
Immune Stimulation in Scleroderma Patients Treated with Thalidomide
12
Immune Stimulation in Scleroderma Patients Treated with Thalidomide Stephen J. Oliver,* Andre Moreira,* , ² and Gilla Kaplan* *The Laboratory of Cellular Physiology and Immunology, Rockefeller University, New York, New York 10021; and ²The Department of Pathology, New York University Medical Center, New York, New York 10016 Scleroderma (SSc) is a fibrosing connective tissue disease that is poorly responsive to any treatment, including immune suppression. SSc shares many char- acteristics with chronic graft-versus-host disease (GVHD). Because the immunomodulatory drug thalid- omide has proven beneficial in chronic GVHD, we studied the immune response and clinical effects of thalidomide in SSc patients. We treated 11 SSc pa- tients with thalidomide in an open label, dose escalat- ing, 12 week study. Histologic comparison of skin bi- opsies showed changes in skin fibrosis and an increase in epidermal and dermal infiltrating CD8 1 T cells with thalidomide treatment. In thalidomide-treated SSc pa- tients, plasma levels of IL-12 and TNF-a increased, while plasma IL-5 and IL-10 levels remained un- changed. These changes were associated with clinical effects, including dry skin, dermal edema, transient rashes, decreased gastroesophageal reflux symptoms, and healing of digital ulcers. When SSc PBMCs acti- vated by anti-CD3 mAb were exposed to thalidomide, increases in both production of IL-2, IL-3, GM-CSF, and IFN-g and T cell expression of CD40L were ob- served. Thalidomide therefore appears to induce im- mune stimulation in SSc patients in association with clinical changes. However, it remains to be shown whether long-term enhancement of immune responses in SSc patients is clinically beneficial. © 2000 Academic Press Key Words: scleroderma; thalidomide; ulcer healing; IL-12; GM-CSF; immune modulation; Th1/Th2 cyto- kines. INTRODUCTION Progressive systemic sclerosis (scleroderma; SSc) is a connective tissue disorder of unknown etiology which affects about 36,000 persons in the United States (re- viewed in 1, 2). The disease is characterized by fibrosis of skin, resulting in progressive restriction of joint range of motion. Fibrosis of internal organs also occurs, leading to cardiac dysrhythmias, gastroesophageal re- flux, and respiratory problems. The vasculature is af- fected by the fibrotic process, resulting in ischemia due to narrowing of the arterial lumen. Recurrent digital ulcers are also characteristic of the disease. The pro- gressive fibrosis causes marked disability and in- creased mortality. Because it is believed to be an au- toimmune disease, SSc is often treated with immunosuppressive and cytotoxic drugs, none of which have been shown to modify the disease course. Recently, investigators have begun to characterize the immune parameters of this disease. Results sug- gest that a T helper 2 (Th2)-type immune response predominates (3–5). The affected skin of SSc patients has been shown to contain mRNA for interleukin-4 (IL-4), but not for interferon-gamma (IFN-g). CD4 1 T cell clones generated from SSc skin biopsies have been shown to express this Th2-type cytokine profile (6). Also, defective IFN-g production by in vitro stimulated peripheral blood cells from SSc patients has been re- ported and abnormally low IFN-g levels have been observed in SSc sera (3). During the early inflamma- tory stage of SSc, activated T cells infiltrate the perivascular and interstitial spaces of the skin (7, 8). This infiltrating cell population contains both CD4 1 and CD8 1 T cells, with a predominance of CD4 1 cells. Systemic T cell and monocyte activation are indicated by the elevated serum levels of soluble IL-2 receptor (sIL-2R), soluble intercellular adhesion molecule-1 (sI- CAM-1), tumor necrosis factor-alpha (TNF-a), IL-2, IL-4, IL-6, and IL-10 (3, 4, 9 –11). SSc shares many characteristics with chronic graft- versus-host disease (GVHD), including fibrosis of skin and visceral organs with involvement of the salivary glands, gastrointestinal tract, liver, and lung (12–14). Chronic GVHD is also associated with a Th2 immune response (15). In patients with chronic GVHD refrac- tory to conventional therapy, thalidomide treatment has resulted in clinical improvement (16). In animal models of both acute and chronic GVHD, a progressive reduction of cutaneous fibrosis and a return of normal dermal appendages has been observed with thalido- mide treatment, with further and possibly synergistic efficacy seen in combination therapy with cyclosporin A (CSA) (17, 18). Investigators in this laboratory have previously shown that thalidomide is a partial inhibitor of TNF-a production by LPS-stimulated monocytes both in vitro Clinical Immunology Vol. 97, No. 2, November, pp. 109 –120, 2000 doi:10.1006/clim.2000.4920, available online at http://www.idealibrary.com on 1521-6616/00 $35.00 Copyright © 2000 by Academic Press All rights of reproduction in any form reserved. 109
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Immune Stimulation in Scleroderma Patients Treated with Thalidomide

smcwiP
Clinical ImmunologyVol. 97, No. 2, November, pp. 109–120, 2000doi:10.1006/clim.2000.4920, available online at http://www.idealibrary.com on
Immune Stimulation in Scleroderma Patients Treated with Thalidomide
Stephen J. Oliver,* Andre Moreira,*,† and Gilla Kaplan*
*The Laboratory of Cellular Physiology and Immunology, Rockefeller University, New York, New York 10021; and
†The Department of Pathology, New York University Medical Center, New York, New York 10016Scleroderma (SSc) is a fibrosing connective tissuedisease that is poorly responsive to any treatment,including immune suppression. SSc shares many char-acteristics with chronic graft-versus-host disease(GVHD). Because the immunomodulatory drug thalid-omide has proven beneficial in chronic GVHD, westudied the immune response and clinical effects ofthalidomide in SSc patients. We treated 11 SSc pa-tients with thalidomide in an open label, dose escalat-ing, 12 week study. Histologic comparison of skin bi-opsies showed changes in skin fibrosis and an increasein epidermal and dermal infiltrating CD81 T cells withthalidomide treatment. In thalidomide-treated SSc pa-tients, plasma levels of IL-12 and TNF-a increased,while plasma IL-5 and IL-10 levels remained un-changed. These changes were associated with clinicaleffects, including dry skin, dermal edema, transientrashes, decreased gastroesophageal reflux symptoms,and healing of digital ulcers. When SSc PBMCs acti-vated by anti-CD3 mAb were exposed to thalidomide,increases in both production of IL-2, IL-3, GM-CSF,and IFN-g and T cell expression of CD40L were ob-erved. Thalidomide therefore appears to induce im-une stimulation in SSc patients in association with
linical changes. However, it remains to be shownhether long-term enhancement of immune responses
n SSc patients is clinically beneficial. © 2000 Academic
ress
Key Words: scleroderma; thalidomide; ulcer healing;IL-12; GM-CSF; immune modulation; Th1/Th2 cyto-kines.
