
Precautionary advice about mobile phones: public understandings and intended responses
Upload
independentCategory
view
3download
0
Biomaterials 23 (2002) 3113–3122
In vitro bioactivity and degradation behavior of silica xerogelsintended as controlled release materials
Shula Radina,*, Sylvie Falaizea, Mark H. Leea, Paul Ducheynea,b
a Department of Bioengineering, Center for Bioactive Materials and Tissue Engineering, University of Pennsylvania, Philadelphia, PA 19104, USAb Department of Orthopaedic Surgery, Center for Bioactive Materials and Tissue Engineering, University of Pennsylvania, Philadelphia, PA 19104, USA
Received 12 July 2001; received in revised form 17 January 2002; accepted 4 February 2002
Abstract
Room temperature-processed silica-based sol–gel, termed silica xerogel, is a novel type of controlled release material. As shown
previously, these materials are porous, degradable and can release biologically functional molecules in a controlled manner. It was
also demonstrated that these materials are biocompatible in vivo. Herein we report on the ability of silica-based xerogels to form a
bioactive, apatite-like (AP) surface and the effect of AP surface on the xerogel stability in vitro. Formation of a crystalline,
carbonated AP (c-AP) was found on all silica xerogels studied, with or without Ca- and P-oxides. Calcium and phosphate (Ca–P)
free xerogels showed long times to Ca–P precipitation and to formation of a detectable AP-layer (up to 2 weeks). In contrast, the
times to precipitation were reduced by 2–3 orders of magnitude, and the c-AP layer was formed within 24 h on all Ca–P containing
xerogels. Mechanisms of the c-AP formation on these xerogels were similar to those typical for Ca–P based ceramics: dissolution of
calcium and phosphate ions, solution oversaturation with respect to AP and subsequent precipitation of bone-like minerals. The
presence of the c-AP surface film produced a remarkable surface stabilizing effect: the rates and the amounts of Si release were
significantly reduced in comparison to those for xerogels without the film. This evidence of in vitro bioactivity and controlled
degradation, combined with previous in vitro and in vivo reports, suggests that silica xerogel is a promising controlled release
material. r 2002 Elsevier Science Ltd. All rights reserved.
Keywords: Sol–gels; In vitro; Bioactivity; Degradation; Controlled release material
1. Introduction
Room temperature-processed silica-based sol–gels,termed silica xerogels, are novel controlled releasematerials. As shown in vitro, these materials can releasefunctional molecules, including antibiotics and growthfactors, in a controlled manner [1–5]. It was alsoreported that these xerogels are biocompatible in vivo[6]. In addition to these properties, desirable propertiesof controlled release materials for orthopedic applica-tions include bone bioactivity and controlled resorption.Bone bioactive behavior of melt-derived silica-basedbioactive glass (BG) is associated with in vivo surfacetransformation. A sandwich-like structure composed ofunderlying polymerized silica gel and upper Ca–P-rich
layers, forms at the glass surface [7]. As been reported inour previous works, silica gel formation and Ca–Paccumulation are intermingled [8]. Among theories, theformation of polymerized silica gel plays a major role innucleation of biological apatite at the BG surface [9].Since silica xerogels are inherently composed of poly-merized silica gel, we hypothesize that the xerogels caninduce formation of a biological type of apatite, i.e.carbonated apatite (c-AP), and thereby display bone bio-active behavior. We further hypothesize that the addi-tion of Ca- and P-oxides to xerogels will facilitate thec-AP formation. We also reason that the AP surfacelayer could have a protective effect on the surfacereactivity of xerogels and its resorption behavior in vivo.
In this paper, we focus on the effect of xerogelstructure and composition on the AP formation, byvarying the CaO from 0% to 10% and P2O5 contentfrom 0% to 5%. In addition, we also describe the effectof AP presence at the surface on xerogel stability.Dissolution behavior of AP-coated and uncoated
*Corresponding author. Hayden Hall, Suite 120, 3320 Smith Walk,
Philadelphia, PA 19104 6315, USA. Tel.: +1-215-898-5140; fax: +1-
215-573-2071.
E-mail address: [email protected] (S. Radin).
0142-9612/02/$ - see front matter r 2002 Elsevier Science Ltd. All rights reserved.
PII: S 0 1 4 2 - 9 6 1 2 ( 0 2 ) 0 0 0 5 1 - 0

xerogels was determined using a mode of immersion inwhich the solutions were continuously exchanged. Post-immersion, the solutions were analyzed for changes incalcium, phosphate and silicon concentrations. Thexerogels were characterized using gas sorption analysis,Fourier transform infrared spectroscopy (FTIR) andscanning electron microscopy (SEM) in combinationwith energy dispersive X-ray analysis (EDXA).
2. Materials and methods
All xerogels were synthesized at room temperature.Tetramethylorthosilane (TMOS, Aldrich, MilwaukeeWI), CaCl2 and triethyl phosphate (TEP) were used asprecursors of Si-, Ca- and P-oxides, respectively.Compositions of synthesized xerogels are indicated inTable 1.
Both Ca–P free and Ca–P containing gels weresynthesized using a one-step acid catalyzed hydrolysis.TMOS and deionized (DI) water were mixed at a 1:10mratio and stirred until a single-phase sol was formed.Corresponding amounts of CaCl2 and TEP were addedto the sol to form SiO2–CaO–P2O5 gels with varyingCaO and P2O5 contents (Table 1). Sol–gels denominatedas S100, S95, S90, and S85 contained (wt%) 0%, 3%,5%, and 10% CaO. The P2O5 content was 2% in S95and 5% in the other Ca–P containing xerogels.
