
Identification of RNA ligands that bind hepatitis C virus polymerase selectively and inhibit its RNA...
16
Identification of RNA ligands that bind hepatitis C virus polymerase selectively and inhibit its RNA synthesis from the natural viral RNA templates Nam Viet Vo, a Jong-Won Oh, a,b,1 and Michael M.C. Lai a,b, * a Department of Molecular Microbiology and Immunology, University of Southern California, Keck School of Medicine, Los Angeles, CA 90033-1054, USA b Howard Hughes Medical Institute, University of Southern California, Keck School of Medicine, Los Angeles, CA 90033-1054, USA Received 15 August 2002; returned to author for revision 4 October 2002; accepted 6 November 2002 Abstract To identify the potential RNA inhibitors of HCV polymerase, we have isolated high-affinity RNA ligands specific to hepatitis C virus (HCV) NS5B protein from a combinatorial RNA library using the Systematic Evolution of Ligands by EXponential enrichment (SELEX) procedure. Thirty-seven selected ligands were classified into eight groups on the basis of their sequence homologies. Most (60%) of the ligands carry the conserved YGUAGR hexamer (Y pyrimidine, R purine) at the 5 end of the 40-nt randomized region, and 74% of the ligands end in (A/C)U at the 3end. However, strong binding to NS5B required the whole RNA ligand including the flanking conserved nucleotides at both ends. The binding of the selected ligands to NS5B is highly specific and strong, as reflected in their low dissociation rate constants (k d 10 4 s 1 ). Analysis of secondary structure by computer program and RNase footprints of the two different aptamers from two most conserved groups revealed RNA structures containing three stem loops with internal bulges. NS5B bound these RNA at a region between the two stem loops from the 5 -end. Some of these RNA aptamers could serve as a template for the HCV polymerase, but some interfered with the activity of the viral enzyme. These RNA ligands will be useful for further characterization of NS5B-binding properties and, with further modifications, may have potential therapeutic value. © 2003 Elsevier Science (USA). All rights reserved. Introduction Hepatitis C virus (HCV) is a major human pathogen afflicting nearly 1–2% of the world population, causing both acute and chronic hepatitis, which often leads to liver cir- rhosis and hepatocellular carcinoma. HCV is a member of the Flaviviridae family and possesses a ()-strand RNA genome of about 9.5 kb in length that contains a single open reading frame (ORF) encoding a polyprotein of 3000 amino acids (De Francesco, 1999; Grakoui et al., 1993). The viral polyprotein is processed by cellular and viral proteases into 10 structural and nonstructural proteins (De Francesco, 1999; Grakoui et al., 1993). Among these, NS5B is a 66- kDa membrane-associated viral nonstructural protein (Hwang et al., 1997; Schmidt-Mende et al., 2001), derived from the C-terminal end of the polyprotein. NS5B was initially identified as an RNA-dependent RNA polymerase (RdRp) by the presence of the hallmark GDD sequence, a motif involved in binding the Mg 2 ions essential for all polymerase functions (Koonin, 1991; O’Reilly and Kao, 1998). More direct evidence supporting NS5B RdRp enzy- matic activity came from in vitro studies using purified recombinant NS5B expressed in insect and bacterial cells (Behrens et al., 1996; Lohmann et al., 1997; Oh et al., 1999). Thus, NS5B is the key replicative enzyme of the HCV genetic material. X-ray crystallographic structure of NS5B reveals the canonical right-handed-like structure with domains representing fingers, palm, and thumb as seen in other polymerases (Bressanelli et al., 1999; Lesburg et al., * Corresponding author. Molecular Microbiology and Immunology, University of Southern California, Investigator, Howard Hughes Medical Institute, 2011 Zonal Avenue, HMR-401, Los Angeles, California 90033- 1054. Fax: 1-323-442-1721. E-mail address: [email protected] (M.M.C. Lai). 1 Present address: Department of Biotechnology, Yonsei University, 134 Shinchon-Dong, Sodaemoon-Ku, Seoul, 120-749, Korea. R Available online at www.sciencedirect.com Virology 307 (2003) 301–316 www.elsevier.com/locate/yviro 0042-6822/03/$ – see front matter © 2003 Elsevier Science (USA). All rights reserved. doi:10.1016/S0042-6822(02)00095-8
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Identification of RNA ligands that bind hepatitis C virus polymerase selectively and inhibit its RNA...

Identification of RNA ligands that bind hepatitis C virus polymeraseselectively and inhibit its RNA synthesis from the natural
viral RNA templates
Nam Viet Vo,a Jong-Won Oh,a,b,1 and Michael M.C. Laia,b,*a Department of Molecular Microbiology and Immunology, University of Southern California, Keck School of Medicine, Los Angeles, CA 90033-1054, USA
b Howard Hughes Medical Institute, University of Southern California, Keck School of Medicine, Los Angeles, CA 90033-1054, USA
Received 15 August 2002; returned to author for revision 4 October 2002; accepted 6 November 2002
Abstract
To identify the potential RNA inhibitors of HCV polymerase, we have isolated high-affinity RNA ligands specific to hepatitis C virus(HCV) NS5B protein from a combinatorial RNA library using the Systematic Evolution of Ligands by EXponential enrichment (SELEX)procedure. Thirty-seven selected ligands were classified into eight groups on the basis of their sequence homologies. Most (60%) of theligands carry the conserved YGUAGR hexamer (Y � pyrimidine, R � purine) at the 5� end of the 40-nt randomized region, and 74% ofthe ligands end in (A/C)U at the 3�end. However, strong binding to NS5B required the whole RNA ligand including the flanking conservednucleotides at both ends. The binding of the selected ligands to NS5B is highly specific and strong, as reflected in their low dissociationrate constants (kd � 10�4 s�1). Analysis of secondary structure by computer program and RNase footprints of the two different aptamersfrom two most conserved groups revealed RNA structures containing three stem loops with internal bulges. NS5B bound these RNA at aregion between the two stem loops from the 5� -end. Some of these RNA aptamers could serve as a template for the HCV polymerase, butsome interfered with the activity of the viral enzyme. These RNA ligands will be useful for further characterization of NS5B-bindingproperties and, with further modifications, may have potential therapeutic value.© 2003 Elsevier Science (USA). All rights reserved.
Introduction
Hepatitis C virus (HCV) is a major human pathogenafflicting nearly 1–2% of the world population, causing bothacute and chronic hepatitis, which often leads to liver cir-rhosis and hepatocellular carcinoma. HCV is a member ofthe Flaviviridae family and possesses a (�)-strand RNAgenome of about 9.5 kb in length that contains a single openreading frame (ORF) encoding a polyprotein of �3000amino acids (De Francesco, 1999; Grakoui et al., 1993). Theviral polyprotein is processed by cellular and viral proteases
into 10 structural and nonstructural proteins (De Francesco,1999; Grakoui et al., 1993). Among these, NS5B is a 66-kDa membrane-associated viral nonstructural protein(Hwang et al., 1997; Schmidt-Mende et al., 2001), derivedfrom the C-terminal end of the polyprotein. NS5B wasinitially identified as an RNA-dependent RNA polymerase(RdRp) by the presence of the hallmark GDD sequence, amotif involved in binding the Mg2� ions essential for allpolymerase functions (Koonin, 1991; O’Reilly and Kao,1998). More direct evidence supporting NS5B RdRp enzy-matic activity came from in vitro studies using purifiedrecombinant NS5B expressed in insect and bacterial cells(Behrens et al., 1996; Lohmann et al., 1997; Oh et al.,1999). Thus, NS5B is the key replicative enzyme of theHCV genetic material. X-ray crystallographic structure ofNS5B reveals the canonical right-handed-like structure withdomains representing fingers, palm, and thumb as seen inother polymerases (Bressanelli et al., 1999; Lesburg et al.,
* Corresponding author. Molecular Microbiology and Immunology,University of Southern California, Investigator, Howard Hughes MedicalInstitute, 2011 Zonal Avenue, HMR-401, Los Angeles, California 90033-1054. Fax: �1-323-442-1721.
E-mail address: [email protected] (M.M.C. Lai).1 Present address: Department of Biotechnology, Yonsei University,
134 Shinchon-Dong, Sodaemoon-Ku, Seoul, 120-749, Korea.
R
Available online at www.sciencedirect.com
Virology 307 (2003) 301–316 www.elsevier.com/locate/yviro
0042-6822/03/$ – see front matter © 2003 Elsevier Science (USA). All rights reserved.doi:10.1016/S0042-6822(02)00095-8

