
Valerian extract and valerenic acid are partial agonists of the 5-HT5a receptor in vitro

Modification on Ursodeoxycholic Acid (UDCA) Scaffold. Discovery ofBile Acid Derivatives As Selective Agonists of Cell-Surface G‑ProteinCoupled Bile Acid Receptor 1 (GP-BAR1)Valentina Sepe,† Barbara Renga,‡ Carmen Festa,† Claudio D’Amore,‡ Dario Masullo,† Sabrina Cipriani,‡
Francesco Saverio Di Leva,† Maria Chiara Monti,§ Ettore Novellino,† Vittorio Limongelli,†
Angela Zampella,*,† and Stefano Fiorucci‡
†Department of Pharmacy, University of Naples “Federico II”, Via D. Montesano, 49, 80131 Napoli, Italy‡Department of Clinical and Experimental Medicine, Nuova Facolta di Medicina, Via Gambuli 1 06132 Perugia, Italy§Department of Pharmacy, University of Salerno, Via Giovanni Paolo II, 132, 84084, Fisciano (Salerno), Italy
*S Supporting Information
ABSTRACT: Bile acids are signaling molecules interactingwith the nuclear receptor FXR and the G-protein coupledreceptor 1 (GP-BAR1/TGR5). GP-BAR1 is a promisingpharmacological target for the treatment of steatohepatitis,type 2 diabetes, and obesity. Endogenous bile acids andcurrently available semisynthetic bile acids are poorly selectivetoward GP-BAR1 and FXR. Thus, in the present study we haveinvestigated around the structure of UDCA, a clinically usedbile acid devoid of FXR agonist activity, to develop a largefamily of side chain modified 3α,7β-dihydroxyl cholanoids thatselectively activate GP-BAR1. In vivo and in vitro pharmaco-logical evaluation demonstrated that administration ofcompound 16 selectively increases the expression of pro-glucagon 1, a GP-BAR1 target, in the small intestine, while ithad no effect on FXR target genes in the liver. Further, compound 16 results in a significant reshaping of bile acid pool in arodent model of cholestasis. These data demonstrate that UDCA is a useful scaffold to generate novel and selective steroidalligands for GP-BAR1.
■ INTRODUCTION
Bile acids (BAs) are signaling molecules interacting with twotypes of dedicated cellular receptors, intracellular nuclearreceptors, and cell-surface receptors. Nuclear receptors includefarnesoid X receptor (FXR),1,2 pregnane X receptor (PXR),3
vitamin D3 receptor (VDR),4 and constitutive androstanereceptor (CAR),5 with FXR identified as the endogenous bileacid sensor.Highly expressed in enterohepatic tissues (liver and
intestine), FXR regulates bile acid homeostasis, preventingtheir accumulation and associated toxicity through theinduction of gene expression for conjugating enzymes andexport pumps with the final aim to provide bile acid eliminationfrom the body. Apart from this pivotal role, FXR controlsfurther important metabolic pathways6 regulating also lipid7
and glucose homeostasis.8 Additionally, FXR agonists wereproved to exert anti-inflammatory9 and antifibrotic effects,10
making this nuclear receptor an appealing pharmacologicaltarget in the treatment of common human diseases rangingfrom metabolic syndrome to cancer.11
Cell-surface G-protein coupled bile acid receptor 1 (GP-BAR1, TGR5, M-BAR1) belongs to the rhodopsin-likesuperfamily of G protein coupled receptors (GPCRs). GP-BAR1 activation results in the elevation of intracellular cAMPlevels, which in turn activates specific intracellular signalingcascades. Similarly to FXR, GP-BAR1 is highly expressed in theliver and in the intestine, but differently from FXR it is alsolocated in tissues and cells not participating in the enter-ohepatic circulation such as muscle, brain, adipose tissue,macrophages, and endothelial cells. In muscle and brownadipose tissue, GP-BAR1 activation increases energy expendi-ture and oxygen consumption,12 while in entero-endocrine Lcells, it stimulates the secretion of glucagon-like peptide (GLP)-1, an incretin that improves insulin release from pancreas, thusregulating glucose blood levels, gastrointestinal motility, andappetite.13
All these data underlay the role of GP-BAR1 agonists inpreventing obesity, atherosclerosis, and hepatic steatosis in
Received: June 11, 2014Published: August 27, 2014
Article
pubs.acs.org/jmc
© 2014 American Chemical Society 7687 dx.doi.org/10.1021/jm500889f | J. Med. Chem. 2014, 57, 7687−7701

mice on a high-fat diet, thus pointing toward GP-BAR1 as apromising target in the pharmacotherapy of enterohepatic andmetabolic disorders.14−16
Thus, today, several efforts are focused toward the discoveryand development of potent and selective GP-BAR1 smallmolecule agonists belonging to bile acid or nonbile acid classesas promising therapeutic strategy in lipid and glucose diseasessuch as in the treatment of nonalcoholic steatohepatitis,hypercholesterolaemia, hypertriglyceridaemia, and type 2diabetes mellitus (T2DM).14−16
In this context, the identification of potent GP-BAR1 smallmolecule agonists with improved selectivity over FXR remainsthe aim of the medicinal chemists.From a chemical point of view, BAs are truncated cholesterol
side chain derivatives (Figure 1). Their molecular repertoire is
generated first in the liver with the production of primary bileacids, cholic acid (CA), and chenodeoxycholic acid (CDCA),successively subjected to microbial transformation in the gut,affording secondary bile acids, deoxycholic acid (DCA), andlithocholic acid (LCA) and their glycine and taurine conjugates.Interestingly, the activity toward the two BAs receptors isstructure dependent, with CDCA and its conjugated forms themost potent endogenous FXR activators and LCA and tauro-lithocholic acid (TLCA) the strongest natural agonists of GP-BAR1.Among BAs, ursodeoxycholic acid (UDCA) represents a
puzzling molecule. Present in human bile at low concentrations,
about 3% of total bile acids, UDCA has been shown effective inbiliary and liver diseases and is now considered as the first-linetreatment for primary biliary cirrhosis (PBC), primarysclerosing cholangitis (PSC), intrahepatic cholestasis ofpregnancy (ICP), and several less common adult and pediatriccholestatic conditions.17 UDCA is a molecule with a goodsafety profile and a clinical history with minimal side effects,also when used in large doses. Additionally, UDCA is endowedwith several beneficial effects such as neuroprotective andantiapoptotic actions, chemopreventative effects in colon cancerwith attenuation of carcinogenic signaling cascades induced bytoxic bile acids.18−20
UDCA is the 7β-hydroxy epimer of CDCA and, despite itspharmacological profile resembling a modulation of FXR,UDCA is not a FXR agonist and its mechanism of actionremains unclear and still subjected to intense scientific debates.In this context we have decided to manipulate UDCA
chemical scaffold to obtain selective GP-BAR1 agonists. Thus, aseries of side chain modified 3α,7β-dihydroxyl cholanoids(Figure 2) has been prepared and screened in their ability tomodulate FXR/GP-BAR1. This research work resulted in theidentification of several UDCA analogues endowed with GP-BAR1 selective agonistic profile. Computational studies havebeen performed to elucidate their binding mechanism to GP-BAR1 and the pharmacological activity of derivative 16, themost potent compound generated in this study, has beencharacterized through a series of in vitro and in vivoexperiments.
■ RESULTS
The steroidal nucleus of both CA and CDCA adopts a bentshape due to the A/B cis ring juncture that forces ring A to lieoutside of the plane of the BCD ring system. As a result, twowell distinct faces are generated: (i) the hydrophobic area (β-site) of the cholanoic acid nucleus and (ii) the α-hydrophilicregion defined by the two hydroxyl groups at C3 and C7. Thesestructural features allow primary BAs (CDCA and CA) to bindthe FXR ligand binding domain (FXR-LBD) and activate thisnuclear receptor.21 If compared with CDCA, the reduced FXR
Figure 1. Endogenus bile acids.
Figure 2. Ursodeoxycholane derivatives generated in this study.
Journal of Medicinal Chemistry Article
dx.doi.org/10.1021/jm500889f | J. Med. Chem. 2014, 57, 7687−77017688

activation of UDCA can be explained by the lower hydro-phobicity of the β-site due to the presence of the β-orientedhydroxyl group at C7. This stresses the pharmacophoric role ofthe configuration at C7 hydroxyl methine carbon in bile acidderivatives to achieve FXR agonism. On the other hand,according to structural studies, the BAs alkyl side chain pointstoward a rather large and polar cavity both in FXR and GP-BAR1.21,22 Consequently, this part of the BAs can befunctionalized by producing derivatives endowed with different
pharmacological profiles, ranging from full potent dualagonism22 to selective FXR or GP-BAR1 modulation.23,24
To get more insights into the structural requisites ruling theactivity of bile acid derivatives on GP-BAR1 and FXR, we havedeveloped a library of compounds introducing on the UDCAscaffold: (i) side chains of different length (C23, C24, and C26)and flexibility, (ii) side chains functionalized with a number ofdiverse polar functional groups such as carboxylic, alcoholic,and sulfate moieties. In fact, we have recently identified a
Scheme 1. nor-Ursodeoxycholane Derivativesa
aReagents and conditions: (a) HCOOH, HClO4, 96%; (b) TFA, trifluoroacetic anhydride, NaNO2, 96%; (c) KOH 30% in MeOH/H2O 1:1 v/v,97%; (d) p-TsOH, MeOH dry, 97%; (e) DMT-MM, Et3N, taurine, DMF dry, 28%; (f) LiBH4, MeOH dry, THF, 0 °C, quantitative yield; (g) Et3N·SO3, DMF, 95 °C.
Scheme 2. Ursodeoxycholane and Bis-homo Derivativesa
aReagents and conditions: (a) LiBH4, MeOH dry, THF, 0 °C, 93%; (b) 2,6-lutidine, t-butyldimethylsilyl trifluoromethanesulfonate, CH2Cl2, 0 °C;(c) LiBH4, MeOH dry, THF, 0 °C, quantitative yield over two steps; (d) DMSO, oxalyl chloride, TEA dry, CH2Cl2, −78 °C, thenmethyl(triphenylphosphoranylidene)acetate, 76%; (e) HCl 37%, MeOH, quantitative yield; (f) NaOH 5% in MeOH/H2O 1:1 v/v, 93%; (g) DMT-MM, Et3N, taurine, DMF dry, 26%; (h) H2, Pd(OH)2/C Degussa type, THF/MeOH 1:1, then HCl 37%, MeOH, quantitative yield; (i) NaOH 5%in MeOH/H2O 1:1 v/v, 60%; (j) DMT-MM, Et3N, taurine, DMF dry, 45%; (k) LiBH4, MeOH dry, THF, 0 °C, 77%.
