quan-tify ethanol in the culture medium to conWrm the ethanolconcentrations used in their studies. However, the scien-tiWc community must be aware that when 70% ethanol isused on cell culture as a surface sanitization process, ifcautions are not taken, the cells will be exposed to highethanol concentrations. These high concentrations areachieved because, under normal cell culture conditions,the cell culture plates have appropriate venting systems
(as they should have, otherwise the cells will not be oxy-genated) and generally, both cabinets and incubatorshave an ethanol-saturated atmosphere due to thefrequent sanitization procedures. Without additionalprecautions, ethanol concentrations of 0.5 mM are easilyattained, and depending on the ethanol-saturation of thecabinets or incubators, these concentrations can reachvalues of 5 mM as illustrated in Fig. 1, which representsthe ethanol concentrations detected on 14 independentexperiments where no precautions were taken to avoidthe exposure of the medium to ethanol. Both theseconcentrations have proven eVects on cell function(Charness et al. 1994; Smith and Gong 2007). In fact, itis well described that ethanol, as a microsomal and mito-chondrial enzymatic inducer (CYP2E1, ADH-I), caninXuence the metabolism of several endogenous andexogenous compounds causing eventual antagonisms,synergisms or potentiations that will necessarily aVectthe Wnal results of the experiments (Jang and Harris2007).
In addition, by itself, ethanol can directly or indirectlyalter the normal cell function, inXuencing cell survival,redox status, receptor functions, mitochondrial homeosta-sis, cell signaling, among others (Wu et al. 2006), and alsoaVecting the activity of infectious agents such as hepatitic Cvirus (Trujillo-Murillo et al. 2007).
Thus, some precautions must be taken to minimize this“contamination” with ethanol: 70% ethanol used to sanitizesurface and objects should be completely dry before theXasks or plates are opened and these should not be sprayedwith 70% ethanol between coming out of the cabinet andgoing into the incubator (but obviously, in between, contactwith other surfaces is forbidden).
These concerns are crucial for every lab working withcell culture and must be taken into account during the data
H. Pontes (&) · M. Carvalho · P. G. de Pinho · H. Carmo · F. Remião · F. Carvalho · M. L. Bastos (&)REQUIMTE, Toxicology Department, Faculty of Pharmacy, University of Porto, Rua Aníbal Cunha 164, 4099-030 Porto, Portugale-mail: [email protected]
M. L. Bastose-mail: [email protected]
M. CarvalhoFaculty of Health Sciences, University Fernando Pessoa, Rua Carlos da Maia 296, 4200-150 Porto, Portugal
123
Helena
Text Box
257

198 Arch Toxicol (2008) 82:197–198
interpretation to avoid erroneous analysis due to the eVectof ethanol on cell physiology or its inXuence on other com-pounds’ eVects.
Acknowledgments Helena Pontes thanks FCT for her Ph.D. grant(SFRH/BD/18527/2004).
References
Charness ME, Safran RM, Perides G (1994) Ethanol inhibits neuralcell-cell adhesion. J Biol Chem 269:9304–9309
Jang GR, Harris RZ (2007) Drug interactions involving ethanol andalcoholic beverages. Expert Opin Drug Metab Toxicol 3:719–731
Smith SS, Gong QH (2007) Ethanol eVects on GABA-gated current ina model of increased alpha4betadelta GABAA receptor expres-sion depend on time course and preexposure to low concentra-tions of the drug. Alcohol 41:223–231
Trujillo-Murillo K, Alvarez-Martínez O, Garza-Rodríguez L,Martínez-Rodríguez H, Bosques-Padilla F, Ramos-Jiménez J,Barrera-Saldaña H, Rincón-Sánchez AR, Rivas-Estilla AM(2007) Additive eVect of ethanol and HCV subgenomic repliconexpression on COX-2 protein levels and activity. J Viral Hepat14:608–617
Wu D, Zhai Q, Shi X (2006) Alcohol-induced oxidative stress and cellresponses. J Gastroenterol Hepatol 21:S26–S29
Fig. 1 Ethanol concentrations detected in blank culture medium whenmanipulated without precautions to avoid ethanol exposure (n = 14)
Experiments0
1
2
3
4
5
6
EtO
H C
on
cen
trat
ion
(m
M)
123
Helena
Rectangle
Helena
Text Box
258

PARTE III
DISCUSSÃO INTEGRADA E CONCLUSÕES


______________________________________________________ Discussão
261
4. DISCUSSÃO INTEGRADA
Os trabalhos apresentados nesta dissertação correspondem aos
primeiros estudos publicados sobre a avaliação dos mecanismos subjacentes à
toxicidade hepática resultante da exposição simultânea ao EtOH e à MDMA.
Esta discussão tem como objectivo fornecer uma visão integrada dos
resultados obtidos nos estudos realizados, necessária à concepção de uma
tese, não pretendendo assim substituir a discussão específica de cada um dos
trabalhos, necessariamente mais pormenorizada.
A importância destes estudos é facilmente constatada pela análise dos
dados epidemiológicos relativos aos padrões de consumo de ecstasy
apresentados na introdução da presente dissertação e que mostram que o
consumo concomitante de ecstasy e EtOH apresenta uma elevada prevalência
e parece estar associado, directa ou indirectamente, a episódios de morbilidade
e mortalidade entre os consumidores.
Até à data, os estudos realizados sobre os efeitos desta associação
centraram-se sobretudo na influência do EtOH na farmacocinética da MDMA
em ratos 682 e em humanos 683 e em alguns dos efeitos induzidos por este
estimulante anfetamínico, nomeadamente a resposta hipertérmica 633, 657, 682, 695,
711, 713, as alterações subjectivas 314, 683, 719 e psicomotoras 633, 657, 682-684, 711, 712,
715, 719, 737, a neurotoxicidade 712, 716 e as alterações imunológicas 728. Outros
estudos centraram-se ainda na capacidade da MDMA influenciar o padrão de
consumo de EtOH 694, 695, 738, 739.
Tendo em conta os conhecimentos pré-exitentes acerca da toxicidade do
EtOH e da MDMA quando consumidos isoladamente, seria de esperar que o
consumo simultâneo de ambos resultasse num sinergismo das respostas
hepatotóxicas provocadas por cada um dos compostos. Era, no entanto,
imprescindível verificar se este sinergismo realmente ocorria e quais as suas
consequências a nível hepático, uma vez que o fígado é o órgão
maioritariamente envolvido no metabolismo de ambas as substâncias, estando
especialmente sujeito à acção tóxica dos metabolitos formados a esse nível 193,
736.
Para verificar a existência de uma interacção toxicológica entre o EtOH e
a MDMA foi realizado um ensaio in vivo em ratinhos CD-1 que foram expostos

Discussão ______________________________________________________
262
a uma dose única de 10 mg/kg de MDMA administrada após uma prévia
exposição repetida a EtOH a 12% durante 8 semanas (Trabalho II), no sentido
de simular o padrão de consumo mais comum que geralmente se caracteriza
por uma exposição continuada ao EtOH anterior ao primeiro contacto com a
MDMA 657. De referir que o aumento da toxicidade da MDMA pelo EtOH foi
evidente em ensaios preliminares nos quais se expuseram ratinhos CD-1,
previamente expostos a EtOH, a doses de MDMA de 20 mg/kg (Trabalho I).
Nestes ensaios, a percentagem de mortes do grupo EtOH+MDMA foi muito
elevada (67% dos animais) e fez com que os estudos subsequentes fossem
realizados com doses de MDMA de 10 mg/kg. Posteriormente, de forma a
esclarecer o(s) mecanismo(s) subjacentes à toxicidade sinérgica da mistura de
MDMA e EtOH que tinha sido, anteriormente, observada nos ensaios in vivo,
foram também realizados ensaios ex vivo e in vitro. Nos ensaios ex vivo
(Trabalho III) foram utilizadas suspensões de hepatócitos isolados de ratinhos
sujeitos ao mesmo padrão de exposição ao EtOH que os utilizados no ensaio in
vivo. Os hepatócitos obtidos destes ratinhos foram, posteriormente, expostos a
diferentes concentrações de MDMA. Nos ensaios in vitro (Trabalhos IV e V)
foram utilizadas culturas primárias de hepatócitos de rato que foram incubadas
com diferentes concentrações de MDMA na presença, ou na ausência, de 300
mM de EtOH (valor aproximado do LC50 do EtOH após 24 horas de incubação
a 40,5ºC, neste modelo experimental). Quer os hepatócitos em suspensão quer
os hepatócitos em cultura foram incubados em condições normotérmicas
(36,5ºC) e hipertérmicas (40,5ºC) com o objectivo de simular e avaliar o efeito
do aumento da temperatura corporal induzido in vivo pela MDMA.
4.1. Efeitos da exposição a EtOH e MDMA no comportamento e na temperatura corporal de ratinhos CD-1
Nos ensaios in vivo uma clara interacção entre o EtOH e a MDMA foi
evidente logo após a administração da MDMA, uma vez que os animais
simultaneamente expostos às duas substâncias ficaram marcadamente
prostrados quando comparados com os animais unicamente expostos à
MDMA. Este fenómeno poderá ter sido causado pelo facto de os animais

______________________________________________________ Discussão
263
expostos a EtOH sofrerem um aumento e um prolongamento significativos do
efeito hipertérmico induzido pela MDMA, comparativamente à exposição
apenas a MDMA (Trabalho II, Figura 1). De facto, os animais expostos a
MDMA e EtOH apresentaram temperaturas corporais significativamente mais
elevadas do que os animais expostos apenas a MDMA, um efeito que se
prolongou por um período de 7 horas. Este agravamento da resposta
hipertérmica induzida pela MDMA poderá, potencialmente, predispor os
animais afectados para o desenvolvimento dos efeitos deletérios associados à
hipertermia. Apesar do efeito hipertérmico da MDMA ser uma reacção
esperada 142, o sinergismo exercido neste efeito pela prévia exposição repetida
ao EtOH foi surpreendente, uma vez que o EtOH está normalmente associado
a indução de vasodilatação e hipotermia. Este efeito é, no entanto, rapidamente
afectado por fenómenos de tolerância 710 e, assim, a acção do EtOH que se
esperaria que contrariasse o efeito hipertérmico da MDMA deixa de existir. O
efeito sinérgico da prévia exposição repetida ao EtOH na hipertermia induzida
pela MDMA poderá dever-se à acção directa do EtOH na libertação de DA 701,
uma vez que a libertação de DA foi já associada ao desenvolvimento de
hipertermia após exposição à MDMA 80, tal como amplamente referido na
introdução.
4.2. Efeitos da exposição de hepatócitos em cultura a EtOH e MDMA na produção de ROS e RNS
Os estudos realizados no âmbito desta dissertação demonstraram, pela
primeira vez, um sinergismo no aumento da produção de ROS e RNS quando
culturas primárias de hepatócitos de rato foram expostas simultaneamente a
EtOH 300 mM e MDMA 1,6 mM (Trabalho IV, Figura 3). De facto, em
condições hipertérmicas, a co-incubação com EtOH e MDMA levou a um
aumento significativo da formação de espécies reactivas de aproximadamente
100%, quando comparado com o efeito da exposição apenas a MDMA (p <
0,001).

Discussão ______________________________________________________
264
Este sinergismo está de acordo com o facto de quer o EtOH quer a
MDMA serem capazes de aumentar a formação de ROS em vários modelos
experimentais 674, 740.
O aumento na formação de ROS e RNS pode surgir como consequência
(i) da indução da CYP2E1 pelo EtOH, observada num modelo in vivo de
patologia hepática alcoólica de administração intra-gástrica de EtOH 741, (ii) da
disfunção mitocondrial subsequente à diminuição dos níveis de GSH e da
produção de ATP provocadas pelo EtOH, já observadas em vários estudos,
incluindo em hepatócitos em cultura obtidos de ratos expostos a EtOH através
da dieta 742, 743, ou (iii) dos ciclos redox associados ao metabolismo da MDMA,
já descritos após administração desta substância (15 mg/kg, i.p.) a ratos Dark
Agouti (fenótipo metabolizador pobre para as CYP2D) 53 e (iv) da conjugação
dos seus metabolitos com a GSH, fenómeno já observado, por exemplo, em
microssomas de fígado de rato 61 e em ratos Sprague-Dawley machos que
foram expostos a MDMA através de uma administração intra-cerebroventricular 62.
4.3. Efeitos da exposição a EtOH e MDMA nos mecanismos de defesa antioxidantes
A hipertermia mais intensa e mais prolongada resultante da co-
exposição de ratinhos CD-1 a EtOH (12% durante 8 semanas) e MDMA (10
mg/kg) foi acompanhada por maior activação, a nível hepático, do factor de
transcrição HSF1 (heat shock factor-1) (Trabalho II, Figura 1). De facto,
observou-se um aumento significativo da intensidade óptica das bandas
correspondentes ao HSF1 activado, de 189,7 ± 29,1 unidades de absorvância
no grupo apenas exposto a MDMA, para 317,9 ± 48,6 no grupo exposto a
EtOH+MDMA. Esta activação ocorre, normalmente, como resposta à exposição
a altas temperaturas e a agressões pró-oxidantes e corresponde a um
mecanismo de defesa contra a agressão, aumentando a síntese das proteínas
de choque térmico (HSPs ou heat shock proteins). Estas proteínas contribuem
para a homeostasia celular 744, contrariam os efeitos da agressão térmica e

______________________________________________________ Discussão
265
favorecem a adaptação a fenómenos de stress oxidativo para evitar a morte
celular 156 (Figura 18).
Figura 18. Mecanismo de resposta ao choque térmico. O HSF1 (principal HSF envolvido na resposta à agressão térmica) existe no citosol na forma inactiva em heterodímeros com a HSP70 ou a HSP90. Perante um estímulo agressivo (ex.: stress oxidativo, choque térmico, etc) ocorre a formação de proteínas não nativas que resultam da desnaturação de proteínas membranares e citoplasmáticas 745, 746 e que vão activar a resposta ao choque térmico que leva à síntese de HSPs. Estas proteínas vão competir com o HSF para as HSPs heterodiméricas. Uma vez que as HSPs têm mais afinidade para as proteínas desnaturadas do que para o HSF, este é libertado no citoplasma. O HSF é depois translocado para o núcleo onde vai formar homotrímeros que são activados por fosforilação e vão iniciar a transcrição de genes de choque térmico por ligação à região promotora da transcrição destes genes denominada elemento de choque térmico (HSE) e que vai levar a um aumento na síntese das HSPs 744, 747. Para além do referido, a activação do HSF1 é regulada por fosforilação reversível na qual podem estar envolvidas várias cinases como as Akt, ERK e JNK activadas pela agressão térmica 748. Por exemplo, a actividade do HSF1 é aumentada pela activação da Akt induzida pelo calor 749 no entanto, a activação da ERK e JNK induzida pelo calor resulta numa diminuição da actividade do HSF1 750. JNK, c-Jun N-terminal cinase; Akt, serina/treonina cinase ou PKB; ERK, cinase regulada por sinais extracelulares.
Por outro lado, o aumento da activação do NF-κB hepático (nuclear factor
kappa-B) nos animais expostos a 10 mg/kg de MDMA após 8 semanas de
exposição repetida a EtOH a 12% (Trabalho II, Figura 2), um dos factores de
transcrição envolvidos na activação de genes de resposta imediata a estímulos
deletérios e inflamatórios 195, indica que a co-exposição a etanol e MDMA
exerce um efeito pró-inflamatório, confirmado pela observação, por microscopia
electrónica de cortes histológicos de tecido hepático, de células Kupffer

Discussão ______________________________________________________
266
activadas (Trabalho II, Figura 3). De facto, observou-se um aumento
significativo da intensidade óptica das bandas correspondentes ao NF-κB
activado, de 45,44 ± 5,97 unidades de absorvância no grupo apenas exposto a
MDMA, para 88,79 ± 4,53 no grupo exposto a EtOH+MDMA. As consequências
de uma maior activação do NF-κB podem ser variadas, uma vez que a sua
activação pode estar relacionada com o desenvolvimento de processos
inflamatórios, bem como na regulação da expressão de enzimas antioxidantes 751 (Figura 19).
Figura 19. Mecanismo de activação do NF-κB. Quando as células são atingidas por estímulos deletérios e inflamatórios (ex.: ROS e outros radicais livres, IL-1, TNF-α, LPS, luz UV, RNA de cadeia dupla, sepsis, hemorragia, isquemia 752 o inibidor do NF-κB é fosforilado pela IκB cinase (IκK) e a forma ativa do NF-κB é libertada 753. O IκB fosforilado é poliubiquitinado e degradado por um mecanismo dependente do proteossoma e o NF-κB activado é translocado para o núcleo, liga-se ao DNA e regula a transcrição de vários mediadores pró-inflamatórios como as citoquinas e as quimioquinas, factores de crescimento, inibidores da apoptose, moléculas de adesão celular endotelial, moléculas do complexo MHC, moléculas de adesão leucocitária (E-selectina, VCAM-1, ICAM-1), outros factores de transcrição como o p-53, o c-myc ou o ras 754, enzimas antioxidantes como a MnSOD e a GPX 751, 755, enzimas pró-inflamatórias efectoras como a COX-2 756, outras enzimas como a NOS 757 e IκBα 758. O IκBα bloqueia as acções do NF-κB ligando-se ao NF-κB e restabelecendo a sua inibição 759. Adaptado de Luedde e Trautwein 2006 760.

______________________________________________________ Discussão
267
No mesmo estudo, verificou-se um aumento significativo da actividade
da SOD (Trabalho II, Tabela 3), provavelmente como um mecanismo de defesa
contra a agressão pró-oxidante habitualmente associada ao consumo de EtOH
e de MDMA. De facto, um aumento da actividade da SOD como resultado da
exposição aguda (dose oral única : 5, 10, 20 ou 40 mg/kg) e subaguda (5, 10,
ou 20 mg/kg/dia durante 14 dias) de ratos Wistar a MDMA foi já descrito no rim 761. Este é, no entanto, o primeiro estudo que mostra um aumento da SOD
hepática após exposição de ratinhos CD-1 a EtOH e MDMA. A exposição a
EtOH+MDMA afectou quer a MnSOD quer a Cu/ZnSOD, provocando aumentos
de, aproximadamente, 42 e 28%, respectivamente, quando comparadas com
as actividades da SOD nos animais expostos apenas a EtOH.
Contudo, a activação do NF-κB e do HSF1 não foi suficiente para
proteger as células da toxicidade da MDMA e do EtOH, apesar de ter sido
eficiente em evitar modificações marcadas em parâmetros de stress oxidativo
como a razão GSH/GSSG, o conteúdo em carbonilos proteicos e o grau de
peroxidação lipídica (Trabalho II, Tabela 2). A ausência de alterações nestes
indicadores do estado redox do fígado foi inesperada, e poderá eventualmente
dever-se a mecanismos de compensação contra a agressão oxidativa
desenvolvidos pelo organismo durante o pré-tratamento com EtOH, ou mesmo
a uma eficaz biossíntese das defesas antioxidantes que, segundo alguns
autores, é suficiente para resistir a uma determinada agressão, em animais
com um regime alimentar equilibrado 762. Esta ausência de alterações nos
níveis de GSH após exposição ao EtOH está em consonância com os
resultados obtidos anteriormente por Oh 762 e por Deaciuc 763 em ensaios com
ratos alimentados durante 6 e 7 semanas, respectivamente, com uma dieta
líquida contendo EtOH. O agravamento da diminuição dos níveis de GSH
produzidos pela MDMA devido ao EtOH foi, no entanto, observado em
suspensões de hepatócitos isolados de ratinhos previamente expostos ao
EtOH (Trabalho III, Figura 2: os níveis de GSH atingiram, nas células obtidas
de animais expostos ao etanol durante 8 semanas, e incubadas em condições
normotérmicas com MDMA 1,6 mM, ao final de 3 horas, cerca de 45% dos
níveis das células controlo) e quando hepatócitos de rato em cultura primária
foram expostos simultaneamente aos dois compostos (Trabalho IV, Figura 4:
os níveis de GSH atingiram, nas células expostas, em condições

Discussão ______________________________________________________
268
normotérmicas a EtOH 300 mM + MDMA 1,6 mM, em 24 horas, cerca de 50%
dos níveis controlo). Esta diminuição dos níveis de GSH pode ser explicada
pela maior oxidação da GSH a GSSG pelas ROS e RNS cuja formação
também se encontra aumentada pela exposição conjunta aos dois compostos
(Trabalho IV, Figura 3), pela formação de aductos entre a GSH e os
metabolitos catecóis da MDMA 192, mas também pela diminuição da síntese da
GSH como resultado do decréscimo na actividade da γ-GCS (γ-glutamil cisteína
sintetase, enzima limitante da síntese da glutationa) (Trabalho IV, Figura 6).
Este facto foi já anteriormente descrito como resultado da exposição ao EtOH 764. Estes efeitos, nomeadamente o aumento da produção de ROS e de RNS e
a diminuição da actividade da γ-GCS, foram significativamente agravados
quando as células foram co-incubadas com EtOH 300 mM e MDMA 1,6 mM,
em condições hipertérmicas. Por um lado, a hipertermia, por si só, já leva a um
aumento do fluxo de ROS nas células por um mecanismo ainda não
esclarecido, mas que parece envolver a cadeia de transporte de electrões
mitocondrial 765. Por outro lado, a diminuição de actividade da γ-GCS pode
corresponder a um fenómeno secundário a alterações dos níveis de cálcio que
ocorrem durante a lesão celular 766, as quais já foram descritas após exposição
a MDMA 767, EtOH 768, ou condições hipertérmicas 769. Sendo a GSH um
importante antioxidante endógeno, a sua depleção pode aumentar a
susceptibilidade do fígado aos efeitos deletérios das ROS e RNS e dos
compostos reactivos formados durante o metabolismo do EtOH (acetaldeído) e
da MDMA (MDA, N-metil-α-MeDA, α-MeDA, metabolitos conjugados com a
GSH). Curiosamente, este aumento de susceptibilidade não se verificou no
ensaio em que se utilizaram suspensões de hepatócitos isolados de ratinhos
expostos ao EtOH durante 8 semanas, no qual a diminuição dos níveis de GSH
produzida pelo EtOH (Trabalho III, Figura 2) não se traduziu num aumento da
morte celular induzida pela MDMA (Trabalho III, Figura 4). Este fenómeno
poderá, em parte, ser explicado por uma diminuição da disponibilidade da GSH
para a formação de conjugados de metabolitos da MDMA com a GSH, os quais
já demonstraram ser mais tóxicos que o composto pai em vários sistemas de
células, como em culturas de neurónios corticais de rato 386 ou de células dos
túbulos proximais de renais 203.

______________________________________________________ Discussão
269
Outras enzimas afectadas pela exposição conjunta a EtOH e a MDMA
foram a catalase e a glutationa peroxidase (GPx), envolvidas na destoxificação
do peróxido de hidrogénio (Trabalho II, Tabela 3), as quais apresentaram um
decréscimo significativo de actividade, mais marcado no grupo EtOH+MDMA.
De facto, a catalase e a GPx evidenciaram reduções de, respectivamente, 19%
e 30% no grupo EtOH+MDMA, comparativamente ao grupo controlo. Tal é
indicativo de uma maior agressão pró-oxidante no fígado neste grupo, já que
uma actividade diminuída da GPx pode levar a uma maior acumulação de H2O2
e pode reflectir o envolvimento de ROS na toxicidade da mistura
etanol+MDMA. Esta diminuição pode dever-se à capacidade de metabolitos da
MDMA (ex.: a α-MeDA), de catecolaminas e dos aminocromos resultantes da
sua oxidação inibirem enzimas contendo grupos tiol através da oxidação ou
alquilação dos grupos sulfidrilo localizados no seu local activo. As orto-
quinonas e os aminocromos podem ainda inibir enzimas que possuam locais
de ligação à GSH (como é o caso da GPx) e/ou resíduos de cisteína que são
essenciais ao correcto funcionamento da enzima 51. Para além da MDMA, o
EtOH pode contribuir também para o decréscimo significativo verificado na
actividade desta enzima nos animais expostos a MDMA e a EtOH, uma vez
que foi já descrito em ratos que o consumo repetido de EtOH durante 90 dias
leva a um decréscimo na actividade da GPx 770.
Também a GST foi afectada pela exposição conjunta à MDMA e ao
EtOH. A GST cataliza o ataque nucleofílico da GSH a substractos electrofílicos,
protegendo os componentes celulares dos efeitos potencialmente lesivos
destes compostos 771. Um decréscimo na actividade desta enzima foi
observado pela co-exposição às duas substâncias quer no ensaio in vivo
(Trabalho II, Tabela 3), no qual a actividade da GST sofreu uma diminuição de
20% no grupo EtOH+MDMA relativamente ao grupo controlo, quer no ensaio
de exposição de culturas primárias de hepatócitos de rato à mistura
EtOH+MDMA (Trabalho IV, Figura 7), onde se verificou uma redução da
actividade da GST até, aproximadamente, 4% do controlo nas células
expostas, em condições hipertérmicas, a EtOH 300 mM + MDMA 1,6 mM
durante 24 horas. Esta alteração na actividade da GST pode ser explicada pela
diminuição da actividade da referida enzima, previamente associado à
exposição in vivo ao EtOH (7,9 g/kg/dia, durante 45 dias 772), que é agravada

Discussão ______________________________________________________
270
pela co-exposição à MDMA. Tal como no caso da GPx, a diminuição da
actividade da GST por acção da MDMA pode dever-se à oxidação ou
alquilação de grupos hidroxilo e sulfidrilo do local activo da enzima pelos
metabolitos da MDMA 192 ou, no caso dos ensaios in vivo, também pelas
catecolaminas libertadas por acção deste derivado anfetamínico 51.
Qualquer um destes efeitos da acção conjunta do EtOH e da MDMA na
homeostase da GSH e das enzimas antioxidantes pode contribuir para o
sinergismo dos seus efeitos hepatotóxicos.
4.4. Efeitos da exposição a EtOH e MDMA na mitocôndria e nos níveis energéticos da célula
Os ensaios ex vivo e in vitro realizados em células (Trabalhos III e IV)
permitiram ainda constatar que a exposição conjunta a EtOH e MDMA leva a
uma depleção dos níveis de ATP, evidenciando danos mitocondriais. Estes
danos na mitocôndria podem resultar de uma perturbação da homeostase dos
grupos tiol, e da formação de ROS, RNS e metabolitos tóxicos a partir da
MDMA e/ou do EtOH que podem afectar os processos energéticos da
mitocôndria (ciclo de Krebs e fosforilação oxidativa ou cadeia transportadora de
electrões) ao danificarem as membranas e/ou proteínas mitocondriais (ex.:
NADH desidrogenase, citocromo c redutase, e citocromo c oxidase da cadeia
transportadora de electrões) 773. Esta danificação mitocondrial foi confirmada
pela diminuição da quantidade de citocromo c e do factor indutor de apoptose
(AIF) no extracto mitocondrial em culturas primárias de hepatócitos de rato
expostos a uma mistura de EtOH 300 mM com MDMA 1,6 mM (Trabalho IV,
Figura 8). A agressão mitocondrial foi também evidente na observação do
tecido hepático por microscopia electrónica (Trabalho II, Figura 3) de
mitocôndrias turgescentes (swelling). Para além disso, a disfunção mitocondrial
subjacente à diminuição de ATP observada pode ser directamente promovida
pela exposição a condições hipertérmicas, como descrito anteriormente em
linhas celulares tumorais e em cardiomiócitos 774-776.
Todos os factores referidos nos parágrafos supracitados podem estar na
base dos efeitos tóxicos sinérgicos observados entre o EtOH e a MDMA.