1999). However, NS5B possesses two unusual features.One is the extensive interaction between the finger andthumb domains, resulting in the O structure rather than theU structure observed for other polymerases (Lesburg et al.,1999). The only other known polymerase with thisO-shaped architecture is the �6 viral RdRp (Butcher et al.,2001). The other novel structural feature of NS5B is a�-hairpin motif protruding from the thumb domain towardthe active site that locates at the base of the palm domain.This structure has been shown to modulate NS5B initiationsite selection in RNA synthesis (Hong et al., 2001).
Specific template recognition by replicase is one of theessential requirements for faithful genome replication. Formost RNA viruses, the mechanisms of binding specificity ofpolymerases and the initiation of RNA synthesis remainpoorly understood. HCV genomic RNA contains two highlyconserved untranslated regions (UTRs) at its 5� and 3� ends.The 5�UTR of about 341 nt contains an internal ribosomalentry site (IRES), which consists of four stem-loop struc-tures followed by a translational initiation codon (Brown etal., 1992; Wang et al., 1994). The 3�UTR varies from 200 to300 nt in length, which consists of three subregions, a shortvariable sequence of approximately 40 nt, a poly(U-U/C)sequence containing a variable poly(U) run and a polypyri-midine poly(U/C) tract, and a highly conserved 98-nt Xregion consisting of three stable stem-loops (Kolykhalov etal., 1996; Tanaka et al., 1996; Yamada et al., 1996). Infec-tivity assays in chimpanzee have shown that the X regionand the U-U/C-rich region are required for viral infectivity,indicating that these sequence elements contain importantcis-acting signals for replication (Yanagi et al., 1999).
Purified HCV NS5B has been shown to interact with the3�UTR by binding to both the poly (U-U/C)-rich and the Xregions (Oh et al., 2000). UV cross-linking and competitionstudies by Cheng and co-workers, however, showed thatNS5B binds with a higher affinity to a 3�-coding region ofNS5B gene, which contains a stem-loop structure, than tothe 3�UTR or the 5�UTR (Cheng et al., 1999). With un-structured RNA, NS5B binds more tightly to poly(U) andpoly(G) over poly(A) or poly(C) (Lohmann et al., 1997). Sofar, NS5B by itself exhibits little binding specificity to thevarious RNA templates examined. Consistent with this ob-servation, NS5B appears to carry out RNA synthesis usingvarious structured and unstructured RNA from both HCVand non-HCV RNA sources (Hagedorn et al., 2000; Loh-mann et al., 1998; Oh et al., 1999; Zhong et al., 2000).
To gain an understanding of how NS5B interacts withRNA, we decided to examine the interaction and bindingspecificity of NS5B to certain RNA structures and identifythe essential structural features of such RNA sites. To thisend, we used the Systematic Evolution of Ligands by EX-ponential enrichment (SELEX) procedure to isolate high-affinity RNA ligands from a randomized RNA repertoire byHCV NS5B. In this article, we report the identification ofseveral groups of RNAs to which NS5B binds tightly andselectively, and we demonstrate how this binding inhibits
the ability of NS5B to carry out RNA synthesis on thenatural HCV minimal RNA templates. A similar study wasrecently reported (Biroccio et al., 2002); however, the struc-tural requirement of NS5B–RNA binding was significantlydifferent. Thus, NS5B appears to have considerable latitudein its RNA-binding specificity. These SELEX RNAs willprovide useful tools for studying the molecular mechanismof the interactions between NS5B and RNA.
Results
Rationale
The objective of these studies was to explore whether theHCV NS5B enzyme is capable of binding selectively to aparticular RNA sequence or structure and to determine theessential features of such an RNA motif. To address thesequestions, we employed the SELEX method (Gold et al.,1995) to screen for RNA structures that possess the opti-mized binding surfaces for NS5B from a large library ofrandom RNA sequences. Successful isolation of high-affin-ity RNA molecules would provide useful templates forfurther binding and enzymatic characterization of NS5B, aswell as potential therapeutic application if these RNAs in-hibit NS5B RdRp activity. The SELEX cycle in these stud-ies consisted of three major steps. First, NS5B was allowedto interact with a pool of randomized RNAs, and the boundRNAs were partitioned from the unbound RNA. Then, therecovered bound RNAs were converted by RT-PCR intoDNA containing the bacteriophage T7 promoter. Finally,RNA was transcribed from the PCR DNA to give an am-plified pool of a subset of RNAs for the next SELEX cycle.
A random RNA library in these studies was made asoutlined in Fig. 1. A pool of randomized DNA products wasfirst prepared from a PCR reaction using three oligonucle-otide primers, one of which (oligo 2a) contains a degenerateregion of 40 nucleotides. The resulting PCR DNA consistsof a bacteriophage T7 promoter fused to an 87-bp tran-scribed region. A library of RNA was then prepared bytranscription under extensive synthesis condition using T7RNA polymerase (Chamberlin et al., 1983). The 87-ntSELEX RNA molecules comprises three regions, CR1,CR2, and VR. CR1 and CR2 (constant region) are 23 and 24nt in length, respectively, and they flank the VR (variableregion) of 40 nt. For optimal RNA synthesis, CR1 wasdesigned to carry at its 5� end a 5-nt conserved initialtranscribing sequence of the T7 promoter. CR2 contains aBamHI restriction site at the 3� end and a SphI restrictionsite at the 5� end. The BamHI site was to be used forsubsequent cloning of the PCR product into a plasmidvector. For the purpose of future SELEX studies, the SphIsite was introduced so that the constant CR2 sequence canbe removed from the RNA library to generate differentsequences at the 3� end. The choice of the sequences in bothCR1 and CR2 was also based on their success as RT-PCR
302 N.V. Vo et al. / Virology 307 (2003) 301–316

