
Inhibition of the glycolytic enzymes in the trypanosome: An approach in the development of new leads...
19
Pergamon Pharmac. Ther. Vol.60, pp. 347-365. 1993 © 1994Elsevier ScienceLtd Printed in Great Britain.All rights reserved 0163-7258/93 $24.00 Associate Editor: D. SHUGAR INHIBITION OF THE GLYCOLYTIC ENZYMES IN THE TRYPANOSOME: AN APPROACH IN THE DEVELOPMENT OF NEW LEADS IN THE THERAPY OF PARASITIC DISEASES JACQUES PERIE,*~ ISABELLE RIVIERE-ALRIC,* CASIMIR BLONSKI,* THIERRY GEFFLAUT,* NANCY LAUTH DE VIGUERIE,* MURIELLE TRINQUIER,* MICI-II~LE WILLSON,* FRED R. OPPERDOES t and MIA CALLENSt *Groupe de Chimie Organique Biologique, URA au CNRS 470, B,~t IIR 1, Universit~ Paul Sabatier, 118 route de Narbonne, 31062 Toulouse Cedex, France tResearch Unit for Tropical Diseases, International Institute of Cellular and Molecular Pathology, Avenue Hippocrate 74, B-1200, Brussels, Belgium Abstraet---Glycolysis in the trypanosome represents an important target for the development of new therapeutic agents due to the fact that this metabolism is essential for the parasite, glucose being its sole source of energy. In addition, different features of this metabolism and those associated with glycolytic enzymes offer opportunities for the development of efficient and selective compounds. Examples are given in this work of inhibitors directed to the enzymes aldolase and glyceraldehyde-phosphate-dehydrogenase and also of molecules acting specifi- cally on the clusters of basic amino-acids present at the surfaces of the glycolytic enzymes in the parasite. Keywords---Glycolytic enzymes, inhibition, suicide inhibitors, allosteric inhibitors, aldolase, Trypanosoma brucei. CONTENTS 1. Introduction 2. Inhibitors for the Enzyme Aldolase 3. Inhibitors for the Enzyme Glyceraldehyde-phosphate-dehydrogenase 4. Towards more Selective Inhibitors 5. Conclusion Acknowledgements References 347 348 355 359 363 364 364 1. INTRODUCTION Among the many diseases that afflict humans, those caused by parasites occupy an important place due to the number of victims, the lack of efficient therapy and their continuing spread. As an example, the World Health Organization (1985) indicated that 25,000 new cases of the African type of trypanosomiasis appear every year in the Sub-Saharian part of the continent. Parasitic infections also significantly affect human nutrition through their impact on food animals; 3 million cattle die every year from trypanosomiasis. Up to the early 1980s, the chemotherapy employed relied on a very limited number of drugs, :~Corresponding author. Abbreviations--DHAP, dihydroxy-acetone-phosphate; FDP, fructose 1.6-diphosphate; GAP, glyceralde- hyde-3-phosphate; GAPDH, glyceraldehyde-phosphate-dehydrogenase. Jm- 60/2--N 347
Transcript of Inhibition of the glycolytic enzymes in the trypanosome: An approach in the development of new leads...

Pergamon Pharmac. Ther. Vol. 60, pp. 347-365. 1993
© 1994 Elsevier Science Ltd Printed in Great Britain. All rights reserved
0163-7258/93 $24.00
Associate Editor: D. SHUGAR
INHIBITION OF THE GLYCOLYTIC ENZYMES IN THE TRYPANOSOME: AN APPROACH IN THE
DEVELOPMENT OF NEW LEADS IN THE THERAPY OF PARASITIC DISEASES
JACQUES PERIE,*~ ISABELLE RIVIERE-ALRIC,* CASIMIR BLONSKI,* THIERRY GEFFLAUT,* NANCY LAUTH DE VIGUERIE,* MURIELLE TRINQUIER,* MICI-II~LE
WILLSON,* FRED R. OPPERDOES t and MIA CALLENSt
*Groupe de Chimie Organique Biologique, URA au CNRS 470, B,~t IIR 1, Universit~ Paul Sabatier, 118 route de Narbonne, 31062 Toulouse Cedex, France
tResearch Unit for Tropical Diseases, International Institute of Cellular and Molecular Pathology, Avenue Hippocrate 74, B-1200, Brussels, Belgium
Abstraet---Glycolysis in the trypanosome represents an important target for the development of new therapeutic agents due to the fact that this metabolism is essential for the parasite, glucose being its sole source of energy. In addition, different features of this metabolism and those associated with glycolytic enzymes offer opportunities for the development of efficient and selective compounds. Examples are given in this work of inhibitors directed to the enzymes aldolase and glyceraldehyde-phosphate-dehydrogenase and also of molecules acting specifi- cally on the clusters of basic amino-acids present at the surfaces of the glycolytic enzymes in the parasite.
Keywords---Glycolytic enzymes, inhibition, suicide inhibitors, allosteric inhibitors, aldolase, Trypanosoma brucei.
C O N T E N T S
1. Introduction 2. Inhibitors for the Enzyme Aldolase 3. Inhibitors for the Enzyme Glyceraldehyde-phosphate-dehydrogenase 4. Towards more Selective Inhibitors 5. Conclusion Acknowledgements References
347 348 355 359 363 364 364
1. I N T R O D U C T I O N
Among the many diseases that afflict humans, those caused by parasites occupy an important place due to the number of victims, the lack of efficient therapy and their continuing spread. As an example, the World Health Organization (1985) indicated that 25,000 new cases of the African type of trypanosomiasis appear every year in the Sub-Saharian part of the continent. Parasitic infections also significantly affect human nutrition through their impact on food animals; 3 million cattle die every year from trypanosomiasis.
Up to the early 1980s, the chemotherapy employed relied on a very limited number of drugs,
:~Corresponding author. Abbreviations--DHAP, dihydroxy-acetone-phosphate; FDP, fructose 1.6-diphosphate; GAP, glyceralde-
hyde-3-phosphate; GAPDH, glyceraldehyde-phosphate-dehydrogenase.
Jm- 60/2--N 347