______________________________________________________ Discussão
271
Contudo, outros factores estão também envolvidos e o efeito hipertérmico
associado ao consumo de MDMA 595 contribui significativamente para o
agravamento da sua toxicidade 387 e da toxicidade da mistura.
A diminuição dos níveis de ATP observada nestes trabalhos confirmou
os resultados obtidos previamente pelo nosso grupo em suspensões de
hepatócitos de rato que provaram uma diminuição dos níveis de ATP por acção
da MDMA 193. Contudo, esta é a primeira referência a um sinergismo na
depleção de ATP em hepatócitos induzido pela MDMA em co-exposição com
EtOH.
4.5. Indicadores de dano hepático
Da acção conjunta de todos os efeitos descritos nas alíneas anteriores,
da co-exposição a EtOH e a MDMA resulta um marcado agravamento do dano
hepático induzido pela MDMA isoladamente, evidenciado pelo aumento das
transaminases plasmáticas (a GOT aumentou 50% e a GPT aumentou 90% no
grupo EtOH+MDMA, relativamente ao controlo) nos ratinhos expostos a 10
mg/kg de MDMA após exposição repetida a EtOH a 12% (Trabalho II, Figura
4), decréscimo do peso relativo do fígado [de 5,582 ± 0,293% no grupo exposto
apenas a EtOH para 4,222 ± 0,261% no grupo exposto a EtOH+MDMA (p <
0,01)] (Trabalho II, Tabela 1) e pela observação de indicadores de lesão
tecidular por técnicas de microscopia óptica e electrónica (Trabalho II, Figura 3)
que incluem aumento de deposição de colagénio, diminuição das
microvilosidades dos hepatócitos, activação lisossómica e necrose celular. O
aumento da GOT e da GPT foi já anteriormente associado à exposição quer a
EtOH (em ratos 777 e em humanos 778), quer a MDMA (em ratos 779 e em
humanos 186), mas o aumento destes marcadores de lesão hepática ainda não
tinha sido descrito como resultado da exposição simultânea a ambos os
compostos. No entanto, este agravamento do dano hepático induzido pela
exposição simultânea a MDMA e EtOH não foi confirmado no trabalho
realizado com suspensões de hepatócitos frescos, o que pode ser explicado
por uma limitação da técnica usada, uma vez que durante o processo de
isolamento dos hepatócitos, apenas aqueles hepatócitos mais resistentes que

Discussão ______________________________________________________
272
toleraram o pré-tratamento dos animais com EtOH é que sobreviveram ao
processo de isolamento, tendo os restantes sido eliminados nos vários
processos de lavagem inerentes à correcta realização da técnica. Para além
disso, os hepatócitos tolerantes ao EtOH continham níveis mais baixos de
GSH, o que poderá resultar numa menor formação de conjugados da GSH com
metabolitos catecóis da MDMA, conjugados esses que já demonstraram ser
mais tóxicos que a própria MDMA em culturas de células renais humanas 203 e
de neurónios corticais de rato 386, bem como após administração intra-
cerebroventricular de MDMA a ratos 62. No entanto, quando se utilizaram
culturas primárias de hepatócitos de rato, a maior hepatotoxicidade da mistura
EtOH+MDMA, relativamente à MDMA isoladamente, foi claramente
demonstrada por uma maior necrose celular induzida pela mistura, avaliada por
uma maior libertação de LDH para o meio de cultura [a qual correspondeu a um
decréscimo de cerca de 25% na viabilidade celular, quando as células foram
co-incubadas com EtOH 300 mM e MDMA 1,6 mM, em condições
normotérmicas, durante 24 horas (comparativamente à exposição apenas a
MDMA, nas mesmas condições)] e pela coloração com brometo de etídio e
laranja de acridina (Trabalho IV, Figuras 1 e 2). Esta hepatotoxicidade foi ainda
agravada em condições hipertérmicas, de acordo com os resultados
previamente obtidos pelo nosso grupo em suspensões de hepatócitos de
ratinho que demonstraram que a exposição continuada a condições
hipertérmicas leva a um aumento da morte celular provavelmente por um
aumento sustentado da produção de ROS e consequente desenvolvimento de
stress oxidativo 156.
A via de morte celular apoptótica parece não ser predominante devido à
ausência de translocação da proteína pró-apoptótica Bax para a mitocôndria e
ausência de activação dos mecanismos efectores das vias apoptóticas
mediadas pelas caspases, apesar de se assistir à libertação de citocromo c a
partir da mitocôndria.
Estes resultados surgem em oposição a estudos anteriores que
atribuíram um mecanismo apoptótico à morte celular induzida pela MDMA em
vários modelos experimentais como culturas de neurónios corticais de rato 60,
de células Ito hepáticas 59 e de células retinianas 780. Esta discrepância poderá
provavelmente ser explicada pelo grave declínio das reservas energéticas das

______________________________________________________ Discussão
273
células expostas simultaneamente a EtOH e a MDMA que faz com que as vias
de morte apoptótica (que envolvem gasto de ATP) estejam comprometidas.
4.6. Importância da hipertermia na toxicidade da mistura de EtOH e MDMA A hipertermia tem sido já amplamente implicada na expressão da
toxicidade resultante da exposição a MDMA, inclusivamente em trabalhos
publicados pelo nosso grupo 387. De facto, a exposição das células a condições
hipertérmicas parece despelotar vários mecanismos de sinalização celular que
poderão, ou não, culminar na morte das células, dependendo da perturbação
da homeostasia e da prevalência dos mecanismos celulares de sobrevivência
ou de morte 748.
A importância da hipertermia estende-se para além da exposição
unicamente a MDMA, e mostra-se também um importante condicionante da
toxicidade da mistura de EtOH e MDMA, como demonstrado nos trabalhos que
são parte integrante desta dissertação. De facto, a exposição a condições
hipertérmicas agravou a depleção dos níveis de glutationa reduzida e de ATP
em suspensões de hepatócitos isolados de ratinho (Trabalho III, Figuras 2 e 3)
e em culturas primárias de hepatócitos de rato (Trabalho IV, Figuras 4 e 5). A
incubação das células a temperatura elevada provocou ainda um aumento da
formação de ROS e RNS (Trabalho IV, Figura 3), uma maior depleção da
actividade de enzimas envolvidas na homeostasia da glutationa (γ-GCS e GST)
(Trabalho IV, Figuras 6 e 7) e um agravamento da lesão mitocondrial (Trabalho
IV, Figura 8) resultantes da co-exposição a EtOH e MDMA.
Todos estes efeitos da mistura de EtOH e MDMA que foram agravados
em condições hipertérmicas resultaram numa maior percentagem de morte
celular em hipertermia (Trabalho III, Figura 4 e Trabalho IV, Figuras 1 e 2),
ilustrando, assim, a relevância que o desenvolvimento de uma resposta
hipertérmica in vivo poderá ter na maior ou menor toxicidade exercida pela co-
exposição a EtOH e MDMA.

Discussão ______________________________________________________
274
4.7. Interacção metabólica entre EtOH e MDMA
Uma vez que o EtOH e a MDMA partilham algumas vias metabólicas
que envolvem o sistema do CYP450 e que o EtOH tem a capacidade de induzir
algumas dessas isoenzimas 665, incluindo aquelas envolvidas no metabolismo
da MDMA, o aumento da toxicidade da MDMA pela co-exposição ao EtOH
pode também dever-se a uma interacção metabólica entre os dois compostos
que parece envolver a indução das isoenzimas CYP2E1 e CYP3A1 pelo EtOH,
comprovada pela pré-incubação das células com inibidores específicos de
ambas as isoformas (Trabalho V), e a bioactivação de ambos os compostos a
metabolitos de maior toxicidade 5, 736. De facto, em condições hipertérmicas, o
EtOH aumentou significativamente a metabolização de MDMA a MDA, HMMA
e HMA, resultando num aumento de 6 vezes na formação de MDA, de 16
vezes na formação de HMMA e de 10 vezes na formação de HMA. É,
sobretudo, preocupante o aumento da formação dos metabolitos catecóis da
MDMA (HMA e HMMA), uma vez que demonstraram ser mais tóxicos do que a
MDMA e a MDA em vários modelos, incluindo hepatócitos isolados 192 e
culturas de neurónios corticais de rato 386.
Embora o EtOH não tenha sido ainda associado a uma indução da
CYP3A1 ele provou já induzir a isoforma do CYP450 que no homem recebe a
designação de CYP3A4 e que equivale à CYP3A1 (testosterona 6β-hidroxilase)
no rato 676. Por outro lado, a indução da CYP2E1 pelo EtOH tem já sido
amplamente descrita 665. No entanto, não se pode descartar a contribuição de
outras vias metabólicas nesta interacção.
Aspectos técnicos aperfeiçoados ao longo do trabalho que validam os resultados obtidos
Como se pode verificar a partir dos resultados obtidos em culturas
primárias de hepatócitos de rato, a utilização de técnicas de cultura celular no
estudo dos efeitos do EtOH é de extrema utilidade, uma vez que permite utilizar
tempos de incubação mais longos do que as 4 horas que correspondem ao
limite máximo de incubação das suspensões de hepatócitos frescos. No

______________________________________________________ Discussão
275
entanto, sempre que se optar por este tipo de modelos experimentais, é
importante garantir que as amostras são mantidas isentas de contaminação
externa pelo álcool utilizado como agente esterilizante de superfícies. Este
facto é relevante não só para os estudos dos efeitos do EtOH, mas também
para qualquer tipo de estudo realizado em cultura celular, uma vez que existem
estudos que indicam que o EtOH, mesmo em baixas concentrações, pode
alterar a actividade de várias enzimas 665, o normal funcionamento celular, a
sobrevivência celular, a funcionalidade de receptores, a homeostase
mitocondrial, a sinalização celular 781, a actividade de agentes infecciosos como
o vírus da hepatite C 782, etc (Trabalho VII).
Para se conseguir garantir a obtenção de controlos realmente isentos de
EtOH é indispensável a validação de métodos sensíveis, precisos, exactos, e
de fácil integração na rotina laboratorial que permitam rapidamente excluir a
possibilidade de exposição das células ao EtOH quando esta exposição não é
pretendida. Para proceder a esse controlo da isenção de EtOH no meio de
cultura foi, então, validado o método de cromatografia gasosa, com detecção
por ionização em chama, apresentado no Trabalho VI.

Discussão ______________________________________________________
276

_____________________________________________________ Conclusões
277
5. CONCLUSÕES FINAIS
Os resultados obtidos nos trabalhos realizados no âmbito desta
dissertação permitiram-nos concluir:
(i) a pré-exposição repetida de ratinhos Charles River CD-1 a 12% de
EtOH, na água de bebida, potencia os efeitos hipertérmicos e hepatotóxicos
induzidos in vivo por uma exposição aguda a MDMA, originando um quadro
com sinais aparentes de necrose hepática e de resposta inflamatória;
(ii) a exposição repetida de ratinhos Charles River CD-1 a 12% de EtOH,
na água de bebida, compromete a obtenção de hepatócitos viáveis através da
perfusão com colagenase e aumenta a vulnerabilidade dos hepatócitos
isolados a agressões pró-oxidantes por um mecanismo que engloba uma
diminuição do conteúdo celular em GSH e um aumento do conteúdo em
GSSG;
(iii) a co-exposição de hepatócitos de rato Wistar em cultura primária a
EtOH+MDMA resulta numa toxicidade sinérgica relativamente aos efeitos
induzidos pela MDMA ou pelo EtOH isoladamente, através de uma diminuição
das defesas antioxidantes, diminuição das reservas energéticas das células,
aumento da formação de espécies reactivas e aumento da morte celular por
necrose, numa extensão dependente da temperatura de incubação;
(iv) a exposição aguda in vitro ao EtOH aumenta o metabolismo da
MDMA a MDA, HMA e HMMA, sendo estes metabolitos dotados de maior
toxicidade do que a própria MDMA
(v) os efeitos hepatotóxicos da mistura EtOH+MDMA são marcadamente
aumentados em condições hipertérmicas.
Os resultados obtidos e discutidos na presente dissertação podem ser,
assim, compilados no seguinte esquema-resumo (Figura 20).

Conclusões______________________________________________________
278
Figura 20. Esquema-resumo dos resultados obtidos no estudo das interacções hepatotóxicas entre etanol e MDMA.

_____________________________________________________ Conclusões
279
Como conclusão final, o co-consumo de EtOH aumenta a hepatotoxicidade da
MDMA através de um aumento da resposta hipertérmica associada ao
consumo deste derivado anfetamínico, aumento da formação de metabolitos
hepatotóxicos, diminuição das defesas antioxidantes, depleção das reservas
energéticas das células e aumento da morte celular por necrose.

Conclusões______________________________________________________
280

PARTE IV
REFERÊNCIAS BIBLIOGRÁFICAS


__________________________________________ Referências Bibliográficas
283
6. REFERÊNCIAS 1. Hall, A.P. and Henry, J.A., Acute toxic effects of 'Ecstasy' (MDMA) and related
compounds: overview of pathophysiology and clinical management. Br J Anaesth, 2006. 96: p. 678-85.
2. Baylen, C.A. and Rosenberg, H., A review of the acute subjective effects of MDMA/ecstasy. Addiction, 2006. 101: p. 933-47.
3. Giroud, C., Augsburger, M., Sadeghipour, F., Varesio, E., Veuthey, J.L., and Rivier, L., Ecstasy--the status in French-speaking Switzerland. Composition of seized drugs, analysis of biological specimens and short review of its pharmacological action and toxicity. Praxis, 1997. 86: p. 510-23.
4. Steele, T.D., McCann, U.D., and Ricaurte, G.A., 3,4-Methylenedioxymethamphetamine (MDMA, "Ecstasy"): pharmacology and toxicology in animals and humans. Addiction, 1994. 89: p. 539-51.
5. Green, A.R., Mechan, A.O., Elliott, J.M., O'Shea, E., and Colado, M.I., The pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA, "ecstasy"). Pharmacol Rev, 2003. 55: p. 463-508.
6. Freudenmann, R.W., Oxler, F., and Bernschneider-Reif, S., The origin of MDMA (ecstasy) revisited: the true story reconstructed from the original documents. Addiction, 2006. 101: p. 1241-5.
7. Shulgin, A.T. and Nichols, D.E., Characterization of three new psychotomimetics., in The psychopharmacology of hallucinogens, (R.C. Stillman and Willette, R.E., ed.), 1978, New York: Pergamon.
8. Nichols, D.E., Hoffman, A.J., Oberlender, R.A., Jacob, P., 3rd, and Shulgin, A.T., Derivatives of 1-(1,3-benzodioxol-5-yl)-2-butanamine: representatives of a novel therapeutic class. J Med Chem, 1986. 29: p. 2009-15.
9. Greer, G. and Strassman, R.J., Information on "Ecstasy". Am J Psychiatry, 1985. 142: p. 1391.
10. Grinspoon, L. and Bakalar, J.B., Can drugs be used to enhance the psychotherapeutic process? Am J Psychother, 1986. 40: p. 393-404.
11. ONU Organização das Nações Unidas, Convention on psychotropic substances. 1971, Viena: Unations vienna.
12. Ricaurte, G., Bryan, G., Strauss, L., Seiden, L., and Schuster, C., Hallucinogenic amphetamine selectively destroys brain serotonin nerve terminals. Science, 1985. 229: p. 986-8.
13. Cole, J.C. and Sumnall, H.R., Altered states: the clinical effects of Ecstasy. Pharmacol Ther, 2003. 98: p. 35-58.
14. Gahlinger, P.M., Club drugs: MDMA, gamma-hydroxybutyrate (GHB), Rohypnol, and ketamine. Am Fam Physician, 2004. 69: p. 2619-26.
15. Klein, M. and Kramer, F., Rave drugs: pharmacological considerations. AANA J, 2004. 72: p. 61-7.
16. Glennon, R.A., Arylalkylamine drugs of abuse: an overview of drug discrimination studies. Pharmacol Biochem Behav, 1999. 64: p. 251-6.
17. Nichols, D.E. and Oberlender, R., Structure-activity relationships of MDMA-like substances. NIDA Res Monogr, 1989. 94: p. 1-29.
18. Caldwell, J., The metabolism of amphetamines and related stimulants in animals and man., in Amphetamines and related stimulants: chemical, biological, clinical and sociological aspects., (J. Caldwell, ed.), 1980, Boca Raton: CRC Press.
19. UNODC United Nations Office on Drugs and Crime, World Drug Report. 2007, Organização das Nações Unidas: Viena.
20. OEDT Observatório Europeu da Droga e da Toxicodependência, Relatório Anual 2007: A evolução do fenómeno da droga na europa. 2007, Luxemburgo. pp. 50-57.
21. Ramsey, J.D., Butcher, M.A., Murphy, M.F., Lee, T., Johnston, A., and Holt, D.W., A new method to monitor drugs at dance venues. BMJ, 2001. 323: p. 603.

Referências Bibliográficas __________________________________________
284
22. Solowij, N., Hall, W., and Lee, N., Recreational MDMA use in Sydney: a profile of 'Ecstacy' users and their experiences with the drug. Br J Addict, 1992. 87: p. 1161-72.
23. de la Torre, R., Farré, M., Navarro, M., Pacifici, R., Zuccaro, P., and Pichini, S., Clinical pharmacokinetics of amfetamine and related substances: monitoring in conventional and non-conventional matrices. Clin Pharmacokinet, 2004. 43: p. 157-85.
24. Mas, M., Farré, M., de la Torre, R., Roset, P.N., Ortuño, J., Segura, J., and Camí, J., Cardiovascular and neuroendocrine effects and pharmacokinetics of 3, 4-methylenedioxymethamphetamine in humans. J Pharmacol Exp Ther, 1999. 290: p. 136-45.
25. Ramcharan, S., Meenhorst, P.L., Otten, J.M., Koks, C.H., de Boer, D., Maes, R.A., and Beijnen, J.H., Survival after massive ecstasy overdose. J Toxicol Clin Toxicol, 1998. 36: p. 727-31.
26. de la Torre, R., Farré, M., Ortuño, J., Mas, M., Brenneisen, R., Roset, P.N., Segura, J., and Camí, J., Non-linear pharmacokinetics of MDMA ('ecstasy') in humans. Br J Clin Pharmacol, 2000. 49: p. 104-9.
27. Wu, D., Otton, S.V., Inaba, T., Kalow, W., and Sellers, E.M., Interactions of amphetamine analogs with human liver CYP2D6. Biochem Pharmacol, 1997. 53: p. 1605-12.
28. Delaforge, M., Jaouen, M., and Bouille, G., Inhibitory metabolite complex formation of methylenedioxymethamphetamine with rat and human cytochrome P450. Particular involvement of CYP 2D. Env Toxicol Pharmacol, 1999. 7: p. 153-158.
29. Heydari, A., Yeo, K.R., Lennard, M.S., Ellis, S.W., Tucker, G.T., and Rostami-Hodjegan, A., Mechanism-based inactivation of CYP2D6 by methylenedioxymethamphetamine. Drug Metab Dispos, 2004. 32: p. 1213-7.
30. Yang, J., Jamei, M., Heydari, A., Yeo, K.R., de la Torre, R., Farré, M., Tucker, G.T., and Rostami-Hodjegan, A., Implications of mechanism-based inhibition of CYP2D6 for the pharmacokinetics and toxicity of MDMA. J Psychopharmacol, 2006. 20: p. 842-9.
31. de la Torre, R., Farré, M., Roset, P.N., Pizarro, N., Abanades, S., Segura, M., Segura, J., and Camí, J., Human pharmacology of MDMA: pharmacokinetics, metabolism, and disposition. Ther Drug Monit, 2004. 26: p. 137-44.
32. Farré, M., de la Torre, R., Mathúna, B.O., Roset, P.N., Peiró, A.M., Torrens, M., Ortuño, J., Pujadas, M., and Camí, J., Repeated doses administration of MDMA in humans: pharmacological effects and pharmacokinetics. Psychopharmacology (Berl), 2004. 173: p. 364-75.
33. Mechan, A., Yuan, J., Hatzidimitriou, G., Irvine, R.J., McCann, U.D., and Ricaurte, G.A., Pharmacokinetic profile of single and repeated oral doses of MDMA in squirrel monkeys: relationship to lasting effects on brain serotonin neurons. Neuropsychopharmacology, 2006. 31: p. 339-50.
34. Fallon, J.K., Kicman, A.T., Henry, J.A., Milligan, P.J., Cowan, D.A., and Hutt, A.J., Stereospecific analysis and enantiomeric disposition of 3, 4-methylenedioxymethamphetamine (Ecstasy) in humans. Clin Chem, 1999. 45: p. 1058-69.
35. Pizarro, N., Farré, M., Pujadas, M., Peiró, A.M., Roset, P.N., Joglar, J., and de la Torre, R., Stereochemical analysis of 3,4-methylenedioxymethamphetamine and its main metabolites in human samples including the catechol-type metabolite (3,4-dihydroxymethamphetamine). Drug Metab Dispos, 2004. 32: p. 1001-7.
36. Fitzgerald, R.L., Blanke, R.V., and Poklis, A., Stereoselective pharmacokinetics of 3,4-methylenedioxymethamphetamine in the rat. Chirality, 1990. 2: p. 241-8.
37. De Letter, E.A., Bouche, M.P., Van Bocxlaer, J.F., Lambert, W.E., and Piette, M.H., Interpretation of a 3,4-methylenedioxymethamphetamine (MDMA) blood level: discussion by means of a distribution study in two fatalities. Forensic Sci Int, 2004. 141: p. 85-90.
38. De Letter, E.A., Piette, M.H., Lambert, W.E., and Cordonnier, J.A., Amphetamines as potential inducers of fatalities: a review in the district of Ghent from 1976-2004. Med Sci Law, 2006. 46: p. 37-65.

__________________________________________ Referências Bibliográficas
285
39. Garcia-Repetto, R., Moreno, E., Soriano, T., Jurado, C., Gimenez, M.P., and Menendez, M., Tissue concentrations of MDMA and its metabolite MDA in three fatal cases of overdose. Forensic Sci Int, 2003. 135: p. 110-4.
40. Maurer, H.H., Bickeboeller-Friedrich, J., Kraemer, T., and Peters, F.T., Toxicokinetics and analytical toxicology of amphetamine-derived designer drugs ('Ecstasy'). Toxicol Lett, 2000. 112-113: p. 133-42.
41. Lim, H.K. and Foltz, R.L., Identification of metabolites of 3,4-(methylenedioxy)methamphetamine in human urine. Chem Res Toxicol, 1989. 2: p. 142-3.
42. Maurer, H.H., Kraemer, T., Springer, D., and Staack, R.F., Chemistry, pharmacology, toxicology, and hepatic metabolism of designer drugs of the amphetamine (ecstasy), piperazine, and pyrrolidinophenone types: a synopsis. Ther Drug Monit, 2004. 26: p. 127-31.
43. Johnson, M., Letter, A.A., Merchant, K., Hanson, G.R., and Gibb, J.W., Effects of 3,4-methylenedioxyamphetamine and 3,4-methylenedioxymethamphetamine isomers on central serotonergic, dopaminergic and nigral neurotensin systems of the rat. J Pharmacol Exp Ther, 1988. 244: p. 977-82.
44. Maurer, H.H., On the metabolism and the toxicological analysis of methylenedioxyphenylalkylamine designer drugs by gas chromatography-mass spectrometry. Ther Drug Monit, 1996. 18: p. 465-70.
45. de la Torre, R., Farré, M., Roset, P.N., Lopez, C.H., Mas, M., Ortuño, J., Menoyo, E., Pizarro, N., Segura, J., and Cami, J., Pharmacology of MDMA in humans. Ann N Y Acad Sci, 2000. 914: p. 225-37.
46. Kraemer, T. and Maurer, H.H., Toxicokinetics of amphetamines: metabolism and toxicokinetic data of designer drugs, amphetamine, methamphetamine, and their N-alkyl derivatives. Ther Drug Monit, 2002. 24: p. 277-89.
47. Kreth, K., Kovar, K., Schwab, M., and Zanger, U.M., Identification of the human cytochromes P450 involved in the oxidative metabolism of "Ecstasy"-related designer drugs. Biochem Pharmacol, 2000. 59: p. 1563-71.
48. Tucker, G.T., Lennard, M.S., Ellis, S.W., Woods, H.F., Cho, A.K., Lin, L.Y., Hiratsuka, A., Schmitz, D.A., and Chu, T.Y., The demethylenation of methylenedioxymethamphetamine ("ecstasy") by debrisoquine hydroxylase (CYP2D6). Biochem Pharmacol, 1994. 47: p. 1151-6.
49. Kumagai, Y., Lin, L.Y., Schmitz, D.A., and Cho, A.K., Hydroxyl radical mediated demethylenation of (methylenedioxy)phenyl compounds. Chem Res Toxicol, 1991. 4: p. 330-4.
50. Lin, L.Y., Kumagai, Y., and Cho, A.K., Enzymatic and chemical demethylenation of (methylenedioxy)amphetamine and (methylenedioxy)methamphetamine by rat brain microsomes. Chem Res Toxicol, 1992. 5: p. 401-6.
51. Bindoli, A., Rigobello, M.P., and Deeble, D.J., Biochemical and toxicological properties of the oxidation products of catecholamines. Free Radic Biol Med, 1992. 13: p. 391-405.
52. Zhang, F. and Dryhurst, G., Effects of L-cysteine on the oxidation chemistry of dopamine: new reaction pathways of potential relevance to idiopathic Parkinson's disease. J Med Chem, 1994. 37: p. 1084-98.
53. Colado, M.I., O'Shea, E., Granados, R., Murray, T.K., and Green, A.R., In vivo evidence for free radical involvement in the degeneration of rat brain 5-HT following administration of MDMA ('ecstasy') and p-chloroamphetamine but not the degeneration following fenfluramine. Br J Pharmacol, 1997. 121: p. 889-900.
54. Bolton, J.L., Trush, M.A., Penning, T.M., Dryhurst, G., and Monks, T.J., Role of quinones in toxicology. Chem Res Toxicol, 2000. 13: p. 135-60.
55. Chipana, C., García-Ratés, S., Camarasa, J., Pubill, D., and Escubedo, E., Different oxidative profile and nicotinic receptor interaction of amphetamine and 3,4-methylenedioxy-methamphetamine. Neurochem Int 2008. 52: p. 401-10.

Referências Bibliográficas __________________________________________
286
56. Felim, A., Urios, A., Neudörffer, A., Herrera, G., Blanco, M., and Largeron, M., Bacterial plate assays and electrochemical methods: an efficient tandem for evaluating the ability of catechol-thioether metabolites of MDMA ("ecstasy") to induce toxic effects through redox-cycling. Chem Res Toxicol, 2007. 20: p. 685-93.
57. Jiménez, A., Jordà, E.G., Verdaguer, E., Pubill, D., Sureda, F.X., Canudas, A.M., Escubedo, E., Camarasa, J., Camins, A., and Pallàs, M., Neurotoxicity of amphetamine derivatives is mediated by caspase pathway activation in rat cerebellar granule cells. Toxicol Appl Pharmacol, 2004. 196: p. 223-34.
58. Jones, D.C., Lau, S.S., and Monks, T.J., Thioether metabolites of 3,4-methylenedioxyamphetamine and 3,4-methylenedioxymethamphetamine inhibit human serotonin transporter (hSERT) function and simultaneously stimulate dopamine uptake into hSERT-expressing SK-N-MC cells. J Pharmacol Exp Ther, 2004. 311: p. 298-306.
59. Montiel-Duarte, C., Ansorena, E., López-Zabalza, M.J., Cenarruzabeitia, E., and Iraburu, M.J., Role of reactive oxygen species, glutathione and NF-kappaB in apoptosis induced by 3,4-methylenedioxymethamphetamine ("Ecstasy") on hepatic stellate cells. Biochem Pharmacol, 2004. 67: p. 1025-33.
60. Capela, J.P., Macedo, C., Branco, P.S., Ferreira, L.M., Lobo, A.M., Fernandes, E., Remião, F., Bastos, M.L., Dirnagl, U., Meisel, A., and Carvalho, F., Neurotoxicity mechanisms of thioether ecstasy metabolites. Neuroscience, 2007. 146: p. 1743-57.
61. Hiramatsu, M., Kumagai, Y., Unger, S.E., and Cho, A.K., Metabolism of methylenedioxymethamphetamine: formation of dihydroxymethamphetamine and a quinone identified as its glutathione adduct. J Pharmacol Exp Ther, 1990(254).
62. Monks, T.J., Jones, D.C., Bai, F., and Lau, S.S., The role of metabolism in 3,4-(+)-methylenedioxyamphetamine and 3,4-(+)-methylenedioxymethamphetamine (ecstasy) toxicity. Ther Drug Monit, 2004. 26: p. 132-6.
63. Lanz, M., Brenneisen, R., and Thormann, W., Enantioselective determination of 3,4-methylene-dioxymethamphetamine and two of its metabolites in human urine by cyclodextrin-modified capillary zone electrophoresis. Electrophoresis, 1997. 18: p. 1035-43.
64. Pizarro, N., Ortuño, J., Farré, M., Hernández-López, C., Pujadas, M., Llebaria, A., Joglar, J., Roset, P.N., Mas, M., Segura, J., Camí, J., and de la Torre, R., Determination of MDMA and its metabolites in blood and urine by gas chromatography-mass spectrometry and analysis of enantiomers by capillary electrophoresis. J Anal Toxicol, 2002. 26: p. 157-65.
65. Meyer, A., Mayerhofer, A., Kovar, K.A., and Schmidt, W.J., Enantioselective metabolism of the designer drugs 3,4-methylenedioxymethamphetamine ('ecstasy') and 3,4-methylenedioxyethylamphetamine ('eve') isomers in rat brain and blood. Neurosci Lett, 2002. 330: p. 193-7.
66. Ingelman-Sundberg, M., Oscarson, M., and McLellan, R.A., Polymorphic human cytochrome P450 enzymes: an opportunity for individualized drug treatment. Trends Pharmacol Sci, 1999. 20.
67. Oesterheld, J.R., Armstrong, S.C., and Cozza, K.L., Ecstasy: pharmacodynamic and pharmacokinetic interactions. Psychosomatics, 2004. 45: p. 84-7.
68. Samyn, N., De Boeck, G., Wood, M., Lamers, C.T., De Waard, D., Brookhuis, K.A., Verstraete, A.G., and Riedel, W.J., Plasma, oral fluid and sweat wipe ecstasy concentrations in controlled and real life conditions. Forensic Sci Int, 2002. 128: p. 90-7.
69. Helmlin, H.J., Bracher, K., Bourquin, D., Vonlanthen, D., and Brenneisen, R., Analysis of 3,4-methylenedioxymethamphetamine (MDMA) and its metabolites in plasma and urine by HPLC-DAD and GC-MS. J Anal Toxicol, 1996. 20: p. 432-40.
70. Navarro, M., Pichini, S., Farré, M., Ortuño, J., Roset, P.N., Segura, J., and de la Torre, R., Usefulness of saliva for measurement of 3,4-methylenedioxymethamphetamine and its metabolites: correlation with plasma drug concentrations and effect of salivary pH. Clin Chem, 2001. 47: p. 1788-95.