primers in an earlier SELEX study (K. Carroll, personalcommunication). We arbitrarily chose to randomize a re-gion of 40 nt in the VR; therefore, a library of 1024 differentRNA species would be required to represent all the possiblesequence combinations. However, due to practical experi-mental constraint, a starting library of an estimated 1012
RNA species was used in our SELEX procedure. Hence,only a very small fraction of the possible RNA sequenceswas tested.
Maximum enrichment of high-binding-affinity RNAreached at cycle 18–20
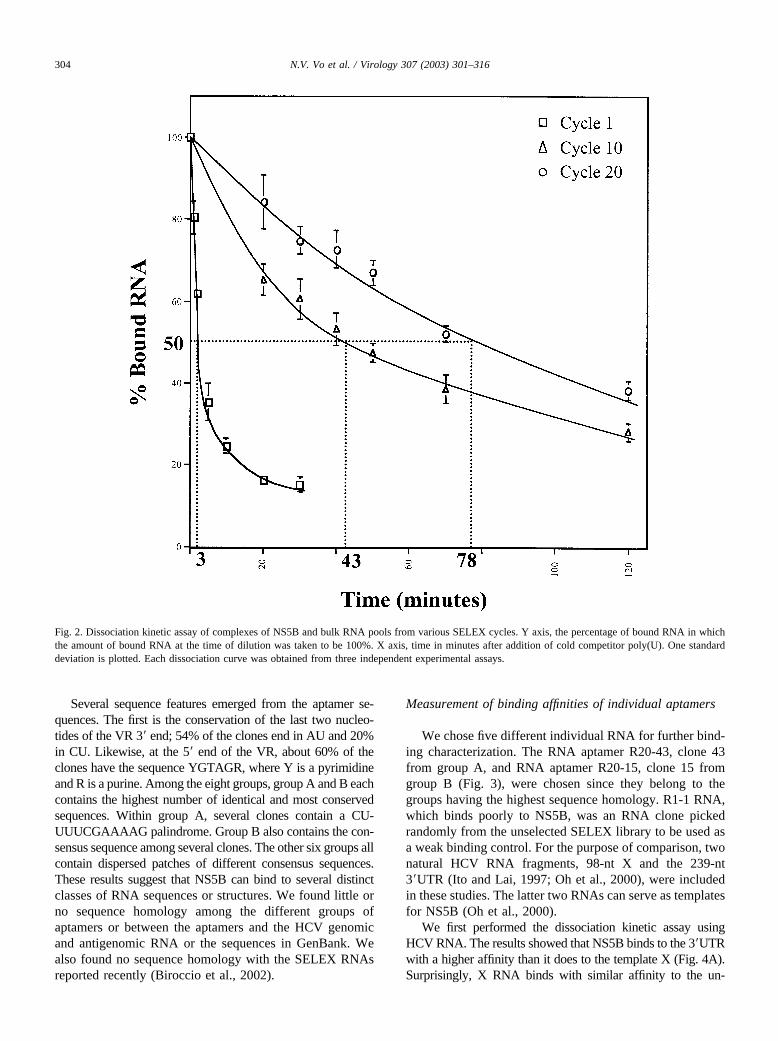
To test for enrichment of high-affinity RNA, dissociationrate constants of NS5B–RNA complex were measured forbulk RNA from different SELEX cycles. We experimen-tally measured the dissociation rate constant, kd (Hinkle andChamberlin, 1972a,b). First, the NS5B protein and the 32P-labeled SELEX RNAs were mixed and allowed to reachbinding equilibrium. The reaction volume was then diluted10-fold in the presence of 60-fold excess, by mass, of coldcompetitor poly(U) RNA (100–200 nt). This mimicked anirreversible first-order dissociation kinetic since any disso-ciated NS5B was expected to be prevented from reassoci-ating with the SELEX RNA by the excess poly(U). Indeed,in a control reaction in which the poly(U) was mixed withthe SELEX RNA prior to addition of the NS5B enzyme,only background binding of SELEX RNA was noted (datanot shown). Aliquots of the reaction were filtered through anitrocellulose filter at various time points to separate thebound complexes from the unbound labeled RNA. Thedissociation curves from various cycles of the SELEX pro-
cedure showed a gradual increase of T50 with increasingnumbers of cycle (Fig. 2). T50 reached a maximum at cycle18 (data not shown); T50 of cycle 18 and cycle 20 wereessentially identical (T50 of about 80 min corresponding toa kd of 1.4 � 10�4 s�1). The estimated rate constants ofdissociation for the different rounds of SELEX are summa-rized in Table 1. Based on the kd values, an estimate of30-fold enrichment was obtained at the end of cycle 20. It isnotable that in the early rounds of selection, the dissociationcurve was biphasic, suggesting the presence of heteroge-neous complexes of different stabilities. The fact that afairly high number of SELEX rounds resulted in only amodest improvement in affinity (�30-fold) suggests a highdegree of nonspecific RNA binding by HCV NS5B.
Sequences of the enriched RNA aptamers
Individual clones from the SELEX cycle 20 were iso-lated and sequenced. Thirty-seven reliable sequences werearranged into eight groups, A–H, by the ClustalW alignmentalgorithm (MacVector; Thompson et al., 1994) (Fig. 3). Themajority of the clones, 22 of 37, or about 60%, retained theoriginal VR length of 40 nt. Thirteen of 37, or 35% of theclones, acquired additional nucleotides, resulting in VRlengths ranging from 41 to 49 nt. The clones with unusuallylong VR often possessed homopolymeric stretch of A’s(clones 53, 20) or C’s (clone 39). Several clones also har-bored runs of U’s (clones 12, 46). The homopolymeric runswere probably due in part to the slippage synthesis by theThermus aquaticus DNA polymerase during PCR (Burge etal., 1992). Only two clones had shorter VR (39 nt), but noclones have a VR shorter than 39 nt.
Fig. 1. A schematic diagram of template construction and preparation of an RNA library. An initial library of PCR templates was made using threeoligonucleotides. The Oligo 2a is 87-nt long and contains a 40-nt degenerate sequence (denoted by the wiggly line) flanked by two constant regions. Theresulting library of PCR templates contains a T7 promoter that drives the synthesis of 87-nt RNA products. The restriction BamHI and HindIII sites wereintroduced for cloning purposes. The SphI restriction site was engineered to remove the CR2 segment from the RNA library in subsequent SELEX studies.The RNA templates consist of a 40-nt variable region (VR) flanked by an upstream 23nt constant region (CR1) and a downstream 24nt constant region (CR2).An estimated pool of 1012 RNA species was used as the starting library in the SELEX procedure in this study.
303N.V. Vo et al. / Virology 307 (2003) 301–316
Several sequence features emerged from the aptamer se-quences. The first is the conservation of the last two nucleo-tides of the VR 3� end; 54% of the clones end in AU and 20%in CU. Likewise, at the 5� end of the VR, about 60% of theclones have the sequence YGTAGR, where Y is a pyrimidineand R is a purine. Among the eight groups, group A and B eachcontains the highest number of identical and most conservedsequences. Within group A, several clones contain a CU-UUUCGAAAAG palindrome. Group B also contains the con-sensus sequence among several clones. The other six groups allcontain dispersed patches of different consensus sequences.These results suggest that NS5B can bind to several distinctclasses of RNA sequences or structures. We found little orno sequence homology among the different groups ofaptamers or between the aptamers and the HCV genomicand antigenomic RNA or the sequences in GenBank. Wealso found no sequence homology with the SELEX RNAsreported recently (Biroccio et al., 2002).
Measurement of binding affinities of individual aptamers
We chose five different individual RNA for further bind-ing characterization. The RNA aptamer R20-43, clone 43from group A, and RNA aptamer R20-15, clone 15 fromgroup B (Fig. 3), were chosen since they belong to thegroups having the highest sequence homology. R1-1 RNA,which binds poorly to NS5B, was an RNA clone pickedrandomly from the unselected SELEX library to be used asa weak binding control. For the purpose of comparison, twonatural HCV RNA fragments, 98-nt X and the 239-nt3�UTR (Ito and Lai, 1997; Oh et al., 2000), were includedin these studies. The latter two RNAs can serve as templatesfor NS5B (Oh et al., 2000).
We first performed the dissociation kinetic assay usingHCV RNA. The results showed that NS5B binds to the 3�UTRwith a higher affinity than it does to the template X (Fig. 4A).Surprisingly, X RNA binds with similar affinity to the un-
Fig. 2. Dissociation kinetic assay of complexes of NS5B and bulk RNA pools from various SELEX cycles. Y axis, the percentage of bound RNA in whichthe amount of bound RNA at the time of dilution was taken to be 100%. X axis, time in minutes after addition of cold competitor poly(U). One standarddeviation is plotted. Each dissociation curve was obtained from three independent experimental assays.
304 N.V. Vo et al. / Virology 307 (2003) 301–316

selected R1-1 RNA (Fig. 4A and B). Both of these RNA havean estimated T50 of about 1 min, corresponding to a kd of�10�2 s�1 (Table 1). In contrast, R20-15 and R20-43 RNAaptamers had much higher binding affinities (Fig. 4B and Cand Table 1). Two other aptamers, R20-54 from group D andR20-5 from group H, also had a kd comparable to that ofR20-43 (data not shown). These results indicate that theaptamer RNAs bind NS5B with much higher affinities than theunselected random RNA or the X or 3�UTR RNA.
To examine the binding specificity of NS5B to theSELEX aptamers, two other RNA-binding polymerases,M-MLV and AMV reverse transcriptases, were tested.AMV RT binds to R1-1, R20-15, and R20-43 RNA withvirtually identical affinity (Fig. 4D, data not shown forR20-15), suggesting that the SELEX aptamers, which wereselected for the HCV NS5B, do not have enhanced bindingaffinity for the AMV RT. Similar results were obtained forM-MLV RT (data not shown). These results demonstratethat the SELEX aptamers bind specifically to HCV NS5B.
To determine whether CR1 and CR2 are important for thestrong binding, we tested the binding of the VR only of R20-15. There was no detectable difference between R1-1 and theVR sequence alone of R20-15, both of which had a T50 of 1–2min (Table 1). Thus, the high affinity of R20-15 binding toNS5B requires the constant CR1 and CR2 regions in additionto the selected VR region, probably for the formation of thecorrect structure necessary for optimal binding.
The SELEX aptamers interact with NS5B through itstemplate-binding site
X-ray structural studies revealed that NS5B protein hasseveral basic patches in addition to the RNA template-binding groove (Bressanelli et al., 1999). The SELEXaptamers may interact with NS5B at its template-bindinggroove or at one of the basic patches. To determine the siteof binding of the SELEX aptamer on NS5B, we performedthe electrophoretic mobility shift assay (EMSA). The NS5Bwas incubated with a mixture of labeled X or 3�UTR RNA
and increasing amounts of unlabeled SELEX RNA; thereaction mixtures were separated by native 4% PAGE. Anaptamer will disrupt the complexes if it competes for thesame binding site on NS5B. Since the HCV X and 3�UTRRNA can be used by NS5B as templates for RNA synthesis,at least a fraction of these RNAs is predicted to bind NS5Bin the RNA-binding pocket of the enzyme. NS5B/3�UTRcomplex can be disrupted by an excess of R20-15 RNA(Fig. 5A). At six-fold molar excess, R20-15 aptamer re-duces the 3�UTR–NS5B complex to 27%. In contrast, R1-1RNA disrupted the NS5B/3�UTR complex only slightly.Similar results were obtained for the complex of NS5B andlabeled X RNA (Fig. 5B). R20-15 RNA readily disruptedthe X-NS5B complex at much lower amounts than R1-1RNA. Conversely, complex of NS5B and R20-15 washighly stable and could only be disrupted by the coldR20-15 RNA, but not by the X RNA (Fig. 5D). Further-more, NS5B-R1-1 complex could easily be disrupted byeither X or R20-15 RNA (Fig. 5C). These findings, takentogether with the observation that R20-15 RNA can be usedas a template by NS5B (see below, Fig. 7B), suggest thatR20-15 most likely binds to the same binding pocket ofNS5B as HCV X or 3�UTR RNA, but at a much strongeraffinity. However, we cannot formally exclude the possibil-ity that binding of these aptamers at a secondary site ofNS5B may induce a conformational change to eject thenative RNA from the template-binding pocket. In contrastto our results, the SELEX aptamers to HCV NS5B isolatedrecently by Biroccio and coworkers appear to bind to NS5Bnot at its template-binding site but rather at the basic patchnear the C-terminus of NS5B (Biroccio et al., 2002).
RNase footprinting revealed specific RNA motifsrecognized by NS5B
To determine the NS5B-binding site on R20-15 andR20-43 RNA aptamers, RNase footprinting was carried outusing single-strand-specific RNases (A and T1) and a dou-ble-strand (ds)-specific RNase V1. Comparison of the di-gestion patterns of free RNA or NS5B–RNA complex withRNase A or T1 or both revealed that NS5B protected aregion that overlaps the CR1 and VR regions on bothaptamers (Fig. 6A). For R20-15, a 25-nt region, extendingfrom 15th to 40th positions, with the first position being the5�-end residue, was protected. A slightly larger NS5B-bind-ing site on R20-43 RNA was observed, extending from 13thto 45th positions. The protected region includes the con-served YGUAGR motif, which resides at the CR1 and VRjunction and is present on both of these RNAs. Treatmentwith the RNase V1 also showed a similar protected regionon the RNA (Fig. 6A, lane V1). In addition, binding ofNS5B appeared to alter the secondary structure of RNA, asis evident by the enhanced digestion by V1 in the regionnear CR2 when NS5B was present. The best matched sec-ondary structures predicted by the Mulfold computer pro-gram (Zuker, 1989) for the two RNA aptamers are shown in
Table 1Summary of dissociation rate constants of different RNA
RNA T50 (min) kd � 104 (1/s)
Round 1 3.0 � 0.2 38 � 3Round 10 43 � 3 2.7 � 0.2Round 20 80 � 4 1.4 � 0.1X 0.7 � 0.1 179 � 193� UTR 5 � 1 24 � 7R1-1 1.2 � 0.1 100 � 6R20-43 84 � 16 1.4 � 0.3R20-15 115 � 10 1.0 � 0.1VR20-15 1 120
Note. Average values and their standard deviations for T50 and kd werecalculated from three to five independent experiments. T50, time requiredfor 50% of the initial complexes to dissociate; kd, dissociation rate constantin s�1.
305N.V. Vo et al. / Virology 307 (2003) 301–316

