
Insights in the regulation of cholesterol 7α-hydroxylase gene reveal a target for modulating bile...
13
Insights in the Regulation of Cholesterol 7- Hydroxylase Gene Reveal a Target for Modulating Bile Acid Synthesis Nico Mitro,* Cristina Godio,* Emma De Fabiani, Elena Scotti, Andrea Galmozzi, Federica Gilardi, Donatella Caruso, Ana Belen Vigil Chacon, and Maurizio Crestani The transcription of the gene (CYP7A1) encoding cholesterol 7-hydroxylase, a key enzyme in cholesterol homeostasis, is repressed by bile acids via multiple mechanisms involving members of the nuclear receptor superfamily. Here, we describe a regulatory mechanism that can be exploited for modulating bile acid synthesis. By dissecting the mechanisms of CYP7A1 transcription, we found that bile acids stimulate the sequential recruitment of the histone deacetylases (HDACs) 7, 3, and 1, and of the corepressor SMRT (silencing mediator of retinoid and thyroid receptors-) and the nuclear core- pressor. Bile acids, but not the farnesoid X receptor–selective agonist GW4064, increase the nuclear concentration of HDAC7, which promotes the assembly of a repressive complex that ultimately represses CYP7A1 transcription. Interestingly, despite its high basal expression level, small heterodimer partner (SHP) is associated with the CYP7A1 promoter only at a later stage of bile acid repression. Gene silencing with small inter- fering RNA confirms that HDAC7 is the key factor required for the repression of CYP7A1 transcription, whereas knockdown of SHP does not prevent the down-regula- tion of CYP7A1. Administration of the HDAC inhibitors valproic acid or trichostatin A to genetically hypercholesterolemic mice increases Cyp7a1 messenger RNA and bile acid synthesis and consequently markedly reduces total plasma and low-density lipoprotein cholesterol. Conclusion: By using a combination of molecular, cellular, and animal models, our study highlights the importance of HDACs in the feedback regulation of CYP7A1 transcription and identifies these enzymes as potential targets to modulate bile acid synthesis and for the treatment of hypercholesterolemia. (HEPATOLOGY 2007;46: 885-897.) Abbreviations: CA, cholic acid; CDCA, chenodeoxycholic acid; CYP7A1, cholesterol 7-hydroxylase; ChIP, chromatin immunoprecipitation; FXR, farnesoid X receptor; FGF-19, fibroblast growth factor-19; HDAC, histone deacetylase; mRNA, messenger RNA; PBS, phosphate-buffered saline; SHP, small heterodimer partner; SMRT-, silencing mediator of retinoid and thyroid receptors-; siRNA, short interfering RNA; TSA, trichostatin A; VPA, valproic acid. From the Laboratorio “Giovanni Galli” di Biochimica e Biologia Molecolare dei Lipidi e di Spettrometria di Massa, Dipartimento di Scienze Farmacologiche, Facolta ` di Farmacia, Universita ` degli Studi di Milano, Milan, Italy. Received February 19, 2007; accepted May 14, 2007. Supported by grants from Telethon (GGP04252), the European Commission (NORTh, QLG1-CT2001-01513), the Italian Ministry of University and Research (MIUR, COFIN 2002062991, COFIN 2002064429, COFIN 2004067491). A.B.V.C. was a Marie Curie training fellow supported by the European Commission (ATHERODIS, QLG1-CT2001-60031). *These authors contributed equally to this work. N.M. and C.G. are currently affiliated with the Department of Cell Biology, The Scripps Research Institute, Stein Research Building, Room 212, 10666 North Torrey Pines Road, La Jolla, CA 92037. Address reprint requests to: Maurizio Crestani, Ph.D., Laboratorio “Giovanni Galli” di Biochimica e Biologia Molecolare dei Lipidi e di Spettrometria di Massa, Dipartimento di Scienze Farmacologiche, Universita ` degli Studi di Milano, via Balzaretti, 9, 20133 Milan, Italy. E-mail: [email protected]; fax: (39) 02-50318391. Copyright © 2007 by the American Association for the Study of Liver Diseases. Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/hep.21819 Potential conflict of interest: Nothing to report. Supplementary material for this article can be found on the HEPATOLOGY website (http://interscience.wiley.com/jpages/0270-9139/suppmat/index.html). 885
Transcript of Insights in the regulation of cholesterol 7α-hydroxylase gene reveal a target for modulating bile...

Insights in the Regulation of Cholesterol 7�-Hydroxylase Gene Reveal a Target for Modulating Bile
Acid Synthesis
Nico Mitro,* Cristina Godio,* Emma De Fabiani, Elena Scotti, Andrea Galmozzi, Federica Gilardi, Donatella Caruso,
Ana Belen Vigil Chacon, and Maurizio Crestani
The transcription of the gene (CYP7A1) encoding cholesterol 7�-hydroxylase, a keyenzyme in cholesterol homeostasis, is repressed by bile acids via multiple mechanismsinvolving members of the nuclear receptor superfamily. Here, we describe a regulatorymechanism that can be exploited for modulating bile acid synthesis. By dissecting themechanisms of CYP7A1 transcription, we found that bile acids stimulate the sequentialrecruitment of the histone deacetylases (HDACs) 7, 3, and 1, and of the corepressorSMRT� (silencing mediator of retinoid and thyroid receptors-�) and the nuclear core-pressor. Bile acids, but not the farnesoid X receptor–selective agonist GW4064, increasethe nuclear concentration of HDAC7, which promotes the assembly of a repressivecomplex that ultimately represses CYP7A1 transcription. Interestingly, despite its highbasal expression level, small heterodimer partner (SHP) is associated with the CYP7A1promoter only at a later stage of bile acid repression. Gene silencing with small inter-fering RNA confirms that HDAC7 is the key factor required for the repression ofCYP7A1 transcription, whereas knockdown of SHP does not prevent the down-regula-tion of CYP7A1. Administration of the HDAC inhibitors valproic acid or trichostatin Ato genetically hypercholesterolemic mice increases Cyp7a1 messenger RNA and bile acidsynthesis and consequently markedly reduces total plasma and low-density lipoproteincholesterol. Conclusion: By using a combination of molecular, cellular, and animalmodels, our study highlights the importance of HDACs in the feedback regulation ofCYP7A1 transcription and identifies these enzymes as potential targets to modulate bileacid synthesis and for the treatment of hypercholesterolemia. (HEPATOLOGY 2007;46:885-897.)
Abbreviations: CA, cholic acid; CDCA, chenodeoxycholic acid; CYP7A1, cholesterol 7�-hydroxylase; ChIP, chromatin immunoprecipitation; FXR, farnesoid X receptor;FGF-19, fibroblast growth factor-19; HDAC, histone deacetylase; mRNA, messenger RNA; PBS, phosphate-buffered saline; SHP, small heterodimer partner; SMRT-�,silencing mediator of retinoid and thyroid receptors-�; siRNA, short interfering RNA; TSA, trichostatin A; VPA, valproic acid.
From the Laboratorio “Giovanni Galli” di Biochimica e Biologia Molecolare dei Lipidi e di Spettrometria di Massa, Dipartimento di Scienze Farmacologiche, Facoltadi Farmacia, Universita degli Studi di Milano, Milan, Italy.
Received February 19, 2007; accepted May 14, 2007.Supported by grants from Telethon (GGP04252), the European Commission (NORTh, QLG1-CT2001-01513), the Italian Ministry of University and Research
(MIUR, COFIN 2002062991, COFIN 2002064429, COFIN 2004067491). A.B.V.C. was a Marie Curie training fellow supported by the European Commission(ATHERODIS, QLG1-CT2001-60031).
*These authors contributed equally to this work.N.M. and C.G. are currently affiliated with the Department of Cell Biology, The Scripps Research Institute, Stein Research Building, Room 212, 10666 North Torrey
Pines Road, La Jolla, CA 92037.Address reprint requests to: Maurizio Crestani, Ph.D., Laboratorio “Giovanni Galli” di Biochimica e Biologia Molecolare dei Lipidi e di Spettrometria di Massa,
Dipartimento di Scienze Farmacologiche, Universita degli Studi di Milano, via Balzaretti, 9, 20133 Milan, Italy. E-mail: [email protected]; fax: (39)02-50318391.
Copyright © 2007 by the American Association for the Study of Liver Diseases.Published online in Wiley InterScience (www.interscience.wiley.com).DOI 10.1002/hep.21819Potential conflict of interest: Nothing to report.Supplementary material for this article can be found on the HEPATOLOGY website (http://interscience.wiley.com/jpages/0270-9139/suppmat/index.html).