__________________________________________ Referências Bibliográficas
287
71. Segura, M., Ortuño, J., Farré, M., McLure, J.A., Pujadas, M., Pizarro, N., Llebaria, A., Joglar, J., Roset, P.N., Segura, J., and de La Torre, R., 3,4-Dihydroxymethamphetamine (HHMA). A major in vivo 3,4-methylenedioxymethamphetamine (MDMA) metabolite in humans. Chem Res Toxicol, 2001. 14: p. 1203-8.
72. Moore, K.A., Mozayani, A., Fierro, M.F., and Poklis, A., Distribution of 3,4-methylenedioxymethamphetamine (MDMA) and 3,4-methylenedioxyamphetamine (MDA) stereoisomers in a fatal poisoning. Forensic Sci Int, 1996. 83: p. 111-9.
73. Peroutka, S.J., Newman, H., and Harris, H., Subjective effects of 3,4-methylenedioxymethamphetamine in recreational users. Neuropsychopharmacology, 1988. 1: p. 273-7.
74. Davison, D. and Parrott, A.C., Ecstasy (MDMA) in recreational users: self-reported psychological and physiological effects. Human Psychopharmacol, 1997. 12: p. 221-6.
75. Parrott, A.C. and Stuart, M., Ecstasy (MDMA), amphetamine and LSD: comparative mood profiles in recreational poly-drug users. Human Psychopharmacol, 1997. 12: p. 501-4.
76. Hegadoren, K.M., Baker, G.B., and Bourin, M., 3,4-Methylenedioxy analogues of amphetamine: defining the risks to humans. Neurosci Biobehav Rev, 1999. 23: p. 539-53.
77. Liechti, M.E. and Vollenweider, F.X., Acute psychological and physiological effects of MDMA ("Ecstasy") after haloperidol pretreatment in healthy humans. Eur Neuropsychopharmacol, 2000. 10: p. 289-95.
78. Morgan, M.J., Ecstasy (MDMA): a review of its possible persistent psychological effects. Psychopharmacology (Berl), 2000. 152: p. 230-48.
79. Bankson, M.G. and Cunningham, K.A., 3,4-Methylenedioxymethamphetamine (MDMA) as a unique model of serotonin receptor function and serotonin-dopamine interactions. J Pharmacol Exp Ther, 2001. 297: p. 846-52.
80. Mechan, A.O., Esteban, B., O'Shea, E., Elliott, J.M., Colado, M.I., and Green, A.R., The pharmacology of the acute hyperthermic response that follows administration of 3,4-methylenedioxymethamphetamine (MDMA, 'ecstasy') to rats. Br J Pharmacol, 2002. 135: p. 170-80.
81. Sabol, K.E. and Seiden, L.S., Reserpine attenuates D-amphetamine and MDMA-induced transmitter release in vivo: a consideration of dose, core temperature and dopamine synthesis. Brain Res, 1998. 806: p. 69-78.
82. Yamamoto, B.K., Nash, J.F., and Gudelsky, G.A., Modulation of methylenedioxymethamphetamine-induced striatal dopamine release by the interaction between serotonin and gamma-aminobutyric acid in the substantia nigra. J Pharmacol Exp Ther, 1995. 273: p. 1063-70.
83. Berger, U.V., Gu, X.F., and Azmitia, E.C., The substituted amphetamines 3,4-methylenedioxymethamphetamine, methamphetamine, p-chloroamphetamine and fenfluramine induce 5-hydroxytryptamine release via a common mechanism blocked by fluoxetine and cocaine. Eur J Pharmacol, 1992. 215: p. 153-60.
84. Crespi, D., Mennini, T., and Gobbi, M., Carrier-dependent and Ca(2+)-dependent 5-HT and dopamine release induced by (+)-amphetamine, 3,4-methylendioxymethamphetamine, p-chloroamphetamine and (+)-fenfluramine. Br J Pharmacol, 1997. 121: p. 1735-43.
85. Nichols, D.E., Lloyd, D.H., Hoffman, A.J., Nichols, M.B., and Yim, G.K., Effects of certain hallucinogenic amphetamine analogues on the release of [3H]serotonin from rat brain synaptosomes. J Med Chem, 1982. 25: p. 530-5.
86. Verrico, C.D., Miller, G.M., and Madras, B.K., MDMA (Ecstasy) and human dopamine, norepinephrine, and serotonin transporters: implications for MDMA-induced neurotoxicity and treatment. Psychopharmacology (Berl), 2007. 189: p. 489-503.
87. Rudnick, G. and Wall, S.C., The molecular mechanism of "ecstasy" [3,4-methylenedioxy-methamphetamine (MDMA)]: serotonin transporters are targets for MDMA-induced serotonin release. Proc Natl Acad Sci U S A, 1992. 89: p. 1817-21.

Referências Bibliográficas __________________________________________
288
88. Simantov, R., Multiple molecular and neuropharmacological effects of MDMA (Ecstasy). Life Sci, 2004. 74: p. 803-14.
89. Carmo, H., Estudo da influência do metabolismo na toxicidade de derivados anfetamínicos: 4-MTA, 2C-B e MDMA (tese de Doutoramento). in Faculdade de Farmácia. 2007, Universidade do Porto, Porto.
90. Gudelsky, G.A. and Nash, J.F., Carrier-mediated release of serotonin by 3,4-methylenedioxymethamphetamine: implications for serotonin-dopamine interactions. J Neurochem, 1996. 66: p. 243-9.
91. Morley, K.C., Arnold, J.C., and McGregor, I.S., Serotonin (1A) receptor involvement in acute 3,4-methylenedioxymethamphetamine (MDMA) facilitation of social interaction in the rat. Prog Neuropsychopharmacol Biol Psychiatry, 2005. 29: p. 648-57.
92. Nash, J.F., Meltzer, H.Y., and Gudelsky, G.A., Effect of 3,4-methylenedioxymethamphetamine on 3,4-dihydroxyphenylalanine accumulation in the striatum and nucleus accumbens. J Neurochem, 1990. 54: p. 1062-7.
93. Sprague, J.E., Everman, S.L., and Nichols, D.E., An integrated hypothesis for the serotonergic axonal loss induced by 3,4-methylenedioxymethamphetamine. Neurotoxicology, 1998. 19: p. 427-41.
94. Bankson, M.G. and Yamamoto, B.K., Serotonin-GABA interactions modulate MDMA-induced mesolimbic dopamine release. J Neurochem, 2004. 91: p. 852-9.
95. Daza-Losada, M., Ribeiro Do Couto, B., Manzanedo, C., Aguilar, M.A., Rodríguez-Arias, M., and Miñarro, J., Rewarding effects and reinstatement of MDMA-induced CPP in adolescent mice. Neuropsychopharmacology, 2007. 32: p. 1750-9.
96. O'Shea, E., Esteban, B., Camarero, J., Green, A.R., and Colado, M.I., Effect of GBR 12909 and fluoxetine on the acute and long term changes induced by MDMA ('ecstasy') on the 5-HT and dopamine concentrations in mouse brain. Neuropharmacology, 2001. 40: p. 65-74.
97. Saadat, K.S., Elliott, J.M., Colado, M.I., and Green, A.R., The acute and long-term neurotoxic effects of MDMA on marble burying behaviour in mice. J Psychopharmacol, 2006. 20: p. 264-71.
98. Vander, A.J., Sherman, J., and Luciano, D.S., Human physiology: the mechanism of body function. 8th ed. 2001, Boston: McGraw−Hill.
99. Klingler, W., Heffron, J.J., Jurkat-Rott, K., O'sullivan, G., Alt, A., Schlesinger, F., Bufler, J., and Lehmann-Horn, F., 3,4-Methylenedioxymethamphetamine (ecstasy) activates skeletal muscle nicotinic acetylcholine receptors. J Pharmacol Exp Ther, 2005. 314: p. 1267-73.
100. Garcia-Ratés, S., Camarasa, J., Escubedo, E., and Pubill, D., Methamphetamine and 3,4-methylenedioxymethamphetamine interact with central nicotinic receptors and induce their up-regulation. Toxicol Appl Pharmacol, 2007. 223: p. 195-205.
101. Ripoll, N., Bronnec, M., and Bourin, M., Nicotinic receptors and schizophrenia. Curr Med Res Opin, 2004. 20: p. 1057-74.
102. Maskos, U., Molles, B.E., Pons, S., Besson, M., Guiard, B.P., Guilloux, J.P., Evrard, A., Cazala, P., Cormier, A., Mameli-Engvall, M., Dufour, N., Cloëz-Tayarani, I., Bemelmans, A.P., Mallet, J., Gardier, A.M., David, V., Faure, P., Granon, S., and Changeux, J.P., Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature, 2005. 436: p. 103-7.
103. Chipana, C., Camarasa, J., Pubill, D., and Escubedo, E., Protection against MDMA-induced dopaminergic neurotoxicity in mice by methyllycaconitine: involvement of nicotinic receptors. Neuropharmacology, 2006. 51: p. 885-95.
104. Chipana, C., Camarasa, J., Pubill, D., and Escubedo, E., Memantine prevents MDMA-induced neurotoxicity. Neurotoxicology, 2008. 29: p. 179-83.
105. Callaway, C.W., Wing, L.L., and Geyer, M.A., Serotonin release contributes to the locomotor stimulant effects of 3,4-methylenedioxymethamphetamine in rats. J Pharmacol Exp Ther, 1990. 254: p. 456-64.

__________________________________________ Referências Bibliográficas
289
106. Bankson, M.G. and Cunningham, K.A., Pharmacological studies of the acute effects of (+)-3,4-methylenedioxymethamphetamine on locomotor activity: role of 5-HT(1B/1D) and 5-HT(2) receptors. Neuropsychopharmacology, 2002. 26: p. 40-52.
107. Fletcher, P.J., Korth, K.M., Robinson, S.R., and Baker, G.B., Multiple 5-HT receptors are involved in the effects of acute MDMA treatment: studies on locomotor activity and responding for conditioned reinforcement. Psychopharmacology (Berl), 2002. 162: p. 282-91.
108. Herin, D.V., Liu, S., Ullrich, T., Rice, K.C., and Cunningham, K.A., Role of the serotonin 5-HT2A receptor in the hyperlocomotive and hyperthermic effects of (+)-3,4-methylenedioxymethamphetamine. Psychopharmacology (Berl), 2005. 178: p. 505-13.
109. Kehne, J.H., Ketteler, H.J., McCloskey, T.C., Sullivan, C.K., Dudley, M.W., and Schmidt, C.J., Effects of the selective 5-HT2A receptor antagonist MDL 100,907 on MDMA-induced locomotor stimulation in rats. Neuropsychopharmacology, 1996. 15: p. 116-24.
110. McCreary, A.C., Bankson, M.G., and Cunningham, K.A., Pharmacological studies of the acute and chronic effects of (+)-3, 4-methylenedioxymethamphetamine on locomotor activity: role of 5-hydroxytryptamine(1A) and 5-hydroxytryptamine(1B/1D) receptors. J Pharmacol Exp Ther, 1999. 290: p. 965-73.
111. Scearce-Levie, K., Viswanathan, S.S., and Hen, R., Locomotor response to MDMA is attenuated in knockout mice lacking the 5-HT1B receptor. Psychopharmacology (Berl), 1999. 141: p. 154-61.
112. Bubar, M.J., Pack, K.M., Frankel, P.S., and Cunningham, K.A., Effects of dopamine D1- or D2-like receptor antagonists on the hypermotive and discriminative stimulus effects of (+)-MDMA. Psychopharmacology (Berl), 2004. 173: p. 326-36.
113. Risbrough, V.B., Masten, V.L., Caldwell, S., Paulus, M.P., Low, M.J., and Geyer, M.A., Differential contributions of dopamine D1, D2, and D3 receptors to MDMA-induced effects on locomotor behavior patterns in mice. Neuropsychopharmacology, 2006. 31: p. 2349-58.
114. Ball, K.T., Budreau, D., and Rebec, G.V., Acute effects of 3,4-methylenedioxymethamphetamine on striatal single-unit activity and behavior in freely moving rats: differential involvement of dopamine D(1) and D(2) receptors. Brain Res, 2003. 994: p. 203-15.
115. Bexis, S. and Docherty, J.R., Effects of MDMA, MDA and MDEA on blood pressure, heart rate, locomotor activity and body temperature in the rat involve alpha-adrenoceptors. Br J Pharmacol, 2006. 147: p. 926-34.
116. Selken, J. and Nichols, D.E., Alpha1-adrenergic receptors mediate the locomotor response to systemic administration of (+/-)-3,4-methylenedioxymethamphetamine (MDMA) in rats. Pharmacol Biochem Behav, 2007. 86: p. 622-30.
117. Easton, N., Fry, J., O'Shea, E., Watkins, A., Kingston, S., and Marsden, C.A., Synthesis, in vitro formation, and behavioural effects of glutathione regioisomers of alpha-methyldopamine with relevance to MDA and MDMA (ecstasy). Brain Res, 2003. 987: p. 144-54.
118. Leonardi, E.T. and Azmitia, E.C., MDMA (ecstasy) inhibition of MAO type A and type B: comparisons with fenfluramine and fluoxetine (Prozac). Neuropsychopharmacology, 1994. 10: p. 231-8.
119. Alves, E., Summavielle, T., Alves, C.J., Gomes-da-Silva, J., Barata, J.C., Fernandes, E., Bastos, M.L., Tavares, M.A., and Carvalho, F., Monoamine oxidase-B mediates ecstasy-induced neurotoxic effects to adolescent rat brain mitochondria. J Neurosci, 2007. 27: p. 10203-10.
120. White, S.R., Obradovic, T., Imel, K.M., and Wheaton, M.J., The effects of methylenedioxymethamphetamine (MDMA, "Ecstasy") on monoaminergic neurotransmission in the central nervous system. Prog Neurobiol, 1996. 49: p. 455-79.
121. Navarro, J.F., Rivera, A., Maldonado, E., Cavas, M., and de la Calle, A., Anxiogenic-like activity of 3,4-methylenedioxy-methamphetamine ("Ecstasy") in the social

Referências Bibliográficas __________________________________________
290
interaction test is accompanied by an increase of c-fos expression in mice amygdala. Prog Neuropsychopharmacol Biol Psychiatry, 2004. 28: p. 249-54.
122. Sprague, J.E., Banks, M.L., Cook, V.J., and Mills, E.M., Hypothalamic-pituitary-thyroid axis and sympathetic nervous system involvement in hyperthermia induced by 3,4-methylenedioxymethamphetamine (Ecstasy). J Pharmacol Exp Ther, 2003. 305: p. 159-66.
123. Mills, E.M., Banks, M.L., Sprague, J.E., and Finkel, T., Pharmacology: uncoupling the agony from ecstasy. Nature, 2003. 426: p. 403-4.
124. Harris, D.S., Baggott, M., Mendelson, J.H., Mendelson, J.E., and Jones, R.T., Subjective and hormonal effects of 3,4-methylenedioxymethamphetamine (MDMA) in humans. Psychopharmacology (Berl), 2002. 162: p. 396-405.
125. Grob, C.S., Poland, R.E., Chang, L., and Ernst, T., Psychobiologic effects of 3,4-methylenedioxymethamphetamine in humans: methodological considerations and preliminary observations. Behav Brain Res, 1996. 73: p. 103-7.
126. Henry, J.A., Fallon, J.K., Kicman, A.T., Hutt, A.J., Cowan, D.A., and Forsling, M., Low-dose MDMA ("ecstasy") induces vasopressin secretion. Lancet, 1998. 351: p. 1784.
127. Gerra, G., Zaimovic, A., Ferri, M., Zambelli, U., Timpano, M., Neri, E., Marzocchi, G.F., Delsignore, R., and Brambilla, F., Long-lasting effects of (+/-)3,4-methylenedioxymethamphetamine (ecstasy) on serotonin system function in humans. Biol Psychiatry, 2000. 47: p. 127-36.
128. Gerra, G., Zaimovic, A., Giucastro, G., Maestri, D., Monica, C., Sartori, R., Caccavari, R., and Delsignore, R., Serotonergic function after (+/-)3,4-methylene-dioxymethamphetamine ('Ecstasy') in humans. Int Clin Psychopharmacol, 1998. 13: p. 1-9.
129. McCann, U.D., Eligulashvili, V., Mertl, M., Murphy, D.L., and Ricaurte, G.A., Altered neuroendocrine and behavioral responses to m-chlorophenylpiperazine in 3,4-methylenedioxymethamphetamine (MDMA) users. Psychopharmacology (Berl), 1999. 147: p. 56-65.
130. Price, L.H., Ricaurte, G.A., Krystal, J.H., and Heninger, G.R., Neuroendocrine and mood responses to intravenous L-tryptophan in 3,4-methylenedioxymethamphetamine (MDMA) users. Preliminary observations. Arch Gen Psychiatry, 1989. 46: p. 20-2.
131. Forsling, M.L., Fallon, J.K., Shah, D., Tilbrook, G.S., Cowan, D.A., Kicman, A.T., and Hutt, A.J., The effect of 3,4-methylenedioxymethamphetamine (MDMA, 'ecstasy') and its metabolites on neurohypophysial hormone release from the isolated rat hypothalamus. Br J Pharmacol, 2002. 135: p. 649-56.
132. Wolff, K., Tsapakis, E.M., Winstock, A.R., Hartley, D., Holt, D., Forsling, M.L., and Aitchison, K.J., Vasopressin and oxytocin secretion in response to the consumption of ecstasy in a clubbing population. J Psychopharmacol, 2006. 20: p. 400-10.
133. Clemens, K.J., McGregor, I.S., Hunt, G.E., and Cornish, J.L., MDMA, methamphetamine and their combination: possible lessons for party drug users from recent preclinical research. Drug Alcohol Rev, 2007. 26: p. 9-15.
134. McGregor, I.S., Callaghan, P.D., and Hunt, G.E., From ultrasocial to antisocial: a role for oxytocin in the acute reinforcing effects and long-term adverse consequences of drug use? Br J Pharmacol, 2008. 154: p. 358-68.
135. Thompson, M.R., Callaghan, P.D., Hunt, G.E., Cornish, J.L., and McGregor, I.S., A role for oxytocin and 5-HT(1A) receptors in the prosocial effects of 3,4 methylenedioxymethamphetamine ("ecstasy"). Neuroscience, 2007. 146: p. 509-14.
136. Dickerson, S.M., Walker, D.M., Reveron, M.E., Duvauchelle, C.L., and Gore, A.C., The Recreational Drug Ecstasy Disrupts the Hypothalamic-Pituitary-Gonadal Reproductive Axis in Adult Male Rats. Neuroendocrinology, 2008.
137. Allott, K., Canny, B.J., Broadbear, J.H., Stepto, N.K., Murphy, B., and Redman, J., Neuroendocrine and subjective responses to pharmacological challenge with citalopram: a controlled study in male and female ecstasy/MDMA users. J Psychopharmacol, 2008.

__________________________________________ Referências Bibliográficas
291
138. Broening, H.W., Bowyer, J.F., and Slikker, W., Jr, Age-dependent sensitivity of rats to the long-term effects of the serotonergic neurotoxicant (+/-)-3,4-methylenedioxymethamphetamine (MDMA) correlates with the magnitude of the MDMA-induced thermal response. J Pharmacol Exp Ther, 1995. 275: p. 325-33.
139. Malberg, J.E. and Seiden, L.S., Small changes in ambient temperature cause large changes in 3,4-methylenedioxymethamphetamine (MDMA)-induced serotonin neurotoxicity and core body temperature in the rat. J Neurosci, 1998. 18: p. 5086-94.
140. Nash, J.F., Jr, Meltzer, H.Y., and Gudelsky, G.A., Elevation of serum prolactin and corticosterone concentrations in the rat after the administration of 3,4-methylenedioxymethamphetamine. J Pharmacol Exp Ther, 1988. 245: p. 873-9.
141. Schmidt, C.J., Black, C.K., Abbate, G.M., and Taylor, V.L., Methylenedioxymethamphetamine-induced hyperthermia and neurotoxicity are independently mediated by 5-HT2 receptors. Brain Res, 1990. 529: p. 85-90.
142. Carvalho, M., Carvalho, F., Remiao, F., Pereira, M.L., Pires-das-Neves, R., and Bastos, M.L., Effect of 3,4-methylenedioxymethamphetamine ("ecstasy") on body temperature and liver antioxidant status in mice: influence of ambient temperature. Arch Toxicol, 2002. 76: p. 166-72.
143. Saadat, K.S., Elliott, J.M., Colado, M.I., and Green, A.R., Hyperthermic and neurotoxic effect of 3,4-methylenedioxymethamphetamine (MDMA) in guinea pigs. Psychopharmacology (Berl), 2004. 173: p. 452-3.
144. Taffe, M.A., Lay, C.C., Von Huben, S.N., Davis, S.A., Crean, R.D., and Katner, S.N., Hyperthermia induced by 3,4-methylenedioxymethamphetamine in unrestrained rhesus monkeys. Drug Alcohol Depend, 2006. 82: p. 276-81.
145. Fantegrossi, W.E., Godlewski, T., Karabenick, R.L., Stephens, J.M., Ullrich, T., Rice, K.C., and Woods, J.H., Pharmacological characterization of the effects of 3,4-methylenedioxymethamphetamine ("ecstasy") and its enantiomers on lethality, core temperature, and locomotor activity in singly housed and crowded mice. Psychopharmacology (Berl), 2003. 166: p. 202-11.
146. Pedersen, N.P. and Blessing, W.W., Cutaneous vasoconstriction contributes to hyperthermia induced by 3,4-methylenedioxymethamphetamine (ecstasy) in conscious rabbits. J Neurosci, 2001. 21: p. 8648-54.
147. Green, A.R., Sanchez, V., O'Shea, E., Saadat, K.S., Elliott, J.M., and Colado, M.I., Effect of ambient temperature and a prior neurotoxic dose of 3,4-methylenedioxymethamphetamine (MDMA) on the hyperthermic response of rats to a single or repeated ('binge' ingestion) low dose of MDMA. Psychopharmacology (Berl), 2004. 173: p. 264-9.
148. Colado, M.I., O'Shea, E., and Green, A.R., Acute and long-term effects of MDMA on cerebral dopamine biochemistry and function. Psychopharmacology (Berl), 2004. 173: p. 249-63.
149. Saadat, K.S., O'shea, E., Colado, M.I., Elliott, J.M., and Green, A.R., The role of 5-HT in the impairment of thermoregulation observed in rats administered MDMA ('ecstasy') when housed at high ambient temperature. Psychopharmacology (Berl), 2005. 179: p. 884-90.
150. Beveridge, T.J., Mechan, A.O., Sprakes, M., Pei, Q., Zetterstrom, T.S., Green, A.R., and Elliott, J.M., Effect of 5-HT depletion by MDMA on hyperthermia and Arc mRNA induction in rat brain. Psychopharmacology (Berl), 2004. 173: p. 346-52.
151. Alex, K.D. and Pehek, E.A., Pharmacologic mechanisms of serotonergic regulation of dopamine neurotransmission. Pharmacol Ther, 2007. 113: p. 296-320.
152. Bexis, S. and Docherty, J.R., Role of alpha2A-adrenoceptors in the effects of MDMA on body temperature in the mouse. Br J Pharmacol, 2005. 146: p. 1-6.
153. Bexis, S., Phillis, B.D., Ong, J., White, J.M., and Irvine, R.J., Baclofen prevents MDMA-induced rise in core body temperature in rats. Drug Alcohol Depend, 2004. 74: p. 89-96.

Referências Bibliográficas __________________________________________
292
154. Mills, E.M., Rusyniak, D.E., and Sprague, J.E., The role of the sympathetic nervous system and uncoupling proteins in the thermogenesis induced by 3,4-methylenedioxymethamphetamine. J Mol Med, 2004. 82: p. 787-99.
155. Sprague, J.E., Yang, X., Sommers, J., Gilman, T.L., and Mills, E.M., Roles of norepinephrine, free Fatty acids, thyroid status, and skeletal muscle uncoupling protein 3 expression in sympathomimetic-induced thermogenesis. J Pharmacol Exp Ther, 2007. 320: p. 274-80.
156. Santos-Marques, M.J., Carvalho, F., Sousa, C., Remião, F., Vitorino, R., Amado, F., Ferreira, R., Duarte, J.A., and Bastos, M.L., Cytotoxicity and cell signalling induced by continuous mild hyperthermia in freshly isolated mouse hepatocytes. Toxicology, 2006. 224: p. 210-8.
157. Escobedo, I., Peraile, I., Orio, L., Colado, M.I., and O'Shea, E., Evidence for a role of Hsp70 in the neuroprotection induced by heat shock pre-treatment against 3,4-methylenedioxymethamphetamine toxicity in rat brain. J Neurochem, 2007. 101: p. 1272-83.
158. Mechan, A.O., O'Shea, E., Elliott, J.M., Colado, M.I., and Green, A.R., A neurotoxic dose of 3,4-methylenedioxymethamphetamine (MDMA; ecstasy) to rats results in a long-term defect in thermoregulation. Psychopharmacology (Berl), 2001. 155: p. 413-8.
159. Henry, J.A., Jeffreys, K.J., and Dawling, S., Toxicity and deaths from 3,4-methylenedioxymethamphetamine ("ecstasy"). Lancet, 1992. 340: p. 384-7.
160. Mallick, A. and Bodenham, A.R., MDMA induced hyperthermia: a survivor with an initial body temperature of 42.9 degrees C. J Accid Emerg Med, 1997. 14: p. 336-8.
161. Chang, L., Grob, C.S., Ernst, T., Itti, L., Mishkin, F.S., Jose-Melchor, R., and Poland, R.E., Effect of ecstasy [3,4-methylenedioxymethamphetamine (MDMA)] on cerebral blood flow: a co-registered SPECT and MRI study. Psychiatry Res, 2000. 98: p. 15-28.
162. Jones, A.W., Driving under the influence of drugs in Sweden with zero concentration limits in blood for controlled substances. Traffic Inj Prev, 2005. 6: p. 317-22.
163. Jones, A.W. and Holmgren, A., Abnormally high concentrations of amphetamine in blood of impaired drivers. J Forensic Sci, 2005. 50: p. 1215-20.
164. Burgess, C., O'Donohoe, A., and Gill, M., Agony and ecstasy: a review of MDMA effects and toxicity. Eur Psychiatry, 2000. 15: p. 287-94.
165. Eifinger, F., Roth, B., Kröner, L., and Rothschild, M.A., Severe Ecstasy poisoning in an 8-month-old infant. Eur J Pediatr, 2008. DOI 10.1007/s00431-007-0609-6.
166. Bedford Russell, A.R., Schwartz, R.H., and Dawling, S., Accidental ingestion of 'Ecstasy' (3,4-methylenedioxymethylamphetamine). Arch Dis Child, 1992. 67: p. 1114-5.
167. Chang, Y.J., Lai, M.W., Kong, M.S., and Chao, H.C., Accidental ingestion of Ecstasy in a toddler. J Formos Med Assoc, 2005. 104: p. 946-7.
168. Hall, A.P., Lyburn, I.D., Spears, F.D., and Riley, B., An unusual case of Ecstasy poisoning. Intensive Care Med, 1996. 22: p. 670-1.
169. Chadwick, I.S., Curry, P.D., Linsley, A., Freemont, A.J., and Doran, B., Ecstasy, 3-4 methylenedioxymethamphetamine (MDMA), a fatality associated with coagulopathy and hyperthermia. J R Soc Med, 1991. 84: p. 371.
170. Shannon, M., Methylenedioxymethamphetamine (MDMA, "Ecstasy"). Pediatr Emerg Care, 2000. 16: p. 377-80.
171. Roberts, L. and Wright, H., Survival following intentional massive overdose of 'Ecstasy'. J Accid Emerg Med, 1994. 11: p. 53-4.
172. Green, A.R., MDMA: fact and fallacy, and the need to increase knowledge in both the scientific and popular press. Psychopharmacology (Berl), 2004. 173: p. 231-233.
173. Cole, J.C., Sumnall, H.R., Smith, G.W., and Rostami-Hodjegan, A., Preliminary evidence of the cardiovascular effects of polysubstance misuse in nightclubs. J Psychopharmacol, 2005. 19: p. 67-70.
174. Redfearn, P.J., Agrawal, N., and Mair, L.H., An association between the regular use of 3,4 methylenedioxy-methamphetamine (ecstasy) and excessive wear of the teeth. Addiction, 1998. 93: p. 745-8.