Fig. 6B. Both RNA possess three stem-loop structures thathave identical 5� stem-loop and no free 3� end. NS5B bindsto the region between the first and second stem-loops ofboth RNA, encompassing part of the loop as well as part ofthe stem. It is notable that NS5B does not protect the 3� endof these two aptamers. As controls, BSA and AMV reversetranscriptase did not give any footprints under the samereaction condition (data not shown), further supporting thebinding specificity of NS5B to the aptamers.
Binding of the high-affinity RNA interferes with RNAsynthesis by NS5B on its natural HCV RNA templates
Previous studies using homopolymeric RNA suggest thattight RNA binding is inhibitory to the NS5B polymeraseactivity (Lohmann et al., 1997). We, therefore, examinedwhether NS5B could use the SELEX aptamers as templatesfor RNA synthesis, and whether the RNA aptamers couldinterfere with NS5B RdRp activity. NS5B was incubated
Fig. 3. RNA sequences from the 20th cycle of SELEX. The sequences of the two constant regions, CR1 and CR2, are shown at the bottom. The sequencesof the variable region of 37 individual clones are organized into eight groups, from A to H, by their sequence homologies. The length, in number ofnucleotides, of the variable region of each clone is shown on the right. The group and clone numbers are shown on the left. Shaded boxes highlight sequencehomologies within each group. White letters within shaded boxes denote nonhomologous residues. Dashes, gaps introduced for purpose of alignment. AnRNA clone from cycle 1, R1-1, is included.
306 N.V. Vo et al. / Virology 307 (2003) 301–316

with a mixture of RNA template and various concentrationsof competitor RNA for RdRp assays. First, we testedwhether NS5B was able to use the SELEX aptamers as
templates for RNA synthesis. Both R20-43 and R20-15aptamers yielded a major RNA product migrating just be-low the 81-nt RNA marker (Fig. 7A). The weak binder R1-1
Fig. 4. Dissociation kinetics of complexes of NS5B or the AMV reverse transcriptase and various aptamers. (A) NS5B-X RNA (solid circle) or NS5B-3�UTRRNA (solid square). (B) NS5B-R20-43 aptamer. Inlet, NS5B and R1-1 RNA. (C) NS5B-R20-15 aptamer. (D) AMV reverse transcriptase (AMV RT), andR20-43 aptamer (circle) or AMV RT and R1-1 complex (square). Y axis, percentage of bound RNA; X axis, time in minutes after addition of cold competitorpoly(U). Boldfaced numbers on the X axis, T50, or time required for 50% of the complex to dissociate. One standard deviation is plotted. Each dissociationcurve was obtained from three independent assays.
307N.V. Vo et al. / Virology 307 (2003) 301–316

RNA also gave rise to a similar RNA product, indicatingthat the SELEX aptamers can serve as templates for RNAsynthesis. However, the template activities of these aptamerRNAs were lower than both X and, particularly, 3�UTRRNA. Similar to the previous reports (Oh et al., 1999,2000), the X template gave rise to a major product migratingbelow the 81-nt RNA marker, and 3�UTR template pro-duced many RNA products that range from 70 to over 500nt, including the ladders at the UC-rich sequences. Thenature and possible origins of these products have beenpreviously discussed (Oh et al., 2000).
Next, we investigated the effect of the SELEX aptamers onthe ability of NS5B to synthesize RNA from the natural HCVRNA templates. Addition of an equal molar amount of R20-15RNA resulted in over 90% reduction of RNA synthesis fromthe X template (Fig. 7B). At 3 or 6 molar excess, the R20-15aptamer completely inhibited NS5B RNA synthesis from theX template. Interestingly, the template activity of R20-15 itself
was also reduced. The inhibition was not due to contaminantsin the RNA preparation since R1-1 RNA, an RNA prepared inthe same manner as other RNA aptamers, did not significantlyinhibit NS5B RNA synthesis. Even at 6 molar excess of R1-1,25% of RNA synthesis still occurred from the X template.Notably, RNA synthesis from the R1-1 template increasedwith increased RNA concentration. R20-15, but not R1-1, alsoinhibited RNA synthesis from the 3�UTR RNA template (Fig.7C). At higher concentrations, R20-43 aptamer also showedsimilar inhibition of RNA synthesis on the X and 3�UTRtemplates (data not shown).
Discussion
To ensure faithful transmission of genetic information,viral polymerases must be able to specifically replicate theirown RNA while ignoring the vast numbers of cellular RNA.
Fig. 5. Binding competition between the SELEX RNAs for NS5B using gel mobility shift assay. NS5B was incubated with a mixture of two RNAs, onelabeled and the other unlabeled, and the reaction mixture resolved by 4% native PAGE. (A) Labeled 3�UTR and the unlabeled R1-1 and R20-15. (B) LabeledX and the unlabeled R1-1 and R20-15. (C) Labeled R1-1 and the unlabeled X and R20-15. (D) Labeled R20-15 and the unlabeled X and R20-15. Ratio, themolar ratio of cold RNA to labeled RNA; f, probe only; F, free probe; C, complex of NS5B and labeled RNA. The relative percentages of the complexesare shown at the bottom of the gel figures. Each percentage value represented an average of two to three experiments. The extra shifted band observed isindicated by the star.
308 N.V. Vo et al. / Virology 307 (2003) 301–316