885

Hepatic bile acid biosynthesis is quantitatively themost important metabolic pathway to maintaincholesterol balance in mammals.1 Cholesterol
7�-hydroxylase (CYP7A1) is a liver-specific enzyme thatcatalyzes the rate-limiting reaction of the so-called “clas-sic” pathway of bile acid synthesis and represents a majorcheckpoint of cholesterol homeostasis.2-4 CYP7A1 istranscriptionally regulated by bile acids returning to theliver via the portal vein5-7 through multiple mechanisms.In particular, bile acids trigger the farnesoid X receptor(FXR, NR1H4)/small heterodimer partner (SHP,NR0B2) regulatory cascade8,9 that leads to CYP7A1 re-pression.10,11 Additionally, bile acids initiate the c-JunN-terminal kinase–dependent cascade that ultimatelysuppresses CYP7A1 expression via FXR-mediated induc-tion of fibroblast growth factor-19 (FGF-19)12 or proteinkinase C.13,14 We also demonstrated an FXR/SHP-independent mechanism of feedback regulation ofCYP7A115,16 whereby bile acids cause the dissociation ofcoactivators from hepatocyte nuclear factor-4� (NR2A1).Finally, studies in Shp null mice show that bile acids re-press gene transcription in a SHP-independent fash-ion.17,18 However, the contribution of the FXR/SHP-independent pathway in the regulation of CYP7A1transcription in response to bile acids is still unclear. Thefull understanding of these mechanisms may be exploitedto develop new therapies that decrease plasma cholesteroland possibly treat patients affected by gallstone disease.
Hypercholesterolemia is a well-known risk factor for ath-erosclerosis and cardiovascular disease. Several therapeuticapproaches have been shown to reduce the rate of morbidityand mortality associated with hypercholesterolemia and ath-erosclerosis. For example, inhibitors of cholesterol synthesis(statins) and absorption (ezetimibe) have shown several ben-eficial effects related to their cholesterol-lowering activi-ty.19,20 However, a more significant decrease of plasmacholesterol levels would be desirable, particularly for patientsat high risk of coronary artery disease, such as those withgenetic defects in lipoprotein and lipid metabolism (for ex-ample, familial hypercholesterolemia).21
It has been shown that adenovirus-mediated overex-pression of CYP7A1 in genetically hypercholesterolemic(Ldl-r�/�) mice lowers plasma low-density lipoproteincholesterol levels,22 indicating that the CYP7A1 gene rep-resents an ideal target for treatment of hypercholesterol-emia. However, to date, ion exchange resins are the onlyagents that can increase bile acid synthesis and, conse-quently, decrease plasma cholesterol levels.
In this study, we performed a detailed dissection of theevents underlying the bile acid–mediated feedback regu-lation of CYP7A1, which indicates that bile acids repressCYP7A1 transcription rapidly via an FXR-independent
pathway, whereas the FXR-dependent regulation occursonly after several hours. In particular, we found that his-tone deacetylase 7 (HDAC7) is the critical factor for thebile acid–mediated repression of CYP7A1 gene transcrip-tion. Finally, we show that valproic acid (VPA) or tricho-statin A (TSA), two known and structurally unrelatedinhibitors of HDAC activity,23-26 prevent the feedbackregulation by bile acids in vitro and de-repress Cyp7a1 invivo, leading to a dramatic reduction of blood cholesterolin Ldl-r�/� mice.27 Our analysis highlights a complemen-tary mechanism for the suppression of bile acid biosyn-thesis and identifies HDACs as possible targets for thetreatment of hypercholesterolemia.
Materials and Methods
Reagents and Plasmids. Chenodeoxycholic acid(CDCA), cholic acid (CA), calyculin A, TSA, and VPAwere obtained from Sigma. The GW4064 agonist waskindly donated by Dr. K. Bamberg (Astra-Zeneca, Moln-dal, Sweden). The plasmid pcDNA3HA–peroxisomeproliferator-activated receptor-� coactivator 1� was pro-vided by Dr. Anastasia Kralli (The Scripps Research In-stitute, La Jolla, CA).
Cell Cultures, RNA Extraction, and QuantitativeAnalysis. HepG2 cells (American Type Culture Collec-tion, Manassas, VA; no. CRL-10741) were cultured asdescribed.16 HepG2 cells were treated with either 25 �MCDCA, 25 �M CA, 5 �M GW4064, 1.5 mM VPA, 200nM TSA, or 10 nM calyculin A at the indicated combi-nations for different times. Total RNA was extracted, andspecific messenger RNA (mRNA) was quantitated via re-al-time PCR, using the Sybr Green kit (Bio-Rad Labora-tories, Milan, Italy) as described.16 Primer sequences areavailable in Table 1. We monitored the specificity of theamplified products by melting curve analysis. Experi-ments were performed in triplicate and repeated twicewith different cell preparations.
Chromatin Immunoprecipitations. Chromatin im-munoprecipitation (ChIP) assays were performed as de-scribed.16 Immunoprecipitations were performed withantibodies against hemagglutin (HA)-tag (Roche) to immu-noprecipitate the transfected HA-tagged peroxisome prolif-erator-activated receptor-� coactivator 1� (PGC-1), whereasthe following antibodies were used to immunoprecipitatethe endogenously expressed proteins: cyclic adenosinemonophosphate response element binding–protein bindingprotein (CBP), acetyl-H3, acetyl-H4 (Upstate Biotechnol-ogy, Charlottesville, VA), HDAC1 to HDAC8, RNA poly-merase II, SHP, silencing mediator of retinoid and thyroidreceptors-� (SMRT�), and nuclear corepressor N-CoR(Santa Cruz Biotechnology, Santa Cruz, CA). Specificity of
886 MITRO, GODIO, ET AL. HEPATOLOGY, September 2007
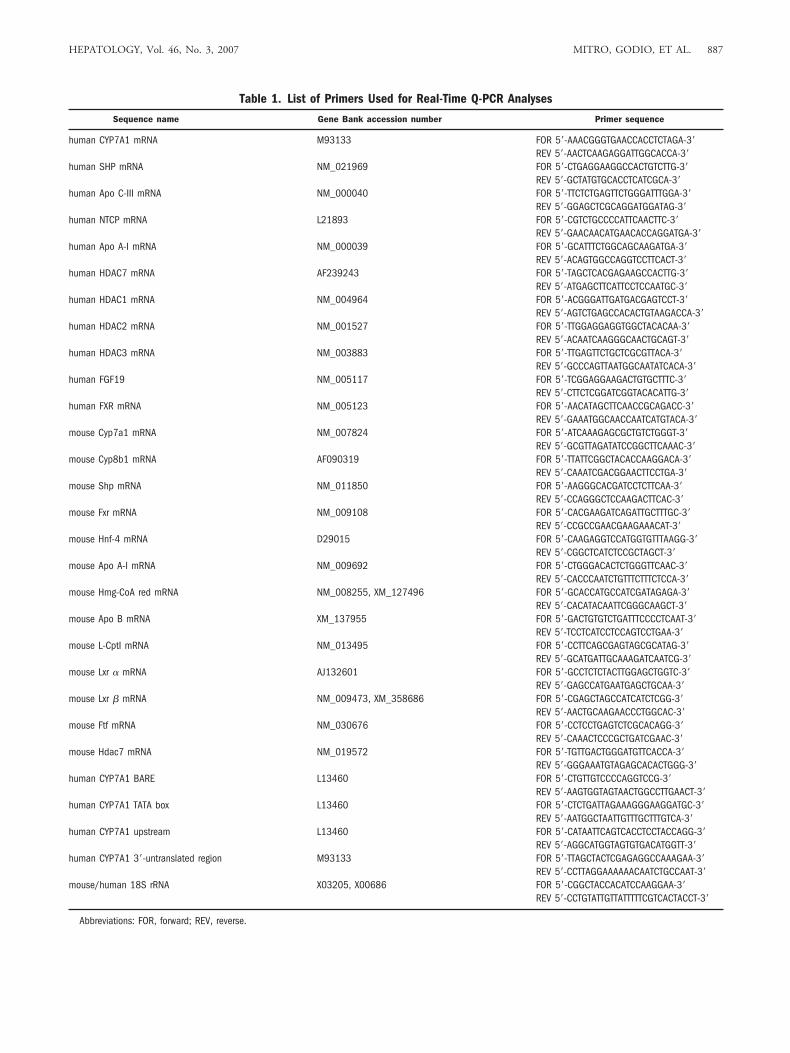
Table 1. List of Primers Used for Real-Time Q-PCR Analyses
Sequence name Gene Bank accession number Primer sequence
human CYP7A1 mRNA M93133 FOR 5�-AAACGGGTGAACCACCTCTAGA-3�REV 5�-AACTCAAGAGGATTGGCACCA-3�
human SHP mRNA NM_021969 FOR 5�-CTGAGGAAGGCCACTGTCTTG-3�REV 5�-GCTATGTGCACCTCATCGCA-3�
human Apo C-III mRNA NM_000040 FOR 5�-TTCTCTGAGTTCTGGGATTTGGA-3�REV 5�-GGAGCTCGCAGGATGGATAG-3�
human NTCP mRNA L21893 FOR 5�-CGTCTGCCCCATTCAACTTC-3�REV 5�-GAACAACATGAACACCAGGATGA-3�
human Apo A-I mRNA NM_000039 FOR 5�-GCATTTCTGGCAGCAAGATGA-3�REV 5�-ACAGTGGCCAGGTCCTTCACT-3�
human HDAC7 mRNA AF239243 FOR 5�-TAGCTCACGAGAAGCCACTTG-3�REV 5�-ATGAGCTTCATTCCTCCAATGC-3�
human HDAC1 mRNA NM_004964 FOR 5�-ACGGGATTGATGACGAGTCCT-3�REV 5�-AGTCTGAGCCACACTGTAAGACCA-3�
human HDAC2 mRNA NM_001527 FOR 5�-TTGGAGGAGGTGGCTACACAA-3�REV 5�-ACAATCAAGGGCAACTGCAGT-3�
human HDAC3 mRNA NM_003883 FOR 5�-TTGAGTTCTGCTCGCGTTACA-3�REV 5�-GCCCAGTTAATGGCAATATCACA-3�
human FGF19 NM_005117 FOR 5�-TCGGAGGAAGACTGTGCTTTC-3�REV 5�-CTTCTCGGATCGGTACACATTG-3�
human FXR mRNA NM_005123 FOR 5�-AACATAGCTTCAACCGCAGACC-3�REV 5�-GAAATGGCAACCAATCATGTACA-3�
mouse Cyp7a1 mRNA NM_007824 FOR 5�-ATCAAAGAGCGCTGTCTGGGT-3�REV 5�-GCGTTAGATATCCGGCTTCAAAC-3�
mouse Cyp8b1 mRNA AF090319 FOR 5�-TTATTCGGCTACACCAAGGACA-3�REV 5�-CAAATCGACGGAACTTCCTGA-3�
mouse Shp mRNA NM_011850 FOR 5�-AAGGGCACGATCCTCTTCAA-3�REV 5�-CCAGGGCTCCAAGACTTCAC-3�
mouse Fxr mRNA NM_009108 FOR 5�-CACGAAGATCAGATTGCTTTGC-3�REV 5�-CCGCCGAACGAAGAAACAT-3�
mouse Hnf-4 mRNA D29015 FOR 5�-CAAGAGGTCCATGGTGTTTAAGG-3�REV 5�-CGGCTCATCTCCGCTAGCT-3�
mouse Apo A-I mRNA NM_009692 FOR 5�-CTGGGACACTCTGGGTTCAAC-3�REV 5�-CACCCAATCTGTTTCTTTCTCCA-3�
mouse Hmg-CoA red mRNA NM_008255, XM_127496 FOR 5�-GCACCATGCCATCGATAGAGA-3�REV 5�-CACATACAATTCGGGCAAGCT-3�
mouse Apo B mRNA XM_137955 FOR 5�-GACTGTGTCTGATTTCCCCTCAAT-3�REV 5�-TCCTCATCCTCCAGTCCTGAA-3�
mouse L-CptI mRNA NM_013495 FOR 5�-CCTTCAGCGAGTAGCGCATAG-3�REV 5�-GCATGATTGCAAAGATCAATCG-3�
mouse Lxr � mRNA AJ132601 FOR 5�-GCCTCTCTACTTGGAGCTGGTC-3�REV 5�-GAGCCATGAATGAGCTGCAA-3�
mouse Lxr � mRNA NM_009473, XM_358686 FOR 5�-CGAGCTAGCCATCATCTCGG-3�REV 5�-AACTGCAAGAACCCTGGCAC-3�
mouse Ftf mRNA NM_030676 FOR 5�-CCTCCTGAGTCTCGCACAGG-3�REV 5�-CAAACTCCCGCTGATCGAAC-3�
mouse Hdac7 mRNA NM_019572 FOR 5�-TGTTGACTGGGATGTTCACCA-3�REV 5�-GGGAAATGTAGAGCACACTGGG-3�
human CYP7A1 BARE L13460 FOR 5�-CTGTTGTCCCCAGGTCCG-3�REV 5�-AAGTGGTAGTAACTGGCCTTGAACT-3�
human CYP7A1 TATA box L13460 FOR 5�-CTCTGATTAGAAAGGGAAGGATGC-3�REV 5�-AATGGCTAATTGTTTGCTTTGTCA-3�
human CYP7A1 upstream L13460 FOR 5�-CATAATTCAGTCACCTCCTACCAGG-3�REV 5�-AGGCATGGTAGTGTGACATGGTT-3�
human CYP7A1 3�-untranslated region M93133 FOR 5�-TTAGCTACTCGAGAGGCCAAAGAA-3�REV 5�-CCTTAGGAAAAAACAATCTGCCAAT-3�
mouse/human 18S rRNA X03205, X00686 FOR 5�-CGGCTACCACATCCAAGGAA-3�REV 5�-CCTGTATTGTTATTTTTCGTCACTACCT-3�
Abbreviations: FOR, forward; REV, reverse.
HEPATOLOGY, Vol. 46, No. 3, 2007 MITRO, GODIO, ET AL. 887

antibodies used for ChIP assays was assessed by western blot-ting. Specific genomic DNA regions were quantitated viareal-time PCR.16 Primer sequences are available in Table 1.Each time point was replicated in three separate dishes. Tocheck the specificity of immunoprecipitated DNA regions,we also amplified distal regions, which confirmed lack ofinteraction with the analyzed nuclear factors. The specificityof amplified regions was assessed by performing a meltingcurve at the end of each real-time PCR reaction. Data areexpressed as fold above background and were calculated ac-cording to the following formula: (Ct ChIP � Ct Input) attn � (Ct no antibody � Ct Input) at tn � ��Ct Recruit-ment (fold above background) � 2���Ct, where “Ct ChIP”is the threshold cycle obtained with samples immunoprecipi-tated with a given antibody at the indicated time (tn), “CtInput” is the value obtained with total genomic DNA, and“Ct no antibody” refers to the controls with no antibodyadded for each immunoprecipitation as a background value.The background levels from a control antibody (anti-mouseIgG) was comparable to the values obtained with no anti-body.
Immunocytochemistry. HepG2 cells were culturedon coverslips with or without 25 �M CDCA. Cells werefixed, washed, permeabilized, and blocked with phos-phate-buffered saline (PBS) containing 10% serum, 1%bovine serum albumin, and 0.5% Tween 20 for 30 min-utes at room temperature. Cells were incubated with apolyclonal antibody against HDAC7 (diluted 1:20 inPBS containing 1% serum) overnight at 4°C. The AlexaFluor 488 goat anti-rabbit IgG (diluted 1:500 in PBScontaining 1% serum) (Invitrogen) was added as a sec-ondary antibody for 1 hour at room temperature. AlexaFluor 546 phalloidin (diluted 1:200 in PBS) and TO-PRO-3 iodide (diluted 1:500) (Invitrogen) were addedfor 15 minutes at room temperature to stain the cytoskel-eton and nucleus, respectively. Coverslips were mountedand analyzed with a confocal microscope (Nikon EclipseTE2000-S, Radiance 2100, Bio-Rad Laboratories). Im-age acquisition was performed with Laser Sharp software(Bio-Rad Laboratories). For the quantitation of HDAC7-containing nuclei, we counted at least 3 different fields ineach coverslip in 2 distinct coverslips.
Inhibition of HDACs with Short Interfering RNA.Predesigned short interfering RNA (siRNA) oligonucleo-tides to knock down HDAC1, HDAC3, HDAC7,SHP, and glyceraldehyde 3-phosphate dehydrogenase(GAPDH) (siGenome siRNA SMART pools) (Dharma-con, Lafayette, CO) were transfected into HepG2 cellsaccording to the manufacturer’s instructions. Dharma-con’s siControl nontargeting siRNA pool, which is a poolof four nontargeting siRNAs, and siControl GAPDH(Supplementary Fig. 4), which is a pool of siRNA oligo-
nucleotides targeting GAPDH, were used as negativecontrols. In cells transfected with siControl siRNA, theresponse of CYP7A1 to bile acids did not differ to those ofmock-transfected cells (data not shown). Thirty hoursafter transfection, HepG2 cells were treated with 25 �MCDCA, and at the end of the incubation total RNA wasextracted and analyzed via real-time quantitative PCR(Q-PCR). To rule out unspecific silencing (that is, off-target effects), we tested the effect of knockdown withsiRNA by monitoring the mRNA levels of differentHDACs and the response to bile acids of other bile acid–regulated genes (Supplementary Figs. 1-3).