__________________________________________ Referências Bibliográficas
293
175. Logan, A.S., Stickle, B., O'Keefe, N., and Hewitson, H., Survival following 'Ecstasy' ingestion with a peak temperature of 42 degrees C. Anaesthesia, 1993. 48: p. 1017-8.
176. Britt, G.C. and McCance-Katz, E.F., A Brief Overview of the Clinical Pharmacology of “Club Drugs”. Subst Use Misuse, 2005. 40: p. 1189-1201.
177. Brown, C. and Osterloh, J., Multiple severe complications from recreational ingestion of MDMA ('Ecstasy'). JAMA, 1987. 258: p. 780-1.
178. Dowling, G.P., McDonough, E.T., 3rd, and Bost, R.O., 'Eve' and 'Ecstasy'. A report of five deaths associated with the use of MDEA and MDMA. JAMA, 1987. 257: p. 1615-7.
179. McCann, U.D., Slate, S.O., and Ricaurte, G.A., Adverse reactions with 3,4-methylenedioxymethamphetamine (MDMA; 'ecstasy'). Drug Saf, 1996. 15: p. 107-15.
180. Screaton, G.R., Singer, M., Cairns, H.S., Thrasher, A., Sarner, M., and Cohen, S.L., Hyperpyrexia and rhabdomyolysis after MDMA ("ecstasy") abuse. Lancet, 1992. 339: p. 677-8.
181. Suarez, R.V. and Riemersma, R., "Ecstasy" and sudden cardiac death. Am J Forensic Med Pathol, 1988. 9: p. 339-41.
182. Milroy, C.M., Clark, J.C., and Forrest, A.R., Pathology of deaths associated with "ecstasy" and "eve" misuse. J Clin Pathol, 1996. 49: p. 149-53.
183. Rutty, G.N. and Milroy, C.M., The pathology of the ring-substituted amphetamine analogue 3,4-methylenedioxymethylamphetamine (MDMA, 'Ecstasy'). J Pathol, 1997. 181: p. 255-6.
184. Rojas, R., Riascos, R., Vargas, D., Cuellar, H., and Borne, J., Neuroimaging in drug and substance abuse part I: cocaine, cannabis, and ecstasy. Top Magn Reson Imaging, 2005. 16: p. 231-8.
185. Dykhuizen, R.S., Brunt, P.W., Atkinson, P., Simpson, J.G., and Smith, C.C., Ecstasy induced hepatitis mimicking viral hepatitis. Gut, 1995. 36: p. 939-41.
186. Ellis, A.J., Wendon, J.A., Portmann, B., and Williams, R., Acute liver damage and ecstasy ingestion. Gut, 1996. 38: p. 454-8.
187. Brauer, R.B., Heidecke, C.D., Nathrath, W., Beckurts, K.T., Vorwald, P., Zilker, T.R., Schweigart, U., Hölscher, A.H., and Siewert, J.R., Liver transplantation for the treatment of fulminant hepatic failure induced by the ingestion of ecstasy. Transpl Int, 1997. 10: p. 229-33.
188. Caballero, F., Lopez-Navidad, A., Cotorruelo, J., and Txoperena, G., Ecstasy-induced brain death and acute hepatocellular failure: multiorgan donor and liver transplantation. Transplantation, 2002. 74: p. 532-7.
189. Milroy, C.M., Ten years of 'ecstasy'. J R Soc Med, 1999. 92: p. 68-72. 190. Liechti, M.E., Kunz, I., and Kupferschmidt, H., Acute medical problems due to Ecstasy
use. Case-series of emergency department visits. Swiss Med Wkly, 2005. 135: p. 652-7. 191. Nakagawa, Y., Suzuki, T., Tayama, S., Ishii, H., and Ogata, A., Cytotoxic effects of 3,4-
methylenedioxy-N-alkylamphetamines, MDMA and its analogues, on isolated rat hepatocytes. Arch Toxicol, 2008.
192. Carvalho, M., Milhazes, N., Remião, F., Borges, F., Fernandes, E., Amado, F., Monks, T.J., Carvalho, F., and Bastos, M.L., Hepatotoxicity of 3,4-methylenedioxyamphetamine and alpha-methyldopamine in isolated rat hepatocytes: formation of glutathione conjugates. Arch Toxicol, 2004. 78: p. 16-24.
193. Carvalho, M., Remião, F., Milhazes, N., Borges, F., Fernandes, E., Carvalho, F., and Bastos, M.L., The toxicity of N-methyl-alpha-methyldopamine to freshly isolated rat hepatocytes is prevented by ascorbic acid and N-acetylcysteine. Toxicology, 2004. 200: p. 193-203.
194. Jones, A.L. and Simpson, K.J., Review article: mechanisms and management of hepatotoxicity in ecstasy (MDMA) and amphetamine intoxications. Aliment Pharmacol Ther, 1999. 13: p. 129-33.
195. Chen, F. and Shi, X., Signaling from toxic metals to NF-kappaB and beyond: not just a matter of reactive oxygen species. Environ Health Perspect, 2002. 110: p. 807-11.
196. Tiangco, D.A., Lattanzio, F.A.J., Osgood, C.J., Beebe, S.J., Kerry, J.A., and Hargrave, B.Y., 3,4-Methylenedioxymethamphetamine activates nuclear factor-kappaB, increases

Referências Bibliográficas __________________________________________
294
intracellular calcium, and modulates gene transcription in rat heart cells. Cardiovasc Toxicol, 2005. 5: p. 301-10.
197. Pacifici, R., Zuccaro, P., Farré, M., Pichini, S., Di Carlo, S., Roset, P.N., Lopez, C.H., Ortuño, J., Segura, J., Camí, J., and de la Torre, R., Immunomodulating activity of MDMA. Ann N Y Acad Sci, 2000. 914: p. 215-24.
198. Pacifici, R., Zuccaro, P., Farre, M., Pichini, S., Di Carlo, S., Roset, P.N., Ortuno, J., Segura, J., and de la Torre, R., Immunomodulating properties of MDMA alone and in combination with alcohol: a pilot study. Life Sci, 1999. 65: p. PL309-16.
199. Pacifici, R., Zuccaro, P., Farré, M., Pichini, S., Di Carlo, S., Roset, P.N., Ortuño, J., Pujadas, M., Bacosi, A., Menoyo, E., Segura, J., and de la Torre, R., Effects of repeated doses of MDMA ("ecstasy") on cell-mediated immune response in humans. Life Sci, 2001. 69: p. 2931-41.
200. Pacifici, R., Zuccaro, P., Farre, M., Pichini, S., Di Carlo, S., Roset, P.N., Palmi, I., Ortuno, J., Menoyo, E., Segura, J., and de la Torre, R., Cell-mediated immune response in MDMA users after repeated dose administration: studies in controlled versus noncontrolled settings. Ann N Y Acad Sci, 2002. 965: p. 421-33.
201. Miranda, M., Bosch-Morell, F., Johnsen-Soriano, S., Barcia, J., Almansa, I., Asensio, S., Araiz, J., Messeguer, A., and Romero, F.J., Oxidative stress in rat retina and hippocampus after chronic MDMA ('ecstasy') administration. Neurochem Res, 2007. 32: p. 1156-62.
202. Cunningham, M., Ecstasy-induced rhabdomyolysis and its role in the development of acute renal failure. Intensive Crit Care Nurs, 1997. 13: p. 216-23.
203. Carvalho, M., Hawksworth, G., Milhazes, N., Borges, F., Monks, T.J., Fernandes, E., Carvalho, F., and Bastos, M.L., Role of metabolites in MDMA (ecstasy)-induced nephrotoxicity: an in vitro study using rat and human renal proximal tubular cells. Arch Toxicol, 2002. 76(10): p. 581-8.
204. Henry, J.A. and Hill, I.R., Fatal interaction between ritonavir and MDMA. Lancet, 1998. 352: p. 1751-2.
205. Holden, R. and Jackson, M.A., Near-fatal hyponatraemic coma due to vasopressin over-secretion after "ecstasy" (3,4-MDMA). Lancet, 1996. 347: p. 1052.
206. Holmes, S.B., Banerjee, A.K., and Alexander, W.D., Hyponatraemia and seizures after ecstasy use. Postgrad Med J, 1999. 75: p. 32-3.
207. Kessel, B., Hyponatraemia after ingestion of ecstasy. BMJ, 1994. 308: p. 414. 208. Lehmann, E.D., Thom, C.H., and Croft, D.N., Delayed severe rhabdomyolysis after
taking 'ecstasy'. Postgrad Med J, 1995. 71: p. 186-7. 209. Matthai, S.M., Davidson, D.C., Sills, J.A., and Alexandrou, D., Cerebral oedema after
ingestion of MDMA ("ecstasy") and unrestricted intake of water. BMJ, 1996. 312: p. 1359.
210. Maxwell, D.L., Polkey, M.I., and Henry, J.A., Hyponatraemia and catatonic stupor after taking "ecstasy". BMJ, 1993. 307: p. 1399.
211. Satchell, S.C. and Connaughton, M., Inappropriate antidiuretic hormone secretion and extreme rises in serum creatinine kinase following MDMA ingestion. Br J Hosp Med, 1994. 51: p. 495.
212. Kalantar-Zadeh, K., Nguyen, M.K., Chang, R., and Kurtz, I., Fatal hyponatremia in a young woman after ecstasy ingestion. Nat Clin Pract Nephrol, 2006. 2: p. 283-8.
213. Fallon, J.K., Shah, D., Kicman, A.T., Hutt, A.J., Henry, J.A., Cowan, D.A., and Forsling, M., Action of MDMA (ecstasy) and its metabolites on arginine vasopressin release. Ann N Y Acad Sci, 2002. 965: p. 399-409.
214. Finch, E., Sell, L., and Arnold, D., Cerebral oedema after MDMA ("ecstasy") and unrestricted water intake. Drug workers emphasise that water is not an antidote to drug. BMJ, 1996. 313: p. 690.
215. Wilkins, B., Cerebral oedema after MDMA ("ecstasy") and unrestricted water intake. Hyponatraemia must be treated with low water input. BMJ, 1996. 313: p. 689-90.
216. Bryden, A.A., Rothwell, P.J., and O'Reilly, P.H., Urinary retention with misuse of "ecstasy". BMJ, 1995. 310: p. 504.

__________________________________________ Referências Bibliográficas
295
217. Forsling, M., Fallon, J.K., Kicman, A.T., Hutt, A.J., Cowan, D.A., and Henry, J.A., Arginine vasopressin release in response to the administration of 3,4-methylenedioxymethamphetamine ("ecstasy"): is metabolism a contributory factor? J Pharm Pharmacol, 2001. 53: p. 1357-63.
218. Falck, R.S., Wang, J., and Carlson, R.G., Depressive symptomatology in young adults with a history of MDMA use: a longitudinal analysis. J Psychopharmacol, 2008. 22: p. 47-54.
219. Schifano, F., Di Furia, L., Forza, G., Minicuci, N., and Bricolo, R., MDMA ('ecstasy') consumption in the context of polydrug abuse: a report on 150 patients. Drug Alcohol Depend, 1998. 52: p. 85-90.
220. Bull, E.J., Hutson, P.H., and Fone, K.C., Reduced social interaction following 3,4-methylenedioxymethamphetamine is not associated with enhanced 5-HT 2C receptor responsivity. Neuropharmacology, 2003. 44: p. 439-48.
221. Khakoo, S.I., Coles, C.J., Armstrong, J.S., and Barry, R.E., Hepatotoxicity and accelerated fibrosis following 3,4-methylenedioxymetamphetamine ("ecstasy") usage. J Clin Gastroenterol, 1995. 20: p. 244-7.
222. Varela-Rey, M., Montiel-Duarte, C., Beitia, G., Cenarruzabeitia, E., and Iraburu, M.J., 3,4-methylenedioxymethamphetamine ("Ecstasy") stimulates the expression of alpha1(I) procollagen mRNA in hepatic stellate cells. Biochem Biophys Res Commun, 1999. 259: p. 678-82.
223. Yuan, J., Cord, B.J., McCann, U.D., Callahan, B.T., and Ricaurte, G.A., Effect of depleting vesicular and cytoplasmic dopamine on methylenedioxymethamphetamine neurotoxicity. J Neurochem, 2002. 80: p. 960-9.
224. McCann, U.D., Eligulashvili, V., and Ricaurte, G.A., (+/-)3,4-Methylenedioxymethamphetamine ('Ecstasy')-induced serotonin neurotoxicity: clinical studies. Neuropsychobiology, 2000. 42: p. 11-6.
225. Ricaurte, G.A., Yuan, J., and McCann, U.D., (+/-)3,4-Methylenedioxymethamphetamine ('Ecstasy')-induced serotonin neurotoxicity: studies in animals. Neuropsychobiology, 2000. 42: p. 5-10.
226. Ricaurte, G.A., McCann, U.D., Szabo, Z., and Scheffel, U., Toxicodynamics and long-term toxicity of the recreational drug, 3, 4-methylenedioxymethamphetamine (MDMA, 'Ecstasy'). Toxicol Lett, 2000. 112-113: p. 143-6.
227. Gouzoulis-Mayfrank, E. and Daumann, J., Neurotoxicity of methylenedioxyamphetamines (MDMA; ecstasy) in humans: how strong is the evidence for persistent brain damage? Addiction, 2006. 101: p. 348-61.
228. Stone, D.M., Johnson, M., Hanson, G.R., and Gibb, J.W., Role of endogenous dopamine in the central serotonergic deficits induced by 3,4-methylenedioxymethamphetamine. J Pharmacol Exp Ther, 1988. 247: p. 79-87.
229. Parrott, A.C., Recreational Ecstasy/MDMA, the serotonin syndrome, and serotonergic neurotoxicity. Pharmacol Biochem Behav, 2002. 71: p. 837-44.
230. O'Shea, E., Orio, L., Escobedo, I., Sanchez, V., Camarero, J., Green, A.R., and Colado, M.I., MDMA-induced neurotoxicity: long-term effects on 5-HT biosynthesis and the influence of ambient temperature. Br J Pharmacol., 2006. 148: p. 778-85.
231. McCann, U.D., Szabo, Z., Seckin, E., Rosenblatt, P., Mathews, W.B., Ravert, H.T., Dannals, R.F., and Ricaurte, G.A., Quantitative PET studies of the serotonin transporter in MDMA users and controls using [11C]McN5652 and [11C]DASB. Neuropsychopharmacology, 2005. 30: p. 1741-50.
232. McCann, U.D., Szabo, Z., Scheffel, U., Dannals, R.F., and Ricaurte, G.A., Positron emission tomographic evidence of toxic effect of MDMA ("Ecstasy") on brain serotonin neurons in human beings. Lancet, 1998. 352: p. 1433-7.
233. Reneman, L., Booij, J., de Bruin, K., Reitsma, J.B., de Wolff, F.A., Gunning, W.B., den Heeten, G.J., and van den Brink, W., Effects of dose, sex, and long-term abstention from use on toxic effects of MDMA (ecstasy) on brain serotonin neurons. Lancet, 2001. 358: p. 1864-9.

Referências Bibliográficas __________________________________________
296
234. Reneman, L., Booij, J., Lavalaye, J., de Bruin, K., Reitsma, J.B., Gunning, B., den Heeten, G.J., and van Den Brink, W., Use of amphetamine by recreational users of ecstasy (MDMA) is associated with reduced striatal dopamine transporter densities: a [123I]beta-CIT SPECT study--preliminary report. Psychopharmacology (Berl), 2002. 159: p. 335-40.
235. Semple, D.M., Ebmeier, K.P., Glabus, M.F., O'Carroll, R.E., and Johnstone, E.C., Reduced in vivo binding to the serotonin transporter in the cerebral cortex of MDMA ('ecstasy') users. Br J Psychiatry, 1999. 175: p. 63-9.
236. Scheffel, U., Szabo, Z., Mathews, W.B., Finley, P.A., Dannals, R.F., Ravert, H.T., Szabo, K., Yuan, J., and Ricaurte, G.A., In vivo detection of short- and long-term MDMA neurotoxicity--a positron emission tomography study in the living baboon brain. Synapse, 1998. 29: p. 183-92.
237. Schmidt, C.J., Neurotoxicity of the psychedelic amphetamine, methylenedioxymethamphetamine. J Pharmacol Exp Ther, 1987. 240: p. 1-7.
238. Ricaurte, G.A., Forno, L.S., Wilson, M.A., DeLanney, L.E., Irwin, I., Molliver, M.E., and Langston, J.W., (+/-)3,4-Methylenedioxymethamphetamine selectively damages central serotonergic neurons in nonhuman primates. JAMA, 1988. 260: p. 51-5.
239. Ricaurte, G.A., Martello, A.L., Katz, J.L., and Martello, M.B., Lasting effects of (+-)-3,4-methylenedioxymethamphetamine (MDMA) on central serotonergic neurons in nonhuman primates: neurochemical observations. J Pharmacol Exp Ther, 1992. 261: p. 616-22.
240. Hatzidimitriou, G., McCann, U.D., and Ricaurte, G.A., Altered serotonin innervation patterns in the forebrain of monkeys treated with (+/-)3,4-methylenedioxymethamphetamine seven years previously: factors influencing abnormal recovery. J Neurosci, 1999. 19: p. 5096-107.
241. Commins, D.L., Vosmer, G., Virus, R.M., Woolverton, W.L., Schuster, C.R., and Seiden, L.S., Biochemical and histological evidence that methylenedioxymethylamphetamine (MDMA) is toxic to neurons in the rat brain. J Pharmacol Exp Ther, 1987. 241: p. 338-45.
242. Insel, T.R., Battaglia, G., Johannessen, J.N., Marra, S., and De Souza, E.B., 3,4-Methylenedioxymethamphetamine ("ecstasy") selectively destroys brain serotonin terminals in rhesus monkeys. J Pharmacol Exp Ther, 1989. 249: p. 713-20.
243. Miller, D.B. and O'Callaghan, J.P., The role of temperature, stress, and other factors in the neurotoxicity of the substituted amphetamines 3,4-methylenedioxymethamphetamine and fenfluramine. Mol Neurobiol, 1995. 11: p. 177-92.
244. Kish, S.J., How strong is the evidence that brain serotonin neurons are damaged in human users of ecstasy? Pharmacol Biochem Behav, 2002. 71: p. 845-55.
245. Reed, L.J., Winstock, A., Cleare, A.J., and McGuire, P., Toxic effect of MDMA on brain serotonin neurons. Lancet, 1999. 353: p. 1268.
246. Pallanti, S. and Mazzi, D., MDMA (Ecstasy) precipitation of panic disorder. Biol Psychiatry, 1992. 32: p. 91-5.
247. Henry, J.A., Ecstasy and the dance of death. BMJ, 1992. 305: p. 5-6. 248. Baumgarten, H.G. and Lachenmayer, L., Serotonin neurotoxins--past and present.
Neurotox Res, 2004. 6: p. 589-614. 249. Davidson, C., Gow, A.J., Lee, T.H., and Ellinwood, E.H., Methamphetamine
neurotoxicity: necrotic and apoptotic mechanisms and relevance to human abuse and treatment. Brain Res Brain Res Rev, 2001. 36: p. 1-22.
250. Seiden, L.S. and Sabol, K.E., Methamphetamine and methylenedioxymethamphetamine neurotoxicity: possible mechanisms of cell destruction. NIDA Res Monogr, 1996. 163: p. 251-76.
251. Brown, J., Edwards, M., McKone, E., and Ward, J., A long-term ecstasy-related change in visual perception. Psychopharmacology (Berl), 2007. 193: p. 437-46.
252. Wareing, M., Murphy, P.N., and Fisk, J.E., Visuospatial memory impairments in users of MDMA ('ecstasy'). Psychopharmacology (Berl), 2004. 173: p. 391-7.

__________________________________________ Referências Bibliográficas
297
253. Boot, B.P., McGregor, I.S., and Hall, W., MDMA (Ecstasy) neurotoxicity: assessing and communicating the risks. Lancet, 2000. 355: p. 1818-21.
254. Dafters, R.I., Duffy, F., O'Donnell, P.J., and Bouquet, C., Level of use of 3,4-methylenedioxymethamphetamine (MDMA or Ecstasy) in humans correlates with EEG power and coherence. Psychopharmacology (Berl), 1999. 145: p. 82-90.
255. Allen, R.P., McCann, U.D., and Ricaurte, G.A., Persistent effects of (+/- )3,4-methylenedioxymethamphetamine (MDMA, "ecstasy") on human sleep. Sleep, 1993. 16: p. 560-4.
256. McCann, U.D. and Ricaurte, G.A., Effects of (+/-) 3,4-methylenedioxymethamphetamine (MDMA) on sleep and circadian rhythms. ScientificWorldJournal, 2007. 7: p. 231-8.
257. McCann, U.D., Peterson, S.C., and Ricaurte, G.A., The effect of catecholamine depletion by alpha-methyl-para-tyrosine on measures of cognitive performance and sleep in abstinent MDMA users. Neuropsychopharmacology, 2007. 32: p. 1695-706.
258. Brody, S., Krause, C., Veit, R., and Rau, H., Cardiovascular autonomic dysregulation in users of MDMA ("Ecstasy"). Psychopharmacology (Berl), 1998. 136: p. 390-3.
259. Bingham, C., Beaman, M., Nicholls, A.J., and Anthony, P.P., Necrotizing renal vasculopathy resulting in chronic renal failure after ingestion of methamphetamine and 3,4-methylenedioxymethamphetamine ('ecstasy'). Nephrol Dial Transplant, 1998. 13: p. 2654-5.
260. McElhatton, P.R., Bateman, D.N., Evans, C., Pughe, K.R., and Thomas, S.H., Congenital anomalies after prenatal ecstasy exposure. Lancet, 1999. 354: p. 1441-2.
261. Plessinger, M.A., Prenatal exposure to amphetamines. Risks and adverse outcomes in pregnancy. Obstet Gynecol Clin North Am, 1998. 25: p. 119-38.
262. Ho, E., Karimi-Tabesh, L., and Koren, G., Characteristics of pregnant women who use ecstasy (3, 4-methylenedioxymethamphetamine). Neurotoxicol Teratol, 2001. 23: p. 561-7.
263. van Tonningen, M.R., Garbis, H., and Reuvers, M., Ecstasy exposure during pregnancy. Teratology, 1998. 58: p. 33.
264. Chesher, G., Designer drugs--the "whats" and the "whys". Med J Aust, 1990. 153: p. 157-61.
265. Beck, J. and Rosenbaum, M., Pursuit of Ecstasy: The MDMA Experience. 1994, Albany: State University of New York Press.
266. Winstock, A.R., Griffiths, P., and Stewart, D., Drugs and the dance music scene: a survey of current drug use patterns among a sample of dance music enthusiasts in the UK. Drug Alcohol Depend, 2001. 64: p. 9-17.
267. Greene, S.L., Dargan, P.I., O'connor, N., Jones, A.L., and Kerins, M., Multiple toxicity from 3,4-methylenedioxymethamphetamine ("ecstasy"). Am J Emerg Med, 2003. 21: p. 121-4.
268. OEDT Observatório Europeu da Droga e da Toxicodependência, Relatório Anual 2005: A evolução do fenómeno da droga na europa. 2005, Luxemburgo. pp. 45-54.
269. Schifano, F., Corkery, J., Deluca, P., Oyefeso, A., and Ghodse, A.H., Ecstasy (MDMA, MDA, MDEA, MBDB) consumption, seizures, related offences, prices, dosage levels and deaths in the UK (1994-2003). J Psychopharmacol, 2006. 20: p. 456-63.
270. Kalant, H., The pharmacology and toxicology of "ecstasy" (MDMA) and related drugs. CMAJ, 2001. 165: p. 917-28.
271. Arimany, J., Medallo, J., Pujol, A., Vingut, A., Borondo, J.C., and Valverde, J.L., Intentional overdose and death with 3,4-methylenedioxyethamphetamine (MDEA; "Eve"): case report. Am J Forensic Med Pathol, 1998. 19: p. 148-51.
272. Hooft, P.J. and van de Voorde, H.P., Reckless behaviour related to the use of 3,4-methylenedioxymethamphetamine (ecstasy): apropos of a fatal accident during car-surfing. Int J Legal Med, 1994. 106: p. 328-9.
273. Cadier, M.A. and Clarke, J.A., Ecstasy and Whizz at a rave resulting in a major burn plus complications. Burns, 1993. 19: p. 239-40.

Referências Bibliográficas __________________________________________
298
274. Mørland, J., Toxicity of drug abuse--amphetamine designer drugs (ecstasy): mental effects and consequences of single dose use. Toxicol Lett, 2000. 112-113: p. 147-52.
275. Lora-Tamayo, C., Tena, T., and Rodríguez, A., Amphetamine derivative related deaths. Forensic Sci Int, 1997. 85: p. 149-57.
276. Verschraagen, M., Maes, A., Ruiter, B., Bosman, I.J., Smink, B.E., and Lusthof, K.J., Verschraagen M, Maes A, Ruiter B, Bosman IJ, Smink BE, Lusthof KJ. Forensic Sci Int, 2007. 170: p. 163-70.
277. Brookhuis, K.A., de Waard, D., and Samyn, N., Effects of MDMA (ecstasy), and multiple drugs use on (simulated) driving performance and traffic safety. Psychopharmacology (Berl), 2004. 173: p. 440-5.
278. Kłys, M., Rojek, S., Woźniak, K., and Rzepecka-Woźniak, E., Fatality due to the use of a designer drug MDMA (Ecstasy). Leg Med (Tokyo), 2007. 9: p. 185-91.
279. Libiseller, K., Pavlic, M., Grubwieser, P., and Rabl, W., An announced suicide with ecstasy. Int J Legal Med, 2007. 121: p. 40-3.
280. Cheng, J.Y., Chan, D.T., and Mok, V.K., An epidemiological study on alcohol/drugs related fatal traffic crash cases of deceased drivers in Hong Kong between 1996 and 2000. Forensic Sci Int, 2005. 153: p. 196-201.
281. Libiseller, K., Pavlic, M., Grubwieser, P., and Rabl, W., Ecstasy--deadly risk even outside rave parties. Forensic Sci Int, 2005. 153: p. 227-30.
282. Dams, R., De Letter, E.A., Mortier, K.A., Cordonnier, J.A., Lambert, W.E., Piette, M.H., Van Calenbergh, S., and De Leenheer, A.P., Fatality due to combined use of the designer drugs MDMA and PMA: a distribution study. J Anal Toxicol, 2003. 27: p. 318-22.
283. Schifano, F., Oyefeso, A., Corkery, J., Cobain, K., Jambert-Gray, R., Martinotti, G., and Ghodse, A.H., Death rates from ecstasy (MDMA, MDA) and polydrug use in England and Wales 1996-2002. Hum Psychopharmacol, 2003. 18: p. 519-24.
284. Vuori, E., Henry, J.A., Ojanperä, I., Nieminen, R., Savolainen, T., Wahlsten, P., and Jäntti, M., Death following ingestion of MDMA (ecstasy) and moclobemide. Addiction, 2003. 98: p. 365-8.
285. De Letter, E.A., Clauwaert, K.M., Lambert, W.E., Van Bocxlaer, J.F., De Leenheer, A.P., and Piette, M.H., Distribution study of 3,4-methylenedioxymethamphetamine and 3,4-methylenedioxyamphetamine in a fatal overdose. J Anal Toxicol, 2002. 26: p. 113-8.
286. Gill, J.R., Hayes, J.A., deSouza, I.S., Marker, E., and Stajic, M., Ecstasy (MDMA) deaths in New York City: a case series and review of the literature. J Forensic Sci, 2002. 47: p. 121-6.
287. Fineschi, V., Centini, F., Mazzeo, E., and Turillazzi, E., Adam (MDMA) and Eve (MDEA) misuse: an immunohistochemical study on three fatal cases. Forensic Sci Int, 1999. 104: p. 65-74.
288. O'Connor, A., Cluroe, A., Couch, R., Galler, L., and Lawrence, J., Death from hyponatraemia-induced cerebral oedema associated with MDMA (“ecstasy”) use. N Z Med J, 1999. 112: p. 255-6.
289. Walubo, A. and Seger, D., Fatal multi-organ failure after suicidal overdose with MDMA, 'ecstasy': case report and review of the literature. Hum Exp Toxicol, 1999. 18: p. 119-25.
290. Byard, R.W., Gilbert, J., James, R., and Lokan, R.J., Amphetamine derivative fatalities in South Australia--is "Ecstasy" the culprit? Am J Forensic Med Pathol, 1998. 19: p. 261-5.
291. Mueller, P.D. and Korey, W.S., Death by "ecstasy": the serotonin syndrome? Ann Emerg Med, 1998. 32: p. 377-80.
292. Parr, M.J., Low, H.M., and Botterill, P., Hyponatraemia and death after "ecstasy" ingestion. Med J Aust, 1997. 166: p. 136-7.
293. Coore, J.R., A fatal trip with ecstasy: a case of 3,4-methylenedioxymethamphetamine/3,4- methylenedioxyamphetamine toxicity. J R Soc Med, 1996. 89: p. 51P-2P.