All sols were cast into polystyrene vials. Addition ofCaCl2 significantly reduced the time to gelation: whereasit took 20 or more hours for the S100 composition togel, the times to gelation (as evidenced by the pour test)were 6, 3 and 1 h for the S95, S90 and S85, respectively.All gels were aged in sealed containers for 3 days andthen were dried to constant weight. Resulting monolithswere crushed and sieved to produce granules of500–710 mm. The granules were kept refrigerated insealed containers until in vitro testing.
After synthesis, the granules were characterized usinggas (nitrogen) sorption analysis (BET method, Quanta-chrome, Boynton Beach, FL), FTIR spectroscopy(diffuse reflectance mode, 5DXC, Nicolet, Madison,WI), and X-ray diffraction (XRD) (Rigaku diffract-ometer, CuKa radiation) analysis.
Two sequential immersion experiments were per-formed. In experiment I, the effect of xerogel composi-tion on the AP formation was determined. Xerogelgranules of various compositions were immersed in tris
buffered solution supplemented with electrolytes typicalfor plasma (TE, pH 7.4 at 371C) [10]. These immersionexperiments were conducted without solution exchange.Post-immersion, concentrations of calcium and phos-phate ions were measured using atomic absorptionspectrophotometry (AAS, 5100, Perkin-Elmer, Norwalk,CT) and colorimetry at 400 nm (UV/visible spectro-photometer, Pharmacia LKB, Ultraspec Plus, Piscat-away, NJ), respectively. Immersion-induced changes atthe granule surface were determined using FTIR andSEM/EDX (JEOL 6400, Tokyo, Japan) analyses.
Experiment II was intended to study the effect of AP-coating on the xerogel stability in vitro. Granules of twoxerogel compositions, S100 and S90, were used.Formation of AP-coating was performed in TE solutionto which 2.5 mm of silicon was added. As was reportedbefore, this Si concentration corresponds to the solubi-lity of S100 xerogel in TE at 371C [11]. Resultingsolution saturation with Si species prevented xerogeldissolution during formation of the coating. It was alsoassumed that the use of Si-saturated solutions wouldprevent changes in xerogel structure (such as an increasein pore size and decrease in surface area) that wereobserved on immersion in undersaturated solutions [11].The granule weight per solution volume (W/V) was2 mg ml�1. The duration of immersion to form the APsurface layer on the S100 and S90 granules was 7 and 1days, respectively. Formation of the AP layer wasconfirmed using FTIR analysis. In addition, in order todetermine the effect of pre-treatment in Ca–P freesolution, a control group of S100 granules was pre-treated in tris buffered solution (T) supplemented with2.5 mm of silicon. Subsequently, all groups of granules(uncoated, AP-coated, and pre-treated in T) weresubjected to a dissolution test in TE using a W/Vparameter of 0.5 mg ml�1. The solutions were exchangedat 3, 6, 10 and 24 h and then once every day up to 1week. The silicon release from the xerogel granules wasmeasured using AAS. Surface composition and mor-phology of the AP-coated granules before and after thedissolution experiments were studied using SEM/EDXanalysis.
3. Results
3.1. Materials characterization
Xerogel properties were determined using gas sorp-tion, FTIR, and XRD analyses. Nitrogen adsorption–desorption isotherms of S100 (Ca–P free) and S90 (Ca–Pcontaining) xerogels are shown in Figs. 1a and b.
Table 1
Composition of synthesized xerogels
Denomination Composition (wt%)
SiO2 CaO P2O5
S100 100 — —
S95 95 3 2
S90 90 5 5
S85 85 10 5
S. Radin et al. / Biomaterials 23 (2002) 3113–31223114
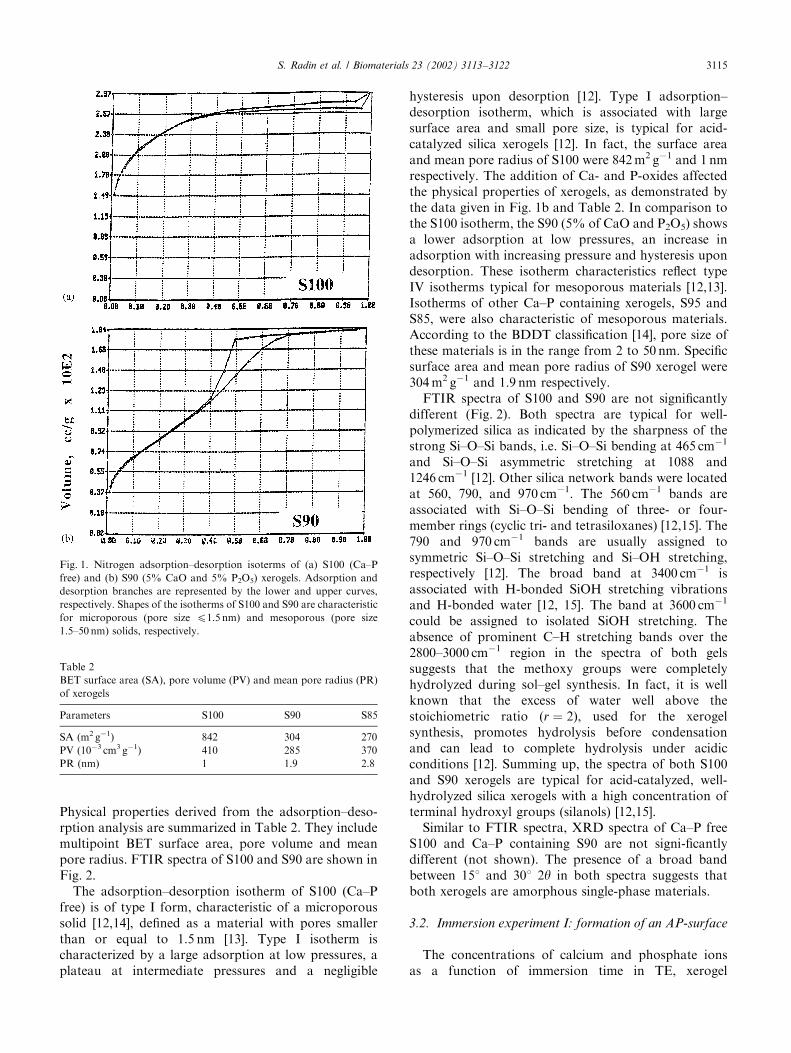
Physical properties derived from the adsorption–deso-rption analysis are summarized in Table 2. They includemultipoint BET surface area, pore volume and meanpore radius. FTIR spectra of S100 and S90 are shown inFig. 2.