Animal Studies. Male 10-week-old Ldl-r�/� mice(genetic background C57BL/6J), 7-9 animals/group,were injected intraperitoneally for 7 days with 205 mgvalproic acid/kg body weight twice a day, 1 mg trichosta-tin A/kg body weight once a day, or vehicle (0.9% NaCl).Plasma alanine aminotransferase and aspartate amino-transferase levels were determined with an enzymatic kit(Sentinel, Milan, Italy) at the end of the treatment, and nostatistically significant difference between control andtreated groups was observed. Animals had free access tofood and water and the light cycle was from 7:00 A.M. to7:00 P.M. Total RNA was extracted from livers, reverse-transcribed, and analyzed via real-time Q-PCR. Primersequences are available in Table 1. All animal experimentswere approved by the University of Milan and the ItalianMinistry of Health and were conducted while strictly fol-lowing European Community (EEC Directive no. 609/86) and local regulations for animal care (ItalianLegislative Decree no. 116, January 27, 1992).
Fecal Bile Acid Excretion. Mice were placed in met-abolic cages for the collection of stools over a 72-hourperiod. Dried feces (0.2 g) were extracted in 4 mL of 75%ethanol at 50°C for 2 hours. The extracts were centrifugedand supernatants were diluted 20-fold with 65 mM phos-phate buffer (pH 7.0). Bile acid concentration was mea-sured with an enzymatic kit (Sentinel). The daily fecesoutput (g/day per 100 g body weight) and fecal bile acidcontent (�mol/g) were used to calculate the rate of bileacid excretion (�mol/day/100 g body weight).28
Liver Bile Acid and Cholesterol Content. Two hun-dred milligrams of livers were homogenized in 2 mL PBS,and 2 � 105 disintegrations per minute of [3H]cholesterolwere used to normalize for recovery. Total lipids wereextracted by adding 10 mL chloroform/methanol (2:1).The organic phase was recovered and dried, and radioac-tivity was counted via liquid scintillation. For the deter-mination of bile acid and cholesterol content in liverextracts, samples were diluted 20-fold with 65 mM phos-phate buffer (pH 7.0) and bile acid and cholesterol con-centration was measured with an enzymatic kit (Sentinel).
888 MITRO, GODIO, ET AL. HEPATOLOGY, September 2007
After correcting for the percentage of recovery, valueswere expressed as the mean � standard deviation (SD) ofnanograms total bile acids per gram of liver.
Determination of Plasma Cholesterol. Total plasmacholesterol was determined with an enzymatic kit (Sentinel).Cholesterol distribution in plasma lipoprotein fractions wasdetermined via fast protein liquid chromatography (FPLC)using a Superose 6 column (Amersham Biosciences, Milan,Italy). One-milliliter fractions were collected and assayed forcholesterol with an enzymatic kit (Sentinel).
Statistical Analyses. Statistical analyses were per-formed via 1-way analysis of variance, using GraphPadPrism version 4.0 (GraphPad Software, San Diego, CA).
Results
Bile Acids Affect CYP7A1 Transcription via Chro-nologically Distinct Mechanisms. To distinguish be-
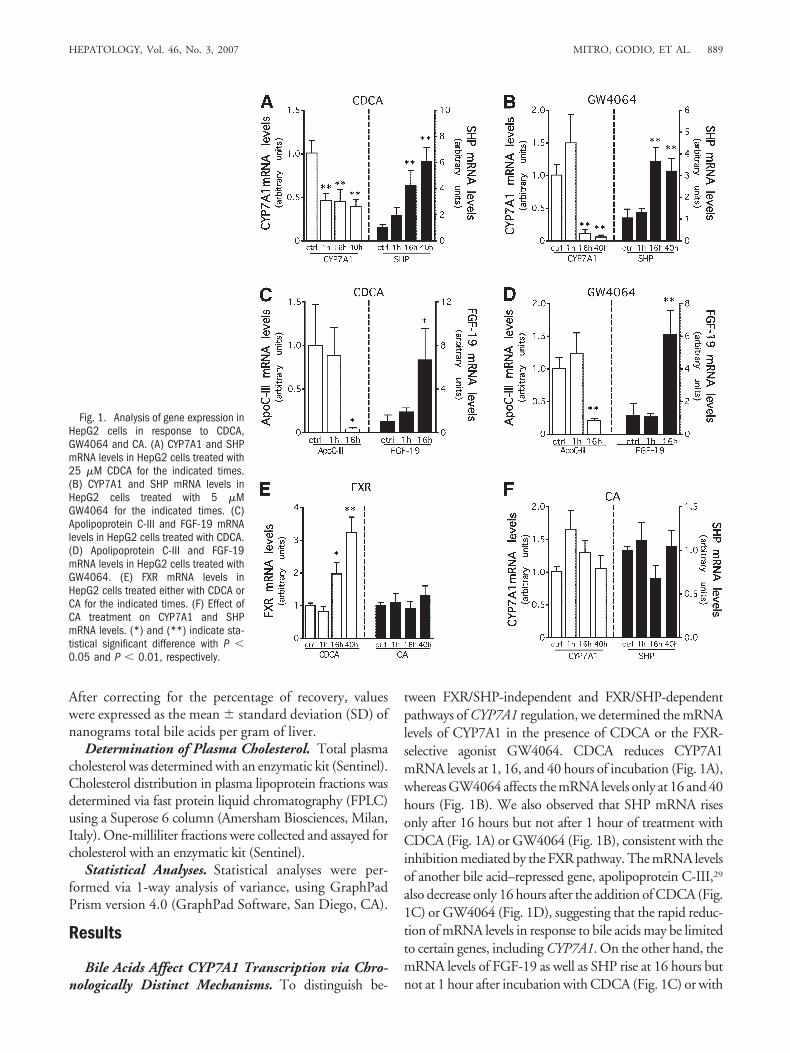
tween FXR/SHP-independent and FXR/SHP-dependentpathways of CYP7A1 regulation, we determined the mRNAlevels of CYP7A1 in the presence of CDCA or the FXR-selective agonist GW4064. CDCA reduces CYP7A1mRNA levels at 1, 16, and 40 hours of incubation (Fig. 1A),whereas GW4064 affects the mRNA levels only at 16 and 40hours (Fig. 1B). We also observed that SHP mRNA risesonly after 16 hours but not after 1 hour of treatment withCDCA (Fig. 1A) or GW4064 (Fig. 1B), consistent with theinhibition mediated by the FXR pathway. The mRNA levelsof another bile acid–repressed gene, apolipoprotein C-III,29
also decrease only 16 hours after the addition of CDCA (Fig.1C) or GW4064 (Fig. 1D), suggesting that the rapid reduc-tion of mRNA levels in response to bile acids may be limitedto certain genes, including CYP7A1. On the other hand, themRNA levels of FGF-19 as well as SHP rise at 16 hours butnot at 1 hour after incubation with CDCA (Fig. 1C) or with
Fig. 1. Analysis of gene expression inHepG2 cells in response to CDCA,GW4064 and CA. (A) CYP7A1 and SHPmRNA levels in HepG2 cells treated with25 �M CDCA for the indicated times.(B) CYP7A1 and SHP mRNA levels inHepG2 cells treated with 5 �MGW4064 for the indicated times. (C)Apolipoprotein C-III and FGF-19 mRNAlevels in HepG2 cells treated with CDCA.(D) Apolipoprotein C-III and FGF-19mRNA levels in HepG2 cells treated withGW4064. (E) FXR mRNA levels inHepG2 cells treated either with CDCA orCA for the indicated times. (F) Effect ofCA treatment on CYP7A1 and SHPmRNA levels. (*) and (**) indicate sta-tistical significant difference with P �0.05 and P � 0.01, respectively.