Journal of Medicinal Chemistry Article
dx.doi.org/10.1021/jm500889f | J. Med. Chem. 2014, 57, 7687−77017689

potent GP-BAR1 agonist showing the CDCA scaffold with asulfate group on the side chain.22 Thus, it is worth investigatingthe effect of this substituent on the GP-BAR1 activity of UDCAderivatives.nor-Ursodeoxycholane Derivatives. A key step in the
synthetic protocol toward nor-ursodeoxycholane derivatives wasBeckmann one-carbon degradation at C24 on the UDCAperfomate derivative 21, generated through UDCA Fisher’sesterification with formic acid and acetic anhydride,25 bytreatment with sodium nitrite in a mixture of trifluoroaceticanhydride and trifluoroacetic acid (Scheme 1).26,27
Alkaline hydrolysis of the resulting C23-nitrile (22) furnished11 in 97% chemical yield, which in a small aliquot wassubjected to the reaction of amidation with taurine in thepresence of the versatile coupling agent, 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM),28 giving the amide derivative as ammonium sulfate salt.Purification on RP-18 column followed by HPLC furnished 12in pure form and as sodium salt (28% chemical yield).Methanol/p-toluenesulfonic acid treatment on 11 afforded
C23 methyl ester 13 that in turn was transformed in thecorresponding C23 alcohol 4 by treatment with lithiumborohydride (88% chemical yield over two steps). Sulfation(Et3N·SO3 10 equiv) afforded a crude reaction product(Scheme 1) containing trisulfate derivative 10, the mixture ofdisulfate derivatives 8−9, and the mixture of monosulfatederivatives 5−7. RP-18 column separation with a gradienteluting system from water to methanol followed by HPLC ofthe enriched fractions furnished pure derivatives 5−10, differingin the sulfation pattern on nor-ursodeoxycholane scaffold.Ursodeoxycholane and Bis-homo-ursodeoxycholane
Derivatives. First, treatment of ursodeoxycholic methyl esterwith LiBH4 afforded triol 3 in high chemical yield (Scheme 2).A four-step reaction sequence on UDCA methyl ester,
including protection of alcoholic functions at C3 and C7 (2,6-lutidine, t-butyldimethylsilyl trifluoromethanesulfonate,CH2Cl2), reduction of the side chain methyl ester (LiBH4,MeOH/THF), and subsequent one-pot Swern oxidation/Wittig C2 homologation gave the protected conjugated methylester 24 in a total 76% chemical yield. Silyl deprotectionfurnished pure Δ24,25 ester 18 that was subjected to methylester hydrolysis to give the corresponding bis-homo conjugatedacid 19. Finally, amidation with taurine and RP18/HPLC
purification allowed us to obtain the corresponding tauroconjugated derivative 20.Double bond hydrogenation on 24 gave methyl ester 15,
which was used as starting material in the preparation ofcarboxyl acid 16 and its corresponding alcohol 14, throughtreatment with LiOH and LiBH4, respectively. Finally, 16 waseasily transformed in the corresponding tauro-conjugated(taurine, DMT-MM, 45% yield) derivative 17.In this study, several derivatives were proved selective GP-
BAR1 agonists (see below in In Vitro PharmacologicalEvaluation), e.g., compounds 4, 14, and 16. These resultsrepresent an interesting evidence of the hierarchical role playedby the length and the functionalization of the side chain ratherthan the configuration at C7 in BA recognition of theirreceptors. To give further support to this hypothesis, also bis-homoCDCA (26) and corresponding alcohol 27 were prepared(Scheme 3) starting from CDCA methyl ester and followingthe synthetic protocol already described for the correspondingbis-homo ursodeoxycholane derivatives (Scheme 2).
In Vitro Pharmacological Evaluation. Ursodeoxycholanederivatives (3−20, Figure 2) and chenodeoxycholane analogues26 and 27 were tested in the luciferase reporter assays onHepG2 and HEK-293T cells transfected with FXR and GP-BAR1, respectively. Our pharmacological data suggest that thestereochemistry at C7 is a determinant factor for the activity onFXR and not on GP-BAR1. In fact, as shown in Figures 3 and 4,all the newly synthesized UDCA derivatives, which have the 7β-OH group, are inactive on FXR, while compounds like CDCA,which presents a 7α-OH group, activates this receptor. Atvariance with FXR, in GP-BAR1 the presence of the 7α-OH asin CDCA, the 7β-OH as in 16 and even the absence of thehydroxyl group at C7 as in TLCA are tolerated.Additionally as previously demonstrated,29 side chain
elongation on CDCA scaffold produce positive effects towardFXR when an hydroxyl function is placed as end-terminus (27),whereas the corresponding carboxyl acid derivative 26 wasalmost inactive when tested at 10 μM (Figure 3A). Moreover,when compound 26 was tested at 50 μM in the presence ofCDCA (Figure 3B), the ratio RLU/RRU was higher than thatof CDCA, indicating that 26 might be an FXR modulator.Indeed, 26 was demonstrated cytotoxic on HEK-293T cells(Figure 4B) at 50 μM, thus ruling out the pharmacologicalpotential of this compound.
Scheme 3. Bis-homo Chenodeoxycholane Derivativesa
aReagents and conditions: (a) LiBH4, MeOH dry, THF, 0 °C; (b) 2,6-lutidine, t-butyldimethylsilyl trifluoromethanesulfonate, CH2Cl2, 0 °C; (c)LiBH4, MeOH dry, THF, 0 °C; (d) DMSO, oxalyl chloride, TEA dry, CH2Cl2, −78 °C; (e) LiOH, TEPA, THF dry, reflux; (e) HCl 37%, MeOH,60% yield over five steps; (f) NaOH 5% in MeOH/H2O 1:1 v/v, 78% yield; (g) LiBH4, MeOH dry, THF, 0 °C, 73% yield.
Journal of Medicinal Chemistry Article
dx.doi.org/10.1021/jm500889f | J. Med. Chem. 2014, 57, 7687−77017690

None of the tested compounds turned out to be FXRantagonist (Figure 3B).Results of transactivations of CREB-responsive elements in
HEK-293T cells transiently transfected with the membrane bileacid receptor GP-BAR1 (Figure 4) revealed that allursodeoxycholane alcohols generated in this study, compounds3, 4, and 14, transactivate GP-BAR1, thus demonstrating thatthe introduction of an hydroxyl group on the side chain ofursodeoxycholane scaffold produces, independently by sidechain length (C23 in 4, C24 in 3 and C26 in 14), derivativesendowed with membrane bile acid receptor agonism.
Interestingly, also 16 and the corresponding Δ24,25
conjugated analogue 19 were demonstrated inducers ofcAMP-luciferase reporter gene, with 16 showing a potencysimilar to that of TLCA, the most potent endogenous GP-BAR1 agonist (Figure 4). This result demonstrates that sidechain elongation on the ursodeoxycholane scaffold producepositive effects toward GP-BAR1 activation also when acarboxyl function is placed as end-terminus. The above activitydeclines in the corresponding tauro-conjugates (compare 16with 17 and 19 with 20 in Figure 4), thus demonstrating thatfurther elongation of UDCA side chain is not tolerated by the
Figure 3. Transactivation assays on FXR. (A) HepG2 cells were transfected with pSG5-FXR, pSG5-RXR, pCMV-βgal, and p(hsp27)TKLUCvectors. Cells were stimulated with compounds 3−20, 26, and 27 (10 μM). CDCA (1, 10 μM) was used as a positive control, UDCA (2, 10 μM) asnegative control. Results are expressed as mean ± standard error; *p < 0.05 versus not treated cells (NT). (B) HepG2 cells were transfected withpSG5-FXR, pSG5-RXR, pCMV-βgal, and p(hsp27)TKLUC vectors. Cells were stimulated with CDCA (1) 10 μM in combination with UDCA (2,50 μM) as negative control and with 3−20, 26, and 27, 50 μM. Results are expressed as mean ± standard error. *p < 0.05 versus NT; #p < 0.05versus CDCA; °cytotoxic at 50 μM.
Journal of Medicinal Chemistry Article
dx.doi.org/10.1021/jm500889f | J. Med. Chem. 2014, 57, 7687−77017691

receptor. Of interest is also the behavior of chenodeoxycholanederivatives 26 and 27, with the alcohol 27 almost inactive andthe carboxylic acid 26 less potent than the correspondingUDCA derivative 16.Notably, 3, 4, 14, 16, and 19 are selective GP-BAR1 agonists
devoid of any activity toward FXR (Figure 3). Moreover, noneof tested compounds was able to induce cAMP-luciferasereporter gene in absence of GP-BAR1 (data not shown), thusindicating that the observed cAMP induction is GP-BAR1mediated.Among all tested compounds, our attention was focused on 4
and 16, both selective GP-BAR1 agonists, but quite different intheir chemical scaffold. As shown in Figure 5, compounds 4 and16 exerted a concentration-dependent effect on activation of
cAMP responsive element in HEK-293T cells transfected withGP-BAR1 with an EC50 of 24.4 and 17.2 μM, respectively.
Computational Studies. To elucidate the structuralrequisites of the activity of the UDCA derivatives on GP-BAR1, we performed docking simulations on the most activecompounds of the series (3, 4, 14, and 16). In thesecalculations, we used the tridimensional structure of GP-BAR1 that we have very recently reported22 (see ExperimentalSection for details). The best-scored docking solution showsthe most potent derivative of the series, compound 16, H-bonding with the Glu169 and Asn93 side chains through the3α- and 7β-hydroxyl groups, respectively (Figure 6).On the other side of the molecule, the flexible side chain
points toward the transmembrane (TM) helices 1, 2, and 7,where the ligand carboxylate group engages H-bonds with the
Figure 4. Transactivation assays on GP-BAR1. (A) HEK-293T cells were cotransfected with GP-BAR1 and a reporter gene containing a cAMPresponsive element in front of the luciferase gene. Then 24 h post transfection, cells were stimulated with CDCA (1), UDCA (2), and compounds3−20, 26, and 27 (10 μM). Luciferase activity served as a measure of the rise in intracellular cAMP following activation of GP-BAR1. TLCA (10μM) was used as a positive control. Results are expressed as mean ± standard error. *p < 0.05 versus NT cells. (B) Antagonistic activity ofcompounds 1−27 on transactivation of cAMP responsive element induced by TLCA in HEK-293T cells transfected with GP-BAR1. Experimentalconditions were the same described in (A). None of the compound had an antagonistic activity. Concentrations used were: TLCA 10 μM, while allthe other compounds were tested at the concentration of 50 μM. Results are expressed as mean ± standard error. *p < 0.05 versus NT cells;°cytotoxic at 50 μM.
Journal of Medicinal Chemistry Article
dx.doi.org/10.1021/jm500889f | J. Med. Chem. 2014, 57, 7687−77017692

side chains of Ser21 and Ser270. This ligand binding mode isfurther stabilized by a series of hydrophobic interactionsengaged by the steroidal scaffold with residues such as Leu71,Phe83, Leu174, and Trp237. It is interesting to note that thecomputed binding mode of 16 is very similar to that of otherpotent GP-BAR1 agonists previously reported by our group.22
Furthermore, mutagenesis data showed a reduced activity ofbile acid derivatives in the Asn93Ala, Glu169Ala, and Ser270AlaGP-BAR1 mutants if compared to the wild-type receptor.30 Theinvolvement of these residues in the binding mode of 16 is inline with these experimental observations. The substitution ofthe carboxylate group with a hydroxyl one does not
substantially alter the ligand binding mode. In fact, the alcoholderivative 14 shows a binding mode very similar to that of 16(Figure 6). In particular, the steroidal scaffold of 14 engages thesame interactions established by 16, while its C26 side chainpoints toward the polar region formed by Ser21, Ser267, andSer270, where the hydroxyl group H-bonds with the Ser270side chain.Our results show that in this pocket the presence of a
negatively charged group, as in compound 16, is desirable butnot necessary, while polar groups, able to form H-bondinteractions, are important for GP-BAR1 activation. Our insilico predictions explain the pharmacological data, which show
Figure 5. Concentration−response curves on GP-BAR1 activation by compounds 4 and 16. HEK-293T cells were transfected with GP-BAR1 andstimulated with increasing concentrations of 4 and 16 (0.1, 1, and 10 μM). Results are mean ± standard error of at least three determination induplicate.
Figure 6. Binding mode of 16 (upper left), 14 (upper right), 3 (lower right), and 4 (lower left) in the GP-BAR1 homology model. Ligands arerepresented as cyan (16), green (14), orange (3), and yellow (4) sticks. The receptor is shown as gray cartoons and sticks. Extracellular loops andnonpolar hydrogens are omitted for clarity.