Using the SELEX strategy to isolate high-affinity RNAligands to the HCV NS5B, we have demonstrated thatNS5B does bind certain RNA selectively and tightly but notirreversibly. These aptamers are specific for NS5B, sincethey bind poorly to AMV and M-MLV reverse transcrip-tases. There is not a single common RNA sequence motifamong the eight different groups of RNA aptamers, orbetween the aptamers identified in this study and those inthe other study (Biroccio et al., 2002), suggesting that NS5Bcan bind several different RNA motifs. The most noticeableconsensus sequence observed among the RNA aptamers inour study is the YGUAGR (60% of the isolated aptamers) atthe 5� end of the variable region and the A/UC (74%) at the3� end of the variable region. Both are adjacent to the CR1and CR2 constant sequences. In the absence of CR1 andCR2, the VR sequence containing the two conserved ele-ments alone did not bind strongly to NS5B. Thus, thehigh-affinity apatmers isolated from our SELEX require theintermolecular interactions among the CR1, VR, and CR2sequences to form optimal RNA structure for NS5B bind-ing. The nature of these CR1 and CR2 sequences likely hasplayed an important role in the final outcome of the selectedaptamers.
Comparison of the primary sequences of various RNAaptamers to HCV genomic and antigenomic sequencesyielded no statistically significant matches. This is in con-trast to the SELEX RNA aptamers using T4 DNA polymer-ase (Brown et al., 1997). The lack of HCV-like sequenceamong the aptamers suggests that other HCV proteins orhost factors, or compartmentalization of HCV polymeraseand RNA, are the specificity-determining factors of theHCV replicase machinery. This has been observed in manyviral replications (Lai, 1998). For instance, specific bindingof the replicase to the RNA M-site and S-site on the Q�(�)-strand RNA is mediated by the cofactor ribosomalprotein S1 (Miranda et al., 1997). Alternatively, in vitroSELEX, unlike genetic evolution, only maximizes affinityrather than demands biological fitness (Gold, 1995). Hence,SELEX needs not produce RNA ligands with similar se-quence to the natural specific RNA recognition motif ofNS5B even if NS5B itself is the actual specificity-determin-ing factor. Indeed, many reported SELEX studies haveyielded nucleic acid ligands with no structural or sequencesimilarity to that of the naturally occurring site, with theselected ligands having much higher binding affinities. Suchexamples include the Q� replicase and the reverse transcrip-tases of AMV and M-MLV (Brown and Gold, 1995a; Chenand Gold, 1994). Due to the practical constraint in ourSELEX studies, we only sampled a very small subset (1012)of the total number (1024) of theoretically possible RNAsequences in the library. This could well be another reasonfor not observing any sequence homology between theaptamers and HCV sequence. It is interesting to note thatHCV X and 3�UTR RNA themselves are weak binders.Thus, the NS5B-binding affinity may not be the majorspecificity-determining factors.
RNA structural motifs of SELEX aptamers
Several earlier SELEX studies reported the finding ofselected RNA ligands having structural rather than sequencehomology to the natural RNA-binding sites, such as a 32-ntcontiguous specific RNA pseudoknot structure for theHIV-1 reverse transcriptase (Tuerk et al., 1992), a bulgedstructure preferred by the HIV-1 rev protein (Tuerk andMacDougal-Waugh, 1993), and a hairpin structure similarto the coat protein binding site in the bacteriophage R17genome (Schneider et al., 1992). Structural analysis of thetwo most enriched aptamers, R20-15 and R20-43, revealeda three-stem-loop structure with internal bulges. While theyshare no primary sequence similarity to our aptamers, theaptamers with highest affinity to HCV NS5B recently iden-tified by SELEX by Biroccio and co-workers also contain astable stem-loop structure with an internal bulge. TheR20-15 and R20-43 structures contain an identical 5� stem-loop and no free 3� end, similar to the 3�-most X region ofHCV RNA (Ito and Lai, 1997) (Fig. 6C). Thus, it is con-ceivable that some common structural features exist be-tween the strong-binding aptamers and the HCV RNA.However, the NS5B-binding sites on these RNA aptamersand X region are distinct: NS5B binds to X RNA at a regionincluding the second and the third stem-loops from the 5�end (Oh et al., 2000), and to R20-43 and R20-15 RNA at aregion including the first and the second stem-loops (Fig.6C). Also, NS5B binds much more tightly to R20-43 andR20-15 aptamers than the X RNA.
The nature of interaction
The binary complexes of NS5B protein and SELEXaptamers are highly stable to dissociation. Several hours at30°C are required for complete dissociation upon dilutionwith excess competitor RNA. In contrast, the free NS5Breadily becomes inactive in minutes even when it is kept onice. Therefore, interaction with nucleic acid likely stabilizesNS5B, as is the case with other polymerase enzymes (Arndtand Chamberlin, 1990). Based on the assumption that ka, theassociation rate constant, is diffusion-limited (Hinkle andChamberlin, 1972b) and the kd, the dissociation rate con-stant, is a value obtained from the kinetic dissociation assay,we estimated the Kd (Kd � kd/ka) of the SELEX aptamers tobe on the order of 1–5 nM. Several previous SELEX studiesyielded RNA aptamers with similar apparent Kd values. Forexample, Kd is 1 nM for Escherichia coli termination factorrho (Schneider et al., 1993), 5 nM for the T4 DNA poly-merase (Tuerk and Gold, 1990), and 10 nM for HCV NS3protease domain (Fukuda et al., 2000). Using the sameassumption, we obtained a Kd of 120 nM for X RNA andNS5B. Oh et al. reported an estimated Kd of 170 nM forNS5B–X interaction (Oh et al., 2000), an affinity lower thanthat calculated for the PB1 influenza virus polymerase sub-unit and the viral complementary RNA (70 nM) (Gonzalezand Ortin, 1999) or Q� replicase to in vitro selected RNAs
309N.V. Vo et al. / Virology 307 (2003) 301–316

Fig. 6. RNase footprinting analysis and predicted secondary RNA structure of aptamers. (A) RNase footprinting analysis. U, untreated; P, piperidine-treatedRNA to give single nucleotide ladder; f, free RNA treated with RNases; c, complex of NS5B and RNA (4:1 protein to RNA molar ratio) treated with RNasesA, T1, or V1 singly or in combination. The lengths of RNA fragments are denoted by the numbers on the left side of the gels. Dotted lines represent theregion protected by NS5B from RNase digestion. (B) Predicted aptamer RNA secondary structure verified by RNase digestion pattern. Black dots, strongcut sites by RNase T1; gray dots, weak cut sites by RNase T1; black stars, strong cut sites by RNase A; gray stars, weak cut sites by RNase A. The threestem-loop modules are designated as SL I, SL II, and SL III. Numbers denote the length of RNA starting from the 5� end. The NS5B-protected region ishighlighted in outlined letters. The conserved sequences at the 5� and 3� ends of the variable region VR are boxed in by dotted lines. The X structure withits proposed NS5B-binding site is included for comparison between the aptamers and X RNA.
310 N.V. Vo et al. / Virology 307 (2003) 301–316

(20–30 nM) (Brown and Gold, 1995a). The selected aptam-ers, in contrast, bind NS5B with affinities that were twoorders of magnitude higher than that of the X RNA. Itshould be emphasized that the Kd values calculated for ouraptamers represent only estimates of binding affinities sincethe binding between NS5B and some aptamers is complex(not 1:1 stoichiometry) and cannot be accurately treated asa simple binary binding reaction.
NS5B binding appears to perturb the secondary structureof the aptamers, as is evident by the changes in the RNaseV1 digest patterns. It is interesting to note that, in theSELEX studies both by us and by Biroccio et al. (2002),NS5B binds strongly to an internal stem-loop structurerather than the 3� end, and yet it preferentially initiates RNAsynthesis from the 3� end of RNA (Oh et al., 2000). By gelshift studies, it was not possible to establish with certaintythe binding stoichiometry of NS5B and the aptamers. How-ever, NS5B–RNA aptamer complexes yielded two shiftedbands in some gel-shift experiments (Fig. 5), suggesting thatmore than one NS5B molecule binds to each aptamer. Oli-gomerization of polymerases has been shown to be impor-
tant for poliovirus polymerase function (Hobson et al.,2001) and HCV NS5B (Qin et al., 2002; Wang et al., 2002).
The binding of high-affinity RNA aptamers inhibits RNAsynthesis of NS5B on the natural HCV RNA templates
In in vitro studies, we found that NS5B could use theSELEX aptamers as templates for RNA synthesis. How-ever, when the aptamer RNA were used together with thenatural HCV RNA, such as the X region, the aptamers actedas inhibitors of RNA synthesis. The degree of inhibitioncorrelated with the binding affinity of the aptamer, suggest-ing that strong binding of RNA to NS5B restricts the move-ment of NS5B along the template. A number of high-affinitynucleic acid ligands isolated by SELEX have been shown toinhibit their target proteins, including Q� replicase, AMVreverse transcriptase, and HCV NS3 helicase, via this pro-posed mechanism (Brown and Gold, 1995b; Chen and Gold,1994; Kumar et al., 1997). Interestingly, the aptamers them-selves can serve as templates for NS5B, but the templateactivities of the strong aptamers decrease with increasingconcentrations of RNA, further suggesting that strong RNA
Fig. 6 (continued)
311N.V. Vo et al. / Virology 307 (2003) 301–316