__________________________________________ Referências Bibliográficas
299
294. Cox, D.E. and Williams, K.R., "ADAM' or "EVE'?--a toxicological conundrum. Forensic Sci Int, 1996. 77: p. 101-8.
295. Crifasi, J. and Long, C., Traffic fatality related to the use of methylenedioxymethamphetamine. J Forensic Sci, 1996. 41: p. 1082-4.
296. Dar, K.J. and McBrien, M.E., MDMA induced hyperthermia: report of a fatality and review of current therapy. Intensive Care Med, 1996. 22: p. 995-6.
297. Fineschi, V. and Masti, A., Fatal poisoning by MDMA (ecstasy) and MDEA: a case report. Int J Legal Med, 1996. 108: p. 272-5.
298. Squier, M.V., Jalloh, S., Hilton-Jones, D., and Series, H., Death after ecstasy ingestion: neuropathological findings. J Neurol Neurosurg Psychiatry, 1995. 58: p. 756.
299. Forrest, A.R., Galloway, J.H., Marsh, I.D., Strachan, G.A., and Clark, J.C., A fatal overdose with 3,4-methylenedioxyamphetamine derivatives. Forensic Sci Int, 1994. 64: p. 57-9.
300. Watson, J.D., Ferguson, C., Hinds, C.J., Skinner, R., and Coakley, J.H., Exertional heat stroke induced by amphetamine analogues. Does dantrolene have a place? Anaesthesia, 1993. 48: p. 1057-60.
301. Campkin, N.T. and Davies, U.M., Another death from Ecstacy. J R Soc Med, 1992. 85: p. 61.
302. Rohrig, T.P. and Prouty, R.W., Tissue distribution of methylenedioxymethamphetamine. J Anal Toxicol, 1992. 16: p. 52-3.
303. Bellis, M.A., Hughes, K., Calafat, A., Juan, M., Ramon, A., Rodriguez, J.A., Mendes, F., Schnitzer, S., and Phillips-Howard, P., Sexual uses of alcohol and drugs and the associated health risks: A cross sectional study of young people in nine European cities. BMC Public Health, 2008. 8: p. 155.
304. Cole, J.C., Goudie, A.J., Field, M., Loverseed, A., Charlton, S., and Sumnall, H.R., The effects of perceived quality on the behavioural economics of alcohol, amphetamine, cannabis, cocaine, and ecstasy purchases. Drug Alcohol Depend, 2008. 94: p. 183-90.
305. Mendes, F.J. and Lomba, L., Positive and negative social representation of ecstasy among consumers in Coimbra (Portugal). Adicciones, 2008. 20: p. 81-8.
306. Pisetsky, E.M., Chao, Y.M., Dierker, L.C., May, A.M., and Striegel-Moore, R.H., Disordered Eating and Substance Use in High-School Students: Results from the Youth Risk Behavior Surveillance System. Int J Eat Disord, 2008. 41: p. 464-70.
307. Wilkins, C. and Sweetsur, P., Trends in population drug use in New Zealand: findings from national household surveying of drug use in 1998, 2001, 2003, and 2006. N Z Med J, 2008. 121: p. 61-71.
308. Chinet, L., Stéphan, P., Zobel, F., and Halfon, O., Party drug use in techno nights: a field survey among French-speaking Swiss attendees. Pharmacol Biochem Behav, 2007. 86: p. 284-9.
309. Dresen, S., Kempf, J., and Weinmann, W., Prevalence of gamma-hydroxybutyrate (GHB) in serum samples of amphetamine, metamphetamine and ecstasy impaired drivers. Forensic Sci Int, 2007. 173: p. 112-116.
310. Goudie, A.J., Sumnall, H.R., Field, M., Clayton, H., and Cole, J.C., The effects of price and perceived quality on the behavioural economics of alcohol, amphetamine, cannabis, cocaine, and ecstasy purchases. Drug Alcohol Depend, 2007. 89: p. 107-15.
311. Halkitis, P.N., Palamar, J.J., and Mukherjee, P.P., Poly-club-drug use among gay and bisexual men: a longitudinal analysis. Drug Alcohol Depend, 2007. 89: p. 153-60.
312. Medina, K.L. and Shear, P.K., Anxiety, depression, and behavioral symptoms of executive dysfunction in ecstasy users: contributions of polydrug use. Drug Alcohol Depend, 2007. 87: p. 303-11.
313. Yen, C., Hsu, S., and Cheng, C., Polysubstance use and its correlates in adolescent ecstasy users in Taiwan. Addict Behav, 2007. 32: p. 2286-91.
314. Breen, C., Degenhardt, L., Kinner, S., Bruno, R., Jenkinson, R., Matthews, A., and Newman, J., Alcohol use and risk taking among regular ecstasy users. Subst Use Misuse, 2006. 41(8): p. 1095-109.

Referências Bibliográficas __________________________________________
300
315. Chou, L., Ho, C., Chen, C., and Chen, W.J., Truancy and illicit drug use among adolescents surveyed via street outreach. Addict Behav, 2006. 31: p. 149-54.
316. Copeland, J., Dillon, P., and Gascoigne, M., Ecstasy and the concomitant use of pharmaceuticals. Addict Behav, 2006. 31: p. 367-70.
317. Fang, Y., Wang, Y., Shi, J., Liu, Z., and Lu, L., Recent trends in drug abuse in China. Acta Pharmacol Sin, 2006. 27: p. 140-4.
318. Halkitis, P.N. and Palamar, J.J., GHB use among gay and bisexual men. Addict Behav, 2006. 31: p. 2135-9.
319. Hopfer, C., Mendelson, B., Van Leeuwen, J.M., Kelly, S., and Hooks, S., Club drug use among youths in treatment for substance abuse. Am J Addict, 2006. 15: p. 94-9.
320. Parsons, J.T., Kelly, B.C., and Wells, B.E., Differences in club drug use between heterosexual and lesbian/bisexual females. Addict Behav, 2006. 31: p. 2344-49.
321. Pavarin, R.M., Substance use and related problems: a study on the abuse of recreational and not recreational drugs in Northern Italy. Ann Ist Super Sanità, 2006. 42: p. 477-484.
322. Wish, E.D., Fitzelle, D.B., O'Grady, K.E., Hsu, M.H., and Arria, A.M., Evidence for significant polydrug use among ecstasy-using college students. J Am Coll Health, 2006. 55: p. 99-104.
323. Wu, Z.H., Holzer, C.E., 3rd, Breitkopf, C.R., Grady, J.J., and Berenson, A.B., Patterns and perceptions of ecstasy use among young, low-income women. Addict Behav, 2006. 31: p. 676-85.
324. Wu, L., Schlenger, W.E., and Galvin, D.M., Concurrent Use of Methamphetamine, MDMA, LSD, Ketamine, GHB, and Flunitrazepam among American Youths. Drug Alcohol Depend, 2006. 84: p. 102-113.
325. Barrett, S.P., Gross, S.R., Garand, I., and Pihl, R.O., Patterns of Simultaneous Polysubstance Use in Canadian Rave Attendees. Subst Use Misuse, 2005. 40: p. 1525-37.
326. Clatts, M.C., Goldsamt, L.A., and Yi, H., Club Drug Use Among Young Men Who Have Sex with Men in NYC: A Preliminary Epidemiological Profile. Subst Use Misuse, 2005. 40: p. 1317-1330.
327. Fernández, M.I., Perrino, T., Collazo, J.B., Varga, L.M., Marsh, D., Hernandez, N., Rehbein, A., and Bowen, G.S., Surfing new territory: club-drug use and risky sex among Hispanic men who have sex with men recruited on the Internet. J Urban Health, 2005. 82: p. i79-88.
328. Goldsamt, L.A., O'Brien, J., Clatts, M.C., and McGuire, L.S., The relationship between club drug use and other drug use: a survey of New York City middle school students. Subst Use Misuse, 2005. 40: p. 1539-55.
329. Ompad, D.C., Galea, S., Fuller, C.M., Edwards, V., and Vlahov, D., Ecstasy Use Among Hispanic and Black Substance Users in New York City. Subst Use Misuse, 2005. 40: p. 1399-1407.
330. Crosby, R. and Diclemente, R.J., Use of recreational Viagra among men having sex with men. Sex Transm Infect, 2004. 80: p. 466-8.
331. Lora-Tamayo, C., Tena, T., Rodríguez, A., Moreno, D., Sancho, J.R., Enseñat, P., and Muela, F., The designer drug situation in Ibiza. Forensic Sci Int, 2004. 140: p. 195-206.
332. Scholey, A.B., Parrott, A.C., Buchanan, T., Heffernan, T.M., Ling, J., and Rodgers, J., Increased intensity of Ecstasy and polydrug usage in the more experienced recreational Ecstasy/MDMA users: a WWW study. Addict Behav, 2004. 29: p. 743-52.
333. Van Leeuwen, J.M., Hopfer, C., Hooks, S., White, R., Petersen, J., and Pirkopf, J., A snapshot of substance abuse among homeless and runaway youth in Denver, Colorado. J Community Health, 2004. 29: p. 217-29.
334. Turner, C., Russell, A., and Brown, W., Prevalence of illicit drug use in young Australian women, patterns of use and associated risk factors. Addiction, 2003. 98: p. 1419-26.
335. Fendrich, M., Wislar, J.S., Johnson, T.P., and Hubbell, A., A contextual profile of club drug use among adults in Chicago. Addiction, 2003. 98: p. 1693-703.

__________________________________________ Referências Bibliográficas
301
336. Gross, S.R., Barrett, S.P., Shestowsky, J.S., and Pihl, R.O., Ecstasy and drug consumption patterns: a Canadian rave population study. Can J Psychiatry, 2002. 47: p. 546-551.
337. NIDA National Institute on Drug Abuse, Monitoring the Future. National Survey Results on Drug Use, 1975–2001. NIH Publication 02-5106. Vol. Volume I: Secondary School Students. 2002, Bethesda, MD: U.S. Department of Health and Human Services, National Institutes of Health.
338. Strote, J., Lee, J.E., and Wechsler, H., Increasing MDMA use among college students: results of a national survey. J Adolesc Health, 2002. 30: p. 64-72.
339. Parrott, A.C., Milani, R.M., Parmar, R., and Turner, J.D., Recreational ecstasy/MDMA and other drug users from the UK and Italy: psychiatric symptoms and psychobiological problems. Psychopharmacology (Berl), 2001(159).
340. Riley, S.C., James, C., Gregory, D., Dingle, H., and Cadger, M., Patterns of recreational drug use at dance events in Edinburgh, Scotland. Addiction, 2001. 96: p. 1035-47.
341. Boys, A., Marsden, J., Griffiths, P., Fountain, J., Stillwell, G., and Strang, J., Substance use among young people: the relationship between perceived functions and intentions. Addiction, 1999. 94: p. 1043-50.
342. Topp, L., Hando, J., Dillon, P., Roche, A., and Solowij, N., Ecstasy use in Australia: patterns of use and associated harm. Drug Alcohol Depend, 1999. 55(Drug and Alcohol Dependence ): p. 105-15.
343. Christophersen, A.S., Amphetamine designer drugs - an overview and epidemiology. Toxicol Lett, 2000. 112-113: p. 127-31.
344. Lenton, S., Boys, A., and Norcross, K., Raves, drugs and experience: drug use by a sample of people who attend raves in Western Australia. Addiction, 1997. 92: p. 1327-37.
345. Boys, A., Lenton, S., and Norcross, K., Polydrug use at raves by a Western Australian sample. Drug Alcohol Rev, 1997. 16: p. 227-34.
346. Williamson, S., Gossop, M., Powis, B., Griffiths, P., Fountain, J., and Strang, J., Adverse effects of stimulant drugs in a community sample of drug users. Drug Alcohol Depend, 1997. 44: p. 87-94.
347. Miller, P.M. and Plant, M., Drinking, smoking, and illicit drug use among 15 and 16 year olds in the United Kingdom. BMJ, 1996. 313: p. 394-7.
348. Webb, E., Ashton, C.H., Kelly, P., and Kamali, F., Alcohol and drug use in UK university students. Lancet, 1996. 348: p. 922-5.
349. Forsyth, A.J.M., Ecstasy and illegal drug design: a new concept in drug use. International Journal of Drug Policy, 1995. 6: p. 193-209.
350. Cuomo, M.J., Dyment, P.G., and Gammino, V.M., Increasing use of "Ecstasy" (MDMA) and other hallucinogens on a college campus. J Am Coll Health, 1994. 42: p. 271-4.
351. Peroutka, S.J., Incidence of recreational use of 3,4-methylenedimethoxymethamphetamine (MDMA, "ecstasy") on an undergraduate campus. N Engl J Med, 1987. 317: p. 1542-3.
352. IDT Instituto da Droga e da Toxicodependência, Relatório Anual 2006: A Situação do País em Matéria de Drogas e Toxicodependências. 2006, Lisboa. pp. 81-82.
353. McCambridge, J., Mitcheson, L., Winstock, A., and Hunt, N., Five-year trends in patterns of drug use among people who use stimulants in dance contexts in the United Kingdom. Addiction, 2005. 100: p. 1140-9.
354. de Sola, S., Tarancón, T., Peña-Casanova, J., Espadaler, J.M., Langohr, K., Poudevida, S., Farré, M., Verdejo-García, A., and De la Torre, R., Auditory event-related potentials (P3) and cognitive performance in recreational ecstasy polydrug users: evidence from a 12-month longitudinal study. Psychopharmacology (Berl), 2008. DOI 10.1007/s00213-008-1217-5.
355. Reid, L.W., Elifson, K.W., and Sterk, C.E., Ecstasy and gateway drugs: initiating the use of ecstasy and other drugs. Ann Epidemiol, 2007. 17: p. 74-80.

Referências Bibliográficas __________________________________________
302
356. NDIC National Drug Intelligence Center, MDMA (Ecstasy): fast facts. Questions and answers. NDIC Product No. 2003-L0559-001, 2003.
357. Gamma, A., Jerome, L., Liechti, M.E., and Sumnall, H.R., Is ecstasy perceived to be safe? A critical survey. Drug Alcohol Depend, 2005. 77: p. 185-93.
358. Gowing, L., Henry-Edwards, S.M., Irvine, R.J., and Ali, R.L., Ecstasy: MDMA and other ring-substituted amphetamines. 2001, Geneva: World Health Organization.
359. Zhao, H., Brenneisen, R., Scholer, A., McNally, A.J., ElSohly, M.A., Murphy, T.P., and Salamone, S.J., Profiles of urine samples taken from Ecstasy users at Rave parties: analysis by immunoassays, HPLC, and GC-MS. J Anal Toxicol, 2001. 25: p. 258-69.
360. Morley-Fletcher, S., Bianchi, M., Gerra, G., and Laviola, G., Acute and carryover effects in mice of MDMA ("ecstasy") administration during periadolescence. Eur J Pharmacol, 2002. 448: p. 31-8.
361. Morley, K.C. and McGregor, I.S., (+/-)-3,4-methylenedioxymethamphetamine (MDMA, 'Ecstasy') increases social interaction in rats. Eur J Pharmacol, 2000. 408: p. 41-9.
362. Navarro, J.F. and Maldonado, E., Behavioral profile of 3,4-methylenedioxy-methamphetamine (MDMA) in agonistic encounters between male mice. Prog Neuropsychopharmacol Biol Psychiatry, 1999. 23: p. 327-34.
363. Nagilla, R., Newland, M.C., Snyder, J., and Bronson, M.E., Effect of once weekly treatment with 3,4-methylenedioxymethamphetamine on schedule-controlled behavior in rats. Eur J Pharmacol, 1998. 358: p. 1-8.
364. Banks, M.L., Sprague, J.E., Czoty, P.W., and Nader, M.A., Effects of ambient temperature on the relative reinforcing strength of MDMA using a choice procedure in monkeys. Psychopharmacology (Berl), 2006. 196: p. 63-70.
365. Cheng, J.Y.K., Chan, M.F., Chan, T.W., and Hung, M.Y., Impurity profiling of ecstasy tablets seized in Hong Kong by gas chromatography–mass spectrometry. Forensic Sci Int, 2006. 162: p. 87-94.
366. Tanner-Smith, E.E., Pharmacological content of tablets sold as "ecstasy": results from an online testing service. Drug Alcohol Depend, 2006. 83: p. 247-54.
367. Teng, S., Wu, S., Liu, C., Li, J., and Chien, C., Characteristics and trends of 3,4-methylenedioxymethamphetamine (MDMA) tablets found in Taiwan from 2002 to February 2005. Forensic Sci Int, 2006. 161: p. 202-8.
368. EACD Expert Advisory Committee on Drugs, Advice to the associate minister of health on MDMA. 2004, New Zealand Ministry of Health.
369. Cheng, W.C., Poon, N.L., and Chan, M.F., Chemical profiling of 3,4-methylenedioxymethamphetamine (MDMA) tablets seized in Hong Kong. J Forensic Sci, 2003. 48: p. 1249-59.
370. Cole, J.C., Bailey, M., Sumnall, H.R., Wagstaff, G.F., and King, L.A., The content of ecstasy tablets: implications for the study of their long-term effects. Addiction, 2002. 97: p. 1531-6.
371. Spruit, I.P., Monitoring synthetic drug markets, trends, and public health. Subst Use Misuse, 2001. 36: p. 23-47.
372. Baggott, M., Heifets, B., Jones, R.T., Mendelson, J., Sferios, E., and Zehnder, J., Chemical analysis of ecstasy pills. JAMA, 2000. 284: p. 2190.
373. Sherlock, K., Wolff, K., Hay, A.W., and Conner, M., Analysis of illicit ecstasy tablets: implications for clinical management in the accident and emergency department. J Accid Emerg Med, 1999. 16: p. 194-197.
374. Renfroe, C.L., MDMA on the street: Analysis Anonymous. J Psychoactive Drugs, 1986. 18: p. 363-9.
375. Dafters, R.I., Hyperthermia following MDMA administration in rats: effects of ambient temperature, water consumption, and chronic dosing. Physiol Behav, 1995. 58: p. 877-82.
376. Bellis, M.A., Hughes, K., and Lowey, H., Healthy nightclubs and recreational substance use. From a harm minimisation to a healthy settings approach. Addict Behav, 2002. 27: p. 1025-35.

__________________________________________ Referências Bibliográficas
303
377. Gordon, C.J., Watkinson, W.P., O'Callaghan, J.P., and Miller, D.B., Effects of 3,4-methylenedioxymethamphetamine on autonomic thermoregulatory responses of the rat. Pharmacol Biochem Behav, 1991. 38: p. 339-44.
378. Green, A.R., O'Shea, E., Saadat, K.S., Elliott, J.M., and Colado, M.I., Studies on the effect of MDMA ('ecstasy') on the body temperature of rats housed at different ambient room temperatures. Br J Pharmacol, 2005. 146: p. 306-12.
379. Sanchez, V., O'shea, E., Saadat, K.S., Elliott, J.M., Colado, M.I., and Green, A.R., Effect of repeated ('binge') dosing of MDMA to rats housed at normal and high temperature on neurotoxic damage to cerebral 5-HT and dopamine neurones. J Psychopharmacol, 2004. 18: p. 412-6.
380. Banks, M.L., Sprague, J.E., Kisor, D.F., Czoty, P.W., Nichols, D.E., and Nader, M.A., Ambient temperature effects on 3,4-methylenedioxymethamphetamine-induced thermodysregulation and pharmacokinetics in male monkeys. Drug Metab Dispos, 2007. 35: p. 1840-5.
381. Von Huben, S.N., Lay, C.C., Crean, R.D., Davis, S.A., Katner, S.N., and Taffe, M.A., Impact of ambient temperature on hyperthermia induced by (+/-)3,4-methylenedioxymethamphetamine in rhesus macaques. Neuropsychopharmacology, 2007. 32: p. 673-81.
382. Rusyniak, D.E., Ootsuka, Y., and Blessing, W.W., When administered to rats in a cold environment, 3,4-methylenedioxymethamphetamine reduces brown adipose tissue thermogenesis and increases tail blood flow: Effects of pretreatment with 5-HT(1A) and dopamine D(2) antagonists. Neuroscience, 2008.
383. Cornish, J.L., Shahnawaz, Z., Thompson, M.R., Wong, S., Morley, K.C., Hunt, G.E., and McGregor, I.S., Heat increases 3,4-methylenedioxymethamphetamine self-administration and social effects in rats. Eur J Pharmacol, 2003. 482: p. 339-41.
384. Di Chiara, G. and Imperato, A., Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A, 1988. 85: p. 5274-8.
385. O'Shea, E., Escobedo, I., Orio, L., Sanchez, V., Navarro, M., Green, A.R., and Colado, M.I., Elevation of ambient room temperature has differential effects on MDMA-induced 5-HT and dopamine release in striatum and nucleus accumbens of rats. Neuropsychopharmacology, 2005. 30: p. 1312-23.
386. Capela, J.P., Meisel, A., Abreu, A.R., Branco, P.S., Ferreira, L.M., Lobo, A.M., Remiao, F., Bastos, M.L., and Carvalho, F., Neurotoxicity of Ecstasy metabolites in rat cortical neurons, and influence of hyperthermia. J Pharmacol Exp Ther, 2006. 316(1): p. 53-61.
387. Carvalho, M., Carvalho, F., and Bastos, M.L., Is hyperthermia the triggering factor for hepatotoxicity induced by 3,4-methylenedioxymethamphetamine (ecstasy)? An in vitro study using freshly isolated mouse hepatocytes. Arch Toxicol, 2001. 74: p. 789-93.
388. Brown, P.L. and Kiyatkin, E.A., Brain hyperthermia induced by MDMA (ecstasy): modulation by environmental conditions. Eur J Neurosci, 2004. 20: p. 51-8.
389. Gesi, M., Ferrucci, M., Giusiani, M., Lenzi, P., Lazzeri, G., Alessandrì, M.G., Salvadorini, A., Fulceri, F., Pellegrini, A., Fornai, F., and Paparelli, A., Loud noise enhances nigrostriatal dopamine toxicity induced by MDMA in mice. Microsc Res Tech, 2004. 64: p. 297-303.
390. Gesi, M., Soldani, P., Lenzi, P., Ferrucci, M., Giusiani, A., Fornai, F., and Paparelli, A., Ecstasy during loud noise exposure induces dramatic ultrastructural changes in the heart. Pharmacol Toxicol, 2002. 91: p. 29-33.
391. Gesi, M., Lenzi, P., Soldani, P., Ferrucci, M., Giusiani, A., Fornai, F., and Paparelli, A., Morphological effects in the mouse myocardium after methylenedioxymethamphetamine administration combined with loud noise exposure. Anat Rec, 2002. 267: p. 37-46.
392. Johnson, E.A., O'Callaghan, J.P., and Miller, D.B., Brain concentrations of d-MDMA are increased after stress. Psychopharmacology (Berl), 2004. 173: p. 278-86.

Referências Bibliográficas __________________________________________
304
393. Piazza, P.V. and Le Moal, M.L., Pathophysiological basis of vulnerability to drug abuse: role of an interaction between stress, glucocorticoids, and dopaminergic neurons. Annu Rev Pharmacol Toxicol, 1996. 36: p. 359-78.
394. Parrott, A.C., MDMA (3,4-Methylenedioxymethamphetamine) or ecstasy: the neuropsychobiological implications of taking it at dances and raves. Neuropsychobiology, 2004. 50: p. 329-35.
395. Meyer, A., Mayerhofer, A., Kovar, K.A., and Schmidt, W.J., Rewarding effects of the optical isomers of 3,4-methylenedioxy-methylamphetamine ('Ecstasy') and 3,4-methylenedioxy-ethylamphetamine ('Eve') measured by conditioned place preference in rats. Neurosci Lett, 2002. 330: p. 280-4.
396. Vistisen, K., Loft, S., and Poulsen, H.E., Cytochrome P450 IA2 activity in man measured by caffeine metabolism: effect of smoking, broccoli and exercise. Adv Exp Med Biol, 1991. 283: p. 407-11.
397. Sotaniemi, E.A., Arranto, A.J., Pelkonen, O., and Pasanen, M., Age and cytochrome P450-linked drug metabolism in humans: an analysis of 226 subjects with equal histopathologic conditions. Clin Pharmacol Ther, 1997. 61: p. 331-9.
398. Parkinson, A., Mudra, D.R., Johnson, C., Dwyer, A., and Carroll, K.M., The effects of gender, age, ethnicity, and liver cirrhosis on cytochrome P450 enzyme activity in human liver microsomes and inducibility in cultured human hepatocytes. Toxicol Appl Pharmacol, 2004. 199: p. 193-209.
399. Marchesini, G., Bua, V., Brunori, A., Bianchi, G., Pisi, P., Fabbri, A., Zoli, M., and Pisi, E., Galactose elimination capacity and liver volume in aging man. Hepatology, 1988. 8: p. 1079-83.
400. Wynne, H.A., Cope, L.H., Mutch, E., Rawlins, M.D., Woodhouse, K.W., and James, O.F., The effect of age upon liver volume and apparent liver blood flow in healthy man. Hepatology, 1989. 9: p. 297-301.
401. Schmucker, D.L., Age-related changes in liver structure and function: Implications for disease ? Exp Gerontol, 2005. 40: p. 650-9.
402. Wiley, J.L., Evans, R.L., Grainger, D.B., and Nicholson, K.L., Age-dependent differences in sensitivity and sensitization to cannabinoids and 'club drugs' in male adolescent and adult rats. Addict Biol, 2007.
403. Broening, H.W., Bacon, L., and Slikker, W., Jr, Age modulates the long-term but not the acute effects of the serotonergic neurotoxicant 3,4-methylenedioxymethamphetamine. J Pharmacol Exp Ther, 1994. 271: p. 285-93.
404. Pirker, W., Asenbaum, S., Hauk, M., Kandlhofer, S., Tauscher, J., Willeit, M., Neumeister, A., Praschak-Rieder, N., Angelberger, P., and Brücke, T., Imaging serotonin and dopamine transporters with 123I-beta-CIT SPECT: binding kinetics and effects of normal aging. J Nucl Med, 2000. 41: p. 36-44.
405. Morley-Fletcher, S., Puopolo, M., Gentili, S., Gerra, G., Macchia, T., and Laviola, G., Prenatal stress affects 3,4-methylenedioxymethamphetamine pharmacokinetics and drug-induced motor alterations in adolescent female rats. Eur J Pharmacol, 2004. 489: p. 89-92.
406. Laviola, G., Adriani, W., Terranova, M.L., and Gerra, G., Psychobiological risk factors for vulnerability to psychostimulants in human adolescents and animal models. Neurosci Biobehav Rev, 1999. 23: p. 993-1010.
407. Spear, L.P., The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev, 2000. 24: p. 417-63.
408. Fone, K.C., Beckett, S.R., Topham, I.A., Swettenham, J., Ball, M., and Maddocks, L., Long-term changes in social interaction and reward following repeated MDMA administration to adolescent rats without accompanying serotonergic neurotoxicity. Psychopharmacology (Berl), 2002. 159: p. 437-44.
409. Relling, M.V., Lin, J.S., Ayers, G.D., and Evans, W.E., Racial and gender differences in N-acetyltransferase, xanthine oxidase, and CYP1A2 activities. Clin Pharmacol Ther, 1992. 52: p. 643-58.

__________________________________________ Referências Bibliográficas
305
410. Chang, L., Ernst, T., Strickland, T., and Mehringer, C.M., Gender effects on persistent cerebral metabolite changes in the frontal lobes of abstinent cocaine users. Am J Psychiatry, 1999. 156: p. 716-22.
411. Hommer, D., Momenan, R., Kaiser, E., and Rawlings, R., Evidence for a gender-related effect of alcoholism on brain volumes. Am J Psychiatry, 2001. 158: p. 198-204.
412. Kawas, C., Resnick, S., Morrison, A., Brookmeyer, R., Corrada, M., Zonderman, A., Bacal, C., Lingle, D.D., and Metter, E., A prospective study of estrogen replacement therapy and the risk of developing Alzheimer's disease: the Baltimore Longitudinal Study of Aging. Neurology, 1997. 48: p. 1517-21.
413. Swaab, D.F. and Fliers, E., A sexually dimorphic nucleus in the human brain. Science, 1985. 228: p. 1112-5.
414. Liechti, M.E., Gamma, A., and Vollenweider, F.X., Gender differences in the subjective effects of MDMA. Psychopharmacology (Berl), 2001. 154: p. 161-8.
415. McCann, U.D., Ridenour, A., Shaham, Y., and Ricaurte, G.A., Serotonin neurotoxicity after (+/-)3,4-methylenedioxymethamphetamine (MDMA; "Ecstasy"): a controlled study in humans. Neuropsychopharmacology, 1994. 10: p. 129-38.
416. Cadet, J.L., Ladenheim, B., Baum, I., Carlson, E., and Epstein, C., CuZn-superoxide dismutase (CuZnSOD) transgenic mice show resistance to the lethal effects of methylenedioxyamphetamine (MDA) and of methylenedioxymethamphetamine (MDMA). Brain Res, 1994. 655: p. 259.62.
417. Hoshi, R., Pratt, H., Mehta, S., Bond, A.J., and Curran, H.V., An investigation into the sub-acute effects of ecstasy on aggressive interpretative bias and aggressive mood - are there gender differences? J Psychopharmacol, 2006. 20: p. 291-301.
418. Farah, R. and Farah, R., Ecstasy (3,4-methylenedioxymethamphetamine)-induced inappropriate antidiuretic hormone secretion. Pediatr Emerg Care, 2008. 24: p. 615-7.
419. Wyeth, R.P., Mills, E.M., Ullman, A., Kenaston, M.A., Burwell, J., and Sprague, J.E., The hyperthermia mediated by 3,4-methylenedioxymethamphetamine (MDMA, Ecstasy) is sensitive to sex differences. Toxicol Appl Pharmacol, 2008: p. doi:10.1016/j.taap.2008.12.003.
420. Johnson, J.A., Pharmacogenetics: potential for individualized drug therapy through genetics. Trends Genet, 2003. 19: p. 660-6.
421. Meisel, C., Gerloff, T., Kirchheiner, J., Mrozikiewicz, P.M., Niewinski, P., Brockmöller, J., and Roots, I., Implications of pharmacogenetics for individualizing drug treatment and for study design. J Mol Med, 2003. 81: p. 154-67.
422. Ramamoorthy, Y., Yu, A.M., Suh, N., Haining, R.L., Tyndale, R.F., and Sellers, E.M., Reduced (+/-)-3,4-methylenedioxymethamphetamine ("Ecstasy") metabolism with cytochrome P450 2D6 inhibitors and pharmacogenetic variants in vitro. Biochem Pharmacol, 2002. 63: p. 2111-9.
423. Wolff, K., Hay, A.W., Sherlock, K., and Conner, M., Contents of "ecstasy". Lancet, 1995. 346(8982): p. 1100-1.
424. Ingelman-Sundberg, M., Pharmacogenetics of cytochrome P450 and its applications in drug therapy: the past, present and future. Trends Pharmacol Sci, 2004. 25: p. 193-200.
425. Johansson, I., Oscarson, M., Yue, Q.Y., Bertilsson, L., Sjöqvist, F., and Ingelman-Sundberg, M., Genetic analysis of the Chinese cytochrome P4502D locus: characterization of variant CYP2D6 genes present in subjects with diminished capacity for debrisoquine hydroxylation. Mol Pharmacol, 1994. 46: p. 452-9.
426. Ramamoorthy, Y., Tyndale, R.F., and Sellers, E.M., Cytochrome P450 2D6.1 and cytochrome P450 2D6.10 differ in catalytic activity for multiple substrates. Pharmacogenetics, 2001. 11: p. 477-87.
427. Masimirembwa, C., Persson, I., Bertilsson, L., Hasler, J., and Ingelman-Sundberg, M., A novel mutant variant of the CYP2D6 gene (CYP2D6*17) common in a black African population: association with diminished debrisoquine hydroxylase activity. Br J Clin Pharmacol, 1996. 42: p. 713-9.

