
Inhibition of Chemoattractant N-Formyl Peptide Receptor Trafficking by Active Arrestins
13
Inhibition of Chemoattractant N-Formyl Peptide Receptor Trafficking by Active Arrestins T. Alexander Key 1,2,3 , Charlotte M. Vines 1,3 , Brant M. Wagener 1,3 , Vsevolod V. Gurevich 4 , Larry A. Sklar 2,3 and Eric R. Prossnitz 1,3, * 1 Department of Cell Biology and Physiology, 2 Department of Pathology, and 3 Cancer Research and Treatment Center, University of New Mexico Health Sciences Center, Albuquerque, NM 87131, USA 4 Department of Pharmacology, Vanderbilt University Medical Center, Nashville, TN 37232, USA *Corresponding author: Eric R. Prossnitz, [email protected] Recent studies have highlighted the emergence of a class of G protein-coupled receptors that are internalized in an arrestin-independent manner. In addition to demonstrating that the N-formyl peptide receptor belongs in this family, we have recently shown that recycling of the receptor requires the presence of arrestins. To further elucidate mechanisms of arrestin-dependent regulation of G protein- coupled receptor processing, we examined the effects of altering the receptor–arrestin complex on ternary complex formation and cellular trafficking of the N-formyl peptide receptor by studying two active arrestin-2 mutants (trun- cated arrestin-2 [1–382], and arrestin-2 I386A, V387A, F388A). Complexes between the N-formyl peptide receptor and active arrestins exhibited higher affinity in vitro than the complex between the N-formyl peptide receptor and wild-type arrestin and furthermore were observed in vivo by colocalization studies using confocal microscopy. To assess the effects of these altered interactions on receptor trafficking, we demonstrated that active, but not wild-type, arrestin expression retards N-formyl peptide receptor inter- nalization. Furthermore, expression of arrestin-2 I386A/ V387A/F388A but not arrestin-2 [1–382] inhibited recycling of the N-formyl peptide receptor, reflecting an expanded role for arrestins in G protein-coupled receptor processing and trafficking. Whereas the extent of N-formyl peptide receptor phosphorylation had no effect on the inhibition of internalization, N-formyl peptide receptor recycling was restored when the receptor was only partially phosphory- lated. These results indicate not only that a functional interaction between receptor and arrestin is required for recycling of certain G protein-coupled receptors, such as the N-formyl peptide receptor, but that the pattern of receptor phosphorylation further regulates this process. Key words: arrestin, desensitization, endocytosis, formyl peptide receptor, G protein-coupled receptor, internalization, phosphorylation, recycling Received 15 June 2004, revised and accepted for publication 14 October 2004 The modulation of G protein-coupled receptor (GPCR) activity, including that of the b 2 -adrenergic (b 2 AR), angio- tensin II type 1 A, adenosine A 2 , D2 dopamine, CCR-5, and vasopressin V2 receptors, is effected through the con- certed actions of kinases, arrestins, and endocytic proteins (reviewed in (1–3)). Agonist activation induces the rapid phosphorylation of serines and threonines in the carboxyl- terminus and/or intracellular loops of GPCRs by a G protein-coupled receptor kinase (GRK). Arrestins then physically prevent the signaling of activated, phospho- rylated receptors by sterically precluding heterotrimeric G protein binding. For many GPCRs, arrestin binding also results in the simultaneous recruitment of endocytic pro- teins to the plasma membrane followed by translocation of the receptor–arrestin complex to clathrin-coated pits. As receptors spatially segregate from both agonists and G proteins upon internalization, they are further desensitized. Internalized GPCRs can re-sensitize after ligand and arrestin dissociation, phosphatase activity, and efferent trafficking (4). Arrestins therefore likely play a broad range of roles in the processing of GPCRs, with effects on signaling, internalization, trafficking and recycling. In recent years, however, GPCRs have been described that internalize in an arrestin-independent manner. These include the m2-muscarinic (5,6), protease-activated-1 (7), gonado- tropin-releasing hormone (8), and N-formyl peptide recep- tors (FPR (9–12)). Following agonist binding, these receptors are phosphorylated by a GRK (13,14). However, unlike class- ical GPCRs, phosphorylation in some cases may be suffi- cient for partial desensitization (15,16). Thus, arrestin binding may not be absolutely required to quench the ago- nist-stimulated signaling of such receptors. Furthermore, studies have revealed the presence of a novel endocytic pathway in the internalization of some of these receptors, since dominant-negative clathrin, dynamin and arrestin (or a subset thereof) do not affect receptor kinetics of endocyto- sis or recycling (5,9). Despite these dissimilarities, as with classical GPCRs, arrestins do in fact translocate to the mem- brane following agonist stimulation, bind to these GPCRs and can traffic with internalized receptors (15). Furthermore, in vitro studies with liganded, phosphorylated receptors in this class suggest complex interactions of the two proteins (15–17). Nevertheless, a clear-cut role for arrestins in the biology of certain receptors remains uncertain (18). The formyl peptide receptor (FPR) is a chemoattractant GPCR that couples to a pertussis toxin-sensitive G protein in leukocytes, where it mediates superoxide formation, degranulation, and chemotaxis (19). The FPR may be Traffic 2005; 6: 87–99 Copyright # Blackwell Munksgaard 2005 Blackwell Munksgaard doi: 10.1111/j.1600-0854.2004.00248.x 87
Transcript of Inhibition of Chemoattractant N-Formyl Peptide Receptor Trafficking by Active Arrestins

Inhibition of Chemoattractant N-Formyl PeptideReceptor Trafficking by Active Arrestins
T. Alexander Key1,2,3, Charlotte M. Vines1,3,Brant M. Wagener1,3, Vsevolod V. Gurevich4,Larry A. Sklar2,3 and Eric R. Prossnitz1,3,*1Department of Cell Biology and Physiology,2Department of Pathology, and3Cancer Research and Treatment Center, University ofNew Mexico Health Sciences Center, Albuquerque, NM87131, USA4Department of Pharmacology, Vanderbilt UniversityMedical Center, Nashville, TN 37232, USA*Corresponding author: Eric R. Prossnitz,[email protected]
Recent studies have highlighted the emergence of a class ofG protein-coupled receptors that are internalized in anarrestin-independent manner. In addition to demonstratingthat the N-formyl peptide receptor belongs in this family,we have recently shown that recycling of the receptorrequires the presence of arrestins. To further elucidatemechanisms of arrestin-dependent regulation of G protein-coupled receptor processing, we examined the effects ofaltering the receptor–arrestin complex on ternary complexformation and cellular trafficking of the N-formyl peptidereceptor by studying two active arrestin-2 mutants (trun-cated arrestin-2 [1–382], and arrestin-2 I386A, V387A,F388A). Complexes between the N-formyl peptide receptorand active arrestins exhibited higher affinity in vitro thanthe complex between the N-formyl peptide receptor andwild-type arrestin and furthermore were observed in vivoby colocalization studies using confocal microscopy. Toassess the effects of these altered interactions on receptortrafficking, we demonstrated that active, but not wild-type,arrestin expression retards N-formyl peptide receptor inter-nalization. Furthermore, expression of arrestin-2 I386A/V387A/F388A but not arrestin-2 [1–382] inhibited recyclingof the N-formyl peptide receptor, reflecting an expandedrole for arrestins in G protein-coupled receptor processingand trafficking. Whereas the extent of N-formyl peptidereceptor phosphorylation had no effect on the inhibitionof internalization, N-formyl peptide receptor recycling wasrestored when the receptor was only partially phosphory-lated. These results indicate not only that a functionalinteraction between receptor and arrestin is requiredfor recycling of certain G protein-coupled receptors, suchas the N-formyl peptide receptor, but that the pattern ofreceptor phosphorylation further regulates this process.
Key words: arrestin, desensitization, endocytosis,formyl peptide receptor, G protein-coupled receptor,internalization, phosphorylation, recycling
Received 15 June 2004, revised and accepted forpublication 14 October 2004
The modulation of G protein-coupled receptor (GPCR)
activity, including that of the b2-adrenergic (b2AR), angio-tensin II type 1A, adenosine A2, D2 dopamine, CCR-5, and
vasopressin V2 receptors, is effected through the con-
certed actions of kinases, arrestins, and endocytic proteins
(reviewed in (1–3)). Agonist activation induces the rapid
phosphorylation of serines and threonines in the carboxyl-
terminus and/or intracellular loops of GPCRs by a G
protein-coupled receptor kinase (GRK). Arrestins then
physically prevent the signaling of activated, phospho-
rylated receptors by sterically precluding heterotrimeric G
protein binding. For many GPCRs, arrestin binding also
results in the simultaneous recruitment of endocytic pro-
teins to the plasma membrane followed by translocation of
the receptor–arrestin complex to clathrin-coated pits. As
receptors spatially segregate from both agonists and G
proteins upon internalization, they are further desensitized.
Internalized GPCRs can re-sensitize after ligand and
arrestin dissociation, phosphatase activity, and efferent
trafficking (4). Arrestins therefore likely play a broad
range of roles in the processing of GPCRs, with effects
on signaling, internalization, trafficking and recycling.