The adsorption–desorption isotherm of S100 (Ca–Pfree) is of type I form, characteristic of a microporoussolid [12,14], defined as a material with pores smallerthan or equal to 1.5 nm [13]. Type I isotherm ischaracterized by a large adsorption at low pressures, aplateau at intermediate pressures and a negligible
hysteresis upon desorption [12]. Type I adsorption–desorption isotherm, which is associated with largesurface area and small pore size, is typical for acid-catalyzed silica xerogels [12]. In fact, the surface areaand mean pore radius of S100 were 842 m2 g�1 and 1 nmrespectively. The addition of Ca- and P-oxides affectedthe physical properties of xerogels, as demonstrated bythe data given in Fig. 1b and Table 2. In comparison tothe S100 isotherm, the S90 (5% of CaO and P2O5) showsa lower adsorption at low pressures, an increase inadsorption with increasing pressure and hysteresis upondesorption. These isotherm characteristics reflect typeIV isotherms typical for mesoporous materials [12,13].Isotherms of other Ca–P containing xerogels, S95 andS85, were also characteristic of mesoporous materials.According to the BDDT classification [14], pore size ofthese materials is in the range from 2 to 50 nm. Specificsurface area and mean pore radius of S90 xerogel were304 m2 g�1 and 1.9 nm respectively.
FTIR spectra of S100 and S90 are not significantlydifferent (Fig. 2). Both spectra are typical for well-polymerized silica as indicated by the sharpness of thestrong Si–O–Si bands, i.e. Si–O–Si bending at 465 cm�1
and Si–O–Si asymmetric stretching at 1088 and1246 cm�1 [12]. Other silica network bands were locatedat 560, 790, and 970 cm�1. The 560 cm�1 bands areassociated with Si–O–Si bending of three- or four-member rings (cyclic tri- and tetrasiloxanes) [12,15]. The790 and 970 cm�1 bands are usually assigned tosymmetric Si–O–Si stretching and Si–OH stretching,respectively [12]. The broad band at 3400 cm�1 isassociated with H-bonded SiOH stretching vibrationsand H-bonded water [12, 15]. The band at 3600 cm�1
could be assigned to isolated SiOH stretching. Theabsence of prominent C–H stretching bands over the2800–3000 cm�1 region in the spectra of both gelssuggests that the methoxy groups were completelyhydrolyzed during sol–gel synthesis. In fact, it is wellknown that the excess of water well above thestoichiometric ratio (r ¼ 2), used for the xerogelsynthesis, promotes hydrolysis before condensationand can lead to complete hydrolysis under acidicconditions [12]. Summing up, the spectra of both S100and S90 xerogels are typical for acid-catalyzed, well-hydrolyzed silica xerogels with a high concentration ofterminal hydroxyl groups (silanols) [12,15].
Similar to FTIR spectra, XRD spectra of Ca–P freeS100 and Ca–P containing S90 are not signi-ficantlydifferent (not shown). The presence of a broad bandbetween 151 and 301 2y in both spectra suggests thatboth xerogels are amorphous single-phase materials.
3.2. Immersion experiment I: formation of an AP-surface
The concentrations of calcium and phosphate ionsas a function of immersion time in TE, xerogel
Fig. 1. Nitrogen adsorption–desorption isoterms of (a) S100 (Ca–P
free) and (b) S90 (5% CaO and 5% P2O5) xerogels. Adsorption and
desorption branches are represented by the lower and upper curves,
respectively. Shapes of the isotherms of S100 and S90 are characteristic
for microporous (pore size p1.5 nm) and mesoporous (pore size
1.5–50 nm) solids, respectively.
Table 2
BET surface area (SA), pore volume (PV) and mean pore radius (PR)
of xerogels
Parameters S100 S90 S85
SA (m2 g�1) 842 304 270
PV (10�3 cm3 g�1) 410 285 370
PR (nm) 1 1.9 2.8
S. Radin et al. / Biomaterials 23 (2002) 3113–3122 3115

composition (S85, S90, S95, and S100) and immersionparameters are shown in Figs. 3a and b: (a) the effect ofxerogel composition at fixed W/V ratio (2 mg ml�1), (b)the effect of W/V parameter for one composition (S85).The time to PO4 uptake for some of the xerogels isincluded in Table 3.