Referências Bibliográficas __________________________________________
306
428. Shen, H., He, M.M., Liu, H., Wrighton, S.A., Wang, L., Guo, B., and Li, C., Comparative metabolic capabilities and inhibitory profiles of CYP2D6.1, CYP2D6.10, and CYP2D6.17. Drug Metab Dispos, 2007. 35: p. 1292-300.
429. Segura, M., Farré, M., Pichini, S., Peiró, A.M., Roset, P.N., Ramírez, A., Ortuño, J., Pacifici, R., Zuccaro, P., Segura, J., and de la Torre, R., Contribution of cytochrome P450 2D6 to 3,4-methylenedioxymethamphetamine disposition in humans: use of paroxetine as a metabolic inhibitor probe. Clin Pharmacokinet, 2005. 44: p. 649-60.
430. Carmo, H., Brulport, M., Hermes, M., Oesch, F., Silva, R., Ferreira, L.M., Branco, P.S., Boer, D., Remião, F., Carvalho, F., Schön, M.R., Krebsfaenger, N., Doehmer, J., Bastos, M.L., and Hengstler, J.G., Influence of CYP2D6 polymorphism on 3,4-methylenedioxymethamphetamine ('Ecstasy') cytotoxicity. Pharmacogenet Genomics, 2006. 16: p. 789-99.
431. Schwab, M., Seyringer, E., Brauer, R.B., Hellinger, A., and Griese, E.U., Fatal MDMA intoxication. Lancet, 1999. 353: p. 593-4.
432. Carvalho, M., Remião, F., Milhazes, N., Borges, F., Fernandes, E., Monteiro, M.C., Goncalves, M.J., Seabra, V., Amado, F., Carvalho, F., and Bastos, M.L., Metabolism is required for the expression of ecstasy-induced cardiotoxicity in vitro. Chem Res Toxicol, 2004. 17: p. 623-32.
433. Ingelman-Sundberg, M., Human drug metabolising cytochrome P450 enzymes: properties and polymorphisms. Naunyn Schmiedebergs Arch Pharmacol, 2004. 369: p. 89-104.
434. McKinnon, R.A., Burgess, W.M., Hall, P.M., Roberts-Thomson, S.J., Gonzalez, F.J., and McManus, M.E., Characterisation of CYP3A gene subfamily expression in human gastrointestinal tissues. Gut, 1995. 36: p. 259-67.
435. Shimada, T., Yamazaki, H., Mimura, M., Inui, Y., and Guengerich, F.P., Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther, 1994. 270: p. 414-23.
436. Weinshilboum, R.M., Otterness, D.M., and Szumlanski, C.L., Methylation pharmacogenetics: catechol O-methyltransferase, thiopurine methyltransferase, and histamine N-methyltransferase. Annu Rev Pharmacol Toxicol, 1999. 39: p. 19-52.
437. McLeod, H.L., Fang, L., Luo, X., Scott, E.P., and Evans, W.E., Ethnic differences in erythrocyte catechol-O-methyltransferase activity in black and white Americans. J Pharmacol Exp Ther, 1994. 270: p. 26-9.
438. Mattay, V.S., Goldberg, T.E., Fera, F., Hariri, A.R., Tessitore, A., Egan, M.F., Kolachana, B., Callicott, J.H., and Weinberger, D.R., Catechol O-methyltransferase val158-met genotype and individual variation in the brain response to amphetamine. Proc Natl Acad Sci U S A, 2003. 100: p. 6186-91.
439. Miners, J.O., McKinnon, R.A., and Mackenzie, P.I., Genetic polymorphisms of UDP-glucuronosyltransferases and their functional significance. Toxicology, 2002. 181-182: p. 453-6.
440. Felton, J.S. and Malfatti, M.A., What Do Diet-Induced Changes in Phase I and II Enzymes Tell Us about Prevention from Exposure to Heterocyclic Amines? J Nutr, 2006. 136: p. 2683S-84S.
441. Nagata, K. and Yamazoe, Y., Pharmacogenetics of sulfotransferase. Annu Rev Pharmacol Toxicol, 2000. 40: p. 159-76.
442. Masellis, M., Paterson, A.D., Badri, F., Lieberman, J.A., Meltzer, H.Y., Cavazzoni, P., and Kennedy, J.L., Genetic variation of 5-HT2A receptor and response to clozapine. Lancet, 1995. 346: p. 1108.
443. Cichon, S., Nöthen, M.M., Rietschel, M., and Propping, P., Pharmacogenetics of schizophrenia. Am J Med Genet, 2000. 97: p. 98-106.
444. Steen, V.M., Løvlie, R., MacEwan, T., and McCreadie, R.G., Dopamine D3-receptor gene variant and susceptibility to tardive dyskinesia in schizophrenic patients. Mol Psychiatry, 1997. 2: p. 139-45.

__________________________________________ Referências Bibliográficas
307
445. Cravchik, A. and Gejman, P.V., Functional analysis of the human D5 dopamine receptor missense and nonsense variants: differences in dopamine binding affinities. Pharmacogenetics, 1999. 9: p. 199-206.
446. Persico, A.M., Bird, G., Gabbay, F.H., and Uhl, G.R., D2 dopamine receptor gene TaqI A1 and B1 restriction fragment length polymorphisms: enhanced frequencies in psychostimulant-preferring polysubstance abusers. Biol Psychiatry, 1996. 40: p. 776-84.
447. Li, T., Chen, C.K., Hu, X., Ball, D., Lin, S.K., Chen, W., Sham, P.C., Loh, e.-W., Murray, R.M., and Collier, D.A., Association analysis of the DRD4 and COMT genes in methamphetamine abuse. Am J Med Genet B Neuropsychiatr Genet, 2004. 129B: p. 120-4.
448. Lesch, K.P. and Gutknecht, L., Pharmacogenetics of the serotonin transporter. Prog Neuropsychopharmacol Biol Psychiatry, 2005. 29: p. 1062-73.
449. Roiser, J.P., Cook, L.J., Cooper, J.D., Rubinsztein, D.C., and Sahakian, B.J., Association of a functional polymorphism in the serotonin transporter gene with abnormal emotional processing in ecstasy users. Am J Psychiatry, 2005. 162: p. 609-12.
450. Roiser, J.P., Rogers, R.D., Cook, L.J., and Sahakian, B.J., The effect of polymorphism at the serotonin transporter gene on decision-making, memory and executive function in ecstasy users and controls. Psychopharmacology (Berl), 2006. 188: p. 213-27.
451. Reneman, L., Schilt, T., de Win, M.M., Booij, J., Schmand, B., van den Brink, W., and Bakker, O., Memory function and serotonin transporter promoter gene polymorphism in ecstasy (MDMA) users. J Psychopharmacol, 2006. 20: p. 389-99.
452. Degenhardt, L., Drug use and risk behaviour among regular ecstasy users: Does sexuality make a difference? Culture, Health & Sexuality, 2005. 7: p. 599-614.
453. Skelton, M.R., Williams, M.T., and Vorhees, C.V., Developmental effects of 3,4-methylenedioxymethamphetamine: a review. Behav Pharmacol, 2008. 19: p. 91-111.
454. Draper, E.S., Rankin, J., Tonks, A.M., Abrams, K.R., Field, D.J., Clarke, M., and Kurinczuk, J.J., Recreational drug use: a major risk factor for gastroschisis? Am J Epidemiol, 2008. 167: p. 485-91.
455. Pichini, S., Puig, C., Zuccaro, P., Marchei, E., Pellegrini, M., Murillo, J., Vall, O., Pacifici, R., and García-Algar, O., Assessment of exposure to opiates and cocaine during pregnancy in a Mediterranean city: preliminary results of the "Meconium Project". Forensic Sci Int, 2005. 153: p. 59-65.
456. Maccari, S., Piazza, P.V., Kabbaj, M., Barbazanges, A., Simon, H., and Le Moal, M., Adoption reverses the long-term impairment in glucocorticoid feedback induced by prenatal stress. J Neurosci, 1995. 15: p. 110-6.
457. Vallée, M., Mayo, W., Maccari, S., Le Moal, M., and Simon, H., Long-term effects of prenatal stress and handling on metabolic parameters: relationship to corticosterone secretion response. Brain Res, 1996. 712: p. 287-92.
458. Morley-Fletcher, S., Rea, M., Maccari, S., and Laviola, G., Environmental enrichment during adolescence reverses the effects of prenatal stress on play behaviour and HPA axis reactivity in rats. Eur J Neurosci, 2003. 18: p. 3367-74.
459. Field, T., Diego, M., and Hernandez-Reif, M., Prenatal depression effects on the fetus and newborn: a review. Infant Behav Dev, 2006. 29: p. 445-55.
460. Zahradnik, H.P., Schäfer, W., Wetzka, B., and Breckwoldt, M., Hypertensive disorders in pregnancy. The role of eicosanoids. Eicosanoids, 1991. 4: p. 123-36.
461. Escher, G. and Mohaupt, M., Role of aldosterone availability in preeclampsia. Mol Aspects Med, 2007. 28: p. 245-54.
462. Wadelius, M., Darj, E., Frenne, G., and Rane, A., Induction of CYP2D6 in pregnancy. Clin Pharmacol Ther, 1997. 62: p. 400-7.
463. Casazza, J.P., Sohn, D.H., Park, K.S., and Song, B.J., Serum acetone and liver acetone monooxygenase activity in pregnant rats, fetuses, and neonates: reversible pretranslational reduction of cytochrome P450IIE1 (P450IIE1) during pregnancy. Arch Biochem Biophys, 1994. 309: p. 111-6.

Referências Bibliográficas __________________________________________
308
464. Anderson, G.D., Using pharmacokinetics to predict the effects of pregnancy and maternal-infant transfer of drugs during lactation. Expert Opin Drug Metab Toxicol, 2006. 2: p. 947-60.
465. Kubota, M., Ohno, J., Shiina, Y., and Suda, T., Vitamin D metabolism in pregnant rabbits: differences between the maternal and fetal response to administration of large amounts of vitamin D3. Endocrinology, 1982. 110: p. 1950-6.
466. Heikkinen, T., Ekblad, U., Palo, P., and Laine, K., Pharmacokinetics of fluoxetine and norfluoxetine in pregnancy and lactation. Clin Pharmacol Ther, 2003. 73: p. 330-7.
467. Banks, M.L., Czoty, P.W., Sprague, J.E., and Nader, M.A., Influence of thyroid hormones on 3,4-methylenedioxymethamphetamine-induced thermogenesis and reinforcing strength in monkeys. Behav Pharmacol, 2008. 19: p. 167-70.
468. Peterson, A.L., Gilman, T.L., Banks, M.L., and Sprague, J.E., Hypothyroidism alters striatal dopamine release mediated by 3,4-methylenedioxymethamphetamine (MDMA, ecstasy). Synapse, 2006. 59: p. 317-9.
469. Silva, J.E., Thermogenic mechanisms and their hormonal regulation. Physiol Rev, 2006. 86: p. 435-64.
470. Ginsberg, J., Diagnosis and management of Graves' disease. CMAJ, 2003. 168: p. 575-85.
471. Martin, T.L., Chiasson, D.A., and Kish, S.J., Does hyperthyroidism increase risk of death due to the ingestion of ecstasy? J Forensic Sci, 2007. 52: p. 951-3.
472. Kahraman, A., Miller, M., Gieseler, R.K., Gerken, G., Scolaro, M.J., and Canbay, A., Non-alcoholic fatty liver disease in HIV-positive patients predisposes for acute-on-chronic liver failure: two cases. Eur J Gastroenterol Hepatol, 2006. 18: p. 101-5.
473. Adedoyin, A., Arns, P.A., Richards, W.O., Wilkinson, G.R., and Branch, R.A., Selective effect of liver disease on the activities of specific metabolizing enzymes: investigation of cytochromes P450 2C19 and 2D6. Clin Pharmacol Ther, 1998. 64: p. 8-17.
474. Pichette, V. and Leblond, F.A., Drug metabolism in chronic renal failure. Curr Drug Metab, 2003. 4: p. 91-103.
475. Dreisbach, A.W. and Lertora, J.J., The effect of chronic renal failure on hepatic drug metabolism and drug disposition. Semin Dial, 2003. 16: p. 45-50.
476. Rege, B., Krieg, R., Gao, N., and Sarkar, M.A., Down-regulation of hepatic CYP3A in chronic renal insufficiency. Pharm Res, 2003. 20: p. 1600-6.
477. Kévorkian, J.P., Michel, C., Hofmann, U., Jacqz-Aigrain, E., Kroemer, H.K., Peraldi, M.N., Eichelbaum, M., Jaillon, P., and Funck-Brentano, C., Assessment of individual CYP2D6 activity in extensive metabolizers with renal failure: comparison of sparteine and dextromethorphan. Clin Pharmacol Ther, 1996. 59: p. 583-92.
478. Leblond, F.A., Giroux, L., Villeneuve, J.P., and Pichette, V., Decreased in vivo metabolism of drugs in chronic renal failure. Drug Metab Dispos, 2000. 28: p. 1317-20.
479. Leblond, F., Guévin, C., Demers, C., Pellerin, I., Gascon-Barré, M., and Pichette, V., Downregulation of hepatic cytochrome P450 in chronic renal failure. J Am Soc Nephrol, 2001. 12: p. 326-32.
480. Upreti, V.V. and Eddington, N.D., Fluoxetine pretreatment effects pharmacokinetics of 3,4-methylenedioxymethamphetamine (MDMA, ECSTASY) in rat. J Pharm Sci, 2008. 97: p. 1593-605.
481. Morgan, E.T., Regulation of cytochromes P450 during inflammation and infection. Drug Metab Rev, 1997. 29: p. 1129-88.
482. Kim, S.K. and Novak, R.F., The role of intracellular signaling in insulin-mediated regulation of drug metabolizing enzyme gene and protein expression. Pharmacol Ther, 2007. 113: p. 88-120.
483. Ioannides, C., Effect of diet and nutrition on the expression of cytochromes P450. Xenobiotica, 1999. 29: p. 109-54.
484. Barnett, C.R., Rudd, S., Flatt, P.R., and Ioannides, C., Sex differences in the diabetes-induced modulation of rat hepatic cytochrome P450 proteins. Biochem Pharmacol, 1993. 45: p. 313-9.

__________________________________________ Referências Bibliográficas
309
485. Corcoran, G.B. and Wong, B.K., Obesity as a risk factor in drug-induced organ injury: increased liver and kidney damage by acetaminophen in the obese overfed rat. J Pharmacol Exp Ther, 1987. 241: p. 921-7.
486. Bentley, J.B., Vaughan, R.W., Gandolfi, A.J., and Cork, R.C., Halothane biotransformation in obese and nonobese patients. Anesthesiology, 1982. 57: p. 94-7.
487. O'Shea, D., Davis, S.N., Kim, R.B., and Wilkinson, G.R., Effect of fasting and obesity in humans on the 6-hydroxylation of chlorzoxazone: a putative probe of CYP2E1 activity. Clin Pharmacol Ther, 1994. 56: p. 359-67.
488. Roe, A.L., Howard, G., Blouin, R., and Snawder, J.E., Characterization of cytochrome P450 and glutathione S-transferase activity and expression in male and female ob/ob mice. Int J Obes Relat Metab Disord, 1999. 23: p. 48-53.
489. Cheymol, G., Effects of obesity on pharmacokinetics implications for drug therapy. Clin Pharmacokinet, 2000. 39: p. 215-31.
490. Ketabi-Kiyanvash, N., Weiss, J., Haefeli, W.E., and Mikus, G., P-glycoprotein modulation by the designer drugs methylenedioxymethamphetamine, methylenedioxyethylamphetamine and paramethoxyamphetamine. Addict Biol, 2003. 8: p. 413-8.
491. Sternbach, H., The serotonin syndrome. Am J Psychiatry, 1991. 148: p. 705-13. 492. Teter, C.J. and Guthrie, S.K., A comprehensive review of MDMA and GHB: two
common club drugs. Pharmacotherapy, 2001. 21: p. 1486-513. 493. Breslau, K., The 'sextasy' craze. Clubland's dangerous party mix: Viagra and ecstasy.
Newsweek, 2002. 139: p. 30. 494. Shin, J.G., Kane, K., and Flockhart, D.A., Potent inhibition of CYP2D6 by haloperidol
metabolites: stereoselective inhibition by reduced haloperidol. Br J Clin Pharmacol, 2001. 51: p. 45-52.
495. Oerther, S. and Ahlenius, S., Atypical antipsychotics and dopamine D(1) receptor agonism: an in vivo experimental study using core temperature measurements in the rat. J Pharmacol Exp Ther, 2000. 292: p. 731-6.
496. Heh, C.W., Herrera, J., DeMet, E., Potkin, S., Costa, J., Sramek, J., Hazlett, E., and Buchsbaum, M.S., Neuroleptic-induced hypothermia associated with amelioration of psychosis in schizophrenia. Neuropsychopharmacology, 1988. 1: p. 149-56.
497. Blessing, W.W., Seaman, B., Pedersen, N.P., and Ootsuka, Y., Clozapine and olanzapine, by interactions with 5-HT receptors or by other mechanisms, could reverse potentially fatal hyperthermia and cutaneous vasoconstriction occurring in some humans after ingestion of MDMA. J Neurosci, 2003. 23: p. 6385-91.
498. Blessing, W.W., Zilm, A., and Ootsuka, Y., Clozapine reverses increased brown adipose tissue thermogenesis induced by 3,4-methylenedioxymethamphetamine and by cold exposure in conscious rats. Neuroscience, 2006. 141: p. 2067-73.
499. Kurling, S., Kankaanpää, A., and Seppälä, T., Sub-chronic nandrolone treatment modifies neurochemical and behavioral effects of amphetamine and 3,4-methylenedioxymethamphetamine (MDMA) in rats. Behav Brain Res, 2008. 189: p. 191-201.
500. Johnson, E.A., O'Callaghan, J.P., and Miller, D.B., Chronic treatment with supraphysiological levels of corticosterone enhances D-MDMA-induced dopaminergic neurotoxicity in the C57BL/6J female mouse. Brain Res, 2002. 933: p. 130-8.
501. Liechti, M.E., Saur, M.R., Gamma, A., Hell, D., and Vollenweider, F.X., Psychological and physiological effects of MDMA ("Ecstasy") after pretreatment with the 5-HT(2) antagonist ketanserin in healthy humans. Neuropsychopharmacology, 2000. 23: p. 396-404.
502. Wang, X., Baumann, M.H., Dersch, C.M., and Rothman, R.B., Restoration of 3,4-methylenedioxymethamphetamine-induced 5-HT depletion by the administration of L-5-hydroxytryptophan. Neuroscience, 2007. 148: p. 212-20.
503. Bomsien, S. and Skopp, G., An in vitro approach to potential methadone metabolic-inhibition interactions. Eur J Clin Pharmacol, 2007. 63: p. 821-7.

Referências Bibliográficas __________________________________________
310
504. Müller, W.E., Current St John's wort research from mode of action to clinical efficacy. Pharmacol Res, 2003. 47: p. 101-9.
505. Butterweck, V., Mechanism of action of St John's wort in depression : what is known? CNS Drugs, 2003. 17: p. 539-62.
506. Hellum, B.H. and Nilsen, O.G., The in vitro inhibitory potential of trade herbal products on human CYP2D6-mediated metabolism and the influence of ethanol. Basic Clin Pharmacol Toxicol, 2007. 101: p. 350-8.
507. Leuner, K., Kazanski, V., Müller, M., Essin, K., Henke, B., Gollasch, M., Harteneck, C., and Müller, W.E., Hyperforin--a key constituent of St. John's wort specifically activates TRPC6 channels. FASEB J, 2007. 21: p. 4101-11.
508. Lieberman, J.A., Mailman, R.B., Duncan, G., Sikich, L., Chakos, M., Nichols, D.E., and Kraus, J.E., Serotonergic basis of antipsychotic drug effects in schizophrenia. Biol Psychiatry, 1998. 44: p. 1099-117.
509. Pal, D. and Mitra, A.K., MDR- and CYP3A4-mediated drug-drug interactions. J Neuroimmune Pharmacol, 2006. 1: p. 323-39.
510. Gerber, J.G., Rhodes, R.J., and Gal, J., Stereoselective metabolism of methadone N-demethylation by cytochrome P4502B6 and 2C19. Chirality, 2004. 16: p. 36-44.
511. Gorman, A.L., Elliott, K.J., and Inturrisi, C.E., The d- and l-isomers of methadone bind to the non-competitive site on the N-methyl-D-aspartate (NMDA) receptor in rat forebrain and spinal cord. Neurosci Lett, 1997. 223: p. 5-8.
512. Armstrong, S.C. and Cozza, K.L., Pharmacokinetic drug interactions of morphine, codeine, and their derivatives: theory and clinical reality, Part II. Psychosomatics, 2003. 44: p. 515-20.
513. Yamreudeewong, W., DeBisschop, M., Martin, L.G., and Lower, D.L., Potentially significant drug interactions of class III antiarrhythmic drugs. Drug Saf, 2003. 26: p. 421-38.
514. Urbina, A. and Jones, K., Crystal methamphetamine, its analogues, and HIV infection: medical and psychiatric aspects of a new epidemic. Clin Infect Dis, 2004. 38: p. 890-4.
515. Hesse, L.M., von Moltke, L.L., Shader, R.I., and Greenblatt, D.J., Ritonavir, efavirenz, and nelfinavir inhibit CYP2B6 activity in vitro: potential drug interactions with bupropion. Drug Metab Dispos, 2001. 29: p. 100-2.
516. Dahl, M.L., Cytochrome p450 phenotyping/genotyping in patients receiving antipsychotics: useful aid to prescribing? Clin Pharmacokinet, 2002. 41: p. 453-70.
517. Stalker, D.J. and Jungbluth, G.L., Clinical pharmacokinetics of linezolid, a novel oxazolidinone antibacterial. Clin Pharmacokinet, 2003. 42: p. 1129-40.
518. Walsky, R.L., Astuccio, A.V., and Obach, R.S., Evaluation of 227 drugs for in vitro inhibition of cytochrome P450 2B6. J Clin Pharmacol, 2006. 46: p. 1426-38.
519. Hyland, R., Roe, E.G., Jones, B.C., and Smith, D.A., Identification of the cytochrome P450 enzymes involved in the N-demethylation of sildenafil. Br J Clin Pharmacol, 2001. 51: p. 239-48.
520. Hamelin, B.A., Bouayad, A., Méthot, J., Jobin, J., Desgagnés, P., Poirier, P., Allaire, J., Dumesnil, J., and Turgeon, J., Significant interaction between the nonprescription antihistamine diphenhydramine and the CYP2D6 substrate metoprolol in healthy men with high or low CYP2D6 activity. Clin Pharmacol Ther, 2000. 67: p. 466-77.
521. Sharma, A. and Hamelin, B.A., Classic histamine H1 receptor antagonists: a critical review of their metabolic and pharmacokinetic fate from a bird's eye view. Curr Drug Metab, 2003. 4: p. 105-29.
522. Siu, A. and Drachtman, R., Dextromethorphan: a review of N-methyl-d-aspartate receptor antagonist in the management of pain. CNS Drug Rev, 2007. 13: p. 96-106.
523. Shin, E.J., Nah, S.Y., Kim, W.K., Ko, K.H., Jhoo, W.K., Lim, Y.K., Cha, J.Y., Chen, C.F., and Kim, H.C., The dextromethorphan analog dimemorfan attenuates kainate-induced seizures via sigma1 receptor activation: comparison with the effects of dextromethorphan. Br J Pharmacol, 2005. 144: p. 908-18.