In recent years, however, GPCRs have been described that
internalize in an arrestin-independent manner. These include
the m2-muscarinic (5,6), protease-activated-1 (7), gonado-
tropin-releasing hormone (8), and N-formyl peptide recep-
tors (FPR (9–12)). Following agonist binding, these receptors
are phosphorylated by a GRK (13,14). However, unlike class-
ical GPCRs, phosphorylation in some cases may be suffi-
cient for partial desensitization (15,16). Thus, arrestin
binding may not be absolutely required to quench the ago-
nist-stimulated signaling of such receptors. Furthermore,
studies have revealed the presence of a novel endocytic
pathway in the internalization of some of these receptors,
since dominant-negative clathrin, dynamin and arrestin (or a
subset thereof) do not affect receptor kinetics of endocyto-
sis or recycling (5,9). Despite these dissimilarities, as with
classical GPCRs, arrestins do in fact translocate to themem-
brane following agonist stimulation, bind to these GPCRs
and can traffic with internalized receptors (15). Furthermore,
in vitro studies with liganded, phosphorylated receptors in
this class suggest complex interactions of the two proteins
(15–17). Nevertheless, a clear-cut role for arrestins in the
biology of certain receptors remains uncertain (18).
The formyl peptide receptor (FPR) is a chemoattractant
GPCR that couples to a pertussis toxin-sensitive G protein
in leukocytes, where it mediates superoxide formation,
degranulation, and chemotaxis (19). The FPR may be
Traffic 2005; 6: 87–99Copyright # Blackwell Munksgaard 2005
Blackwell Munksgaard doi: 10.1111/j.1600-0854.2004.00248.x
87

considered a nonclassical GPCR, since both desensiti-
zation (at least in vitro) and internalization can proceed
through arrestin-independent mechanisms (9,10,12,15,16).
Furthermore, MAPK signaling, granule exocytosis and
chemotaxis can function through arrestin-independent
processes (9,14,20). Despite these findings, recent stud-
ies with arrestins and phosphorylation-deficient receptors
have illustrated key hallmarks of arrestin-FPR assemblies,
as well as critical determinants underlying ternary complex
formation (15–17). In addition, the FPR binds arrestin-2 and
-3 with near equal affinity (16,21) and associates with
arrestins for prolonged time periods after agonist stimula-
tion and receptor internalization (15). Notwithstanding
these findings, the scope of functions served by arrestins
in the biology of the FPR has remained unclear until very
recently. A new finding from our lab, however, has
revealed that FPR recycling is absent in cells lacking arrest-
ins, providing the first clear evidence for a role for arrestins
in FPR trafficking and a novel role for arrestins in general
(12,18). Furthermore, FPR stimulation in the absence of
arrestins leads to the rapid induction of apoptosis, suggest-
ing additional roles for arrestins (22).
Previous investigations focusing on mechanisms of
arrestin activation have relied on the development and
characterization of activating mutations (23–25), given the
high basal inactivity of the protein (26). In general, these
studies have focused on in vitro receptor binding studies in
the context of various states of GPCR activation and phos-
phorylation, as well as on rates of receptor desensitization.
From that work, an appreciation of the significant intra-
molecular interactions involving the carboxy-terminus and
the phosphate-sensing polar core has emerged. Recently,
however, the study of active arrestin mutants and specifi-
cally their effects on GPCR function and trafficking has
begun to shed light on the mechanisms involved in
arrestin-mediated regulation of certain GPCRs (27–29). In
the case of the b2AR, receptor activation in the presence
of two active arrestin mutants had no effect on internaliza-
tion but accelerated the recycling of the receptor, and in so
doing decreased its long-term degradation (27).
In the current report, we sought to extend our recent
studies that demonstrate a requirement for arrestin in FPR
recycling (12). We initially employed a well-characterized
spectrofluorometric assay of ligand–receptor–arrestin inter-
actions to examine the differences between two activating
mutations in arrestins: arrestin-2–3A, which contains alter-
ations of three bulky hydrophobic residues in the tail of
arrestin, and a truncated form of arrestin (amino acids
1–382). We then utilized a flow cytometric assay to explore
the effects of activated arrestin expression on FPR internal-
ization and recycling. Active arrestins significantly delayed
the internalization of the FPR, including the wild-type and
phosphorylation-deficient receptors. Most surprisingly,
however, we show that arrestin-2–3A, but not truncated
arrestin-2 [1–382], inhibits the recycling of the FPR.
Results
High agonist affinity complexes of arrestin-2–3A with
phosphorylated forms of the FPR
Arrestin-2 contains a series of consecutive amino acids in the
carboxy-terminus with bulky, hydrophobic side chains (Ile386
Val387 Phe388). These residues are hypothesized to bind to the
core of arrestin in the resting state and have been demon-
strated to contribute significantly toward basal inactivity of
the protein (30). Removal of the bulky hydrophobic side
chains of these residues, as with the mutant arrestin-2–3A,
has been shown to lead to phosphorylation-independent
binding to the b2AR, as well as more rapid desensitization
of the b2AR and d-opioid receptor (23). As the FPR can
internalize in an arrestin-independent manner (12), as
opposed to the b2AR (31), we sought to determine the
effects of the activation state of arrestins in the context of
the FPR biology.
In order to determine the affinity of arrestin binding to the
FPR as well as the effect of arrestin binding on ligand
affinity, we employed a soluble spectrofluorometric recon-
stitution assay which assesses reconstitution between a
fluorescent ligand, the FPR and either G proteins or arrest-
ins (16). The system operates on the following bases.
First, the antifluorescein antibody specifically binds to and
quenches the fluorescence of unbound ligand, providing a
direct measurement of the ligand dissociation rate. Second,
ligand dissociation rates depend on the receptor assemblies
formed. Ternary complexes of G proteins and wild-type
arrestins with the nonphosphorylated and phosphorylated
FPR, respectively, display high affinity for agonist. Isolated
receptor, on the contrary, displays low affinity. Lastly, add-
ition of nonhydrolyzable guanine nucleotides induces the
rapid dissociation of G proteins from receptors, which
consequently results in the rapid dissociation of ligand,
since uncoupled receptors exhibit low affinity for agonist.
Conversely, guanine nucleotide addition has no effect on
the stability of FPR-arrestin complexes.
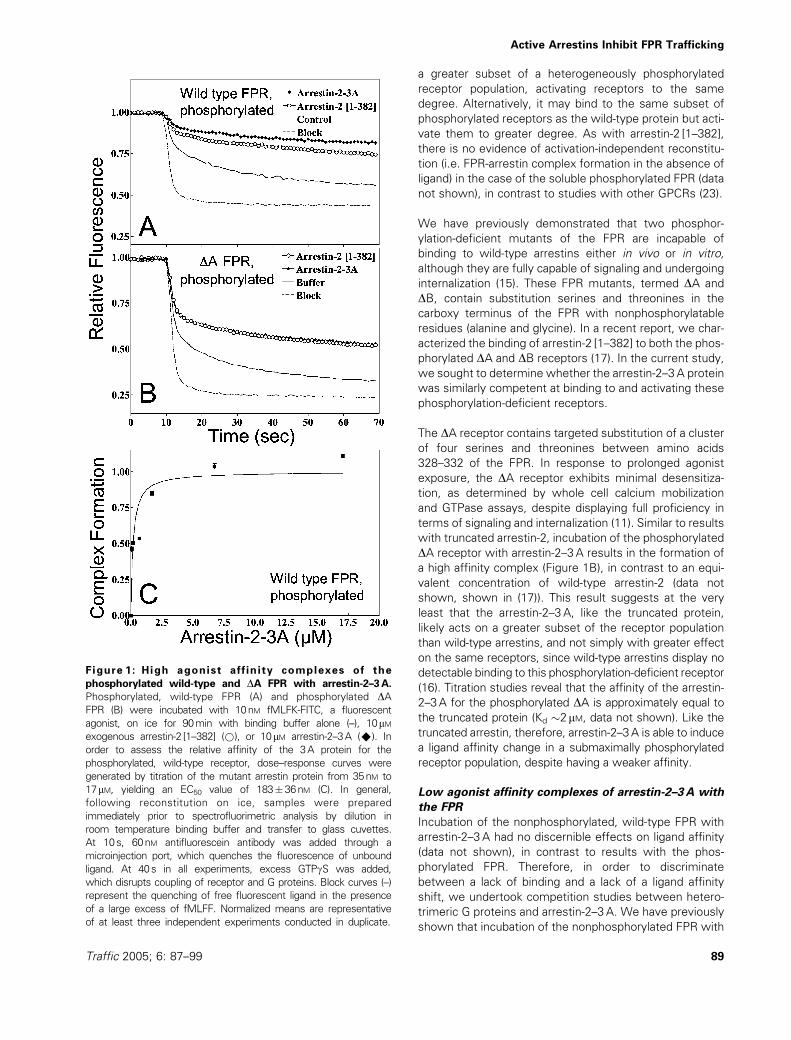
We initially examined the effects of the 3A mutation on
the receptor-ligand dynamics of the phosphorylated, wild-
type FPR. Incubation of the phosphorylated, wild-type
receptor with 10 mM arrestin-3A and fluorescent ligand
leads to the formation of a slowly dissociating complex
(Figure 1A), as previously described for arrestin-2 [1–382],
a truncated form of arrestin-2 that also displays constitu-
tive activation (17). Arrestin-2–3A reconstitution at an
equivalent concentration results in a similar fraction of
slowly dissociating species (Figure 1A). Titration studies
of arrestin-2–3A with the phosphorylated FPR demon-
strate an EC50 of 183� 32 nM (Figure 1C) that closely
resembles binding of arrestin-2 [1–382] (EC50�220nM
(17)) and wild-type arrestin-2 (EC50�600nM (16), see
table 1). Similar to results with truncated arrestins, where
the Bmax of the ligand affinity shift is significantly higher
than for wild-type arrestins, the arrestin-2–3A may bind to
Key et al.