Journal of Medicinal Chemistry Article
dx.doi.org/10.1021/jm500889f | J. Med. Chem. 2014, 57, 7687−77017693
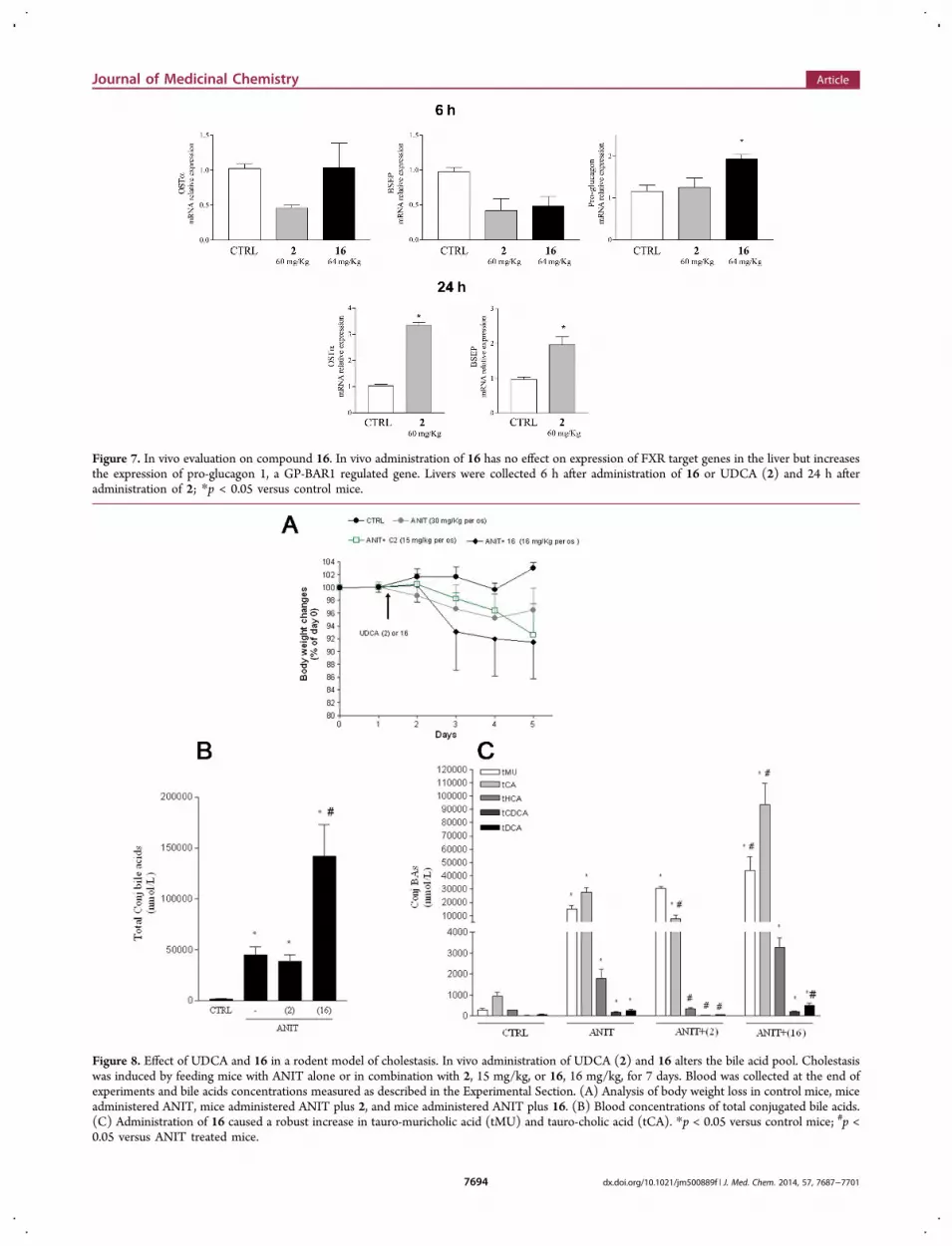
Figure 7. In vivo evaluation on compound 16. In vivo administration of 16 has no effect on expression of FXR target genes in the liver but increasesthe expression of pro-glucagon 1, a GP-BAR1 regulated gene. Livers were collected 6 h after administration of 16 or UDCA (2) and 24 h afteradministration of 2; *p < 0.05 versus control mice.
Figure 8. Effect of UDCA and 16 in a rodent model of cholestasis. In vivo administration of UDCA (2) and 16 alters the bile acid pool. Cholestasiswas induced by feeding mice with ANIT alone or in combination with 2, 15 mg/kg, or 16, 16 mg/kg, for 7 days. Blood was collected at the end ofexperiments and bile acids concentrations measured as described in the Experimental Section. (A) Analysis of body weight loss in control mice, miceadministered ANIT, mice administered ANIT plus 2, and mice administered ANIT plus 16. (B) Blood concentrations of total conjugated bile acids.(C) Administration of 16 caused a robust increase in tauro-muricholic acid (tMU) and tauro-cholic acid (tCA). *p < 0.05 versus control mice; #p <0.05 versus ANIT treated mice.
Journal of Medicinal Chemistry Article
dx.doi.org/10.1021/jm500889f | J. Med. Chem. 2014, 57, 7687−77017694

a decreased activity of the hydroxyl compound 14 if comparedwith 16, while the more hydrophobic ester derivative 15 showsan even lower activity.To provide further structural insights on the binding
mechanism to GP-BAR1 of this series of compounds, westudied also the binding mode of the alcohol derivatives 3 and4. These ligands bear a shorter side chain with respect to 14and 16, specifically 3 and 4 are C24 and C23 derivatives,respectively. Docking calculations show that these compoundsoccupy the binding site similarly to 14 and 16, demonstratingthat shorter side chains are allowed if they present H-bondinggroups which can interact with the neighboring serine residuesthrough direct or water mediated H-bonds (Figure 6). Thisfinding is in agreement with the pharmacological data showingthat 3 and 4 are both able to activate GP-BAR1 intransactivation assays. It is important to stress that oursimulations highlight the high conformational flexibility of theligand side chain, which allows the proper binding of the ligandin the binding pocket and the formation of the interactions withthe receptor. Compounds with a more rigid side chain, such asthe Δ24,25 compound 19 (see Supporting Information, FigureS1), show indeed a lower activity. A reduced activity wasobserved also for the tauro-conjugated compounds 12 and 20,where the presence of the amide group decreases theconformational flexibility of the ligand side chain. Finally, thepresence of bulky groups on rings A and B, like sulfate incompounds 6−10, hampers the proper orientation of the ligandin the binding site to interact with residues such as Asn93 andGlu169, causing a drop in the activity.On the basis of our pharmacological assays, all the UDCA
derivatives are inactive on FXR. These data prompted us toinvestigate the binding mechanism of UDCA to FXR in theattempt to elucidate the structural bases of its inactivity towardthis receptor. Docking calculations on UDCA in the FXR-LBDindicates that it is able to form a salt-bridge with the Arg328side chain through its C24 carboxylate group and two hydrogenbonds with the Tyr358 and His444 side chains through its 3α-OH (see Supporting Information, Figure S2). Similarinteractions are engaged by the potent FXR agonist 6-ECDCA,21 however, at variance with 6-ECDCA, UDCA isnot able to form hydrogen bonds with the Tyr366 and Ser329side chains due to the presence of the β-oriented 7-hydroxylgroup (see Supporting Information, Figure S2). The lack ofthese interactions might explain the inactivity of UDCA towardFXR as also previously reported by Fujino et al.31 Thisconclusion is further supported by experimental data, showingthat the interaction with Tyr366 and Ser329 is determinant forthe agonistic activity on FXR. In fact, compounds such as 3-deoxyCDCA, which interacts with Tyr366 and Ser329 and notwith His444 and Trp466, have an agonist profile in FXRtransactivation assays.21
In Vivo Evaluation on Compound 16. Compound 16,the most potent GP-BAR1 agonist generated in this study, wasfurther investigated in vivo and its effects on FXR and GP-BAR1 target genes in the liver and intestine assessed by RT-PCR. In these experiments CD1, 11 week old male mice wereorally administered with compound 16 (64 mg/kg) or itsparent compound UDCA (60 mg/kg) and liver, intestine, andblood collected after 3, 6, and 24 h (n = 3).As illustrated in Figure 7, compound 16 failed to induce the
expression of OST and BSEP mRNA in the liver at 6 and 24 h(data not shown) after administration. UDCA increased theexpression of these two FXR-target genes although that the
effect became apparent only 24 h after its administration (p <0.05 versus control mice).In contrast, expression of pro-glucagon-1 gene, a GP-BAR1
regulated target, in the intestine was increased by 2-fold inresponse to 6 h treatment with compound 16 (Figure 7). Insummary, compound 16 is an effective and selective GP-BAR1ligand in vivo and in vitro, and its activity is different fromUDCA, its parent compound.To further confirm the role of compound 16 as a GP-BAR1
agonist, we also performed a cholestatic model by administer-ing mice with α-naphthylisothiocyanate (ANIT). As shown inFigure 8, exposure to ANIT caused cholestasis as confirmed bythe analysis of plasma bile acid concentration in ANIT treatedanimals which revealed that these animals had an increase inthe total conjugated bile acids in comparison with not treatedanimals.In addition, ANIT treated animals had a significant lost
weight in comparison with not treated animals, while nosignificant differences among the groups ANIT and ANIT plusUDCA was observed in terms of both weight loss and bile acidconcentration. By the contrast, administration of compound 16significantly increased total amount of plasma bile acids incomparison with ANIT treated mice (p < 0.05 versus ANITtreated mice). It is noteworthy that mice administered withcompound 16 had a significant increase in plasma levels oftauro-muricholic acid (tMU) and tauro-cholic acid (tCA)compared with ANIT treated mice (p < 0.05 versus ANITtreated mice).
■ CONCLUSIONIn conclusion, in this study we demonstrated that manipulatingUDCA side chain represents a promising strategy to generatebile acid derivatives as selective GP-BAR1 agonists. Pharmaco-logical evaluation on 16 demonstrated this compound as apromising lead for the treatment of diabetes. Administration ofcompound 16 selectively increase the expression of pro-glucagon 1, a GP-BAR1 target, in the small intestine, while ithad no effect on FXR target genes in the liver. Further,compound 16 results in a significant reshaping of bile acid poolin a rodent model of cholestasis. This study provides newopportunities for the treatment of enterohepatic and metabolicdisorders.