348 J. PERIE et al.
mainly suramin, pentamidin and arsenicals. However, severe drawbacks are associated with their use: chemo-resistance and kidney damage in the use of suramin, hypoglycemia sometimes leading to death with lomidin and high toxicity linked to arsenicals (Howells, 1985). However, new opportunities have appeared following the publication of more advanced knowledge of the biology of the trypanosome. Indeed, on the basis of several features of the parasite metabolism, specific targets can be considered (Fairlamb, 1982; Englund et al., 1982; Wang, 1984). Included among them are kinetoplast DNA (Fairlamb et al., 1978), polyamine biosynthesis (Bacchi et al., 1980), the detoxification system (Shames et al., 1986) and glycolysis (Visser and Opperdoes, 1980; Fairlamb, 1982; Englund et al., 1982; Wang, 1984). As a first result, ~t-difluoro-methyl-ornithine, an ornithine decarboxylase inhibitor, was initially developed as an anticancer drug (Sjoerdsma, 1980); it subsequently was found to be active against trypanosome and developed as an anti-parasitic agent (Sjoerdsma and Schechter, 1984). However, even if cleared of any toxicity, this compound manifests a weak activity and large amounts of the drug are required in the treatment of a patient, in the range of 400 g for an adult over a 2-week period (Van Nieuwenhove, 1985). In such conditions, the development of this compound cannot be considered for large populations. The need for inexpensive, highly efficient and nontoxic drugs, therefore, remains a priority.
With this in mind, we consider that glycolysis should be thought of as eminently suitable for various reasons: glycolysis is essential to the trypanosome since it represents its only source of energy (Visser and Opperdoes, 1980); it follows that any compound that can efficiently block it can be regarded as a possible antiparasitic lead since the damage will be much more significant in the trypanosome than in the host. This was exactly why difluoro-methyl-ornithine could be developed as a drug being more damaging for the parasite than for mammalian cells. Subsequently, it has been shown that trypanosomal glycolysis contains several features distinct from mammalian glycolysis, which should allow the development of compounds acting selectively. Indeed, try- panosome glycolysis occurs in a specific microbody called glycosome (see Fig. 1) (Opperdoes et al., 1984) where all enzymes except pyruvate kinase are packed together.
In addition, in the parasite, NADH regeneration occurs through a coupling between a glyceraldehyde-phosphate-dehydrogenase (GAPDH) and a glycerol-phosphate dehydrogenase rather than a lactate-dehydrogenase; one of the ATP molecules is then produced by a glycerol kinase transferring one phosphate group from glycerol-phosphate to ADP and glycerol is subsequently extruded from the glycosome. This process takes place in anaerobic conditions (Visser and Opperdoes, 1980; Opperdoes, 1987).
Moreover, enzymes in trypanosome reveal differences in the amino acid sequence as compared with corresponding mammalian enzymes, the sequence identity being 40-50% (Michels, 1986). Finally, insertions of highly basic amino acids that generate clusters of positive charges with a definite topology at the enzyme surfaces have been characterized (Wierenga et al., 1987).
From these features, different strategies were developed directed at specifically blocking glycolysis in trypanosome (Scheme 1). Approaches considered included blocking the glucose import, inhibiting one or several glycolytic enzymes with compounds acting at the active site and possibly interfering with the mechanism of the reactions catalyzed by these enzymes, or disrupting enzyme function with compounds acting at the enzyme surface to obstruct catalytic conformational changes. Considering that such compounds should have to cross several membranes and that their concentration within the glycosome would be lower than those of the various metabolites, we thought that priority should be given to molecules acting at low concentration and, therefore, as irreversible or possibly suicide inhibitors. Examples of this situation are in Sections 2 and 3, with inhibitors directed to the enzymes aldolase and GAPDH. In Section 4, the results of the development of more specifically acting molecules will be given.
2. INHIBITORS OF THE ENZYME ALDOLASE
Fructose 1.6 biphosphate aldolase (EC 4.1.2.13) occupies a key position in glycolysis (Scheme 1) as it provides the link between the hexose and the triose. Inhibitors similar in structure to the substrates in the two directions, dihydroxy-acetone phosphate (DHAP) and fructose-l.6-diphos- phate (FDP) should, therefore, be considered.

Inhibition of glycolytic enzymes in the trypanosome 3 4 9
In Scheme 2, the general mechanism for aldolase is indicated; this includes the formation of an immonium ion between a lysyl group in the active site and the carbonyl at position 2 on the substrate (FDP in the glycolysis pathway, DHAP for the reverse reaction). Subsequently, proton abstraction from the O-H group at position 4 occurs, followed by a retro-aldolization reaction from the corresponding oxyanion, the immonium ion playing the role of an electron sink.
\ \\
) ..' ' I
v<~j .~:-. .,~. --.. . _
" ' < - ' i ~ : :~ }a ! :~7~ !FP5+ • ~ . . . , -.. .::.. " C:( . ~ : : % : ~ : ~ F . + : : . , "
- I-- : . ~ : ! ; ~ " - ~7".:~ ~ ~- : ~ ~ " - :
.2
; i
~.~2 . i
. . . .
~ 'l'DiT,.++ J . A " ~ ".: ~ ; ~ : ; ._.:' - "
:. -+::+,!r:+;~,.
+.f / . . . . +,.~,
< : ~ . 1.+.
• -.. : .
~ " ~ .
;)+-.
• - . • .;.
.,.---.
f
"'<] • N
f
t~
" + k ' ' " R . . . . t.. ,ra,,~...*i:/..,,.
~ " ' : ; " ~ k ~ ; - I / " % 2 " " ~'
.1 "+ L , ~
, , .~ . . . . : - , . -~ . ~ - ~ ~ "
.. . ,-;..,..t,~.:,.~.~¢?,,,.~/..o ~=.
F I G . 1. Electron-microscopy picture of a trypanosome. FP, flagelle pocket; F, flageile; G L ,
glycosome.

350 J. PEtriE et al.
glucose GLYCOSOME
o H 1 glucose
H g ~ATP
G-6-P
PGI I ~_J oH
PFK ~(~ATP ~'~.J~DP
(B°'~°~ °O FDP y_., o.
F-oH 1 ~NAD' J "'-"-'--~I oyo® I EOH G-3-P 1,3-DPGA I--OH I o® IfADP I#'ATP Loel
aK~I'~ATP PaK~K~'~ADoP o" I 0' i ro'
glycdrol 3-PGA
It" ADP Py.K ~'~AT P
wruvate
SCHEME 1. Giycolysis in the trypanosome.
Glyceraldehyde-3-phosphate (GAP) is the first product, the second being DHAP, formed after protonation of the enamine and hydrolysis of the second intermediate immonium ion (Horecker et al., 1976). In addition, it is known from previous studies, where rabbit muscle aldolase was used for the synthesis of FDP analogs, that the P~O-CH2-C(O)- moiety is essential for recognition at the active site (Bednarski et al., 1989). On this basis, possible inhibitors were designed containing this group, linked in the same molecule to a part susceptible of generating an electrophilic center following a proton abstraction induced by the enzyme. Scheme 3 illustrates the corresponding expected reaction, using sulfinyl and sulfonic esters, tetrahedral structures liable to be recognized at the binding site of the 6-phosphate group in the substrate FDP.
Proton abstraction at position 3,~ to the immonium ion, should generate an ~-enone on which a nucleophile in the protein could react later since this has been proven to occur for other inhibitors (Magnien et al., 1984) and has also been observed for the pro-S proton exchange in DHAP (Pratt, 1977). It has also been proven (Motiu-Degrood et al., 1979) that ~-enones inhibit aldolase; thus, if the reaction occurs as indicated in Scheme 3, sulfinic ester 1 should behave as a suicide inhibitor. In addition, compound 2 was synthesized (Scheme 3) where proton abstraction can occur either at position 3 as in 1, or at position 4 as for FDP. In the latter case, proton abstraction and

Inhibition of glycolytic enzymes in the trypanosome 351
elimination of a phenate group, once again by means of an E1CB type elimination, should lead to another type of electrophile, that is to say, a sulfene. Suicide inhibitors, where the reactive intermediate is a sulfene, have previously been described (Baltas et al., 1988).
In Scheme 4, the corresponding synthetic routes for preparing compounds 1 and 2 are given. Inhibition studies are given in Scheme 5. They were performed in the cleavage direction; GAP
formed was isomerized by triose phosphate isomerase to DHAP and the total DHAP then oxidized to glycerol-3-phosphate using glycerol-3-phosphate dehydrogenase. The progress of the reaction was followed by N A D H absorbance at 340 nm. Irreversible inactivation was demonstrated by the time dependence of inactivation and also the non-restoration of enzyme activity on dilution.
It was first shown that compounds 1 and 2 behave as irreversible inhibitors and that the whole structure is required for inhibition. For example, in compound 1, it was shown that the two parts,
A .OH 0 . ®o-y %A,/~ ®°" T .%,A,/o@ -~ .. o. o
o OH
GAP DHAP FOP
E + D H A P
H20 d ~H20
" o H
QAP h ~ GAP i .o / / e...
"."00 ,o
-- " - ' % . .
.odho E + FDP
SCHEME 2. General mechanism for aldolase; both routes for aidolisation and retro-aldolisation are indicated. The H, proton in DHAP's and the - O H group in FDP immonium are
indicated.