88 Traffic 2005; 6: 87–99
a greater subset of a heterogeneously phosphorylated
receptor population, activating receptors to the same
degree. Alternatively, it may bind to the same subset of
phosphorylated receptors as the wild-type protein but acti-
vate them to greater degree. As with arrestin-2 [1–382],
there is no evidence of activation-independent reconstitu-
tion (i.e. FPR-arrestin complex formation in the absence of
ligand) in the case of the soluble phosphorylated FPR (data
not shown), in contrast to studies with other GPCRs (23).
We have previously demonstrated that two phosphor-
ylation-deficient mutants of the FPR are incapable of
binding to wild-type arrestins either in vivo or in vitro,
although they are fully capable of signaling and undergoing
internalization (15). These FPR mutants, termed DA and
DB, contain substitution serines and threonines in the
carboxy terminus of the FPR with nonphosphorylatable
residues (alanine and glycine). In a recent report, we char-
acterized the binding of arrestin-2 [1–382] to both the phos-
phorylated DA and DB receptors (17). In the current study,
we sought to determine whether the arrestin-2–3A protein
was similarly competent at binding to and activating these
phosphorylation-deficient receptors.
The DA receptor contains targeted substitution of a cluster
of four serines and threonines between amino acids
328–332 of the FPR. In response to prolonged agonist
exposure, the DA receptor exhibits minimal desensitiza-
tion, as determined by whole cell calcium mobilization
and GTPase assays, despite displaying full proficiency in
terms of signaling and internalization (11). Similar to results
with truncated arrestin-2, incubation of the phosphorylated
DA receptor with arrestin-2–3A results in the formation of
a high affinity complex (Figure 1B), in contrast to an equi-
valent concentration of wild-type arrestin-2 (data not
shown, shown in (17)). This result suggests at the very
least that the arrestin-2–3A, like the truncated protein,
likely acts on a greater subset of the receptor population
than wild-type arrestins, and not simply with greater effect
on the same receptors, since wild-type arrestins display no
detectable binding to this phosphorylation-deficient receptor
(16). Titration studies reveal that the affinity of the arrestin-
2–3A for the phosphorylated DA is approximately equal to
the truncated protein (Kd �2mM, data not shown). Like the
truncated arrestin, therefore, arrestin-2–3A is able to induce
a ligand affinity change in a submaximally phosphorylated
receptor population, despite having a weaker affinity.
Low agonist affinity complexes of arrestin-2–3A with
the FPR
Incubation of the nonphosphorylated, wild-type FPR with
arrestin-2–3A had no discernible effects on ligand affinity
(data not shown), in contrast to results with the phos-
phorylated FPR. Therefore, in order to discriminate
between a lack of binding and a lack of a ligand affinity
shift, we undertook competition studies between hetero-
trimeric G proteins and arrestin-2–3A. We have previously
shown that incubation of the nonphosphorylated FPR with
Figure 1: High agonist affinity complexes of the
phosphorylated wild-type and DA FPR with arrestin-2–3A.
Phosphorylated, wild-type FPR (A) and phosphorylated DAFPR (B) were incubated with 10nM fMLFK-FITC, a fluorescent
agonist, on ice for 90min with binding buffer alone (–), 10mMexogenous arrestin-2 [1–382] (*), or 10mM arrestin-2–3A (^). In
order to assess the relative affinity of the 3A protein for the
phosphorylated, wild-type receptor, dose–response curves were
generated by titration of the mutant arrestin protein from 35nM to
17mM, yielding an EC50 value of 183�36nM (C). In general,
following reconstitution on ice, samples were prepared
immediately prior to spectrofluorimetric analysis by dilution in
room temperature binding buffer and transfer to glass cuvettes.
At 10s, 60nM antifluorescein antibody was added through a
microinjection port, which quenches the fluorescence of unbound
ligand. At 40s in all experiments, excess GTPgS was added,
which disrupts coupling of receptor and G proteins. Block curves (–)
represent the quenching of free fluorescent ligand in the presence
of a large excess of fMLFF. Normalized means are representative
of at least three independent experiments conducted in duplicate.
Active Arrestins Inhibit FPR Trafficking
Traffic 2005; 6: 87–99 89

G proteins results in the formation of a high affinity,
nucleotide-sensitive complex (32), in contrast to ligand–
receptor–arrestin complexes and some ligand–phosphory-
lated receptor–arrestin complexes that exhibit low agonist
affinity (15–17). Since we have also demonstrated that G
protein binding can be blocked by truncated arrestin-2, we
incubated the FPR with arrestin-2–3A and G protein to
assess competition. As shown in Figure 2A, minimal
inhibition of high agonist affinity complex formation was
observed at 10 mM arrestin-2–3A, despite discernible
(� 50%) inhibition by truncated arrestin-2 at the same
concentration. This suggests that the 3A mutant can com-
pete for the nonphosphorylated FPR carboxyl-terminus,
though with lower affinity than the truncated protein.
The DB receptor contains targeted substitution of a cluster
of four serines and threonines between amino acids 334–
339 of the FPR. Despite normal signaling and internaliza-
tion, the DB receptor exhibits partial phosphorylation and
desensitization. Similar to recent results with truncated
arrestin-2 (17), incubation of the phosphorylated DB recep-
tor with arrestin-2–3A had no discernible effects on ago-
nist affinity (data not shown). Since truncated arrestin-2
can bind the carboxyl-terminus of the DB receptor, as
revealed by competition studies with exogenous G pro-
teins (17), we performed competition studies to examine
binding of arrestin-2–3A. Competition of G protein with
arrestin-2–3A did result in the conversion of the receptor
from a high affinity to low agonist affinity state (Figure 2B).
However, the level of competition at 10mM arrestin-2–3A
was somewhat less than with arrestin-2 [1–382] at an
equivalent concentration. Thus, the arrestin-2–3A in vitro
appears to display less phosphorylation-independence for
the FPR than the truncated arrestin protein, consistent with
the results above for the unphosphorylated wild-type recep-
tor. These results suggest that the interaction of the two
forms of activated arrestins with the FPR is distinct, leading
to the possibility that the effects of the two proteins on FPR
function may also be distinct. The affinities of wild-type and
mutant arrestins as well as ligand for the various states of
the FPR are summarized in Table1.
In vivo colocalization of arrestin–GFP with the FPR
To establish the binding characteristics of the arrestin-2–
3A with the FPR in vivo, we used U937 cells stably expres-
sing various forms of the FPR and transiently expressing
arrestin-2–GFP, arrestin-2 [1–382]–GFP, or arrestin-2–3A–
GFP. After a 24-h recovery period, cells expressed
arrestin–GFPs at 2–3 times the level of endogenous
arrestin, as determined by flow cytometric analysis of
anti-arrestin antibody stained cells (data not shown). Trans-
fected cells were stimulated with Alexa-546-labeled formyl
hexapeptide, fixed, and analyzed by confocal microscopy.
As shown in Figure 3, stimulation of the wild-type FPR for
3 and 10min at 37 �C resulted in the colocalization of
arrestin-2–3A and receptors into punctate, internal struc-
tures, consistent with prior results using wild-type
arrestin-2. Stimulation on ice produced neither receptor
clustering nor arrestin colocalization. In agreement with
in vitro binding results, both the DA and DB receptors
exhibited colocalization with the arrestin-2–3A, but not
with the wild-type arrestin, into punctate, endosomal
structures (Figure 4), similar to results with truncated
arrestin-2 (17). Interestingly, a significant fraction of the
arrestin-2–3A–GFP protein seemed to associate with the
plasma membrane in the wild-type, DA and DB FPR
expressing cells. This has not been observed to the
same extent with the wild-type arrestin–GFP (see Figure
3) or arrestin-2 [1–382]–GFP (17). These results confirm
the interaction of the arrestin-2–3A mutant with
Figure 2: Low agonist affinity complexes of the wild-type
and phosphorylated DB FPR with arrestin-2–3A. Given the lack
of discernible reconstitution between arrestins and low agonist
affinity states of putative receptor–arrestin complexes,
competition studies were undertaken between heterotrimeric G
proteins and arrestin-2–3A in order to assess coupling. Solubilized
samples of the nonphosphorylated, wild-type (A) and
phosphorylated DB FPR (B) were incubated with fluorescent
ligand (10nM), 1.0mM bovine brain G proteins, and either buffer
alone (&), 10mM truncated arrestin-2 (*), or 10 mM arrestin-2–3A
(^) for 120min prior to spectrofluorimetric analysis. Addition of
the 3A protein at high concentrations only minimally inhibited the
formation of the high agonist affinity, nucleotide-sensitive G
protein-coupled complexes, in comparison with the truncated
protein, suggesting a greater dependence on the phosphorylation
status of the FPR. Injections of both antifluorescein antibody and
GTPgS were made at 10 and 40 s, respectively. Normalized
means are representative of at least three independent
experiments conducted in duplicate.