■ EXPERIMENTAL SECTIONChemistry. General. Specific rotations were measured on a Jasco
P-2000 polarimeter. High-resolution ESI-MS spectra were performedwith a Micromass Q-TOF mass spectrometer. NMR spectra wereobtained on Varian Inova 400 and Varian Inova 500 NMRspectrometers (1H at 400 and 500 MHz, 13C at 100 and 125 MHz)equipped with Sun hardware and recorded in CDCl3 (δH = 7.26 andδC = 77.0 ppm) and CD3OD (δH = 3.30 and δC = 49.0 ppm). J are inhertz, and chemical shifts (δ) are reported in ppm and referred toCHCl3 and CHD2OD as internal standards. HPLC was performedusing a Waters model 510 pump equipped with Waters Rheodineinjector and a differential refractometer, model 401. Reaction progresswas monitored via thin-layer chromatography (TLC) on Alugramsilica gel G/UV254 plates. Silica gel MN Kieselgel 60 (70−230 mesh)from Macherey-Nagel Company was used for column chromatog-raphy. All chemicals were obtained from Sigma-Aldrich, Inc. Solventsand reagents were used as supplied from commercial sources with thefollowing exceptions. Tetrahydrofuran, dichloromethane, diisopropyl-amine, and triethylamine were distilled from calcium hydrideimmediately prior to use. Methanol was dried from magnesiummethoxide as follows. Magnesium turnings (5 g) and iodine (0.5 g)were refluxed in a small (50−100 mL) quantity of methanol until all of
Journal of Medicinal Chemistry Article
dx.doi.org/10.1021/jm500889f | J. Med. Chem. 2014, 57, 7687−77017695

the magnesium had reacted. The mixture was diluted (up to 1 L) withreagent grade methanol, refluxed for 2−3 h, then distilled undernitrogen. All reactions were carried out under argon atmosphere usingflame-dried glassware.The purity of tested compounds was determined to be always
greater than 95% by analytical HPLC analysis using a Nucleodur 100−5 C18 (5 μm; 4.6 mm i.d. × 250 mm) column eluting with the solventsystem (flow rate 1 mL/min) reported below in the sectioncorresponding to each individual compound.3α,7β-Diformyloxy-5β-cholan-24-oic Acid (21). A solution of
ursodeoxycholic acid 2 (550 mg, 1.3 mmol) in 10 mL of 90% formicacid containing 25 μL of 70% perchloric acid was stirred at 47−50 °Cfor 12 h. The temperature of the heating bath was lowered to 40 °C,then 5 mL of acetic anhydride was added over 10 min and the mixturewas stirred for 10 min more. The solution was cooled to roomtemperature, poured into 50 mL of water, and extracted with diethylether. The organic layers were washed with water to neutrality, driedover Na2SO4, and evaporated to give 570 mg of 21 (96%). An analyticsample was obtained by silica gel chromatography eluting withCH2Cl2/MeOH 9:1. Selected 1H NMR (400 MHz CD3OD): δ 8.03(1H, s), 7.98 (1H, s), 4.89 (1H, m), 4.80 (1H, m), 0.98 (3H, s), 0.93(3H, d, J = 6.5 Hz), 0.69 (3H, s). 13C NMR (100 MHz CD3OD): δ178.1, 163.0, 162.5, 74.9, 74.8, 56.6, 56.4, 44.8, 43.5, 41.3, 41.2, 40.7,36.6, 35.4 (2C), 35.1, 34.1, 34.0, 32.3, 32.0, 29.5, 27.6, 23.6, 22.3, 18.9,12.5. HRMS-ESI m/z 449.2907 [M + H]+, C26H41O6 requires449.2903.3α,7β-Diformyloxy-24-nor-5β-cholan-23-nitrile (22). Crude 21
(500 mg, 1.1 mmol), 1.7 mL of cold trifluoroacetic acid, and 466μL (3.3 mmol) of trifluoroacetic anhydride were stirred at 0−5 °Cuntil dissolution. Sodium nitrite (83 mg, 1.21 mmol) was added insmall portions. After the addition was complete, the reaction mixturewas stirred first at 0−5 °C for 1 h, then at 38−40 °C for 8 h. Oncompletion, the reaction was neutralized with NaOH 2 N, then theproduct was extracted with 50 mL of diethyl ether (3 × 50 mL),followed by washing with brine and dried over anhydrous Na2SO4.The ether was removed under reduced pressure to afford 440 mg of 22(96%) that was subjected to next step without any purification. Ananalytic sample was obtained by silica gel chromatography eluting withn-hexane/ethyl acetate 8:2. Selected 1H NMR (400 MHz CD3OD): δ8.04 (1H, s), 7.99 (1H, s), 4.87 (1H, m), 4.73 (1H, m), 1.12 (3H, d, J= 6.5 Hz), 0.98 (3H, s), 0.71 (3H, s). 13C NMR (100 MHz CD3OD):δ 163.1, 162.6, 120.3, 74.8 (2C), 56.4, 55.5, 44.8, 43.2, 41.2, 40.9, 40.6,35.4, 35.1, 34.6, 34.1, 34.0, 30.9, 29.3, 27.6, 25.1, 23.6, 22.3, 19.8, 12.5.HRMS-ESI m/z 416.2807 [M + H]+, C25H38NO4 requires 416.2801.24-nor-Ursodeoxycholic Acid (11). Crude compound 22 (400 mg,
0.96 mmol) was refluxed in ca. 50 mL of methanol/water 1:1 with 30%KOH. After stirring for 48 h, the basic aqueous solution wasneutralized with HCl 6 N. Then methanol was evaporated, and theresidue was extracted with AcOEt (3 × 50 mL). The combinedorganic layers were washed with brine, dried, and evaporated todryness to give white solid residue, which was purified by silica gelchromatography, eluting with CH2Cl2/MeOH 9:1 (355 mg, 97%). Ananalytic sample was purified by HPLC on a Nucleodur 100−5 C18 (5μm; 4.6 mm i.d. × 250 mm) with MeOH/H2O (70:30) as eluent (flowrate 1 mL/min), to give compound 11 (tR = 20.7 min); [α]25
D = +11.0(c 0.41, CH3OH). Selected
1H NMR (400 MHz CD3OD): δ 3.44(2H, m), 0.98 (3H, d, J = 6.5 Hz), 0.93 (3H, s), 0.71 (3H, s). 13CNMR (100 MHz CD3OD): δ 177.3, 72.1, 71.9, 57.5, 56.6, 44.8, 44.5,44.0, 42.5, 41.4, 40.7, 38.6, 38.0, 36.1, 35.2, 34.9, 31.0, 29.7, 27.9, 23.9,22.4, 20.2, 12.7. HRMS-ESI m/z 379.2844 [M + H]+, C23H39O4requires 379.2848.3α,7β-Dihydroxy-24-nor-5β-cholan-23-oyl Taurine Sodium Salt
(12). Carboxylic acid 11 (20 mg, 0.05 mmol) in DMF dry (2 mL) wastreated with DMT-MM (41 mg, 0.15 mmol) and triethylamine (174μL, 1.25 mmol), and the mixture was stirred at room temperature for10 min. Then to the mixture was added taurine (37 mg, 0.3 mmol).After 1 h, the reaction mixture was concentrated under vacuo anddissolved in water (5 mL). The solution was poured over a C18 silicagel column. Fraction eluted with H2O/MeOH 99:1 gave a mixture thatwas further purified by HPLC on a Nucleodur 100−5 C18 (5 μm; 4.6
mm i.d. × 250 mm) with MeOH/H2O (60:40) as eluent (flow rate 1mL/min) to give 7 mg (28%) of compound 12 (tR = 12.4 min); [α]25
D
= −4.8 (c 0.15, CH3OH). Selected1H NMR (400 MHz CD3OD): δ
3.58 (2H, t, J = 6.6 Hz), 3.48 (2H, m), 2.95 (2H, t, J = 6.6 Hz), 0.97(3H, d, ovl), 0.96 (3H, s), 0.71 (3H, s). HR ESIMS m/z 484.2735 [M− Na]−, C25H42NO6S requires 484.2732.
Methyl 3α,7β-Dihydroxy-24-nor-5β-cholan-23-oate (13). Com-pound 11 (100 mg, 0.26 mmol) was dissolved in 30 mL of drymethanol and treated with p-toluenesulfonic acid (251 mg, 1.3 mmol).The solution was left to stand at room temperature for 5 h. Themixture was quenched by addition until neutrality of NaHCO3saturated solution. Most of the solvent was evaporated, and theresidue was extracted with EtOAc. The combined extract was washedwith brine, dried with Na2SO4, and evaporated to give 13 asamorphous solid (100 mg, 97% yield). An analytic sample was purifiedby HPLC on a Nucleodur 100−5 C18 (5 μm; 4.6 mm i.d. × 250 mm)with MeOH/H2O (9:1) as eluent (flow rate 1 mL/min) to givecompound 13 (tR = 9.5 min); [α]25
D = +19.8 (c 0.22, CHCl3).Selected 1H NMR (400 MHz CDCl3): δ 3.60 (3H, s), 3.51 (2H, m),0.92 (3H, d, J = 6.3 Hz), 0.88 (3H, s), 0.65 (3H, s). 13C NMR (100MHz CDCl3): δ 173.8, 71.0, 70.9, 55.7, 54.9, 51.3, 43.6, 43.4, 42.4,41.3, 39.9, 39.1, 37.1, 37.0, 34.8, 33.9, 33.6, 30.0, 28.6, 26.7, 23.3, 21.0,19.5, 12.0. HRMS-ESI m/z 393.3009 [M + H]+, C24H41O4 requires393.3005.
3α,7β-Dihydroxy-24-nor-5β-cholan-23-ol (4). To a solution of 13(90 mg, 0.23 mmol) in dry THF (5 mL) at 0 °C under argon wereadded LiBH4 (0.8 mL, 2 M in THF, 1.6 mmol) and dry methanol (65μL, 1.6 mmol), and the resulting mixture was stirred for 3 h at 0 °C.The mixture was quenched by addition of NaOH (1 M, 1.4 mL) andthen allowed to warm to room temperature. Ethyl acetate was added,and the separated aqueous phase was extracted with ethyl acetate (3 ×30 mL). The combined organic phases were washed with water, dried(Na2SO4), and concentrated. Purification by silica gel (CH2Cl2/MeOH 97:3) gave compound 4 as a colorless oil (85 mg, quantitativeyield). An analytic sample was purified by HPLC on a Nucleodur100−5 C18 (5 μm; 4.6 mm i.d. × 250 mm) with MeOH/H2O (8:2) aseluent (flow rate 1 mL/min) to give compound 4 (tR = 17.8 min);[α]25
D = +23.7 (c 0.18, CH3OH).1H NMR (400 MHz CD3OD): δ
3.60 (1H, m ovl), 3.51 (1H, m), 3.50 (2H, m), 0.97 (3H, d, ovl), 0.96(3H, s), 0.72 (3H, s). 13C NMR (100 MHz CD3OD): δ 72.1, 71.9,60.8, 57.5, 57.1, 44.8, 44.5, 44.0, 41.6, 40.7, 39.9, 38.6, 38.0, 36.1, 35.2,34.1, 31.0, 29.8, 27.9, 23.9, 22.4, 19.5, 12.6. HRMS-ESI m/z 365.3053[M + H]+, C23H41O3 requires 365.3056.
3α,7β-Dihydroxy-24-nor-5β-cholan-23-yl-23-sodium Sulfate (5),7β,23-Dihydroxy-24-nor-5β-cholan-3α-yl-3-sodium Sulfate (6),3α,23-Dihydroxy-24-nor-5β-cholan-7β-yl-7-sodium Sulfate (7), 7β-Hydroxy-24-nor-5β-cholan-3α,23-diyl-3,23-sodium Disulfate (8),3α-Hydroxy-24-nor-5β-cholan-7β,23-diyl-7,23-sodium Disulfate(9), and 24-nor-5β-Cholan-3α,7β,23-tryl-3,7,23-sodium Trisulfate(10). The triethylamine−sulfur trioxide complex (253 mg, 1.4 mmol)was added to a solution of compound 4 (50 mg, 0.14 mmol) in DMFdry (5 mL) under an argon atmosphere, and the mixture was stirred at95 °C for 24 h. The solution was then concentrated under vacuum. Tothe solid dissolved in methanol (15 mL) was added three drops of HCl37% v/v, and the mixture was stirred for 3 h at room temperature. Atthe end of reaction, silver carbonate was added to precipitate chloride.Then the reaction mixture was centrifuged, and the supernatant wasconcentrated in vacuo. The residue was poured over a RP18 column.
Fraction eluted with H2O/MeOH 99:1 gave a mixture that wasfurther purified by HPLC on a Nucleodur 100−5 C18 (5 μm; 4.6 mmi.d. × 250 mm) with MeOH/H2O (35:65) as eluent (flow rate 1 mL/min) to give 3.7 mg (4%) of compound 10 (tR = 5 min); fractioneluted with H2O/MeOH 95:5 and with H2O/MeOH 9:1 gave amixture that was purified as previously to afford respectively 8.7 mg(11%) of compound 8 (tR = 15 min) and 7 mg (9%) of compound 9(tR = 14 min). Fraction eluted with H2O/MeOH 75:25 gave a mixturethat was further purified by HPLC on a Nucleodur 100−5 C18 (5 μm;4.6 mm i.d. × 250 mm) with MeOH/H2O (65:35) as eluent (flow rate1 mL/min), to give 15 mg (23%) of compound 7 (tR = 7 min).Fraction eluted with H2O/MeOH 7:3 gave a mixture that was further
Journal of Medicinal Chemistry Article
dx.doi.org/10.1021/jm500889f | J. Med. Chem. 2014, 57, 7687−77017696

purified as previously to give 5 mg (8%) of compound 5 (tR = 8.4 min)and 4 mg (6%) of compound 6 (tR = 6.4 min).3α,7β-Dihydroxy-24-nor-5β-cholan-23-yl-23-sodium Sulfate (5).