A significant increase in [Ca] and some increase in[PO4] over control levels were observed after immersionof all Ca–P containing xerogels. The relative increase in[Ca], DCa, varied either with CaO content in the xerogelor with the W/V ratio. The initial increase in [Ca] and[PO4] was followed by uptake of these ions. This
Fig. 2. FTIR spectra of S100 (Ca–P free) and S90 (5% CaO and 5% P2O5) xerogels. Both spectra are typical for xerogels synthesized by acid-
catalyzed hydrolysis with an excess of water above the stoichiometric ratio [12]. Both spectra reveal a complete hydrolysis and the presence of surface
silanol groups.
Fig. 3. [Ca] and [PO4] as a function of immersion time in TE: (a) for various xerogel compositions and (b) for various weight-to-solution volume
(W/V) values. A fixed W/V value of 2 mg ml�1 was used for testing the compositional effect (a), and one composition, S85, was tested with varying
W/V values (b). Note that after immersion of S100 (Ca–P free), no changes in the [Ca] and [PO4] over the control levels occurred up to 72 h. For all
Ca–P containing xerogels, the initial increase in the [Ca] and [PO4] was followed by ion uptake.
S. Radin et al. / Biomaterials 23 (2002) 3113–31223116

suggested precipitation of a Ca–P phase. The inductiontime to precipitation, expressed as the time to ameasurable PO4 uptake, decreased with an increase inDCa. For instance, for the DCa values of 0.65, 1.3 and3.5 mm the time to precipitation was 24, 3, and 1 h,respectively. The uptake profiles also depend on the DCavalues.
In contrast, no change in the concentrations of Caand PO4 ions in comparison to the control level could bemeasured upon immersion of S100 (Ca–P free) xerogelfor duration up to 72 h (Fig. 3a). Precipitation of a Ca–Pphase on S100 did occur eventually, but after muchlonger time periods. The time to precipitation variedfrom 4 to 14 days (depending on the W/V ratio)(Table 3). The lag time to Ca–P precipitation on S100was two orders of magnitude greater than that on Ca–Pcontaining xerogels (Table 3).
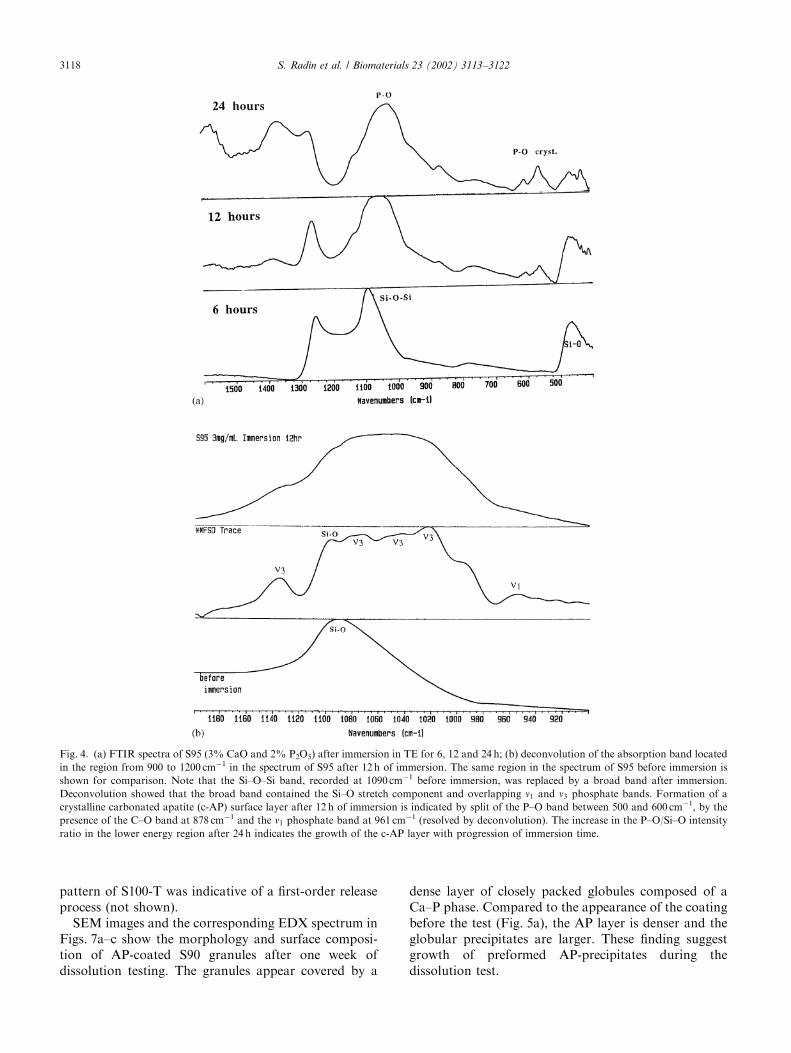
FTIR analysis revealed the presence of a crystallineCa–P layer on all Ca–P containing xerogels within 1 dayof immersion (Table 3). For some immersion conditions,formation of the crystalline Ca–P layer was detectedeven earlier, as illustrated by the spectrum of S95 after12 h of immersion at (W/V ratio of 3 mg ml�1) (Fig. 4).Formation of a crystalline carbonated Ca–P phase isindicated by the presence of the divided lower energyP–O band and the C–O band at 878 cm�1. However, theoccurrence of prominent Si–O bending vibration at470 cm�1 suggested that the Ca–P layer, after 12 h ofimmersion, did not cover the silica sufficiently. Thebroad band, recorded at 12 h of immersion in the regionfrom 900 to 1200 cm�1, replaced the Si–O band, locatedat 1090 cm�1 before immersion (Figs. 4a and b).Deconvolution of this broad band (Fig. 4b) showedthat the large width was associated with overlapping ofthe Si–O band (1090 cm�1) and P–O bands. The P–Obands, resolved by deconvolution, were assigned asfollows: the bands at 1140, 1080, 1040 and 1020 cm�1 toP–O asymmetric stretching vibrations (n3) [16] and theband at 960 cm�1 to P–O symmetric stretching (n1),characteristic of hydroxyapatite. Taken together, thespectral analysis revealed the formation of crystallinecarbonated apatite (c-AP) on the surface of S95 after12 h of immersion. In comparison to the spectrum of
S95 after 12 h of immersion, the P–O/Si–O intensityratio in the lower energy region increased after 24 h ofimmersion. This suggests further growth of the c-APsurface layer.