INTRODUCTION
Progressive systemic sclerosis (scleroderma; SSc) is aconnective tissue disorder of unknown etiology whichaffects about 36,000 persons in the United States (re-viewed in 1, 2). The disease is characterized by fibrosisof skin, resulting in progressive restriction of jointrange of motion. Fibrosis of internal organs also occurs,leading to cardiac dysrhythmias, gastroesophageal re-flux, and respiratory problems. The vasculature is af-fected by the fibrotic process, resulting in ischemia due
to narrowing of the arterial lumen. Recurrent digital109
ulcers are also characteristic of the disease. The pro-gressive fibrosis causes marked disability and in-creased mortality. Because it is believed to be an au-toimmune disease, SSc is often treated withimmunosuppressive and cytotoxic drugs, none of whichhave been shown to modify the disease course.
Recently, investigators have begun to characterizethe immune parameters of this disease. Results sug-gest that a T helper 2 (Th2)-type immune responsepredominates (3–5). The affected skin of SSc patientshas been shown to contain mRNA for interleukin-4(IL-4), but not for interferon-gamma (IFN-g). CD41 Tcell clones generated from SSc skin biopsies have beenshown to express this Th2-type cytokine profile (6).Also, defective IFN-g production by in vitro stimulatedperipheral blood cells from SSc patients has been re-ported and abnormally low IFN-g levels have beenobserved in SSc sera (3). During the early inflamma-tory stage of SSc, activated T cells infiltrate theperivascular and interstitial spaces of the skin (7, 8).This infiltrating cell population contains both CD41
and CD81 T cells, with a predominance of CD41 cells.Systemic T cell and monocyte activation are indicatedby the elevated serum levels of soluble IL-2 receptor(sIL-2R), soluble intercellular adhesion molecule-1 (sI-CAM-1), tumor necrosis factor-alpha (TNF-a), IL-2,IL-4, IL-6, and IL-10 (3, 4, 9–11).
SSc shares many characteristics with chronic graft-versus-host disease (GVHD), including fibrosis of skinand visceral organs with involvement of the salivaryglands, gastrointestinal tract, liver, and lung (12–14).Chronic GVHD is also associated with a Th2 immuneresponse (15). In patients with chronic GVHD refrac-tory to conventional therapy, thalidomide treatmenthas resulted in clinical improvement (16). In animalmodels of both acute and chronic GVHD, a progressivereduction of cutaneous fibrosis and a return of normaldermal appendages has been observed with thalido-mide treatment, with further and possibly synergisticefficacy seen in combination therapy with cyclosporinA (CSA) (17, 18).
Investigators in this laboratory have previouslyshown that thalidomide is a partial inhibitor of TNF-a
production by LPS-stimulated monocytes both in vitro1521-6616/00 $35.00Copyright © 2000 by Academic Press
All rights of reproduction in any form reserved.

i
cfw
fuT
110 OLIVER, MOREIRA, AND KAPLAN
and in vivo (19, 20). More recently, we have shown thatthalidomide is also a potent costimulator of human Tcells in vitro (21). Addition of a specific T cell stimulus(anti-CD3 mAb) to peripheral blood cells in vitro in thepresence of thalidomide results in increased productionof IFN-g, IL-2, and IL-12 as well as enhanced T cellexpression of CD40 ligand (CD40L) (22). Thus, thalid-omide appears to stimulate a Th1-type cellular im-mune response.
We hypothesized that thalidomide, an effective im-mune-modulating treatment in chronic GVHD, mightsimilarly modify the immune status of SSc patients,thereby altering scleroderma disease activity. To testthis hypothesis we carried out a pilot study in which8/11 SSc patients completed a 3-month course of tha-lidomide treatment. Immunologic status, clinicalsymptoms, and skin biopsy histology were evaluated.Because of the possibility of disease exacerbation by ashift toward a Th1-type immune response in SSc pa-tients, we chose a relatively short treatment protocolwith dose escalation.
MATERIALS AND METHODS
Study patients. Four male and 7 female subjects,aged 18 to 70, who met the American College of Rheu-matology criteria (23) for either diffuse (9/11) or limited(2/11) scleroderma were enrolled into the study over a3-year period. Enrolled subjects were not on any im-munomodulating treatment other than a stable dose of5 mg/day prednisone (patients 1, 2, 7) in the 4 weeksprior to the protocol. Patients were allowed to remainat a stable corticosteroid dose and no additional immu-nomodulating agents were introduced during the pro-tocol. Patient 8, who had been taking both prednisoneand CSA for the previous year, was allowed to remainon stable doses of prednisone (20 mg/day) and of CSA(150 mg/day) during thalidomide treatment. Patientexclusion criteria included systemic sclerosing-like ill-nesses associated with known environmental, in-gested, or injected agents or with other connective tis-sue diseases. This study was conducted according toestablished ACR guidelines for clinical trials in sclero-derma (24). Patients gave informed consent for thestudy protocol, which had been approved by the Rocke-feller University Institutional Review Board.
All patients studied were required to practice effec-tive contraception for at least 1 month prior to studyentry and to use at least two forms of contraception forthe duration of the study and for 60 days post comple-tion of the study. All women of child bearing potentialwere required to have a negative pregnancy test (se-rum b-HCG) at the time of screening and at eachclinical evaluation time point of the protocol.
Protocol. All patient evaluations took place in the
npatient and outpatient facilities of The RockefellerUniversity Hospital and were performed by the samephysician throughout the protocol. Subjects meetingentry criteria were admitted to the Hospital for a 36-hvisit at week 0 for measurement of baseline studyparameters. Initial dosing of thalidomide (generouslyprovided by Celgene, Warren, NJ) was 50 mg/dayorally. Repeat study parameter evaluations were per-formed during inpatient visits at weeks 2, 4, 8, and 12.At each visit the thalidomide dose was doubled. Thefinal thalidomide dose, administered between weeks 8and 12, was 400 mg/day. This is the maximum doseused to treat erythema nodosum leprosum in leprosypatients (25).
Blood collection. EDTA anticoagulated blood sam-ples were collected from 8 of the 11 study patients atbaseline (week 0) and protocol time points (weeks 2, 4,8, and 12). Within 30 min of phlebotomy, the plasmawas removed by centrifugation at 4°C and stored at270°C until analysis. Similarly, blood samples wereobtained from 21 normal, healthy laboratory personnel(11 males, 10 females), with a median age of 35 yearsand a range of 23 to 70 years. In addition, at least 1year after completion of the study, blood samples for invitro studies were obtained from patients 2, 5, 6, and 7at a time when they had not been on any immune-modulating treatment, including thalidomide, duringthe previous year. Blood for in vitro studies was alsoollected from 4 additional patients with diffuse SSc: 3emales, ages 47, 57, and 59, and 1 male, aged 68, whoere not on any immune-modulating therapies.
Cytokine determination. Cytokine and soluble sur-ace protein levels were determined according to man-facturer’s instructions using commercial ELISA kits.he following kits were utilized: TNF-a EASIA, IL-3,
IL-4, IL-5 (Biosource International, Camarillo, CA),IFN-g, GM-CSF, sIL-2R, IL-10, IL-12, and sICAM-1(Endogen, Woburn, MA). Samples were assayed in du-plicate and compared to a standard curve, using amicrotiter plate reader (Dynatech, Chantilly, VA).