__________________________________________ Referências Bibliográficas
311
524. Hernandez, S.C., Bertolino, M., Xiao, Y., Pringle, K.E., Caruso, F.S., and Kellar, K.J., Dextromethorphan and its metabolite dextrorphan block alpha3beta4 neuronal nicotinic receptors. J Pharmacol Exp Ther, 2000. 293: p. 962-7.
525. Kamei, J., Mori, T., Igarashi, H., and Kasuya, Y., Serotonin release in nucleus of the solitary tract and its modulation by antitussive drugs. Res Commun Chem Pathol Pharmacol, 1992. 76: p. 371-4.
526. Liechti, M.E. and Vollenweider, F.X., The serotonin uptake inhibitor citalopram reduces acute cardiovascular and vegetative effects of 3,4-methylenedioxymethamphetamine ('Ecstasy') in healthy volunteers. J Psychopharmacol, 2000. 14: p. 269-74.
527. Liechti, M.E., Baumann, C., Gamma, A., and Vollenweider, F.X., Acute psychological effects of 3,4-methylenedioxymethamphetamine (MDMA, "Ecstasy") are attenuated by the serotonin uptake inhibitor citalopram. Neuropsychopharmacology, 2000. 22: p. 513-21.
528. Tancer, M. and Johanson, C.E., The effects of fluoxetine on the subjective and physiological effects of 3,4-methylenedioxymethamphetamine (MDMA) in humans. Psychopharmacology (Berl), 2007. 189: p. 565-73.
529. Lauerma, H., Wuorela, M., and Halme, M., Interaction of serotonin reuptake inhibitor and 3,4-methylenedioxymethamphetamine? Biol Psychiatry, 1998. 43: p. 929.
530. Sanchez, V., Camarero, J., Esteban, B., Peter, M.J., Green, A.R., and Colado, M.I., The mechanisms involved in the long-lasting neuroprotective effect of fluoxetine against MDMA ('ecstasy')-induced degeneration of 5-HT nerve endings in rat brain. Br J Pharmacol, 2001. 2001: p. 46-57.
531. Thompson, M.R., Li, K.M., Clemens, K.J., Gurtman, C.G., Hunt, G.E., Cornish, J.L., and McGregor, I.S., Chronic fluoxetine treatment partly attenuates the long-term anxiety and depressive symptoms induced by MDMA ('Ecstasy') in rats. Neuropsychopharmacology, 2004. 29: p. 694-704.
532. Farré, M., Abanades, S., Roset, P.N., Peiró, A.M., Torrens, M., O'Mathúna, B., Segura, M., and de la Torre, R., Pharmacological interaction between 3,4-methylenedioxymethamphetamine (ecstasy) and paroxetine: pharmacological effects and pharmacokinetics. J Pharmacol Exp Ther, 2007. 323: p. 954-62.
533. Hashimoto, K., Maeda, H., Hirai, K., and Goromaru, T., Drug effects on distribution of [3H]3,4-methylenedioxymethamphetamine in mice. Eur J Pharmacol, 1993. 228: p. 247-56.
534. Pacifici, R., Pichini, S., Zuccaro, P., Farré, M., Segura, M., Ortuño, J., Di Carlo, S., Bacosi, A., Roset, P.N., Segura, J., and de la Torre, R., Paroxetine inhibits acute effects of 3,4-methylenedioxymethamphetamine on the immune system in humans. J Pharmacol Exp Ther, 2004. 309: p. 285-92.
535. Kaskey, G.B., Possible interaction between an MAOI and "ecstasy". Am J Psychiatry, 1992. 149: p. 411-2.
536. Smilkstein, M.J., Smolinske, S.C., and Rumack, B.H., A case of MAO inhibitor/MDMA interaction: agony after ecstasy. J Toxicol Clin Toxicol, 1987. 25: p. 149-59.
537. Hewton, R., Salem, A., and Irvine, R.J., Potentiation of 3,4-methylenedioxymethamphetamine-induced 5-HT release in the rat substantia nigra by clorgyline, a monoamine oxidase A inhibitor. Clin Exp Pharmacol Physiol, 2007. 34: p. 1051-7.
538. Freezer, A., Salem, A., and Irvine, R.J., Effects of 3,4-methylenedioxymethamphetamine (MDMA, 'Ecstasy') and para-methoxyamphetamine on striatal 5-HT when co-administered with moclobemide. Brain Res, 2005. 1041: p. 48-55.
539. Stanley, N., Salem, A., and Irvine, R.J., The effects of co-administration of 3,4-methylenedioxymethamphetamine ("ecstasy") or para-methoxyamphetamine and moclobemide at elevated ambient temperatures on striatal 5-HT, body temperature and behavior in rats. Neuroscience, 2007. 146: p. 321-9.
540. Scorza, C., Silveira, R., Nichols, D.E., and Reyes-Parada, M., Effects of 5-HT-releasing agents on the extracellullar hippocampal 5-HT of rats. Implications for the

Referências Bibliográficas __________________________________________
312
development of novel antidepressants with a short onset of action. Neuropharmacology, 1999. 38: p. 1055-61.
541. Sprague, J.E. and Nichols, D.E., The monoamine oxidase-B inhibitor L-deprenyl protects against 3,4-methylenedioxymethamphetamine-induced lipid peroxidation and long-term serotonergic deficits. J Pharmacol Exp Ther, 1995. 273: p. 667-73.
542. Hrometz, S.L., Brown, A.W., Nichols, D.E., and Sprague, J.E., 3,4-methylenedioxymethamphetamine (MDMA, ecstasy)-mediated production of hydrogen peroxide in an in vitro model: the role of dopamine, the serotonin-reuptake transporter, and monoamine oxidase-B. Neurosci Lett, 2004. 367: p. 56-9.
543. Falk, E.M., Cook, V.J., Nichols, D.E., and Sprague, J.E., An antisense oligonucleotide targeted at MAO-B attenuates rat striatal serotonergic neurotoxicity induced by MDMA. Pharmacol Biochem Behav, 2002. 72: p. 617-22.
544. Fornai, F., Giorgi, F.S., Gesi, M., Chen, K., Alessrì, M.G., and Shih, J.C., Biochemical effects of the monoamine neurotoxins DSP-4 and MDMA in specific brain regions of MAO-B-deficient mice. Synapse, 2001. 39: p. 213-21.
545. Antoniou, T. and Tseng, A.L., Interactions between recreational drugs and antiretroviral agents. Ann Pharmacother, 2002. 36: p. 1598-613.
546. von Moltke, L.L., Durol, A.L., Duan, S.X., and Greenblatt, D.J., Potent mechanism-based inhibition of human CYP3A in vitro by amprenavir and ritonavir: comparison with ketoconazole. Eur J Clin Pharmacol, 2000. 56: p. 259-61.
547. von Moltke, L.L., Greenblatt, D.J., Duan, S.X., Daily, J.P., Harmatz, J.S., and Shader, R.I., Inhibition of desipramine hydroxylation (Cytochrome P450-2D6) in vitro by quinidine and by viral protease inhibitors: relation to drug interactions in vivo. J Pharm Sci, 1998. 87: p. 1184-9.
548. Harrington, R.D., Woodward, J.A., Hooton, T.M., and Horn, J.R., Life-threatening interactions between HIV-1 protease inhibitors and the illicit drugs MDMA and gamma-hydroxybutyrate. Arch Intern Med, 1999. 159: p. 2221-4.
549. Pritzker, D., Kanungo, A., Kilicarslan, T., Tyndale, R.F., and Sellers, E.M., Designer drugs that are potent inhibitors of CYP2D6. J Clin Psychopharmacol, 2002. 22: p. 330-2.
550. Chen, C., Biller, J., Willing, S.J., and Lopez, A.M., Ischemic stroke after using over the counter products containing ephedra. J Neurol Sci, 2004. 217: p. 55-60.
551. Thull, U. and Testa, B., Screening of unsubstituted cyclic compounds as inhibitors of monoamine oxidases. Biochem Pharmacol, 1994. 47: p. 2307-10.
552. Urbain, A., Marston, A., Grilo, L.S., Bravo, J., Purev, O., Purevsuren, B., Batsuren, D., Reist, M., Carrupt, P.A., and Hostettmann, K., Xanthones from Gentianella amarella ssp. acuta with acetylcholinesterase and monoamine oxidase inhibitory activities. J Nat Prod, 2008. 71: p. 895-7.
553. Kall, M.A. and Clausen, J., Dietary effect on mixed function P450 1A2 activity assayed by estimation of caffeine metabolism in man. Hum Exp Toxicol, 1995. 14: p. 801-7.
554. Moore, L.B., Goodwin, B., Jones, S.A., Wisely, G.B., Serabjit-Singh, C.J., Willson, T.M., Collins, J.L., and Kliewer, S.A., St. John's wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc Natl Acad Sci U S A, 2000. 97: p. 7500-2.
555. Thummel, K.E. and Wilkinson, G.R., In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol, 1998. 38: p. 389-430.
556. Manjgaladze, M., Chen, S., Frame, L.T., Seng, J.E., Duffy, P.H., Feuers, R.J., Hart, R.W., and Leakey, J.E., Effects of caloric restriction on rodent drug and carcinogen metabolizing enzymes: implications for mutagenesis and cancer. Mutat Res, 1993. 295: p. 201-22.
557. Mote, P.L., Grizzle, J.M., Walford, R.L., and Spindler, S.R., Influence of age and caloric restriction on expression of hepatic genes for xenobiotic and oxygen metabolizing enzymes in the mouse. J Gerontol, 1991. 46: p. B95-100.

__________________________________________ Referências Bibliográficas
313
558. Parrott, A.C., Is ecstasy MDMA? A review of the proportion of ecstasy tablets containing MDMA, their dosage levels, and the changing perceptions of purity. Psychopharmacology (Berl), 2004. 173: p. 234-41.
559. Stoové, M., Jenkinson, R., Cvetkovski, S., Matthews, S., Quinn, B., Dietze, P., and McElwee, P., The Victorian drug statistics handbook 2006: Patterns of drug use and related harm in Victoria. 2006, Melbourne, Victoria: Victorian Government Publishing Service.
560. Chen, C.K., Lin, S.K., Sham, P.C., Ball, D., Loh, E.W., Hsiao, C.C., Chiang, Y.L., Ree, S.C., Lee, C.H., and Murray, R.M., Pre-morbid characteristics and co-morbidity of methamphetamine users with and without psychosis. Psychol Med, 2003. 33: p. 1407-14.
561. Shulgin, A.T., The background and chemistry of MDMA. J Psychoactive Drugs, 1986. 18: p. 291-304.
562. Renton, R.J., Cowie, J.S., and Oon, M.C., A study of the precursors, intermediates and reaction by-products in the synthesis of 3,4-methylenedioxymethylamphetamine and its application to forensic drug analysis. Forensic Sci Int, 1993. 60: p. 189-202.
563. Marnell, T., Drug identification bible. 2002, Grand Junction: Amera-Chem Inc. 564. Schifano, F., Potential human neurotoxicity of MDMA ('Ecstasy'): subjective self-
reports, evidence from an Italian drug addiction centre and clinical case studies. Neuropsychobiology, 2000. 42: p. 25-33.
565. Swist, M., Wilamowski, J., and Parczewski, A., Determination of synthesis method of ecstasy based on the basic impurities. Forensic Sci Int, 2005. 152: p. 175-84.
566. Swist, M., Wilamowski, J., and Parczewski, A., Basic and neutral route specific impurities in MDMA prepared by different synthesis methods. Comparison of impurity profiles. Forensic Sci Int, 2005. 155: p. 100-11.
567. Crean, R.D., Davis, S.A., Von Huben, S.N., Lay, C.C., Katner, S.N., and Taffe, M.A., Effects of (+/-)3,4-methylenedioxymethamphetamine, (+/-)3,4-methylenedioxyamphetamine and methamphetamine on temperature and activity in rhesus macaques. Neuroscience, 2006. 142: p. 515-25.
568. Milhazes, N., Cunha-Oliveira, T., Martins, P., Garrido, J., Oliveira, C., Rego, A.C., and Borges, F., Synthesis and cytotoxic profile of 3,4-methylenedioxymethamphetamine ("ecstasy") and its metabolites on undifferentiated PC12 cells: A putative structure-toxicity relationship. Chem Res Toxicol, 2006. 19: p. 1294-304.
569. Phillips, D.H., DNA adducts derived from safrole, estragole and related compounds, and from benzene and its metabolites. IARC Sci Publ, 1994. 125: p. 131-40.
570. Moody, D.E., The effect of tetrahydrofuran on biological systems: does a hepatotoxic potential exist? Drug Chem Toxicol, 1991. 14: p. 319-42.
571. Lanigan, S., Final report on the safety assessment of Methyl Alcohol. Int J Toxicol, 2001. 20: p. 57-85.
572. Clagett-Carr, K., Sarosy, G., Plowman, J., Hoth, D.F., and Leyland-Jones, B., N-methylformamide: cytotoxic, radiosensitizer, or chemosensitizer. J Clin Oncol, 1988. 6: p. 906-18.
573. IARC International Agency for Research on Cancer, Pyridine. 2000. 77: p. 503. 574. Pifl, C., Nagy, G., Berényi, S., Kattinger, A., Reither, H., and Antus, S.,
Pharmacological characterization of ecstasy synthesis byproducts with recombinant human monoamine transporters. J Pharmacol Exp Ther, 2005. 314: p. 346-54.
575. Kalasinsky, K.S., Hugel, J., and Kish, S.J., Use of MDA (the "love drug") and methamphetamine in Toronto by unsuspecting users of ecstasy (MDMA). J Forensic Sci, 2004. 49: p. 1106-12.
576. Clemens, K.J., Van Nieuwenhuyzen, P.S., Li, K.M., Cornish, J.L., Hunt, G.E., and McGregor, I.S., MDMA ("ecstasy"), methamphetamine and their combination: long-term changes in social interaction and neurochemistry in the rat. Psychopharmacology (Berl), 2004. 173: p. 318-25.

Referências Bibliográficas __________________________________________
314
577. Clemens, K.J., Cornish, J.L., Hunt, G.E., and McGregor, I.S., Repeated weekly exposure to MDMA, methamphetamine or their combination: long-term behavioural and neurochemical effects in rats. Drug Alcohol Depend, 2007. 86: p. 183-90.
578. Kuwayama, K., Inoue, H., Kanamori, T., Tsujikawa, K., Miyaguchi, H., Iwata, Y., Miyauchi, S., Kamo, N., and Kishi, T., Interactions between 3,4-methylenedioxymethamphetamine, methamphetamine, ketamine, and caffeine in human intestinal Caco-2 cells and in oral administration to rats. Forensic Sci Int, 2007. 170: p. 183-8.
579. Lin, L.Y., Di Stefano, E.W., Schmitz, D.A., Hsu, L., Ellis, S.W., Lennard, M.S., Tucker, G.T., and Cho, A.K., Oxidation of methamphetamine and methylenedioxymethamphetamine by CYP2D6. Drug Metab Dispos, 1997. 25: p. 1059-64.
580. Itzhak, Y., Achat-Mendes, C.N., Ali, S.F., and Anderson, K.L., Long-lasting behavioral sensitization to psychostimulants following p-chloroamphetamine-induced neurotoxicity in mice. Neuropharmacology, 2004. 46: p. 74-84.
581. Wolff, K. and Winstock, A.R., Ketamine : from medicine to misuse. CNS Drugs, 2006. 20: p. 199-218.
582. Grant, I.S., Nimmo, W.S., and Clements, J.A., Pharmacokinetics and analgesic effects of i.m. and oral ketamine. Br J Anaesth, 1981. 53: p. 805-10.
583. Nishimura, M., Sato, K., Okada, T., Yoshiya, I., Schloss, P., Shimada, S., and Tohyama, M., Ketamine inhibits monoamine transporters expressed in human embryonic kidney 293 cells. Anesthesiology, 1998. 88: p. 768-74.
584. Winstock, A.R., Wolff, K., and Ramsey, J., Ecstasy pill testing: harm minimization gone too far? Addiction, 2001. 96: p. 1139-48.
585. Hijazi, Y. and Boulieu, R., Contribution of CYP3A4, CYP2B6, and CYP2C9 isoforms to N-demethylation of ketamine in human liver microsomes. Drug Metab Dispos, 2002. 30: p. 853-8.
586. Yanagihara, Y., Kariya, S., Ohtani, M., Uchino, K., Aoyama, T., Yamamura, Y., and Iga, T., Involvement of CYP2B6 in n-demethylation of ketamine in human liver microsomes. Drug Metab Dispos, 2001. 29: p. 887-90.
587. Hiratsuka, A., Chu, T.Y., Distefano, E.W., Lin, L.Y., Schmitz, D.A., and Cho, A.K., Inactivation of constitutive hepatic cytochromes P450 by phencyclidine in the rat. Drug Metab Dispos, 1995. 23: p. 201-6.
588. Yates, K.M., O'Connor, A., and Horsley, C.A., "Herbal Ecstasy": a case series of adverse reactions. N Z Med J, 2000. 113: p. 315-7.
589. Rothman, R.B., Baumann, M.H., Dersch, C.M., Romero, D.V., Rice, K.C., Carroll, F.I., and Partilla, J.S., Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse, 2001. 39: p. 32-41.
590. Soni, M.G., Carabin, I.G., Griffiths, J.C., and Burdock, G.A., Safety of ephedra: lessons learned. Toxicol Lett, 2004. 150: p. 97-110.
591. Daly, J.W. and Fredholm, B.B., Caffeine--an atypical drug of dependence. Drug Alcohol Depend, 1998. 51: p. 199-206.
592. Okada, M., Kiryu, K., Kawata, Y., Mizuno, K., Wada, K., Tasaki, H., and Kaneko, S., Determination of the effects of caffeine and carbamazepine on striatal dopamine release by in vivo microdialysis. Eur J Pharmacol, 1997. 321: p. 181-8.
593. Camarasa, J., Pubill, D., and Escubedo, E., Association of caffeine to MDMA does not increase antinociception but potentiates adverse effects of this recreational drug. Brain Res, 2006. 1111: p. 72-82.
594. McNamara, R., Kerans, A., O'Neill, B., and Harkin, A., Caffeine promotes hyperthermia and serotonergic loss following co-administration of the substituted amphetamines, MDMA ("Ecstasy") and MDA ("Love"). Neuropharmacology, 2006. 50: p. 69-80.

__________________________________________ Referências Bibliográficas
315
595. Green, A.R., O'shea, E., and Colado, M.I., A review of the mechanisms involved in the acute MDMA (ecstasy)-induced hyperthermic response. Eur J Pharmacol, 2004. 500: p. 3-13.
596. McNamara, R., Maginn, M., and Harkin, A., Caffeine induces a profound and persistent tachycardia in response to MDMA ("Ecstasy") administration. Eur J Pharmacol, 2007. 555: p. 194-8.
597. Derlet, R.W., Tseng, J.C., and Albertson, T.E., Potentiation of cocaine and d-amphetamine toxicity with caffeine. Am J Emerg Med, 1992. 10: p. 211-6.
598. Plaisance, K.I., Toxicities of drugs used in the management of fever. Clin Infect Dis, 2000. 31: p. S219-23.
599. Shin, E.J., Lee, P.H., Kim, H.J., Nabeshima, T., and Kim, H.C., Neuropsychotoxicity of abused drugs: potential of dextromethorphan and novel neuroprotective analogs of dextromethorphan with improved safety profiles in terms of abuse and neuroprotective effects. J Pharmacol Sci, 2008. 106: p. 22-7.
600. Milani, R.M., Parrott, A.C., Schifano, F., and Turner, J.J., Pattern of cannabis use in ecstasy polydrug users: moderate cannabis use may compensate for self-rated aggression and somatic symptoms. Hum Psychopharmacol, 2005. 20: p. 249-61.
601. Hernandez López, C., Interacciones farmacológicas entre la 3,4-metilenodioximetanfetamina (MDMA, Éxtasis) y el alcohol administrados a dosis únicas en humanos (tese de Doutoramento). in Departament de Farmacologia, de Terapèutica i Toxicologia. 2002, Universitat Autònoma de Barcelona: Barcelona.
602. Pedersen, W. and Skrondal, A., Ecstasy and new patterns of drug use: a normal population study. Addiction, 1999. 94: p. 1695-706.
603. Merrill, J., Ecstasy and neurodegeneration. Advice is that "less is more". BMJ, 1996. 313: p. 423.
604. Parrott, A.C., Human psychopharmacology of Ecstasy (MDMA): a review of 15 years of empirical research. Hum Psychopharmacol, 2001. 16: p. 557-577.
605. Reay, J.L., Hamilton, C., Kennedy, D.O., and Scholey, A.B., MDMA polydrug users show process-specific central executive impairments coupled with impaired social and emotional judgement processes. J Psychopharmacol, 2006. 20: p. 385-8.
606. Kopelman, M.D., Reed, L.J., Marsden, P., Mayes, A.R., Jaldow, E., Laing, H., and Isaac, C., Amnesic syndrome and severe ataxia following the recreational use of 3,4-methylene-dioxymethamphetamine (MDMA, 'ecstasy') and other substances. Neurocase, 2001. 7: p. 423-32.
607. Gerevich, J., Fatal combination of ecstasy and heroin. Psychosomatics, 2005. 46: p. 189.
608. Williams, H., Dratcu, L., Taylor, R., Roberts, M., and Oyefeso, A., "Saturday night fever": ecstasy related problems in a London accident and emergency department. J Accid Emerg Med, 1998. 15: p. 322-6.
609. Parrott, A.C., Milani, R.M., Gouzoulis-Mayfrank, E., and Daumann, J., Cannabis and Ecstasy/MDMA (3,4-methylenedioxymethamphetamine): an analysis of their neuropsychobiological interactions in recreational users. J Neural Transm, 2007. 114: p. 959-68.
610. Rodgers, J., Cognitive performance amongst recreational users of "ecstasy". Psychopharmacology (Berl), 2000. 151: p. 19-24.
611. Sala, M. and Braida, D., Endocannabinoids and 3,4-methylenedioxymethamphetamine (MDMA) interaction. Pharmacol Biochem Behav, 2005. 81: p. 407-16.
612. Gouzoulis-Mayfrank, E. and Daumann, J., The confounding problem of polydrug use in recreational ecstasy/MDMA users: a brief overview. J Psychopharmacol, 2006. 20(2): p. 188-93.
613. Parrott, A.C., MDMA in humans: factors which affect the neuropsychobiological profiles of recreational ecstasy users, the integrative role of bioenergetic stress. J Psychopharmacol, 2006. 20: p. 147-63.

Referências Bibliográficas __________________________________________
316
614. Dafters, R.I., Hoshi, R., and Talbot, A.C., Contribution of cannabis and MDMA ("ecstasy") to cognitive changes in long-term polydrug users. Psychopharmacology (Berl), 2004. 173: p. 405-10.
615. Fisk, J.E., Montgomery, C., Wareing, M., and Murphy, P.N., The effects of concurrent cannabis use among ecstasy users: neuroprotective or neurotoxic? Hum Psychopharmacol, 2006. 21: p. 355-66.
616. Lamers, C.T., Bechara, A., Rizzo, M., and Ramaekers, J.G., Cognitive function and mood in MDMA/THC users, THC users and non-drug using controls. J Psychopharmacol, 2006. 20: p. 302-11.
617. Daumann, J., Hensen, G., Thimm, B., Rezk, M., Till, B., and Gouzoulis-Mayfrank, E., Self-reported psychopathological symptoms in recreational ecstasy (MDMA) users are mainly associated with regular cannabis use: further evidence from a combined cross-sectional/longitudinal investigation. Psychopharmacology (Berl), 2004. 173: p. 398-404.
618. Young, J.M., McGregor, I.S., and Mallet, P.E., Co-administration of THC and MDMA ('ecstasy') synergistically disrupts memory in rats. Neuropsychopharmacology, 2005. 30: p. 1475-82.
619. Robledo, P., Trigo, J.M., Panayi, F., de la Torre, R., and Maldonado, R., Behavioural and neurochemical effects of combined MDMA and THC administration in mice. Psychopharmacology (Berl), 2007. 195: p. 255-64.
620. Chan, G.C., Hinds, T.R., Impey, S., and Storm, D.R., Hippocampal neurotoxicity of Delta9-tetrahydrocannabinol. J Neurosci, 1998. 18: p. 5322-32.
621. Morley, K.C., Li, K.M., Hunt, G.E., Mallet, P.E., and McGregor, I.S., Cannabinoids prevent the acute hyperthermia and partially protect against the 5-HT depleting effects of MDMA ("Ecstasy") in rats. Neuropharmacology, 2004. 46: p. 954-65.
622. Curran, H.V. and Travill, R.A., Mood and cognitive effects of +/-3,4-methylenedioxymethamphetamine (MDMA, 'ecstasy'): week-end 'high' followed by mid-week low. Addiction, 1997. 92: p. 821-31.
623. Parrott, A.C. and Lasky, J., Ecstasy (MDMA) effects upon mood and cognition: before, during and after a Saturday night dance. Psychopharmacology (Berl), 1998. 139: p. 261-8.
624. Pacifici, R., Zuccaro, P., Farré, M., Poudevida, S., Abanades, S., Pichini, S., Langohr, K., Segura, J., and de la Torre, R., Combined immunomodulating properties of 3,4-methylenedioxymethamphetamine (MDMA) and cannabis in humans. Addiction, 2007. 102: p. 931-6.
625. Fergusson, D.M. and Horwood, L.J., Early onset cannabis use and psychosocial adjustment in young adults. Addiction, 1997. 92: p. 279-96.
626. Wagner, F.A. and Anthony, J.C., Into the world of illegal drug use: exposure opportunity and other mechanisms linking the use of alcohol, tobacco, marijuana, and cocaine. Am J Epidemiol, 2002. 155: p. 918-25.
627. Matsunaga, T., Kishi, N., Higuchi, S., Watanabe, K., Ohshima, T., and Yamamoto, I., CYP3A4 is a major isoform responsible for oxidation of 7-hydroxy-Delta(8)-tetrahydrocannabinol to 7-oxo-delta(8)-tetrahydrocannabinol in human liver microsomes. Drug Metab Dispos, 2000. 28: p. 1291-6.
628. Hanson, G.R., Rau, K.S., and Fleckenstein, A.E., The methamphetamine experience: a NIDA partnership. Neuropharmacology, 2004. 47(Suppl 1): p. 92-100.
629. McCann, U.D. and Ricaurte, G.A., Amphetamine neurotoxicity: accomplishments and remaining challenges. Neurosci Biobehav Rev, 2004. 27: p. 821-6.
630. Partilla, J.S., Dempsey, A.G., Nagpal, A.S., Blough, B.E., Baumann, M.H., and Rothman, R.B., Interaction of amphetamines and related compounds at the vesicular monoamine transporter. J Pharmacol Exp Ther, 2006. 319: p. 237-46.
631. Morley, K.C., Cornish, J.L., Li, K.M., and McGregor, I.S., Preexposure to MDMA ("Ecstasy") delays acquisition but facilitates MDMA-induced reinstatement of amphetamine self-administration behavior in rats. Pharmacol Biochem Behav, 2004. 79: p. 331-42.

__________________________________________ Referências Bibliográficas
317
632. Clemens, K.J., Cornish, J.L., Hunt, G.E., and McGregor, I.S., Intravenous methamphetamine self-administration in rats: effects of intravenous or intraperitoneal MDMA co-administration. Pharmacol Biochem Behav, 2006. 85: p. 454-63.
633. Hamida, S.B., Plute, E., Cosquer, B., Kelche, C., Jones, B.C., and Cassel, J.C., Interactions between ethanol and cocaine, amphetamine, or MDMA in the rat: thermoregulatory and locomotor effects. Psychopharmacology (Berl), 2008. 197: p. 67-82.
634. Han, D.D. and Gu, H.H., Comparison of the monoamine transporters from human and mouse in their sensitivities to psychostimulant drugs. BMC Pharmacol, 2006. 6: p. 6.
635. Jaehne, E.J., Salem, A., and Irvine, R.J., Effects of 3,4-methylenedioxymethamphetamine and related amphetamines on autonomic and behavioral thermoregulation. Pharmacol Biochem Behav, 2005. 81: p. 485-96.
636. Daza-Losada, M., Rodríguez-Arias, M., Aguilar, M.A., and Miñarro, J., Effect of adolescent exposure to MDMA and cocaine on acquisition and reinstatement of morphine-induce CPP. Prog Neuropsychopharmacol Biol Psychiatry, 2008. 32: p. 701-9.
637. Fletcher, P.J., Robinson, S.R., and Slippoy, D.L., Pre-exposure to (+/-)3,4-methylenedioxy-methamphetamine (MDMA) facilitates acquisition of intravenous cocaine self-administration in rats. Neuropsychopharmacology, 2001. 25: p. 195-203.
638. Diller, A.J., Rocha, A., Cardon, A.L., Valles, R., Wellman, P.J., and Nation, J.R., The effects of concurrent administration of +/-3,4-methylenedioxymethamphetamine and cocaine on conditioned place preference in the adult male rat. Pharmacol Biochem Behav, 2007. 88: p. 165-70.
639. Laviola, G., Wood, R.D., Kuhn, C., Francis, R., and Spear, L.P., Cocaine sensitization in periadolescent and adult rats. J Pharmacol Exp Ther, 1995. 275: p. 345-57.
640. Aberg, M., Wade, D., Wall, E., and Izenwasser, S., Effect of MDMA (ecstasy) on activity and cocaine conditioned place preference in adult and adolescent rats. Neurotoxicol Teratol, 2007. 29: p. 37-46.
641. Morgan, A.E., Horan, B., Dewey, S.L., and Ashby, C.R., Jr, Repeated administration of 3,4-methylenedioxymethamphetamine augments cocaine's action on dopamine in the nucleus accumbens: a microdialysis study. Eur J Pharmacol, 1997. 331: p. R1-3.
642. Ansah, T.A., Wade, L.H., and Shockley, D.C., Changes in locomotor activity, core temperature, and heart rate in response to repeated cocaine administration. Physiol Behav, 1996. 60: p. 1261-7.
643. Gonzalez, L.P., Cocaine alters body temperature and behavioral thermoregulatory responses. Neuroreport, 1993. 4: p. 106-8.
644. Schiff, P.L., Ergot and its alkaloids. Am J Pharm Educ, 2006. 70: p. 98. 645. Schechter, M.D., 'Candyflipping': synergistic discriminative effect of LSD and MDMA.
Eur J Pharmacol, 1998. 341: p. 131-4. 646. Gill, J.R. and Stajíc, M., Ketamine in non-hospital and hospital deaths in New York
City. J Forensic Sci, 2000. 45: p. 655-8. 647. Uys, J.D. and Niesink, R.J., Pharmacological aspects of the combined use of 3,4-
methylenedioxymethamphetamine (MDMA, ecstasy) and gamma-hydroxybutyric acid (GHB): a review of the literature. Drug Alcohol Rev, 2005. 24: p. 359-68.
648. Freese, T.E., Miotto, K., and Reback, C.J., The effects and consequences of selected club drugs. J Subst Abuse Treat, 2002. 23: p. 151-6.
649. Nimmerrichter, A.A., Walter, H., Gutierrez-Lobos, K.E., and Lesch, O.M., Double-blind controlled trial of gamma-hydroxybutyrate and clomethiazole in the treatment of alcohol withdrawal. Alcohol Alcohol, 2002. 37: p. 67-73.
650. Addolorato, G., Balducci, G., Capristo, E., Attilia, M.L., Taggi, F., Gasbarrini, G., and Ceccanti, M., Gamma-hydroxybutyric acid (GHB) in the treatment of alcohol withdrawal syndrome: a randomized comparative study versus benzodiazepine. Alcohol Clin Exp Res, 1999. 23: p. 1596-604.
651. Drasbek, K.R., Christensen, J., and Jensen, K., Gamma-hydroxybutyrate--a drug of abuse. Acta Neurol Scand, 2006. 114: p. 145-56.