The data of FTIR analysis for S100 were in agreementwith solution chemical analysis, which showed asignificant delay of Ca–P precipitation on this Ca–Pfree xerogel. In fact, the time to formation of a c-APlayer on S100 was significantly longer than that on theCa–P containing xerogels (Table 3). A crystalline c-APlayer was not observed till 7 to 14 days of immersion,depending on the immersion conditions.
The morphology and the surface composition ofCa–P containing granules were analyzed by SEM/EDXafter 24 h of immersion (Figs. 5a and b). The SEMimage and corresponding EDX spectrum of a S90granule show that the granule was uniformly coveredwith very fine (0.5–1 mm) globular precipitates, com-posed of a Ca–P phase.
3.3. Immersion experiment II: effect of a preformed
AP-coating on the dissolution behavior of xerogels in vitro
Granules of two compositions, S100 (Ca–P free) andS90 (Ca–P containing), both uncoated and AP-coated,were used. Granules of S100 pre-treated in Ca–P free Twere used as a control group. Immersion in TE wasperformed with solution exchange at designated timeperiods. The cumulative Si release, expressed as apercentage of the original Si-content in the xerogelsamples, is shown as a function of immersion time inFigs. 6a and b. The effect of AP-coating and pre-treatment in T on the dissolution rate of S100 isdisplayed in Table 4.
A 100% Si release from uncoated granules of bothcompositions, i.e. full dissolution, was observed afterone week of immersion with solution exchange. Thepresence of AP-coating dramatically altered the dissolu-tion behavior of both xerogel compositions. In compar-ison to a 100% Si release from uncoated granules, onlyabout 25% Si was released from AP-coated granulesafter 1 week of immersion. The presence of the AP-coating also affected the Si release pattern. In compar-ison to a fast, first-order release from uncoated xerogels,a slower, near zero-order release was observed in thepresence of AP-coating. In fact, the silica dissolutionrates of uncoated S100 xerogel were 7.49 mg mg�1 h�1 inthe time range between 0 and 24 h and 3.62 mg mg�1 h�1
between 24 and 72 h. In contrast, the dissolution rate ofAP-coated S100 was 2.57 mg mg�1 h�1 over the entireduration of the experiment.
In comparison to AP-coating, control pre-treatmentof S100 in Ca–P free T (S100-T) showed an initialreduction in the dissolution rate (0–24 h), but didnot show any noticeable effect at further immersion(Table 4). Similar to uncoated S100, the Si release
Table 3
Time to measurable PO4 uptake and to AP layer formation (as
detected by FTIR)
Composition Time to PO4
uptake (h)
Time to AP
formation (days)
S100 96a–240b 7a–14b
S95 3a 0.5a–1b
S90 2b 0.5a–1b
S85 o1b 0.5a–1b
a Immersion in TE at 3 mg ml�1.b Immersion in TE at 2 mg ml�1.
S. Radin et al. / Biomaterials 23 (2002) 3113–3122 3117
pattern of S100-T was indicative of a first-order releaseprocess (not shown).
SEM images and the corresponding EDX spectrum inFigs. 7a–c show the morphology and surface composi-tion of AP-coated S90 granules after one week ofdissolution testing. The granules appear covered by a
dense layer of closely packed globules composed of aCa–P phase. Compared to the appearance of the coatingbefore the test (Fig. 5a), the AP layer is denser and theglobular precipitates are larger. These finding suggestgrowth of preformed AP-precipitates during thedissolution test.
Fig. 4. (a) FTIR spectra of S95 (3% CaO and 2% P2O5) after immersion in TE for 6, 12 and 24 h; (b) deconvolution of the absorption band located
in the region from 900 to 1200 cm�1 in the spectrum of S95 after 12 h of immersion. The same region in the spectrum of S95 before immersion is
shown for comparison. Note that the Si–O–Si band, recorded at 1090 cm�1 before immersion, was replaced by a broad band after immersion.
Deconvolution showed that the broad band contained the Si–O stretch component and overlapping n1 and n3 phosphate bands. Formation of a
crystalline carbonated apatite (c-AP) surface layer after 12 h of immersion is indicated by split of the P–O band between 500 and 600 cm�1, by the
presence of the C–O band at 878 cm�1 and the n1 phosphate band at 961 cm�1 (resolved by deconvolution). The increase in the P–O/Si–O intensity
ratio in the lower energy region after 24 h indicates the growth of the c-AP layer with progression of immersion time.
S. Radin et al. / Biomaterials 23 (2002) 3113–31223118

4. Discussion
In this study we focused on the ability of silica xerogelto form a bioactive AP surface in vitro and its effect onthe xerogel degradability.