HEPATOLOGY, Vol. 46, No. 3, 2007 MITRO, GODIO, ET AL. 889
GW4064 (Fig. 1D), indicating that the early response ofCYP7A1 to bile acids and the induction of FGF-19 and SHPare dissociated. As reported,30,31 CDCA increases the expres-sion of FXR (Fig. 1E), whereas CA does not affect themRNA levels of FXR, CYP7A1, and SHP (Fig. 1E,F). Noneof the compounds used in these experiments affect the via-bility of HepG2 cells (data not shown). Because GW4064affects CYP7A1 and SHP mRNA levels only after 16 hours,we infer that the initial inhibition elicited by CDCA does notinvolve FXR/SHP inhibitory pathway.10,11
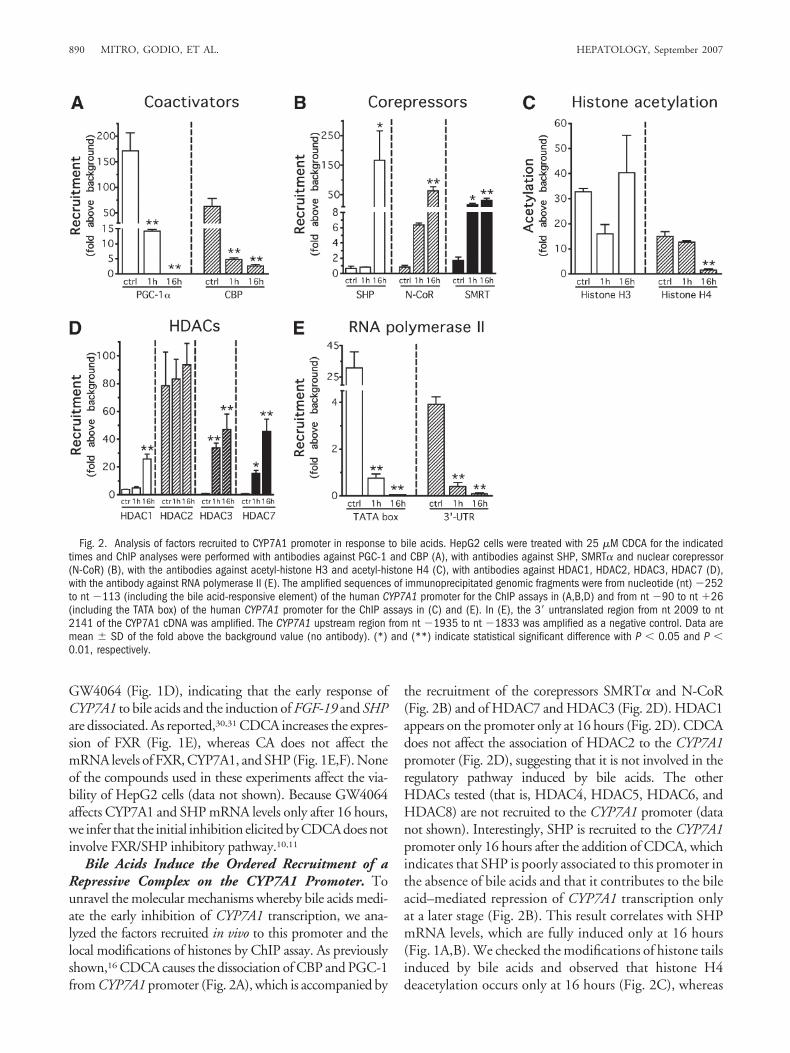
Bile Acids Induce the Ordered Recruitment of aRepressive Complex on the CYP7A1 Promoter. Tounravel the molecular mechanisms whereby bile acids medi-ate the early inhibition of CYP7A1 transcription, we ana-lyzed the factors recruited in vivo to this promoter and thelocal modifications of histones by ChIP assay. As previouslyshown,16 CDCA causes the dissociation of CBP and PGC-1from CYP7A1 promoter (Fig. 2A), which is accompanied by
the recruitment of the corepressors SMRT� and N-CoR(Fig. 2B) and of HDAC7 and HDAC3 (Fig. 2D). HDAC1appears on the promoter only at 16 hours (Fig. 2D). CDCAdoes not affect the association of HDAC2 to the CYP7A1promoter (Fig. 2D), suggesting that it is not involved in theregulatory pathway induced by bile acids. The otherHDACs tested (that is, HDAC4, HDAC5, HDAC6, andHDAC8) are not recruited to the CYP7A1 promoter (datanot shown). Interestingly, SHP is recruited to the CYP7A1promoter only 16 hours after the addition of CDCA, whichindicates that SHP is poorly associated to this promoter inthe absence of bile acids and that it contributes to the bileacid–mediated repression of CYP7A1 transcription onlyat a later stage (Fig. 2B). This result correlates with SHPmRNA levels, which are fully induced only at 16 hours(Fig. 1A,B). We checked the modifications of histone tailsinduced by bile acids and observed that histone H4deacetylation occurs only at 16 hours (Fig. 2C), whereas
Fig. 2. Analysis of factors recruited to CYP7A1 promoter in response to bile acids. HepG2 cells were treated with 25 �M CDCA for the indicatedtimes and ChIP analyses were performed with antibodies against PGC-1 and CBP (A), with antibodies against SHP, SMRT� and nuclear corepressor(N-CoR) (B), with the antibodies against acetyl-histone H3 and acetyl-histone H4 (C), with antibodies against HDAC1, HDAC2, HDAC3, HDAC7 (D),with the antibody against RNA polymerase II (E). The amplified sequences of immunoprecipitated genomic fragments were from nucleotide (nt) �252to nt �113 (including the bile acid-responsive element) of the human CYP7A1 promoter for the ChIP assays in (A,B,D) and from nt �90 to nt �26(including the TATA box) of the human CYP7A1 promoter for the ChIP assays in (C) and (E). In (E), the 3� untranslated region from nt 2009 to nt2141 of the CYP7A1 cDNA was amplified. The CYP7A1 upstream region from nt �1935 to nt �1833 was amplified as a negative control. Data aremean � SD of the fold above the background value (no antibody). (*) and (**) indicate statistical significant difference with P � 0.05 and P �0.01, respectively.
890 MITRO, GODIO, ET AL. HEPATOLOGY, September 2007

the acetylation state of histone H3 does not change (Fig.2C).
Finally, we monitored the kinetics of RNA polymeraseII on the core promoter and at the 3� untranslated regionof CYP7A1 and found that the total amount of RNApolymerase II had already decreased at 1 hour on both theTATA box and the 3� untranslated region of the CYP7A1
promoter (Fig. 2E), which probably reflects the early tran-scriptional repression elicited by bile acids.
Comparative studies on apolipoprotein C-III promotershow that CDCA inhibits only after 16 hours (data notshown). These results suggest that bile acids promote, at anearly stage, the recruitment of a repressive complex contain-ing corepressors and HDACs on CYP7A1 promoter.
Fig. 3. HDAC7 is the key factor for the bileacid-mediated repression of CYP7A1 gene tran-scription. (A,B) Specific siRNA oligonucleotidesto silence HDAC7 were transfected into HepG2cells, which were treated with 25 �M CDCA orwith 0.1% ethanol as vehicle (ctrl) for theindicated times. Cells were harvested for RNAextraction and quantitation of CYP7A1 (A) andHDAC7 (B) mRNA by real time Q-PCR. (C,D)Specific siRNA oligonucleotides to silence SHPwere transfected into HepG2 cells, which weretreated with 25 �M CDCA or ethanol for theindicated times and analyzed for specific mRNAcontent as indicated. (E,F) Specific siRNA oli-gonucleotides to silence HDAC1 were trans-fected into HepG2 cells, which were treatedwith 25 �M CDCA or ethanol for the indicatedtimes and analyzed for specific mRNA contentas indicated. (G,H) Specific siRNA oligonucle-otides to silence HDAC3 were transfected intoHepG2 cells, which were treated with 25 �MCDCA or ethanol for the indicated times andanalyzed for specific mRNA content as indi-cated. Scrambled oligonucleotides were usedas negative controls in each RNA interferenceexperiment (CTRLsiRNA). Data are expressed asmean � SD. (*) and (**) indicate statisticalsignificance at P � 0.05 and P � 0.01,respectively.
HEPATOLOGY, Vol. 46, No. 3, 2007 MITRO, GODIO, ET AL. 891

Inhibition of HDAC7 Expression Prevents the Neg-ative Effects of Bile Acids on CYP7A1 Transcription.We next sought to confirm the role of specific HDACs inthe bile acid–induced repression of CYP7A1 transcrip-tion. When HDAC7 is silenced in HepG2 cells, bile acidsdo not decrease CYP7A1 mRNA (Fig. 3A,B). The basalmRNA levels of SHP are not affected in HDAC7-de-pleted cells and are normally induced upon treatmentwith CDCA (Supplementary Fig. 1), indicating that de-spite the normal expression and induction of SHP, bileacids cannot inhibit CYP7A1 transcription whenHDAC7 is silenced. The mRNA levels of HDAC1,HDAC2, and HDAC3 and of other genes relevant tolipid metabolism and transport are not affected inHDAC7-silenced cells (Supplementary Fig. 1). In con-trast, siRNA against SHP (Fig. 3C,D), HDAC1 (Fig.3E,F), HDAC3 (Fig. 3G,H), and GAPDH (Supplemen-tary Fig. 4) do not affect the response of HepG2 cells toCDCA. Notably, knockdown of HDCA7 does not blockCYP7A1 repression by GW4064 (Supplementary Fig. 5).
In summary, these results underscore the key role ofHDAC7 in the feedback regulation of CYP7A1 gene tran-scription by bile acids.