Histology and immunohistology. Four- to six-milli-meter punch biopsy samples from disease-affected ar-eas were obtained at week 0 from four patients (5, 6, 9,10). After 12 weeks of thalidomide treatment, a secondbiopsy was obtained from a site immediately adjacentto the first biopsy site. Tissue samples were fixed in10% buffered Formalin, embedded in paraffin, andthen sectioned and stained with H&E for microscopicviewing. Sections for immunostaining were pretreatedwith Antigen Unmasking Solution (Vector Laborato-ries, Burlingame, CA) and then incubated with a pri-mary mouse anti-human mAb against CD8 (DakoCorp., Carpinteria, CA) at 1:25. Control sections wereincubated with whole mouse IgG (Jackson Immunore-
search Laboratories, Inc., West Grove, PA). The sec-
111THALIDOMIDE IMMUNOMODULATION IN SCLERODERMA
tions were then incubated with a secondary goat anti-mouse mAb conjugated with Texas Red at 1:300(Jackson Lab., PA). Tissue samples from patients 9 and10 were divided in two at the time of biopsy, with halfprocessed as above and the other half embedded inOCT and frozen. The frozen sections were later fixed inacetone for 10 min. Prepared sections were stainedwith chromophore-labeled mAbs (FITC-CD4, RPE-CD8) or isotype controls (IgG1-FITC, IgG1-RPE)(Dako, CA). Specimens were examined using an Olym-pus epifluorescence microscope, equipped with a CCDcamera (Hamamatsu) and Metamorph deconvolutionsoftware (Universal Imaging, West Chester, PA).Counting of CD81 cells was done directly from thesections using the 40X objective.
In vitro cell cultures. Peripheral blood mononuclearcells (PBMCs) (1 3 106 cells/ml) were stimulated bycross-linking of the T cell receptor with immobilized
TABCharacterization
Patient Age Sex
DiseaseOrgan systinvolvemenType Duration (y)
1 69 M Diffuse 2 P2 45 F Diffuse 2 GI3 42 F Limited 4 P/GI4 24 F Diffuse 1 P/GI5 29 F Diffuse 16 62 F Diffuse 1 GI7 23 F Diffuse 18 29 M Diffuse 3 GI9 55 F Diffuse 1.5 P/GI
10 43 M Diffuse 0.5 P/GI11 28 M Limited 0.5 P
a P, pulmonary; GI, gastrointestinal.b Modified Rodnan skin score system (26) (maximum score 51).c Prior treatment with immune active therapy.d Patient 8 remained on CSA and prednisone during the protocol.
TABLE 2Peripheral Blood
Median cell counta (cells/ml)
0 2 4 8 12
Total leucocytes 9450 7850 7200 6650c 6850Neutrophils 6249 5500 4566 3750 4343Lymphocytes 1772 1461 1736 1512 1436Monocytes 606 552 614 584 544Eosinophils 172 214 297 314d 312c
Basophils 61 45 90b 103d 84c
a Calculated by multiplying the total leukocyte count by percent-age from the blood count differential.
Significance vs baseline values: b P 0.0173. c P 0.0357. d P 0.0117.
FIG. 1. The effect of thalidomide on digital ulcers. A depicts thehand of patient 9 at the beginning of thalidomide treatment. Im-paired digital circulation is indicated by blanching of the distalsecond digit. B shows complete healing of the ulcers after 8 weeks ofthalidomide treatment. However, blanching of the distal digits re-
LE 1of Study Patients
emta
Antibody titersBaseline
skin scoreb Prior treatmentcANA Scl 70
1:160 Negative 46 d-pcn/prednisone1:80 Negative 28 d-pcn/prednisone
1:160 Negative 12 None1:2560 Positive 29 Prednisone
1:80 Negative 17 Prednisone1:80 Negative 27 None
Negative Negative 36 Prednisone1:80 Positive 17 CSA/prednisoned
1:640 Negative 26 None1:320 Positive 21 Prednisone/colchicine1:640 Positive 6 None
mains present.

112 OLIVER, MOREIRA, AND KAPLAN

E4(aS
ppdpwS
rd
s
113THALIDOMIDE IMMUNOMODULATION IN SCLERODERMA
monoclonal mouse anti-CD3 (Orthoclone OKT3; a giftof Dr. Robert Zivin, OrthoBiotech Inc., Raritan, NJ) aspreviously described (21). PBMCs were isolated fromblood of scleroderma patients by Ficoll–Hypaque den-sity gradient centrifugation. The recovered leukocyteswere washed three times with cold PBS prior to platingin triplicate in RPMI 1640 media (GIBCO, Grand Is-land, NY) supplemented with 10% AB1 human serum,2 mM L-glutamine, 100 units/ml penicillin, and 100mg/ml streptomycin (GIBCO) in anti-CD3-coated 48-well plates. A stock solution of thalidomide was pre-pared by dissolving the drug in DMSO at a stock con-centration of 20 mg/ml. Cultures were treated dailywith the appropriate dilutions of thalidomide orDMSO. At the indicated times, supernatants were har-vested and frozen immediately at 270°C until assay by
LISA as described above. Cells were harvested at8 h and analyzed with chromophore-labeled mAbCD4-FITC, CD8-FITC, CD154-PE, and CD3-APC) on
FACStar three-color cytometer (Becton–Dickinson,an Jose, CA).
Statistical analysis. This study, with a small sam-le size, was designed as a feasibility study. Individualatient data are graphically presented along with me-ian group values unless otherwise indicated. Non-arametric comparisons of clinical and laboratory dataere performed using SPSS software (version 7.0,PSS Inc., Chicago, IL) employing the Wilcoxon signed
TABLE 3CD81 T Cells in Skin Lesions
Patient Week Epidermisa Subepidermisa,b
5 0 0 1412 1 58
6 0 8 2912 29 180
9 0 0 412 8 88
10 0 2 512 35 95
a Number of cells per 4 mm of epidermis from paraffin-embeddedections.
b Subepidermis is defined as an area starting at the dermal/epi-dermal border and extending into the dermis approximately 400 mm(1 high-powered field diameter).
FIG. 2. The effect of thalidomide on the histopathology of the skshown (A, B, patient 6; C, D, patient 10; E–H, patient 9) before thalcolumn). A and C show a thickened epidermis and scattered mononucompact epidermis after 12 weeks of thalidomide treatment (40X).fibers (10X). Note the presence of mononuclear inflammatory infiltrfibers and scattered cell nuclei (40X). F shows decreased skin fibrosis
In H, note the thinner collagen fibers and increased cellularity (40X obanks test and Mann–Whitney test for paired and in-ependent samples, respectively.
RESULTS
Response to thalidomide and effects on clinical symp-toms. Eleven patients were enrolled into the studyover a 3-year period (Table 1). Nine patients had dif-fuse disease and 2 patients had limited disease at thetime of enrollment. The median age was 42 years, withfemales comprising 64% of the study group. Ten pa-tients had detectable antinuclear antibodies, 4 hadantibodies to topoisomerase-1 (SCL 70), and 1 hadanticentromere antibodies. The median disease dura-tion was 1 year (range 0.5–4 years).