352 J. PERLE e t al.
CH2--O--(~ ?H2--O--(~ ~H2--O-- (~ ~:0 ~ /pC~k~H__ En z c--N~.4-- Enz
o = s _ - o
C= ~H-- Enz C'- N_H- Enz
j CH 2 H2 I ~ N~ S Enz
( ~ - - s o o CH2--O-- Q ~H2-C~
C~O (~=O I CH 2 CH2
I CH2 ~H2 O=~ =O O~S~O
? f CH3
2 3
SCHEME 3. Expected suicide inhibition mechanism with compound 1.
CH 2 t
O~S=O I O
~ H2-C ~
C = O I CH 2
namely 3-phenylsulfinyl propionic acid and mono-hydroxy-acetone phosphate, produce no inhi- bition.
From the secondary plot obtained from the first graph in Scheme 5, inactivation constants Ki and ki,act could be determined. The corresponding values are Ki = 1.65 mM, kinac t = 0.201 min- ~ for the rabbit muscle enzyme. For the Trypanosoma brucei enzyme, the same experiment gives Ki = 3 mM, kina¢ t = 0.842 min-J; these results correspond to similar effects, since k inac t /g i values are 0.12 and 0.28 min-~ . M -~, respectively. The second plot in Scheme 5 indicates that protection by the substrate DHAP is effective and, therefore, that inhibition may occur at the active site. However, cysteine assay using 5,5'-dithiobis 2-nitrobenzoic acid indicates that for the rabbit muscle enzyme, the four cysteine groups of each subunit are no longer accessible and may have reacted with the inhibitor. This indicates that the reactive Michael acceptor (~-enone) intermediate is expelled into the medium and reacts on all available cysteines; this intermediate is formed either in the active site, or in both the active site and in the buffer (triethanolamine). UV spectroscopic analysis of the base-catalyzed decomposition of compound 1 (pH = 10.8) indicates the formation of the corre- sponding sulfinate anion C6H5- -5OO-- (E215 = 104 ; E264 = 103) and that of the phosphorylated ~-enone. At pH 7.6, the pH at which kinetic experiments were done, the decomposition of 1 becomes much slower. The comparison of decomposition rates at pH 7.6 with and without enzyme is hard to follow, because the enzyme and buffer have high absorbance at wavelenghts below 245 nm and also because decomposition produces only slight changes in absorbance. Even though the formation of the reactive intermediate ct-enone is catalyzed by the enzyme, meaning that 1 behaves partially as a suicide inhibitor, a large part of the decomposition of the inhibitor also occurs in the buffer.
Rate comparison is still more difficult with inhibitor 2, which inactivates the rabbit muscle aldolase irreversibly, but also decomposes readily in the buffer. To circumvent these limitations, two other inhibitors, 3 and 4 (Scheme 3), were synthesized and assayed; compound 3 is similar in structure to compound 1, but was expected to show more selectivity in active-site decomposition, since its CH3SO:- group is a weaker leaving group (Stirling, 1979). In fact, it turned out that in this case, too, generation of the reactive ~t-enone is catalyzed both by the enzyme and by the buffer. An alternative route was looked for in compound 4, where proton abstraction at position 3 could

Inhibition of glycolytic enzymes in the trypanosome 353
only lead to a non-productive process, whereas proton abstraction at position 4 could only produce a reactive sulfene. Unfortunately, this compound is not recognized at the active site by aldolase, probably due to its too large size, and, therefore, inhibition through the expected process cannot occur .
With this in mind, it turns out that even though precursors 1--4 of the =-enone, 2-keto-3 butenyl phosphate are not the most appropriate, since they do not allow the selective formation of this reactive species within the active site, such studies deserve to be pursued. Indeed, when assayed on cultures of Trypanosoma equiperdum, compound 1 displayed a lethal dose of 10 pM, most likely through the ~-enone.
Experiments are now in progress to design more selective structures along the following lines: First, the affinity of the inhibitor for the enzyme should be increased: the comparison of the
affinity of DHAP (50/~M) compared with that of compound 5 (Scheme 6), which is 6.7 mM (Rose et al., 1969), indicates that the -OH group at position 3 is of importance for affinity. Assays of compounds 6 and 7 (Scheme 6) confirms this point, as well as the importance of chirality. These two compounds have first affinities in the same order of magnitude as the substrate DHAP and also behave as slow-binding inhibitors (Schloss, 1988), a feature that reflects a significant binding. The following scheme corresponds to this process:
kl k3 E + I ~ E . I ~ E . I *
k 2 k4
with K, [ E ] [ I ]
Cl CI OH OH I I I I
I CrO, I CH2 ~H2 ?H2 CH2 I BzEt=NCl HMPA H,SO. I
OH 2 "~ 01-,12 I= CH 2 . CH 2 I NaOH I H=O H20
0 ~- S : O HzO O:=== S = O ~ S-':-~O (GH3)z cO ~ I S = O I CH,Ci, I I I ° ? ? ?-
?H (COCI) z CI CH=N= C : O ,~ C----O == I CH2CI = I Et=O R R
/OBz / .OH N O =- ~'---OB z O = ~-~.OH II N O O II I I
dibenzyl ?H 2 ?H 2 ~.H phosphate H z Pd/C
C : O ~ C : O C = O Et=O I GH3OH I R benzene R R
R = (~ "SO2"CH2"C H 2" 1
R - C - O-SOa-CHa-CH 2- 2
S C n E ~ 4. Synthetic routes for compounds 1 and 2.