Bile Acids Induce the Nuclear Localization ofHDAC7. To assess if CDCA affects the intracellular lo-calization of HDAC7, we used immunocytochemistryand found that HDAC7 is mostly localized in the cyto-plasm in control HepG2 cells32 (Fig. 4A), whereasHDAC7 is mainly localized in the nuclear compartmentin HepG2 cells exposed to CDCA for 1 hour and for 16hours (Fig. 4A,B). In contrast, GW4064 does not affectthe intracellular localization of HDAC7 (GW4064) (Fig.4A). This experiment shows that bile acids induce therapid nuclear localization of HDAC7 in an FXR-inde-pendent fashion, an event that most likely facilitates itsrecruitment on the CYP7A1 promoter.
To further support the role of HDAC7 in bile acid–mediated down-regulation of CYP7A1 transcription, weasked whether the inhibition of the nuclear import ofHDAC7 could prevent the repressive effect of bile acids.
Fig. 4. Bile acids induce the nuclear localization of HDAC7 via an FXR-independent pathway. (A) HepG2 cells were cultured on coverslips and treatedwith 25 �M CDCA or 5 �M GW4064 for the indicated times. Coverslips were incubated with a polyclonal antibody against HDAC7 and then with Alexa Fluor488 goat anti-rabbit IgG secondary antibody. Alexa Fluor 546 phalloidin and TOPRO-3 iodide were added to stain the cytoskeleton and the nucleus,respectively. Images were finally merged into a single layer and analyzed by confocal microscopy (magnification was 40�). (B) For the quantitation ofHDAC7-containing nuclei, we counted at least 3 different fields in each coverslip in 2 distinct coverslips. Data are expressed as mean � SD of the percentageof nuclei positive to HDAC7 staining. (**) indicates statistical significance at P � 0.01.
892 MITRO, GODIO, ET AL. HEPATOLOGY, September 2007

For this purpose, we used calyculin A, a phosphatase in-hibitor previously shown to inhibit the nuclear import ofHDAC7 due to the interaction with cytosolic adaptorproteins 14-3-3.33 HepG2 cells were incubated for 1 hourin the absence or presence of combinations of calyculin Aand CDCA. In the presence of calyculin A, CDCA can-not promote the nuclear import of HDAC7 (Fig. 5A).Calyculin A also prevents the CDCA-mediated decreaseof CYP7A1 expression (Fig. 5B).
HDAC Inhibitors Prevent the Feedback Regulationof CYP7A1 Transcription. The sequential recruit-ment of HDAC7, HDAC3, and HDAC1 on theCYP7A1 promoter led us to hypothesize that HDACinhibitors may prevent this negative regulation. There-fore, we measured CYP7A1 mRNA levels in HepG2cells incubated with CDCA, the HDAC inhibitors
VPA and TSA, and a combination of CDCA and VPAor TSA. As expected, CDCA decreased CYP7A1 tran-script levels but VPA and TSA completely preventedthe negative effect of bile acids (Fig. 6A). Possible toxiceffects of VPA and TSA were ruled out by a cytotoxic-ity test (data not shown). The same experiment wasalso performed with GW4064 but in this case theHDAC inhibitors do not prevent the repression elic-ited by GW4064 (Fig. 6B).
HDAC inhibitors reduce low-density lipoproteincholesterol in vivo by increasing the expression ofCyp7a1 and bile acid synthesis. The detailed analysis ofthe molecular mechanisms underlying the regulation ofCYP7A1 transcription suggests that targeting HDACswith an appropriate inhibitor may release Cyp7a1 fromthe physiological repression caused by bile acids in vivo
Fig. 5. Phosphatase inhibitors prevent the translocation of HDAC7 to the nucleus and the transcriptional repression of CYP7A1 by bile acids. HepG2cells were treated with combinations of 10 nM calyculin A and 25 �M CDCA as indicated, for 1 hour and analyzed for the subcellular localizationof HDAC7 by confocal microscopy (magnification was 20�) (A) and CYP7A1 mRNA by real-time Q-PCR (B). Data are expressed as mean � SD. (**)indicates statistical significance at P � 0.01.
HEPATOLOGY, Vol. 46, No. 3, 2007 MITRO, GODIO, ET AL. 893

and, consequently, may also decrease blood cholesterol asa result of its enhanced conversion to bile acids. To testthis hypothesis and to verify the effects of HDAC inhib-itors in vivo, we performed experiments with the Ldl-r�/�
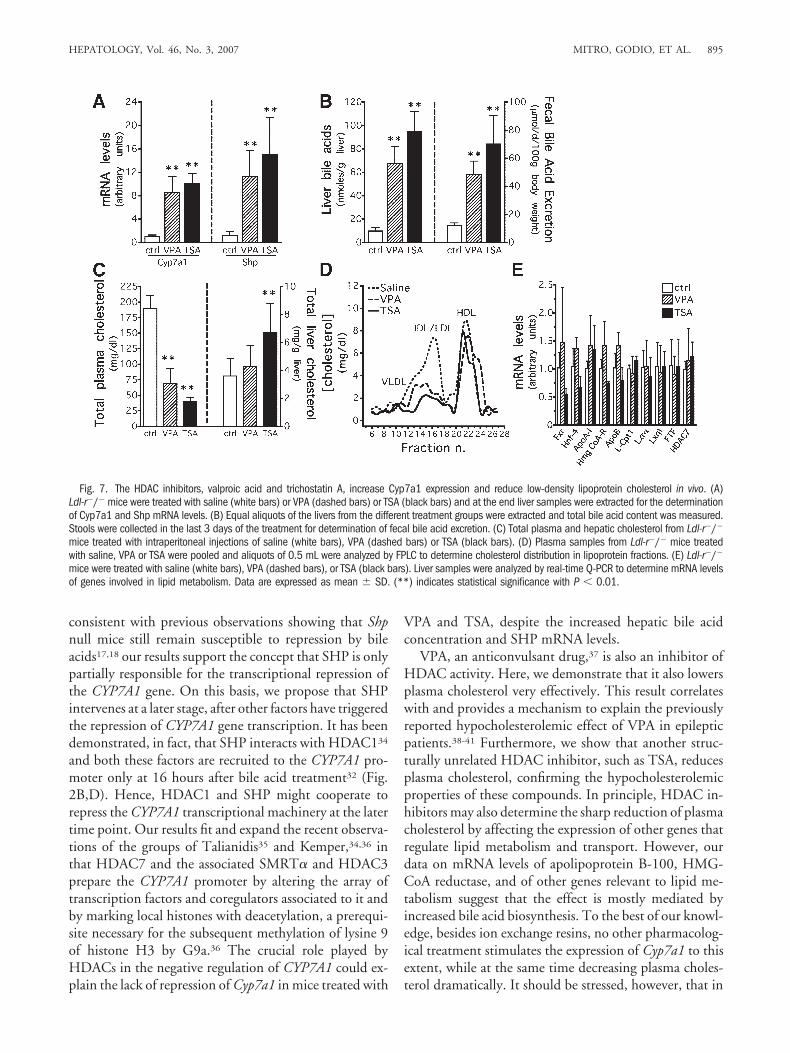
mouse, an animal model of hypercholesterolemia causedby the genetic deficiency of the low-density lipoproteinreceptor. We chose VPA and TSA because they are welltolerated in animals even at high doses.24,26 VPA and TSAelevate Cyp7a1 mRNA levels dramatically in Ldl-r�/�
mice (Fig. 7A). Interestingly, we found that the mRNAlevels of Shp are also elevated in mice treated with bothHDAC inhibitors (Fig. 7A), as a consequence of the in-creased total bile acid concentration in the liver and fecal
bile acid excretion (Fig. 7B), the latter considered an in-dex of bile acid synthesis.28 Notably, both VPA and TSAreduce total plasma cholesterol levels very effectively,whereas hepatic cholesterol does not decrease (Fig. 7C).FPLC analysis of lipoprotein fractions shows that the ad-ministration of VPA or TSA decreases low-density li-poprotein cholesterol in Ldl-r�/� mice whereas the high-density lipoprotein peak is not altered (Fig. 7D). Finally,to make sure that the decrease of plasma cholesterol is dueprimarily to the enhanced expression of the Cyp7a1 gene,we also measured the expression of a subset of genes rele-vant to lipid metabolism and found no changes (Fig. 7E)that could contribute to the hypocholesterolemic effect ofVPA and TSA. Altogether, these results reveal for the firsttime that the inhibition of HDACs, by enhancing theconversion of cholesterol to bile acids, may be a therapeu-tic strategy for the treatment of hypercholesterolemia.
DiscussionThe kinetic analysis of mRNA levels and of cofactors
recruited on the CYP7A1 promoter allowed us to showthat bile acids repress CYP7A1 transcription in two waves,the first one involving a repressive complex containingHDAC3 and HDAC7, the latter the FXR/SHP cascade.The combination of results obtained by ChIP, RNA in-terference, and immunocytochemistry analysis suggeststhat the early stage of CYP7A1 transcriptional repressionis due to the recruitment of a repressor complex on thispromoter containing HDAC7, HDAC3, the corepressorsSMRT�, and N-CoR that promotes the dissociation ofRNA polymerase II.