It is well-known that synthetic bone-bioactive materi-als such as Ca–P based ceramics and Ca–P containingsilicate glasses form a carbonated, biologically equiva-lent carbonate apatite (c-AP) on their surfaces uponimmersion into physiological solutions [7,8,10,17–20].Reactions leading to the c-AP formation includedissolution of calcium and phosphate ions, solutionoversaturation with respect to AP and reprecipitation ofnew, bone-like minerals on the ceramic surfaces [10,17–21]. It has also been suggested that bioactive materialsdo not need to contain the calcium and phosphate ionssince these ions are present in body fluids and can beadsorbed on the materials surface when polar functionalgroups, such as OH� groups, are present at the surface[9]. In this regard, we compared kinetics and mechan-isms of AP formation on silica xerogels synthesized withand without Ca- and P-oxides. All xerogels tested werehighly porous materials with abundant OH� groups attheir surface.
Immersion in simulated physiological solution (TE)induced c-AP formation at the surface of all xerogelsstudied (Table 3). However, the kinetics and mechan-isms of the c-AP formation on Ca–P free and Ca–Pcontaining xerogels were different. Ca–P free xerogels(S100) required long lag times to Ca–P precipitation(4–10 days) and to formation of a detectable c-APsurface layer (7–14 days). In contrast, all Ca–P contain-ing xerogels showed a reduction of the time toprecipitation by 2–3 orders of magnitude, and a c-APlayer was formed within 24 h. The remarkable enhance-ment of the c-AP formation was observed even uponaddition of minor amounts of Ca- and P-oxides (as little
Fig. 5. (a) Backscattered electron image and (b) corresponding EDX
spectrum of a S90 granule, immersed in TE+2.5 mm Si for 24 h to
form a AP-coating. The granule surface is uniformly covered with fine,
globular precipitates composed of a Ca–P phase (confirmed to be
c-HA by FTIR).
Fig. 6. Cumulative Si release from AP-coated versus uncoated (a) S100 and (b) S90 during immersion in TE for up to one week. The solution was
totally exchanged at the indicated points. Whereas uncoated granules showed a 100% Si release after one week of immersion, the overall release from
HA-coated granules did not exceed 25%.
S. Radin et al. / Biomaterials 23 (2002) 3113–3122 3119
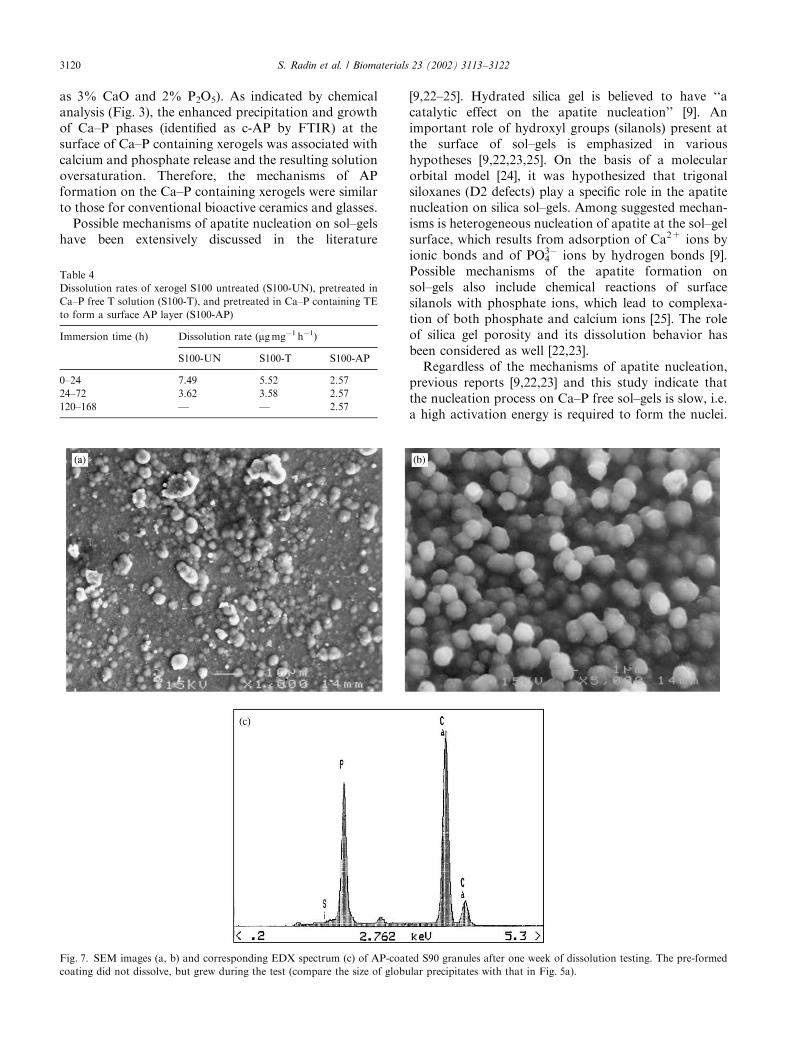
as 3% CaO and 2% P2O5). As indicated by chemicalanalysis (Fig. 3), the enhanced precipitation and growthof Ca–P phases (identified as c-AP by FTIR) at thesurface of Ca–P containing xerogels was associated withcalcium and phosphate release and the resulting solutionoversaturation. Therefore, the mechanisms of APformation on the Ca–P containing xerogels were similarto those for conventional bioactive ceramics and glasses.