Eight patients completed the 3-month treatmentprotocol. Three patients dropped out of the protocolwithin 1 month of enrollment: two patients (Nos. 4 and7) for non-drug-related reasons and one patient (No 1)for an apparent allergic reaction to thalidomide thatconsisted of abdominal cramping, skin rash, and mark-edly increased levels of circulating and dermal infil-trating eosinophils.
Two specific side effects of thalidomide, pruritus anddry skin, were noted in seven of the eight patients by 4weeks into the study. Nonpitting extremity edema wasseen in seven patients during the protocol period. Thismay have contributed to our finding that skin scores(26) remained unchanged during the 12-week treat-ment period. Macular, erythematous rashes occurredin two patients; one of these had a persistent rashduring weeks 8–12 and the second patient experienceda diffuse rash at week 2, with complete spontaneousresolution within 48 h. One patient noted transientmorning paresthesias in his extremities throughouthis treatment. Symptoms of dysphagia, constipation,and sedation due to thalidomide were minimal andwell tolerated.
Consistent changes in absolute peripheral blood leu-kocyte counts were noted in patients receiving thalid-omide (Table 2). A reduction in the overall leukocytecount due to decreased neutrophils was observed in allpatients while on thalidomide. Eosinophil and basophilcounts increased in all patients, with peak mediancounts observed at week 8. Numbers of lymphocytesand monocytes were unaffected.
Gastroesophageal reflux (GER), as defined by the
The histology of skin biopsies obtained from three SSc patients aremide treatment (left column) and after 12 weeks of treatment (rightr cells infiltrating the superficial dermis (40X). B and D show a morehows atrophy of skin appendages in the dermis and dense collagens around vessels and within collagen fibers. G shows dense collagenh thinner collagen fibers (10X) after 12 weeks of thalidomide therapy.
in.idocleaE satewit
jective).
114 OLIVER, MOREIRA, AND KAPLAN
subjective presence of nocturnal and postprandialheartburn symptoms, improved progressively and re-solved by week 12 in 6/8 patients. No changes or addi-tions were made in previously established anti-reflux
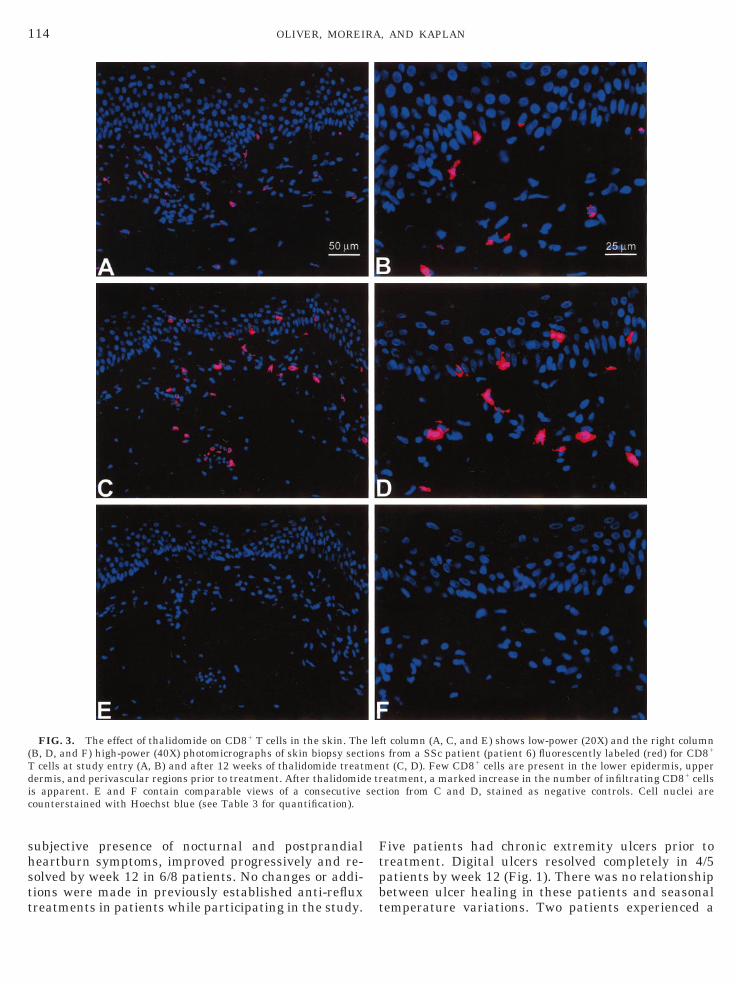
FIG. 3. The effect of thalidomide on CD81 T cells in the skin. The(B, D, and F) high-power (40X) photomicrographs of skin biopsy sectT cells at study entry (A, B) and after 12 weeks of thalidomide treatdermis, and perivascular regions prior to treatment. After thalidomidis apparent. E and F contain comparable views of a consecutivecounterstained with Hoechst blue (see Table 3 for quantification).
treatments in patients while participating in the study.
Five patients had chronic extremity ulcers prior totreatment. Digital ulcers resolved completely in 4/5patients by week 12 (Fig. 1). There was no relationshipbetween ulcer healing in these patients and seasonal
ft column (A, C, and E) shows low-power (20X) and the right columns from a SSc patient (patient 6) fluorescently labeled (red) for CD81
nt (C, D). Few CD81 cells are present in the lower epidermis, uppereatment, a marked increase in the number of infiltrating CD81 cellstion from C and D, stained as negative controls. Cell nuclei are
leionmee trsec
temperature variations. Two patients experienced a

cmC
a
115THALIDOMIDE IMMUNOMODULATION IN SCLERODERMA
return of their digital ulcers within a year of discon-tinuing thalidomide. A third patient remained contin-uously on thalidomide and did not experience furtherulcer formation during 1 year of treatment. A fourthpatient had not experienced further digital ulcers at 4months after having stopped treatment with thalido-mide. In a fifth patient, who had been on a stable doseof CSA during the previous year, complete healing of achronic malleolus ulcer was seen at week 2 of thalido-mide treatment. This patient, who was able to continuehis occupation as a manual laborer, experienced newextremity ulcers while on thalidomide that ultimatelyresponded to antibiotic therapy initiated after comple-tion of the study.