[α]25D = +35.8 (c 0.69, CH3OH).
1H NMR (400 MHz CD3OD): δ4.04 (2H, m), 3.48 (2H, m), 1.00 (3H, d, J = 6.5 Hz), 0.97 (3H, s),0.72 (3H, s). HR ESIMS m/z 443.2469 [M − Na]−, C23H39O6Srequires 443.2467.7β,23-Dihydroxy-24-nor-5β-cholan-3α-yl-3-sodium Sulfate (6).
[α]25D = +11.4 (c 0.04, CH3OH).
1H NMR (400 MHz CD3OD): δ4.23 (1H, m), 3.48 (3H, m), 0.98 (3H, d, J = 6.4 Hz), 0.97 (3H, s),0.72 (3H, s). HR ESIMS m/z 443.2462 [M − Na]−, C23H39O6Srequires 443.2467.3α,23-Dihydroxy-24-nor-5β-cholan-7β-yl-7-sodium Sulfate (7).
[α]25D = +7.8 (c 0.19, CH3OH).
1H NMR (400 MHz CD3OD): δ4.29 (1H, m), 3.61 (1H, m), 3.51 (2H, m), 0.97 (3H, s), 0.96 (3H, d,ovl), 0.70 (3H, s). HR ESIMS m/z 443.2465 [M − Na]−, C23H39O6Srequires 443.2467.7β-Hydroxy-24-nor-5β-cholan-3α,23-diyl-3,23-sodium Disulfate
(8). [α]25D = +0.3 (c 0.23, CH3OH). 1H NMR (400 MHz
CD3OD): δ 4.23 (1H, m), 4.04 (2H, m), 3.48 (1H, m), 1.00 (3H,d, J = 6.6 Hz), 0.97 (3H, s), 0.72 (3H, s). HR ESIMS m/z 545.1859[M − Na]−, C23H38NaO9S2 requires 545.1855.3α-Hydroxy-24-nor-5β-cholan-7β,23-diyl-7,23-sodium Disulfate
(9). [α]25D = +0.5 (c 0.15, CH3OH). 1H NMR (400 MHz
CD3OD): δ 4.29 (1H, m), 4.05 (2H, m), 3.50 (1H, m), 0.99 (3H,d, J = 6.5 Hz), 0.97 (3H, s), 0.71 (3H, s). HR ESIMS m/z 545.1857[M − Na]−, C23H38NaO9S2 requires 545.1855.24-nor-5β-Cholan-3α,7β,23-tryl-3,7,23-sodium Trisulfate (10).
[α]25D = +13.3 (c 0.03, CH3OH).
1H NMR (400 MHz CD3OD): δ4.31 (1H, m), 4.23 (1H, m), 4.04 (2H, m), 1.00 (3H, d, J = 6.4 Hz),0.98 (3H, s), 0.71 (3H, s). HR ESIMS m/z 647.1247 [M − Na],C23H37Na2O12S3 requires 647.1243.3α,7β-Dihydroxy-5β-cholan-24-ol (3). At a solution of methyl
3α,7β-dihydroxy-5β-cholan-24-oate (50 mg, 0.12 mmol) in THF dry(10 mL) were added at 0 °C dry methanol (51 μL, 1.255 mmol) andLiBH4 (300 μL, 2 M in THF, 1.25 mmol). After 1 h, the mixture wasquenched by addition of NaOH (1 M, 240 μL) and then allowed towarm to room temperature. Ethyl acetate was added, and the separatedaqueous phase was extracted with ethyl acetate (3 × 30 mL). Thecombined organic phases were washed with water, dried (Na2SO4),and concentrated. Purification by silica gel (CH2Cl2/methanol 9:1)gave compound 3 as colorless oil (42 mg, 93%). An analytic samplewas purified by HPLC on a Nucleodur 100−5 C18 (5 μm; 4.6 mm i.d.× 250 mm) with MeOH/H2O (85:15) as eluent (flow rate 1 mL/min), to give compound 3 (tR = 5.8 min); [α]25
D = +54.7 (c 0.36,CHCl3). Selected
1H NMR (400 MHz CD3OD): δ 3.51 (4H, m), 0.97(3H, d, ovl), 0.97 (3H, s), 0.72 (3H, s). 13C NMR (100 MHzCD3OD): δ 72.1, 71.9, 63.6, 57.6, 56.8, 44.8, 44.5, 44.0, 41.6, 40.7,38.6, 38.0, 37.0, 36.1, 35.2, 33.3, 31.0, 30.3, 29.7, 27.9, 23.9, 22.4, 19.4,12.6. HRMS-ESI m/z 379.3215 [M + H]+, C24H43O3 requires379.3212.3α,7β-Di(tert-butyldimethylsilyloxy)-5β-cholan-24-ol (23). 2,6-
Lutidine (2.8 mL, 20 mmol) and tert-butyldimethylsilyltrifluorome-thanesulfonate (1.7 mL, 6 mmol) were added at 0 °C to a solution ofmethyl 3α,7β-dihydroxy-5β-cholan-24-oate (800 mg, 2 mmol) in 30mL of CH2Cl2. After 2 h stirring at 0 °C, the reaction was quenched byaddition of aqueous NaHSO4 (1 M, 100 mL). The layers wereseparated, and the aqueous phase was extracted with CH2Cl2 (3 × 100mL). The combined organic layers were washed with NaHSO4, water,saturated aqueous NaHCO3, and brine and evaporated in vacuo togive 1.3 g of methyl 3α,7β-di(tert-butyldimethylsilyloxy)-5β-cholan-24-oate in the form of colorless needles that was subjected to next stepwithout any purification. To a solution of methyl ester (1 g, 1.6 mmol)in dry THF (30 mL), at 0 °C dry methanol (453 μL, 11.2 mmol) andLiBH4 (5.6 mL, 2 M in THF, 11.2 mmol) were added. The resultingmixture was stirred for 2 h at 0 °C. The mixture was quenched byaddition of 1 M NaOH (3.2 mL) and then ethyl acetate. The organicphase was washed with water, dried (Na2SO4), and concentrated.Purification by silica gel (hexane/ethyl acetate 99:1 and 0.5% TEA)gave 23 as a white solid (1.2 g, quantitative yield over two steps).
Methyl 3α,7β-Di(tert-butyldimethylsilyloxy)-25,26-bis-homo-5β-chol-24-en-26-oate (24). DMSO (2 mL, 28 mmol) was addeddropwise for 15 min to a solution of oxalyl chloride (7 mL, 14 mmol)in dry dichloromethane (50 mL) at −78 °C under argon atmosphere.After 30 min, a solution of alcohol 23 (1.2 g, 2 mmol) in dry CH2Cl2was added via cannula and the mixture was stirred at −78 °C for 30min. Et3N (2.8 mL, 20 mmol) was added dropwise. After 1 h,methyl(triphenylphosphoranylidene)acetate (2.1 g, 3 mmol) wasadded and the mixture was allowed to warm to room temperature.NaCl saturated solution was added, and the aqueous phase wasextracted with diethyl ether (3 × 100 mL). The combined organicphases were washed with water, dried (Na2SO4), and concentrated.Purification by silica gel (hexane/ethyl acetate 95:5 and 0.5% TEA)gave compound 24 as a colorless oil (1 g, 76%); [α]25
D = +45.0 (c0.70, CH3OH).
1H NMR (400 MHz CDCl3): δ 6.94 (1H, dt, J = 7.3,15.5 Hz), 5.78 (1H, d, J = 15.5 Hz), 3.66 (3H, s), 3.64 (2H, m), 0.92(3H, d, J = 6.5 Hz), 0.90 (3H, s), 0.86 (18H, s), 0.62 (3H, s), 0.04(6H, s), 0.03 (6H, s). 13C NMR (100 MHz CDCl3): δ 167.0, 150.0,120.9, 72.7, 72.5, 55.5, 54.9, 51.3, 43.9, 43.7, 42.8, 39.9, 38.8, 37.9,37.8, 35.3 (2C), 35.1, 34.3, 34.1, 30.9 (2C), 30.8, 28.9, 27.3, 26.4 (3C),25.9 (3C), 23.5, 21.2, 18.6, 12.1, −2.8, −3.4, −4.6 (2C). HRMS-ESIm/z 661.5050 [M + H]+, C39H73O4Si2 requires 661.5047.
Methyl 3α,7β-Dihydroxy-25,26-bis-homo-5β-cholan-26-oate(15). A solution of compound 24 (500 mg, 0.76 mmol) in THFdry/EtOH dry (5 mL/5 mL, v/v) was hydrogenated in the presence ofPd(OH)2 5 wt % on activated carbon Degussa type (5 mg). The flaskwas evacuated and flushed first with argon and then with hydrogen.After 12 h, the reaction was complete. The catalyst was filteredthrough Celite, and the recovered filtrate was concentrated undervacuum to give the methyl ester, which was dissolved in methanol (40mL). At the solution was added 1 mL of HCl 37% v/v. After 1 h, silvercarbonate was added to the solution to precipitate chloride. Then thereaction mixture was centrifuged, and the supernatant wasconcentrated in vacuo to give compound 15 as colorless amorphoussolids (330 mg, quantitative yield). An analytic sample was purified byHPLC on a Nucleodur 100−5 C18 (5 μm; 4.6 mm i.d. × 250 mm)with MeOH/H2O (92:8) as eluent (flow rate 1 mL/min) to givecompound 15 (tR = 13 min); [α]25
D = +41.8 (c 0.15, CH3OH).Selected 1H NMR (400 MHz CDCl3): δ 3.67 (3H, s), 3.59 (2H, m),2.30 (2H, dt, J = 2.5, 7.5 Hz), 0.95 (3H, s), 0.91 (3H, d, J = 6.5 Hz),0.67 (3H, s). 13C NMR (100 MHz CDCl3): 167.1, 71.3 (2C), 55.7,55.1, 51.4, 43.8, 43.7, 42.4, 40.1, 39.2, 37.3, 36.8, 35.5 (2C), 34.9, 34.2,34.1, 30.4, 28.7, 26.9, 25.7, 25.4, 23.4, 21.2, 18.7, 12.1. HRMS-ESI m/z435.3479 [M + H]+, C27H47O4 requires 435.3474.
3α,7β-Dihydroxy-25,26-bis-homo-5β-cholan-26-oic Acid (16). Aportion of compound 15 (200 mg, 0.46 mmol) was hydrolyzed withNaOH (184 mg, 4.6 mmol) in a solution of MeOH/H2O 1:1 v/v (20mL). The mixture was stirred for 4 h at reflux. The resulting solutionwas then acidified with HCl 6 N and extracted with ethyl acetate (3 ×50 mL). The collected organic phases were washed with brine, driedover Na2SO4 anhydrous, and evaporated under reduced pressure togive compound 16 (115 mg, 60%). An analytic sample was purified byHPLC on a Nucleodur 100−5 C18 (5 μm; 4.6 mm i.d. × 250 mm)with MeOH/H2O (95:5) as eluent (flow rate 1 mL/min), to givecompound 16 (tR = 5 min); [α]25
D = −9.0 (c 0.04, CH3OH). Selected1H NMR (500 MHz CD3OD): δ 3.47 (2H, m), 2.27 (2H, dt, J = 3.4,7.4 Hz), 0.96 (3H, s), 0.94 (3H, d, J = 6.5 Hz), 0.70 (3H, s). 13C NMR(125 MHz CD3OD): δ 178.2, 72.1, 71.9, 57.6, 56.7, 44.8, 44.5, 44.0,41.6, 40.7, 38.6, 38.0, 36.9, 36.8, 36.1, 35.3, 35.2, 30.9, 29.8, 27.9, 26.7,26.6, 23.9, 22.4, 19.3, 12.7. HRMS-ESI m/z 421.3315 [M + H]+,C26H45O4 requires 421.3318.