Referências Bibliográficas __________________________________________
318
652. Passie, T., Seifert, J., Schneider, U., and Emrich, H.M., The pharmacology of psilocybin. Addict Biol, 2002. 7: p. 357-64.
653. Benowitz, N.L., Clinical pharmacology of nicotine: implications for understanding, preventing, and treating tobacco addiction. Clin Pharmacol Ther, 2008. 83: p. 531-41.
654. Picciotto, M.R., Zoli, M., Rimondini, R., Léna, C., Marubio, L.M., Pich, E.M., Fuxe, K., and Changeux, J.P., Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature, 1998. 391: p. 173-7.
655. Bonomo, Y., Coffey, C., Wolfe, R., Lynskey, M., Bowes, G., and Patton, G., Adverse outcomes of alcohol use in adolescents. Addiction, 2001. 96: p. 1485-96.
656. Colfax, G., Vittinghoff, E., Husnik, M.J., McKirnan, D., Buchbinder, S., Koblin, B., Celum, C., Chesney, M., Huang, Y., Mayer, K., Bozeman, S., Judson, F.N., Bryant, K.J., and Coates, T.J., Substance use and sexual risk: a participant- and episode-level analysis among a cohort of men who have sex with men. Am J Epidemiol, 2004. 159: p. 1002-12.
657. Hamida, S.B., Bach, S., Plute, E., Jones, B.C., Kelche, C., and Cassel, J.C., Ethanol-ecstasy (MDMA) interactions in rats: preserved attenuation of hyperthermia and potentiation of hyperactivity by ethanol despite prior ethanol treatment. Pharmacol Biochem Behav, 2006. 84: p. 162-168.
658. Karch, S.B., Drug abuse handbook. Alcohol, ed. C.S. Martin. 1998, Washington DC: CRC Press.
659. Eckardt, M.J., File, S.E., Gessa, G.L., Grant, K.A., Guerri, C., Hoffman, P.L., Kalant, H., Koob, G.F., Li, T.K., and Tabakoff, B., Effects of moderate alcohol consumption on the central nervous system. Alcohol Clin Exp Res, 1998. 22: p. 998-1040.
660. Holford, N.H., Clinical pharmacokinetics of ethanol. Clin Pharmacokinet, 1987. 13: p. 273-92.
661. Zakhari, S., Overview: how is alcohol metabolized by the body? Alcohol Res Health, 2006. 29: p. 245-54.
662. Parkinson, A., Biotransformations of xenobiotics, in Casarett & Doull's Toxicology, the Basic Science of Poisons, (C.D. Klaassen, ed.), 1996, New York: McGraw-Hill.
663. Lieber, C.S., Relationships between nutrition, alcohol use, and liver disease. Alcohol Res Health, 2003. 27(3): p. 220-231.
664. Park, B.K., Pirmohamed, M., and Kitteringham, N.R., The role of cytochrome P450 enzymes in hepatic and extrahepatic human drug toxicity. Pharmacol Ther, 1995. 68: p. 385-424.
665. Jang, G.R. and Harris, R.Z., Drug interactions involving ethanol and alcoholic beverages. Expert Opin Drug Metab Toxicol, 2007. 3: p. 719-31.
666. Hu, Y., Ingelman-Sundberg, M., and Lindros, K.O., Induction mechanisms of cytochrome P450 2E1 in liver: interplay between ethanol treatment and starvation. Biochem Pharmacol, 1995. 50: p. 155-61.
667. Lieber, C.S., Mechanism of ethanol induced hepatic injury. Pharmacol Ther, 1990. 46: p. 1-41.
668. Lieber, C.S., Interaction of ethanol with drugs, hepatotoxic agents, carcinogens and vitamins. Alcohol Alcohol, 1990. 25: p. 157-76.
669. Djordjević, D., Nikolić, J., and Stefanović, V., Ethanol interactions with other cytochrome P450 substrates including drugs, xenobiotics, and carcinogens. Pathol Biol (Paris), 1998. 46: p. 760-70.
670. Maher, J.J., Exploring alcohol’s effects on liver function. Alcohol Res Health, 1997. 21: p. 5-12.
671. Handler, J.A. and Thurman, R.G., Redox interactions between catalase and alcohol dehydrogenase pathways of ethanol metabolism in the perfused rat liver. J Biol Chem, 1990. 265: p. 1510-5.
672. Eriksson, C.J.P., The role of acetaldehyde in the actions of alcohol (update 2000). Alcohol Clin Exp Res, 2001. 25(5): p. 15S-32S.

__________________________________________ Referências Bibliográficas
319
673. Sastre, J., Serviddio, G., Pereda, J., Minana, J.B., Arduini, A., Vendemiale, G., Poli, G., Pallardo, F.V., and Vina, J., Mitochondrial function in liver disease. Front Biosci, 2007. 12: p. 1200-9.
674. Das, S.K. and Vasudevan, D.M., Alcohol-induced oxidative stress. Life Sci, 2007. 81: p. 177–187.
675. Lu, Y. and Cederbaum, A.I., CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med, 2008. 44: p. 723–738.
676. Feierman, D.E., Melinkov, Z., and Nanji, A.A., Induction of CYP3A by ethanol in multiple in vitro and in vivo models. Alcohol Clin Exp Res, 2003. 27: p. 981-8.
677. Sinclair, J.F., McCaffrey, J., Sinclair, P.R., Bement, W.J., Lambrecht, L.K., Wood, S.G., Smith, E.L., Schenkman, J.B., Guzelian, P.S., and Park, S.S., Ethanol increases cytochromes P450IIE, IIB1/2, and IIIA in cultured rat hepatocytes. Arch Biochem Biophys, 1991. 284: p. 360-5.
678. de Waziers, I., Bouguet, J., Beaune, P.H., Gonzalez, F.J., Ketterer, B., and Barouki, R., Effects of ethanol, dexamethasone and RU 486 on expression of cytochromes P450 2B, 2E, 3A and glutathione transferase pi in a rat hepatoma cell line (Fao). Pharmacogenetics, 1992. 2: p. 12-8.
679. Louis, C.A., Wood, S.G., Kostrubsky, V., Sinclair, P.R., and Sinclair, J.F., Synergistic increases in rat hepatic cytochrome P450s by ethanol and isopentanol. J Pharmacol Exp Ther, 1994. 269: p. 838-45.
680. Eisenburg, B., Schütz, C.G., Kuss, J.P., Heumüller, M., and Soyka, M., Increased O-demethylation rate of dextromethorphan in alcohol dependent patients. A possible role of ethanol in changing cytochrome P450 2D6 activity. Eur Neuropsychopharmacology, 2000. 10: p. 379-380.
681. Busby, W.F., Jr, Ackermann, J.M., and Crespi, C.L., Effect of methanol, ethanol, dimethyl sulfoxide, and acetonitrile on in vitro activities of cDNA-expressed human cytochromes P-450. Drug Metab Dispos, 1999. 27: p. 246-9.
682. Ben Hamida, S., Plute, E., Bach, S., Lazarus, C., Tracqui, A., Kelche, C., de Vasconcelos, A.P., Jones, B.C., and Cassel, J.C., Ethanol-MDMA interactions in rats: the importance of interval between repeated treatments in biobehavioral tolerance and sensitization to the combination. Psychopharmacology (Berl), 2007. 192: p. 555-69.
683. Hernández-López, C., Farré, M., Roset, P.N., Menoyo, E., Pizarro, N., Ortuño, J., Torrens, M., Camí, J., and de La Torre, R., 3,4-Methylenedioxymethamphetamine (ecstasy) and alcohol interactions in humans: psychomotor performance, subjective effects, and pharmacokinetics. J Pharmacol Exp Ther, 2002. 300: p. 236-44.
684. Ramaekers, J.G. and Kuypers, K.P., Acute effects of 3,4-methylenedioxymethamphetamine (MDMA) on behavioral measures of impulsivity: alone and in combination with alcohol. Neuropsychopharmacology, 2006. 31: p. 1048-55.
685. Schmitt, G., Aderjan, R., Keller, T., and Wu, M., Ethyl glucuronide: an unusual ethanol metabolite in humans. Synthesis, analytical data, and determination in serum and urine. J Anal Toxicol, 1995. 19: p. 91-4.
686. Nevo, I. and Hamon, M., Neurotransmitter and neuromodulatory mechanisms involved in alcohol abuse and alcoholism. Neurochem Int, 1995. 26: p. 305-36.
687. Oscar-Berman, M., Shagrin, B., Evert, D.L., and Epstein, C., Impairments of brain and behavior. The neurological effects of alcohol. Alcohol Res Health, 1997. 21: p. 65-75.
688. Apte, M.V., Wilson, J.S., and Korsten, M.A., Alcohol-related pancreatic damage: mechanisms and treatment. Alcohol Health Res World, 1997. 21: p. 13-20.
689. Chastain, G., Alcohol, neurotransmitter systems, and behavior. J Gen Psychol, 2006. 133: p. 329-35.
690. Yan, Q.S., Reith, M.E., Jobe, P.C., and Dailey, J.W., Focal ethanol elevates extracellular dopamine and serotonin concentrations in the rat ventral tegmental area. Eur J Pharmacol, 1996. 301: p. 49-57.
691. Holdstock, L. and de Wit, H., Individual differences in the biphasic effects of ethanol. Alcohol Clin Exp Res, 1998. 22: p. 1903-11.

Referências Bibliográficas __________________________________________
320
692. Doty, P. and de Wit, H., Effect of setting on the reinforcing and subjective effects of ethanol in social drinkers. Psychopharmacology (Berl), 1995. 118: p. 19-27.
693. Schaffer, A. and Naranjo, C.A., Recommended drug treatment strategies for the alcoholic patient. Drugs, 1998. 56: p. 571-85.
694. Bilsky, E.J., Hui, Y.Z., Hubbell, C.L., and Reid, L.D., Methylenedioxymethamphetamine's capacity to establish place preferences and modify intake of an alcoholic beverage. Pharmacol Biochem Behav, 1990. 37: p. 633-8.
695. Rezvani, A.H., Garges, P.L., Miller, D.B., and Gordon, C.J., Attenuation of alcohol consumption by MDMA (ecstasy) in two strains of alcohol-preferring rats. Pharmacol Biochem Behav, 1992. 43: p. 103-10.
696. Grant, K.A., Colombo, G., and Gatto, G.J., Characterization of the ethanol-like discriminative stimulus effects of 5-HT receptor agonists as a function of ethanol training dose. Psychopharmacology (Berl), 1997. 133: p. 133-41.
697. Risinger, F.O., Bormann, N.M., and Oakes, R.A., Reduced sensitivity to ethanol reward, but not ethanol aversion, in mice lacking 5-HT1B receptors. Alcohol Clin Exp Res, 1996. 20: p. 1401-5.
698. Crabbe, J.C., Phillips, T.J., Feller, D.J., Hen, R., Wenger, C.D., Lessov, C.N., and Schafer, G.L., Elevated alcohol consumption in null mutant mice lacking 5-HT1B serotonin receptors. Nat Genet, 1996. 14: p. 98-101.
699. Pandey, S.C., Davis, J.M., and Pandey, G.N., Phosphoinositide system-linked serotonin receptor subtypes and their pharmacological properties and clinical correlates. J Psychiatry Neurosci, 1995. 20: p. 215-25.
700. Pandey, S.C., Piano, M.R., Schwertz, D.W., Davis, J.M., and Pandey, G.N., Effect of ethanol administration and withdrawal on serotonin receptor subtypes and receptor-mediated phosphoinositide hydrolysis in rat brain. Alcohol Clin Exp Res, 1992. 16: p. 1110-6.
701. Yoshimoto, K., Ueda, S., Kato, B., Takeuchi, Y., Kawai, Y., Noritake, K., and Yasuhara, M., Alcohol enhances characteristic releases of dopamine and serotonin in the central nucleus of the amygdala. Neurochem Int, 2000. 37: p. 369-76.
702. Yim, H.J., Schallert, T., Randall, P.K., Bungay, P.M., and Gonzales, R.A., Effect of ethanol on extracellular dopamine in rat striatum by direct perfusion with microdialysis. J Neurochem, 1997. 68: p. 1527-33.
703. Campbell, A.D., Kohl, R.R., and McBride, W.J., Serotonin-3 receptor and ethanol-stimulated somatodendritic dopamine release. Alcohol, 1996. 13: p. 569-74.
704. Samson, H.H., Hodge, C.W., Erickson, H.L., Niehus, J.S., Gerhardt, G.A., Kalivas, P.W., and Floyd, E.A., The effects of local application of ethanol in the n. accumbens on dopamine overflow and clearance. Alcohol, 1997. 14: p. 485-92.
705. Wang, Y., Palmer, M.R., Cline, E.J., and Gerhardt, G.A., Effects of ethanol on striatal dopamine overflow and clearance: an in vivo electrochemical study. Alcohol, 1997. 14: p. 593-601.
706. Rodd, Z.A., Bell, R.L., Zhang, Y., Goldstein, A., Zaffaroni, A., McBride, W.J., and Li, T.K., Salsolinol produces reinforcing effects in the nucleus accumbens shell of alcohol-preferring (P) rats. Alcohol Clin Exp Res, 2003. 27: p. 440-9.
707. Huttunen, P., Sämpi, M., and Myllylä, R., Ethanol-induced hypothermia and thermogenesis of brown adipose tissue in the rat. Alcohol, 1998. 15: p. 315-8.
708. Deng, X.S. and Deitrich, R.A., Ethanol metabolism and effects: nitric oxide and its interaction. Curr Clin Pharmacol, 2007. 2: p. 145-53.
709. Altura, B.M. and Altura, B.T., Peripheral and cerebrovascular actions of ethanol, acetaldehyde, and acetate: relationship to divalent cations. Alcohol Clin Exp Res, 1987. 11: p. 99-111.
710. Khanna, J.M., Chau, A., and Shah, G., Characterization of the Phenomenon of rapid tolerance to ethanol. Alcohol, 1996. 13: p. 621-8.
711. Cassel, J.C., Jeltsch, H., Koenig, J., and Jones, B.C., Locomotor and pyretic effects of MDMA-ethanol associations in rats. Alcohol, 2004. 34: p. 285-9.

__________________________________________ Referências Bibliográficas
321
712. Cassel, J.C., Riegert, C., Rutz, S., Koenig, J., Rothmaier, K., Cosquer, B., Lazarus, C., Birthelmer, A., Jeltsch, H., Jones, B.C., and Jackisch, R., Ethanol, 3,4-methylenedioxymethamphetamine (ecstasy) and their combination: long-term behavioral, neurochemical and neuropharmacological effects in the rat. Neuropsychopharmacology, 2005. 30: p. 1870-82.
713. Cassel, J.C., Ben Hamida, S., and Jones, B.C., Attenuation of MDMA-induced hyperthermia by ethanol in rats depends on ambient temperature. Eur J Pharmacol, 2007. 571: p. 152-5.
714. NIAAA National Institute on Alcohol Abuse and Alcoholism, Health risks and benefits of alcohol consumption. Alcohol Res Health, 2000. 24: p. 5-11.
715. Riegert, C., Wedekind, F., Hamida, S.B., Rutz, S., Rothmaier, A.K., Jones, B.C., Cassel, J.C., and Jackisch, R., Effects of ethanol and 3,4-methylenedioxymethamphetamine (MDMA) alone or in combination on spontaneous and evoked overflow of dopamine, serotonin and acetylcholine in striatal slices of the rat brain. Int J Neuropsychopharmacol, 2008: p. 1-21.
716. Izco, M., Orio, L., O'Shea, E., and Colado, M.I., Binge ethanol administration enhances the MDMA-induced long-term 5-HT neurotoxicity in rat brain. Psychopharmacology (Berl), 2007. 189: p. 459-70.
717. Colado, M.I., Granados, R., O'Shea, E., Esteban, B., and Green, A.R., Role of hyperthermia in the protective action of clomethiazole against MDMA ('ecstasy')-induced neurodegeneration, comparison with the novel NMDA channel blocker AR-R15896AR. Br J Pharmacol, 1998. 124: p. 479-84.
718. O'Shea, E., Granados, R., Esteban, B., Colado, M.I., and Green, A.R., The relationship between the degree of neurodegeneration of rat brain 5-HT nerve terminals and the dose and frequency of administration of MDMA ('ecstasy'). Neuropharmacology, 1998. 37: p. 919-26.
719. Kuypers, K.P., Samyn, N., and Ramaekers, J.G., MDMA and alcohol effects, combined and alone, on objective and subjective measures of actual driving performance and psychomotor function. Psychopharmacology (Berl), 2006. 187: p. 467-75.
720. Carmen del Río, M., Gómez, J., Sancho, M., and Alvarez, F.J., Alcohol, illicit drugs and medicinal drugs in fatally injured drivers in Spain between 1991 and 2000. Forensic Sci Int, 2002. 127: p. 63-70.
721. Inder, W.J., Joyce, P.R., Wells, J.E., Evans, M.J., Ellis, M.J., Mattioli, L., and Donald, R.A., The acute effects of oral ethanol on the hypothalamic-pituitary-adrenal axis in normal human subjects. Clin Endocrinol (Oxf), 1995. 42: p. 65-71.
722. Emanuele, N. and Emanuele, M.A., The endocrine system. Alcohol alters critical hormonal balance. Alcohol Res Health, 1997. 21: p. 53-64.
723. Gianoulakis, C., Guillaume, P., Thavundayil, J., and Gutkowska, J., Increased plasma atrial natriuretic peptide after ingestion of low doses of ethanol in humans. Alcohol Clin Exp Res, 1997. 21: p. 162-70.
724. Farré, M., de la Torre, R., González, M.L., Terán, M.T., Roset, P.N., Menoyo, E., and Camí, J., Cocaine and alcohol interactions in humans: neuroendocrine effects and cocaethylene metabolism. J Pharmacol Exp Ther, 1997. 283: p. 164-76.
725. Schuckit, M.A., Gold, E., and Risch, C., Serum prolactin levels in sons of alcoholics and control subjects. Am J Psychiatry, 1987. 144: p. 854-9.
726. Zakhari, S., Alcohol and the cardiovascular system. Molecular mechanisms for beneficial and harmful action. Alcohol Res Health, 1997. 21: p. 21-9.
727. Kawano, Y., Abe, H., Kojima, S., Yoshimi, H., Sanai, T., Kimura, G., Matsuoka, H., Takishita, S., and Omae, T., Different effects of alcohol and salt on 24-hour blood pressure and heart rate in hypertensive patients. Hypertens Res, 1996. 19: p. 255-61.
728. Pacifici, R., Zuccaro, P., Hernandez López, C., Pichini, S., Di Carlo, S., Farré, M., Roset, P.N., Ortuño, J., Segura, J., and Torre, R.L., Acute effects of 3,4-methylenedioxymethamphetamine alone and in combination with ethanol on the immune system in humans. J Pharmacol Exp Ther, 2001. 296: p. 207-15.

Referências Bibliográficas __________________________________________
322
729. Bode, C. and Bode, J.C., Activation of the innate immune system and alcoholic liver disease: effects of ethanol per se or enhanced intestinal translocation of bacterial toxins induced by ethanol? Alcohol Clin Exp Res, 2005. 29: p. 166S-71S.
730. Zima, T. and Kalousova, M., Oxidative stress and signal tranduction pathways in alcoholic liver disease. Alcohol Clin Exp Res, 2005. 29(11 Suppl): p. 110S-115S.
731. Epstein, M., Alcohol’s impact on kidney function. Alcohol Res Health, 1997. 21: p. 84-93.
732. Seitz, H.K. and Becker, P., Alcohol metabolism and cancer risk. Alcohol Res Health, 2007. 30: p. 38-47.
733. Schifano, F., A bitter pill. Overview of ecstasy (MDMA, MDA) related fatalities. Psychopharmacology (Berl), 2004. 173: p. 242-8.
734. Schuckit, M.A., Alcoholism: An Introduction. Drug and Alcohol Abuse: A Clinical Guide to Diagnosis and Treatment, ed. M.A. Schuckit. 2006, San Diego: Springer Science+Business Media, Inc.
735. Grobin, A.C., Matthews, D.B., Devaud, L.L., and Morrow, A.L., The role of GABAA receptors in the acute and chronic effects of ethanol. Psychopharmacology, 1998. 139: p. 2-19.
736. Gemma, S., Vichi, S., and Testai, E., Individual susceptibility and alcohol effects:biochemical and genetic aspects. Ann Ist Super Sanita, 2006. 42: p. 8-16.
737. Dumont, G.J., Wezenberg, E., Valkenberg, M.M., de Jong, C.A., Buitelaar, J.K., van Gerven, J.M., and Verkes, R.J., Acute neuropsychological effects of MDMA and ethanol (co-)administration in healthy volunteers. Psychopharmacology (Berl), 2008. 197: p. 465-74.
738. Meehan, S.M., Gordon, T.L., and Schechter, M.D., MDMA (Ecstasy) substitutes for the ethanol discriminative cue in HAD but not LAD rats. Alcohol, 1995. 12: p. 569-72.
739. Izco, M., Marchant, I., Escobedo, I., Peraile, I., Delgado, M., Higuera-Matas, A., Olias, O., Ambrosio, E., O'Shea, E., and Colado, M.I., Mice with decreased cerebral dopamine function following a neurotoxic dose of MDMA (3,4-methylenedioxymethamphetamine, "Ecstasy") exhibit increased ethanol consumption and preference. J Pharmacol Exp Ther, 2007. 322: p. 1003-12.
740. Quinton, M.S. and Yamamoto, B.K., Causes and consequences of methamphetamine and MDMA toxicity. AAPS J, 2006. 8: p. E337-47.
741. Morimoto, M., Hagbjörk, A.L., Nanji, A.A., Ingelman-Sundberg, M., Lindros, K.O., Fu, P.C., Albano, E., and French, S.W., Role of cytochrome P4502E1 in alcoholic liver disease pathogenesis. Alcohol, 1993. 10: p. 459-64.
742. Cunningham, C.C., Coleman, W.B., and Spach, P.I., The effects of chronic ethanol consumption on hepatic mitochondrial energy metabolism. Alcohol Alcohol, 1990. 25: p. 127-36.
743. Colell, A., García-Ruiz, C., Miranda, M., Ardite, E., Marí, M., Morales, A., Corrales, F., Kaplowitz, N., and Fernández-Checa, J.C., Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology, 1998. 115: p. 1541-51.
744. Pirkkala, L., Nykänen, P., and Sistonen, L., Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J, 2001. 15: p. 1118-31.
745. Sapareto, S.A., Thermal isoeffect dose: addressing the problem of thermotolerance. Int. J. Hyperthermia, 1987. 3: p. 297-305.
746. Lepock, J.R., Cellular effects of hyperthermia: relevance to the minimum dose for thermal damage. Int J Hyperthermia, 2003. 19: p. 252-66.
747. Park, H.G., Han, S.I., Oh, S.Y., and Kang, H.S., Cellular responses to mild heat stress. Cell Mol Life Sci, 2005. 62: p. 10-23.
748. Gabai, V.L. and Sherman, M.Y., Interplay between molecular chaperones and signaling pathways in survival of heat shock. J Appl Physiol, 2002. 92: p. 1743-8.
749. Chu, B., Soncin, F., Price, B.D., Stevenson, M.A., and Calderwood, S.K., Sequential phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase 3

__________________________________________ Referências Bibliográficas
323
represses transcriptional activation by heat shock factor-1. J Biol Chem, 1996. 271: p. 30847-57.
750. Dai, R., Frejtag, W., He, B., Zhang, Y., and Mivechi, N.F., c-Jun NH2-terminal kinase targeting and phosphorylation of heat shock factor-1 suppress its transcriptional activity. J Biol Chem, 2000. 275: p. 18210-8.
751. Lenart, J., Dombrowski, F., Görlach, A., and Kietzmann, T., Deficiency of manganese superoxide dismutase in hepatocytes disrupts zonated gene expression in mouse liver. Arch Biochem Biophys, 2007. 462: p. 238-44.
752. Morel, Y. and Barouki, R., Repression of gene expression by oxidative stress. Biochem J, 1999. 342(Pt 3): p. 481-96.
753. Bowie, A. and O'Neill, L.A., Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol, 2000. 59: p. 13-23.
754. Ryan, K.M., Ernst, M.K., Rice, N.R., and Vousden, K.H., Role of NF-kappaB in p53-mediated programmed cell death. Nature, 2000. 404: p. 892-7.
755. Zhou, L.Z., Johnson, A.P., and Rando, T.A., NF kappa B and AP-1 mediate transcriptional responses to oxidative stress in skeletal muscle cells. Free Radic Biol Med, 2001. 31: p. 1405-16.
756. Maiuri, M.C., De Stefano, D., Mele, G., Fecarotta, S., Greco, L., Troncone, R., and Carnuccio, R., Nuclear factor kappa B is activated in small intestinal mucosa of celiac patients. J Mol Med, 2003. 81: p. 373-9.
757. Spitzer, J.A., Zheng, M., Kolls, J.K., Vande Stouwe, C., and Spitzer, J.J., Ethanol and LPS modulate NF-kappaB activation, inducible NO synthase and COX-2 gene expression in rat liver cells in vivo. Front Biosci, 2002. 7: p. a99-108.
758. Bonizzi, G. and Karin, M., The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol, 2004. 25: p. 280-8.
759. Senftleben, U. and Karin, M., The IKK/NF-kappaB pathway. Crit Care Med, 2002. 30: p. S18-S26.
760. Luedde, T. and Trautwein, C., Intracellular survival pathways in the liver. Liver Int, 2006. 26: p. 1163-74.
761. Ninković, M., Selaković, V., Ethukić, M., Milosavljević, P., Vasiljević, I., Jovanović, M., and Malicević, Z., Oxidative stress in rat kidneys due to 3,4-methylenedioxymetamphetamine (ecstasy) toxicity. Nephrology (Carlton), 2008. 13: p. 33-7.
762. Oh, S.I., Kim, C.I., Chun, H.J., and Park, S.C., Chronic ethanol consumption affects glutathione status in rat liver. J Nutr, 1998. 128: p. 758-63.
763. Deaciuc, I.V., Fortunato, F., D'Souza, N.B., Hill, D.B., Schmidt, J., Lee, E.Y., and McClain, C.J., Modulation of caspase-3 activity and Fas ligand mRNA expression in rat liver cells in vivo by alcohol and lipopolysaccharide. Alcoholism: Clinical and Experimental Research, 1999. 23: p. 349-56.
764. Lauterburg, B.H., Davies, S., and Mitchell, J.R., Ethanol suppresses hepatic glutathione synthesis in rats in vivo. J Pharmacol Exp Ther, 1984. 230: p. 7-11.
765. Flanagan, S.W., Moseley, P.L., and Buettner, G.R., Increased flux of free radicals in cells subjected to hyperthermia: detection by electron paramagnetic resonance spin trapping. FEBS Lett, 1998. 431: p. 285-6.
766. Sun, W.M., Huang, Z.Z., and Lu, S.C., Regulation of gamma-glutamylcysteine synthetase by protein phosphorylation. Biochem J, 1996. 320: p. 321-28.
767. Beitia, G., Cobreros, A., Sainz, L., and Cenarruzabeitia, E., 3,4-Methylenedioxymethamphetamine (ecstasy)-induced hepatotoxicity: effect on cytosolic calcium signals in isolated hepatocytes. Liver, 1999. 19: p. 234-41.
768. Yan, M., Zhu, P., Liu, H.M., Zhang, H.T., and Liu, L., Ethanol induced mitochondria injury and permeability transition pore opening: role of mitochondria in alcoholic liver disease. World J Gastroenterol, 2007. 13: p. 2352-6.
769. Vidair, C.A. and Dewey, W.C., Evaluation of a role for intracellular Na+, K+, Ca2+, and Mg2+ in hyperthermic cell killing. Radiat Res, 1986. 105: p. 187-200.

Referências Bibliográficas __________________________________________
324
770. Yao, P., Li, K., Jin, Y., Song, F., Zhou, S., Sun, X., Nussler, A.K., and Liu, L., Oxidative damage after chronic ethanol intake in rat tissues: Prophylaxis of Ginkgo biloba extract. Food Chemistry, 2006. 99: p. 305-14.
771. Mannervik, B. and Danielson, U.H., Glutathione transferases--structure and catalytic activity. CRC Crit Rev Biochem, 1988. 23: p. 283-337.
772. Pari, L. and Suresh, A., Effect of grape (Vitis vinifera L.) leaf extract on alcohol induced oxidative stress in rats. Food Chem Toxicol, 2008. 46: p. 1627-34.
773. Seitz, H.K. and Stickel, F., Risk factors and mechanisms of hepatocarcinogenesis with special emphasis on alcohol and oxidative stress. Biological Chemistry, 2006. 387: p. 349-60.
774. Qian, L., Song, X., Ren, H., Gong, J., and Cheng, S., Mitochondrial mechanism of heat stress-induced injury in rat cardiomyocyte. Cell Stress & Chaperones, 2004. 9(3): p. 281-93.
775. Yuen, W.F., Fung, K.P., Lee, C.Y., Choy, Y.M., Kong, S.K., Ko, S., and Kwok, T.T., Hyperthermia and tumour necrosis factor-alpha induced apoptosis via mitochondrial damage. Life Sciences, 2000. 67(6): p. 725-32.
776. Zhao, Q.L., Fujiwara, Y., and Kondo, T., Mechanism of cell death induction by nitroxide and hyperthermia. Free Radical Biology and Medicine, 2006. 40(7): p. 1131-43.
777. Montet, A.M., Oliva, L., Beaugé, F., and Montet, J.C., Bile salts modulate chronic ethanol-induced hepatotoxicity. Alcohol Alcohol, 2002. 37: p. 25-9.
778. Yue, M., Ni, Q., Yu, C.H., Ren, K.M., Chen, W.X., and Li, Y.M., Transient elevation of hepatic enzymes in volunteers after intake of alcohol. Hepatobiliary Pancreat Dis Int, 2006. 5: p. 52-5.
779. Beitia, G., Cobreros, A., Sainz, L., and Cenarruzabeitia, E., Ecstasy-induced toxicity in rat liver. Liver, 2000. 20: p. 8-15.
780. Alvaro, A.R., Martins, J., Costa, A.C., Fernandes, E., Carvalho, F., Ambrósio, A.F., and Cavadas, C., Neuropeptide Y protects retinal neural cells against cell death induced by ecstasy. Neuroscience, 2008. 152: p. 97-105.
781. Wu, D., Zhai, Q., and Shi, X., Alcohol-induced oxidative stress and cell responses. J Gastroenterol Hepatol, 2006. 21: p. S26-9.
782. Trujillo-Murillo, K., Alvarez-Martínez, O., Garza-Rodríguez, L., Martínez-Rodríguez, H., Bosques-Padilla, F., Ramos-Jiménez, J., Barrera-Saldaña, H., Rincón-Sánchez, A.R., and Rivas-Estilla, A.M., Additive effect of ethanol and HCV subgenomic replicon expression on COX-2 protein levels and activity. J Viral Hepat, 2007. 14: p. 608-17.




