Key et al.
90 Traffic 2005; 6: 87–99

phosphorylation-deficient mutants of the FPR but suggest
that the spatial and/or kinetic aspects of active arrestin
binding to the FPR may differ from that of the wild-type
arrestin.
Effects of active arrestins on FPR internalization
A previous study of the b2AR study has suggested that the
truncated ‘active’ form of arrestin translocates to liganded
receptors more quickly and stays associated for longer
than wild-type arrestins (33). To fully assess the effects
of active arrestin expression on FPR trafficking and proces-
sing, we employed a multiparameter flow cytometric
assay, with simultaneous gates for live cells, arrestin–
GFP expression, and a novel Alexa-labeled FPR agonist.
As shown in Figure 5A, overexpression of wild-type
arrestin-2–GFP had no significant effect on the rate of
wild-type FPR internalization in comparison with nontrans-
fected control cells, or non–GFP-expressing transfected
cells (data not shown). We therefore considered nontrans-
fected cells, which represented a significant fraction of our
electroporated samples, as valid, internal controls for the
expression of wild-type arrestins.
In contrast to wild-type arrestins, both arrestin-2–3A–GFP
and arrestin-2 [1–382]–GFP significantly inhibited the rate of
wild-type FPR internalization (Figure 5B). The most dramatic
inhibition was observed at early time points, with 94% and
80% percent inhibition at 3min for the truncated and 3A
mutant arrestin proteins, respectively. The observed inhibi-
tion was dose dependent, since gating on cell populations
with differential levels of arrestin–GFP expression yielded a
positive correlation with the extent of inhibition (Figure 5C).
Active arrestin–GFP chimerae, furthermore, also markedly
inhibited the rate of internalization of both phosphory-
lation-deficient, DA and DB, receptors, in comparison to
nontransfected controls (Figure 6A,B). Wild-type arrestin
overexpression, on the other hand, again had no effect
(data not shown). It therefore appears that in vitro binding
interactions of phosphorylation-deficient forms of the FPR
with activated arrestins can predict FPR internalization
dynamics, as in every case of demonstrable in vitro binding
interactions, inhibitory in vivo effects were observed.
Recent studies have suggested that active arrestins can
mediate the internalization of the b2AR in the absence of
significant receptor phosphorylation (27). In order to test
whether the reduced rate of internalization of the FPR
could be due to active arrestins mediating internalization
of the unphosphorylated FPR, we examined the effects of
active arrestin–GFP chimerae on the internalization of an
unphosphorylatable mutant form of the FPR (designated
DST, in which all the serine and threonine residues of the
carboxy terminus have been replaced with alanine and
glycine residues). This form of the FPR undergoes neither
ligand-mediated internalization nor desensitization in
the presence of endogenous arrestins (13,14). Our results
demonstrate that expression of active arrestin
mutants (arrestin-2–3A and arrestin-2 [1–382]) does not
induce internalization of this active unphosphorylated
form of the FPR (Figure 6C). These results are consistent
with previous conclusions that FPR internalization
Table 1: Arrestin and ligand affinities of FPR complexes a
FPR form Binding partner
G
proteinbWT
arrestin-2
arrestin affinityc
WT arrestin-2
ligand
affinityd
Arrestin-2 [1-382]
arrestin
affinity
Arrestin-2
[1-382]
ligand affinity
Arrestin-2–3A
arrestin
affinity
Arrestin-2 3A
ligand
affinity
WT FPR yes — NDe þ low þ low
Phosphorylated
WT FPR no þþþ med þþþ high þþþ high
(0.6 mM) (0.22 mM) (0.18 mM)DA yes — ND þ low þ low
Phosphorylated
DA yes — ND þþ high þþ high
(2.2 mM) (� 2 mM)DB yes — ND þ low þ low
Phosphorylated
DB yes — ND þþþ low þ/þþ low
(0.34 mM) (�10 mM)
aCompiled from this work and refs. (15–17).bRefers to the ability of the indicated FPR state to form a complex with G protein in vitro and initiate cell signaling in vivo.c— indicates an affinity >> 30 mM and not measurable; þ indicates an affinity >10 mM and weakly measurable; þþ indicates an affinity
between 1 and 10mM; þþþ indicates an affinity < 1mM. Where accurate values are known, they are shown in parentheses.dA qualitative determination inferred from both the direction and magnitude of the shift in the ligand dissociation curve of the indicated
complex. See also footnote a.eNot determinable due to the lack of observable complex formation. By default the receptors exist in a low affinity state for ligand.
Active Arrestins Inhibit FPR Trafficking
Traffic 2005; 6: 87–99 91

requires receptor phosphorylation yet is not mediated by
arrestins (12), in contrast to the b2AR.
Effects of active arrestins on FPR recycling
Given the prior results, one might hypothesize that the
apparent inhibition of internalization could be the result of
an enhanced recycling rate such that active arrestin
expression would lead to more rapid trafficking of internal-
ized receptors back to the cell surface, as has recently
been described for the b2AR (27). Additional support for
this hypothesis lies in the fact that the kinetics of arrestin
binding to the carboxyl-terminus of many GPCRs dictates
their kinetics of recycling (34). However, belying this
hypothesis, high affinity complexes are less likely to
dissociate than low affinity ones. In that sense, ‘active’
arrestins could stay associated with the FPR for longer
time periods, since they are of higher affinity, and there-
fore could retard, not enhance, recycling.
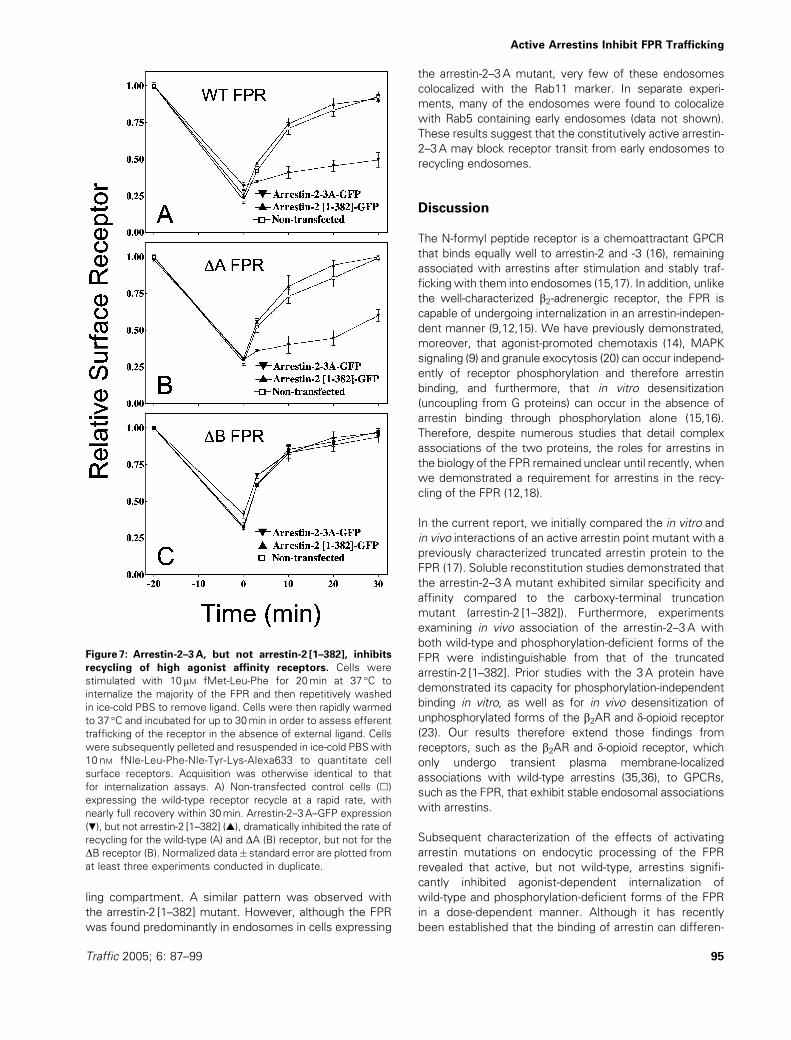
To assess directly the effects of active arrestin expression
on FPR recycling, we used the same flow cytometric
system as employed for internalization assays. However,
after agonist stimulation at 37 �C and washing on ice, cells
were rewarmed to 37 �C in the absence of ligand for vary-
ing times in order to allow trafficking from endosomes
back to the cell surface. As shown in Figure 7, overexpres-
sion of arrestin [1–382]–GFP had no observable effect on
the rate of wild-type FPR recycling in comparison with
nontransfected control cells. Surprisingly, however,
arrestin-2–3A–GFP transfected cells exhibited diminished
recycling rates with the wild-type receptor. Thus, the basis
of retarded internalization rates associated with active
arrestin expression is not accounted for in terms of
Figure 3: Co-localization of
arrestin-2–3A with Alexa546-
labeled wild-type FPR. Wild-type
FPR-expressing U937 cells were
electroporated with DNA for either
arrestin-2–GFP or arrestin-2–3A–
GFP. After 24–48h of recovery,
cells were treated with an Alexa-
568-labeled formyl hexapeptide
for 10min, fixed with parafor-
maldehyde, and observed by
confoca l mic roscopy . The
trafficking of arrestin-2–3A after
stimulation on ice (top row) or at
37 �C for 3min (2nd row from top)
and 10min (3rd row from top) and
wild-type arrestin for 10min at
37 �C (bottom row) was assessed
by imaging of ligand-receptor
complexes (red) and arrestins
(green). Co-localization (yellow) of
arrestins and receptors after
stimulation at 37 �C confirmed the
activation-dependent trafficking of
the proteins into punctate endo-
somes. Trafficking of arrestin-2–3A
to both the plasma membrane and
endosomes was observed for both
theDAandDB receptors, in contrast
to results with wild-type arrestins.
