
Transplantation of endothelial cells corrects the phenotype in hemophilia A mice

H-00076-2002.R1 1
Increasing IKs corrects abnormal repolarization in rabbit models of
acquired LQT2 and ventricular hypertrophy
Xiaoping Xu,1 Joseph J. Salata,2 Jixin Wang,2 Ying Wu,1 Gan-Xin Yan,1
Tengxian Liu,1 Roger A. Marinchak,1,3 Peter R. Kowey1,3
1Main Line Health Heart Center, Wynnewood, PA 19096
2Department of Pharmacology, Merck Research Laboratories, West Point, PA 19486
3Jefferson Medical College, Philadelphia, PA 19107
Running head: Normalizing repolarization by enhancing IKs
Mailing proofs: Xiaoping Xu, PhDMain Line Health Heart CenterSuite 558, Medical Office Building East100 Lancaster AvenueWynnewood, PA 19096Phone: (610)-645-2687Fax: (610)-896-0643E-mail: [email protected]
Copyright 2002 by the American Physiological Society.
AJP-Heart Articles in PresS. Published on May 2, 2002 as DOI 10.1152/ajpheart.00076.2002

H-00076-2002.R1 2
Excessive action potential (AP) prolongation and early after-depolarizations (EAD) are
triggers of malignant ventricular arrhythmias. Slowly activating delayed rectifier K+
current (IKs) is important for repolarization of ventricular AP. We examined the effects of
IKs activation by a novel benzodiazepine (L3), on AP of control, dofetilide-treated, and
hypertrophied rabbit ventricular myocytes. In both control and hypertrophied myocytes,
L3 activated IKs via a negative shift in the voltage-dependence of activation and a
slowing of deactivation. L3 had no effect on L-type Ca2+ current or other cardiac K+
currents tested. L3 shortened AP of control, dofetilide-treated and hypertrophied
myocytes more at 0.5 Hz than 2 Hz. Selective activation of IKs by L3 attenuates
prolonged AP and eliminated EAD induced by IKr inhibition in control myocytes at 0.5 Hz
and spontaneous EAD in hypertrophied myocytes at 0.2 Hz. Pharmacological activation
of IKs is a promising new strategy to suppress arrhythmias resulting from excessive AP
prolongation in patients with certain forms of long-QT syndrome or cardiac hypertrophy
and failure.
Keywords: action potential; ion channel; early-afterdepolarization; arrhythmia

H-00076-2002.R1 3
Cardiac hypertrophy and failure cause more than 200,000 deaths annually in the
US. As many as 50% are sudden and due to the onset of polymorphic ventricular
tachycardia (VT) or torsades de pointes, associated with Long-QT syndrome (LQTS)
(7). LQTS can be congenital or acquired. Congenital LQTS most commonly develops
from mutations in genes encoding delayed rectifier K+ channels that cause functional
decreases in repolarizing K+ currents (4, 12, 27). Acquired LQTS is associated with
administration of certain antiarrhythmics, antihistamines, antibiotics and other drugs
(http://georgetowncert.org/qtdrugs_torsades.asp). Many of these agents can cause
excessive action potential (AP) prolongation via inhibition of the rapidly activating
delayed rectifier K+ current (IKr), resulting in a specific form of acquired LQTS (LQT2)
(20). Heart failure may also be considered a common acquired form of LQTS (16).
Functional down-regulation of K+ currents is a recurring theme in hypertrophied and
failing ventricular myocardium (26).
At the cellular level, excessive AP prolongation and unstable repolarization is
consistently observed in ventricular myocytes from hypertrophied and failing hearts,
animal models or patients with LQTS (2, 7, 31). Early after-depolarizations (EAD) occur
in the setting of prolonged AP due commonly to diminished K+ currents or in some
cases to slowly- or non-inactivating components of Ca2+ or Na+ currents. EAD have
been identified as the triggering mechanisms for polymorphic VT (2, 4, 35). Ventricular
myocytes from hypertrophied and failing hearts are highly susceptible to EAD (3, 16, 17,
31). No effective or safe drugs are available for treatment of acquired LQT2 or
ventricular arrhythmias associated with cardiac hypertrophy and failure.
We hypothesized that selective activation of the slowly activating delayed rectifier
K+ current (IKs) could provide an effective antiarrhythmic action in certain forms of LQTS
and heart failure. Recently, we identified a novel benzodiazepine (L3), a selective
activator of IKs, which shortened AP of guinea pig ventricular myocytes (22). In this
study of rabbits, we investigated if L3-induced activation of IKs could abbreviate AP and
suppress EAD resulting from IKr inhibition in acquired LQT2 or diminished IKs in left
ventricular hypertrophy (LVH).