3α,7β-Dihydroxy-25,26-bis-homo-5β-cholan-26-oyl Taurine So-dium Salt (17). Carboxylic acid 16 (20 mg, 0.047 mmol) in DMF dry(5 mL) was treated with DMT-MM (39 mg, 0.14 mmol) andtriethylamine (164 μL, 1.18 mmol), and the mixture was stirred atroom temperature for 10 min. Then to the mixture was added taurine(35 mg, 0.28 mmol). After 3 h, the reaction mixture was concentratedunder vacuo and dissolved in water (5 mL). The solution was pouredover a C18 silica gel column. Fraction eluted with H2O/MeOH 95:5gave a mixture that was further purified by HPLC on a Nucleodur
Journal of Medicinal Chemistry Article
dx.doi.org/10.1021/jm500889f | J. Med. Chem. 2014, 57, 7687−77017697

100−5 C18 (5 μm; 4.6 mm i.d. × 250 mm) with MeOH/H2O (45:55)as eluent (flow rate 1 mL/min) to give 12 mg (45%) of compound 17(tR = 12.4 min); [α]25
D = +108.3 (c 0.06, CH3OH). Selected1H NMR
(400 MHz CD3OD): δ 3.59 (2H, t, J = 6.5 Hz), 3.47 (2H, m), 2.96(2H, t, J = 6.5 Hz), 2.18 (2H, t, J = 7.4 Hz), 0.96 (3H, s), 0.94 (3H, d,J = 6.2 Hz), 0.71 (3H, s). HRMS-ESI m/z 526.3200 [M − Na]−,C28H48NO6S requires 526.3202.3α,7β-Dihydroxy-25, 26-bis-homo-5β-cholan-26-ol (14). At a
solution of compound 15 (50 mg, 0.11 mmol) in THF dry (10mL) were added at 0 °C dry methanol (31 μL, 0.77 mmol) and LiBH4(382 μL, 2 M in THF, 0.77 mmol). After 1 h, the mixture wasquenched by addition of NaOH (1 M, 220 μL) and then allowed towarm to room temperature. Ethyl acetate was added, and the separatedaqueous phase was extracted with ethyl acetate (3 × 30 mL). Thecombined organic phases were washed with water, dried (Na2SO4),and concentrated. Purification by silica gel (CH2Cl2/methanol 9:1)gave compound 14 as a colorless oil (35 mg, 77%). An analytic samplewas purified by HPLC on a Nucleodur 100−5 C18 (5 μm; 4.6 mm i.d.× 250 mm) with MeOH/H2O (85:15) as eluent (flow rate 1 mL/min), to give compound 14 (tR = 9 min); [α]25
D = +41.8 (c 0.21,CH3OH). Selected
1H NMR (400 MHz CD3OD): δ 3.53 (2H, t, J =6.5 Hz), 3.48 (2H, m), 0.95 (3H, s), 0.94 (3H, d, ovl), 0.70 (3H, s).13C NMR (100 MHz CD3OD): δ 72.1, 71.9, 63.0, 57.5, 56.7, 44.7,44.4, 44.0, 41.6, 40.7, 38.5, 37.9, 37.2, 37.0, 36.1, 35.2, 33.7, 30.9, 29.8,27.9, 27.4, 27.1, 23.9, 22.4, 19.4, 12.7. HRMS-ESI m/z 407.3520 [M +H]+, C26H47O3 requires 407.3525.Methyl 3α,7β-Dihydroxy-25,26-bis-homo-5β-chol-24-en-26-oate
(18). To the compound 24 (180 mg, 0.27 mmol) dissolved inmethanol (30 mL) was added 1 mL of HCl 37% v/v, and the mixturewas stirred for 2 h at room temperature. At the end of reaction, silvercarbonate was added to precipitate chloride. Then the reaction mixturewas centrifuged and the supernatant was concentrated in vacuo to give120 mg (quantitative yield) of the desired compound 18 as colorlessamorphous solids. An analytic sample was purified by HPLC on aNucleodur 100−5 C18 (5 μm; 4.6 mm i.d. × 250 mm) with MeOH/H2O (92:8) as eluent (flow rate 1 mL/min), to give compound 18 (tR= 14 min); [α]25
D = +45.0 (c 0.70, CH3OH).1H NMR (400 MHz
CDCl3): δ 6.93 (1H, dt, J = 6.6, 15.7 Hz), 5.79 (1H, d, J = 15.7 Hz),3.64 (3H, s), 3.58 (2H, m), 0.92 (3H, s), 0.91 (3H, d, J = 6.4 Hz), 0.65(3H, s). 13C NMR (100 MHz CDCl3): δ 167.2, 150.1, 120.4, 71.1,71.0, 55.9, 55.1, 51.4, 43.7, 43.6, 42.5, 40.2, 39.3, 37.3, 37.1, 35.4, 34.9,34.3, 34.0, 30.2, 29.0, 28.7, 26.9, 23.4, 21.2, 18.5, 12.2. HRMS-ESI m/z433.3323 [M + H]+, C27H45O4 requires 433.3318.3α,7β-Dihydroxy-25,26-bis-homo-5β-chol-24-en-26-oic Acid
(19). A portion of compound 18 (100 mg, 0.23 mmol) was hydrolyzedwith NaOH (93 mg, 2.31 mmol) in a solution of MeOH/H2O 1:1 v/v(20 mL). The mixture was stirred for 1 h at reflux. The resultingsolution was then acidified with HCl 6 N and extracted with ethylacetate (3 × 50 mL). The collected organic phases were washed withbrine, dried over Na2SO4 anhydrous, and evaporated under reducedpressure to give compound 19 (89 mg, 93% yield). An analytic samplewas purified by HPLC on a Nucleodur 100−5 C18 (5 μm; 4.6 mm i.d.× 250 mm) with MeOH/H2O (95:5) as eluent (flow rate 1 mL/min)to give compound 19 (tR = 5 min); [α]25
D = +203.0 (c 0.39, CH3OH).Selected 1H NMR (400 MHz CDCl3): δ 7.07 (1H, dt, J = 7.0, 15.0Hz), 5.81 (1H, d, J = 15.0 Hz), 3.60 (2H, m), 0.94 (3H, s), 0.94 (3H,d, ovl), 0.67 (3H, s). 13C NMR (100 MHz CD3OD): δ 170.7, 151.6,122.7, 72.1, 71.9, 57.5, 56.6, 44.8, 44.5, 44.0, 41.6, 40.7, 38.6, 38.0,36.8, 36.1, 35.6, 35.2, 31.0, 29.9, 29.7, 27.9, 23.9, 22.4, 19.1, 12.7.HRMS-ESI m/z 419.3163 [M + H]+, C26H43O4 requires 419.3161.3α,7β-Dihydroxy-25,26-bis-homo-5β-chol-24-en-26-oyl Taurine
Sodium Salt (20). Carboxylic acid 19 (10 mg, 0.024 mmol) inDMF dry (2 mL) was treated with DMT-MM (20 mg, 0.075 mmol)and triethylamine (87 μL, 0.63 mmol), and the mixture was stirred atroom temperature for 10 min. Then to the mixture was added taurine(18 mg, 0.14 mmol). After 2 h, the reaction mixture was concentratedunder vacuo and dissolved in water (5 mL). The solution was pouredover a C18 silica gel column. Fraction eluted with H2O/MeOH 95:5gave a mixture that was further purified by HPLC on a Nucleodur100−5 C18 (5 μm; 4.6 mm i.d. × 250 mm) with MeOH/H2O (45:55)
as eluent (flow rate 1 mL/min), to give 3.5 mg (26%) of compound 20(tR = 15.8 min); [α]25
D = +52.1 (c 0.08, CH3OH). Selected1H NMR
(400 MHz CD3OD): δ 6.73 (1H, dt, J = 6.5, 14.0 Hz), 5.89 (1H, d, J =14.0 Hz), 3.65 (2H, t, J = 7.0 Hz), 3.47 (2H, m), 2.98 (2H, t, J = 7.0Hz), 0.97 (3H, d, ovl), 0.96 (3H, s), 0.71 (3H, s). HRMS-ESI m/z524.3043 [M − Na]−, C28H46NO6S requires 524.3046.
Ethyl 25,26-Bis-homo-3α,7α-dihydroxy-5β-cholan-26-oate (25).Compound 25 (67 mg, 60% over five steps) was synthesized, startingfrom methyl 3α,7α-dihydroxy-5β-cholan-24-oate (100 mg, 0.25mmol), as previously reported;22 [α]25
D = +6.72 (c 0.25, CHCl3).Selected 1H NMR (400 MHz CDCl3): δ 4.14 (2H, q, J = 7.0 Hz), 3.80(1H, br s), 3.38 (1H, m), 2.29 (2H, t, J = 6.7 Hz), 1.24 (3H, t, J = 7.0Hz), 0.94 (3H, d, J = 6.6 Hz), 0.92 (3H, s), 0.68 (3H, s). 13C NMR(100 MHz CDCl3): δ 175.4, 72.8, 69.0, 61.4, 57.4, 51.5, 43.6, 43.1,41.0, 40.7, 40.3, 37.0, 36.7, 36.5, 35.8, 35.2, 34.8, 34.0, 31.3, 29.3, 26.6,26.5, 24.6, 23.5, 21.8, 19.3, 14.6, 12.3. HRMS-ESI m/z 449.3635 [M +H]+, C28H49O4 requires 449.3631.
25,26-Bis-homo-3α,7α-dihydroxy-5β-cholan-26-oic Acid (26). Aportion of compound 25 (15 mg, 0.033 mmol) was hydrolyzed withNaOH (13.2 mg, 0.33 mmol) in a solution of MeOH/H2O 1:1 v/v(10 mL). The mixture was stirred for 2 h at reflux. The resultingsolution was then acidified with HCl 6 N and extracted with ethylacetate (3 × 50 mL). The collected organic phases were washed withbrine, dried over Na2SO4 anhydrous, and evaporated under reducedpressure to give compound 26 (10.8 mg, 78%); [α]25
D = +6.6 (c 0.38,CH3OH). Selected
1H NMR (400 MHz CD3OD): δ 3.79 (1H, br s),3.40 (1H, m), 2.25 (2H, t, J = 7.5 Hz), 0.94 (3H, d, J = 6.7 Hz), 0.92(3H, s), 0.68(3H, s). 13C NMR (100 MHz CD3OD): δ 179.6, 72.8,68.9, 57.5, 51.5, 43.6, 43.1, 41.0, 40.7, 40.4, 37.0, 36.8, 36.6, 36.2, 35.9(2C), 34.0, 31.3, 29.4, 26.8 (2C), 24.6, 23.5, 21.8, 19.3, 12.3. HRMS-ESI m/z 421.3315 [M + H]+, C26H45O4 requires 421.3318.