The FXR/SHP-dependent repression occurs only afterseveral hours, as demonstrated by the delayed increase ofSHP mRNA. One may argue that the basal intracellularconcentrations of SHP may be already sufficient to sus-tain the repression of CYP7A1 gene transcription elicitedby bile acids. However, ChIP assay demonstrates thatSHP does not associate to the CYP7A1 gene promoterduring the initial exposure of liver cells to bile acids. Theindispensable role of HDAC7 at early time points, dem-onstrated in the knockdown experiments (Fig. 3A) and inthe experiments with calyculin A (Fig. 5), suggests thatthe initial events triggered on CYP7A1 gene promoter byHDAC7 in response to bile acids are strictly required. Wealso showed that the silencing of SHP in cell culture doesnot prevent the repression of CYP7A1 by bile acids.Moreover, even if SHP mRNA is normally induced bybile acids in HDAC7-silenced HepG2 cells (Supplemen-tary Fig. 1) as well as in mice with elevated bile acidconcentration in the liver after treatment with HDACinhibitors (Fig. 7A), this does not seem to be sufficient toachieve the repression of CYP7A1 transcription. Thus,
Fig. 6. HDAC inhibitors prevent the feedback regulation of CYP7A1 bybile acids. (A) HepG2 cells were cultured in the presence of differentcombinations of 25 �M CDCA and 1.5 mM VPA or 200 nM TSA for 16hours as indicated (C�T� CDCA�TSA; C�V� CDCA�VPA). CYP7A1mRNA levels are expressed relative to the control wells treated withvehicle (ethanol, EtOH) and are expressed as mean � SD. (B) HepG2cells were cultured in the presence of different combinations of 5 �MGW4064 (GW) and 1.5 mM VPA or 200 nM TSA for 16 hours asindicated (GW�T� GW4064�TSA; GW�V� GW4064�VPA). CYP7A1mRNA levels are expressed relative to the control wells treated withvehicle (ethanol, EtOH) and are expressed as mean � SD. (*) and (**)indicate statistical significance versus control (EtOH) at P � 0.05 andP � 0.01, respectively.
894 MITRO, GODIO, ET AL. HEPATOLOGY, September 2007
consistent with previous observations showing that Shpnull mice still remain susceptible to repression by bileacids17,18 our results support the concept that SHP is onlypartially responsible for the transcriptional repression ofthe CYP7A1 gene. On this basis, we propose that SHPintervenes at a later stage, after other factors have triggeredthe repression of CYP7A1 gene transcription. It has beendemonstrated, in fact, that SHP interacts with HDAC134
and both these factors are recruited to the CYP7A1 pro-moter only at 16 hours after bile acid treatment32 (Fig.2B,D). Hence, HDAC1 and SHP might cooperate torepress the CYP7A1 transcriptional machinery at the latertime point. Our results fit and expand the recent observa-tions of the groups of Talianidis35 and Kemper,34,36 inthat HDAC7 and the associated SMRT� and HDAC3prepare the CYP7A1 promoter by altering the array oftranscription factors and coregulators associated to it andby marking local histones with deacetylation, a prerequi-site necessary for the subsequent methylation of lysine 9of histone H3 by G9a.36 The crucial role played byHDACs in the negative regulation of CYP7A1 could ex-plain the lack of repression of Cyp7a1 in mice treated with
VPA and TSA, despite the increased hepatic bile acidconcentration and SHP mRNA levels.
VPA, an anticonvulsant drug,37 is also an inhibitor ofHDAC activity. Here, we demonstrate that it also lowersplasma cholesterol very effectively. This result correlateswith and provides a mechanism to explain the previouslyreported hypocholesterolemic effect of VPA in epilepticpatients.38-41 Furthermore, we show that another struc-turally unrelated HDAC inhibitor, such as TSA, reducesplasma cholesterol, confirming the hypocholesterolemicproperties of these compounds. In principle, HDAC in-hibitors may also determine the sharp reduction of plasmacholesterol by affecting the expression of other genes thatregulate lipid metabolism and transport. However, ourdata on mRNA levels of apolipoprotein B-100, HMG-CoA reductase, and of other genes relevant to lipid me-tabolism suggest that the effect is mostly mediated byincreased bile acid biosynthesis. To the best of our knowl-edge, besides ion exchange resins, no other pharmacolog-ical treatment stimulates the expression of Cyp7a1 to thisextent, while at the same time decreasing plasma choles-terol dramatically. It should be stressed, however, that in
Fig. 7. The HDAC inhibitors, valproic acid and trichostatin A, increase Cyp7a1 expression and reduce low-density lipoprotein cholesterol in vivo. (A)Ldl-r�/� mice were treated with saline (white bars) or VPA (dashed bars) or TSA (black bars) and at the end liver samples were extracted for the determinationof Cyp7a1 and Shp mRNA levels. (B) Equal aliquots of the livers from the different treatment groups were extracted and total bile acid content was measured.Stools were collected in the last 3 days of the treatment for determination of fecal bile acid excretion. (C) Total plasma and hepatic cholesterol from Ldl-r�/�
mice treated with intraperitoneal injections of saline (white bars), VPA (dashed bars) or TSA (black bars). (D) Plasma samples from Ldl-r�/� mice treatedwith saline, VPA or TSA were pooled and aliquots of 0.5 mL were analyzed by FPLC to determine cholesterol distribution in lipoprotein fractions. (E) Ldl-r�/�
mice were treated with saline (white bars), VPA (dashed bars), or TSA (black bars). Liver samples were analyzed by real-time Q-PCR to determine mRNA levelsof genes involved in lipid metabolism. Data are expressed as mean � SD. (**) indicates statistical significance with P � 0.01.
HEPATOLOGY, Vol. 46, No. 3, 2007 MITRO, GODIO, ET AL. 895

this study VPA and TSA were used mainly as tools todemonstrate that pharmacological inhibition ofHDAC activity may be a promising approach tostrongly up-regulate CYP7A1 and consequently lowerplasma cholesterol. Future investigations should beaimed at identifying more selective HDAC inhibitorsthat decrease plasma cholesterol by stimulating its con-version to bile acids.
In conclusion, we define here the molecular events andthe factors that determine the rapid bile acid–mediated re-pression of CYP7A1 transcription. The relevant conse-quence of this study is the identification of HDACs, and inparticular HDAC7, as possible targets for the design of hy-pocholesterolemic agents that may help in the prevention ofthe associated risk of atherosclerosis and coronary artery dis-ease, especially in those patients that do not respond very wellto other hypocholesterolemic drugs. Moreover, modulationof bile acid synthesis by targeting these mechanisms may alsobe exploited for the treatment of liver diseases.
Acknowledgment: We thank Drs. Enrique Saez(The Scripps Research Institute, La Jolla, CA), IannisTalianidis (Biomedical Sciences Research Center “Al-exander Fleming”, Vari, Greece), Walter Wahli (Cen-ter for Integrative Genomics, Universite de Lausanne,Lausanne, Switzerland), Ronald Evans (The Salk Insti-tute for Biological Studies, San Diego, CA) and LauraCalabresi (Dipartimento di Scienze Farmacologiche,Universita degli Studi di Milano, Italia) for criticallyreading the manuscript and for helpful discussion. Weare grateful to Drs. E. Verdin (The Gladstone Instituteof Virology and Immunology, San Francisco, CA), E.Seto (Moffitt Cancer Center, Tampa, FL), R. M. Evans(The Salk Institute, San Diego, CA), I. Talianidis (Bio-medical Sciences Research Center “Alexander Flem-ing”, Vari, Greece), and K. Bamberg (Astra-Zeneca,Molndal, Sweden) for kindly providing plasmids, an-tibodies, and other reagents. We also thank Drs. LauraCalabresi, Giulio Simonutti, and Danilo Norata (Di-partimento di Scienze Farmacologiche, Universita diMilano) for help with FPLC analyses of plasma li-poproteins and immunocytochemistry and Miss EldaDesiderio Pinto for administrative assistance.
References1. Russell DW, Setchell KDR. Bile acid biosynthesis. Biochemistry 1992;31:
4737-4749.2. Javitt NB. Bile acid synthesis from cholesterol: regulatory and auxiliary
pathways. FASEB J 1994;8:1308-1311.3. Russell DW. Nuclear orphan receptors control cholesterol catabolism. Cell
1999;97:539-542.4. Pullinger CR, Eng C, Salen G, Shefer S, Batta AK, Erickson SK, et al.
Human cholesterol 7�-hydroxylase (CYP7A1) deficiency has a hypercho-lesterolemic phenotype. J Clin Invest 2002;110:109-117.