H-00076-2002.R1 4
METHODS
Experimental Animals
Male, New Zealand rabbits (1.4-2.0 kg) underwent unilateral nephrectomy and
contralateral renovascular banding to produce LVH as reported previously (19). Banded
rabbits were studied three months after surgery when documented LVH had developed.
Data were collected from 15 control and 9 LVH rabbits.
Myocyte Isolation
Single ventricular myocytes were isolated using a method described previously
(19). After enzyme perfusion, a thin layer (<1.5 mm) of tissue was dissected from
epicardial (Epi) and endocardial (Endo) surface of the left ventricular free wall
(excluding extreme apex and base) and myocytes were dispersed.
Action Potential Recording
AP were recorded from Epi and Endo myocytes using standard microelectrodes
(25-40 MΩ) filled with 3 M KCl. Cells were superfused with a solution containing (mM):
137 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES (pH=7.4) at 36±0.3oC.
AP were recorded at steady state using various stimulus frequencies (0.2, 0.5 and 2
Hz). Action potential duration (APD) was measured at 90% repolarization (APD90).
Because of the beat-to-beat variation of APD, especially at low frequencies, APD90 was
averaged from 10 consecutive AP.
Membrane Current Recording
Membrane ionic currents were recorded at 36±0.5oC using the whole-cell patch-
clamp technique. Solutions (mM): IKs and IKr-Pipette: 119 K-gluconate, 15 KCl, 3.2
MgCl2, 5 HEPES, 5 EGTA, and 5 K2ATP, pH=7.2, (electrode resistance=3-4 MΩ); IKs-
Bath: 132 NaCl, 2 KCl, 1.8 CaCl2, 1.2 MgCl2, 10 HEPES, 10 glucose, 1 µM dofetilide,
0.6 µM nisoldipine, pH=7.2; IKr-Bath: 132 NaCl, 5 KCl, 1.2 MgCl2, 10 HEPES, 10
glucose, 0.4 µM nisoldipine, 0.1 µM L-768,673 (a specific IKs blocker, 32), pH=7.2; ICa,L-

H-00076-2002.R1 5
Pipette: 151 CsOH, 10 L-aspartic acid, 20 taurine, 20 TEA chloride, 5 glucose, 10
EGTA, pH=7.5 with H3PO4, 5 MgATP and 0.4 Na2GTP were added before use, final
pH=7.3 (electrode resistance=1-2 MΩ); ICa,L-Bath: 140 NaCl, 5 CsCl, 1 MgCl2, 2 CaCl2,
10 glucose, 10 HEPES, pH=7.4. Series resistance was compensated electronically 70-
80%. Liquid junction potential was compensated in the bath but not under whole-cell
conditions.
Epi myocytes were chosen for IKs recording due to their relatively larger IKs
density than in Endo myocytes (32). IKs was recorded with 1-s depolarizing voltage
steps applied at 12-s inter-pulse intervals from a holding potential (Vh) of -40 mV to test
potentials (Vt) of -20 to +60 mV in 20-mV increments (32). Isochronal activation curves
for IKs were determined from peak tail current amplitudes during return to a Vh of -40 mV
following 5-s test pulses. Tail currents (I) were normalized to the maximal tail current
(Imax) obtained following a step to Vt of +70 mV. A Boltzmann function, I/Imax=1/1+exp
[(V0.5-V)/k] (V0.5=half-maximal activation potential, k=slope factor), was fit to the data.
Current-voltage relations were plotted for averaged data normalized to cell capacitance.
A second-order exponential function was fit to the deactivating tail current on return to –
40 mV following a 5-s test pulse to +30 mV to derive the time constants of IKs
deactivation.
IKr was induced by 500-ms pulses to Vt of –10 mV from a Vh of –50 mV. Six
consecutive pulses were delivered at 0.2 Hz and the current traces were averaged. IKr
was quantified as dofetilide sensitive tail current. ICa,L was recorded with 180-ms test
pulses applied at a 5-s interpulse interval from a Vh of -40 mV to Vt of -30 to +50 mV in
10-mV increments.
Data Analysis
Data are expressed as mean ± SEM. Statistical analyses were performed using
GraphPad Prism 2.0 (GraphPad Software, Inc). A paired t-test was used to compare
control and L3 treatment means. Two-way analysis of variance was used to compare
means involving two different categorical variables. P<0.05 was considered statistically
significant.

H-00076-2002.R1 6
Drugs
L3 (R-L3) was synthesized at Merck Research Laboratories, West Point, PA as
previously described (22). Dofetilide and nisoldipine were gifts from Pfizer and Bayer
AG, respectively.
RESULTS
The effects of L3 on IKs in ventricular myocytes of control rabbits are shown in
Fig. 1. L3 throughout these studies was tested at its maximally effective concentration of
1 µM unless otherwise noted (22). Compared to control (Fig. 1A), L3 increased IKs
amplitude and slowed deactivation (Fig. 1B). L3 increased IKs significantly at all Vt (Fig.
1D). At a Vt of +60 mV, L3 increased IKs density from 2.31±0.35 to 3.05±0.46 pA/pF
(n=10). Percentage increases in IKs declined with increasing membrane potential,
286±20% at -20 mV versus 32±2% at +60 mV (n=10, Fig. 1E). L3 shifted the midpoint
(V0.5) of the voltage-dependence of IKs activation by -17.6 mV (Fig. 1F). In control,
deactivation of IKs was best described by a second order exponential function. L3
increased the fast (τf) and slow (τs) time constants of IKs deactivation from 95±7 to
116±14 ms (n=6, n.s.), and 279±25 to 712±127 ms (n=6, P<0.05), respectively.
Fig. 1C shows the ratio of IKs in the presence of L3 to the control (Fig. 1B/1A) for
a test pulse to +60 mV. During the test pulse, L3 increased activating IKs by a constant
ratio of ~1.4 for the entire duration of the pulse, analogous to the increase in current
density measured at the end of the pulse. Upon repolarization to –40 mV, the initial
deactivating tail current is increased to a degree similar to that during the pulse.
Importantly, however, because L3 slowed IKs deactivation, the relative increase in IKs
was greater at later times in the deactivation process, such that at 1 s after
repolarization, the relative current was increased by ~10 fold.
L3 had no effect on ICa,L at all Vt . There was also no evidence for use-dependent
block of ICa,L during repetitive 500-ms pulses from a Vh of -40 mV to a Vt of +20 mV at
0.5 Hz (data not shown). L3 reduced IKr tail current slightly but not significantly from
86±9 to 82±10 pA (n=6, n.s.), and had no significant effect on inward rectifier K+ current