354 J. PERIE et al.
SCHEME 5.
a
m [l]=lmM
l [I] = 2mM
II [I] = 4mM
10 20 30
time (min)
b
q [OHAP]=OpM
. IDHAP]= O$d
q [DHAPJSOFM
* [DHAP]=l OOpM
10 I I , 0 10 20 30
time (mln)
Kinetics of irreversible inactivation of aldolase by compound la: of inactivation; b: protection experiments.
Time dependence

Inhibition of glycolytic enzymes in the trypanosome 355
and [E][I]
K~'- [El + [El*]
It should be noted that the best K~* value is obtained with the compound of R stereochemistry, which has an abstractable pro S proton as in the substrate; new possible suicide inhibitors are now being designed on this data.
Second, design of high affinity molecules for the enzyme in which a retro-aldolisation reaction cannot take place.
Compounds 8 and 9 (Scheme 7) correspond to this condition: (a) with compound 8, the formation of the immonium structure (proven by reduction with sodium
borohydride) then leads to an enol. Proton abstraction at C4 in the corresponding enol cannot be followed by a retro-aldolisation reaction and, therefore, inhibition should be observed. This turns out to be the case and a slow-binding inhibition is observed with K~* = 134 # i . If the two carbonyl groups are vicinal, as in compound 9', an irreversible inhibition is obtained with K~ = 6.2 mM and ki,act = 0.11 min- ~ as inhibition values;
(b) with compound 9, immonium formation at carbon 2 after ring opening cannot be followed by retro-aldolization since the -OH group at position 4 is absent. This compound behaves as a good competitive inhibitor (Ki = 33//M, for a K m value of 12 ~M for FDP). The significant affinity of this unreactive analog should allow this compound to be cocrystallized with aldolase. If the structure of the corresponding crystal can be solved, this should give a suitable image of the active site for the design of new more efficient inhibitors. These experiments are in progress.
DHAP
CH20 Q CH20 Q I I C=O C--O I I CH2OH CH3
K M = 50 ~Ml K i = 6.7 mM
CH20 Q CH20 Q i i
6 C=O 7 C----O I I CH-OH CH-OH
o~) t (s) I CH 3 CH 3
K i = 220 p.M K i = 800 laM
(K* i = 155 laM) (K* i = 390 I~M)
SCHEME 6. S t r u c t u r e s a n d i n a c t i v a t i o n c o n s t a n t s f o r c o m p o u n d s 5, 6 a n d 7.
3. INHIBITORS FOR TH E ENZYME G L Y C E R A L D E H Y D E - P H O S P H A T E - D E H Y D R O G E N A S E
G A P D H catalyses the transformation of GAP into 1,3-diphosphoglycerate according to the mechanism indicated on Scheme 8.
This reaction includes several steps: formation of a thio-hemiacetal resulting from the reaction of cysteine 149 on GAP (165 in the T. brucei enzyme), oxidation by NAD'- into a reactive thio-ester, followed by nucleophilic attack of an inorganic phosphate on the latter. This step corresponds to an unusual phosphorylation reaction and requires conditions where a phosphate can become an efficient nucleophile. Corresponding to the first step described above, we considered that analogs of substrates bearing a reactive group could also undergo a similar reaction by the same essential cysteine. We were

356 J. PEP.IE et al.
© o
8 K*i = 134 laM
g '
©
©
Ki = 6.2 m M
kinact = 0.11 min-1
B I H
/ o
9 Ki = 33 I~M
SCrmME 7. Structures and inactivation constants for compounds 9, 9" and 10.
encouraged to follow this direction by published results showing that two naturally occurring molecules, pentalenolactone (Duszenko and Mecke, 1986) and koningic acid (Kato et al., 1991) bind covalently to this enzyme; koningic acid, at least, binds through cysteine 149.
Observation of these two molecules prompts some comments: (a) neither one contains any similarity with the substrate GAP; (b) both have a high hydrophobic character and (c) two reactive groups, an epoxide and an ~-enone, are present in each of them. Based on this, different sets of molecules shown in Scheme 8 were synthesized with a chain similar to that of the substrate and bearing one of the reactive groups, epoxide or ~-enone. In these compounds, two factors were considered: chain length (consequently, both phosphates and phosphonates were prepared) and hydrophobic character. To increase hydrophobicity, the oxygen atom on phosphorus was replaced by sulfur and the negative charges in the phosphates or phosphonates were replaced by alkyl groups.
These compounds were then assayed on the enzyme GAPDH, both from rabbit muscle and from T. brucei, to characterize a possible selectivity.
From preliminary assays (percentage of enzyme inhibition after 5 min of incubation with the inhibitor, the concentration of which varied from 3--0.3 mM), kinetic studies were then performed to determine the type of inhibition. In Table 1, the results corresponding to irreversible inhibitions are presented; these cases were characterized by pseudo-first order kinetics of inactivation vs time, with no restoration of the enzyme activity upon dilution and cysteine assay using 5,5'-dithio-bis-(2- nitrobenzoic acid). This assay indicated that one cysteine group per subunit is blocked by the inhibitor. In addition, from the secondary plot of the reciprocal of the half-time of inactivation

Inhibition of glycolytic enzymes in the trypanosome 357
as a function of the inhibitor concentration, the different kinetic parameters in Table 1 could be determined. Two different cases were observed: (1) those where the secondary plot has an intercept and that correspond to the general scheme:
E + 1 ~ E.1 ~ E-1 covalent complex
non-covalent complex
with the two parameters Ki and kina¢ t and (2) those where the secondary plot goes through the origin and that correspond to the scheme: E + I --. E-I covalent complex with the constant k2,a. In fact, the two different types can be compared, since the values k~/K~ and k2na are expressed in the same units.
The following comments can be made: Irreversible inhibitions correspond to phosphates in the first series and to phosphonates in the
second. For example, phosphonates corresponding to compounds 10 or 11 (oxygen atom between phosphorus and the carbon chain being suppressed) or phosphates related to 13 or 14 have no activity. Corresponding molecular modelling (MM2) indicates that for the first set of compounds, the distance between the reactive CH2 carbon atom in the epoxide and the phosphorus atom can be closed to the same distance as between the reactive carbon atom in the carbonyl group and the
H y O H -Enz O y S - E n z O y O - (~)
H-C--OH ~ - H-C- -OH ~ H-C--OH ~ H-C--OH I I ~ I CH20_(~ Enz-SH CH20-( ~ NAD + I CH20- ® P' CH20- ®
Glyc~raldehyde-3-phosphate Thiohemiacetal Thioester 1,3-diphosphoglycdrate GAP . 1,3-.GAP
Y y tl o II
, c = c .3
Y = O , S Y = O , S
Z = O, (CH2) n . (n=l ou O) R1 = H, CH a
R = OH, OCH3, OiPr R2 = H, CH a
Z = O, (CH2)n (n= 1 ou 13)
R2 = Ra = OCH3, OiPr
1:12 = OCH3 ; R3 = NiPr2
P,2 = P ~ = N i P r 2
SCI-mM~ 8. General mechanism for glyceraldehyde-phosphate-dehydrogenase and structures of the synthesized inhibitors.