Patient 10 was allowed to continue treatment underour supervision after having completed the 12-weekprotocol period. After 1 year of thalidomide treatment,
FIG. 4. The effect of thalidomide on plasma cytokine levels. Percshown. Individual patient data and median values for each time poin4, bP 0.0251) and IL-12 (weeks 2, 4, *P 0.0117, week 8, aP 0.0499) co
TABLE 4Plasma Cytokine and Soluble Ligand Levels in Normal
Volunteers and Scleroderma Patients at Baseline
Normals Scleroderma P valueb
TNF-a (pg/ml) 8 (4–12)a 22 (1–90) 0.055IL-12 (pg/ml) 111 (63–175) 197 (102–1120) 0.0001IL-5 (pg/ml) 6 (0–140) 9 (0–91) 0.92IL-10 (pg/ml) 10 (4–16) 10 (9–19) 0.44sIL-2R (U/ml) 264 (214–493) 703 (438–2003) 0.049sICAM-1 (ng/ml) 199 (145–303) 466 (213–653) 0.023
a Median (range).b Calculated for the difference between normal individuals and
SSc patients.
ctual plasma levels).
he had experienced changes, including regrowth of hairand softening of the skin on his forearms, suggestingclinical improvement. However, we cannot rule outthat these clinical changes occurred as a natural con-sequence of his disease course.
Changes in skin fibrosis and skin adnexia. To de-termine the effect of thalidomide treatment on thefibrotic skin of SSc patients, punch biopsies of involvedskin at adjacent locations were obtained from four pa-tients at weeks 0 and 12 of the protocol. Histologicsections of pretreatment skin showed the typical find-ings of scleroderma, including a marked increase in thedeposition of compact collagen in the dermis associatedwith atrophy of skin appendages (Fig. 2). Mononuclearinflammatory cell infiltration around blood vessels inthe collagen was noted. Skin biopsies obtained after 12weeks of thalidomide treatment in these individualsconsistently showed loosening of the collagen fibers inthe dermis and decreased thickness of the epidermallayer.
Changes in CD41 and CD81 T cells in the skin.Thalidomide has previously been shown to increase thenumber of CD81 T cells in the circulation of HIV1
individuals (27). Immunohistologic staining of bothparaffin-embedded and frozen (data not shown) tissuebiopsy samples obtained from SSc patients revealedconsistent increases in the numbers of CD81 lympho-ytes in the skin after 12 weeks of thalidomide treat-ent compared to pretreatment biopsies (Table 3). TheD81 lymphocytes were predominantly located in
ages of increase over baseline plasma levels of TNF-a and IL-12 arere presented. Significant increases in plasma levels of TNF-a (weekared to baseline levels were observed (statistical analysis based on
entt amp
m
Tl
ao
116 OLIVER, MOREIRA, AND KAPLAN
perivascular and periadnexal areas of the superficialdermis (Fig. 3). No obvious change in the numbers ofCD41 cells in the dermis was noted following treat-
ent with thalidomide.
Changes in plasma cytokines and soluble ligands.halidomide treatment also affected plasma cytokine
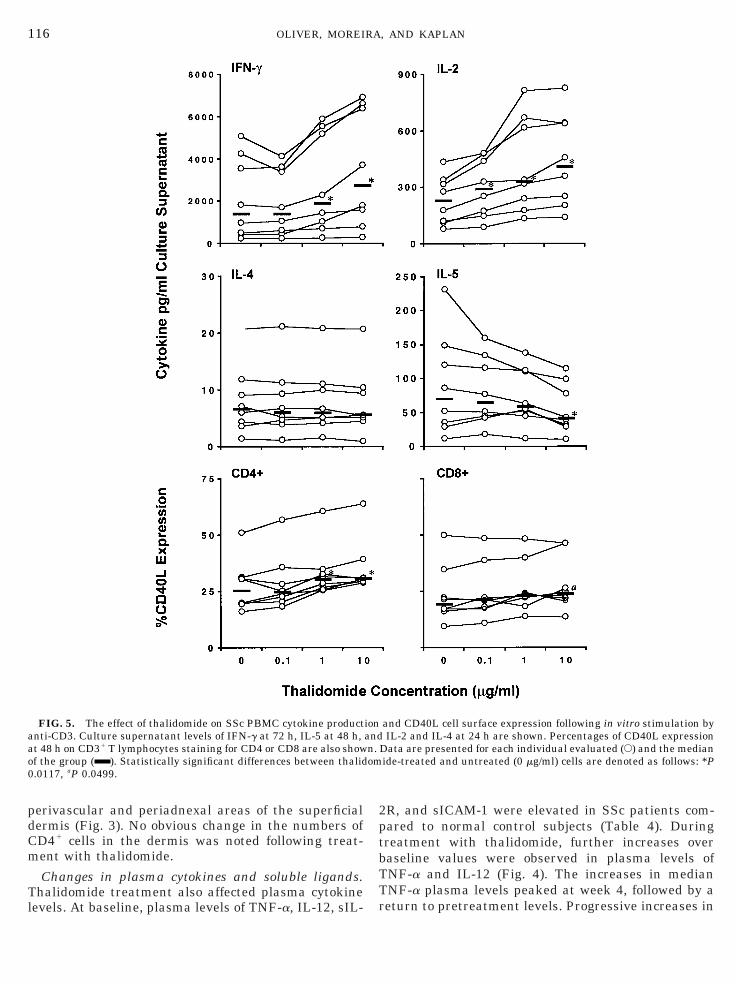
FIG. 5. The effect of thalidomide on SSc PBMC cytokine productanti-CD3. Culture supernatant levels of IFN-g at 72 h, IL-5 at 48 h,t 48 h on CD31 T lymphocytes staining for CD4 or CD8 are also showf the group ( ). Statistically significant differences between thalid
0.0117, aP 0.0499.
evels. At baseline, plasma levels of TNF-a, IL-12, sIL-
2R, and sICAM-1 were elevated in SSc patients com-pared to normal control subjects (Table 4). Duringtreatment with thalidomide, further increases overbaseline values were observed in plasma levels ofTNF-a and IL-12 (Fig. 4). The increases in medianTNF-a plasma levels peaked at week 4, followed by a
and CD40L cell surface expression following in vitro stimulation byIL-2 and IL-4 at 24 h are shown. Percentages of CD40L expression
Data are presented for each individual evaluated (E) and the medianide-treated and untreated (0 mg/ml) cells are denoted as follows: *P
ionandn.om
return to pretreatment levels. Progressive increases in

a
s(
117THALIDOMIDE IMMUNOMODULATION IN SCLERODERMA
median plasma IL-12 levels peaked at week 8. Plasmalevels of IL-5 and IL-10 were low at baseline in SScpatients and were comparable to levels found in thenormal control group. No changes in the levels ofplasma IL-10 or sIL-2R occurred during thalidomidetreatment, while sICAM-1 levels exhibited a slightdownward trend with treatment (data not shown). Nei-ther IL-4 nor IFN-g was detectable in plasma fromnormal controls or from SSc patients.