binding potentially interferes with the movement of poly-merase on the template necessary for transcript elongation.It is also possible that the ratio of aptamer RNA and NS5Bmay affect the number of oligomeric NS5B molecules oneach RNA molecule. RNA elements important for specificrecognition for replication have been identified for a numberof RNA viruses (Miranda et al., 1997; Sivakumaran et al.,1999; Teterina et al., 2001). However, for most viruses,specific initiation of viral RNA synthesis often involvesprotein cofactors other than the polymerase itself (Lai,1998). For HCV, the natural viral RNA binds to NS5B moreweakly than the strong aptamers. Furthermore, many cellu-lar proteins have been found to bind the 5�- and 3�UTR ofHCV RNA. Thus, the specificity-determining factor forHCV RNA synthesis may not be NS5B–RNA binding.Nevertheless, the SELEX RNA isolated in this study shouldprove useful in the further characterization of the binding
and enzymatic properties of NS5B and prove in principlethat RNA aptamers may be used to inhibit viral RNAreplication.
Materials and methods
Materials
Reagents were purchased from the following sources:Nickel-nitrilotriacetic acid (Ni-NTA) agarose, Qiagen Plas-mid Purification Kit (Qiagen); [�32P] UTP and [�32P]GTP(NEN); HPLC-grade nucleoside triphosphates and glycogen(Pharmacia); RNase A, RNase T1, and T7 MEGAscript InVitro Transcription Kit (Ambion); betaine (Sigma); oligo-nucleotides (USC/Norris Microchemical Core Facility); T7RNA polymerase (Promega); Moloney murine leukemia
Fig. 7. NS5B RdRp assays using the SELEX RNA aptamers and natural HCV RNAs. (A) NS5B RdRp assays using SELEX RNA or various HCV RNAas single templates. The lengths of the RNA markers are numbered. (B) NS5B RdRp assay using a mixture of the X template and the R20-15 or R1-1 aptamer.(C) NS5B RdRp using a mixture of the 3�UTR and the R20-15 or R1-1 aptamer. Lanes 1, 3, 6 represent the molar ratios of SELEX RNA to X RNA. OnlyX or 3�UTR was present in the reaction in lane 0. (a) The major RNA product generated from the X template; (b) the major RNA product from the R1-1template; (c) the major RNA product from the R20-15 template; (d) the major RNA product from the 3�UTR template. The percentages of inhibition of HCVRNA synthesis are shown at the bottom.
312 N.V. Vo et al. / Virology 307 (2003) 301–316

virus (M-MLV); and avian myeloblastosis virus (AMV)reverse transcriptases (Life Science).
Buffers
TBE for gel electrophoresis contains 89 mM Tris–bo-rate, pH 8.3, 2.5 mM EDTA. Formamide loading buffer(FLB) contains TBE, 0.05% xylene cyanol and bromphenolblue dyes, 10 mM EDTA, and 80% freshly deionized for-mamide. NS5B transcription buffer contains 40 mM Tris–HCl, pH 8, 5 mM MgCl2, 10 mM �-mercaptoethanol(�ME), 5% glycerol, 200 mM Betaine, 0.01% Tween 20, 10�g/ml acetylated bovine serum albumin. SELEX bindingbuffer (SBB) contains 40 mM Tris–HCl, pH 8, 5 mMMgCl2, 5% glycerol. PCR buffer contains 10 mM Tris–HCl,pH 8.3, 50 mM KCl, 2.5 mM MgCl2. Buffers for NS5Bprotein purification were prepared as follows: extractionbuffer (EB) contains 10 mM Tris–HCl, pH 8, 50% glycerol,10 mM �ME, 500 mM NaCl, 5 mM MgCl2, 2% TritonX-100, 10 mM imidazole, 130 �g/ml hen egg white ly-sozyme, and 23 �g/ml fresh phenylmethylsulfonyl fluoride,an inhibitor of serine proteases. Wash buffer NWB1 con-tains 10% glycerol, 50 mM sodium phosphate, pH 8, 10 mM�ME, 20 mM imidazole, 2M NaCl, 2% Nonidet NP-40.Buffer NWB2 is similar to buffer NWB1 except that itcontains 1 M NaCl and 1% Nonidet NP-40. Buffer NWB3is similar to buffer NWB1 except that it contains 100 mMNaCl and no NP-40. Buffer NEB is similar to buffer NWB3except that it additionally contains 300 mM imidazole.Buffer P50 contains 40 mM potassium phosphate, pH 8, 1mM dithiothreitol (DTT), 0.1 mM EDTA, and 50% glyc-erol. Buffer P50/50 is the same as buffer P50 except that itadditionally contains 50 mM KCl. Buffer P50/250 is P50with 250 mM KCl. Buffer 50/500 is P50 with 500 mM KCl.
Purification of HCV NS5B
The recombinant NS5B derived from NS5B NIH1bstrain was purified from the E. coli strain BL21 using amodified protocol of Oh et al. (1999). This full-length NS5Bprotein contains a tag of six histidines at the N-terminus.The cell lysate was prepared by sonication of the recombi-nant BL21 cells resuspended in buffer EB, followed bycentrifugation at 30,000 g for 45 min at 4°C, and chromato-graphed on a Ni-NTA column as described previously (Ohet al., 1999). This column was washed in succession withthree different buffers: NWB1, NWB2, and NWB3, andfinally the NS5B protein was eluted using buffer NEB. Theeluted NS5B fraction was dialyzed against buffer P50 over-night at 4°C, and the dialyzed mixture was further purifiedby phosphocellulose chromatography (Uptain and Cham-berlin, 1997). The column was washed with P50 to removecontaminant proteins, followed by the P50/250 wash toremove additional contaminant proteins and truncated formsof NS5B. The full-length NS5B protein was then elutedfrom the column using the buffer P50/500. The fractions
were stored at �80°C. The purified NS5B was �90% pureas estimated from SDS–PAGE and Coomassie blue R250staining.
Preparation of RNA templates
The X region and the 3�UTR of HCV RNA derived fromthe HCV NIH 1b strain were prepared as described previ-ously (Oh et al., 2000). SELEX RNA were made using theT7 promoter-containing DNA templates under extensivesynthesis condition using the reagents from the Ambion’sT7 MEGAscript In Vitro Transcription Kit. For labeledRNA, the RNA was either body-labeled using [�32P]UTP or5� end-labeled using [�32P]GTP. The initial SELEX RNAlibrary was created using the SELEX PCR library, preparedfrom a large PCR reaction containing a combination of threeoligonucleotides 1a, 2a, and 3a (Fig. 1). The sequence ofoligo 1a is as follows: 5� accgaagcttaata cgactcactatagg-gagctcagaataaacgctcaa, oligo 2a: 5�gccggatccgggc ctcatgc-atgcN40ttg agcgtttattctgagctccc, and oligo 3a: 5�gccggatc-cgggcctcatgcatgc. The oligo 2a contained a stretch of 40randomized nucleotides represented by N40 in the sequence.PCR reactions were carried out for 25 cycles, each cycleconsisting of three steps: 94°C 1 min, 53°C 1 min, and 72°Cfor 0.5 min. The RNA products of the expected size werepurified by PAGE on a 7.5% acrylamide gel containing 8 Murea. RNA concentration was determined spectrophoto-metrically.
SELEX procedure
In Round 1, high-affinity RNA ligands were selectedfrom a starting library of 1012 unique species. This numberwas estimated from the quantity of the randomized oligo 2sequences used in the first binding reaction. Binding wascarried out in a 500-�l reaction volume by mixing 100 pmolof purified NS5B with 2000 pmol of the labeled RNA (1200cpm/pmol) in the SSB buffer at a final KCl concentration of20 mM. The Ni-NTA resin slurry was equilibrated with SSBand the binding reaction mix was added to the washed resinand incubated at 30°C for 10 min. For achieving optimalbinding of the His-tagged NS5B to the Ni-NTA resin, themixture was pipetted every 2 min. The reaction resin wasthen washed three times with 1 ml SBB each time, andNS5B protein was eluted from the resin with 100 �l SBB �2 M imidazole. The eluted mixture was extracted once withequilibrated phenol, and the bound RNA was precipitatedby ethanol using 200 �g/ml glycogen as carrier. The per-centages of bound and unbound RNA were estimated byscintillation counting and the known radioactive specificactivity of the labeled RNA. The bound RNA was convertedinto DNA by RT-PCR. For RT reaction, 4 �M oligo 3a andthe bound RNA mixture was incubated at 70°C for 5 minand placed on ice. Added to this mixture were 2 mM dNTPand 2 U/�l M-MLV reverse transcriptase in the RT buffer,and the reaction was incubated at 42°C for 15 min, followed
313N.V. Vo et al. / Virology 307 (2003) 301–316