358
,,OH
c,- o
0
J. PERLE et al.
o
OH ,C=O
SCHEME 9. Structures of pentaleno-lactone (a) koningic acid (b).
phosphorus atom in the second set, namely 4.2 A. This distance in the epoxide series accords with a non-ground state conformation, but at only 1.5 kcal from the minimum (which is associated with the extended conformation). As for the epoxide series, we checked the possible effect of chirality and found a negligible one.
TABLE 1. Inactivation Values
0 II
OH
0 II
O II
OPh
(raM)
Rm 22,22
To 12s
Rm 1,36
To 0,9
Rm 7,e2
To s
for Inhibitors 10-17
(M'hnn "~) K i / K m ki
(rnn -I )
278 0,28
893 1,73
19 0,69
6,5 0,41
98 1,86
35 1,15
0,130(102
0,13X10 2
5,O7XlO= i
4,55xl 02
2,37x102
2,3xl 02
O II
H ~ ~CH = CH" P~'~'OH
T 1.,1 OH 0
3,33
4,54
42 0,33
4,6
lx102
10x102
0 II Rm
H. ~ CH =CH~ P~"~'OCH3
Y 14 To OCH 3
O
II H - . . - . ." P~."" OiP r y-°-; ,o,H,
0
0,06 0,75 0,73 121X102
792Xl 021
116](102
30x10 = "r
O II
-H~.OiPr C H a y C H ' = ~;iPr
O
O II
/CH = CH* P~" OCH3 NIIC OCHa
17
lk2na
Rm
Tb 0,55
Rm 4,44
To 1,as
6,87
55
23
0,81 15xi02 1
1,06 2,4X10 "2
0,66 3,56 xl 0 "2

Inhibition of glycolytic enzymes in the trypanosome 359
For example, with compound 10 at 8 mM concentration, the following inactivation rates were found: for the racemic, 0.126 + 0.015 min-~, for the R, 0.115 min- ' and for the S, 0.106 min- ' . A similar result was obtained with an ester comparable in structure to 11 (phenyl instead of ethyl).
The second point is that in both series, neutral esters are more reactive than charge- bearing compounds. Thus, in rabbit muscle enzyme, phosphoric ester 11 is 40 times more reactive than compound 10 (from ki/K~ ratios). Similarly, compound 14 is 120 times more reactive than reference compound 13. This result is a priori surprising since compounds 10 and 13, more similar in structure to the substrate GAP, should have better affinities than compounds 11 and 14, respectively. A possible explanation of this phenomenon may come from the peculiarities of the binding site of these inhibitors. Indeed, several experimental studies indicate that the inhibitor glycidol phosphate binds at the inorganic phosphate binding site Pi and not at that of the substrate GAP (Corbier et al., 1989). This should also be true for the other inhibitors presented here and molecular modelling has been performed on these grounds with the T. brucei enzyme. This study confirms the binding at the Pi's site; by positioning the phosphorus atom in the inhibitors at the Ps site for GAP, the required stretching for formation of a covalent bond between the CH2 group in the epoxide (inhibitor type I), or the carbon atom on the carbonyl (inhibitor type II) and the essential cysteine would be prohibitive. Conversely, the energy barrier to making the same bond when the inhibitor binds at the inorganic Pi site is much lower. Moreover, the conformational change associated with the binding brings the phosphorus head of the inhibitor into close proximity with two rather hydrophobic amino acids of the protein, alanine 135 and threonine 225.
This conformational change induced by the inhibitor could account for the fact that neutral esters 11 and 12, 14-17 in Table 1 have better affinities for the enzyme than polar compounds 10 and 13 (see, for example, the 125 mM Ki value for 10 with the T. brucei enzyme, compared with value 0.9 for compound 11 with the same enzyme). This may also account for the irreversible inhibition of the two hydrophobic compounds mentioned above, pentalenolactone and koningic acid.
The last point concerns the selectivity factor for the inhibition of the T. brucei enzyme compared with rabbit muscle enzyme. Comparison of the k~/K~ values (or k2nd for compounds 14, 15 and 16) for the two enzymes indicates that if inhibition is similar in both enzymes for compounds of the epoxide series, then a selectivity is observed for compounds 13 and 14. Indeed, the T. brucei enzyme reveals itself as being approximately 10 times more sensitive to inhibition than the mammalian enzyme.
Interestingly enough, when these different compounds were assayed on cultures of trypanosome in conditions defined earlier (Baltz et al., 1985), activities for the epoxides ranged 200-300 #M in value, whereas for compounds 13-17, a significant effect was found, since lethal doses were in the range of 0.5 pM (see Table 2). Expressed differently, the best activity is also observed with the most selective compounds.
Activities of the compounds could also be compared with the two drugs difluoro-methyl-or- nithine and lomidin and it turned out that compounds 15 and 16 (where the hydrophobic character is increased by the presence of two isopropyl groups) reveal themselves to be more active than the two reference drugs. However, it should be noted that activities in culture, not in vivo, are considered here.
4. TOWARDS MORE SELECTIVE INHIBITORS
The inhibitors described so far correspond to situations where selectivity is weak, a factor of 10 in the best case for compounds in Table 1. This is already significant since, as emphasized above, the damage in the trypanosome, where glycolysis is essential, will be higher than in the host cells, particularly if the uptake of the compound by the parasite is fast. However, as a drug generally has some toxicity, the best therapeutic indexes will be obtained with the most selective

360 J. PERLE et al.
TABLE 2. Lethal Doses on Cultures of Trypanosoma equiperdum of Inhibitors 141-17 and Comparison with Commercially Available Drugs
N ° Structures DLloo I~M
O II
14 H CH = CH/P~'~'OCHs 10
O
O U
y OiPr 0,3
O
O II
1 S CHa~ ~CHaCH \ OiPr T OiPr 0,4
O
O II
17 ,,CH, CH~ ~'~ OC1% 9OU N==C OCH~ 4(+DFMO)
~ HF z
H3N*(CHt)~I - - COO"
NH s,
OFMO 100
compounds. Two approaches have been developed concerning compounds acting on glycolysis in the trypanosome:
(a) One is based on the differences observed from the X-ray structures between human and T. brucei GAPDH' enzymes (Vellieux et al., 1993). The comparison of the two structures indicated major differences at the NAD + co-factor binding site. In Scheme 10, the differences are indicated in the boxes. In the left box, the residue for the T. brucei enzyme is given and in the right, the residue in the human enzyme. These were determined by Hol et al. (1991).
Based on these differences, several types of inhibitors were designed and assayed. Among them, adenosines modified at position 2' show a selectivity factor of 50, the half-inactivation concen- tration of such compounds on the T. brucei enzyme being in the range of 0.3 mM. n
Another approach developed by our group (Willson et al., 1993) is based on the fact that glycolytic enzymes in T. brucei have clusters of basic amino acids at their surfaces that determine zones of positive charges at physiological pH. It was first confirmed that the spatial organization of these clusters, as expected from molecular modelling (Wierenga et al., 1987), corresponds to paired clusters, each pair being 40/~ apart (Opperdoes et al., 1990). It was also predicted that because of the large conformational changes associated with the catalytic cycle of most glycolytic enzymes, particularly kinases, molecules of proper length bearing negative charges (and, therefore, able to react with the positive clusters) should strongly inhibit those enzymes and should do so selectively, since such organized clusters only exist in T. brucei enzymes. In Scheme 11, a
)Herdejin, P. Selective inhibitors for 7". brucei. 21st Symposium on Glycosomes, Bruxelles, Belgium, May 14, 1993.