H-00076-2002.R1 7
(IK1) or transient outward K+ current (Ito) (data not shown). Thus, by all these measures,
L3 was a selective activator of IKs.
L3 concentration-dependently shortened AP in rabbit control ventricular
myocytes (Fig. 2). L3 (1 µM) decreased APD90 at 0.5 and 2 Hz by 23±2% and 17±2% in
Epi and by 21±2% and 14±2% in Endo, respectively (n=8). Percentage decreases in
APD90 were similar between Epi and Endo myocytes, but were more pronounced at 0.5
Hz than 2 Hz.
L3 attenuated AP prolongation and eliminated EAD induced by the specific IKr
blocker, dofetilide, in myocytes isolated from control rabbits (Fig. 3). At a stimulus
frequency of 0.5 Hz, 0.1 µM dofetilide induced EAD in 4/11 Endo myocytes. L3
eliminated EAD in all 4 cells in the continuous presence of 0.1 µM dofetilide (Fig. 3A).
The effects of L3 were reversible; e.g., EAD re-appeared during washout of L3 (data not
shown). In the presence of 0.1 µM dofetilide, L3 decreased APD90 from 315±41 to
245±32 ms in Epi myocytes and 627±135 to 388±70 ms in Endo myocytes at 0.5 Hz
(n=8). L3 effects on APD were frequency-dependent. L3 decreased APD90 by 15±1%
and 13±1% at 2 Hz, and 24±2% and 35±3% at 0.5 Hz in Epi and Endo, respectively
(n=8). AP prolonged by 0.1 µM dofetilide in myocytes from both Epi and Endo were
shortened to a lesser extent at 2 Hz than 0.5 Hz by L3. In the presence of dofetilide, L3
decreased APD90 more in Endo (239±69 ms) than Epi myocytes (70±11 ms) at 0.5 Hz
(n=8, P<0.05).
Three months after renal artery banding, rabbits developed LVH with
characteristic changes. LVH significantly increased heart to body weight ratio from
2.10±0.04 (n=15) to 2.53±0.07 g/kg (n=9, P<0.05) and myocyte size, measured as cell
membrane capacitance, from 158±7 (n=20) to 200±12 pF (n=16, P<0.05). Consistent
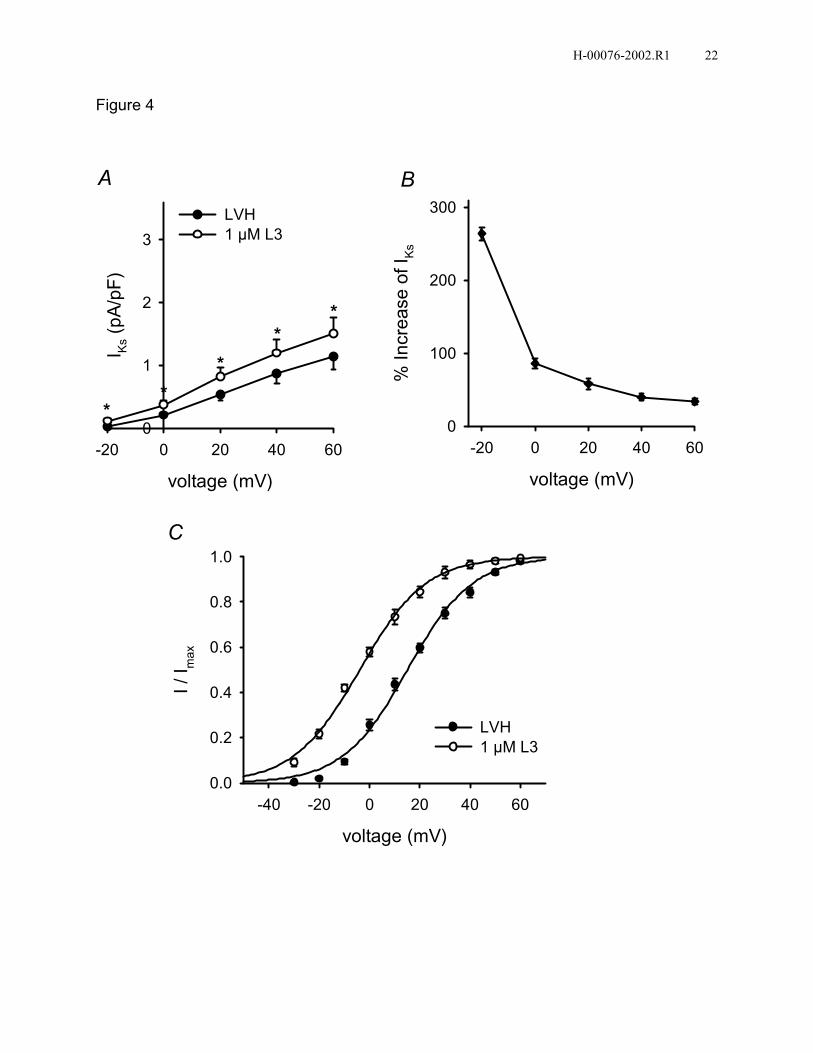
with our previous findings (32), IKs density of rabbit LVH myocytes was significantly
lower than that of controls, 1.15±0.21 versus 2.31±0.35 pA/pF (n=10, P<0.05). L3 had
similar effects on IKs of myocytes from both control and LVH rabbits (Figs. 1B & 4A). At
a Vt of +60 mV, L3 increased IKs density from 2.31±0.35 to 3.05±0.46 pA/pF, and from
1.15±0.21 to 1.51±0.26 pA/pF, respectively (n=10). L3 increased IKs by 264±9% at -20
mV and 34±4% at +60 mV in myocytes from LVH rabbits (n=10, Fig. 4B). L3 shifted the