by 15 min at 70°C to inactivate the enzyme. Next, a PCRmixture containing 2 �M oligo 1a and 2 mM dNTP and 2U/100 �l T. aquaticus DNA polymerase was combined withthe RT reaction mixture and 25 cycles of PCR were done,each cycle consisting of 94°C incubation for 1 min, 53°C 1min, and 72°C for 0.5 min. The PCR products were used fortranscription by T7 RNA polymerase and the RNA productswere purified as described above.
For subsequent SELEX cycles, 20 pmol NS5B and 200pmol SELEX RNA were used in each cycle. The SELEXPCR DNA from cycle 20 was cloned into the pBluescriptSK(�) vector via the HindIII and BamHI sites. Individualclones were isolated, and the SELEX cDNA sequences weredetermined by the USC Microchemical Core Facility.
Kinetic dissociation assay
An 80-�l reaction mixture, containing 2 pmol of NS5Band 0.4 pmol labeled SELEX RNA in SBB at a final KClconcentration of 20 mM, was incubated at 30°C for 5 min.Unlabeled poly(U) RNA competitor (750 �l 1 mg/ml) wasthen added to the reaction. At various time points, 100 �laliquots were removed and mixed with 1 ml SBB andfiltered under gentle suction through nitrocellulose mem-branes presoaked in SBB. The membranes were air driedand counted in a scintillation counter. The resulting cpmrepresents the amount of NS5B/RNA complex. Assays weredone in triplicate for each RNA sample and a dissociationcurve, represented by percentage of complex versus timeafter dilution, was plotted. The filter-bound cpm at t � 0 aredefined as 100%. These data were applied to the equationfor first-order rates of decay: ln(%bound) � ln(%bound atstart � 100) � tkd. This equation can be rearranged to givekd � 0.693/T50, where T50 is the time required for 50% ofthe complexes to dissociate. Thus kd was determined fromT50, a value obtained from the dissociation curve.
Gel shift assay
The NS5B–RNA complex was formed by incubating 1pmol NS5B with 0.5 pmol labeled RNA in SBB and 20 mMKCl at 30°C for 5 min. The free RNA and NS5B RNAcomplex in the reaction were then resolved on a native 4%(39:1 acrylamide:bisacrylamide) PAGE in 1� TBE (Friedand Crothers, 1984). For competition studies, NS5B wasadded to a mixture containing labeled RNA and unlabeledcompetitor RNA present at one-, three-, and sixfold molarexcess over the labeled RNA. The relative amounts ofbound complex were determined by analysis of the ImageQuant software (Molecular Dynamic PhosphoImager).
RNase footprinting assay
R20-15 and R20-43 RNA were labeled at their 5�-terminias described above. Free RNA (2 pmol) and NS5B/RNAcomplexes (12 pmol NS5B and 2 pmol RNA) in SBB and
200 mM KCl in 20 �l reaction volume were digested byribonucleases at 30°C for 1 min. Five separate digestionswere done: RNase A (0.01 �g/ml), RNase T1(10 U/�l),RNase V1 (10 U/ml), RNase A (0.01 �g/ml) plus RNaseT1(10 U/�l), and a combination of all three RNases. Thereactions were stopped by phenol extraction, and the RNAproducts were recovered by ethanol precipitation using gly-cogen as carrier. Piperidine ladders of 5�- end-labeled RNAwere generated by incubating 1 pmol 5�- end-labeled RNAplus carrier tRNA (0.3 mg/ml) in 20 �l volume with 1%piperidine at 92°C for 2 min. The digested RNA productswere resolved on a denaturing 8 M urea, 20% PAGE.
NS5B RdRp assay
In a standard 20 �l reaction, 500 nM NS5B was incu-bated with 250 nM RNA template together with 5 �M NTP,1 �l [�32P]UTP (3000 Ci/mmol), and 200 mM potassiumglutamate in NS5B transcription buffer. To test for theinhibitory effect on the NS5B RpRd activity on the X or3�UTR template, the SELEX RNA template was added in 1,3, or 6 molar excess to that of the HCV RNA template. Thereactions were incubated at 30°C for 2 h, and the RNAproducts were precipitated using ethanol in the presence of0.5 mg/ml glycogen as a carrier. The precipitated pellet wasdissolved in 7 �l FLB, heated at 100°C for 3 min, and runon a denaturing 6% PAGE containing 8 M urea. The gel wasdried and subjected to autoradiography. The RNA productsformed were quantified using the Image Quant software(Molecular Dynamic PhosphoImager).
Acknowledgments
We thank Drs. Michael Chamberlin and Caroline Kane,and Kate Carroll and Linda D’Ari of University of Califor-nia, Berkeley for helpful suggestions and some technicalassistance, and Dr. Lilian Hsu of Mt. Holyoke College forcritically reading the manuscript. This work was partiallysupported by an NIH Grant AI 47348. M.M.C.L. is anInvestigator of the Howard Hughes Medical Institute.
References
Arndt, K.M., Chamberlin, M.J., 1990. RNA chain elongation by Esche-richia coli RNA polymerase. Factors affecting the stability of elongat-ing ternary complexes. J. Mol. Biol. 213, 79–108.
Behrens, S.E., Tomei, L., De Francesco, R., 1996. Identification andproperties of the RNA-dependent RNA polymerase of hepatitis C virus.EMBO J. 15, 12–22.
Biroccio, A., Hamm, J., Incitti, I., De Francesco, R., Tomei, L., 2002.Selection of RNA aptamers that are specific and high-affinity ligands ofthe hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 76,3688–3696.
Bressanelli, S., Tomei, L., Roussel, A., Incitti, I., Vitale, R.L., Mathieu, M.,De Francesco, R., Rey, F.A., 1999. Crystal structure of the RNA-
314 N.V. Vo et al. / Virology 307 (2003) 301–316