Inhibition of glycolytic enzymes in the trypanosome
lsm~19*l ~ 1 7 9 I _=
o ~-~:1~ m N - f l " - T ~ o o ]+ '"".! A~S9 lo,.'TS
II fl \ ! + . , , + t %;,'V o , - - o - P - o - p - o - - , o -%
,o- ,o- , :+ ,+++, Mo ~oH
I '=P 2°91 re". tg+ I I Oly2t21Asp1971 [ Sex223 I ",1"20m.I
,, HO OH
,,,++"'~ I ran39 i he37 ~=+8 I M=45 I
SCHEME 10. Comparison of the amino-residues at the NAD ÷ co-factor binding site in GAPDH. Left box: in the T. brucei enzyme, right box: in the human enzyme.
361
representation of these clusters is given as well as the structure of the inhibitors, in which the chain length is varied by modifying the number n of CH2 groups in the linker.
Inhibition measurements on enzymes from reference enzymes (yeast, Candida or rabbit muscle) and T. brucei indicate that in most cases, the inhibition is rather selective, particularly for the
o o II II
RO-C C-OR
C-NH-ICH=)n-HN-C
RO-C C-OR II II 0 o
R=H or CH 3
SCHEME 1 1. Clusters of basic amino-acids at the surface of the T. brucei enzyme and general formula of the inhibitors.
362
100
[ I ] = 5 0 I.JM R.M.
a -
0 i 6 11 14 20
J . PERLE et al.
100
50
[ I ] =50 pM
~ R~TM 100
A ~ ' 50 ,b.
0 6 8 11 14 20
[ I ] =50 uM 100
T.b. o\/o i i ~ 1 0
6 8 11 14 20
[ I ] = 5 0 pM R.M.
o-..o--o.-o..-o T.b.
I I I ,
+ 8 ,,, 2o
[ I ] =100 pM [ I ] =100 pM [ I ] =100 pM [ I ] =100 pM , ° °
50 50 50 50
0 I I I I I l 0 ~ i i T I 0 i ~ i qP I 0 | T T T I 6 8 11 14 20 6 8 11 14 20 6 8 11 14 20 6 8 11 14 20
Spacer Length (n)
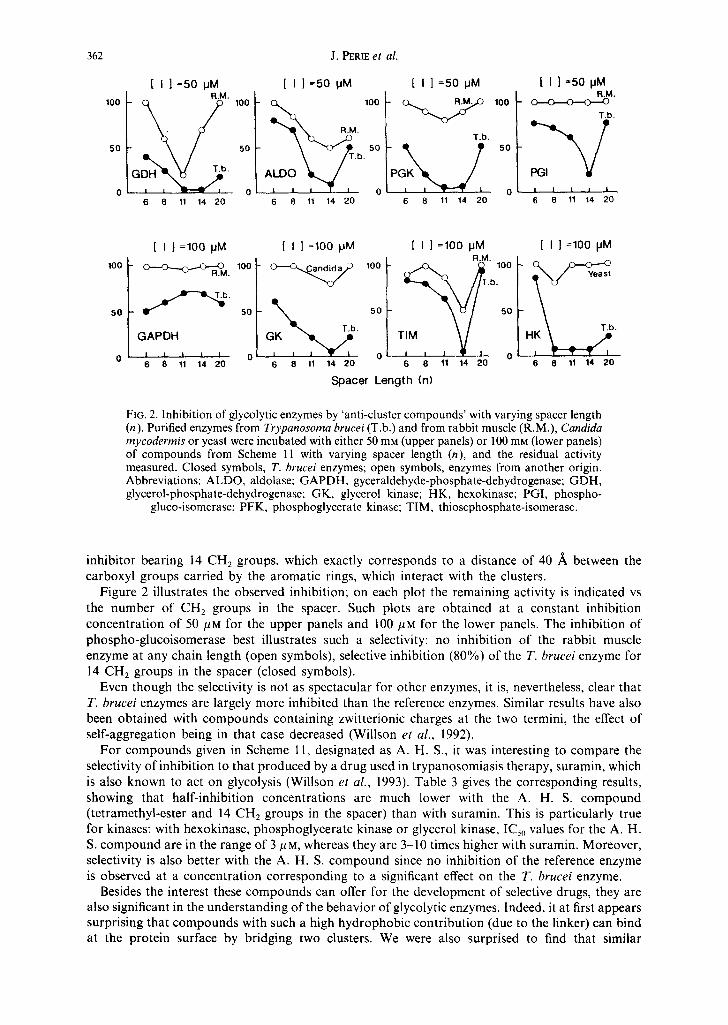
FIG. 2. Inhibition of glycolytic enzymes by 'anti-cluster compounds' with varying spacer length (n). Purified enzymes from Trypanosoma brucei (T.b.) and from rabbit muscle (R.M.), Candida mycodermis or yeast were incubated with either 50 mM (upper panels) or 100 mm (lower panels) of compounds from Scheme 11 with varying spacer length (n), and the residual activity measured. Closed symbols, T. brucei enzymes; open symbols, enzymes from another origin. Abbreviations: ALDO, aldolase; GAPDH, gyceraldehyde-phosphate-dehydrogenase; GDH, glycerol-phosphate-dehydrogenase; GK, glycerol kinase; HK, hexokinase; PGI, phospho-
gluco-isomerase; PFK, phosphoglycerate kinase; TIM, thiosephosphate-isomerase.
inhibitor bearing 14 CH 2 groups, which exactly corresponds to a distance of 40 /~ between the carboxyl groups carried by the aromatic rings, which interact with the clusters.
Figure 2 illustrates the observed inhibition; on each plot the remaining activity is indicated vs the number of CH2 groups in the spacer. Such plots are obtained at a constant inhibition concentration of 50/~m for the upper panels and 100 #M for the lower panels. The inhibition of phospho-glucoisomerase best illustrates such a selectivity: no inhibition of the rabbit muscle enzyme at any chain length (open symbols), selective inhibition (80%) of the T. brucei enzyme for 14 C H 2 groups in the spacer (closed symbols).
Even though the selectivity is not as spectacular for other enzymes, it is, nevertheless, clear that T. brucei enzymes are largely more inhibited than the reference enzymes. Similar results have also been obtained with compounds containing zwitterionic charges at the two termini, the effect of self-aggregation being in that case decreased (Willson et al., 1992).
For compounds given in Scheme 11, designated as A. H. S., it was interesting to compare the selectivity of inhibition to that produced by a drug used in trypanosomiasis therapy, suramin, which is also known to act on glycolysis (Willson et al., 1993). Table 3 gives the corresponding results, showing that half-inhibition concentrations are much lower with the A. H. S. compound (tetramethyl-ester and 14 CH2 groups in the spacer) than with suramin. This is particularly true for kinases: with hexokinase, phosphoglycerate kinase or glycerol kinase, ICs0 values for the A. H. S. compound are in the range of 3/~r~, whereas they are 3-10 times higher with suramin. Moreover, selectivity is also better with the A. H. S. compound since no inhibition of the reference enzyme is observed at a concentration corresponding to a significant effect on the T. brucei enzyme.
Besides the interest these compounds can offer for the development of selective drugs, they are also significant in the understanding of the behavior of glycolytic enzymes. Indeed, it at first appears surprising that compounds with such a high hydrophobic contribution (due to the linker) can bind at the protein surface by bridging two clusters. We were also surprised to find that similar