Possible mechanisms of apatite nucleation on sol–gelshave been extensively discussed in the literature
[9,22–25]. Hydrated silica gel is believed to have ‘‘acatalytic effect on the apatite nucleation’’ [9]. Animportant role of hydroxyl groups (silanols) present atthe surface of sol–gels is emphasized in varioushypotheses [9,22,23,25]. On the basis of a molecularorbital model [24], it was hypothesized that trigonalsiloxanes (D2 defects) play a specific role in the apatitenucleation on silica sol–gels. Among suggested mechan-isms is heterogeneous nucleation of apatite at the sol–gelsurface, which results from adsorption of Ca2+ ions byionic bonds and of PO4
3� ions by hydrogen bonds [9].Possible mechanisms of the apatite formation onsol–gels also include chemical reactions of surfacesilanols with phosphate ions, which lead to complexa-tion of both phosphate and calcium ions [25]. The roleof silica gel porosity and its dissolution behavior hasbeen considered as well [22,23].
Regardless of the mechanisms of apatite nucleation,previous reports [9,22,23] and this study indicate thatthe nucleation process on Ca–P free sol–gels is slow, i.e.a high activation energy is required to form the nuclei.
Table 4
Dissolution rates of xerogel S100 untreated (S100-UN), pretreated in
Ca–P free T solution (S100-T), and pretreated in Ca–P containing TE
to form a surface AP layer (S100-AP)
Immersion time (h) Dissolution rate (mg mg�1 h�1)
S100-UN S100-T S100-AP
0–24 7.49 5.52 2.57
24–72 3.62 3.58 2.57
120–168 — — 2.57
Fig. 7. SEM images (a, b) and corresponding EDX spectrum (c) of AP-coated S90 granules after one week of dissolution testing. The pre-formed
coating did not dissolve, but grew during the test (compare the size of globular precipitates with that in Fig. 5a).
S. Radin et al. / Biomaterials 23 (2002) 3113–31223120

In addition, the nucleation process depends on a numberof parameters (such as surface chemistry, porosity,dissolution rate, etc.) that are not easily controlled. Aswas demonstrated in this study, the nucleation process issignificantly faster when Ca- and P-oxides are present insol–gels. In comparison to Ca–P free sol–gels, the lagtime to Ca–P precipitation can be reduced by 2–3 ordersof magnitude with addition of minor amounts ofCa- and P-oxides. The findings of this study suggestthat the kinetics of nucleation, precipitation and growthof apatite at the surface of Ca–P containing sol–gel aredetermined by the ion content in the gel, their releaseupon immersion and the resulting solution oversatura-tion. This indicates that formation of c-AP, i.e. bone-like mineral, on the surface of sol–gels could be wellmonitored via their compositional design.
As far as controlled degradability of silica xerogels isconcerned, we hypothesize that the presence of a APsurface film formed upon immersion can play a dualrole, i.e. it provides a bioactive surface and protectsthe xerogel from dissolution. Such a dual effect of aCa–P-rich surface was at the basis of formulation ofbioactive glasses (BG) [26]. In the case of melt-derivedBG, the stabilizing effect is produced by a two-layersurface, composed of subsurface polymerized silica geland a Ca–P-rich surface, which are formed upon BGimmersion. Since silica xerogel is already composed ofpolymerized silica, we believed that the formation of asingle-layer Ca–P surface upon xerogel immersionwould produce a stabilizing effect. In fact, the dissolu-tion rates of silica xerogels and the overall amount of Sirelease were remarkably reduced in the presence of anAP surface layer. Compared to a 100% Si release, i.e.total dissolution of uncoated xerogels, the amount of Sirelease from AP-coated granules was reduced to about25% (Fig. 6). Moreover, in comparison to a fast, first-order Si release from uncoated xerogels, a slower, zero-order release process was observed for AP-coated gels(Fig. 6a and b). The first-order dissolution processobserved for untreated xerogels is typical for diffusion-controlled dissolution of solid silica [27]. As demon-strated in our previous work [11], structural changes,which may occur on xerogel immersion, do not lead toa change in the mechanism of xerogel dissolution. Incontrast, the presence of AP-coating produces a majorchange in the mechanism of dissolution, as suggestedby observed zero-order dissolution process. Thesedata demonstrate a protective effect of the AP surfacelayer on the dissolution of silica xerogels. As indicatedby SEM analysis, the preformed AP-coating, com-posed of fine precipitates (Fig. 5a), did not dissolve,but continued to grow during the dissolution test(Figs. 7a and b). Therefore, the presence of apreformed AP-coating, which grows further withimmersion, allows for a control of the surface reactivityof silica xerogels.
5. Conclusions
Formation of a bioactive, crystalline carbonateapatite (c-AP) surface was observed on all silica xerogelsstudied, with or without Ca- and P-oxides. Ca–P freexerogels showed long times to precipitation and toformation of a detectable c-AP surface layer (up to 2weeks). In contrast, Ca–P containing xerogels showed areduction of the time to precipitation by 2–3 orders ofmagnitude, and the c-AP layer was formed within 24 h.The presence of a preformed AP surface layer has astabilizing effect on the surface reactivity of silicaxerogel.
These properties, in vitro bioactivity and controlleddegradation, combined with previously reported bio-compatibility in vivo and sustained release of functionalbiological molecules in vitro, suggest that silica xerogelis a promising controlled release material.
Acknowledgements
The participation of K. Reicin and V. Fan in theexperimentation is gratefully acknowledged. This workwas supported by NSF BCS-93-09053 and NIHDE-10693 grants.
References
[1] Nicoll SB, Radin S, Santos EM, Ducheyne P, Tuan RS. In vitro
release kinetics of biologically active transforming growth factor-
b1 from a novel porous glass carrier. Biomaterials 1997;18:853–9.
[2] Santos EM, Ducheyne P, Radin S, Shenker B, Shapiro IM.