H-00076-2002.R1 8
midpoint (V0.5) of the voltage-dependence of IKs activation in LVH rabbits by -19.0 mV
(Fig. 4C).
As observed previously (32), APD90 of Epi and Endo ventricular myocytes in LVH
rabbits was increased by 21% and 22% at 2 Hz, respectively, compared with controls.
At 0.5 Hz, APD90 increased from 175±15 to 212±27 ms by LVH in Epi, and from 231±18
to 296±35 ms in Endo myocytes, respectively (n≥11). Spontaneous EAD were recorded
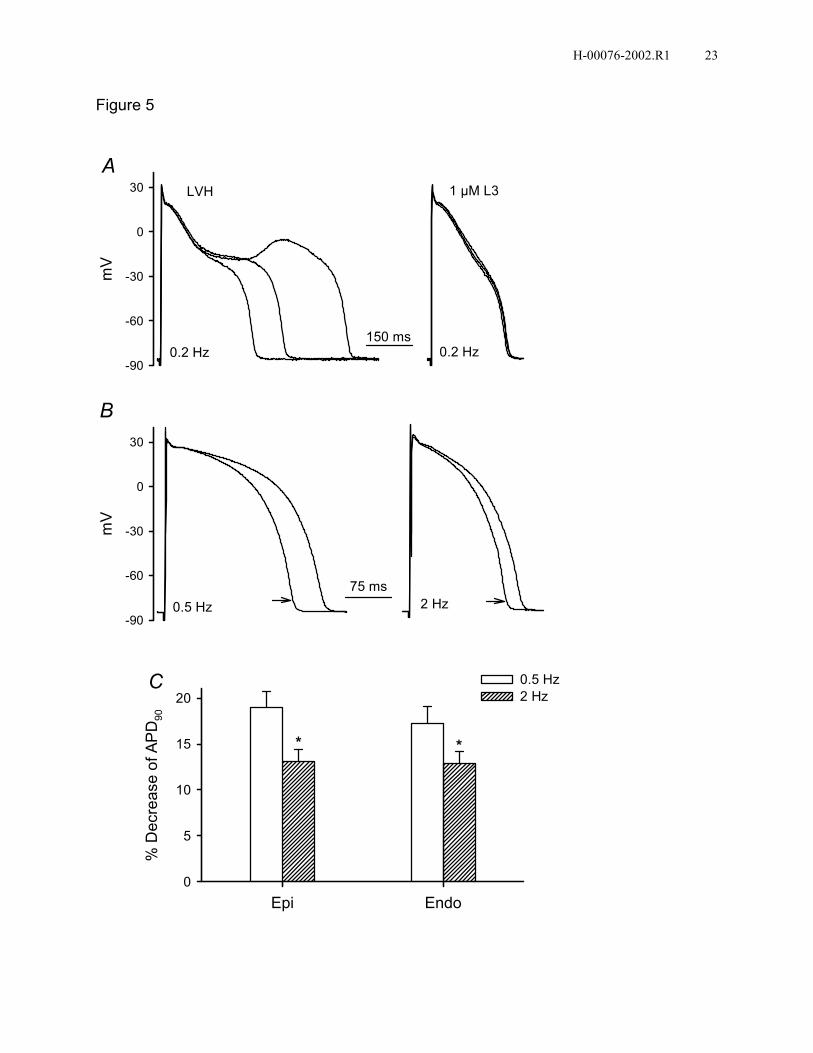
in 3/13 myocytes from Endo of LVH rabbits at a stimulus frequency of 0.2 Hz. L3
eliminated EAD in all 3 cells (Fig. 5A). In LVH myocytes, 1 µM L3 decreased APD90 by
19±2% and 13±1% in Epi and by 17±2% and 13±1% in Endo at 0.5 and 2 Hz,
respectively (n=8). Percentage decreases of APD90 by L3 were similar between Epi and
Endo at both 0.5 and 2 Hz. However, L3 decreased APD90 more at 0.5 Hz than 2 Hz.
DISCUSSION
This study provides the first direct evidence that IKs activation can be useful in
reversing excessive AP prolongation and suppressing arrhythmogenic EAD in certain
forms of LQTS. We examined this utility in rabbit models of acquired LQTS using the
selective activator of IKs, L3, described previously in detail (22). In a model of LQT2, IKr
inhibition with dofetilide produced typical reverse frequency-dependent increases in
APD and EAD (25). In a model of LVH induced by renovascular hypertension, APD
were prolonged, spontaneous EAD occurred in 23% of Endo myocytes at 0.2 Hz, and
IKs density was significantly reduced, consistent with our previous findings (32). These
profiles of abnormal repolarization reflect deficiencies in two major repolarizing currents,
IKr and IKs, respectively.
Importantly, in control, dofetilide-treated and LVH myocytes, L3 shortened APD
in a concentration-dependent manner and eliminated EAD in the latter two conditions.
AP mapping across the left ventricular free wall using arterially perfused wedge
preparations shows that endocardium has the longest and epicardium the shortest APD
in both control and LVH rabbits (33). Across the left ventricular free wall, there is a
gradual increase in APD from epicardial side to endocardial side. This differs from
canine left ventricle where the longest APD is recorded in subendocardium (M-cells)