In vitro responses of PBMCs. To determine whetherthalidomide exerted a direct immunostimulatory effecton SSc T cells, PBMCs were obtained from SSc patientswho were not receiving thalidomide. When PBMCsfrom these untreated individuals were activated invitro with anti-CD3 mAb, IL-2, IL-4, IL-5, and IFN-gwere detectable in the supernatants (Fig. 5). The addi-tion of thalidomide to these cultures did not affect thelevels of IL-4 detected in supernatants. However, IL-2and IFN-g levels were significantly enhanced in a dose-dependent manner in response to thalidomide. CD40ligand (CD40L; CD154), expressed on activated T cells,is a costimulatory molecule that is a critical componentin many aspects of the immune response, includingT-cell-dependent activation of antigen-presenting cells(APCs) and the subsequent production of IL-12 by theAPCs. We therefore examined the in vitro effect ofthalidomide on T cell expression of CD40L (Fig. 5).When T cells purified from the peripheral blood of SScpatients were stimulated with anti-CD3 mAb in thepresence of thalidomide, we observed a significant in-crease in the surface expression of CD40L on CD41 andCD81 T cells. Similar increases in cytokine production
FIG. 6. The effect of thalidomide on IL-3 and GM-CSF produupernatant levels of GM-CSF at 72 h and IL-3 at 24 h are shown. D
). Statistically significant differences between thalidomide-treat0.0173.
nd CD40L expression were seen in thalidomide-
treated PBMCs obtained from normal volunteers (datanot shown).
Changes in hematopoietic factors. To address theobserved increases in circulating eosinophils and ba-sophils in these patients during thalidomide treat-ment, we also measured hematopoietic factors associ-ated with these leukocyte subsets. When PBMCs wereactivated by immobilized anti-CD3 mAbs, dose-depen-dent increases were observed in supernatant levels ofboth GM-CSF and IL-3 (Fig. 6), while a decrease oc-curred in IL-5 levels (Fig. 5). IL-5 plasma levels in SScpatients did not show any appreciable change in re-sponse to thalidomide treatment (data not shown). Nei-ther IL-3 nor GM-CSF was detectable by ELISA inplasma from patients or normal volunteers.
DISCUSSION
The lack of any effective treatment for the progres-sive autoimmune disease SSc underscores the impor-tance of understanding the disease pathogenesis andidentifying new therapeutic interventions. Our obser-vations, although preliminary and derived from asmall group of patients, suggest that immune activa-tion with the drug thalidomide may provide a newtreatment option. However, a larger, placebo-con-trolled study of longer duration will be necessary todetermine whether the drug has clinical efficacy inSSc. With the exception of one patient who had anapparent allergic reaction to the drug, thalidomide waswell tolerated in this group without evidence of SSc
n by SSc PBMCs stimulated in vitro by anti-CD3 mAb. Culturea are presented for each individual (E) and the median of the groupand untreated (0 mg/ml) cells are denoted as follows: *P 0.0117, bP
ctioat
ed
disease exacerbation.

imoCt
pcmsld
va
pmvbioI
118 OLIVER, MOREIRA, AND KAPLAN
A striking clinical effect observed in these patientswas the healing of digital and other extremity ulcerswhile being treated with thalidomide. Surprisingly,signs and symptoms of digital vasospasm in these pa-tients did not change during the 3-month course oftreatment, suggesting that improved cutaneous bloodcirculation was not responsible for these clinical bene-fits. However, improved ulcer healing by thalidomidein other disease processes, including pyoderma gangre-nosum (28–30) and aphthous ulcers (31, 32), suggeststhat healing of SSc digital ulcers may not be dependenton changes in blood circulation. Recently, thalidomidewas shown to increase both proliferation and migrationof keratinocytes in vitro (33), which may contribute toits effects on ulcer healing. In addition, the thalido-mide-associated increases in GM-CSF observed in vitro(Fig. 6) in PBMCs of SSc patients provide a possiblemechanism for the enhanced ulcer healing observed.Our laboratory has previously shown that GM-CSFstimulates keratinocyte proliferation both in vitro andin vivo and enhances wound healing in lepromatousleprosy patients (34–36). More recently, topical GM-CSF has been reported effective in healing chronicvenous ulcers (37) as well as the healing of aphthousulcers in HIV1 patients (38). The thalidomide-inducedincrease in PBMC production of GM-CSF also providesa possible explanation for the thalidomide-associatedincreases in peripheral circulating eosinophils; a leu-kocyte subset whose specific growth factors includeGM-CSF. The relationship between thalidomide andGM-CSF will be explored in further studies.
The immune stimulatory effect of thalidomide ob-served in this study is noteworthy. In these patients,we observed that thalidomide induces an increase inplasma IL-12 and TNF-a levels. The observation ofmmune stimulation in vivo was confirmed by experi-
ents in vitro, in which SSc PBMCs exposed to thalid-mide and anti-CD3 mAb showed upregulation ofD40L expression and release of increased amounts of
he Th1-type cytokines IL-2 and IFN-g.IL-12 is an important immunoregulatory cytokine
roduced by APCs , including macrophages, dendriticells, and B cells (reviewed in 39). IL-12 production byacrophages can be triggered both directly via LPS
timulation and indirectly by activated T cells throughigation of CD40 (40). This cytokine is a powerful in-ucer of IFN-g production by T and NK cells, leading to
the development of Th1-type immune responses impor-tant for protection against intracellular pathogens,such as mycobacteria and viruses. IL-12 has also beenimplicated in animal models of autoimmune disease. Ina murine model, administration of IL-12 has beenshown to inhibit acute GVHD and delay mortality ofchronic GVHD (41). Unexpected benefits from IL-12treatment were also observed in a rat model of T-cell-
mediated uveitis (42). Taken together, these observa-tions suggest that in the SSc patients on thalidomide,increased IL-12 production may have contributed tothe clinical effects noted.
Our present results suggest that skin differentia-tion, including collagen biosynthesis and the organiza-tion of dermal adnexia, may be affected by systemicimmune activation and cytokine production. Indeed, ithas been shown that cytokine production following se-lective activation of either Th1 or Th2 T cell subsetscan exert opposing effects on collagen synthesis bydermal fibroblasts. IL-4, produced by Th2-type lym-phocytes, is an activator of collagen production (43).Also, anti-IL-4 treatment in the “tight skin” mousemodel of scleroderma has been shown to prevent der-mal collagen deposition (44). In contrast, IFN-g is apotent inhibitor of collagen biosynthesis (45, 46) andhas been shown to completely suppress IL-4-inducedcollagen gene expression by both normal and SSc der-mal fibroblasts (47). In SSc patients, a selective defi-ciency in IFN-g production has been observed in cul-tured dermal (6) and blood (3) lymphocytes. Clinicaltrials using IFN-g treatment in SSc patients haveshown mild beneficial effects on skin sclerosis param-eters (48, 49). In the present study, the thalidomide-induced increases in IL-12 levels may up-regulateIFN-g production by T cells in the skin, as suggested byin vitro studies, potentially leading to a reduction incollagen biosynthesis. This possibility will be examinedin future studies.