Images are representative of the
majority of transfected cells from
three separate experiments.
Key et al.
92 Traffic 2005; 6: 87–99

enhanced recycling rates. In fact, it is likely that the
diminished recycling rate mitigates the inhibitory inter-
nalization effect. To determine whether the extent of
FPR phosphorylation affected the inhibition of recycling
seen with the wild-type FPR, we tested both the DAand DB FPR mutants for their recycling kinetics upon
expression of activated arrestins. As with the wild-type
FPR, arrestin-2–3A–GFP markedly inhibited the rate of
recycling of the DA receptor, in contrast to nontrans-
fected controls (Figure 7B), unlike truncated arrestin
(Figure 7B) or wild-type arrestin (data not shown)
overexpression. Surprisingly, however, neither active
arrestin inhibited the recycling of the DB receptor
(Figure 7C).
Effects of active arrestins on intracellular localization
of the FPR
Given that the arrestin-2–3A inhibited recycling of the wild-
type and DA forms of the FPR, we sought to determine
whether the intracellular localization of the FPR was
altered in the presence of the arrestin-2–3A mutant. We
have recently demonstrated that arrestins are required for
FPR recycling (12). In addition, we observed that, in
arrestin-replete cells under equilibrium conditions repre-
senting continuous internalization and recycling, the FPR,
its ligand and arrestin display significant, though partial,
colocalization with recycling endosomes, as defined by
the marker Rab11. However, in the absence of arrestins,
the FPR and ligand accumulate in this compartment over
time, suggesting that exit from this compartment requires
arrestin. To examine the effects of active arrestins, we
cotransfected FPR-expressing U937 cells with Rab11–
GFP and monomeric red fluorescent protein chimeras of
wild-type and mutant arrestins and stimulated the cells
with an Alexa-633 derivative of the formylated hexapeptide
(Figure 8). Stimulation of cells expressing wild-type
arresin-2 and Rab11 for 30min yielded FPR in somewhat
enlarged endosomes compared to the 10min point (cf.
Figures 3 and 4). As we have observed before, a fraction
of these endosomes colocalize with the perinuclear recyc-
Figure 4: Co-localization ofGFP-
arrestins with Alexa546-labeled
FPR. DA (top two rows) and DB(bottom two rows) FPR-expressing
U937 cells were electroporated
with DNA for either arrestin-2–
GFP (rows 1 and 3) or arrestin-2–
3A–GFP (rows 2 and 4). After 24h
of recovery, cells were stimulated
with 10 nM Alexa-568-labeled
formyl hexapeptide for 10min,
fixed with paraformaldehyde, and
observed by confocal microscopy.
Colocalization of arrestins and the
phosphorylation-deficient forms of
the FPR was observed only with
the arrestin-2–3A mutant and not
thewild-type arrestin. Trafficking of
arrestin-2–3A to both the plasma
membrane and endosomes was
observed for both the DA and DBreceptors, in contrast to results
with wild-type arrestins. Images
are representative of the majority
of transfected cells from three
separate experiments.
Active Arrestins Inhibit FPR Trafficking
Traffic 2005; 6: 87–99 93

Figure 5: Active arrestins inhibit internalization of the wild-
type FPR. U937 cells stably expressing the wild-type FPR were
transiently transfected with arrestin–GFP constructs. Cells in
serum-free RPMI were stimulated with 10mM fMet-Leu-Phe for
up to 30min at 37 �C. Samples were repetitively washed in ice-cold
PBS and resuspended in PBS with 10nM fNle-Leu-Phe-Nle-Tyr-Lys-
Alexa633. A) Arrestin-2–GFP (*) expression had no discernible
effects on the internalization rate of the FPR, in comparison to
nontransfected control cells (&). We therefore used
nontransfected cells within the electroporated samples as
internal controls representative of the effects of wild-type
arrestin-2 for all subsequent experiments. B) Both arrestin-2 [1–
382]–GFP (~) and arrestin-2–3A–GFP ( .) expression significantly
delay internalization of the wild-type FPR. C) Plot of the extent of
internalization inhibition at the 3min point for the wild-type FPR as a
function of arrestin-2–3A–GFP fluorescence levels, based on gating
of FL1 events. A direct relationship between the level of arrestin-2–
3A–GFP expression and endocytic inhibition is evident. Data are
plotted as normalized means� standard error from at least three
experiments conducted in duplicate.
Figure 6: Active arrestins inhibit internalization of
phosphorylation deficient forms of the FPR. U937 cells
stably expressing the DA (A), the DB (B) or the DST (C) form of
the FPR were transiently transfected with arrestin-2–GFP,
arrestin-2–3 A–GFP, arrestin-2 [1–382]–GFP constructs or
vector only as indicated. Cells were stimulated with 10 mMfMet-Leu-Phe for up to 30min at 37 �C, repetitively washed in
ice-cold PBS and resuspended in PBS with 10 nM fNle-Leu-Phe-
Nle-Tyr-Lys-Alexa633 for analysis. Non-transfected cells
represent those in the transfected sample that did not
express green fluorescence above the level of control
untransfected cells. Data are plotted as normalized
means� standard error from at least three experiments.
Key et al.
94 Traffic 2005; 6: 87–99
ling compartment. A similar pattern was observed with
the arrestin-2 [1–382] mutant. However, although the FPR
was found predominantly in endosomes in cells expressing
the arrestin-2–3A mutant, very few of these endosomes
colocalized with the Rab11 marker. In separate experi-
ments, many of the endosomes were found to colocalize
with Rab5 containing early endosomes (data not shown).
These results suggest that the constitutively active arrestin-
2–3A may block receptor transit from early endosomes to
recycling endosomes.
Discussion
The N-formyl peptide receptor is a chemoattractant GPCR
that binds equally well to arrestin-2 and -3 (16), remaining
associated with arrestins after stimulation and stably traf-
ficking with them into endosomes (15,17). In addition, unlike
the well-characterized b2-adrenergic receptor, the FPR is
capable of undergoing internalization in an arrestin-indepen-
dent manner (9,12,15). We have previously demonstrated,
moreover, that agonist-promoted chemotaxis (14), MAPK
signaling (9) and granule exocytosis (20) can occur independ-
ently of receptor phosphorylation and therefore arrestin
binding, and furthermore, that in vitro desensitization
(uncoupling from G proteins) can occur in the absence of
arrestin binding through phosphorylation alone (15,16).
Therefore, despite numerous studies that detail complex
associations of the two proteins, the roles for arrestins in
the biology of the FPR remained unclear until recently, when
we demonstrated a requirement for arrestins in the recy-
cling of the FPR (12,18).
In the current report, we initially compared the in vitro and
in vivo interactions of an active arrestin point mutant with a
previously characterized truncated arrestin protein to the
FPR (17). Soluble reconstitution studies demonstrated that
the arrestin-2–3A mutant exhibited similar specificity and
affinity compared to the carboxy-terminal truncation
mutant (arrestin-2 [1–382]). Furthermore, experiments
examining in vivo association of the arrestin-2–3A with
both wild-type and phosphorylation-deficient forms of the
FPR were indistinguishable from that of the truncated
arrestin-2 [1–382]. Prior studies with the 3A protein have
demonstrated its capacity for phosphorylation-independent
binding in vitro, as well as for in vivo desensitization of
unphosphorylated forms of the b2AR and d-opioid receptor
(23). Our results therefore extend those findings from
receptors, such as the b2AR and d-opioid receptor, which
only undergo transient plasma membrane-localized
associations with wild-type arrestins (35,36), to GPCRs,
such as the FPR, that exhibit stable endosomal associations
with arrestins.
Subsequent characterization of the effects of activating
arrestin mutations on endocytic processing of the FPR
revealed that active, but not wild-type, arrestins signifi-
cantly inhibited agonist-dependent internalization of
wild-type and phosphorylation-deficient forms of the FPR
in a dose-dependent manner. Although it has recently
been established that the binding of arrestin can differen-
Figure 7: Arrestin-2–3A, but not arrestin-2 [1–382], inhibits
recycling of high agonist affinity receptors. Cells were
stimulated with 10 mM fMet-Leu-Phe for 20min at 37 �C to
internalize the majority of the FPR and then repetitively washed
in ice-cold PBS to remove ligand. Cells were then rapidly warmed
to 37 �C and incubated for up to 30min in order to assess efferent
trafficking of the receptor in the absence of external ligand. Cells
were subsequently pelleted and resuspended in ice-cold PBS with
10 nM fNle-Leu-Phe-Nle-Tyr-Lys-Alexa633 to quantitate cell
surface receptors. Acquisition was otherwise identical to that
for internalization assays. A) Non-transfected control cells (&)
expressing the wild-type receptor recycle at a rapid rate, with
nearly full recovery within 30min. Arrestin-2–3A–GFP expression
(.), but not arrestin-2 [1–382] (~), dramatically inhibited the rate of
recycling for the wild-type (A) and DA (B) receptor, but not for the
DB receptor (B). Normalized data� standard error are plotted from
at least three experiments conducted in duplicate.