H-00076-2002.R1 9
(34). L3 decreased APD to a similar degree in Epi and Endo myocytes in both control
and LVH (Figs. 2B & 5C), indicating that IKs activation by L3 in rabbits should not
increase transmural dispersion of repolarization (TDR). In dofetilide-treated myocytes,
L3 decreased APD more significantly in Endo than Epi at 0.5 Hz, due partly to
elimination of EAD in Endo. Thus, in acquired LQT2 or abnormally prolonged
repolarization due to IKr inhibition, IKs activation should actually decrease TDR.
L3 decreased APD more at 0.5 Hz than 2 Hz in control, dofetilide-treated and
LVH. Because excessive AP prolongation, EAD and torsades de pointes are associated
with bradycardia, this characteristic of L3 could be particularly beneficial. Because of the
complicated interaction of many time and voltage dependent currents in controlling AP
morphology, it is oversimplified to extrapolate changes in a single current to effects on
APD. Nevertheless, the L3 induced slowing of deactivation may provide a basis for the
greater shortening of APD observed at slower rates. Normally, due to its slow
deactivation, IKs can accumulate more at higher frequencies and thereby contribute to
rate dependent shortening of APD (11, 13, 24). L3 slows IKs deactivation and increases
relative deactivating current more at later times or longer intervals (Fig. 1C). The larger
relative increases in deactivating IKs at longer intervals may lead to greater than normal
accumulation at the slower rates and explain the greater decreases in APD by L3 at 0.5
Hz than at 2 Hz. However, the mechanism underlying the frequency dependent effects
of L3 on APD still requires further study.
L3 concentration-dependently activated IKs and shortened APD in guinea pig
ventricular myocytes (22). In this study, L3 effects on rabbit IKs were studied at a
maximally effective concentration of 1 µM as determined previously for guinea pig. L3
increased IKs in rabbit via a negative shift in the voltage-dependence of activation and a
slowing of deactivation. L3 shifted the V0.5 of rabbit IKs activation by -17.6 mV in control
and by -19.0 mV in LVH, comparable to the shift of -24 mV found previously for guinea
pig. This shift accounts for the larger percent increases of IKs at more negative test
potentials (Figs. 1E & 4B). Deactivation of IKs was best described by a second-order
exponential function. L3 increased both τf (22%) and τs (155%) of deactivation of rabbit
IKs, consistent with the changes in guinea pig (70% and 190%, respectively).

H-00076-2002.R1 10
Our experimental results indicate that L3 (up to 1 µM) is a selective activator of
IKs in rabbit ventricular myocytes. L3 also slows deactivation of IKs. These selective
actions on the gating of IKs are likely the predominant if not only mechanisms underlying
the APD shortening and EAD elimination by L3. Both the negative shift in the voltage
dependence of activation and the slowing of deactivation can increase the contribution
of IKs to AP repolarization.
A reduction of repolarizing K+ currents in LQTS and hypertrophied hearts has
been widely observed. Reductions in the density of Ito and IK1 are commonly observed in
cardiac hypertrophy and failure (26). Reductions in IKs and IKr, which play dominant roles
in ventricular AP repolarization of many mammalian species including rabbit (21), are
also common. In cats, delayed rectifier K+ current density was decreased in right
ventricular hypertrophy induced by pressure overload (14), and LVH induced by aortic
stenosis (10). In rabbits, LVH induced by renovascular hypertension significantly
reduced IKs but not IKr density (32), whereas pacing-induced heart failure reduced both
IKs and IKr (28). In dogs with chronic complete atrioventricular block, IKr density of
mid-myocardial cells was decreased in right ventricle and IKs density was decreased in
both ventricles (30). In failing human hearts, IKs density was decreased by 60% (15).
The reduction in the delayed rectifier K+ currents predisposes ventricular myocytes to
potentially arrhythmogenic EAD (9, 17, 32). Computer models of the mammalian
ventricular AP predict that reducing IKs induces EAD in the endocardial cells during
normal cell-to-cell coupling (29).
Other studies aimed at treating arrhythmias resulting from excessive AP
prolongation and LQTS have explored various strategies for augmenting repolarizing K+
currents. These include, increasing IKr by elevating serum K+ concentrations (5, 6),
administration of ATP-sensitive potassium channel openers (PCO) (1, 8, 23), and over-
expression of HERG K+ channels (IKr) (18).
Study limitations: Since excessive APD prolongation and EAD are associated
with bradycardia, the pacing rates used in this study are below the normal physiologic
rates in the rabbit. The rabbit models used in this study only address the down
regulation of IKr amplitude (acquired LQT2) or IKs density (LVH). In patients with
congenital LQTS, not only is current density affected, but also other properties of IKr or