In previous studies, we had focused on the ability ofthalidomide to inhibit TNF-a production in vivo and initro (19, 20). More recently, we found that thalidomideffects TNF-a in a differential manner. The drug in-
hibits TNF-a production by monocytes but does notinhibit TNF-a production by T cells (19, 21, 22). It ispossible that the TNF-a observed in the plasma ofthese patients is produced by T cells and therefore notblocked by thalidomide. Alternatively, T cell activationduring thalidomide treatment may have stimulatedincreased TNF-a production. We have also investi-gated the effect of the drug on IL-12 production. Tha-lidomide was shown to inhibit IL-12 production bymonocytes stimulated in vitro by LPS (a T-cell-inde-
endent stimulus), but to increase IL-12 production byonocytes in vitro in the presence of T cells activated
ia the T cell receptor (22). Furthermore, these effectsy thalidomide on T cells could be blocked by antibod-es to IL-2 but not by antibodies to IL-4. The presentbservation that thalidomide induces an increase inL-12 and TNF-a in vivo suggests that the drug may be
preferentially affecting T cell activation and T-cell-dependent cytokine production in SSc patients.
This is the first study in which the clinical and im-mune effects of thalidomide have been reported in SScpatients. However, these clinical results are primarily
descriptive and are derived from a small group of pa-
1
1
1
2
2
2
119THALIDOMIDE IMMUNOMODULATION IN SCLERODERMA
tients treated for a short period of time. The in vivo andin vitro results suggest that immune stimulation bythalidomide may be a potential therapeutic interven-tion in SSc. Longer term, placebo-controlled studieswill be necessary to show whether thalidomide-inducedimmune changes can lead to sustained beneficial clin-ical effects in SSc patients and others with relatedautoimmune disorders.
ACKNOWLEDGMENTS
We thank P. Clements for helpful discussions, K. Mahnke forassistance in immunohistology preparation and interpretation, P.Haslett for critical review of the manuscript, V. Freedman for assis-tance in manuscript preparation, and J. Adams for help in figurepreparation. This work was supported in part by Celgene Corp.(Warren, NJ), NIH Grant AI22626, Direct Effect AIDS ResearchFund (New York, NY), and the United Scleroderma Foundation.S.J.O. was supported by General Clinical Research Center GrantM01-RR001012 from the National Center for Research Resources atthe National Institutes of Health.
REFERENCES
1. van den Hoogen, F. H., and de Jong, E. M., Clinical aspects ofsystemic and localized scleroderma. Curr. Opin. Rheumatol. 7,546–550, 1995.
2. White, B., Immunopathogenesis of systemic sclerosis. Rheum.Dis. Clin. North Am. 22, 695–708, 1996.
3. Kantor, T. V., Friberg, D., Medsger, T. A., Jr., Buckingham,R. B., and Whiteside, T. L., Cytokine production and serumlevels in systemic sclerosis. Clin. Immunol. Immunopathol. 65,278–285, 1992.
4. Needleman, B. W., Wigley, F. M., and Stair, R. W., Interleukin-1,interleukin-2, interleukin-4, interleukin-6, tumor necrosis factoralpha, and interferon-gamma levels in sera from patients withscleroderma. Arthritis Rheum. 35, 67–72, 1992.
5. Prior, C., and Haslam, P. L., In vivo levels and in vitro produc-tion of interferon-gamma in fibrosing interstitial lung diseases.Clin. Exp. Immunol. 88, 280–287, 1992.
6. Mavalia, C., Scaletti, C., and Romagnani, P., et al., Type 2 helperT-cell predominance and high CD30 expression in systemic scle-rosis. Am. J. Pathol. 151, 1751–1758, 1997.
7. Prescott, R. J., Freemont, A. J., Jones, C. J., Hoyland, J., andFielding, P., Sequential dermal microvascular and perivascularchanges in the development of scleroderma. J. Pathol. 166, 255–263, 1992.
8. Roumm, A. D., Whiteside, T. L., Medsger, T. A., Jr., and Rodnan,G. P., Lymphocytes in the skin of patients with progressivesystemic sclerosis. Quantification, subtyping, and clinical corre-lations. Arthritis Rheum. 27, 645–653, 1984.
9. Famularo, G., Procopio, A., and Giacomelli, R., et al., Solubleinterleukin-2 receptor, interleukin-2 and interleukin-4 in seraand supernatants from patients with progressive systemic scle-rosis. Clin. Exp. Immunol. 81, 368–372, 1990.
10. Hasegawa, M., Fujimoto, M., Kikuchi, K., and Takehara, K.,Elevated serum levels of interleukin 4 (IL-4), IL-10, and IL-13 inpatients with systemic sclerosis. J. Rheumatol. 24, 328–332,1997.
1. Sfikakis, P. P., Tesar, J., Baraf, H., Lipnick, R., Klipple, G., andTsokos, G. C., Circulating intercellular adhesion molecule-1 inpatients with systemic sclerosis. Clin. Immunol. Immunopathol.
68, 88–92, 1993.2. Graham-Brown, R. A., and Sarkany, I., Scleroderma-likechanges due to chronic graft-versus-host disease. Clin. Exp. Der-matol. 8, 531–538, 1983.
3. Lawley, T. J., Peck, G. L., Moutsopoulos, H. M., Gratwohl, A. A.,and Deisseroth, A. B., Scleroderma, Sjogren-like syndrome, andchronic graft-versus-host disease. Ann. Intern. Med. 87, 707–709, 1977.
14. Nelson, J. L., Microchimerism and the pathogenesis of systemicsclerosis. Curr. Opin. Rheumatol. 10, 564–571, 1998.
15. Hakim, F. T., and Mackall, C. L., The immune system: effectorand target of graft-versus-host disease. In “Graft-vs-Host Diseases”(J. L. Ferrara et al., Eds.), 2nd ed., Dekker, New York, 1997).
16. Vogelsang, G. B., Farmer, E. R., and Hess, A. D., et al., Thalid-omide for the treatment of chronic graft-versus-host disease.N. Engl. J. Med. 326, 1055–1058, 1992.
17. Vogelsang, G. B., Hess, A. D., Gordon, G., and Santos, G. W.,Treatment and prevention of acute graft-versus-host diseasewith thalidomide in a rat model. Transplantation 41, 644–647,1986.
18. Vogelsang, G. B., Hess, A. D., Friedman, K. J., and Santos,G. W., Therapy of chronic graft-v-host disease in a rat model.Blood 74, 507–511, 1989.
19. Moreira, A. L., Sampaio, E. P., Zmuidzinas, A., Frindt, P., Smith,K. A., and Kaplan, G., Thalidomide exerts its inhibitory action ontumor necrosis factor alpha by enhancing mRNA degradation. J.Exp. Med. 177, 1675–1680, 1993.
20. Corral, L. G., Muller, G. W., and Moreira, A. L., et al., Selectionof novel analogs of thalidomide with enhanced tumor necrosisfactor alpha inhibitory activity. Mol. Med. 2, 506–515, 1996.
21. Haslett, P. A., Corral, L. G., Albert, M., and Kaplan, G., Thalid-omide costimulates primary human T lymphocytes, preferen-tially inducing proliferation, cytokine production, and cytotoxicresponses in the CD81 subset. J. Exp. Med. 187, 1885–1892,1998.
2. Corral, L. G., Haslett, P. A. J., and Muller, G. W., et al., Differ-ential cytokine modulation and T cell activation by two distinctclasses of thalidomide analogues that are potent inhibitors ofTNF-alpha. J. Immunol. 163, 380–386, 1999.