3α,7α-Dihydroxy-25,26-bis-homo-5β-cholan-26-ol (27). Methylester 25 (20 mg, 0.044 mmol) was reduced with LiBH4 (2 M inTHF dry, 156 μL, 0.31 mmol) and MeOH dry (12 μL, 0.31 mmol) inTHF dry at 0 °C for 5 h. The mixture was quenched by addition of 1M NaOH solution (100 μL), and then ethyl acetate was added. Theseparated aqueous phase was extracted with ethyl acetate (3 × 30 mL).The combined organic phases were washed with water, dried(Na2SO4), and concentrated. Purification by silica gel (CH2Cl2/MeOH 9:1) gave compound 27 as a white solid (13 mg, 73%); [α]25
D
= +15.0 (c 0.32, CH3OH). Selected1H NMR (400 MHz CD3OD): δ
3.82 (1H, br s), 3.61 (2H, t, J = 6.6 Hz), 3.43 (1H, m), 0.89 (3H, d, J =6.0 Hz), 0.88 (3H, s), 0.64 (3H, s). 13C NMR (100 MHz CDCl3): δ71.8, 68.4, 62.8, 56.0, 50.4, 42.5, 41.4, 39.6 (2C), 39.4, 35.8, 35.6, 35.2,35.0, 34.5, 32.8 (2C), 30.5, 28.2, 26.1, 25.8, 23.6, 22.7, 20.5, 18.6, 11.6.HRMS-ESI m/z 407.3523 [M + H]+, C26H47O4 requires 407.3525.
Animal Studies. All animal experimental procedures wereapproved by the Ethics Committee of the University of Perugia andby the Italian Health Ministry, according to the Italian guideline forcare and use of laboratory animals. The ID for this project is no. 245/2013-B. The authorization was released to Prof. Stefano Fiorucci, as aprincipal investigator, on October 10, 2013. The 6−8 week old CD1male mice were obtained from Harlan Laboratories Srl (San Pietro alNatisone, Udine, Italy). Mice were maintained in a temperaturecontrolled facility with a 12 h light/dark cycle and were given freeaccess to food and water. Mice (n = 5, 6) were orally administered for5 days with 30 mg/kg dose of α-naphthylisothiocyanate (ANIT) inolive oil, a chemical agent that causes specific damage to biliaryepithelial cells leading to bile duct injury and cell proliferation,ultimately causing intrahepatic cholestasis.32,33 Animals were admin-istered with UDCA (15 mg/kg) or 16 (16 mg/kg). At the end of theexperiment, serum and liver samples were collected. During theexperiment, mice were daily weight.
In another experimental section, mice (n = 3, 4) were treated 6 (or24) hours with UDCA (60 mg/kg, os) or with 16 (64 mg/kg, os). Atthe end of the treatments, mice were sacrificed and liver and ileumsamples were collected to perform real-time PCR analysis.
Transactivation Assay. For FXR mediated transactivation,HepG2 cells were plated in a 24 well-plate and transfected with 100ng of pSG5-FXR, 100 ng of pSG5-RXR, 200 ng of the reporter vector
Journal of Medicinal Chemistry Article
dx.doi.org/10.1021/jm500889f | J. Med. Chem. 2014, 57, 7687−77017698

p(hsp27)-TK-LUC containing the FXR response element IR1 clonedfrom the promoter of heat shock protein 27 (hsp27), and with 100 ngof pGL4.70 (Promega), a vector encoding the human Renilla gene.For GP-BAR1 mediated transactivation, HEK-293T cells were platedin a 24-well plate and transfected with 200 ng of pGL4.29 (Promega),a reporter vector containing a cAMP response element (CRE) thatdrives the transcription of the luciferase reporter gene luc2P, with 100ng of pCMVSPORT6-human GP-BAR1 and with 100 ng of pGL4.70.At 24 h post-transfection, cells were stimulated 18 h with 10 μMCDCA, TLCA, and compounds 2−20 and 26−27. After treatments,cells were lysed in 100 μL of lysis buffer (25 mM Tris-phosphate, pH7.8; 2 mM DTT; 10% glycerol; 1% Triton X-100), and 20 μL ofcellular lysate was assayed for luciferase activity using the luciferaseassay system (Promega). Luminescence was measured using Glomax20/20 luminometer (Promega).Real-Time PCR. Real-time PCR was performed as previously
described.32 Briefly, total RNA was isolated using the TRIzol reagentaccording to the manufacturer’s specifications (Invitrogen). First, 1 μgof RNA was purified of the genomic DNA by DNase I treatment(Invitrogen) and random reverse-transcribed with Superscript II(Invitrogen) in a 20 μL reaction volume. Then 10 ng of template wasadded to the PCR mixture (final volume 25 μL) containing thefollowing reagents: 0.2 μM of each primer and 12.5 μL of 2× SYBRFAST Universal ready mix (Invitrogen). All reactions were performedin triplicate and the thermal cycling conditions were: 2 min at 95 °C,followed by 40 cycles of 95 °C for 20 s, 60 °C for 30 s in iCycler iQinstrument (Biorad). The relative mRNA expression was calculatedand expressed as 2−(ΔΔCt). Forward and reverse primer sequences werethe following: mouse GAPDH, ctgagtatgtcgtggagtctac andgttggtggtgcaggatgcattg; mOSTα, cagtggacatagccctcacc and gaccaaag-cagcagaacaca; mBSEP, aaatcggatggtttgactgc and tgacagcgagaatcaccaag;mPro-glucagon, tgaagacaaacgccactcac and caatgttgttccggttcctc.Bile Acids Determination. Stock solutions of bile acids (tMu,
tCA, tDCA, tCDCA, tHCA) were prepared separately in methanol atthe concentration of 10 mmol/L and stored at −20 °C. Theappropriate amounts of each bile acid stock solution was added intopooled samples ranging in a five-points calibration curve from 5 nM to50 μM.Aliquots of 100 μL of each cell lysate were deproteinized with 1 mL
of cold acetonitrile with 5% of NH4OH vortexing for 1 min.34 Aftercentrifugation at 16000g for 10 min, the clear supernatant wastransferred to a new vial, snap frozen, and lyophilized. The sampleswere then redissolved in methanol−water (2:1, v/v) for tauro-conjugated bile acids determination. A bile acids extraction yield of95% was measured using bile acids standards addition in blank samplesbefore and after deproteinization procedure.Liquid Chromatography and Mass Spectrometry. For LC-
MS/MS analysis, chromatographic separation was carried out on theHPLC−MS system LTQ XL ThermoScientific equipped with Accelera600 pump and Accelera AutoSampler system. The mixture wasseparated on a Jupiter 5u C18 column from Phenomenex (150 mm ×2.00 mm), and the column flow rate was set at 200 μL/min. Tauro-conjugated bile acids were separated using a methanol−aqueousammonium acetate (NH4OAc) gradient.35 Mobile phase A was 5%methanol in water containing 2 mM ammonium acetate at pH 7,mobile phase B was methanol, containing ammonium acetate at 2 mM.The gradient started at 30% B and increased to 100% B in 20 min,kept at 100% B for 5 min, then decreased to 30% B in 1 min and keptat 30% B for 10 min. ESI was performed in negative ion mode, and theion source temperature was set at 280 °C. The tune page parameterswere automatically optimized by injecting taurocholic acid at 1 μM asstandard. The MS/MS detection was operated in MRM mode using acollision energy of 20 (arbitrary units), the observed transitions were:tauromuricholic acid (t-MCA) at 13.5 min MRM of 514.28 Th →514.28 Th, taurohyocholic acid (t-HCA) at 15.6 min MRM of 498.29Th → 498.29 Th, taurocholic acid (t-CA) at 16.6 min MRM of 514.28Th → 514.28 Th, taurochenodeoxycholic acid (t-CDCA) at 18.5 minMRM of 498.29 Th→ 498.29 Th, and taurodeoxycholic acid (t-DCA)at 18.9 min MRM of 498.29 Th → 498.29 Th.
Molecular Docking. The AutoDock4.2 software package36 wasused to perform molecular docking calculations in the three-dimensional model of hGP-BAR1.22 Ligands tridimensional structureswere generated with the Maestro Build Panel.37 For each ligand, anextensive ring conformational sampling was performed with the OPLS-AA force field38 and a 2.0 Å rmsd cutoff using MacroModel (version9.9)39 as implemented in Maestro 9.3.37 All conformers were thenrefined using LigPrep40 as implemented in Maestro 9.3.37 Protonationstates at pH 7.0 were assigned using Epik.41 Protein structure wasprepared through the Protein Preparation Wizard through thegraphical user interface of Maestro 9.3.37 Water molecules wereremoved, and hydrogen atoms were added and minimized using theOPLS-2005 force field.38 Ligands and receptor structures wereconverted to AD4 format files using ADT, and the Gesteiger−Marsilipartial charges were then assigned. Grid points of 65 × 80 × 55 with a0.375 Å spacing were calculated around the ligand binding site of GP-BAR1 using AutoGrid4.2. Thus, 100 separate docking calculationswere performed for each run. Each docking run consisted of 10 millionenergy evaluations using the Lamarckian genetic algorithm local search(GALS) method, otherwise, default docking parameters were applied.Docking conformations were clustered on the basis of their rmsd(tolerance = 2.0 Å) and were ranked based on the AutoDock scoringfunction.36
All the residue labels were taken from the wild-type amino acidicsequence of human GP-BAR1.
Docking calculations of UDCA in the FXR-LBD were performedusing the previously reported protocol.42
All figures were rendered using PyMOL (http://www.pymol.org).Statistical Analysis. All values are expressed as mean ± SD of n
values per group. Comparisons of more than two groups were madewith a one-way ANOVA with posthoc Tukey’s test. Comparison oftwo groups was made using Student’s t test for unpaired data whenappropriate. Differences were considered statistically significant if pwas <0.05.
■ ASSOCIATED CONTENT
*S Supporting InformationTabulated analytical data for compounds 3−20 (HPLC k’s, MSdata) and NMR spectra for all tested compounds. Bindingmode of 19 in the GP-BAR1 homology model, binding modeof UDCA in the crystal structure of the rFXR-LBD, andsuperposition between 2 and 6-ECDCA in the rFXR-LBD.CSV file. This material is available free of charge via theInternet at http://pubs.acs.org.
■ AUTHOR INFORMATION
Corresponding Author*Phone: (0039) 081-678525. Fax: (0039) 081-678552. E-mail:[email protected].
Author ContributionsS. Fiorucci and A. Zampella designed the project, analyzed theresults, organized and wrote the manuscript. V. Sepe, C. Festa,and D. Masullo performed whole synthetic protocols, structuralcharacterization of intermediates and final products, andcontributed to the organization of the manuscript. B. Renga,C. D’Amore and S. Cipriani performed whole pharmacologicalexperiments, analyzed the results, and contributed to theorganization of the manuscript. M. C. Monti performed bileacid determination and contributed to the organization of themanuscript. F. S. Di Leva, E. Novellino, and V. Limongellidesigned, performed and analyzed the computational studiesand wrote the manuscript.
NotesThe authors declare no competing financial interest.
Journal of Medicinal Chemistry Article
dx.doi.org/10.1021/jm500889f | J. Med. Chem. 2014, 57, 7687−77017699

■ ACKNOWLEDGMENTS
This work was supported by grants from PSC Partners (5237South Kenton Way, Englewood, Colorado 80111 UnitedStates) and grants from BAR Pharmaceuticals, Srl, Italy.