Active Arrestins Inhibit FPR Trafficking
Traffic 2005; 6: 87–99 95

tially dictate the kinetics of GPCR recycling as a function of
the density of phosphorylation sites in the carboxy-termi-
nus of the receptor (34), this is the first study that links the
rate of GPCR internalization to the activation state of
arrestin. Furthermore, we demonstrate an inverse correla-
tion between the state of arrestin activity and the rate of
receptor internalization, since active arrestins actually
retarded FPR internalization.
Prior studies of a constitutively active arrestin point mutant,
arrestin-2-R169E, demonstrated that the active arrestin
could augment in an agonist-dependent, GRK-independent
manner, the rate of desensitization of the slowly desensitiz-
ing m-opioid receptor so that it was indistinguishable from
the rapidly desensitizing d-opioid receptor (37). This sug-
gested that it is the activation of arrestin, rather than its
binding, which is the rate limiting step in m-opioid receptor
desensitization. Since both the 3A and truncated arrestin-2
proteins used in our studies are preactivated, we can
hypothesize that the timing of arrestin association with acti-
vated states of the FPR is altered significantly in comparison
with wild-type arrestins. Furthermore, because the phos-
phorylation requirements for internalization of the FPR are
in general more relaxed than for desensitization and arrestin
binding (i.e. partially phosphorylated forms of the FPR, such
as DA and DB, internalize, although they do not bind, wild-
type arrestin), it follows that the process of FPR internaliza-
tion may be initiated temporally before the binding of endo-
genous arrestins. As we have recently shown, arrestin may
bind to the wild-type FPR only after initiation of the inter-
nalization process (45). It is therefore possible that the
activated forms of arrestin, which bind with high affinity to
the partially phosphorylated forms of the FPR, inhibit inter-
nalization by precluding the access of factors that must
interactwith the FPR prior to arrestin binding. In the absence
of specific knowledge regarding the mechanisms of FPR
internalization, however, it is difficult to propose what this
disrupted timing may be altering.
It is also possible that the activated arrestins are binding to
the ligand-bound, yet still unphosphorylated, FPR, thereby
inhibiting internalization. In the case of the b2AR, such an
interaction was demonstrated to permit phosphorylation-
independent internalization of this arrestin-dependent
receptor with unaltered kinetics (27). However, this possi-
bility seems unlikely for the FPR, as we have demon-
strated that an active unphosphorylated form of the FPR
can not be internalized by active arrestins (Figure 6C), that
the FPR internalizes in the absence of arrestins (12) and
furthermore that activated arrestins bind to fully and par-
tially phosphorylated forms of the FPR with 50–100-fold
higher affinity than to the unphosphorylated FPR (this
study and (17)). Although the b2AR preferentially interna-
lizes using arrestin-3 (31), it has been demonstrated to
associate with truncated arrestin-2 more quickly than
with wild-type arrestin and subsequently to traffic intra-
cellularly for prolonged times (33). However, in the case
of the b2AR, expression of active arrestins mediates much
more rapid recycling of the receptor (27). The likelihood
therefore that the inhibition of FPR internalization is a
consequence of decreased phosphorylation, despite an
inextricable link between these two processes, is greatly
diminished. Lastly, since internalization of the low agonist
affinity DB receptor was also significantly impaired, similar
to results with high agonist affinity receptors, we can
conclude that the resulting ligand affinity of the ternary
complex per se does not underlie the inhibitory internaliza-
tion effect. The same conclusion can be drawn with
Figure8: The FPR fails to traffic
t o the Rab11 r e cy c l i ng
compartment in the presence of
arrestin-2–3 A. U937 cel ls
express ing the FPR were
electroporated with Rab11–GFP
(green) and arrestin-2-mRFP1,
arrestin-2–3A-mRFP1 or arrestin-
2 [1–382]-mRFP1 constructs (red).
After 16–24h of recovery, cells
were treated with fNle-Leu-
Phe-Nle-Tyr-Lys-Alexa633 (blue)
f o r 3 0 m i n , f i x e d w i t h
paraformaldehyde, and examined
by confocal microscopy. Whereas
colocalization between a significant
fraction of the FPR with the Rab11
compartment is seenwithwild-type
arrestin-2 and arrestin-2 [1–382],
little colocalization is evident in
cells expressing the non-recycling
arrestin-2–3A mutant. In all cases,
arrestin is seen to colocalize with
the receptor. Images are repre-
sentative of transfected cells from
two separate experiments.
Key et al.
96 Traffic 2005; 6: 87–99

respect to the affinity of the mutant arrestin for the recep-
tor, since the DA mutant displays approximately 10-fold
lower affinity for both arrestin-2–3 A and arrestin-2 [1–382]
compared to the WT FPR. Finally, since the DA and DB
mutants are capable of recycling in the absence of WT
arrestin binding but the WT FPR requires arrestin for recy-
cling (12), we must presume that the extent of phospho-
rylation of the FPR dictates the requirement for arrestin in
recycling.
Perhaps the most puzzling result of our study is that
arrestin-2–3 A, but not wild-type arrestin-2 or arrestin-
2 [1–382], inhibits the recycling of the wild-type and DA,
but not DB, form of the FPR. It has been suggested that
the stability of a receptor–arrestin interaction dictates the
fate of an internalized GPCR (34). In that regard, the for-
mation of a transient receptor–arrestin complex on the
plasma membrane only favors rapid dephosphorylation
and return to the plasma membrane, whereas the forma-
tion of a stable receptor–arrestin complex (as defined
by colocalization on endosomes) retards resensitization
and may favor targeting of the receptor for eventual degra-
dation. Presumably, receptor resensitization requires
endosomal arrestin dissociation in addition to receptor
dephosphorylation. Since the principal difference between
the DA and DB receptors in terms of ternary complex
formation is agonist affinity, it is possible that differences
in complex stability (i.e. ligand affinity) account for the
differences in re-sensitization kinetics. That is, since the
DA receptor binds to arrestin-2–3 A and forms a high ago-
nist affinity complex, such assemblies are on average less
likely to dissociate in an acidic endosomal environment
than low agonist affinity assemblies composed of DB
receptors and are, concomitantly, less likely to recycle. It
should also be noted, however, that the somewhat lower
affinity of the arrestin-2–3 A mutant for the DB receptor
compared with the WT and DA FPR, could allow the for-
mer receptor but not the latter receptors to recycle.
A further explanation may lie in the loss of key regulatory
sites as a result of carboxy-terminal truncation of the
arrestin. The distal tail, which is intact in the 3 A, but not
the truncated arrestin-2 mutant, has been demonstrated to
contain several domains through which arrestins interact
with protein factors, as well as critical sites of regulation.
These protein factors include the b2-adaptin subunit of the
adaptor protein AP-2 (38), which has been shown to func-
tion in classical GPCR internalization (39), as well as
N-ethylmaleimide-sensitive factor, an ATPase which
binds to the distal tip of arrestin and functions in receptor
transport and sorting (40). Furthermore, it has been shown
that dephosphorylation of serine 412 in the tail of arrestin
is necessary for classical receptor–arrestin complexes to
engage the endocytic machinery (41). The truncated pro-
tein also lacks this important phosphorylation site. And
finally, there may be unknown conformational differences,
including sites of ubiquitination (42), between the two
proteins that would account for the differences in recy-
cling. The presence or lack of a critical domain may affect
not only the kinetics of trafficking, but also the endosomal
compartmentalization of internalized assemblies. Our
results suggest that the inhibition of recycling may in fact
result from a defect in the ability of the FPR to traffic
properly from early endosomes to recycling endosomes
in the presence of the arrestin-2–3 A mutant.
In summary, our recent results have demonstrated that
the presence of arrestin is required for FPR recycling (12).
The results presented here extend this observation to
reveal how the activation state of arrestin is a critical
determinant in regulating receptor recycling. Thus, an
arrestin mutant that is perhaps incapable of returning to
its basal inactive state (i.e. dissociating from the receptor)
is capable of blocking intracellular trafficking and thus pre-
vent the recycling of the FPR. Our results suggest that the
carboxy-terminus of arrestin as well as the ability of the
receptor to attain a conformation upon arrestin binding
displaying high affinity for the ligand are required for this
inhibition. These studies further suggest that dissociation
of the GPCR–arrestin complex is a critical determinant in
receptor recycling. It will be of interest to examine
whether disruptions in the timing and quality of arrestin
associations will similarly disrupt the endocytic profile of
other GPCRs that internalize in an arrestin-independent
manner. Lastly, our results underscore the fundamental
differences in the mechanisms involved in the internaliza-
tion and subsequent trafficking of the FPR as compared to
receptors such as classic b2AR.
Materials and Methods
ReagentsfNle-Leu-Phe-Nle-Tyr-Lys-Alexa546 and -Alexa633 were synthesized by
incubating Alexa Fluor-546 and -633 carboxylic acid succinimidyl esters
(Molecular Probes, Eugene, OR) in anhydrous DMSO (containing 100 mM
triethylamine as catalyst) with equimolar fNle-Leu-Phe-Nle-Tyr-Lys (Sigma,
St. Louis, MO) at room temperature overnight (17). GTPgS (guanosine 50-3-
O-(thio)triphosphate) was from Sigma. Frozen stocks were thawed imme-
diately prior to use. Monomeric RFP (mRFP1) was a generous gift from
Dr. Roger Tsien (43).