5. Stravitz RT, Hylemon PB, Heuman DM, Hagey LR, Schteingart CD,Ton-Nu H-T, et al. Transcriptional regulation of cholesterol 7�-hydrox-
ylase mRNA by conjugated bile acids in primary cultures of rat hepato-cytes. J Biol Chem 1993;268:13987-13993.
6. Stroup D, Crestani M, Chiang JY. Identification of a bile acid responseelement in the cholesterol 7�-hydroxylase gene (CYP7A). Am J Physiol1997;273:G508-G517.
7. Chiang JYL. Bile acid regulation of hepatic physiology: III. Bile acids andnuclear receptors. Am J Physiol Gastrointest Liver Physiol 2003;284:G349-G356.
8. Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al.Identification of a nuclear receptor for bile acids. Science 1999;284:1362-1365.
9. Chiang JYL, Kimmel R, Weinberger C, Stroup D. Farnesoid X receptorresponds to bile acids and represses cholesterol 7�-hydroxylase gene(CYP7A1) transcription. J Biol Chem 2000;275:10918-10924.
10. Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, etal. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1represses bile acid biosynthesis. Mol Cell 2000;6:517-526.
11. Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, et al.Molecular basis for feedback regulation of bile acid synthesis by nuclearreceptors. Mol Cell 2000;6:507-515.
12. Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, et al. Defi-nition of a novel growth factor-dependent signal cascade for the suppres-sion of bile acid biosynthesis. Genes Dev 2003;17:1581-1591.
13. Stravitz RT, Vlahcevic ZR, Gurley EC, Hylemon PB. Repression of cho-lesterol 7�-hydroxylase transcription by bile acids is mediated throughprotein kinase C in primary cultures of rat hepatocytes. J Lipid Res 1995;36:1359-1369.
14. Gupta S, Stravitz RT, Dent P, Hylemon PB. Down-regulation of choles-terol 7�-hydroxylase (CYP7A1) gene expression by bile acids in primary rathepatocytes is mediated by the c-Jun N-terminal kinase pathway. J BiolChem 2001;276:15816-15822.
15. De Fabiani E, Mitro N, Anzulovich AC, Pinelli A, Galli G, Crestani M.The negative effects of bile acids and tumor necrosis factor-� on the tran-scription of cholesterol 7�-hydroxylase gene (CYP7A1) converge to hepa-tocyte nuclear factor-4. A novel mechanism of feedback regulation of bileacid synthesis mediated by nuclear receptors. J Biol Chem 2001;276:30708-30716.
16. De Fabiani E, Mitro N, Gilardi F, Caruso D, Galli G, Crestani M. Coor-dinated control of cholesterol catabolism to bile acids and of gluconeogen-esis via a novel mechanism of transcription regulation linked to the fasted-to-fed cycle. J Biol Chem 2003;278:39124-39132.
17. Kerr T, Saeki S, Schneider M, Schaefer K, Berdy S, Redder T, et al. Loss ofnuclear receptor SHP impairs but does not eliminate negative feedbackregulation of bile acid synthesis. Dev Cell 2002;2:713-720.
18. Wang L, Lee Y, Bundman D, Han Y, Thevananther S, Kim C, et al.Redundant pathways for negative feedback regulation of bile acid produc-tion. Dev Cell 2002;2:721-731.
19. Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circu-lation 2004;109(23 Suppl 1):III39–43.
20. Bruckert E, Giral P, Tellier P. Perspectives in cholesterol-lowering therapy:the role of ezetimibe, a new selective inhibitor of intestinal cholesterolabsorption. Circulation 2003;107:3124-3128.
21. Kwiterovich PO Jr, Fredrickson DS, Levy RI. Familial hypercholesterol-emia (one form of familial type II hyperlipoproteinemia). A study of itsbiochemical, genetic and clinical presentation in childhood. J Clin Invest1974;53:1237-1249.
22. Spady DK, Cuthbert JA, Willard MN, Meidell RS. Overexpression ofcholesterol 7�-hydroxylase (CYP7A) in mice lacking the low density li-poprotein (LDL) receptor gene. J Biol Chem 1998;273:126-132.
23. Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. his-tone deacetylase is a direct target of valproic acid, a potent anticonvulsant,mood stabilizer, and teratogen. J Biol Chem 2001;276:36734-36741.
24. Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, et al.Valproic acid defines a novel class of HDAC inhibitors inducing differen-tiation of transformed cells. EMBO J 2001;20:6969-6978.
896 MITRO, GODIO, ET AL. HEPATOLOGY, September 2007

25. Minucci S, Horn V, Bhattacharyya N, Russanova V, Ogryzko VV, Gabriele L,et al. A histone deacetylase inhibitor potentiates retinoid receptor action inembryonal carcinoma cells. Proc Natl Acad Sci U S A 1997;94:11295-11300.
26. Vigushin DM, Ali S, Pace PE, Mirsaidi N, Ito K, Adcock I, et al. Tricho-statin A is a histone deacetylase inhibitor with potent antitumor activityagainst breast cancer in vivo. Clin Cancer Res 2001;7:971-976.
27. Crestani M, Mitro N, Godio C, inventors; Inhibition of histone deacetylasesfor the treatment of hyperlipidaemias and prevention of atherosclerosis andcardiovascular diseases. Patent no. WO2005105066. April 29, 2005.
28. Turley SD, Spady DK, Dietschy JM. Regulation of fecal bile acid excretionin male golden Syrian hamsters fed a cereal-based diet with and withoutadded cholesterol. HEPATOLOGY 1997;25:797-803.
29. Claudel T, Inoue Y, Barbier O, Duran-Sandoval D, Kosykh V, Fruchart J,et al. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIIIexpression. Gastroenterology 2003;125:544-555.
30. Xu G, Pan L-x, Erickson SK, Forman BM, Shneider BL, Ananthanaray-anan M, et al. Removal of the bile acid pool upregulates cholesterol 7�-hydroxylase by deactivating FXR in rabbits. J Lipid Res 2002;43:45-50.
31. Xu G, Pan L-x, Li H, Forman BM, Erickson SK, Shefer S, et al. Regulationof the farnesoid X receptor (FXR) by bile acid flux in rabbits. J Biol Chem2002;277:50491-50496.
32. Kao H-Y, Verdel A, Tsai C-C, Simon C, Juguilon H, Khochbin S. Mech-anism for nucleocytoplasmic shuttling of histone deacetylase 7. J BiolChem 2001;276:47496-47507.
33. Li X, Song S, Liu Y, Ko S-H, Kao H-Y. Phosphorylation of the histonedeacetylase 7 modulates its stability and association with 14-3-3 proteins.J Biol Chem 2004;279:34201-34208.
34. Kemper JK, Kim H, Miao J, Bhalla S, Bae Y. Role of an mSin3A-Swi/Snfchromatin remodeling complex in the feedback repression of bile acidbiosynthesis by SHP. Mol Cell Biol 2004;24:7707-7719.
35. Boulias K, Talianidis I. Functional role of G9a-induced histone methyl-ation in small heterodimer partner-mediated transcriptional repression.Nucl Acids Res 2004;32:6096-6103.
36. Fang S, Miao J, Xiang L, Ponugoti B, Treuter E, Kemper JK. Coordinatedrecruitment of histone methyltransferase G9a and other chromatin-mod-ifying enzymes in SHP-mediated regulation of hepatic bile acid metabo-lism. Mol Cell Biol 2007;27:1407-1424.
37. Loscher W. Valproate: a reappraisal of its pharmacodynamic propertiesand mechanisms of action. Prog Neurobiol 1999;58:31-59.
38. Heldenberg D, Harel S, Holtzman M, Levtow O, Tamir I. The effect ofchronic anticonvulsant therapy on serum lipids and lipoproteins in epilep-tic children. Neurology 1983;33:510-513.
39. Eiris JM, Lojo S, Del Rio MC, Novo I, Bravo M, Pavon P, et al. Effects oflong-term treatment with antiepileptic drugs on serum lipid levels in chil-dren with epilepsy. Neurology 1995;45:1155-1157.
40. Calandre EP, Rodriquez-Lopez C, Blazquez A, Cano D. Serum lipids,lipoproteins and apolipoproteins A and B in epileptic patients treated withvalproic acid, carbamazepine or phenobarbital. Acta Neurol Scand 1991;83:250-253.
41. Nikolaos T, Stylianos G, Chryssoula N, Irini P, Christos M, Dimitrios T,et al. The effect of long-term antiepileptic treatment on serum cholesterol(TC, HDL, LDL) and triglyceride levels in adult epileptic patients onmonotherapy. Med Sci Monit 2004;10:MT50-MT52.
HEPATOLOGY, Vol. 46, No. 3, 2007 MITRO, GODIO, ET AL. 897



