dependent RNA polymerase of hepatitis C virus. Proc. Natl. Acad. Sci.USA 96, 13034–13039.
Brown, D., Brown, J., Kang, C., Gold, L., Allen, P., 1997. Single-strandedRNA recognition by the bacteriophage T4 translational repressor, regA.J. Biol. Chem. 272, 14969–14974.
Brown, D., Gold, L., 1995a. Selection and characterization of RNAsreplicated by Q beta replicase. Biochemistry 34, 14775–14782.
Brown, D., Gold, L., 1995b. Template recognition by an RNA-dependentRNA polymerase: identification and characterization of two RNA bind-ing sites on Q beta replicase. Biochemistry 34, 14765–14774.
Brown, E.A., Zhang, H., Ping, L.H., Lemon, S.M., 1992. Secondary struc-ture of the 5� nontranslated regions of hepatitis C virus and pestivirusgenomic RNAs. Nucleic Acids Res. 20, 5041–5045.
Burge, C., Campbell, A.M., Karlin, S., 1992. Over- and under-representa-tion of short oligonucleotides in DNA sequences. Proc. Natl. Acad. Sci.USA 89, 1358–1362.
Butcher, S.J., Grimes, J.M., Makeyev, E.V., Bamford, D.H., Stuart, D.I.,2001. A mechanism for initiating RNA-dependent RNA polymeriza-tion. Nature 410, 235–240.
Chamberlin, M., Kingston, R., Gilman, M., Wiggs, J., Devera, A., 1983.Isolation of bacterial and bacteriophage RNA polymerases and theiruse in synthesis of RNA in vitro. Methods Enzymol. 101, 540–568.
Chen, H., Gold, L., 1994. Selection of high-affinity RNA ligands to reversetranscriptase: inhibition of cDNA synthesis and RNase H activity.Biochemistry 33, 8746–8756.
Cheng, J.C., Chang, M.F., Chang, S.C., 1999. Specific interaction betweenthe hepatitis C virus NS5B RNA polymerase and the 3� end of the viralRNA. J. Virol. 73, 7044–7049.
De Francesco, R., 1999. Molecular virology of the hepatitis C virus.J. Hepatol. 31, 47–53.
Fried, M.G., Crothers, D.M., 1984. Kinetics and mechanism in the reactionof gene regulatory proteins with DNA. J. Mol. Biol. 172, 263–282.
Fukuda, K., Vishnuvardhan, D., Sekiya, S., Hwang, J., Kakiuchi, N., Taira,K., Shimotohno, K., Kumar, P.K., Nishikawa, S., 2000. Isolation andcharacterization of RNA aptamers specific for the hepatitis C virusnonstructural protein 3 protease. Eur. J. Biochem. 267, 3685–3694.
Gold, L., 1995. Conformational properties of oligonucleotides. NucleicAcids Symp. Ser. 33, 20–22.
Gold, L., Polisky, B., Uhlenbeck, O., Yarus, M., 1995. Diversity of oligo-nucleotide functions. Annu. Rev. Biochem. 64, 763–797.
Gonzalez, S., Ortin, J., 1999. Distinct regions of influenza virus PB1polymerase subunit recognize vRNA and cRNA templates. EMBO J.18, 3767–3775.
Grakoui, A., Mccourt, D.W., Wychowski, C., Feinstone, S.M., Rice, C.M.,1993. Characterization of the hepatitis C virus-encoded serine protein-ase: determination of proteinase-dependent polyprotein cleavage sites.J. Virol. 67, 2832–2843.
Hagedorn, C.H., Van Beers, E.H., De Staercke, C., 2000. Hepatitis C virusRNA- dependent RNA polymerase (NS5B polymerase). Curr. Top.Microbiol. Immunol. 242, 225–260.
Hinkle, D.C., Chamberlin, M.J., 1972a. Studies of the binding of Esche-richia coli RNA polymerase to DNA. I. The role of sigma subunit insite selection. J. Mol. Biol. 70, 157–185.
Hinkle, D.C., Chamberlin, M.J., 1972b. Studies of the binding of Esche-richia coli RNA polymerase to DNA. II. The kinetics of the bindingreaction. J. Mol. Biol. 70, 187–195.
Hobson, S.D., Rosenblum, E.S., Richards, O.C., Richmond, K., Kirke-gaard, K., Schultz, S.C., 2001. Oligomeric structures of polioviruspolymerase are important for function. EMBO J. 20, 1153–1163.
Hong, Z., Cameron, C.E., Walker, M.P., Castro, C., Yao, N., Lau, J.Y.,Zhong, W., 2001. A novel mechanism to ensure terminal initiation byhepatitis C virus NS5B polymerase. Virology 285, 6–11.
Hwang, S.B., Park, K.J., Kim, Y.S., Sung, Y.C., Lai, M.M., 1997. HepatitisC virus NS5B protein is a membrane-associated phosphoprotein with apredominantly perinuclear localization. Virology 227, 439–446.
Ito, T., Lai, M.M., 1997. Determination of the secondary structure of andcellular protein binding to the 3�-untranslated region of the hepatitis Cvirus RNA genome. J. Virol. 71, 8698–8706.
Kolykhalov, A.A., Feinstone, S.M., Rice, C.M., 1996. Identification of ahighly conserved sequence element at the 3� terminus of hepatitis Cvirus genome RNA. J. Virol. 70, 3363–3371.
Koonin, E.V., 1991. The phylogeny of RNA-dependent RNA polymerasesof positive- strand RNA viruses. J. Gen. Virol. 72, 2197–2206.
Kumar, P.K., Machida, K., Urvil, P.T., Kakiuchi, N., Vishnuvardhan, D.,Shimotohno, K., Taira, K., Nishikawa, S., 1997. Isolation of RNAaptamers specific to the NS3 protein of hepatitis C virus from a pool ofcompletely random RNA. Virology 237, 270–282.
Lai, M.M., 1998. Cellular factors in the transcription and replication ofviral RNA genomes: a parallel to DNA-dependent RNA transcription.Virology 244, 1–12.
Lesburg, C.A., Cable, M.B., Ferrari, E., Hong, Z., Mannarino, A.F., Weber,P.C., 1999. Crystal structure of the RNA-dependent RNA polymerasefrom hepatitis C virus reveals a fully encircled active site. Nat. Struct.Biol. 6, 937–943.
Lohmann, V., Korner, F., Herian, U., Bartenschlager, R., 1997. Biochem-ical properties of hepatitis C virus NS5B RNA-dependent RNA poly-merase and identification of amino acid sequence motifs essential forenzymatic activity. J. Virol. 71, 8416–8428.
Lohmann, V., Roos, A., Korner, F., Koch, J.O., Bartenschlager, R., 1998.Biochemical and kinetic analyses of NS5B RNA-dependent RNA poly-merase of the hepatitis C virus. Virology 249, 108–118.
Miranda, G., Schuppli, D., Barrera, I., Hausherr, C., Sogo, J.M., Weber, H.,1997. Recognition of bacteriophage Qbeta plus strand RNA as a tem-plate by Qbeta replicase: role of RNA interactions mediated by ribo-somal proteins S1 and host factor. J. Mol. Biol. 267, 1089–1103.
Oh, J.W., Ito, T., Lai, M.M., 1999. A recombinant hepatitis C virus RNA-dependent RNA polymerase capable of copying the full-length viralRNA. J. Virol. 73, 7694–7702.
Oh, J.W., Sheu, G.T., Lai, M.M., 2000. Template requirement and initia-tion site selection by hepatitis C virus polymerase on a minimal viralRNA template. J. Biol. Chem. 275, 17710–17717.
O’reilly, E.K., Kao, C.C., 1998. Analysis of RNA-dependent RNA poly-merase structure and function as guided by known polymerase struc-tures and computer predictions of secondary structure. Virology 252,287–303.
Qin, W., Luo, H., Nomura, T., Hayashi, N., Yamashita, T., Murakami, S.,2002. Oligomeric interaction of hepatitis C virus NS5B is critical forcatalytic activity of RNA-dependent RNA polymerase. J. Biol. Chem.277, 2132–2137.
Ranjith-Kumar, C.T., Gajewski, J., Gutshall, L., Maley, D., Sarisky, R.T.,Kao, C.C., 2001. Terminal nucleotidyl transferase activity of recombi-nant Flaviviridae RNA-dependent RNA polymerases: implication forviral RNA synthesis. J. Virol. 75, 8615–8623.
Schmidt-Mende, J., Bieck, E., Hugle, T., Penin, F., Rice, C.M., Blum,H.E., Moradpour, D., 2001. Determinants for membrane association ofthe hepatitis C virus RNA-dependent RNA polymerase. J. Biol. Chem.276, 44052–44063.
Schneider, D., Gold, L., Platt, T., 1993. Selective enrichment of RNAspecies for tight binding to Escherichia coli rho factor. FASEB J. 7,201–207.
Schneider, D., Tuerk, C., Gold, L., 1992. Selection of high affinity RNAligands to the bacteriophage R17 coat protein. J. Mol. Biol. 228,862–869.
Sivakumaran, K., Kim, C.H., Tayon Jr., R., Kao, C.C., 1999. RNA se-quence and secondary structural determinants in a minimal viral pro-moter that directs replicase recognition and initiation of genomic plus-strand RNA synthesis. J. Mol. Biol. 294, 667–682.
Tanaka, T., Kato, N., Cho, M.J., Sugiyama, K., Shimotohno, K., 1996.Structure of the 3� terminus of the hepatitis C virus genome. J. Virol.70, 3307–3312.
315N.V. Vo et al. / Virology 307 (2003) 301–316

Teterina, N.L., Egger, D., Bienz, K., Brown, D.M., Semler, B.L., Ehrenfeld,E., 2001. Requirements for assembly of poliovirus replication complexesand negative-strand RNA synthesis. J. Virol. 75, 3841–3850.
Thompson, J.D., Higgins, D.G., Gibson, T.J., 1994. CLUSTAL W: im-proving the sensitivity of progressive multiple sequence alignmentthrough sequence weighting, position-specific gap penalties and weightmatrix choice. Nucleic Acids Res. 22, 4673–4680.
Tuerk, C., Gold, L., 1990. Systematic evolution of ligands by exponentialenrichment: RNA ligands to bacteriophage T4 DNA polymerase. Sci-ence 249, 505–510.
Tuerk, C., Macdougal, S., Gold, L., 1992. RNA pseudoknots that inhibithuman immunodeficiency virus type 1 reverse transcriptase. Proc. Natl.Acad. Sci. USA 89, 6988–6992.
Tuerk, C., Macdougal, S., 1993. In vitro evolution of functional nucleicacid: high-affinity RNA ligands of HIV-1 proteins. Gene 137,33–39.
Uptain, S.M., Chamberlin, M.J., 1997. Escherichia coli RNA polymeraseterminates transcription efficiently at rho-independent terminators onsingle-stranded DNA templates. Proc. Natl. Acad. Sci. USA 94,13548–13553.
Wang, C., Sarnow, P., Siddiqui, A., 1994. A conserved helical element is
essential for internal initiation of translation of hepatitis C virus RNA.J. Virol. 68, 7301–7307.
Wang, Q.M., Hockman, M.A., Staschke, K., Johnson, R.B., Case, K.A.,Lu, J., Parsons, S., Zhang, F., Rathnachalam, R., Kirkegaard, K.,Colacino, J.M., 2002. Oligomerization and cooperative RNA synthesisactivity of hepatitis C virus RNA-dependent RNA polymerase. J. Virol.76, 3865–3872.
Yamada, N., Tanihara, K., Takada, A., Yorihuzi, T., Tsutsumi, M., Shi-momura, H., Tsuji, T., Date, T., 1996. Genetic organization and diver-sity of the 3� noncoding region of the hepatitis C virus genome.Virology 223, 255–261.
Yanagi, M., St Claire, M., Emerson, S.U., Purcell, R.H., Bukh, J., 1999. Invivo analysis of the 3� untranslated region of the hepatitis C virus afterin vitro mutagenesis of an infectious cDNA clone. Proc. Natl. Acad.Sci. USA 96, 2291–2295.
Zhong, W., Ferrari, E., Lesburg, C.A., Maag, D., Ghosh, S.K., Cameron,C.E., Lau, J.Y., Hong, Z., 2000. Template/primer requirements andsingle nucleotide incorporation by hepatitis C virus nonstructural pro-tein 5B polymerase. J. Virol. 74, 9134–9143.
Zuker, M., 1989. Computer prediction of RNA structure. Methods Enzy-mol. 180, 262–288.
316 N.V. Vo et al. / Virology 307 (2003) 301–316




