Cell stimulation and membrane preparationAs previously described, U937 cells stably expressing the FPR were cul-
tured in RPMI at 37 �C with 5% CO2 and passaged every 3–4 days by
reseeding at 2�105 cells/mL. For membrane harvest, cells were expanded
in sealed Pyrex spinner flasks, stimulated for 8 min at 37 �C with 10 mM
fMLF, which results in near maximal receptor phosphorylation, and trans-
ferred to ice (16).
Stimulated cells were collected by centrifugation, resuspended in ice-cold
cavitation buffer (10 mM HEPES, 100 mM KCl, 30 mM NaCl, 3.5 mM MgCl2,
600 mg/mL ATP, pH 7.3), and bombed by nitrogen cavitation for 20 min at
500 psi. Following centrifugation, the membrane fraction was resuspended
in HEPES sucrose buffer (200 mM sucrose, 25 mM HEPES, pH 7.0) with
protease inhibitor and phosphatase inhibitor cocktails (Calbiochem,
San Diego, CA) and flash frozen. Aliquots were stored until use at � 80 �C.
Detergent solubilizationThawed membranes were diluted in an intracellular binding buffer (BB:
30 mM HEPES, 100 mM KCl, 20 mM NaCl, 1 mM EGTA, 0.1% w/v BSA,
Active Arrestins Inhibit FPR Trafficking
Traffic 2005; 6: 87–99 97

0.5mM MgCl2), isolated by centrifugation, and resuspended in BB contain-
ing protease inhibitor cocktail set I, phosphatase inhibitor cocktail, and 1%
n-dodecyl b-D-maltoside (Calbiochem/EMD Biosciences, San Diego, CA).
Following solubilization for 90min at 4 �C, the soluble fraction was collected
by centrifugation for immediate experimentation.
Receptor reconstitutionSolubilized FPR was incubated with either bovine brain Gi/Go heterotrimer,
purified arrestins, or BB, as well as with fluorescent agonist, N-formyl-met-
leu-phe-lys-fluorescein 5-isothiocyanate (10 nM). The liganded samples
were gently mixed at 4 �C for up to 120min. Blocked samples received a
large excess of unlabeled peptide, N-formyl-met-leu-phe-phe (fMLFF), prior
to fluorescent ligand addition.
Spectrofluorometric analysisFluorescence was measured with an SLM 8000 spectrofluorometer (Spec-
tronics, Urbana IL) in time acquisition, photon-counting mode. Excitation
was fixed at 490 nm with a 490 nm band pass filter and the excitation
monochromater. Emission was monitored using a 520 nm band pass inter-
ference filter and a 500-nm-long pass filter. Antibody and nucleotide add-
itions were made through a microinjection port above the sample holder.
Following reconstitution at 4 �C, samples were diluted with room tem-
perature BB with 0.1% n-dodecyl b-D-maltoside and inhibitors, transferred
to glass cuvettes, and placed into the spectrofluorometer with gentle stirring.
For the first 10s, equilibrium levels were obtained. At 10s, excess antifluor-
escein antibodies, as previously described, were added to the sample. The
antibodies rapidly quench the fluorescence of the unbound, fluorescein-con-
jugated ligand. For some samples, excess GTPgS (guanosine 50-3-O-(thio)tri-
phosphate) was added at 40 s in order to disrupt G protein-receptor coupling.
ElectroporationsThe arrestin-2–GFP construct in pEGFPN1 was a generous gift from
Dr. Jeffrey Benovic (Thomas Jefferson University). Mutant arrestin
constructs were generated through PCR amplification using arrestin-specific
primers and verified in their entirety by dideoxy sequencing. For
transfections, DNA (25mg) was added to 8� 106 FPR-expressing U937
cells in serum-free RPMI. Cells were electroporated at 200V, 2000O, with
a 50ms pulse time on a BTX (Holliston, MA) electroporator and transferred to
T25 flasks with serum-containing RPMI. After 24–48h, cells were collected
by centrifugation, and resuspended in serum-free RPMI for assay.
Confocal microscopyTransfected cells were incubated with 10nM fNle-Leu-Phe-Nle-Tyr-Lys-
Alexa546 or 633 for the indicated time at either 0 �C or 37 �C. Stimulated
samples were immediately fixed with ice-cold, 2% paraformaldehyde for
30min. Cells werewashed, resuspended in Vectashield (Vector Laboratories,
Burlingame, CA), and mounted onto glass slides. Fluorescence images were
acquired on a Zeiss LSM510 confocal microscope (Carl Zeiss Inc., Thornwood,
NY, USA) to localize both the FPR (red) and arrestin (green) signals.
Internalization and recyclingTransfected cells in serum-free RPMI were stimulated with 10 mMfMet-Leu-Phe (FPR) or buffer for up to 30min at 37 �C. Samples were
repetitively washed in ice-cold PBS and resuspended in PBS with either
10 nM fNle-Leu-Phe-Nle-Tyr-Lys-Alexa633. Samples were kept on ice until
analysis. Fluorescence measurements were acquired on a Beckman Dick-
inson FACS Calibur. Live cells were initially gated by forward scatter vs.
side scatter plots. GFP-expressing cells were then gated by FL1 histogram
in comparison to untransfected control cells. Finally, the receptor-asso-
ciated fluorescence was resolved FL4 histogram analysis. The mean chan-
nel fluorescence for 50 000 events per sample was collected and analyzed.
For recycling assays, prior to fluorescent ligand/antibody addition, cells
were warmed for up to 30min at 37 �C to allow for receptor recycling,
quenched on ice, and collected by centrifugation. Acquisition was other-
wise identical to that for internalization assays.
Data analysisData were analyzed using FCSQuery (44) and Prism software (Graphpad,
San Diego, CA). In general, data were plotted as normalized fluorescent
intensity for receptor reconstitution or mean channel fluorescence for
internalization and recycling as a function of time. Non-linear regression
analyses were performed to generate dose–response curves and normal-
ized to Bmax values. For repetitive spectrofluorometric reconstitution
assays, sample data were pooled prior to analysis.
Acknowledgments
ThisworkwassupportedbyNIHgrantAI36357 toE.R.P.;NIHgrantsGM60799/
EB00265 and RR01315 to L.A.S., theNewMexico Cancer Research Fund; and
NIH grants EY11500 andGM63097 to V.V.G. Flow cytometry data and confocal
images in this paper were generated in the Flow Cytometry and Fluorescence
Microscopy Facilities, respectively, at the University of New Mexico Health
Sciences Center, which received support from NCRR 1S10 RR14668, NSF
MCB9982161, NCRR P20 RR11830, NCI R24 CA88339, the University of
NewMexicoHealthSciencesCenter, and theUniversity ofNewMexicoCancer
Center. C.M.V. received support from the University of New Mexico Cancer
Research and Treatment Center and is a recipient of an NIH post-doctoral
training fellowship (T32 AI007538). B.M.W. received support from an NIH pre-
doctoral training fellowship (T32 AI007538) and Department of Defense Breast
Cancer Research Program pre-doctoral award (BC030217).
Note added in Proofs
At the time of publication of this article, Dinh et al. (DinhDT, QianH, Seeber R,
Lim E, Pfleger K, Eidne KA, Thomas WG. Helix I of b-arrestin is involved in
post-endocytic trafficking, but is not required for membrane transloca-
tion, receptor binding and internalization. Mol Pharmacol 2004 (in press))
demonstrated that residues L100/L104/L108 in helix I of arrestin, which inter-
acts directly with I386/V387/F388 (the residues mutated in arrestin-2-3A), are
required for trafficking of type 1 Angiotensin II receptors to deep-core (likely
recycling) endosomes. This result is consistent with our conclusion that
disruption of the L100/L104/L108-I386/V387/F388 prevents the FPR from traf-
ficking to recycling endosomes and thus recycling.
References
1. Ferguson SS. Evolving concepts in G protein-coupled receptor endocy-
tosis: the role in receptor desensitization and signaling. Pharmacol Rev
2001;53:1–24.
2. Luttrell LM, Lefkowitz RJ. The role of b-arrestins in the termination
and transduction of G protein-coupled receptor signals. J Cell Sci
2002;115:455–465.
3. Pierce KL, Premont RT, Lefkowitz RJ. Signalling: Seven-transmem-
brane receptors. Nat Rev Mol Cell Biol 2002;3:639–650.
4. Pitcher JA, Payne ES, Csortos C, DePaoli-Roach AA, Lefkowitz RJ. The
G-protein-coupled receptor phosphatase: a protein phosphatase type
2A with a distinct subcellular distribution and substrate specificity. Proc
Natl Acad Sci U S A 1995;92:8343–8347.
5. Roseberry AG, Hosey MM. Internalization of the M2 muscarinic acety-
lcholine receptor proceeds through an atypical pathway in HEK293 cells
that is independent of clathrin and caveolae. J Cell Sci 2001;114:739–746.
6. Pals-Rylaarsdam R, Gurevich VV, Lee KB, Ptasienski JA, Benovic JL,
Hosey MM. Internalization of the m2 muscarinic acetylcholine recep-
tor. Arrestin-independent and -dependent pathways. J Biol Chem
1997;272:23682–23689.