Inhibition of glycolytic enzymes in the trypanosome
TABLE 3. Half-inactivation Concentrations (ICso) in Micromolar for Suramin and the C14
Anti-clusters
IC5o tetramethyl Suramine A.H.S.
T.b 24 3 HK Y 220 - - T.b 60 27 PGI R.m 120 - - T.b 3 4.5 PFK R.m 0.6 16 T.b 100 60 TIM R.m 100 120 T.b 17 3 ALDO R.m 44 10 T.b 10 8 GDH R.m 40 - -
T.b 8 2.5 PGK R.m 55 - - T.b 35 3.5 GK R.m 140 - -
-- , inactive at the same concentration; HK, hexokinase; PGI, phospho-gluco-isomerase; PFK, phosphoglycerate kinase; TIM, triose-phosphate- isomerase; ALDO, aldolase; GDH, glycerol-phos- phate dehydrogenase; PGK, phosphoglycerol kinase; GK, glycerol kinase; T.b, Trypanosoma brucei; R.m, rabbit muscle; Y, yeast.
363
compounds with three -OH groups in the linker have weaker affinities than the compound in Scheme 11. For a better understanding on the interaction between the protein and these inhibitors, two approaches are being developed. The first is kinetic. From nonlinear Dixon plots and Scatchard determinations, it was possible to demonstrate two modes of binding: one corresponding to a bridging effect, which is a minor component and a second corresponding to the binding of two inhibitor molecules per enzyme molecule. The two inhibitor molecules interact. hydrophobically through the spacer chains and possibly also through the two aromatic rings. ~
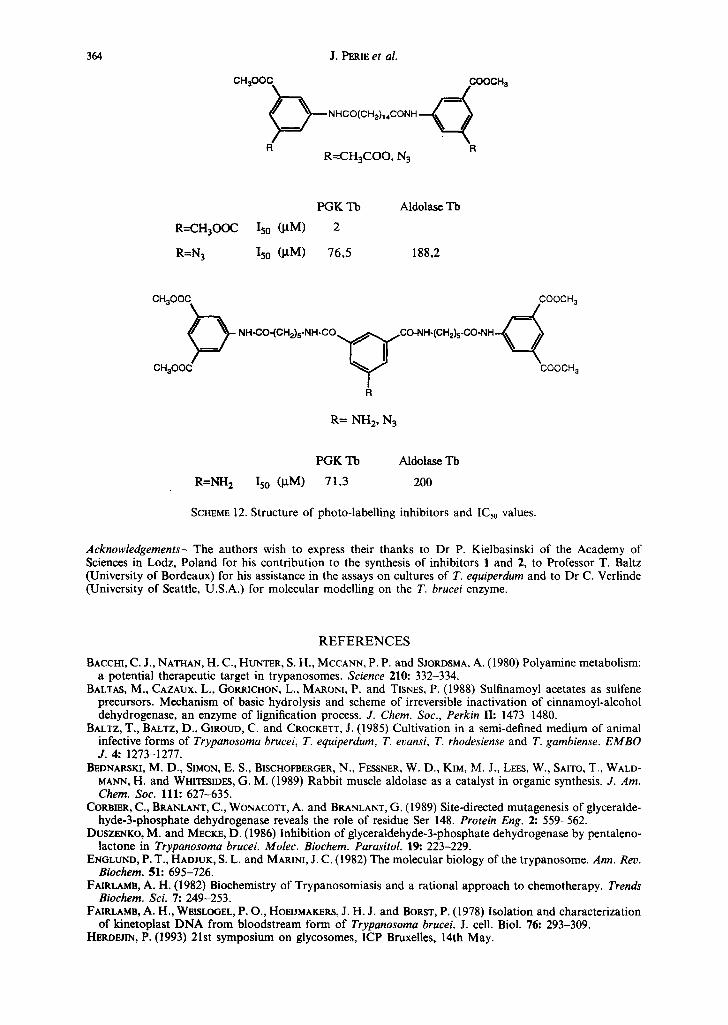
The second approach is the use of photo-labelling inhibitors; compounds in Scheme 12 illustrate this route. Inhibitors were synthesized bearing azido-groups either on the aromatic rings at the two termini or in the middle of the chain; these inhibitors, upon irradiation, free an electrophilic nitrene. Such nitrenes then bind covalently to the nucleophile on the protein in close proximity, therefore, allowing the determination of the binding site after sequencing and peptides analysis.
From the results obtained using these two methods, it should become possible to determine the exact binding mode of these inhibitors and subsequently, to design new compounds of higher selective affinities.
5. CONCLUSION
It can be said that the production of a therapeutic drug still remains a distant possibility with all the problems as yet to be solved, from transport to chemo-resistance. It is clear from this work that glycolysis represents a real opportunity for the development of new anti-trypanosomiasis agents. Owing to the essential role and the specific features of glycolysis in trypanosomes, in the future, it should be possible to obtain efficient and selective compounds. Strategies demonstrated here illustrate such possibilities.
JPuech, J., Willson, M. and Callens, M. Kinetic evidence for the binding mode ofinhibitors at the hot-spots on glycolytic enzymes in Trypanosoma brucei. Manuscript in preparation.
$PT 60/2--0
364 J. PERLE et al.
CH300C COOCH a
F1 R R=CH3COO, N 3
PGK Tb Aldolase Tb
R--CH3OOC Iso (IXM) 2
R=N 3 15o (l, tM) 76,5 188,2
CHaOOC COOCH 3
CHaOOC ~ COOCHa
R
R= NH 2, N 3
PGK Tb Aldolase Tb
R=NH 2 I5o (gM) 71,3 200
SCHEME 12. Structure of photo-labelling inhibitors and IC50 values.
Acknowledgements--The authors wish to express their thanks to Dr P. Kielbasinski of the Academy of Sciences in Lodz, Poland for his contribution to the synthesis of inhibitors 1 and 2, to Professor T. Baltz (University of Bordeaux) for his assistance in the assays on cultures of T. equiperdum and to Dr C. Verlinde (University of Seattle, U.S.A.) for molecular modelling on the T. brucei enzyme.
REFERENCES
BACCH[, C. J., NATHAN, H. C., HUNTER, S. H., MCCANN, P. P. and SJORDSMA, A. (1980) Polyamine metabolism: a potential therapeutic target in trypanosomes. Science 210: 332-334.
BALTAS, M., CAZAUX, L., GORRICHON, L., MARONI, P. and TISNES, P. (1988) Sulfinamoyl acetates as sulfene precursors. Mechanism of basic hydrolysis and scheme of irreversible inactivation of cinnamoyl-alcohol dehydrogenase, an enzyme of lignification process. J. Chem. Soc., Perkin II: 1473-1480.
BALTZ, T., BALTZ, D., GIROUD, C. and CROCKETT, J. (1985) Cultivation in a semi-defined medium of animal infective forms of Trypanosoma brucei, T. equiperdum, T. evansi, T. rhodesiense and T. gambiense. EMBO J. 4: 1273-1277.
BEDNARSKI, M. D., SIMON, E. S., BISCHOEBERGER, N., FESSNER, W. D., KIM, l~d. J., LEES, W., SAITO, T., WALD- MANN, H. and WHITESIDES, G. M. (1989) Rabbit muscle aldolase as a catalyst in organic synthesis. J. Am. Chem. Soc. 111: 627-635.
CORBIER, C., BRANLANT, C., WONACOTT, A. and BRANLANT, G, (1989) Site-directed mutagenesis of glyceralde- hyde-3-phosphate dehydrogenase reveals the role of residue Ser 148. Protein Eng. 2: 559-562.
DUSZENKO, M. and MECKE, D. (1986) Inhibition of glyceraldehyde-3-phosphate dehydrogenase by pentaleno- lactone in Trypanosoma brucei. Molec. Biochem. Parasitol. 19: 223-229.
ENGLUND, P. T., HADJUK, S. L. and MARINI, J. C. (1982) The molecular biology of the trypanosome. Ann. Rev. Biochem. 51: 695-726.
FAIRLAMB, A. H. (1982) Biochemistry of Trypanosomiasis and a rational approach to chemotherapy. Trends Biochem. Sci. 7: 249-253.
FAIRLAMB, A. H., WEISLOGEL, P. O., HOEIJMAKERS, J. H. J. and BORST, P. (1978) Isolation and characterization of kinetoplast DNA from bloodstream form of Trypanosoma brucei. J. cell. Biol. 76: 293-309.
HEll,IN, P. (1993) 21St symposium on glycosomes, ICP Bruxelles, 14th May.