3. Preliminary criteria for the classification of systemic sclerosis(scleroderma). Subcommittee for scleroderma criteria of theAmerican Rheumatism Association Diagnostic and TherapeuticCriteria Committee. Arthritis Rheum. 23, 581–590, 1980.
4. White, B., Bauer, E. A., and Goldsmith, L. A., et al., Guidelinesfor clinical trials in systemic sclerosis (scleroderma). I. Disease-modifying interventions. The American College of RheumatologyCommittee on Design and Outcomes in Clinical Trials in Sys-temic Sclerosis. Arthritis Rheum. 38, 351–360, 1995.
25. Jacobson, R. R., In “Leprosy” (R. C. Hastings, Ed.), pp. 193–222,Churchill Livingston, Edinburgh, 1985.
26. Clements, P., Lachenbruch, P., and Siebold, J., et al., Inter andintraobserver variability of total skin thickness score (modifiedRodnan TSS) in systemic sclerosis. J. Rheumatol. 22, 1281–1285, 1995.
27. Haslett, P., Hempstead, M., and Seidman, C., et al., The meta-bolic and immunologic effects of short-term thalidomide treat-ment of patients infected with the human immunodeficiencyvirus. AIDS Res. Hum. Retroviruses 13, 1047–1054, 1997.
28. Hecker, M. S., and Lebwohl, M. G., Recalcitrant pyoderma gan-grenosum: treatment with thalidomide. J. Am. Acad. Dermatol.38, 490–491, 1998.
29. Rustin, M. H., Gilkes, J. J., and Robinson, T. W., Pyodermagangrenosum associated with Behcet’s disease: treatment with
thalidomide. J. Am. Acad. Dermatol. 23, 941–944, 1990.
3
3
3
3
3
3
3
3
4
4
4
4
4
4
4
4
4
4
R
120 OLIVER, MOREIRA, AND KAPLAN
30. Munro, C. S., and Cox, N. H., Pyoderma gangrenosum associatedwith Behcet’s syndrome—response to thalidomide. Clin. Exp.Dermatol. 13, 408–410, 1988.
1. Hamuryudan, V., Mat, C., and Saip, S., et al., Thalidomide in thetreatment of the mucocutaneous lesions of the Behcet syndrome.A randomized, double-blind, placebo-controlled trial. Ann. In-tern. Med. 128, 443–450, 1998.
2. Jacobson, J. M., Greenspan, J. S., and Spritzler, J., et al., Tha-lidomide for the treatment of oral aphthous ulcers in patientswith human immunodeficiency virus infection. National Insti-tute of Allergy and Infectious Diseases AIDS Clinical TrialsGroup. N. Engl. J. Med. 336, 1487–1493, 1997.
33. Nasca, M. R., O’Toole, E. A., Palicharla, P., West, D. P., andWoodley, D. T., Thalidomide increases human keratinocyte mi-gration and proliferation. J. Invest. Dermatol. 113, 720–724,1999.
4. Hancock, G. E., Kaplan, G., and Cohn, Z. A., Keratinocytegrowth regulation by the products of immune cells. J. Exp. Med.168, 1395–1402, 1988.
5. Kaplan, G., Walsh, G., and Guido, L. S., et al., Novel responses ofhuman skin to intradermal recombinant granulocyte/macro-phage-colony-stimulating factor: Langerhans cell recruitment,keratinocyte growth, and enhanced wound healing. J. Exp. Med.175, 1717–1728, 1992.
6. Braunstein, S., Kaplan, G., and Gottlieb, A. B., et al., GM-CSFactivates regenerative epidermal growth and stimulates kera-tinocyte proliferation in human skin in vivo. J. Invest. Dermatol.103, 601–604, 1994.
7. Da Costa, R. M., Ribeiro Jesus, F. M., Aniceto, C., and Mendes,M., Randomized, double-blind, placebo-controlled, dose- rangingstudy of granulocyte-macrophage colony stimulating factor inpatients with chronic venous leg ulcers. Wound Repair Regen. 7,17–25, 1999.
8. Herranz, P., Arribas, J. R., and Navarro, A., et al., Successfultreatment of aphthous ulcerations in AIDS patients using topicalgranulocyte-macrophage colony-stimulating factor. Br. J. Der-matol. 142, 171–176, 2000.
9. Trinchieri, G., Interleukin-12: A proinflammatory cytokine with
immunoregulatory functions that bridge innate resistance andantigen-specific adaptive immunity. Annu. Rev. Immunol. 13,251–276, 1995.
0. DeKruyff, R. H., Gieni, R. S., and Umetsu, D. T., Antigen-drivenbut not lipopolysaccharide-driven IL-12 production in macro-phages requires triggering of CD40. J. Immunol. 158, 359–366,1997.
1. Sykes, M., Szot, G. L., Nguyen, P. L., and Pearson, D. A., Inter-leukin-12 inhibits murine graft-versus-host disease. Blood 86,2429–2438, 1995.
2. Tarrant, T. K., Silver, P. B., and Wahlsten, J. L., et al., Inter-leukin 12 protects from a T helper type 1-mediated autoimmunedisease, experimental autoimmune uveitis, through a mecha-nism involving interferon gamma, nitric oxide, and apoptosis. J.Exp. Med. 189, 219–230, 1999.
3. Fertin, C., Nicolas, J. F., Gillery, P., Kalis, B., Banchereau, J.,and Maquart, F. X., Interleukin-4 stimulates collagen synthesisby normal and scleroderma fibroblasts in dermal equivalents.Cell. Mol. Biol. 37, 823–829, 1991.
4. Ong, C., Wong, C., Roberts, C. R., Teh, H. S., and Jirik, F. R.,Anti-IL-4 treatment prevents dermal collagen deposition in thetight-skin mouse model of scleroderma. Eur. J. Immunol. 28,2619–2629, 1998.
5. Duncan, M. R., and Berman, B., Gamma interferon is the lym-phokine and beta interferon the monokine responsible for inhi-bition of fibroblast collagen production and late but not earlyfibroblast proliferation. J. Exp. Med. 162, 516–527, 1985.
6. Jimenez, S. A., Freundlich, B., and Rosenbloom, J., Selectiveinhibition of human diploid fibroblast collagen synthesis by in-terferons. J. Clin. Invest. 74, 1112–1116, 1984.
7. Serpier, H., Gillery, P., and Salmon-Ehr, V., et al., Antagonisticeffects of interferon-gamma and interleukin-4 on fibroblast cul-tures. J. Invest. Dermatol. 109, 158–162, 1997.
8. Varga, J., Recombinant cytokine treatment for scleroderma. Canthe antifibrotic potential of interferon-gamma be realized clini-cally? Arch. Dermatol. 133, 637–642, 1997.
9. Grassegger, A., Schuler, G., and Hessenberger, G., et al., Inter-feron-gamma in the treatment of systemic sclerosis: A random-ized controlled multicentre trial. Br. J. Dermatol. 139, 639–648,
1998.eceived February 28, 2000; accepted with revision July 24, 2000




