H-00076-2002.R1 11
IKs channels may change. It is uncertain whether IKs activation will have antiarrhythmic
effects in these patients. Use of IKs activators may not be void of adverse effects in
some situations. Shortening APD by IKs activation may facilitate the development of
reentrant arrhythmia under certain circumstances, particularly in myocardial ischemia
and infarction. The effects of L3 on action potentials under ischemic conditions are
unknown, however, we expect the APD shortening by IKs activation would be very
modest and possibly overwhelmed by activation of the large conductance of the KATP
current.
In conclusion, selective activation of IKs by pharmacological agents is a promising
new strategy to suppress arrhythmias resulting from excessive AP prolongation in
patients with certain forms of LQTS or cardiac hypertrophy and failure.
Acknowledgements
This work was supported in part by the Sharpe Foundation and the Fourjay
Foundation.

H-00076-2002.R1 12
REFERENCES
1. Aizawa Y, Uchiyama H. Yamaura M, Nakayama T, and Arita M. Effects of the
ATP-sensitive K channel opener nicorandil on the QT interval and the effective
refractory period in patients with congenital long QT syndrome. J Electrocardiol
31:117-123, 1998.
2. Aronson RS, and Ming Z. Cellular mechanisms of arrhythmias in hypertrophied
and failing myocardium. Circulation 87:VII-76-VII-83, 1993.
3. Ben-David J, Zipes DP, Ayers GM, and Pride HP. Canine left ventricular
hypertrophy predisposes to ventricular tachycardia induction by phase 2 early
afterdepolarizations after administration of BAY K 8644. J Am Coll Cardiol
20:1576-1584, 1992.
4. Chiang C-E, and Roden DM. The long QT syndromes: genetic basis and clinical
implications. J Am Coll Cardiol 36:1-12, 2000.
5. Choy AM, Lang CC, Chomsky DM, Rayos GH, Wilson JR, and Roden DM.
Normalization of acquired QT prolongation in humans by intravenous potassium.
Circulation 96:2149-2154, 1997.
6. Compton SJ, Lux RL, Ramsey MR, Strelich KR, Sanguinetti MC, Green LS,
Keating MT, and Mason JW. Genetically defined therapy of inherited long-QT
syndrome. Correction of abnormal repolarization by potassium. Circulation
94:1018-1022, 1996.
7. Eckardt L, Haverkamp W, Johna R, Bocker D, Deng MC, Breithardt G, and
Borggrefe M. Arrhythmias in Heart Failure: current concepts of mechanisms and
therapy. J Cardiovasc Electrophysiol 11:106-117, 2000.

H-00076-2002.R1 13
8. Fish FA, Prakash C, and Roden DM. Suppression of repolarization-related
arrhythmias in vitro and in vivo by low-dose potassium channel activator.
Circulation 82:1362-1369, 1990.
9. Furukawa T, Bassett AL, Furukawa N, Kimura S, and Myerburg RJ. The ionic
mechanism of reperfusion-induced early afterdepolarizations in feline left
ventricular hypertrophy. J Clin Invest 91:1521-1531, 1993.
10. Furukawa T, Myerburg RJ, Furukawa N, Kimura S, and Bassett AL. Metabolic
inhibition of ICa,L and IK differs in feline left ventricular hypertrophy. Am J Physiol
266:H1121-H1131, 1994.
11. Gintant GA. Two components of delayed rectifier current in canine atrium and
ventricle. Does IKs play a role in the reverse rate dependence of class III agents?
Circ Res 78:26-37, 1996.
12. January CT, Gong Q, and Zhou Z. Long QT syndrome: cellular basis and
arrhythmia mechanism in LQT2. J Cardiovasc Electrophysiol 11:1413-1418,
2000.
13. Jurkiewicz NK, and Sanguinetti MC. Rate-dependent prolongation of cardiac
action potentials by a methanesulfonanilide Class III antiarrhythmic agent:
specific block of rapidly activating delayed rectifier K+ current by dofetilide. Circ
Res 72: 75-83, 1993.
14. Kleiman RB, and Houser SR. Outward currents in normal and hypertrophied
feline ventricular myocytes. Am J Physiol 256:H1450-H1461, 1989.
15. Li G-R, Sun H, Feng J, and Nattel S. Ionic mechanisms of the action potential
prolongation in failing human ventricular cells. PACE 21:877(Abstract), 1998.