Inhibition of glycolytic enzymes in the Trypanosome 365
HOL, W. G. J., VELLIEUX, F. M. D., VERLINDE, C. L. M., WIERENGA, R. K., NOBLE, M. E. N. and RANDY, J. R. (1991) Crystallographic investigations of glycolytic enzymes from Trypanosoma brucei: potential starting points for the design of new sleeping sickness drugs. In: Molecular Conformation and Biological Interactions, pp. 215-244, BALARAM, B. and RAMASESHAN, S. (eds) Indian Academy of Sciences, Bangalore.
HORECKER, B. L., TSOLAS, O. and LAI, C. Y. (1976) Aldolases. In: The Enzymes, 3rd edn, Vol. 7, pp. 213-258, BOYER, P. D. (ed.) J. Wiley, New York.
HOWELLS, R. E. (1985) The modes of action of anti-protazoal drugs. Parasitology 90: 687-703. KATO, M., SAKAI, K. and ENDO, A. (1991) Identification of koningic acid-modified site in rabbit muscle
glyceraldehyde-3-phosphate dehydrogenase. Biochem. biophys. Acta 1077: 192-196. MAGNIEN, A., LE CLEF, B. and BIELLMANN, J. F. (1984) Suicide inactivation of fructose 1-6 biphosphate
aldolase. Biochemistry 23: 6858-6862. MICHELS, P. A. M. (1986) Evolutionary aspects of trypanosomes; analysis of genes, d. Molec. Evol. 24: 45-52. MOTIU-DE GROOD, R., HUNT, W., WILDE, J. and HUPE, D. J. (1979) Rates and equilibria of the inactivation of
muscle aldolase by an active site directed Michael reaction. 3". Am. Chem. Soc. 101: 2182-2190. OPPERDOES, F. R. (1987) Compartimentation of carbohydrate metabolism in Trypanosomes. Ann. Rev. Microb.
41: 127-151. OPPERDOES, F. R., BAUDHIN, P., COPPENS, J., DE ROE, C., EDWARDS, S. W., WEIJERS, P. J. and MISSET, O. (1984)
Purification, morphometric analysis and characterization of the glycosomes of the protozoan hemoflagallate Trypanosoma brucei. J. Cell Biol. 98: 1178-1184.
OPPERDOES, F. R., WIERENGA, R. K., NOBLE, M. E. M., HOL, W. G. J., WILLSON, i . , KUNTZ, D. A., CALLENS, M. and PERIE, J. (1990) Unique properties of glycosomal enzymes. In: Parasites, Molecular Biology, Drug and Vaccine Design, pp. 233-246, AGABIAN, N. and CERAMI, A. (eds) Wiley-Liss, New-York.
PRATT, R. W. (1977) Rabbit muscle aldolase catalyzed proton exchange of hydroxy-acetone-phosphate with solvent. Biochemistry 16: 3988-3994.
ROSE, I. A. and O'CONNELL, E. L. (1969) Studies of interaction of aldolase with substrates analogs, o r. biol. Chem. 244: 126-134.
SCHLOSS, J. V. (1988) Significance of slow-binding enzyme inhibition and its relationship to reaction intermediate analogs. Acc. Chem. Res. 21: 348-353.
SHAMES, S. L., FAIRLAMB, A. H., CERAMI, A. and WALSCH, C. T. (1986) Purification and characterization of trypanothione reductase from Crithidia fasciculata, a newly discovered member of the family of disulfide containing flavoprotein reductases. Biochemistry 25: 3519-3526.
SJOERDSMA, A. (1980) Suicide enzyme inhibitors as potential drugs. Clin. Pharmac. Ther. 30: 3-22. SJOERDSMA, A. and SCHECHTER, P. J. (1984) Chemotherapeutic implications of polyamine biosynthesis
inhibition. Clin. Pharmac. Ther. 35: 287-300. STmLING, C. J. M. (1979) Leaving groups and nucleofugality in elimination and other organic reactions. Acc.
Chem. Res. 12: 198-203. VAN NmUWENHOVE, S. (1985) Clinical evaluations ofeflornithine in trypanosomiasis. Trans. R. Soc. Trop. Med.
Hyg. 79: 692-696. VELLIEUX, F. M. D., HADJU, J., VERLINDE, C. L. M., GROENDJIK, H., READ, R. J., GREENHOUGH, T. J., CAMPBELL,
J. W., KALK, K. H., LITTLECHILD, J. A., WATSON, H. C. and HOL, W. G. J. (1993) Structure of glycosomai glyceraldehyde-3-phosphate dehydrogenase from Trypanosoma brucei determined from Laue data. Proc. narD. Acad. Sci. U.S.A. 90: 2355-2359.
VISSER, N. and OPPERDOES, F. R. (1980) Glycolysis in Trypanosoma brucei. Eur. d. Biochem. 103: 623-632. WANG, C. C. (1984) Parasite enzymes as potential targets for anti-parasitic chemotherapy, d. Med. Chem. 27:
1-9. WIERENGA, R. K., SWINKELS, B., MICHELS, P. A. M., OSINGA, K., MISSET, 0., VAN BEEMEN, J., GIBSON, W. C.,
POSTMA, J. P. M., BORST, P., OPPERDOES, F. R. and HOL, W. G. J. (1987) Common elements on the surface of glycolytic enzymes from Trypanosoma brucei may serve as topogenic signals for import into glycosomes. EMBO J. 6: 215-221.
WILLSON, M., PERLE, J. J., MALECAZE, F., OPPERDOES, F. R. and CALLENS, M. (1992) Biological properties of amidinium sulfinic and sulfonic acid derivatives: inhibition of glycolytic enzymes of Trypanosoma brucei and protective effects on cell growth. Fur. or. Med. Chem. 27: 799-808.
WILLSON, M., CALLENS, M., KUNTZ, D. A., PERLE, J. and OPPERDOES, F. R. (1993) Synthesis and activity of inhibitors highly specific for the glycolytic enzymes from Trypanosoma brucei. Molec. Biochem. Parasitol. 59: 201-210.
WORLD HEALTH ORGANIZATION (1985) 7th Report on Tropical Diseases Research (TDR) Program. Geneva.




