Si–Ca–P xerogels and morphogenetic protein act synergistically
on rat marrow stromal cell differentiation in vitro. J Biomed
Mater Res 1998;41:87–94.
[3] Santos EM, Radin S, Ducheyne P. Sol–gel derived carrier for the
controlled release of proteins. Biomaterials 1999;20:1695–700.
[4] Radin S, Ducheyne P, Kamplain T, Tan BH. Silica sol–gel for the
controlled release of antibiotics. I. Synthesis, characterization and
in vitro release. J Biomed Mater Res 2001;57:313–20.
[5] Aughenbaugh W, Radin S, Ducheyne P. Silica sol–gel for the
controlled release of antibiotics. II. The effect of synthesis
parameters on the in vitro release kinetics of vancomycin.
J Biomed Mater Res 2001;57:321–6.
[6] Radin S, El-Bassyouni G, Vreslovic EJ, Schepers E, Ducheyne P.
Tissue reactions to controlled release silica xerogel carriers. In:
LeGeros RZ, LeGeros JP, editors. Bioceramics, vol. 11. New
York: World Scientific, 1998. p. 529–32.
[7] Hench LL. Bioceramics: from concept to clinic. J Am Ceram Soc
1991;74(7):1487–510.
[8] Radin S, Ducheyne P, Rothman B, Conti A. The effect of in vitro
modeling conditions on the surface reactions on bioactive glass.
J Biomed Mater Res 1997;37:363–75.
[9] Li P, Ohtsuki C, Kokubo T, Nakanishi K, Soga N. Apatite
formation induced by silica gel in simulated body fluid. J Am
Ceram Soc 1992;75:2094–7.
[10] Radin S, Ducheyne P. The effect of calcium phosphate ceramic
composition and structure on in vitro behavior. II. Precipitation.
J Biomed Mater Res 1993;27:35–45.
S. Radin et al. / Biomaterials 23 (2002) 3113–3122 3121

[11] Falaize S, Radin S, Ducheyne P. In vitro behavior of silica-based
xerogels intended as controlled release carriers. J Am Ceram Soc
1999;82(4):969–76.
[12] Brinker CJ, Scherer GW. Sol–gel science: the physics and
chemistry of sol–gel processing. Boston: Academic Press, 1990.
[13] Lowell S, Shields JE. Powder surface and porosity. London:
Chapman & Hall, 1991.
[14] Brunauer S, Deming LS, Deming WS, Teller E. J Amer Chem Soc
1940;62:1723.
[15] Ying JY, Benziger JB. Structural evolution of alkoxide silica gels
to glass: effect of catalyst pH. Am Ceram Soc 1993;76:2571–82.
[16] Pleshko N, Mendelsohn R, Boskey A. Novel infrared spectro-
scopic method for the determination of crystallinity of hydro-
xyapatite minerals. Biophys J 1991;60:780–93.
[17] LeGeros RZ, Daculsi G, Orly I, Gregoire M, Heughebaert M,
Gineste M, Kijkowska R. Formation of carbonate apatite on
calcium phosphate materials: dissolution/precipitation processes.
In: Ducheyne P, Kokubo T, van Blitterswijk CA, editors. Bone-
bonding biomaterials. Leiderdorp, The Netherlands: Reed
Healthcare Communications, 1993. p. 201–12.
[18] Kokubo T. Surface chemistry of bioactive glass-ceramics. J Non-
Cryst Solids 1990;120:138–51.
[19] Jarcho M. Calcium phosphate ceramics as hard tissue prosthetics.
Clin Orthop 1981;157:259–78.
[20] Ducheyne P, Bianco P, Radin S, Schepers E. Bioactive materials:
mechanisms and bioengineering considerations. In: Ducheyne P,
Kokubo T, van Blitterswijk CA, editors. Bone-bioactive bioma-
terials. Leiderdorp, The Netherlands: Reed Healthcare Commu-
nications, 1993. p. 1–12.
[21] Ducheyne P, Radin S, King L. The effect of calcium-phosphate
ceramic composition and structure on in vitro behavior; I.
Dissolution. J Biomed Mater Res 1993;27:25–34.
[22] Kokubo T, Cho SB, Nakanishi K, Soga N, Ohtsuki C, Kitsugi T,
Yamamuro T, Nakamura T. Dependence of bone apatite
formation on structure of silica gel. In: Andersson OH and Yli-
Urpo A, editors. Bioceramics, vol. 7. Butterworth-Heinemann,
1994. p. 49–54.
[23] Cho SB, Miyaji F, Kokubo T, Nakanishi K, Soga N, Nakamura
T. Apatite formation on silica gel in simulated body fluid: effects
of structural modification with solvent exchange. J Mater Sci:
Mater Med 1998;9:279–84.
[24] West JH, Hench LL. Reaction kinetics of bioactive ceramics. Part
Y: molecular orbital modeling of bioactive glass surface reactions.
In: Yamamuro T, Kokubo T, Nakamura T, editors. Bioceramics,
vol. 5. Kyoto: Kobunshi Kankokai, 1991. p. 75–86.
[25] Andersson OH, Karlsson KH. On the bioactivity of silicate glass.
J Non-Cryst Solids 1991;129:145–51.
[26] Hench LL, Clark DE. Physical chemistry of glass surfaces. J Non-
Cryst Solids 1978;28:83–105.
[27] O’Connor TL, Greenberg SA. The kinetics for the solution
of silica in aqueous solutions. J Phys Chem 1958;62:
1195–8.
S. Radin et al. / Biomaterials 23 (2002) 3113–31223122
Copyright © 2022 FDOKUMEN




