H-00076-2002.R1 14
16. Marban E. Heart failure: the electrophysiologic connection. J Cardiovasc
Electrophysiol 10:1425-1428, 1999.
17. Nuss HB, Kaab S, Kass DA, Tomaselli GF, and Marban E. Cellular basis of
ventricular arrhythmias and abnormal automaticity in heart failure. Am J Physiol
277:H80-H91, 1999.
18. Nuss HB, Marban E, and Johns DC. Overexpression of a human potassium
channel suppresses cardiac hyperexcitability in rabbit ventricular myocytes. J
Clin Invest 103:889-896, 1999.
19. Rials SJ, Wu Y, Xu X, Filart RA, Marinchak RA, and Kowey PR. Regression of
left ventricular hypertrophy with captopril restores normal ventricular action
potential duration, dispersion of refractoriness, and vulnerability to inducible
ventricular fibrillation. Circulation 96:1330-1336, 1997.
20. Roden DM. Mechanisms and management of proarrhythmia. Am J Cardiol
82:49I-57I, 1998.
21. Salata JJ, Jurkiewicz NK, Jow B, Folander K, Guinosso PJ, Jr., Raynor B,
Swanson R, and Fermini B. IK of rabbit ventricle is composed of two currents:
evidence for IKs. Am J Physiol 271:H2477-H2489, 1996.
22. Salata JJ, Jurkiewicz NK, Wang J, Evans BE, Orme HT, and Sanguinetti MC. A
novel benzodiazepine that activates cardiac slow delayed rectifier K+ currents.
Molecular Pharmacol 53:220-230, 1998.
23. Shimizu W, Kurita T, Matsuo K, Suyama K, Aihara N, Kamakura S, Towbin JA,
and Shimomura K. Improvement of repolarization abnormalities by a K+ channel

H-00076-2002.R1 15
opener in the LQT1 form of congenital long-QT syndrome. Circulation 97:1581-
1588, 1998.
24. Stengl M, Vos MA, Spatjens RLHMG, Sipido KR, and Volders PGA.
Accumulation of slow delayed rectifier K+ current (IKs) in canine ventricular
myocytes. Biophysical J 82: 606a (Abstract), 2002.
25. Tande PM, Bjornstad H, Yang T, and Refsum H. Rate-dependent Class III
antiarrhythmic action, negative chronotropy and positive inotropy of a novel IK
blocking drug, UK-68,798: potent in guinea pig but no effect in rat myocardium. J
Cardiovasc Pharmacol 16:401-410, 1990.
26. Tomaselli GF, and Marban E. Electrophysiological remodeling in hypertrophy and
heart failure. Cardiovasc Res 42:270-283, 1999.
27. Tristani-Firouzi M, Chen J, Mitcheson JS, and Sanguinetti MC. Molecular Biology
of K+ channels and their role in cardiac arrhythmias. Am J Med 110:50-59, 2001.
28. Tsuji Y, Opthof T, Kamiya K, Yasui K, Liu W, Lu Z, and Kodama I. Pacing-
induced heart failure causes a reduction of delayed rectifier potassium currents
along with decreases in calcium and transient outward currents in rabbit
ventricle. Cardiovasc Res 48:300-309, 2000.
29. Viswanathan PC, and Rudy Y. Cellular arrhythmogenic effects of congenital and
acquired long-QT syndrome in the heterogeneous myocardium. Circulation
101:1192-1198, 2000.
30. Volders PGA, Sipido KR, Vos MA, Spatjens RLHMG, Leunissen JDM, Carmeliet
E, and Wellens HJJ. Downregulation of delayed rectifier K+ currents in dogs with

H-00076-2002.R1 16
chronic complete atrioventricular block and acquired Torsades de Pointes.
Circulation 100:2455-2461, 1999.
31. Vos MA, de Groot SHM, Verduyn SC, van der Zande J, Leunissen HDM,
Cleutjens JPM, van Bilsen M, Daemen MJAP, Schreuder JJ, Allessie, MA and
Wellens HJJ. Enhanced susceptibility for acquired torsade de pointes
arrhythmias in the dog with chronic, complete AV block is related to cardiac
hypertrophy and electrical remodeling. Circulation 98:1125-1135, 1998.
32. Xu X, Rials SJ, Wu Y, Salata JJ, Liu T, Bharucha DB, Marinchak RA, and Kowey
PR. Left ventricular hypertrophy decreases slowly, but not rapidly activating
delayed rectifier K+ currents of epicardial and endocardial myocytes in rabbits.
Circulation. 103:1585-1590, 2001.
33. Yan G-X, Rials SJ, Wu Y, Liu T, Xu X, Marinchak RA, and Kowey PR. Ventricular
hypertrophy amplifies transmural dispersion of repolarization and induces phase
2 early afterdepolarization. Am J Physiol. 281:H1968-H1975, 2001.
34. Yan G-X, Shimizu W, and Antzelevitch C. Characteristics and distribution of M
cells in arterially perfused canine left ventricular wedge preparations. Circulation.
98:1921-1927, 1998.
35. Yan G-X, Wu Y, Liu T, Wang J, Marinchak RA, and Kowey PR. Phase 2 early
afterdepolarization as a trigger of polymorphic ventricular tachycardia in acquired
long-QT syndrome: direct evidence from intracellular recordings in the intact left
ventricular wall. Circulation. 103:2851-2856, 2001.