■ ABBREVIATIONS USED
BAs, bile acids; BSEP, bile salt export pump; CDCA,chenodeoxycholic acid; FXR, farnesoid-X-receptor; GP-BAR-1, G protein-coupled bile acid receptor 1; HEK-293T, humanembryonic kidney 293 cells; HepG2, human hepatoma cell line;LBD, ligand binding domain; LCA, lithocholic acid; M-BAR/TGR5, membrane-type bile acid receptor/Takeda G-protein-coupled receptor; OSTα, organic solute transporter α; RT-PCR, real-time polymerase chain reaction; T2DM, type 2diabetes mellitus; TLCA, tauro-lithocolic acid; UDCA,ursodeoxycholic acid
■ REFERENCES(1) Makishima, M.; Okamoto, A. Y.; Repa, J. J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M. V.; Lustig, K. D.; Mangelsdorf, D. J.; Shan, B.Identification of a nuclear receptor for bile acids. Science 1999, 284,1362−1365.(2) Parks, D. J.; Blanchard, S. G.; Bledsoe, R. K.; Chandra, G.;Consler, T. G.; Kliewer, S. A.; Stimmel, J. B.; Willson, T. M.; Zavacki,A. M.; Moore, D. D.; Lehmann, J. M. Bile acids: natural ligands for anorphan nuclear receptor. Science 1999, 284, 1365−1368.(3) Baes, M.; Gulick, T.; Choi, H. S.; Martinoli, M. G.; Simha, D.;Moore, D. D. A new orphan member of the nuclear hormone receptorsuperfamily that interacts with a subset of retinoic acid responseelements. Mol. Cell. Biol. 1994, 14, 1544−1552.(4) Makishima, M.; Lu, T. T.; Xie, W.; Whitfield, G. K.; Domoto, H.;Evans, R. M.; Haussler, M. R.; Mangelsdorf, D. J. Vitamin D receptoras an intestinal bile acid sensor. Science 2002, 296, 1313−1316.(5) Choi, H. S.; Chung, M.; Tzameli, I.; Simha, D.; Lee, Y. K.; Seol,W.; Moore, D. D. Differential transactivation by two isoforms of theorphan nuclear hormone receptor CAR. J. Biol. Chem. 1997, 272,23565−23571.(6) Wang, Y. D.; Chen, W. D.; Moore, D. D.; Huang, W. FXR: ametabolic regulator and cell protector. Cell Res. 2008, 18, 1087−1095.(7) Zhang, Y.; Lee, F. Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T. M.; Edwards, P. A. Activation of the nuclear receptorFXR improves hyperglycemia and hyperlipidemia in diabetic mice.Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 1006−1011.(8) Renga, B.; Mencarelli, A.; D’Amore, C.; Cipriani, S.; Baldelli, F.;Zampella, A.; Distrutti, E.; Fiorucci, S. Glucocorticoid receptormediates the gluconeogenic activity of the farnesoid X receptor inthe fasting condition. FASEB J. 2012, 26, 3021−3031.(9) Wang, Y. D.; Chen, W. D.; Wang, M.; Yu, D.; Forman, B. M.;Huang, W. Farnesoid X receptor antagonizes nuclear factor kappaB inhepatic inflammatory response. Hepatology 2008, 48, 1632−1643.(10) Fiorucci, S.; Rizzo, G.; Antonelli, E.; Renga, B.; Mencarelli, A.;Riccardi, L.; Orlandi, S.; Pruzanski, M.; Morelli, A.; Pellicciari, R. Afarnesoid X receptor−small heterodimer partner regulatory cascademodulates tissue metalloproteinase inhibitor-1 and matrix metal-loprotease expression in hepatic stellate cells and promotes resolutionof liver fibrosis. J. Pharmacol. Exp. Ther. 2005, 314, 584−595.(11) Baptissart, M.; Vega, A.; Maqdasy, S.; Caira, F.; Baron, S.;Lobaccaro, J. M.; Volle, D. H. Bile acids: from digestion to cancers.Biochimie 2013, 95, 504−517.(12) Watanabe, M.; Houten, S. M.; Mataki, C.; Christoffolete, M. A.;Kim, B. W.; Sato, H.; Messaddeq, N.; Harney, J. W.; Ezaki, O.;Kodama, T.; Schoonjans, K.; Bianco, A. C.; Auwerx, J. Bile acidsinduce energy expenditure by promoting intracellular thyroid hormoneactivation. Nature 2006, 439, 484−489.(13) Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.;Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.;
Pellicciari, R.; Auwerx, J.; Schoonjans, K. TGR5-mediated bile acidsensing controls glucose homeostasis. Cell. Metab. 2009, 10, 167−177.(14) Fiorucci, S.; Mencarelli, A.; Palladino, G.; Cipriani, S. Bile-acid-activated receptors: targeting TGR5 and farnesoid-X-receptor in lipidand glucose disorders. Trends Pharmacol. Sci. 2009, 30, 570−580.(15) Fiorucci, S.; Cipriani, S.; Baldelli, F.; Mencarelli, A. Bile acid-activated receptors in the treatment of dyslipidemia and relateddisorders. Prog. Lipid Res. 2010, 49, 171−185.(16) Tiwari, A.; Maiti, P. TGR5: an emerging bile acid G-protein-coupled receptor target for the potential treatment of metabolicdisorders. Drug Discovery Today 2009, 14, 523−530.(17) Schaap, F. G.; Trauner, M.; Jansen, P. L. Bile acid receptors astargets for drug development. Nature Rev. Gastroenterol. Hepatol. 2014,1, 55−67.(18) Azzaroli, F.; Mehal, W.; Soroka, C. J.; Wang, L.; Lee, J.; Crispe,N.; Boyer, J. L. Ursodeoxycholic acid diminishes Fas-ligand-inducedapoptosis in mouse hepatocytes. Hepatology 2002, 36, 49−54.(19) Keene, C. D.; Rodrigues, C. M.; Eich, T.; Chhabra, M. S.; Steer,C. J.; Low, W. C. Tauroursodeoxycholic acid, a bile acid, isneuroprotective in a transgenic animal model of Huntington’s disease.Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 10671−10676.(20) Alberts, D. S.; Martinez, M. E.; Hess, L. M.; Einspahr, J. G.;Green, S. B.; Bhattacharyya, A. K.; Guillen, J.; Krutzsch, M.; Batta, A.K.; Salen, G.; Fales, L.; Koonce, K.; Parish, D.; Clouser, M.; Roe, D.;Lance, P. Phase III trial of ursodeoxycholic acid to prevent colorectaladenoma recurrence. J. Natl. Cancer Inst. 2005, 97, 846−853.(21) Mi, L.-Z.; Devarakonda, S.; Harp, J. M.; Han, Q.; Pellicciari, R.;Willson, T. M.; Khorasanizadeh, S.; Rastinejad, F. Structural basis forbile acid binding and activation of the nuclear receptor FXR. Mol. Cell2003, 11, 1093−1100.(22) D’Amore, C.; Di Leva, F. S.; Sepe, V.; Renga, B.; Del Gaudio,C.; D’Auria, M. V.; Zampella, A.; Fiorucci, S.; Limongelli, V. Design,synthesis, and biological evaluation of potent dual agonists of nuclearand membrane bile acid receptors. J. Med. Chem. 2014, 57, 937−954.(23) Pellicciari, R.; Sato, H.; Gioiello, A.; Costantino, G.;Macchiarulo, A.; Sadeghpour, B. M.; Giorgi, G.; Schoonjans, K.;Auwerx, J. Nongenomic actions of bile acids. Synthesis and preliminarycharacterization of 23- and 6,23-alkyl-substituted bile acid derivativesas selective modulators for the G-protein coupled receptor TGR5. J.Med. Chem. 2007, 50, 4265−4268.(24) Pellicciari, R.; Costantino, G.; Camaioni, E.; Sadeghpour, B. M.;Entrena, A.; Willson, T. M.; Fiorucci, S.; Clerici, C.; Gioiello, A. Bileacid derivatives as ligands of the farnesoid X receptor. Synthesis,evaluation, and structure−activity relationship of a series of body andside chain modified analogues of chenodeoxycholic acid. J. Med. Chem.2004, 47, 4559−4569.(25) Sepe, V.; Ummarino, R.; D’Auria, M. V.; Renga, B.; Fiorucci, S.;Zampella, A. The first total synthesis of solomonsterol B, a marinepregnane X receptor agonist. Eur. J. Org. Chem. 2012, 5187−5194.(26) Schteingart, C. D.; Hofmann, A. F. Synthesis of 24-nor-5 beta-cholan-23-oic acid derivatives: a convenient and efficient one-carbondegradation of the side chain of natural bile acids. J. Lipid. Res. 1988,29, 1387−1395.(27) Tserng, K. Y.; Klein, P. D. Formylated bile acids: improvedsynthesis, properties, and partial deformylation. Steroids 1977, 29,635−648.(28) Sepe, V.; Ummarino, R.; D’Auria, M. V.; Lauro, G.; Bifulco, G.;D’Amore, C.; Renga, B.; Fiorucci, S.; Zampella, A. Modification in theside chain of solomonsterol A: discovery of cholestan disulfate as apotent pregnane-X-receptor agonist. Org. Biomol. Chem. 2012, 10,6350−6362.(29) Iguchia, Y.; Kihira, K.; Nishimaki-Mogami, T.; Une, M.Structure−activity relationship of bile alcohols as human farnesoid Xreceptor agonist. Steroids 2010, 75, 95−100.(30) Macchiarulo, A.; Gioiello, A.; Thomas, C.; Pols, T. W. H.; Nuti,R.; Ferrari, C.; Giacche, N.; De Franco, F.; Pruzanski, M.; Auwerx, J.;Schoonjans, K.; Pellicciari, R. Probing the binding site of bile acids inTGR5. ACS Med. Chem. Lett. 2013, 4, 1158−1162.
Journal of Medicinal Chemistry Article
dx.doi.org/10.1021/jm500889f | J. Med. Chem. 2014, 57, 7687−77017700

(31) Fujino, T.; Une, M.; Imanaka, T.; Inoue, K.; Nishimaki-Mogami,T. Structure−activity relationship of bile acids and bile acid analogs inregard to FXR activation. J. Lipid Res. 2004, 45, 132−138.(32) Renga, B.; Migliorati, M.; Mencarelli, A.; Cipriani, S.; D’Amore,C.; Distrutti, E.; Fiorucci, S. Farnesoid X receptor suppressesconstitutive androstane receptor activity at the multidrug resistanceprotein-4 promoter. Biochim. Biophys. Acta 2011, 1809, 157−165.(33) Fiorucci, S.; Baldelli, F. Farnesoid X receptor agonists in biliarytract disease. Curr. Opin. Gastroenterol. 2009, 25, 252−259.(34) Mencarelli, A.; Renga, B.; D’Amore, C.; Santorelli, C.; Graziosi,L.; Bruno, A.; Monti, M. C.; Distrutti, E.; Cipriani, S.; Donini, A.;Fiorucci, S. Dissociation of intestinal and hepatic activities of FXR andLXRa supports metabolic effects of terminal ileum interposition inrodents. Diabetes 2013, 62, 3384−3393.(35) Bobeldijk, I.; Hekman, M.; de Vries-van derWeij, J.; Couliera, L.;Ramakera, R. Quantitative profiling of bile acids in biofluids andtissues based on accurate mass high resolution LC-FT-MS: compoundclass targeting in a metabolomics workflow. J. Chromatogr., B 2008,871, 306−313.(36) Huey, R.; Morris, G. M.; Olson, A. J.; Goodsell, D. S. Asemiempirical free energy force field with charge-based desolvation. J.Comput. Chem. 2007, 28, 1145−1152.(37) Maestro, version 9.3; Schrodinger, LLC: New York, 2012.(38) Jorgensen, W. L.; Maxwell, D. S.; Tirado-Rives, J. Developmentand testing of the OPLS all-atom force field on conformationalenergetics and properties of organic liquids. J. Am. Chem. Soc. 1996,118, 11225−11236.(39) MacroModel, version 9.9; Schrodinger, LLC: New York, 2012.(40) LigPrep, version 2.5; Schrodinger, LLC: New York, 2012.(41) Epik, version 2.3; Schrodinger, LLC: New York, 2012.(42) Di Leva, F. S.; Festa, C.; D’Amore, C.; De Marino, S.; Renga, B.;D’Auria, M. V.; Novellino, E.; Limongelli, V.; Zampella, A.; Fiorucci, S.Binding mechanism of the farnesoid X receptor marine antagonistsuvanine reveals a strategy to forestall drug modulation on nuclearreceptors. Design, synthesis, and biological evaluation of novel ligands.J. Med. Chem. 2013, 56, 4701−4717.
Journal of Medicinal Chemistry Article
dx.doi.org/10.1021/jm500889f | J. Med. Chem. 2014, 57, 7687−77017701
Copyright © 2022 FDOKUMEN




