7. Paing MM, Stutts AB, Kohout TA, Lefkowitz RJ, Trejo J. b-arrestins
regulate protease-activated receptor-1 desensitization but not internal-
ization or down-regulation. J Biol Chem 2002;277:1292–1300.
8. Heding A, Vrecl M, Hanyaloglu AC, Sellar R, Taylor PL, Eidne KA. The
rat gonadotropin-releasing hormone receptor internalizes via a b-
Key et al.
98 Traffic 2005; 6: 87–99

arrestin-independent, but dynamin-dependent, pathway: addition of a
carboxyl-terminal tail confers b-arrestin dependency. Endocrinology
2000;141:299–306.
9. Gilbert TL, Bennett TA, Maestas DC, Cimino DF, Prossnitz ER. Intern-
alization of the human N-formyl peptide and C5a chemoattractant
receptors occurs via clathrin-independent mechanisms. Biochemistry
2001;40:3467–3475.
10. Bennett TA, Maestas DC, Prossnitz ER. Arrestin binding to the G
protein-coupled N-formyl peptide receptor is regulated by the con-
served ‘DRY’ sequence. J Biol Chem 2000;275:24590–24594.
11. Maestes DC, Potter RM, Prossnitz ER. Differential phosphorylation
paradigms dictate desensitization and internalization of the N-formyl
peptide receptor. J Biol Chem 1999;274:29791–29795.
12. Vines CM, Revankar CM, Maestas DC, Larusch LL, Cimino DF, Kohout
TA, Lefkowitz RJ, Prossnitz ER. N-formyl peptide receptors internalize
but do not recycle in the absence of arrestins. J Biol Chem
2003;278:41581–41584.
13. Prossnitz ER. Desensitization of N-formylpeptide receptor-mediated
activation is dependent upon receptor phosphorylation. J Biol Chem
1997;272:15213–15219.
14. Hsu MH, Chiang SC, Ye RD, Prossnitz ER. Phosphorylation of the N-
formyl peptide receptor is required for receptor internalization but not
chemotaxis. J Biol Chem 1997;272:29426–29429.
15. Bennett TA, Foutz TD, Gurevich VV, Sklar LA, Prossnitz ER.
Partial phosphorylation of the N-formyl peptide receptor inhibits G
protein association independent of arrestin binding. J Biol Chem
2001;276:49195–49203.
16. Key TA, Bennett TA, Foutz TD, Gurevich VV, Sklar LA, Prossnitz ER.
Regulation of formyl peptide receptor agonist affinity by reconstitution
with arrestins and heterotrimeric G proteins. J Biol Chem
2001;276:49204–49212.
17. Key TA, Foutz TD, Gurevich VV, Sklar LA, Prossnitz ER. N-formyl
peptide receptor phosphorylation domains differentially regulate
arrestin and agonist affinity. J Biol Chem 2003;278:4041–4047.
18. Prossnitz ER. Novel roles for arrestins in the post-endocytic trafficking
of G protein-coupled receptors. Life Sci 2004;75:893–899.
19. Prossnitz ER, Ye RD. The N-formyl peptide receptor: a model for the
study of chemoattractant receptor structure and function. Pharmacol
Ther 1997;74:73–102.
20. Vines CM, XueM, Cimino DF, Maestas DC, Prossnitz ER. Regulation of
N-formyl peptide-mediated degranulation by receptor phosphorylation.
J Immunol 2002;169:6760–6766.
21. Potter RM, Key TA, Gurevich VV, Sklar LA, Prossnitz ER. Arrestin
variants display differential binding characteristics for the phosphory-
lated N-formyl peptide receptor carboxyl terminus. J Biol Chem
2002;277:8970–8978.
22. Revankar CM, Vines CM, Cimino DF, Prossnitz ER. Arrestins block
G protein-coupled receptor-mediated apoptosis. J Biol Chem
2004;279:24578–24584.
23. Celver J, Vishnivetskiy SA, Chavkin C, Gurevich VV. Conservation of
the phosphate-sensitive elements in the arrestin family of proteins.
J Biol Chem 2002;277:9043–9048.
24. Kovoor A, Celver J, Abdryashitov RI, Chavkin C, Gurevich VV. Targeted
construction of phosphorylation-independent b-arrestin mutants with
constitutive activity in cells. J Biol Chem 1999;274:6831–6834.
25. Vishnivetskiy SA, Paz CL, Schubert C, Hirsch JA, Sigler PB,
Gurevich VV. How does arrestin respond to the phosphorylated state
of rhodopsin? J Biol Chem 1999;274:11451–11454.
26. Schleicher A, Kuhn H, Hofmann KP. Kinetics, binding constant, and
activation energy of the 48-kDa protein- rhodopsin complex by extra-
metarhodopsin II. Biochemistry 1989;28:1770–1775.
27. Pan L, Gurevich EV, Gurevich VV. The nature of the arrestin-receptor
complex determines the ultimate fate of the internalized receptor.
J Biol Chem 2003;278:11623–11632.
28. Gray JA, Bhatnagar A, Gurevich VV, Roth BL. The interaction of a
constitutively active arrestin with the arrestin-insensitive 5-HT (2A)
receptor induces agonist-independent internalization. Mol Pharmacol
2003;63:961–972.
29. Penn RB, Pascual RM, Kim YM, Mundell SJ, Krymskaya VP,
Panettieri RA Jr, Benovic JL. Arrestin specificity for G protein-coupled
receptors in human airway smooth muscle. J Biol Chem
2001;276:32648–32656.
30. Vishnivetskiy SA, Schubert C, Climaco GC, Gurevich YV, Velez MG,
Gurevich VV. An additional phosphate-binding element in arrestin mol-
ecule. Implications for the mechanism of arrestin activation. J Biol
Chem 2000;275:41049–41057.
31. Kohout TA, Lin FS, Perry SJ, Conner DA, Lefkowitz RJ. b-arrestin 1 and
2 differentially regulate heptahelical receptor signaling and trafficking.
Proc Natl Acad Sci U S A 2001;98:1601–1606.
32. Bennett TA, Key TA, Gurevich VV, Neubig R, Prossnitz ER, Sklar LA.
Real-time analysis of G protein-coupled receptor reconstitution in a
solubilized system. J Biol Chem 2001;276:22453–22460.
33. Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Molecular
determinants underlying the formation of stable intracellular G pro-
tein-coupled receptor-b-arrestin complexes after receptor endocytosis.
J Biol Chem 2001;276:19452–19460.
34. Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Association of b-
arrestin with G protein-coupled receptors during clathrin-mediated
endocytosis dictates the profile of receptor resensitization. J Biol
Chem 1999;274:32248–32257.
35. Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential
affinities of visual arrestin, b arrestin1, and b arrestin2 for G protein-
coupled receptors delineate two major classes of receptors.
J Biol Chem 2000;275:17201–17210.
36. Zhang J, Barak LS, Anborgh PH, Laporte SA, Caron MG, Ferguson SS.
Cellular trafficking of G protein-coupled receptor/b-arrestin endocytic
complexes. J Biol Chem 1999;274:10999–11006.
37. Celver JP, Lowe J, Kovoor A, Gurevich VV, Chavkin C. Threonine 180
is required for G-protein-coupled receptor kinase 3- and b-arrestin
2-mediated desensitization of the mu-opioid receptor in Xenopus
oocytes. J Biol Chem 2001;276:4894–4900.
38. Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SS, Caron MG,
Barak LS. The b2-adrenergic receptor/b arrestin complex recruits the
clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci U S A
1999;96:3712–3717.
39. Laporte SA, Miller WE, Kim KM, Caron MG. b-arrestin/AP-2 interaction
in G protein-coupled receptor internalization: identification of a
b-arrestin binging site in b2-adaptin. J Biol Chem 2002;277:
9247–9254.
40. McDonald PH, Cote NL, Lin FT, Premont RT, Pitcher JA, Lefkowitz RJ.
Identification of NSF as a b-arrestin1-binding protein. Implications for b2-
adrenergic receptor regulation. J Biol Chem 1999;274:10677–10680.
41. Lin FT, Krueger KM, Kendall HE, Daaka Y, Fredericks ZL, Pitcher JA,
Lefkowitz RJ. Clathrin-mediated endocytosis of the b-adrenergic recep-
tor is regulated by phosphorylation/dephosphorylation of b-arrestin1.
J Biol Chem 1997;272:31051–31057.
42. Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of
receptor fate by ubiquitination of activated b2-adrenergic receptor and
b-arrestin. Science 2001;294:1307–1313.
43. Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias
DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci
U S A 2002;99:7877–7882.
44. Edwards BS, Kuckuck FW, Prossnitz ER, RansomJT, Sklar LA. HTPS flow
cytometry: a novel platform for automated high throughput drug discov-
ery and characterization. J Biomol Screen 2001;6:83–90.
45. Xue M, Vines CM, Buranda T, Cimino DF, Bennett TA, Prossnitz ER. N-
formyl peptide receptors cluster in an active raft-associated state prior
to phosphorylation. J Biol Chem 2004;279:45175–45184.
Active Arrestins Inhibit FPR Trafficking
Traffic 2005; 6: 87–99 99




