H-00076-2002.R1 17
FIGURE LEGENDS
Figure 1. L3 increases IKs in rabbit control ventricular myocytes. IKs traces shown for
control (A) and 1 µM L3 (B) at Vt of -20, 0, +20, +40, and +60 mV. C: Ratio of IKs in the
presence of L3 to the control (B/A) for a test pulse to +60 mV (top traces). D:
Normalized current-voltage (I-V) relation for IKs in control and after 1 µM L3 (n=10,
*P<0.05, L3 versus control). E: Percentage increases of IKs by 1 µM L3 at various Vt. F:
Negative shift of the voltage-dependence of IKs activation by 1 µM L3 (n=6). Control:
V0.5=14.1 mV, k=12.0 mV; L3: V0.5=-3.5 mV, k=13.9 mV.
Figure 2. L3 concentration-dependently shortens AP in rabbit control ventricular
myocytes. A: Superimposed AP traces recorded in Epi (top) and Endo (bottom)
myocytes at stimulus frequencies of 0.5 and 2 Hz. L3 at 0.1 µM (arrows) shortened AP.
B: Concentration-dependent decrease of APD90 in Epi (top) and Endo (bottom)
myocytes by L3 (n=8, *P<0.05, 2 Hz versus 0.5 Hz).
Figure 3. L3 attenuates AP prolongation and eliminates EAD induced by IKr blockade in
rabbit control ventricular myocytes. A: AP recorded in Epi (top) and Endo (bottom)
myocytes at a stimulus frequency of 0.5 Hz for control, 0.1 µM dofetilide; and 0.1 µM
dofetilide + 1 µM L3. Three superimposed traces are shown for each condition. B:
Decreases of APD90 in Epi and Endo myocytes by 1 µM L3 in the presence of 0.1 µM
dofetilide (n=8, *P<0.05 2 Hz versus 0.5 Hz; +P<0.05 Endo versus Epi).

H-00076-2002.R1 18
Figure 4. L3 increases IKs in rabbit LVH ventricular myocytes. A: Normalized I-V relation
for IKs in LVH and after 1 µM L3 (n=10, *P<0.05, L3 versus LVH). B: Percentage
increases of IKs by 1 µM L3 at various Vt. C: Negative shift of the voltage-dependence of
IKs activation by 1 µM L3 (n=6). Control: V0.5=15.1 mV, k=12.9 mV; L3: V0.5=-3.9 mV,
k=13.2 mV.
Figure 5. L3 normalizes APD and eliminates spontaneous EAD in rabbit LVH
ventricular myocytes. A: L3 at 1 µM eliminated spontaneous EAD in a LVH Endo
myocyte at a stimulus frequency of 0.2 Hz. Three superimposed traces are shown for
each condition. B: L3 at 1 µM (arrows) shortened AP of a LVH Endo myocyte at 0.5 and
2 Hz. C: Average percentage decreases of APD90 by 1 µM L3 in LVH Epi and Endo
myocytes (n=8, *P<0.05, 2 Hz versus 0.5 Hz).

H-00076-2002.R1 19
Figure 1
0
200
400
0
200
400
1 µM L3
voltage (mV)
-20 0 20 40 60
I Ks
(pA
/pF
)
0
1
2
3
Control1 µM L3*
*
*
*
*
voltage (mV)
-20 0 20 40 60
% In
cre
ase
of I K
s
0
100
200
300
A
C
B
Control
F
voltage (mV)
-40 -20 0 20 40 60
I / I m
ax
0.0
0.2
0.4
0.6
0.8
1.0
Control1 µM L3
0
4
8
12
16
pA
pA
ratio
500 ms
D
E

H-00076-2002.R1 20
Figure 2
0.03 0.30.1 1
% D
ecre
ase
of A
PD
90
0
5
10
15
20
25Epi
[L3] (µM)
0.03 0.30.1 1
% D
ecre
ase
of A
PD
90
0
5
10
15
20
25
0.5 Hz 2 Hz
Endo
*
*
*
*
*
*
mV
-90
-60
-30
0
30
mV
-90
-60
-30
0
30
50 ms
A B
0.5 Hz
0.5 Hz
2 Hz
2 Hz
Epi
Endo

H-00076-2002.R1 21
Figure 3m
V
-90
-60
-30
0
30
150 ms
mV
-90
-60
-30
0
30 Control Dofetilide Dofetilide+L3A
B
Epi Endo
% D
ecre
ase
of A
PD
90
0
10
20
30
40 0.5 Hz 2 Hz
+Dofetilide
**
+
H-00076-2002.R1 22
Figure 4
voltage (mV)
-40 -20 0 20 40 60
I / I m
ax
0.0
0.2
0.4
0.6
0.8
1.0
voltage (mV)
-20 0 20 40 60
% In
cre
ase
of I
Ks
0
100
200
300
C
BA
voltage (mV)
-20 0 20 40 60
I Ks
(pA
/pF
)
0
1
2
3
LVH1 µM L3
*
*
*
*
*
LVH1 µM L3
H-00076-2002.R1 23
Figure 5m
V
-90
-60
-30
0
30
150 ms
mV
-90
-60
-30
0
30
75 ms
A
C
B
Epi Endo
% D
ecr
ease
of A
PD
90
0
5
10
15
20
0.5 Hz 2 Hz
* *
LVH 1 µM L3
0.2 Hz 0.2 Hz
0.5 Hz 2 Hz
Copyright © 2022 FDOKUMEN











![Ca]i elevation and oxidative stress induce KCNQ1 translocation from cytosol to cell surface and increase IKs in cardiac myocytes](https://static.fdokumen.com/doc/165x107/6313ba673ed465f0570ace55/cai-elevation-and-oxidative-stress-induce-kcnq1-translocation-from-cytosol-to-cell.jpg)








![465 fnYyh] 'kqØokj] fnlEcj 22] 2017@ikS"k 1] 1939 ¹jk-jk](https://static.fdokumen.com/doc/165x107/63203ca9b71aaa142a03b884/465-fnyyh-kqookj-fnlecj-22-2017iksk-1-1939-jk-jk.jpg)